Азаполициклические соединения, конденсированные с арилом






Формула / Реферат
1. Соединение формулы

где R1 представляет собой водород, (C1-C6)алкил, неконъюгированный (C3-C6)алкенил, XC(=O)R13, бензил или -CH2CH2-O-(C1-C4) алкил;
R2 и R3 независимо выбирают из водорода, (C2-C6)алкенила, (C2-C6)алкинила, гидрокси, нитро, амино, гало, циано, -SOq(C1-C6)алкила, где q равно нулю, одному или двум, (C1-C6)алкиламино-, [(C1-C6)алкил]2амино-, -CO2R4, CONR5R6, -SO2NR7R8, -C(=O)R13, -XC(=O)R13, арил-(C0-C3)алкила- или арил-(C0-C3)алкил-O-, где указанный арил выбирают из фенила и нафтила, гетероарил-(С0-C3)алкила- или гетероарил-(C0-C3)алкил-O-, где указанный гетероарил выбирают из 5-7-членных ароматических колец, содержащих от одного до четырех гетероатомов, выбранных из кислорода, азота и серы, X2(C0-C6)алкила- и X2(C1-C6)алкокси-(C0-C6)алкила-, где X2 отсутствует или X2 представляет собой (C1-C6)алкиламино- или [(C1-C6)алкил]2амино-, и где (C0-C6)алкильная или (C1-C6)алкокси-(C0-C6)алкильная части указанных X2(C0-C6)алкила- или X2(C1-C6)алкокси-(С0-C6)алкила- содержат, по меньшей мере, один атом углерода и где от одного до трех атомов углерода указанных (C0-C6)алкильной или (C1-C6)алкокси-(C0-C6)алкильной частей необязательно могут быть заменены атомом кислорода, азота или серы, с тем условием, что любые два таких гетероатома должны быть разделены, по меньшей мере, двумя атомами углерода, и где любая из алкильных частей указанных (C0-C6)алкила- или (C1-C6)алкокси-(C0-C6)алкила- могут быть необязательно замещены от двух до семи атомами фтора, и где один из атомов углерода каждой из алкильных частей указанного арил-(C0-C6)алкила- и указанного гетероарил-(C0-C6)алкила- могут быть необязательно заменены атомом кислорода, азота или серы, и где каждая из указанных выше арильных и гетероарильных групп может быть необязательно замещена одним или более заместителями, предпочтительно от нуля до двух заместителей, независимо выбранных из (C1-C6)алкила, необязательно замещенного от одного до семи атомами фтора, (C1-C6)алкокси, необязательно замещенного от двух до семи атомами фтора, гало, (C2-C6)алкенила, (C2-C6)алкинила, гидрокси, нитро, циано, амино, (C1-C6)алкиламино-, [(C1-C6)алкил]2амино-, -CO2R4, -CONR5R6, -SO2NR7R8, -C(=O)R13 и -XC(=O)R13;
или R2 и R3 вместе с углеродами, к которым они присоединены, образуют 4-7-членное моноциклическое или 10-14-членное бициклическое карбоциклическое кольцо, которое может быть насыщенным или ненасыщенным, где от одного до трех неконденсированных атомов углерода указанных моноциклических колец и от одного до пяти атомов углерода указанных бициклических колец, которые не являются частью бензокольца, показанного в формуле I, могут быть необязательно и независимо заменены азотом, кислородом или серой, и где указанные моноциклические и бициклические кольца могут быть необязательно замещены одним или более заместителями, предпочтительно от нуля до двух заместителей для моноциклических колец и от нуля до трех заместителей для бициклических колец, которые независимо выбирают из (C1-C6)алкила, необязательно замещенного от одного до семи атомами фтора, (C1-C6)алкокси, необязательно замещенного от одного до семи атомами фтора, нитро, циано, гало, (C2-C6)алкенила, (C2-C6)алкинила, гидрокси, амино, (C1-C6)алкиламино и [(C1-C6)алкил]2амино, -CO2R4, -CONR5R6, -SO2NR7R8, -C(=O)R13 и -XC(=O)R13;
где R4, R5, R6, R7, R8 и R13 независимо выбирают из водорода и (C1-C6)алкила, или R5 и R6, или R7 и R8 вместе с азотом, к которому они присоединены, образуют пирролидиновое, пиперидиновое, морфолиновое, азетидиновое, пиперазиновое, N-(C1-C6)алкилпиперазиновое или тиоморфолиновое кольцо, или тиоморфолиновое кольцо, в котором атом серы кольца заменен сульфоксидом или сульфоном; и каждый X независимо представляет собой (C1-C6)алкилен;
при условии, что (a) по меньшей мере, один из R1, R2 и R3 должен отличаться от водорода и (b) когда R2 и R3 оба являются водородом, R1 не может быть водородом, (C1-C6)алкилом или неконъюгированным (C3-C6)алкенилом; или его фармацевтически приемлемая соль.
2. Соединение по п.1, где R2 и R3 вместе с бензокольцом формулы I образуют бициклическую кольцевую систему, выбранную из следующих структур:





где R10 и R17 независимо выбирают из (C0-C6)алкила- и (C1-C6)алкокси-(C0-C6)алкила-, где общее количество атомов углерода не превышает шести и где любая из алкильных частей может быть необязательно замещена от одного до семи атомами фтора; нитро, циано, гало, амино, (C1-C6)алкиламино-, [(C1-C6)алкил]2амино-, CO2R4, -CONR5R6, -SO2NR7R8, -C(=O)R13, -XC(=O)R13, фенила и моноциклического гетероарила, где указанный гетероарил выбирают из 5-7-членных ароматических колец, содержащих от одного до четырех гетероатомов, выбранных из кислорода, азота и серы и где R4, R5, R6, R7, R8 и R13 имеют значения, определенные в п.1.
3. Соединение по п.1, в котором R2 и R3 вместе с бензокольцом формулы I не образуют бициклическую или трициклическую кольцевую систему.
4. Соединение по п.1, в котором один или оба из R2 и R3 представляют собой -C(=O)R13, где R13 является (C1-C6)алкилом.
5. Соединение по п.1, в котором один из R2 и R3 представляет собой -COR13, где R13 является (C1-C6)алкилом или (C1-C3)алкилом, необязательно замещенным от одного до семи атомами фтора.
6. Соединение по п.1, в котором один из R2 и R3 представляет собой CF3, фтор, цианогруппу или C2F5.
7. Фармацевтическая композиция для снижения никотиновой зависимости или для помощи в прекращении или снижеэшш употребления табака у млекопитающего, включающая количество соединения по п.1, которое эффективно для снижения никотиновой зависимости или для помощи в прекращении или снижении употребления табака, и фармацевтически приемлемый носитель.
8. Способ снижения никотиновой зависимости или помощи в прекращении или снижении употребления табака у млекопитающего, включающий введение указанному млекопитающему количества соединения по п.1, которое эффективно для снижения никотиновой зависимости или для помощи в прекращении или снижении употребления табака.
9. Фармацевтическая композиция для лечения заболевания или состояния, выбранного из воспалительного заболевания кишечника, язвенного колита, гангренозной пиодермии, болезни Крона, кишечного синдрома с болями, спастической дистонии, хронической боли, острой боли, синдрома брюшной мальабсорбции, пучита (воспаления хирургически образованного кармана кишечника), вазоконстрикции, тревожности, панического состояния, депрессии, биполярного расстройства, аутизма, расстройств сна, расстройства биоритмов, амилотропного латерального склероза (ALS), когнитивной дисфункции, гипертензии, булимии, анорексии, ожирения, сердечных аритмий, желудочной гиперсекреции кислоты, язв, феохромоцитомы, прогрессирующего супрамышечного паралича, зависимости от химических веществ и наркомании, зависимости от или привыкания к никотину и/или табачным продуктам, алкоголю, бензодиазепинам, барбитуратам, опиоидам или кокаину; головной боли, удара, травматической черепно-мозговой травмы (TBI), обсессивно-компульсивного расстройства (OCD), психоза, хореи Гентингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении, мультиинфарктного слабоумия, ассоциированного с возрастом снижения когнитивной функции, эпилепсии, включая малый эпилептический припадок, старческого слабоумия типа Альцгеймера (AD), болезни Паркинсона (PD), заболевания с дефицитом внимания и гиперактивностью (ADHD) и синдрома Туретта у млекопитающего, включающая количество соединения по п.1, которое эффективно для лечения такого нарушения или состояния, и фармацевтически приемлемый носитель.
10. Способ лечения заболевания или состояния, выбираемого из воспалительного заболевания кишечника, язвенного колита, гангренозной пиодермии, болезни Крона, кишечного синдрома с болями, спастической дистонии, хронической боли, острой боли, синдрома брюшной мальабсорбции, пучита (воспаления хирургически образованного кармана кишечника), вазоконстрикции, тревожности, панического состояния, депрессии, биполярного расстройства, аутизма, расстройств сна, расстройства биоритмов, амилотропного латерального склероза (ALS), когнитивной дисфункции, гипертензии, булимии, анорексии, ожирения, сердечных аритмий, желудочной гиперсекреции кислоты, язв, феохромоцитомы, прогрессирующего супрамышечного паралича, зависимости от химических веществ и наркомании; зависимости от или привыкания к никотину и/или табачным продуктам, алкоголю, бензодиазепинам, барбитуратам, опиоидам или кокаину; головной боли, удара, травматической черепно-мозговой травмы (TBI), обсессивно-компульсивного расстройства (OCD), психоза, хореи Гентингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении, мультиинфарктного слабоумия, ассоциированного с возрастом снижения когнитивной функции, эпилепсии, включая малый эпилептический припадок, старческого слабоумия типа Альцгеймера (AD), болезни Паркинсона (PD), заболевания с дефицитом внимания и гиперактивностью (ADHD) и синдрома Туретта у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, количества соединения по п.1, которое эффективно для лечения такого заболевания или состояния.
11. Соединение формулы

где Р представляет собой водород, метил, COOR16, где R16 является (C1-C6)алкилом, аллилом или 2,2,2-трихлорэтилом; -C(=O)NR5R6, где R5 и R6 имеют значения, определенные в п.1; -C(=O)H, -C(=O)(C1-C6)алкил, где алкильная часть может быть необязательно замещена от 1 до 3 атомами галогена, предпочтительно от 1 до 3 атомами фтора или хлора; бензил или трет-бутоксикарбонил (t-Boc) или трифторацетил, и R14 и R15 независимо выбирают из водорода, (C1-C6)алкила, необязательно замещенного от одного до семи атомами фтора; -C(=O)(C1-C6)алкила, циано, гидрокси, нитро, амино, -O(C1-C6)алкила и гало; с тем условием, что R14 и R15 не могут быть оба водородом, когда Р является водородом, (C1-C6)алкилом или неконъюгированным (C3-C6)алкенилом.
12. Способ снижения никотиновой зависимости или помощи в прекращении или снижении употребления табака у млекопитающего, включающий введение указанному млекопитающему количества соединения формулы

или его фармацевтически приемлемой соли, которое эффективно для снижения никотиновой зависимости или для помощи в прекращении или снижении употребления табака.
13. Способ лечения заболевания или состояния, выбираемого из воспалительного заболевания кишечника, язвенного колита, гангренозной пиодермии, болезни Крона, кишечного синдрома с болями, спастической дистонии, хронической боли, острой боли, синдрома брюшной мальабсорбции, пучита (воспаления хирургически образованного кармана кишечника), вазоконстрикции, тревожности, панического состояния, депрессии, биполярного расстройства, аутизма, расстройств сна, расстройства биоритмов, амилотропного латерального склероза (ALS), когнитивной дисфункции, гипертензии, булимии, анорексии, ожирения, сердечных аритмий, желудочной гиперсекреции кислоты, язв, феохромоцитомы, прогрессирующего супрамышечного паралича, зависимости от химических веществ и наркомании; зависимости от или привыкания к никотину и/или табачным продуктам, алкоголю, бензодиазепинам, барбитуратам, опиоидам или кокаину; головной боли, удара, травматической черепно-мозговой травмы (TBI), обсессивно-компульсивного расстройства (OCD), психоза, хореи Гентингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении, мультиинфарктного слабоумия, ассоциированного с возрастом снижения когнитивной функции, эпилепсии, включая малый эпилептический припадок, старческого слабоумия типа Альцгеймера (AD), болезни Паркинсона (PD), заболевания с дефицитом внимания и гиперактивностью (ADHD) и синдрома Туретта у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, количества соединения формулы

или его фармацевтически приемлемой соли;
которое эффективно для лечения такого заболевания или состояния.
14. Соединение формулы

где R2 и R3 определены также, как в п.1; и P' представляет собой COOR16, где R16 является аллилом, 2,2,2-трихлорэтилом или (C1-C6)алкилом; -C(=O)NR5R6, где R5 и R6 являются теми же, что и определенные в п.1; -C(=O)H, C(=O)(C1-C6)алкил, где алкильная часть может быть необязательно замещена от 1 до 3 атомами галогена, предпочтительно от 1 до 3 атомами фтора или хлора; бензил или трет-бутоксикарбонил (t-Boc).
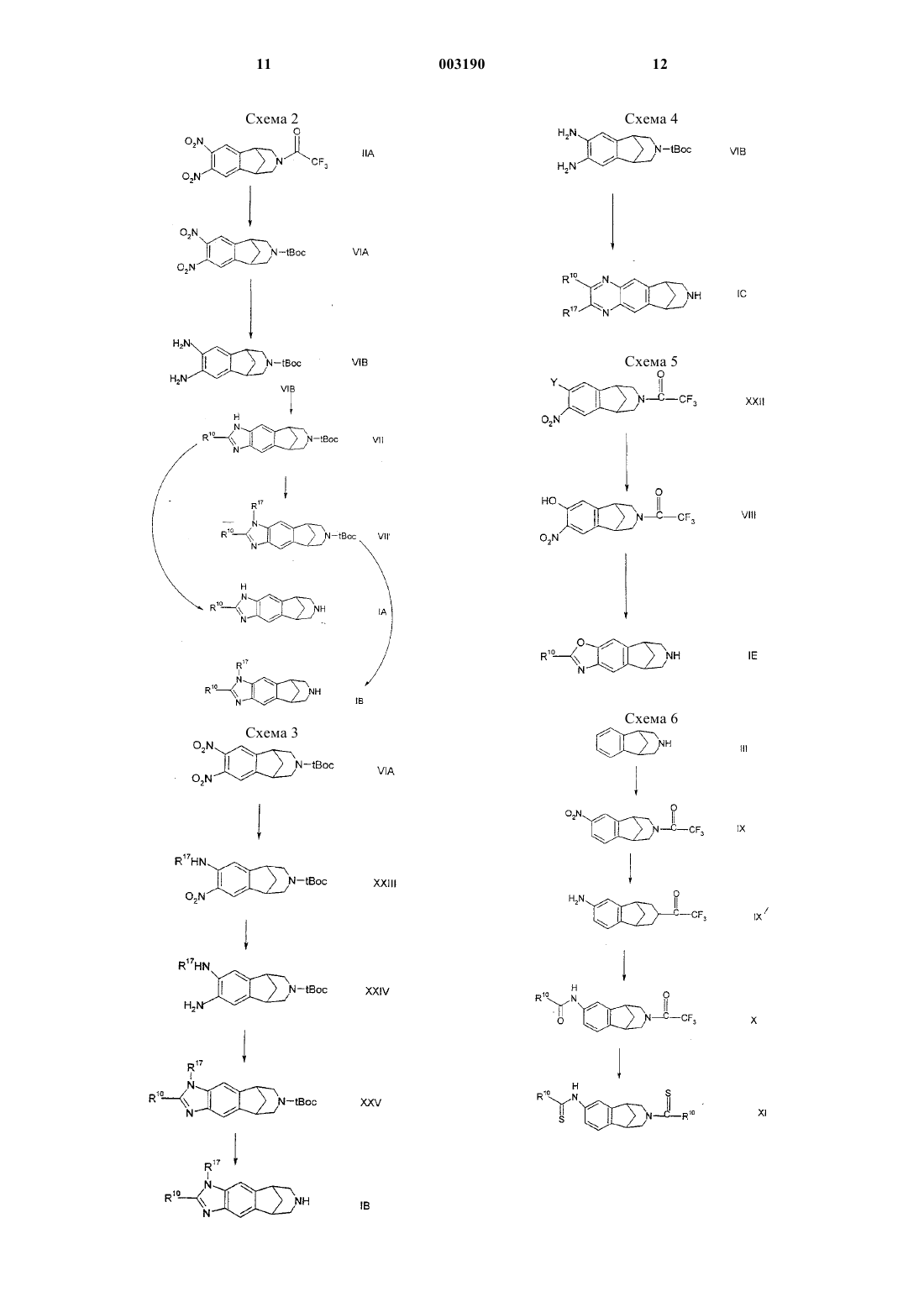
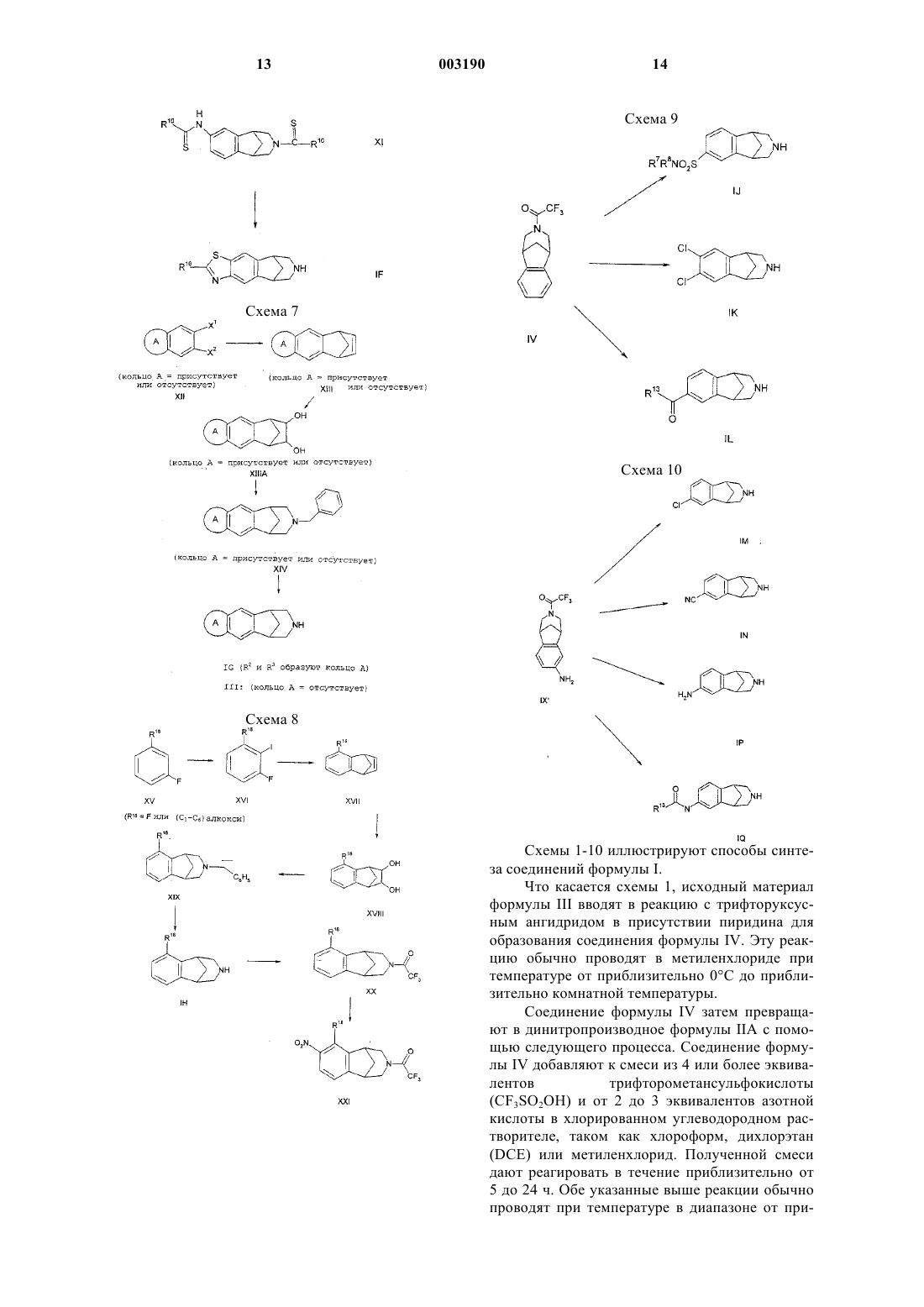
Текст
1 Известный уровень техники Это изобретение относится к конденсированным с арилом азаполициклическим соединениям, определенным более конкретно формулойI ниже. Соединения формулы I связывают специфические сайты нейронов рецепторов никотинового ацетилхолина и полезны для модуляции холинергической функции. Такие соединения полезны для лечения воспалительного заболевания кишечника (включая, но не ограничиваясь этим, язвенный колит, гангренозную пиодермию и болезнь Крона), кишечного синдрома с болями, спастической дистонии, хронической боли, острой боли, синдрома брюшной мальабсорбции, пучита (воспаления хирургически образованного кармана кишечника), вазоконстрикции, тревожности, панического состояния,депрессии, биполярного расстройства, аутизма,расстройств сна, расстройства биоритмов, амилотропного латерального склероза (ALS), когнитивной дисфункции, гипертензии, булимии,анорексии, ожирения, сердечных аритмий, желудочной гиперсекреции кислоты, язв, феохромоцитомы, прогрессирующего супрамышечного паралича, зависимости от химических веществ и наркомании (например, зависимости от или привыкания к никотину (и/или табачным продуктам), алкоголю, бензодиазепинам, барбитуратам, опиоидам или кокаину), головной боли,удара, травматической черепно-мозговой травмы (TBI), обсессивно-компульсивного расстройства, психоза, хореи Гентингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении, мультиинфарктного слабоумия типа Альцгеймера (AD), болезни Паркинсона (PD),заболевания с дефицитом внимания и гиперактивностью (ADHD) и синдрома Туретта. Соединения этого изобретения могут также применяться в сочетании с антидепрессантом, таким, например, как трициклический антидепрессант или антидепрессант, ингибирующий обратный захват серотонина (SRI), для лечения сразу снижения когнитивной функции и депрессии, ассоциированных с AD, PD, ударом,хореей Гентингтона или травматической черепно-мозговой травмой (TBI); в сочетании с мускариновыми агонистами, для того, чтобы стимулировать как центральные мускариновые, так и никотиновые рецепторы для лечения, например, ALS, когнитивной дисфункции, ассоциированного с возрастом снижения когнитивной функции, AD, PD, удара, хореи Гентингтона иTBI; в сочетании с нейротропными факторами,такими как NGF (фактор роста нервов), для максимального холинергического увеличения при лечении, например, ALS, когнитивной дисфункции, ассоциированного с возрастом снижения когнитивной функции, AD, PD, удара, хореи Гентингтона и TBI; или в сочетании с агентами,замедляющими или подавляющими AD, такими как усилители когнитивной функции, или с ингибиторами агрегации амилоида, ингибиторами 2 секретазы, ингибиторами таукиназы, нейрональными противовоспалительными агентами и с терапией эстрогеноподобными соединениями. Другие соединения, которые связываются с сайгами нейронов никотинового рецептора,указаны в заявке на патент США 08/963852,которая была зарегистрирована 4 ноября 1997 г. Указанная заявка признана совместной с настоящей заявкой и полностью включена здесь в качестве ссылки. Краткое изложение существа изобретения Данное изобретение относится к конденсированным с арилом азаполициклическим соединениям формулыR2 и R3 выбирают независимо из водорода,(С 2-С 6)алкенила, (С 2-С 6)алкинила, гидрокси,нитро, амино, гало, циано, -SOq(C1-C6)алкила,где q равно нулю, единице или двум, (C1C6)алкиламино-, [(C1-С 6)алкил]2 амино-, -CO2R4,-CONR5R6, -SO2NR7R8, -C(=O)R13, -XC(=O)R13,арил-(С 0-С 3)алкила- или арил-(С 0-С 3)алкил-O-,где указанный арил выбирают из фенила и нафтила, гетероарил-(С 0-С 3)алкила- или гетероарил(С 0-С 3)алкил-O-, где указанный гетероарил выбирают из 5-7-членных ароматических колец,содержащих от одного до четырех гетероатомов, выбранных из кислорода, азота и серы, иX2(С 0-С 6)алкила- и X2(C1-С 6)алкокси-(С 0-С 6) алкила- содержат, по меньшей мере, один атом углерода и где от одного до трех атомов углерода указанных (С 0-С 6)алкильной или (C1-С 6) алкокси-(С 0-С 6)алкильной частей необязательно могут быть заменены атомом кислорода, азота или серы, с тем условием, что любые два таких гетероатома должны быть разделены, по меньшей мере, двумя атомами углерода, и где любая из алкильных частей указанных (С 0-С 6)алкилаили (C1-С 6)алкокси-(С 0-С 6)алкила- могут быть необязательно замещены от двух до семи атомами фтора и где один из атомов углерода каждой из алкильных частей указанного арил-(С 0 С 3)алкила- и указанного гетероарил-(С 0-С 3) алкила- могут быть необязательно заменены атомом кислорода, азота или серы, и где каждая из указанных выше арильных и гетероарильных групп может быть необязательно замещена одним или более заместителями, предпочтительно от нуля до двух заместителей, независимо вы 3 бираемых из (C1-С 6)алкила, необязательно замещенного от одного до семи атомами фтора,(C1-С 6)алкокси, необязательно замещенного от одного до семи атомами фтора, гало (например,хлор, фтор, бром или иод), (С 2-С 6)алкенила, (С 2 С 6)алкинила, гидрокси, нитро, циано, амино,(C1-C6)алкиламино-,[(C1-C6)алкил]2 амино-,-CO2R4, -CONR5R6, SO2NR7R8, -С(=O)R13 и-ХС(=O)R13; или R2 и R3 вместе с углеродами, к которым они присоединены, образуют 4-7-членное моноциклическое или 10-14-членное бициклическое карбоциклическое кольцо, которое может быть насыщенным или ненасыщенным,где от одного до трех неконъюгированных атомов углерода указанных моноциклических колец и от одного до пяти атомов углерода указанных бициклических колец, которые не являются частью бензокольца, показанного в формуле I, могут быть необязательно и независимо заменены азотом, кислородом или серой, и где указанные моноциклические и бициклические кольца могут быть необязательно замещены одним или более заместителями, предпочтительно от нуля до двух заместителей для моноциклических колец и от нуля до трех заместителей для бициклических колец, которые независимо выбирают из (C0-C6)алкила- или (C1C6)алкокси-(С 0-С 6)алкила-, где общее количество атомов углерода не превышает шести и где любая из алкильных частей может быть необязательно замещена от одного до семи атомами фтора; нитро, оксо, циано, гало, (С 2 С 6)алкенила, (C2-C6)алкинила, гидрокси, амино,(C1-С 6)алкиламино-,[(C1-С 6)алкил]2 амино-,-CO2R4, -CONR5R6, -SO2NR7R8, -C(=O)R13 и-XC(=O)R13; каждый из R4, R5, R6, R7, R8 и R13 независимо выбирают из водорода и (C1-С 6)алкила,или R5 и R6, или R7 и R8 вместе с азотом, к которому они присоединены, образуют пирролидиновое, пиперидиновое, морфолиновое, азетидиновое, пиперазиновое, -N-(C1-C6)алкилпиперазиновое или тиоморфолиновое кольцо, или тиоморфолиновое кольцо, в котором сера кольца заменена сульфоксидом или сульфоном; и каждый Х независимо представляет собой(C1-С 6)алкилен; при условии, что: (а) по меньшей мере,один из R1, R2 и R3 должен отличаться от водорода и (b) когда R2 и R3 являются водородом, R1 не может быть водородом, (C1-C6)алкилом или неконъюгированным (С 3-С 6)алкенилом; и к фармацевтически приемлемым солям таких соединений. Примерами гетероарильных групп, в которых могут быть каждый из R2 и R3, являются следующие: тиенильная, оксазоильная, изоксазолильная, пиридильная, пиримидильная, тиазолильная, тетразолильная, изотиазолильная,триазолильная, имидазолильная, тетразолильная, пирроильная и следующие группы: где один из R9 и R18 является водородом или(C1-C6)алкилом, а другой является связью с бензокольцом формулы I. Примерами соединений настоящего изобретения являются соединения формулы I и их фармацевтически приемлемые соли, где R2 и R3 вместе с бензокольцом формулы I образуют бициклическую кольцевую систему, выбранную из следующих: где R10 и R17 независимо выбирают из (С 0 С 6)алкила- и (C1-C6)алкокси-(C0-C6)алкила-, где общее количество атомов углерода не превышает шести и где любая из алкильных частей может быть необязательно замещена от одного до семи атомами фтора; нитро, циано, гало, амино,(C1-C6)алкиламино-,[(C1-C6)алкил]2 амино-,-CO2R4, -CONR5R6, SO2NR7R8, -C(=O)R13,-XC(=O)R13, фенила и моноциклического гетероарила, где указанный гетероарил определен как R2 и R3, которые указаны в определении соединений формулы I выше; другие воплощения данного изобретения относятся к соединениям формулы I и их фармацевтически приемлемым солям, где R2 и R3 вместе с бензокольцом формулы I образуют бициклическую или трициклическую кольцевую систему, выбранную из следующих: где R10 и R17 те же, что и указанные выше, и m равно нулю, единице или двум и где один из атомов углерода кольца А может быть необязательно замещен кислородом или -N(C1-C6) алкилом. 5 Другие воплощения данного изобретения относятся к соединениям формулы I и их фармацевтически приемлемым солям, где ни R2, ниR3 не присоединены к бензокольцу формулы I через атом кислорода. Другие воплощения данного изобретения относятся к соединениям формулы I и их фармацевтически приемлемым солям, где R2 и R3 не образуют вместе с бензокольцом формулы I бициклическую или трициклическую кольцевую систему. Другие воплощения данного изобретения относятся к соединениям формулы I, где один или оба из R2 и R3 представляют собой-C(=O)R13, где R13 является (С 1-С 6)алкилом. Другие осуществления данного изобретения относятся к соединениям формулы I, где один или оба из R2 и R3 представляют собой(C1-С 3)алкилом, необязательно замещенным от одного до семи атомами фтора. Другие осуществления относятся к соединениям формулы I,где один из R2 и R3 представляет собой СF3,фтор, циано или C2F5-; другие воплощения данного изобретения относятся к соединениям формулы I, где R1 не является метилом. Примерами конкретных соединений формулы I являются следующие: гидрохлорид 6-метил-5,7-диоксо-6,13-диазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,8 триена; гидрохлорид 6-метил-5-оксо-6,13-диазатетрацикло[9.3.1.02,10.04,8]пeнтaдeкa-2(10),3,8 триена; гидрохлорид 5,7-диметил-6-оксо-5,7,13 триазатетрацикло[9.3.1.02,10.04,8]пeнтaдeкa-2(10),3,8-триена; гидрохлорид 5,7-диоксо-6,13-диазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,8-триена; гидрохлорид 5-оксо-6,13-диазатетрацикло 6 гидрохлорид 4-хлор-5-трифторметил-10 азатрицикло[6.3.1.02,7]додека-2(7),3,5-триена; гидрохлорид 5-трифторметил-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-4-карбонитрила; гидрохлорид 4-этинил-5-трифторметил-10 азатрицикло[6.3.1.02,7]додека-2(7),3,5-триена; гидрохлорид 6-метил-5-тиа-5-диокса-6,13 диазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,8-триена; гидрохлорид 7-диметиламино-5-тиа-5-диокса-6,13-диазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,8-триена; гидрохлорид 6,7-диокса-5,8,14-триазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,9 триена; гидрохлорид 5,8-диметил-6,7-диокса 5,8,14-триазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,9-триена. Это изобретение относится также к соединениям формулы где Р представляет собой водород, метил,COOR16, где R16 является (C1-C6)алкилом, аллилом, 2,2,2-трихлороэтилом или -C(=O)NR5R6,где R5 и R6 являются теми же, что и определенные в формуле I выше; -С(=O)Н, -С(=O)(C1C6)алкил, где алкильная часть может быть необязательно замещена от 1 до 3 атомами галогена, предпочтительно от 1 до 3 атомами фтора или хлора; бензил или трет-бутоксикарбонил (tBoc); и R14 и R15 выбирают независимо из водорода, (C1-C6)алкила, необязательно замещенного от одного до семи атомами фтора; -С(=O)(C1C6)алкил, циано, гидрокси, нитро, амино, -O(C1C6)алкил или гало; с тем условием, что R14 и R15 не могут быть оба водородом, когда Р является водородом, (C1-С 6)алкилом или неконъюгированным (С 3-С 6)алкенилом. Такие соединения полезны в качестве промежуточных продуктов при синтезе соединений формулы I. Изобретение также относится к соединению формулы-С(=O)Н, -С(=O)(C1-C6)алкил, где алкильная часть может быть необязательно замещена от 1 до 3 атомами галогена, предпочтительно от 1 до 3 атомами фтора или хлора; бензил или третбутоксикарбонил (t-Boc). 7 Если это специально не указано, термин"гало", как здесь используется, включает фтор,хлор, бром и иод. Если это специально не указано, термин"алкил", как здесь используется, включает неразветвленные, разветвленные или циклические алкилы и может включать неразветвленные и циклические алкильные части, а также разветвленные и циклические части. Термин "алкокси", как здесь используется,обозначает "алкил-O-", где "алкил" является тем же, что определено выше. Термин "алкилен", как здесь используется,обозначает алкильный радикал, содержащий два доступных связывающих места (т.е. -алкил-),где "алкил" является тем же, что определено выше. Если это специально не указано, термин"один или более заместителей", как здесь используется, относится к заместителям, от одного до максимального количества, возможного на основе имеющегося числа доступных связывающих участков. Термин "лечение", как здесь используется,относится к аннулированию, облегчению, торможению развития или предотвращению заболевания или состояния, в отношении которого этот термин применяют, или к одному или более симптомам таких состояний или заболеваний. Термин "лечение", как здесь используется,относится к акту лечения, причем "лечение" определено здесь только что выше. Соединения формулы I могут содержать оптические центры и, следовательно, могут существовать в различных энантиомерных конфигурациях. Изобретение включает все энантиомеры, диастереомеры и другие стереоизомеры таких соединений формулы I, a также рацемические и другие их смеси. Настоящее изобретение относится также к радиоактивно меченым формам соединений формулы I. Предпочтительными радиоактивно мечеными соединениями формулы I являются те, в которых радиоактивную метку выбирают из 3H, 11 С, 14 С, 18F, 123I и 125I. Такие радиоактивно меченые соединения полезны в качестве исследовательских и диагностических инструментов в исследованиях метаболической фармакокинетики и анализа связывания как у животных, так и у человека. Настоящее изобретение относится также к фармацевтической композиции, применяемой для снижения никотиновой зависимости или для помощи в прекращении или снижении употребления табака у млекопитающего, включая человека, включающей количество соединения формулы I или его фармацевтически приемлемой соли, которое эффективно для снижения никотиновой зависимости или для помощи в прекращении или снижении употребления табака и фармацевтически приемлемому носителю. 8 Настоящее изобретение относится также к способу снижения никотиновой зависимости или помощи в прекращении или снижении употребления табака у млекопитающего, включая человека, включающему введение указанному млекопитающему количества соединения формулы I или его фармацевтически приемлемой соли, которое эффективно для снижения никотиновой зависимости или для помощи в прекращении или снижении употребления табака. Настоящее изобретение также относится к способу лечения заболевания или состояния,выбираемого из воспалительного заболевания кишечника (включая, но не ограничиваясь этим,язвенный колит, гангренозную пиодермию и болезнь Крона), кишечного синдрома с болями,спастической дистонии, хронической боли, острой боли, синдрома брюшной мальабсорбции,пучита (воспаления хирургически образованного кармана кишечника), вазоконстрикции, тревожности, панического состояния, депрессии,биполярного расстройства, аутизма, расстройств сна, расстройства биоритмов, амилотропного латерального склероза (ALS), когнитивной дисфункции, гипертензии, булимии, анорексии,ожирения, сердечных аритмий, желудочной гиперсекреции кислоты, язв, феохромоцитомы,прогрессирующего супрамышечного паралича,зависимости от химических веществ и наркомании (например, зависимости от или привыкания к никотину (и/или табачным продуктам), алкоголю, бензодиазепинам, барбитуратам, опиоидам или кокаину), головной боли, удара, травматической черепно-мозговой травмы (TBI),обсессивно-компульсивного расстройства(OCD), психоза, хореи Гентингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении, мультиинфарктного слабоумия, ассоциированного с возрастом снижения когнитивной функции, эпилепсии, включая малый эпилептический припадок, старческого слабоумия типа Альцгеймера (AD), болезни Паркинсона (PD),заболевания с дефицитом внимания и гиперактивностью (ADHD) и синдрома Туретта у млекопитающего, включающему введение млекопитающему, нуждающемуся в таком лечении,количества соединения формулы I или его фармацевтически приемлемой соли, которое эффективно для лечения такого заболевания или состояния. Настоящее изобретение также относится к фармацевтической композиции для лечения заболевания или состояния, выбираемого из воспалительного заболевания кишечника (включая,но не ограничиваясь этим, язвенный колит, гангренозную пиодермию и болезнь Крона), кишечного синдрома с болями, спастической дистонии, хронической боли, острой боли, синдрома брюшной мальабсорбции, пучита (воспаления хирургически образованного кармана кишечника), вазоконстрикции, тревожности, па 9 нического состояния, депрессии, биполярного расстройства, аутизма, расстройств сна, расстройства биоритмов, амилотропного латерального склероза (ALS), когнитивной дисфункции,гипертензии, булимии, анорексии, ожирения,сердечных аритмий, желудочной гиперсекреции кислоты, язв, феохромоцитомы, прогрессирующего супрамышечного паралича, зависимости от химических веществ и наркомании (например, зависимости от или привыкания к никотину (и/или табачным продуктам), алкоголю, бензодиазепинам, барбитуратам, опиоидам или кокаину), головной боли, удара, травматической черепно-мозговой травмы (TBI), обсессивнокомпульсивного расстройства (OCD), психоза,хореи Гентингтона, поздней дискинезии, гиперинезии, дислексии, шизофрении, мультиинфарктного слабоумия, ассоциированного с возрастом снижения когнитивной функции, эпилепсии, включая малый эпилептический припадок, старческого слабоумия типа Альцгеймера(ADHD) и синдрома Туретта у млекопитающего, включающей количество соединения формулы I или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя. Настоящее изобретение относится также к способу снижения никотиновой зависимости или помощи в прекращении или снижении использования табака у млекопитающего, включающий введение указанному млекопитающему количества соединения, включающего количество соединения формулы или его фармацевтически приемлемой соли,которое эффективно для снижения никотиновой зависимости или для помощи в прекращении или снижении использования табака. Настоящее изобретение также относится к способу лечения заболевания или состояния,выбираемого из воспалительного заболевания кишечника (включая, но не ограничиваясь этим,язвенный колит, гангренозную пиодермию и болезнь Крона), кишечного синдрома с болями,спастической дистонии, хронической боли, острой боли, синдрома брюшной мальабсорбции,пучита (воспаления хирургически образованного кармана кишечника), вазоконстрикции, тревожности, панического состояния, депрессии,биполярного расстройства, аутизма, расстройств сна, расстройства биоритмов, амилотропного латерального склероза (ALS), когнитивной дисфункции, гипертензии, булимии, анорексии,ожирения, сердечных аритмий, желудочной гиперсекреции кислоты, язв, феохромоцитомы,прогрессирующего супрамышечного паралича,зависимости от химических веществ и наркомании (например, зависимости от или привыкания к никотину (и/или табачным продуктам), алко 003190 10 голю, бензодиазепинам, барбитуратам, опиоидам или кокаину), головной боли, удара, травматической черепно-мозговой травмы (TBI), обсессивно-компульсивного расстройства (OCD),психоза, хореи Гентингтона, малого эпилептического припадка, старческого слабоумия типа Альцгеймера (AD), болезни Паркинсона (PD),заболевания с дефицитом внимания и гиперактивностью (ADHD) и синдрома Туретта у млекопитающего, включающему введение млекопитающему, нуждающемуся в таком лечении,количества соединения формулы или его фармацевтически приемлемой соли,которое эффективно для лечения такого заболевания или состояния. Настоящее изобретение также относится к фармацевтически приемлемым кислым аддитивным солям соединений формулы I. Примерами фармацевтически приемлемых кислых аддитивных солей соединений формулы I являются соли соляной кислоты, паратолуолсульфокислоты, фумаровой кислоты, лимонной кислоты, янтарной кислоты, салициловой кислоты,щавелевой кислоты, бромисто-водородной кислоты, фосфорной кислоты, метансульфокислоты, винной кислоты, соль яблочной кислоты,дипаратолуоилвинной кислоты и миндальной кислоты. Подробное описание изобретения За исключением специально оговоренных случаев, R1-R18, m и Р и структурная формула I в схемах реакций и обсуждениях, которые будут представлены ниже, имеют те же значения, что определены выше. Схема 1 Схемы 1-10 иллюстрируют способы синтеза соединений формулы I. Что касается схемы 1, исходный материал формулы III вводят в реакцию с трифторуксусным ангидридом в присутствии пиридина для образования соединения формулы IV. Эту реакцию обычно проводят в метиленхлориде при температуре от приблизительно 0 С до приблизительно комнатной температуры. Соединение формулы IV затем превращают в динитропроизводное формулы IIА с помощью следующего процесса. Соединение формулы IV добавляют к смеси из 4 или более эквивалентов трифторометансульфокислоты(СF3SО 2 ОН) и от 2 до 3 эквивалентов азотной кислоты в хлорированном углеводородном растворителе, таком как хлороформ, дихлорэтан(DCE) или метиленхлорид. Полученной смеси дают реагировать в течение приблизительно от 5 до 24 ч. Обе указанные выше реакции обычно проводят при температуре в диапазоне от при 15 близительно -78 до приблизительно 0 С в течение приблизительно 2 ч и затем дают нагреться до комнатной температуры в течение остального времени. Восстановление соединения формулы IIА с применением способов, хорошо известных специалистам, дает соединение формулы IIВ. Это восстановление может быть выполнено, например, с помощью водорода и палладиевого катализатора, такого как гидроокись палладия, и проведения реакции в метаноле при приблизительно комнатной температуре. Что касается схемы 2, соединение формулы IIА превращают в соответствующее соединение, где трифторацетильную защитную группу замещают t-Boc защитной группой (VIA) с помощью реакции сначала с гидоокисью или карбонатом щелочного металла или щелочноземельного металла (или аммония) и затем реакции выделенного из предшествующей реакции продукта с дитретбутилдикарбонатом. Реакцию с гидоокисью или карбонатом щелочного металла или щелочно-земельного металла (или аммония) обычно проводят в водном спирте,диоксане или тетрагидрофуране (ТГФ) при температуре от приблизительно комнатной температуры до приблизительно 70 С, предпочтительно при приблизительно 70 С в течение от приблизительно 1 до приблизительно 24 ч. Реакцию выделенного из указанной выше реакции незащищенного амина или аддитивной кислой соли такого амина с дитретбутилдикарбонатом предпочтительно проводят в растворителе, таком как ТГФ, диоксан или метиленхлорид при температуре от приблизительно 0 С до приблизительно комнатной температуры. Эту реакцию можно проводить, а можно и не проводить в присутствии основания. Когда реагентом служит соль амина предпочтительно применение основания. Полученное соединение формулыVIA может быть превращено в соответствующее диаминопроизводное формулы VIB с помощью процедуры, описанной выше для превращения динитросоединения формулы IIA в соответствующее диаминосоединение формулыIIВ. Превращение соединения формулы VIB в желаемое соединение формулы VII может быть произведено с помощью реакции соединения формулы VIB с соединением формулы где R10 представляет собой водород, (С 1 С 6)алкил, необязательно замещенный от одного до семи атомами фтора, арил-(С 0-С 3)алкил, где указанный арил выбирают из фенила и нафтила,или гетероарил-(С 0-С 3)алкил, где указанный гетероарил выбирают из 5-7-членных ароматических колец, содержащих от одного до четырех 16 гетероатомов, выбранных из кислорода, азота и серы, и где каждая из указанных выше арильных и гетероарильных групп может быть необязательно замещена одним или более заместителями, предпочтительно от нуля до двух заместителями, независимо выбранными из (C1C6)алкила, необязательно замещенного от одного до семи атомами фтора, (С 1-С 6)алкокси, необязательно замещенного от одного до семи атомами фтора и циано. Предпочтительным растворителем для этой реакции является смесь 10:1 этанола:уксусной кислоты. Температуру реакции можно варьировать от приблизительно 40 до приблизительно 100 С. Предпочтительна температура приблизительно 60 С. Другие подходящие растворители включают уксусную кислоту, этанол и изопропанол. Другие способы получения соединений формулы VII соединения формулы VIB описаныSegelstein et al., Tetrahedron Lett., 1993, 34, 1897. Удаление t-Вос защитной группы из соединения формулы VII дает соответствующее соединение формулы IA. Защитная группа может быть удалена с помощью способов, хорошо известных специалистам, например, соединение формулы VII может быть обработано безводной кислотой, такой как соляная кислота, бромисто-водородная кислота, метансульфокислота или фторуксусная кислота, предпочтительно соляная кислота в этилацетате, при температуре от приблизительно 0 до приблизительно 100 С,предпочтительно от приблизительно комнатной температуры до приблизительно 70 С, в течение приблизительно от одного до 24 ч. Соединение формулы VII может быть превращено в соответствующее соединение формулы IB с помощью его реакции с соединением формулы R17Z, где R17 определен как R10, как определено выше, и Z является удаляемой группой, такой как гало или сульфонат (например,хлоро, бромо, мезилат или тозилат), в присутствии основания, такого как гидрид, гидроокись или карбонат щелочного металла, предпочтительно гидроокись калия, в полярном растворителе, таком как вода, диметилсульфоксид(ДМСО), ТГФ или ДМФ, предпочтительно смеси ДМСО и воды, и последующего удаления защитной группы, как описано выше. Реакцию сR17Z обычно проводят при температуре от приблизительно комнатной температуры до приблизительно 100 С, предпочтительно при приблизительно 50 С, в течение приблизительно 5 ч. Схема 3 иллюстрирует альтернативный способ получения соединений формулы IB из соединения формулы VIA. Этот способ является предпочтительным способом получения соединений формулы IB, где R17 является объемной группой, такой как группа, содержащая арил или гетероарил, или когда R17 не может быть присоединен, как показано на схеме 2, с помощью методов алкилирования или замещения 17 арилом. Что касается схемы 3, соединение формулы VIA вводят в реакцию с подходящим соединением формулы R17NH2 в полярном растворителе, таком как ТГФ, ДМФ или ДМСО, предпочтительно ТГФ, при температуре от приблизительно комнатной температуры до приблизительно 100 С, предпочтительно при температуре кипения с обратным холодильником, в течение от приблизительно 4 до 18 ч. Полученное соединение формулы XXIII затем превращают в соответствующее соединение формулы XXIV путем восстановления нитрогруппы до аминогруппы, используя способы, хорошо известные специалистам. Такие способы отмечены выше для превращения соединений формулы IIА в соединение формулы IIВ на схеме 1 и представлены в экспериментальных примерах 12 В и 18 В. Замыкание имидазольного кольца с образованием соответствующего соединения формулыXXV может быть затем проведено с помощью реакции соединения формулы XXIV из указанной выше реакции с соединением формулы где R10 является тем же, что и определено выше,как описано выше, для превращения соединений формулы VIB в соединения формулы VII. Удаление защитной группы из соединения формулы XXV дает соответствующее соединение формулы IB. Это может быть осуществлено с помощью способов, хорошо известных в науке, например, как описано выше для образования соединений формулы IA из соответствующих соединений формулы VII. Схема 4 иллюстрирует способ получения соединений формулы IC, где R10 и R17 являются теми же, что определено выше. Что касается схемы 4, соединение формулы VIB вводят в реакцию с соединением формулы(аддукт присоединения натрия бисульфитэтандиона) в воде или другом полярном растворителе, таком как ТГФ, ДМФ или ДМСО, предпочтительно в смеси воды и смешивающегося с водой растворителя, такого как ТГФ, в течение приблизительно от 1 до 4 ч. Температура реакции может варьировать от приблизительно 40 до приблизительно 100 С и предпочтительно при приблизительно температуре кипения с обратным холодильником. Альтернативно, соединение формулы VIB может быть введено в реакцию с соединением формулы(реакция двойной конденсации) в полярном растворителе, таком как ТГФ, вода или уксусная кислота, предпочтительно в смеси воды и ТГФ. Эту реакцию обычно проводят при температуре от приблизительно 40 до приблизительно 100 С,предпочтительно при температуре кипения с обратным холодильником, в течение от приблизительно 2 до 4 ч. Затем может быть получен желаемый хиноксолин формулы IC путем снятия защиты с образованного соединения с помощью одной из предшествующих реакций, применяя способы,описанные выше для превращения соединения формулы VII в соединение формулы IA. Схема 5 иллюстрирует способ получения соединений формулы I, где R2 и R3 вместе с бензольным кольцом, к которому они присоединены, образуют бензоксазольную кольцевую систему. Такое соединение, где R1 является водородом, изображено на схеме 5 в виде химической формулы IE. Что касается схемы 5, соединение формулы XXII, где Y является нитро, гало, трифторометансульфонатом или солью диазония, взаимодействует с ацетатом калия или другим карбоксилатом щелочного или щелочноземельного металла в растворителе, таком как диметилсульфоксид (ДМСО), ДМФ или ацетонитрил, предпочтительно ДМСО. Этой реакции обычно дают проходить в течение приблизительно 12-24 ч. Подходящие температуры реакции лежат в диапазоне от приблизительно 70 до приблизительно 140 С. Предпочтительна температура около 100 С. Указанная выше реакция дает соединение формулы VIII, которое затем может быть превращено в желаемое соединение, имеющее формулу IE, с помощью следующей процедуры. Сначала соединение формулы VIII восстанавливают в реакции с водородом и палладиевым или платиновым катализатором, таким как гидроокись палладия в метаноле, при температуре от приблизительно 0 до приблизительно 70 С,предпочтительно при приблизительно комнатной температуре, для образования соответствующего аминопроизводного. Продукт этой реакции затем вводят в реакцию с хлорангидридом формулы R10COCl или кислотным ангидридом формулы (R10CO)2 О, где R10 является (C1 С 6)алкилом, или с соединением формулыR10C(OC2H5)3 в подходящем инертном растворителе, таком как декалин, хлорбензол или ксилолы. Смесь ксилолов предпочтительна. Эту реакцию обычно проводят при температуре приблизительно 120-150 С, предпочтительно при приблизительно 140 С. Когда в качестве реагента используют R10COCl, предпочтительно добавлять к реакционной смеси стехиометрическое количество триэтиламина (TEA) или другого 19 органического третичного аминооснования и каталитическое количество паратолуолсульфокислоты пиридиния или паратолуолсульфоната пиридиния (PPTs). Когда в качестве реагента используют R10C(OC2H5)3, предпочтительно добавлять к реакционной смеси каталитическое количество PPTs. Удаление трифтороацетильной защитной группы азота дает желаемое соединение формулы IE. Это можно осуществить с помощью способов, хорошо известных специалистам, например, с помощью реакции защищенного соединения с низшим алканолом и водной гидроокисью или карбонатом щелочного или щелочноземельного металла (или аммония), водным карбонатом натрия при температуре от приблизительно 50 С до приблизительно 100 С, предпочтительно при приблизительно 70 С, в течение приблизительно от 2 до 6 ч. Схема 6 иллюстрирует получение соединений формулы I, где R1 является водородом, аR2 и R3 вместе с бензольным кольцом, к которому они присоединены образуют бензотиазольную кольцевую систему. Что касается схемы 6, соединение формулы III вводят в реакцию с трифторуксусным ангидридом для образования соответствующего соединения, где азот кольца защищен трифторацетильной группой, и полученное соединение с защищенным азотом затем вводят в реакцию с двумя эквивалентами ангидрида трифторометансульфокислоты и одним эквивалентом азотной кислоты для образования соответствующего соединения формулыIX, где имеется единственный нитрозаместитель в бензокольце. Реакцию с трифторуксусной кислотой обычно проводят в присутствии пиридина. Обе указанные выше реакции обычно проводят в реакционно-инертном растворителе,таком как хлорированный углеводородный растворитель, предпочтительно метиленхлорид,при температуре от приблизительно 0 С до приблизительно комнатной температуры, предпочтительно при приблизительно комнатной температуре. Указанная выше трансформация может быть также произведена с помощью других методов азотирования, известных специалистам. Восстановление нитрогруппы до аминогруппы может быть проведено, как описано выше для получения соединения формулы IX'. Соединение формулы IX' затем вводят в реакцию с галоангидридом карбоновой кислоты или ангидридом формулы R10COX или[R10CO)2O, где Х является гало, и R10 является водородом или (C1-С 6)алкилом, и пиридином,TEA или другим третичным аминооснованием для образования соединения формулы X, которое затем может быть превращено в желаемое соединение, имеющее формулу XI, путем его взаимодействия с реагентом Лавессона, который изображен ниже Реакцию с R10COX, где Х является гало,или (R10CO)2O обычно проводят при температуре от приблизительно 0 С до приблизительно комнатной температуры, предпочтительно при приблизительно комнатной температуре. Реакцию с реагентом Лавессона обычно проводят в реакционно-инертном растворителе, таком как бензол или толуол, предпочтительно толуол,при температуре от приблизительно комнатной температуры до приблизительно температуры кипения реакционной смеси с обратным холодильником, предпочтительно при температуре кипения с обратным холодильником. Замыкание бензотиазольного кольца и снятие защиты с азота для образования желаемого соединения формулы IF может быть выполнено путем взаимодействия соединения формулы XI с феррицианидом калия и гидроокисью натрия в смеси воды и метанола (NаОН/Н 2O/СН 3 ОН) при температуре от приблизительно 50 до приблизительно 70 С, предпочтительно при приблизительно 60 С в течение приблизительно 1,5 ч. Схема 7 иллюстрирует способ получения соединения формулы III, которое используется в качестве исходного материала для процесса схемы 1, или соединения формулы IG, где R2 и"А"), как определено выше в определении соединений формулы I. Что касается схемы 7, соединение формулы XII, где X1 и X2 выбирают независимо из хлора, фтора, брома и иода, но где, по меньшей мере, один из X1 и X2, является Вr- или I-, вводят в реакцию с циклопентадиеном в присутствии металлического магния в ТГФ, диоксане или другом эфирном растворителе при температуре от приблизительно 40 до приблизительно 100 С, предпочтительно при приблизительно температуре кипения с обратным холодильником, для образования соединения формулы XIII. Реакция полученного соединения формулы XIII с N-метилморфолин-Nоксидом (NMO) и четырехокисью осмия в ацетоне при приблизительно комнатной температуре дает соответствующее соединение формулыXIIIА. Соединение, имеющее формулу XIIIА, затем превращают в соответствующее соединение формулы XIV с помощью следующей процедуры. Сначала соединение формулы XIIIА вводят в реакцию с перииодатом натрия в смеси хлорированного углеводорода, предпочтительно дихлорэтана (DCE), и воды, или с тетраацетатом свинца в хлорированном углеводородном растворителе при температуре от приблизительно 0C до приблизительно комнатной температуры,для образования диальдегидного или гликального производного. Продукт этой реакции затем 21 вводят в реакцию с бензиламином и триацетоксиборогидридом натрия в хлорированном углеводородном растворителе при температуре от приблизительно 0 С до приблизительно комнатной температуры, предпочтительно при приблизительно комнатной температуре, для образования желаемого соединения формулы XIV. Удаление бензильной группы из соединения формулы XIV дает соединение формулы III (где кольцо А отсутствует) или IG (где кольцо А присутствует). Это может быть выполнено с применением методов, хорошо известных специалистам, например, необязательно путем реакции свободного основания с одним эквивалентом кислоты, например, соляной кислоты(для образования соответствующей кислой аддитивной соли), с последующей реакцией с водородом и гидроокисью палладия в метаноле при приблизительно комнатной температуре. На стадии восстановительного аминирования, описанной выше, и на протяжении всего этого документа в качестве альтернативных бензиламину могут быть также использованы такие амины, как аммоний, гидроксиламин, алкоксиамины, метиламин, аллиламин, и замещенные бензиламины (например, дифенилметиламин и 2- и 4-алкоксизамещенные бензиламины). Их можно использовать в виде свободных оснований или их солей, предпочтительно их уксуснокислых солей, и их можно впоследствии удалять способами, описанными для каждого из них T.W. Greene and G.M. Wuts, "ProtectiveSons, New York, NY. Процедура схемы 7 может быть также применена для получения соединений формулыI, где R2 и R3 не образуют кольцо и не являются оба водородом, заменой исходного материала формулы XII подходящим соединением, имеющим формулу Схемы 8, 9 и 10 иллюстрируют способы получения соединений формулы I, где R1 является водородом, а R2 и R3 представляют множество различных заместителей как определено выше, но не образуют кольцо. Схема 8 иллюстрирует вариацию процесса,показанного на схеме 7, которая может быть применена для создания соединения, идентичного таковому формулы III, за исключением того, что бензокольцо замещено фторогруппой или алкоксигруппой (R18 на схеме 8). Это соединение изображено на схеме 8 в виде химической структуры 1 Н. Что касается схемы 8, где,например, R18 является F, 1,3-дифторобензол вводят в реакцию с сильным основанием, таким как диалкиламин щелочного металла или алкил 22 творителе, таком как этиловый эфир или ТГФ,при температуре ниже -50 С с последующим гашением иодом или N-иодосукцинамидом для образования 1,3-дифторо-2-иодобензола. Соединение 1,3-дифторо-2-иодобензол (структурная формула XVI на схеме 8) затем превращают в соединение формулы IH с помощью серии реакций (представленных на схеме 8 в видеXVIXVIIXVIIIXIXIH), которая аналогична серии реакций, описанной выше и иллюстрированной на схеме 7 для превращения соединений формулы XIII в таковые формулы IG или III. Превращение соединения формулы XVI в соединение формулы XVII также может быть проведено с помощью обработки смеси соединения формулы XVI и циклопентадиена реагентом алкиллития,предпочтительно нбутиллитием в инертном углеводородном растворителе, таком как петролейный эфир или метилциклогексан, при температуре от приблизительно -20 С до приблизительно комнатной температуры, предпочтительно при приблизительно 0 С. Соединение формулы IH может быть затем превращено в соответствующее производное с защищенным азотом формулы XX с применением способов, описанных выше для синтеза соединения формулы IV на схеме 1. Азотирование соединения формулы XX с применением способа, описанного выше для получения соединения формулы IX на схеме 6, дает соединение формулы XXI, где бензокольцо замещено как фтором, так и нитрогруппой или алкоксигруппой и нитрогруппой. Соединение формулы XXI может быть использовано для создания разнообразных соединений формулы I, где один из R2 и R3 является фтором, с помощью способов, хорошо известных специалистам, например, путем превращения сначала нитрогруппы в аминогруппу,превращения аминогруппы во множество других заместителей, как проиллюстрировано на схеме 10, и затем удаления защитной группы азота. Соединение формулы XXI действует как региоизомерный функциональный эквивалент соединений, имеющих формулы IIА, VIA иXXII, в том отношении, что атом фтора формулы XXI взаимодействует сходным образом с нитро и Y группами формул IIА, VIA и XXII, и тем самым может быть подвержено той же самой серии реакций, что и описанная выше для трех последних соединений, обеспечивая альтернативные способы получения продуктов таких реакций. Сходным образом алкоксигруппа формулы XXI (R18=aлкoкcи) может быть превращена в гидроксильную группу перед или после введения нитрогруппы и затем превращена в изомерные продукты, как описано выше. Также трифторометансульфонатная соль такого гидроксипроизводного может действовать как 23 Получение соединений формулы I, где R2=-О(C1-С 6)алкил, (C1-С 6)алкил или арил, где арил представляет собой то же самое, что и определено выше в определении формулы I, и R3 является Н или одним из других заместителей, определенных выше в определении формулы I, может быть проведено как описано выше и проиллюстрировано на схеме 8, путем замены одного из атомов фтора соединения формулы XV -О(C1-С 6)алкилом, (C1-C6)алкилом или арилом,соответственно. Схема 9 иллюстрирует способы получения соединений формулы I, где (а) R1 является водородом, а R2 является R7R8NO2S-; (b) R1 и R2 оба являются хлором; и (с) R1 является водородом, а R2 является R13C(=O)-. Эти соединения обозначены на схеме 9, соответственно как соединения формул IG, IK и IL. Что касается схемы 9, соединения формулы IG могут быть получены с помощью взаимодействия соединения формулы IV с двумя или более эквивалентами галосульфокислоты, предпочтительно хлоросульфокислоты, при температуре от приблизительно 0 С до приблизительно комнатной температуры. Реакция образованного таким образом хлоросульфокислотного производного с амином, имеющим формулу R7R8NH, где R7 и R8 определены выше, с последующим удалением защитной группы азота дает желаемое соединение, имеющее формулуIG. Соединения формулы IК могут быть получены с помощью реакции соединения формулыIV с треххлористым иодом в хлорированном углеводородном растворителе с последующим удалением защитной группы азота. Реакцию с треххлористым иодом обычно проводят при температуре от приблизительно 0 С до приблизительно комнатной температуры и предпочтительно проводят при приблизительно комнатной температуре. Сходным образом аналогичные моно- или дибромированные или моно- или дииодированные соединения могут быть получены с помощью реакции соединения IV с Nиодосукцинимидом или N-бромосукцинимидом в трифторометансульфокислотном растворителе с последующим удалением защитной группы азота, как описано выше. Реакция соединения IV с галоидангидридом формулы R13COCl или ангидридом кислоты формулы (R13CO)2O с или без реакционноинертного растворителя, такого как хлорированный углеводородный растворитель предпочтительно метиленхлорид, в присутствии кислоты Льюиса, такой как хлористый алюминий, при температуре от приблизительно 0 С до приблизительно 100 С с последующим удалением защиты азота дает соединение формулы IL. Реакцию с галоидангидридом или ангидридом можно проводить, применяя другие известные кислоты Льюиса или другие способы ацилирования Фриделя-Крафта, которые известны науке. 24 Описанные здесь реакции, в которых NO2,-SO2NR7R8, -COR13, I, Br или Сl вводят в соединение формулы IV, как обозначено на схеме 9 и описано выше, могут быть проведены с любым аналогичным соединением, где R2 является водородом, (C1-С 6)алкилом, гало, (C1-С 6)алкокси или NHCONR7R8, с образованием соединений формулы I, где R2 и R3 определены как в определении соединений формулы I выше. Соединения, которые идентичны таковым формулы IL, но которые сохраняют защитную группу азота, могут быть превращены в соответствующие O-ацилзамещенные соединения,т.е. такие, в которых -C(=O)R13 группа формулыIL замещена -O-C(=O)R13 группой, с применением процессов Бейера-Виллигера, хорошо известных специалистам. Полученные соединения могут быть частично гидролизованы, как описано в примере 35, для получения соответствующих гидроксизамещенных соединений и затем алкилированы для образования соответствующих алкоксизамещенных соединений. Также,как описано в примере 36, такие O-ацилзамещенные соединения могут быть использованы для получения различным образом замещенных бензизоксазолов. Схема 10 иллюстрирует способы создания соединений формулы I, где (a) R1 является водородом, a R2 является хлором; (b) R1 является водородом, а R2 является циано; (с) R1 является водородом, а R2 является амино; и (d) R1 является водородом, а R2 является R13C(=O)N(Н)-. Эти соединения обозначены на схеме 10, соответственно как соединения формул IM, IN, IP и IQ. Соединения формулы IM могут быть получены из соединений формулы IX' с помощью образования соли диазония с, например, нитритом щелочного металла и сильной неорганической кислотой (например, соляной кислотой,серной кислотой, бромисто-водородной кислотой) в воде с последующей реакцией с солью галоида меди, такой как хлористая медь (I). Удаление защиты азота с помощью описанных выше способов дает желаемое соединение формулы IM. Также могут быть применены альтернативные способы получения солей диазония,как известно и практикуется специалистами. Указанную выше реакцию обычно проводят в диапазоне температур от приблизительно 0 до приблизительно 60 С, предпочтительно при приблизительно 60 С в течение приблизительно от 15 мин до 1 ч. Реакция соли диазония, полученной как описано выше, с йодистым калием в водной среде дает аналогичное иодидное производное. Эту реакцию обычно проводят при температуре от приблизительно 0 С до приблизительно комнатной температуры, предпочтительно при приблизительно комнатной температуре. Полученное соединение или его аналогичная N-третбутилкарбонатная защищенная форма может быть использовано для получения соответст 25 вующего цианопроизводного с помощью реакции с цианидом меди(I) и цианидом натрия в ДМФ, N,N-диметилпропилмочевине (DMPU) или ДМСО, предпочтительно ДМФ, при температуре от приблизительно 50 до приблизительно 180 С, предпочтительно при приблизительно 150 С. Снятие защиты азота, как описано выше,дает желаемое соединение формулы IM. Описанное выше иодидное производное может быть также использовано для получения множества других заместителей, таких как арильные, ацетиленовые и винильные заместители, а также соответствующих карбонильных эфиров и амидов, с помощью катализируемых палладием и никелем процессов, известных специалистам, таких как присоединения Хека,Сузуки и Стилля и карбонилирований Хека. Удаление защиты азота соединения формулы IX' дает соединение формулы IP. Соединение формулы IX' может быть введено в реакцию с ацильной группой, имеющей формулу R13COCl или (R13CO)2O, с использованием способов, описанных выше, с последующим снятием защиты азота для получения соединений формулы IQ. Сходным образом обработка защищенного амина соединением, имеющим формулу R13SO2X, где Х является хлоро или бромо, с последующим снятием защиты азота дает соответствующее сульфонамидное производное. В процедурах, описанных на протяжении всего этого документа, могут быть альтернативно использованы другие подходящие защитные группы для амина, которые включают - СОСF3,-СОССl3, -COOCH2CCl3, -COO(C1-C6)алкил и-СООСН 2 С 6 Н 5. Эти группы стабильны в описанных здесь условиях и могут быть удалены способами, описанными для каждой из них вGreene's "Protective groups in Organic Chemistry",цитированной выше. В каждой из реакций, обсуждавшихся выше и иллюстрированных на схемах 1-10 выше,давление не является решающим, если это специально не отмечено. Обычно приемлемо давление от приблизительно 0,5 до приблизительно 5 атм, причем окружающее давление, т.е. приблизительно 1 атм, является предпочтительным с точки зрения удобства. Соединения формулы I и их фармацевтически приемлемые соли (здесь и далее "активные соединения") пероральным, чрескожным(например, с помощью применения пластыря),интраназальным, подъязычным, ректальным,парентеральным или местным способами. Чрескожное и пероральное введение предпочтительны. Наиболее желательно, чтобы эти соединения вводили в дозах в диапазоне от приблизительно 0,25 до приблизительно 1500 мг в день,предпочтительно от приблизительно 0,25 до приблизительно 300 мг в день, в виде единственной или дробной дозы, хотя необходимы вариации в зависимости от веса и состояния 26 субъекта, подвергаемого лечению, и выбранного конкретного пути введения. Однако наиболее желательно применять уровень дозы, лежащий в диапазоне от приблизительно 0,01 до приблизительно 10 мг на кг веса тела в день. Тем не менее, могут иметь место вариации в зависимости от веса и состояния субъектов, подвергаемых лечению, и их индивидуальных ответов на указанное лекарство, а также от типа выбранного фармацевтического состава и периода времени и интервала, в течение которого проводят такое введение. В некоторых случаях могут быть более предпочтительны уровни доз ниже упомянутого выше диапазона, в то время как в других случаях могут быть применены еще большие дозы без оказания вредных побочных эффектов,при условии, что такие большие дозы сначала делят на несколько малых доз для введения на протяжении дня. Активные соединения можно вводить сами по себе или в сочетании с фармацевтически приемлемыми носителями или разбавителями с помощью любого из ранее указанных нескольких путей. Более конкретно, активные соединения можно вводить в широко варьирующихся различных формах дозировки, например, их можно объединять с различными фармацевтически приемлемыми инертными носителями в форме таблеток, капсул, чрескожных пластырей, лепешек, пастилок, твердых свечей, порошков, спреев, кремов, целебных мазей, суппозиториев, желе, гелей, паст, лосьонов, мазей, водных суспензий, растворов для инъекций, эликсиров, сиропов и тому подобное. Такие носители включают твердые разбавители или наполнители, стерильные водные среды и различные нетоксичные органические растворители. Кроме того, пероральные фармацевтические композиции могут быть подходящим образом подслащены и/или ароматизированы. В целом активные соединения в таких формах доз присутствуют в концентрациях в диапазоне от приблизительно 5,0 до приблизительно 70% по весу. Для перорального введения могут быть применены таблетки, содержащие различные наполнители, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция,вторичный кислый фосфорно-кислый кальций и глицин, совместно с различными разрыхлителями, такими как крахмал (предпочтительно кукурузный, картофельный крахмал или крахмал тапиоки), альгиновой кислотой и определенными комплексными силикатами, совместно с гранулирующими связующими агентами, такими как поливинилпирролидон, сахароза, желатин и камедь. Дополнительно для целей таблетирования могут быть использованы смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции сходного типа могут быть также применены в качестве наполнителей в желатиновых капсулах; предпочтительные материалы в этой связи 27 также включают лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Когда для перорального введения желательны водные суспензии и/или эликсиры, активный ингредиент можно сочетать с различными подсластителями или отдушками, окрашивающим материалом и, если это желательно, эмульгаторами и/или суспендирующими агентами, вместе с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин и их различными сочетаниями. Для парентерального введения может быть применен раствор активного соединения либо в кунжутном, либо в арахисовом масле, или в водном пропиленгликоле. Водные растворы должны быть при необходимости подходящим образом забуферены (предпочтительно, чтобы рН был выше 8), и жидкий разбавитель сначала должен быть сделан изотоническим. Эти водные растворы пригодны для целей внутривенного введения. Масляные растворы пригодны для целей внутрисуставного, внутримышечного и подкожного введения. Получение всех этих растворов в стерильных условиях легко проводится с помощью стандартных фармацевтических методов, хорошо известных специалистам. Возможно также местное введение активных соединений и это может быть достигнуто с помощью кремов, пластыря, желе, гелей, паст,мазей и тому подобное в соответствии со стандартной фармацевтической практикой. Биологическое тестирование Эффективность активных соединений в подавлении связывания никотина со специфическими рецепторными сайтами определяют с помощью следующей процедуры, которая представляет собой модификацию способов Lippiello, Р. М. and Fernandes, К. G. (в Binding of L[3H] Nicotine to A Single Class of High-AffinityRiver содержали в группах в подвесных проволочных клетках из нержавеющей стали с 12 часовым циклом чередования темноты и света(световой период с 7 утра до 7 вечера). Они получали стандартный корм Purina Rat Chow и воду ad libitum. Крыс забивали декапитацией. Мозг удаляли немедленно после декапитации. Мембраны получали из ткани мозга в соответствии со способами Lippiello and Fernandez (Molec Pharmacol, 29, 448-454, (1986) с некоторыми модификациями. Удаляли весь мозг, промывали в охлажденном на льду буфере и гомогенизировали при 0 С в 10 объемах буфера (вес./об.), применяя Brinkmann Polytron, 6 заходов по 30 с. 28 Буфер состоял из 50 мМ трис-НСl с рН 7,5 при комнатной температуре. Гомогенат осаждали центрифугированием (10 мин; 50000 х g; от 0 до 4 С). Супернатант сливали и мембраны осторожно ресуспендировали политроном и вновь центрифугировали (10 мин; 50000 х g; от 0 до 4 С). После второго центрифугирования мембраны ресуспендировали в буфере для определения в концентрации 1,0 г/100 мл. Стандартный буфер для определения имел состав: 50 мМ трис-HCl, 120 мМ NaCl, 5 мМ КСl, 2 мМ MgCl2,2 мМ CaCl2, и рН 7,4 при комнатной температуре. Обычные тесты выполняли в пробирках для тестирования из боросиликатного стекла. Смесь для тестирования обычно состояла из 0,9 мг белка мембран в 1,0 мл конечного инкубационного объема. Готовили три серии пробирок,где пробирки каждой серии содержали 50 мкл растворителя, пустую пробу или раствор тестируемого соединения, соответственно. К каждой пробирке добавляли 200 мкл [3 Н]-никотина в буфере для тестирования, затем 750 мкл суспензии мембран. Конечная концентрация никотина в каждой пробирке составляла 0,9 нМ. Конечная концентрация цитизина в пустой пробе составляла 1 мкМ. Растворитель состоял из деионизированной воды, содержащей 30 мкл 1 н. уксусной кислоты на 50 мл воды. Тестируемые соединения и цитизин растворяли в растворителе. Тестирования начинали с помощью перемешивания после добавления в пробирку суспензии мембран. Образцы инкубировали при от 0 до 4 С в перемешиваемой ледяной бане. Инкубации заканчивали путем быстрой фильтрации через фильтры из стекловолокна WhatmanGF/B под вакуумом с применением многоячеистого коллектора ткани Brandel. После первоначальной фильтрации тестируемой смеси фильтры отмывали два раза охлажденном на льду буфером для тестирования (5 мин каждый раз). Затем фильтры помещали в сосуды для счета и энергично смешивали с 20 мл ReadySafe (Beckman) перед подсчетом радиоактивности. Радиоактивность образцов подсчитывали в жидкостном сцинтилляционном счетчике LKBWallach Rackbeta с эффективностью 40-50%. Все определения проводили в трех параллельных пробах. Расчеты Специфическое связывание (С) с мембранами представляет собой разницу между общим связыванием в образцах, содержащих только растворитель и мембраны (А) и неспецифическим связыванием в образцах, содержащих мембраны и цитизин (В), т.е. Специфическое связывание = (С)=(А)-(В). Специфическое связывание в присутствии тестируемого соединения (Е) представляет собой разницу между общим связыванием в присутствии тестируемого соединения (D) и неспецифическим связыванием (В), т.е. (Е) = (D)-(В).% ингибирования = (1-Е)/(С раз на 100. Соединения изобретения, которые протестированы в указанном выше тесте, проявляют величины IC50 менее 10 мкМ. Последующие экспериментальные примеры иллюстрируют, но не ограничивают объем настоящего изобретения. Пример 1. 10-Азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен. А) 1,4-Дигидро-1,4-метанонафталин. (Основан целиком или частично на a) Wittig, G.;Stothers, J. В. Can. J. Chem. 1993, 71, 1290.). Стружку магния (36,5 г, 1,5 М) перемешивали в безводном ТГФ (250 мл) в сухой 2 литровой 3-горлой круглодонной колбе, снабженной неуравновешивающей дополнительной воронкой с адаптером для потока азота (N2),механической мешалкой и эффективным конденсатором, снабженным адаптером для потока азота (N2). Колбу перемешивали и нагревали до температуры кипения с обратным холодильником с помощью удаляемого нагревающего кожуха. Добавляли 2-фторбромбензол (2 г), затем 1 мл 3 н. бромистого этилмагния (EtMgBr в ТГФ). Дополнительную воронку нагружали смесью циклопентадиена (94,4 г, 1,43 М, полученного способом, описанным в Org. Syn. Col.Vol. V, 414-418) и бромфторобензола (250 г,1,43 М), которую поддерживали при 0 С в отдельном сосуде с помощью ледяной бани, и переносили в дополнительную воронку через канюлю. Для поддержки инициации вводили маленькие порции (1 мл) тщательно перемешанной смеси (4 х). Через 15 мин инициировали реакцию (экзотермически и паровой конденсацией), нагревающий кожух удаляли и содержимое дополнительной воронки добавляли по каплям с такой скоростью, чтобы поддержать кипение с обратным холодильником (1,5 ч). Заново надевали нагревающий кожух и температуру кипения с обратным холодильником поддерживали в течение 1,5 ч. (ТСХ 100% гексаны, отн. подв. 0,67). Реакционную смесь охлаждали до комнатной температуры и гасили Н 2O (500 мл) и осторожно 1 н. НСl (200 мл, продуцирует выделение Н 2 из неиспользованного Mg). К этим 50 мл добавляли концентрированную НСl для растворения твердых соединений. Общее время добавления/гашения 1 час. Добавляли насыщенный водный раствор хлористого натрия (NaCl) (300 мл) и продукт экстрагировали гексанами до тех пор, пока переставал удаляться активный продукт перманганата калия (KMnO4). (4 х 250 мл). Объединенный органический слой промывали насыщенным раствором NаНСО 3 (250 мл),бикарбонатом натрия, сушили над безводнымNa2SO4 и концентрировали до масла (200 г). Продукт перегоняли при 78-83 С 15 мм (131 г, 64%). (Альтернативный способ описан наB) 1,2,3,4-Тетрагидро-1,4-метанонафталин 2,3-диол. (За исключением рабочего метода и количества применяемого OsO4 основан наSyn. 1988, 6, 342.). В 2-литровую 3-горлую круглодонную колбу, снабженную адаптером для потока N2 и механической мешалкой, помещали 1,4 дигидро-1,4-метанонафталин (79,5 г, 560 ммоль), перемешивали в ацетоне (800 мл) и Н 2 О(100 мл) и N-оксиде N-метилморфолина (67,5 г,576 ммоль). Туда добавляли четырехокись осмия (OsO4) (15 мл 15 моль% раствор в t-BuOH,1,48 ммоль, 0,26 моль%) и смесь тщательно перемешивали. Через 60 ч реакционную смесь фильтровали и белый продукт промывали в ацетоне и сушили на воздухе (60,9 г). Маточный раствор концентрировали до твердого масла: обеспечивали растирание в ацетоне, фильтрацию и промывку в ацетоне (27,4 г, суммарно 88,3 г, 89%). (ТСХ 50% EtOAc/гексаны, отн. подв. 0,5). Т. пл. 176-177,5 С.Shah, R. D. J. Org. Chem. 1996, 61, 3849; и Mazzocchi, P.H.; Stahly, B.C. J. Med. Chem. 1979, 22,455.). 1,2,3,4-Тетрагидро-1,4-метанонафталин 2,3-диол (40 г, 227,3 ммоль) перемешивали в Н 2 О (1050 мл) и 1,2-дихлорэтане (DCE) (420 мл) в 2-литровой круглодонной колбе под азотом в бане с холодной водой (10 С). Туда добавляли периодат натрия (NaIO4) (51 г, 239 ммоль) и солянокислый триэтилбензиламмоний (EtBnNCl)(50 мг). Полученную смесь перемешивали в течение 1 ч (легкая начальная экзотермическая реакция), затем слои разделяли и водный слой экстрагировали DСЕ (200 мл). Органический слой промывали Н 2 О (4200 мл или до прекращения реакции иодида с крахмалом в промывной воде, затем сушили с помощью ватной пробки. Туда добавляли бензиламин (25,5 г,238,6 ммоль) и смесь перемешивали в течение 2 мин, затем немедленно переносили в триацетоксиборгидрид натрия NaHB(OAc)3/DCE (см. ниже) на протяжении 10 мин. В отдельной круглодонной колбе под азотом перемешивали с помощью магнитной мешалки при 0 С (ледяная баня) NaHB(OAc)3 (154 г, 0,727 ммоль) в DCE (800 мл). Туда добавляли полученную выше смесь на протяжении 10 мин,не прекращая после смешивания диальдегида и амина. Полученной оранжевой смеси давали нагреться до комнатной температуры и ее перемешивали в течение 30-60 мин. Реакцию гасили добавлением раствора насыщенного карбоната натрия (Nа 2 СО 3) (300 мл),в первый момент осторожным, и смесь перемешивали в течение 1 ч (рН 9). Слои разделяли и 31 водный слой экстрагировали CH2Cl2 (2300 мл). Органический слой промывали насыщенным водным раствором NaCl (200 мл), сушили с помощью ватной пробки, затем упаривали до красного масла. Его растворяли в минимуме EtO и фильтровали через подложку из двуокиси кремния (34 дюймов (1 дюйм = 25,410-3 м),элюируя 15% этилацетатом (EtOAc)/гексанами+ от 1 до 37% раствором водной гидроокиси аммония (NH4OH) для удаления основного красного цвета. Концентрирование дает светложелтое масло (48,5 г, 194,8 ммоль, 85,7%). (ТСХ 10% EtOAc/гексаны, отн. подв. 0,75). 1D) 10-Азатрицикло [6,3.1.02,7]дoдeкa-2(7),3,5-триен. (Альтернативный синтез, см. Mazzocchi, P. H.; Stahly, В. С. J. Med. Chem. 1979, 22,455.). 10-Бензил-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен (70,65 г, 284 ммоль) перемешивали в EtOAc (250 мл) и обрабатывали 3N НСl EtOAc (1,03 экв.), медленно при охлаждении (ледяная баня). Полученный осадок фильтровали и промывали EtOAc. Твердые остатки растворяли в МеОН (250 мл) в сосуде Парра. Туда добавляли Pd(OH)2 (7 г 20% вес./конц.) и смесь встряхивали при 50-40 psi (фунтов на кв. дюйм, 1 psi = 6,89103 н/м 2) Н 2 в течение 24 ч или до выполнения ТСХ. Реакционную смесь фильтровали через целитную подложку и концентрировали до маслянистого твердого вещества. Создавали его азеотропную смесь с метанолом (МеОН) (3 х), затем растирали с ацетоном, обрабатывали этиловым эфиром (Et2O) для преципитации продукта и фильтровали. Концентрирование маточных жидкостей и вторая обработка дала беловатое твердое веществоSoc. 1976, 98, 753-761. Paquette, L. A.; Cottrell,D. М.; Snow, R. A. J. Amer. Chem. Soc. 1977, 99,3723-3733). Стружку магния (0,66 г, 27,2 ммоль) перемешивали в безводном ТГФ (10 мл) в высушенной под пламенем 3-горлой круглодонной колбе, снабженной неуравновешивающей дополнительной воронкой с адаптером для потока N2,магнитной мешалкой и эффективным конденсатором, снабженным адаптером для потока N2. 32 Колбу перемешивали и нагревали до температуры кипения с обратным холодильником с помощью удаляемого нагревающего кожуха. Добавляли 2,5-дифторобромобензол (0,1 г), затем 3 н. EtMgBr в ТГФ (0,1 мл). Дополнительную воронку нагружали тщательно перемешанной смесью циклопентадиена (1,71 г, 25,9 ммоль) и 2,5-дифторбромбензола (5,0 г, 25,9 ммоль). Для способствования инициации вводили маленькие порции (0,2 мл) тщательно перемешанной смеси (4 х). Через 15 мин инициировали реакцию(экзотермически и паровой конденсацией) и нагревание поддерживали в необходимой степени в течение добавления содержимого дополнительной воронки. Реакцию затем поддерживали при кипении с обратным холодильником в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и гасили Н 2 О (20 мл) и затем водным раствором 1 н. НСl (20 мл) для растворения твердых соединений. Добавляли насыщенный водный раствор NaCl (30 мл) и продукт экстрагировали гексанами (425 мл). Объединенный органический слой промывали насыщенным водным раствором NaHCO3 (25 мл),сушили (Na2SO4), фильтровали через подложку из двуокиси кремния, промывая гексанами, и концентрировали до масла. Хроматография на силикагеле с элюцией гексанами дала масло(680 мг, 4,22 ммоль) и N-оксид Nметилморфолина (599 мг, 4,43 ммоль) перемешивали в ацетоне (50 мл) и Н 2 О (5 мл). Туда добавляли раствор OsO4 (0,2 мл 2,5 вес.% раствор в t-BuOH, 0,02 ммоль). Флоризил фильтровали и фильтрат концентрировали для получения кристаллического продукта, который растирали в ацетоне и фильтровали (524 мг, 64%). 1(15 мл) и H2O (45 мл) затем обрабатывали периодатом натрия (0,603 мг, 2,82 ммоль). Через 1,5 ч слои разделяли и водный слой экстрагировали DCE (220 мл). Объединенный органический слой промывали H2O (420 мл) до пре 33 кращения реакции иодида с крахмальным фильтром, затем промывали насыщенным водным раствором NaCl (20 мл). Органический слой сушили с помощью ватной пробки и обрабатывали бензиламином (0,308 мл, 2,82 ммоль),и перемешивали в течение 2 мин, затем переносили в дополнительную воронку. Этот раствор добавляли на протяжении 10 мин к осторожно перемешиваемой охлажденной смесиNаНВ(ОАс)3 (1,82 г, 8,58 ммоль) в DСЕ (50 мл). После окончания добавления смесь перемешивали без охлаждения в течение 2 ч. Смесь гасили насыщенным водным раствором Nа 2 СО 3 (100 мл) и перемешивали в течение 1 ч, затем слои разделяли и водный слой экстрагировали CH2Cl2(330 мл). Объединенный органический слой промывали насыщенным водным растворомNaCl (50 мл), сушили с помощью ватной пробки и концентрировали. Хроматография на силикагеле дала масло (520 мг, 80%). (ТСХ 2% ацетон/СН 2 Сl2, отн. подв. 0,40). 1 Н ЯМР (400 МГц, СDСl3)7,18 (м, 1 Н),6,88 (м, 2 Н), 3,48 (с, 2 Н), 3,06 (м, 2 Н), 2,78 (м,2 Н), 2,41 (м, 2 Н), 2,27 (м, 1 Н), 1,69 (д, J=10,5 Гц,1 Н).(50 мл) и давали кипеть с обратным холодильником под N2 в течение 1,5 ч. Добавляли муравьино-кислый аммоний (1,0 г) и кипение с обратным холодильником продолжали в течение 0,5 ч. Реакционную смесь фильтровали через целитную подложку, которую промывали МеОН. Фильтрат концентрировали. Остатки обрабатывали насыщенным водным растворомNa2 СО 3 (30 мл), и продукт экстрагировали метиленхлоридом (СН 2 Сl2) (325 мл). Органический слой промывали насыщенным водным раствором NaCl (50 мл), сушили с помощью ватной пробки и концентрировали. Остаток обрабатывали 2 н. НСl МеОН (5 мл) и концентрировали,затем переносили в минимальном количестве МеОН и насыщали Et2O. После перемешивания в течение 18 ч белые кристаллы собирали фильтрацией (86 мг, 28%). (ТСХ 5%-78 С раствору н-бутиллития (n-BuLi) (200 мл,2,5 М/гексаны, 0,5 М) и ТГФ (500 мл) под N2. С помощью контролирования скорости добавления внутреннюю температуру поддерживали ниже -70 С. Суммарное время добавления составляло 1,2 ч. Полученный шлам перемеши 35 вали дополнительно 1,2 ч, затем дисперсную смесь обрабатывали раствором иода (126,9 г,0,5 М) в ТГФ (300 мл) при скорости, которая поддерживала внутреннюю температуру ниже-70 С. После завершения добавления смеси давали нагреться до комнатной температуры, обрабатывали Н 2 О (100 мл) и 10% водным раствором Nа 2S2O3 (100 мл) и перемешивали. Слои разделяли и водный слой экстрагировали гексанами (2250 мл). Объединенный органический слой отмывали 10% водным раствором Nа 2S2O3(100 мл), Н 2 О (100 мл), насыщенным водным раствором NaCl (100 мл), сушили (Na2SO4),фильтровали и концентрировали для получения желтого масла (106,5 г). Перегонка при 1-5 мм и при 80 С дала светло-желтое масло (89,5 г,75%). 1B) 5-Фтор-1,4-дигидро-1,4-метанонафталин. Раствор 2,6-дифториодбензола (5,0 г, 20,8 ммоль) и циклопентандиена (2,07 г, 31,3 ммоль) перемешивали при 0 С в перт. эфире (70 мл, 4060 С) под N2 и обрабатывали, добавляя по каплям n-BuLi (8,74 мл, 2,5 М в гексанах, 21,8 ммоль) на протяжении 10 мин. Реакционную смесь гасили через 15 мин путем добавления водного раствора 1 н. НСl и продукт экстрагировали гексанами (350 мл). Объединенный органический слой промывали Н 2 О (50 мл), насыщенным водным раствором NaCl (50 мл), сушили (MgSO4), фильтровали и упаривали. Хроматография на силикагеле дала продукт в виде масла (1,5 г, 45%). (ТСХ гексаны, отн. подв. 0,55). 1 36 Хлористо-водородную соль 10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триена (12,4 г,63,9 ммоль) перемешивали в CH2Cl2 (200 мл). Эту смесь охлаждали (ледяная баня) и обрабатывали пиридином (12,65 г, 160 ммоль) с последующей обработкой трифторуксусным ангидридом (TFAA) (16,8 г, 11,3 мл, 80 ммоль) из дополнительной воронки на протяжении 10 мин. После 3 ч раствор вливали в 0,5 н. водную НСl(200 мл) и слои разделяли. Водный слой экстрагировали CH2Cl2 (350 мл) и объединенный органический слой промывали 0,5 н. водной НСl(50 мл), Н 2 О (250 мл) и насыщенным водным раствором NaHCO3 (50 мл). Этот раствор сушили с помощью ватной пробки, затем разводили 3% EtOAc и фильтровали через 2-дюймовую (1 дюйм = 25,410-3 м) подложку из двуокиси кремния, элюируя 3% EtOAc)/СН 2 Сl2. Концентрирование дало прозрачное масло, которое,кристаллизуясь, дает белые иглы (15,35 г, 60,2 ммоль, 94%). (ТСХ 30% EtOAc/гексаны, отн. подв. 0,53). 1 Н ЯМР (400 МГц, СDСl3)7,18 (м, 4 Н),4,29 (уш.д, J=12,6 Гц, 1 Н), 3,84 (уш.д, J=12,6 Гц,1 Н), 3,51 (дд, J=12,6, 1,5 Гц, 1 Н), 3,21 (уш.с, 1 Н),3,10 (уш.с, 1 Н), 3,10 (уш.д, J=12,6 Гц, 1 Н), 2,37(2,4 мл, 13,7 ммоль) в СН 2 Сl2 (10 мл), перемешиваемому при 0 С, медленно добавляли азотную кислоту (0,58 мл, 27,4 ммоль), что давало белый осадок. Через 10 мин полученную смесь охлаждали до -78 С и обрабатывали добавлением по каплям через дополнительную воронку в течение 5 мин 1-(10-азатрицикло[6.3.1.02,7] додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанона (3,5 г, 13,7 ммоль) в СН 2 Сl2 (15 мл). Реакционную смесь перемешивали при -78 С в течение 30 мин и затем нагревали до 0 С в течение 1 ч. Реакционную смесь выливали в тщательно перемешиваемый лед (100 г). Слои разделяли и водный слой экстрагировали СН 2 Сl2 (330 мл). Органический слой объединяли и промывали Н 2O (330 мл). Объединенный органический слой промывали насыщенным водным раствором NaHCO3 (20 мл) и H2O (20 мл), затем сушили с помощью ватной пробки и концентрировали для получения оранжевого масла, которое затвердевало при стоянии (4,2 г). Хроматография дала чистый продукт в виде кристаллического твердого вещества (3,2 г, 78%). (ТСХ 30%CH2Cl2. Органический слой экстрагировали 1 н. водной НСl (320 мл) и кислотный слой промывали CH2Cl2 (220 мл). Водный слой защелачивали до рН 10 с помощью Nа 2 СО 3 (р-р) и продукт экстрагировали СН 2 Сl2 (330 мл). Органический слой сушили с помощью ватной пробки и концентрировали до масла. Его растворяли в МеОН и обрабатывали 1 н. НСl в МеОН, концентрировали до твердого вещества,которое перекристаллизовывали из MeOH/Et2O для получения продукта в виде белого твердого вещества (73 мг, 50%). (ТСХ 5% MeOH/CH2Cl2APCI MS m/e 205,1 [(М+1)+]. Т.пл. 265270 С. Пример 8. Гидрохлорид 4-амино-10 азатрицикло[6.3.1.02,7]додека-2(7),3,5-триена. 4-Нитро-10-азатрицикло[6.3.1.02,7]додека 2(7),3,5-триен (500 мг, 2,08 ммоль) перемешивали в 1,4-диоксане (40 мл) и обрабатывали насыщенным водным раствором Nа 2 СО 3 (15 мл). Туда добавляли ди-трет-бутилдикарбонат (1,8 г,8,31 ммоль). После перемешивания в течение 18 ч реакционную смесь обрабатывали H2O (50 мл), экстрагировали CH2Cl2 (430 мл), сушили с помощью ватной пробки и концентрировали для получения масла (500 мг, 91%). Полученное масло (500 мг, 1,64 ммоль) растворяли в МеОН (30 мл), обрабатывали 10%Pd/C (50 мг) и гидрогенизировали под атмосферой Н 2 (45 psi) (фунтов на кв. дюйм, 1 psi = 6,89103 н/м 2) в течение 1 ч. Смесь фильтровали через подложку целита и концентрировали до прозрачного масла (397 мг, 88%). Это масло (50 мг, 0,18 ммоль) перемешивали в 3 н. HCl в EtOAc (3 мл) в течение 2 ч, затем концентрировали до белого твердого вещества (25 мг, 56%). 1A) 1-(4-Амино-10-азатрицикло[6.3.1.02,7] додека-2(7),3,5-триен-10-ил)-2,2,2-трифтороэтанон. Гидрогенизация 1-(4-нитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2 трифторэтанона (2,0 г, 6,66 ммоль) под атмосферой Н 2 (40 psi) (фунтов на кв.дюйм, 1 psi = 6,89103 н/м 2) и в присутствии 10% Pd/C (200 мг) в МеОН на протяжении 1,5 ч, фильтрация через целит и концентрирование дают желтое масло (1,7 г). (ТСХ 50% EtOAc/гексаны, отн. подв. 0,27). 1(850 мг, 3,14 ммоль) перемешивали в СН 2 Сl2 (5 мл) и обрабатывали триэтиламином (0,53 мл,3,76 ммоль) и ацетилхлоридом (0,23 мл, 3,2 ммоль), затем перемешивали в течение 18 ч. Стандартная обработка NаНСО 3 дала масло,которое хроматографировали для получения прозрачного масла (850 мг, 87%). (50%EtOAc. Органический слой экстрагировали 1 н. водной НСl (320 мл) и кислотный слой промывали EtOAc (220 мл). Водный слой защелачивали до рН 10 Nа 2 СО 3 (р-р) и продукт экстрагировали EtOAc (320 мл). Органический слой сушили (сульфат натрия (Na2SO4 и концентрировали до масла. Этот материал растворяли в МеОН и обрабатывали 3 н. НСl в EtOAc(3 мл), концентрировали и перекристаллизовывали из MeOH/Et2O для получения твердого вещества (40 мг, 50%). 1(850 мг, 2,72 ммоль) и 2,4-бис(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфид (реагент Ловессона) (1,1 г, 2,72 ммоль) объединяли в толуоле (10 мл) и давали кипеть с обратным холодильником в течение 1,5 ч. После охлаждения реакционную смесь обрабатывалиNаНСО 3. Органический слой сушили (Na2SO4),фильтровали, концентрировали и хроматографировали на силикагеле для получения продукта (410 мг, 44%). (50% EtOAc/гексаны, отн. подв. 0,38).B) Гидрохлорид 6-метил-5-тиа-7,13-диазатетрацикло[6.3.1.02,10.04,8]пeнтaдeкa-2(10),3,6,8 тетраена. Указанное выше масло, 2,2,2-трифтор-N(10-трифтортиоацетил-10-азатрицикло[6.3.1. 02,7]дoдeкa-2(7),3,5-триен-4-ил)тиоацетамид,(360 мг, 1,05 ммоль) растворяли в МеОН (10 мл) и 1 н. NaOH (5 мл) и добавляли к феррицианиду калия (К 3Fе(СN)6) (1,72 г, 5,23 ммоль) в Н 2O (10 мл). Эту смесь нагревали до 60 С в течение 1,5 ч, охлаждали, концентрировали и обрабатывалиEtOAc/H2O. Этот материал перемешивали в диоксане (20 мл) и обрабатывали Н 2 О (50 мл) иNa2CO3 до достижения рН 10. Туда добавляли ди-трет-бутилдикарбонат (436 мг, 2,0 ммоль) и смесь перемешивали в течение 18 ч. Реакционную смесь концентрировали, обрабатывали Н 2 О и экстрагировали CH2Cl2. Продукт хроматографировалиEtOAc/гексаны, отн. подв. 0,41) для получения масла (100 мг). Указанный выше продукт обрабатывали 3 н. HCl/EtOAc (3 мл) и нагревали до кипения с обратным холодильником в течение 15 мин,затем концентрировали до твердого вещества,которое азеотропировали отгонкой с СН 2 Сl2(2 х). Эти твердые вещества растворяли в минимальном количестве МеОН, затем насыщалиEt2O и перемешивали. Полученный белый кристаллический порошок собирали фильтрацией[6.3.1.02,7]додека-2(7),3,5-триен. А) 1-(4,5-Динитpo-10-aзaтpициклo[6.3.1. 02,7]дoдeкa-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон. (Основано на методе, описанном у Coon,С.L; Blucher, W.G.; Hill, М.Е.J. Org. Chem. 1973,25, 4243. В качестве дополнительного относящегося к этому примера введения двух нитро 003190(79,8 мл, 902,1 ммоль) в CH2Cl2 (550 мл), перемешиваемому при 0 С, медленно добавляли азотную кислоту (19,1 мл, 450,9 ммоль), вызывая образование белого преципитата. Через 10 мин по каплям из дополнительной воронки на протяжении 10 мин добавляли 1-(10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)2,2,2-трифторэтанон (50 г, 196 ммоль) в CH2Cl2(300 мл). Реакционную смесь перемешивали при 0 С в течение 2,5 ч и затем перемешивали при комнатной температуре в течение 24 ч. Реакционную смесь вливали в тщательно перемешиваемую смесь Н 2 О (500 мл) и льда (400 г). Слои разделяли и водный слой опять экстрагировали СН 2 Сl2 (3300 мл). Органический слой объединяли и отмывали Н 2 О (3300 мл). Объединенные водные слои опять экстрагировалиCH2Cl2 (2100 мл). Органический слой объединяли и насыщенным водным растворомNаНСО 3 (200 мл) и Н 2O (200 мл) затем сушили с помощью ватной пробки и концентрировали до твердых веществ. Растирание с EtOAc/гексанами дало белые твердые вещества, которые фильтровали и сушили (52 г, 151 ммоль, 77%). Маточную жидкость хроматографировали, что дало дополнительные 4,0 г из суммарных 56,0 гGCMS m/e 345 (М+). В) 4,5-Динитро-10-азатрицикло[6.3.1.02,7] додека-2(7),3,5-триен. 1-(4,5-Динитpo-10-aзaтpициклo[6.3.1.02,7] дoдeкa-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (3,7 г, 10,7 ммоль) и Nа 2 СО 3 (2,3 г, 21,4 ммоль) объединяли в МеОН (50 мл) и H2O (20 мл), затем нагревали до кипения с обратным холодильником в течение 18 ч. Реакционную смесь охлаждали, концентрировали, обрабатывали Н 2O и экстрагировали CH2Cl2 (350 мл),затем сушили с помощью ватной пробки. После концентрирования остаток хроматографировали для получения коричневых твердых соединений 41 А) трет-Бутиловый эфир 4,5-динитро-10 азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10 карбоновой кислоты. 4,5-Динитро-10-азатрицикло[6.3.1.02,7] додека-2(7),3,5-триен (1,9 г, 7,6 ммоль), перемешивали в 1,4-диоксане (75 мл) и обрабатывали насыщенным водным раствором Nа 2 СО 3 (10 мл). Туда добавляли ди-трет-бутилдикарбонат(3,31 г, 15,2 ммоль). После перемешивания в течение 6 ч реакционную смесь обрабатывали Н 2O (50 мл) и экстрагировали EtOAc (425 мл),сушили (Na2SO4), фильтровали, концентрировали и хроматографировали для получения продукта (1,9 г, 71%). (ТСХ 30% EtOAc/гексаныB) трет-Бутиловый эфир 4,5-диамино-10 азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10 карбоновой кислоты. трет-Бутиловый эфир 4,5-динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (1,9 г, 5,44 ммоль) гидрогенизировали в МеОН в атмосфере Н 2 (40 psi) (фунтов на кв.дюйм, 1 psi = 6,89103 н/м 2) в присутствии 10% Pd/C (100 мг) на протяжении 1,5 ч,затем фильтровали через целитную подложку и концентрировали до белых твердых веществPost, R.J. Tetrahedron Lett. 1993, 34, 1897). трет-Бутиловый эфир 4,5-диамино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (700 мг, 2,42 ммоль) растворяли в EtOH (10 мл) и уксусной кислоте (НОАс) (1 мл) и обрабатывали 1-этоксиэтиленмалононитрилом (329 мг, 2,42 ммоль). Полученную смесь нагревали до 60 С и перемешивали 18 ч. Реакционную смесь охлаждали, концентрировали, обрабатывали Н 2 О и насыщенным водным раствором Nа 2 СО 3 и экстрагировали EtOAc (350 мл), затем сушили(Na2SO4). После фильтрации и концентрирования остаток хроматографировали для получения коричневых твердых веществ (247 мг,36%). (ТСХ 5% MeOH/CH2Cl2 (NН 3) отн. подв. 0,28). 42 0,267 ммоль) перемешивали в 50% водном растворе NaOH (3 мл) и ДМСО (1 мл), затем обрабатывали 1-иодпропаном (0,03 мл, 0,321 ммоль). Эту смесь нагревали до 40 С (3 х), затем сушили(Na2SO4), фильтровали и концентрировали до масла (90 мг, 0,253 ммоль). (ТСХ 5%EtOAc (5 мл) и нагревали до 100 С в течение 1,2 ч. Смесь охлаждали, концентрировали, получали шлам в EtOAc и фильтровали для получения белого твердого вещества (25 мг, 34%). 1Segelstein, В.Е.; Chenard, В.L.; Macor, J.Е,; Post,R.J. Tetrahedron Lett. 1993, 34. 1897). трет-Бутиловый эфир 4,5-диамино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (1,0 г, 3,45 ммоль) растворяли в EtOH (10 мл) и НОАс (1 мл) и обрабатывали этоксиметиленмалононитрилом (421 мг, 3,45 ммоль). Полученную смесь нагревали до 60 С и перемешивали 18 ч. Реакционную смесь охлаждали, концентрировали, обрабатывали Н 2 О и насыщенным водным раствором Na2CO3 и экстрагировали EtOAc (350 мл), затем сушили(Na2SO4). После фильтрации и концентрирования остаток хроматографировали для получения коричневых твердых веществ (580 мг, 56%). 43 Используя методы, описанные в примере 12D, трет-бутиловый эфир 5,7,13-триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты превращали в соединение, указанное в заголовке, путем взаимодействия с иодометаном с последующим снятием защитных групп, как описано в примере 12 Е. 1GCMS m/e 213,5 (М+). Пример 16. Гидрохлорид 6,7-диметил 5,7,13-триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраена. Используя методы, описанные в примере 12D, трет-бутиловый эфир 6-метил-5,7,13 триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты превращали в соединение, указанное в заголовке, путем взаимодействия с иодметаном с последующим снятием защитных групп, как описано в примере 12 Е. 1APCI MS m/e 228,2 [(М+1)+]. Т.пл. 225230 С. Пример 17. Гидрохлорид 7-пропил-5,7,13 триазатетрацикло[9.3.1.02,10.04,8]пентадeкa-2(10),3,5,8-тетраена. Используя методы, описанные в примере 12D, трет-бутиловый эфир 5,7,13-триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты превращали в соединение, указанное в заголовке, путем взаимодействия с иодпропаном с последующим снятием защитных групп, как описано в примере 12 Е. 1Youngs, W.J. Tetrahedron Lett. 1995, 36, 6217). трет-Бутиловый эфир 4,5-динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10 карбоновой кислоты (500 мг, 1,43 ммоль) и 1 бутиламин (1,42 мл, 14,3 ммоль) объединяли в ТГФ (5 мл) и перемешивали 4 ч. Смесь разводили EtOAc (50 мл) и промывали Н 2 О (330 мл) затем сушили (Na2SO4), фильтровали и концентрировали до масла. Это масло пропускали через колонку с силикагельным фильтром для удаления основных примесей, элюируя 30%B) трет-Бутиловый эфир 4-бутиламино-5 амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5 триен-10-карбоновой кислоты. трет-Бутиловый эфир 4-бутиламино-5 нитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5 триен-10-карбоновой кислоты (460 мг, 1,27 ммоль) обрабатывали муравьино-кислым аммонием (850 мг, 12,7 ммоль) и 10% Pd(OH)2/C (50 мг) в МеОН (20 мл) и давали кипеть с обратным холодильником в течение 1 ч, затем фильтровали через подложку целита и концентрировали. Твердые вещества обрабатывали насыщенным СН 2 Сl2 (330 мл) и сушили с помощью ватной пробки для получения масла (440 мг, 100%).C) трет-Бутиловый эфир 7-бутил 5,7,13 триазатетрацикло-[9.3.1.02,10.04,8]пентадека 2(10),3,5,8-тетраен-13-карбоновой кислоты. трет-Бутиловый эфир 4-бутиламино-5 амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5 триен-10-карбоновой кислоты (440 мг, 1,27 ммоль) растворяли в EtOH (20 мл) и НОАс (2 мл) и обрабатывали этоксиметиленмалононитрилом (186 мг, 1,52 ммоль). Полученную смесь нагревали до 60 С и перемешивали 18 ч. Реакционную смесь охлаждали, концентрировали,обрабатывали Н 2 О и насыщенным водным раствором Nа 2 СО 3 и экстрагировали EtOAc (350 мл) и сушили (Na2SO4). После фильтрации и концентрирования остаток хроматографировали для получения желтого масла. (400 мг, 89%).A) трет-Бутиловый эфир 6-метил-7 изобутил-5,7,13-триазатетрацикло[9.3.1.02,10.04,8] пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты. трет-Бутиловый эфир 4-амино-5-бутиламино-10-азатрицикло[6. 3.1.02,7]додека-2(7),3,5 триен-10-карбоновой кислоты (250 мг, 0,74 ммоль) из примера 19 В растворяли в EtOH (10 мл) и НОАс (2 мл) и обрабатывали 1 этоксиэтиленмалононитрилом (118 мг, 0,87 ммоль). Реакцию проводили как в примере 18 С(18 ч) и обрабатывали аналогично для получения продукта. (ТСХ 3% MeOH/CH2Cl2 (NН 3), Rf 0,57).APCI MS m/e 270,3 [(М +1)+]. Т.пл. 129130 С (субл.). Пример 21. Гидрохлорид 7-фенил-5,7,13 триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраена. С помощью методов, описанных в примере 18 А, трет-бутиловый эфир 4,5-динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты и анилин превращали в третбутиловый эфир 4-фениламино-5-нитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10 карбоновой кислоты при 75 С в течение 4 ч на стадии присоединения. Этот продукт затем превращали в соединение, указанное в заголовке, с помощью методов, описанных в примере 18B,C, D.J. Org. Chem. 1947, 522). трет-Бутиловый эфир 4,5-динитро-10-азатрицикло[6.3.1.02,7]дoдeкa-2(7),3,5-триен-10-карбоновой кислоты (100 мг, 0,35 ммоль) нагревали до 80 С в Н 2 О (5 мл). Туда добавляли бутан-2,3 дион (0,034 мл, 0,38 ммоль) под N2 в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и экстрагировали EtOAc (340 мл). Объединенный органический слой промывали Н 2 О (230 мл), сушили (Na2SO4), фильтровали, концентрировали и хроматографировали на силикагеле для получения масла (120 мг,100%). Масло растворяли в 2 н. HCl в МеОН (5 мл) и нагревали до кипения с обратным холо 47 дильником в течение 30 мин, затем концентрировали. Перекристаллизация из MeOH/Et2O дала белый порошок (50 мг, 43%). (ТСХ EtOAc, отн. подв. 0,14). 1[(M+1)+]. Пример 26. Гидрохлорид 5,8,14-триазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7,9 пентаена. А) 1-(4,5-Диамино-10-азатрицикло[6.3.1. 02,7]дoдeкa-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон. 1-(4,5-динитро-10-азатрицикло[6.3.1.02,7] дoдeкa-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (3,0 г, 8,70 ммоль) гидрогенизировали в МеОН (30 мл) в атмосфере Н 2 (45 psi) (фунтов на кв.дюйм, 1 psi = 6,89103 н/м 2) в присутствии 10% Pd(OH)2 (300 мг 20% вес.%/конц. 10 вес.%). Через 2,5 ч реакционную смесь фильтровали через целитную подложку и промывали МеОН (30 мл). Раствор концентрировали до светло-коричневого масла, которое кристаллизовали (2,42 г, 96%). (ТСХ 10% MeOH,CH2Cl2,отн. подв. 0,56).(2 мл). Эту смесь обрабатывали Н 2 О (2 мл) и продуктом присоединения гидратом бисульфит натрия глиоксаля (931 мг, 3,50 ммоль), затем перемешивали при 55 С в течение 2,5 ч. Реакционную смесь охлаждали до комнатной температуры и экстрагировали EtOAc (340 мл). Объединенный органический слой промывали Н 2 О (230 мл), сушили (Na2SO4), фильтровали,концентрировали и хроматографировали на силикагеле для получения беловатого порошка[10.3.1.02,11.04,9]гексадека-2(11),3,5,7,9-пентаен)2,2,2-трифторэтанона (320 мг, 1,04 ммоль) в МеОН (2,0 мл) и обрабатывали Nа 2 СО 3 (221 мг,2,08 ммоль) в Н 2 О (2,0 мл). Смесь нагревали до 70 С в течение 2 ч, затем концентрировали, обрабатывали Н 2 О (20 мл) и экстрагировалиCH2Cl2 (310 мл). Органический слой сушили с помощью ватной пробки и концентрировали для получения светло-желтого масла (183 мг, 83%),которое отвердевало при стоянии (т.пл. 138 003190 48 140 С). Этот материал растворяли в МеОН (10 мл), обрабатывали 3 М HCl/EtOAc (3 мл), концентрировали и азеотропной отгонкой с МеОН(220 мл) получали твердые вещества, которые перекристаллизовывали из MeOH/Et2O для получения соединения в виде белого твердого вещества (208 мг, 97%). (ТСХ 5% МеОН/ СН 2 Сl2(NН 3), Rf 0,26). 1GCMS m/e 211 (М+). Т.пл. 225-230 С. Пример 27. Гидрохлорид 14-метил-5,8,14 триазатетрацикло[10.3.1.02,11.04,9]гексадека 2(11),3,5,7,9-пентаена. 5,8,14-Триазатетрацикло[103.1.02,11.04,9] гексадека-2(11),3,5,7,9-пентаен (207 мг, 0,98 ммоль) обрабатывали 37% водным раствором формалина (1 мл) и муравьиной кислотой (1 мл), затем нагревали до 80 С в течение 1 ч. Реакционную смесь вливали в воду, подщелачивали (NaOH, pH 11) и экстрагировали EtOAc. Органический слой сушили (Na2SO4), концентрировали и хроматографировали на силикагеле для получения желтого твердого вещества. Его перемешивали в МеОН (2 мл) и обрабатывали 3 н. НСl в EtOAc (2 мл). После концентрирования твердые вещества перекристаллизовывали из MeOH/Et2O для получения продукта в виде белого твердого вещества (70 мг, 27%). (2% МеОН/СН 2 Сl2 (NН 3), Rf 0,47). 1(2,6 г, 26,1 ммоль) растворяли в ДМСО (10 мл) и нагревали при перемешивании до 100 С в течение 16 ч. Смесь охлаждали и разбавляли Н 2 ОEtOAc/гексанами (625 мл). Органический слой промывали Н 2 О (320 мл), сушили(Na2SO4), фильтровали и концентрировали, а также очищали хроматографией для получения масла (575 мг, 70%). (ТСХ 50% 35 EtOAc/ гексаны (NН 3), отн. подв. 0,56).B) 2,2,2-Трифтор-1-(4-гидрокси-5-амино 10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен 10-ил)этанон. 2,2,2-Трифтор-1-(4-гидрокси-5-нитро-10 азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10 49 ил)этанон (575 мг, 1,82 ммоль) гидрогенизировали в МеОН в атмосфере Н 2 при (45 psi) (фунтов на кв.дюйм, 1 psi = 6,89103 н/м 2) в присутствии 10% Pd/C (80 мг) в течение 1,5 ч, затем фильтровали через целитную подложку и концентрировали до белых твердых веществ (450 мг, 86%). (ТСХ 5% MeOH/CH2Cl2 (NН 3), отн. подв. 0,6). 1Het. Chem. 1990, 27, 335). 2,2,2-Трифтор-1-(4-гидрокси-5-амино-10 азатрицикло[6.3.1.02,7]дoдекa-2(7),3,5-триен-10 ил)этанон (150 мг, 0,524 ммоль), триметилортоформат (0,19 мл, 1,73 ммоль), пиридиний-паратолуолсульфоновую кислоту (PPTS, 18 мг, 0,07 ммоль) и ксилены (10 мл) объединяли под азотом и перемешивали при 135 С в течение 18 ч. Смесь охлаждали, обрабатывали Н 2 О и экстрагировали EtOAc. Экстракты сушили (Na2SO4,фильтровали, концентрировали и очищали хроматографией для получения масла (110 мг,71%). (ТСХ 20% EtOAc/гексаны, Rf 0,40).D) Гидрохлорид 5-окса-7,13-диазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,6,8-тетраена. 2,2,2-Трифтор-1-(5-окса-7,13-диазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,6,8-тетраен)этанон (110 мг, 0,37 ммоль) перемешивали в МеОН (5 мл) и обрабатывали Nа 2 СО 3 (78 мг,0,74 ммоль) в Н 2 О (2 мл). Перемешиваемую смесь нагревали до 80 С в течение 2 ч, концентрировали до твердых веществ, разводили H2O и экстрагировали EtOAc (340 мл). Продукт экстрагировали в водном растворе 1 н. НСl (240 мл), который промывали EtOAc, затем нейтрализовали насыщенным водным растворомEtOAc (340 мл), сушили (Na2SO4), концентрировали и хроматографировали на силикагеле для получения масла. (ТСХ 5% MeOH/CH2Cl2CH2Cl2 и насыщали гексанами. Через 18 ч продукт собирали фильтрацией (55 мг, 63%). 1A) 2,2,2-Трифтор-1-(6-метил-5-окса-7,13 диазатетрацикло[9.3.1.02,10.04,8]пeнтaдeка-2(10),3,6,8-тетраен)этанон. 2,2,2-Трифтор-1-(4-гидрокси-5-амино-10 азатрицикло[6.3.1. 02,7]додека-2(7),3,5-триен-10 ил)этанон (150 мг, 0,524 ммоль), триэтилортоацетат (0,34 мл, 1,83 ммоль), пиридиний-паратолуолсульфоновую кислоту (PPTS, 20 мг, 0,08 ммоль) и ксилены (10 мл) объединяли под азотом и перемешивали при 135 С в течение 18 ч. Обработка, выделение и очистка как в примере 28 С дала соединение, поименованное в заголовке (90 мг, 55%).Nа 2 СО 3 (61 мг, 0,58 ммоль) в Н 2O (2 мл). Перемешиваемую смесь нагревали до 80 С в течение 2 ч, концентрировали до твердых веществ, разводили H2O и экстрагировали EtOAc (340 мл). Раствор сушили (Na2SO4), концентрировали и хроматографировали на силикагеле для получения масла. (ТСХ 10% MeOH/CH2Cl2, (NH3) отн. подв. 0,18). 1H ЯМР (свободное основание) (400 МГц,CDCl3)7,40 (с, 1 Н), 7,26 (с, 1H), 3,05-2,98 (м,4 Н), 2,72 (д, J=12,8 Гц, 2 Н), 2,59 (с, 3 Н), 2,46 (м,1H), 1,98 (д, J=10,5 Гц, 1H). Масло растворяли в МеОН и обрабатывали 3 н. НСl в EtOAc (4 мл), затем концентрировали,перемешивали в минимальном количестве СН 2 Сl2 и насыщали гексанами. Через 18 ч продукт собирали фильтрацией (10 мг, 13%). APCI(PPTS, 20 мг, 0,08 ммоль), пиридин (0,046 мл,0,576 ммоль) и ксилены (5 мл) объединяли под азотом и перемешивали при 135 С в течение 18 ч. Через 24 ч дополнительно добавляли PPTS(50 мг) и материал перемешивали при 135 С в течение дополнительных 24 ч. Обрабатывали,как и указанный выше сырой продукт (145 мг,0,375 ммоль), который объединяли с Nа 2 СО 3 (рр) (80 мг, 0,75 ммоль) в МеОН (5 мл) и Н 2O (2 мл) и нагревали до кипения с обратным холодильником. Через 3 ч реакционную смесь охлаждали и разводили водой, затем экстрагировалиCH2Cl2 (440 мл), сушили с помощью ватной пробки, затем хроматографировали для удаления основных примесей (5% МеОН/СН 2 Сl2(NН 3. Сырой материал обрабатывали избытком 3 н. НСl в EtOAc и концентрировали, затем растворяли в минимальном количестве МеОН и раствор насыщали Et2O и перемешивали. После перемешивания в течение 4 ч продукт собирали фильтрацией (85 мг, 68%). 1(NaOH) (690 мг) растворяли в H2O (7 мл) и добавляли к горячему кислотному раствору на протяжение 5 мин. Выпавшие белые твердые вещества фильтровали и промывали водой. 1-(4-Амино-10-азатрицикло[6.3.1.02,7] додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (460 мг, 1,7 ммоль) растворяли в H2O (2 мл) и концентрированном растворе НСl (1 мл), затем охлаждали до 0 С и обрабатывали, добавляя по каплям раствор нитрита натрия (NaNO2) (275 мг) в Н 2 О (1 мл). К полученному раствору добавляли CuCl (202 мг, полученный как описано выше, 2,04 ммоль) в концентрированном растворе НСl (2 мл) на протяжении 10 мин (наблюдалось образование газа). Полученный раствор нагревали до 60 С в течение 15 мин, затем охлаждали до комнатной температуры и экстрагировали EtOAc (430 мл). После сушки над Na2SO4, раствор фильтровали и концентрировали до масла, которое фильтровали через подложку из двуокиси кремния для удаления основного материала, элюируя 50% EtOAc/гексанами с получением масла (470 мг, 95%). В) Гидрохлорид 4-хлор-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триена. 1-(4-Хлоро-10-азатрицикло[6.3.1.02,7] додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (470 мг, 1,62 ммоль) и Nа 2 СО 3 (344 мг, 3,24 ммоль) в МеОН (30 мл) и Н 2 О (10 мл) нагревали до кипения с обратным холодильником. Через 2 ч реакционную смесь охлаждали и разводили водой, затем экстрагировали EtOAc (440 мл),сушили (Na2SO4), фильтровали и концентрировали до желтого масла. Сырой материал обрабатывали избытком 3 н. НСl в EtOAc и концентрировали, затем растворяли в минимальном количестве СН 2 Сl2, раствор насыщали гексанами и перемешивали. После перемешивания в течение 52 4 ч продукт собирали фильтрацией (155 мг,42%). 1(500 мг, 1,85 ммоль) растворяли в Н 2 О (5 мл) и концентрированном растворе Н 2SO4 (0,5 мл),затем охлаждали до 0 С и обрабатывали по каплям раствором нитрита натрия (NaNO2) (140 мг,2,04 ммоль) в Н 2 О (2 мл). Иодид калия (460 мг,2,78 ммоль) в 1 н. растворе Н 2SO4 (0,5 мл) добавляли на протяжении 10 мин (реакционная смесь становится темно-красной). Полученный раствор нагревали до комнатной температуры и перемешивали в течение 18 ч. Реакцию гасилиEtOAc (430 мл). После сушки (Na2SO4) раствор фильтровали и концентрировали до желтого масла, которое хроматографировали на силикагеле для получения желтого масла (260 мг,37%). (ТСХ 30% EtOAc/гексаны, отн. подв. 0,70). (Реакция с использованием 5,4 г, выполненная как указано выше, дала выход 5 г, 67%).NH4OH (50 мл) перемешивали в МеОН (250 мл) в течение 2 ч, затем концентрировали и осуществляли азеотропную отгонку с МеОН (250 мл). Полученный продукт перемешивали в 1,4 диоксане (75 мл) и обрабатывали насыщенным раствором Nа 2 СО 3 (15 мл). Туда добавляли дитрет-бутилдикарбонат (5,71 г, 26,2 ммоль). После перемешивания в течение 18 ч реакционную смесь обрабатывали Н 2 О (50 мл) и экстрагировали CH2Cl2 (430 мл), сушили (Na2SO4),фильтровали, концентрировали и хроматографировали на силикагелеEtOAc/гексаны) для получения продукта в виде масла (4,9 г, 98%).CuCN (108 мг, 1,21 ммоль) и NaCN (59 мг,1,21 ммоль) объединяли в сухом ДМФ (6 мл) и нагревали до 150 С под N2. В течение 20 мин образовывался раствор. Туда добавляли третбутиловый эфир 4-иод-10-азатрицикло[6.3.1.02,7] додека-2(7),3,5-триен-10-карбоновой кислоты(232 мг, 0,6 ммоль) в ДМФ (3,5 мл) и смесь перемешивали в течение 18 ч при 150 С. Реакционную смесь охлаждали и разводили 50% насыщенным водным раствором NaCl и экстрагировали 50% EtOAc/гексанами (330 мл). После сушки (Na2SO4), фильтрации и концентрирования продукт выделяли хроматографией (86 мг,50%). (ТСХ 20% EtOAc/гексаны, отн. подв. 0,28).(6 мл) и нагревали до кипения с обратным холодильником в течение 2 ч, затем концентрировали, растворяли в минимальном количестве МеОН, который насыщали Et2O и перемешивали в течение 18 ч. Продукт собирали фильтрациейGCMS m/e 184 (М+). Т.пл. 268-273 С. Пример 33. Гидрохлорид 3-(10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-4-ил)-5 метил-1,2,4-оксадиазола. трет-Бутиловый эфир 4-циано-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (300 мг, 1,1 ммоль) перемешивали в EtOH (10 мл). Туда добавляли гидрохлорид гидроксиламина (382 мг, 5,5 ммоль) и NaOH(242 мг, 6,05 ммоль) и смесь нагревали до кипения с обратным холодильником. Через 45 мин реакционную смесь охлаждали, разводили H2O и экстрагировали EtOAc. Органический слой сушили (Na2SO4) и концентрировали для получения желтого твердого вещества (110 мг, 0,35 ммоль). Это твердое вещество растворяли в пиридине (1 мл) и обрабатывали ацетилхлоридом(0,03 мл, 0,415 ммоль) и нагревали до 100 С в течение 18 ч. Реакционную смесь охлаждали,обрабатывали H2O и экстрагировали EtOAc. Органические экстракты промывали водой и насыщенным водным раствором NaCl, сушили(Na2SO4) и концентрировали. Хроматография на силикагеле дала продукт (50 мг, 0,15 ммоль)(25% EtOAc/гексаны, отн. подв. 0,18). Этот продукт обрабатывали 2 н. НСl в МеОН (10 мл),нагревали до 70 С в течение 1 ч, охлаждали,концентрировали и перекристаллизовывали изMeOH/Et2O для получения продукта (15 мг).A) 1-(4-Aцeтил-10-aзaтpициклo[6.3.1.02,7] дoдeкa-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон. 1-(10-Азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (253 мг,1,0 ммоль) и AcCl (0,68 мл, 10 ммоль) растворяли в DCE (3 мл) и обрабатывали хлоридом алюминия (АlСl3) (667 мг, 50 ммоль). Полученную желтую смесь перемешивали в течение 30 мин,затем выливали на лед и насыщенный водный раствор NаНСО 3. После перемешивания в течение 20 мин смесь экстрагировали CH2Cl2 (330 мл). Органический слой сушили с помощью ватной пробки, затем концентрировали до оранжево-желтого масла (255 мг, 86%).NH4OH (10 мл) перемешивали в МеОН (30 мл) в течение 3 ч, затем концентрировали и проводили азеотропную отгонку с МеОН (250 мл).(Этот продукт может быть прямо превращен в соль НСl: см. следующий пример). Полученный продукт перемешивали в 1,4-диоксане (20 мл) и обрабатывали насыщенным водным растворомNа 2 СО 3 (5 мл). Туда добавляли ди-третбутилдикарбонат (1,91 г, 8,74 ммоль). После перемешивания в течение 2 ч реакционную смесь обрабатывали Н 2 О (50 мл), экстрагировали CH2Cl2 (430 мл), сушили (Nа 2SO4), фильтровали, концентрировали и хроматографировали для получения масла (1,3 г, 100%). (ТСХ 40%C) Гидрохлорид 1-(10-азатрицикло[6.3.1. 02,7]додека-2(7),3,5-триен-4-ил)-1-этанона. трет-Бутиловый эфир 4-ацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (190 мг, 0,63 ммоль) обрабатывали 3 н. НСl в EtOAc и нагревали до 70 С в течение 1 ч, затем концентрировали и растворяли в минимальном количестве МеОН. Полученный раствор насыщали Et2O и перемешивали. Через 18 ч белый кристаллический продукт собирали фильтрацией (81 мг, 54%). 1A) Сложный эфир уксусной кислоты 10 трифторацетил-10-азатрицикло[6.3.1.02,7]додека 2(7),3,5-триен-4-ила.(2,5 г, 8,41 ммоль) и 3-хлорпероксибензойную кислоту (m-СРВА) (7,5 г, 42 ммоль) перемешивали в СН 2 Сl2 (20 мл) и нагревали до 40 С в течение 18 ч. Смесь охлаждали до комнатной температуры, затем обрабатывали диметилсульфидом (Ме 2S) (3 мл, 40,8 ммоль) и перемешивали в течение 24 ч. Полученную смесь выливали на лед и насыщенный водный растворNаНСО 3 (100 мл), затем экстрагировали Et2O (440 мл). Органический слой промывали насыщенным водным раствором Nа 2 СО 3 (340 мл),затем сушили (Na2SO4), фильтровали и концентрировали для получения масла (1,83 г, 69%).B) 2,2,2-Трифтор-1-(4-гидрокси-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил) этанон. Сложный эфир 10-трифторацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-4-ила уксусной кислоты (900 мг, 2,87 ммоль) перемешивали в МеОН (20 мл) и насыщенном водном растворе NаНСО 3 (15 мл) в течение 48 ч. Смесь концентрировали, разводили Н 2O и экстрагировали СН 2 Сl2 (320 мл), затем сушили с помощью ватной пробки. Хроматография на силикагеле дала чистый продукт (420 мг, 54%). (ТСХ 5% MeOH/CH2Cl2 Rf 0,44). 1(3/1, 5 мл), обрабатывали Nа 2 СО 3 (р-р) (40 мг,0,369 ммоль) и нагревали до 65 С в течение 2 ч. Смесь концентрировали, разводили H2O и экстрагировали CH2Cl2 (320 мл), затем сушили с помощью ватной пробки. Фильтрация через силикагельную подложку дала масло (10%EtOAc (3 мл), затем концентрировали, растворяли в минимальном количестве МеОН, который был насыщен Et2O, и перемешивали. Через 18 ч белый кристаллический продукт собирали фильтрацией (10 мг, 26%). 1EtOAc и сушили (Na2SO4). При хроматографии получено масло (190 мг, 24%) (ТСХ EtOAc Rf 0,75). 1(Na2SO4), фильтровали и концентрировали для получения желтого масла (177 мг, 93%).(0,3 мл. 2,8 ммоль), после чего перемешивали в течение 18 ч. Реакционную смесь обрабатывали Н 2 О и экстрагировали EtOAc. Экстракты сушили (Na2SO4), фильтровали и концентрировали до желтого масла, которое растворяли в безводном ДМФ (3 мл) и обрабатывали 60% NaH в масле(32 мг, 1,08 ммоль). После перемешивания в течение 18 ч вносили дополнительный 60% NaH в масле (33 мг) и смесь перемешивали в течение 2 ч. Реакцию гасили Н 2 О (5 мл) и экстрагировали 80% EtOAc/гексаны (330 мл). Органический слой промывали Н 2O (320 мл), сушили(Nа 2SO4), фильтровали, концентрировали и хроматографировали для получения масла (40%EtOAc (3 мл), концентрировали, растворяли в минимальном количестве CH2Cl2, насыщенного гексанами, и перемешивали. Через 18 ч белый кристаллический продукт собирали фильтрацией (18 мг, общий выход 13%). 1APCI MS m/e 215,2 [(М+1)+]. Пример 37. Гидрохлорид 4-(2-метил-2 Нпиразол-3-ил)-10-азатрицикло[6.3.1.02,7]додека 2(7),3,5-триена и гидрохлорид 4-(1-метил-1 Нпиразол-3-ил)-10-азатрицикло[6.3.1.02,7]додека 2(7),3,5-триена. 1-(4-Ацетил-10-азатрицикло[6.3.1.02,7] додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (1,0 г, 3,3 ммоль) и диметилформамиддиметилацеталь (DMF-DMA) (4,0 г, 33,6 ммоль) нагревали до 140 С в течение 18 ч. После охлаждения кристаллический осадок отфильтровывали и промывали EtOAc (690 мг, 58%). Указанное выше твердое вещество, 3 диметиламино-1-(10-трифторацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-4-ил) пропенон, (200 мг, 0,56 ммоль) растворяли вEtOH (2 мл), обрабатывали 5 н. НСl в EtOH (0,1 мл) и затем метилгидразином (0,6 ммоль). Полученную смесь нагревали до 70 С в течение 4 ч. Смесь охлаждали, разводили водой, экстрагировали EtOAc, сушили (Na2SO4) и концентрировали. Хроматография на силикагеле дала 3,1 смесь региоизомерных продуктов (130 мг, 68%).(ТСХ 50% EtOAc/гексаны Rf 0,40). Указанное выше масло (130 мг, 0,388 ммоль) и Na2 СО 3 (р-р) (82 мг, 0,775 ммоль) перемешивали в МеОН (10 мл) и H2O (5 мл) в течение 18 ч. После охлаждения реакционную смесь разводили водой, экстрагировали CH2Cl2,сушили с помощью ватной пробки и концентрировали. Продукт очищали хроматографией на силикагеле и концентрировали до масла. Соль получали с помощью 2 н. НСl в МеОН, концентрировали и перекристаллизовывали изMeOH/EtOAc для получения смеси 3,1 региоизомерных пирразолов (85 мг, 58%). (5%Org. Chem. 1950, 72, 629). 1-(10-Азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (539 мг,2,1 ммоль) перемешивали в CH2Cl2 (5 мл) и обрабатывали IСl3 (p-p) (982 мг, 4,21 ммоль). Полученный оранжевый раствор перемешивали в течение 0,5 ч, выливали в насыщенный водный раствор NаНSО 3 (25 мл), экстрагировали CH2Cl2(325 мл), сушили с помощью хлопковой пробки и концентрировали до масла (570 мг,84%) (ТСХ 50% EtOAc/гексаны Rf 0,62).(25 мл) и обрабатывали Nа 2 СО 3 (р-р) (5 г, 47 ммоль) в Н 2 О (5 мл). Перемешиваемую смесь нагревали до 70 С в течение 4 ч, концентрировали до твердого состояния, разводили Н 2 О и экстрагировали EtOAc (340 мл). Продукт экстрагировали 1 н. водным раствором НСl (240 мл), который промывали EtOAc и затем нейтрализовывали насыщенным водным растворомNа 2 СО 3 до рН 10. Продукт экстрагировали СН 2 Сl2 (340 мл), фильтровали через хлопковую пробку и концентрировали до масла (400 мг, 100%). Масло растворяли в МеОН, обрабатывали 3 н. НСl в EtOAc (4 мл) и концентрировали, после чего растворяли в минимальном количестве МеОН, насыщенного Et2O, и перемешивали в течение 18 ч. Продукт собирали фильтрациейEtOAc, сушили (Na2SO4), фильтровали и концентрировали для получения масла (640 мг,87%). (ТСХ 30% EtOAc/гексаны Rf 0,15).(320 мг, 0,9 ммоль) перемешивали в ТГФ (10 мл) и обрабатывали 40% Me2NH/H2O (15 мл). Через 10 мин смесь концентрировали и хроматографировали на силикагеле (ТСХ 30%EtOAc/гексаны отн. подв. 0,31) для получения масла (256 мг, 78%). Этот материал растворяли в МеОН (6 мл) и NH4OH (2 мл) и перемешивали в течение 18 ч. Смесь концентрировали и проводили азеотропную отгонку с МеОН (3 х). По 59 лученное масло растворяли в МеОН, обрабатывали 3 н. НСl в EtOAc (4 мл), концентрировали,растворяли в минимальном количестве МеОН,насыщенного Et2O, и перемешивали в течение 18 ч. Продукт в виде белого порошка собирали фильтрацией (163 мг, 59%). (ТСХ 10% МеОН,CH2Cl2 (NН 3) Rf 0,54). 1 Н ЯМР (данные для свободного основания) (400 МГц, СDСl3)7,64 (м, 2 Н), 7,41 (д,J=8,0 Гц, 1 Н), 3,30 (м, 2 Н), 3,20 (д, J=12,5 Гц,2 Н), 3,07 (дд, J=12,5, 2,2 Гц, 2 Н), 2,69 (с, 6 Н),2,45 (м, 1 Н), 2,00 (д, J=11,0 Гц, 1 Н). 13 С ЯМР (100 МГц, CDCl3)128,43,124,16, 122,75, 46,67, 46,55, 42,11, 39,44, 37,81,GCMS m/e 266 (М+). (Данные для соли НСl) 1GCMS m/e 266 (М+). Анал. Вычисл. для С 13 Н 18N2O2 НСl: С,51,56; Н, 6,32; N, 9,25. Обнаружено С, 51,36; Н,6,09; N, 9,09. Пример 40. Гидрохлорид (1-пирролидинилсульфонил)-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триена. Аналог пирролидина получали из 10 трифторацетил-10-азатрицикло[6.3.1.02,7]додека 2(7),3,5-триен-4-сульфонилхлорида (320 мг, 0,9 ммоль) как при замещении пирролина на этапе присоединения, описанного в примере 39 В. Продукт TFA выделяли в виде масла (314 мг,89%). Снятие защиты и превращение в соль, как в примере 39 В, дает белый порошок (189 мг,63%). (ТСХ 10% MeOH/CH2Cl2 (NH3) Rf 0,60). 60 карбоновой кислоты как эквивалент ортофторфенильной части). 1THE (10 мл), обрабатывали карбонилдиимидазолом (269 мг, 1,66 ммоль) и нагревали до 60 С в течение 18 ч. Смесь концентрировали, разбавляли CH2Cl2 (50 мл) и промывали 1 н. водным раствором НСl (310 мл). Органический слой сушили с помощью ватной пробки, концентрировали и хроматографировали на силикагеле(50% EtOAc/гексаны) для получения масла (130 мг). Этот материал превращали в соединение,поименованное в заголовке, с помощью способов, описанных в примере 9 С. 1
МПК / Метки
МПК: A61P 25/00, C07D 221/22, A61K 31/439
Метки: арилом, конденсированные, соединения, азаполициклические
Код ссылки
<a href="https://eas.patents.su/30-3190-azapoliciklicheskie-soedineniya-kondensirovannye-s-arilom.html" rel="bookmark" title="База патентов Евразийского Союза">Азаполициклические соединения, конденсированные с арилом</a>
Четырехциклические конденсированные соединения с гетероатомами, замещенные арилом, промежуточные соединения, способы получения, композиции и методы лечения

Номер патента: 1649
Опубликовано: 25.06.2001
Автор: Гриз Тимоти А.
МПК: A61P 5/30, A61K 31/35, C07D 471/00...
Метки: замещенные, получения, конденсированные, арилом, соединения, методы, гетероатомами, четырехциклические, лечения, способы, композиции, промежуточные
Формула / Реферат:
1. Соединение формулы I или II где Y обозначает -О-, -S-, -СН2-, -СН2СН2-, -СН=СН- или -NR4-; В обозначает -СН2- или -СО-; R1 и R2 обозначают каждый независимо -Н, -ОН, -O(C1-C4алкил), -ОСОС6Н5, -ОСО(С1-С6алкил), -ОSО2(С4-С6алкил), OSO2СF3, Сl или F; n равно 1 или 2; W обозначает -СН2- или >С=O; R3 обозначает 1-пиперидинил, 2-оксо-1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 2-оксо-1-пирролидинил,...
Конденсированные производные имидазола в качестве модуляторов множественной лекарственной устойчивости

Номер патента: 1004
Опубликовано: 28.08.2000
Авторы: Сюрлеро Доминик Луи Нестор Гилейн, Жансенс Франс Эдуард, Ленартс Йозеф Элизабет, Соммен Франсуа Мария
МПК: C07D 487/04, A61P 35/00, A61K 31/55...
Метки: производные, модуляторов, конденсированные, множественной, имидазола, устойчивости, лекарственной, качестве
Формула / Реферат:
1. Соединение формулы (I) его N-оксидная форма, фармацевтически приемлемая аддитивная соль или стереохимически изомерная форма, где пунктирная линия обозначает простую связь, n равно 1 или 2; R1 обозначает водород; галоген; формил; C1-4 алкил; C1-4 алкил, замещенный 1 или 2 заместителями, каждый из которых независимо выбран из гидрокси, C1-4 алкокси, C1-4 алкилкарбонилокси, имидазолила, тиазолила или оксазолила; или радикал формулы ...
Фармкомпозиция, содержащая соединения с анти-ха активностью и соединения антагониста агрегации тромбоцитов, их применение, набор, содержащий эти соединения, способ лечения или профилактики заболеваний, сопутствующих тромбообразованию

Номер патента: 2475
Опубликовано: 27.06.2002
Авторы: Кюродо Алан Х., Лидли Роберт Дж., Юзан Андре, Перроне Марк Х., Данвидди Кристофер Т.
МПК: A61P 7/02, A61K 31/715
Метки: содержащий, эти, сопутствующих, лечения, анти-ха, способ, тромбоцитов, фармкомпозиция, тромбообразованию, набор, соединения, активностью, содержащая, профилактики, агрегации, применение, антагониста, заболеваний
Формула / Реферат:
1. Фармацевтическая композиция, содержащая фармацевтически пригодный носитель и фармацевтически эффективные количества соединения, обладающего анти-Ха активностью, и соединения антагониста агрегации тромбоцитов. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что соединение, обладающее анти-Ха активностью, является низкомолекулярно весовым гепарином. 3. Фармацевтическая композиция по п.2, отличающаяся тем, что низкомолекулярно весовой...
Хиральные бифосфиновые соединения, комплексы на их основе и способ получения хирального бифосфинового соединения

Номер патента: 1377
Опубликовано: 26.02.2001
Авторы: Пай Филип, Россен Кай, Воланте Ральф П.
МПК: C07F 9/50, C07B 53/00, C07C 25/22...
Метки: хирального, основе, хиральные, бифосфиновые, соединения, комплексы, способ, бифосфинового, получения
Формула / Реферат:
1. Хиральные бифосфины формулы где R представляет C1-4-алкил, С3-6-циклоалкил или арил, незамещенный или замещенный -F, -СН3, -СF3 или СН3О-; и Х1 и Х2 связывают два R2Р-замещенные бензола и независимо образуют 2-4-членную связывающую группу, состоящую из 2-4 атомов углерода и одного незамещенного или замещенного гетероатома, выбранных из О, S, SO, SO2 или 2. Соединение по п.1, в котором число атомов в связи X1 является таким же, как и...
Нафтильные соединения, промежуточные соединения для их получения, применение нафтильных соединений, способ снижения холестерина

Номер патента: 1600
Опубликовано: 25.06.2001
Авторы: Кроуелл Томас А., Палковиц Алан Д., Джонс Чарльз Д., Брайант Генри У.
МПК: C07C 47/546, A61P 19/10, A61K 31/33...
Метки: соединения, способ, получения, нафтильные, применение, снижения, соединений, нафтильных, промежуточные, холестерина
Формула / Реферат:
1. Соединение формулы I где R1 является -Н, -ОН, -O(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr; где Аr является фенилом или замещенным фенилом, OCO(С1-С6-алкилом), -O(СО)O(С1-С6-алкилом) или -ОSО2(С4-С6-алкилом); R2 является -Н, -F, -Cl, -ОН, -О(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr, где Аr является фенилом или замещенным фенилом, -OCO(С1-С6-алкилом),...
Предыдущий патент: Производные триазола, обладающие противогрибковой активностью, предназначенные для человека и животных
Следующий патент: Кристаллическая форма кальций (3s)тетрагидро-3-фуранил(1s,2r)-3-[[(4-аминофенил)сульфонил](изобу- тил)амино]-1-бензил-2-(фосфоноокси)пропилкарбамата
Случайный патент: Способ транспортировки и упаковки пищевых продуктов