Производные алкинарилнафтиридин-4(1н)-онов в качестве ингибитора фосфодиэстеразы типа iv
Номер патента: 6800
Опубликовано: 28.04.2006
Авторы: Амель Пьер, Жирар Марио, Гай Даниель, Лалиберте Себастьен, Фриезен Ришар















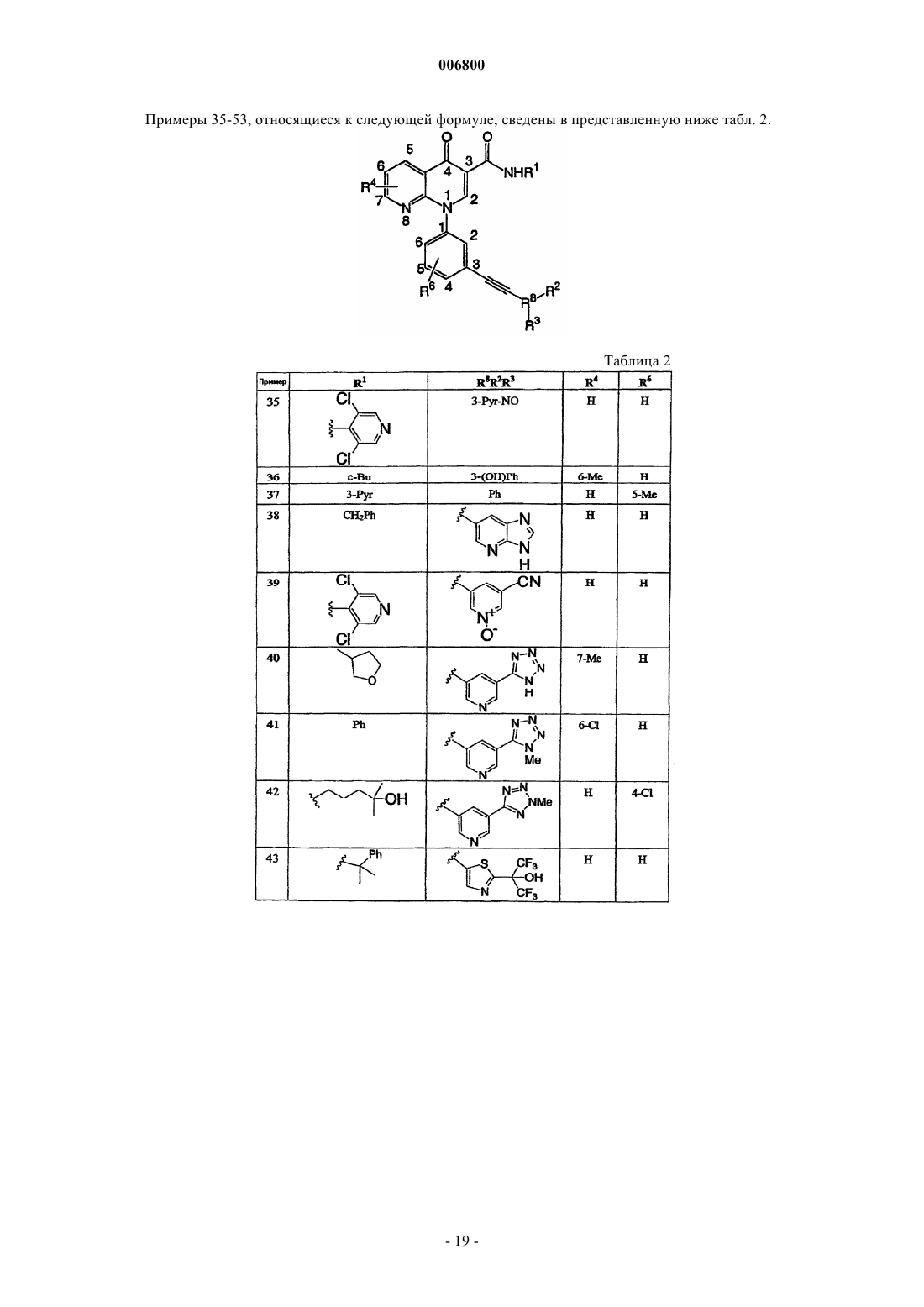









Формула / Реферат
1. Соединение, представленное формулой (I)

или его фармацевтически приемлемая соль, где R означает Н;
R1 означает Н или группу -C1-6алкил, -С3-6циклоалкил;
R2 отсутствует либо означает Н, -C1-6алкил, -С3-6циклоалкил или фенил, и любой из алкилов необязательно замещен 1-6 независимыми заместителями, означающими галоген;
R3 отсутствует либо означает Н, ОН, -NH2 или C1-6алкил, где любой из алкилов необязательно замещен 1-6 независимыми заместителями, означающими галоген;
каждый из R4, R5, R6 и R7 независимо означает Н и
R8 означает необязательно замещенный пиридилом фенил, пиридил, хинолинил, тиенил или тиазолил или оксиды пиридила или хинолинила; где указанный пиридил, хинолинил, тиенил или тиазолил необязательно замещен группой, означающей 1-гидрокси-1-метилэтил или NH2; или R8 означает Н, -C1-6 алкил или -С3-6циклоалкил, где любой из алкилов необязательно замещен 1-6 независимыми заместителями, означающими галоген, -NH2 или -ОН.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где R2 и R3 отсутствуют и R8 означает Н.
3. Соединение по п.1 или его фармацевтически приемлемая соль, где R8 означает фенил, пиридил, хинолинил, тиенил или тиазолил; или оксиды пиридила или хинолинила.
4. Соединение по п.3 или фармацевтически приемлемая соль, где R8 означает фенил.
5. Соединение по п.3 или его фармацевтически приемлемая соль, где R8 означает пиридил или его оксиды.
6. Соединение по п.1 или его фармацевтически приемлемая соль, где R8 означает хинолинил, тиенил, тиазолил, или оксид хинолинила; -С3-6циклоалкил, необязательно замещенный 1-6 независимыми заместителями, означающими галоген, -NH2 или -ОН; -C1-6алкил, необязательно замещенный 1-6 независимыми заместителями, означающими галоген, NH2 или -ОН.
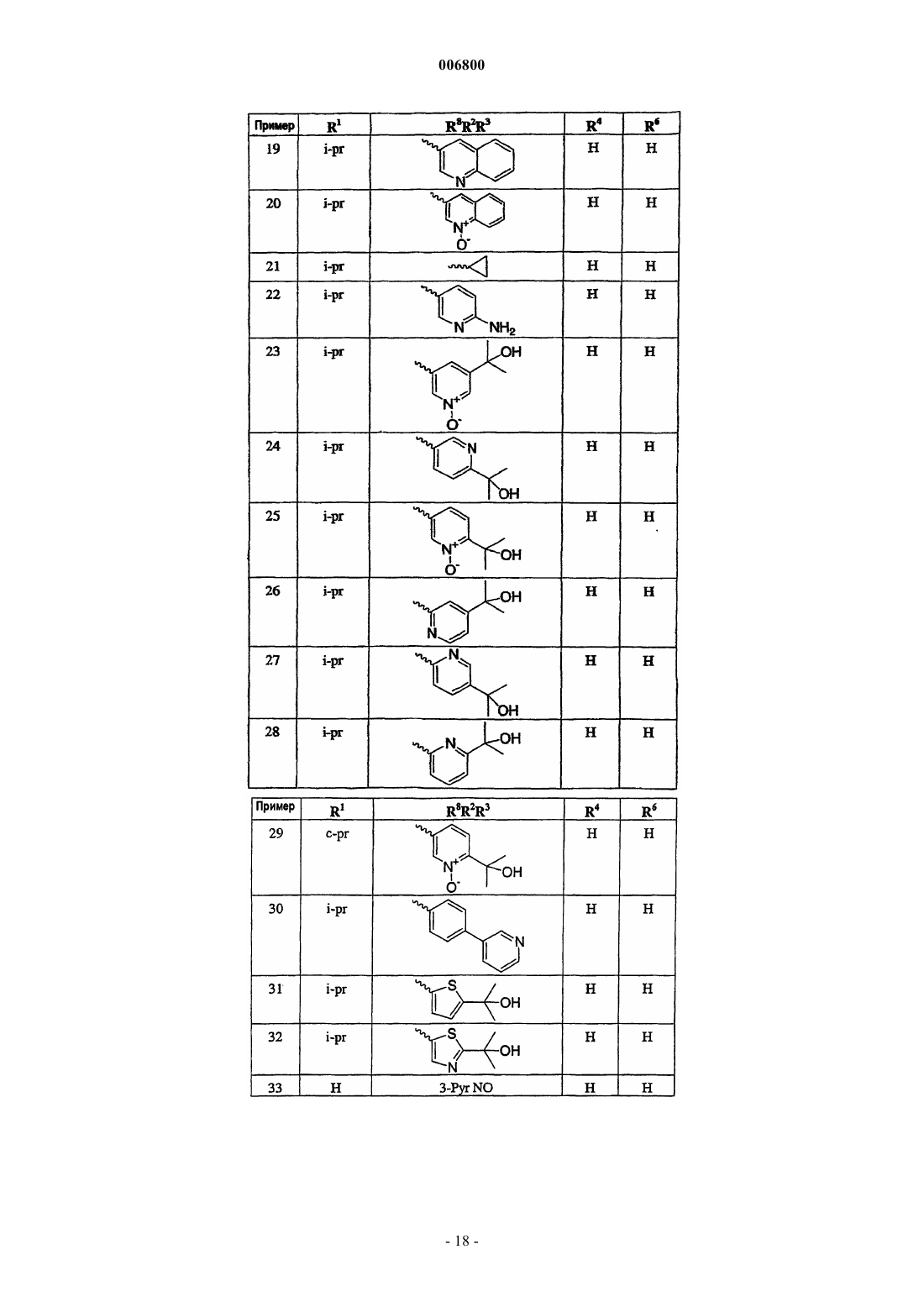
7. Соединение по п.1, выбираемое из группы, включающей
N-изопропил-1-(3-этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-циклопропил-1-(3-этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(фенилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-{3-[(4-пиридин-3-илфенил)этинил]фенил}-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(2-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(4-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(1-оксидо-4-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-циклопропил-1-[3-(3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-циклопропил-1-[3-(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(6-амино-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-{3-[5-(1-гидрокси-1-метилэтил)-1-оксидо-3-пиридинилэтинил]фенил}-1,4-дигидро [1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-{3-[6-(1-гидрокси-1-метилэтил)-3-пиридинилэтинил]фенил}-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-{3-[6-(1-гидрокси-1-метилэтил)-1-оксидо-3-пиридинилэтинил]фенил}-1,4-дигидро [1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-{3-[4-(1-гидрокси-1-метилэтил)-2-пиридинилэтинил]фенил}-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-{3-[5-(1-гидрокси-1-метилэтил)-2-пиридинилэтинил]фенил}-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-{3-[6-(1-гидрокси-1-метилэтил)-2-пиридинилэтинил]фенил}-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-циклопропил-1-{3-[6-(1-гидрокси-1-метилэтил)-1-оксидо-3-пиридинилэтинил]фенил}-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
1-[3-(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
1-[3-(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоновую кислоту;
N-изопропил-1-[3-(3-хинолинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(1-оксидо-3-хинолинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-(3-{[5-(1-гидрокси-1-метилэтил)тиен-2-ил]этинил}фенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-(3-{[2-(1-гидрокси-1-метилэтил)-1,3-тиазол-5-ил]этинил}фенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(1-гидроксициклопентил)этинилфенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(1-гидроксициклопропил)этинилфенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(циклопропилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(3-гидрокси-3-метилбут-1-инил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-циклопропил-1-[3-(3-гидрокси-3-метилбут-1-инил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-{3-[4,4,4-трифтор-3-гидрокси-3-(трифторметил)бут-1-инил]фенил}-1,4-дигидро [1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(3-гидрокси-3-фенилбут-1-инил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
N-изопропил-1-[3-(3-амино-3-этилпент-1-инил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид или
N-циклопропил-1-[3-(3-амино-3-этилпент-1-инил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид;
или его фармацевтически приемлемая соль.
8. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по одному из пп.1-7 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
9. Способ лечения или профилактики заболеваний, таких как астма; хронические бронхиты; хроническое обструктивное заболевание легких; респираторный дистресс-синдром взрослых; респираторный дистресс-синдром новорожденных; кашель; хроническое обструктивное заболевание легких у животных; неспецифический язвенный колит; болезнь Крона; гиперсекреция желудочного сока; сепсис, вызванный бактериями, грибами или вирусом или септический шок; эндотоксический шок; воспаление копыт и колики у лошадей; травма спинного мозга; травма головы; нейрогенное воспаление; боль; реперфузионное повреждение головного мозга; псориатический артрит; ревматоидный артрит; анкилозирующий спондилит; остеоартрит; воспаление или цитокин-опосредованная хроническая дегенерация тканей; аллергический ринит, аллергический конъюнктивит, эозинофильная гранулема, остеопороз, артериальный рестеноз, атеросклероз, реперфузионное нарушение миокарда, хронический гломерулонефрит, весенний конъюнктивит, кахексия, отторжение трансплантата или реакция "трансплантат против хозяина", депрессия, нарушение памяти, монополярная депрессия, болезнь Паркинсона, болезнь Альцгеймера, острый и хронический рассеянный склероз, псориаз, доброкачественные или злокачественные пролиферативные кожные заболевания, атопический дерматит, крапивница, рак, рост опухоли или раковое прорастание нормальных тканей, включающий стадию введения терапевтически эффективного количества или профилактически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
10. Применение соединения по одному из пп.1-7 или его фармацевтически приемлемой соли при изготовлении лекарственного средства для лечения или профилактики заболевания, указанного в п.9.
11. Применение соединения по одному из пп.1-7 или его фармацевтически приемлемой соли в качестве ингибитора фосфодиэстеразы-4.
12. Применение соединения по одному из пп.1-7 или его фармацевтически приемлеьющ соли при лечении или профилактике заболевания, указанного в п.9.
13. N-Циклопропил-1-[3-(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид или его фармацевтически приемлемая соль.
14. Фармацевтическая композиция, содержащая терапевтически эффективное количество карбоксамида или его соли по п.13 в сочетании с фармацевтически приемлемым носителем.
15. Применение карбоксамида или его соли по п.13 в качестве ингибитора фосфодиэстеразы-4.
16. Применение карбоксамида по п.13 или его соли при изготовлении лекарственного средства для лечения или профилактики заболевания, указанного в п.9.
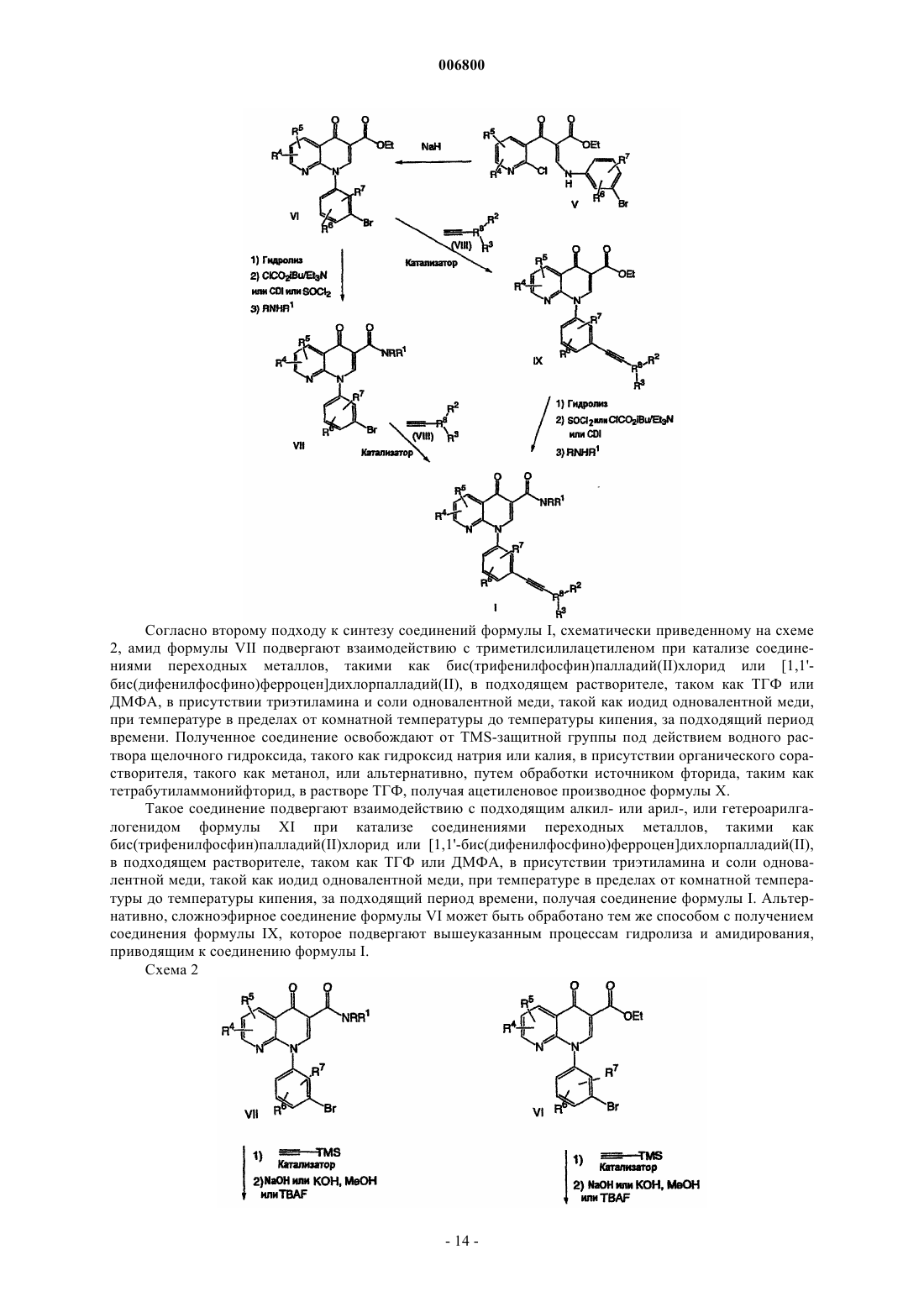
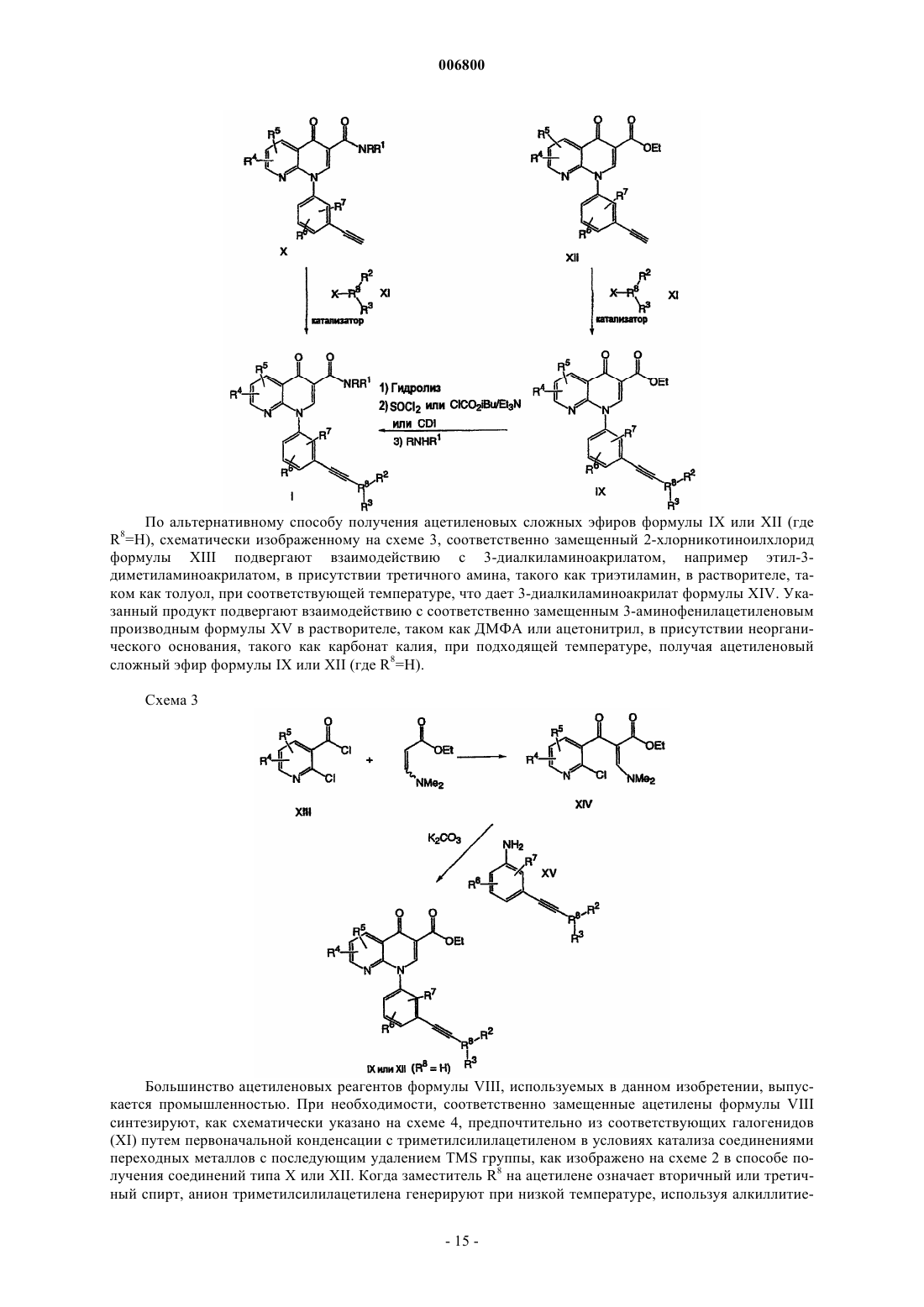
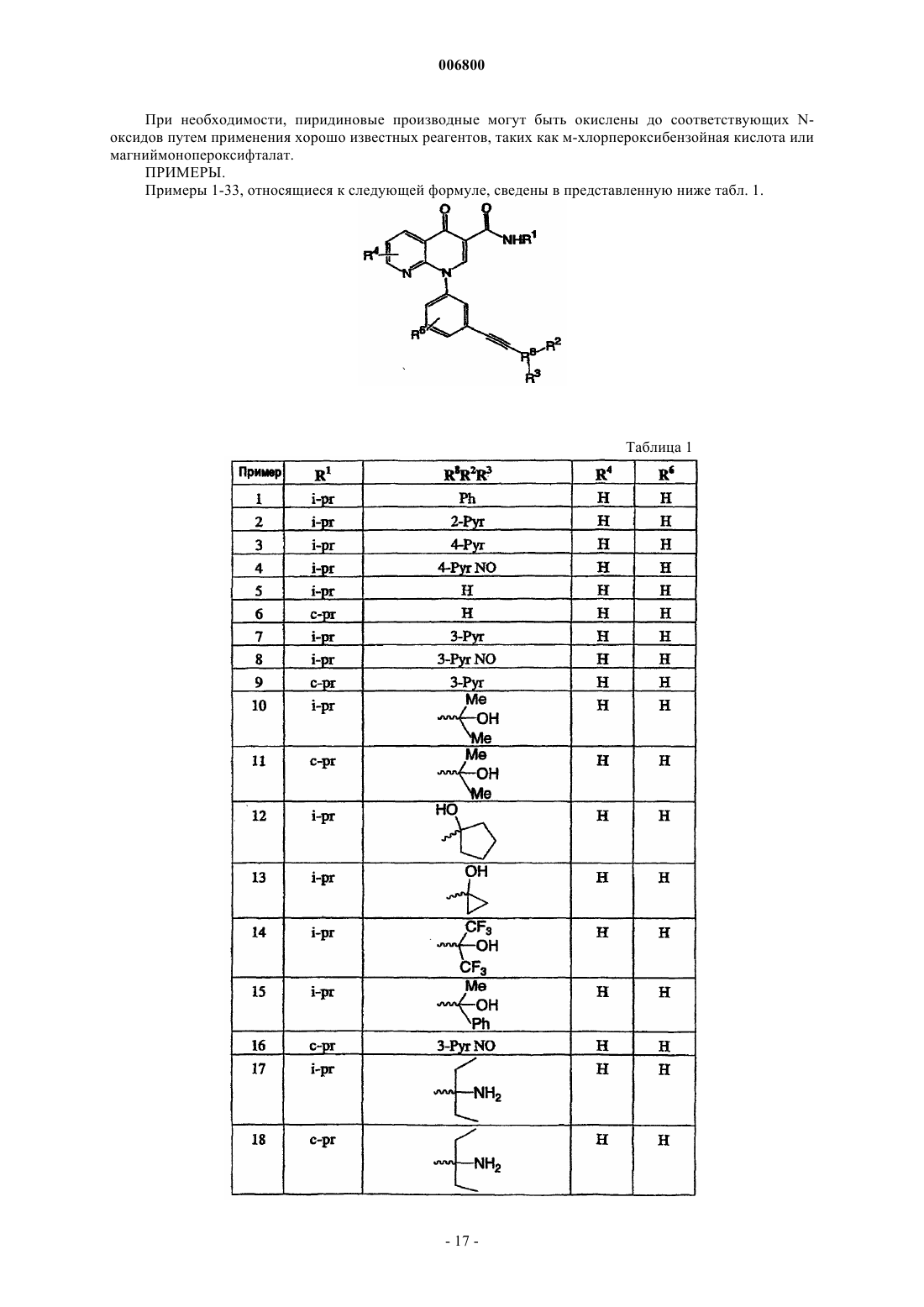
Текст
006800 Предпосылки изобретения Область изобретения Настоящее изобретение касается соединений, которыми являются алкинарилзамещенные 1,8 нафтиридин-4(1 Н)-оны. В частности, настоящее изобретение касается фенилзамещенных 1,8 нафтиридин-4(1 Н)-онов, являющихся ингибиторами фосфодиэстеразы-4, в которых фенильная группа находится в 1-положении и содержит 2-алкинзамещающую группу, необязательно, дополнительно замещенную. Предшествующий уровень техники Гормоны являются соединениями, которые оказывают различное воздействие на клеточную активность. Во многих отношениях гормоны действуют как мессенджеры, инициируя специфические клеточные реакции и активности. Многие производимые гормонами действия, однако, не вызваны сингулярным действием именно этого гормона. Вместо этого гормон сначала связывается с рецептором, тем самым, запуская высвобождение второго соединения, которое продолжает влиять на клеточную активность. В этом случае гормон называют первым мессенджером, тогда как второе соединение называют вторым мессенджером. Циклический аденозинмонофосфат (аденозин 3',5'-циклический монофосфат, "сАМР" или "циклический АМР") известен как второй мессенджер по отношению к гормонам,включающим эпинефрин, глюкагон, кальцитонин, кортикотропин, липотропин, лютеинизирующий гормон, норэпинефрин, паратиреоидный гормон, тиреотропный гормон и вазопрессин. Таким образом,сАМР опосредует клеточные реакции на гормоны. Циклический АМР также опосредует клеточные реакции на различные нейромедиаторы. Фосфодиэстеразы ("PDE") составляют семейство ферментов, которые превращают в ходе обмена 3',5'-циклические нуклеотиды в 5'-нуклеотидмонофосфаты, тем самым прерывая активность второго мессенджера сАМР. Особая фосфодиэстераза, фосфодиэстераза-4 ("PDE4", известная также как "PDE-IV"),обладающая высокой аффинностью, специфический сАМР, PDE тип IV, вызывает интерес в качестве потенциального объекта для создания новых противоастматических и противовоспалительных соединений. Известно, что PDE4 существует, по меньшей мере, в виде четырех изоферментов, каждый из которых кодируется особым геном. Считается, что каждый из четырех известных генных продуктов PDE4 играет различную роль в аллергической и/или воспалительной реакциях. Таким образом, по всей видимости, ингибирование PDE4, в частности специфических изоформ PDE4, которые вызывают различные вредные реакции, может положительно влиять на аллергические и воспалительные симптомы. Желательно получить новые соединения и композиции, ингибирующие активность PDE4. Основное беспокойство при использовании ингибиторов PDE4 заключается в побочном действии,приводящем к рвоте, наблюдаемом для некоторых, рассматриваемых в качестве возможных кандидатов,соединений, как описано у C.Burnouf et al., ("Burnouf"), Ann. Rep. In Med. Chem., 33:91-109(1998).(1998); S.B.Christensen et al., J.Med. Chem., 41:821-835 (1998) и Burnouf описывают широкий ряд различных по тяжести нежелательных побочных действий, производимых различными соединениями. Как показано у M.D.Houslay et al., Adv. In Pharmacol., 44:225-342 (1998) и D.Spina et al., Adv. In Pharmacol.,44:33-89 (1998) существует большой интерес к исследованию терапевтических ингибиторов PDE4. Международная патентная заявка WO9422852 описывает хинолины в качестве ингибиторов PDE4. Международная патентная заявка WO9907704 описывает производные 1-арил-1,8-нафтилидин-4-она в качестве ингибиторов PDE4. А.Н.Cook, et al., J.Chem. Soc, 413-417 (1943) описывает гамма-пиридилхинолины. Другие соединения хинолина описаны у Kei Manabe et al., J.Org. Chem., 58(24): 6692-6700 (1993); Kei Manabe et al., J.(1992). Соединения, включающие кольцевые системы, описываются различными исследователями как эффективные для различных терапий и комплексных программ. Например, международная патентная заявкаWO 98/25883 описывает кетобензамиды в качестве ингибиторов кальпаина, европейская патентная заявкаЕР 811610 и патенты США 5679712, 5693672 и 5747541 описывают замещенные бензоилгуанидиннатриевые блокаторы каналов, патент США 5736297 описывает кольцевые системы, полезные в качестве светочувствительной композиции. Патенты США 5491147, 5608070, 5622977, 5739144, 5776958, 5780477, 5786354, 5798373,5849770, 5859034, 5866593, 5891896 и международная патентная заявка WO 95/35283 описывают ингибиторы PDE4, которыми являются тризамещенные арил или гетероарилфенильные производные. Патент США 5580888 описывает ингибиторы PDE4, которые являются стирил-производными. Патент США 5550137 описывает ингибиторы PDE4, которыми являются фениламинокарбонильные производные. Патент США 5340827 описывает ингибиторы PDE4, которыми являются фенилкарбоксамидные соединения. Патент США 5780478 описывает ингибиторы PDE4, которыми являются тетразамещенные фенильные производные. Международная патентная заявка WO 96/00215 описывает замещенные оксимпроизводные, полезные в качестве ингибиторов PDE4. Патент США 5633257 описывает ингибиторы-1 006800 Однако сохраняется потребность в новых соединениях и композициях, которые терапевтически ингибируют PDE4 при минимальных побочных эффектах. Краткое описание изобретения Настоящее изобретение касается алкинарилзамещенных 1,8-нафтиридин-4(1 Н)-онов, представленных формулой (I) или их фармацевтически приемлемых солей, которые являются ингибиторами фосфодиэстеразы-4. Настоящее изобретение также касается фармацевтической композиции, которая содержит эффективное количество новых алкинарилзамещенных 1,8-нафтиридин-4(1 Н)-онов и фармацевтически приемлемый носитель. Кроме того, настоящее изобретение представляет способ лечения млекопитающих, например, при i) легочных нарушениях, таких как астма, хронические бронхиты, хроническое обструктивное заболевание легких (COPD), респираторный дистресс-синдром взрослых, респираторный дистресссиндром новорожденных, кашель, хроническое обструктивное заболевание легких у животных, ii) желудочно-кишечных расстройствах, таких как неспецифический язвенный колит, болезнь Крона и гиперсекреция желудочного сока, iii) инфекционных заболеваниях, таких как индуцированный сепсис или септический шок, вызванный бактериями, грибами или вирусом, эндотоксический шок (и вызванные эндотоксическим шоком состояния, такие как воспаление копыт и колики у лошадей) и септический шок, iv) неврологических нарушениях, таких как травма спинного мозга, травма головы, нейрогенное воспаление, боль и реперфузионное повреждение головного мозга, v) воспалительных нарушениях, таких как псориатический артрит, ревматоидный артрит, анкилозирующий спондилит, остеоартрит, воспаление и цитокинопосредованная хроническая дегенерация тканей, vi) аллергических нарушениях, таких как аллергический ринит, аллергический конъюнктивит и эозинофильная гранулема, vii) психических расстройствах, таких как депрессия, нарушение памяти и монополярная депрессия, viii) нейродегенеративных нарушениях, таких как болезнь Паркинсона, болезнь Альцгеймера, острый и хронический рассеянный склероз, ix) дерматологических нарушениях, таких как псориаз и другие доброкачественные и злокачественные пролиферативные кожные заболевания, атопический дерматит и крапивница, х) онкологических нарушениях, таких как рак, рост опухолей и раковое прорастание нормальных тканей, xi) нарушениях обмена веществ, таких как несахарный диабет, xii) нарушениях костей, таких как остеопороз,xiii) сердечно-сосудистых нарушениях, таких как артериальный рестеноз, атеросклероз, реперфузионное нарушение миокарда и xiv) других нарушениях, таких как хронический гломерулонефрит, весенний конъюнктивит, отторжение трансплантата и реакция "трансплантат против хозяина", и кахексия - состояниях, поддающихся улучшению путем ингибирования изофермента PDE4 и возникающих в результате повышенных уровней сАМР - путем введения эффективного количества новых алкинарилзамещенных 1,8-нафтиридин-4(1 Н)-онов или соединений-предшественников, образующих in vivo новые алкинарил-замещенные 1,8-нафтиридин-4(1 Н)-оны, являющиеся ингибиторами фосфодиэстеразы-4. Подробное описание изобретения Соединение по настоящему изобретению представлено формулой (I)-2 006800 или его фармацевтически приемлемой солью,где R означает Н;R2 отсутствует либо означает Н, -C1-6 алкил, -С 3-6 циклоалкил или фенил, и любой из алкилов необязательно замещен 1-6 независимыми заместителями, означающими галоген;R3 отсутствует либо означает Н, ОН, -NH2 или C1-6 алкил, где любой из алкилов необязательно замещен 1-6 независимыми заместителями, означающими галоген; каждый из R4, R5, R6 и R7 независимо означает Н; иR8 означает необязательно замещенный пиридилом фенил; пиридил, хинолинил, тиенил или тиазолил, или оксиды пиридила или хинолинила; где указанный пиридил, хинолинил, тиенил или тиазолил необязательно замещен группой, означающей 1-гидрокси-1-метилэтил или NH2; или R8 означает Н, -C1-6 алкил или -С 3-6 циклоалкил, где любой из алкилов необязательно замещен 1-6 независимыми заместителями, означающими галоген, -NH2 или -ОН. По одному из аспектов соединение по настоящему изобретению представлено формулой (I) или фармацевтически приемлемой солью,где R2 и R3 отсутствуют и и R8 означает Н. Согласно следующему аспекту соединение по настоящему изобретению представлено формулой (I) или фармацевтически приемлемой солью,где R8 означает фенил, пиридил, хинолинил, тиенил или тиазолил; или оксиды пиридила или хинолинила. По одному из вариантов изобретения, отражающих указанный аспект, соединение по данному изобретению представлено формулой (I) или фармацевтически приемлемой солью, где R8 означает фенил. По другому варианту изобретения, отражающих указанный аспект, соединение по данному изобретению представлено формулой (I) или фармацевтически приемлемой солью, где R8 означает пиридил или его оксиды. По еще одному варианту изобретения, отражающему указанный аспект, соединение по данному изобретению представлено формулой (I) или фармацевтически приемлемой солью, где R8 означает хинолинил, тиенил, тиазолил, или оксид хинолинила; -С 3-6 циклоалкил, необязательно замещенный 1-6 независимыми заместителями, означающими галоген, -NH2 или -ОН; -C1-6 алкил, необязательно замещенный 1-6 независимыми заместителями, означающими галоген, NH2 или -ОН. По еще одному варианту изобретения соединение формулы (I) по данному изобретению представляет собой соединение, выбираемое из группы, включающейN-циклопропил-1-[3-(3-амино-3-этилпент-1-инил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид; или его фармацевтически приемлемую соль. Еще один аспект настоящего изобретения касается соединения формулы (I), представляющего собойN-циклопропил-1-[3-(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3 карбоксамид или его фармацевтически приемлемую соль. Как использовано здесь, "алкил", а также другие группы с приставкой "алк", такие как, например,алкокси, алканоил, алкенил, алкинил и тому подобные, означают углеродные цепи, которые могут быть линейными или разветвленными, либо представлять собой комбинацию указанных цепей. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил,гептил и тому подобное. "Алкенил", "алкинил" и другие подобные термины включают углеродные цепи,содержащие по меньшей мере одну ненасыщенную С-С связь. Термин "циклоалкил" означает карбоциклы, не содержащие гетероатомы, и включают моно-, би- и трициклические насыщенные карбоциклы, а также конденсированные кольцевые системы. Такие конденсированные кольцевые системы могут включать одно кольцо, являющееся частично или полностью ненасыщенным, такое как бензольное кольцо, образующее конденсированные кольцевые системы, такие как бензоконденсированные карбоциклы. Циклоалкил включает такие конденсированные кольцевые системы, как спироконденсированные кольцевые системы. Примеры циклоалкила включают циклопропил,циклобутил, циклопентил, циклогексил, декагидронафталинил, адамантил, инданил, инденил, флуоренил, 1,2,3,4-тетрагидронафталинил и тому подобное. Подобным образом, "циклоалкенил" означает карбоциклы, не содержащие гетероатомы и содержащие по меньшей мере одну неароматическую двойную С-С связь, и включают моно-, би- и трициклические частично насыщенные карбоциклы, а также бензоконденсированные циклоалкены. Примеры циклоалкенила включают циклогексенил, инденил и тому подобное. Термин "циклоалкилокси", если не оговорено особо, включает циклоалкильную группу, соединенную с оксисвязующим атомом. Термин "алкокси", если не оговорено особо, включает алкильную группу, соединенную с оксисвязующим атомом. Термин "арил", если не оговорено особо, включает полициклические системы, а также системы с одним кольцом, такие как, например, фенил или нафтил. Термин "арилокси", если не оговорено особо, включает полициклические системы, а также системы с одним кольцом, такие как, например, фенил или нафтил, присоединенные через оксисвязующий атом по участку связывания. Термин "С 0-6 алкил" включает алкилы, содержащие 6, 5, 4, 3, 2, 1 атомов углерода, либо не содержащие атомы углерода. Алкил, не содержащий атомы углерода, означает заместитель в виде атома водо-4 006800 рода, когда алкил является концевой группой. Алкил, не содержащий атомы углерода, означает простую связь, когда алкил является связующей группой. Термин "гетеро", если не оговорено особо, включает один или более О, S или N атомов. Например,гетероциклоалкил и гетероарил включают кольцевые системы, содержащие один или более О, S или N атомов в кольце, включая смеси указанных атомов. Гетероатомы заменяют атомы углерода в кольце. Так,например, гетероциклоС 5 алкил означает пятичленное кольцо, содержащее от 5 до 0 атомов углерода. Примеры гетероарила включают, например, пиридинил, хинолинил, изохинолинил, пиридазинил,пиримидинил, пиразинил, хиноксалинил, фурил, бензофурил, дибензофурил, тиенил, бензотиенил, пирролил, индолил, пиразолил, индазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, имидазолил, бензимидазолил, оксадиазолил, тиадиазолил, триазолил, тетразолил. Термин "гетероарилокси", если не оговорено особо, означает гетероарильную группу, присоединенную через окси-связующий атом по участку связывания. Примеры гетероарил(C1-6)алкила включают, например, фурилметил, фурилэтил, тиенилметил, тиенилэтил, пиразолилметил, оксазолилметил, оксазолилэтил, изоксазолилметил, тиазолилметил, тиазолилэтил, имидазолилметил, имидазолилэтил, бензимидазолилметил, оксадиазолилметил, оксадиазолилэтил,тиадиазолилметил, тиадиазолилэтил, триазолилметил, триазолилэтил, тетразолилметил, тетразолилэтил,пиридинилметил, пиридинилэтил, пиридазинилметил, пиримидинилметил, пиразинилметил, хинолинилметил, изохинолинилметил и хиноксалинилметил. Примеры гетероциклоС 3-7 алкила включают, например, азетидинил, пирролидинил, пиперидинил,пергидроазепинил, пиперазинил, морфолинил, тетрагидрофуранил, имидазолинил, пиролидин-2-он, пиперидин-2-он и тиоморфолинил. Термин "N-гетероциклоС 4-7 алкил" описывает неарильные гетероциклические соединения, где цикл образован 3-6 атомами углерода и одним атомом азота. Примеры включают азетидинил, пирролидинил,пиперидинил и пергидроазепинил. Примеры арил(С 1-6)алкила включают, например, фенил(C1-6)алкил и нафтил(C1-6)алкил. Примеры гетероциклоС 3-7 алкилкарбонил(C1-6)алкила включают, например, азетидинилкарбонил(C1-6) алкил, пирролидинилкарбонил(C1-6)алкил, пиперидинилкарбонил(C1-6)алкил, пиперазинилкарбонил(C1-6)алкил, морфолинилкарбонил(C1-6)алкил и тиоморфолинилкарбонил(C1-6)алкил. Термин "амин", если не оговорено особо, включает первичный, вторичный и третичный амины. Если не оговорено особо, используемый термин "карбамоил" включает -NHC(O)ОС 1-С 4 алкил и-ОС(O)NНС 1-C4 алкил. Термин "галоген" включает атомы фтора, хлора, брома и иода. Подразумевается, что термин "необязательно замещенный" включает как замещенный, так и незамещенный. Так, например, необязательно замещенный арил может означать пентафторфенильный или фенильный цикл. Кроме того, замещение может быть осуществлено по любой группе. Например, замещенный арил(С 1-6)алкил включает замещение по арильной группе, а также замещение по алкильной группе. Термин "оксид" гетероарильных групп использован в обычном хорошо известном химическом значении и включает, например, N-оксиды азотных гетероатомов. Описанные здесь соединения содержат одну или более двойных связей и могут, таким образом, давать цис/транс-изомеры, а также другие конформационные изомеры. Настоящее изобретение включает все такие возможные изомеры, а также смеси указанных изомеров. Описанные здесь соединения могут содержать один или более асимметрических центров и могут,таким образом, давать диастереомеры и оптические изомеры. Настоящее изобретение включает все такие возможные диастереомеры, а также их рацемические смеси, их, по существу, чистые разделенные энантиомеры, все возможные геометрические изомеры и их фармацевтически приемлемые соли. Вышеуказанная формула I приведена без указания точной стереохимии на определенных участках. Настоящее изобретение включает все стереоизомеры формулы I и их фармацевтически приемлемые соли. Кроме того, включены смеси стереоизомеров, а также выделенные специфические стереоизомеры. При осуществлении способов синтеза, используемых для получения указанных соединений, или при использовании способов рацемизации или эпимеризации, известных специалистам в данной области, полученные такими способами продукты могут представлять собой смеси стереоизомеров. Термин "фармацевтически приемлемые соли" означает соли, полученные с фармацевтически приемлемыми нетоксичными основаниями или кислотами. Когда соединение по настоящему изобретению проявляет кислотные свойства, соответствующую соль удобно получать с фармацевтически приемлемыми нетоксичными основаниями, включающими неорганические основания и органические основания. Соли, полученные с такими неорганическими основаниями, включают соли алюминия, аммония, кальция, меди (двухвалентной и одновалентной), трехвалентного железа, двухвалентного железа, лития, магния, марганца (трехвалентного и двухвалентного), калия, натрия, цинка и тому подобные. В особенности предпочтительны соли аммония, кальция, магния, калия и натрия. Соли, полученные с фармацевтически приемлемыми органическими нетоксичными основаниями, включают соли первичных, вторичных и третичных аминов, а также циклических аминов и замещенных аминов, таких как замещенные амины при-5 006800 родного и синтетического происхождения. Другие фармацевтически приемлемые органические нетоксичные основания, из которых могут быть получены соли, включают ионообменные смолы, такие как,например, аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2 диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, Nэтилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин,морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин,триметиламин, трипропиламин, трометамин и тому подобное. Когда соединение по настоящему изобретению проявляет основные свойства, соответствующую соль удобно получать с фармацевтически приемлемыми нетоксичными кислотами, включающими неорганические и органические кислоты. В число таких кислот входят, например, уксусная, бензолсульфоновая, бензойная, камфорсульфоновая, лимонная, этансульфоновая, фумаровая, глюконовая, глутаминовая,бромисто-водородная, хлористо-водородная, изэтионовая, молочная, малеиновая, яблочная, миндальная,метансульфоновая, слизевая, азотная, памовая, пантотеновая, фосфорная, янтарная, серная, винная, птолуолсульфоновая кислота и тому подобное. В особенности предпочтительными являются бензолсульфоновая, лимонная, бромисто-водородная, хлористо-водородная, малеиновая, фосфорная, серная и винная кислоты. Фармацевтические композиции по настоящему изобретению содержат в качестве активного ингредиента соединение, представленное формулой I (или фармацевтически приемлемые соли указанного соединения), фармацевтически приемлемый носитель и, необязательно, другие терапевтические ингредиенты или адъюванты. Такие дополнительные терапевтические ингредиенты включают, например, i) антагонисты рецепторов лейкотриенов, ii) ингибиторы биосинтеза лейкотриенов, iii) кортикостероиды, iv) антагонисты рецепторов H1, v) адреномиметики бета-2, vi) селективные ингибиторы COX-2, vii) статины, viii) нестероидные противовоспалительные лекарственные средства ("NSAID") и ix) антагонисты М 2/М 3. Композиции включают композиции, подходящие для перорального, ректального, местного и парентерального (включая подкожное, внутримышечное и внутривенное) введения, хотя большинство подходящих способов в любом отдельно взятом случае зависят от конкретного хозяина и природы и тяжести состояний, применительно к которым вводится активный ингредиент. Фармацевтические композиции обычно могут быть представлены в единичной дозированной форме и получены любыми способами, хорошо известными в фармацевтической области. Для местного применения могут использоваться кремы, мази, гели, растворы или суспензии, содержащие соединение формулы I. Полоскания для рта и горла относятся к области местного применения в соответствии с назначением по изобретению. Уровни доз приблизительно от 0,001 мг/кг до 140 мг/кг массы тела в день полезны для лечения таких состояний, как i) легочные нарушения, такие как астма, хронические бронхиты, хроническое обструктивное заболевание легких (COPD), респираторный дистресс-синдром взрослых, респираторный дистресс-синдром новорожденных, кашель, хроническое обструктивное заболевание легких у животных,ii) желудочно-кишечные расстройства, такие как неспецифический язвенный колит, болезнь Крона и гиперсекреция желудочного сока, iii) инфекционные заболевания, такие как индуцированный сепсис или септический шок, вызванный бактериями, грибами или вирусом, эндотоксический шок (и вызванные эндотоксическим шоком состояния, такие как воспаление копыт и колики у лошадей) и септический шок, iv) неврологические нарушения, такие как травма спинного мозга, травма головы, нейрогенное воспаление, боль и реперфузионное повреждение головного мозга, v) воспалительные нарушения, такие как псориатический артрит, ревматоидный артрит, анкилозирующий спондилит, остеоартрит, воспаление и цитокинопосредованная хроническая дегенерация тканей, vi) аллергические нарушения, такие как аллергический ринит, аллергический конъюнктивит и эозинофильная гранулема, vii) психические расстройства, такие как депрессия, нарушение памяти и монополярная депрессия, viii) нейродегенеративные нарушения, такие как болезнь Паркинсона, болезнь Альцгеймера, острый и хронический рассеянный склероз, ix) дерматологические нарушения, такие как псориаз и другие доброкачественные и злокачественные пролиферативные кожные заболевания, атопический дерматит и крапивница, х) онкологические нарушения, такие как рак, рост опухолей и раковое прорастание нормальных тканей, xi) нарушения обмена веществ, такие как несахарный диабет, xii) нарушения костей, такие как остеопороз, xiii) сердечнососудистые нарушения, такие как артериальный рестеноз, атеросклероз, реперфузионное нарушение миокарда и xiv) другие нарушения, такие как хронический гломерулонефрит, весенний конъюнктивит,отторжение трансплантата и реакция "трансплантат против хозяина", и кахексия - состояния, реагирующие на ингибирование PDE4, либо альтернативно, приблизительно от 0,05 мг до 7 г на пациента в день. Например, воспаление эффективно можно лечить путем введения приблизительно от 0,01 мг до 50 мг соединения на килограмм массы тела в день, либо альтернативно, приблизительно от 0,5 мг до 2,5 г на пациента в день. Кроме того, понятно, что ингибирующие PDE4 соединения по настоящему изобретению могут быть введены на уровнях профилактически эффективных доз в целях предупреждения вышеуказанных состояний. Количество активного ингредиента, которое может быть объединено с несущими материалами для получения единичной дозированной формы, может варьироваться в зависимости от нуждающегося в-6 006800 лечении хозяина и конкретного способа введения. Например, композиция, предназначенная для перорального введения человеку, может обычно содержать приблизительно от 0,5 мг до 5 г активного агента,смешиваемого с соответствующим и удобным количеством несущего вещества, которое может варьироваться в пределах приблизительно от 5 до 95% от общей композиции. Единичные дозированные формы обычно содержат приблизительно от 0,01 мг до 1000 мг активного ингредиента, обычно 0,01, 0,05, 0,25,1, 5, 25, 50, 100, 200, 300, 400, 500, 600, 800 или 1000 мг. Однако понятно, что конкретный уровень дозы для каждого конкретного пациента определяется множеством факторов, включающих возраст, массу тела, общее состояние здоровья, пол, диету, время введения, способ введения, скорость выведения из организма, комбинацию лекарственных средств и тяжесть конкретного заболевания, подвергаемого лечению. На практике, соединения, представленные формулой I, или фармацевтически приемлемые соли указанных соединений могут быть объединены в качестве активного ингредиента в однородную смесь с фармацевтически приемлемым носителем в соответствии с общепринятыми способами получения фармацевтических смесей. Носитель может принимать множество разнообразных форм в зависимости от требуемой формы введения препарата, например пероральной или парентеральной (включая внутривенную). Так, фармацевтические композиции по настоящему изобретению могут быть представлены в дискретных дозированных формах, подходящих для перорального введения, таких как капсулы, крахмальные облатки или таблетки, каждая из которых содержит заранее рассчитанное количество активного ингредиента. Кроме того, композиции могут быть представлены в виде порошка, гранул, раствора, суспензии в водной жидкости или неводной жидкости, в виде эмульсии по типу масло-в-воде или эмульсии по типу вода-в-масле. Вдобавок к вышеуказанным общепринятым дозированным формам соединение, представленное формулой I, или фармацевтически приемлемая соль указанного соединения могут также быть введены способами контролируемого высвобождения и/или с помощью доставляющих средств. Композиции могут быть получены любыми фармацевтическими способами. Обычно такие способы включают стадию объединения активного ингредиента с носителем, состоящим из одного или более необходимых ингредиентов. Обычно композиции получают путем равномерного и однородного смешивания активного ингредиента с жидкими носителями или тонко измельченными твердыми носителями, либо с теми и другими. Затем продукту может быть придана удобная для выпуска форма. Итак, фармацевтические композиции по настоящему изобретению могут включать фармацевтически приемлемый носитель и соединение, или фармацевтически приемлемую соль, формулы I. Соединения формулы I или фармацевтически приемлемые соли указанных соединений могут также быть включены в фармацевтические композиции в комбинации с одним или более другими терапевтически активными соединениями. Используемый фармацевтический носитель может быть, например, твердым, жидким или газообразным. Примерами твердых носителей являются лактоза, каолин, сахароза, тальк, желатин, агар, пектин,акация, стеарат магния и стеариновая кислота. Примерами жидких носителей являются сахарный сироп,арахисовое масло, оливковое масло и вода. Примерами газообразных носителей являются диоксид углерода и азот. При получении композиций для пероральной дозированной формы может быть использована любая удобная фармацевтическая среда. Например, вода, гликоли, масла, спирты, корригенты, консерванты,красители и тому подобное могут быть использованы для получения пероральных жидких препаратов,таких как суспензии, эликсиры и растворы; тогда как носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие средства, лубриканты, связывающие средства,дезинтегрирующие средства и тому подобное могут быть использованы для получения пероральных твердых препаратов, таких как порошки, капсулы и таблетки. По причине удобства их приема таблетки и капсулы являются предпочтительными пероральными дозированными единицами, в которых используются твердые фармацевтические носители. Необязательно, на таблетки может быть нанесено покрытие путем применения стандартных водных и неводных способов. Таблетка, содержащая композицию по настоящему изобретению, может быть получена путем прессования или формования, необязательно, с одним или более дополнительными ингредиентами или адъювантами. Прессованные таблетки могут быть получены прессованием, в подходящей машине, активного ингредиента в свободно-сыпучей форме, такой как порошок или гранулы, необязательно, смешанного со связывающим веществом, лубрикантом, инертным разбавителем, поверхностно-активным или диспергирующим средством. Формованные таблетки могут быть получены формованием в подходящей машине смеси, состоящей из порошкообразного соединения, увлажненного инертным жидким разбавителем. Каждая таблетка предпочтительно содержит приблизительно от 0,1 мг до 500 мг активного ингредиента, и каждая крахмальная облатка или капсула предпочтительно содержит от 0,1 до 500 мг активного ингредиента. Фармацевтические композиции по настоящему изобретению, подходящие для парентерального введения, могут быть получены в виде растворов или суспензий активных соединений в воде. Может быть включено подходящее поверхностно-активное вещество, такое как, например, гидроксипропилцеллюлоза. Дисперсии могут также быть получены в глицерине, жидких полиэтиленгликолях и их смесях в-7 006800 маслах. Кроме того, может быть включен консервант для предупреждения вредного роста микроорганизмов. Фармацевтические композиции по настоящему изобретению, подходящие для применения в виде инъекций, включают стерильные водные растворы или дисперсии. Кроме того, композиции могут быть в форме стерильных порошков для немедленного приготовления таких стерильных растворов или дисперсий для инъекций. Во всех случаях конечная инъецируемая форма должна быть стерильной и в достаточной степени жидкой для удобства введения с помощью шприца. Фармацевтические композиции должны быть устойчивыми в условиях изготовления и хранения; поэтому, предпочтительно, должны предохраняться от разлагающего действия микроорганизмов, таких как бактерии и грибы. Носитель может быть растворяющей или диспергирующей средой, содержащей, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), растительные масла и подходящие смеси указанных соединений. Фармацевтические композиции по настоящему изобретению могут быть в форме, подходящей для местного применения, такой как, например, аэрозоль, крем, мазь, лосьон, присыпка или тому подобное. Кроме того, композиции могут быть в форме, подходящей для применения в трансдермальных средствах. Такие композиции могут быть получены с применением соединения, представленного формулой I по настоящему изобретению, или фармацевтически приемлемых солей указанного соединения, общепринятыми способами переработки. Например, крем или мазь получают смешиванием гидрофильного материала и воды приблизительно с 5-10 мас.% соединения, что приводит к получению крема или мази требуемой консистенции. Фармацевтические композиции по настоящему изобретению могут быть в форме, подходящей для ректального введения, где носитель является твердым веществом. Предпочтительно смеси придают форму суппозиториев, содержащих единичную дозу. Подходящие носители включают масло какао и другие вещества, обычно используемые в данной области. Суппозитории удобно формовать путем первоначального смешивания композиции с размягченным или расплавленным носителем (носителями) с последующим охлаждением и формованием в пресс-формах. В дополнение к вышеуказанным несущим ингредиентам описанные выше фармацевтические композиции могут включать, по обстоятельствам, один или более дополнительных несущих ингредиентов,таких как разбавители, буферы, корригенты, связывающие вещества, поверхностно-активные вещества,загустители, лубриканты, консерванты (включая антиоксиданты) и тому подобное. Кроме того, другие адъюванты могут быть включены для того, чтобы сделать композицию изотонической по отношению к крови предполагаемого реципиента. Композиции, содержащие соединение, описываемое формулой I,или фармацевтически приемлемые соли указанного соединения, могут также быть получены в форме порошка или жидкого концентрата. Найдено, что соединения и фармацевтические композиции по настоящему изобретению проявляют биологическую активность в качестве ингибиторов PDE4. Поэтому еще один аспект настоящего изобретения составляет лечение млекопитающих, например, при i) легочных нарушениях, таких как астма, хронические бронхиты, хроническое обструктивное заболевание легких (COPD), респираторный дистресс-синдром взрослых, респираторный дистресс-синдром новорожденных, кашель, хроническое обструктивное заболевание легких у животных, ii) желудочно-кишечных расстройствах, таких как неспецифический язвенный колит,болезнь Крона и гиперсекреция желудочного сока, iii) инфекционных заболеваниях, таких как индуцированный сепсис или септический шок, вызванный бактериями, грибами или вирусом, эндотоксический шок (и вызванные эндотоксическим шоком состояния, такие как воспаление копыт и колики у лошадей) и септический шок, iv) неврологических нарушениях, таких как травма спинного мозга, травма головы, нейрогенное воспаление, боль и реперфузионное повреждение головного мозга, v) воспалительных нарушениях, таких как псориатический артрит, ревматоидный артрит, анкилозирующий спондилит, остеоартрит, воспаление и цитокинопосредованная хроническая дегенерация тканей, vi) аллергических нарушениях, таких как аллергический ринит, аллергический конъюнктивит и эозинофильная гранулема, vii) психических расстройствах, таких как депрессия, нарушение памяти и монополярная депрессия, viii) нейродегенеративных нарушениях, таких как болезнь Паркинсона, болезнь Альцгеймера, острый и хронический рассеянный склероз, ix) дерматологических нарушениях, таких как псориаз и другие доброкачественные и злокачественные пролиферативные кожные заболевания, атопический дерматит и крапивница, х) онкологических нарушениях, таких как рак, рост опухолей и раковое прорастание нормальных тканей, xi) нарушениях обмена веществ, таких как несахарный диабет, xii) нарушениях костей, таких как остеопороз, xiii) сердечно-сосудистых нарушениях, таких как артериальный рестеноз, атеросклероз, реперфузионное нарушение миокарда и xiv) других нарушениях, таких как хронический гломерулонефрит, весенний конъюнктивит, отторжение трансплантата и реакция "трансплантат против хозяина", и кахексия - состояниях, поддающихся улучшению путем ингибирования изоферментаPDE4 и возникающих в результате повышенных уровней сАМР - путем введения эффективного количества соединений по настоящему изобретению. Термин "млекопитающие" включает человека, а также других животных, таких как, например, собаки, кошки, лошади, свиньи и коровы. Таким образом, очевидно, что лечение млекопитающих, отличных от человека, состоит в лечении заболеваний, клинически соотносящихся с вышеуказанными примерами заболеваний человека.-8 006800 Далее, как указано выше, соединения по настоящему изобретению могут быть использованы в комбинации с другими терапевтическими соединениями. В частности, удобно использовать комбинации ингибирующего PDE4 соединения по настоящему изобретению в сочетании с i) антагонистами рецепторов лейкотриенов, ii) ингибиторами биосинтеза лейкотриенов, iii) селективными ингибиторами COX-2, iv) статинами, v) NSAIDами, vi) антагонистами М 2/М 3, vii) кортикостероидами, viii) антагонистами рецепторов H1 (гистамина) и ix) адреномиметиками бета-2. Так, например, легочные нарушения, такие как астма, хронические бронхиты, хроническое обструктивное заболевание легких (COPD), респираторный дистресс-синдром взрослых, респираторный дистресс-синдром новорожденных, кашель, хроническое обструктивное заболевание легких у животных,удобно лечить с помощью капсул, крахмальных облаток или таблеток, каждая из которых содержит 1, 5,25, 50, 100, 200, 300, 400 или 500 мг активного ингредиента, представленного соединением по настоящему изобретению или фармацевтически приемлемой солью указанного соединения, вводимых один, два или три раза в день. Желудочно-кишечные расстройства, такие как неспецифический язвенный колит, болезнь Крона и гиперсекреция желудочного сока, удобно лечить с помощью капсул, крахмальных облаток или таблеток,каждая из которых содержит 1, 5, 25, 50, 100, 200, 300, 400 или 500 мг активного ингредиента, представленного соединением по настоящему изобретению или фармацевтически приемлемой солью указанного соединения, вводимых один, два или три раза в день. Инфекционные заболевания, такие как индуцированный сепсис или септический шок, вызванный бактериями, грибами или вирусом, эндотоксический шок (и вызванные эндотоксическим шоком состояния, такие как воспаление копыт и колики у лошадей) и септический шок, удобно лечить с помощью капсул, крахмальных облаток или таблеток, каждая из которых содержит 1, 5, 25, 50, 100, 200, 300, 400 или 500 мг активного ингредиента, представленного соединением по настоящему изобретению или фармацевтически приемлемой солью указанного соединения, вводимых один, два или три раза в день. Неврологические нарушения, такие как травма спинного мозга, травма головы, нейрогенное воспаление, боль и реперфузионное повреждение головного мозга, удобно лечить с помощью капсул, крахмальных облаток или таблеток, каждая из которых содержит 1, 5, 25, 50, 100, 200, 300, 400 или 500 мг активного ингредиента, представленного соединением по настоящей заявке или фармацевтически приемлемой солью указанного соединения, вводимых один, два или три раза в день. Воспалительные нарушения, такие как псориатический артрит, ревматоидный артрит, анкилозирующий спондилит, остеоартрит, воспаление и цитокинопосредованная хроническая дегенерация тканей, удобно лечить с помощью капсул, крахмальных облаток или таблеток, каждая из которых содержит 1, 5, 25, 50, 100,200, 300, 400 или 500 мг активного ингредиента, представленного соединением по настоящей заявке или фармацевтически приемлемой солью указанного соединения, вводимых один, два или три раза в день. Аллергические нарушения, такие как аллергический ринит, аллергический конъюнктивит и эозинофильная гранулема, удобно лечить с помощью капсул, крахмальных облаток или таблеток, каждая из которых содержит 1, 5, 25, 50, 100, 200, 300, 400 или 500 мг активного ингредиента, представленного соединением по настоящему изобретению или фармацевтически приемлемой солью указанного соединения, вводимых один, два или три раза в день. Психические расстройства, такие как депрессия, нарушение памяти и монополярная депрессия,удобно лечить с помощью капсул, крахмальных облаток или таблеток, каждая из которых содержит 1, 5,25, 50, 100, 200, 300, 400 или 500 мг активного ингредиента, представленного соединением по настоящему изобретению или фармацевтически приемлемой солью указанного соединения, вводимых один, два или три раза в день. Нейродегенеративные нарушения, такие как болезнь Паркинсона, болезнь Альцгеймера, острый и хронический рассеянный склероз, удобно лечить с помощью капсул, крахмальных облаток или таблеток,каждая из которых содержит 1, 5, 25, 50, 100, 200, 300, 400 или 500 мг активного ингредиента, представленного соединением по настоящей заявке или фармацевтически приемлемой солью указанного соединения, вводимых один, два или три раза в день. Дерматологические нарушения, такие как псориаз и другие доброкачественные и злокачественные пролиферативные кожные заболевания, атопический дерматит и крапивница, удобно лечить с помощью капсул, крахмальных облаток или таблеток, каждая из которых содержит 1, 5, 25, 50, 100, 200, 300, 400 или 500 мг активного ингредиента, представленного соединением по настоящему изобретению или фармацевтически приемлемой солью указанного соединения, вводимых один, два или три раза в день. Онкологические нарушения, такие как рак, рост опухолей и раковое прорастание нормальных тканей, удобно лечить с помощью капсул, крахмальных облаток или таблеток, каждая из которых содержит 1, 5, 25, 50, 100, 200, 300, 400 или 500 мг активного ингредиента, представленного соединением по настоящей заявке или фармацевтически приемлемой солью указанного соединения, вводимых один, два или три раза в день. Нарушения обмена веществ, такие как несахарный диабет, удобно лечить с помощью капсул, крахмальных облаток или таблеток, каждая из которых содержит 1, 5, 25, 50, 100, 200, 300, 400 или 500 мг-9 006800 активного ингредиента, представленного соединением по настоящей заявке или фармацевтически приемлемой солью указанного соединения, вводимых один, два или три раза в день. Нарушения костей, такие как остеопороз, сердечно-сосудистые нарушения, такие как артериальный рестеноз, атеросклероз, реперфузионное нарушение миокарда и другие нарушения, такие как хронический гломерулонефрит, весенний конъюнктивит, отторжение трансплантата и реакция "трансплантат против хозяина", и кахексия, удобно лечить с помощью капсул, крахмальных облаток или таблеток, каждая из которых содержит 1, 5, 25, 50, 100, 200, 300, 400 или 500 мг активного ингредиента, представленного соединением по настоящей заявке или фармацевтически приемлемой солью указанного соединения,вводимых один, два или три раза в день. Используемые здесь сокращения имеют приведенные в таблице значения. Сокращения, не указанные в приведенной ниже таблице, имеют свои общепринятые значения, если не оговорено особо. Испытания, демонстрирующие биологическую активность Анализы на LPS- и FМLР-индуцированные TNF- и LTB4 в цельной крови человека Цельная кровь обеспечивает белок и обогащенную клетками среду, подходящие для изучения биохимической эффективности противовоспалительных соединений, таких как РDЕ 4-селективные ингибиторы. Нормальная, нестимулированная кровь человека не содержит определяемых уровней TNF- иLTB4. При стимуляции с помощью LPS активированные моноциты экспрессируют и секретируют TNFдо 8 ч и уровни в плазме остаются устойчивыми в течение 24 ч. Опубликованные исследования показывают, что ингибирование TNF- путем увеличения внутриклеточного сАМР через РDЕ 4-ингибирование и/или повышенную активность аденилилциклазы осуществляется на транскрипционном уровне. СинтезLTB4 также чувствителен к уровням внутриклеточного сАМР и может быть полностью подавлен PDE4 селективными ингибиторами. Поскольку существует незначительное количество LTB4, продуцируемого за 24 ч LPS-стимуляции цельной крови, необходима дополнительная LPS-стимуляция цельной крови человека с последующим введением fMLP для синтеза LTB4 активированными нейтрофилами. Таким образом, используя одну и ту же пробу крови возможно оценить эффективность соединения на двух маркерах-имитаторах активности PDE4 в цельной крови по следующей методике. Свежую кровь забирают у здоровых добровольцев (мужчин и женщин) в гепаринизированные пробирки путем венопункции. У этих субъектов не наблюдалось воспалительных состояний и указанные субъекты не принимали никаких NSAIDов, по меньшей мере, в течение 4 дней до начала забора крови. Аликвотные пробы крови 500 мкл предварительно инкубируют либо с 2 мкл растворителя (ДМСО), либо 2 мкл испытуемого соединения при ряде концентраций в течение 15 мин при 37 С. После чего добавляют либо 10 мкл растворителя (PBS), в качестве контролей, либо 10 мкл LPS (конечная концентрация 1 мкг/мл, L-2630 (Sigma Chemical Co., St. Louis, МО) из Е.coli, серотип 0111:В 4; разбавленный 0,1% мас./об. BSA (в PBS. После 24 ч инкубации при 37 С к крови добавляют еще 10 мкл PBS (контроль) или 10 мкл LPS (конечная концентрация 1 мкг/мл) и инкубируют в течение 30 мин при 37 С. Затем осуществляют сенсибилизацию крови либо 10 мкл PBS (контроль), либо 10 мкл fMLP (конечная концентрация 1 мкМ, F-3506 (Sigma); разбавленный 1% мас./об. BSA (в PBS в течение 15 мин при 37 С. Образцы крови центрифугируют при 1500xg в течение 10 мин при 4 С, получая плазму. Аликвотную пробу плазмы в 50 мкл смешивают с 200 мкл метанола для осаждения белка и центрифугируют, как указано выше. Супернатант анализируют на LTB4, используя стандартный набор для иммуноферментного анализа (520111 от Cayman Chemical Co., Ann Arbor, MI) согласно инструкции производителя. TNF- анализируют в разбавленной плазме (в PBS), используя стандартный набор ELISA (Cistron Biotechnology, PineBrook, NJ) согласно инструкции производителя. Значения IC50 должны быть ниже приблизительно 5 мкМ, лучше ниже 2,5 мкМ. Значения IC50 для примеров 1-33 находятся в пределах от 0,01 до 2,4 мкМ. Противоаллергическая активность in vivo Соединения по изобретению исследованы на эффективность при IgE-опосредованном аллергическом воспалении легких, вызванном у сенсибилизированных морских свинок путем ингаляции антигена. Морских свинок первоначально сенсибилизируют относительно овальбумина при слабой циклофосфамидиндуцируемой иммуносупрессии путем внутрибрюшинной инъекции антигена в комбинациях с гидроксидом алюминия и коклюшной вакциной. Повторные дозы антигена вводят спустя две и четыре недели. По прошествии шести недель животных сенсибилизируют овальбумином в аэрозольном состоянии,осуществляя при этом терапию в виде внутрибрюшинно вводимого антигистаминного средства (мепирамина). Спустя еще 48 ч выполняют бронхиальный альвеолярный лаваж (BAL) и подсчитывают число эозинофилов и других лейкоцитов в BAL-смывных жидкостях. Легкие также извлекают для гистологического исследования на воспалительное нарушение. Введение соединений по примерам (0,001-10 мг/кгi.p. или р.о.), до трех раз в течение 48 ч после антигенной стимуляции, приводит к значительному снижению в эозинофилах и аккумуляции других воспалительных лейкоцитов.- 11006800 Протокол исследования активности PDE на SPA-основе Скрининг соединений, ингибирующих гидролиз cAMP до АМР посредством сАМР-специфической фосфодиэстеразы типа-IV, проводят в 96-луночном планшете следующим образом. На 96-луночный планшет при 30 С наносят испытуемое соединение (растворенное в 2 мкл ДМСО),188 мкл субстратного буфера, содержащего [2,8-3H]-аденозин-3',5'-циклический фосфат (cAMP, 100 нМ 50 мкМ), 10 MM MgCl2, 1 мМ EDTA, 50 мМ Tris, рН 7,5. Взаимодействие инициируют добавлением рекомбинантного PDE4 человека (количество регулируют таким образом, чтобы 10% продукта образовывалось за 10 мин). Взаимодействие останавливают спустя 10 мин путем добавления 1 мг PDE-SPAгранул (Amersham Pharmacia Biotech, Inc., Piscataway, NJ). Генерируемый АМР-продукт определяют количественно на Wallac Microbeta счетчике для 96-луночных планшетов (EGG Wallac Co., Gaithersburg, MD). Сигнал в отсутствие фермента принимают за фоновый. За 100% активность принимают сигнал, регистрируемый в присутствии фермента и ДМСО за вычетом фона. Процент ингибирования рассчитывают соответственно. Значение IC50 получено путем приближения по методу нелинейной регрессии к стандартному уравнению, связывающему 4-параметра/места множественного связывания, на основании десятиточечного титрования. Значения IC50 для примеров 1-33 определяют с 100 нМ cAMP, используя очищенный слитый белокGST рекомбинантной фосфодиэстеразы IVa человека (met-248), продуцируемый системой экспрессии бакуловирус/Sf-9. Значения IC50 должны быть приблизительно ниже 1000 нМ, лучше, приблизительно ниже 250 нМ и, еще лучше, приблизительно ниже 100 нМ. Значения IC50 для примеров 1-33 находятся в пределах от 0,1 нМ до 90,0 нМ. Подразумевается, что следующие примеры предназначены для иллюстрации некоторых предпочтительных вариантов выполнения изобретения, но не для ограничения изобретения. Если не оговорено особо, экспериментальные методики осуществляют в следующих условиях. Все операции выполняют при комнатной температуре или температуре окружающей среды, то есть при температуре в пределах 18-25 С. Выпаривание растворителя осуществляют, используя роторный испаритель при пониженном давлении (600-4000 паскаль: 4,5-30 мм Нg) и температуру бани до 60 С. Протекание реакций контролируют тонкослойной хроматографией (ТСХ) и приведенное время реакций является только иллюстративным. Температуры плавления приведены без поправок и "разл." означает разложение. Приведенные температуры плавления установлены для соединений, полученных согласно описанию. В некоторых препаративных примерах полиморфизм может приводить к выделению соединений с различными температурами плавления. Структура и чистота всех конечных продуктов доказаны по меньшей мере одним из следующих методов: ТСХ, масс-спектрометрией, спектрометрией ядерного магнитного резонанса (ЯМР) или данными микроанализа. Выходы, когда указаны, приведены только для иллюстрации. Данные ЯМР, когда указаны, приведены в форме дельтазначений для основных характеристических протонов, выраженных в миллионных долях (м.д.), по отношению к тетраметилсилану(ТМС) в качестве внутреннего стандарта, установленных при 300, 400 или 500 МГц с применением указанного растворителя. Способы синтеза Соединения по настоящему изобретению могут быть получены следующими способами. Заместители являются такими, как указаны для формулы I, за исключением особо оговоренных случаев. По первому способу, схематически представленному схемой 1, соответственно замещенное производное этил-2-хлорникотиноилацетата формулы II подвергают взаимодействию с 1,5 эквивалентами триэтилортоформиата и 5 эквивалентами уксусного ангидрида при 130 С и после удаления летучих компонентов сырой 2-хлорникотиноилакрилат формулы III сразу же подвергают взаимодействию с 1,2 эквивалентами соответственно замещенного галогенариламина формулы IV, такого как, например, 3 броманилин, в галогенированном углеводородном растворителе, таком как метиленхлорид, при температуре от 0 С до комнатной температуры. По прошествии соответствующего реакционного времени в пределах от 2 до 24 ч образующийся 3-ариламиноакрилат формулы V может быть получен путем выпаривания растворителя и дополнительно очищен хроматографией на силикагеле или кристаллизацией из подходящего растворителя. Альтернативно, соединение формулы V может быть использовано на следующей стадии без дополнительной очистки. Циклизацию соединения формулы V до 1-галогенарил-1,4 дигидро[1,8]нафтиридин-4-онкарбоксилата формулы VI осуществляют путем обработки небольшим избытком сильного основания, такого как гидрид щелочного металла, например гидрид натрия, в подходящем растворителе, таком как тетрагидрофуран, с исходной температурой 0 С при нагревании до комнатной температуры, по необходимости, до завершения процесса. Продукт формулы VI выделяют в неочищенной форме путем разбавления большим объемом воды с последующими фильтрованием или экстракцией подходящим органическим растворителем, таким как диэтиловый эфир, этилацетат или галогенированный углеводородный растворитель, такой как хлороформ или метиленхлорид. Продукт может быть дополнительно очищен хроматографией на силикагеле, кристаллизацией или продолжительным перемешиванием в подходящем растворителе с последующим фильтрованием. Полученный таким образом сложноэфирный продукт формулы VI может быть гидролизован до соответствующего производного карбоновой кислоты в основных условиях с применением водного рас- 12006800 твора щелочного основания, такого как щелочной карбонат или, предпочтительно, гидроксид натрия или калия, с органическим сорастворителем, таким как тетрагидрофуран или первичный, вторичный или третичный спирт, такой как метанол или этанол, или комбинацией указанных растворителей при температурах в пределах от комнатной температуры до температуры кипения за подходящий период времени. Полученную карбоновую кислоту выделяют в неочищенной форме путем подкисливания с применением водного раствора неорганической кислоты, такой как соляная, серная или подобные кислоты, и фильтрования или экстракции подходящим органическим растворителем, таким как диэтиловый эфир, этилацетат или галогенированный углеводородный растворитель, такой как хлороформ или метиленхлорид. Продукт может быть дополнительно очищен хроматографией на силикагеле, кристаллизацией или продолжительным перемешиванием в подходящем растворителе с последующим фильтрованием. Затем карбоновую кислоту превращают в соответствующий аналог первичного, вторичного или третичного амида формулы VII по любой общепринятой методике, хорошо известной химику-органику, предпочтительно, через первоначальное превращение в смешанный ангидрид путем обработки небольшим избытком, обычно 1,25 эквивалента, соответствующего алкилхлорформиата, такого как этил- или изобутилхлорформиат, в присутствии большего избытка, обычно 2,5 эквивалента, третичного органического амина, такого как триэтиламин или N,N-диизопропилэтиламин, в органическом растворителе, таком как тетрагидрофуран, при низкой температуре, предпочтительно 0 С, за период времени от 30 мин до 3 ч. Альтернативно, кислота может быть превращена в хлорангидрид путем действия, например, тионилхлорида. Затем добавляют избыток, обычно 5 или более эквивалентов, соответствующего первичного или вторичного амина или водного раствора гидроксида аммония и взаимодействию дают осуществляться при температуре в интервале от 0 С до комнатной температуры в течение достаточно длительного времени,обычно 1-24 ч. Требуемый амид формулы VII затем выделяют в неочищенной форме путем осаждения водой и фильтрования или экстракции подходящим органическим растворителем, таким как диэтиловый эфир, этилацетат или галогенированнный углеводородный растворитель, такой как хлороформ или метиленхлорид. Продукт может быть дополнительно очищен хроматографией на силикагеле, кристаллизацией или продолжительным перемешиванием в подходящем растворителе с последующим фильтрованием. Альтернативно, амид формулы VII может быть получен из кислоты и амина с помощью подходящего связывающего реагента, такого как карбонилдиимидазол (CDI). В случаях, когда амидная группа означает 2,6-дихлорпиридин-4-ил, используют другую методику, по которой анион 4-амино-3,5 дихлорпиридина получают при низкой температуре, предпочтительно, при 0 С, используя сильный щелочной гидрид, такой как гидрид натрия, в растворителе, таком как тетрагидрофуран, и подвергают взаимодействию с хлорангидридом карбоновой кислоты (от гидролиза сложного эфира формулы VI),получаемым по соответствующей известной методике, обычно под действием оксалилхлорида, активированного каталитическим количеством N,N-диметилформамида, в растворителе, таком как тетрагидрофуран. Для синтеза соединений формулы I, амидное соединение формулы VII подвергают взаимодействию с соответственно замещенным ацетиленом формулы VIII при катализе соединениями переходных металлов, такими как бис(трифенилфосфин)-палладий(II)хлорид или [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II), в подходящем растворителе, таком как ТГФ или ДМФА, в присутствии триэтиламина и соли одновалентной меди, такой как иодид одновалентной меди, при температуре в пределах от комнатной температуры до температуры кипения, за подходящий период времени. Альтернативно,сложноэфирное соединение формулы VI может быть подвергнуто взаимодействию тем же способом,приводящим к образованию соединения формулы IX, которое подвергают вышеуказанным процессам гидролиза и амидирования, приводящим к соединению формулы I. Схема 1 Согласно второму подходу к синтезу соединений формулы I, схематически приведенному на схеме 2, амид формулы VII подвергают взаимодействию с триметилсилилацетиленом при катализе соединениями переходных металлов, такими как бис(трифенилфосфин)палладий(II)хлорид или [1,1'бис(дифенилфосфино)ферроцен]дихлорпалладий(II), в подходящем растворителе, таком как ТГФ или ДМФА, в присутствии триэтиламина и соли одновалентной меди, такой как иодид одновалентной меди,при температуре в пределах от комнатной температуры до температуры кипения, за подходящий период времени. Полученное соединение освобождают от ТМS-защитной группы под действием водного раствора щелочного гидроксида, такого как гидроксид натрия или калия, в присутствии органического сорастворителя, такого как метанол, или альтернативно, путем обработки источником фторида, таким как тетрабутиламмонийфторид, в растворе ТГФ, получая ацетиленовое производное формулы X. Такое соединение подвергают взаимодействию с подходящим алкил- или арил-, или гетероарилгалогенидом формулы XI при катализе соединениями переходных металлов, такими как бис(трифенилфосфин)палладий(II)хлорид или [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II),в подходящем растворителе, таком как ТГФ или ДМФА, в присутствии триэтиламина и соли одновалентной меди, такой как иодид одновалентной меди, при температуре в пределах от комнатной температуры до температуры кипения, за подходящий период времени, получая соединение формулы I. Альтернативно, сложноэфирное соединение формулы VI может быть обработано тем же способом с получением соединения формулы IX, которое подвергают вышеуказанным процессам гидролиза и амидирования,приводящим к соединению формулы I. Схема 2 По альтернативному способу получения ацетиленовых сложных эфиров формулы IX или XII (гдеR8=H), схематически изображенному на схеме 3, соответственно замещенный 2-хлорникотиноилхлорид формулы XIII подвергают взаимодействию с 3-диалкиламиноакрилатом, например этил-3 диметиламиноакрилатом, в присутствии третичного амина, такого как триэтиламин, в растворителе, таком как толуол, при соответствующей температуре, что дает 3-диалкиламиноакрилат формулы XIV. Указанный продукт подвергают взаимодействию с соответственно замещенным 3-аминофенилацетиленовым производным формулы XV в растворителе, таком как ДМФА или ацетонитрил, в присутствии неорганического основания, такого как карбонат калия, при подходящей температуре, получая ацетиленовый сложный эфир формулы IX или XII (где R8=H). Схема 3 Большинство ацетиленовых реагентов формулы VIII, используемых в данном изобретении, выпускается промышленностью. При необходимости, соответственно замещенные ацетилены формулы VIII синтезируют, как схематически указано на схеме 4, предпочтительно из соответствующих галогенидов(XI) путем первоначальной конденсации с триметилсилилацетиленом в условиях катализа соединениями переходных металлов с последующим удалением TMS группы, как изображено на схеме 2 в способе получения соединений типа Х или XII. Когда заместитель R8 на ацетилене означает вторичный или третичный спирт, анион триметилсилилацетилена генерируют при низкой температуре, используя алкиллитие- 15006800 вое основание, такое как н-бутиллитий, и подвергают взаимодействию с соответственно замещенным альдегидом или кетоном, получая требуемый реагент формулы VIII. Схема 4 Далее приведены примеры синтеза арил- и гетероарилгалогенидов, соответствующих соединениям формулы XI, несущим вторичный или третичный спирт в качестве заместителя. Что касается пиридиновых производных (схема 5), галогензамещенный пиридилкарбоксилат формулы XVI может быть подвергнут взаимодействию с металлоорганическими соединениями, такими как реактив Гриньяра, что приводит к третичному спирту формулы XVII. Альтернативно, дибромпиридиннесущее соединение формулы XVIII может быть монометаллировано с применением алкиллитиевых соединений, таких как нбутиллитий, с последующим присоединением альдегида или кетона, приводящим к соединению формулы XVII. Схема 5 Тиофеновое производное формулы XX получают путем взаимодействия галогензамещенного тиофенальдегида или кетона формулы XIX (схема 6) с металлоорганическими соединениями, такими как реактив Гриньяра. Схема 6 Что касается синтеза тиазолпроизводных формулы XXII, описываемого схемой 7, первоначальное металлирование тиазола с применением алкиллитиевых соединений, таких как н-бутиллитий, с последующим присоединением альдегида или кетона, дает 2-тиазолил- вторичный или третичный спирт, который удобно защищен, как например, SEM-простой эфир формулы XXI. Последующее бромирование приводит к введению атома брома в 5-положение при сопутствующем удалении защитной группы, что дает соединение формулы XXII. Схема 7- 16006800 При необходимости, пиридиновые производные могут быть окислены до соответствующих Nоксидов путем применения хорошо известных реагентов, таких как м-хлорпероксибензойная кислота или магниймонопероксифталат. ПРИМЕРЫ. Примеры 1-33, относящиеся к следующей формуле, сведены в представленную ниже табл. 1. Пример 1. N-Изопропил-1-[3-(фенилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. Этил-3-(3-броманилино)-2-(2-хлорникотиноил)акрилат Смесь этил-2-хлорникотиноилацетата (41,1 г, 180,5 ммоль), триэтилортоформиата (40,12 г, 271 ммоль) и уксусного ангидрида (92,05 г, 902,5 ммоль) нагревают при 130 С в течение 2,5 ч. Летучие компоненты отгоняют и полученный остаток дважды совместно упаривают с ксилолом. Маслянистый остаток растворяют в метиленхлориде (250 мл) и медленно добавляют 3-броманилин (37,25 г, 216,6 ммоль). Полученный раствор перемешивают при комнатной температуре в течение 18 ч и растворитель выпаривают. Полученное сырое соединение используют как таковое на последующей стадии. Стадия 2. Этил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксилат. Неочищенное соединение со стадии 1 растворяют в тетрагидрофуране (500 мл), раствор охлаждают до 0 С и добавляют порциями гидрид натрия (в виде 60% дисперсии в масле, 9,4 г, 235 ммоль). После перемешивания при 0 в течение 1 ч полученной смеси дают нагреться до комнатной температуры. Спустя 2 ч к образовавшейся суспензии добавляют воду (400 мл) и нерастворимый твердый продукт фильтруют и обильно промывают водой. После высыхания твердый продукт перемешивают в диэтиловом эфире (150 мл) при комнатной температуре в течение 24 ч и фильтруют, получая соединение, этил-1(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксилат, в виде твердого вещества с кремовой окраской. 1(с, 1 Н), 8,66-8,71 (м, 3H). Стадия 3. 1-(3-Бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоновая кислота Суспензию этил 1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксилата со стадии 2(52,5 г, 140,7 ммоль) в смеси тетрагидрофурана (400 мл), метанола (400 мл) и 1 н водного гидроксида натрия (280 мл) нагревают приблизительно до 50 С при перемешивании в течение 20 мин. После охлаждения смесь разбавляют водой (300 мл) и добавляют 1 н водный НСl (325 мл). После перемешивания в течение 45 мин осадок фильтруют, хорошо промывают водой и сушат, получая 1-(3-бромфенил)-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоновую кислоту в виде твердого вещества с кремовой окраской. 1- 20006800 Стадия 4. N-изопропил-1-(3-бромфенил)-1,4-дигидро[1,8]-нафтиридин-4-он-3-карбоксамид К суспензии 1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоновой кислоты со стадии 3(26,3 г, 76 ммоль) и триэтиламина (23,2 г, 230 ммоль) в тетрагидрофуране (1000 мл) при 0 С добавляют изобутилхлорформиат (18,85 г, 138 ммоль). После перемешивания при 0 С в течение 2 ч добавляют изопропиламин (23 г, 390 ммоль) и смеси дают нагреться до комнатной и перемешивают в течение ночи. Затем смесь распределяют между этилацетатом и водой, органическую фазу сушат и упаривают до твердого вещества, которое размешивают в диэтиловом эфире при комнатной температуре в течение 3 ч и фильтруют, получая N-изопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид в виде белого твердого вещества. 1H-ЯМР (Ацетон-d6)1,25 (д, 6 Н), 4,17 (м, 1 Н), 7,59-7,63 (м, 2 Н), 7,70 (д, 1 Н), 7,80 (д, 1 Н), 7,94 (с,1 Н), 8,73 (м, 1 Н), 8,78 (д, 1 Н), 8,85 (с, 1 Н), 9,61 (ушир, NH). Стадия 5. N-изопропил-1-[(3-фенилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Смесь амида со стадии 4, фенилацетилена (1,9 экв.), триэтиламина (1,6 экв.), трифенилфосфина(0,06 экв.) и бис(трифенилфосфин)палладий(II)хлорида (0,05 экв.) в ТГФ (16 мл/ммоль) перемешивают при комнатной температуре в течение 20 мин. Иодид меди (I) (5 мг/ммоль) добавляют к смеси и перемешивают при температуре кипения с обратным холодильником в течение 18 ч. После охлаждения смесь гасят насыщенным водным раствором хлорида аммония и распределяют между этилацетатом и водой. Органическую фазу сушат над сульфатом магния и сырой продукт очищают хроматографией на силикагеле, элюируя смесью 1:9 диэтилового эфира и метиленхлорида, что дает твердый продукт, который перемешивают в диэтиловом эфире при комнатной температуре и фильтруют, получая соединение, Nизопропил-1-[(3-фенилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид, в виде твердого вещества. 1H-ЯМР (Ацетон-d6)1,24 (д, 6 Н), 4,18 (м, 1 Н) 7,42 (т, 3H), 7,56-7,61 (м, 3H), 7,69 (м, 2 Н), 7,76 (м,1 Н), 7,85 (с, 1 Н), 8,73 (м, 1 Н), 8,77 (дд, 1 Н), 8,88 (с, 1 Н), 9,62 (ушир, NH). Пример 2. N-Изопропил-1-[3-(2-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Следуя методике для стадии 5 примера 1, но заменяя фенилацетилен на 2-этинилпиридин, указанное в заголовке соединение получают в виде коричневого твердого вещества. 1H-ЯМР (Ацетон-d6)1,25 (д, 6 Н), 4,18 (м, 1 Н), 7,38 (м, 1 Н), 7,59-7,64 (м, 2 Н), 7,71-7,76 (м, 2 Н),7,81-7,85 (м, 2 Н), 7,92 (с, 1 Н), 8,61 (м, 1 Н), 8,74 (м, 1 Н) 8,78 (дд, 1 Н), 8,89 (с, 1 Н), 9,62 (ушир, NH). Пример 3. N-Изопропил-1-[3-(4-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Следуя методике для стадии 5 примера 1, но заменяя фенилацетилен на 4-этинилпиридин(J.Org.Chem. 1996, 61, 6535), указанное в заголовке соединение получают в виде твердого вещества. 1H-ЯМР (Ацетон-d6)1,24 (д, 6 Н), 4,18 (м, 1 Н), 7,49 (м, 2 Н), 7,61 (м, 1 Н), 7,71-7,78 (м, 2 Н), 7,81 (м,1 Н), 7,92 (с, 1 Н), 8,62 (м, 2 Н), 8,73 (м, 1 Н), 8,78 (дд, 1 Н), 8,87 (с, 1 Н), 9,62 (ушир, NH). Пример 4. N-Изопропил-1-[3-(1-оксидо-4-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4 он-3-карбоксамид. К раствору N-изопропил-1-[3-(4-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3 карбоксамида примера 3 в метиленхлориде (36 мл/ммоль) и метаноле (3 мл/ммоль) добавляют гексагидрат магниймонопероксифталата (ММРР, 3,6 экв.) и смесь перемешивают при комнатной температуре в течение ночи. Добавляют дополнительное количество ММРР (2 экв.) и перемешивание продолжают в течение 24 ч. Полученную смесь фильтруют через слой целита, фильтрат разбавляют метиленхлоридом и промывают водным бикарбонатом натрия и водой. После высушивания органическую фазу упаривают и сырой продукт очищают хроматографией на силикагеле, элюируя смесью 10% метанола в метиленхлориде, что дает указанное в заголовке соединение в виде твердого вещества. 1(д, 2 Н), 8,69 (м, 1 Н), 8,81 (дд, 1 Н), 8,99 (с, 1 Н), 9,62 (ушир, NH). Пример 5. N-Изопропил-1-(3-этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. N-изопропил-1-[3-(триметилсилилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3 карбоксамид Следуя методике для стадии 5 примера 1, но заменяя фенилацетилен на триметилсилилацетилен, Nизопропил-1-[3-(триметилсилилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид получают и используют на следующей стадии без дополнительной очистки. Стадия 2. N-изопропил-1-(3-этинилфенил)-1,4-дигидро[1,8]-нафтиридин-4-он-3-карбоксамид Сырой продукт со стадии 1 растворяют в метаноле (12 мл/ммоль) и добавляют 1 н водный гидроксид натрия (3 экв.), получая суспензию. Суспензионную смесь перемешивают при комнатной температуре в течение 2 ч и метанол выпаривают. Полученную водную суспензию разбавляют водой и продукт экстрагируют этилацетатом. Сырой продукт очищают хроматографией на силикагеле, элюируя смесью 10% диэтилового эфира в метиленхлориде, что дает соединение, N-изопропил-1-(3-этинилфенил)-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксамид, в виде твердого вещества.H-ЯМР (Ацетон-d6)1,24 (д, 6 Н), 3,81 (с, 1 Н), 4,17 (м, 1H), 7,59 (м, 1H), 7,64-7,71 (м, 3H), 7,81 (с,1 Н), 8,72 (м, 1H), 8,76 (дд, 1 Н), 8,84 (с, 1 Н), 9,61 (ушир, NH). Пример 6. N-Циклопропил-1-(3-этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. N-циклопропил-1-(3-бромфенил)-1,4-дигидро[1,8]-нафтиридин-4-он-3-карбоксамид Следуя методике для стадии 4 примера 1, но заменяя изопропиламин на циклопропиламин, Nциклопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид получают в виде пушистого белого твердого вещества. 1(дд, 1H), 7,97 (с, 1H), 8,72-8,81 (м, 2 Н), 8,89 (с, 1H), 9,70 (ушир, NH). Стадии 2 и 3. N-циклопропил-1-(3-этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике для стадий 1 и 2 примера 5, но заменяя N-изопропил-1-(3-бромфенил)-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксамид продуктом со стадии 1, получают соединение, Nциклопропил-1-(3-этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид, в виде твердого вещества. 1H-ЯМР (CDCl3)0,66 (м, 2 Н), 0,85 (м, 2 Н), 2,97 (м, 1H), 3,18 (с, 1H), 7,42 (д, 1H), 7,47 (м, 1H), 7,527,58 (м, 2 Н), 7,65 (д, 1H), 8,70 (м, 1H), 8,80 (дд, 1H), 8,98 (с, 1H), 9,74 (ушир, NH). Пример 7. N-Изопропил-1-[3-(3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике для стадии 5 примера 1, но заменяя фенилацетилен на N-изопропил-1-(3 этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид из примера 5 и N-изопропил-1-(3 бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид на 3-бромпиридин, получают указанное в заголовке соединение в виде светло-коричневого твердого вещества. 1H-ЯМР (Ацетон-d6)1,24 (д, 6 Н), 4,18 (м, 1H), 7,43 (м, 1H), 7,61 (м, 1H), 7,70-7,75 (м, 2 Н), 7,80 (д,1 Н), 7,90 (с, 1H), 7,94 (д, 1 Н), 8,58 (м, 1 Н), 8,74-8,79 (м, 3H), 8,88 (с, 1 Н), 9,62 (ушир, NH). Пример 8. N-Изопропил-1-[3-(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4 он-3-карбоксамид. Следуя методике примера 4, но заменяя N-изопропил-1-[3-(4-пиридинилэтинил)фенил]-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксамид на N-изопропил-1-[3-(3-пиридинилэтинил)фенил]-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксамид из примера 1, получают указанное в заголовке соединение в виде твердого вещества. 1H-ЯМР (CDCl3)1,28 (д, 6 Н), 4,28 (м, 1H), 7,26 (м, 1H), 7,36 (д, 1H), 7,45-7,49 (м, 2 Н), 7,57-7,62 (м,2 Н), 7,69 (д, 1H), 8,16 (д, 1H), 8,31 (с, 1H), 8,69 (м, 1H), 8,81 (дд, 1H), 8,99 (с, 1H), 9,63 (ушир, NH). Пример 9. N-Циклопропил-1-[3-(3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3 карбоксамид. Следуя методике стадии 5 примера 1, но заменяя фенилацетилен на N-циклопропил-1-(3 этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид из примера 6 и N-изопропил-1-(3 бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид на 3-бромпиридин, получают указанное в заголовке соединение в виде твердого вещества. 1 Н-ЯМР (CDCl3)0,66 (м, 2 Н), 0,85 (м, 2 Н), 2,97 (м, 1H), 7,28 (м, 1H), 7,43-7,48 (м, 2 Н), 7,57 (т, 1H),7,62 (с, 1H), 7,70 (д, 1H), 7,79 (д, 1H), 8,55 (м, 1H), 8,70 (м, 1H), 8,75 (с, 1H), 8,79 (дд, 1H), 9,01 (с, 1H),9,74 (ушир, NH). Пример 10. N-Изопропил-1-[3-(3-гидрокси-3-метилбут-1-инил)фенил]-1,4-дигидро[1,8]нафтиридин 4-он-3-карбоксамид. Следуя методике стадии 5 примера 1, но заменяя фенилацетилен на 2-метил-3-бутин-2-ол, получают указанное в заголовке соединение в виде белого твердого вещества. 1(с, 1 Н), 8,72 (м, 1 Н), 8,77 (дд, 1 Н), 8,84 (с, 1 Н), 9,62 (ушир, NH). Пример 11. N-Циклопропил-1-[3-(3-гидрокси-3-метилбут-1-инил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Следуя методике примера 10, но заменяя N-изопропил-1-(3-этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид на N-циклопропил-1-(3-этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3 карбоксамид из примера 6, получают указанное в заголовке соединение в виде белого твердого вещества. 1(м, 4 Н), 7,67 (с, 1 Н), 8,72 (м, 1 Н), 8,76 (дд, 1 Н), 8,85 (с, 1 Н), 9,69 (ушир, NH). Пример 12. N-Изопропил-1-[3-(1-гидроксициклопентил)этинилфенил]-1,4-дигидро[1,8]нафтиридин 4-он-3-карбоксамид. Следуя методике стадии 5 примера 1, но заменяя фенилацетилен на 1-этинилциклопентанол, получают указанное в заголовке соединение в виде белого твердого вещества. 1- 22006800 Пример 13. N-Изопропил-1-[3-(1-гидроксициклопропил)этинилфенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. 1-этинилциклопропанол 1-Этинилциклопропанол получают по методике, описанной в J. Org. Chem. 1976, 41, 1237, из [(1 этоксициклопропил)окси]триметилсилана и этинилмагнийбромида и выделяют в виде жидкого вещества. Стадия 2. N-изопропил-1-[3-(1-гидроксициклопропил)-этинилфенил]-1,4-дигидро[1,8]нафтиридин 4-он-3-карбоксамид Следуя методике стадии 5 примера 1, но заменяя фенилацетилен на продукт приведенной выше стадии 1, соединение, N-изопропил-1-[3-(1-гидроксициклопропил)этинилфенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид, получают в виде твердого вещества. 1H-ЯМР (CDCl3)1,09 (м, 2 Н), 1,17 (м, 2 Н), 1,28 (д, 6 Н), 2,57 (с, 1 Н, ОН), 4,28 (м, 1 Н), 7,35 (д, 1 Н),7,44-7,50 (м, 3H), 7,54 (д, 1 Н), 8,68 (м, 1 Н), 8,79 (дд, 1 Н), 8,96 (с, 1 Н), 9,63 (ушир, NH). Пример 14. N-Изопропил-1-3-[4,4,4-трифтор-3-гидрокси-3-(трифторметил)бут-1-инил]фенил-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. 1,1,1-трифтор-2-(трифторметил)-4-(триметилсилил)-бут-3-ин-2-ол К раствору триметилсилилацетилена (4 мл) в ТГФ (30 мл) при -78 С добавляют 2,5 М н-бутиллития в гексане (14 мл) и полученную смесь перемешивают в течение 1 ч. Избыток гексафторацетона барботируют через охлажденную смесь и перемешивание продолжают в течение 4 ч. После нагревания до комнатной температуры смесь гасят насыщенным водным раствором хлорида аммония и распределяют между диэтиловым эфиром и водой. Органическую фазу сушат и упаривают, получая 1,1,1-трифтор-2(трифторметил)-4-(триметилсилил)бут-3-ин-2-ол в виде жидкого вещества. Стадия 2. N-изопропил-1-3-[4,4,4-трифтор-3-гидрокси-3-(трифторметил)бут-1-инил]фенил-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксамид К раствору 1,1,1-трифтор-2-(трифторметил)-4-(триметилсилил)бут-3-ин-2-ола по приведенной выше стадии 1 (6,8 ммоль) в 10 мл ТГФ добавляют 1 М тетрабутиламмонийфторид (8,5 мл) и полученную смесь нагревают при кипении с обратным холодильником в течение 30 мин для удаления TMS-защитной группы. Затем применяют методику стадии 5 примера 1, но заменяя фенилацетилен данным раствором,что приводит к образованию соединения,N-изопропил-1-3-[4,4,4-трифтор-3-гидрокси-3(трифторметил)бут-1-инил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамида, в виде твердого вещества. 1H-ЯМР (Ацетон-d6)1,24 (д, 6 Н), 4,17 (м, 1 Н), 7,60 (м, 1H), 7,72-7,79 (м, 2 Н), 7,83 (д, 1 Н), 7,90 (с,1 Н), 8,14 (с, 1 Н, ОН), 8,72 (м, 1 Н), 8,77 (дд, 1 Н), 8,85 (с, 1 Н), 9,62 (ушир, NH). Пример 15. N-Изопропил-1-[3-(3-гидрокси-3-фенилбут-1-инил)фенил]-1,4-дигидро[1,8]нафтиридин 4-он-3-карбоксамид. Смесь N-изопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамида со стадии 4 примера 1, 2-фенил-3-бутин-2-ола (2 экв.), триэтиламина (1,66 экв.), бис(дифенилфосфино)ферроцендихлорпалладия(II) (0,05 экв.) и иодида меди(I) (5 мг/ммоль) в ДМФА (20 мл/ммоль) нагревают при 85 С в течение 18 ч. После охлаждения до комнатной температуры полученную смесь гасят насыщенным водным раствором хлорида аммония и распределяют между этилацетатом и водой. Сырой продукт из органической фазы очищают хроматографией на силикагеле, элюируя смесью 20% диэтилового эфира в метиленхлориде. Очищенный продукт перемешивают в диэтиловом эфире при комнатной температуре в течение 3 ч и фильтруют, получая указанное в заголовке соединение в виде белого твердого вещества. 1(ушир, NH). Пример 16. N-Циклопропил-1-[3-(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. 3-Этинилпиридин-N-оксид К раствору 3-этинилпиридина в метиленхлориде (5 мл/ммоль) при комнатной температуре добавляют м-хлорпероксибензойную кислоту (m-СРВА, 70% чистоты, 1,2 экв.) и полученную смесь перемешивают в течение 2 ч. Добавляют дополнительное количество m-СРВА (0,25 экв.) и перемешивание продолжают в течение 1 ч. Добавляют гидроксид кальция (2 экв.) и спустя 15 мин смесь фильтруют через целит и фильтрат упаривают. Твердый остаток перемешивают в диэтиловом эфире в течение 3 ч и фильтруют, получая соединение, 3-этинилпиридин-N-оксид, в виде белого твердого вещества. Стадия 2. N-циклопропил-1-[3-(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин 4-он-3-карбоксамид Следуя методике примера 15, но заменяя N-изопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид на N-циклопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3 карбоксамид со стадии 1 примера 6 и 2-фенил-3-бутин-2-ол на 3-этинилпиридин-N-оксид со стадии 1,получают соединение, N-циклопропил-1-[3-(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид, в виде белого твердого вещества.(ушир, NH). Пример 17. N-Изопропил-1-[3-(3-амино-3-этилпент-1-инил)фенил]-1,4-дигидро[1,8]нафтиридин-4 он-3-карбоксамид. Следуя методике примера 15, но заменяя 2-фенил-3-бутин-2-ол на 1,1-диэтилпропаргиламин, указанное в заголовке соединение получают в виде твердого вещества. 1H-ЯМР (CDCl3)1,05 (т, 6 Н), 1,28 (д, 6 Н), 1,57 (м, 2 Н), 1,69 (м, 2 Н), 4,27 (м, 1 Н), 7,33 (д, 1 Н), 7,447,49 (м, 3H), 7,53 (д, 1 Н), 8,69 (м, 1 Н), 8,79 (дд, 1 Н), 8,97 (с, 1 Н), 9,63 (ушир, NH). (NH2 не наблюдается). Пример 18. N-Циклопропил-1-[3-(3-амино-3-этилпент-1-инил)фенил]-1,4-дигидро[1,8]нафтиридин 4-он-3-карбоксамид. Следуя методике примера 17, но заменяя N-изопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид на N-циклопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3 карбоксамид со стадии 1 примера 6, указанное в заголовке соединение получают в виде твердого вещества. 1(д, 1 Н), 7,44-7,49 (м, 3H), 7,54 (д, 1 Н), 8,69 (м, 1 Н), 8,77 (дд, 1 Н), 8,97 (с, 1 Н), 9,75 (ушир, NH). (NH2 не наблюдается). Пример 19. N-Изопропил-1-[3-(3-хинолинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Следуя методике примера 15, но заменяя 2-фенил-3-бутин-2-ол на N-изопропил-1-(3-этинилфенил)1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид со стадии 2 примера 5 и N-изопропил-1-(3-бромфенил)1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид на 3-бромхинолин, указанное в заголовке соединение получают в виде твердого вещества. 1H-ЯМР (CDCl3)1,32 (д, 6 Н), 4,32 (м, 1 Н), 7,48-7,51 (м, 2 Н), 7,58-7,65 (м, 2 Н), 7,71 (с, 1 Н), 7,737,80 (м, 2 Н), 7,83 (д, 1 Н), 8,12 (д, 1 Н), 8,35 (с, 1 Н), 8,75 (м, 1 Н), 8,85 (дд, 1 Н), 9,02 (с, 1 Н), 9,06 (с, 1 Н),9,65 (шир, NH). Пример 20. N-Изопропил-1-[3-(1-оксидо-3-хинолинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин 4-он-3-карбоксамид. Следуя методике примера 19, но заменяя 3-бромхинолин на 3-бромхинолин-N-оксид, указанное в заголовке соединение получают в виде твердого вещества. 1H-ЯМР (CDCl3)1,33 (д, 6 Н), 4,32 (м, 1 Н), 7,49-7,53 (м, 2 Н), 7,63 (т, 1 Н), 7,68-7,73 (м, 2 Н), 7,757,83 (м, 2 Н), 7,88-7,92 (м, 2 Н), 8,63 (с, 1 Н), 8,73-8,78 (м, 2 Н), 8,86 (дд, 1 Н), 9,05 (с, 1 Н), 9,67 (ушир, NH). Пример 21. N-Изопропил-1-[3-(циклопропилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3 карбоксамид. Следуя методике примера 15, но заменяя 2-фенил-3-бутин-2-ол на циклопропилацетилен (Tetrahedron letters 2000, 41, 4007), указанное в заголовке соединение получают в виде серого твердого вещества. 1H-ЯМР (CDCl3)0,83 (м, 2 Н), 0,90 (м, 2 Н), 1,31 (д, 6 Н), 1,48 (м, 1H), 4,31 (м, 1 Н), 7,33 (м, 1 Н),7,45-7,51 (м, 3H), 7,55 (д, 1 Н), 8,72 (м, 1 Н), 8,83 (дд, 1 Н), 9,01 (с, 1 Н), 9,68 (ушир, NH). Пример 22. N-Изопропил-1-[3-(6-амино-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4 он-3-карбоксамид. Следуя методике примера 19, но заменяя 3-бромхинолин на 5-бром-2-аминопиридин, указанное в заголовке соединение получают в виде твердого вещества. 1H-ЯМР (СDCl3)1,33 (д, 6 Н), 4,31 (м, 1 Н), 4,71 (ушир, NH2), 6,49 (д, 1 Н), 7,40 (м, 1 Н), 7,48 (м, 1H),7,54-7,60 (м, 3H), 7,68 (д, 1 Н), 8,28 (с, 1 Н), 8,72 (м, 1 Н), 8,83 (дд, 1 Н), 9,04 (с, 1 Н), 9,67 (ушир, NH). Пример 23. N-Изопропил-1-3-[5-(1-гидрокси-1-метилэтил)-1-оксидо-3-пиридинилэтинил]фенил 1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. 3-Бром-5-(1-гидрокси-1-метилэтил)пиридин К раствору этил 5-бромникотината (1,02 г, 4,4 ммоль) в диэтиловом эфире (15 мл) при -30 С добавляют 3 М раствор метилмагнийбромида в диэтиловом эфире (4 мл, 12 ммоль). Полученную суспензию нагревают при температуре кипения с обратным холодильником в течение 2 ч, затем охлаждают и гасят избытком 0,5 М водного одноосновного фосфата натрия и распределяют между диэтиловым эфиром и водой. Продукт из органической фазы очищают хроматографией на силикагеле, элюируя смесью 2:1:2 диэтилового эфира, пентана и насыщенного аммиаком метиленхлорида, получая соединение, 3-бром-5(1-гидрокси-1-метилэтил)пиридин, в виде желтого масла. Стадия 2. 3-Бром-5-(1-гидрокси-1-метилэтил)пиридин-N-оксид К раствору 3-бром-5-(1-гидрокси-1-метилэтил)пиридина со стадии 1 (3,1 ммоль) в хлороформе (10 мл) добавляют м-хлорпероксибензойную кислоту 70% (1,5 экв.) и полученную смесь перемешивают при комнатной температуре в течение 18 ч. Добавляют избыток гидроксида кальция и после перемешивания в течение 5 мин смесь фильтруют через целит и фильтрат упаривают. Сырой продукт очищают хромато- 24006800 графией на силикагеле, элюируя 10% этанолом в метиленхлориде (насыщенном аммиаком), что дает соединение, 3-бром-5-(1-гидрокси-1-метилэтил)пиридин-N-оксид, в виде твердого вещества. Стадия 3. N-Изопропил-1-3-[5-(1-гидрокси-1-метилэтил)-1-оксидо-3-пиридинилэтинил]фенил 1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике примера 15, но заменяя 2-фенил-3-бутин-2-ол на N-изопропил-1-(3-этинилфенил)1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид из примера 5 и N-изопропил-1-(3-бромфенил)-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксамид на 3-бром-5-(1-гидрокси-1-метилэтил)пиридин-N-оксид со стадии 2, соединение, N-изопропил-1-3-[5-(1-гидрокси-1-метилэтил)-1-оксидо-3-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид, получают в виде твердого вещества. 1(ушир, NH). Пример 24. N-Изопропил-1-3-[6-(1-гидрокси-1-метилэтил)-3-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. 5-Бром-2-(1-гидрокси-1-метилэтил)пиридин К суспензии 2,5-дибромпиридина в толуоле (12 мл/ммоль), охлажденной до -78 С, добавляют 2,5 М н-бутиллитий в гексане (1,05 экв.) и полученную смесь перемешивают при охлаждении в течение 2,5 ч. Добавляют ацетон (2 экв.) и перемешивание продолжают в течение 1,5 ч. После гашения насыщенным водным раствором хлорида аммония, смесь нагревают до комнатной температуры и распределяют между этилацетатом и водой. Продукт из органической фазы очищают хроматографией на силикагеле, элюируя смесью 20% этилацетата в гексане, что дает соединение, 5-бром-2-(1-гидрокси-1-метилэтил)пиридин, в виде сиропа. Стадия 2. 5-Бром-2-(1-метил-1-[2-(триметилсилил)-этокси]метоксиэтил)пиридин К раствору 5-бром-2-(1-гидрокси-1-метилэтил)пиридина со стадии 1 (14 ммоль) в метиленхлориде(50 мл) при 0 С добавляют N,N-диизопропилэтиламин (37,3 ммоль) и 2-(триметилсилил)этоксиметилхлорид (15,3 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 18 ч,затем нагревают при температуре кипения с обратным холодильником в течение 24 ч. После охлаждения до комнатной температуры смесь гасят насыщенным водным раствором хлорида аммония и распределяют между метиленхлоридом и водой. Сырой продукт из органической фазы очищают хроматографией на силикагеле, элюируя смесью 6% этилацетата в гексане, что дает соединение, 5-бром-2-(1-метил-1-[2(триметилсилил)этокси]метоксиэтил)пиридин. Стадия 3. 2-(1-Метил-1-[2-(триметилсилил)этокси]метоксиэтил)-5-[(триметилсилил)этинил]пиридин Следуя методике стадии 5 примера 1, но заменяя N-изопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид на продукт приведенной выше стадии 2 и фенилацетилен на триметилсилилацетилен, получают соединение, 2-(1-метил-1-[2-(триметилсилил)этокси]метоксиэтил)-5[(триметилсилил)этинил]пиридин. Стадия 4. 5-Этинил-2-(1-метил-1-[2-(триметилсилил)этокси]метоксиэтил)пиридин Следуя методике стадии 2 примера 5, но заменяя N-изопропил-1-[3-(триметилсилилэтинил)фенил]1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид на продукт приведенной выше стадии 3, получают соединение, 5-этинил-2-(1-метил-1-[2-(триметилсилил)этокси]метоксиэтил)пиридин. Стадия 5. N-Изопропил-1-(3-[6-(1-метил-1-(2-триметилсилил)этокси]метоксиэтил)пиридин-3-ил] этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике стадии 5 примера 1, но заменяя фенилацетилен на продукт приведенной выше стадии 4, получают соединение, N-изопропил-1-(3-[6-(1-метил-1-[(2-триметилсилил)этокси]метокси этил)пиридин-3-ил]этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 6. N-Изопропил-1-3-[6-(1-гидрокси-1-метилэтил)-3-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид К раствору N-изопропил-1-(3-[6-(1-метил-1-[(2-триметилсилил)этокси]метоксиэтил)пиридин-3 ил]этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамида, продукта вышеуказанной стадии 5,в метиленхлориде (32 мл/ммоль) при 0 С добавляют трифторуксусную кислоту (3,2 мл/ммоль). Полученную смесь перемешивают при 0 С в течение 2 ч, затем при комнатной температуре в течение 2 ч. Смесь нейтрализуют, медленно добавляя насыщенный водный раствор бикарбоната натрия, и распределяют между метиленхлоридом и водой. Сырой продукт из органической фазы очищают хроматографией на силикагеле, элюируя смесью 40% диэтилового эфира в метиленхлориде, очищенный продукт перемешивают в диэтиловом эфире при комнатной температуре в течение 2 ч и фильтруют, получая соединение, N-изопропил-1-3-[6-(1-гидрокси-1-метилэтил)-3-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид, в виде твердого вещества. 1- 25006800 Пример 25. N-Изопропил-1-3-[6-(1-гидрокси-1-метилэтил)-1-оксидо-3-пиридинилэтинил]фенил 1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Следуя методике стадии 2 примера 23, но заменяя 3-бром-5-(1-гидрокси-1-метилэтил)пиридин наN-изопропил-1-3-[6-(1-гидрокси-1-метилэтил)-3-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид из примера 24, указанное в заголовке соединение получают в виде твердого вещества. 1H-ЯМР (Ацетон-d6)1,25 (д, 6 Н), 1,60 (с, 6 Н), 4,18 (м, 1 Н), 7,24 (с, 1 Н, ОН), 7,60-7,63 (м, 3H), 7,727,78 (м, 2 Н), 7,82 (д, 1 Н), 7,91 (с, 1 Н), 8,46 (с, 1 Н), 8,74 (м, 1 Н), 8,78 (дд, 1 Н), 8,87 (с, 1 Н), 9,62 (ушир,NH). Пример 26. N-Изопропил-1-3-[4-(1-гидрокси-1-метилэтил)-2-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. Метил-2-бромизоникотинат К раствору 2-бромизоникотиновой кислоты (Chem. Pharm. Bull. 1990, 38, 2446) (2,0 г) в тетрагидрофуране (100 мл) добавляют избыток эфирного диазометана и полученную смесь перемешивают при комнатной температуре в течение 1 ч. Смесь упаривают и продукт очищают хроматографией на силикагеле,элюируя смесью 1:3 этилацетата и гексана, что дает сложный эфир метил-2-бромизоникотината в виде бесцветной жидкости. Стадия 2. 2-Бром-4-(1-гидрокси-1-метилэтил)пиридин Следуя методике стадии 1 примера 23, но заменяя этил-5-бромникотинат на метил-2-бромизоникотинат из приведенной выше стадии 1 соединение, 2-бром-4-(1-гидрокси-1-метилэтил)пиридин, получают в виде белого твердого вещества. Стадия 3. N-Изопропил-1-3-[4-(1-гидрокси-1-метилэтил)-2-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике примера 19, но заменяя 3-бромхинолин на 2-бром-4-(1-гидрокси-1 метилэтил)пиридин из приведенной выше стадии 2, соединение, N-изопропил-1-3-[4-(1-гидрокси-1 метилэтил)-2-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид, получают в виде желтого пенистого вещества. 1(ушир, NH). Пример 27. N-Изопропил-1-3-[5-(1-гидрокси-1-метилэтил)-2-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. 2-Бром-5-(1-гидрокси-1-метилэтил)пиридин Раствор 2,5-дибромпиридина в диэтиловом эфире (5 мл/ммоль) охлаждают до -78 С и медленно добавляют 2,5 М н-бутиллития в гексане (1,05 экв.). После охлаждения в течение 2 ч добавляют ацетон (1,3 экв.) и перемешивание продолжают в течение 1 ч. Полученную смесь гасят насыщенным водным раствором хлорида аммония, нагревают до комнатной температуры и распределяют между диэтиловым эфиром и водой. Сырой продукт из органической фазы растирают в смеси 1:1 диэтиловый эфир-гексан и фильтруют, получая соединение, 2-бром-5-(1-гидрокси-1-метилэтил)пиридин, в виде твердого вещества. Стадия 2. 5-(1-Гидрокси-1-метилэтил)-2-[(триметилсилил)этинил]пиридин Следуя методике примера 15, но заменяя N-изопропил-1-(3-бромфенил)-1,4-дигидро[1,8] нафтиридин-4-он-3-карбоксамид на продукт приведенной выше стадии 1, 2-бром-5-(1-гидрокси-1 метилэтил)пиридин, и 2-фенил-3-бутин-2-ол на триметилсилилацетилен, получают соединение, 5-(1 гидрокси-1-метилэтил)-2-[(триметилсилил)этинил]пиридин. Стадия 3. 2-Этинил-5-(1-гидрокси-1-метилэтил)пиридин Следуя методике стадии 2 примера 5, но заменяя N-изопропил-1-[3-(триметилсилилэтинил)фенил]1,4-дигидро[1,8]-нафтиридин-4-он-3-карбоксамид на продукт приведенной выше стадии 2, 5-(1 гидрокси-1-метилэтил)-2-[(триметилсилил)этинил]пиридин, получают соединение, 2-этинил-5-(1 гидрокси-1-метилэтил)пиридин. Стадия 4. N-изопропил-1-3-[5-(1-гидрокси-1-метилэтил)-2-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике примера 15, но заменяя 2-фенил-3-бутин-2-ол на продукт приведенной выше стадии 3, 2-этинил-5-(1-гидрокси-1-метилэтил)пиридин, получают соединение, N-изопропил-1-3-[5-(1 гидрокси-1-метилэтил)-2-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. 1- 26006800 Стадия 1. 2-Бром-6-(1-гидрокси-1-метилэтил)пиридин Следуя методике стадии 1 примера 27, но заменяя 2,5-дибромпиридин на 2,6-дибромпиридин, получают соединение, 2-бром-6-(1-гидрокси-1-метилэтил)пиридин, в виде твердого вещества. Стадия 2. N-Изопропил-1-3-[6-(1-гидрокси-1-метилэтил)-2-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике примера 19, но заменяя 3-бромхинолин на продукт приведенной выше стадии 1, 2 бром-6-(1-гидрокси-1-метилэтил)пиридин, получают соединение, N-изопропил-1-3-[6-(1-гидрокси-1 метилэтил)-2-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. 1H-ЯМР (CDCl3)1,31 (д, 6 Н), 1,58 (с, 6 Н), 4,32 (м, 1 Н), 4,83 (с, 1 Н, ОН), 7,38 (д, 1 Н), 7,43-7,52 (м,3H), 7,60 (т, 1 Н), 7,70-7,75 (м, 2 Н), 7,79 (д, 1 Н), 8,74 (м, 1 Н), 8,84 (дд, 1 Н), 9,03 (с, 1 Н), 9,66 (ушир, NH). Пример 29. N-Циклопропил-1-3-[6-(1-гидрокси-1-метилэтил)-1-оксидо-3-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Стадия 1. 5-Бром-2-(1-гидрокси-1-метилэтил)пиридин-N-оксид Следуя методике стадии 2 примера 23, но заменяя 3-бром-5-(1-гидрокси-1-метилэтил)пиридин на 5 бром-2-(1-гидрокси-1-метилэтил)пиридин со стадии 1 примера 24, получают соединение, 5-бром-2-(1 гидрокси-1-метилэтил)пиридин-N-оксид. Стадия 2. N-Циклопропил-1-3-[6-(1-гидрокси-1-метилэтил)-1-оксидо-3-пиридинилэтинил]фенил 1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике примера 15, но заменяя 2-фенил-3-бутин-2-ол на N-циклопропил-1-(3 этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид из примера 6 и N-изопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид на 5-бром-2-(1-гидрокси-1-метилэтил)пиридинN-оксид из приведенной выше стадии 1, соединение, N-циклопропил-1-3-[6-(1-гидрокси-1-метилэтил)1-оксидо-3-пиридинилэтинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид, получают в виде твердого продукта. 1(1,1 экв.), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (0,05 экв.) и 2 М водного карбоната натрия (5 экв.) в N,N-диметилформамиде (2 мл/ммоль) перемешивают при 85 С в течение 4 ч. После гашения насыщенным водным раствором хлорида аммония смесь распределяют между этилацетатом и водой и сырой продукт из органической фазы очищают хроматографией на силикагеле, элюируя смесью 1:9 этилацетата и гексана, что дает соединение, 3-(4-бромфенил)пиридин, в виде твердого вещества. Стадия 2. N-Изопропил-1-3-[(4-пиридин-3-илфенил)этинил]фенил-1,4-дигидро[1,8]нафтиридин 4-он-3-карбоксамид Следуя методике примера 19, но заменяя 3-бромхинолин на продукт приведенной выше стадии 1,получают соединение, N-изопропил-1-3-[(4-пиридин-3-илфенил)этинил]фенил-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. 1[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. 2-Бром-5-(1-гидрокси-1-метилэтил)тиофен К раствору 2-ацетил-5-бромтиофена в ТГФ (2,5 мл/ммоль) при -30 С добавляют 1,4 М метилмагнийбромид в 3:1 смеси толуол-ТГФ (1,5 экв.) и полученную смесь нагревают до -10 С и перемешивают в течение 1,5 ч. После гашения насыщенным водным раствором хлорида аммония, смесь распределяют между диэтиловым эфиром и водой. Органическую фракцию сушат и упаривают и сырой продукт очищают хроматографией на силикагеле, элюируя смесью 1:4 диэтилового эфира и гексана, что дает соединение, 2-бром-5-(1-гидрокси-1-метилэтил)тиофен. Стадия 2. 2-(1-Гидрокси-1-метилэтил)-5-триметилсилилэтинилтиофен Следуя методике примера 15, но заменяя N-изопропил-1-(3-бромфенил)-1,4-дигидро[1,8] нафтиридин-4-он-3-карбоксамид на продукт приведенной выше стадии 1 и 2-фенил-3-бутин-2-ол на триметилсилилацетилен, получают соединение, 2-(1-гидрокси-1-метилэтил)-5-триметилсилилэтинилтиофен. Стадия 3. 2-Этинил-5-(1-гидрокси-1-метилэтил)тиофен Следуя методике стадии 2 примера 5, но заменяя N-изопропил-1-[3-(триметилсилилэтинил)фенил]1,4-дигидро[1,8]-нафтиридин-4-он-3-карбоксамид на продукт приведенной выше стадии 2, получают соединение, 2-этинил-5-(1-гидрокси-1-метилэтил)тиофен.[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике примера 15, но заменяя 2-фенил-3-бутин-2-ол на продукт приведенной выше стадии 3, 2-этинил-5-(1-гидрокси-1-метилэтил)тиофен, соединение, N-изопропил-1-(3-[5-(1-гидрокси-1 метилэтил)тиен-2-ил]этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид, получают в виде твердого вещества. 1H-ЯМР (CDCl3)1,31 (д, 6 Н), 1,70 (с, 6 Н), 2,42 (с, 1 Н, ОН), 4,31 (м, 1 Н), 6,87 (д, 1 Н), 7,16 (д, 1 Н),7,42 (д, 1 Н), 7,48 (м, 1 Н), 7,59 (т, 1 Н), 7,63 (с, 1 Н), 7,68 (д, 1 Н), 8,73 (м, 1 Н), 8,84 (дд, 1 Н), 9,02 (с, 1 Н),9,68 (ушир, NH). Пример 32. N-Изопропил-1-(3-[2-(1-гидрокси-1-метилэтил)-1,3-тиазол-5-ил]этинилфенил)-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. 2-(1-Гидрокси-1-метилэтил)тиазол К раствору тиазола в диэтиловом эфире (1 мл/ммоль) при -78 С добавляют 2,2 М н-бутиллитий в гексане (1,1 экв.) и полученную смесь перемешивают в течение 30 мин. Добавляют ацетон (1,2 экв.) и смесь перемешивают при -78 С в течение еще 30 мин. Смесь гасят охлажденным насыщенным водным раствором хлорида аммония и нагревают до комнатной температуры, затем распределяют между диэтиловым эфиром и водой. Органическую фазу сушат и упаривают, получая сырой продукт в виде оранжево-коричневого масла, которое используют как таковое на следующей стадии. Стадия 2. 2-(1-Метил-1-[2-(триметилсилил)этокси]метоксиэтил)тиазол Следуя методике стадии 2 примера 24, но заменяя 5-бром-2-(1-гидрокси-1-метилэтил)пиридин на продукт приведенной выше стадии 1, соединение, 2-(1-метил-1-[2-(триметилсилил)этокси]метокси этил)тиазол, получают в виде масла. Стадия 3. 5-Бром-2-(1-гидрокси-1-метилэтил)тиазол К раствору 2-(1-метил-1-[2-(триметилсилил)этокси]метоксиэтил)тиазола со стадии 2 в хлороформе (2 мл/ммоль) при комнатной температуре добавляют бром (2 молярных экв.) и полученную смесь перемешивают в течение 1 ч. Добавляют бикарбонат натрия (0,55 экв.) и смесь перемешивают в течение 5 ч. Добавляют дополнительное количество бикарбоната натрия (0,55 экв.) и перемешивание продолжают в течение 18 ч. После заключительного добавления бикарбоната натрия (0,55 экв.) смесь перемешивают еще 5 ч, разбавляют хлороформом и органическую фазу промывают насыщенным водным раствором бикарбоната натрия, затем водой, сушат и упаривают. Сырой продукт очищают хроматографией, элюируя смесью 3:7 этилацетата и гексана, что дает требуемый продукт. Стадия 4. N-Изопропил-1-(3-[2-(1-гидрокси-1-метилэтил)-1,3-тиазол-5-ил]этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике примера 19, но заменяя 3-бромхинолин на продукт приведенной выше стадии 3,соединение, N-изопропил-1-(3-[2-(1-гидрокси-1-метилэтил)-1,3-тиазол-5-ил]этинилфенил)-1,4-дигидроH-ЯМР (CDCl3)1,29 (д, 6 Н), 1,68 (с, 6 Н), 2,90 (с, 1 Н, ОН), 4,28 (м, 1 Н), 7,42 (д, 1 Н), 7,46 (м, 1 Н),7,54-7,60 (м, 2 Н), 7,66 (д, 1 Н), 7,82 (с, 1 Н), 8,70 (м, 1 Н), 8,80 (дд, 1 Н), 8,99 (с, 1 Н), 9,63 (ушир, NH). Пример 33. 1-[3-(1-Оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид. Стадия 1. 1-(3-Бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике стадии 4 примера 1, но заменяя изопропиламин на 28% водный гидроксид аммония, соединение, 1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид, получают в виде твердого вещества. Стадия 2. 1-[3-(Триметилсилилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике стадии 5 примера 1, но заменяя N-изопропил-1-(3-бромфенил)-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксамид на 1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3 карбоксамид приведенной выше стадии 1 и фенилацетилен на триметилсилилацетилен, продукт, 1-[3(триметилсилилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид, получают в виде твердого вещества. Стадия 3. 1-(3-Этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид К раствору 1-[3-(триметилсилилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамида со стадии 2 в ТГФ (30 мл/ммоль) при 0 С добавляют 1 М тетрабутиламмонийфторид в ТГФ (1,5 экв.) и полученную смесь перемешивают при 0 С в течение 30 мин. Смесь распределяют между метиленхлоридом и водой и органическую фазу сушат и упаривают. Сырой продукт, 1-(3-этинилфенил)-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксамид, используют как таковой на следующей стадии. Стадия 4. 1-[3-(1-Оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид Следуя методике примера 19, но заменяя N-изопропил-1-(3-этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид на 1-(3-этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3 карбоксамид со стадии 3 и 3-бромхинолин на 3-бромпиридин-N-оксид, 1-[3-(1-оксидо-3-пиридинил- 28006800 этинил)фенил]-1,4-дигидро-[1,8]нафтиридин-4-он-3-карбоксамид получают в виде белого твердого вещества. 1H-ЯМР (CDCl3)5,84 (ушир, 1 Н, NH), 7,30 (м, 1 Н), 7,41 (д, 1 Н), 7,53 (м, 2 Н), 7,64 (т, 1 Н), 7,67 (с,1 Н), 7,74 (д, 1 Н), 8,21 (д, 1 Н), 8,35 (с, 1 Н), 8,75 (м, 1 Н), 8,88 (дд, 1 Н), 9,05 (с, 1 Н), 9,52 (ушир, 1 Н, NH). Пример 34. 1-[3-(1-Оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3 карбоновая кислота Стадия 1. Этил-1-(3-этинилфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксилат. Следуя методикам стадий 1 и 2 примера 5, но заменяя исходный продукт, N-изопропил-1-(3 бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид, на этил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксилат со стадии 2 примера 1, соединение, этил-1-(3-этинилфенил)-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксилат, получают в виде твердого вещества. Стадия 2. Этил-1-[3-(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3 карбоксилат Следуя методике примера 15, но заменяя 2-фенил-3-бутин-2-ол на этил 1-(3-этинилфенил)-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксилат из приведенной выше стадии 1, и N-изопропил-1-(3 бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид на 3-бромпиридин-N-оксид, соединение этил-1-[3-(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксилат получают в виде твердого вещества. Стадия 3. 1-[3-(1-Оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоновая кислота Следуя методике стадии 3 примера 1,но заменяя этил-1-(3-бромфенил)-1,4 дигидро[1,8]нафтиридин-4-он-3-карбоксилат на сложный эфир из приведенной выше стадии 2, этил-1-[3(1-оксидо-3-пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксилат, 1-[3-(1-оксидо-3 пиридинилэтинил)фенил]-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоновую кислоту получают в виде белого твердого вещества. 1H-ЯМР (DMSO-d6)7,46 (т, 1 Н), 7,51 (д, 1 Н), 7,70 (т, 1 Н), 7,75 (м, 2 Н), 7,80 (д, 1 Н), 7,92 (с, 1 Н),8,26 (д, 1 Н), 8,47 (с, 1 Н), 8,81 (дд, 1 Н), 8,89 (м, 1 Н), 8,97 (с, 1 Н). Прочие изменения и модификации, очевидные для специалиста в данной области, входят в объем настоящего изобретения и формулировок приложенных пунктов. Предполагается, что настоящее изобретение ограничивается только приложенными пунктами. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, представленное формулой (I) или его фармацевтически приемлемая соль, где R означает Н;R2 отсутствует либо означает Н, -C1-6 алкил, -С 3-6 циклоалкил или фенил, и любой из алкилов необязательно замещен 1-6 независимыми заместителями, означающими галоген;R3 отсутствует либо означает Н, ОН, -NH2 или C1-6 алкил, где любой из алкилов необязательно замещен 1-6 независимыми заместителями, означающими галоген; каждый из R4, R5, R6 и R7 независимо означает Н иR8 означает необязательно замещенный пиридилом фенил, пиридил, хинолинил, тиенил или тиазолил или оксиды пиридила или хинолинила; где указанный пиридил, хинолинил, тиенил или тиазолил необязательно замещен группой, означающей 1-гидрокси-1-метилэтил или NH2; или R8 означает Н, -C1-6 алкил или -С 3-6 циклоалкил, где любой из алкилов необязательно замещен 1-6 независимыми заместителями, означающими галоген, -NH2 или -ОН. 2. Соединение по п.1 или его фармацевтически приемлемая соль, где R2 и R3 отсутствуют и R8 означает Н. 3. Соединение по п.1 или его фармацевтически приемлемая соль, где R8 означает фенил, пиридил,хинолинил, тиенил или тиазолил; или оксиды пиридила или хинолинила.- 29006800 4. Соединение по п.3 или фармацевтически приемлемая соль, где R8 означает фенил. 5. Соединение по п.3 или его фармацевтически приемлемая соль, где R8 означает пиридил или его оксиды. 6. Соединение по п.1 или его фармацевтически приемлемая соль, где R8 означает хинолинил, тиенил, тиазолил, или оксид хинолинила; -С 3-6 циклоалкил, необязательно замещенный 1-6 независимыми заместителями, означающими галоген, -NH2 или -ОН; -C1-6 алкил, необязательно замещенный 1-6 независимыми заместителями, означающими галоген, NH2 или -ОН. 7. Соединение по п.1, выбираемое из группы, включающей
МПК / Метки
МПК: A61K 31/435, C07D 519/00, C07D 471/04
Метки: типа, качестве, производные, ингибитора, алкинарилнафтиридин-4(1н)-онов, фосфодиэстеразы
Код ссылки
<a href="https://eas.patents.su/30-6800-proizvodnye-alkinarilnaftiridin-41n-onov-v-kachestve-ingibitora-fosfodiesterazy-tipa-iv.html" rel="bookmark" title="База патентов Евразийского Союза">Производные алкинарилнафтиридин-4(1н)-онов в качестве ингибитора фосфодиэстеразы типа iv</a>
Производные индазола и их использование в качестве ингибиторов фосфодиэстеразы (фдэ) типа iv и продуцирования фактора некроза опухоли (фно)

Номер патента: 2113
Опубликовано: 24.12.2001
Автор: Марфат Энтони
МПК: A61K 31/415, C07D 401/12, A61P 11/06...
Метки: типа, качестве, фосфодиэстеразы, производные, фдэ, некроза, фактора, фно, использование, опухоли, ингибиторов, продуцирования, индазола
Формула / Реферат:
1. Соединение формулы (I) или их фармацевтически приемлемые соли, в которых R является Н, C1-C9 алкилом, -(СН2)m (5-10 членным гетероциклилом), где m равно от 0 до 2, или (Z1)b(Z2)с(С6-С10 арилом), где b и с независимо равны от 0 до 1, Z1 является C1-С6 алкиленом или C2-C8 алкениленом и Z2 является О, S, SO2 или NR12; и где указанные R группы необязательно замещены от 1 до 3 заместителями, независимо выбранными из группы, включающей галоген,...
Производные замешенных индазолов, их применение в качестве ингибиторов фосфодиэстеразы (фдэ) типа iv и продукции фактора некроза опухолей (фно)

Номер патента: 2272
Опубликовано: 28.02.2002
Автор: Марфэт Энтони
МПК: C07D 231/56, A61P 35/00, A61K 31/415...
Метки: фактора, производные, типа, фдэ, фно, ингибиторов, продукции, применение, замешенных, опухолей, качестве, индазолов, фосфодиэстеразы, некроза
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемые соли, где R представляет собой водород, -(СН2)n(С3-С7циклоалкил), где n равно 0, или фенил, возможно замещенный 1-3 галогено; R1 представляет собой С1-С7алкил; R2 выбран из группы, состоящей из где пунктирные линии в формулах (Iа) и (Iб) представляют простую связь; m равно от 0 до 1; R3 представляет собой циано или CH2CN; R4 представляет собой Н, CO2R14, C(Y)NR17R14, CN,...
Призводные замещенного индазола и их применение в качестве ингибиторов фосфодиэстеразы (фдэ) типа iv и фактора некроза опухоли (фно)

Номер патента: 2274
Опубликовано: 28.02.2002
Автор: Марфэт Энтони
МПК: C07D 403/04, A61K 31/416, A61P 37/00...
Метки: фактора, типа, индазола, призводные, фно, фосфодиэстеразы, применение, ингибиторов, фдэ, качестве, замещенного, опухоли, некроза
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль, где R представляет собой циклопентил или циклогексил; R1 представляет собой С1-С2алкил; и R2 представляет собой группировку, выбранную из группы, состоящей из группировок формул с (1.1) по (1.12): 2. Соединение по п.1, в котором R2 представляет собой группировку формулы (1.1). 3. Соединение по п.1, в котором R2 представляет собой группировку формулы (1.10). 4. Соединение по п.1,...
Производные фталазина в качестве ингибиторов фосфодиэстеразы 4

Номер патента: 3702
Опубликовано: 28.08.2003
Авторы: Наполетано Мауро, Мораццони Габриеле, Гранчини Джанкарло, Норчини Габриеле, Пеллачини Франко
МПК: A61K 31/502, C07D 237/30
Метки: фталазина, производные, качестве, ингибиторов, фосфодиэстеразы
Формула / Реферат:
1. Соединение формулы I где ---- представляет одинарную или двойную связь; B представляет метилен; A представляет пиридин, замещенный одним заместителем или большим количеством заместителей; R представляет два атома водорода или C = O группу, когда ---- представляет одинарную связь, или, когда ---- представляет двойную связь, R представляет водород, необязательно замещенный арил или гетероцикл, (C1-8)алкил, (C2-8)алкенил или (C2-8)алкинил,...
Производные пирролидина в качестве ингибиторов фосфодиэстеразы, специфичной к циклическому амф

Номер патента: 5686
Опубликовано: 28.04.2005
Авторы: Кесицки Эдвард А., Джоунс Закари С., Фаулер Керри У., Оливер Эйми, Бэрджесс Лоренс И., Годино Джон Дж., Ньюхаус Брэдли Дж., Одинго Джошуа, Шлахтер Стивен, Мартинс Тимоти Дж.
МПК: C07D 295/20, A61K 31/40
Метки: производные, фосфодиэстеразы, качестве, циклическому, ингибиторов, специфичной, амф, пирролидина
Формула / Реферат:
1. Соединение, имеющее формулу где R1 выбран из группы, включающей водород, C1-6алкил, мостиковый алкил, выбранный из группы, включающей норборнил, адамантил, бицикло[2.2.2]октил, бицикло[3.2.1]гептил, бицикло[3.2.1]октил, бицикло[4.1.0]гептил, бицикло[3.1.0]гексил и декагидронафтил, замещенный или незамещенный арил, выбранный из группы включающей фенил, нафтил, бифенил, тетрагидронафтил, инданил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил,...
Предыдущий патент: Фунгицидная смесь
Следующий патент: Смеси гербицидов, содержащие специальные сульфонилмочевины
Случайный патент: Антибактериальные аналоги аминогликозида