Имидазоламины в качестве модуляторов киназной активности
Номер патента: 25098
Опубликовано: 30.11.2016
Авторы: Банкстон Доналд, Сяо Юфан, Диселм Лизбет Селест, Хак Бейард Р., Чэнь Сяолин, Неагу Константин, Джонс Кристофер Чарлз Виктор, Лань Руокси, Цю Сюй

























Формула / Реферат
1. Соединение формулы (I)

и его фармацевтически приемлемые соли,
в которой R1 представляет собой фенил, возможно замещенный 1-2 радикалами, выбранными из фтора, хлора, метила, трифторметила, метокси, трифторметокси, циано, гидроксиметила, аминометила, 2-гидроксипроп-2-ила, гидрокси, карбамоила, метоксикарбонила, карбокси; или
R1 представляет собой пиридинил, возможно замещенный одним радикалом, выбранным из метила, метокси, хлора, фтора, амино, морфолин-4-ила, пиперазин-1-ила; пиримидин-5-ил, возможно замещенный в положении 2 радикалом амино или диметиламино; индол-6-ил, 2-метилтиазол-5-ил, пиразол-4-ил, изооксазол-4-ил, 4,5-дигидроизоксазол-4-ил, пиррол-3-ил; или
R1 представляет собой фтор, хлор, бром, циано, формил, C1-3алкокси, 2,2,2-трифторэтокси, этил(метил)аминометил, гидроксиметил, карбамоил, 2-метоксикарбонилэтенил, 2-карбамоилэтенил, 3-метоксипроп-1-енил, этенил, 1-метилэтенил, С1-3алкил, 2-циклопропилэтил, нитро, амино, ацетиламино, карбокси, 2-этоксиэтил, 2-циклопропилэтенил, С3-4циклоалкил, циклопент-1-енил;
R2 представляет собой Н, NH2 или MeNH;
R3 представляет собой N;
R4 представляет собой Н, Me, пиперидин-4-ил, пирролидин-3-ил, азетидин-3-ил азетидин-2-ил, пирролидин-2-илметил, азетидин-3-илметил, азетидин-2-илметил, 1-метилазетидин-3-ил, 1-гидроксиметилазетидин-3-илметил, 1-метилазетидин-3-илметил, 2-аминоэтил, 2-(диметиламино)этил, 2-метиламиноэтил, трет-бутиламиноэтил, этиламиноэтил, диэтиламиноэтил, (2-метоксиэтил)метиламиноэтил, (3-хлорпропил)аминоэтил, изопропилэтиламиноэтил, (2-диметиламиноэтил)метиламиноэтил, 2-(бензилметиламино)этил, 2-изобутиламиноэтил, 2-гидроксиэтил, 2-изопропиламиноэтил, 2-(2-метокси-1-метилэтиламино)этил, 2-изобутиламиноэтил, 2-метиламиноэтил, 2-трет-бутиламиноэтил, 2-(циклопропилметиламино)этил, 2-(изопропилметиламино)этил, 2-циклопентиламиноэтил, 2-(1,1-диметилпропиламино)этил, 2-(1,1-диметилпропиламино)этил, пирролидин-1-илэтил, необязательно замещенный одним или двумя атомами фтора; пиперидин-1-илэтил, необязательно замещенный одним или двумя атомами фтора; азетидин-1-илэтил, необязательно замещенный метилом или одним или двумя атомами фтора; морфолин-4-илэтил, 2-окса-6-азаспиро[3.3]гепт-6-илэтил, 2-окса-6-азаспиро[3.4]окт-6-илэтил, 6-окса-1-азаспиро[3.3]гепт-1-илэтил;
R5 представляет собой фенил, возможно замещенный 1-2 радикалами, выбранными из хлора, фтора, трифторметила, дифторметокси, метила, сульфамоила, мезила; или
R5 представляет собой пиридинил, замещенный двумя галогенами или одним радикалом, выбранным из хлора, фтора, С1-4алкила, трифторметила, метокси, циклопропила; или
R5 представляет собой циклогексил, фуран-3-ил, тиен-3-ил, изоксазол-4-ил, пиразол-4-ил, бензопиразол-5-ил, 1-оксо-1Н-пиридин-4-ил, 2-диметиламинопиримидин-5-ил.
2. Соединения по п.1, которые соответствуют формуле (II)

и их фармацевтически приемлемые соли,
в которой R1, R2, R3, R4 и R5 имеют значение, указанное для формулы (I).
3. Соединение в соответствии с п.1 или 2, где подробно не определенные остатки имеют значение, указанное для формулы (I), в соответствии с п.1, но
в подформуле 1
R1 представляет собой фтор, хлор, бром, циано, формил, С1-3алкокси, 2,2,2-трифторэтокси, карбамоил, 3-метоксипроп-1-енил, этенил, 1-метилэтенил, C1-3алкил, 2-этоксиэтил, фенил, пиридинил, пиримидин-5-ил, пиразол-4-ил, изооксазол-4-ил, 4,5-дигидроизоксазол-4-ил, пиррол-3-ил;
в подформуле 2
R2 представляет собой NH2;
в подформуле 3
R3 представляет собой N;
в подформуле 4
R4 представляет собой Me, пиперидин-4-ил, пирролидин-3-ил, азетидин-3-ил, азетидин-2-ил, пирролидин-2-илметил, азетидин-3-илметил, азетидин-2-илметил, 1-метилазетидин-3-ил, 1-метилазетидин-3-илметил, 2-аминоэтил, 2-(диметиламино)этил, 2-метиламиноэтил, трет-бутиламиноэтил, этиламиноэтил, диэтиламиноэтил, (2-метоксиэтил)метиламиноэтил, изопропилэтиламиноэтил, (2-диметиламиноэтил)метиламиноэтил, 2-(бензилметиламино)этил, 2-изобутиламиноэтил, 2-изопропиламиноэтил, 2-(2-метокси-1-метилэтиламино)этил, 2-изобутиламиноэтил, 2-метиламиноэтил, 2-трет-бутиламиноэтил, 2-(циклопропилметиламино)этил, 2-(изопропилметиламино)этил, 2-циклопентиламиноэтил, 2-(1,1-диметилпропиламино)этил, 2-(1,1-диметилпропиламино)этил, пирролидин-1-илэтил, пиперидин-1-илэтил, азетидин-1-илэтил, необязательно замещенный метилом; морфолин-4-илэтил;
в подформуле 5
R5 представляет собой циклогексил, фенил или пиридил, который не замещен или моно- или дизамещен галогеном, трифторметилом, дифторметокси, сульфамоилом, мезилом, С1-4алкилом, метокси, циклопропилом;
в подформуле 6
R1 представляет собой фтор, хлор, бром, циано, формил, С1-3алкокси, 2,2,2-трифторэтокси, карбамоил, 3-метоксипроп-1-енил, этенил, 1-метилэтенил, С1-3алкил, 2-этоксиэтил, фенил, пиридинил, пиримидин-5-ил, пиразол-4-ил, изооксазол-4-ил, 4,5-дигидроизоксазол-4-ил, пиррол-3-ил;
R2 представляет собой NH2,
R3 представляет собой N;
в подформуле 7
R2 представляет собой NH2,
R3 представляет собой N,
R4 представляет собой Me, пиперидин-4-ил, пирролидин-3-ил, азетидин-3-ил, азетидин-2-ил, пирролидин-2-илметил, азетидин-3-илметил, азетидин-2-илметил, 1-метилазетидин-3-ил, 1-метилазетидин-3-илметил, 2-аминоэтил, 2-(диметиламино)этил, 2-метиламиноэтил, трет-бутиламиноэтил, этиламиноэтил, диэтиламиноэтил, (2-метоксиэтил)метиламиноэтил, изопропилэтиламиноэтил, (2-диметиламиноэтил)метиламиноэтил, 2-(бензилметиламино)этил, 2-изобутиламиноэтил, 2-изопропиламиноэтил, 2-(2-метокси-1-метилэтиламино)этил, 2-изобутиламиноэтил, 2-метиламиноэтил, 2-трет-бутиламиноэтил, 2-(циклопропилметиламино)этил, 2-(изопропилметиламино)этил, 2-циклопентиламиноэтил, 2-(1,1-диметилпропиламино)этил, 2-(1,1-диметилпропиламино)этил, пирролидин-1-илэтил, пиперидин-1-илэтил, азетидин-1-илэтил, необязательно замещенный метилом; морфолин-4-илэтил;
в подформуле 8
R2 представляет собой NH2,
R3 представляет собой N,
R5 представляет собой циклогексил, фенил или пиридил, который не замещен или моно- или дизамещен галогеном, трифторметилом, дифторметокси, сульфамоилом, мезилом, С1-4алкилом, метокси, циклопропилом;
в подформуле 9
R1 представляет собой фтор, хлор, бром, циано, формил, С1-3алкокси, 2,2,2-трифторэтокси, карбамоил, 3-метоксипроп-1-енил, этенил, 1-метилэтенил, С1-3алкил, 2-этоксиэтил, фенил, пиридинил, пиримидин-5-ил, пиразол-4-ил, изооксазол-4-ил, 4,5-дигидроизоксазол-4-ил, пиррол-3-ил,
R2 представляет собой NH2,
R3 представляет собой N,
R5 представляет собой циклогексил, фенил или пиридил, который не замещен или моно- или дизамещен галогеном, трифторметилом, дифторметокси, сульфамоилом, мезилом, С1-4алкилом, метокси, циклопропилом;
в подформуле 10
R1 представляет собой фтор, хлор, бром, циано, формил, С1-3алкокси, 2,2,2-трифторэтокси, карбамоил, 3-метоксипроп-1-енил, этенил, 1-метилэтенил, C1-3алкил, 2-этоксиэтил, фенил, пиридинил, пиримидин-5-ил, пиразол-4-ил, изооксазол-4-ил, 4,5-дигидроизоксазол-4-ил, пиррол-3-ил,
R2 представляет собой NH2,
R3 представляет собой N,
R4 представляет собой Me, пиперидин-4-ил, пирролидин-3-ил, азетидин-3-ил, азетидин-2-ил, пирролидин-2-илметил, азетидин-3-илметил, азетидин-2-илметил, 1-метилазетидин-3-ил, 1-метилазетидин-3-илметил, 2-аминоэтил, 2-(диметиламино)этил, 2-метиламиноэтил, трет-бутиламиноэтил, этиламиноэтил, диэтиламиноэтил, (2-метоксиэтил)метиламиноэтил, изопропилэтиламиноэтил, (2-диметиламиноэтил)метиламиноэтил, 2-(бензилметиламино)этил, 2-изобутиламиноэтил, 2-изопропиламиноэтил, 2-(2-метокси-1-метилэтиламино)этил, 2-изобутиламиноэтил, 2-метиламиноэтил, 2-трет-бутиламиноэтил, 2-(циклопропилметиламино)этил, 2-(изопропилметиламино)этил, 2-циклопентиламиноэтил, 2-(1,1-диметилпропиламино)этил, 2-(1,1-диметилпропиламино)этил, пирролидин-1-илэтил, пиперидин-1-илэтил, азетидин-1-илэтил, необязательно замещенный метилом, морфолин-4-илэтил;
в подформуле 11
R2 представляет собой NH2,
R3 представляет собой N,
R4 представляет собой Me, пиперидин-4-ил, пирролидин-3-ил, азетидин-3-ил, азетидин-2-ил, пирролидин-2-илметил, азетидин-3-илметил, азетидин-2-илметил, 1-метилазетидин-3-ил, 1-метилазетидин-3-илметил, 2-аминоэтил, 2-(диметиламино)этил, 2-метиламиноэтил, трет-бутиламиноэтил, этиламиноэтил, диэтиламиноэтил, (2-метоксиэтил)метиламиноэтил, изопропилэтиламиноэтил, (2-диметиламиноэтил)метиламиноэтил, 2-(бензилметиламино)этил, 2-изобутиламиноэтил, 2-изопропиламиноэтил, 2-(2-метокси-1-метилэтиламино)этил, 2-изобутиламиноэтил, 2-метиламиноэтил, 2-трет-бутиламиноэтил, 2-(циклопропилметиламино)этил, 2-(изопропилметиламино)этил, 2-циклопентиламиноэтил, 2-(1,1-диметилпропиламино)этил, 2-(1,1-диметилпропиламино)этил, пирролидин-1-илэтил, пиперидин-1-илэтил, азетидин-1-илэтил, необязательно замещенный метилом, морфолин-4-илэтил,
R5 представляет собой циклогексил, фенил или пиридил, который не замещен или моно- или дизамещен галогеном, трифторметилом, дифторметокси, сульфамоилом, мезилом, С1-4алкилом, метокси, циклопропилом;
в подформуле 12
R1 представляет собой фтор, хлор, бром, циано, формил, C1-3алкокси, 2,2,2-трифторэтокси, карбамоил, 3-метоксипроп-1-енил, этенил, 1-метилэтенил, С1-3алкил, 2-этоксиэтил, фенил, пиридинил, пиримидин-5-ил, пиразол-4-ил, изооксазол-4-ил, 4,5-дигидроизоксазол-4-ил, пиррол-3-ил,
R2 представляет собой NH2,
R3 представляет собой N,
R4 представляет собой Me, пиперидин-4-ил, пирролидин-3-ил, азетидин-3-ил, азетидин-2-ил, пирролидин-2-илметил, азетидин-3-илметил, азетидин-2-илметил, 1-метилазетидин-3-ил, 1-метилазетидин-3-илметил, 2-аминоэтил, 2-(диметиламино)этил, 2-метиламиноэтил, трет-бутиламиноэтил, этиламиноэтил, диэтиламиноэтил, (2-метоксиэтил)метиламиноэтил, изопропилэтиламиноэтил, (2-диметиламиноэтил)метиламиноэтил, 2-(бензилметиламино)этил, 2-изобутиламиноэтил, 2-изопропиламиноэтил, 2-(2-метокси-1-метилэтиламино)этил, 2-изобутиламиноэтил, 2-метиламиноэтил, 2-трет-бутиламиноэтил, 2-(циклопропилметиламино)этил, 2-(изопропилметиламино)этил, 2-циклопентиламиноэтил, 2-(1,1-диметилпропиламино)этил, 2-(1,1-диметилпропиламино)этил, пирролидин-1-илэтил, пиперидин-1-илэтил, азетидин-1-илэтил, необязательно замещенный метилом, морфолин-4-илэтил,
R5 представляет собой циклогексил, фенил или пиридил, который не замещен или моно- или дизамещен галогеном, трифторметилом, дифторметокси, сульфамоилом, мезилом, С1-4алкилом, метокси, циклопропилом;
в подформуле 13
R1 представляет собой Cl, CN, CONH2, изопропил, изопропилокси, этенил,
R2 представляет собой NH2,
R3 представляет собой N;
в подформуле 14
R2 представляет собой NH2,
R3 представляет собой N,
R4 представляет собой пирролидин-3-ил, азетидин-3-ил, азетидин-2-ил, пирролидин-2-илметил, азетидин-3-илметил, азетидин-2-илметил, 1-метилазетидин-3-ил, 1-метилазетидин-3-илметил, 2-(диметиламино)этил, трет-бутиламиноэтил, этиламиноэтил, диэтиламиноэтил, 2-изобутиламиноэтил, 2-изопропиламиноэтил, 2-метиламиноэтил, 2-трет-бутиламиноэтил, 2-(циклопропилметиламино)этил, 2-(изопропилметиламино)этил, пирролидин-1-илэтил, азетидин-1-илэтил, необязательно замещенный метилом;
в подформуле 15
R2 представляет собой NH2,
R3 представляет собой N,
R5 представляет собой фенил или пиридил, который замещен в пара-положении галогеном и/или замещен в мета-положении галогеном или трифторметилом, мезилом, С1-4алкилом;
в подформуле 16
R1 представляет собой Cl, CN, CONH2, изопропил, изопропилокси, этенил,
R2 представляет собой NH2,
R3 представляет собой N,
R4 представляет собой пирролидин-3-ил, азетидин-3-ил, азетидин-2-ил, пирролидин-2-илметил, азетидин-3-илметил, азетидин-2-илметил, 1-метилазетидин-3-ил, 1-метилазетидин-3-илметил, 2-(диметиламино)этил, трет-бутиламиноэтил, этиламиноэтил, диэтиламиноэтил, 2-изопропиламиноэтил, 2-изобутиламиноэтил, 2-метиламиноэтил, 2-трет-бутиламиноэтил, 2-(циклопропилметиламино)этил, 2-(изопропилметиламино)этил, пирролидин-1-илэтил, азетидин-1-илэтил, необязательно замещенный метилом;
в подформуле 17
R1 представляет собой Cl, CN, CONH2, изопропил, изопропилокси, этенил,
R2 представляет собой NH2,
R3 представляет собой N,
R5 представляет собой фенил или пиридил, который замещен в пара-положении галогеном и/или замещен в мета-положении галогеном или трифторметилом, мезилом, С1-4алкилом;
в подформуле 18
R2 представляет собой NH2,
R3 представляет собой N,
R4 представляет собой пирролидин-3-ил, азетидин-3-ил, азетидин-2-ил, пирролидин-2-илметил, азетидин-3-илметил, азетидин-2-илметил, 1-метилазетидин-3-ил, 1-метилазетидин-3-илметил, 2-(диметиламино)этил, трет-бутиламиноэтил, этиламиноэтил, диэтиламиноэтил, 2-изобутиламиноэтил, 2-изопропиламиноэтил, 2-изобутиламиноэтил, 2-метиламиноэтил, 2-трет-бутиламиноэтил, 2-(циклопропилметиламино)этил, 2-(изопропилметиламино)этил, пирролидин-1-илэтил, азетидин-1-илэтил, необязательно замещенный метилом,
R3 представляет собой фенил или пиридил, который замещен в пара-положении галогеном и/или замещен в мета-положении галогеном или трифторметилом, мезилом, С1-4алкилом;
в подформуле 19
R1 представляет собой Cl, CN, CONH2, изопропил, изопропилокси, этенил,
R2 представляет собой NH2,
R3 представляет собой N,
R4 представляет собой пирролидин-3-ил, азетидин-3-ил, азетидин-2-ил, пирролидин-2-илметил, азетидин-3-илметил, азетидин-2-илметил, 1-метилазетидин-3-ил, 1-метилазетидин-3-илметил, 2-(диметиламино)этил, трет-бутиламиноэтил, этиламиноэтил, диэтиламиноэтил, 2-изопропиламиноэтил, 2-изобутиламиноэтил, 2-метиламиноэтил, 2-трет-бутиламиноэтил, 2-(циклопропилметиламино)этил, 2-(изопропилметиламино)этил, пирролидин-1-илэтил, азетидин-1-илэтил, необязательно замещенный метилом,
R5 представляет собой фенил или пиридил, который замещен в пара-положении галогеном и/или замещен в мета-положении галогеном или трифторметилом, мезилом, С1-4алкилом,
и его фармацевтически приемлемые соли.
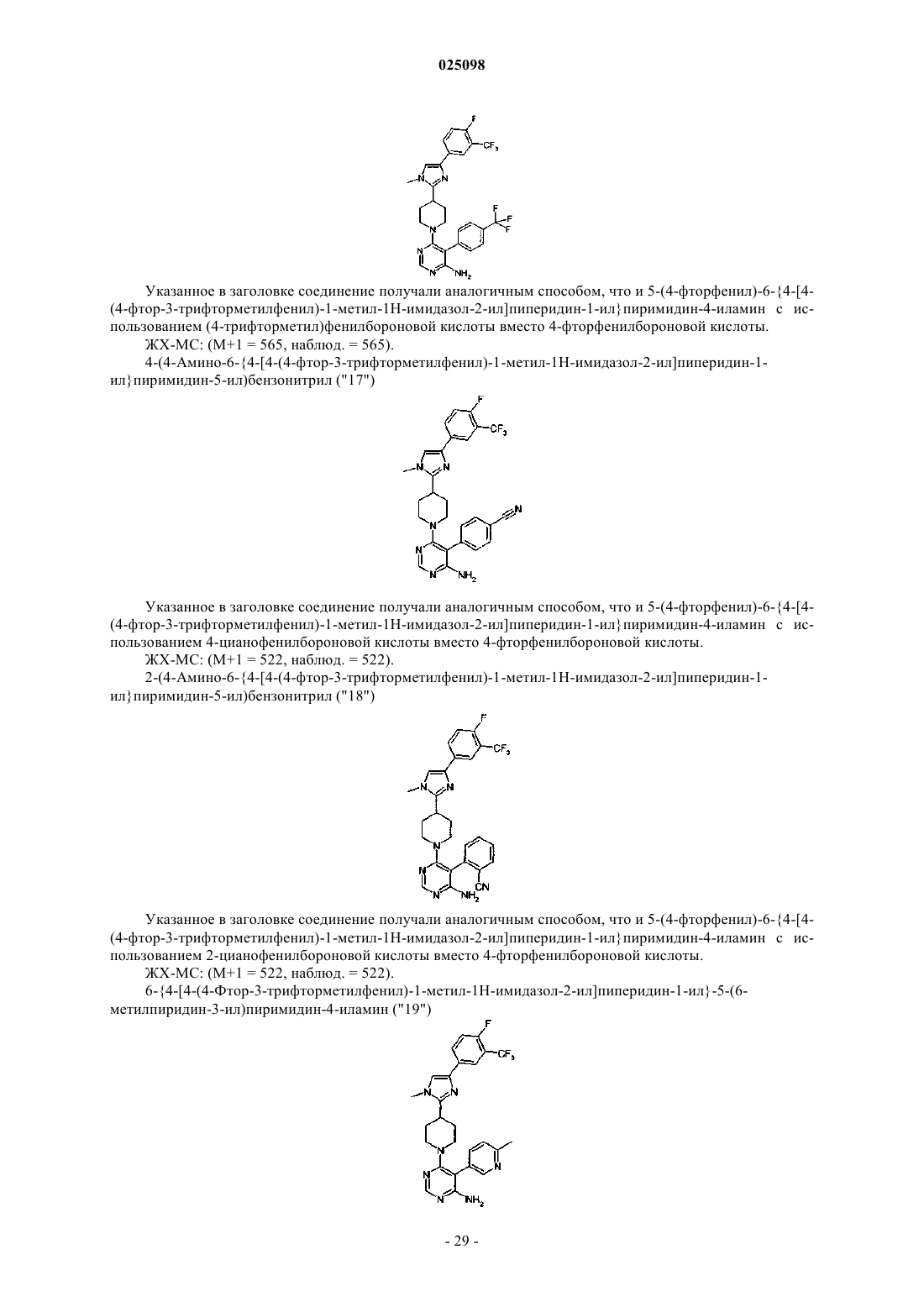
4. Соединения в соответствии с п.1, выбранные из группы, которая включает следующие соединения:
амид 4-амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1Н-имидазол-2-ил]пиперидин-1-ил}пиримидин-5-карбоновой кислоты;
6-(4-(1-(2-(азетидин-1-ил)этил)-4-(2-(трифторметил)пиридин-4-ил)-1Н-имидазол-2-ил)пиперидин-1-ил)-5-хлорпиримидин-4-амин;
6-(4-(1-(2-(азетидин-1-ил)этил)-4-(4-фтор-3-(трифторметил)фенил)-1Н-имидазол-2-ил)пиперидин-1-ил)-5-изопропоксипиримидин-4-амин;
амид 4-амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(3-хлор-4-фторфенил)-1Н-имидазол-2-ил]пиперидин-1-ил}пиримидин-5-карбоновой кислоты;
6-(4-(1-(2-аминоэтил)-4-(4-фтор-3-(трифторметил)фенил)-1Н-имидазол-2-ил)пиперидин-1-ил)-5-этилпиримидин-4-амин;
4-амино-6-(4-{4-(4-фтор-3-трифторметилфенил)-1-[2-(2-метоксиэтиламино)этил]-1Н-имидазол-2-ил}пиперидин-1-ил)пиримидин-5-карбонитрил;
4-амино-6-(4-(1-(2-(азетидин-1-ил)этил)-4-(4-фтор-3-метилфенил)-1Н-имидазол-2-ил)пиперидин-1-ил)пиримидин-5-карбоксамид;
4-амино-6-{4-[1-(2-азетидин-1-илэтил)-4-циклогексил-1Н-имидазол-2-ил]пиперидин-1-ил}пиримидин-5-карбонитрил;
амид 4-амино-6-{4-[4-(4-фтор-3-трифторметилфенил)-1-(2-изопропиламиноэтил)-1Н-имидазол-2-ил]пиперидин-1-ил}пиримидин-5-карбоновой кислоты;
амид 4-амино-6-{4-[1-[2-(циклопропилметиламино)этил]-4-(4-фтор-3-метилфенил)-1Н-имидазол-2-ил]пиперидин-1-ил}пиримидин-5-карбоновой кислоты;
4-амино-6-(4-(4-(4-фтор-3-(трифторметил)фенил)-1-(пирролидин-3-ил)-1Н-имидазол-2-ил)пиперидин-1-ил)пиримидин-5-карбоксамид;
5-этил-6-(4-(4-(4-фтор-3-(трифторметил)фенил)-1-(пиперидин-4-ил)-1Н-имидазол-2-ил)пиперидин-1-ил)пиримидин-4-амин;
амид 4-амино-6-{4-[1-(2-циклопентиламиноэтил)-4-(4-фтор-3-метилфенил)-1Н-имидазол-2-ил]пиперидин-1-ил}пиримидин-5-карбоновой кислоты;
амид 4-амино-6-{4-[4-(4-фтор-3-метилфенил)-1-(2-метиламиноэтил)-1Н-имидазол-2-ил]пиперидин-1-ил}пиримидин-5-карбоновой кислоты;
5-этокси-6-{4-[4-(4-фтор-3-трифторметилфенил)-1-(2-изопропиламиноэтил)-1Н-имидазол-2-ил]пиперидин-1-ил}пиримидин-4-иламин;
5-хлор-6-(4-(1-(2-(циклопропиламино)этил)-4-(2-(трифторметил)пиридин-4-ил)-1Н-имидазол-2-ил)пиперидин-1-ил)пиримидин-4-амин;
6-(4-(1-(2-(этиламино)этил)-4-(4-фтор-3-(трифторметил)фенил)-1Н-имидазол-2-ил)пиперидин-1-ил)-5-изопропоксипиримидин-4-амин;
6-(4-(1-(2-(диметиламино)этил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил)пиперидин-1-ил)-5-(1Н-пиразол-4-ил)пиримидин-4-амин;
6-(4-(1-(2-(азетидин-1-ил)этил)-4-(2-изопропилпиридин-4-ил)-1Н-имидазол-2-ил)пиперидин-1-ил)-5-винилпиримидин-4-амин;
6-(4-(1-(азетидин-3-илметил)-4-(4-фтор-3-метилфенил)-1Н-имидазол-2-ил)циклогексил)-5-изопропилпиримидин-4-амин
и их фармацевтически приемлемые соли.
5. Соединение в соответствии с п.1, выбранное из группы, которая включает следующие соединения:
амид 4-амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1Н-имидазол-2-ил]пиперидин-1-ил}пиримидин-5-карбоновой кислоты;
4-амино-6-(4-(1-(2-(азетидин-1-ил)этил)-4-(4-фтор-3-метилфенил)-1Н-имидазол-2-ил)пиперидин-1-ил)пиримидин-5-карбоксамид;
6-(4-(1-(2-(диметиламино)этил)-4-(4-фтор-3-метилфенил)-1Н-имидазол-2-ил)пиперидин-1-ил)-5-(1Н-пиразол-4-ил)пиримидин-4-амин;
6-(4-(1-(2-(азетидин-1-ил)этил)-4-(2-изопропилпиридин-4-ил)-1Н-имидазол-2-ил)пиперидин-1-ил)-5-этилпиримидин-4-амин
и их фармацевтически приемлемые соли.
6. Фармацевтическая композиция, которая содержит соединение в соответствии с любым из пп.1-5 или его фармацевтически приемлемую соль в качестве активного ингредиента вместе с фармацевтически приемлемым носителем.
7. Применение соединения в соответствии с любым из пп.1-5 или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения злокачественного новообразования.
8. Способ для лечения злокачественного новообразования, который включает введение субъекту соединения в соответствии с любым из пп.1-5 или его фармацевтически приемлемой соли.
9. Способ в соответствии с п.8, в котором указанное злокачественное новообразование выбрано из группы, которая включает рак мозга, рак легкого, рак толстой кишки, эпидермоидный рак, плоскоклеточный рак, рак мочевого пузыря, рак желудка, рак поджелудочной железы, рак молочной железы, рак головы, рак шеи, ренальный рак, рак почки, рак печени, рак яичек, рак предстательной железы, колоректальный рак, рак матки, ректальный рак, рак пищевода, тестикулярный рак, гинекологический рак, рак щитовидной железы, меланому, гематологические злокачественные опухоли, такие как острая миелогенная лейкемия, множественная миелома, хронический миелоидный лейкоз, миелоидный клеточный лейкоз, глиома и саркома Капоши.
10. Способ получения соединений формулы (I) в соответствии с п.1 и их фармацевтически пригодных солей, который отличается тем, что:
соединение формулы, где LG представляет собой уходящую группу, обычно применяемую в нуклеофильных замещениях:

вводят в реакцию в основных условиях с промежуточным соединением формулы

с получением соединения формулы

11. Способ в соответствии с п.10, где LG представляет собой Hal.
Текст
ИМИДАЗОЛАМИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ КИНАЗНОЙ АКТИВНОСТИ Изобретение обеспечивает новые соединения имидазоламина в соответствии с формулами (I) и (II): Хак Бейард Р., Чэнь Сяолин, Сяо Юфан, Лань Руокси, Диселм Лизбет Селест, Цю Сюй, Неагу Константин,Банкстон Доналд, Джонс Кристофер Чарлз Виктор (US) их изготовление и применение для лечения гиперпролиферативных заболеваний, таких как злокачественное новообразование.(71)(73) Заявитель и патентовладелец: МЕРК ПАТЕНТ ГМБХ (DE) Область техники изобретения Изобретение относится к ряду соединений имидазоламинов, пригодных для лечения гиперпролиферативных заболеваний, таких как злокачественное новообразование, у млекопитающих. Также настоящее изобретение охватывает применение таких соединений в лечении гиперпролиферативных заболеваний у млекопитающих, особенно людей, и фармацевтических композиций, которые содержат такие соединения. Краткое изложение известного уровня техники Протеинкиназы составляют большое семейство структурно сходных ферментов, которые отвечают за регулирование многих путей передачи сигналов в клетках (Hardie, G. и Hanks, S. (1995), The ProteinKinase Facts Book. I and II, Academic Press, Сан-Диего, Калифорния). Киназы могут подразделяться на семейства согласно субстратам, которые они фосфорилируют (например, протеин-тирозин, протеинсерин/треонин, липиды и т.д.). Были идентифицированы последовательности, которые в целом соответствуют каждому из этих семейств киназ (например, Hanks, S.K., Hunter, Т., FASEB J., 9:576-596 (1995);Knighton, et al., Science, 253:407-414 (1991); Hiles, et al., Cell, 70:419-429 (1992); Kunz, et al., Cell, 73:585596 (1993); Garcia-Bustos, et al., EMBO, J., 13:2352-2361 (1994. Протеинкиназы могут быть охарактеризованы с помощью механизмов, регулирующих их активность. Эти механизмы включают, например, аутофосфорилирование, трансфосфорилирование с помощью других киназ, белок-белковые взаимодействия, белок-липидные взаимодействия и белокполинуклеотидные взаимодействия. Некоторые протеинкиназы могут регулироваться более чем одним механизмом. Киназы регулируют различные процессы в клетках, включая, но не ограничиваясь только ими, пролиферацию, апоптоз, подвижность, транскрипцию, трансляцию и другие сигнальные процессы, путем добавления фосфатных групп к белкам-мишеням. Такое фосфорилирование действует в качестве молекулярных "включающих"/"выключающих" "переключателей", которые могут модулировать или регулировать биологическую функцию белка-мишени. Фосфорилирование белков-мишеней происходит в ответ на действие различных внеклеточных сигналов (гормонов, нейромедиаторов, факторов роста и дифференциации и т.д.), событий клеточного цикла, факторов окружающей среды или пищевых стрессов и т.д. Характерной функцией протеинкиназ в путях передачи сигналов является активирование или инактивирование (непосредственно или косвенно), например, метаболического фермента, регуляторного белка,рецептора, белка цитоскелета, ионного канала или насоса или фактора транскрипции. Неконтролируемая передача сигналов вследствие нарушения контроля фосфорилирования белков вовлечена в различные заболевания, включая, например, воспаление, злокачественное новообразование, аллергию/астму, заболевания и состояния иммунной системы, заболевания и состояния центральной нервной системы и ангиогенез. Протеинкиназа 70S6K, киназа 70 кДа рибосомного белка p70S6K (также известна как SK6, р 70/р 85S6 киназа, р 70/р 85 рибосомная S6 киназа и pp70S6K), является представителем AGC субсемейства протеинкиназ. p70S6K является серин-треонин киназой, которая является компонентом фосфатидилинозитол 3 киназа (PI3K)/Akt пути. p70S6K расположена ниже PI3K, и активация происходит путем фосфорилирования в различных сайтах в ответ на действие различных митогенов, гормонов и факторов роста. Активность p70S6K также находится под контролем mTOR-содержащего комплекса (TORC1), поскольку рапамицин действует путем ингибирования p70S6K активности. p70S6K регулируется с помощью PI3K нижерасположенных мишеней Akt и PKC. Akt непосредственно фосфорилирует и инактивирует TSC2,таким образом активируя mTOR. Дополнительно, исследования с мутантными аллелями p70S6K, которая ингибируется вортманнином, но не рапамицином, свидетельствует от том, что PI3K путь может проявлять влияния на p70S6K независимо от регуляции активности mTOR. Фермент p70S6K модулирует синтез белка путем фосфорилирования S6 рибосомного белка. S6 фосфорилирование коррелирует с повышенной трансляцией мРНК, кодирующих компоненты трансляционного аппарата, включая рибосомные белки и факторы элонгации трансляции, повышенная экспрессия которых является важной для роста и пролиферации клеток. Эти мРНК содержат олигопиримидиновый участок на их 5' транскрипционном начале (обозначаемом 5'ТОР), который, как было показано, является важным для их регуляции на трансляционном уровне. Дополнительно к его вовлечению в трансляцию, активация p70S6K также задействована в контролирование клеточного цикла, дифференциацию нейронных клеток, регуляцию подвижности клеток и клеточную ответную реакцию, что является важным при метастазировании опухолей, иммунном ответе и восстановлении ткани. Антитела к p70S6K отменяют митогенный ответ, запускающий вхождение фибробластов крыс в S фазу, указывая на то, что p70S6K функция является важной для прохождения из G1 вS фазу в клеточном цикле. Кроме того, ингибирование пролиферации клеточного цикла с G1 в S фазу клеточного цикла с помощью рапамицина было идентифицировано в виде следствия ингибирования продукции гиперфосфорилированной активированной формы p70S6K. Роль p70S6K в пролиферации опухолевых клеток и защите клеток от апоптоза основывается на его участии в передаче сигналов рецепторов факторов роста, сверхэкспрессии и активации в опухолевых тканях. Например, при нозерн- и вестерн-анализах было установлено, что амплификация PS6K гена со-1 025098 провождается соответствующими повышениями мРНК и экспрессии белка соответственно (Cancer Res.Amplification in Breast Cancer). Хромосома 17q23 амплифицирована во вплоть до 20% первичных опухолей молочной железы, в 87% опухолей молочной железы, содержащей BRCA2 мутации и в 50% опухолей, содержащих BRCA1 мутаций, а также при других типах злокачественных новообразований, таких как поджелудочной железы, мочевого пузыря и нейробластомы (см. М. Barlund, О. Monni, J. Kononen, R. Cornelison, J. Torhorst,G. Sauter, O.-P. Kallioniemi и A. Kallioniemi, Cancer Res., 2000, 60:5340-5346). Было показано, что 17q23 амфлификации при раке молочной железы вовлечены РАТ 1, RAD51C, PS6K, и SIGMA1B гены (CancerRes. (2000): 60, p. 5371-5375). Ген p70S6K был идентифицирован в качестве мишени амплификации и сверхэкспрессии в этом участке, и наблюдается статистически достоверная связь между амплификацией и плохим прогнозом. Клиническое ингибирование p70S6K активации наблюдается у пациентов с раком почки, которых лечили с помощью CCI-779 (сложный эфир рапамицина), ингибитора вышерасположенной киназыmTOR. Описана достоверная линейная ассоциация между прогрессированием заболевания и ингибированием активности p70S6K. В ответ на энергетический стресс, опухолевый супрессор LKB1 активирует AMPK, который фосфорилирует TSC1/2 комплекс и предоставляет ему возможность инактивировать mTOR/p70S6K путь. Мутации в LKB1 вызывают синдром Пейтца-Егерса (PJS), где у пациентов PJS в 15 раз больше вероятность развития рака, чем у общей популяции. Дополнительно, 1/3 аденокарциномы легких заякоривают инактивирующие LKB1 мутации.p70S6K защищает от возрастного и индуцированного питанием ожирения, в то время как усиливает чувствительность к инсулину. Роль для p70S6K при метаболических заболеваниях и нарушениях, таких как ожирение, диабет, метаболический синдром, резистентность к инсулину, гипергликемия, гипераминоацидемия, и гиперлипидемия подтверждается этими наблюдениями. Соединения, описанные в качестве пригодных для ингибирования p70S6K, раскрыты вPCT/US10/000313. Соединения, описанные в качестве пригодных для ингибирования p70S6K, раскрыты вWO 03/064397, WO 04/092154, WO 05/054237, WO 05/056014, WO 05/033086, WO 05/117909,WO 05/039506, WO 06/120573, WO 06/136821, WO 06/071819, WO 06/131835, WO 08/140947, WO 10/056563, WO 10/093419, WO 12/013282, WO 12/016001 и WO 12/069146. Описание изобретения Объектом настоящего изобретения является обеспечение новых соединений, которые модулируют киназную активность. Эта модуляция протеинкиназы включает, но не ограничивается следующим, ингибирование p70S6K и ингибирование Akt, что является полезным для лечения гиперпролиферативных заболеваний, в особенности тех, которые связаны с гиперактивностью вышеуказанных протеинкиназ,таких как злокачественное новообразование у млекопитающих, с улучшенными фармакологическими свойствами как в отношении их активности, так и характеристик растворимости, метаболического клиренса и биодоступности. В результате данное изобретение обеспечивает новые соединения гетероциклических пиримидинил- и пиридиниламинов и их фармацевтически приемлемые соли, сольватов или пролекарств, которые являются ингибиторами киназ и пригодны для лечения вышеуказанных заболеваний. Соединения определены формулой (I) и их фармацевтически приемлемые соли,где R1 представляет собой фенил, возможно замещенный 1-2 радикалами, выбранными из фтора,хлора, метила, трифторметила, метокси, трифторметокси, циано, гидроксиметила, аминометила,2-гидроксипроп-2-ила, гидрокси, карбамоила, метоксикарбонила, карбокси; илиR1 представляет собой пиридинил, возможно замешенный одним радикалом, выбранным из метила,метокси, хлора, фтора, амино, морфолин-4-ила, пиперазин-1-ила; пиримидин-5-ил, возможно замещен-2 025098 ный в положении 2 радикалом амино или диметиламино; индол-6-ил, 2-метилтиазол-5-ил, пиразол-4-ил,изооксазол-4-ил, 4,5-дигидроизоксазол-4-ил, пиррол-3-ил; илиR4 представляет собой Н, Me, пиперидин-4-ил, пирролидин-3-ил, азетидин-3-ил, азетидин-2-ил,пирролидин-2-илметил, азетидин-3-илметил, азетидин-2-илметил, 1-метилазетидин-3-ил, 1-гидроксиметилазетидин-3-илметил, 1-метилазетидин-3-илметил, 2-аминоэтил, 2-(диметиламино)этил, 2-метиламиноэтил, трет-бутиламиноэтил, этиламиноэтил, диэтиламиноэтил, (2-метоксиэтил)метиламиноэтил,(3-хлорпропил)аминоэтил,изопропилэтиламиноэтил,(2-диметиламиноэтил)метиламиноэтил,2-(бензилметиламино)этил, 2-изобутиламиноэтил, 2-гидроксиэтил, 2-изопропиламиноэтил, 2-(2-метокси 1-метилэтиламино)этил,2-изобутиламиноэтил. 2-метиламиноэтил,2-трет-бутиламиноэтил,2-(циклопропилметиламино)этил,2-(изопропилметиламино)этил,2-циклопентиламиноэтил,2-(1,1-диметилпропиламино)этил, 2-(1,1-диметилпропиламино)этил, пирролидин-1-илэтил, необязательно замещенный одним или двумя атомами фтора; пиперидин-1-илэтил, необязательно замещенный одним или двумя атомами фтора; азетидин-1-илэтил, необязательно замещенный метилом или одним или двумя атомами фтора; морфолин-4-илэтил,2-окса-6-азаспиро[3.3]гепт-6-илэтил,2-окса-6-азаспиро[3.4]окт-6-илэтил, 6-окса-1-азаспиро[3.3]гепт-1-илэтил;R5 представляет собой пиридинил, замещенный двумя галогенами или одним радикалом, выбранным из хлора, фтора, С 1-4 алкила, трифторметила, метокси, циклопропила; илиR5 представляет собой циклогексил. фуран-3-ил, тиен-3-ил, изоксазол-4-ил, пиразол-4-ил, бензопиразол-5-ил, 1-оксо-1 Н-пиридин-4-ил, 2-диметиламинопиримидин-5-ил. В предпочтительном варианте осуществления соединения согласно изобретению соответствуют формуле (II), в которой все заместители имеют значения, указанные для формулы (I): В дополнительном предпочтительном варианте осуществления соединения согласно изобретению соответствуют подформулам 1-19 формул (I) или (II), где в подформуле 1R5 представляет собой циклогексил, фенил или пиридил, который не замещен или моно- или дизамещен галогеном, трифторметилом, дифторметокси, сульфамоилом, мезилом, С 1-4 алкилом, метокси,-3 025098R4 представляет собой собой Me, пиперидин-4-ил, пирролидин-3-ил, азетидин-3-ил, азетидин-2-ил,пирролидин-2-илметил,азетидин-3-илметил,азетидин-2-илметил,1-метилазетидин-3-ил,1 метилазетидин-3-илметил, 2-аминоэтил. 2-(диметиламино)этил, 2-метиламиноэтил, трет-бутиламиноэтил. этиламиноэтил, диэтиламиноэтил, (2-метоксиэтил)метиламиноэтил, изопропилэтиламиноэтил,(2-диметиламиноэтил)метиламиноэтил,2-(бензилметиламино)этил,2-изобутиламиноэтил,2-изопропиламиноэтил, 2-(2-метокси-1-метилэтиламино)этил, 2-изобутиламиноэтил, 2-метиламиноэтил,2-трет-бутиламиноэтил,2-(циклопропилметиламино)этил,2-(изопропилметиламино)этил,2-циклопентиламиноэтил, 2-(1,1-диметилпропиламино)этил, 2-(1,1-диметилпропиламино)этил, пирролидин-1-илэтил, пиперидин-1-илэтил, азетидин-1-илэтил, необязательно замещенный метилом; морфолин 4-илэтил; в подформуле 8R5 представляет собой циклогексил, фенил или пиридил, который не замещен или моно- или дизамещен галогеном, трифторметилом, дифторметокси, сульфамоилом, мезилом, С 1-4 алкилом, метокси,циклопропилом; в подформуле 9R5 представляет собой циклогексил, фенил или пиридил, который не замещен или моно- или дизамещен галогеном, трифторметилом, дифторметокси, сульфамоилом, мезилом, С 1-4 алкилом, метокси,циклопропилом; в подформуле 10R5 представляет собой циклогексил, фенил или пиридил, который не замещен или моно- или диза-4 025098R5 представляет собой циклогексил, фенил или пиридил, который не замещен или моно- или дизамещен галогеном, трифторметилом, дифторметокси, сульфамоилом, мезилом, С 1-4 алкилом, метокси,циклопропилом,в подформуле 13R5 представляет собой фенил или пиридил, который замещен в пара-положении галогеном и/или замещен в мета-положении галогеном или трифторметилом, мезилом, С 1-4 алкилом; в подформуле 16R5 представляет собой фенил или пиридил, который замещен в пара-положении галогеном и/или замещен в мета-положении галогеном или трифторметилом, мезилом, С 1-4 алкилом; в подформуле 18R5 представляет собой фенил или пиридил, который замещен в пара-положении галогеном и/или замещен в мета-положении галогеном или трифторметилом, мезилом, С 1-4 алкилом; в подформуле 19R5 представляет собой фенил или пиридил, который замещен в пара-положении галогеном и/или замещен в мета-положении галогеном или трифторметилом, мезилом, С 1-4 алкилом, и их фармацевтически приемлемые соли. Даже в более предпочтительных вариантах осуществления заместители, обозначенные R1, R4 и R5 в формуле (I) выше, определены, как указано ниже [с неизмененными заместителями формулы (I) с определениями, указанными выше]:R5 представляет собой 4-фтор-3-трифторметилфенил; 4-фтор-3-метилфенил; 2-трифторметилпиридин-4-ил; 2-изопропилпиридин-4-ил; 3-хлор-4-фторфенил; циклогексил; 3 трифторметилфенил; 3-хлор-4-метоксифенил; 3-хлор-4-метилфенил; 3-фтор-4-метоксифенил; 3-фтор-4 метилфенил; 2-трет-бутилпиридин-4-ил; 2-циклопропилпиридин-4-ил; 2-этилпиридин-4-ил; 4-метил-3 трифторметилфенил; 3-дифторметокси-4-фторфенил; 1H-пиразол-4-ил; изоксазол-4-ил; 4-дифторметоксифенил; фенил; тиофен-3-ил; фуран-3-ил; 6-хлорпиридин-2-ил; 2-фторпиридин-4-ил; 6-трифторметилпиридин-2-ил; 3-хлорофенил; 3-фгорфенил; 4-фтор-3-метоксифенил; 3,4-дифторфенил; 4-фторфенил; 4-хлорфенил; 2-фторфенил; 5-хлор-6-фторпиридин-3-ил; 2-метилпиридин-4-ил и 3-трифторметоксифенил. В другом предпочтительном варианте осуществления заместители, обозначенные R1 в формуле (I),представлены в табл. 1. Таблица 1 Предпочтительные заместители для R1 в формуле (I) В другом предпочтительном варианте осуществления заместители, обозначенные R4 в формуле (I),представлены в табл. 2. Таблица 2 Предпочтительные заместители для R4 в формуле (I) В другом предпочтительном варианте осуществления заместители, обозначенные R5 в формуле (I),представлены в табл. 3. Таблица 3 Предпочтительные заместители для R5 в формуле (I) Как правило, все остатки, которые встречаются больше одного раза, могут быть идентичными или различными, т е. независимыми друг от друга. Соединения настоящего изобретения могут быть в форме соединений-пролекарств. "Соединениепролекарство" означает производное, которое превращено в биологически активное соединение в соответствии с настоящим изобретением в физиологических условиях в живом организме, например, путем окисления, восстановления, гидролиза или т.п., каждое из которых осуществляют ферментативным путем или без участия ферментов. Примерами пролекарств являются соединения, где аминогруппа в соединении настоящего изобретения ацилирована, алкилирована или форфорилирована, например, эйкозаноиламино, аланиламино, пивалоилоксиметиламино, или где гидроксильная группа ацилирована, алкилирована, форфорилирована или превращена в борат, например, ацетилокси, палмитоилокси, пивалоилокси, сукцинилокси, фумарилокси, аланилокси, или где карбоксильная группа эстерифицирована или амидирована, или где сульфгидрильная группа образует дисульфидный мостик с молекулой-носителем,например пептид, который доставляет лекарство селективно к цели и/или к цитозолю клетки. Эти соединения можно получить из соединений настоящего изобретения в соответствии с хорошо известными способами. Другими примерами пролекарств являются соединения, где карбоксилат в соединении настоящего изобретения превращен, например, в алкил-, арил-, холин-, амино, ацилоксиметиловый сложный эфир, линоленоиловый сложный эфир. Соединения настоящего изобретения могут быть в форме метаболитов. Там, где может иметь место таутомерия, например кетоенольная таутомерия, соединений настоящего изобретения или их пролекарств, индивидуальные формы, например кето- или енольная форма, заявляются отдельно или вместе в виде смесей в любом соотношении. То же самое касается стереоизомеров,например энантиомеров, цис/транс-изомеров, конформеров и т.п. При желании, изомеры можно отделить хорошо известными в уровне техники способами, например с помощью жидкостной хроматографии. То же самое касается энантиомеров, например, с использованием хиральных неподвижных фаз. Дополнительно, энантиомеры можно выделять посредством их превращения в диастереомеры, т.е. путем сочетания с энантиомерно чистым вспомогательным соединением, с последующим отделением полученных в результате диастереомеров и расщеплением вспомогательного остатка. Альтернативно, любой энантиомер соединения настоящего изобретения можно получить из стереоселективного синтеза с использованием оптически чистых исходных веществ. Соединения настоящего изобретения могут быть в форме фармацевтически приемлемой соли или сольвата. Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические основания или кислоты и органические основания или кислоты. В случаях, где соединения настоящего изобретения содержат одну или несколько кислотных или основных групп, изобретение также включает их соответствующие фармацевтически или токсикологически приемлемые соли, в частности их фармацевтически пригодные для использования соли. Таким образом, соединения настоящего изобретения, которые содержат кислотные группы, могут присутствовать в форме соли и могут применяться в соответствии с изобретением,например, в виде солей щелочных металлов, соли щелочно-земельных металлов или в виде аммониевых солей. Более точные примеры таких солей включают натриевые соли, калиевые соли, кальциевые соли,магниевые соли или соли с аммиаком или органическими аминами, такими как, например, этиламин,этаноламин, триэтаноламин, или с аминокислотами. Соединения настоящего изобретения, которые содержат одну или несколько основных групп, т.е. групп, которые могут быть протонированы, присутствовать в форме соли и могут применяться в соответствии с изобретением в виде их аддитивных солей с неорганическими или органическими кислотами. Примеры пригодных кислот включают хлорид водорода, бромид водорода, фосфорную кислоту, серную кислоту, азотную кислоту, метансульфоновую кислоту, п-толуолсульфоновую кислоту, нафталиндисульфоновые кислоты, щавелевую кислоту, уксусную кислоту, винную кислоту, молочную кислоту, салициловую кислоту, бензойную кислоту, муравьиную кислоту, пропионовую кислоту, пивалевую кислоту, диэтилуксусную кислоту, малоновую кислоту, янтарную кислоту, пимелиновую кислоту, фумаровую кислоту, малеиновую кислоту, яблочную кислоту,сульфаминовую кислоту, фенилпропионовую кислоту, глюконовую кислоту, аскорбиновую кислоту,изониксотиновую кислоту, лимонную кислоту, адипиновую кислоту и другие кислоты, известные специалисту в данной области техники. Если соединения настоящего изобретения одновременно содержат кислотные и основные группы в молекуле, изобретение также включает, в дополнение к указанным солевым формам, внутренние соли или бетаины (цвиттерионы). Соответствующие соли можно получить обычными способами, известными специалисту в данной области техники, например путем введения их в контакт с органической или неорганической кислотой или основанием в растворителе или диспергаторе или путем анионного или катионного обмена с другими солями. Настоящее изобретение также включает все соли соединений настоящего изобретения, которые из-за низкой физиологической совместимости не являются пригодными для непосредственного применения в фармацевтических средствах, но которые можно использовать, например, в качестве промежуточных соединений для химических реакций или для получения фармацевтически приемлемых солей. Термин "замещенный" предпочтительно относится к замещению вышеуказанными заместителями,где возможно множество степеней замещения, если не указано иначе. Все физиологически приемлемые соли являются также в соответствии с изобретением. Соединения формулы (I) могут иметь один или несколько центров хиральности. Соответственно,они могут встречаться в различных энантиомерных формах и быть рацемическими или оптически активными формами. Поэтому изобретение также относится к оптически активным формам (стереоизомеры),энантиомерам, рацематам, диастереомерам и гидратам и сольватам этих соединений. Поскольку фармацевтическая активность рацематов или стереоизомеров соединений в соответствии с изобретением может отличаться, может быть желательным применение энантиомеров. В этих случаях конечный продукт или даже промежуточные соединения могут разделяться на энантиомерные соединения с помощью химических или физических методов, известных специалисту в данной области техники, или даже использоваться как таковые в синтезе. В случае рацемических аминов диастереомеры образовываются из смеси путем реакции с оптически активным расщепляющим агентом. Примерами пригодных расщепляющих агентов являются оптически активные кислоты, такие как R- и S-формы винной кислоты, диацетилвинной кислоты, дибензоилвинной кислоты, миндальной кислоты, яблочной кислоты, молочной кислоты, соответственноN-защищенной аминокислоты (например, N-бензоилпролин или N-бензолсульфонилпролин), или различных оптически активных камфорсульфоновых кислот. Также благоприятным является хроматографическое расщепление энантиомеров с помощью оптически активного расщепляющего агента (например, динитробензоилфенилглицин, триацетат целлюлозы или другие производные карбогидратов или хирально производные метакрилатные полимеры, иммобилизированные на силикагеле). Пригодными элюентами для этого являются водные или спиртовые смеси растворителей, такие как,например, гексан/изопропанол/ацетонитрил, например, в соотношении 82:15:3. Лучшим способом растворения рацематов, содержащих сложноэфирные группы (например, ацетиловые сложные эфиры), является применение ферментов, в частности эстераз. Кроме того, настоящее изобретение относится к фармацевтическим композициям, которые содержат соединение настоящего изобретения или фармацевтически приемлемую соль, в качестве активного ингредиента вместе с фармацевтически приемлемым носителем."Фармацевтическая композиция" означает один или несколько активных ингредиентов и один или несколько инертных ингредиентов, которые составляют носитель, а также любой полученный в резуль- 10025098 тате продукт, непосредственно или опосредованно, из комбинаций, комплексообразования или агрегации любых двух или более ингредиентов или из разложения одного или нескольких ингредиентов или из других типов реакций или взаимодействий одного или нескольких ингредиентов. Соответственно, фармацевтические композиции настоящего изобретения охватывают любую композицию, составленную путем смешивания соединения настоящего изобретения и фармацевтически приемлемого носителя. Фармацевтическая композиция настоящего изобретения может дополнительно содержать одно или несколько других соединений в качестве активных ингредиентов, таких как одно или несколько дополнительных соединений настоящего изобретения, или соединение-пролекарство, или другие ингибиторыp70S6K. Фармацевтические композиции включают композиции, пригодные для перорального, ректального,местного, парентерального (включая подкожное, внутримышечное и внутривенное), окулярного (глазного), пульмонального (назальная или буккальная ингаляция) или назального введения, хотя наиболее подходящий путь введения в любом вышеуказанном случае будет зависеть от природы и тяжести состояния,которое лечат, и от природы активного ингредиента. Они могут удобным образом присутствовать в стандартной лекарственной форме и быть приготовлены любым хорошо известным в области фармации способом. В одном из вариантов осуществления указанные соединения и фармацевтическая композиция предназначены для лечения злокачественного новообразования, такого как рак мозга, легкого, толстой кишки, эпидермоидный рак, плоскоклеточный рак, рак мочевого пузыря, рак желудка, поджелудочной железы, молочной железы, головы, шеи, ренальный рак, рак почки, печени, яичника, предстательной железы,колоректальный рак, рак матки, ректальный рак, рак пищевода, рак яичка, гинекологический рак, рак щитовидной железы, меланома, гематологических злокачественных опухолей, таких как острая миелоцитарная лейкемия, множественная миелома, хронический миелолейкоз, миелоидный клеточный лейкоз,глиома, саркома Капоши или любых других видов солидных или так называемых "жидких" опухолей. Предпочтительно злокачественное новообразование, которое подвергается лечению, выбрано из рака молочной железы, колоректального рака, рака легкого, предстательной железы или поджелудочной железы или глиобластомы. Изобретение также относится к применению соединений в соответствии с изобретением для получения лекарственного средства для лечения гиперпролиферативных заболеваний, связанных с гиперактивностью p70S6K, a также заболеваний, модулированных каскадом p70S6K, у млекопитающих, или расстройств, опосредованных аберрантной пролиферацией, таких как злокачественное новообразование и воспаление. Изобретение также относится к соединению или фармацевтической композиции для лечения заболевания, связанного с васкулогенезом или ангиогенезом у млекопитающего, которые включают терапевтически эффективное количество соединения настоящего изобретения или его фармацевтически приемлемой соли, пролекарства или гидрата и фармацевтически приемлемый носитель. В одном из вариантов осуществления указанное соединение или фармацевтическая композиция предназначены для лечения заболевания, выбранного из группы, которая включает ангиогенез опухоли,хроническое воспалительное заболевание, такое как ревматоидный артрит, воспалительные заболевания кишечника, атеросклероз, кожные заболевания, такие как псориаз, экзема, и склеродерма, диабет, диабетическую ретинопатию, ретинопатию недоношенных и возрастную дегенерацию макулы. Соединения или фармацевтическая композиция, определенные выше, могут использоваться для ингибирования атипичного клеточного роста у млекопитающего, которая включает количество соединения настоящего изобретения или его фармацевтически приемлемой соли или сольвата или пролекарства, в комбинации с количеством другого противоракового терапевтического средства, где количества соединения, соли, сольвата или пролекарства и химиотерапевтического средства вместе являются эффективными для ингибирования атипичного клеточного роста. Многие противораковые терапевтические средства в настоящее время известны в данной области техники. В одном из вариантов осуществления противораковое терапевтическое является химиотерапевтическим средством, выбранным из группы, которая включает ингибиторы митоза, алкилирующие агенты, антиметаболиты, интеркалирующие антибиотики,ингибиторы факторов роста, ингибиторы клеточного цикла, фермента, ингибиторы типоизомеразы, модификаторы биологического отклика, антигормональные средства, ингибиторы ангиогенеза и антиандрогены. В другом варианте осуществления противораковое терапевтическое средство представляет собой антитело, выбранное из группы, которая включает бевацизумаб, CD40-специфические антитела,chTNT-1/B, денозумаб, занолимумаб, IGF1R-специфические антитела, линтузумаб, эдреколомаб,WX G250, ритуксимаб, тицилимумаб, трастузумаб и цетуксимаб. В еще другом варианте осуществления противораковое терапевтическое средство представляет собой ингибитор другой протеинкиназы, такой как Akt, Axl, Aurora A, Aurora B, dyrk2, epha2, fgfr3, igf1r, IKK2, JNK3, Vegfr1, Vegfr2, Vegfr3 (также известная как Flt-4), KDR, MEK, MET, Plk1, RSK1, Src, TrkA, Zap70, cKit, bRaf, EGFR, Jak2, PI3K,NPM-Alk, c-Abl, BTK, FAK, PDGFR, TAK1, LimK, Flt-3, PDK1 и Erk. Соединения по изобретению могут использоваться в способе ингибирования атипичного клеточного роста у млекопитающего или лечения гиперпролиферативного нарушения, который включает введе- 11025098 ние млекопитающему количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, или сольвата, или пролекарства в комбинации с лучевой терапией, где количества соединения, соли, сольвата или пролекарства, представленные в комбинации с лучевой терапией, эффективны для ингибирования атипичного клеточного роста или лечения гиперпролиферативного нарушения у млекопитающего. Техники введения лучевой терапии известны из уровня техники, и эти техники можно использовать в комбинированной терапии, описанной в настоящем документе. Введение соединения согласно изобретению в этой комбинированной терапии можно определить, как описано в настоящем документе. Полагают, что соединения в соответствии с настоящим изобретением могут придавать атипичным клеткам большую чувствительность к лечению с применением лучевой терапии для уничтожения и/или ингибирования роста таких клеток. Соединения по изобретению могут использоваться в способе сенсибилизации атипичных клеток у млекопитающего к лечению с применением лучевой терапии, который включает введение млекопитающему количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, или сольвата, или пролекарства, где количество является эффективным для сенсибилизации атипичных клеток к лечению с помощью лучевой терапии. Количество соединения, соли или сольвата в этом методе можно определить в соответствии с методами для установления эффективных количеств таких соединений, описанных в настоящем документе. Изобретение также относится к способу ингибирования атипичного клеточного роста у млекопитающего, который включает применение количества соединения согласно настоящему изобретению, или его фармацевтически приемлемой соли или сольвата, его пролекарства, или его производного, меченного радиоактивным изотопом, и количества одного или нескольких веществ, выбранных из антиангиогенных агентов, ингибиторов передачи сигналов и антипролиферативных агентов. При практическом использовании соединения в соответствии с настоящим изобретением можно комбинировать в качестве активного ингредиента в тесной смеси с фармацевтическим носителем в соответствии с общепринятыми методиками приготовления лекарственных средств. Носитель может иметь различные формы в зависимости от формы препарата, желательного для введения, например перорального или парентерального (включая внутривенное). Для приготовления композиций для пероральных дозированных форм можно использовать любые обычные фармацевтические среды, такие как, например,вода, гликоли, масла, спирты, ароматизаторы, консерванты, красители и др. В случае пероральных жидких препаратов можно использовать любые обычные фармацевтические среды, такие как, например,суспензии, эликсиры и растворы; или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, замасливатели, связующие, дезинтеграторы и др. В случае твердых пероральных препаратов композиция может находиться в таких формах, как, например, порошки, твердые и мягкие капсулы и таблетки, при этом твердые пероральные препараты являются предпочтительными относительно жидких препаратов. В связи с простотой их введения таблетки и капсулы является наиболее благоприятными пероральными дозируемыми единичными формами, в этих случаях обязательно используются твердые фармацевтические носители. Если это является желательным, то таблетки могут быть покрыты оболочкой с помощью стандартных водных или неводных техник. Такие композиции и препараты должны содержать по меньшей мере 0,1% активного соединения. Процент активного соединения в этих композициях, очевидно, может изменяться и подходяще может составлять от приблизительно 2 до приблизительно 60% от массы единицы. Количество активного соединения в таких терапевтически пригодных композициях будет таким, чтобы получить эффективную дозу. Активные соединения также можно вводить интраназально, например, в виде жидких капель или спрея. Таблетки, пилюли, капсулы и другие формы также могут содержать связующее, такое как трагакантовую камедь, гуммиарабик, кукурузный крахмал или желатин; наполнители, такие как дикальций фосфат; дезинтегрирующее средство, такое как кукурузный крахмал, картофельный крахмал, альгиновую кислоту; замасливатель, такой как стеарат магния; и подсластитель, такой как сахароза, лактоза или сахарин. Если единичная дозированная форма представляет собой капсулу, то она может содержать, дополнительно к веществам вышеописанного типа, жидкий носитель, такой как жирное масло. Различные другие вещества могут присутствовать в виде покрытий или для модификации физической формы дозированной единицы. Например, таблетки могут быть покрыты с помощью шеллака, сахара или обоих веществ. Сироп или эликсир может содержать, дополнительно к активному компоненту,сахарозу в качестве подсластителя, метил и пропилпарабены в качестве консервантов, краситель и ароматизатор, такой как вишневый или апельсиновый ароматизатор. Соединения согласно настоящему изобретению также могут вводиться парентерально. Растворы или суспензии этих активных соединений могут быть приготовлены в воде, подходяще смешаны с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии также могут быть приготовлены в глицерине, жидких полиэтиленгликолях и их смесях в маслах. При обычных условиях хранения и применения эти препараты содержат консервант для предотвращения роста микроорганизмов. Фармацевтические формы, подходящие для инъекций, включают стерильные водные растворы или дисперсии и стерильные порошки для экстемпоральных препаратов стерильных инъекционных растворов или дисперсий. Во всех случаях форма должна быть стерильной и должна быть жидкой до такой степени, чтобы ее легко можно было вводить с помощью шприца. Она должна быть стабильной в условиях изготовления и хранения и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла. Любой подходящий путь введения может применяться для обеспечения млекопитающего, в особенности человека, эффективной дозой соединения согласно настоящему изобретению. Например, можно применять пероральный, ректальный, местный, парентеральный, глазной, легочный, назальный и другие пути. Дозированные формы включают таблетки, лепешки, дисперсии, суспензии, растворы, капсулы,кремы, мази, аэрозоли и др. Предпочтительно соединения согласно настоящему изобретению вводят перорально. Эффективная дозировка применяемого активного компонента может изменяться в зависимости от конкретного применяемого соединения, способа введения, состояния, подвергаемого лечению, и тяжести состояния, подвергаемого лечению. Такие дозировки легко могут быть установлены специалистом в данной области техники. При лечении или предотвращении злокачественного новообразования, воспаления или других пролиферативных заболеваний, для которых показаны соединения согласно настоящему изобретению,обычно удовлетворительные результаты получают, если соединения в соответствии с настоящим изобретением вводят в суточной дозе от приблизительно 0,01 до приблизительно 100 мг/кг веса тела животного, предпочтительно представленных в виде единичной суточной дозы. Для наиболее крупных млекопитающих общая суточная доза составляет от приблизительно 0,2 до приблизительно 2000 мг, предпочтительно от приблизительно 0,5 до приблизительно 1000 мг. Для взрослого человека весом 70 кг общая суточная доза обычно будет составлять от приблизительно 0,5 до приблизительно 1000 мг. Эта схема дозирования может регулироваться для обеспечения оптимального терапевтического ответа. Соединения по изобретению могут использоваться в наборе (комплекте), который состоит из отдельных пакетов: а) эффективного количества в соответствии с изобретением или его физиологически приемлемой соли, сольвата или пролекарства; б) эффективного количества дополнительного активного ингредиента лекарственного средства. Набор включает подходящие контейнеры, такие как коробочки, индивидуальные флаконы, пакеты или ампулы. Набор может, например, включать отдельные ампулы, каждая из которых содержит эффективное количество соединения в соответствии с изобретением и/или их фармацевтически пригодных производных, сольватов и стереоизомеров, включая их смеси во всех соотношениях, и эффективное количество дополнительного активного ингредиента лекарственного средства в растворенной или лиофилизированной форме. Экспериментальный раздел Некоторые сокращения, которые можно встретить в данном описании, указаны ниже. Сокращения. Соединения согласно настоящему изобретению можно получить в соответствии с методиками следующих схем и примеров, с использованием подходящих веществ, и они дополнительно проиллюстрированы с помощью следующих специфических примеров. Кроме того, используя методики, описанные в данном документе, в сочетании с обычными в уровне техники, можно легко получить дополнительные соединения настоящего изобретения, заявленные в данном документе. Однако соединения, проиллюстрированные в примерах, не следует толковать как формирующие единственный вид, рассматриваемый как изобретение. Примеры дополнительно иллюстрируют детали для получения соединений настоящего изобретения. Специалисты в данной области техники легко поймут, что известные вариации условий и способов следующих препаративных технологий могут быть использованы для получения этих соединений. Настоящие соединения, как правило, выделяют в форме их фармацевтически приемлемых солей,таких как описанные выше. Амины в форме свободных оснований, соответствующие выделенным солям,можно получить путем нейтрализации пригодным основанием, таким как водный раствор гидрокарбоната натрия, карбоната натрия, гидроксида натрия и гидроксида калия, и экстракции освобожденного амина в форме свободного основания в органический растворитель с последующим упариванием. Амин в форме свободного основания, выделенный таким способом, можно дополнительно превратить в другую фармацевтически приемлемую соль посредством растворения в органическом растворителе с последующим добавлением подходящей кислоты и последующим упариванием, осаждением или кристаллизацией. Изобретение будет проиллюстрировано, но не ограничено, исходя из специфических вариантов осуществления, описанных в следующих схемах и примерах. Если на схемах не указано иначе, переменные имеют то же значение, как описано выше. Если не описано иначе, все исходные вещества получают от коммерческих поставщиков и используют без дополнительной очистки. Если не описано иначе, все температуры выражают в C и все реакции осуществляют при комнатной температуре. Соединения очищали с помощью либо хроматографии на силикагеле, либо препаративной ВЭЖХ. Настоящее изобретение также относится к способам получения соединений формулы (I) и формулы(II) в соответствии с нижеописанными схемами и демонстрационными примерами. Схемы синтеза, описывающие промежуточные соединение и соединения конечных продуктов Аминопиримидинхлоридные промежуточные соединения были коммерчески доступны или получены в соответствии с путями синтезы, приведенными на схемах 1-3. Схема 1 Формамидацетат вводили в реакцию с 1 а в присутствии этоксида натрия в сухом этаноле с получением 1b, которое превращали в 1 с с помощью РОС 13 в присутствии TEA в толуоле. Затем 1 с вводили в реакцию с водным аммиаком в н-бутаноле при 100C с получением 1d. Схема 2 4-Амино-6-хлорпиримидин-5-ол 2 а вводили в реакцию с алкилированными реагентами для получения желаемых аминопиримидинхлоридных промежуточных соединений 2b. Схема 3 4,5,6-трихлорпиримидин 3 а вводили в реакцию с водным метиламином в 2-пропанолом с получением 5,6-дихлор-N-метилпиримидин-4-амина 3b. 4-Имидазол-2-илпиперидиновые промежуточные соединения получали в соответствии с путями синтеза, представленными на схемах 4-6. трет-Бутил 4-формилпиперидин-1-карбоксилат 4 а вводили в реакцию с водным глиоксалем в присутствии водного гидроксида аммония в метаноле с получением трет-бутил 4-(1H-имидазол-2 ил)пиперидин-1-карбоксилата 4b, который бромировали с получением трет-бутил 4-(4-бром-1 Нимидазол-2-ил)пиперидин-1-карбоксилата 4 с; 4 с алкилировали 2-(2-бромэтокси)тетрагидро-2 Н-пираном с последующим удалением группы THP в кислотных условиях с получением трет-бутил 4-(4-бром-1-(2 гидроксиэтил)-1 Н-имидазол-2-ил)пиперидин-1-карбоксилата 4d. Реакцию сочетания Сузуки осуществляли с помощью бромидного промежуточного соединения 4d с получением 4 е; 4 е вводили в реакцию с метансульфонилхлоридом или 4-метилбензолсульфонилхлоридом, после чего гасили с помощью амина с получением 4f, которое обрабатывали хлороводородом в метаноле или трифторуксусной кислотой в ДХМ для получения промежуточных соединений аминов 4g. Схема 5 трет-Бутил 4-(4-бром-1-(2-гидроксиэтил)-1 Н-имидазол-2-ил)пиперидин-1-карбоксилат 4d превращали в трет-бутил 4-(1-(2-(азетидин-1-ил)этил)-4-бром-1 Н-имидазол-2-ил)пиперидин-1-карбоксилат 5 а. Реакцию сочетания Сузуки осуществляли с помощью промежуточного соединения бромида 5 а с последующим снятием защиты Boc в кислотных условиях с получением 4-(4-Ar-1-(2-(азетидин-1-ил)этил)-1 Нимидазол-2-ил)пиперидина 5b. Бромкетон 6 а вводили в реакцию с 1,3,5,7-тетраазаадамантаном с получением 6b, которое подвергали амидному сочетанию с образованием 6 с; 6 с циклизировали в присутствии ацетата аммония с образованием 6d, которое затем было превращено в Cbz-защищенное 6 е; 6 е алкилировали 2-(бромметил)-1,3 диоксоланом в присутствии сильного основания с образованием 6f с последующим кислотным гидролизом с получением альдегидом 6g. Восстановительное аминирование 6g с помощью соответствующих вторичных аминов давало 6h. Снятие с защиты с 6h давало промежуточные соединения 4-имидазол-2 илпиперидинов 6i. Соединения со структурой, как в формуле (II), получают следующими путями синтеза (схемы 7-9). Схема 7 Аминопиримидинхлориды 7 а (коммерчески доступные или полученные в соответствии со схемами 1-3) вводили в реакцию со вторичным амином 7b (полученным в соответствии со схемами 4-6) в присутствии основания для получения соединений 7 с. Затем реакцию сочетания Сузуки осуществляли с соединениями 7 с, в то время как R1 являет собой бромид и бороновую кислоту или сложный эфир с получением соединений 7d. Гидролиз соединений 7 с при R1 = нитрил обеспечивает соединения 7 е. В некоторых вариантах осуществления настоящего изобретения аминопиримидинхлориды 7 а могут быть более в общем представлены как: где "LG" представляет собой любую уходящую группу, как правило, используемую в нуклеофильных замещениях, более предпочтительно Hal, как, например, Cl или Br. Схема 8 Аминопиримидинхлориды 7 а (коммерчески доступные или полученные в соответствии со схемами 1-3) вводили в реакцию с аминами 8 а (полученными путем снятия защиты с промежуточных соединений 4 е (схема 4 в присутствии основания для получения соединений 8b. Реакцию мезилирования осуществляли на соединениях 8b с образованием соединения 8 с, которое концентрировали затем алкилировали желаемым амином с получением соединений 8d. Гидролиз соединений 8d при R1 = нитрил обеспечивало соединения 8 е. Схема 9 Промежуточные соединения 6 е обрабатывали NaH и затем сочетали с алкильными реагентами 9 а с образованием 9b. Cbz-группу 9b удаляли с помощью каталитического гидрирования с получением 9 с. Аминопиримидинхлориды 7 а сочетали с 9 с с получением 9d, а снятие защиты Вос с 9d давало 9 е. Аналитические методики. Аналитическую ЖХ/МС осуществляли, используя три следующих метода. Метод А. Колонку Discovery С 18, 5 мкм, 330 мм, использовали при скорости потока 400 мкл/мин, пробоотборная петля 5 мкл, подвижная фаза: (А) вода с 0.1% муравьиной кислоты, подвижная фаза: (В) метанол с 0.1% муравьиной кислоты; время удержания приведено в минутах. Подробности метода: (I) работали с использованием насоса G1311A (Agilent) для четырхкомпонентных смесей с УФ/ВИД, детектором на диодной матрице G1315B (Agilent) и МС-детектором FinniganLCQ Duo в ESI + режим с УФ-детектированием при 254 и 280 нм с градиентом 15-95% (В) за 3,2 мин линейный градиент, (II) удерживали в течение 1,4 мин при 95% (В), (III) уменьшали от 95-15% (В) за 0,1 мин линейный градиент, (IV) удерживали в течение 2.3 мин при 15% (В). Метод В. Колонку Waters Symmetry С 18, 3,5 мкм, 4,675 мм, использовали при скорости потока 1 мл/мин,пробоотборная петля 10 мкл, подвижной фазой (А) является вода с 0.05% ТФУ, подвижной фазой (В) является ACN с 0.05% ТФУ; время удержания приведено в минутах. Подробности метода: (I) работали с использованием двойного насоса G1312A (Agilent) с УФ/ВИД,детектором на диодной матрице G1315B (Agilent) и МС-детектором Agilent G1956B (SL) в ESI + режим с УФ-детектированием при 254 и 280 нм с градиентом 20-85% (В) за 10 мин линейный градиент, (II) удерживали в течение 1 мин при 85% (В), (III) уменьшали от 20-85% (В) за 0.2 мин линейный градиент, (IV) удерживали в течение 3,8 мин при 20% (В). Метод С. Градиент: 4.2 мин/поток: 2 мл/мин 99:01 - 0:100 вода + 0.1 об.% ТФУ; Ацетонитрил + 0.1 об.% ТФУ; 0.0-0.2 мин: 99:01; 0.2-3.8 мин: 99:010:100; 3.8-4.2 мин: 0:100; Колонка: Chromolith PerformanceRP18e; длиной 100 мм, диаметром 3 мм; длина волны: 220 нм. Аналитическая хиральная ВЭЖХ. Аналитическую хиральную ВЭЖХ осуществляли с использованием колонки ChiralPak AD-H(2504.6 мм) компании Daicel Chemical Industries, Ltd. на системе Agilent 1100 Series. Для метода использовали инжекцию 5.0 мкл, со скоростью потока 1 мл/мин 100% метанола в течение 15 мин при 25C и УФ-детектирование при 254 и 280 нм. Препаративная ВЭЖХ. Препаративную ВЭЖХ осуществляли с использованием колонки Waters Atlantis dC18 OBD 10 мкм(30250 мм) или колонки Waters Sunfire Prep C18 OBD 10 мкм (30250 мм). Колонки использовали при скорости потока 60 мл/мин на системе Waters Prep LC 4000, оснащенной пробоотборной петлей (10 мл) и УФ/ВИД-детектором ISCO UA-6. Подвижную фазу извлекали из двух резервуаров с растворителями,содержащими (А) воду и (В) ацетонитрил квалификации "для ВЭЖХ". Для типичной препаративной работы использовали линейный градиент (например, 0-60%растворитель В в течение 60 мин). Примеры Демонстрационные примеры, представленные ниже, предназначены для иллюстрации отдельных вариантов осуществления изобретения и не предназначены для ограничения рамок объема описания или формулы изобретения каким-либо образом. Получение промежуточных соединений. Образец синтеза промежуточного соединения А (схема 1) 6-Хлор-5-этилпиримидин-4-амин Стадия 1. 5-Этилпиримидин-4,6-диол. К перемешиваемому раствору этоксида натрия (21% в этаноле, 349 мл, 1.06 моль) в сухом этаноле(500 мл) партиями добавляли формамид-ацетат (30.4 г, 0.29 моль) при 0C в атмосфере азота. Реакционную смесь перемешивали при этой температуре в течение 15 мин и затем добавляли раствор диэтилэтилмалоната (50.0 г, 0.26 моль) в этаноле (200 мл) по каплям при 0C в атмосфере азота. Реакционную смесь перемешивали при КТ в течение 14 ч. После завершения реакции реакционную смесь концентрировали при пониженном давлении. Сырой осадок растворяли в воде (250 мл) и подкисляли (рН 3-4) водным раствором HCl (1.5 н., 100 мл). Полученное твердое вещество отфильтровывали, промывали водой(100 мл) и сушили в вакууме с получением указанного в заголовке соединения (27.0 г, 72.5%) в виде твердого вещества белого цвета. ЖХМС: Найденная масса (М+, 141.0),1 Н ЯМР (400 МГц, ДМСО-d6) 11.54 (bs, 2H), 7.86 (s, 1H), 2.28-2.22 (m, 2H), 0.95-0.91 (m, 3 Н). Стадия 2. 4,6-Дихлор-5-этилпиримидин. К суспензии 5-этилпиримидин-4, 6-диола (20.0 г, 0.14 моль) в сухом толуоле (200 мл) добавляли триэтиламин (20.0 мл, 0.004 моль) при КТ в атмосфере азота. Реакционную смесь нагревали до 115C и затем по каплям добавляли POCl3 (52.3 мл, 0.57 моль). Реакционную смесь перемешивали при такой же температуре в течение 3 ч. После завершения реакции реакционную смесь охлаждали до 0C и гасили ледяной водой (50 мл). Органический слой отделяли, промывали насыщенным бикарбонатом натрия(100 мл) и сушили над натрия сульфат. Органический слой концентрировали в вакууме с получением указанного в заголовке соединения (22.5 г, 89%) в виде жидкости желтого цвета. 1 Н ЯМР (400 МГц, CDCl3) 8.61 (bs, 1H), 2.95-2.89 (m, 2H), 1.25-1.22 (m, 3 Н). Стадия 3. 6-Хлор-5-этилпиримидин-4-амин. К смеси 4,6-дихлор-5-этилпиримидина (22.5 г, 0.12 моль) в 1-бутаноле (70 мл) в 1 л стеклянную толстостенную пробирку добавляли водный аммиак (26%, 150 мл) при КТ. Сосуд запечатывали и нагревали до 100C в течение 12 ч. Реакционную смесь охлаждали до КТ, полученное твердое вещество фильтровали, промывали водой (50 мл) и сушили в вакууме с получением указанного в заголовке соединения(11.0 г, 55%) в виде твердого вещества белого цвета. ЖХМС: Найденная масса (М+, 158.0),1 Н ЯМР (400 МГц, CDCl3) 8.02 (s, 1H), 7.10 (bs, 2H), 2.57-2.50 (m, 2H), 1.03-0.99 (m, 3 Н). Образец синтеза промежуточного соединения В (схема 2). 6-Хлор-5-этоксипиримидин-4-амин Реакционную смесь 4-амино-6-хлорпиримидин-5-ола (1000.0 мг; 6.8 ммоль; 1.0 экв.), цезия карбоната (2686.3 мг; 8.24 ммоль; 1.2 экв.) и йодэтана (1285.9 мг; 8.24 ммоль; 1.2 экв.) в ацетоне (10 мл) перемешивали при 50C в течение ночи. Реакционный раствор разбавляли этилацетатом (50 мл), промывали водой, затем соляным раствором, сушили и концентрировали. Осадок обрабатывали эфиром, перемешивали в течение 30 мин, фильтровали, собирали не совсем белое твердое вещество в качестве указанного в заголовке соединения (1165 мг с выходом 98%). Промежуточное соединение С. 5,6-Дихлор-N-метилпиримидин-4-амин (схема 3). К перемешиваемому суспендирующему раствору 4,5,6-трихлорпиримидина (300.00 мг; 1.00 экв.) в изопропаноле (1 мл) добавляли 1.0 мл 40% водн. раствора метиламина и перемешивали при КТ в течение 1 ч. Указанное в заголовке соединение собирали путем фильтрации и промывали водой (175 мг, выход 60%) Образец синтеза промежуточного соединения D (схема 4). 2-[4-(4-Фтор-3-метилфенил)-2-пиперидин-4-ил-имидазол-1-ил]этилдиметиламин Стадия 1. трет-Бутил 4-(1 Н-имидазол-2-ил)пиперидин-1-карбоксилат. Раствор трет-бутил 4-формилпиперидин-1-карбоксилата (99.0 г, 464.20 ммоль) в метаноле (200 мл) обрабатывали 30% водным гидроксидом аммония (500 мл, 3.85 моль), после чего 40% водным глиоксалем (53.50 мл, 466.42 ммоль). Содержимому позволяли перемешаться при комнатной температуре в течение 1 ч, перед тем как его роторно упаривали для удаления метанола. Остатки обрабатывали соляным раствором (500 мл) и экстрагировали дихлорметаном (1500 мл), органические вещества сушили над сульфатом натрия и концентрировали до получения желтого масла. В масло вносили затравку автентичного вещества с получением не совсем белого твердого вещества в качестве указанного в заголовке соединения, котрое сушили в вакууме в течение 6 ч (110.37 г, 439.16 ммоль, 95%). Стадия 2. трет-Бутил 4-(4-бром-1 Н-имидазол-2-ил)пиперидин-1-карбоксилат. Раствор трет-бутил 4-(1 Н-имидазол-2-ил)пиперидин-1-карбоксилата (20.04 г, 79.74 ммоль) в ТГФ(450 мл) охлаждали до -46C (баня) перед тем, как колбу нагружали NBS (14.27 г) в равных порциях(10-мин отдельно). Данные ЖХМС показывали завершение реакции через 15 мин, таким образом, содержимое концентрировали до получения желтой твердой пены, которую далее сушили в вакууме в течение ночи. Сырое вещество (монобромид: дибромид = 40:27) очищали с помощью хроматографии с получением чистого белого твердого вещества (8.08 г, 24.47 ммоль, 31%) в качестве указанного в заголовке соединения.(36.0 г,109.02 ммоль) в сухом ДМСО (400 мл) обрабатывали KOH (36.0 г, 641.60 ммоль, 4.5 экв.) и содержимое становилось чисто-желтым раствором после 20-30 мин. Спустя 90 мин раствор обрабатывали 2-(2-бромэтокси)тетрагидропираном (36,0 г, 172,18 ммоль, 1.2 экв.). Реакцию считали завершенной через 4 ч с помощью данных ЖХМС, так содержимое разбавляли этилацетатом (800 мл) и промывали соляным раствором (3800 мл). Органические вещества сушили над сульфатом натрия и концентрировали до получения масла с приблизительным соотношением 8:1 региоизомеров, причем главный изомер желательно присутствует один. Масло повторно растворяли в метаноле (400 мл) и обрабатывали моногидратом п-толуолсульфокислоты (32.11 г, 168.80 ммоль, 1.5 экв.). Содержимое перемешивали при комнатной температуре в течение 20 мин, выливали в этилацетат (500 мл) и промывали водным карбонатом калия(400 мл), после чего - соляным раствором (2400 мл). Органические вещества сушили над сульфатом натрия и концентрировали до получения бесцветного масла, которое далее сушили в вакууме в течение 60 мин. Полученное густое масло суспендировали в диэтиловом эфире (200 мл) и вращали в соляной бане со льдом в течение 90 мин. Осаждали белое твердое вещество, которое фильтровали и сушили в вакууме в течение 30 мин с получением указанного в заголовке соединения (25.15 г, 67.20 ммоль, 62%,96% с помощью ЯМР и данных ЖХМС). Стадия 4. трет-Бутиловый эфир 4-[4-(4-фтор-3-метилфенил)-1-(2-гидроксиэтил)-1 Н-имидазол-2 ил]пиперидин-1-карбоновой кислоты. Смесь трет-бутилового эфира 4-[4-бром-1-(2-гидроксиэтил)-1 Н-имидазол-2-ил]пиперидин-1 карбоновой кислоты (1600.00 мг; 4.27 ммоль; 1.00 экв.), 3-фтор-4-метилфенилбороновой кислота(855.55 мг; 5.56 ммоль; 1.30 экв.), бис-(трибутилфосфина)палладия (436.95 мг; 0.85 ммоль; 0.20 экв.) и карбоната цезия (4178.60 мг; 12.82 ммоль; 3.00 экв.) в 1,4-диоксане (10.0 мл) и воде (1.50 мл) в запечатанной пробирке перемешивали при 70C в течение 5 ч. Сырой продукт очищали с помощью флэшхроматографии (EtOAc в гексанах 30-80%) для получения указанного в заголовке соединения (1.2 г,70%). Стадия 5. 2-[4-(4-Фтор-3-метилфенил)-2-пиперидин-4-ил-имидазол-1-ил]этилдиметиламин. К раствору трет-бутилового эфира 4-[4-(4-фтор-3-метилфенил)-1-(2-гидроксиэтил)-1 Н-имидазол-2 ил]пиперидин-1-карбоновой кислоты (1500.00 мг; 3.72 ммоль; 1.00 экв.) в ДХМ (15 мл) при -78C добавляли 4-метилбензолсульфонилхлорид (850.50 мг; 4.46 ммоль; 1.20 экв.) и TEA (1128.54 мг; 11.15 ммоль; 3.00 экв.). Смесь перемешивали при -78C в течение 1 ч, позволяли ей нагреться до комнатной температуры и перемешивали в течение ночи. ДХМ удаляли и осадок растворяли в эфире. Эфирный раствор промывали дважды водой, сушили над MgSO4 и концентрировали. Сырой продукт использовали на следующей стадии реакции без очистки. Смесь вышеуказанного промежуточного соединения и диметиламина (3352.05 мг; 74.35 ммоль; 20.00 экв., 2.0 М в ТГФ) в запечатанной пробирке перемешивали в течение нескольких часов. ЖХ/МС и ВЭЖХ использовали для мониторинга хода реакции. После завершения реакции растворитель удаляли,смесь перемешивали в течение 20 мин с ТФУ для удаления Вос-группы. Указанное в заголовке соединение получали путем очистки с помощью препаративной ВЭЖХ. Образец синтеза промежуточного соединения Е (схема 5).(22.0 г) и Et3N (24.5 мл) в ДХМ (250 мл) охлаждали до 0C. По каплям добавляли мезилхлорид (6.8 мл) и полученную смесь перемешивали в течение 1 ч. Реакционную смесь гасили насыщенным воднымNaHCO3 и экстрагировали ДХМ. Органический слой концентрировали с получением трет-бутил 4-(4-бром-1-(2-метилсульфонил)окси)этил)-1 Н-имидазол-2-ил)пиперидин-1-карбоксилата (28.0 г). Реакционную смесь трет-бутил 4-(4-бром-1-(2-метилсульфонил)окси)этил)-1H-имидазол-2 ил)пиперидин-1-карбоксилата (28.0 г) и азетидина (11 мл) в ДМФА (200 мл) перемешивали при 50C в течение ночи. Реакционную смесь разбавляли ЭА, промывали водой, после чего - насыщенным водным хлоридом натрия, сушили и концентрировали. Осадок очищали с помощью колоночной хроматографии с получением трет-бутил 4-(1-(2-(азетидин-1-ил)этил)-4-бром-1 Н-имидазол-2-ил)пиперидин-1 карбоксилата в виде не совсем белого твердого вещества (17.2 г).(3932.91 мг; 25.55 ммоль; 1.20 экв.) и Cs2CO3 (13872.97 мг; 42.58 ммоль; 2.00 экв.) в диоксане (80 мл) и воде (8 мл) продували аргоном в течение 10 мин, затем бис-(три-т-бутилфосфин)палладий(0) (435.20 мг; 0.85 ммоль; 0.04 экв.) добавляли под потоком аргона и продували аргоном в течение 1 мин. Полученную смесь перемешивали при 50 С в течение ночи, реакционный раствор разбавляли этилацетатом 250 мл,промывали соляным раствором 2100 мл. Органический слой сушили и концентрировали с получением сырого продукта, который очищали с помощью колонки biotage Ki-Sil с получением трет-бутилового эфира 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1 Н-имидазол-2-ил]пиперидин-1-карбоновой кислоты (9370.00 мг; 99% выход). К раствору трет-бутилового эфира 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1Hимидазол-2-ил]пиперидин-1-карбоновой кислоты в 55 мл метанола медленно добавляли 4.0 М HCl в диоксане (52.87 мл; 211.49 ммоль; 10.00 экв.). Реакционную смесь перемешивали при КТ в течение 4 ч. ЖХМС показывала, что реакция была осуществлена. Не совсем белый осадок образовывался после 1 ч перемешивания. Твердое вещество собирали путем фильтрации и промывали трижды эфиром с получением белого твердого вещества (8.78 г) в качестве чистого указанного в заголовке соединения, маточную жидкость концентрировали досуха и добавляли эфир (30 мл) и 5 мл метанола. Твердое вещество собирали путем фильтрации и промывали трижды эфиром для получения ещ 0,82 г указанного в заголовке соединения в виде не совсем белого твердого вещества (общий выход: 100%) Образец синтеза промежуточного соединения F (схема 6). 4-(4-(4 Фтор-3-(трифторметил)фенил)-1-(2-(3-фторазетидин-1-ил)этил)-1 Н-имидазол-2-ил)пиперидин Стадия 1. 2-Амино-1-(4-фтор-3-(трифторметил)фенил)этанон гидрохлорид. 2-Бром-1-(4-фтор-3-трифторметилфенил)этанон (10.00 г; 35.08 ммоль; 1.00 экв.) добавляли к раствору 1,3,5,7-тетраазатрицикло[3.3.1.13,7]декана (5.80 г; 41.37 ммоль; 1.18 экв.) в CCl4 100 мл, перемешивали в течение ночи. Осадок фильтровали и собирали в виде твердого вещества белого цвета. Вышеуказанное твердое вещество добавляли в этанол 100 мл и затем 28 мл концентрированного 36.5% HCl (водн.) и перемешивали при КТ в течение выходных, отфильтровывали твердое вещество. Фильтрат концентрировали и получалось твердое вещество. 10 мл изопропанол (содержащийся 1% HCl) добавляли, фильтровали и собирали белое твердое вещество в качестве указанного в заголовке соединения 5.22 г, 57.8% Стадия 2. трет-Бутил 4-2-(4-фтор-3-(трифторметил)фенил)-2-оксоэтил)карбамоил)пиперидин-1 карбоксилат. К раствору моно-трет-бутилового эфира пиперидин-1,4-дикарбоновой кислоты (9611.92 мг; 41.92 ммоль; 1.08 экв.) в ТГФ 100 мл добавляли 4-метилморфолин (14.51 мл; 131.98 ммоль; 3.40 экв.),реакционную смесь охлаждали до -10C, по каплям добавляли 3-метилбутирилхлорид (5.03 мл; 38.82 ммоль; 1.00 экв.) и поддерживали температуру ниже -5C. После перемешивания в течение 30 мин от -5 до 10C добавляли хлорид 2-(4-фтор-3-трифторметилфенил)-2-оксоэтиламмония (10.00 г; 38.82 ммоль; 1.00 экв.) при -5C и перемешивали смесь в течение 20 мин и затем при КТ в течение 1 ч. Добавляли соляной раствор, экстрагировали с помощью ЭА, промывали соляным раствором, сушили и концентрировали. Сырое масло обрабатывали эфиром. Белое твердое вещество собирали в качестве указанного в заголовке соединения (12 г, выход 71.5%). Стадия 3. трет-Бутил 4-(4-(4-фтор-3-(трифторметил)фенил)-1 Н-имидазол-2-ил)пиперидин-1 карбоксилат. Смесь трет-бутилового эфира 4-[2-(4-фтор-3-трифторметилфенил)-2-оксоэтилкарбамоил]пиперидин-1-карбоновой кислоты (2900.00 мг; 6.71 ммоль; 1.00 экв.), триэтиламина(0.94 мл; 6.71 ммоль; 1.00 экв.) и ацетата аммония (3618.76 мг; 46.95 ммоль; 7.00 экв.) в 20 мл н-бутанола помещали в микроволновую печь (150C) в течение 20 мин. После охлаждения до КТ реакционную смесь разбавляли 150 мл этилацетата и промывали водой, водн. 5% NaHCO3, затем соляным раствором и концентрировали. Осадок обрабатывали эфиром и собирали белое твердое вещество в качестве указанного в заголовке соединения (1500 мг, 54% выход). Стадия 4. Бензил 4-(4-(4-фтор-3-(трифторметил)фенил)-1 Н-имидазол-2-ил)пиперидин-1 карбоксилат. К трет-бутиловому эфиру 4-[4-(4-фтор-3-трифторметилфенил)-1 Н-имидазол-2-ил]пиперидин-1- 22025098 карбоновой кислоты (6.00 г; 14.51 ммоль; 1.00 экв.) в 10 мл метанола добавляли 4.0 М HCl в диоксане 15 мл. Реакционную смесь перемешивали при КТ в течение ночи и концентрировали с получением гидрохлоридной соли 4-(4-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-2-ил)пиперидина в виде твердого вещества белого цвета. К гидрохлоридной соли 4-(4-(4-фтор-3-(трифторметил)фенил)-1 Н-имидазол-2-ил)пиперидина(1510.00 мг; 3.91 ммоль; 1.00 экв.) в ДХМ добавляли этилдиизопропиламин (2.81 мл; 15.64 ммоль; 4.00 экв.) и перемешивали в течение 5 мин. Затем добавляли N-(бензилоксикарбонилокси)сукцинимид(1169.26 мг; 4.69 ммоль; 1.20 экв.). Реакционную смесь перемешивали при КТ в течение ночи. Реакционную смесь разбавляли ДХМ, промывали водой, водн. 5% NaHCO3 и соляным раствором. Органический слой сушили и концентрировали. Осадок подвергали разделению на колонке SNAP (100 г), элюировали 20-80% ЭА в гексане с получением указанного в заголовке соединения (1310 мг, выход 74.9%). Стадия 5. Бензил 4-(1-1,3-диоксолан-2-ил)метил)-4-(4-фтор-3-(трифторметил)фенил)-1Hимидазол-2-ил)пиперидин-1-карбоксилат. Раствор бензилового сложного эфира 4-[4-(4-фтор-3-трифторметилфенил)-1 Н-имидазол-2 ил]пиперидин-1-карбоновой кислоты (1100.00 мг; 2.46 ммоль; 1.00 экв.) в 2.0 мл ТГФ охлаждали до-20C, по каплям добавляли NaHMDS (541.00 мг; 2.95 ммоль; 1.20 экв.) и полученную смесь перемешивали при КТ в течение 30 мин. Добавляли 2-бромметил-[1,3]диоксолан (0.76 мл; 7.38 ммоль; 3.00 экв.) и перемешивали при КТ в течение 5 мин. Добавляли ДМСО (10 мл) и смесь помещали в микроволновую печь при 100C в течение 2 ч. Реакционную смесь охлаждали и разбавляли водой и экстрагировали с помощью ЭА. Органический слой промывали соляным раствором, отделяли, сушили и концентрировали,осадок подвергали разделению на колонке SNAP (100 г), элюировали 20-80% ЭА в гексане с получением указанного в заголовке соединения (1000 мг, выход 76.2%). Стадия 6. Бензил 4-(4-(4-фтор-3-(трифторметил)фенил)-1-(2-оксоэтил)-1 Н-имидазол-2 ил)пиперидин-1-карбоксилат. Реакционную смесь бензил 4-(1-1,3-диоксолан-2-ил)метил)-4-(4-фтор-3-(трифторметил)фенил)1 Н-имидазол-2-ил)пиперидин-1-карбоксилата (1450.00 мг; 2.72 ммоль; 1.00 экв.) и моногидрата толуол 4-сульфоновой кислоты (5169.79 мг; 27.18 ммоль; 10.00 экв.) в 30 мл ацетона и 2.5 мл воды перемешивали при 50C в течение одной недели. ЖХ-МС показывала некоторую смесь желаемого продукта, 30% исходного вещества и некоторые побочные продукты. Реакционную смесь концентрировали и добавляли 50 мл воды, экстрагировали ЭА. Объединенные органические слои промывали соляным раствором, сушили и концентрировали. Осадок подвергали разделению на колонке SNAP (100 г), элюировали с помощью 0-100% ЭА в гексане, затем элюировали с помощью 10% метанола в ДХМ с получением указанного в заголовке соединения в виде желтого масла (480 мг, выход 36%) с восстановлением некоторого исходного вещества. Стадия 7. Бензил 4-(4-(4-фтор-3-(трифторметил)фенил)-1-(2-(3-фторазетидин-1-ил)этил)-1 Нимидазол-2-ил)пиперидин-1-карбоксилат. К 3-фторазетидин гидрохлориду (66.99 мг; 0.45 ммоль; 1.05 экв.) в 1 мл DCE добавляли этилдиизопропиламин (0.15 мл; 0.86 ммоль; 2.00 экв.). После перемешивания при КТ в течение 10 мин добавляли бензиловый сложный эфир 4-[4-(4-фтор-3-трифторметилфенил)-1-(2-оксоэтил)-1 Н-имидазол-2 ил]пиперидин-1-карбоновой кислоты (211.00 мг; 0.43 ммоль; 1.00 экв.) с последующим добавлением триацетоксиборогидрида натрия (274.09 мг; 1.29 ммоль; 3.00 экв.). Полученную смесь перемешивали в течение ночи при КТ. После обработки, указанное в заголовке соединение получали путем очистки с помощью препаративной ВЭЖХ. Стадия 8. 4-(4-(4-Фтор-3-(трифторметил)фенил)-1-(2-(3-фторазетидин-1-ил)этил)-1 Н-имидазол-2 ил)пиперидин. К бензиловому сложному эфиру 4-[1-[2-(3-f-азетидин-1-ил)этил]-4-(4-фтор-3-трифторметилфенил)1 Н-имидазол-2-ил]пиперидин-1-карбоновой кислоты (200.00 мг; 0.36 ммоль; 1.00 экв.) в 10 мл метанола добавляли формиат аммония (229.90 мг; 3.65 ммоль; 10.00 экв.) и 200 мг 10%Pd/C (влажный). Смесь перемешивали при КТ в течение 1 ч. Отфильтровывали Pd/C. Фильтрат концентрировали и затем растворяли в EtOAc, промывали 5% NaHCO3, затем соляным раствором, сушили и концентрировали. Сырое указанное в заголовке соединение (138 мг, выход 91.3%) непосредственно использовали на следующей стадии реакции без очистки. Смесь 5-бром-6-хлорпиримидин-4-амина (357.2 мг; 1.71 ммоль; 1.02 экв.), 4-[4-(4-фтор-3 трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидина (550.00 мг; 1.68 ммоль; 1.0 экв.), карбоната калия (464.4 мг; 3.36 ммоль; 2.0 экв.) в ДМСО (6.0 мл) перемешивали при 60C в течение ночи. Реакционную смесь выливали в воду. Осадок фильтровали, промывали водой и сушили в вакууме с получением указанного в заголовке соединения с выходом 86%. ЖХ-МС: (М+1 = 499, наблюд. = 499). 5-Бром-6-4-[5-(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1 илпиримидин-4-иламин ("2") Указанное в заголовке соединение получали аналогичным способом, что и 5-бром-6-4-[4-(4-фтор 3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-4-иламин, с использованием 4-[5-(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидина вместо 4-[4-(4-фтор-3 трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидина. ЖХ-МС: (М+1 = 499, наблюд. = 499). 5-Бром-6-4-[4-(3-трифторметилфенил)-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-4-иламин Указанное в заголовке соединение получали аналогичным способом, что и 5-бром-6-4-[4-(4-фтор 3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-4-иламин с использованием 4-[4-(3-трифторметилфенил)-1H-имидазол-2-ил]пиперидина вместо 4-[4-(4-фтор-3 трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидина. ЖХ-МС: (М+1 = 467, наблюд. = 467). Указанное в заголовке соединение получали аналогичным способом, что и 5-бром-6-4-[4-(3 трифторметилфенил)-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-4-иламин с использованием 5-бром-4-хлорпиперидина вместо 5-бром-6-хлорпиримидин-4-амина. ЖХ-МС: (М+1 = 452, наблюд. = 452). 3-Бром-4-(4-4-[3-(трифторметил)фенил]-1 Н-имидазол-2-илпиперидин-1-ил)пиридин ("5") Смесь 3-бром-4-хлорпиридина (100.0 мг; 0.52 ммоль; 1.0 экв.), 4-[4-(3-трифторметилфенил)-1Hимидазол-2-ил]пиперидина (153.4 мг; 0.52 ммоль; 1.0 экв.), N,N-диизопропилэтиламина (0.26 мл; 1.56 ммоль; 3.0 экв.) в NMP (2.0 мл) в микроволновой пробирке нагревали при 160C в течение 4 ч. Реакционную смесь выливали в воду. Осадок фильтровали, промывали водой и сушили в вакууме с получением указанного в заголовке соединения с выходом 64%. ЖХ-МС: (М+1 = 451, наблюд. = 451). 4-Амино-6-4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1 илпиримидин-5-карбальдегид ("6")(0.95 мл; 5.5 ммоль; 3.0 экв.) в ацетонитриле (10.0 мл) нагревали при 40C в течение 1 ч. Реакционную смесь выливали в воду. Осадок фильтровали, промывали водой и сушили в вакууме с получением указанного в заголовке соединения с выходом 85%. ЖХ-МС: (М+1 = 449, наблюд. = 449). Указанное в заголовке соединение получали аналогичным способом, что и 4-амино-6- (4-[4-(4 фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-5-карбальдегид с использованием 4-амино-6-хлорпиримидин-5-карбонитрила вместо 4-амино-6-хлорпиримидин-5 карбальдегида. ЖХ-МС: (М+1 = 446, наблюд. = 446). 4-Амино-6-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1 Н-имидазол-2 ил]пиперидин-1-илпиримидин-5-карбонитрил ("8") Указанное в заголовке соединение получали аналогичным способом, что и 4-амино-6-4-[4-(4-фтор 3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-5-карбонитрил с использованием 2-(4-(4-фтор-3-(трифторметил)фенил)-2-(пиперидин-4-ил)-1 Н-имидазол-1-ил)-N,Nдиметилэтанамина вместо 4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидина. ЖХ-МС: (М+1 = 503, наблюд. = 503). 6-4-[4-(4-Фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-ил-5 метоксипиримидин-4-иламин ("9") Смесь 4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидина (40.00 мг; 0.12 ммоль; 1.0 экв.), 6-хлор-5-метоксипиримидин-4-амина (20.48 мг; 0.13 ммоль; 1.05 экв.), карбоната калия (33.78 мг; 0.24 ммоль; 2.0 экв.) в ДМСО (1.00 мл) в микроволновой пробирке нагревали при 150C в течение 2 ч. ЖХ-МС показывала 40% превращение и продукт, элюированный близко к аминному исходному веществу. Реакционную смесь обрабатывали и сырой продукт, очищали с помощью обращеннофазовой препаративной ВЭЖХ (Waters, ацетонитрил/0.1% ТФУ в воде) с получением желаемого продукта с выходом 22%. ЖХ-МС: (М+1 = 451, наблюд. = 451). Смесь 5-бром-6-(4-4-[4-фтор-3-(трифторметил)фенил]-1-метил-1 Н-имидазол-2-илпиперидин-1 ил)пиримидин-4-амина (100.0 мг; 0.2 ммоль; 1.0 экв.), (4-фторфенил)бороновой кислоты (56.0 мг; 0.4 ммоль; 2.0 экв.), ацетата палладия(II) (2.25 мг; 0.01 ммоль; 0.05 экв.), дициклогексил(2',6'диметоксибифенил-2-ил)фосфина (8.22 мг; 0.02 ммоль; 0.1 экв.) и карбоната цезия (195.7 мг; 0.6 ммоль; 3.0 экв.) в диоксане (4 мл) и воде (0.5 мл) в микроволновой пробирке нагревали при 100C в течение 30 мин. Реакционную смесь обрабатывали и сырой продукт очищали с помощью хроматографии с обращенной фазой (Yamazen, ацетонитрил/0.1% NH4OH в воде). Чистые фракции концентрировали досуха в вакууме. Белое твердое вещество растворяли в воде и ацетонитриле, добавляли 200 мкл 0.5 н. HCl. Избыток HCl удаляли путем концентрации в вакууме в течение 20 мин. Полученный раствор затем лиофилизировали с получением указанного в заголовке соединения в виде соли HCl с выходом 74%. ЖХ-МС: (М+1 = 515, наблюд. = 515). 6-4-[4-(4-Фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-ил-5-(4 метоксифенил)пиримидин-4-иламин ("11") Указанное в заголовке соединение получали аналогичным способом, что и 5-(4-фторфенил)-6-4-[4(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-4-иламин с использованием 4-метоксифенилбороновой кислотой вместо 4-фторфенилбороновой кислоты. ЖХ-МС: (М+1 = 527, наблюд. = 527). 6-4-[4-(4-Фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-4 иламин ("12") Указанное в заголовке соединение получали, как и деброминирующий побочный продукт во время получения 6-4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-ил-5-(4 метоксифенил)пиримидин-4-иламина. ЖХ-МС: (М+1 = 421, наблюд. = 421). Указанное в заголовке соединение получали аналогичным способом, что и 5-(4-фторфенил)-6-4-[4(4-фтор-3-трифторметилфенил)-1-метил-1H-имидазол-2-ил]пиперидин-1-илпиримидин-4-иламин с использованием 4-метилфенилбороновой кислоты вместо 4-фторфенилбороновой кислоты. ЖХ-МС: (М+1 = 511, наблюд. = 511). Указанное в заголовке соединение получали аналогичным способом, что и 5-(4-фторфенил)-6-4-[4(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-4-иламин с использованием пинаколового эфира 4-(гидроксиметил)бензолбороновой кислоты вместо 4-фторфенилбороновой кислоты. ЖХ-МС: (М+1 = 527, наблюд. = 527). 3-(4-Амино-6-4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1 илпиримидин-5-ил)бензонитрил ("15") Указанное в заголовке соединение получали аналогичным способом, что и 5-(4-фторфенил)-6-4-[4(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-4-иламин с использованием 3-цианофенилбороновой кислоты вместо 4-фторфенилбороновйо кислоты. ЖХ-МС: (М+1 = 522, наблюд. = 522). 6-4-[4-(4-Фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-ил-5-(4 трифторметилфенил)пиримидин-4-иламин ("16") Указанное в заголовке соединение получали аналогичным способом, что и 5-(4-фторфенил)-6-4-[4(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-4-иламин с использованием (4-трифторметил)фенилбороновой кислоты вместо 4-фторфенилбороновой кислоты. ЖХ-МС: (М+1 = 565, наблюд. = 565). 4-(4-Амино-6-4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1 илпиримидин-5-ил)бензонитрил ("17") Указанное в заголовке соединение получали аналогичным способом, что и 5-(4-фторфенил)-6-4-[4(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-4-иламин с использованием 4-цианофенилбороновой кислоты вместо 4-фторфенилбороновой кислоты. ЖХ-МС: (М+1 = 522, наблюд. = 522). 2-(4-Амино-6-4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1 илпиримидин-5-ил)бензонитрил ("18") Указанное в заголовке соединение получали аналогичным способом, что и 5-(4-фторфенил)-6-4-[4(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1-илпиримидин-4-иламин с использованием 2-цианофенилбороновой кислоты вместо 4-фторфенилбороновой кислоты. ЖХ-МС: (М+1 = 522, наблюд. = 522). 6-4-[4-(4-Фтор-3-трифторметилфенил)-1-метил-1H-имидазол-2-ил]пиперидин-1-ил-5-(6 метилпиридин-3-ил)пиримидин-4-иламин ("19")
МПК / Метки
МПК: A61K 31/506, C07D 401/14
Метки: киназной, активности, имидазоламины, модуляторов, качестве
Код ссылки
<a href="https://eas.patents.su/30-25098-imidazolaminy-v-kachestve-modulyatorov-kinaznojj-aktivnosti.html" rel="bookmark" title="База патентов Евразийского Союза">Имидазоламины в качестве модуляторов киназной активности</a>
Соединения и композиции в качестве модуляторов активности gpr119

Номер патента: 18703
Опубликовано: 30.10.2013
Авторы: Эппле Роберт, Кау Кристофер
МПК: A61P 3/10, A61P 3/04, A61K 31/497...
Метки: gpr119, соединения, качестве, активности, композиции, модуляторов
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, в которомА может содержать вплоть до 2 кольцевых атомов углерода, замещенных оксогруппой;n выбран из 0, 1 и 2;t1 и t2, каждый независимо, выбраны из 0 и 1;R1 выбран из цианогруппы, -S(O)2R6a, -N(R6a)S(O)2R6a, -S(O)2OR6a, -S(O)2C(O)R6a, -S(O)2C(O)OR6a и -S(O)2NR6aR6b, где R6a выбран из C1-6алкила, где указанный алкил в R6a необязательно замещен 1-3 радикалами, независимо...
4-феноксиметилпиперидины в качестве модуляторов активности gpr119

Номер патента: 17260
Опубликовано: 30.11.2012
Авторы: Никулин Виктор, Уэсткотт-Бейкер Лукас, Леле Жеральд, Эппле Роберт
МПК: C07D 211/30, A61K 31/4523, A61P 3/10...
Метки: 4-феноксиметилпиперидины, качестве, gpr119, активности, модуляторов
Формула / Реферат:
1. Соединение формулы Iгде R1 выбран из фенила, пиридин-2-ила, пиридин-3-ила, пиридин-4-ила, пиримидин-5-ила, пиразол-4-ила, 1H-пиразол-4-ила, 6-оксо-1,6-дигидропиридин-3-ила, 2-оксо-1,2-дигидропиримидин-5-ила, 2-оксо-1,2-дигидропиридин-4-ила и 2-оксо-2,3-дигидрооксазоло[4,5-b]пиридин-6-ила; где указанный фенил, пиридин-2-ил, пиридин-3-ил, пиридин-4-ил, пиримидин-5-ил, пиразол-4-ил, 1H-пиразол-4-ил, 6-оксо-1,6-дигидропиридин-3-ил,...
Соединения и композиции в качестве модуляторов активности tlr

Номер патента: 18068
Опубликовано: 30.05.2013
Авторы: Ву Том Яосянь, Кортес Алекс, Мишра Пранаб, Сингх Манмохан, Зоу Йэфень, Чжан Сяою, Ли Йонкай, Скибински Дейвид, Валианте Николас
МПК: A61K 31/4375, A61P 37/04, C07D 221/12...
Метки: соединения, активности, композиции, качестве, модуляторов
Формула / Реферат:
1. Соединение формулы (I-A) или его фармацевтически приемлемая сольформула (I-A)где X3 обозначает N;X4 обозначает CR3;X5 обозначает -CR4=CR5-;R3 обозначает H;R4 обозначает H;R5 выбирают из галогена, -C(O)OR7, -N(R9)2, -NHN(R9)2, -(CH2)nR7, -LR10, C1-C6-алкила, C1-C6-галогеналкила, C2-C8-алкенила, C2-C8-алкинила и C1-C6-алкоксигруппы, причем указанный C1-C6-алкил, C2-C8-алкенил и C2-C8-алкинил в составе R5, каждый необязательно, замещен 1-3...
N-уреидоалкилпиперидины в качестве модуляторов активности рецепторов хемокинов

Номер патента: 3147
Опубликовано: 27.02.2003
Авторы: Сантелла Джозеф Б.III, Ким Уи Тае, Вэкер Дин А.К., Ко Соо С., Данкиа Джон В., Делукка Джордж В.
МПК: A61P 11/06, A61K 31/445, C07D 211/32...
Метки: активности, хемокинов, n-уреидоалкилпиперидины, качестве, рецепторов, модуляторов
Формула / Реферат:
1. Соединение формулы (I) или его стереоизомеры или фармацевтически приемлемые соли, где M отсутствует или выбран из CH2, CHR5, CHR13, CR13R13 и CR5R13; Q выбран из CH2, CHR5, CHR13, CR13R13 и CR5R13; J и K выбраны из CH2, CHR5, CHR6, CR6R6 и CR5R6; L представляет CHR5; при условии, что если M отсутствует, то J выбран из CH2, CHR5, CHR13 и CR5R13; Z выбран из O и S; E выбран из кольцо A представляет C3-6карбоциклический остаток, при условии,...
Пиперидинильные производные в качестве модуляторов активности хемокинового рецептора

Номер патента: 19192
Опубликовано: 30.01.2014
Авторы: Сантелла Джозеф Б., Кавалларо Куллен Л., Лиу Руи-Кин, Гарднер Дэниел С., Додд Дхармпал С., Хайнс Джон, Дунсиа Джон В., Картер Перси Х.
МПК: A61K 31/454, A61K 31/445, C07D 211/18...
Метки: качестве, производные, хемокинового, пиперидинильные, активности, рецептора, модуляторов
Формула / Реферат:
1. Соединение формулы (Ib)или его стереоизомеры, или фармацевтически приемлемые солевые формы, гдеТ представляет собойR1 представляет собой алкил, фенил или циклоалкил, все из которых могут быть необязательно замещены с помощью 0-5 R1a;R1a при каждом появлении независимо выбран из алкила, галогеналкила, арила, галогена, -С(=O)ОН, -C(=O)O(CR8R8)rR10 и -ОН, где арил может быть необязательно замещен с помощью 0-3 R1b;R1b при каждом появлении...