Конденсированные с арилом азаполициклические соединения






Формула / Реферат
1. Соединение формулы

где Z представляет собой CH2;
R1 представляет собой водород, (C1-C6)алкил или бензил;
R2 и R3 выбраны независимо из водорода, гидрокси, нитро, галогено, -SOq(C1-C6)алкила, где q обозначает 0, 1 или 2, CO2R4, арил-(C0-C3)алкила или арил-(C0-C3)алкил-O-, где указанный арил выбран из фенила и нафтила, гетероарил-(C0-C3)алкила или гетероарил-(C0-C3)алкил-O-, где указанный гетероарил выбран из пиридила и пиримидила, и (C0-C6)алкокси-(C0-C6)алкила, где указанный (C0-C6)алкокси-(C0-C6)алкил содержит, по меньшей мере, один атом углерода, и где от 1 до 3 из атомов углерода указанной (C0-C6)алкокси-(C0-C6)алкильной группировки могут быть возможно заменены атомами кислорода, при условии, что любые два таких гетероатома должны быть разделены, по меньшей мере, двумя атомами углерода и где любая из алкильных группировок указанного (C0-C6)алкокси-(C0-C6)алкила может быть возможно замещена атомами фтора в количестве до двух, и где один из атомов углерода каждой из алкильных группировок указанного арил-(C0-C3)алкила и указанного гетероарил-(C0-C3)алкила может быть возможно заменен атомом кислорода, и где каждая из вышеуказанных арильных групп может быть возможно замещена одним или более чем одним заместителем, независимо выбранным из (C1-C6)алкила, возможно замещенного атомами фтора в количестве от 1 до 3, (C1-C6)алкокси, возможно замещенного атомами фтора в количестве до двух;
либо R2 и R3 вместе с атомами углерода, к которым они присоединены, образуют 4-7-членное моноциклическое карбоциклическое кольцо, причем от 1 до 3 из неузловых атомов углерода указанных моноциклических колец могут быть возможно и независимо заменены кислородом;
R4 выбран независимо из водорода и (C1-C6)алкила;
при условии, что (а) по меньшей мере, один из R1, R2 и R3 должен быть иным, чем водород, (б) когда R2 и R3 представляют собой водород, R1 не может представлять собой метил или водород; и (в) в любой из фторзамещенных алкильных или алкоксигруппировок R2 и R3 атом углерода, соседний с гетероатомом, не может быть монофторированным;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где R2 и R3 вместе с бензольным кольцом формулы I не образуют бициклическую или трициклическую кольцевую систему.
3. Соединение по п.1, где один из R2 и R3 представляет собой фторо.
4. Фармацевтическая композиция для применения при снижении привыкания к никотину или при оказании помощи в прекращении или уменьшении употребления табака у млекопитающего, содержащая количество соединения по п.1, которое является эффективным при снижении привыкания к никотину или при оказании помощи в прекращении или уменьшении употребления табака, и фармацевтически приемлемый носитель.
5. Способ снижения привыкания к никотину или оказания помощи в прекращении или уменьшении употребления табака у млекопитающего, при котором указанному млекопитающему вводят количество соединения по п.1, которое является эффективным при снижении привыкания к никотину или при оказании помощи в прекращении или уменьшении употребления табака.
6. Фармацевтическая композиция для лечения расстройства или состояния, выбранного из воспалительного кишечного заболевания, синдрома раздражения кишечника, спастической дистонии, хронической боли, острой боли, спру-целиакии, анастомозита, вазоконстрикции, тревоги, панического расстройства, депрессии, биполярного расстройства, аутизма, расстройств сна, расстройства биоритмов в связи с быстрым перемещением через часовые пояса, бокового амиотрофического склероза (БАС), дисфункции познавательной способности, гипертензии, булимии, анорексии, ожирения, сердечных аритмий, гиперсекреции желудочной кислоты, язв, феохромоцитомы, прогрессивного супрамускулярного паралича, химических зависимостей и привыкания, головной боли, удара, черепно-мозговой травмы (ЧМТ), обсессивно-компульсивного расстройства (ОКР), психоза, хореи Хантингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении, мультиинфарктной деменции, старческого ухудшения познавательной способности, эпилепсии, включая малую эпилепсию, старческой деменции типа Альцгеймера (болезнь Альцгеймера (БА)), болезни Паркинсона (БП), дефицита внимания с гиперактивностью (ДВГ) и синдрома Туретта, у млекопитающего, содержащая количество соединения по п.1, которое является эффективным при лечении такого расстройства или состояния, и фармацевтически приемлемый носитель.
7. Способ лечения расстройства или состояния, выбранного из воспалительного кишечного заболевания, синдрома раздражения кишечника, спастической дистонии, хронической боли, острой боли, спру-целиакии, анастомозита, вазоконстрикции, тревоги, панического расстройства, депрессии, биполярного расстройства, аутизма, расстройств сна, расстройства биоритмов в связи с быстрым перемещением через часовые пояса, бокового амиотрофического склероза (БАС), дисфункции познавательной способности, гипертензии, булимии, анорексии, ожирения, сердечных аритмий, гиперсекреции желудочной кислоты, язв, феохромоцитомы, прогрессивного супрамускулярного паралича, химических зависимостей и привыкания, головной боли, удара, черепно-мозговой травмы (ЧМТ), обсессивно-компульсивного расстройства (ОКР), психоза, хореи Хантингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении, мультиинфарктной деменции, старческого ухудшения познавательной способности, эпилепсии, включая малую эпилепсию, старческой деменции типа Альцгеймера (болезнь Альцгеймера (БА)), болезни Паркинсона (БП), дефицита внимания с гиперактивностью (ДВГ) и синдрома Туретта, у млекопитающего, при котором указанному млекопитающему, нуждающемуся в таком лечении, вводят количество соединения по п.1, которое является эффективным при лечении такого расстройства или состояния.
8. Соединение формулы

где Z представляет собой CH2; P представляет собой водород; -C(=O)H, -C(=O)(C1-C6)алкил, где алкильная группировка может быть возможно замещена атомами галогенов в количестве от 1 до 3; бензил; и R14 и R15 выбраны независимо из водорода, гидрокси, нитро, -O(C1-C6)алкила или галогено; при условии, что R14 и R15 не могут оба представлять собой водород, когда P представляет собой водород, метил или -C(=O)H.
9. Соединение по п.1, выбранное из группы, состоящей из
5,6-дифтор-11-азатрицикло[7.3.1.02,7]тридека-2,4,6-триена;
11-бензил-6-метокси-11-азатрицикло[7.3.1.02.7]тридека-2(7),3,5-триена;
6-метокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен-6-ола;
6-фтор-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
11-бензил-5-метокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
11-бензил-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен-5-ола;
5-метокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен-6-ола;
11-бензил-5-дифторметокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
5-дифторметокси-11-азатрициклo[7.3.1.02,7]тридека-2(7),3,5-триена;
11-бензил-5-этил-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-трихэр;
5-этил-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
5-изопропокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
11-бензил-4-метокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
4-метокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен-4-ола;
11-бензил-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
4-нитро-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
5-нитро-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
3-нитро-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
11-бензил-5-фтор-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
5-фтор-11-азатрициклo[7.3.1.02,7]тридека-2(7),3,5-триена;
5,7-диокса-14-азатетрациклo[10.3.1.02,10.04,8]гексадека-2(10),3,8-триена;
11-бензил-6-бром-5-метокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
11-бензил-6-гидрокси-5-метокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
6-гидрокси-5-метокси-11-азатрициклo[7.3.1.02,7]тридека-2(7),3,5-триена;
11-бензил-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен-5-илового эфира трифторметансульфоновой кислоты;
5-(4-трифторметилфенил)-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
5-(4-метоксифенил)-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена;
метилового эфира 11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен-5-карбоновой кислоты;
2-(11-азатрициклo[7.3.1.02,7]тридека-2(7),3,5-триен-5-ил)пропан-2-ола;
5-пиридин-3-ил-11-азатрициклo[7.3.1.02,7]тридека-2(7),3,5-триена
и их фармацевтически приемлемых солей и оптических изомеров.
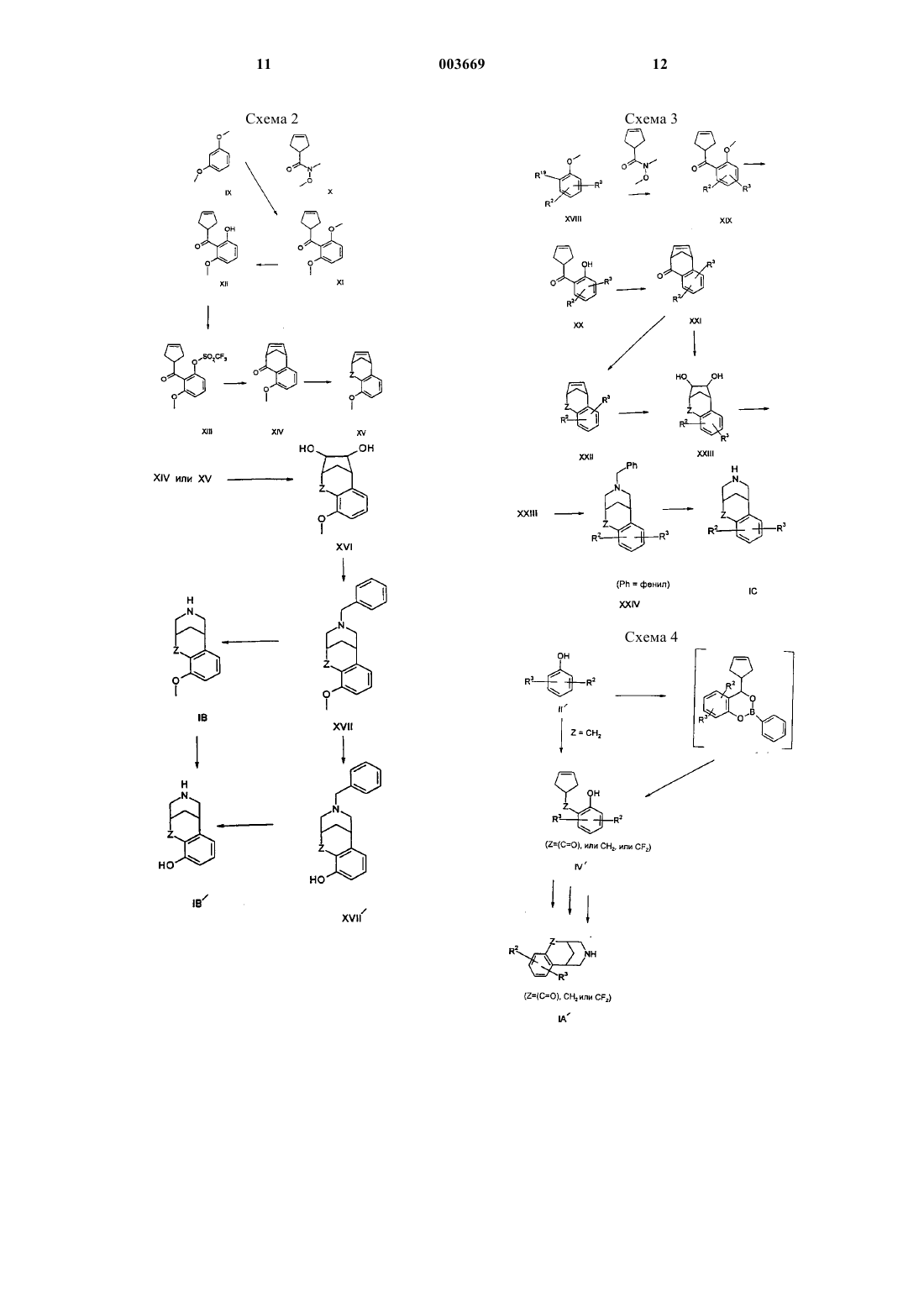
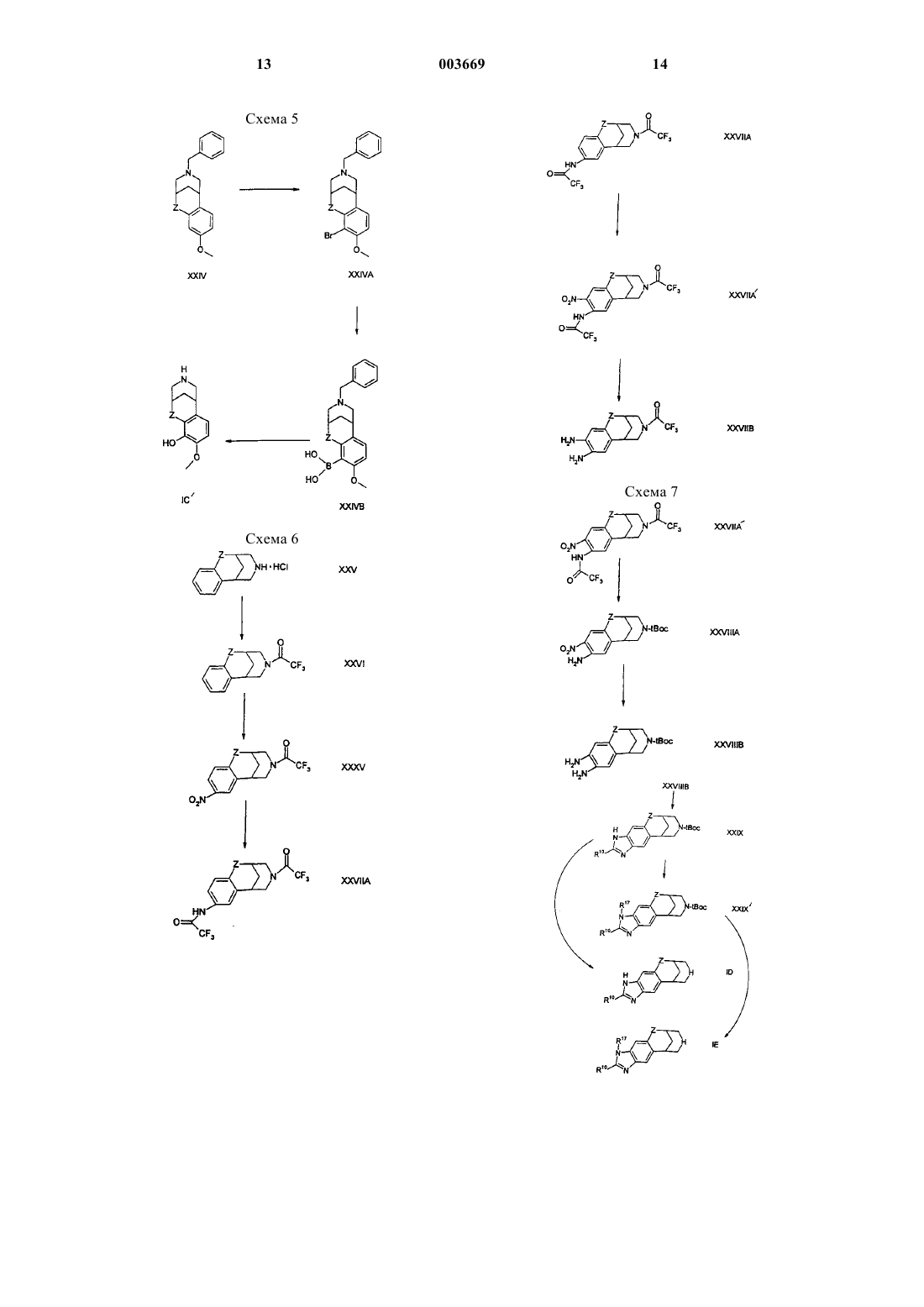
Текст
1 Предпосылки изобретения Данное изобретение относится к конденсированным с арилом азаполициклическим соединениям, как определено более конкретно формулой I ниже. Соединения формулы I связываются с сайтами определенных никотиновых ацетилхолиновых рецепторов нейронов и являются полезными для модулирования холинэргической функции. Такие соединения являются полезными при лечении воспалительного кишечного заболевания (включая неспецифический язвенный колит, гангренозную пиодермию и болезнь Крона, но не ограничиваясь ими),синдрома раздражения кишечника, спастической дистонии, хронической боли, острой боли,спру-целиакии, анастомозита, вазоконстрикции,тревоги, панического расстройства, депрессии,биполярного расстройства, аутизма, расстройств сна, расстройства биоритмов в связи с быстрым перемещением через часовые пояса, бокового амиотрофического склероза (БАС), дисфункции познавательной способности, гипертензии, булимии, анорексии, ожирения, сердечных аритмий, гиперсекреции желудочной кислоты, язв,феохромоцитомы, прогрессивного супрамускулярного паралича, химических зависимостей и привыкания (например, зависимостей или привыкания к никотину (и/или табачным продуктам), алкоголю, бензодиазепинам, барбитуратам, опиоидам или кокаину), головной боли,удара, черепно-мозговой травмы (ЧМТ), обсессивно-компульсивного расстройства, психоза,хореи Хантингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении, мультиинфарктной деменции, старческого ухудшения познавательной способности, эпилепсии, включая малую эпилепсию, старческой деменции типа Альцгеймера (болезнь Альцгеймера (БА,болезни Паркинсона (БП), дефицита внимания с гиперактивностью (ДВГ) и синдрома Туретта. Соединения по данному изобретению можно также использовать в сочетании с антидепрессантом, таким как, например, трициклический антидепрессант или антидепрессант,блокирующий обратный захват серотонина(БОЗС), чтобы лечить как ухудшение познавательной способности, так и депрессии, связанной с БА, БП, ударом, хореей Хантингтона или черепно-мозговой травмой (ЧМТ); в сочетании с мускариновыми агонистами, чтобы стимулировать относящиеся к центральной нервной системе мускариновые, а также никотиновые рецепторы для лечения, например, БАС, дисфункции познавательной способности, старческого ухудшения познавательной способности, БА,БП, удара, хореи Хантингтона и ЧМТ; в сочетании с нейротрофическими факторами, такими как NGF (фактор роста нервной ткани), чтобы максимизировать холинэргическое усиление для лечения, например, БАС, дисфункции познавательной способности, старческого ухудшения познавательной способности, БА, БП, удара, 003669 2 хореи Хантингтона и ЧМТ; либо в сочетании с агентами, которые замедляют или прекращают БА, такими как усилители познавательной способности, ингибиторы агрегации амилоида, ингибиторы секретазы, ингибиторы тау-киназы,нейронные противовоспалительные агенты и эстрогензаместительная терапия. На другие соединения, которые связываются с сайтами никотиновых рецепторов нейронов, есть ссылки в заявке на патент США 08/963852, которая была подана 4 ноября 1997 г., и в предварительной заявке на патент США 60/070245, которая была подана 31 декабря 1997 г. Обе вышеуказанные заявки имеют того же заявителя, что и настоящая заявка, и обе включены сюда в полном объеме путем ссылки. О некоторых других конденсированных с арилом азаполициклических соединениях,включая бензазоцины, структурно родственных соединениям по настоящему изобретению, были сообщения в химической литературе 1960-х и 70-х годов. Chang et al., J. Med. Chem. 14: 10111013 (1971) относится к исследованию соединений 1,5-метано-3-метил-1,2,3,4,5,6-гексагидро 3-бензацина как возможных обезболивающих, о которых, однако, сообщали, что они не проявляют представляющих интерес воздействий на поведение. Kometani et al., Chem. Pharm. Bull.,24: 541-544 (1976) сообщали о получении соединений бензазоцина как побочного продукта при синтезе соединений метанобензазонина. Кроме того, Kitahonoki et al. сообщают в Tetrahedron Letters, 13: 1651-1655 (1968) и с большими подробностями в Tetrahedron, 25: 335-353(1969) о способах получения соединений азиридина, при котором бензазоцины получали в виде второстепенного побочного продукта. О других замещенных соединениях 6,6-диметилом 1,5 метано-3-бензазоцина сообщали в Japanese Kokai Nos. 74-14473 и 74-24968. Краткое изложение сущности изобретения Данное изобретение относится к конденсированным с арилом азаполициклическим соединениям формулыR2 и R3 выбраны независимо из водорода,(С 2-С 6)алкенила, (С 2-С 6)алкинила, гидрокси,нитро, амино, галогено, циано, -SОq(С 1 С 6)алкила, где q обозначает 0, 1 или 2, (С 1 С 6)алкиламино, [(С 1-С 6)алкил]2 амино, CO2R4,CONR5R6, SO2NR7R8, C(=O)R13, XC(=O)R13,арил-(С 0-С 3)алкила или арил-(С 0-С 3)алкил-O-,где указанный арил выбран из фенила и нафти 3 ла, гетероарил-(С 0-С 3)алкила или гетероарил(С 0-С 3)алкил-O-, где указанный гетероарил выбран из 5-7-членных ароматических колец, содержащих от 1 до 4 гетероатомов, выбранных из кислорода, азота и серы, и X2(C0-C6)aлкoкcи(C0-C6)aлкилa, где X2 отсутствует, либо X2 представляет собой (С 1-С 6)алкиламино или [(С 1 С 6)алкил]2 амино, и где (С 0-С 6)алкокси-(С 0 С 6)алкильная группировка указанного Х 2(C0C6)алкокси-(С 0-C6)алкила содержит, по меньшей мере, один атом углерода и где от 1 до 3 из атомов углерода указанной (С 0-С 6)алкокси-(С 0 С 6)алкильной группировки могут быть возможно заменены атомами кислорода, азота или серы при условии, что любые два таких гетероатома должны быть разделены, по меньшей мере,двумя атомами углерода, и где любая из алкильных группировок указанного(С 0 С 6)алкокси-(С 0-С 6)алкила может быть возможно замещена атомами фтора в количестве от 2 до 7,и где один из атомов углерода каждой из алкильных группировок указанного арил-(С 0 С 3)алкила и указанного гетероарил-(С 0 С 3)алкила может быть возможно заменен атомом кислорода, азота или серы, и где каждая из вышеуказанных арильных и гетероарильных групп может быть возможно замещена одним или более чем одним заместителем, предпочтительно заместителями в количестве от 0 до 2,независимо выбранных из (С 1-С 6)алкила, возможно замещенного атомами фтора в количестве от 1 до 7, (С 1-С 6)алкокси, возможно замещенного атомами фтора в количестве от 1 до 7,галогено (например, хлоро, фторо, бромо или иодо), гидрокси, нитро, циано, амино, (С 1 С 6)алкиламино и [(С 1-С 6)алкил]2 амино; либо R2 и R3 вместе с атомами углерода, к которым они присоединены, образуют 4-7 членное моноциклическое или 10-14-членное бициклическое карбоциклическое кольцо, которое может быть насыщенным или ненасыщенным, где от 1 до 3 из неслитых атомов углерода указанных моноциклических колец и от 1 до 5 атомов углерода указанных бициклических колец, которые не составляют часть бензокольца,показанного в формуле I, могут быть возможно и независимо заменены азотом, кислородом или серой, и причем указанные моноциклические и бициклические кольца могут быть возможно замещены одним или более чем одним заместителем, предпочтительно заместителями в количестве от 0 до 2 для моноциклических колец и в количестве от 0 до 3 заместителей для бициклических колец, которые выбраны независимо из(С 0-С 6)алкокси-(С 0-С 6)алкил-, где общее число атомов углерода не превышает шесть и где любая из алкильных группировок может быть возможно замещена атомами фтора в количестве от 1 до 7; нитро, оксо, циано, галогено, гидрокси,амино,(С 1-С 6)алкиламино,[(С 1-С 6)алкил]2 амино, фенила и моноциклического гетероари 003669 4 ла, где указанный гетероарил является таким,как в определении R2 и R3 выше; каждый из R4, R5, R6, R7, R8 и R13 выбран независимо из водорода и (C1-С 6)алкила, либоR5 и R6 или R7 и R8 вместе с атомом азота, к которому они присоединены, образуют пирролидиновое, пиперидиновое, морфолиновое, азетидиновое, пиперазиновое, -N-(С 1-С 6)алкилпиперазиновое, или тиоморфолиновое кольцо, или тиоморфолиновое кольцо, где сера кольца заменена сульфоксидом или сульфоном; и каждый Х представляет собой независимо(С 1-С 6)алкилен; при условии, что (а) по меньшей мере,один из R1, R2 и R3 должен быть иным, чем водород, (б) когда R2 и R3 представляют собой водород, R1 не может представлять собой метил или водород; и (в) ни один из атомов фтора в любой из фторзамещенных алкильных или алкоксигруппировок R2 и R3 не может быть присоединен к атому углерода, который присоединен к гетероатому; а также фармацевтически приемлемые соли таких соединений. Примерами гетероарильных группировок,которые может представлять собой каждый из где один из R9 и R18 представляет собой водород или (С 1-С 6)алкил, а другой представляет собой связь с бензольным кольцом формулыI. Примерами соединений по данному изобретению являются соединения формулы I и их фармацевтически приемлемые соли, где R2 и R3 вместе с бензольным кольцом формулы I образуют бициклическую кольцевую систему, выбранную из следующих: где R10 и R17 выбраны независимо из (С 0 С 6)алкокси-(С 0-С 6)алкила, где общее число атомов углерода не превышает шести и где любая из алкильных группировок может быть возможно замещена атомами фтора в количестве от 1 до 7; (С 1-С 6)алкокси, возможно замещенного 5 атомами фтора в количестве от 1 до 7, нитро,циано, галогено, амино, (C1-С 6)алкиламино,[(С 1-С 6)алкил]2 амино, фенила и моноциклического гетероарила, где указанный гетероарил является таким, как в определении R2 и R3 выше. Другие воплощения данного изобретения относятся к соединениям формулы I и их фармацевтически приемлемым солям, где R2 и R3 вместе с бензольным кольцом формулы I образуют бициклическую или трициклическую кольцевую систему, выбранную из следующих: где R10 и R17 являются такими, как определено выше и m обозначает 0, 1 или 2, и где один из атомов углерода кольца А может быть возможно заменен на кислород или -N(С 1-С 6)алкил. Другие воплощения данного изобретения относятся к соединениям формулы I и их фармацевтически приемлемым солям, где ни R2, ниR3 не присоединен к бензольному кольцу формулы I через атом кислорода. Другие воплощения данного изобретения относятся к соединениям формулы I, где R1 не представляет собой метил. Примерами конкретных соединений формулы I являются следующие: 11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5 триен-5-карбонитрил; 11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5 триен-4-карбонитрил; 1-[11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен-5-ил]-1-этанон; 1-[11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен-5-ил]-1-пропанон; 4-фтop-11-aзaтpициклo[7.3.1.02,7]тpидeкa2(7),3,5-тpиeн-5-кapбoнитpил; 5-фтор-11-азатрицикло[7.3.1.02,7]тридека 2(7),3,5-триен-4-карбонитрил; 1-[11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен-4-ил]-1-этанон; 1-[11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен-4-ил]-1-пропанон;[7.3.1.02,7]тpидeкa-2(7),3,5-тpиeн; и 4,5-диxлop-11-aзaтpициклo[7.3.1.02,7] тpидeкa-2(7),3,5-тpиeн. Данное изобретение также относится к соединениям формулы где Z представляет собой СН 2, С(=O) или-C(=O)NR5R6, где R5 и R6 являются такими, как определено в формуле I выше; -С(=O)Н,-С(=O)(С 1-С 6)алкил, где алкильная группировка может быть возможно замещена атомами галогенов в количестве от 1 до 3, предпочтительно атомами фтора или хлора в количестве от 1 до 3; бензил или трет-бутоксикарбонил (t-Boc); и R14 и R15 выбраны независимо из водорода, гидрокси, нитро, амино, -O(С 1-С 6)алкила или галогено; при условии, что R14 и R15 не могут оба представлять собой водород, когда Р представляет собой водород или метил. Такие соединения являются полезными в качестве промежуточных соединений при синтезе соединений формулы I. Если не указано иначе, термин галогено,как он используется здесь, включает в себя фторо, хлоро, бромо и иодо. Если не указано иначе, термин алкил,как он используется здесь, включает в себя нормальные, разветвленные или циклические, и может включать в себя нормальные и циклические алкильные группировки, а также разветвленные и циклические группировки. 8 Термин алкокси, как он используется здесь, означает алкил-O-, где алкил является таким, как определено выше. Термин алкилен, как он используется здесь, означает алкильный радикал, имеющий два доступных места связывания (например-алкил-), где алкил является таким, как определено выше. Если не указано иначе, термин один или более чем один заместитель, как он используется здесь, относится к числу заместителей от одного до максимального числа, возможного на основании числа доступных мест связывания. Термин лечение, как он используется здесь, относится к возвращению к прежнему состоянию, облегчению, торможению развития или предупреждению расстройства или состояния, к которому применяют данный термин, или одного или более чем одного симптома такого состояния или расстройства. Термин лечение,как он используется здесь, относится к акту лечения, оно определено непосредственно выше. Соединения формулы I могут иметь оптические центры и поэтому могут встречаться в различных энантиомерных конфигурациях. Изобретение включает в себя все энантиомеры,диастереомеры и другие стереоизомеры таких соединений формулы I, а также их рацемические и другие смеси. Настоящее изобретение относится также ко всем меченным радиоактивным изотопом формам соединений формул I. Предпочтительными меченными радиоактивными изотопами соединениями формулы I являются те, где радиоактивные метки выбраны из таких, как 3H,11 С, 14 С, 18F,123I и 125I. Такие меченные радиоактивными изотопами соединения являются полезными в качестве инструментов исследования и диагностики при изучении фармакокинетики метаболизма и в анализах на связывание как у животных, так и у человека. Настоящее изобретение относится также к фармацевтической композиции для применения при снижении привыкания к никотину или при оказании помощи в прекращении или уменьшении употребления табака у млекопитающего,включая человека, содержащей количество соединения формулы I или его фармацевтически приемлемой соли, которое является эффективным при снижении привыкания к никотину или при оказании помощи в прекращении или уменьшении употребления табака, и фармацевтически приемлемый носитель. Настоящее изобретение относится также к способу снижения привыкания к никотину или оказания помощи в прекращении или уменьшении потребления табака у млекопитающего,включая человека, при котором указанному млекопитающему вводят количество соединения формулы I или его фармацевтически приемлемой соли, которое является эффективным при снижении привыкания к никотину или при ока 9 зании помощи в прекращении или уменьшении употребления табака. Настоящее изобретение относится также к способу лечения расстройства или состояния,выбранного из воспалительного кишечного заболевания (включая, но не ограничиваясь ими,неспецифический язвенный колит, гангренозную пиодермию и болезнь Крона), синдрома раздражения кишечника, спастической дистонии, хронической боли, острой боли, спруцелиакии, анастомозита, вазоконстрикции, тревоги, панического расстройства, депрессии, биполярного расстройства, аутизма, расстройств сна, расстройств биоритмов в связи с быстрым перемещением через часовые пояса, бокового амиотрофического склероза (БАС), дисфункции познавательной способности, гипертензии, булимии, анорексии, ожирения, сердечных аритмий, гиперсекреции желудочной кислоты, язв,феохромоцитомы, прогрессивного супрамускулярного паралича, химических зависимостей и привыкания (например, зависимостей или привыкания к никотину (и/или табачным продуктам), алкоголю, бензодиазепинам, барбитуратам, опиоидам или кокаину), головной боли,удара, черепно-мозговой травмы (ЧМТ), обсессивно-компульсивного расстройства (ОКР),психоза, хореи Хантингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении,мультиинфарктной деменции,старческого ухудшения познавательной способности, эпилепсии, включая малую эпилепсию, старческой деменции типа Альцгеймера (БА), болезни Паркинсона (БП), дефицита внимания с гиперактивностью (ДВГ) и синдрома Туретта, у млекопитающего, при котором указанному млекопитающему, нуждающемуся в таком лечении, вводят количество соединения формулы I или его фармацевтически приемлемой соли, которое является эффективным при лечении такого расстройства или состояния. Настоящее изобретение относится также к фармацевтической композиции для лечения расстройства или состояния, выбранного из воспалительного кишечного заболевания(включая неспецифический язвенный колит,гангренозную пиодермию и болезнь Крона, но не ограничиваясь ими), синдрома раздражения кишечника, спастической дистонии, хронической боли, острой боли, спру-целиакии, анастомозита, вазоконстрикции, тревоги, панического расстройства, депрессии, биполярного расстройства, аутизма, расстройств сна, расстройств биоритмов в связи с быстрым перемещением через часовые пояса, бокового амиотрофического склероза (БАС), дисфункции познавательной способности, гипертензии, булимии, анорексии, ожирения, сердечных аритмий,гиперсекреции желудочной кислоты, язв, феохромоцитомы, прогрессивного супрамускулярного паралича, химических зависимостей и привыкания (например, зависимостей или привыка 003669 10 ния к никотину (и/или табачным продуктам),алкоголю, бензодиазепинам, барбитуратам,опиоидам или кокаину), головной боли, удара,черепно-мозговой травмы (ЧМТ), обсессивнокомпульсивного расстройства (ОКР), психоза,хореи Хантингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении, мультиинфарктной деменции, старческого ухудшения познавательной способности, эпилепсии, включая малую эпилепсию, сенильной деменции типа Альцгеймера (БА), болезни Паркинсона (БП),дефицита внимания с гиперактивностью (ГДВ) и синдрома Туретта, у млекопитающего, содержащей количество соединения формулы I или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель. Данное изобретение также относится к фармацевтически приемлемым солям присоединения кислоты соединений формулы I. Примерами фармацевтически приемлемых солей соединений формулы I являются соли соляной кислоты, п-толуолсульфоновой кислоты, фумаровой кислоты, лимонной кислоты, янтарной кислоты, салициловой кислоты, щавелевой кислоты, бромисто-водородной кислоты, фосфорной кислоты, метансульфоновой кислоты, винной кислоты, малата, ди-п-толуол-винной кислоты и миндальной кислоты. Подробное описание изобретения Если не указано иначе, R1-R16, m и Р, а также структурная формула I в реакционных схемах и в обсуждении, которое следует далее,являются такими, как определено выше. Схема 1 18 Схемы 1-13 иллюстрируют способы синтеза соединений формулы I. Схемы 1-4 иллюстрируют такие способы, где замещающие группыR2 и R3 присоединяют до циклизации с образованием трициклического ядра формулы I, которое представлено свободным основанием структурной формулы IA (схема 1) или IC (схема 3),где R2 и R3 представляют собой водород. Схемы 5-13 иллюстрируют способы образования соединений формулы I из исходных веществ, которые содержат такое ядро. Согласно схеме 1 исходное вещество формулы II превращают в соединение формулы II с помощью следующего способа. Исходное вещество формулы II подвергают взаимодействию примерно с 1 экв. сильного основания, такого как н-бутиллитий, в растворителе, таком как безводный ТГФ (тетрагидрофуран), простой эфир или метил-трет-бутиловый эфир, при температуре от примерно -78 до примерно -65 С. Это металлирование осуществляют в течение периода от примерно 10 мин до 5 ч, обычно в течение примерно 2 ч, при температуре, поддерживаемой ниже -65 С. Полученный таким образом анион затем обрабатывают циклопент 3-ен-карбальдегидом в том же растворителе при такой скорости, чтобы температура поддерживалась ниже -65 С. Затем эту реакцию останавливают добавлением реакционной смеси к водной кислой среде и завершают. Затем полученное таким образом соединение формулы III восстанавливают в бензильном положении с помощью действия трифторуксусной кислоты и восстанавливающего агента, такого как триэтилсилан, с образованием соответствующего соединения, имеющего формулу IV. Как правило, эту реакцию проводят в растворителе - хлорсодержащем углеводороде, таком как хлороформ, дихлорэтан (ДХЭ) или метиленхлорид, при примерно комнатной температуре в течение периода от примерно 6 до 24 ч, предпочтительно в течение примерно 18 ч. Затем это соединение формулы IV превращают в соответствующее соединение формулы V с помощью его обработки эквивалентными количествами иодида тетрабутиламмония и трихлорида бора в растворителе - хлорсодержащем углеводороде, таком как хлороформ,дихлорэтан (ДХЭ) или метиленхлорид. Эту реакцию обычно проводят сначала при температуре -78 С, а затем дают этой реакции идти в течение периода времени, составляющего примерно 2 ч, до нагревания до температуры окружающей среды. Полученное в результате соединение формулы V затем подвергают взаимодействию с трифторметансульфоновым ангидридом в растворителе - хлорсодержащем углеводороде, таком как хлороформ, дихлорэтан (ДХЭ) или метиленхлорид, в присутствии основания, такого как пиридин или 3-метилпиридин, с образованием соответствующего эфира трифторметан 19 сульфоновой кислоты формулы VI. Обычно исходная температура реакции составляет примерно -78 С, и для завершения этой реакции ей дают нагреться до комнатной температуры. Затем этот эфир трифторметансульфоновой кислоты формулы VI подвергают взаимодействию в условиях циклизации Хека с получением соответствующего соединения формулыVII. Эту реакцию можно проводить в растворителе или без него. Подходящие растворители включают в себя N,N-диметилформамид(ДМФ), N-метилпирролидон (NМП) и толуол. Подходящими являются температуры, находящиеся в диапазоне от примерно 60 С до примерно 130 С, и эта реакция, как правило, идет в течение периода от примерно 1 до 48 ч. Предпочтительно эту реакцию проводят при температуре примерно 100 С в течение примерно 218 ч. Катализаторы в этой реакции производятin situ обработкой источниками палладия, такими как ацетат палладия (Pd(OAc)2), дихлорид палладия (PdCl2) или палладий в восстановленном состоянии нулевого окисления, таком как палладий на углероде (Pd/C) или трис(дибензилиденацетон)дипалладий(O)(Pd2(dba)3). Можно также использовать аналогичные никелевые катализаторы. Требуемое количество катализатора составляет от примерно 0,1 мол.% до стехиометрического количества. Предпочтительно используют примерно 2-10 мол.% палладиевого или никелевого катализатора. Часто условия, используемые в этих реакциях, включают в себя лиганды, такие как трифенилфосфин или три-о-толилфосфин, либо двузубые лиганды, такие как DPPF, DPPE, DPPB, DPPP(DPP = бис-дифенилфосфин, F = ферроцен, Е = этил, Р = пропан, В = бутан), либо любой из разнообразных хиральных лигандов, таких какBINAP (2,2'-бис(дифенилфосфино)-1,1'-бинафтил), либо арсенатные лиганды, либо бидентантные комбинации этих лигандов с хиральными направляющими группами, такими как,например, оксазолины, хотя включение лигандов не во всех случаях может быть необходимым. Если лиганды используют в сочетании с источниками палладия или никеля, их обычно используют в количествах от примерно 0,5 до примерно 4 мол. экв. палладиевого или никелевого катализатора. Вышеуказанную реакцию проводят в присутствии основания, обычно основания третичного амина, такого как триэтиламин или диизопропилэтиламин. Другие основания, такие как карбонаты или ацетаты (например, карбонат калия, карбонат натрия, ацетат натрия или ацетат калия), могут также обеспечить адекватные или желаемые результаты. В некоторых случаях, проиллюстрированных в экспериментальных примерах, предпочтительно использовать основание третичный амин, как описано выше, в комбинации с каталитической солью ацетатом или карбонатом, как, например, с ацетатом ка 003669 20 лия, в количестве, эквивалентном фосфиновому лиганду, для ускорения реакции. Дополнительной добавкой, которая может быть полезна, является соль галогенид алкиламмония, как, например, хлорид тетрабутиламмония. Эти условия являются общими и основаны на условиях,описанных Jeffrey Т. в J. Chem. Soc., Сhem. Соmmun., 1984, 1287, и Synthesis, 1987, 70. Эти реакции обычно проводят в атмосфере азота или аргона, но может требоваться или не требоваться присутствие кислорода. Взаимодействие соединения формулы VII с тетроксидом осмия и повторным окислителем,таким как N-метилморфолин-N-оксид (NMO) в ацетоне и воде при примерно комнатной температуре дает соответствующее соединение формулы VIII. Затем соединение, имеющее формулу VIII,превращают в желаемое соответствующее соединение формулы IA, используя следующую методику. Во-первых, соединение формулы VIII подвергают взаимодействию с периодатом натрия в смеси хлорированного углеводорода,предпочтительно дихлорэтана (ДХЭ), и воды,или с тетраацетатом свинца в растворителе хлорсодержащем углеводороде при температуре от примерно 0 С до примерно комнатной температуры, с получением диальдегида или промежуточного соединения гликаля. Затем продукт этой реакции подвергают взаимодействию с бензиламином (или аммиаком) и триацетоксиборогидридом натрия. Удалением N-бензильной группы получают желаемое соединение формулы IА. Удаление бензильной группы можно осуществлять, используя способы, хорошо известные специалистам в данной области техники, например, вопервых, возможно подвергая свободное основание взаимодействию с одним эквивалентом кислоты, например соляной кислоты (с образованием соответствующей соли присоединения кислоты), а затем с водородом и гидроксидом палладия в метаноле при примерно комнатной температуре. Альтернативно восстановительное аминирование можно осуществлять in situ следующим образом. Окислительное расщепление диола формулы VIII проводят, используя периодат натрия в водном ТГФ или спирте с образованием промежуточного соединения диальдегида/гликаля, на которое ссылаются выше. Обработкой этого промежуточного соединения избытком бензиламина (или аммиака), гидроксидом палладия и водородом при температуре от примерно комнатной температуры до примерно 70 С получают желаемое соединение формулыIА. Если используемый вышеуказанный способ оставляет бензильную группу на соединении, удаление этой бензильной группы даст желаемое соединение формулы IА. Удаление бензильной группы можно осуществлять, исполь 21 зуя способы, хорошо известные специалистам в данной области техники, например, возможно подвергая свободное основание взаимодействию с одним эквивалентом кислоты, например соляной кислоты (с образованием соответствующей соли присоединения кислоты), а затем с водородом и гидроксидом палладия в метаноле при примерно комнатной температуре. На стадии восстановительного аминирования, описанной выше и повсюду в этом документе, можно также использовать такие альтернативы бензиламину, как аммиак, гидроксиламин, алкоксиамины, метиламин, аллиламин и замещенные бензиламины (например дифенилметиламин и 2- и 4-алкоксизамещенные бензиламины). Их можно использовать в виде свободных оснований или в виде их солей, предпочтительно их солей ацетатов, и можно их последовательно удалять с помощью способов,описанных для каждого из них T.W. Greene andG.M. Wuts Protective Groups in Organic Synthesis, 1991, John WileySons, New York, NY. Методика, описанная выше и проиллюстрированная на схеме 1, является предпочтительной для получения соединений формулы I,где R2 и R3 склонны к взаимодействию с образованием арина, или при другом типе побочной реакции. Описанная выше методика дает соединения формулы IA, где Z представляет собой СН 2. Соединения формулы IA, где Z представляет собой (С=O), можно образовать, используя методику, проиллюстрированную на схеме 1, как описано выше, за исключением того, что соединение формулы III окисляют, а не восстанавливают, в бензильном положении с образованием соединения формулы IV, где Z представляет собой (С=O). Это можно осуществить, используя способы, хорошо известные специалистам в данной области техники, как, например, с помощью обработки реактивом Джонса (раствором хромовой кислоты) в эфире или ацетоне при температуре от примерно 0 С до примерно комнатной температуры. Соединения формулыIA, где Z представляет собой CF2, можно получить подобным образом путем превращения окисленного соединения формулы IV, где Z представляет собой (С=O), в соответствующее соединение формулы IV, где Z представляет собой CF2, а затем продолжения последовательности реакций схемы 1. Это превращение можно осуществить, используя способы, хорошо известные в данной области техники, как, например, с помощью обработки реактивом Лоуссона. Эту реакцию с реактивом Лоуссона обычно проводят в реакционно инертном растворителе, таком как бензол или толуол, при температуре от примерно комнатной до примерно температуры дефлегмации этой реакционной смеси,предпочтительно при примерно температуре дефлегмации. 22 Схема 2 иллюстрирует альтернативный способ получения соединений формулы I. Этот способ является предпочтительным способом для получения таких соединений, где ни R2, ниR3 не склонны к взаимодействию в нежелательной побочной реакции. Согласно схеме 2 соединение формулы IX обрабатывают сильным основанием, таким как н-бутиллитий, при температуре от примерно комнатной до примерно температуры дефлегмации этой реакционной смеси, в растворителе, таком как эфир или третбутилметиловый эфир. Это металлирование происходит в течение периода от примерно 1 до 5 ч, обычно в течение примерно 4 ч, когда реакцию проводят при температуре дефлегмации в простом эфире. Затем полученный анион охлаждают в том же растворителе или в смеси растворителей, как, например, в смеси, содержащей тетрагидрофуран (ТГФ), до температуры примерно -78 С. Затем этот анион можно подвергать взаимодействию с метоксиметиламидом циклопент-3-енкарбоновой кислоты (X) при примерно -78 С, в течение примерно получаса, с завершением реакции, происходящим при нагревании до температуры окружающей среды. Эта реакция дает соединение формулы XI. Затем это соединение формулы XI растворяют в растворителе, таком как метиленхлорид, и обрабатывают трихлоридом бора при примерно-78 С. После периода времени, составляющего примерно 20 мин, эту реакционную смесь оставляют нагреваться до примерно 0 С, и реакция завершена. Затем полученный фенол формулы XII превращают в трифторметансульфоновый эфир с помощью способов, описанных выше для получения соединения формулы XIII. Затем полученный эфир можно превратить в соединение формулы XIV в условиях Хека, как описано выше. Восстановлением соединения формулыXIV при использовании стандартных условий Вольфа-Кишнера получают соединение формулы XV. Эти условия хорошо известны специалистам в данной области техники и включают в себя взаимодействие соединения формулы XIV с гидразином и гидроксидом калия, сначала при температуре примерно 100 С в растворителе,обычно в этиленгликоле или диглиме, а затем при повышении температуры до примерно 180200 С. Можно также использовать восстановления, которые известны в данной области как эквивалентные стандартному восстановлению Вольфа-Кишнера. Соединение формулы XV можно превратить в соединение формулы IB с помощью методики, аналогичной превращению соединений формулы VII в соединения формулы IA в схеме 1. Соответствующее соединение, где оксогруппа заменена на CF2, можно образовать с помощью обработки реактивом Лоуссона или используя другие способы для осуществления этого превращения, которые хорошо известны 23 специалистам в данной области техники, а не восстановлением кетона в соединении формулыXIV. Простые метиловые эфиры можно превратить в соответствующие им фенолы с помощью способов, хорошо известных специалистам в данной области техники. Это можно осуществить с помощью воздействия на соединение формулы IВ или XVII бромисто-водородной кислотой и нагревания полученной смеси до температуры дефлегмации в течение периода времени примерно 1 ч. Этой реакцией получают соответствующий фенол формулы IB' или XVII' соответственно. Альтернативой способам, описанным на схемах 1 и 2 для образования арильных анионов, является использование условий обмена галоген-металл. Например, соединение формулы XVIII, проиллюстрированное на схеме 3, гдеR19 представляет собой бромо или иодо, можно обработать алкиллитиевым основанием, таким как н-бутиллитий, при температуре примерно от-78 до 20 С, обычно при примерно -78 С, с получением арильного аниона формулы Этот анион, полученный в данной реакции,затем можно подвергать взаимодействию с альдегидом, таким как описан на схеме 1, или с подходящим двузамещенным амидом, как описано на схеме 2, с получением соединения формулы XIX. (Скорее чем подвергать взаимодействию соединение формулы XVIII с алкиллитиевым основанием, как описано непосредственно выше, такое соединение можно сначала превратить в реактив Гриньяра (R19MgR19),используя стандартные способы, а затем подвергнуть взаимодействию, как описано выше для соединений формулы XVIII', с получением соединения формулы XIX). Затем полученное соединение формулыIC (схема 3), используя способы, описанные выше для превращения соединений формулы XI в соединения формулы IB (схема 2) и для превращения соединений формулы IV в соединения формулы IA (схема 1). Образование анионов в орто-положении ароматических систем, используемое в методиках синтеза, описанных в данной заявке, включено в общую стратегию синтеза, известную специалистам в данной области техники какDirected Ortho Metalation (DOM) (направленное ортометаллирование). В данной области с этой целью изучен ряд функциональных групп, известных как Directed Metalation Groups (DMGs) 24 зор некоторых из них дан в Snieckus, V. Chem.Rev. 1990, 879. Там, где это применимо, для получения соединений и промежуточных соединений, описанных здесь, можно равно применять иные DMGs, чем те, которые используют в данной работе. Альтернативный способ получения соединений, подобных соединениям формулы V, XII или XX, представлен на схеме 4. При данном способе циклопент-3-енкарбоксальдегид и фенол объединяют с арилборной кислотой и кислотным катализатором, таким как уксусная кислота, возможно замещенная галогенозаместителями в альфа-положении для модулирования кислотности реакции, или с арилбордигалогенидом, который по своей природе в условиях этой реакции будет давать минеральную кислоту, в растворителе, таком как бензол, толуол,диоксан или дихлорметан, предпочтительно в бензоле. Температурой этой реакции обычно является температура дефлегмации или температура, которая дает возможность удаления воды, образующейся при этой реакции, любым из стандартных способов удаления воды, со скоростью, которая дает возможность осуществления желаемой реакции. Удобным является способ,при котором используют ловушки Дина-Старка для удаления воды, образующейся при этой реакции. Обычно эту реакцию проводят в течение периода времени, составляющего 3-48 ч, как правило, 10-24 ч или до тех пор, пока не собирается теоретически рассчитанное количество воды. В это время реакционную смесь освобождают от растворителя, а затем создают для нее такие условия, как описанные выше для восстановления бензильных гидроксильных групп или эфиров, например обработка данного промежуточного соединения трифторуксусной кислотой и восстанавливающим агентом, таким как триэтилсилан. Эту реакцию проводят в растворителе - хлорсодержащем углеводороде, таком как хлороформ, дихлорэтан (ДХЭ) или метиленхлорид, при примерно комнатной температуре в течение периода времени от 6 до 24 ч, предпочтительно 18 ч. Вышеуказанной реакцией получают соединение формулы IV', где Z представляет собой CH2. Соответствующие соединения формулы IV', где Z представляет собой (С=O) и CF2,можно образовать, используя способы, описанные выше для получения соединений формулыIV (схема 1), где Z представляет собой (С=O) или CF2. Полученные соединения формулы IV' (Z представляет собой (С=O), СН 2 или CF2) затем превращают в соответствующее соединение формулы IA', используя способы, описанные выше и изображенные на схеме 1, для получения соединений формулы IA. Схема 5 иллюстрирует способ введения заместителей, таких как бром или кислород, в соединения по изобретению. При обработке 25 соединения формулы XXIV бромом в стандартных условиях, известных специалистам в данной области техники, например в растворителе хлорсодержащем углеводороде, таком как хлороформ, дихлорэтан (ДХЭ) или метиленхлорид,при температуре от примерно 0 С до примерно комнатной температуры, предпочтительно при комнатной температуре, в присутствии основания, такого как ацетат натрия, образуется соответствующее соединение формулы XXIVA. Полученный таким образом бромид (XXIVA) затем можно превратить с помощью способа обмена галоген-металл, описанного выше, в литиевое анионное производное, которое можно затем обработать рядом электрофилов, например триалкилборатами, обычно при температурах, находящихся в диапазоне между -78 и 0 С,с получением соответствующего производного бороновой кислоты формулы XXIVB. Это соединение можно затем превратить в ряд производных, достижимых посредством химии связывания Судзуки (Suzuki), в стандартных условиях, известных специалистам в данной области техники. Альтернативно эти соединения бороновой кислоты можно превратить в соответствующие фенольные производные с помощью взаимодействия с перекисью водорода или N-метилморфолином в растворителе, таком как ТГФ, или с помощью любых других стандартных способов, известных специалистам в данной области техники. Удаление бензильной защитной группы с помощью способов, описанных выше, дает желаемое соединение формулы IС'. Фенолы, полученные, как описано выше, а также в экспериментальном разделе, можно превратить в соответствующие эфиры трифторметансульфоновой кислоты. Эти производные,так же как и бромиды формулы XXIVA, можно использовать для доступа к ряду других заместителей (т.е. других значений R2 и R3), таких как арильные, ацетиленовые и виниловые заместители, а также соответствующие карбонильные эфиры и амиды, с помощью способов, катализируемых палладием и никелем, известных специалистам в данной области, таких как связывания Хека (Heck), Судзуки и Стиля (Stille) и карбонилирования Хека. Кроме того, фенолы можно алкилировать с помощью ряда общепринятых способов с получением эфиров. Кроме того,эфиры можно обрабатывать нуклеофилами, такими как реактивы Гриньяра, с получением соответствующих третичных спиртов. Примеры таких превращений приведены в экспериментальных примерах. Схема 6 иллюстрирует получение некоторых промежуточных соединений, используемых в методике схемы 7. Согласно схеме 6 исходный материал формулы XXV подвергают взаимодействию с трифторуксусным ангидридом в присутствии пиридина с образованием соединения формулы XXVI. Эту реакцию обычно про 003669 26 водят в метиленхлориде при температуре от примерно 0 С до примерно комнатной температуры. Соединение формулы XXVI, когда Z не представляет собой (С=O), затем можно превратить в нитропроизводное формулы XXXV с помощью следующего способа. Соединение формулы XXVI добавляют к смеси 2 или более чем 2 экв. трифторметансульфоновой кислоты(CF3SO2OH) и от 1 до 1,5 экв. азотной кислоты в растворителе хлорсодержащем углеводороде,таком как хлороформ, дихлорэтан (ДХЭ) или метиленхлорид. Полученной смеси дают возможность взаимодействовать в течение примерно от 5 до 24 ч. Обе вышеуказанные реакции,как правило, проводят при температуре, находящейся в диапазоне от примерно -78 до примерно 0 С в течение примерно 2 ч, а затем дают реакционной смеси нагреться до комнатной температуры в течение оставшегося времени. Соединения формулы XXXV, где Z представляет собой (С=O), можно получить окислением аналогичных соединений, где Z представляет собой CH2, как описано Kapur et al., Can. J.XXXV с использованием способов, хорошо известных специалистам в данной области техники, дает соответствующий анилин. Это восстановление можно осуществлять, например, используя водород и палладиевый катализатор,такой как гидроксид палладия, и проводя эту реакцию в метаноле или этаноле при примерно комнатной температуре. Это промежуточное соединение анилин затем превращают в трифторацетамид формулы XXVIIA, как описано выше для получения соединений формулыXXVIIA, как описано выше для получения соединений формулы XXXV, дает соответствующее нитропроизводное формулы XXVIIA'. Обработка этого нитропроизводного формулыXXVIIA' водным бикарбонатом в метаноле или ТГФ при температуре от примерно 20 до примерно 70 С с последующим восстановлением нитрогруппы, как описано выше, дает соответствующее соединение формулы XXVIIB. Согласно схеме 7 соединение формулыXXVIIA' превращают в соответствующее соединение, где защитная группа трифторацетил заменена на защитную группу t-Boc (XXVIIIA),с помощью взаимодействия этого соединения сначала с гидроксидом или карбонатом щелочного металла или щелочно-земельного металла(или аммония), а затем путем взаимодействия выделенного из вышеуказанной реакции продукта с ди-трет-бутилдикарбонатом. Реакцию с гидроксидом или карбонатом щелочного металла или щелочно-земельного металла (или аммония), как правило, осуществляют в водном спирте, диоксане или тетрагидрофуране (ТГФ) 27 при температуре от примерно комнатной температуры до примерно 70 С, предпочтительно при примерно 70 С в течение от примерно 1 до примерно 24 ч. Реакцию выделенного из вышеприведенной реакции с ди-трет-бутилдикарбонатом незащищенного амина или соли такого амина,образованной присоединением кислоты, предпочтительно осуществляют в растворителе, таком как ТГФ, диоксан или метиленхлорид, при температуре от примерно 0 С до примерно комнатной температуры. Эту реакцию можно проводить в присутствии основания или без него. Когда реагентом является соль амина, использование основания является предпочтительным. Полученное соединение формулы XXVIIIА можно превратить в соответствующее диаминопроизводное формулы XXVIIIВ, используя методику, описанную выше для превращения соединений формулы XXVIIA' в соответствующие диаминосоединения формулы XXVIIB. Превращение соединения формулыXXIX можно осуществлять с помощью взаимодействия этого соединения формулы XXVIIIB с соединением формулы где R10 представляет собой водород, (С 1 С 6)алкил, возможно замещенный атомами фтора в количестве от 1 до 7, арил-(С 0-С 3)алкил, где указанный арил выбран из фенила и нафтила,или гетероарил-(С 0-С 3)алкил, где указанный гетероарил выбран из 5-7-членных ароматических колец, содержащих от 1 до 4 гетероатомов,выбранных из кислорода, азота или серы, и где каждая из вышеуказанных арильных и гетероарильных групп может быть возможно замещена одним или более чем одним заместителем,предпочтительно от 0 до 2 заместителей, независимо выбранных из (С 1-С 6)алкила, возможно замещенного атомами фтора в количестве от 1 до 7, (С 1-С 6)алкокси, возможно замещенного атомами фтора в количестве от 1 до 7, и циано. Предпочтительным растворителем для этой реакции является смесь этанола:уксусной кислоты 10:1. Температура этой реакции может находиться в диапазоне от примерно 40 до примерно 100 С. Предпочтительно она составляет примерно 60 С. Другие подходящие растворители включают в себя уксусную кислоту, этанол и изопропанол. Альтернативные способы получения соединений формулы XXIX из соединения формулы XXVIIIB описаны Segelstein et al., Tetrahedron Lett., 1993, 34,1897. Удаление защитной группы t-Вос из соединения формулы XXIX дает соответствующее соединение формулы ID. Эту защитную группу можно удалить, используя способы, хорошо 28 известные специалистам в данной области техники. Например, соединение формулы XXIX можно обработать безводной кислотой, такой как соляная кислота, бромисто-водородная кислота, метансульфоновая кислота или трифторуксусная кислота, предпочтительно соляной кислотой в этилацетате при температуре от примерно 0 до примерно 100 С, предпочтительно от примерно комнатной температуры до примерно 70 С в течение от примерно 1 до 24 ч. Соединение формулы XXIX можно превратить в соответствующее соединение формулы IE с помощью взаимодействия этого соединения с соединением формулы R17Z, где R17 является таким, как выше определен R10, а Z представляет собой уходящую группу, такую как галогено или сульфонат (например хлоро, бромо, иодо, мезилат или тозилат) в присутствии основания, такого как гидрид, гидроксид или карбонат щелочного металла, предпочтительно гидроксида калия, в полярном растворителе,таком как вода, диметилсульфоксид (ДМСО),ТГФ или ДМФ, предпочтительно в смеси ДМСО и воды, а затем удаления защитной группы, как описано выше. Реакцию с R17Z, как правило, проводят при температуре от примерно комнатной температуры до примерно 100 С,предпочтительно при примерно 50 С в течение примерно 5 ч. Последующее удаление защитной группы, как описано выше, дает желаемое соединение формулы IE. Схема 8 иллюстрирует альтернативный способ получения соединений формулы IE из соединения формулы XXVIIIA'. Этот способ является предпочтительным способом получения соединений формулы IE, где R17 представляет собой группу, такую как арил- или гетероарилсодержащая группа, либо, когда R17 невозможно присоединить, как проиллюстрировано на схеме 7, с помощью способов алкилирования или замещения арила. Согласно схеме 8 соединение формулы XXVIIIA' подвергают взаимодействию с подходящим соединением формулыR17NH2 в полярном растворителе, таком как ТГФ, ДМФ или ДМСО, предпочтительно ТГФ,при температуре от примерно комнатной температуры до примерно 100 С, предпочтительно при температуре дефлегмации, от 4 до 18 ч. Эта реакция дает соединение формулы XXX. Затем полученное в результате соединение формулыXXX превращают в соответствующее соединение формулы XXXI восстановлением нитрогруппы до аминогруппы, используя способы,хорошо известные специалистам в данной области техники. На такие способы ссылаются выше при превращении соединений формулыXXVIIA' в соединение формулы XXVIIВ на схеме 6. Закрытие имидазольного кольца с образованием соответствующего соединения формулы XXXII можно затем осуществлять с помощью взаимодействия соединения формулыXXXI из вышеуказанной реакции с соединением формулы(где R10 является таким, как определено выше),как описано выше для превращения соединений формулы XXVIIIB в соединения формулыXXIX. Удаление защитной группы из соединения формулы XXXII дает соответствующее соединение формулы IE. Это можно осуществить,используя способы, хорошо известные специалистам в данной области техники, например как описано выше для образования соединений формулы ID из соответствующих соединений формулы XXIX. Соединения формулы XXVIIIA', которые являются исходными материалами, используемыми в способе схемы 8, можно синтезировать,как изображено на схеме 8 А и описано ниже. Соответствующее соединение IC (схема 3), гдеR2 представляет собой фторо, превращают в его трифторацетамидное производное формулыICTFA, используя способы, описанные выше. Затем такое производное нитруют, как описано выше, или используя другие способы, хорошо известные специалистам в данной области техники, для получения соответствующего нитропроизводного формулы ICTFA'. Последующее удаление трифторацетамидной группы карбонатом или бикарбонатом щелочного металла в метаноле или ТГФ с последующей защитой дитрет-бутилдикарбонатом, как описано выше,дает соответствующее соединение формулыXXVIIIA'. Схема 9 иллюстрирует способ получения соединений формулы IF, где R10 и R17 являются такими, как определены выше. Согласно схеме 9 соединение формулы XXVIIIВ подвергают взаимодействию с соединением формулы(аддукт присоединения этандионбисульфита натрия) в воде или в другом полярном растворителе, таком как ТГФ, ДМФ или ДМСО, предпочтительно в смеси воды и смешиваемого с водой растворителя, такого как ТГФ, в течение примерно 1-4 ч. Температура этой реакции может находиться в диапазоне от примерно 40 до примерно 100 С и предпочтительно составляет примерно температуру дефлегмации. Альтернативно соединение формулыXXVIIIВ можно подвергнуть взаимодействию с соединением формулы(реакция двойной конденсации) в полярном растворителе, таком как ТГФ, вода или уксусная кислота, предпочтительно в смеси воды и ТГФ. Обычно эту реакцию осуществляют при температуре от примерно 40 до примерно 100 С,предпочтительно при температуре дефлегмации,в течение примерно 2-4 ч. Обе вышеописанные методики можно также использовать для превращения соответствующих соединений, где защитная группа t-Boc заменена на другую защитную группу, такую как TFA (например, соединений формулыXXVIIB), в хиноксалины. Затем желаемый хиноксалин формулы IF можно образовать с помощью удаления защиты из соединения, образованного в любой из вышеприведенных реакций, используя способ,описанный выше для превращения соединения формулы XXIX в соединение формулы ID, или способ, описанный выше для удаления TFAгруппы из соединения формулы XXVIIA'. Схема 10 иллюстрирует способ получения соединений формулы I, где R2 и R3 вместе с бензольным кольцом, к которому они присоединены, образуют бензоксазольную кольцевую систему. Такое соединение, где R1 представляет собой водород, изображено на схеме 10 в виде химической формулы IG. Согласно схеме 10 соединение формулы ICTFA', где Y представляет собой нитро или фтор-, подвергают взаимодействию с ацетатом калия или карбоксилатом другого щелочного или щелочно-земельного металла в растворителе, таком как диметилсульфоксид (ДМСО), ДМФ или ацетонитрил,предпочтительно ДМСО. Этой реакции, как правило, дают идти в течение примерно 12-24 ч. Подходящие температуры реакции находятся в диапазоне от примерно 70 до примерно 140 С. Предпочтительно примерно 100 С. Вышеуказанная реакция дает соединение формулы XXXIV, которое можно превратить в желаемое соединение, имеющее формулу IG, с помощью следующей методики. Сначала соединение формулы XXXIV восстанавливают с помощью взаимодействия с водородом и палладиевым или платиновым катализатором, таким как гидроксид палладия в метаноле, при температуре от примерно 0 до примерно 70 С, предпочтительно при примерно комнатной температуре, с образованием соответствующего аминопроизводного. Затем продукт этой реакции подвергают взаимодействию с хлорангидридом кислоты формулы R10COCl или с ангидридом кислоты формулы (R10CO)2O, где R10 представляет собой (С 1-С 6)алкил, либо с соединением формулы R10C(OC2H5)3 в подходящем инертном растворителе, таком как декалин, хлорбензол или 31 ксилолы. Предпочтительной является смесь ксилолов. Эту реакцию обычно проводят при температуре примерно 120-150 С, предпочтительно при примерно 140 С. Когда в качестве реагента используют R10COCl, предпочтительно добавлять в реакционную смесь стехиометрическое количество триэтиламина (ТЭА) или другого основания - органического третичного амина и каталитическое количество пиридинийп-толуолсульфоновой кислоты или пиридинийп-толуолсульфоната (ППТС). Когда в качестве реагента используют R10C(OC2H5)3, предпочтительно добавлять в реакционную смесь каталитическое количество ППТС. Удаление трифторацетильной защитной группы азота дает желаемое соединение формулы IG. Это можно осуществлять, используя способы, хорошо известные специалистам в данной области техники, например, подвергая защищенное соединение взаимодействию с низшим алканолом и водным гидроксидом или карбонатом щелочного или щелочно-земельного металла (или аммония), водным карбонатом натрия,при температуре от примерно 50 до примерно 100 С, предпочтительно при примерно 70 С, в течение примерно 2-6 ч. Схема 11 иллюстрирует получение соединений формулы I, где R1 представляет собой водород, а R2 и R3 вместе с бензольным кольцом, к которому они присоединены, образуют бензотиазольную кольцевую систему. На эти соединения ссылаются в схеме 11 и далее как на соединения формулы IH. Согласно схеме 11 соединение формулы XXV' подвергают взаимодействию с трифторуксусным ангидридом с образованием соответствующего соединения,где атом азота кольца защищен трифторацетильной группой, и полученное в результате соединение, защищенное по атому азота, подвергают затем взаимодействию с 2 экв. трифторметансульфоновой кислоты и 1 экв. азотной кислоты с образованием соответствующего соединения формулы XXXV, где имеется единственный нитрозаместитель на бензольном кольце. Реакцию с трифторуксусной кислотой обычно проводят в присутствии пиридина. Обе вышеприведенные реакции обычно проводят в реакционно инертном растворителе, таком как растворитель хлорсодержащий углеводород,предпочтительно метиленхлорид, при температуре от примерно 0 С до примерно комнатной температуры, предпочтительно при примерно комнатной температуре. Вышеуказанное превращение можно также осуществлять, используя другие способы нитрования, известные специалистам в данной области. Восстановление нитрогруппы до аминогруппы можно осуществлять, как описано выше,с получением соединения формулы XXXV'. Затем соединение формулы XXV' подвергают взаимодействию с галогенангидридом или 32 ангидридом карбоновой кислоты формулыR10COX или (R10CO)2O, где Х представляет собой галогено, и с пиридином, ТЭА или другим основанием - третичным амином, с образованием соединения формулы XXXVI, которое затем можно превратить в желаемое соединение,имеющее формулу XXXVII, с помощью взаимодействия этого соединения с реактивом Лоуссона, который изображен ниже Реакцию с R10COX, где Х представляет собой галогено, или с (R10CO)2O, как правило,осуществляют при температуре от примерно 0 С до примерно комнатной температуры,предпочтительно при примерно комнатной температуре. Реакцию с реактивом Лоуссона, как правило, осуществляют в реакционно инертном растворителе, таком как бензол или толуол,предпочтительно в толуоле, при температуре от примерно комнатной температуры до примерно температуры дефлегмации реакционной смеси,предпочтительно примерно при температуре дефлегмации. Закрытие бензотиазольного кольца и удаление защиты с атома азота с образованием желаемого соединения формулы IН можно осуществлять с помощью взаимодействия соединения формулы XXXVII с феррицианидом калия и гидроксидом натрия в смеси воды и метанола(NaOH/H2O/CH3OH) при температуре от примерно 50 до примерно 70 С, предпочтительно при примерно 60 С, в течение примерно 1,5 ч. Схемы 12 и 13 иллюстрируют способы получения соединений формулы I, где R1 представляет собой водород, a R2 и R3 представляют собой ряд различных заместителей, как определено выше, но не образуют кольцо. Схема 12 иллюстрирует способы получения соединений формулы I, где (а) R1 представляет собой водород, а R2 представляет собойR7R8NO2S-; (б) R1 и R2 оба представляют собой хлоро; и (в) R1 представляет собой водород, а R2 представляет собой R13C(=O)-. Эти соединения указаны на схеме 12 соответственно как соединения формул IJ, IK и IL. Согласно схеме 12 соединения формулы IJ можно получить с помощью взаимодействия соединения формулы XXVI с двумя или более чем двумя эквивалентами галогенсульфоновой кислоты, предпочтительно хлорсульфоновой кислоты, при температуре от примерно 0 С до примерно комнатной температуры. Взаимодействие образованного таким образом производного хлорсульфоновой кислоты с амином, 33 имеющим формулу R7R8NH, где R7 и R8 являются такими, как определены выше, с последующим удалением защитной группы азота,дает желаемое соединение, имеющее формулуIJ. Соединения формулы IK можно получить с помощью взаимодействия соединения формулы XXVI с трихлоридом иода в растворителехлорсодержащем углеводороде с последующим удалением защитной группы азота. Эту реакцию с трихлоридом иода обычно осуществляют при температуре от примерно 0 С до примерно комнатной температуры, и предпочтительно ее осуществляют при примерно комнатной температуре. Подобным образом аналогичные моноили дибромированные или моно- или дииодированные соединения можно получить с помощью взаимодействия соединения XXVI с N-иодсукцинимидом или с N-бромсукцинимидом в растворителе трифторметансульфоновой кислоте с последующим удалением защитной группы атома азота, как описано выше. Взаимодействие соединения XXVI с галогенангидридом кислоты формулы R13COCl или с ангидридом кислоты формулы (R13CO2)O в реакционно инертном растворителе, таком как растворитель - хлорсодержащий углеводород,предпочтительно метиленхлорид, либо без него в присутствии кислоты Льюиса, такой как хлорид алюминия, при температуре от примерно 0 до примерно 100 С с последующим удалением защиты атома азота дает соединение формулыIL. Реакцию с галогенангидридом или с ангидридом кислоты можно осуществлять, используя другие известные кислоты Льюиса или другие способы ацилирований Фриделя-Крафтса, которые известны в данной области техники. Реакции, описанные здесь, в которых NO2,-SO2NR7R8, -COR13, I, Br или Cl вводят в соединение формулы XXVI, как изображено на схеме 12 и описано выше, можно осуществлять с любым аналогичным соединением, где R2 представляет собой водород, (С 1-С 6)алкил, галогено,(С 1-С 6)алкокси или -NHCONR7R8, получая соединения формулы I, где R2 и R3 определены,как в определении соединений формулы I выше. Соединения, которые идентичны таковым формулы IL, но в которых сохраняется защитная группа атома азота, можно превратить в соответствующие O-ацилзамещенные соединения, т.е. те, где группа -C(=O)R13 формулы IL заменена на группу -O-C(=O)R13, используя методы Байера-Виллигера, хорошо известные специалистам в данной области техники. Полученные соединения можно подвергать частичному гидролизу с получением соответствующих гидроксизамещенных соединений, а затем алкилированию с образованием соответствующих алкоксизамещенных соединений. Такие Oацилзамещенные соединения можно также использовать для получения разнообразно замещенных бензизоксазолов, используя способы, 003669 34 хорошо известные специалистам в данной области, такие как последовательное применение перегруппировки Фриса (Fries), образования оксима, ацилирования и обработки основанием. Такой способ сопряжен с осуществлением перегруппировки Фриса соединения формулыXXXIII с помощью обработки кислотой Льюиса, такой как хлорид алюминия (Аl2 Сl3), неразбавленной или в растворителе, таком как хлорбензол, при температуре от примерно 100 до примерно 200 С, предпочтительно при примерно 170 С в течение примерно 1 -2 ч, предпочтительно в течение 2 ч, с получением соединения формулы XXXIX. Расщепление защитной группы дает соответствующее соединение формулыIS. Альтернативно соединение формулы XXXIX можно превратить в его оксим, используя стандартные способы, хорошо известные специалистам в данной области техники, такие как обработка солянокислым гидроксиламином в спирте(например, в метаноле) в присутствии основания, такого как ацетат натрия, при температуре от примерно 20 до примерно 70 С, предпочтительно при примерно 50 С в течение примерно от 5 до 20 ч. Ацилирование этого оскима с использованием способов, хорошо известных специалистам в данной области техники, таких как обработка уксусным ангидридом и пиридином с последующей обработкой выделенного ацилоксима основанием, таким как гидрид натрия, в растворителе, таком как ДМФ, NMП или ДМСО, дает соответствующий защищенный бензизоксазол. Расщепление защитной группы в стандартных условиях, как описано выше, дает желаемое соединение формулы IT. Схема 13 иллюстрирует способы получения соединений формулы I, где (a) R1 представляет собой водород, a R2 представляет собой хлоро; (б) R1 представляет собой водород, а R2 представляет собой циано; (в) R1 представляет собой водород, а R2 представляет собой амино; и (г) R1 представляет собой водород, а R2 представляет собой R13C(=O)N(H)-. Эти соединения указаны на схеме 13 соответственно как соединения формул IM, IN, IP и IQ. Соединения формулы IM можно получить из соединений формулы XXXV' с помощью образования соли диазония, например, с нитритом щелочного металла и сильной минеральной кислотой (например, соляной кислотой, серной кислотой, бромисто-водородной кислотой) в воде с последующим взаимодействием с солью галогенидом меди, такой как хлорид меди (I). Удаление защиты атома азота с помощью способов, описанных выше, дает желаемое соединение формулы IM. Можно использовать также альтернативные способы образования солей диазония, как известные и практикуемые специалистами в данной области техники. Вышеприведенную реакцию, как правило, осуществляют при температурах, находящихся в диапазоне от примерно 0 до примерно 60 С, предпоч 35 тительно примерно 60 С в течение примерно от 15 мин до 1 ч. Взаимодействие этой соли диазония, полученной как описано выше, с иодидом калия в водной среде дает аналогичное производное иодид. Эту реакцию, как правило, осуществляют при температуре от примерно 0 С до примерно комнатной температуры, предпочтительно при примерно комнатной температуре. Полученное в результате соединение или его аналогичную защищенную формуN-третбутилкарбонат можно использовать для получения соответствующего цианопроизводного с помощью взаимодействия с цианидом меди (I) и цианидом натрия в ДМФ, N-метилпирролидоне(NMП), N,N-диметилпропилмочевине (ДМПМ) или ДМСО, предпочтительно в NMП при температуре от примерно 50 до примерно 180 С,предпочтительно при 175 С. Удаление защиты атома азота, как описано выше, дает соответствующее желаемое соединение формулы IN. Вышеуказанное производное иодид, бромид или соль диазония можно также использовать для доступа к ряду других заместителей,таких как арильные, ацетиленовые и виниловые заместители, а также к соответствующим карбонильным эфирам и амидам с помощью процессов, катализируемых палладием или никелем, известных специалистам в данной области техники, таких как Хека, Судзуки и Стилля и карбонилирование Хека. Удаление защиты с атома азота соединения формулы XXXV' дает соединение формулыIP. Соединение формулы XXXV' можно подвергать взаимодействию с ацильной группой,имеющей формулу R13COCl или (R13CO)2O, используя способы, описанные выше, с последующим удалением защиты атома азота с получением соединений формулы IQ. Подобным образом, обработка защищенного амина соединением, имеющим формулу R13SO2X, когда Х представляет собой хлоро или бромо, с последующим удалением защиты атома азота дает соответствующее сульфонамидное производное. Другие подходящие защитные группы амина, которые можно использовать альтернативно в методиках, описанных во всем данном документе, включают в себя -COCF3, -СОССl3,-СООСН 2 СCl3,-СОО(С 1-С 6)алкил и-COOCH2C6H5. В описанных здесь условиях эти группы являются стабильными, и их можно удалить с помощью способов, описанных для каждой из них в Greene Protective Groups inOrganic Chemistry, на которую ссылаются выше. Соединения формулы I, где R1 является иным, чем водород, можно получить, как описано выше, как например образованием кольца восстановительным аминированием, посредством чего образуется соединение XXIV на схеме 3 (R1 = бензил), а также с помощью способов, 003669 36 описанных ниже. Соединения формулы I, где R1 представляет собой водород, можно превратить в соответствующие соединения, где R1 является иным, чем водород, с помощью обработки этих соединений эквивалентным количеством альдегида (R1CHO) или кетона (R1R1CO, где два R1 являются одинаковыми или различными) и восстанавливающего агента, предпочтительно реагента-гидрида, такого как триацетоксиборогидрид натрия или цианоборогидрид натрия, в растворителе, таком как метиленхлорид, тетрагидрофуран или диоксан. В некоторых случаях может быть необходимо добавление кислоты, чтобы способствовать взаимодействию, и обычно используют уксусную кислоту. Температура этой реакции обычно соответствует температуре окружающей среды в течение периода примерно от 0,5 до 24 ч. Используемые обычно способы описаны в J.Org.Chem, 1996, 61, 3849. Соединения формулы I, где R1 является иным, чем водород, можно также получить,подвергая соответствующие соединения, где R1 представляет собой водород, реакции алкилирования, используя способы, хорошо известные специалистам в данной области. Например, соединение, где R1 представляет собой водород,обрабатывают эквивалентным количеством или избытком R1X, где R1 является иным, чем водород, а Х представляет собой галогено, предпочтительно бромо или иодо, или эфир O-сульфата и R1OH. Эту реакцию обычно осуществляют в чистом виде или в полярном растворителе, таком как вода, диметилформамид или диметилсульфоксид, обычно в присутствии основания,такого как, например, не ограничиваясь им, карбонат щелочного металла. Температура этой реакции, как правило, будет находиться в диапазоне от примерно 20 до 120 С (предпочтительно она будет составлять примерно 100 С) в течение периода примерно от 0,1 до 24 ч. Соединения формулы I, где R1 является иным, чем водород, можно также получить с помощью превращения соответствующих соединений, где R1 представляет собой водород, в амиды, подвергая эти соединения взаимодействию с соединением формулы R1C(=O)X, где Х является таким, как определено выше, используя способы, хорошо известные специалистам в данной области техники, а затем восстанавливая полученный в результате амид бором или литий алюмогидридом лития. Стадию восстановления обычно осуществляют в эфирном растворителе,таком как диэтиловый эфир или ТГФ, при температуре от примерно 20 до примерно 70 С в течение примерно от 1 до 20 ч, с получением желаемого амина. В каждой из реакций, обсуждаемых выше или проиллюстрированных на схемах 1-13 выше, давление не является принципиальным, если не указано иначе. Как правило, приемлемым является давление от примерно 0,5 до примерно 5 атм, причем для удобства предпочтительным 37 является давление окружающей среды, т.е. примерно 1 атм. Соединения формулы I и их фармацевтически приемлемые соли (здесь далее активные соединения) можно вводить посредством либо перорального, чрескожного (например, посредством использования пластыря), интраназального, подъязычного, ректального, парентерального, либо местного пути. Предпочтительными являются чрескожное и пероральное введение. Наиболее желательно, чтобы эти соединения вводили в дозировках, находящихся в диапазоне от примерно 0,25 до примерно 1500 мг в сутки,предпочтительно от примерно 0,25 до примерно 300 мг в сутки в виде однократной или дробных доз, хотя варьирование будет неизбежно происходить в зависимости от массы и состояния субъекта, подлежащего лечению, и конкретного выбранного пути введения. Однако наиболее желательно применять уровень дозировки, который находится в диапазоне от примерно 0,01 до примерно 10 мг на кг массы тела в сутки. Тем не менее, возможно варьирование в зависимости от массы и состояния людей, подлежащих лечению, и их индивидуальных ответов на указанное лекарство, а также от типа выбранного фармацевтического препарата и от периода времени и интервала, в течение которых осуществляют такое введение. В некоторых случаях уровни дозировки ниже самого низкого предела вышеуказанного диапазона могут быть более чем адекватны, тогда как в других случаях можно применять еще более высокие дозы, не вызывая каких-либо вредных побочных эффектов, при условии, что такие еще более высокие дозы сначала делят на несколько малых доз для введения на протяжении суток. Активные соединения можно вводить отдельно или в сочетании с фармацевтически приемлемыми носителями или разбавителями посредством любого из нескольких путей, указанных ранее. Более конкретно активные соединения можно вводить в широком разнообразии различных лекарственных форм, например, их можно объединять с различными фармацевтически приемлемыми инертными носителями в форме таблеток, капсул, чрескожных пластырей, лепешек, пастилок, твердых леденцов, порошков, распыляемых растворов, кремов, бальзамов, суппозиториев, желе, гелей, паст, лосьонов, мазей, водных суспензий, инъекционных растворов, эликсиров, сиропов и тому подобного. Такие носители включают в себя твердые разбавители или наполнители, стерильные водные среды и различные нетоксичные органические растворители. Кроме того, пероральные фармацевтические композиции можно подходящим образом подслащивать и/или придавать им вкус и аромат. Как правило, активные соединения присутствуют в таких лекарственных формах на уровнях концентрации, находящихся 38 в диапазоне от примерно 5,0 до примерно 70% мас./мас. Для перорального введения можно применять таблетки, содержащие различные эксципиенты, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, вторичный кислый фосфат кальция и глицин, параллельно с различными разрыхлителями, такими как крахмал (предпочтительно кукурузный, картофельный или маниоковый крахмал), альгиновая кислота и некоторые комплексные силикаты, вместе со связывающими веществами для грануляции типа поливинилпирролидона, сахарозы, желатина и аравийской камеди. Кроме того, для целей прессования можно использовать смазывающие вещества, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердыми композициями подобного типа можно также наполнять желатиновые капсулы; предпочтительные в этом отношении вещества включают в себя также лактозу, или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Когда для перорального введения желательны водные суспензии и/или эликсиры, активный ингредиент можно объединять с различными подслащивающими или корригирующими веществами, красящим веществом и, если желательно, эмульгирующими и/или суспендирующими веществами вместе с такими растворителями, как вода, этанол, пропиленгликоль, глицерин и различные их комбинации. Для парентерального введения можно применять раствор активного соединения либо в кунжутном или арахисовом масле, либо в водном пропиленгликоле. Водные растворы следует подходящим образом забуферить (предпочтительно рН выше 8), если необходимо, и жидкий растворитель сначала сделать изотоническим. Эти водные растворы являются подходящими для целей внутривенных инъекций. Масляные растворы являются подходящими для целей внутрисуставной, внутримышечной и подкожной инъекций. Приготовление всех этих растворов в стерильных условиях легко выполнимо с помощью стандартных фармацевтических методик, хорошо известных специалистам в данной области техники. Активные соединения возможно также вводить местно, и это можно осуществить с помощью кремов, пластыря, желе, гелей, паст,мазей и тому подобного в соответствии со стандартной фармацевтической практикой. Биологический тест Эффективность активных соединений в подавлении связывания никотина с сайтами определенных рецепторов определяли с помощью следующей методики, которая представляет собой модификацию способов Lippello, P.M. иand 3H-Methylcarmbamylcholine In Rat Brain,European J. Pharm., 253, 261-67 (1994. Методика Самцов крыс Sprague-Dawley (200-300 г) из Charles River содержали группами в подвесных проволочных клетках из нержавеющей стали и поддерживали при 12-часовом световом цикле (световой период с 7 ч утра до 7 ч вечера). Они получали стандартный корм Purina RatChow и свободный доступ к воде. Крыс забивали путем декапитации. Мозги извлекали сразу после декапитации. Препараты мембран из ткани головного мозга готовили согласно способам Lippello, P.M. и Femandez,K.G. (Molec Pharmacol. 29, 448-454 (1986 с некоторыми модификациями. Целые головные мозги извлекали, промывали охлажденным во льду буфером и гомогенизировали при 0 С в 10 объемах буфера (мас./об.), используя BrinkmannPolytron, режим 6, в течение 30 с. Буфер состоял из 50 мМ Трис HCl с рН 7,5 при комнатной температуре. Гомогенат осаждали с помощью центрифугирования (10 мин; 50000g; от 0 до 4 С). Супернатант выливали и мембраны мягко ресуспендировали с помощью Polytron и центрифугировали еще раз (10 мин; 50000g; от 0 до 4 С). После второго центрифугирования мембраны ресуспендировали в аналитическом буфере при концентрации 1,0 г/100 мл. В составе стандартного аналитического буфера входили 50 мМ Трис HCl, 120 мМ NaCl, 5 мМ KCl, 2 мМ MgCl2, 2 мМ CaCl2, и буфер имел рН 7,4 при комнатной температуре. Стандартные анализы осуществляли в аналитических пробирках из боросиликатного стекла. Аналитическая смесь обычно состояла из 0,9 мг мембранного белка в конечном инкубационном объеме 1,0 мл. Готовили три серии пробирок, причем пробирки в каждой серии содержали 50 мкл растворителя, контроля или раствора тестируемого соединения, соответственно. В каждую пробирку добавляли 200 мкл[3H]-никотина в аналитическом буфере, а затем 750 мкл мембранной суспензии. Конечная концентрация никотина в каждой пробирке составляла 0,9 нМ. Конечная концентрация цитизина в контроле составляла 1 мкМ. Растворитель состоял из деионизированной воды, содержащей 30 мкл 1 н. уксусной кислоты на 50 мкл воды. Тестируемые соединения и цитизин растворяли в растворителе. Анализы инициировали интенсивным перемешиванием после добавления мембранной суспензии в пробирку. Образцы инкубировали при температуре от 0 до 4 С в ледяной водяной бане-шейкере. Процессы инкубации заканчивали быстрым фильтрованием в вакууме через фильтры из стекловолокна Whatman GF/B, используя многоколенный тканесборник Brandel. После начальной фильтрации аналитической смеси фильтры промывали два 40 раза охлажденным во льду аналитическим буфером (5 мин каждый). Затем эти фильтры помещали в сцинтилляционные флаконы и энергично перемешивали с 20 мл Ready Safe(Beckman) перед подсчетом радиоактивности. Образцы считали в счетчике жидкостной сцинтилляции LKB Wallach Rackbeta при 40-50% эффективности. Все измерения проводили трижды. Вычисления Специфическое связывание (С) с мембраной представляет собой разность между суммарным связыванием в образцах, содержащих только растворитель и мембрану (А) и неспецифическим связыванием в образцах, содержащих мембрану и цитизин (В), то есть,Специфическое связывание =(С)=(А)-(В) Специфическое связывание в присутствии тестируемого соединения (Е) представляет собой разность между суммарным связыванием в присутствии тестируемого соединения (D) и неспецифическим связыванием (В), т.е. (Е) =% ингибирования - (1-(Е)/(С умножить на 100. Соединения по изобретению, которые тестировали в вышеописанном анализе, демонстрировали значения IC50 менее чем 10 мкМ. Следующие примеры иллюстрируют, но не ограничивают объем данного изобретения. Пример 1. 5,6-Дифтор-11-аза-трицикло[7.3.1.02,7]тридека-2,4,6-триена гидрохлорид. А) Циклопент-3-енил-(2,3-дифтор-6-метоксифенил)метанол. (В качестве ведущих ссылок на металлирование см. пример 6 А. Циклопент 3-енкарбальдегид получали в результате восстановления алюмогидридом лития метоксиметиламида циклопент-3-енкарбоновой кислоты, получение которого можно найти в примере 2 А. Условия восстановления см. в Garigipati R.S.;(80 мл) в сухой 250 мл трехгорлой круглодонной колбе (колба 3NRB) при -78 С в атмосфере азота (N2). К этому добавляли н-бутиллитий (nBuLi) (28 мл, 2,5 М/р-р в гексанах, 70 ммоль) в течение 5 мин. После перемешивания при температуре ниже -70 С в течение 4,5 ч раствор циклопент-3-енкарбальдегида (5,7 г, 69,4 ммоль) в безв. ТГФ (30 мл) добавляли через воронку для добавления по стенке реакционного сосуда,поддерживая внутреннюю температуру ниже-70 С. После перемешивания в течение 1/2 ч реакционную смесь наливали в насыщенный водный раствор хлорида аммония (нас. водн. р-рNH4Cl) (100 мл) и эту смесь перемешивали и экстрагировали этиловым эфиром (Et2O) (250 мл). Органический слой промывали рассолом(50 мл), высушивали (Na2SO4), фильтровали,концентрировали и хроматографировали на си 41 ликагеле с получением масла (6,64 г, 40%).(40 мл) при 0 С. К этому раствору добавляли трифторуксусную кислоту (17,3 мл, 224 ммоль). Эту смесь перемешивали при температуре окружающей среды в течение 18 ч. Эту смесь концентрировали до масла, которое растворяли в гексанах (100 мл), промывали водой (Н 2O) (250 мл) и насыщенным водным раствором бикарбоната натрия (нас. водн. р-р NaHCO3) (50 мл), а затем высушивали (сульфат натрия (Na2SO4,фильтровали, концентрировали и хроматографировали на силикагеле с получением масла(N2). К этому добавляли трихлорид бора (ВСl3)(22 мл, 1 М р-р CH2Cl2, 22 ммоль) в течение 2 мин. Через 5 мин этот раствор оставляли нагреваться до комнатной температуры (к.т.) и перемешивали в течение 2 ч. Эту реакцию останавливали Н 2O (100 мл) и перемешивали в течение 1 ч. Слои разделяли и водн. слой экстрагировали метиленхлоридом (CH2Cl2) (230 мл). Объединенный органический слой промывали H2O(250 мл) и нас. водн. р-ром NаНСО 3 (50 мл),высушивали через ватную пробку, концентрировали и хроматографировали на силикагеле с получением масла (3,30 г, 96%). (ТСХ 50% этилацетат (ЕtOАс)/гексаны (hex) Rf 0,70). 1(d, J=8.0 Гц, 2H), 2.61 (m, 1H), 2.39 (AB dd,J=14.0, 5.4 Гц, 2H). ГХМС m/e 210 (M+). Г) 2-Циклопент-3-енилметил-3,4-дифторфениловый эфир трифторметансульфоновой кислоты. (В качестве ведущей ссылки см. Su, 003669-78 С в атмосфере азота (N2) и обрабатывали трифторметансульфоновым ангидридом (6,20 г,22,0 ммоль) по каплям в течение 20 мин. Эту смесь оставляли нагреваться до к.т. и перемешивали в течение 1/2 ч, затем наливали в 1 н. водн. р-р HCl и встряхивали. Слои разделяли и водн. слой экстрагировали CH2Cl2 (230 мл). Объединенный органический слой промывали Н 2O (50 мл) и нас. водн. р-ром NаНСО 3 (50 мл),высушивали через ватную пробку, концентрировали и хроматографировали на силикагеле с получением масла (4,34 г, 81%). (ТСХ 30% ЕtOАс/Нех Rf 0,60). 1H ЯМР (СDСl3)7.13-7.03 (2H), 5.67 (br s,2H), 2.82 (dd, J=7.5, 2.0 Гц, 2H), 2.58 (m, 1H),2.40 (dd, J=14.0, 8.0 Гц, 2H), 2.05 (dd, J=14.0, 5.5 Гц, 2H). ГХМС m/e 342 (M+). Д) 5,6-Дифтортрицикло[7.2.1.02,7]дoдeкa2(7),3,5,10-тетраен. 2-Циклопент-3-енилметил-3,4-дифторфениловый эфир трифторметансульфоновой кислоты (340 мг, 0,99 ммоль) растворяли в ДМФ(5 мл) в атмосфере N2 и обрабатывали диизопропилэтиламином (0,26 мл, 1,5 ммоль), ацетатом калия (981 мг, 10,0 ммоль) и три-отолилфосфином (12 мг, 0,04 ммоль). Эту смесь перемешивали и дегазировали (3 цикла очистки вакуум/N2), а затем обрабатывали ацетатом палладия (5 мг, 0,02 ммоль). Через 20 мин эту смесь нагревали до 100 С в течение 18 ч, охлаждали и наливали в рассол (50 мл). Полученную в результате смесь экстрагировали гексанами (425 мл) и объединенный органический слой промывали нас. водн. р-ром NaHCO3 (10 мл), водой(сульфат магния (MgSO4, фильтровали и хроматографировали на силикагеле с получением масла (110 мг, 60%). (ТСХ гексаны Rf 0,58). 1 Н ЯМР (CDCl3)6.80 (ddd, J=6.6, 8.1, 8.3 Гц, 1H), 6.68 (m, 1H), 6.17 (dd, J=5.5, 2.8 Гц, 1 Н),5.77 (dd, J=5.5, 2.8 Гц, 1 Н), 3.29 (br s, 1H), 2.96[7.2.1.02,7]дoдeкa-2(7),3,5-тpиeн. 5,6-Дифтopтpициклo[7.2.1.02,7]дoдeкa2(7),3,5,10-тeтpaeн (714 мг, 3,72 ммоль) и Nметилморфолин-N-оксид (553 мг, 4,10 ммоль) перемешивали в ацетоне (20 мл) и H2O (3 мл). К этому добавляли раствор тетраоксида осмия(OsO4) (0,2 мл, 2,5% мас./мас. р-р в третбутаноле (t-BuOH), 0,02 ммоль). Через 18 ч эту смесь концентрировали до масла, растворяли в минимуме CH2Cl2 и фильтровали через прокладку из диоксида кремния (33 мм), элюируя 43 20% ЕtOАс/гексанами. Фракции, содержащие продукт, концентрировали до масла (850 мг,100%). (ТСХ 20% ЕtOАс/гексаны Rf 0,37). 1(ЕtOН) (30 мл) и Н 2 О (10 мл). К этому добавляли раствор периодата натрия (NaIO4) (810 мг,3,72 ммоль) в Н 2O (5 мл). Полученную в результате молочно-белую дисперсию перемешивали 15 мин, затем обрабатывали 37% водн. раствором гидроксида аммония (NH4OH) (25 мл) и гидроксидом палладия (Pd(OH)2) (360 мг, 20% мас./С) и перемешивали при давлении Н 2 310,26420 кПа. Через 18 ч эту смесь фильтровали через целитовую прокладку и промывали ЕtOН и смесью этанол: вода (3:1). Фильтрат концентрировали до маслянистого твердого вещества, которое растворяли в ЕtOАс (50 мл) и промывали нас. водн. р-ром карбоната натрия(Nа 2 СО 3) (220 мл). Органический слой высушивали сульфатом натрия (Na2SO4), фильтровали, концентрировали и хроматографировали на силикагеле с получением масла (330 мг, 42%).APCI MC m/e 209.8 [(М+1)+]. Этот продукт растворяли в метаноле(НСl)/ЕtOАс (3 мл). Полученную суспензию концентрировали, растворяли в минимуме МеОН, насыщенного Et2O, и перемешивали в течение 18 ч. Твердые вещества фильтровали с получением белого твердого вещества (335 мг,86%). Тпл 290-305 С. Пример 2. 11-Бензил-6-метокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена гидрохлорид. А) Метоксиметиламид циклопент-3 енкарбоновой кислоты. (Получение циклопент 3-енкарбоновой кислоты см. в Depres, J.-P.;Greene, A.E. J. Org. Chem. 1984, 49, 928-931 и о более поздних методах см. в a) Nugent, W.A.;Feldman, J.; Calabrese, J.C. J. Am. Chem. Soc. 1995, 117, 8992-8998, и б) Marinez, L.E.; Nugent,W.A.; Jacobsen, E.N. J. Org. Chem. 1996, 61,7963-7966. Подобные способы образования амида см. в Nitz, T.J.; Volkots, D.L; Aldous, D.J.; 44 Циклопент-3-енкарбоновую кислоту (65,5 г, 586 ммоль) в CH2Cl2 (1 л) обрабатывали карбонилдиимидазолом (100 г, 617 ммоль) порциями. Через 3/4 ч полученный в результате раствор обрабатывали N,O-диметилгидроксиламином (60,8 г, 623 ммоль), и эту смесь перемешивали в течение 40 ч. Эту реакцию останавливали 1 н. водн. р-ром HCl (600 мл), встряхивали и разделяли слои. Водный слой экстрагировали CH2Cl2 (2100 мл). Объединенный органический слой промывали 1 н. водн. р-ром HClNa2CO3/рассолом (200 мл) и высушивали через ватную пробку. Фильтрат разбавляли ЕtOАс до 10% EtOAc/CH2Cl2 и фильтровали через прокладку из диоксида кремния (1010 мм), элюируя 10% EtOAc/CH2Cl2 до исчезновения основного цвета. Концентрированием получали жидкость (86 г, 95%). (ТСХ 10% EtOAc/CH2Cl2 Rf 0,56). 1H ЯМР (CDCl3)5.64 (br s, 2H), 3.69 (s,3 Н), 3.47 (m, 1H), 3.19 (s, 3 Н), 2.61 (m, 4H). ГХМС m/e 155 (M+). Б) 2-Циклопент-3-енил-(2,6-диметоксифенил)метанон. (В качестве ведущей ссылки см.Koft, E.R.; Smith, A.B. III. J. Am. Chem. Soc. 1982, 104, 2659). 1,3-Диметоксибензол (31,9 г, 231 ммоль) перемешивали в безводном Et2O (200 мл) при 0 С в атмосфере N2 и обрабатывали нбутиллитием (n-BuLi) (92,5 мл, 2,5 М/р-р в гексанах, 231 ммоль) в течение 5 мин. Этот раствор доводили до флегмообразования в течение 4 ч,затем охлаждали до -78 С. Эту суспензию обрабатывали метоксиметиламидом циклопент-3 енкарбоновой кислоты (35,9 г, 231 ммоль) по каплям в течение 1 ч, затем эту смесь перемешивали в течение 18 ч (охлаждающую баню выпаривали в течение ночи). Эту смесь наливали в 1 н. водн. р-р HCl (200 мл) и встряхивали. Слои разделяли и водн. слой экстрагировалиEt2O (2100 мл). Органический слой промывали Н 2 О (50 мл) и нас. водн. р-ром NаНСО 3 (100 мл), высушивали (Na2SO4), фильтровали через прокладку диоксида кремния и концентрировали до масла (52,6 г, 98%). (ТСХ 10% ЕtOАс/гексаны Rf 0,25). 1m/e 232 (M+). В) Циклопент-3-енил-(2-гидрокси-6-метоксифенил)метанон. (В качестве ведущей ссылки см. Nagaoka, Н.; Schmid, G.; lio, H.; Kishi, Y.CH2Cl2 (200 мл) при -78 С в атмосфере N2 и обрабатывали трихлоридом бора (ВСl3) (273 мл, 1 М р-р CH2Cl2, 273 ммоль) в течение 30 мин. Эту смесь оставляли нагреваться до температуры 45 окружающей среды и обрабатывали дополнительным количеством ВСl3 (41,0 мл, 1 М р-рCH2Cl2, 41,0 ммоль). После того как эту смесь перемешивали в течение 20 мин, ее медленно наливали в Н 2O (300 мл) и перемешивали в течение 30 мин. Слои разделяли, и водн. слой экстрагировали СН 2 Сl2 (250 мл). Объединенный органический слой промывали Н 2O (3100 мл),нас. водн. р-ром NаНСО 3 (100 мл), высушивали через ватную пробку и фильтровали через прокладку из диоксида кремния до исчезновения основного цвета. Концентрирование получали масло янтарного цвета (46,0 г, 93%). (ТСХ 10% ЕtOАс/гексаны Rf 0,50). 1(36,0 г, 453 ммоль) перемешивали в CH2Cl2 (250 мл) при -78 С в атмосфере N2. К этому добавляли раствор трифторметансульфонового ангидрида (75,7 г, 268 ммоль) в CH2Cl2 (100 мл) по каплям в течение 1/2 ч. Эту смесь оставляли нагреваться до температуры окружающей среды, перемешивали в течение 1 ч, затем наливали в 1 н. водн. р-р HCl (250 мл). Эту смесь встряхивали, слои разделяли и органический слой промывали 1 н. водн. р-ром HCl (3150 мл), Н 2 О(2100 мл), нас. водн. р-ром NаНСО 3 (100 мл) и наконец рассолом (100 мл). Этот органический слой высушивали через ватную пробку и концентрировали до масла, которое хроматографировали на силикагеле,элюируя 10% ЕtOАс/гексанами с получением после концентрирования масла (62,5 г, 87%). (ТСХ 10% ЕtOАс/гексаны Rf 0,14). 1 Н ЯМР (CDCl3)7.41 (t, J=8.5 Гц, 1H),6.95 (dd, J=8.5, 1.0 Гц, 2 Н), 5.64 (br s, 2H), 3.86(s, 3 Н), 3.73 (m, 1H), 2.70 (m, 2H), 2.57 (m, 2H). ГХМС m/e 350 (М+). Д) 6-Метокситрицикло[7.2.1.02,7]дoдeкa2(7),3,5,10-тетраен-8-он. (В качестве основных ссылок см. Heck, R.F. Оrg. React. (N.Y.) 1982, 27,345 и Cabri, W.; Candiani, I. Acc. Chem. Res. 1995, 28, 2-7). 2-(Циклопент-3-енкарбонил)-3-метоксифениловый эфир трифторметансульфоновой кислоты (45,0 г, 129 ммоль) растворяли в ДМФ(1,89 г, 19,0 ммоль) и 1,3-бис(дифенилфосфино)пропаном (5,30 г, 12,9 ммоль). Эту смесь перемешивали и дегазировали (3 цикла очистки вакуум/N2), затем обрабатывали ацетатом палладия (1,16 г, 5,14 ммоль). Через 20 мин эту смесь нагревали до 130 С в течение 1 ч, охлаж 003669 46 дали и наливали в рассол (300 мл). Полученную в результате смесь экстрагировали ЕtOАс(4100 мл) и объединенный органический слой промывали нас. водн. р-ром NаНСО 3 (100 мл),H2O (100 мл) и рассолом (100 мл), высушивали(55 г). Это масло хроматографировали на силикагеле с получением продукта в виде белого твердого вещества (12,0 г, 47%). (ТСХ 25% ЕtOАс/гексаны Rf 0,27). 1 Н ЯМР (CDCl3)7.29 (t, J=8.0 Гц, 1H),6.84 (d, J=8.0 Гц, 1H), 6.73 (d, J=8.0 Гц, 1H), 6.63and Fieser, Reagents for Organic Synthesis, (N.Y.),1967, 1, p.435). 6-Meтoкcитpициклo[7.2.1.02,7]дoдeкa2(7),3,5,10-тeтpaeн-8-oн (3,0 г, 15 ммоль) и измельченный КОН (5,05 г, 90 ммоль) нагревали в этиленгликоле (40 мл), пока не образовывался раствор. Эту смесь охлаждали до комнатной температуры, обрабатывали гидратом гидразина(3,0 г, 60 ммоль) и нагревали до флегмообразования в течение 2 ч. Дефлегматор заменяли дистилляционной насадкой и дистилляты собирали при температуре 120-190 С. Эти дистилляты разбавляли Н 2O (100 мл) и экстрагировали ЕtOАс (440 мл). Органический слой промывали Н 2O (430 мл) и рассолом (25 мл), высушивали (МgSO4), фильтровали и концентрировали до масла (2,68 г, 96%). (ТСХ 50%[7.2.1.02,7]додека-2(7),3,5,10-триен. 6-Meтoкcитpициклo[7.2.1.02,7]дoдeкa2(7),3,5,10-тeтpaeн (1,5 г, 8.19 ммоль) и Nметилморфолин-N-оксид (1,06 г, 9,03 ммоль) перемешивали в ацетоне (20 мл) и Н 2O (0,16 мл). К этому добавляли раствор тетраоксида осмия (OsO4) (0,2 мл, 2,5% мас./мас. р-р в третбутаноле (t-BuOH), 0,02 ммоль). Через 2 ч эту смесь разбавляли ЕtOАс (50 мл) и промывали 10% водн. р-ром NaHSO3 (30 мл), H2O (230 мл), нас. водн. р-ром NаНСО 3 (30 мл) и рассолом (30 мл). Органический слой высушивали(Для обсуждения окислительного расщепления Рb(ОАс 4) см. Fieser and Fieser, Reagents for Organic Synthesis, (N.Y.), 1967, 1, p. 549. Условия восстановительного аминирования и ссылки см. в Abdel-Magid et al. J. Org. Chem., 1996, 61,3849; и Mazzocchi et al. J. Med. Chem., 1979, 22,455). 1-Метокси-6,7,8,9-тетрагидро-5 Н-5,8-метанобензоциклогептен-6,7-диол (2,40 г, 11,0 ммоль) перемешивали при 0 С в CH2Cl2 (70 мл) и обрабатывали Рb(ОАс)4 (5,08 г, 11,5 ммоль). Через 2 ч эту смесь фильтровали через целитовую прокладку и промывали СН 2 Сl2 (10 мл). К перемешанному фильтрату добавляли уксусную кислоту (АсОН) (1,97 г, 33,0 ммоль) и бензиламин (1,23 г, 11,5 ммоль). Через 15 мин эту смесь обрабатывали триацетоксиборогидридом натрия(NаВН(ОАс)3) (6,94 г, 33,0 ммоль) и перемешивали в течение 18 ч. Эту смесь наливали в нас. водн. р-р NаНСО 3 (100 мл) и перемешивали в течение 1/2 ч. Слои разделяли и экстрагировали СН 2 Сl2 (250 мл). Органический слой промывали насыщенным (нас.) водным (водн.) р-ром бикарбоната натрия (NаНСО 3) (250 мл), Н 2 О(50 мл), рассолом (50 мл), высушивали через ватную пробку, концентрировали и очищали хроматографией на силикагеле, элюируя 10% ЕtOАс/гексанами с получением продукта в виде масла (1,45 г, 45%). (ТСХ 25% ЕtOАс/гексаныd, J=14.2 Гц, 1 Н), 2.87-2.70 (m, 5H), 2.36-2.23 (m,3 Н), 1.85 (br AB d, J=12.1 Гц, 1H), 1.77 (br АВ d,J=12.1 Гц, 1 Н). Это масло растворяли в минимуме метанола (МеОН), перемешивали и насыщали Et2O. Через 18 ч белые твердые вещества отфильтровывали. 1(525 мг, 1,64 ммоль), формиат аммония (2,07 г,32,0 ммоль) и 10% гидроксид палладия на углероде (Pd(OH)2/C) (200 мг) объединяли в МеОН(30 мл) и подвергали дефлегмации в течение 2 ч. Эту смесь горячей фильтровали через целит и фильтрат концентрировали, а затем получали азеотропную смесь из МеОН (550 мл) с полу 003669 48 чением твердого вещества. Это вещество перекристаллизовывали из МеОН/Et2 О с получением белого твердого вещества (306 мг, 81%). 1(d, J=8.0 Гц, 1 Н), 3.82 (s, 3H), 3.34 (br d, J=13.0 Гц, 1H), 3.11-3.02 (m, 4H), 2.94 (AB d, J=18.3 Гц,1H), 2.87 (AB dd, J=18.3, 6.5 Гц, 1H), 2.41 (br s,1H), 1.91 (AB q, 2H). ГХМС m/e 203 (M+). Тпл 272-274C. Пример 4. 11-Азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен-6-ол. 6-Метокси-11-азатрицикло[7.3.1.02,7]тpидeкa-2(7),3,5-тpиeнa гидрохлорид (55 мг, 0,23 ммоль) доводили до дефлегмации в 48% водн. бромисто-водородной кислоте (НВr) (5 мл). Через 1 ч этот раствор охлаждали и наливали в 1 н. водн. р-р NaOH, доводили до рН 10 и экстрагировали продукт ЕtOАс (340 мл). Органический слой промывали рассолом (50 мл), высушивали(MgSO4) и концентрировали до белого твердого вещества, которое перекристаллизовывали из ЕtOАс/гексанов (20 мг, 46%). 1[7.3.1.02,7]тридека-2(7),3,5-триена гидрохлорид. 3-Фторметоксибензол (15,8 г, 125 ммоль) перемешивали при -78 С в безв. ТГФ (100 мл) и обрабатывали n-BuLi (50 мл, 2,5 М/р-р в гексанах, 125 ммоль) в течение 5 мин. После перемешивания при температуре ниже -70 С в течение 4 ч эту смесь обрабатывали метоксиметиламидом циклопент-3-енкарбоновой кислоты(18,4 г, 119 ммоль) по каплям в течение 1/4 ч. Эту смесь перемешивали при температуре ниже-70 С в течение 1 ч, а затем оставляли нагреваться до температуры окружающей среды в течение 1 ч. Эту смесь наливали в 1 н. водн. р-рHCI (200 мл) и встряхивали. Слои разделяли и водный слой экстрагировали ЕtOАс (3100 мл). Органический слой промывали Н 2O (50 мл), нас. водн. р-ром NаНСО 3 (100 мл) и рассолом (50 мл), высушивали (Na2SO4), фильтровали через прокладку из диоксида кремния и концентрировали до масла (21,0 г, 76%). (ТСХ 30% ЕtOАс/гексаны Rf 0,43). ГХМС m/e 220 (M+). Это вещество превращали в соединение,указанное в заголовке, с помощью способов,описанных в примере 2 В-Ж и в примере 1 Ж. 49 Пример 6. 11-Бензил-5-метокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена гидрохлорид. А) Циклопент-3-енил-(2,5-метоксифенил) метанон. (Для обсуждения обмена галогенметалл см. Parham, W.E.; Bradsher, С.К. Асc.Chem. Res. 1982, 15, 300). 2-Бром-1,4-диметоксибензол (42,2 г, 195 ммоль) перемешивали в Et2O (200 мл) в атмосфере N2 при -78 С. Полученный в результате осадок растворяли добавлением ТГФ (50 мл). К полученному в результате раствору добавлялиn-BuLi (78 мл, 2,5 M в гексанах, 195 ммоль) в течение 10 мин. После перемешивания в течение 10 мин этот желтый раствор обрабатывали метоксиметиламидом циклопент-3-енкарбоновой кислоты (29,15 г, 188 ммоль) в Et2O (50 мл) в течение 10 мин, затем эту смесь перемешивали в течение 18 ч (охлаждающую баню выпаривали в течение ночи). Эту смесь наливали в 10% водн. р-р HCl (400 мл) и встряхивали. Слои разделяли, и водн. слой экстрагировали Et2O (350 мл). Органический слой промывали Н 2O (50 мл),нас. водн. р-ром NаНСО 3 (100 мл), высушивали(Na2SO4), фильтровали через прокладку из диоксида кремния и концентрировали до масла(43,0 г, 99%). (В отдельном эксперименте в вышеуказанной реакции ТГФ успешно заменялиN2. К этому добавляли раствор трифторметансульфонового ангидрида (63,8 г, 226 ммоль) вCH2Cl2 (100 мл) по каплям в течение 1/2 ч. Эту смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч, затем наливали в 1 н. водн. р-р HCl (250 мл). Эту смесь встряхивали, слои разделяли и органический слой промывали 1 н. водн. р-ром HCl 50 Органический слой высушивали через ватную пробку и концентрировали до масла, которое хроматографировали через силикагель, элюируя 10% ЕtOАс/гексанами, с получением масла после концентрирования (55,7 г, 93% за 2 стадии). ГХМС m/е 350 (М+). Г) 5-Метокситрицикло[7.2.1.02,7]дoдeкa2(7),3,5,10-тeтpaeн-8-oн. 2-(Циклопент-3-енкарбонил)-4-метоксифениловый эфир трифторметансульфоновой кислоты (19,09 г, 54,5 ммоль) растворяли в ДМФ(100 мл) в атмосфере N2 и обрабатывали диизопропилэтиламином (10,6 г, 82,0 ммоль), ацетатом калия (1,07 г, 11,0 ммоль) и 1,3 бис(дифенилфосфино)пропаном (2,25 г, 5,46 ммоль). Эту смесь перемешивали и дегазировали (3 цикла очистки вакуум/N2), а затем обрабатывали ацетатом палладия (0,49 г, 2,18 ммоль). После перемешивания в течение 20 мин эту смесь нагревали до 120 С в течение 18 ч, охлаждали и наливали в рассол (300 мл). Полученную в результате смесь экстрагировали ЕtOАс(4100 мл) и объединенный органический слой промывали нас. водн. р-ром NаНСО 3 (100 мл),Н 2O (100 мл), рассолом (100 мл), высушивали(МgSO4), фильтровали, концентрировали и хроматографировали на силикагеле с получением масла (10,4 г, 95%) (элюировали водой/7% мас./мас. ЕtOАс/гексаны). 1C ЯМР (CDCl3) 196.11, 158.87, 145.90,140.34, 130.295, 129.94, 126.14, 119.42, 111.90,55.61, 55.48, 49.08, 45.97. ГХМС m/e 200 (M+). Д) 5-Метокситрицикло[7.2.1.02,7]дoдeкa2(7),3,5,10-тeтpaeн. 5-Meтoкcитpициклo[7.2.1.02,7]дoдeкa2(7),3,5,10-тeтpaeн-8-oн (9,41 г, 47 ммоль) и измельченный гидроксид калия (КОН) (6,17 г, 110 ммоль) нагревали в этиленгликоле (50 мл), пока не образовывался раствор. Эту смесь охлаждали до кг, обрабатывали гидратом гидразина (6 мл,190 ммоль) и нагревали до флегмообразования в течение 2 ч. Дефлегматор заменяли дистилляционной насадкой и дистилляты собирали при температуре 120-190 С. Эти дистилляты разбавляли H2O (100 мл) и экстрагировали ЕtOАс(440 мл). Органический слой промывали Н 2O(MgSO4), фильтровали и концентрировали до масла (8,2 г, 94%). (ТСХ 25% ЕtOAс/гексаны Rf 0,68). 1(Рb(ОАс)4) (35,0 г, 79,0 ммоль). Через 30 мин эту смесь фильтровали через целитовую прокладку и промывали CH2Cl2 (50 мл). К перемешанному фильтрату добавляли АсОН (23,7 г,395 ммоль) и бензиламин (8,50 г, 79,0 ммоль). Через 15 мин эту смесь обрабатывалиNаВН(ОАс)3 (50,2 г, 237 ммоль) и перемешивали в течение 18 ч. Эту смесь наливали в нас. водн. р-р Na2CO3 (100 мл) и перемешивали в течение 1/2 ч. Слои разделяли и экстрагировалиCH2Cl2 (2100 мл). Органический слой промывали нас. водн. р-ром Nа 2 СО 3 (250 мл), Н 2 О (50 мл), а затем рассолом (50 мл), высушивали через ватную пробку и концентрировали до масла. Хроматографией на силикагеле, элюируя 5% ЕtOАс/гексанами, получали продукт в виде масла (9,48 г, 41%). (ТСХ 25% ЕtOАс/гексаны Rf 0,69). 1(dd, J=10.5, 2.0 Гц, 1H), 2.27 (dd, J=10.2, 2.0 Гц,1H), 2.15 (br s, 1H), 1.86 (AB d, J=12.3 Гц, 1H),1.78 (AB d, J=12.3 Гц, 1H). ГХМС m/e 293 (M+). Это вещество растворяли в избытке 1 н.HCl МеОН и концентрировали. Твердые вещества растворяли в минимуме МеОН, перемешивали и насыщали Et2O. После перемешивания в течение 18 ч белые твердые вещества фильтровали (900 мг, 58%). 1[7.3.1.02,7]тpидeкa-2(7),3,5-тpиeн (203 мг, 0,62 ммоль) доводили до дефлегмации в 48% НВr (5 мл). Через 1 ч этот раствор охлаждали и наливали в водн. р-р NH4OH, рН доводили до 9 и экстрагировали продукт ЕtOАс (340 мл). Органический слой промывали рассолом (50 мл), высушивали (МgSO4) и концентрировали до масла.(ТСХ 25% ЕtOАс/гексаны (NН 3) Rf 0,37). Это вещество растворяли в избытке 1 н. HCl в МеОН и концентрировали. Перекристаллизацией изMeOH/Et2O получали твердое вещество (154 мг,80%). 1 Н ЯМР (CDCl3)7.42 (m, 5H), 6.90 (d,J=8.0 Гц, 1H), 6.60 (m, 2H), 4.27 (AB d, J=13.0 Гц, 1H), 4.15 (AB d, J=13.0 Гц, 1 Н), 3.47 (d,J=12.2 Гц, 1H), 3.33-3.15 (5H), 2.86 (d, J=18.0 Гц,1H), 2.52 (br s, 1H), 1.99 (AB d, J=12.5 Гц, 1H),1.88 (AB d, J=12.5 Гц, 1H). Тпл 251-253 С. Пример 8. 5-Метокси-11-азатрицикло(206 мг, 0,63 ммоль) превращали в соединение,указанное в заголовке, с помощью способа, описанного в примере 3, с получением белого твердого вещества (122 мг, 81%). (ТСХ 10%(АВ d, J=18.4 Гц, 1 Н), 2.43 (br s, 1 Н), 2.10 (АВ d,J=13.0 Гц, 1 Н), 1.94 (АВ d, J=13.0 Гц, 1 Н). ГХМС m/е 203 (M+). Тпл 253,5-256 С. Пример 9. 11-Азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен-5-ола гидрохлорид. 5-Метокси-11-азатрицикло[7.3.1.02,7]тpидeкa-2(7),3,5-тpиeнa гидрохлорид (187 мг, 0,78 ммоль) доводили до дефлегмации в 48% НВr (5 мл). Через 1 ч этот раствор охлаждали и наливали в водн. р-р NH4OH, рН доводили до 9 и экстрагировали продукт ЕtOАс (340 мл). Органический слой промывали рассолом (50 мл), высушивали (MgSO4) и концентрировали до твердого вещества. (ТСХ 10% MeOH/CH2Cl2 (NH3)Rf 0,13). Это вещество растворяли в избытке 1 н.HCl в МеОН и концентрировали. Перекристаллизацией из MeOH/Et2O получали твердое вещество (70 мг, 40%). 1(В качестве ведущих ссылок см. Langlois, B.R. J. 53 перемешивали в диоксане (5 мл) и Н 2O (1 мл) при температуре дефлегмации с подключенным баллоном фреона (HCF2Cl). К этому по каплям добавляли 3 н. КОН, так чтобы поддерживать рН 12. Контроль потребления исходного материала осуществляли с помощью ТСХ в течение более 2 ч. Эту реакционную смесь охлаждали, разбавляли Н 2O (40 мл) и экстрагировали EtOAc. Органический слой промывали нас. водн. р-ром(MgSO4), фильтровали и концентрировали до масла (620 мг, 92%). ГХМС m/е 329 (M+). Пример 11. 5-Дифторметокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена гидрохлорид. 11-Бензил-5-дифторметокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триен (620 мг, 1,88 ммоль) превращали в соединение, указанное в заголовке, как описано в примере 3. Соль HCl образовывали, как в примере 9, с получением продукта в виде белого порошка (280 мг, 54%). 1N2 при 0 С в ТГФ (2,5 мл). К этому по каплям добавляли диэтилазодикарбоксилат (259 мг,1,49 ммоль). Через 18 ч эту реакционную смесь концентрировали, разбавляли Et2O (20 мл) и экстрагировали 1% водн. р-ром фосфорной кислоты (Н 3 РO4) (320 мл). Объединенный водный слой экстрагировали Et2O (10 мл), а затем подщелачивали до рН 10 1 н. р-ром NaOH. Продукт экстрагировали ЕtOАс (320 мл), и объединенный органический слой промывали 1 н. рром NaOH (20 мл) и рассолом (20 мл). Этот раствор высушивали (MgSO4), фильтровали и выпаривали до масла (170 мг, 74%). (ТСХ 17%s, 2H), 3.03 (dd, J=7.0, 7.0 Гц, 1H), 2.82-2.68 (4 Н),2.18 (2H), 2.12 (br s, 1H), 1.83 (AB d, J=12.0 Гц,1H), 1.75 (АВ d, J=12.0 Гц, 1H), 1.43 (t, J=7.0 Гц,3 Н). ГХМС m/e 307 (М+). Это вещество растворяли в избытке 1 н. HCl МеОН и концентрировали. Перекристаллизацией из CH2Cl2/Et2O получали твердое вещество (185 мг, 97%). Тпл 200203 С. Пример 13. 5-Этокси-11-азатрицикло(160 мг, ммоль), формиат аммония (220 мг, 3,49 ммоль) и 10% Pd(OH)2/C (100 мг) смешивали в метаноле (МеОН) (5 мл) и нагревали до дефлегмации в течение 15 мин. Эту смесь охлаждали,фильтровали, концентрировали, разбавляли нас. водн. р-ром Nа 2 СО 3 и экстрагировали ЕtOАс(320 мл). Экстракты высушивали (МgSO4),фильтровали и концентрировали до масла (94 мг, 83%). (ТСХ 50% ЕtOАс/гексаны (NН 3) Rf 0,20). 1H ЯМР (CDCl3)6.90 (d, J=9.0 Гц, 1H),6.66 (2 Н), 3.97 (m, 2H), 3.08 (dd, J=18.0, 6.0 Гц,1 Н), 2.94 (m, 3H), 2.76-2.65 (3H), 1.96 (m, 2H),1.88 (d, J=11.0 Гц, 1H), 1.38 (t, J=7.0 Гц, 3H). Это вещество растворяли в избытке 1 н. HCl МеОН и концентрировали. Перекристаллизацией изCH2Cl2/Et2O получали твердое вещество (74 мг,68%). Тпл 243-245 С. Пример 14. 5-Изопропокси-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена гидрохлорид. 11-Бензил-11-азатрицикло[7.3.1.02,7]тpидeкa-2(7),3,5-тpиeн-5-oл (208 мг, 0,75 ммоль) и изопропиловый спирт (90 мг, 1,49 ммоль) превращали в соединение, указанное в заголовке,как описано в примерах 12 (ТСХ промежуточного бензильного соединения,17% ЕtOАс/гексаны Rf 0,78). ГХМС m/e 321 (М+). Удалением защиты и превращением в соль, как описано в примере 13, получали твердое вещество (83 мг, 42% общий выход). (ТСХ соединения, указанного в заголовке, ТСХ 50% ЕtOАс/гексаны (NН 3) Rf 0,10). 1A) 2-Циклопент-3-енилметил-5-метоксифенол. (В качестве ведущих ссылок см. a) Nagata, W.; Okada, К.; Aoki, Т. Synthesis 1979, 365368; b) Lau, C.K.; Williams, H.W.R.; Tardiff, S.;Chem. 1989, 67, 1384). 3-Метоксифенол (5,12 г, 42,0 ммоль), циклопент-3-енкарбальдегид (8,00 г, 83,0 ммоль),фенилбороновую кислоту (5,58 г, 46 ммоль) и 1,1,1-трихлоруксусную кислоту (2,04 г, 12,5 ммоль) подвергали дефлегмации в бензоле (150 мл) в течение 18 ч. (ТСХ 5% СН 2 Сl2/гексаны Rf 0,47). Эту смесь концентрировали до масла, которое размешивали при 0 С в СН 2Cl2 (100 мл) и обрабатывали триэтилсиланом (8,87 г, 76,0 ммоль), а затем трифторуксусной кислотой (36,3 г, 318 ммоль). Эту смесь перемешивали в течение 1 ч, затем нагревали до дефлегмации в тече 55 ние 24 ч. Эту смесь концентрировали, растворяли в CH2Cl2 (200 мл) и промывали нас. водн. рром NаНСО 3 (350 мл). Объединенный органический слой высушивали через ватную пробку,концентрировали и хроматографировали на силикагеле с получением масла (3,85 г, 45%).(0,91 г, 8,92 ммоль) и 1,3-бис(дифенилфосфино) пропаном (0,37 г, 0,89 ммоль). Эту смесь перемешивали и дегазировали (3 цикла очистки вакуум/N2), а затем обрабатывали ацетатом палладия (93 мг, 0,42 ммоль). После перемешивания в течение 20 мин эту смесь нагревали до 100 С в течение 18 ч, охлаждали и наливали в рассол (30 мл). Полученную в результате смесь экстрагировали ЕtOАс (410 мл) и объединенный органический слой промывали нас. водн. р-ромNаНСО 3 (10 мл), H2O (10 мл), рассолом (10 мл),высушивали (МgSO4), фильтровали и выпаривали до масла. Это масло хроматографировали на силикагеле (2% СН 2 Сl2/гексаны) с получением продукта в виде масла (1,05 г, 95%). (ТСХ 10% ЕtOАс/гексаны Rf 0,52). 1(ТСХ 50% ЕtOAc/СН 2 Сl2 Rf 0,45) согласно методике, описанной в примере 2 Ж. Это вещество превращали в соединение, указанное в заголовке, согласно методикам, описанным в примере 23, с конечной перекристаллизацией из Еt2O/гексанов (650 мг, 48%). (ТСХ 50%(525 мг, 1,60 ммоль) превращали в соединение,указанное в заголовке, с помощью способов,описанных в примере 3, с получением белого твердого вещества (336 мг, 88%). (ТСХ 40%C ЯМР (CDCl3) 158.47, 136.58, 130.15,127.71, 114.11, 112.61, 54.32, 49.99, 49.47, 32.16,31.97, 27.15, 25.70. Тпл 259-261 С. Пример 17. 11-Aзатрицикло[7.3.1.02,7] тридека-2(7),3,5-триен-4-ол. 4-Метокси-11-азатрицикло[7.3.1.02,7]тpидeкa-2(7),3,5-тpиeнa гидрохлорид (120 мг, 0,50 ммоль) доводили до дефлегмации в 48% НВr (2 мл). Через 1 ч раствор охлаждали и наливали в 1 н. водн. р-р NaOH, pH доводили до 10 и экстрагировали продукт ЕtOАс (340 мл). Органический слой промывали рассолом (50 мл), высушивали (MgSO4) и концентрировали до белого твердого вещества, которое перекристаллизовывали из Еt2O/гексанов (40 мг, 42%). (ТСХ 50% EtOAc/CH2Cl2 Rf 0,15). 1[7.3.1.02,7]тридека-2(7),3,5-триена гидрохлорид. Соединение, указанное в заголовке, получали из фенола согласно методикам, описанным в примере 15. (ТСХ 10% ЕtOАс/гексаны (NН 3)(трифторуксусная кислота) (1,90 мл, 14,2 ммоль). Через 1 ч этот раствор наливали в 0,5 н.HCl (200 мл) и разделяли слои. Водный слой экстрагировали CH2Cl2 (350 мл) и объединенный органический слой промывали 0,5 н. HClNaHCO3 (50 мл). Этот раствор высушивали через ватную пробку, затем разбавляли 3% ЕtOАс и фильтровали через 5,08 см прокладку из диоксида кремния, элюируя 3% EtOAc/CH2Cl2. Концентрированном получали прозрачное масло (1,90 г, 99%). 1(AB d, 2H). ГХМС m/e 269 (М+). Б) Нитро-11-aзaтpициклo[7.3.1.02,7]тpидeкa-2(7),3,5-тpиeнa гидрохлорид. Соединение, указанное в заголовке, получали следующим образом на основании способа,описанного Coon et al., J. Org. Chem., 1973, 25,4243. К раствору трифторметансульфоновой кислоты (0,94 мл, 10,6 ммоль) в CH2Cl2 (10 мл),перемешанному при 0 С, медленно добавляли азотную кислоту (0,60 мл, 14,1 ммоль) в CH2Cl2(10 мл), образуя белый осадок. Через 10 мин полученную в результате смесь охлаждали до(15 мл) по каплям в течение 5 мин. Эту реакционную смесь перемешивали при -78 С в течение 2 ч, затем нагревали до 0 С в течение 1/2 ч. Эту реакционную смесь наливали в перемешанный лед (50 г). Слои разделяли, и водный слой снова экстрагировали CH2Cl2 (330 мл). Органический слой объединяли и промывали Н 2O (330 мл). Объединенный органический слой промывали нас. водн. р-ром NаНСО 3 (20 мл) и H2O (20 мл),затем высушивали через ватную пробку и концентрировали до желтого твердого вещества(1,58 г), которое содержало четыре продукта(ТСХ). Эти твердые вещества суспендировали вEt2O и фильтровали с получением твердого вещества (900 мг, 41%). (ТСХ 30% ЕtOАс/гексаныRf 0,21). Этот фильтрат хроматографировали на силикагеле, элюируя 30% ЕtOАс/гексанами, с получением трех веществ. Rf 0,32 (50 мг, 2%), Rf 0,21 (как вышеуказанные твердые вещества) и[7.3.1.02,7]тридека-2(7),3,5-триен-11-ил)этанон по 4% ЯЭО между Н-3 и Н-1. Это твердое вещество (780 мг, 2,48 ммоль) перемешивали в МеОН (20 мл) и обрабатывали Na2CO3 (650 мг,4,96 ммоль) в H2O (10 мл). Эту перемешанную смесь нагревали до 70 С в течение 6 ч, концентрировали до твердых веществ, разбавляли H2O и экстрагировали CH2Cl2 (340 мл). Этот продукт экстрагировали 1 н. водн. р-ром HCl (340 мл) и экстракт промывали ЕtOАс, затем нейтрализовали нас. водн. р-ром Na2CO3 до рН 10. Этот продукт экстрагировали CH2Cl2 (340 мл),высушивали через ватную пробку, концентрировали до масла. Это масло растворяли в МеОН и обрабатывали 3 н. HCl ЕtOАс (4 мл) и концентрировали, затем растворяли в минимумеCH2Cl2, и этот раствор насыщали гексанами и перемешивали в течение 18 ч. Этот продукт собирали фильтрованием (145 мг, 23%). 1[7.3.1.02,7]тридека 2(7),3,5-триена гидрохлорид. Другой метазамещенный изомер из вышеуказанных, 2,2,2-трифтор-1-(5-нитро-11-азатрицикло[7.3.1.02,7]тpидeкa-2(7),3,5-тpиeн-11-ил) этанон (ТСХ 30% ЕtOАс/гексаны Rf 0,13), превращали в соединение, указанное в заголовке, с помощью способа, описанного в примере 20 В. 1H ЯМР свободного основания (СDСl3)8.01 (d, J=2.0 Гц, 1 Н), 7.95 (dd, J=8.0, 2.0 Гц,1H), 7.17 (d, J=8.0 Гц, 1 Н), 3.16 (dd, J=18.0, 6.5 Гц, 1H), 3.10-2.97 (4 Н), 2.89 (d, J=18.0 Гц, 1H),2.79 (d, J=12.0 Гц, 1H), 2.12 (m, 1H), 2.02 (d,J=12.5 Гц, 1H), 1.88 (d, J=12.5 Гц, 1 Н). Превра 59 щением в соль, как в примере 20 В, получали твердое вещество с Тпл 245-255 С. Пример 22. 3-Нитро-11-азатрицикло[7.3.1.02,7]тридека-2(7),3,5-триена гидрохлорид. Оставшийся выделенный изомер из вышеуказанных,2,2,2-трифтор-1-(3-нитро-11-aзaтpициклo[7.3.1.02,7]тpидeкa-2(7),3,5-тpиeн-11 ил)этанон (ТСХ 30% ЕtOАс/гексаны Rf 0,32) (50 мг), превращали в соединение, указанное в заголовке, с помощью способа, описанного в примере 20 В с получением 25 мг, 64%. Стереохимию данного нитроизомера устанавливали с помощью ГМКК (гетероядерной многоквантовой корреляции) между С-3 и Н-1. 1(310 мг, 0,94 ммоль) превращали в соединение,указанное в заголовке, с помощью способов,описанных в примере 3, с получением белого твердого вещества (140 мг, 65%). 1Chu, P.S.; Lin, C.M.; Hamel, E. J. Med. Chem. 1992, 35, 1058-1067) превращали в соединение,указанное в заголовке, используя способы, описанные в примере 3 и в примере 6, с получением белого твердого вещества (110 мг). 1[7.3.1.02,7]тpидeкa-2(7),3,5-тpиeн (3,00 г, 10,2 ммоль) перемешивали при 0 С в CH2Cl2 (10 мл) и АсОН (5 мл) и обрабатывали бромом (3,21 г,20 ммоль) в CH2Cl2 (10 мл) и АсОН (5 мл). Через 18 ч эту реакцию останавливали 20% водн. р-ром NaHSO3 (100 мл). Продукт экстрагировали CH2Cl2 (340 мл) и промывали нас. водн. рром NaHCO3 (350 мл). Объединенный органический слой высушивали через ватную пробку,концентрировали и хроматографировали на силикагеле с получением масла (1,05 г, 28%).d, J=13.0 Гц, 2H). ГХМС m/e 373, 371 (М+). Пример 27. 11-Бензил-6-гидрокси-5 метокси-11-азатрицикло[7.3.1.02,7]тридека 2(7),3,5-триен. 11-Бензил-6-бром-5-метокси-11-азатрицикло[7.3.1.02,7]тpидeкa-2(7),3,5-триен (1,05 г, 2,70 ммоль) перемешивали при -78 С в безв. ТГФ (10 мл) и обрабатывали n-BuLi (1,08 мл, 2,5 М р-р в гексанах, 2,70 ммоль) по каплям в течение 1 мин. Через 10 мин добавляли триизопропилборат (559 мг, 2,97 ммоль) и эту смесь оставляли нагреваться до температуры окружающей среды. Реакцию останавливали добавлением нас. водн. р-ра NаНСО 3 (50 мл) и экстрагировали продукт ЕtOАс (320 мл). Органический слой высушивали (МgSO4), фильтровали и выпаривали с получением масла (640 мг, 67%). (ТСХ 30% ЕtOАс/гексаны Rf 0,18). Этот материал (640 мг,1,81 ммоль) перемешивали в ТГФ (10 мл) с 30% водн. р-ром перекиси водорода (205 мг, 1,81 ммоль). Через 18 ч эту реакцию останавливали добавлением 20% водн. р-ра NаНSO3 (10 мл). Эту смесь разбавляли нас. водн. р-ром NаНСО 3(50 мл) и продукт экстрагировали CH2Cl2 (340 мл). Органический слой промывали нас. водн. рром NаНСО 3 (350 мл), высушивали через ватную пробку, концентрировали и хроматографировали на силикагеле с получением масла (360 мг, 64%). (ТСХ 40% ЕtOАс/гексаны Rf 0,44). 1 Н ЯМР (CDCl3)7.14 (3 Н), 6.95 (2 Н), 6.67(58 мг, 0,18 ммоль) превращали в соединение, указанное в заголовке, согласно методике, описанной в примере 3, с последующим превращением в соль, как описано в примере 9, с получением белого твердого вещества (15 мг, 32%). (ТСХ 10% MeOH/CH2Cl2 (NН 3) Rf 0,26).
МПК / Метки
МПК: A61K 31/435, A61P 25/00, C07D 221/22
Метки: соединения, арилом, конденсированные, азаполициклические
Код ссылки
<a href="https://eas.patents.su/30-3669-kondensirovannye-s-arilom-azapoliciklicheskie-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Конденсированные с арилом азаполициклические соединения</a>
Азаполициклические соединения, конденсированные с арилом

Номер патента: 3190
Опубликовано: 27.02.2003
Авторы: Коэ Джотэм Водзворт, Брукс Пейдж Роэнн Палмер
МПК: C07D 221/22, A61K 31/439, A61P 25/00...
Метки: азаполициклические, соединения, конденсированные, арилом
Формула / Реферат:
1. Соединение формулы где R1 представляет собой водород, (C1-C6)алкил, неконъюгированный (C3-C6)алкенил, XC(=O)R13, бензил или -CH2CH2-O-(C1-C4) алкил; R2 и R3 независимо выбирают из водорода, (C2-C6)алкенила, (C2-C6)алкинила, гидрокси, нитро, амино, гало, циано, -SOq(C1-C6)алкила, где q равно нулю, одному или двум, (C1-C6)алкиламино-, [(C1-C6)алкил]2амино-, -CO2R4, CONR5R6, -SO2NR7R8, -C(=O)R13, -XC(=O)R13, арил-(C0-C3)алкила- или...
Четырехциклические конденсированные соединения с гетероатомами, замещенные арилом, промежуточные соединения, способы получения, композиции и методы лечения

Номер патента: 1649
Опубликовано: 25.06.2001
Автор: Гриз Тимоти А.
МПК: A61K 31/35, C07D 471/00, A61P 5/30...
Метки: методы, промежуточные, арилом, замещенные, композиции, лечения, соединения, конденсированные, способы, гетероатомами, получения, четырехциклические
Формула / Реферат:
1. Соединение формулы I или II где Y обозначает -О-, -S-, -СН2-, -СН2СН2-, -СН=СН- или -NR4-; В обозначает -СН2- или -СО-; R1 и R2 обозначают каждый независимо -Н, -ОН, -O(C1-C4алкил), -ОСОС6Н5, -ОСО(С1-С6алкил), -ОSО2(С4-С6алкил), OSO2СF3, Сl или F; n равно 1 или 2; W обозначает -СН2- или >С=O; R3 обозначает 1-пиперидинил, 2-оксо-1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 2-оксо-1-пирролидинил,...
Конденсированные производные имидазола в качестве модуляторов множественной лекарственной устойчивости

Номер патента: 1004
Опубликовано: 28.08.2000
Авторы: Ленартс Йозеф Элизабет, Жансенс Франс Эдуард, Сюрлеро Доминик Луи Нестор Гилейн, Соммен Франсуа Мария
МПК: C07D 487/04, A61P 35/00, A61K 31/55...
Метки: множественной, лекарственной, конденсированные, устойчивости, имидазола, производные, модуляторов, качестве
Формула / Реферат:
1. Соединение формулы (I) его N-оксидная форма, фармацевтически приемлемая аддитивная соль или стереохимически изомерная форма, где пунктирная линия обозначает простую связь, n равно 1 или 2; R1 обозначает водород; галоген; формил; C1-4 алкил; C1-4 алкил, замещенный 1 или 2 заместителями, каждый из которых независимо выбран из гидрокси, C1-4 алкокси, C1-4 алкилкарбонилокси, имидазолила, тиазолила или оксазолила; или радикал формулы ...
Фармкомпозиция, содержащая соединения с анти-ха активностью и соединения антагониста агрегации тромбоцитов, их применение, набор, содержащий эти соединения, способ лечения или профилактики заболеваний, сопутствующих тромбообразованию

Номер патента: 2475
Опубликовано: 27.06.2002
Авторы: Перроне Марк Х., Юзан Андре, Кюродо Алан Х., Данвидди Кристофер Т., Лидли Роберт Дж.
МПК: A61P 7/02, A61K 31/715
Метки: сопутствующих, заболеваний, применение, анти-ха, эти, профилактики, лечения, активностью, фармкомпозиция, антагониста, тромбоцитов, соединения, набор, содержащая, содержащий, тромбообразованию, агрегации, способ
Формула / Реферат:
1. Фармацевтическая композиция, содержащая фармацевтически пригодный носитель и фармацевтически эффективные количества соединения, обладающего анти-Ха активностью, и соединения антагониста агрегации тромбоцитов. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что соединение, обладающее анти-Ха активностью, является низкомолекулярно весовым гепарином. 3. Фармацевтическая композиция по п.2, отличающаяся тем, что низкомолекулярно весовой...
Хиральные бифосфиновые соединения, комплексы на их основе и способ получения хирального бифосфинового соединения

Номер патента: 1377
Опубликовано: 26.02.2001
Авторы: Россен Кай, Воланте Ральф П., Пай Филип
МПК: C07C 25/22, C07F 9/50, C07B 53/00...
Метки: соединения, хирального, основе, комплексы, получения, способ, хиральные, бифосфинового, бифосфиновые
Формула / Реферат:
1. Хиральные бифосфины формулы где R представляет C1-4-алкил, С3-6-циклоалкил или арил, незамещенный или замещенный -F, -СН3, -СF3 или СН3О-; и Х1 и Х2 связывают два R2Р-замещенные бензола и независимо образуют 2-4-членную связывающую группу, состоящую из 2-4 атомов углерода и одного незамещенного или замещенного гетероатома, выбранных из О, S, SO, SO2 или 2. Соединение по п.1, в котором число атомов в связи X1 является таким же, как и...