Арилсульфонилпиразолинкарбоксамидиновые производные в качестве антагонистов 5-нт6
Номер патента: 23176
Опубликовано: 31.05.2016
Авторы: Крюсе Корнелис Г., Стойт Аксел, Ван Лувезейн Арнольд, Ивема Баккер Ваутер И., Венхорст Дженнифер, Ван Дер Нэт Мартина А.В., Ренсинк Агата А.М., Де Хан Мартин


Формула / Реферат
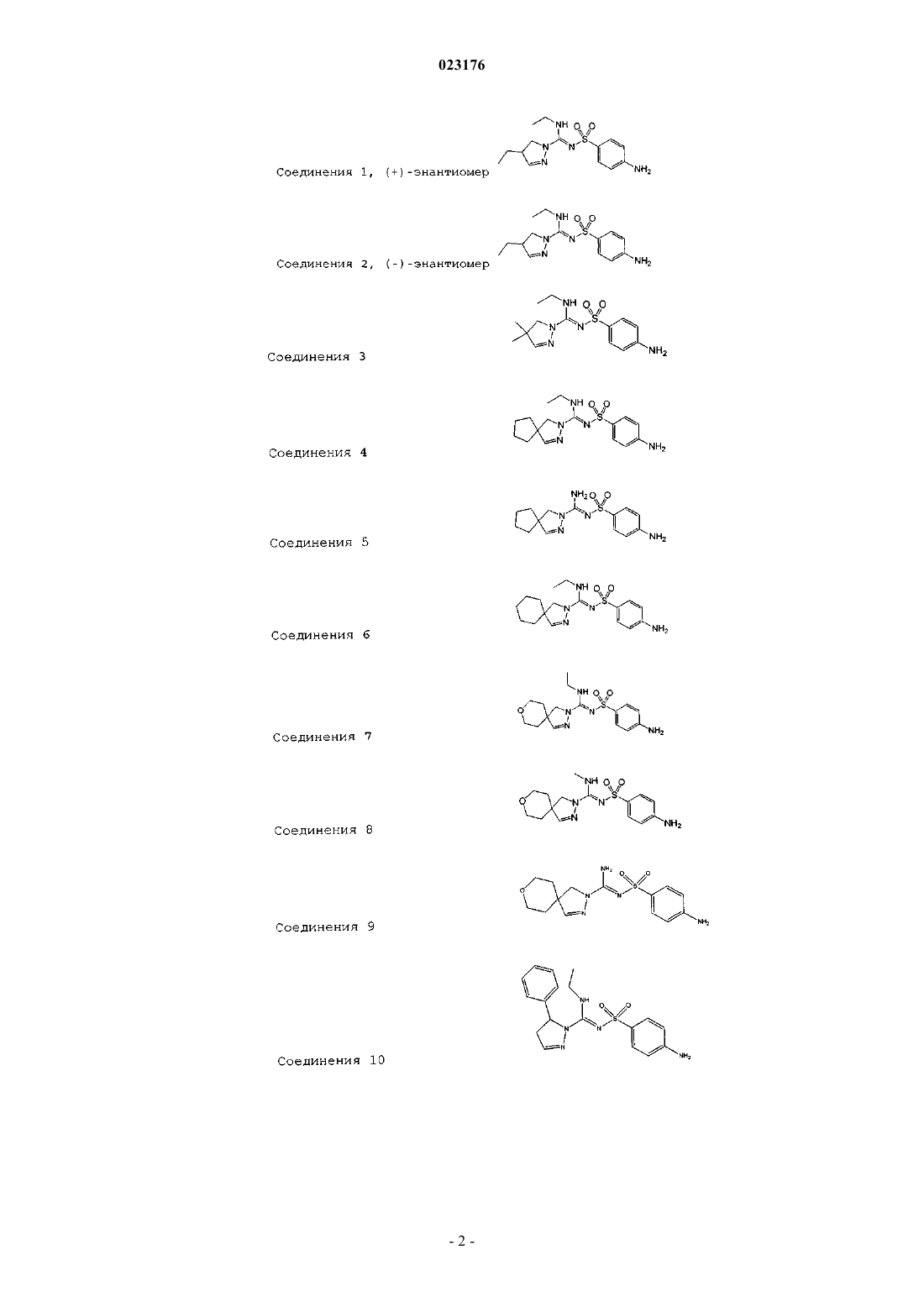
1. Соединение, выбираемое из
Соединения 1, (+)-энантиомер

Соединения 2, (-)-энантиомер

Соединения 3

Соединения 4

Соединения 5

Соединения 6

Соединения 7

Соединения 8

Соединения 9

Соединения 10

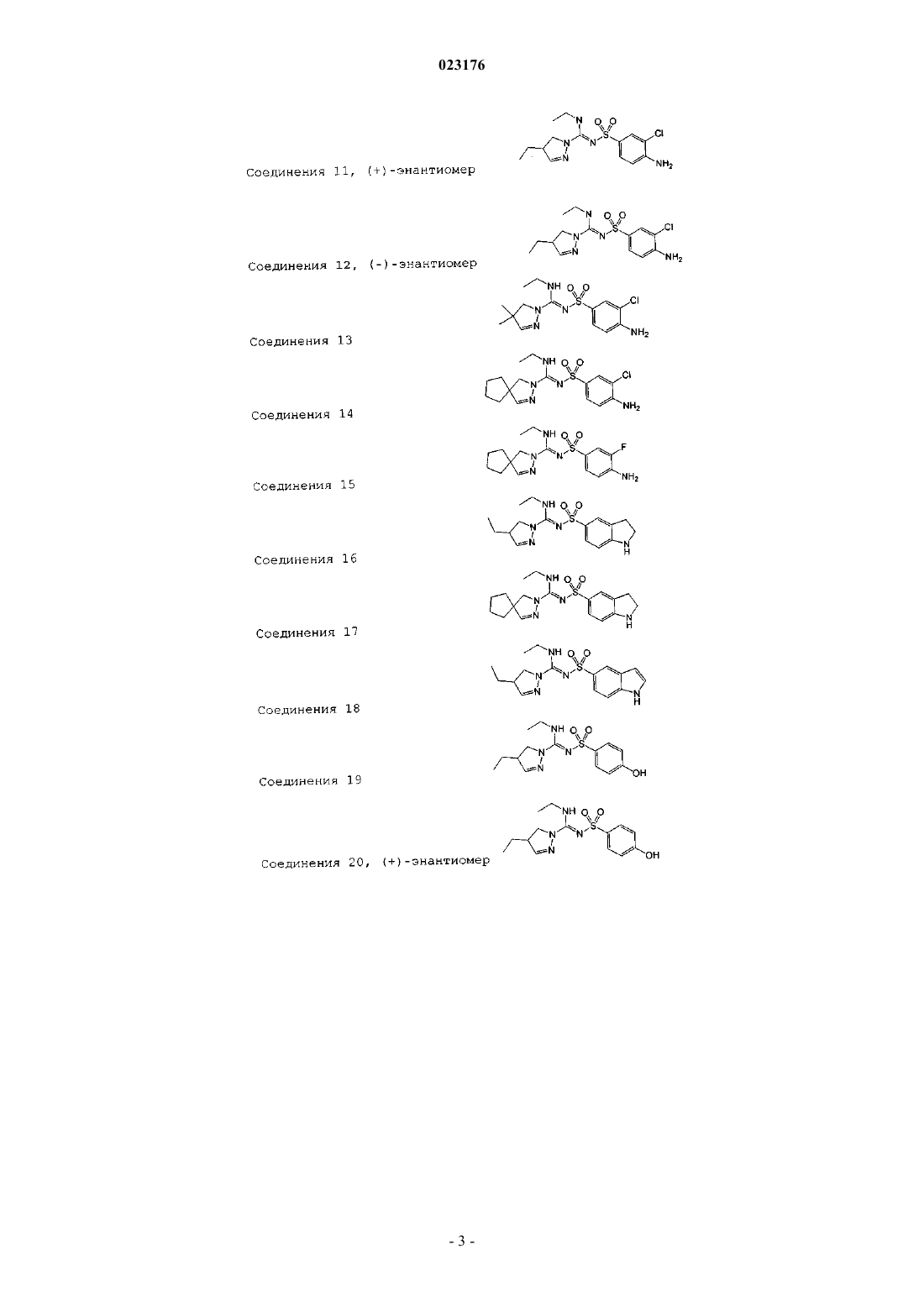
Соединения 11, (+)-энантиомер

Соединения 12, (-)-энантиомер

Соединения 13

Соединения 14

Соединения 15

Соединения 16

Соединения 17

Соединения 18

Соединения 19

Соединения 20, (+)-энантиомер

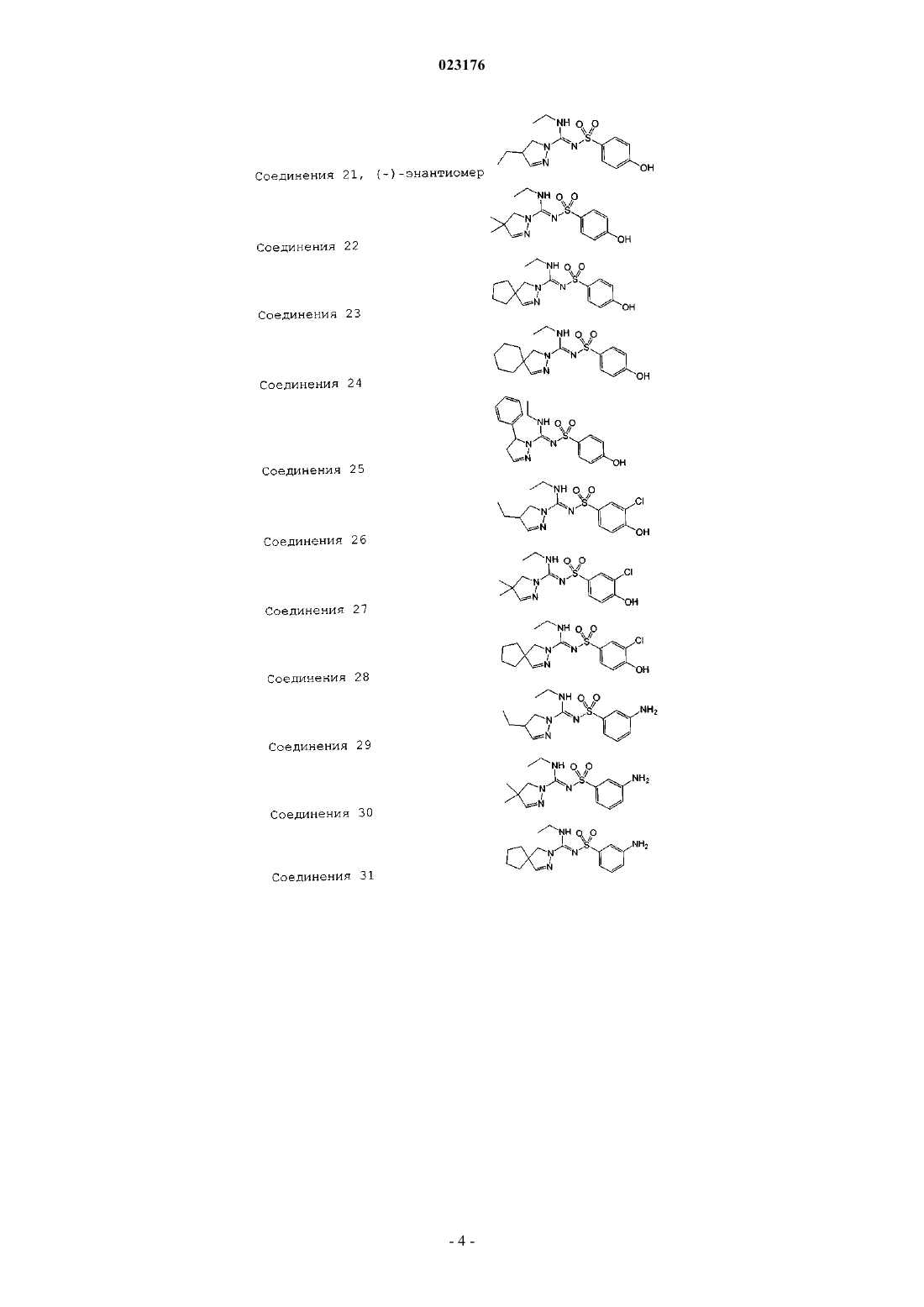
Соединения 21, (-)-энантиомер

Соединения 22

Соединения 23

Соединения 24

Соединения 25

Соединения 26

Соединения 27

Соединения 28

Соединения 29

Соединения 30

Соединения 31

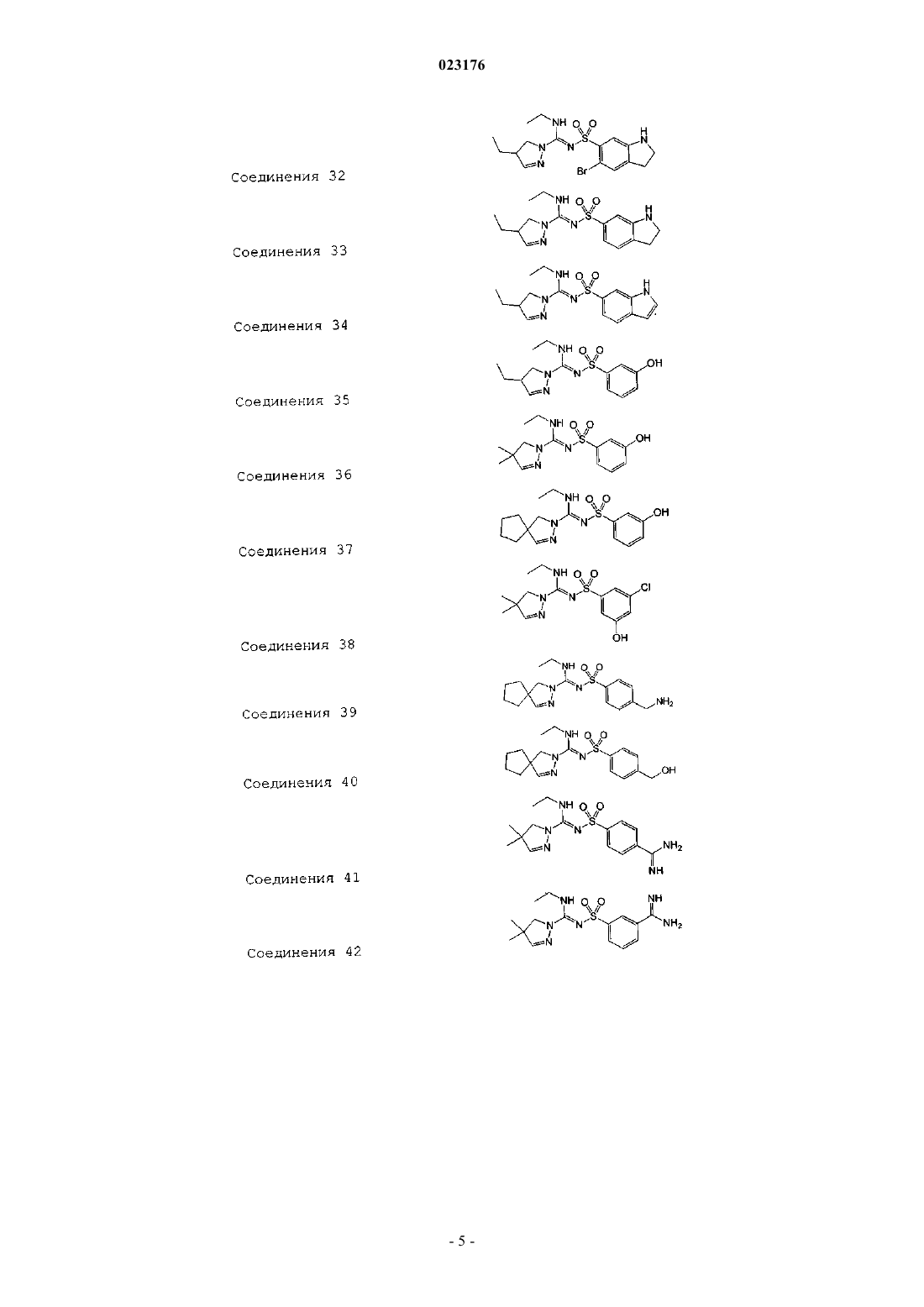
Соединения 32

Соединения 33

Соединения 34

Соединения 35

Соединения 36

Соединения 37

Соединения 38

Соединения 39

Соединения 40

Соединения 41

Соединения 42

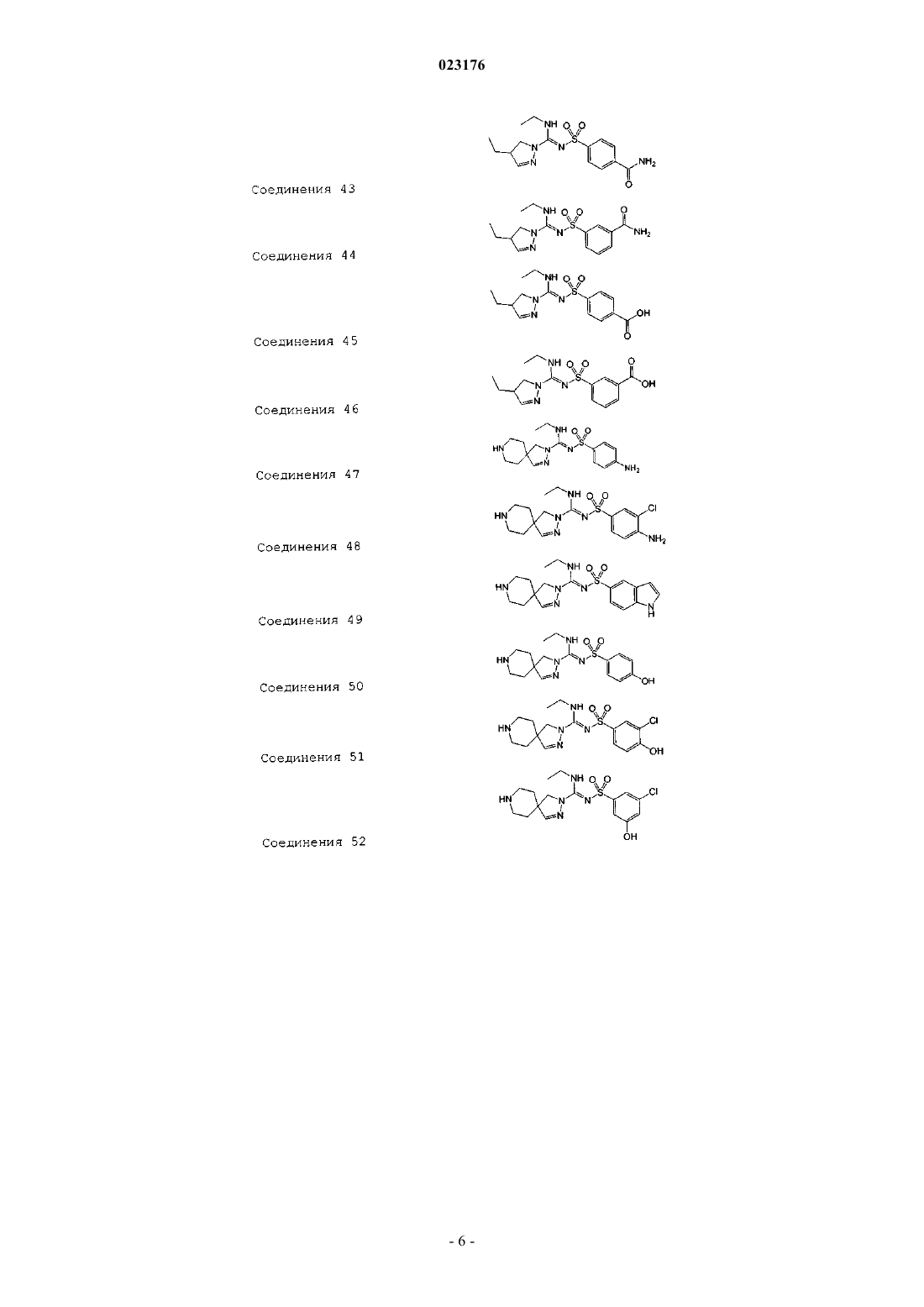
Соединения 43

Соединения 44

Соединения 45

Соединения 46

Соединения 47

Соединения 48

Соединения 49

Соединения 50

Соединения 51

Соединения 52

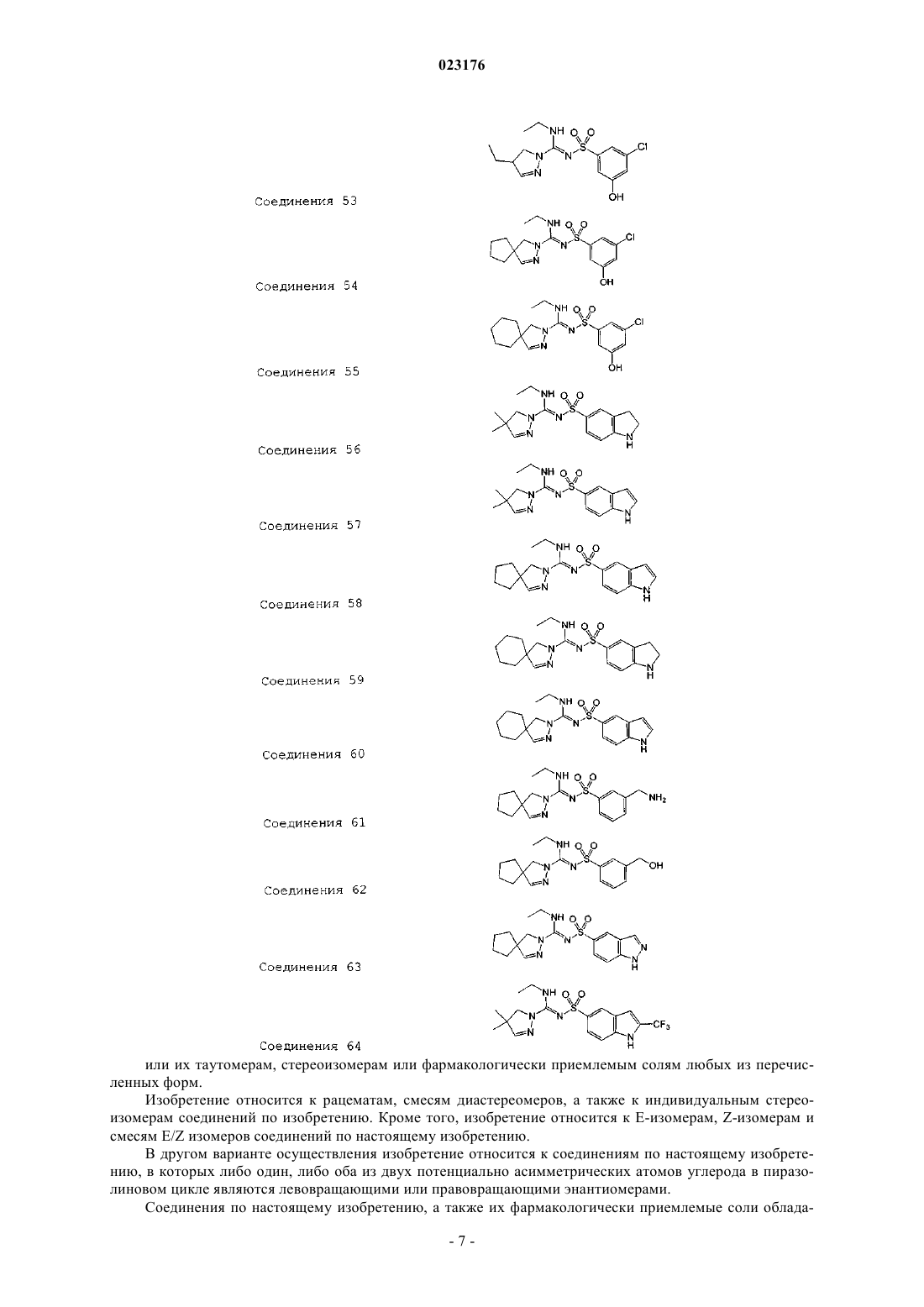
Соединения 53

Соединения 54

Соединения 55

Соединения 56

Соединения 57

Соединения 58

Соединения 59

Соединения 60

Соединения 61

Соединения 62

Соединения 63

Соединения 64

или их фармакологически приемлемых солей.
2. Соединение по п.1 формулы

или его фармакологически приемлемая соль.
3. Соединение по п.1 формулы

или его фармакологически приемлемая соль.
4. Соединение по п.1 формулы

или его фармакологически приемлемая соль.
5. Соединение по п.1 формулы

или его фармакологически приемлемая соль.
6. Соединение по п.1 формулы

или его фармакологически приемлемая соль.
7. Соединение по любому из пп.1-6 или таутомер, стереоизомер или фармакологически приемлемая соль любого вышеперечисленного указанного соединения в виде оптически активного энантиомера.
8. Фармацевтическая композиция для лечения расстройств центральной нервной системы, содержащая фармацевтически эффективное количество соединения по любому из пп.1-7 или его фармакологически приемлемую соль и фармацевтически приемлемый носитель.
9. Лекарственное средство для лечения или профилактики болезни Паркинсона, хореи Хантингтона, шизофрении, тревожности, депрессии, маниакально-депрессивного психоза, психозов, эпилепсии, обсессивно-компульсивных расстройств, аффективных расстройств, мигрени, болезни Альцгеймера, возрастного ослабления когнитивных способностей, умеренных когнитивных нарушений, расстройств сна, расстройств, связанных с питанием, анорексии, булимии, расстройств, связанных с перееданием, приступов панического расстройства, акатизии, синдрома дефицита внимания и гиперактивности, синдрома дефицита внимания, избавления от пагубных пристрастий к кокаину, этанолу, никотину или бензодиазепинам, боли, расстройств, связанных с травмами позвоночника и/или поражениями головы, гидроцефалии, расстройств деятельности кишечника, синдрома раздраженного кишечника, ожирения и диабета 2 типа, содержащее соединение по любому из пп.1-7, или его фармакологически приемлемую соль.
10. Способ получения соединений по любому из пп.1-7 формулы (1')

или таутомера, стереоизомера или фармакологически приемлемой соли любого из вышеперечисленных, где
R1 выбирают из водорода или алкил(С1-4) группы,
R2 и R3 независимо выбирают из водорода или алкил(С1-4) группы или
R2 и R3 вместе с атомом углерода, отмеченным b, образуют С3-8-циклоалкил или С4-8-гетероциклоалкильный цикл, содержащий один атом N или О,
R4 и R5 независимо выбирают из водорода или фенила,
R6 независимо выбирают из водорода или алкил(C1-4) группы,
R8 представляет собой фенил, замещенный группой R" и (R"')p, или группу, выбираемую из

где R представляет собой водород или галоген,
R' представляет собой водород или трифторметил,
R" представляет собой галоген, СООН, ОН, C(NH)NH2, NH2, CH2NH2 или СН2ОН,
R"' представляет собой водород или гидроксил,
р представляет собой 0 или 1,
звездочка (*) обозначает связь к атому S,
включающий стадии:
(i) взаимодействия соединения формулы (X), которое можно получить взаимодействием соединения формулы (IX) с алкилгалогенидом, например метилйодидом, с пиразолином в присутствии основания, с получением соединения формулы (1z),
(ii) взаимодействия соединения формулы (1z) с сульфонилгалогенидом формулы R8-SO2-X, где X представляет собой Br, Cl или F, в апротонном растворителе, например дихлорметане, в присутствии основания, например диизопропилметиламина,

Текст
Изобретение относится к арилсульфонилпиразолинкарбоксамидиновым производным в качестве антагонистов рецепторов 5-НТ 6, к способам получения этих соединений и к новым промежуточным соединениям, применимым в их синтезе. Кроме того, настоящее изобретение относится к применениям указанных соединений и композиций, в частности их применению для введения пациентам с целью достижения терапевтического эффекта при лечении болезни Паркинсона,хореи Хантингтона, шизофрении, тревожности, депрессии, маниакально-депрессивного психоза,психозов, эпилепсии, обсессивно-компульсивных расстройств, аффективных расстройства,мигрени, болезни Альцгеймера, возрастного ослабления когнитивных способностей, умеренных когнитивных нарушений, расстройств сна, расстройств, связанных с питанием, анорексии,булимии, расстройств, связанных с перееданием, приступов панического расстройства, акатизии,синдрома дефицита внимания и гиперактивности, синдрома дефицита внимания, избавления от пагубных пристрастий к кокаину, этанолу, никотину или бензодиазепинам, боли, расстройств,связанных с травмами позвоночника и/или поражениями головы, гидроцефалии, расстройств деятельности кишечника, синдрома раздраженного кишечника, ожирения и диабета 2 типа. Указанные соединения имеют формулы, приведенные в описании. Ван Лувезейн Арнольд, Ивема Баккер Ваутер И., Стойт Аксел, Ренсинк Агата А.М., Венхорст Дженнифер,Ван Дер Нэт Мартина А.В., Де Хан Мартин, Крюсе Корнелис Г. (NL) Медведев В.Н. (RU) Область техники, к которой относится настоящее изобретение Настоящее изобретение относится к областям фармацевтической и органической химии, и в изобретении разработаны арилсульфонилпиразолинкарбоксамидиновые производные, а также связанные с ними промежуточные соединения, составы и способы. Уровень техники Серотонин (5-гидрокситриптамин или 5-НТ), представляющий собой важнейший нейромедиатор периферической и центральной нервной системы, модулирует широкий диапазон физиологических и патологических функций, причем его действие опосредуется рядом семейств рецепторов, именуемых 5HT1, 5-HT2, 5-НТ 3, 5-НТ 4, 5-НТ 5, 5-НТ 6 и 5-НТ 7. Хотя функции трех последних семейств понятны не столь хорошо, как функции остальных семейств, общепризнано, что соединения, которые селективно препятствуют передаче сигнала, опосредованного 5-НТ, представляют собой важные цели для разработки новых лекарственных препаратов. Крысиный рецептор 5-НТ 6 был клонирован двумя различными группами ученых (Ruat, 1993; Sebben, 1994) и вскоре после этого был клонирован аналогичный рецептор человека, имеющий на 89% идентичную последовательность (Kohen, 1996). В настоящее время основной интерес к рецептору 5-НТ 6 вызван тем, что несколько психотропных агентов являются высокоафинными антагонистами человеческого рецептора 5-НТ 6 (Kohen, 1996; Roth, 1994). Эти соединения включают амитриптилин (Ki=65 нМ) и атипичные антипсихотические средства клозапин (Ki=9,5 нМ), оланзапин (Ki=10 нМ) и кветиапин (Ki=33 нМ). Однако ни одно из этих соединений не является селективным. Первыми опубликованными селективными антагонистами рецептора 5-НТ 6 являются Ro 04-6790 и Ro 63-0563. Однако их применимость ограничена умеренным сродством к рецептору (Ki=50 нМ и 12 нМ, соответственно) и плохой фармакокинетикой (Sleight, 1998). Что касается текущих разработок селективных антагонистов рецептора 5-НТ 6Ro-04-6790 и SB-271046, имелось несколько сообщений относительно активности этих соединений в моделях когнитивной функции. SB-271046 улучшал выполнение тестов в водном лабиринте Морриса(Rogers, 1999). Эти результаты согласуются с данными о том, что длительное интрацеребровентрикулярное введение антисмысловых нуклеотидов, направленных на последовательность рецептора 5-НТ 6, приводило к некоторому улучшению выполнения теста в водном лабиринте Морриса (Bentley, 1999b). В последнее время сообщалось о влиянии антагонистов 5-НТ 6 и 5-НТ 6 антисмысловых нуклеотидов на уменьшение потребления пищи крысами (Bentley, 1997; Bentley, 1999a; Woolley, 2001). Ожирение представляет собой состояние, характеризующееся увеличением содержания жира в организме и приводящее к избыточной массе тела по сравнению с принятыми нормами. Ожирение является наиболее важным пищевым расстройством в западном мире и представляет собой значительную проблему для здоровья населения во всех промышленно развитых странах. Это расстройство ведет к повышенной смертности вследствие увеличения частоты возникновения таких заболеваний, как сердечно-сосудистые заболевания, заболевания пищеварительного тракта, заболевания дыхательных путей, рак и диабет 2 типа. Было выявлено, что 5-НТ 6-селективные лиганды потенциально применимы при лечении или профилактике некоторых расстройств центральной нервной системы, таких как болезнь Паркинсона, хорея Хантингтона и/или шизофрения, тревожность, депрессия, маниакально-депрессивный психоз, психозы,эпилепсия, обсессивно-компульсивные расстройства, аффективные расстройства, мигрень, болезнь Альцгеймера (улучшение когнитивной памяти), возрастное ослабление когнитивных способностей, умеренные когнитивные нарушения, нейродегенеративные заболевания, характеризующиеся нарушением роста нейронов, расстройства сна, расстройства, связанные с питанием, такие как анорексия и булимия, расстройства, связанные с перееданием, приступы панического расстройства, акатизия, синдром дефицита внимания и гиперактивности (ADHD), синдром дефицита внимания (ADD), избавление от пагубных пристрастий, например к кокаину, этанолу, никотину и бензодиазепинам, и боли, а также расстройств, связанных с травмами позвоночника и/или поражениями головы, например гидроцефалии. Ожидается также, что селективные лиганды 5-НТ 6 будут применимы при лечении некоторых желудочно-кишечных расстройств, например, расстройств деятельности кишечника и синдрома раздраженного кишечника, а также при лечении или профилактике ожирения и диабета 2 типа, с целью уменьшения массы тела и увеличения массы тела. Уменьшение массы тела и увеличение массы тела (т.е. лечение расстройств, связанных с массой тела) достигается, в том числе, за счет уменьшения потребления пищи. Задача настоящего изобретения заключалась в разработке мощных и селективных антагонистов 5 НТ 6, обладающих большей метаболической стабильностью по сравнению с известными, химически родственными антагонистами 5-НТ 6 (раскрытыми в WO 2008/034863) и применимых для лечения некоторых расстройств ЦНС. Описание настоящего изобретения Неожиданно было обнаружено, что некоторые арилсульфонилпиразолинкарбоксамидиновые производные, у или в арилсульфонильном фрагменте которых имеется функциональная группа, служащая донором водородной связи, являются более эффективными и более метаболически устойчивыми антагонистами рецептора 5-НТ 6, по сравнению с известными, химически родственными антагонистами рецептора 5-НТ 6. Изобретение относится к соединениям, выбираемым из группы: или их таутомерам, стереоизомерам или фармакологически приемлемым солям любых из перечисленных форм. Изобретение относится к рацематам, смесям диастереомеров, а также к индивидуальным стереоизомерам соединений по изобретению. Кроме того, изобретение относится к Е-изомерам, Z-изомерам и смесям E/Z изомеров соединений по настоящему изобретению. В другом варианте осуществления изобретение относится к соединениям по настоящему изобретению, в которых либо один, либо оба из двух потенциально асимметрических атомов углерода в пиразолиновом цикле являются левовращающими или правовращающими энантиомерами. Соединения по настоящему изобретению, а также их фармакологически приемлемые соли облада-7 023176 ют антагонистической активностью в отношении рецептора 5-НТ 6. Они применимы при лечении расстройств, в развитие которых вовлечены рецепторы 5-НТ 6, или поддающихся лечению путем воздействия на эти рецепторы. Например, соединения по настоящему изобретению применимы при лечении болезни Паркинсона, хореи Хантингтона, шизофрении, тревожности, депрессии, маниакальнодепрессивного психоза, психозов, эпилепсии, обсессивно-компульсивных расстройств, аффективных расстройств, мигрени, болезни Альцгеймера, возрастного ослабления когнитивных способностей, умеренных когнитивных нарушений, расстройств сна, расстройств, связанных с питанием, анорексии, булимии, расстройств, связанных с перееданием, приступов панического расстройства, акатизии, синдрома дефицита внимания и гиперактивности, синдрома дефицита внимания, пагубных пристрастий к кокаину,этанолу, никотину и бензодиазепинам, боли, расстройств, связанных с травмами позвоночника или поражениями головы, гидроцефалии, расстройств деятельности кишечника, синдрома раздраженного кишечника, ожирения и диабета 2 типа. Другие варианты осуществления настоящего изобретения включают: фармацевтические композиции для лечения, например, расстройств или состояний, которые поддаются лечению путем блокирования рецепторов 5-НТ 6, где композиции включают соединения по настоящему изобретению или их фармацевтически приемлемые соли, а также фармацевтически приемлемый носитель; способы лечения расстройств или состояний, которые поддаются лечению путем блокирования рецепторов 5-НТ 6, где способы включают введение пациенту, при необходимости такого лечения, соединения по настоящему изобретению или его фармацевтически приемлемой соли; фармацевтические композиции, предназначенные для лечения, например, расстройств или состояний, выбранных из расстройств, перечисленных в настоящей заявке; способы лечения расстройств или состояний, выбранных из расстройств, перечисленных в настоящей заявке, где упомянутые способы включают введение пациенту, при необходимости такого лечения,соединения по настоящему изобретению или его фармацевтически приемлемой соли; фармацевтические композиции, предназначенные для лечения расстройств или состояний, выбранных из расстройств, перечисленных в настоящей заявке, где упомянутые композиции включают соединение по настоящему изобретению или его фармацевтически приемлемую соль, а также фармацевтически приемлемый носитель; способы лечения расстройств или состояний, выбранных из расстройств, перечисленных в настоящей заявке, где упомянутые способы включают введение пациенту, при необходимости такого лечения,соединения по настоящему изобретению или его фармацевтически приемлемой соли; способы противодействия активности рецептора 5-НТ 6, которые включают введение субъекту, при наличии такой необходимости, эффективного количества соединения по настоящему изобретению. Кроме того, изобретение относится к применению соединений или солей по настоящему изобретению для производства лекарственных средств. Помимо этого изобретение относится к соединениям, фармацевтическим композициям, наборам и способам, предназначенным для лечения расстройств или состояний, выбранных из расстройств, перечисленных в настоящей заявке, где упомянутый способ включает введение пациенту, при наличии необходимости такого лечения, соединения по настоящему изобретению или его фармацевтически приемлемой соли. Соединения по настоящему изобретению обладают антагонистической активностью в отношении рецептора 5-НТ 6. Эту активность соединений по настоящему изобретению легко продемонстрировать,например, с использованием одного или нескольких анализов, описанных в настоящей заявке или известных в технике. В настоящем изобретении, помимо перечисленного разработаны способы получения соединений по настоящему изобретению, а также промежуточные соединения, применяемые в этих способах. Более конкретно настоящее изобретение относится к способу получения соединений по настоящему изобретению формулы (1') или таутомера, стереоизомера или фармакологически приемлемой соли любого из вышеперечисленных, гдеR1 выбирают из водорода или алкил(С 1-4) группы,R2 и R3 независимо выбирают из водорода или алкил(С 1-4) группы или,-8 023176R2 и R3 вместе с атомом углерода, отмеченным b, образуют С 3-8-циклоалкил или С 4-8-гетероциклоалкильный цикл, содержащий один атом N или О,R4 и R5 независимо выбирают из водорода или фенила,R6 независимо выбирают из водорода или алкил(С 1-4) группы,R8 представляет собой фенил, замещенный группой R" и (R"')p или группу выбираемую из где R представляет собой водород или галоген,R' представляет собой водород или трифторметил,R" представляет собой галоген, СООН, ОН, C(NH)NH2, NH2, CH2NH2 или СН 2 ОН,R"' представляет собой водород или гидроксил,р представляет собой 0 или 1,звездочкаобозначает связь к атому S,включающий стадии:(i) взаимодействия соединения формулы (X), которое можно получить взаимодействием соединения формулы (IX) с алкилгалогенидом, например метилйодидом, с пиразолином в присутствии основания, с получением соединения формулы (1z),(ii) взаимодействия соединения формулы (1z) с сульфонилгалогенидом формулы R8-SO2-X, где X представляет собой Br, Cl или F, в апротонном растворителе, например дихлорметане, в присутствии основания, например диизопропилметиламина, Выделение и очистка соединений и промежуточных соединений, описанных в настоящей заявке,может быть осуществлена, если это желательно, при помощи любой подходящей методики выделения или очистки, как, например, фильтрования, экстракции, кристаллизации, колоночной хроматографии,тонкослойной хроматографии, толстослойной хроматографии, препаративной жидкостной хроматографии высокого и низкого давления или комбинации перечисленных методик. Конкретные иллюстрации подходящих способов разделения и выделения могут быть взяты из описаний методик синтеза и примеров. Однако помимо указанных могут, разумеется, применяться и другие эквивалентные методики разделения или выделения. Соединения по настоящему изобретению могут содержать один или несколько асимметрических центров и, следовательно, могут существовать в виде рацематов и рацемических смесей, отдельных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. В зависимости от природы различных заместителей молекула может содержать дополнительные асимметрические центры. Каждый такой асимметрический центр будет независимо приводить к появлению двух оптических изомеров. Все возможные оптические изомеры и диастереомеры в форме смесей, а также в виде чистых или частично очищенных соединений, входят в объем настоящего изобретения. Настоящее изобретение включает в себя все такие изомерные формы этих соединений. Независимый синтез упомянутых диастереомеров или их хроматографическое разделение могут быть осуществлены в соответствии с известными в технике способами с помощью подходящих модификаций раскрытой в заявке методологии. Истинное пространственное строение диастереомеров может быть установлено с помощью рентгеноструктурного исследования кристаллических продуктов или кристаллических промежуточных соединений, которые при необходимости обрабатывают реагентом, содержащим асимметрический центр известной абсолютной конфигурации. Рацемические смеси соединений можно разделить на индивидуальные энантиомеры при помощи способов, хорошо известных в технике, например, взаимодействия рацемической смеси соединений с энантиомерно чистым соединением с образованием диастереомерной смеси, с последующим разделением индивидуальных диастереомеров по стандартным методикам, например, фракционной кристаллизации или хроматографии. Указанное взаимодействие часто заключается в образовании солей с использованием энантиомерно чистой кислоты или основания, например, (-)-ди-птолуоил-D-винной кислоты и/или (+)-ди-п-толуоил-L-винной кислоты. Затем эти диастереомерные производные могут быть превращены в чистые энантиомеры путем отщепления присоединенного хирального остатка. Рацемическую смесь соединений можно также разделить непосредственно с помощью хроматографических методик, используя хиральные неподвижные фазы: т.е. способами, хорошо известными в технике. В качестве альтернативы, любой энантиомер соединения можно получить стереоседективным синтезом, используя оптически чистые исходные вещества или реагенты известной конфигурации, с помощью методик, хорошо известных в технике. Цис- и транс-изомеры соединений по настоящему изобретению или их фармацевтически приемлемых солей также входят в объем настоящего изобретения, и сказанное относится также и к таутомерам соединений по настоящему изобретению или их фармацевтически приемлемых солей. Некоторые из кристаллических форм соединений могут существовать в виде полиморфных модификаций, причем имеется в виду, что они входят в объем настоящего изобретения. Кроме того, некоторые соединения могут образовывать сольваты с водой (т.е. гидраты) или обычными органическими растворителями. Эти сольваты также входят в объем настоящего изобретения. Соединения по настоящему изобретению могут также применяться в качестве реагентов или стандартов в биохимических исследованиях неврологической функции, дисфункции и заболеваний. Определения В контексте данного описания термин "антагонист рецептора 5-НТ 6" относится к соединению, демонстрирующему данную активность, измеренную с помощью однозначных и общепризнанных фармакологических анализов, включая методики, описанные в WO 2008/034863, но не проявляющему существенной перекрестной реакционной способности, направленной на другие рецепторы. Основные термины, используемые в описании раскрытых в заявке соединений, имеют их обычный смысл. Термин "алкил" в настоящем описании означает одновалентную насыщенную разветвленную или линейную углеводородную цепь. Если не указано иное, такие цепи могут содержать от 1 до 18 атомов углерода. Типовыми примерами таких алкильных групп являются метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, трет-пентил, гексил, изогексил,гептил, октил, нонил, децил, ундецил, додецил, тридецил, тетрадецил, пентадецил, гексадецил, гептадецил, октадецил и т.п. Если алкильная группа названа "низшей", она будет содержать от 1 до 6 атомов углерода. То же самое определение количества атомов углерода применимо к исходному термину "алкан" и к производным терминам, например "алкокси". Количество атомов углерода в различных углеводород-содержащих фрагментах указывается с помощью префикса, указывающего минимальное и максимальное количество атомов углерода в данном фрагменте, т.е. префикс Сх-у определяет количество имеющихся углеродных атомов от целого числа х до целого числа у включительно. Например "алкил (C1-3)" означает метил, этил, н-пропил или изопропил, и"алкил (C1-4)" означает метил, этил, н-пропил, изопропил, н-бутил, 2-бутил, изобутил или 2-метил-нпропил. Термин "алкенил" означает линейный или разветвленный углеводородный радикал, имеющий одну или несколько двойных углерод-углеродных связей, например, винил, аллил, бутенил и т.д. и, например, представляет собой (С 2-4)алкенил. В "алкинильных" группах линейный или разветвленный углеводородный радикал содержит одну или несколько тройных углерод-углеродных связей, как, например,в этиниле, пропаргиле, 1-бутиниле, 2-бутиниле и т.д., и примером алкинила является (С 2-4) алкинил. Если не указано иное, цепи "алкенилов" и "алкинилов" могут содержать от 1 до 18 атомов углерода. Термин "ацил" означает алкил (C1-3) карбонил, арилкарбонил или арилалкил (C1-3) карбонил. Термин "арил" включает моно- или полициклические ароматические группы, в т.ч. фенил, нафтил, 1,2,3,4 тетрагидронафтил, инденил, флуоренил, антраценил, фенантренил, нафтаценил и азуленил. Термин "гетероарил" охватывает моно- или полициклические гетероароматические группы, в т.ч. фурил, тиенил,пирролил, оксазолил, тиазолил, имидазолил, имидазо[2,1-b][1,3]тиазолил, пиразолил, изоксазолил, изотиазолил, пиридил, пиридазинил, пиримидинил, пиразинил, 1,3,5-триазинил, индазолил, индолил, индолизинил, изоиндолил, бензо[b]фуранил, 1,2,3,4-тетрагидроизохинолинил, инданил, инденил, бензо[b] тиенил, 2,3-дигидро 1,4-бензодиоксин-5-ил, бензимидазолил, циннолинил, карбазолил, акридинил, фена- 10023176 зинил, фенотиазинил, феноксазинил, бензотиазолил, бензо[1,2,5]тиадиазолил, пуринил, хинолинил, изохинолинил, хинолизинил, фталазинил, хиназолинил, хиноксалинил, 1,8-нафтиридинил и птеридинил. Термин "галоген" означает хлор, фтор, бром или йод; приставка "гетеро", как, например, в терминах "гетероалкил", "гетероароматический" и т.д. означает содержащий один или несколько атомов N, O или S. Термин "гетероалкил" включает алкильные группы с гетероатомами в любом положении, охватывая, таким образом, N-, О- или S-связанные алкильные группы. Термин "замещенный" означает, что указанная группа или фрагмент несет на себе один или несколько заместителей. Если какая-либо группа может включать несколько заместителей и в группу можно ввести большое число возможных заместителей, заместители выбирают независимо и они не должны обязательно быть одинаковыми. Термин "незамещенный" означает, что указанная группа не несет на себе заместителей. В отношении заместителей термин "независимо" означает, что когда возможно присутствие более чем одного такого заместителя, они могут быть одинаковыми или отличаться друг от друга."гетероциклоалкил (С 4-8)" относится к циклам, содержащим гетероатомы, включая пиперидинил,морфолинил, азепанил, пирролидинил, тиоморфолинил, пиперазинил, тетрагидрофурил, тетрагидропиранил. Термины "окси", "тио" и "карбо", используемые в настоящей заявке в качестве части наименования другой группы, соответственно относятся к атому кислорода, атому серы и карбонильной (С=O) группе,которые служат линкерами между двумя группами, как, например, в случае гидроксила, оксиалкила,тиоалкила, карбоксиалкила и т.д. Термин "амино" при использовании его в данной заявке индивидуально или в качестве части названия другой группы, относится к атому азота, который может быть либо концевым атомом, либо служить линкером между двумя другими группами, где аминогруппа может быть первичной, вторичной или третичной (два атома водорода связаны с атомом азота, один атом водорода связан с атомом азота и ни одного атома водорода не связано с атомом азота, соответственно) аминогруппой. Термины "сульфинил" и "сульфонил" при использовании в настоящем описании в качестве части названия другой группы относятся соответственно к группам -SO- или -SO2-. Для достижения большей краткости изложения, термины "соединение" или "соединения" также включают таутомеры, стереоизомеры или фармакологически приемлемые соли, в случаях, когда они не упомянуты в явном виде. Термин "форма" охватывает сольваты. Термин "кристаллическая форма" относится к различным твердым формам одного и того же соединения, например сольватам. Общее описание кристаллических форм можно найти у Byrn (1995) и Martin (1995). Для большей краткости описания, некоторые из количественных выражений, приведенных в настоящей заявке, не сопровождаются оговорками типа "примерно" или "приблизительно". Подразумевается, что несмотря на то, используются эти термины в явном виде или нет, каждой приведенной величине приписывается указанное значение, а также значения, приближенные к этому указанному значению, которые могли бы обоснованно подразумеваться на основе типовых знаний в данной области, включая неточности, связанные с условиями эксперимента или измерения этой указанной величины. Термины "селективный" и "селективность" относятся к соединениям, которые демонстрируют реакционную способность в отношении конкретного рецептора (например, рецептора 5-НТ 6), но не проявляют существенной перекрестной реакционной способности в отношении других рецепторов (например,других подтипов рецепторов 5-НТ). Имеется в виду, что в тексте описания и формулы изобретения настоящей заявки слово "включать" и варианты этого слова, такие как "включающий" и "включает", не исключают других добавок, компонентов, целых чисел или стадий. Хотя имеется возможность вводить соединения по настоящему изобретению в виде чистых химических веществ, предпочтительно применять их в форме "фармацевтической композиции". Согласно еще одному аспекту, настоящее изобретение относится к фармацевтической композиции, включающей соединение по настоящему изобретению или его фармацевтически приемлемую соль или сольват, в комбинации с одним или несколькими фармацевтически приемлемыми носителями действующего соединения и, необязательно, одним или несколькими другими терапевтическими ингредиентами. Носитель (носители) должен быть приемлемым в смысле совместимости с другими ингредиентами состава и отсутствия вреда для реципиента. Термин "композиция" в настоящей заявке охватывает продукты, включающие указанные ингредиенты в заранее определенных количествах или пропорциях, а также любые продукты, которые прямо или опосредованно образуются при смешивании указанных ингредиентов в указанных количествах. В отношении фармацевтических композиций этот термин охватывает продукт, включающий один или несколько действующих ингредиентов и, необязательно, носитель, включающий инертные ингредиенты, а также любой продукт, который прямо или опосредованно образуется в результате смешивания, комплексообра- 11023176 зования или агрегирования любых двух или нескольких ингредиентов, или в результате диссоциации одного или нескольких ингредиентов или в результате других типов реакций или взаимодействий одного или нескольких из указанных ингредиентов. В основном фармацевтические композиции получают путем однородного и тесного смешивания действующего ингредиента с жидким носителем или тонкоизмельченным твердым носителем или носителями двух этих типов, и затем, при необходимости, придания полученному составу желаемой формы. Фармацевтическая композициявключает достаточное количество действующего соединения для достижения желаемого воздействия на развитие или состояние болезни. Соответственно, фармацевтическая композиция по настоящему изобретению охватывает любую композицию, полученную смешиванием соединения по настоящему изобретению и фармацевтически приемлемого носителя. Под термином "фармацевтически приемлемый" подразумевается, что носитель, разбавитель или наполнитель должен быть совместим с другими ингредиентами состава и не являться вредным для реципиента. В контексте данной заявки, термин "комбинированный препарат" включает как действительные комбинации, в которых соединение по настоящему изобретению и одно или несколько других лекарственных средств физически объединены в одном препарате, например, таблетке или жидкости, предназначенной для инъекции, так и "наборы компонентов", зключающие соединение по настоящему изобретению и одно или несколько других лекарственных средств в отдельных дозированных лекарственных формах, совместно с инструкцией по применению, необязательно с дополнительными средствами, облегчающими пациенту прием входящих в набор соединений, например, этикетками или рисунками. В случае действительных комбинаций, фармакотерапия является "одновременной" по определению. Содержимое "наборов компонентов" может вводиться либо одновременно, либо через различные промежутки времени. Будет терапия являться одновременной или "последовательной", будет зависеть от характеристик других применяемых лекарственных средств, таких как начало и продолжительность действия, уровни в плазме, клиренс и т.д., а также от заболевания, его стадии и индивидуальных характеристик пациента. Сродство соединений по настоящему изобретению к рецепторам 5-НТ 6 определяли как описано выше. Исходя из измеренного для данного соединения по настоящему изобретению сродства к связыванию можно оценить теоретическую минимальную эффективную "дозу". При концентрации соединения,равной двукратному измеренному значению K1, почти 100% рецепторов 5-НТ 6, вероятно, будут связаны с соединением. Перевод этой концентрации в единицы мг соединения/кг массы тела пациента позволяет вычислить наименьшую теоретическую эффективную дозу, в предположении идеальной биодоступности. Фармакокинетические, фармакодинамические и другие факторы могут изменить реально вводимую дозу в большую или меньшую сторону. Типовая дневная доза действующего ингредиента меняется в широком диапазоне и будет зависеть от различных факторов, таких как соответствующие показания к применению, путь введения, возраст, масса тела и пол пациента, и эту дозу может определить врач. Как правило, общая дневная доза, вводимая пациенту в виде одной или нескольких отдельных доз, может составлять, например, от 0,001 до 10 мг/кг массы тела пациента ежедневно, и чаще от 0,01 до 1,000 мг в день всех действующих ингредиентов. Такие дозы будут вводиться пациенту при необходимости лечения от одного до трех раз каждый день, или так часто, как это необходимо для достижения эффекта, и в течение периода не менее двух месяцев, чаще в течение не менее шести месяцев или постоянно. Термин "терапевтически эффективное количество" относится к количеству терапевтического агента, необходимого для лечения состояния, которое можно лечить введением композиции по настоящему изобретению. Это количество включает количество, достаточное для проявления обнаруживаемого терапевтического или благоприятного отклика системы тканей человека. Эффект может включать лечение состояний, перечисленных в настоящей заявке. Точное фармацевтически эффективное количество для того или иного субъекта будет зависеть от габаритов и состояния здоровья субъекта, природы и степени развития состояния, подвергаемого лечению, рекомендаций врача и выбранных для лечения терапевтических средств или комбинаций терапевтических средств. Таким образом, нецелесообразно заблаговременно устанавливать точное фармацевтически эффективное количество. Термин "фармацевтически приемлемая соль" относится к таким солям, которые, в рамках обоснованного медицинского суждения, подходят для применения в контакте с тканями людей и низших животных, без недопустимой токсичности,раздражения, аллергических реакций и т.п. и соответствуют разумному соотношению польза/риск. Фармацевтически приемлемые соли хорошо известны в технике. Они могут быть получены in situ при окончательном выделении и очистке соединений по настоящему изобретению, или отдельно, путем взаимодействия соединений с фармацевтически приемлемыми нетоксичными основаниями или кислотами, в т.ч. неорганическими или органическими основаниями и неорганическими или органическими кислотами (Berge, 1977). Форма "свободного основания" может быть регенерирована путем введения соли в контакт с основанием или кислотой и выделения исходного соединения стандартным способом. Исходная форма соединения отличается от различных солевых форм некоторыми физическими свойствами, например, растворимостью в полярных растворителях, но в других аспектах для целей настоящего изобретения соли эквивалентны исходной форме соединения. Термин "лечение" относится к любому лечению состояния или заболевания человека и включает (1) подавление заболевания или состояния, т.е. остановку его развития (2) облегчение заболевания или состояния, т.е. приведение состояния к регрессу или (3) прекращение проявления симптомов заболевания. Термины "подавлять", "ингибировать" включают их общепринятые значения, т.е. сдерживание, ослабление, облегчение и замедление, остановка или обращение прогрессирования, тяжести заболевания или результирующих симптомов. Предполагается, что в настоящем описании термин "медицинское лечение" включает диагностические и терапевтические режимы лечения людей, осуществляемые in vivo или exvivo. В настоящем описании термин "расстройства, относящиеся к массе тела" подразумевает расстройства, вызванные дисбалансом между поступлением и расходованием энергии, приводящие к аномальной(Roth, 1994; Sibley, 1993; Sleigh, 1995, 1997). Термин "ожирение" относится к состоянию, при котором человек имеет коэффициент массы тела (BMI), вычисляемый как частное от деления массы тела на квадрат роста (кг/м 2), не менее 25,9. Соответственно, люди с нормальной массой тела имеют значения BMI от 19,9 до 25,9. При этом ожирение может быть вызвано любой причиной, как генетической, так и связанной с окружающей средой. Примеры расстройств, которые могут привести к ожирению или являться причиной ожирения, включают переедание и булимию, поликистозную болезнь яичников, краниофарингиому, синдром Прадера-Вилли, синдром Фрелиха, диабет 2 типа, GH-дефицитных субъектов, нормальный вариант низкорослости, синдром Тернера и другие патологические состояния, при которых наблюдается пониженная метаболическая активность или снижение остаточного расхода энергии как процента от общей безжировой массы, например, детей с острым лимфобластным лейкозом. Сокращения АСЕ-Cl - 1-хлорэтилхлорформиат;ADHD - расстройство дефицита внимания и гиперактивности;API - ионизация при атмосферном давлении; ВЕМР - 2-трет-бутиламино-2-диэтиламино-1,3-диметилпергидро-1,3,2-диазафосфорин;Boc2O - дитрет-бутил дикарбонат; СНО - клетки яичника китайского хомячка;HPLC (ВЭЖХ) - жидкостная хроматография высокого давления (высокоэффективная); 5-НТ - 5-гидрокситриптамин, серотонин;m.p. (т.пл.) - температура плавления либо диапазон плавления;SIM - мониторинг по одному иону;X-Phos - 2-дициклогексилфосфино-2',4'6'-триизопропилбифенил. Пример 1. Аналитические методики. Спектры ядерного магнитного резонанса (1 Н ЯМР) регистрировали в указанном растворителе, используя спектрометры Bruker ARX 400 (1 Н: 400 МГц) или Varian VXR200 (1 Н: 200 МГц) при 300 К, если не указано иное. Спектры регистрировали в дейтерированном хлороформе или ДМСО, которые получали у Cambridge Isotope Laboratories Ltd. Химические сдвиги (5) в миллионных долях в слабое поле относительно тетраметилсилана (1 Н). Константы спин-спинового взаимодействия J приведены в Гц. Формы сигналов в спектрах ЯМР указаны с помощью символов "q" (кв, квартет), "dq" (дкв, дублет квартетов),"t" (т, триплет), "dt" (дт, дублет триплетов), "d" (д, дублет), "dd" (дд, дублет дублетов), "ddd" (ддд, двойной дублет дублетов), "s" (с, синглет), "bs" (уш.с, уширенный синглет) и "m" (м, мультиплет). Сигналы NH и ОН идентифицировали после смешивания образца с каплей D2O. Термин "флэш-хроматография" относится к очистке с использованием указанного элюента и силикагеля (Merck silica gel 60: 0,040-0,063 мм). Температуры плавления регистрировали с помощью прибора для определения температур плавления Buchi B-545. Все реакции, включающие соединения, чувствительные к влаге и/или кислороду, проводили в атмосфере безводного азота. 3 а ходом реакций наблюдали с помощью тонкослойной хроматографии (ТСХ) на стеклянных пластинах, покрытых оксидом кремния(Merck precoated silica gel 60 F254) с использованием указанного элюента. Пятна визуализировали с помощью УФ-света (254 нм) или жидкостная хроматографиямасс-спектроскопия (ЖХ-МС): ЖХ-МС система состояла из двух микронасосов Perkin Elmer series 200. Насосы были соединены друг с другом Тобразным смесителем, соединенным с автодозатором Gilson 215. Использовали следующую методику: А=100% воды с 0,025% НСООН и 10 ммоль NH4HCOO pH 3. В=100% ACN с 0,025% НСООН. Автодозатор имел 2 мкл дозирующий контур и был соединен с колонкой Waters Atlantis C18 304,6 мм, заполненной 3 мкм частицами. Колонку термостатировали в печи для колонок Perkin Elmer series 200 при 40 С. Колонка была присоединена к УФ-измерительному устройству Perkin Elmer series 200 с 2,7 мкл измерительной ячейкой. Была установлена длина волны 254 нм. УФ-измерительное устройство было присоединено к масс-спектрометру Sciex API 150EX. Масс-спектрометр имел следующие параметры: диапазон сканирования: 150-900 ат.ед.массы; полярность: положительная; режим сканирования: профиль; разрешение Q1: UNIT; величина шага: 0,10 ат.ед.массы; время на скан: 0,500 с; NEB: 10; CUR: 10;IS: 5200; ТЕМ: 325; DF: 30; FP: 225 и ЕР:10. Детектор светорассеяния был присоединен к спектрометруSciex API 150. Детектором светорассеяния являлся Sedere Sedex 55, работавший при температуре 50 С и давлении N2 3 бара. Вся система управлялась компьютером power Mac G3. Пример 2. Общие аспекты синтеза. Синтез заявленных соединений и промежуточных соединений, содержащих пиразолиновые фрагменты, удобно осуществлять, следуя способам, аналогичным описанным ранее в WO 2008/034863, используя в качестве структурных элементов производные 4,5-дигидро-1 Н-пиразола или 4,5-дигидро-3 Нпиразола, которые либо являются коммерчески доступными, либо могут быть получены, как показано ниже. Способ синтеза 1 В первом способе синтеза используются N-(бис-алкилсульфанилметилен)сульфонамиды общей формулы (V), которые могут быть получены из сульфонамидов взаимодействием с CS2 в присутствии КОН, и последующим взаимодействием полученного промежуточного соединения с алкилгалогенидом,например, метилйодидом. Две S-алкильные группы в соединении (V) могут быть последовательно заменены на амины, предпочтительно, начиная с производных пиразолиновых фрагментов, с получением соединений общей формулы (VI), и с последующим образованием сульфонилпиразолин карбоксамидиновых производных общей формулы (IV). Способ синтеза 2 Во втором способе синтеза используются фрагменты алкил-изотиомочевины или образованных ей подходящих солей общей формулы (IX), которые удобно получать взаимодействием производных тиомочевины с алкилгалогенидами, например, метилйодидом, причем соединения (IX) могут быть введены во взаимодействие с пиразолинами в присутствии основания с получением пиразолинкарбоксамидиновых производных общей формулы (X). Эти последние можно ввести в реакцию с сульфонилгалогенидами (X=Br, Cl, F, предпочтительно Cl) в присутствии основания с получением сульфонилпиразолин карбоксамидиновых производных общей формулы (IV). Способ синтеза 3 В третьем способе синтеза используются сульфонилкарбаматы общей формулы (I), которые могут быть получены, например, взаимодействием сульфонамидов с метилхлорформиатом или дитрет-бутил дикарбонатом в присутствии основания. Продукты взаимодействия соединений формулы (I) с пиразолинами, имеющие общую формулу (II), могут быть последовательно превращены в хлориминовые промежуточные соединения общей формулы (III), при использовании галогенирующих агентов, например,PCl3, POCl3/DMAP или хлорида 2-хлор-1,3-диметилимидазолиния (DMC), и затем в сульфонилпиразолин карбоксамидиновые производные общей формулы (IV) при реакции с аминами. Выбор конкретных методик синтеза зависит от факторов, известных специалисту в данной области техники, например, от совместимости функциональных групп с используемыми реагентами, возможности применения защитных групп, катализаторов, активации сшивающих реагентов и основных структурных особенностей, имеющихся в конечном соединении, синтез которого предполагается осуществить. Фармацевтически приемлемые соли можно получить с использованием стандартных методик, хорошо известных в технике, например, при смешивании соединения по настоящему изобретению с подходящей кислотой, например неорганической кислотой или органической кислотой. Пример 3. Синтез пиразолиновых промежуточных соединений. Приведенные ниже пиразолиновые промежуточные соединения синтезировали как описано в WO 2008/034863. 100 г сульфаниламида растворяли в 375 мл ДМФА, по каплям добавляли 33,2 мл 50% водного раствора NaOH и продолжали перемешивание в течение 10 мин при комнатной температуре. К полученной белой суспензии по каплям добавляли 19,2 мл дисульфида углерода и перемешивали смесь в течение 30 мин при комнатной температуре. Полученную смесь еще дважды обрабатывали 18,1 мл 50% водного раствора NaOH и 9,6 мл дисульфида углерода, с 10-минутным периодом перемешивания между этими двумя циклами. После заключительного перемешивания смеси в течение 30 мин, оранжевый/красный раствор охлаждали на ледяной бане и по каплям добавляли 72,3 мл йодметана, с такой скоростью, чтобы температура смеси оставалась ниже 25 С. Для сохранения возможности перемешивания смеси добавляли ДМФА в количестве 25 мл, и продолжали перемешивание в течение 1 ч. Продолжая охлаждение, к смеси добавляли 250 мл воды, и затем осуществляли механическое перемешивание суспензии в течение ночи при комнатной температуре. Осадок отделяли фильтрованием и промывали водой и холодным этанолом. Остаток перекристаллизовывали из этилацетата и после высушивания при 50 С в вакууме получали 64,9 г (40%) белого твердого вещества. 1 Н ЯМР (400 МГц, ДМСО-d6)3,38 (с, 6 Н), 6,15 (с, 2 Н), 6,66 (д,J=8,73 Гц, 2 Н), 7,56 (д, J=8,73 Гц, 2 Н). 4-Амино-N-[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)метилсульфанилметилен]бензолсульфонамид В 25 мл микроволновом сосуде растворяли 2,00 г 4-амино-N-(бис-метилсульфанилметилен)бензолсульфонамида и 1,00 г 2,3-диазаспиро[4.4]нон-2-ена в 15 мл пиридина. Сосуд закрывали крышкой и нагревали в течение 2 ч при 180 С в микроволновой печи. Объединяли смеси, полученные в результате 8 последовательных повторений этой методики, и концентрировали при пониженном давлении. Остаток подвергали флэш-хроматографии (DCM/EA 95:590:10) и упаривали чистые фракции, получая 5,20 г К раствору 4,05 г 4-амино-N-[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)метилсульфанилметилен]бензолсульфонамида в 30 мл МеОН добавляли 7,86 мл 70% водного раствора этиламина. Смесь перемешивали в течение 1 ч при комнатной температуре и упаривали досуха. Остаток растворяли в минимальном количестве DCM и растирали с МТВЕ. Осадок отделяли фильтрованием, высушивали в вакууме и затем перекристаллизовывали из н-бутилацетата, получая 2,40 г (67%) 4-амино-N-[(2,3-диазаспиро[4.4]нон-3-ен-2 ил)этиламинометилен]бензолсульфонамида в виде не совсем белого микрокристаллического вещества,после высушивания в вакууме при 80 С; т.пл. 141-142 С. 1 Н ЯМР (400 МГц, CDCl3)1,14 (т, J=7,22 Гц,3 Н), 1,47-1,89 (м, 8 Н), 3,35-3,57 (м, 2 Н), 3,79 (с, 2 Н), 4,02 (уш.с, 2 Н), 6,65 (д, J=8,73 Гц, 2 Н), 6,78 (с, 1 Н),6,91 (уш.с, 1 Н), 7,70 (д, J=8,73 Гц, 2 Н). 4-Амино-N-[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]-3-фторбензолсульфонамид (соединение 15). 3-Фтор-4-нитробензолсульфонамид К раствору 5,00 г 3-фтор-4-нитробензолсульфонилхлорида в 20 мл ацетонитрила, охлажденному на ледяной бане, по каплям прибавляли 4,40 мл 30% раствора гидроксида аммония. После удаления ледяной бани перемешивание продолжали в течение 30 мин при комнатной температуре. Добавляли воду и экстрагировали смесь DCM. Объединенные органические слои высушивали над MgSO4 и упаривали досуха,получая 4,65 г (99%) твердого желтого вещества. 1 Н ЯМР (400 МГц, ДМСО-d6)7,51 (с, 2 Н), 7,89 (д,J=9,33 Гц, 1 Н), 7,92 (дд, J=10,23, 1,81 Гц, 1 Н), 8,14-8,21 (м, 1 Н). 4-Амино-3-фторбензолсульфонамид К раствору 1,00 г 3-фтор-4-нитробензолсульфонамида в 10 мл МеОН добавляли 10 мол.% палладия на угле. Полученную смесь гидрировали в течение 30 мин при давлении Н 2 50 фунтов/кв.дюйм. После фильтрования с применением материала Hyflo и концентрирования в вакууме получали 630 мг (74%) темно-коричневого масла. 1 Н ЯМР (400 МГц, ДМСО-d6)6,83 (т, J=8,43 Гц, 1 Н), 7,42 (дд, J=8,28, 1,96 Гц, 1 Н), 7,47 (дд, J=10,99, 1,96 Гц, 1 Н) [сигнал группы NH2 в спектре невидим]. 4-Амино-N-(бис-метилсульфанилметилен)-3-фторбензолсульфонамид 1,15 г 4-амино-3-фторбензолсульфонамида растворяли в 50 мл ДМФА, по каплям добавляли 0,33 мл 50% водного раствора NaOH и осуществляли перемешивание в течение 30 мин при комнатной температуре. К полученной смеси по каплям добавляли 0,16 мл дисульфида углерода и перемешивали смесь в течение 1 ч при комнатной температуре. Полученную смесь еще два раза обрабатывали 0,16 мл 50% водного раствора NaOH и 0,08 мл дисульфида углерода, с 30 мин периодом перемешивания между этими двумя циклами. В заключении перемешивали реакционную смесь в течение 1 ч и к полученному пурпурному раствору по каплям добавляли 0,72 мл йодметана, после чего осуществляли перемешивание в течение 1 ч. После охлаждения смеси на водяной бане, к ней медленно добавляли 100 мл воды и полученную суспензию перемешивали механической мешалкой в течение ночи при комнатной температуре. Осадок отделяли фильтрованием и высушивали, получая 0,60 г (35%) коричневого твердого вещества. 1 Н ЯМР В 10 мл микроволновом сосуде растворяли 530 мг 4-амино-N-(бис-метилсульфанилметилен)-3 фторбензолсульфонамида и 325 мг 2,3-диазаспиро[4.4]нон-2-ена в 5 мл пиридина и добавляли каплю ионной жидкости (гексафторфосфата 1-бутил-3-метилимидазолия). Сосуд закрывали крышкой и нагревали в течение 2 ч при 180 С в микроволновой печи. Смесь концентрировали при пониженном давлении,высушивали в вакууме и неочищенный продукт использовали в следующей стадии. 1 Н ЯМР (400 МГц,CDCl3)1,60-2,03 (м, 8 Н), 2,24 (с, 3 Н), 3,07 (с, 2 Н), 4,90 (уш.с, 2 Н), 7,00 (с, 1 Н), 7,28-7,33 (м, 1 Н), 7,657,73 (м, 1 Н), 8,62 (д, J=3,91 Гц, 1 Н). К раствору 840 мг (неочищенного) 4-амино-N-[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)метилсульфанилметилен]-3-фторбензолсульфонамида в 25 мл МеОН добавляли 3,43 мл 70% водного раствора этиламина. Полученную смесь перемешивали в течение 1 ч при комнатной температуре, добавляли воду и экстрагировали смесь DCM. Объединенные органические слои высушивали над MgSO4 и упаривали досуха. Ос- 18023176 10,00 г 4-метоксибензолсульфонамида растворяли в 90 мл ДМФА и добавляли 5,16 мл дисульфида углерода. Смесь охлаждали на ледяной бане и затем по каплям добавляли 6,47 мл 50% водного раствораNaOH. Полученную темно-красную смесь перемешивали в течение 30 мин, по каплям добавляли 7,65 мл йодметана, убирали ледяную баню и перемешивали смесь в течение 1 ч при комнатной температуре. Затем к смеси медленно добавляли 33 мл воды и перемешивали полученную суспензию в течение ночи при комнатной температуре. Осадок отделяли фильтрованием, 3 раза промывали водой и высушивали в вакууме. Продукт очищали флэш-хроматографией (DCMDCM/MeOH 95:5), получая 8,00 г (44%) аморфного маслянистого белого вещества. 1 Н ЯМР (400 МГц, CDCl3) д 2,53 (с, 6 Н), 3,88 (с, 3 Н), 6,97 (кв, J=5,12 Гц, 2 Н), 7,93 (кв, J=5, 02 Гц, 2 Н). 4-Метокси-N-[метилсульфанил-(5-фенил-4,5-дигидропиразол-1-ил)метилен]бензолсульфонамид В атмосфере азота растворяли 3,26 г N-(бис-метилсульфанилметилен)-4-метоксибензолсульфонамида и 4,34 г 5-фенил-4,5-дигидро-1 Н-пиразола в 25 мл пиридина и кипятили раствор с обратным холодильником в течение 3 дней. Смесь охлаждали и концентрировали при пониженном давлении. Остаток смешивали с ЕА и экстрагировали 5% водным раствором NaHCO3. Органический слой высушивали над MgSO4, упаривали досуха и остаток очищали флэш-хроматографией (DCMDCM/MeOH 95:5). Упаривание фракций, содержавших чистый продукт, позволяло получить 2,30 г (40%) желтого масла. ТСХ: К раствору 2,30 г 4-метокси-N-[метилсульфанил-(5-фенил-4,5-дигидропиразол-1-ил)метилен]бензолсульфонамида в 50 мл МеОН добавляли 3,80 мл 70% водного раствора этиламина. Смесь перемешивали в течение ночи при комнатной температуре и упаривали досуха. Остаток смешивали с ЕА и экстрагировали 5% водным раствором NaHCO3. Органический слой высушивали над MgSO4, упаривали досуха и остаток очищали препаративной ВЭЖХ, получая 1,20 г (62%) белого аморфного вещества. 1 Н ЯМР В атмосфере N2 к раствору 1,05 г N-[этиламино-(5-фенил-4,5-дигидропиразол-1-ил)метилен]-4 метоксибензолсульфонамида в 25 мл DCM добавляли 12,91 мл 1 М раствора трибромида бора в DCM. Смесь перемешивали в течение ночи при комнатной температуре в атмосфере N2, гасили водой и перемешивали в течение еще 30 мин. Твердые вещества удаляли фильтрованием и фильтрат экстрагировали водой. Органический слой высушивали над MgSO4 и упаривали досуха. Остаток очищали флэшхроматографией (ступенчатый градиент DCMDCM/МеОН 95:5). Фракции, содержащие чистое вещество, концентрировали и растирали с Et2O. Твердое вещество отделяли фильтрованием и высушивали в вакууме, получая 0,34 г (34%) N-[этиламино-(5-фенил-4,5-дигидропиразол-1-ил)метилен]-4-гидроксибензолсульфонамида в виде серого кристаллического вещества, т.пл. 158-160 С. 1H ЯМР (400 МГц,ДМСО-d6)1,07 (т, J=7,2 Гц, 3 Н), 2,69-2,81 (м, 1 Н), 3,36-3,47 (м, 1 Н), 3,49-3,59 (м, 2 Н), 5,40-5,51 (м, 1 Н),6,55 (д, J=8,7 Гц, 2 Н), 7,00 (д, J=8,4 Гц, 2 Н), 7,04-7,12 (м, 3 Н), 7,22-7,29 (м, 3 Н), 9,71 (с, 1 Н) [сигнал гуанидиновой группы NH невидим в спектре]. К смеси 5,16 г 4-карбоксилбензолсульфонамида и 150 г метанола добавляли 6,84 мл серной кислоты. Смесь кипятили с обратным холодильником в течение ночи и охлаждали до комнатной температуры. Полученную смесь упаривали досуха и остаток растирали с Et2O. Полученный осадок отделяли фильтрованием, промывали Et2O и высушивали, получая 5,2 г (92%) белого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3)3,90 (с, 3 Н), 7,59 (с, 2 Н), 7,97 (д, J=5,84 Гц, 2 Н), 8,15 (д, J=5,84 Гц, 2 Н). 4-Гидроксиметилбензолсульфонамид К раствору 5,2 г метилового эфира 4-сульфамоилбензойной кислоты в 100 мл ТГФ и 1,44 мл МеОН,порциями в течение 10 мин добавляли 0,77 г боргидрида лития. Смесь нагревали до кипения с обратным холодильником в течение ночи, охлаждали до комнатной температуры и выливали на лед, смешанный со 100 мл 1 н. HCl. Смесь экстрагировали EtOAc, органический слой высушивали над MgSO4 и концентрировали при пониженном давлении. Остаток очищали с помощью автоматической флэш-хроматографии К смеси 750 мг 4-гидроксиметилбензолсульфонамида и 50 мл ДМФА добавляли 1,55 мл третбутилхлордифенилсилана и 539 мг имидазола. Смесь перемешивали в течение ночи при комнатной температуре, разбавляли EtOAc и экстрагировали водой. Органическую фазу высушивали над MgSO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью автоматической флэш-хроматографии (DCM), получая 0,5 г чистого продукта и 0,6 г вещества, состоящего из загрязненных фракций продукта. 1 Н ЯМР (400 МГц, CDCl3)1,07 (с, 3 Н), 1,11 (с, 6 Н), 4,75 (с, 2 Н), 4,82 (с, 2 Н),7,35-7,47 (м, 6 Н), 7,49 (д, J=5,68 Гц, 2 Н), 7,64-7,74 (м, 4 Н), 7,90 (д, J=5,68 Гц, 2 Н). К смеси 500 мг 4-(трет-бутилдифенилсиланилоксиметил)бензолсульфонамида и 50 мл ДМФА добавляли 0,11 мл дисульфида углерода и охлаждали смесь до 10 С. При перемешивании по каплям добавляли 0,14 мл 50% водного раствора NaOH и перемешивали смесь в течение одного часа при комнатной температуре. Затем по каплям добавляли 0,16 мл подметана и продолжали перемешивать при комнатной температуре в течение 30 мин. После добавления 10 мл воды смесь перемешивали в течение ночи при комнатной температуре. Осадок отделяли фильтрованием, промывали водой и высушивали, получая 0,4 г продукта. 1 Н ЯМР (400 МГц, ДМСО-d6)1,04-1,08 (м, 9 Н), 2,57 (с, 6 Н), 4,88 (с, 2 Н), 7,40-7,50 (м, 6 Н),7,59 (д, J=8,34 Гц, 2 Н), 7,63-7,68 (м, 4 Н), 7,90 (д, J=8,34 Гц, 2 Н). 4-(трет-Бутилдифенилсиланилоксиметил)-N-[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)метилсульфанилметилен]бензолсульфонамид К 15 мл пиридина добавляли 400 мг N-(бис-метилсульфанилметилен)-4-(трет-бутилдифенилсиланилоксиметил)бензолсульфонамида и 111 мг 2,3-диазаспиро[4.4]нон-2-ена. Смесь нагревали до 90 С в течение двух ночей, концентрировали при пониженном давлении и высушивали в вакууме, получая 700 мг продукта (ЖХ-МС Rt 3,91 мин), который использовали в следующей стадии без очистки. 1 Н ЯМР (400 МГц, CDCl3)м.д. 1,10-1,12 (м, 9 Н), 1,63-1,94 (м, 8 Н), 4,82 (с, 2 Н), 7,01 (с, 1 Н), 7,65-7,71 (м, 4 Н), 7,927,96 (м, 2 Н). 4-(трет-бутилдифенилсиланилоксиметил)-N-[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен] бензолсульфонамид К раствору 700 мг 4-(трет-бутилдифенилсиланилоксиметил)-N-[(2,3-диазаспиро[4.4]нон-3-ен-2 ил)метилсульфанилметилен]бензолсульфонамида в 50 мл метанола добавляли 1,84 мл 70% водного раствора этиламина. Смесь перемешивали в течение 1 ч при комнатной температуре и концентрировали при пониженном давлении. Остаток очищали автоматической флэш-хроматографией (DCMDCM/MeOH 97:3), получая 730 мг продукта. 1 Н ЯМР (400 МГц, CDCl3)м.д. 1,10 (с, 9 Н), 1,15 (т, J=7,21 Гц, 3 Н), 1,661,75 (м, 8 Н), 3,48 (дд, J=7,21, 5,38 Гц, 2 Н), 3,85 (с, 2 Н), 4,80-4,81 (м, 2 Н), 6,80 (с, 1 Н), 7,35-7,47 (м, 6 Н),7,65-7,70 (м, 4 Н), 7,65-7,70 (м, 2 Н), 7,88-7,91 (м, 2 Н). 694 мг 4 -(трет-бутилдифенилсиланилоксиметил)-N-[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]бензолсульфонамида смешивали с 40 мл ТГФ и по каплям добавляли 1,04 мл 1 М раствора фторида тетрабутиламмония. Смесь перемешивали при комнатной температуре в течение 4 ч. Полученную смесь разбавляли EtOAc и 3 раза экстрагировали 5% водным раствором NaHCO3. Органическую фазу высушивали над MgSO4 и концентрировали при пониженном давлении. Остаток подвергали автоматической флэш-хроматографии (DCM/MeOH 95:5), полученный неочищенный продукт смешивали сEtOAc и дважды экстрагировали 2 н. водным раствором NaOH. После высушивания и концентрирования остаток перемешивали с 5 мл МТВЕ, полученное твердое вещество отделяли фильтрованием и высушивали, получая 40 мг продукта. 1 Н ЯМР (400 МГц, CDCl3)1,15 (т, J=7,20 Гц, 3 Н), 1,62-1,86 (м, 8 Н), 3,413,52 (м, 2 Н), 3,84 (уш.с, 1 Н), 4,77 (д, J=5,31 Гц, 2 Н), 6,80 (с, 1 Н), 7,45 (д, J=8,34 Гц, 2 Н), 7,93 (д, J=8,34 Гц,2 Н). 4-Амино-N-[этиламино(2,3,8-триазаспиро[4.5]дец-3-ен-2-ил)метилен]бензолсульфонамид (соединение 47). 4-Амино-N-[(8-бензил-2,3,8-триазаспиро[4.5]дец-3-ен-2-ил)метилсульфанилметилен]бензолсульфонамид В 25 мл микроволновом сосуде 1,50 г 4-амино-N-(бис-метилсульфанилметилен)бензолсульфонамида и 1,37 г 8-бензил-2,3,8-триазаспиро[4.5]дец-2-ена суспендировали в 20 мл пиридина. Сосуд закрывали крышкой и нагревали в течение 1 ч при 180 С (6 бар) в микроволновой печи. Реакционную смесь концентрировали на оксиде кремния. Очистка колоночной флэш-хроматографией (DCMDCM/МеОН 99:1DCM/MeOH 98:2) приводила к получению 1,03 г (41%) бежевого аморфного вещества. 1 Н ЯМР К раствору 1,35 г 4-амино-N-[(8-бензил-2,3,8-триазаспиро[4.5]дец-3-ен-2-ил)метилсульфанилметилен]бензолсульфонамида в 30 мл МеОН добавляли 2,26 мл (10 экв.)70% водного раствора зтиламина. Смесь перемешивали в течение выходных дней при комнатной температуре и концентрировали на оксиде кремния. Очистка колоночной флэш-хроматографией (DCMDCM/MeOH 99:1DCM/MeOH 95:5) приводила к получению 1,16 г (87%) бледно-желтого стеклообразного вещества. 1 Н ЯМР (400 МГц,CDCl3)1,14 (т, J=7 Гц, 3 Н), 1,50-1,60 (м, 2 Н), 1,73-1,84 (м, 2 Н), 2,11-2,26 (м, 2 Н), 2,63-2,76 (м, 2 Н), 3,413,53 (м, 2 Н), 3,79 (с, 2 Н), 3,98 (с, 2 Н), 6,62-6,70 (м, 2 Н), 6,76 (с, 1 Н), 6,97 (уш.с, 1 Н), 7,22-7,36 (м, 5 Н),7,67-7,75 (м, 2 Н). трет-Бутиловый эфир (4-[(8-бензил-2,3,8-триазаспиро[4.5]дец-3-ен-2-ил)этиламинометилен]сульфамоилфенил)карбаминовой кислоты К раствору 510 мг 4-амино-N-[(8-бензил-2,3,8-триазаспиро[4.5]дец-3-ен-2-ил)этиламинометилен] бензолсульфонамида в 10 мл 1,4-диоксана добавляли 490 мг (2 экв.) дитрет-бутилдикарбоната. Смесь перемешивали при кипячении с обратным холодильником в течение ночи, охлаждали и концентрировали на оксиде кремния. Очистка колоночной флэш-хроматографией (DCM/MeOH 99:195:5) приводила к получению 550 мг (87%) желтого стеклообразного вещества. 1 Н ЯМР (400 МГц, CDCl3)1,14 (т, J=7 Гц,3 Н), 1,47-1,61 (м, 11 Н), 1,73-1,86 (м, 2 Н), 2,11-2,26 (м, 2 Н), 2,64-2,76 (м, 2 Н), 3,41-3,54 (м, 2 Н), 3,80 (с,2 Н), 6,66 (с, 1 Н), 6,78 (с, 1 Н), 6,94 (уш.с, 1 Н), 7,21-7,37 (м, 5 Н), 7,41-7,48 (м, 2 Н), 7,82-7,89 (м, 2 Н). трет-Бутиловый эфир (4-[этиламино (2,3,8-триазаспиро[4.5]дец-3-ен-2-ил)метилен]сульфамоил фенил)карбаминовой кислоты нометилен]сульфамоилфенил)карбаминовой кислоты в 10 мл 1,2-дихлорэтана охлаждали на ледяной бане и по каплям добавляли 0,12 мл (1,1 экв. ) 1-хлорэтилхлорформиата и 0,03 мл DIPEA. Через 15 мин ледяную баню убирали и смесь перемешивали в течение 30 мин при комнатной температуре. Смесь концентрировали в вакууме и 3 раза упаривали вместе с толуолом. Остаток смешивали с 10 мл МеОН и перемешивали в течение ночи при комнатной температуре. Полученную смесь концентрировали. Остаток смешивали с ЕА и экстрагировали 2 М NaOH. Органический слой высушивали над Na2SO4, фильтровали и концентрировали на оксиде кремния. Очистка колоночной флэш-хроматографией (EtOAc/MeOH/Et3N 50:45:5) позволяла получить 360 мг (72%) оранжевого стеклообразного вещества. 1 Н ЯМР (400 МГц,ДМСО-d6)0,96 (т, J=7 Гц, 3 Н), 1,33-1,43 (м, 2 Н), 1,48 (с, 9 Н), 1,53-1,64 (м, 2 Н), 2,44-2,56 (м, 2 Н), 2,762,88 (м, 2 Н), 3,21-3,33 (м, 2 Н), 3,68 (с, 2 Н), 7,29 (с, 1 Н), 7,50-7,59 (м, 2 Н), 7,60-7,74 (м, 3 Н), 9,70 (с, 1 Н). 360 мг трет-бутилового эфира (4-[этиламино(2,3,8-триазаспиро[4.5]дец-3-ен-2-ил)метилен]сульфамоилфенил) карбаминовой кислоты суспендировали в 10 мл этанола; добавляли 0,44 мл (5 экв.) вератрола и затем 3,49 мл 1 М HCl в этаноле (5 экв.). Полученную смесь перемешивали при 60 С в течение ночи. После охлаждения смесь очищали с помощью SPE (Isolute Flash SCX-2, подготовка, отбор образцов и промывание МеОН, элюирование 1 М NH3 в МеОН), получая 150 мг (53%) желтого стеклообразного вещества. 1 Н ЯМР(400 МГц, ДМСО-d6)0,97 (т, J=7 Гц, 3 Н), 1,33-1,45 (м, 2 Н), 1,52-1,66 (м, 2 Н), 2,46-2,60 (м, 2 Н), 2,76-2,90 (м,2 Н), 3,20-3,40 (м, 2 Н), 3,66 (с, 2 Н), 5,71 (с, 2 Н), 6,50-6,61 (м, 2 Н), 7,26 (с,1 Н), 7,37-7,52 (м, 3 Н). Соединения, полученные по этому способу синтеза, отмечены "способ синтеза 1" в приведенной ниже таблице. 4-Амино-N-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]бензолсульфонамид (соединение 3) гидройодид 1-этил-2-метилизотиомочевины 20,5 г этилтиомочевины растворяли в 100 мл EtOH. Смесь охлаждали на ледяной бане и по каплям добавляли 13,5 мл (1,1 экв.) MeI. Смесь перемешивали в течение 1 ч при комнатной температуре и концентрировали в вакууме, получая 48,3 г светло-желтого масла. 1 Н ЯМР (400 МГц, ДМСО-d6)1,17 (т,J=7, 5 Гц, 3 Н), 2,61 (с, 3 Н), 3,34 (кв, J=7,5 Гц, 2 Н), 9,10 (уш.с, 2 Н). Гидрохлорид N-этил-4,4-диметил-4,5-дигидропиразол-1-карбоксамидина 12,0 г 4,4-диметил-4,5-дигидро-3 Н-пиразола растворяли в 100 мл пиридина. Добавляли раствор 30,0 г гидройодида 1-этил-2-метилизотиомочевины в 50 мл пиридина и кипятили смесь с обратным холодильником в течение 20 ч. Полученную смесь охлаждали до комнатной температуры, концентрировали при пониженном давлении и остаток смешивали с DCM (120 мл). Органическую фазу экстрагировали 2 н. NaOH (2120 мл),промывали водой (120 мл), высушивали над Na2SO4 и упаривали при пониженном давлении, получая 16,3 г(79%) оранжевого масла. Это масло (10,0 г) смешивали с EtOAc (50 мл) и нагревали до 60 С. Убирали источник тепла и в течение 4 мин прибавляли 5-6 н. раствор HCl в изопропаноле (20 мл). После охлаждения до комнатной температуры в течение 4 мин добавляли EtOAc (50 мл), и смесь перемешивали при 20 С в течение 90 мин. Полученные кристаллы собирали фильтрованием и промывали EtOAc (20 мл), после чего высушивали при пониженном давлении и умеренном нагревании, получая 6,52 г (54%) желаемого продукта в виде желтого твердого вещества. 1 Н ЯМР (400 МГц, ДМСО-d6)1,13 (т, J=7 Гц, 3 Н), 1,24 (с, 6 Н), 3,27-3,34 (м, 2 Н), 3,64 (с,2 Н), 7,26 (с, 1 Н), 8,03 (уш.с, 2 Н), 8,13 (уш.с, 1 Н). 500 мг гидрохлорида N-этил-4,4-диметил-4,5-дигидропиразол-1-карбоксамидина суспендировали в 10 мл DCM, добавляли 0,88 мл DIPEA и затем 571 мг 4-ацетиламинобензолсульфонилхлорида. Смесь перемешивали в течение ночи при комнатной температуре. Для улучшения степени превращения добавляли дополнительные 0,44 мл основания и 290 мг сульфонилхлорида и оставляли реакционную смесь на ночь. Затем полученную смесь экстрагировали 5% водным раствором NaHCO3 и 2 М раствором NaOH,органический слой высушивали над Na2SO4, упаривали досуха и неочищенный продукт (900 мг пурпурного масла, содержащего 95% ожидаемого продукта по данным ЖХ-МС) использовали в следующей стадии. ЖХ-МС: Rt 1,34 мин (МН+ 366). 900 мг N-(4-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]сульфамоилфенил)ацетамида растворяли в 5 мл МеОН и добавляли 5 мл концентрированной HCl. Смесь перемешивали в течение ночи при комнатной температуре. Смесь подщелачивали 2 М NaOH и дважды экстрагировали DCM. Объединенные органические слои высушивали над Na2SO4 и упаривали досуха. Остаток очищали флэшхроматографией (DCM/MeOH 99:1), получая 400 мг (50%) 4-амино-N-[(4,4-диметил-4,5-дигидропиразол 1-ил)этиламинометилен]бензолсульфонамида в виде аморфного твердого вещества. 1 Н ЯМР (400 МГц,CDCl3)1,15 (т, J=7 Гц, 3 Н), 1,20 (с, 6 Н), 3,42-3,51 (м, 2 Н), 3,74 (уш.с, 2 Н), 4,00 (уш.с, 2 Н), 6,62-6,68 (м,2 Н), 6,71 (с, 1 Н), 6,90 (уш.с, 1 Н), 7,67-7,73 (м, 2 Н). 4-Амино-3-хлор-N-[(4,4-диметил-4,5-дигидропиразол-1-ил)зтиламинометилен]бензолсульфонамид 500 мг гидрохлорида N-этил-4,4-диметил-4,5-дигидропиразол-1-карбоксамидина суспендировали в 10 мл DCM, добавляли 0,88 мл DIPEA и затем 655 мг 4-ацетиламино-3-хлорбензолсульфонилхлорида. Смесь перемешивали в течение ночи при комнатной температуре. Для улучшения степени превращения добавляли дополнительные 0,44 мл основания и 290 мг сульфонилхлорида и оставляли реакционную смесь на ночь. Полученную смесь последовательно экстрагировали 5% водным раствором NaHCO3 и 2 М раствором NaOH, органический слой высушивали над Na2SO4, упаривали досуха и неочищенный продукт (680 мг, содержащий 85% ожидаемого продукта по данным ЖХ-МС) использовали в следующей стадии. ЖХ-МС: Rt 1,46 мин (МН+ 400). 680 мг N-(2-хлор-4-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]сульфамоилфенил)ацетамида растворяли в 5 мл МеОН и добавляли 5 мл концентрированной HCl. Смесь перемешивали в течение ночи при комнатной температуре. Смесь подщелачивали 2 М NaOH и дважды экстрагировалиDCM. Объединенные органические слои высушивали над Na2SO4 и упаривали досуха. Остаток очищали флэш-хроматографией (DCM/MeOH 99:1), получая 240 мг (40%) 4-амино-3-хлор-N-[(4,4-диметил-4,5 дигидропиразол-1-ил)этиламинометилен]бензолсульфонамида в виде оранжевого масла. 1 Н ЯМР (400 МГц, CDCl3)1,17 (т, J=7 Гц, 3 Н), 1,21 (с, 6 Н), 3,43-3,52 (м, 2 Н), 3,75 (уш.с, 2 Н), 4,37 (уш.с, 2 Н), 6,73 (с,1 Н), 6,76 (д, J=8 Гц, 1 Н), 6,86 (уш.с, 1 Н), 7,62 (дд, J=2 и 8 Гц, 1 Н), 7,83 (д, J=2 Гц, 1 Н). Этиламино-(4-этил-4,5-дигидропиразол-1-ил)метиленамид 2,3-дигидро-1 Н-индол-5-сульфоновой кислоты (соединение 16). 19,36 г 4-этил-4,5-дигидро-1 Н-пиразола растворяли в 100 мл толуола. Добавляли 48,5 г гидрохлорида 1-этил-2-метилизотиомочевины и 33,8 мл DiPEA, и смесь кипятили с обратным холодильником в течение 48 ч. Смесь концентрировали, добавляли 2 М NaOH и затем экстрагировали DCM (три раза). Объединенные органические слои высушивали над Na2SO4 и выпаривали растворитель в вакууме, получая 32,7 г (99%) красного масла, содержащего 75% желаемого продукта, согласно данным ЯМР. Полученное масло растворяли в EtOH и по каплям добавляли 194 мл 1 М раствора HCl в EtOH. Смесь перемешивали при комнатной температуре в течение 30 мин и концентрировали в вакууме. Кристаллизация из смесиCH3CN:MTBE=1:1 позволяла получить 11,52 г (29%) желаемого продукта в виде бежевого твердого вещества. 1 Н ЯМР (400 МГц, ДМСО-d6)0,96 (т, J=7,5 Гц, 3 Н), 1,16 (т, J=7 Гц, 3 Н), 1,46-1,72 (м, 2 Н), 3,32DCM, добавляли 13,10 мл DiPEA и затем 5,00 г 1-ацетил-2,3-дигидро-1 Н-индол-5-сульфонилхлорида. Смесь перемешивали в течение ночи при комнатной температуре. Полученную смесь последовательно экстрагировали 5% водным раствором NaHCO3 и 2 М раствором NaOH, органический слой высушивали над Na2SO4 и упаривали досуха. Остаток очищали флэш-хроматографией (DCM/EA 3:1 ЕА), получая 1,85 г (25%) желтого масла. 1 Н ЯМР (400 МГц, CDCl3)0,97 (т, J=7,52 Гц, 3 Н), 1,15 (т, J=7,37 Гц, 3 Н),1,45-1,69 (м, 2 Н), 2,25 (с, 3 Н), 3,01-3,16 (м, 1 Н), 3,24 (т, J=8,58 Гц, 2 Н), 3,42-3,52 (м, 2 Н), 3,64-3,75 (м,1 Н), 4,02-4,21 (м, 3 Н), 6,90 (д, J=1,20 Гц, 1 Н), 7,72-7,82 (м, 2 Н), 8,24 (д, J=8,43 Гц, 1 Н) [сигнал гуанидиновой группы NH невидим в спектре]. 1,74 г этиламино-(4-этил-4,5-дигидропиразол-1-ил)метиленамида 1-ацетил-2,3-дигидро-1 Н-индол-5 сульфоновой кислоты растворяли в 100 мл EtOH и добавляли 22,2 мл 1 М HCl. Смесь перемешивали в течение 5 ч при кипячении с обратным холодильником. После охлаждения до комнатной температуры смесь подщелачивали 5% раствором NaHCO3 и дважды экстрагировали DCM. Объединенные органические слои высушивали над Na2SO4 и упаривали досуха. Остаток очищали флэш-хроматографией (DCMDCM/EA 4:1), получая 0,66 г (43%) этиламино-(4-этил-4,5-дигидропиразол-1-ил)метиленамида 2,3 дигидро-1 Н-индол-5-сульфоновой кислоты в виде желтого масла. 1 Н ЯМР (400 МГц, CDCl3)0,97 (т,J=7,52 Гц, 3 Н), 1,15 (т, J=7,22 Гц, 3 Н), 1,44-1,68 (м, 2 Н), 2,97-3,13 (м, 3 Н), 3,42-3,54 (м, 2 Н), 3,58-3,72 (м,3 Н), 3,99-4,09 (м, 1 Н), 6,55 (д, J=8,13 Гц, 1 Н), 6,88 (д, J=1,50 Гц, 1 Н), 7,00 (уш.с, 1 Н), 7,55-7,65 (м, 2 Н) 0,42 г этиламино-(4-этил-4,5-дигидропиразол-1-ил)метиленамида 2,3-дигидро-1 Н-индол-5-сульфоновой кислоты растворяли в 25 мл толуола и добавляли 10 мол.% палладия на угле. Смесь перемешивали при 50 С в течение 5 дней, с добавлением через 2 дня порции свежего катализатора (10 мол.%). Смесь охлаждали до комнатной температуры и фильтровали с применением Hyflo. Фильтрат упаривали досуха и остаток очищали флэш-хроматографией (DCMDCM/EA 9:1), получая 0,26 г (66%) зтиламино(4-этил-4,5-дигидропиразол-1-ил)метиленамида 1 Н-индол-5-сульфоновой кислоты в виде синего масла. 1 Н ЯМР (400 МГц, CDCl3)0,87 (т, J=7,37 Гц, 3 Н), 1,08 (т, J=7,22 Гц, 3 Н), 1,32-1,59 (м, 2 Н), 2,89-3,02 (м,1 Н), 3,35-3,50 (м, 2 Н), 3,58 (дд, J=11,44, 7,52 Гц, 1 Н), 3,96 (т, J=11,29 Гц, 1 Н), 6,54-6,58 (м, 1 Н), 6,85 (д,J=1,50 Гц, 1 Н), 6,97 (уш.с, 1 Н) 7,23-7,29 (м, 1 Н), 7,42 (д, J=8,73 Гц, 1 Н), 7,69 (дд, J=8,73, 1,81 Гц, 1 Н), 8,24 В атмосфере N2 0,50 г гидрохлорида 4,N-диэтил-4,5-дигидропиразол-1-карбоксамида суспендировали в 50 мл DCM, добавляли 0,43 мл DiPEA и затем 0,61 г 4-метоксибензолсульфонилхлорида. Эту смесь перемешивали в течение выходных дней при комнатной температуре. Смесь последовательно экстрагировали 5% водным раствором NaHCO3 и 2 М раствором NaOH, органический слой высушивали над 0,28 г N-[этиламино-(4-зтил-4,5-дигидропиразол-1-ил)метилен]-4-метоксибензолсульфонамида растворяли в 20 мл DCM и добавляли 3,32 мл 1 М раствора BBr3 в DCM. Смесь перемешивали в течение ночи при комнатной температуре, экстрагировали 5% водным раствором NaHCO3, высушивали над MgSO4 и упаривали досуха. Остаток очищали флэш-хроматографией (ступенчатый градиент DCM-DCM/MeOH 95:5), получая 0,186 г (59%) N-[этиламино-(4-этил-4,5-дигидропиразол-1-ил)метилен]-4-гидроксибензолсульфонамида. 1 Н ЯМР (400 МГц, CDCl3)0,95 (т, J=7,52 Гц, 3 Н), 1,13 (т, J=7,22 Гц, 3 Н), 1,50 (дкв,J=14,20, 7,00 Гц, 1 Н), 1,60 (дкв, J=14,22, 7,00 Гц, 1 Н), 3,01-3,16 (м, 1 Н), 3,43-3,50 (м, 2 Н), 3,66 (дд,J=11,44, 7,52 Гц, 1 Н), 4,05 (т, J=11,29 Гц, 1 Н), 6,80 (уш.с, 1 Н), 6,87 (д, J=8,73 Гц, 2 Н), 6,91 (д, J=1,50 Гц,1 Н), 7,78 (д, J=8,73 Гц, 2 Н) [гуанидиновая группа NH в спектре невидима]. 3-Хлор-N-[этиламино-(4-этил-4,5-дигидропиразол-1-ил)метилен]-4-гидроксибензолсульфонамид В атмосфере N2 охлаждали на ледяной бане 41,25 мл хлорсульфоновой кислоты и по каплям при перемешивании добавляли 22,26 мл 2-хлоранизола. Смесь нагревали до 55 С; через 10 мин убирали источник тепла и смесь перемешивали в течение ночи при комнатной температуре. Полученную смесь выливали в ледяную воду и дважды экстрагировали DCM. Объединенные органические слои высушивали над MgSO4 и упаривали досуха. Остаток очищали флэш-хроматографией (РА/ЕА 9:1), получая 24,94 гDCM, добавляли 10,76 мл DiPEA и затем 3,79 г 3-хлор-4-метоксибензолсульфонилхлорида. Полученную смесь перемешивали в течение выходных дней при комнатной температуре и затем упаривали досуха. Остаток очищали флэш-хроматографией (ступенчатый градиент DCMDCM/EA 9:1 и затем DCM/ 1,09 г 3-хлор-N-[этиламино-(4-этил-4,5-дигидропиразол-1-ил)метилен]-4-метоксибензолсульфонамида растворяли в 25 мл DCM и добавляли 11,69 мл 1 М раствора BBr3 в DCM. Смесь перемешивали в течение ночи при комнатной температуре, экстрагировали 5% водным раствором NaHCO3, высушивали над MgSO4 и упаривали досуха. Остаток очищали флэш-хроматографией (DCM/EA 95:5), получая 0,89 г 1,50 г гидрохлорида N-этил-4,4-диметил-4,5-дигидропиразол-1-карбоксамидина суспендировали в 50 мл DCM, добавляли 5,02 мл DiPEA и затем 1,95 г. 3-нитробензолсульфонилхлорида. Смесь перемешивали в течение ночи при комнатной температуре и экстрагировали 5% водным раствором NaHCO3. Водный слой подкисляли 1 М HCl и экстрагировали DCM. Органическую фазу высушивали над MgSO4 и упаривали досуха, получая 2,18 г (84%) коричневого масла. 1 Н ЯМР (400 МГц, CDCl3)1,19 (т, J=7,22 Гц, 3 Н), 1,25 (с, 6 Н), 3,44-3,53 (м, 2 Н), 3,83 (уш.с, 2 Н), 6,80 (с, 1 Н), 7,66 (т, J=7,98 Гц, 1 Н), 8,28 (д, J=7,82 Гц, 1 Н), 8,34 (дд, J=8,13, 1,20 Гц, 1 Н). В смеси 50 мл EtOH и 25 мл воды растворяли 1,11 г N-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]-3-нитробензолсульфонамида. Затем добавляли 1,05 г металлического железа и 1,08 мл уксусной кислоты, и смесь кипятили с обратным холодильником в течение 4 ч. После охлаждения до комнатной температуры полученную смесь фильтровали с применением Hyflo и промывали HyfloMeOH. Из фильтрата выпаривали спирты и добавляли 5% водный раствор NaHCO3 и DCM. Вещество,нерастворимое в обеих фазах, удаляли фильтрованием, отделяли органическую фазу и водную фазу еще раз экстрагировали DCM. Объединенные органические слои высушивали над MgSO4 и упаривали досу- 27023176 В атмосфере N2 охлаждали на ледяной бане 25,00 мл хлорсульфоновой кислоты и при перемешивании порциями добавляли 5,00 г 1-ацетил-5-броминдола. Продолжали перемешивание в течение 20 мин,после чего убирали ледяную баню и смесь нагревали до 70 С. После охлаждения до комнатной температуры полученную смесь осторожно выливали в ледяную воду и дважды экстрагировали DCM. Объединенные органические слои высушивали над MgSO4 и упаривали досуха, получая 6,57 г (93%) бежевого твердого вещества. 1 Н ЯМР (400 МГц, ДМСО-d6)2,16 (с, 3 Н), 3,15 (т, J=8,58 Гц, 2 Н), 4,11 (т, J=8,58 Гц,2 Н), 7,37 (с, 1 Н), 8, 66 (с, 1 Н). Этиламино-(4-этил-4,5-дигидропиразол-1-ил)метиленамид 1-ацетил-5-бром-2,3-дигидро-1 Н-индол 6-сульфоновой кислотыDCM, добавляли 4,58 мл DiPEA и затем 2,34 г 1-ацетил-5-бром-2,3-дигидро-1 Н-индол-6-сульфонилхлорида. Смесь перемешивали в течение ночи при комнатной температуре и затем упаривали досуха. Остаток очищали флэш-хроматографией (градиент DCM/EA 95 : 575 : 25), получая 0,65 г (16%) коричневого масла. 1 Н ЯМР (400 МГц, CDCl3)0,98 (т, J=7,37 Гц, 3 Н), 1,13-1,21 (м, 3 Н), 1,43-1,80 (м, 2 Н), 2,22 (с,3 Н), 3,11 (уш.с, 1 Н), 3,17-3,27 (м, 2 Н), 3,48-3,58 (м, 2 Н), 3,73-3,84 (м, 1 Н), 4,04-4,27 (м, 3 Н), 6,91 (с, 1 Н),7,47 (с, 1 Н), 8,99 (с, 1 Н) [сигнал гуанидиновой группы NH невидим в спектре]. 0,65 г этиламино-(4-этил-4,5-дигидропиразол-1-ил)метиленамида 1-ацетил-5-бром-2,3-дигидро-1 Ниндол-6-сульфоновой кислоты растворяли в 20 мл МеОН и добавляли 20,7 мл 1 М раствора HCl в МеОН. Смесь перемешивали в течение ночи при кипячении с обратным холодильником. После охлаждения до комнатной температуры смесь подщелачивали 5% раствором NaHCO3 и дважды экстрагировали DCM. Объединенные органические слои высушивали над Na2SO4 и упаривали досуха. Остаток очищали флэшхроматографией (DCM/EA 9:18:2), получая 0,35 г (64%) этиламино-(4-этил-4,5-дигидропиразол-1-ил) метиленамида 5-бром-2,3-дигидро-1 Н-индол-6-сульфоновой кислоты в виде желтого масла. 1H ЯМР (400 МГц, CDCl3)0,96 (т, J=7,37 Гц, 3 Н), 1,17 (т, J=7,22 Гц, 3 Н), 1,45-1,68 (м, 2 Н), 3,00-3,15 (м, 3 Н), 3,48-3,57 К раствору 0,30 г этиламино-(4-этил-4,5-дигидропиразол-1-ил)метиленамида 5-бром-2,3-дигидро 1 Н-индол-6-сульфоновой кислоты в 50 мл EtOH добавляли 0,94 мл триэтиламина. Смесь тщательно дегазировали и добавляли 10 мол.% палладия на угле. Смесь гидрировали в течение ночи при давлении Н 2 1 атм. Смесь фильтровали с применением Hyflo, Hyflo промывали EtOH и фильтрат концентрировали в вакууме. Остаток очищали флэш-хроматографией (DCMDCM/ЕА 95:5DCM/EA 9:1), получая 0,20 г 0,14 г этиламино-(4-этил-4,5-дигидропиразол-1-ил)метиленамида 2,3-дигидро-1 Н-индол-6-сульфоновой кислоты растворяли в 25 мл толуола, смесь дегазировали и добавляли 10 мол.% палладия на угле. Смесь перемешивали в течение ночи при 50 С. Затем смесь охлаждали до комнатной температуры,фильтровали с применением Hyflo и промывали Hyflo толуолом. Фильтрат упаривали досуха и остаток очищали флэш-хроматографией (DCMUDCM/EA 95:5DCM/EA 8:2), получая 70 мг этиламино-(4-этил 4,5-дигидропиразол-1-ил)метиленамида 1 Н-индол-6-сульфоновой кислоты в виде белого аморфного твердого вещества. 1 Н ЯМР (400 МГц, CDCl3)0,86 (т, J=7,52 Гц, 3 Н), 1,06 (т, J=7,22 Гц, 3 Н), 1,32-1,57 2,5 г гидрохлорида N-этил-4,4-диметил-4,5-дигидропиразол-1-карбоксамидина суспендировали в 20 мл DCM, добавляли 4,39 мл DiPEA и затем 2,52 г 3-метоксибензолсульфонилхлорида. Смесь перемешивали в течение выходных дней при комнатной температуре. Полученную смесь последовательно экстрагировали 5% водным раствором NaHCO3 и 2 М раствором NaOH, органический слой высушивали над Растворяли 2,32 г N-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]-3-метоксибензолсульфонамида в 20 мл DCM и добавляли 13,7 мл 1 М раствора ВВг 3 в DCM. Смесь перемешивали в течение выходных дней при комнатной температуре. Для гашения смеси, которая содержала липкий осадок,добавляли 5% водный раствор NaHCO3; после гашения осадок растворялся при осторожном нагревании смеси. Отделяли органический слой, и водный слой еще раз экстрагировали DCM. Объединенные органические слои высушивали над Na2SO4 и упаривали досуха. Остаток очищали флэш-хроматографией
МПК / Метки
МПК: C07D 403/12, C07D 231/06, A61P 25/00, A61K 31/416, A61K 31/4155, A61K 31/4162, C07D 491/10, C07D 231/54
Метки: арилсульфонилпиразолинкарбоксамидиновые, производные, 5-нт6, качестве, антагонистов
Код ссылки
<a href="https://eas.patents.su/30-23176-arilsulfonilpirazolinkarboksamidinovye-proizvodnye-v-kachestve-antagonistov-5-nt6.html" rel="bookmark" title="База патентов Евразийского Союза">Арилсульфонилпиразолинкарбоксамидиновые производные в качестве антагонистов 5-нт6</a>
Производные пиридилкарбамоилиндолинов в качестве антагонистов 5-нт2с-рецепторов

Номер патента: 1780
Опубликовано: 27.08.2001
Авторы: Форбес Ян Томсон, Бромидж Стивен Марк
МПК: A61P 25/28, C07D 401/14, A61K 31/4439...
Метки: антагонистов, производные, качестве, 5-нт2с-рецепторов, пиридилкарбамоилиндолинов
Формула / Реферат:
1. Соединение формулы (I) или его соль где Х обозначает СН или N; R1 обозначает водород или C1-6-алкил; R2 и R3 обозначают независимо C1-6-алкил или трифторметил. 2. Соединение по п.1, в котором Х обозначает СН. 3. Соединение по п.1 или 2, в котором R1 обозначает метил. 4. Соединение по любому из пп.1-3, в котором R2 обозначает СF3. 5. Соединение по любому из пп.1-4, в котором R3 обозначает C1-6-алкил. 6. Соединение по любому из пп.1-5, в...
Производные циклопентена в качестве антагонистов рецептора мотилина

Номер патента: 3252
Опубликовано: 27.02.2003
Авторы: Биверз Мэри Пэт, Чен Роберт Х., Ксианг Мин, Мур Джон Б.Мл.
МПК: C07C 233/41, A61K 31/5375, A61P 1/00...
Метки: качестве, мотилина, антагонистов, циклопентена, рецептора, производные
Формула / Реферат:
1. Соединение формулы I в которой R1 представляет H, C1-5-алкил, замещенный C1-5-алкил (где заместителями являются один или несколько галогенов), амино-C1-5-алкил, C1-5-алкиламино-C1-5-алкил, ди-C1-5-алкиламино-C1-5-алкил, RaRbN-C1-5-алкил (где Ra и Rb независимо выбраны из H и C1-5-алкила или вместе образуют морфолин, пиперазин, пиперидин или N-замещенный пиперидин, где N-заместитель представляет C1-5-алкил или фенил-C1-5-алкил),...
Азабициклические производные в качестве антагонистов мускаринового рецептора

Номер патента: 9942
Опубликовано: 28.04.2008
Авторы: Кумар Нареш, Кхугх Анита, Каур Кирандип, Дхармараджан Санкаранарайанан, Салман Мохаммад, Сарма Пакала Кумара Савитхру, Мехта Анита
МПК: A61P 11/00, A61P 1/00, A61K 31/403...
Метки: производные, мускаринового, качестве, антагонистов, азабициклические, рецептора
Формула / Реферат:
1. Азабициклические производные в качестве антагонистов мускаринового рецептора, выбранные из следующих соединений: (2R,2S)(1a,5a,6a)-N-{[4-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-бутил]-3-азабицикло[3.1.0]гекс-6-ил-метил}-2-гидрокси-2-циклопентил-2-фенилацетамид (соединение 1), (2R)(1a,5a,6a)-N-(3-бензил-3-азабицикло[3.1.0]гекс-6-ил-метил)-2-гидрокси-2-циклопент-1-енил-2-фенилацетамид (соединение 2),...
Азабициклические производные в качестве антагонистов мускаринового рецептора

Номер патента: 9387
Опубликовано: 28.12.2007
Авторы: Салман Мохаммад, Дхармараджан Санкаранарайанан, Кумар Нареш, Кхугх Анита, Сарма Пакала Кумара Савитхру
МПК: A61P 11/00, A61K 31/403, A61P 1/00...
Метки: мускаринового, рецептора, антагонистов, производные, азабициклические, качестве
Формула / Реферат:
1. 6-Замещенные азабицикло[3,1,0]гексаны, выбранные из следующих соединений: N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-фенил-2-гидрокси-2-(N-метил)фенилацетамид (соединение 1); N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-фенил-2-гидрокси-2-(N-метил)фенилацетамида тартрат (соединение 2); (2R,2S)-N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2-гидрокси-2-фенилацетамид (соединение 3);...
Замещенные производные азабициклогексана в качестве антагонистов мускаринового рецептора

Номер патента: 9059
Опубликовано: 26.10.2007
Авторы: Гупта Джанг Бахадур, Силамкоти Арундутт Висванатхам, Мехта Анита
МПК: A61P 11/00, A61K 31/403, A61P 1/00...
Метки: азабициклогексана, замещенные, рецептора, мускаринового, производные, качестве, антагонистов
Формула / Реферат:
1. Соединения структурной формулы I и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереомеры, N-оксиды, полиморфные формы, где Ar представляет собой арильное или гетероарильное кольцо, имеющее 1-2 гетероатома, выбранных из группы, состоящей из атомов кислорода, серы и азота, арильное или гетероарильное кольца могут быть незамещенными или замещенными от одного до трех заместителями, независимо...
Предыдущий патент: Таргетирование раковых клеток с использованием наночастиц
Следующий патент: Микроэлектрод и пучок микроэлектродов, содержащие устройства для высвобождения лекарственных средств в ткани
Случайный патент: Способ изготовления тепловой ударной трубки и тепловая ударная трубка, изготовленная этим способом