Гетероциклические соединения, агонисты рецептора ip
Номер патента: 22046
Опубликовано: 30.10.2015
Авторы: Маккеоун Стивен Карл, Лебланк Катрин, Чарлтон Стивен Джон





























Формула / Реферат
1. Соединение формулы Ia

или его фармацевтически приемлемая соль, в которой
А обозначает N или CR1;
R' обозначает Н;
R1 обозначает Н или C1-C8-алкил; или
R1 обозначает -X-Y; или
R1 обозначает -W-R7-X-Y; или
R2 обозначает Н или C1-C8-алкил; или
R2 обозначает -X-Y; или
R2 обозначает -W-R7-X-Y;
где R1 или R2 обозначает -X-Y или -W-R7-X-Y;
R2a обозначает водород;
R2 и R2a вместе обозначают оксогруппу;
R3 обозначает Н, С1-С4-алкоксигруппу, ОН, C1-C8-алкил или C1-C8-галогеналкил;
R4 обозначает Н, С1-С4-алкоксигруппу, ОН или С1-С8-алкил;
R5 обозначает С6-С14-арил; -(С0-С4-алкил)-4-14-членный гетероарил, где гетероарил содержит по меньшей мере один гетероатом, выбранный из группы, включающей N, О и S, где арил и гетероарил каждый необязательно содержит один-пять заместителей Z;
R6 обозначает С6-С14-арил; -(С0-С4-алкил)-4-14-членный гетероарил, где гетероарил содержит по меньшей мере один гетероатом, выбранный из группы, включающей N, О и S, где арил и гетероарил каждый необязательно содержит один-пять заместителей Z;
W обозначает C1-C8-алкилен, необязательно замещенный гидроксигруппой, галогенами или С1-С4-алкилом;
X обозначает C1-C8-алкилен, необязательно замещенный гидроксигруппой, галогенами или С1-С4-алкилом;
Y обозначает карбоксигруппу и C1-C8-алкоксикарбонил;
R7 обозначает двухвалентный фрагмент, представляющий собой -О-, -NHC(O)-, -СН2=СН2-, -С6-С14-арил-D-; -3-14-членный гетероциклил-D-, где гетероциклил содержит по меньшей мере один гетероатом, выбранный из группы, включающей N, О и S, где D обозначает О, S, NH или не присутствует;
Z независимо обозначает C1-С6-алкил, C1-С6-галогеналкил, C1-С6-алкоксигруппу или галоген;
R19 и R21, каждый независимо, обозначают Н или C1-C8-алкил.
2. Соединение по п.1, в котором R1 обозначает X-Y или -W-R7-X-Y;
R2 обозначает Н или C1-C8-алкил;
X обозначает C1-С6-алкилен, необязательно замещенный гидроксигруппой, галогеном или С1-С4-алкилом;
Y обозначает -С(О)ОН, -C(O)ORX, где Rx обозначает -С1-С4-алкил;
W обозначает C1-С6-алкилен, необязательно замещенный гидроксигруппой, галогеном или С1-С4-алкилом;
R3 обозначает Н, С1-С4-алкоксигруппу, ОН, С1-С4-алкил или С1-С4-галогеналкил;
R4 обозначает Н, С1-С4-алкоксигруппу, ОН или C1-C8-алкил;
R7 обозначает двухвалентный фрагмент, представляющий собой -С6-С14-арил-D-; -3-14-членный гетероциклил-D-, где гетероциклил содержит по меньшей мере один гетероатом, выбранный из группы, включающей N, О и S, где D обозначает О; и
R19 и R21, каждый независимо, обозначают Н или C1-C8-алкил.
3. Соединение по п.1 или 2, в котором R1 обозначает -(CH2)m-C(O)OR";
R2 обозначает Н или С1-С4-алкил;
R3 обозначает Н, С1-С4-алкоксигруппу, ОН, С1-С4-алкил или С1-С4-галогеналкил;
R4 обозначает Н, С1-С4-алкоксигруппу, ОН или С1-С4-алкил;
R" обозначает Н или С1-С4-алкил;
m равно 1, 2, 3, 4, 5 или 6.
4. Соединение по любому из пп.1-3, в котором
R5 обозначает фенил, необязательно замещенный С1-С4-алкилом, C1-C4-галогеналкилом, С1-С4-алкоксигруппой или галогеном; и
R6 обозначает фенил, необязательно замещенный С1-С4-алкилом, С1-С4-галогеналкилом, С1-С4-алкоксигруппой или галогеном.
5. Соединение по любому из пп.1-4, в котором А обозначает N.
6. Соединение по п.1, выбранное из группы, состоящей из
7-(2-фенил-3-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6Н)-ил)гептановой кислоты;
7-(8-гидрокси-2,3-ди-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6Н)-ил)гептановой кислоты;
7-(7-гидрокси-6-оксо-2,3-ди-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6Н)-ил)гептановой кислоты;
7-(2,3-ди-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6Н)-ил)гептановой кислоты;
2-(3-((2,3-дифенил-7,8-дигидропиридо[3,2-b]пиразин-5(6Н)-ил)метил)фенокси)уксусной кислоты;
7-(3-фенил-2-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6Н)-ил)гептановой кислоты
или их фармацевтически приемлемых солей.
7. Соединение по п.1, которое представляет собой 7-(2,3-ди-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6Н)-ил)гептановую кислоту формулы

или ее фармацевтически приемлемую соль.
8. Соединение по п.1, которое представляет собой 7-(2-фенил-3-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6Н)-ил)гептановую кислоту формулы

или ее фармацевтически приемлемую соль.
9. Соединение по п.1, которое представляет собой 2-(3-((2,3-дифенил-7,8-дигидропиридо[2,3-b]пиразин-5(6Н)-ил)метил)фенокси)уксусную кислоту формулы

или ее фармацевтически приемлемую соль.
10. Соединение по любому из пп.6-9, в котором соединение находится в свободной форме.
11. Фармацевтическая композиция для лечения артериальной легочной гипертензии, нарушений, для которых необходима антитромбоцитарная терапия, атеросклероза, астмы, ХОЗЛ, гипергликемии, воспалительного заболевания или фиброзного заболевания, включающая соединение по любому из пп.1-10 или его фармацевтически приемлемую соль в терапевтически эффективном количестве и один или большее количество фармацевтически приемлемых носителей.
12. Применение соединения по любому из пп.1-10 или его фармацевтически приемлемой соли для лечения артериальной легочной гипертензии, нарушений, для которых необходима антитромбоцитарная терапия, атеросклероза, астмы, ХОЗЛ, гипергликемии, воспалительного заболевания или фиброзного заболевания.
13. Применение по п.12 для лечения артериальной легочной гипертензии, астмы, хронической обструкции нижних дыхательных путей или муковисцидоза.
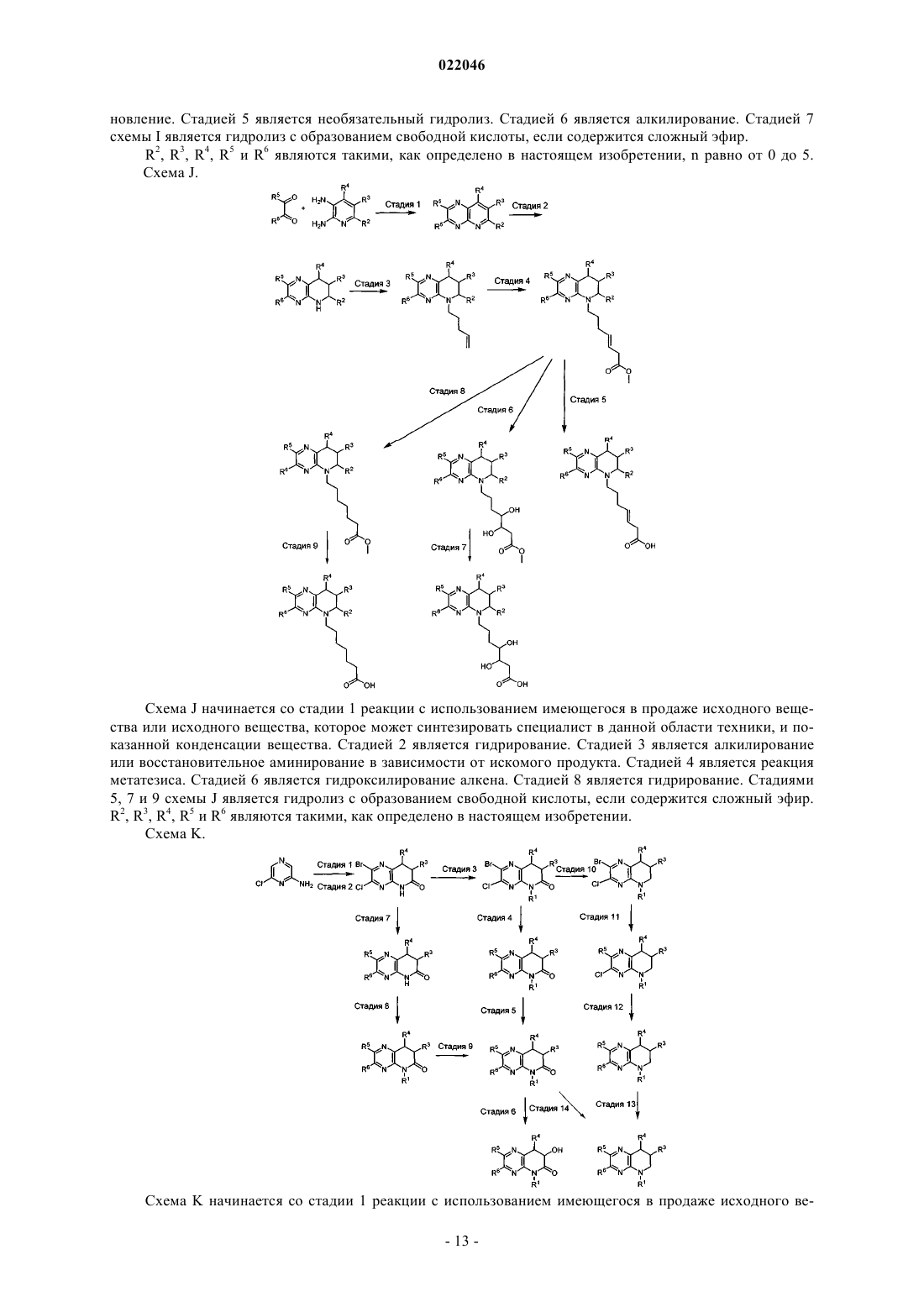
Текст
В изобретении описаны гетероциклические производные, которые активируют рецептор IP. Активация пути передачи сигнала рецептора IP применима для лечения многих форм АЛГ, фиброза легких и оказывает благоприятное воздействие при фиброзных патологических состояниях различных органов в моделях на животных и у пациентов. Также описаны фармацевтические композиции, содержащие такие производные. Чарлтон Стивен Джон, Лебланк Катрин, МакКеоун Стивен Карл (GB) Медведев В.Н., Павловский А.Н. (RU) Уровень техники Простациклин (или PGI2) является представителем семейства липидных молекул, известных, как эйкозаноиды. Он является активным сосудорасширяющим, антипролиферативным, антитромбоцитарным средством, который опосредует свои воздействия, как агонист рецептора IP. Рецептор IP является рецептором, связанным с белком G, который после активации простациклином стимулирует образование циклического аденозинмонофосфата (цАМФ). Простациклин противодействует сосудорасширяющей и протромбоцитарной активности эндотелина. Артериальная легочная гипертензия (АЛГ) является опасным для жизни заболеванием, характеризующимся прогрессирующей легочной васкулопатией, приводящей к гипертрофии левого желудочка. Экзогенное введение агониста рецептора IP стало важной стратегией лечения АЛГ (См., например, Tuderet al., Am. J. Respir. Crit. Care. Med., 1999, 159: 1925-1932; Humbert et al., J. Am. Coll. Cardiol., 2004,43:13S-24S; Rosenzweig, Expert Opin. Emerging Drugs, 2006, 11 :609-619; McLaughlin et al., Circulation,2006, 114:1417-1431; Rosenkranz, Clin. Res. Cardiol., 2007, 96:527-541; Driscoll et al., Expert Opin. Pharmacother., 2008, 9:65-81.). Аналог простациклина эпопростенол (флолан) с точки зрения выживаемости, по меньшей мере,столь же эффективен, как трансплантация. Несмотря на это его не используют в качестве средства первой линии вследствие значительной сопротивляемости, неудобства и высокой стоимости. Вместо этого пациентов, страдающих АЛГ, часто лечат сначала антагонистами эндотелинового рецептора (например,босентаном) и/или ингибиторами PDE5 (например, силденафилом), которые лучше переносятся, но могут обладать ограниченной эффективностью. Аналоги простациклина используют преимущественно в качестве вспомогательных средств лечения, когда тяжесть заболевания прогрессирует и сопротивляемость и неудобства становятся менее важными. Два основных момента препятствуют использованию современных аналогов простациклина в качестве средств первой линии для лечения АЛГ. Во-первых, они очень нестабильны и обладают очень коротким временем полувыведения и это означает, что их необходимо непрерывно вливать с помощью установленного внутривенного (ВВ) катетера, что неудобно для пациента и связано с большой опасностью инфекции и сепсиса. Во-вторых, это связано со значительными побочными эффектами, включая тошноту, боль из-за зажимного устройства, головную боль и другие побочные эффекты, связанные с системной гипертензией. Одним решением этой проблемы является использование илопроста, который выпускается в виде распыляемого препарата, который лучше переносится, но вследствие непродолжительного периода полувыведения его необходимо вводить 6-9 раз в сутки. Недавно исследователи предприняли усилия для получения стабильных, вводимых перорально агонистов рецептора IP. Эти лиганды удобнее и улучшают соблюдение пациентом режима лечения, но для обеспечения фармакодинамического эффекта для легких необходимы большие содержания системного лекарственного препарата; это, возможно, приводит к побочным эффектам, сходным с наблюдающимися при ВВ введении флорана. В настоящем изобретении описаны стабильные, высокоселективные агонисты рецептора IP, которые являются подходящими для пероральной и ингаляционной доставки. Настоящее изобретение обеспечивает значительные преимущества по сравнению с имеющимися аналогами простациклина и позволяет использовать их для пациентов, находящихся в менее тяжелом состоянии. Кроме того, показано, что длительная активация рецептора IP обращает ремоделирование, связанное с АЛГ; поэтому более раннее вмешательство в соответствии с настоящим изобретением может оказать значительное влияние на прогрессирование заболевания и может привести к обращению развития заболевания. Кроме того, исследователи-фармацевты проявляют значительный интерес к разработке агонистов рецептора IP для лечения фиброза легких. Показано, что мыши с дефицитом IP более восприимчивы к вызванному блеомицином фиброзу легких, чем животные дикого типа (Lovgren AK et al. (2006) Am JPhysiol Lung Cell Моля Physiol. 291:L144-56), и агонист рецептора IP илопрост повышает выживаемость мышей, которых лечат блеомицином (Zhu et al (2010) Respir Res. 11(1):34). Кроме того, показано, что передача сигнала рецептора IP благоприятно влияет на фиброзные патологические состояния различных органов в моделях на животных и у пациентов. Преимущества агониста рецептора IP продемонстрированы для фиброза сердца, легких, кожи, поджелудочной железы и печени,и при системном склерозе (Gayraud M (2007) Joint Bone Spine. 74(l):el-8; Hirata Y. et al (2009) Biomed PRehberger P. et al. (2009) Acta Derm Venereol. 89(3):245-9). Фиброзные патологические состояния могут возникнуть в большинстве органов после хронического воспаления по всему организму и, вероятно, обусловлены общими причинами. Поэтому противофиброзные средства, такие как агонисты рецептора IP, предлагаемые в настоящем изобретении, потенциально благоприятны при всех показаниях, которые связаны с фиброзным ремоделированием тканей. Проявляется значительный интерес к разработке агонистов рецептора IP, предназначенных для применения для лечения других заболеваний, таких как атеротромбоз, преэклампсия. Весьма желательна разработка стабильных вводимых путем ингаляции агонистов рецептора IP, которые могут привести к улучшенному лечению АЛГ. Настоящее изобретение относится к соединениям, способам их применения и к их применениям,описанным в настоящем изобретении. Примеры соединений, предлагаемых в настоящем изобретении,включают соединения формулы, Ia или их фармацевтически приемлемую соль и соединения, приведенные в примерах. Поэтому настоящее изобретение относится к соединению формулы Ia или его фармацевтически приемлемой соли, в которой А обозначает N или CR';R5 обозначает С 6-С 14-арил; -(С 0-С 4-алкил)-4-14-членный гетероарил, где гетероарил содержит по меньшей мере один гетероатом, выбранный из группы, включающей N, О и S, где арил, гетероарил каждый необязательно содержат один-пять заместителей Z;R6 обозначает С 6-С 14-арил; -(С 0-С 4-алкил)-4-14-членный гетероарил, где гетероарил содержит по меньшей мере один гетероатом, выбранный из группы, включающей N, О и S, где арил и гетероарил каждый необязательно содержат один-пять заместителей Z;R7 обозначает двухвалентный фрагмент, представляющий собой -О-, -NHC(O)-, -СН 2=СН 2-, -С 6-С 14 арил-D-; -3-14-членный гетероциклил-D-, где гетероциклил содержит по меньшей мере один гетероатом,выбранный из группы, включающей N, О и S, где D обозначает О, S, NH или не присутствует;R19 и R21 каждый независимо обозначают Н или C1-C8-алкил. Ниже описаны различные варианты осуществления настоящего изобретения. Следует понимать,что признаки, указанные для каждого варианта осуществления, можно объединить с другими указанными признаками и получить другие варианты осуществления. В одном варианте осуществления настоящего изобретения, описанном в настоящем изобретении, А обозначает N. В одном варианте осуществления настоящего изобретения, описанном в настоящем изобретении, А обозначает CR'. В одном варианте осуществления настоящего изобретения, описанном в настоящем изобретении, А обозначает CR', где R' обозначает Н. В одном варианте осуществления настоящего изобретения, описанном в настоящем изобретении R1 обозначает -X-Y; или -W-R7-X-Y; R2 обозначает Н или C1-C8-алкил;R7 обозначает двухвалентный фрагмент, представляющий собой -С 6-С 14-арил-D-; -3-14-членный гетероциклил-D-, где гетероциклил содержит по меньшей мере один гетероатом, выбранный из группы,включающей N, О и S, где D обозначает О;R19 и R21 все независимо обозначают Н или C1-C8-алкил. В одном варианте осуществления настоящего изобретения, описанном в настоящем изобретении, R1 обозначает -(CH2)m-C(O)OR"; R2 обозначает Н или С 1-С 4-алкил;R3 обозначает Н, С 1-С 4-алкоксигруппу, ОН, C1-C4-алкил или С 1-С 4-галогеналкил; R4 обозначает Н,С 1-С 4-алкоксигруппу, ОН или С 1-С 4-алкил; m равно 1, 2, 3, 4, 5 или 6; R" обозначает Н или С 1-С 4-алкил. В одном варианте осуществления настоящего изобретения, описанном в настоящем изобретении, R5 обозначает фенил, необязательно замещенный ОН, С 1-С 4-алкилом, С 1-С 4-галогеналкилом, С 1-С 4 алкоксигруппой, или галогеном; иR6 обозначает фенил, необязательно замещенный ОН, С 1-С 4-алкилом, С 1-С 4-галогеналкилом, С 1-С 4 алкоксигруппой или галогеном. В одном варианте осуществления настоящего изобретения А обозначает N. Следует понимать, что любые и все варианты осуществления настоящего изобретения могут использоваться совместно с любым другим вариантом осуществления для описания дополнительных вариантов осуществления настоящего изобретения. Кроме того, любые элементы варианта осуществления комбинируются с любыми и всеми другими элементами любого из вариантов осуществления для описания дополнительных вариантов осуществления. Специалисты в данной области техники должны понимать, что невозможные комбинации заместителей не являются объектами настоящего изобретения. Другой вариант осуществления настоящего изобретения, определенный выше, относится к соединениям формулы 1 а, выбранных из группы, состоящей из 7-(2-фенил-3-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6 Н)-ил)гептановой кислоты; 7-(8-гидрокси-2,3-ди-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6 Н)-ил)гептановой кислоты; 7-(7-гидрокси-6-оксо-2,3-ди-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6 Н)-ил)гептановой кислоты; 7-(2,3-ди-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6 Н)-ил)гептановой кислоты; 2-(3-2,3-дифенил-7,8-дигидропиридо[3,2-b]пиразин-5(6 Н)-ил)метил)фенокси)уксусной кислоты; 7-(3-фенил-2-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6 Н)-ил)гептановой кислоты или их фармацевтически приемлемых солей. В одном варианте осуществления настоящего изобретения представлено соединение, которое представляет собой 7-(2,3-ди-п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6 Н)-ил)гептановую кислоту формулы или ее фармацевтически приемлемую соль. Также настоящее изобретение представляет соединение, которое представляет собой 7-(2-фенил-3 п-толил-7,8-дигидропиридо[2,3-b]пиразин-5(6 Н)-ил)гептановую кислоту формулы или ее фармацевтически приемлемую соль. В другом варианте изобретения представлено соединение, которое представляет собой 2-(3-2,3 дифенил-7,8-дигидропиридо[2,3-b]пиразин-5(6 Н)-ил)гептановую кислоту формулы или ее фармацевтически приемлемую соль. В следующем варианте осуществления изобретения представлено соединение, в котором соединение находится в свободной форме. Особенно предпочтительными соединениями формулы Ia или их фармацевтическими солями являются описанные ниже в настоящем изобретении в разделе "Примеры". Определения Термины, использующиеся в настоящем описании, обладают указанными ниже значениями."Необязательно замещенный" означает группу, которая может быть замещена в одном или большем количестве положений любым радикалом или любой комбинацией радикалов, перечисленных ниже."Необязательно замещенный одной или большим количеством групп Z" означает, что соответствующая группа может включать один или большее количество заместителей, каждый из которых независимо выбран из числа групп, включенных в определение Z. Таким образом, если содержатся две или большее количество замещающих групп Z, они могут быть одинаковыми или разными."Галоген" при использовании в настоящем изобретении может означать фтор, хлор, бром или йод."С 1-С 3-Алкил" при использовании в настоящем изобретении означает обладающий линейной или разветвленной цепью алкил, содержащий 1-8 атомов углерода. Если указано другое количество атомов углерода, такое как С 6 или С 3, то определение следует соответствующим образом изменить, например,"С 1-С 4-алкил" будет означать метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил."C1-C8-Алкоксигруппа" при использовании в настоящем изобретении означает обладающую линейной или разветвленной цепью алкоксигруппу, содержащую 1-8 атомов углерода. Если указано другое количество атомов углерода, такое как С 6 или С 3, то определение следует соответствующим образом изменить, например, "С 1-С 4-алкоксигруппа" будет означать метоксигруппу, этоксигруппу, пропоксигруппу,изопропоксигруппу, бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу и трет-бутоксигруппу."С 1-С 4-Галогеналкил" при использовании в настоящем изобретении означает обладающий линейной или разветвленной цепью алкил, содержащий 1-4 атома углерода, в котором по меньшей мере один атом водорода, замещен атомом галогена. Если указано другое количество атомов углерода, такое как С 6 или С 3, то определение следует соответствующим образом изменить, например, "С 1-С 4-галогеналкил" будет означать метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил, который содержит по меньшей мере один атом водорода, замещенный атомом галогена, например, если галогеном является фтор: CF3CF2-, (CF3)2CH-, CH3-CF2-, CF3CF2-, CF3, CF2H-, CF3CF2CHCF3 или CF3CF2CF2CF2-. Термин "алкилен" означает обладающий линейной или разветвленной цепью алкилен (двухвалентную алкильную цепь), содержащий от 1 до 8 атомов углерода, например, метилен, этилен, 1 метилэтилен, 2-метилэтилен, триметилен, тетраметилен, пентаметилен, гексаметилен, гептаметилен и октаметилен."С 3-С 15-Циклоалкил" при использовании в настоящем изобретении означает карбоциклическую группу, содержащую от 3 до 15 кольцевых атомов углерода, которая является насыщенной или частично насыщенной, такую как С 3-C8-циклоалкил. Примеры С 3-С 15-карбоциклических групп включают, но не ограничиваются только ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил или бициклические группы, такие как бициклооктил, бициклононил, включая инданил и инденил, и бициклодецил. Если указано другое количество атомов углерода, такое как С 6, то определение следует соответствующим образом изменить."Арил" или "С 6-С 15-ароматическая карбоциклическая группа" при использовании в настоящем изобретении означают ароматическую группу, содержащую от 6 до 15 кольцевых атомов углерода. Примеры С 6-С 15-ароматических карбоциклических групп включают, но не ограничиваются только ими, фенил,фенилен, бензолтриил, нафтил, нафтилен, нафталинтриил или антрилен. Если указано другое количество атомов углерода, такое как С 10, то определение следует соответствующим образом изменить."4-8-Членный гетероциклил", "5-6-членный гетероциклил", "3-10-членный гетероциклил", "3-14 членный гетероциклил", "4-14-членный гетероциклил" и "5-14-членный гетероциклил" означают соответственно 4-8-членные, 5-6-членные, 3-10-членные, 3-14-членные, 4-14-членные и 5-14-членные гетероциклические кольца, содержащие по меньшей мере один кольцевой гетероатом, выбранный из группы,включающей азот, кислород и серу, которые могут быть насыщенными, частично насыщенными или ненасыщенными (ароматическими). Гетероциклил включает моноциклические кольцевые группы, конденсированные кольцевые группы и мостиковые группы. Примеры таких гетероциклилов включают, но не ограничиваются только ими, фуран, пиррол, пирролидин, пиразол, имидазол, триазол, изотриазол,-4 022046"Гетероарил" является подгруппой гетероциклилов, которые являются полностью ненасыщенными(ароматическими). Примерами таких групп являются пиридин и пиразин. Термин "гидроксигруппа" или "гидроксил" включает группы, содержащие -ОН. Термин "гетероатом" включает любой элемент кроме углерода или водорода. Предпочтительными гетероатомами являются азот, кислород, сера и фосфор. В одном варианте осуществления "гетероатом" включает азот, серу и кислород. Термин "карбоксигруппа" означает группу карбоновой кислоты. Термин "алкоксикарбоксигруппа" означает сложный эфир. Термин "карбамоил" означает -C(O)NH2. Термины "моноалкилкарбамоил" и "диалкилкарбамоил" означают карбамоил, в которой атом водорода или атомы водорода атома азота заменены на С 1-С 3-алкил,описанный выше. Вторым объектом настоящего изобретения является соединению формулы Ia или его фармацевтические соли, определенные в настоящем изобретении, предназначенные для использования в качестве лекарственного средства. Согласно изобретению показано, что активация рецептора IP оказывает благоприятное воздействие или лечит следующие заболевания или нарушения: АЛГ, выбранную из группы, включающей идиопатическую АЛГ; семейную АЛГ; АЛГ, связанную с коллагеновым заболеванием сосудов, выбранным из группы, включающей: склеродермию, синдромCREST, системную красную волчанку (СКВ), ревматоидный артрит, синдром Такаясу, полимиозит и дерматомиозит; АЛГ, связанную с наследственным заболеванием сердца, выбранным из группы, включающей: дефект межпредсердной перегородки (ДПП), дефект межжелудочковой перегородки (ДЖП) и открытый артериальный проток у индивидуума; АЛГ, связанную с портальной гипертензией; АЛГ, связанную с инфекцией ВИЧ (вирус иммунодефицита человека); АЛГ, связанную с приемом лекарственного препарата или токсина; АЛГ, связанную с наследственной геморрагической телеангиэктазией; АЛГ, связанную со спленэктомией; АЛГ, связанную со значительным участием вен или капилляров; АЛГ, связанную с тромбозом вен легких (ТРВЛ); и АЛГ, связанную с легочным капиллярным гемангиоматозом(ЛКГ); феномен Рейно, включая болезнь Рейно и синдром Рейно; фиброзные заболевания, включая фиброз легких, системный склероз/склеродермию, фиброз/цирроз печени, фиброз почек; тромботические заболевания, связанные с чрезмерной агрегацией тромбоцитов, заболевание коронарной артерии, инфаркт миокарда, преходящий ишемический приступ, стенокардию, удар, ишемически-реперфузионное поражение, рестеноз, фибрилляцию предсердий, тромбообразование, атеросклероз, атеротромбоз, астму,симптом астмы, связанное с диабетом нарушение, диабетическую периферическую невропатию, диабетическую нефропатию, диабетическую ретинопатию, глаукому или другое заболевание глаз с аномальным внутриглазным давлением, гипертензию, преэклампсию, воспаление, профилактику нежелательных побочных эффектов СОХ-1, СОХ-2 и неселективных ингибиторов СОХ, псориаз, псориатический артрит, ревматоидный артрит, болезнь Крона, отторжение трансплантата, рассеянный склероз, системную красную волчанку (СКВ), язвенный колит, ишемически-реперфузионное поражение, рестеноз, атеросклероз, акне, диабет типа 1, диабет типа 2, сепсис и хроническое обструктивное заболевание легких(ХОЗЛ). Другим объектом настоящего изобретения является соединение формулы Ia, или его фармацевтические соли, предназначенные для применения для лечения АЛГ, описанного выше. Другим объектом настоящего изобретения является соединение формулы Ia или его фармацевтические соли, предназначенные для применения для лечения нарушения, выбранного из числа указанных выше заболеваний и нарушений. Еще одним объектом настоящего изобретения является применение соединения формулы Ia, определенного в любом из приведенных выше вариантов осуществления, в свободной форме или в форме фармацевтически приемлемой соли для приготовления лекарственного средства, предназначенного для лечения артериальной легочной гипертензии. Один вариант осуществления настоящего изобретения относится к применению соединения формулы Ia, определенного в любом из приведенных выше вариантов осуществления, в свободной форме или в форме фармацевтически приемлемой соли для приготовления лекарственного средства, предназначенного для лечения АЛГ, выбранной из группы, включающей: идиопатическую АЛГ; семейную АЛГ; АЛГ, связанную с коллагеновым заболеванием сосудов, выбранным из группы, включающей: склеродермию, синдром CREST, системную красную волчанку (СКВ), ревматоидный артрит, синдром Такаясу,полимиозит и дерматомиозит; АЛГ, связанную с наследственным заболеванием сердца, выбранным из группы, включающей: дефект межпредсердной перегородки (ДПП), дефект межжелудочковой перегородки (ДЖП) и открытый артериальный проток у индивидуума; АЛГ, связанную с портальной гипертензией; АЛГ, связанную с инфекцией ВИЧ; АЛГ, связанную с приемом лекарственного препарата или ток-5 022046 сина; АЛГ, связанную с наследственной геморрагической телеангиэктазией; АЛГ, связанную со спленэктомией; АЛГ, связанную со значительным участием вен или капилляров; АЛГ, связанную с тромбозом вен легких (ТРВЛ); и АЛГ, связанную с легочным капиллярным гемангиоматозом (ЛКГ). Один вариант осуществления настоящего изобретения относится к способу предупреждения или лечения опосредуемого рецептором IP патологического состояния или заболевания, включающему введение эффективного количества по меньшей мере одного соединения, описанного в настоящем изобретении, субъекту, нуждающемуся в таком лечении. Такое опосредуемое рецептором IP патологическое состояние или заболевание выбрано из числа АЛГ, выбранных из группы, включающей: идиопатическую АЛГ; семейную АЛГ; АЛГ, связанную с коллагеновым заболеванием сосудов, выбранным из группы,включающей: склеродермию, синдром CREST, системную красную волчанку (СКВ), ревматоидный артрит, синдром Такаясу, полимиозит и дерматомиозит; АЛГ, связанную с наследственным заболеванием сердца, выбранным из группы, включающей: дефект межпредсердной перегородки (ДПП), дефект межжелудочковой перегородки (ДЖП) и открытый артериальный проток у индивидуума; АЛГ, связанную с портальной гипертензией; АЛГ, связанную с инфекцией ВИЧ; АЛГ, связанную с приемом лекарственного препарата или токсина; АЛГ, связанную с наследственной геморрагической телеангиэктазией; АЛГ,связанную со спленэктомией; АЛГ, связанную со значительным участием вен или капилляров; АЛГ, связанную с тромбозом вен легких (ТРВЛ); и АЛГ, связанную с легочным капиллярным гемангиоматозом(ЛКГ). Другое опосредуемое рецептором IP патологическое состояние или заболевание выбрано из группы, включающей агрегацию тромбоцитов, заболевание коронарной артерии, инфаркт миокарда, преходящий ишемический приступ, стенокардию, удар, ишемически-реперфузионное поражение, рестеноз,фибрилляцию предсердий, тромбообразование, атеросклероз, атеротромбоз, астму, симптом астмы, связанное с диабетом нарушение, диабетическую периферическую невропатию, диабетическую нефропатию, диабетическую ретинопатию, глаукому или другое заболевание глаз с аномальным внутриглазным давлением, гипертензию, воспаление, псориаз, псориатический артрит, ревматоидный артрит, болезнь Крона, отторжение трансплантата, рассеянный склероз, системную красную волчанку (СКВ), язвенный колит, ишемически-реперфузионное поражение, рестеноз, атеросклероз, акне, диабет типа 1, диабет типа 2, сепсис и хроническое обструктивное заболевание легких (ХОЗЛ). Один вариант осуществления настоящего изобретения относится к способу предупреждения или лечения опосредуемого рецептором IP патологического состояния или заболевания, включающему введение эффективного количества по меньшей мере одного соединения, описанного в настоящем изобретении, субъекту, нуждающемуся в таком лечении, таким опосредуемым рецептором IP патологическим состоянием или заболеванием является АЛГ. В настоящем описании и в приведенной ниже формуле изобретения, если из контекста не следует иное, термин "включает" и его варианты, такие как "включающий" следует понимать, как означающий включение указанного целого числа или стадии или группы, или группы целых чисел или стадий, но не исключение любого другого целого числа или стадии или группы, или группы целых чисел или стадий. При использовании в настоящем изобретении термин "фармацевтически приемлемые соли" означает соли, которые сохраняют биологическую эффективность и свойства соединений, предлагаемых в настоящем изобретении, и которые обычно не являются нежелательными в биологическом или ином отношении. Во многих случаях соединения, предлагаемые в настоящем изобретении, могут образовать соли с кислотой и/или основанием вследствие наличия аминогруппы и/или карбоксигрупп или сходных с ними групп. Фармацевтически приемлемые соли присоединения с кислотами можно образовать с неорганическими кислотами и с органическими кислотами, например, ацетат, аспартат, бензоат, безилат, бромид/гидробромид, бикарбонат/карбонат, бисульфат/сульфат, камфорсульфонат, хлорид/гидрохлорид,хлортеофиллонат, цитрат, этандисульфонат, фумарат, глюцептат, глюконат, глюкуронат, гиппурат, гидройодид/йодид, изетионат, лактат, лактобионат, лаурилсульфат, малат, малеат, малонат, манделат, мезилат, метилсульфат, нафтоат, напзилат, никотинат, нитрат, октадеканоат, олеат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, полигалактуронат, пропионат, стеарат, сукцинат, сульфосалицилат, тартрат, тозилат, трифторацетат и ксинафоат. Неорганические кислоты, из которых можно образовать соли, включают, например, хлористоводородную кислоту, бромисто-водородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и т. п. Органические кислоты, из которых можно образовать соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, толуолсульфоновую кислоту, 1-гидрокси-2-нафтойную кислоту и сульфосалициловую кислоту. Фармацевтически приемлемые соли присоединения с основанием можно образовать с неорганическими и органическими основаниями. Неорганические основания, из которых можно образовать соли, включают, например, аммоний и основания металлов групп I - XII Периодической системы. В некоторых вариантах осуществления образуются соли натрия, калия, аммония, кальция, магния, железа, серебра, цинка и меди; особенно подходящие соли включают соли аммония, калия, натрия, кальция и магния. Органические основания, из которых можно образовать соли, включают, например, первичные,вторичные и третичные амины, замещенные амины, включая природные замещенные амины, циклические амины, основные ионообменные смолы и т. п. Некоторые органические амины включают изопропиламин, бензатин, холин, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин. Фармацевтически приемлемые соли, предлагаемые в настоящем изобретении, можно синтезировать из исходного соединения, основного или кислотного фрагмента по обычным химическим методикам. Обычно такие соли можно получить по реакции свободных кислотных форм этих соединений со стехиометрическим количеством соответствующего основания (такого как гидроксид, карбонат, бикарбонат и т. п. Na, Ca, Mg или K) или по реакции свободных основных форм этих соединений со стехиометрическим количеством соответствующей кислоты. Такие реакции обычно проводят в воде или в органическом растворителе или в их смеси. Обычно, если это возможно, предпочтительными являются неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Перечни дополнительных подходящих солей приведены, например, в публикациях "Remington's Pharmaceutical Sciences", 20th ed.,Mack Publishing Company, Easton, Pa., (1985); и "Handbook of Pharmaceutical Salts: Properties, Selection,and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002). Кроме того, соединения, предлагаемые в настоящем изобретении, включая их соли, также можно получить в форме их гидратов или с включением других растворителей, использовавшихся для их кристаллизации. Соединения, предлагаемые в настоящем изобретении, т. е. соединения формулы 1 а, которые содержат группы, способные выступать в качестве доноров и/или акцепторов для водородных связей, могут образовывать совместные кристаллы с подходящими образующими совместные кристаллы веществами. Эти совместные кристаллы можно получить из соединений формулы Ia по известным методикам образования совместных кристаллов. Такие методики включают размол, нагревание, совместную сублимацию,совместное плавление или взаимодействие в растворе соединений формулы Ia с образующим совместные кристаллы веществом при условиях кристаллизации и выделение образовавшихся таким образом совместных кристаллов. Подходящие образующие совместные кристаллы вещества включают описанные вWO 2004/078163. Поэтому настоящее изобретение также относится к совместным кристаллам, включающим соединение Ia. При использовании в настоящем изобретении термин "оптический изомер" или "стереоизомер" означает любую из разных стереоизомерных конфигураций, которые могут существовать для данного соединения, предлагаемого в настоящем изобретении, и включает геометрические изомеры. Следует понимать, что заместитель может быть присоединен к атому углерода, являющемуся хиральным центром. Поэтому настоящее изобретение включает энантиомеры, диастереоизомеры и рацематы соединения."Энантиомеры" представляют собой пару стереоизомеров, которые являются зеркальными изображениями, не налагающимися друг на друга. Смесь двух энантиомеров состава 1:1 называется "рацемической" смесью. Этот термин используется для обозначения рацемической смеси, если это является подходящим. "Диастереоизомеры" являются стереоизомерами, которые содержат не менее двух асимметрических атомов, но которые не являются зеркальными изображениями друг друга. Абсолютную стереохимическую конфигурацию описывают с помощью R-S системы Кана-Ингольда-Прелога. Если соединение является чистым энантиомером, то стереохимическую конфигурацию каждого хирального атома углерода можно описать, как R или S. Разделенные соединения, абсолютная конфигурация которых не установлена, можно описать с помощью символов (+) или (-) в зависимости от направления, в котором они вращают плоскость поляризации света при длине волны линии D натрия (право- и левовращающие). Некоторые соединения, описанные в настоящем изобретении, содержат один или большее количество асимметрических центров или осей и поэтому могут образовывать энантиомеры, диастереоизомеры и другие стереоизомерные формы, абсолютную стереохимическую конфигурацию которых можно обозначить,как (R)- или (S)-. В объем настоящего изобретения входят все такие возможные изомеры, включая рацемические смеси, оптически чистые формы и смеси промежуточного состава. Оптически активные (R)- и(S)-изомеры можно получить с помощью хиральных синтонов или хиральных реагентов или выделить по обычным методикам. Если соединение содержит двойную связь, то заместители могут находиться в Е или Z конфигурации. Если соединение представляет собой дизамещенный циклоалкил, то заместители циклоалкила могут находиться в цис- или транс-конфигурации. Предполагается, что в объем настоящего изобретения также входят все таутомерные формы. Любой асимметрический атом (например, углерода и т. п.) соединения (соединений), предлагаемого в настоящем изобретении, может находиться в рацемической или энантиомерно обогащенной форме,например, в (R)-, (S)- или (R,S)-конфигурации. В некоторых вариантах осуществления каждый асимметрический атом характеризуется энантиомерным избытком в (R)- или (S)-конфигурации, составляющим не менее 50%, энантиомерным избытком, составляющим не менее 60%, энантиомерным избытком, составляющим не менее 70%, энантиомерным избытком, составляющим не менее 80%, энантиомерным избытком, составляющим не менее 90%, энантиомерным избытком, составляющим не менее 95%, или энантиомерным избытком, составляющим не менее 99%. Заместители атомов, образующих кратную связь, если это возможно, могут находиться в цис- (Z)- или транс- (Е)-форме. Поэтому при использовании в настоящем изобретении соединение, предлагаемое в настоящем изобретении, может находиться в форме одного из возможных изомеров, поворотных изомеров, атропоизомеров, таутомеров или их смесей, например, в виде в основном чистых геометрических (цис- или транс-) изомеров, диастереоизомеров, оптических изомеров (антиподов), рацематов или их смесей. С использованием различий физико-химических характеристик компонентов все полученные смеси изомеров можно разделить на чистые или в основном чистые геометрические или оптические изомеры,диастереоизомеры, рацематы, например, с помощью хроматографии и/или фракционной кристаллизации. Все полученные рацематы конечных или промежуточных продуктов можно разделить на оптические антиподы по известным методикам, например, путем разделения солей их диастереоизомеров, полученных с оптически активной кислотой или основанием, с последующим выделением оптически активной кислоты или основания. В частности, таким образом можно использовать основной фрагмент,чтобы разделить соединения, предлагаемые в настоящем изобретении, на оптические антиподы, например, с помощью фракционной кристаллизации соли, образованной с оптически активной кислотой, например, винной кислотой, дибензоилвинной кислотой, диацетилвинной кислотой, ди-О,О'-птолуилвинной кислотой, миндальной кислотой, яблочной кислотой или камфор-10-сульфоновой кислотой. Рацемические продукты также можно разделить с помощью хиральной хроматографии, например,высокоэффективной жидкостной хроматографии (ВЭЖХ) с использованием хирального сорбента. Поскольку соединения, предлагаемые в настоящем изобретении, предназначены для применения в фармацевтических композициях, нетрудно понять, что все они предпочтительно предоставляются в основном в чистом виде, например, чистотой не менее 60%, более предпочтительно чистотой не менее 75% и еще более предпочтительно чистотой не менее 85%, еще более предпочтительно чистотой не менее 98% (выраженные в процентах значения являются массовыми). Неочищенные препараты соединений можно использовать для получения более чистых форм, использующихся в фармацевтических композициях; эти менее чистые препараты соединений должны содержать не менее 1%, предпочтительно не менее 5% и более предпочтительно от 10 до 59% соединения, предлагаемого в настоящем изобретении. Соединения, предлагаемые в настоящем изобретении, получают в свободной форме или в виде соли. Если в одной молекуле содержатся одновременно основная группа и кислотная группа, то соединения, предлагаемые в настоящем изобретении, также могут образовывать внутренние соли, например,цвиттерионные молекулы. Любая формула, приведенная в настоящем изобретении, также характеризует немеченые формы, а также изотопно-меченые формы соединений. Изотопно-меченые соединения обладают структурами,описывающимися формулами, приведенными в настоящем изобретении, за исключением того, что один или большее количество атомов заменены на атомы, обладающими указанными атомными массами или массовыми числами. Примеры изотопов, которые можно вводить в соединения, предлагаемые в настоящем изобретении, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2 Н, 3 Н, 11 С, 13 С, 14 С, 15N, 18F 31P, 32P, 35S, 36C1, 125I соответственно. В объем настоящего изобретения входят различные изотопно-меченые соединения, предлагаемые в настоящем изобретении, например, в которые включены радиоактивные изотопы, такие как 3 Н, 13 С, и 14 С. Такие изотопно-меченые соединения применимы для изучения метаболизма (предпочтительно содержащие 14 С), кинетики реакций (содержащие, например, 2 Н или 3 Н), в методологиях детектирования и визуализации, таких как позитронная эмиссионная томография (ПЭТ) или однофотонная эмиссионная компьютерная томография(ОФЭКТ), включая исследование распределения лекарственного средства или субстрата в тканях или лучевую терапию пациентов. В частности, содержащее 18F или меченое им соединение может быть особенно предпочтительным для исследований с помощью ПЭТ или ОФЭКТ. Изотопно-меченые соединения, предлагаемые в настоящем изобретении, обычно можно получить по методикам, раскрытым в схемах или в примерах и синтезах, описанных ниже, путем замены не содержащего изотопа реагента на легко доступный изотопно-меченый реагент. Кроме того, замещение более тяжелыми изотопами, предпочтительно дейтерием (т.е. 2 Н или D),может обеспечить некоторые терапевтические преимущества, обусловленные их более высокой метаболической стабильностью, например, увеличенной длительностью полувыведения in vivo или возможностью использования меньших доз, или улучшением терапевтического индекса. Следует понимать, что в этом контексте дейтерий рассматривается в качестве заместителя в соединении формулы (Ia). Концентрацию такого более тяжелого изотопа, в частности, дейтерия, можно определить с помощью коэффициента изотопного обогащения. Термин "коэффициент изотопного обогащения" при использовании в настоящем изобретении означает отношение содержания изотопа к содержанию конкретного изотопа в природе. Если заместитель в соединении, предлагаемом в настоящем изобретении, означает дейтерий, то такое соединение характеризуется коэффициентом изотопного обогащения для каждого обозначенного атома дейтерия, равным не менее 3500 (содержание дейтерия для каждого обозначенного атома дейтерия равно 52,5%), не менее 4000 (содержание дейтерия равно 60%), не менее 4500 (содержание дейтерия равно 67,5%), не менее 5000 (содержание дейтерия равно 75%), не менее 5500 (содержание дейтерия равно 82,5%), не менее 6000 (содержание дейтерия равно 90%), не менее 6333,3 (содержание дейтерия равно 95%), не менее 6466,7 (содержание дейтерия равно 97%), не менее 6600 (содержание дейтерия равно 99%) или не менее 6633,3 (содержание дейтерия равно 99,5%). Изотопно-меченые соединения формулы (Ia) обычно можно получить по стандартным методикам,известным специалистам в данной области техники, или по методикам, аналогичным описанным в приведенных ниже примерах и в разделе, посвященном синтезу, с использованием подходящего изотопномеченого реагента вместо использовавшегося ранее не содержащего изотопа реагента. Синтез Обычно соединения формулы Ia или их фармацевтические соли можно синтезировать путями, описанными на схемах А-М и в примерах. Схема А. Схема А начинается со стадии 1 реакции с использованием имеющегося в продаже исходного вещества или исходного вещества, которое может синтезировать специалист в данной области техники, и показанной конденсации вещества. Стадией 2 является гидрирование. Стадией 3 является алкилирование или восстановительное аминирование в зависимости от искомого продукта. Стадией 4 схемы А является гидролиз с образованием свободной кислоты, если содержится сложный эфир. R1, R2, R3, R4, R5 и R6 являются такими, как определено в настоящем изобретении. Схема В. Схема В начинается со стадии 1 реакции с использованием имеющегося в продаже исходного вещества или исходного вещества, которое может синтезировать специалист в данной области техники, и показанной конденсации вещества. Стадией 2 является гидрирование. Стадией 3 является алкилирование или восстановительное аминирование в зависимости от искомого продукта. Стадией 4 схемы В является гидролиз с образованием свободной кислоты, если содержится сложный эфир. R1, R2, R3, R4, R5 и R6 являются такими, как определено в настоящем изобретении. Схема С. Схема С начинается со стадии 1 реакции с использованием имеющегося в продаже исходного веще-9 022046 ства или исходного вещества, которое может синтезировать специалист в данной области техники, и показанной конденсации вещества. Стадией 2 является образование N-оксида. Стадией 3 является селективное введение хлора. Стадией 4 является перекрестное сочетание по Негиши по атому хлора кольца. Стадией 5 является гидрирование. Стадией 6 является алкилирование или восстановительное аминирование в зависимости от искомого продукта. Стадией 7 является хиральное разделение смеси соединений с использованием сверхкритической жидкостной хроматографии с получением отдельных энантиомеров. Стадией 8 схемы С является гидролиз с образованием свободной кислоты, если содержится сложный эфир. R1, R2, R3, R4, R5 и R6 являются такими, как определено в настоящем изобретении. Схема D. Схема D начинается со стадии 1 реакции с использованием имеющегося в продаже исходного вещества или исходного вещества, которое может синтезировать специалист в данной области техники, и показанной конденсации вещества. Стадией 2 является образование N-оксида. Стадией 3 является стереоселективное присоединение реагента Гриньяра к N-оксидному производному. Стадией 4 является гидрирование. Стадией 5 является алкилирование или восстановительное аминирование в зависимости от искомого продукта. Стадией 6 является хиральное разделение смеси соединений с использованием сверхкритической жидкостной хроматографии с получением отдельных энантиомеров. Стадией 7 схемыD является гидролиз с образованием свободной кислоты, если содержится сложный эфир. R1, R2, R3, R4,R5 и R6 являются такими, как определено в настоящем изобретении. Схема Е. Схема Е начинается со стадии 1 реакции с использованием имеющегося в продаже исходного вещества или исходного вещества, которое может синтезировать специалист в данной области техники, и показанной конденсации вещества. Стадией 2 является хлорирование. Стадией 3 и стадией 4 являются реакции перекрестного сочетания по Судзуки. Стадией 5 является гидрирование. Стадией 6 является алкилирование или восстановительное аминирование в зависимости от искомого продукта. Стадией 7 схемы Е является гидролиз с образованием свободной кислоты, если содержится сложный эфир. R1, R2, R3, R4,- 10022046 Схема F начинается со стадии 1 реакции с использованием имеющегося в продаже исходного вещества или исходного вещества, которое может синтезировать специалист в данной области техники, и показанной конденсации вещества. Стадией 2 является хлорирование. Стадией 3 является реакция перекрестного сочетания по Судзуки. Стадией 4 является гидрирование. Стадией 5 является алкилирование или восстановительное аминирование в зависимости от искомого продукта. Стадией 6 схемы F является гидролиз с образованием свободной кислоты, если содержится сложный эфир. R1, R2, R3, R4, R5 и R6 являются такими, как определено в настоящем изобретении. Схема G. Схема G начинается со стадии 1 реакции с использованием имеющегося в продаже исходного вещества или исходного вещества, которое может синтезировать специалист в данной области техники, и показанной конденсации вещества. Стадией 2 является гидрирование. Стадией 3 является введение защитной группы (PG). Стадией 4 является бромирование. Стадией 5 является реакция с металлоорганическим соединением или нуклеофильное замещение галогенидного производного в зависимости от искомого продукта. Стадией 6 является необязательное удаление защитной группы. Стадией 7 является алкилирование или восстановительное аминирование в зависимости от искомого продукта. Стадией 8 схемыG является необязательная стадия удаления защитной группы и гидролиз с образованием свободной кислоты, если содержится сложный эфир. Хиральное разделение можно провести в качестве стадии 7 а или стадии 8 а. R1, R2, R3, R4, R5 и R6 являются такими, как определено в настоящем изобретении. Схема Н. Схема Н начинается со стадии 1 реакции с использованием имеющегося в продаже исходного вещества или исходного вещества, которое может синтезировать специалист в данной области техники, и показанной конденсации вещества. Стадией 2 является восстановление. Стадией 3 является введение защитной группы (PG). Стадией 4 является гидроксилирование алкена. Стадией 5 является введение защитной группы гидроксигруппы. Стадией 6 является селективное удаление защитной группы. Стадией 7 является алкилирование или восстановительное аминирование в зависимости от искомого продукта. Стадией 8 является стадия удаления защитной группы и гидролиз с образованием свободной кислоты,если содержится сложный эфир. Стадией 9 схемы Н является хиральное разделение смеси соединений с использованием сверхкритической жидкостной хроматографии с получением отдельных энантиомеров. Схема I начинается со стадии 1 реакции с использованием имеющегося в продаже исходного вещества или исходного вещества, которое может синтезировать специалист в данной области техники, и показанной конденсации вещества. Стадией 2 является гидрирование. Стадией 3 является алкилирование или восстановительное аминирование в зависимости от искомого продукта. Стадией 4 является восста- 12022046 новление. Стадией 5 является необязательный гидролиз. Стадией 6 является алкилирование. Стадией 7 схемы I является гидролиз с образованием свободной кислоты, если содержится сложный эфир. Схема J начинается со стадии 1 реакции с использованием имеющегося в продаже исходного вещества или исходного вещества, которое может синтезировать специалист в данной области техники, и показанной конденсации вещества. Стадией 2 является гидрирование. Стадией 3 является алкилирование или восстановительное аминирование в зависимости от искомого продукта. Стадией 4 является реакция метатезиса. Стадией 6 является гидроксилирование алкена. Стадией 8 является гидрирование. Стадиями 5, 7 и 9 схемы J является гидролиз с образованием свободной кислоты, если содержится сложный эфир. Схема K начинается со стадии 1 реакции с использованием имеющегося в продаже исходного ве- 13022046 щества или исходного вещества, которое может синтезировать специалист в данной области техники, и дибромирование соединения. Стадией 2 является реакция перекрестного сочетания по Негиши с сопутствующей внутримолекулярной циклизацией. Стадиями 3, 8 является алкилирование. Стадиями 4, 7, 11 и 12 являются реакции перекрестного сочетания по Судзуки. Стадиями 5, 9 и 13 является гидролиз с образованием свободной кислоты. Стадиями 10 и 14 является восстановление. Стадией 6 схемы K является гидроксилирование. R1, R3, R4 , R5 и R6 являются такими, как определено в настоящем изобретении. Схема L. Схема L начинается со стадии 1 реакции с использованием имеющегося в продаже исходного вещества или исходного вещества, которое может синтезировать специалист в данной области техники, и показанной конденсации вещества. Стадией 2 является восстановление. Стадией 3 является алкилирование или восстановительное аминирование в зависимости от искомого продукта. Стадией 4 является гидроборирование алкена. Стадией 5 является хиральное разделение смеси соединений с использованием сверхкритической жидкостной хроматографии с получением отдельных энантиомеров. Стадией 6 схемы L является гидролиз с образованием свободной кислоты, если содержится сложный эфир. R1, R2, R5 и R6 являются такими, как определено в настоящем изобретении. Схема М. Схема М начинается со стадии 1 реакции с использованием имеющегося в продаже исходного вещества или исходного вещества, которое может синтезировать специалист в данной области техники, и показанной конденсации вещества. Стадией 2 является гидрирование. Стадией 3 является алкилирование или восстановительное аминирование в зависимости от искомого продукта. Стадией 4 является необязательное удаление защитной группы. Стадией 5 является образование амидной связи. Стадией 6 схемы М является гидролиз с образованием свободной кислоты, если содержится сложный эфир. R2, R3 , R4 , R5 иR6 являются такими, как определено в настоящем изобретении, n равно от 1 до 5. PG является подходящей защитной группой. Специалист в данной области техники должен понимать, что на общих путях синтеза, подробно описанных выше, представлены обычные реакции необходимых превращений исходных веществ. Конкретные условия проведения реакций не указаны, но они хорошо известны специалистам в данной области техники и подходящие условия специалист в данной области техники может выбрать в соответствии со своей подготовкой. Исходные вещества являются имеющимися в продаже соединениями или являются известными соединениями и их можно получить по методикам, описанным в области органической химии. Соединения формулы Ia в свободной форме можно превратить в форму соли и наоборот обычным образом, известным специалистам в данной области техники. Соединения, находящиеся в свободной форме или в форме соли можно получить в форме гидратов или сольватов, содержащих растворитель,использовавшийся для кристаллизации. Соединения формулы Ia можно извлечь из реакционных смесей и очистить обычным образом. Изомеры, такие как стереоизомеры, можно получить обычным образом,например, с помощью фракционной кристаллизации или асимметрического синтеза из соответствующим образом асимметрически замещенных, например, оптически активных исходных веществ. Соединения формулы Ia или их фармацевтические соли можно получить, например, по реакциям и методикам, описанным ниже и в примерах. Реакции можно проводить в растворителе, подходящем для использующихся реагентов и веществ и подходящем для проводимых превращений. Специалисты в области органического синтеза должны понимать, что функциональные группы, содержащиеся в молекуле,должны соответствовать предполагаемым превращениям. Для этого иногда необходимо принять решение об изменении порядка стадий синтеза или выбора схемы реакций, более предпочтительной, чем другая, с получением искомого соединения, предлагаемого в настоящем изобретении. Различные заместители в промежуточных продуктах синтеза и в конечных продуктах, указанных на схемах реакций, могут находиться в соответствующем необходимом для проведения реакций виде, если необходимо, то с подходящими защитными группами, как это понимает специалист в данной области техники, или в виде предшественников, которые затем можно превратить в соответствующие конечные формы по методикам, известным специалисту в данной области техники, заместители также можно ввести на разных стадиях последовательности синтеза или после завершения последовательности синтеза. Во многих случаях обычные операции с функциональными группами можно использовать для превращения одного промежуточного продукта в другой промежуточный продукт или одного соединения формулы Ia в другое соединение формулы Ia. Примерами таких операций являются превращение сложного эфира или кетона в спирт; превращение сложного эфира в кетон; взаимопревращения сложных эфиров,кислот и амидов; алкилирование, ацилирование и сульфонирование спиртов и аминов; и многие другие. Заместители также можно вводить по обычным реакциям, таким как алкилирование, ацилирование, галогенирование или окисление. Такие операции хорошо известны в данной области техники и в многих публикациях описаны процедуры и методики таких операций. Некоторыми справочными публикациями,в которых приведены примеры и ссылки на оригинальную литературу по органическому синтезу, в которой описаны многочисленные операции с функциональными группами, а также другие превращения,обычно использующиеся в области органического синтеза, являются March's Organic Chemistry, 5 Edition,Wiley and Chichester, Eds. (2001); Comprehensive Organic Transformations, Larock, Ed., VCH (1989); Comprehensive Organic Functional Group Transformations, Katritzky et al. (series editors), Pergamon (1995); иComprehensive Organic Synthesis, Trost and Fleming (series editors), Pergamon (1991). Также следует понимать, что другим важным фактором при планировании любого пути синтеза является продуманный выбор защитной группы, использующейся для защиты реакционноспособных функциональных групп, содержащихся в соединениях, описанных в настоящем изобретении. Для одной и той же молекулы можно выбрать множество защитных групп так, чтобы в зависимости от необходимого результата каждую из этих защитных групп можно было бы удалить без удаления других защитных групп этой молекулы, или несколько защитных групп можно было бы удалить на одной стадии реакции. Авторитетной публикацией, в которой для подготовленного исследователя описаны многие альтернативы, является Greene andWuts, Protective Groups in Organic Synthesis, Wiley and Sons, 4 Edition (2006). Фармакологическая активность Соединения, раскрытые в настоящем изобретении, активируют рецептор IP и применимы для лечения различных заболеваний и нарушений и для облегчения их симптомов. Без наложения ограничений они включают следующие. Артериальная легочная гипертензия (АЛГ) АЛГ обладает многофакторной патобиологией. Сужение сосудов, ремоделирование стенки сосуда легких и тромбоз способствуют повышению сопротивления сосудов легких при АЛГ (Humbert et al, J.Am. Coll. Cardiol., 2004, 43:13S-24S.). Соединения, предлагаемые в настоящем изобретении, раскрытые в настоящем изобретении, применимы для лечения артериальной легочной гипертензии (АЛГ) и ее симптомов. АЛГ включает следующие формы артериальной легочной гипертензии, описанные в клинической классификации артериальной легочной гипертензии Всемирной организации здравоохранения(ВОЗ) 2003 г.: идиопатическая АЛГ (ВРАН); семейная АЛГ (FPAH); АЛГ, связанная с другими патологическими состояниями (АРАН), такие как АЛГ, связанная с коллагеновым заболеванием сосудов, АЛГ,связанная с наследственными системно-легочными шунтами, АЛГ, связанная с портальной гипертензией, АЛГ, связанная с инфекцией HTV, АЛГ, связанная с лекарственными препаратами или токсинами,или АЛГ, связанная с другими причинами; и АЛГ, связанная со значительным участием вен или капилляров. Идиопатическая АЛГ означает АЛГ неустановленной этиологии. Семейная АЛГ означает АЛГ,для которой предполагается или документирована передача по наследству. АЛГ, связанная с коллагеновым заболеванием сосудов, включает АЛГ, связанную со склеродермией, АЛГ, связанную с синдромомCREST (кальциноз кожи, феномен Рейно, дисфункция пищевода, склеродактилия и телеангиэктазия),АЛГ, связанную с системной красной волчанкой (СКВ), АЛГ, связанную с ревматоидным артритомом,АЛГ, связанную с синдром Такаясу, АЛГ, связанную с полимиозитом, и АЛГ, связанную с дерматомиозитом. АЛГ, связанная с наследственными системно-легочными шунтами, включает АЛГ, связанную с дефектом межпредсердной перегородки (ДПП), АЛГ, связанную с дефектом межжелудочковой перегородки (ДЖП), и АЛГ, связанную с открытым артериальным протоком. АЛГ, связанная с лекарственными препаратами или токсинами, включает АЛГ, связанную с приемом аминорекса, АЛГ, связанную с приемом фенфлурамина (например, АЛГ, связанную с приемом фенфлурамина, или АЛГ, связанную с приемом дексфенфлурамина), АЛГ, связанную с приемом некоторых токсичных масел (например, АЛГ, связанную с приемом рапсового масла), АЛГ, связанную с приемом пирролидизиновых алкалоидов (например, АЛГ, связанную с приемом чая Робойс) и АЛГ, связанную с приемом монокроталина. АЛГ, связанная с другими причинами, включает АЛГ, связанную с нарушением щитовидной железы, АЛГ, связанную с болезнью накопления гликогена, АЛГ, связанную с болезнью Гоше, АЛГ, связанную с наследственной геморрагической телеангиэктазией, АЛГ, связанную с гемоглобинопатией, АЛГ, связанную с миелопролиферативным нарушением, и АЛГ, связанную со спленэктомией. АЛГ, связанная со значительным участием вен или капилляров, включает АЛГ, связанную с тромбозом вен легких (ТРВЛ), и АЛГ, связанную с легочным капиллярным гемангиоматозом (ЛКГ). (См., например, Simonneau et al., J. Am. Coll. Cardiol., 2004, 43:5S-12S; McGoon et al, Chest, 2004, 126:14S-34S;Rabinovitch, Annu. Rev. Pathol. Mech. Dis., 2007, 2:369-399; McLaughlin et al., Circulation, 2006, 114:14171431; Strauss et al., Clin. Chest. Med., 2007, 28:127-142; Taichman et al., Clin. Chest. Med., 2007, 28:1-22.). Данные по связи АЛГ со склеродермией и благоприятному воздействию агониста рецептора IP на АЛГ приведены в публикации Badesch et al. (Badesch et al., Ann. Intern. Med., 2000, 132:425-434). Данные по связи АЛГ со смешанным коллагеновым заболеванием сосудов и заболеванием соединительной ткани(MCTD), системной красной волчанкой (СКВ), синдромом Шегрена и синдромом CREST и благоприятному воздействию агониста рецептора IP на АЛГ приведены в публикации Humbert et al. (Eur. Respir. J.,1999, 13:1351-1356). Данные по связи АЛГ с синдромом CREST и благоприятному воздействию агониста рецептора IP на АЛГ приведены в публикации Miwa et al. (Int. Heart J., 2007, 48:417-422). Данные по связи АЛГ с СКВ и благоприятному воздействию агониста рецептора IP на АЛГ приведены в публикацииRobbins et al (Chest, 2000, 117:14-18). Данные по связи АЛГ с инфекцией ВИЧ и благоприятному воздействию агониста рецептора IP на АЛГ приведены в публикации Aguilar et al. (Am. J. Respir. Crit. CareMed., 2000, 162:1846-1850). Данные по связи АЛГ с наследственными дефектами сердца (включая ДПП,ДЖП и открытый артериальный проток) и благоприятному воздействию агониста рецептора IP на АЛГ приведены в публикации Rosenzweig et al. (Circulation, 1999, 99:1858-1865). Данные по связи АЛГ с фенфлурамином и с дексфенфлурамином, анорексигенами, приведены в публикации Archer et al. (Am. J. Respir. Crit. Care Med., 1998, 158: 1061-1067). Данные по связи АЛГ с наследственной геморрагической телеангиэктазией приведены в публикации McGoon et al. (Chest, 2004,126:14-34). Данные по связи АЛГ со спленэктомией приведены в публикации Hoeper et al. (Ann. Intern.Med., 1999, 130:506-509). Данные по связи АЛГ с портальной гипертензией и благоприятному воздействию агониста рецептора IP на АЛГ приведены в публикации Hoeper et al. (Eur. Respir. J., 2005, 25:502508). Симптомы АЛГ включают одышку, стенокардию, синкопе и отек (McLaughlin et al., Circulation,2006, 114:1417-1431). Соединения, предлагаемые в настоящем изобретении, раскрытые в настоящем изобретении, применимы для лечения симптомов АЛГ. Антитромбоцитарные терапии (патологические состояния, связанные с агрегацией тромбоцитов) Антитромбоцитарные средства назначают при различных патологических состояниях. Например,при заболевании коронарной артерии их используют для содействия предупреждению инфаркта миокарда или удара у пациентов, у которых существует опасность развития обструктивных тромбов (например,тромбоза коронарных артерий). При инфаркте миокарда сердечная мышца не получает достаточного количества обогащенной кислородом крови вследствие закупорки коронарных кровеносных сосудов. В случае приема при протекании приступа или сразу после него (предпочтительно не позже, чем через 30 мин) антитромбоцитарные средства могут уменьшить поражение сердца. Преходящий ишемический приступ ("ПИП" или "мини-удар") представляет собой кратковременное прерывание потока кислорода в головной мозг вследствие уменьшенного кровотока через артерии,- 16022046 обычно вследствие закупорки тромбом. Установлено, что антитромбоцитарные лекарственные средства эффективны для предупреждения ПИП. Стенокардия является временной и часто рецидивирующей болью в груди, сдавливанием или дискомфортом, обусловленным недостаточным обогащенным кислородом кровотоком (ишемией) в некоторые области сердца. У пациентов, страдающих стенокардией, антитромбоцитарная терапия может ослабить проявления стенокардии и уменьшить опасность инфаркта миокарда. Удар является проявлением, при котором головной мозг не получает достаточного количества обогащенной кислородом крови, обычно вследствие закупорки кровеносного сосуда головного мозга тромбом. У пациентов, для которых существует высокая опасность, установлено, что регулярный прием антитромбоцитарных средств предупреждает образование тромбов, который вызывают первый или второй удары. Ангиопластика является основанной на использовании катетера методикой, использующейся для восстановления кровотока через артерии, закупоренные тромбом. Независимо от того, проводится ли стентирование, сразу после этой процедуры, предназначенной для восстановление кровотока, антитромбоцитарные средства могут уменьшить опасность образования дополнительных тромбов после этой процедуры (процедур). Операция коронарного шунтирования является хирургической методикой, при которой из какого-то участка тела берут артерию или вену и трансплантируют к закупоренной коронарной артерии, перенаправляя кровоток вокруг закупорки и через новый добавленный сосуд. После этой процедуры антитромбоцитарные средства могут уменьшить опасность образования вторичных тромбов. Фибрилляция предсердий является самым обычным типом стойкого нарушенного сердечного ритма(аритмии). Фибрилляция предсердий ежегодно поражает примерно два миллиона американцев. При фибрилляции предсердий предсердие (верхние камеры сердца) быстро получает электрические сигналы, которые вынуждают их трепетать, а не сокращаться обычным образом. Результатом является аномально частое и сильно нерегулярное сердцебиение. Введении после приступа фибрилляции предсердий антитромбоцитарных средствха может уменьшить опасность образования тромбов в сердце и их попадания в головной мозг (эмболии). Имеются данные о том, что агонист рецептора IP подавляет агрегацию тромбоцитов и таким образом перспективен для использования при лечении в качестве антитромбоцитарного средства (см., например, Moncada et al., Lancet, 1977, 1: 18-20). Показано, что генетический недостаток рецептора IP у мышей приводит к увеличенной склонности к тромбозу (Murata et al., Nature, 1997, 388:678-682). Агонисты рецептора IP можно использовать для лечения, например, перемежающейся хромоты или заболевания периферической артерии, а также сердечно-сосудистых осложнений, тромбоза артерии, атеросклероза, сужения сосудов, вызванного серотонином, ишемически-реперфузионного поражения и рестеноза артерий после ангиопластики или установки стента. (См., например, Fetalvero et al, ProstaglandinsEngl. J. Med., 2007, 357:2482-2494; Fetalvero et al., Am. J. Physiol. Heart. Circ. Physiol., 2006, 290:H1337H1346; Murata et al., Nature, 1997, 388:678-682; Wang et al., Proc. Natl. Acad. Sci. USA, 2006, 103:1450714512; Xiao et al., Circulation, 2001, 104:2210-2215; McCormick et al., Biochem. Soc. Trans., 2007, 35:910911; Arehart et al., Circ. Res., 2008, Mar 6.). Агонисты рецептора IP также можно использовать по отдельности или в комбинации с тромболитическим средством, например, тканевым активатором плазминогена (t-PA) с обеспечением кардиозащитного действия после инфаркта миокарда или постишемической дисфункции миокарда или защиты от ишемического поражения во время чрескожной коронарной ангиопластики и т.п., включая вызванные ими осложнения. Агонисты рецептора IP также можно использовать в антитромбоцитарных средствах в комбинации, например, с альфа-токоферолом (витамин Е), эхистатином (дезинтегрин) или, в состояниях гиперкоагуляции, гепарином. (См., например, Chan., J. Nutr., 1998, 128:1593-1596; Mardla et al., Platelets,2004, 15:319-324; Bernabei et al., Ann. Thorac. Surg., 1995, 59:149-153; Gainza et al., J. Nephrol., 2006,19:648-655.) Агонисты рецептора IP, раскрытые в настоящем изобретении, обеспечивают эффективное улучшение микроциркуляции у пациентов, нуждающихся в антитромбоцитарной терапии, путем антагонистического воздействия, например, на сосудосуживающие продукты агрегации тромбоцитов и не ограничиваются показаниями, описанными выше. Поэтому в некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения агрегации тромбоцитов у нуждающегося в нем пациента, включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении. В других вариантах осуществления настоящее изобретение относится к способам лечения заболевания коронарной артерии, инфаркта миокарда, преходящего ишемического приступа, стенокардии, удара, фибрилляции предсердий, или симптома любого из указанных выше заболеваний у пациента, нуждающегося в лечении,включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении. В других вариантах осуществления настоящее изобретение относится к способам уменьшения опасности тромбообразования при ангиопластике или операции коронарного шунтирования у пациента или у пациента, страдающего фибрилляцией предсердий, включающим введение пациенту композиции,содержащей агонист рецептора IP, раскрытый в настоящем изобретении, в момент, когда существует такая опасность. Атеросклероз Атеросклероз является сложным заболеванием, характеризующимся воспалением, накоплением липидов, гибелью клеток и фиброзом. Он является главной причиной смерти во многих странах, включая США. Атеросклероз, как термин, использующийся в настоящем изобретении, включает нарушения крупных и средних артерий, которые приводят к прогрессирующему накоплению гладкомышечных клеток и липидов в интиме. Показано, что агонист рецептора IP может обеспечить защиту от атеросклероза, такого как атеротромбоз (Arehart et al., Curr. Med. Chem., 2007, 14:2161-2169; Stitham et al., Prostaglandins Other Lipid Mediat., 2007, 82:95-108; Fries et al., Hematology Am. Soc. Hematol. Educ. Program, 2005, :445-451; Egan et al.,Science, 2004, 306:1954-1957; Kobayashi et al., J. Clin. Invest, 2004, 114:784-794; Arehart et al., Circ. Res.,2008, Mar 6). Показано, что сигналы дефектного рецептора IP, видимо, ускоряют атеротромбоз у людей,т.е. что агонист рецептора IP может обеспечить защиту от атеротромбоза у людей (Arehart et al., Circ.Res., 2008, Mar 6.) Соединения, предлагаемые в настоящем изобретении, раскрытые в настоящем изобретении, применимы для лечения атеросклероза и лечения его симптомов. Поэтому в некоторых вариантах осуществления настоящее изобретение относится к способам лечения атеросклероза у пациента, нуждающегося в лечении, включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении. В других вариантах осуществления настоящее изобретение относится к способам лечения симптома атеросклероза у пациента, нуждающегося в лечении, включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении. Астма Астма представляет собой опосредуемое лимфоцитами воспалительное нарушение дыхательных путей, характеризующимся эозинофилией дыхательных путей, усиленным продуцированием слизи бокаловидными клетками и структурным ремоделированием стенок дыхательных путей. В последние десятилетия распространенность астмы в мире очень резко выросла. Показано, что генетически обусловленная недостаточность рецептора IP у мышей усиливает аллергическое воспаление дыхательных путей (Takahashi et al, Br J Pharmacol, 2002, 137:315-322). Показано, что агонист рецептора IP может подавлять не только развитие астмы при введении в фазе сенсибилизации, но и основные проявления экспериментальной астмы при введении в фазе стимуляции (Idzko et al, J. Clin. Invest., 2007, 117:464-72, Nagao et al, Am.J. Respir. Cell Mol. Biol., 2003, 29:314-320), по меньшей мере, частично вследствие заметного мешающего влияния на функцию содержащих антиген дендритных клеток, находящихся в дыхательных путях (Idzkoet al., J. Clin. Invest., 2007, 117:464-472; Zhou et al, J. Immunol., 2007, 178:702-710; Jaffar et al., J. Immunol.,2007, 179:6193-6203; Jozefowski et al, Int. Immunopharmacol., 2003, 3:865-878). Эти клетки являются критически важными на стадиях инициирования и поддержания аллергической астмы, поскольку уменьшение содержания дендритных клеток в дыхательных путях при вторичной стимуляции у сенсибилизированных мышей устраняет все характеристические признаки астмы и этот эффект можно полностью восстановить с помощью адоптивного переноса дендритных клеток дикого типа (van Rijt et al., J. Exp. Med.,2005, 201:981-991). Также показано, что агонист рецептора IP может подавлять секрецию провоспалительных цитокинов альвеолярными макрофагами человека (Raychaudhuri et al., J. Biol. Chem., 2002,277:33344-33348). Соединения, предлагаемые в настоящем изобретении, раскрытые в настоящем изобретении, применимы для лечения астмы и лечения ее симптомов. Поэтому в некоторых вариантах осуществления настоящее изобретение относится к способам лечения астмы у пациента, нуждающегося в лечении, включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении. Другие варианты осуществления относятся к способам лечения симптома астмы у пациента, нуждающегося в лечении, включающим введение пациенту композиции, содержащей агонист рецептора IP,раскрытый в настоящем изобретении. Хроническое обструктивное заболевание легких Активация рецептора IP также может быть полезной при хроническом обструктивном заболевании легких (ХОЗЛ). Тапростен, агонист рецептора IP, in vitro подавляет выработку хемоаттрактантов CXCL9 и CXCL10 для CD8+ Т-клеток эпителиальными клетками дыхательных путей человека. (Ayer, L. M., S. М.Wilson, S. L. Traves, D. Proud, M. A. Giembycz. 2008. J. Pharmacol. Exp. Ther. 324: 815-826). Берапрост,агонист рецептора IP, защищает мышей от развития вызванной дымом сигарет экспериментальной эмфиземы, вероятно, вследствие согласованного подавляющего влияния на апоптоз альвеолярных эпителиальных клеток, окислительную нагрузку, экспрессию матричной маталлопротеиназы и генерацию провоспалительных цитокинов (Chen Y., M. Hanaoka, P. Chen, Y. Droma N. F. Voelkel, К. Kubo. 2009. Am. J.Physiol. 296: L648-L656). Другие варианты осуществления относятся к способам лечения ХОЗЛ у пациента, нуждающегося в лечении, включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении. Гипергликемия Хотя гипергликемия является главной причиной патогенеза осложнений при диабете, таких как диабетическая периферическая невропатия (ДПН), диабетическая нефропатия (ДН) и диабетическая ретинопатия (ДР), усиленное сужение сосудов и агрегация тромбоцитов у страдающих диабетом пациентов также играет роль в прогрессировании заболевания (Cameron et al., Naunyn Schmiedebergs Arch. Pharmacol., 2003, 367:607-614). Агонисты рецептора IP стимулируют сужение сосудов и подавляют агрегацию тромбоцитов. Улучшение капиллярного кровообращения также благоприятно в случае осложнений при диабете (Cameron, Diabetologia, 2001, 44:1973-1988). Показано, что у крыс, страдающих диабетом, вызванным стрептозотоцином, агонист рецептора IP может предупреждать и обращать аномалии проводимости двигательных и периферических нервов (Cotter et al., Naunyn Schmiedebergs Arch. Pharmacol., 1993, 347:534-540). Дополнительные данные о благоприятном воздействии агониста рецептора IP при лечении диабетической периферической невропатии приведены в публикациях Hotta et al. (Diabetes, 1996, 45:361-366), Ueno et al. (Jpn. J. Pharmacol., 1996,70:177-182), Ueno et al. (Life Sci., 1996, 59:PLlO5-PL110), Hotta et al. (Prostaglandins, 1995, 49:339-349),Shindo et al. (Prostaglandins, 1991, 41:85-96), Okuda et al. (Prostaglandins, 1996, 52:375-384) и Koike et al.(FASEB J., 2003, 17:779-781). Данные о благоприятном воздействии агониста рецептора IP при лечении диабетической нефропатии приведены в публикациях Owada et al. (Nephron, 2002, 92:788-796) и Yamashita et al. (Diabetes Res.Clin. Pract., 2002, 57:149-161). Данные о благоприятном воздействии агониста рецептора IP при лечении диабетической ретинопатии приведены в публикациях Yamagishi et al. (Mol. Med., 2002, 8:546-550), Burnette et al. (Exp. Eye Res., 2006, 83: 1359-1365), и Hotta et al. (Diabetes, 1996, 45:361-366). Показано, что агонист рецептора IP может снижать повышенные содержания фактора некроза опухоли-[альфа] (TNF[альфа]) у пациентов, страдающих диабетом, и это означает, что агонист рецептора IP может способствовать предупреждению прогрессирования осложнений при диабете (Fujiwara et al., Exp. Clin. Endocrinol.Diabetes, 2004, 112:390-394). Данные о том, что местное введение агониста рецептора IP может привести к снижению внутриглазного давления (ВГД) у кроликов и собак и тем самым оказать благоприятное воздействие при лечении глаукомы, приведены в публикации Hoyng et al. (Hoyng et al., Invest. Ophthalmol. Vis. Sci., 1987,28:470-476). Показано, что агонисты рецептора IP проявляют активность при регуляции тонуса сосудов, сужения сосудов и облегчении легочной гипертензии (см., например, Strauss et al., Clin Chest Med, 2007,28:127-142; Driscoll et al., Expert Opin. Pharmacother., 2008, 9:65-81). Данные о благоприятном воздействии агониста рецептора IP при лечении гипертензии приведены в публикации Yamada et al. (Peptides,2008, 29:412-418). Данные о том, что агонист рецептора IP может защищать от ишемии головного мозга,приведены в публикациях Dogan et al (Gen. Pharmacol., 1996, 27:1163-1166) и Fang et al. (J. Cereb. BloodFlow Metab., 2006, 26:491-501). Борьба с воспалением Противовоспалительные средства назначают при различных патологических состояниях. Например,при воспалительном заболевании их используют для противодействия причинам вредных воздействий и тем самым для их ослабления. Имеются данные о том, что агонист рецептора IP может подавлять и тем самым являться возможным средством лечения в качестве противовоспалительной терапии. Показано, что агонист рецептора IP может подавлять продуцирование провоспалительных цитокинов и хемокинов (интерлейкин-12 (IL-12),фактора некроза опухоли-[альфа] (TNF-[альфа]), DL-1[альфа], EL-6, макрофагального воспалительного белка-1-альфа (MIP-1-[альфа]), моноцитарного хемоаттрактантного белка-1 (MCP-I и способность дендритных клеток стимулировать Т-клетки (Jozefowski et al., Int. Immunopharmacol., 2003, 865-878; Zhou etal., J. Immunol., 2007, 178:702-710; Nagao et al., Am. J. Respir. Cell Mol. Biol., 2003, 29:314-320; Idzko et al.,J. Clin. Invest., 2007, 117:464-472). Показано, что агонист рецептора IP может подавлять продуцирование провоспалительных цитокинов (TNF-[альфа], IL-1/3, EL-6, гранулоцитарного макрофагального стимулирующего фактора (GM-CSF макрофагами (Raychaudhuri et al., J. Biol. Chem., 2002, 277:33344-33348;Acids, 2005, 73:405-410; Shinomiya et al., Biochem. Pharmacol., 2001, 61:1153-1160). Показано, что агонист рецептора IP может стимулировать продуцирование провоспалительных цитокинов (DL-IO) дендритными клетками (Jozefowski et al., Int. Immunopharmacol., 2003, 865-878; Zhou et al., J. Immunol., 2007,178:702-710). Показано, что агонист рецептора IP может стимулировать продуцирование провоспалительных цитокинов (DL-IO) макрофагами (Shinomiya et al., Biochem. Pharmacol., 2001, 61: 1153-1160). Показано, что агонист рецептора IP может подавлять индуцированный хемокинами (CCL 17) хемотаксис лейкоцитов (CD4+ Th2 Т-клеток) (Jaffar et al., J. Immunol., 2007, 179:6193-6203). Показано, что агонист рецептора IP может обеспечить защиту от атеросклероза, такого как атеротромбоз (Arehart et al., Curr.Kobayashi et al., J. Clin. Invest., 2004, 114:784-794; Arehart et al., Circ. Res., 2008, Mar 6). Показано, что агонист рецептора IP может ослаблять астму (Idzko et al, J. Clin. Invest., 2007, 117:464-472; Jaffar et al., J.Immunol., 2007, 179:6193-6203; Nagao et al., Am. J. Respir. Cell. Mol. Biol., 2003, 29:314-320). Показано,что при диабете типа 2 агонист рецептора IP может снижать продуцирование TNF-[альфа] у пациентов(Fujiwara et al., Exp. Clin. Endocrinol. Diabetes, 2004, 112:390-394; Goya et al., Metabolism, 2003, 52: 192198). Показано, что агонист рецептора IP может подавлять ишемически-реперфузионное поражение(Xiao et al., Circulation, 2001, 104:2210-2215). Показано, что агонист рецептора IP может подавлять рестеноз (Cheng et al, Science, 2002, 296:539-541). Показано, что агонист рецептора IP может ослаблять поражение сосудов легких и шок и в модели септического шока на крысах (Harada et al, Shock, 2008, Feb 21). Показано, что агонист рецептора IP может in vivo уменьшать содержание TNF-[альфа] в сыворотке у пациентов, страдающих ревматоидным артритам, и это связано с улучшением клинического протекания заболевания (Gao et al, Rheumatol. Int., 2002, 22:45-51; Boehme et al., Rheumatol. Int., 2006, 26:340-347). Соединения, предлагаемые в настоящем изобретении, раскрытые в настоящем изобретении, обеспечивают благоприятное уменьшение воспаления. Соединения, предлагаемые в настоящем изобретении, раскрытые в настоящем изобретении, обеспечивают благоприятное уменьшение вредного воспалительного ответа, связанного с воспалительным заболеванием. Поэтому в некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения воспаления у нуждающегося в нем пациента, включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении. В некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения продуцирования IL-12,ТКР-[альфа], IL-1[альфа], IL-IjS, BL-6, MIP-Ia или MCP-I у нуждающегося в нем пациента, включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении. В некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения продуцирования TNF-[альфа] у нуждающегося в нем пациента, включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении. В некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения продуцирования EL-IO у нуждающегося в нем пациента, включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении. В некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения вредного воспалительного ответа, связанного с воспалительным заболеванием у нуждающегося в нем пациента, включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении. В некоторых вариантах осуществления настоящее изобретение относится к способам лечения воспалительного заболевания или его симптома у пациента, нуждающегося в лечении, включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении. В некоторых вариантах осуществления настоящее изобретение относится к способам лечения воспалительного заболевания или его симптома у пациента,нуждающегося в лечении, включающим введение пациенту композиции, содержащей агонист рецептораIP, раскрытый в настоящем изобретении. В некоторых вариантах осуществления настоящее изобретение относится к способам лечения воспалительного заболевания или его симптома у пациента, нуждающегося в лечении, включающим введение пациенту композиции, содержащей агонист рецептора IP, раскрытый в настоящем изобретении, где воспалительное заболевание выбрано из группы, включающей псориаз, псориатический артрит, ревматоидный артрит, болезнь Крона, отторжение трансплантата, рассеянный склероз, системную красную волчанку (СКВ), язвенный колит, ишемически-реперфузионное поражение,рестеноз, атеросклероз, акне, диабет (включая диабет типа 1 и диабет типа 2), сепсис, хроническое обструктивное заболевание легких (ХОЗЛ) и астму. Фиброз Показано, что передача сигналов PGI2 играет благоприятную роль при фиброзных заболеваниях различных органов, включая почки, сердце, легкие, кожу, поджелудочную железу и печень, а также при системном склерозе и связанных с ним патологиях. Показано, что агонист рецептора IP может облегчать фиброз сердца (Chan EC et al. (2010) J Mol Cell Cardiol. Apr 18; Hirata Y et al. (2009) Biomed Pharmacother. 63(10):781-6; Kaneshige T et al. (2007) J Vet Med Sci. 69(12): 1271-6). Показано, что агонист рецептора IP может уменьшать фиброз почек (Takenaka M et al. (2009) Prostaglandins Leukot Essent Fatty Acids. 80(56):263-7). Показано, что агонист рецептора IP может защищать от фиброза легких в модели с использованием блеомицина (Zhu Y et al. (2010) Respir Res. 20; 11(1): 34). Показано, что у пациентов, страдающих склеродермией, агонист рецептора IP может подавлять продуцирование фактора роста соединительной ткани, ключевого медиатора фиброза (Stratton R et al (2001) J Clin Invest. 108(2):241-50). Показано, что агонист рецептора IP может уменьшать частоту пальцеобразных изъязвлений у пациентов, страдающих системным склерозом М. Vayssairat (1999) J Rheumatol 26:2173-2178. Показано, что агонист рецептора IP может уменьшать некроз кончиков пальцев у младенцев, страдающих стойким феноменом Рейно (Shouval DS et al. (2008) Clin Exp Rheumatol. 26 (3 Suppl 49):S 105-7). Показано, что агонист рецептора IP может уменьшить содержание маркеров эндотелиальной активации у пациентов, страдающих системным склерозом (Rehberger P et al. (2009) Acta Derm Venereol. 89 (3):245-9.). Показано, что агонист рецептораIP может уменьшить тяжесть, частоту и длительность приступов Рейно у пациентов, страдающих сис- 20022046 темным склерозом (Torlay et al. (1991) Ann Rheum Dis 50, 800-804). Показано, что агонист рецептора IP может улучшить динамику портального кровообращения у пациентов, страдающих системным склерозом и феноменом Рейно (Zardi et al. (2006), In Vivo 20 (3):377-80). Показано, что агонист рецептора IP может подавлять прогрессирование фиброза поджелудочной железы у страдающих ожирением крысZucker (Sato et al. (2010) Diabetes 59 (4):1092-100). Агонисты рецептора IP, раскрытые в настоящем изобретении, обеспечивают благоприятные антифиброзные воздействия для пациентов, страдающих фиброзом почек, легких, сердца, кожи, поджелудочной железы и печени, которые могут быть идиопатическими или вторичными, например, после хронического воспаления и системного склероза, и не ограничиваются показаниями, описанными выше. Кроме того, имеется значительное количество данных о том, что агонист рецептора IP может улучшать функцию почек при острой и хронической почечной недостаточности. Показано, что агонист рецептора IP может восстанавливать функцию почек при связанной с эндотоксикозом острой почечной недостаточности (Johannes T. et al. (2009) Crit Care Med. 37(4): 1423-32). Показано, что агонист рецептораIP может улучшать функцию почек в модели ишемического/реперфузионного поражения почек (Sahsivar МО et al. (2009) Shock 32(5):498-502). Показано, что агонист рецептора IP может предупреждать вызванную контрастным веществом нефропатию у пациентов, страдающих нарушением функции почек, которым проводили операцию на сердце (Spargias К et al. (2009) Circulation 3; 120(18): 1793-9.) Показано, что агонист рецептора IP может улучшать функцию почек, уменьшать воспаление и склеротические изменения в почках в модели диабетической нефропатии (Watanabe M et al. (2009) Am J Nephrol. 2009; 30 (1):111). Агонисты рецептора IP, раскрытые в настоящем изобретении, обеспечивают благоприятное улучшение функции почек у пациентов, страдающих, например, острым и хроническим поражением почек и нефтопатиями после введения красителей-контрастных веществ, ишемически-реперфузионным поражением, системным воспалением и диабетом и не ограничиваются показаниями, описанными выше. Имеется значительное количество данных о причинном влиянии дефицита простациклина на развитие преэклампсии (Mills JL et al. (1999) JAMA 282: 356-362; Walsh SW (2004) Prostaglandins Leukot EssentFatty Acids 70: 223-232). Показано, что в модели преэклампсии на крысах введение агониста рецептора IP снижает артериальное давление (Zlatnik MG et al (1999) Am J Obstet Gynecol. 180(5):1191-5). Агонисты рецептора IP, раскрытые в настоящем изобретении, обеспечивают благоприятное улучшение гемодинамики у пациентов, страдающих преэклампсией. Агонист рецептора IP, раскрытые в настоящем изобретении, могут обеспечить превосходное лечение муковисцидоза. Агонисты рецептора IP, раскрытые в настоящем изобретении, могут обеспечить химиотерапевтическую профилактику. Химиотерапевтическая профилактика представляет собой практику использования лекарственных средств, витаминов или питательных добавок для уменьшения опасности развития или рецидива рака. Пероральное введение илопроста (Ventavis), аналога простациклина, представляется перспективным для использования в качестве химиотерапевтического средства при раке легких. Данные в пользу использования агониста рецептора IP для химиотерапевтической профилактики приведены в публикации Paul Bunn Jr. MD, который является исполнительным директором Международной ассоциации исследования рака легких Американской ассоциации научных исследований в области раковых заболеваний, и на 102-ом Ежегодном собрании сообщил, что он значительно улучшает протекание эндобронхиальной дисплазии у тех, кто раньше курил. Агонист PGI2, включая соединения формулы Ia, также используется в качестве применяющегося совместно терапевтического средства в комбинации со вторыми средствами, такими как органические нитраты и доноры NO, такие как нитропруссид натрия, нитроглицерин, изосорбидмононитрат, изосорбиддинитрат, молсидомин или SIN-1 и ингаляция NO; соединения, которые ингибируют разложение циклического гуанозинмонофосфата (цГМФ) и/или циклического аденозинмонофосфата (цАМФ), такие как ингибиторы фосфодиэстераз (PDE) 1, 2, 3, 4 и/или 5, предпочтительно ингибиторы PDE 5, такие как силденафил, варденафил и тадалафил; независимые от NO, но зависимые от гема стимуляторы гуанилатциклазы, такие как, в частности, соединения, описанные в WO 00/06568, WO 00/06569, WO 02/42301 иWO 03/095451; независимые от NO и гема активаторы гуанилатциклазы, такие как, в частности, соединения, описанные в WO 01/19355, WO 01/19776, WO 01/19778, WO 01/19780, WO 02/070462 и WO 02/070510; соединения, которые ингибируют нейтрофильную эластазу человека, такие как сивелестат или DX-890 (Reltran); соединения, ингибирующие каскад передачи сигналов, такие как ингибиторы тирозинкиназы и/или серин/треонинкиназы, в частности, иматиниб, гефитиниб, эрлотиниб, сорафениб и сунитиниб; соединения, влияющие на метаболизм энергии сердца, например и предпочтительно этомоксир,дихлорацетат, ранолазин или триметазидин; противотромботические средства, например и предпочтительно выбранные из группы, включающей ингибиторы агрегации тромбоцитов, антикоагулянты или профибринолитические соединения; активные соединения, предназначенные для снижения артериального давления, например и предпочтительно выбранные из группы, включающей антагонисты кальция,антагонисты ангиотензина II, ингибиторы АСЕ, антагонисты эндотелина, ингибиторы ренина, ингибито- 21022046 ры альдоестеронсинтазы, блокаторы рецептора альфа, блокаторы рецептора бета, антагонисты минералокортикоидного рецептора, ингибиторы Rho-киназы и диуретики; и/или активные соединения, которые модифицируют метаболизм липидов, например и предпочтительно выбранные из группы, включающей агонисты рецептора щитовидной железы, ингибиторы синтеза холестерина, например и предпочтительно ингибиторы HMG-CoA-редуктазы или ингибиторы синтеза сквалена, ингибиторы АСАТ, ингибиторы СЕТР, ингибиторы МТР, агонисты PPAR-альфа, PPAR-гамма и/или PPAR-дельта, ингибиторы всасывания холестерина, ингибиторы липазы, адсорбенты полимерной желчной кислоты, ингибиторы повторного всасывания желчной кислоты и антагонисты липопротеина (липопротеинов), в особенности предназначенные для лечения АЛГ или заболеваний и нарушений, таких как указанные выше в настоящем изобретении, например, предназначенные для использования в качестве активаторов терапевтической активности таких лекарственных средств или в качестве средств для снижения необходимой дозы или возможных побочных эффектов таких лекарственных средств. В частности, одним вариантом осуществления настоящего изобретения является фармацевтическая комбинация, содержащая соединения формулы Ia или их фармацевтические соли и второе средство, где вторым средством является ингибитор PDEV или ингибитор нейтральной эндопептидазы. Соединения формулы Ia или их фармацевтические соли можно смешать со вторым средством в фиксированной фармацевтической композиции или их можно вводить по отдельности до, одновременно или после второго лекарственного вещества. В соответствии с этим другим объектом настоящего изобретения является комбинация рецептора IP с осмотическими средствами (гипертонический солевой раствор, декстран, маннит, ксилит), блокаторыENaC, противовоспалительное, бронхолитическое, антигистаминное, противокашлевое, антибиотическое и/или ДНКазное лекарственное вещество, где агонист рецептора IP и другое лекарственное вещество могут находиться в одной или разных фармацевтических композициях. Подходящие антибиотики включают макролидные антибиотики, например, тобрамицин (TOBI). Подходящие ДНКазные лекарственные вещества включают дорназу альфа (пульмозим), высокоочищенный раствор рекомбинантной дезоксирибонуклеазы I человека (rhDNase), который селективно расщепляет ДНК. Дорназу альфа используют для лечения муковисцидоза. Другими полезными комбинациями агониста рецептора IP с противовоспалительными лекарственными средствами являются комбинации с антагонистами хемокиновых рецепторов, например, CCR-1,CCR-2, CCR-3, CCR-4, CCR-5, CCR-6, CCR-7, CCR-8, CCR-9 и CCR10, CXCR1, CXCR2, CXCR3,CXCR4, CXCR5, в особенности с антагонистами CCR-5, такими как выпускающиеся фирмой ScheringPlough антагонисты SC-351125, SCH-55700 и SCH-D; антагонистами, выпускающимися фирмой Takeda,такими как N-4-6,7-дигидро-2-(4-метилфенил)-5H-бензоциклогептен-8-ил]карбонил]амино]фенил]метил]тетрагидро-N,N-диметил-2H-пиран-4-аминийхлорид (ТАК-770); и CCR-5 антагонисты, описанные в патенте US 6166037 (в особенности в пп.18 и 19), WO 00/66558 (в особенности в п.8), WO 00/66559 (в особенности в п.9), WO 04/018425 и WO 04/026873. Подходящие противовоспалительные лекарственные средства включают стероиды, например, кортикостероиды. Подходящие стероиды включают будезонид, бекламетазон (например, дипропионат), бутиксокорт (например, пропионат), CHF5188, циклезонид, дексаметазон, флунизолид, флутиказон (например, пропионат или фуроат), GSK-685698, GSK-870086, LAS40369, метилпреднизолон, мометазон(например, фуроат), преднизолон, рофлепонид и триамцинолон (например, ацетонид). В некоторых предпочтительных вариантах осуществления стероидом является кортикостероид кратковременного действия, такой как будезонид, циклезонид, флутиказон или мометазон. Подходящие вторые активные ингредиенты включают 2-агонисты. Подходящие 2-агонисты включают арформотерол (например, тартрат), албутерол/салбутамол (например, рацемат или один энантиомер, такой как R-энантиомер, или его соль, предпочтительно сульфат), AZD3199, бамбутерол, BI171800, бутолтерол (например, мезилат), кармотерол, кленбутерол, этантерол, фенотерол (например, рацемат или один энантиомер, такой как R-энантиомер, или его соль, предпочтительно гидробромид),флербутерол, формотерол (например, рацемат или один диастереоизомер, такой как R,Rдиастереоизомер, или его соль, предпочтительно фумарат или дигидрат фумарата), GSK-159802, GSK597901, GSK-678007, индакатерол (например, рацемат или один энантиомер, такой как R-энантиомер,или его соль, предпочтительно малеат, ацетат или ксинафоат), LAS-100977, метапротеренол, милветерол(например, гидрохлорид), наминтерол, олодатерол (например, рацемат или один энантиомер, такой какR-энантиомер, или его соль, предпочтительно гидрохлорид), PF-610355, пирбутерол (например, ацетат),прокатерол, репротерол, салмефамол, салметерол (например, рацемат или один энантиомер, такой как Rэнантиомер, или его соль, предпочтительно ксинафоат), тербуталин (например, сульфат) и вилантерол(или его соль, предпочтительно трифенатат. В некоторых предпочтительных вариантах осуществления 2-агонистом является 2-агонист сверхдлительного действия, такой как индакатерол или потенциально кармотерол, LAS-100977, милветерол, олодатерол, PF-610355 или вилантерол. Предпочтительным вариантом осуществления одного из вторых активных ингредиентов является индакатерол (т.е. (R)-5-[2-(5,6 диэтилиндан-2-иламино)-1-гидроксиэтил]-8-гидрокси-1 Н-хинолин-2-он) или его соль. Он представляет собой агонист 2-адренорецептора, который обладает особенно продолжительным действием (т. е. более 24 ч) и быстрым началом действия (т. е. примерно 10 мин). Это соединение получают способами, описанным в заявках на международные патенты WO 2000/75114 и WO 2005/123684. Он может образовывать соли присоединения с муравьиной кислотой, в частности, фармацевтически приемлемые соли присоединения с кислотами. Предпочтительной солью (R)-5-[2-(5,6-диэтилиндан-2-иламино)-1 гидроксиэтил]-8-гидрокси-1 Н-хинолин-2-она является малеат. Другой предпочтительной солью является ацетат (R)-5-[2-(5,6-диэтилиндан-2-иламино)-1-гидроксиэтил]-8-гидрокси-1 Н-хинолин-2-она. Другой предпочтительной солью является ксинафоат (R)-5-[2-(5,6-диэтилиндан-2-иламино)-1-гидроксиэтил]-8 гидрокси-1 Н-хинолин-2-она. Подходящие бронхолитические лекарственные средства включают антихолинергические или антимускариновые средства, такие как аклидиний (например, бромид), ВЕА-2108 (например, бромид), ВЕА 2180 (например, бромид), CHF-5407, дарифенацин (например, бромид), даротропий (например, бромид),гликопирролат (например, рацемат или один энантиомер или его соль, предпочтительно бромид), декспирроний (например, бромид), iGSK-202405, GSK-203423, GSK-573719, GSK-656398, ипратропий (например, бромид), LAS35201, LAS 186368, отилоний (например, бромид), окситропий (например, бромид), оксибутинин, PF-3715455, PF-3635659, пирензепин, реватропат (например, гидробромид), солифенацин (например, сукцинат), SVT-40776, TD-4208, теродилин, тиотропий (например, бромид), толтеродин (например, тартрат) и троспий (например, хлорид). В некоторых предпочтительных вариантах осуществления мускариновые антагонисты представляют собой мускариновые антагонисты длительного действия, такие как даротропийбромид, гликопирролат или тиотропийбромид. Подходящие противовоспалительные и бронхолитические лекарственные средства двойного действия включают агонисты бета-2 адренорецептора/мускариновые антагонисты двойного действия, такие как GSK-961081 (например, сукцинат) и раскрытые в патенте US 2004/0167167, WO 04/74246 и WO 04/74812. Подходящие антигистаминные лекарственные вещества включают цетиризингидрохлорид, ацетаминофен, клемастинфумарат, прометазин, лоратидин, деслоратидин, дифенгидрамин и фексофенадингидрохлорид, активастин, астемизол, азеластин, эбастин, эпинастин, мизоластин и тефенадин, а также раскрытые в JP 2004107299, WO 03/099807 и WO 04/026841. В соответствии с этим, другим объектом настоящего изобретения является комбинация агониста рецептора IP со средствами, которые ингибируют фосфорилирование Smad2 и Smad3 с помощью ALK5 и/или ALK4. В соответствии с этим, другим объектом настоящего изобретения является комбинация агониста рецептора IP со вторыми средствами, которыми являются ингибиторы Rho-киназы. В соответствии с этим, другим объектом настоящего изобретения является комбинация агониста рецептора IP со вторыми средствами, которыми являются ингибиторы триптофангидролазы 1 (ТРН 1). В соответствии с этим, другим объектом настоящего изобретения является комбинация агониста рецептора IP со вторыми средствами, которыми являются ингибиторы нескольких киназ, такие как иматинибмезилат, глеевек. Иматиниб действует как специфический ингибитор целого ряда ферментов тирозинкиназ (ТК). Он занимает активные центры ТК, что приводит к снижению активности. Ферменты ТК в организме включают рецептор инсулина. Иматиниб является специфическим по отношению к домену ТК в фотоонкогене Абельсона, c-kit и PDGF-R (рецептор тромбоцитарного фактора роста). В одном варианте осуществления настоящего изобретения агонист рецептора IP, предлагаемый в настоящем изобретении, вводят в комбинации со вторым активным средством, выбранным из группы,включающей ингибиторы фосфодиэстеразы V, ингибиторы нейтральной эндопептидазы 1, ингибиторы ТНР 1, ингибиторы нескольких киназ, антагонист эндотелина, диуретик, блокатор альдостеронового рецептора и блокатор эндотелинового рецептора. В одном варианте осуществления настоящего изобретения агонист рецептора IP, предлагаемый в настоящем изобретении, вводят в комбинации со вторым активным средством, выбранным из группы,включающей ингибиторы фосфодиэстеразы V, ингибиторы нейтральной эндопептидазы 1, ингибиторы ТНР 1 и ингибиторы нескольких киназ, такие как PDGFR или c-Kit. Другим объектом настоящего изобретения является соединение формулы Ia в свободной форме или в форме фармацевтически приемлемой соли, предназначенное для применения для приготовления лекарственного средства, предназначенного для лечения патологического состояния, реагирующего на активность агониста рецептора IP, предпочтительно АЛГ. Средства, предлагаемые в настоящем изобретении, можно вводить любым подходящим путем, например, перорально, например, в виде таблетки или капсулы; парентерально, например, внутривенно; путем ингаляции, например, для лечения обструктивного заболевания дыхательных путей; назально, например, для лечения аллергического ринита; местно на кожу; или ректально. Другим объектом настоящего изобретения также является фармацевтическая композиция, включающая соединения формул формулы Ia в свободной форме или в форме фармацевтически приемлемой соли, необязательно совместно с фармацевтически приемлемым для нее разбавителем или носителем. Композиция может содержать совместно применяющееся терапевтическое средство, такое как противовоспалительное, бронхорасши- 23022046 ряющее, антигистаминное или противокашлевое лекарственное средство, как это описано выше в настоящем изобретении. Такие композиции можно получить с использованием обычных разбавителей и инертных наполнителей и методик, известных в области фармацевтики. Так, дозированные формы для перорального введения могут представлять собой таблетки или капсулы. Препараты для местного применения могут находиться в виде кремов, мазей, гелей или систем чрескожной доставки, например, пластырей. Композиции для ингаляции могут представлять собой аэрозоль или другие распыляемые препараты или сухие порошкообразные препараты. Если композиция представляет собой аэрозольный препарат, то он предпочтительно содержит пропеллент, например, гидрофторалкан (HFA), такой как HFA134a или HFA227 или их смесь, и может содержать один или большее количество сорастворителей, известных в данной области техники, таких как этанол (до 20 мас.%); и/или одно или большее количество поверхностно-активных веществ, таких как олеиновая кислота или сорбитантриолеат; и/или один или большее количество наполнителей, таких как лактоза. Если композиция представляет собой сухой порошкообразный препарат, то она предпочтительно содержит, например, соединение формулы Ia или его фармацевтические соли, обладающее частицами диаметром до 10 мкм, необязательно совместно с разбавителем или носителем, таким как лактоза, обладающим желательным распределением частиц по размерам, и соединение, которое способствует защите от ухудшения характеристик продукта вследствие влажности, например, стеарат магния. Если композиция представляет собой препарат для распыления, то она предпочтительно содержит, например, соединение формулы Ia,или его фармацевтические соли, растворенное или суспендированное в разбавителе,содержащем воду, сорастворитель, такой как этанол или пропиленгликоль, и стабилизатор, которым может являться поверхностно-активное вещество. Другими объектами настоящего изобретения являются:(a) соединение формулы Ia или его фармацевтические соли в пригодной для вдыхания форме, например, в виде аэрозоля или другой распыляемой композиции, или в виде вдыхаемого измельченного вещества, например, в микронизированной форме;(b) предназначенное для вдыхания лекарственное средство, включающее соединение формулы Ia или его фармацевтические соли в пригодной для вдыхания форме;(c) фармацевтический продукт, включающий соединение формулы (Ia) в пригодной для вдыхания форме вместо с устройством для ингаляции; и(d) устройство для ингаляции, содержащее соединение формулы Ia или его фармацевтические соли в пригодной для вдыхания форме. Дозировки соединений формулы Ia или их фармацевтических солей, использующиеся при практическом применении настоящего изобретения, разумеется, будут меняться в зависимости, например, от конкретного подвергающегося лечению патологического состояния, необходимого эффекта и пути введения. Обычно суточные дозы, подходящие для введения путем ингаляции, составляют порядка 0,005-10 мг, а для перорального введения подходящие суточные дозы обычно составляют порядка 0,05-100 мг. Применение в фармацевтике и анализ Соединения и их фармацевтически приемлемые соли, ниже в настоящем изобретении альтернативно называющиеся, как "средства, предлагаемые в настоящем изобретении", применимы в качестве лекарственных средств. В частности, соединения являются подходящими агонистами рецептора IP и могут быть исследованы с помощью приведенных ниже анализов. Активность соединений по отношению к рецептору IP (рецептор IP) исследуют путем определения накопления цАМФ в клетках СНО (клетки яичников китайского хомячка), стабильно экспрессирующих рецептор IP (CHO-IP), с помощью анализа PerkinElmer AlphaScreen. В этой технологии определяется эндогенное продуцирование цАМФ с помощью использующего люминесценцию нерадиоактивных веществ проксимального люминесцентного однородного анализа. Между покрытыми стрептавидином донорными гранулами, биотинилированным цАМФ и анти-цАМФ акцепторными гранулами происходит биологическая реакция, в результате которой донорные и акцепторные гранулы сближаются, так что при возбуждении вырабатывается сигнал флуоресценции. После образования эндогенного цАМФ конкуренция между биотинилированным цАМФ и образовавшимся в клетках цАМФ приводит к ослаблению сигнала флуоресценции. Это ослабление сигнала пропорционально количеству образовавшегося цАМФ, что делает возможным определение количества цАМФ, образовавшегося в результате стимулирования агонистом. Исследуемые и эталонные соединения готовят в разведении 100[конечное] в 100% ДМСО и разводят в отношении 1:3 с использованием Biomek Fx (Beckman Coulter). Затем разводят промежуточный продукт и получают 5 [конечное разведение] в буфере для анализа (HBSS (сбалансированный солевой раствор Хенкса), содержащий 5 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота),0,1% (мас./об.) БСА (бычий сывороточный альбумин). Затем 5 мкл 5 х [конечное разведение] исследуемых соединений, эталонных соединений контроля и буфер/ДМСО переносят в 384-луночный белый планшет OptiPlate, содержащий 20 мкл суспензии клеток CHO-IP (15000 клеток/лунка, получают из замороженного продукта), и планшет инкубируют при комнатной температуре в течение 1 ч. Стандартную зависимость для цАМФ строят для каждого эксперимента (диапазон концентраций от 10000 до 0,001 нМ в буфере для анализа) и 25 мкл раствора каждой концентрации добавляют в последние два столбца планшета для анализа. Инкубацию останавливают путем добавления литического буфера (dH2O; 0,3%(об./об.) Tween-20), содержащего 20 Ед/мл покрытых стрептавидином донорных гранул и биотинилированный цАМФ (предварительно инкубированный в течение 30 мин) и 20 Ед/мл акцепторных гранул анти-цАМФ, которые добавляют к литическому буферу непосредственно перед добавлением в планшет для анализа. Затем планшет для анализа инкубируют при комнатной температуре в темноте в течение 60 мин при осторожном встряхивании и считывают на считывающем устройстве для планшетов Envision (PerkinElmer). Необработанные данные для эталонных соединений, исследуемых соединений и контролей пересчитывают в концентрации цАМФ с использованием стандартной зависимости для цАМФ с помощью программного обеспечения GraphPadPrism (GraphPad Software Inc). Значения ЕС 50, a также максимальные значения для зависимостей для агониста определяют с использованием 4-параметрического логистического уравнения. Выраженные в % максимальные значения ответов для всех исследуемых соединений определяют по максимальным значениям зависимости концентрация трепростинила - ответ. Соединения примеров, приведенных ниже в настоящем изобретении, по данным описанных выше исследований обычно обладают значениями ЕС 50, равными менее 5 мкМ. В табл.1 приведен перечень типичных соединений и полученных для них значений EC50. Таблица 1 Соединения, перечисленные ниже, входят в объем самого широкого пункта формулы изобретения; однако значения ЕС 50 в результатах описанных выше измерений, превышают 10 мкМ: этил-6-(2,3-бис(4-пропилфенил)-7,8-дигидропиридо[2,3-b]пиразин-5(6 Н)-ил)гексаноат; этил-7-(2-(м-толил)-3-(п-толил)-7,8-дигидропиридо[2,3-b]пиразин-5(6 Н)-ил)гептаноат и 5-(6,7-дифенил-3,4-дигидро-1,8-нафтиридин-1(2 Н)-ил)пентановая кислота. Настоящее изобретение иллюстрируется приведенными ниже примерами. Примеры Общие положения: Масс-спектры снимали с помощью систем ЖХ-МС с использованием ионизации электрораспылением. Использовали комбинации Agilent 1100 HPLC/масс-спектрометр Micromass Platform или WatersAcquity UPLC с масс-спектрометром SQD. [M+H]+ означает моноизотопную молекулярную массу. Спектры ЯМР снимали на находящихся в общем доступе спектрометрах Bruker AVANCE 400 ЯМР с использованием ICON-ЯМР. Все спектры снимали при 298 К и для сравнения использовали пик растворителя. Приведенные ниже примеры предназначены для иллюстрации настоящего изобретения и не предназначены для его ограничения. Температуры приведены в градусах стоградусной шкалы. Если не указа- 25022046 но иное, выпаривание всегда проводили при пониженном давлении, предпочтительно примерно от 15 до 100 мм Hg (= 20-133 мбар). Структуру конечных продуктов, промежуточных продуктов и исходных веществ подтверждали с помощью стандартных методик анализа, например микроанализа и спектроскопических характеристик, например МС, ИК (инфракрасная спектроскопия), ЯМР. Использующиеся аббревиатуры являются обычными в данной области техники. Не определенные термины обладают общепринятыми значениями. Аббревиатуры: АсОН - уксусная кислотаd - дублет ДБУ -1,8-диазабицикло[5.4.0]ундец-7-ен ДХМ - дихлорметан ДХЭ - 1,2-дихлорэтан ДИПЭА - диизопропилэтиламин ДМФ - N,N-диметилформамид ДМИ - 1,3-диметил-2-имидазолидинон ДМСО - диметилсульфоксид ДСК - дифференциальная сканирующая калориметрия ЭДКД - 1-этил-3-(3'-диметиламинопропил)карбодиимидEtOH - этанол ч - час(ы) катализатор Граббса 2-го (1,3-бис(2,4,6-триметилфенил)-2-имидазолидинилипоколения ден)дихлор(фенилметилен)(трициклогексилфосфин)ру тений,бензилиден[1,3-бис(2,4,6-триметилфенил)-2-имидазолидинилиден]дихлор(трициклогексилфосфин)рутений,[1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден]дихлор(фенилметилен)(трициклогексилфосфин)ру тений ВЭЖХ - высокоэффективная жидкостная хроматография ЖХ-МС - жидкостная хроматография и масс-спектрометрия МеОН - метанолPEPPSi-iPr - усиленный пиридином предварительный катализатор получения, стабилизации и инициирования - 2,6-диизопропилфенилимидазолийхлорид част./млн частей на миллион ПП -на полимерной подложке РЕАХ - РЕ-анионообменная смола (например, Isolute, колонки РЕ-AX, выпускающиеся фирмойRt - время удерживания КТ - комнатная температураSCX-2 - сильная катионообменная смола (например, Isolute SCX-2 колонки, выпускающиеся фирмой Biotage) ТГФ - тетрагидрофуран В приведенных ниже примерах соединения предпочтительных вариантов осуществления синтезировали по методикам, описанным в настоящем изобретении, или по другим методикам, которые известны в данной области техники. Различные исходные вещества, промежуточные продукты и соединения предпочтительных вариантов осуществления можно выделить и очистить, если это необходимо, по обычным методикам, таким как осаждение, фильтрование, кристаллизация, выпаривание, перегонка и хроматография. Если не указано иное, все исходные вещества получали от коммерческих поставщиков и использовали без дополнительной очистки. Соли можно получить из соединений по известным методикам получения солей. Следует понимать, что органические соединения, предлагаемые в предпочтительных вариантах осуществления, могут обладать таутомерией. Поскольку химические структуры в настоящей заявке могут описывать только одну из возможных таутомерных форм, следует понимать, что предпочтительные варианты осуществления включают любую таутомерную форму представленной структуры. Если не указано иное, условия проведения аналитической ВЭЖХ являются следующими: Методика 2minLCv001 Колонка Waters ВЕН С 18 1002,1 мм, 1,7 мкм Температура колонки 50 С Элюенты А: Н 2 О, В: ацетонитрил, оба содержат 0,1% ТФК (трифторуксусная кислота) Скорость потока 0,7 мл/мин Градиентный режим 0,25 мин 5% В; от 5 до 95% В за 1,00 мин, 0,25 мин 95% В Методика 2 minLCv002 Колонка Waters ВЕН С 18 502,1 мм, 1,7 мкм Температура колонки 50 С Элюенты А: Н 2 О, В: метанол, оба содержат 0,1% ТФК Скорость потока 0,8 мл/мин Градиентный режим 0,20 мин 5% В; от 5 до 95% В за 1,30 мин, 0,25 мин 95% В Методика 2minLCv003 Колонка Waters ВЕН С 18 502,1 мм, 1,7 мкм Температура колонки 50 С Элюенты А: Н 2 О, В: ацетонитрил, оба содержат 0,1% ТФК Скорость потока 0,8 мл/мин Градиентный режим 0,20 мин 5% В; от 5 до 95% В за 1,30 мин, 0,25 мин 95% В Методика LowpH30v001 Колонка Phenomenex Gemini C18 504,6 мм, 3,0 мкм Температура колонки 40 С Элюенты А: Н 2 О, В: ацетонитрил, оба содержат 0,1% ТФК Скорость потока 1,2 мл/мин Градиентный режим от 30 до 95% В за 2,0 мин, 0,2 мин 95% В Методика 2minLC30v003 Колонка Waters ВЕН С 18 502,1 мм, 1,7 мкм Температура колонки 50 С Элюенты А: Н 2 О, В: ацетонитрил, оба содержат 0,1% ТФК Скорость потока 0,8 мл/мин Градиентный режим 0,25 мин 30% В; от 30 до 95% В за 1,00 мин, 0,25 мин 95% В Методика 2minLowpH Колонка: Waters Acquity CSH 1,7 мкм, 2,150 мм Температура: 50 С Подвижная фаза: А: Вода + 0,1% муравьиной кислоты В: ацетонитрил +0,1 % муравьиной кислоты Скорость потока: 1,0 мл/мин Градиентный режим: 0,0 мин 5%В, 0,2-1,3 мин 5-98%В, 1,3-1,55 мин 98%В, 1,55-1,6 мин 98-5%В Методика 10 minLCv003 Колонка Waters ВЕН С 18 502,1 мм, 1,7 мкм Температура колонки 50 С Элюенты А: Н 2 О, В: ацетонитрил, оба содержат 0,1% ТФК Скорость потока 0,8 мл/мин Градиентный режим 0,20 мин 5% В; от 5 до 95% В за 7,80 мин, 1,00 мин 95% В Методика А. Колонка: HSS Т 3 1,8 мкм 2,150 мм Температура колонки: 50 С Элюенты: А: Н 2 О + 0,05% муравьиной кислоты + 3,75 мМ ацетат аммония, В: ацетонитрил + 0,04% муравьиной кислоты Скорость потока: 1,2 мл/мин Градиентный режим: 0,0 мин 2% В, 2-98% В за 1,40 мин, 1,40 мин-2,15 мин 98% В Методика OJ20MEOH Колонка: Chiralcel OJ-H 25010 мм, 5 мкм Подвижная фаза: 20% метанол/80% СО 2 Скорость потока: 10 мл/мин Детектирование: УФ при 220 нм Методика AS25IPA Колонка: Chirapak AS-H 25010 мм, 5 мкм Подвижная фаза: 25% ИПС (изопропиловый спирт)/75% СО 2 Скорость потока: 10 мл/мин Детектирование: УФ при 220 нм Методика AD40IPA Колонка Chirapak AD-H 25010 мм внутренний диаметр, 5 мкм Подвижная фаза: 10% метанол/90% СО 2 Скорость потока 10 мл/мин Детектирование: УФ при 220 нм Методика В. Колонка Zorbax Eclipse XDB-C18 4,650 мм, 1,8 мкм Температура колонки 35 С Элюенты А: Н 2 О + 0,1% ТФК, В: ацетонитрил + 0,1% ТФК Скорость потока 1 мл/мин Градиентный режим 5-100% MeCN (6 мин), 100 MeCN (1,5 мин), 100-5% MeCN (0,5 мин) Методика С. Колонка: Chiralcel OJ-H 25010 мм, 5 мкм Подвижная фаза: 15% метанол/85% СО 2 Скорость потока: 10 мл/мин Детектирование: УФ при 220 нм Примеры соединений, предлагаемых в настоящем изобретении, включают: Получение конечных соединений Пример 1.1 7-(6,7-Дифенил-3,4-дигидро-1,8-нафтиридин-1(2 Н)-ил)гептановая кислота Стадия 1: Этил-7-(6,7-дифенил-3,4-дигидро-1,8-нафтиридин-1(2 Н)-ил)гептаноат Раствор 6,7-дифенил-1,2,3,4-тетрагидро-[1,8]нафтиридина (промежуточный продукт В) (200 мг,0,698 ммоль) в сухом NMP (1 мл) в атмосфере N2 обрабатывали карбонатом цезия (910 мг, 2,79 ммоль) и этил-7-бромгептаноатом (0,544 мл, 2,79 ммоль). Реакционную смесь перемешивали при 120 С в течение 1 ч и в течение еще 3 ч при 140 С. После охлаждения до комнатной температуры смесь подвергали распределению между EtOAc и водой. Органическую порцию промывали рассолом, сушили над MgSO4,фильтровали и концентрировали в вакууме. Очистка неочищенного продукта с помощью хроматографии на диоксиде кремния при элюировании смесью 4:1 изогексан/EtOAc давала остаток в виде розового масла. Остаток загружали в картридж Isolute SCX-2 и элюировали с помощью МеОН, затем с помощью 2 М NH3 в МеОН. Метанольно-аммиачные фракции концентрировали в вакууме и сушили в вакууме при 40 С и получали искомое соединение в виде бесцветного масла. ЖХ-МС Rt =1,54 мин; [М+Н]+443,4, Методика 2minLCv001. 1 Н ЯМР (400 МГц, ДМСО-d6)7,3 (9 Н, m), 7,0 (2 Н, m), 4,0 (2 Н, q), 3,6 (2 Н, m), 3,4 ( 2 Н, m), 2,75(2 Н, m), 2,2 (2 Н, t), 1,9 (2 Н, т ), 1,6 (2 Н, m), 1,45 (2 Н, m), 1,3 (4 Н,т), 1,1 (3 Н, t). Стадия 2: 7-(6,7-Дифенил-3,4-дигидро-1,8-нафтиридин-1(2 Н)-ил)гептановая кислота Раствор этил-7-(6,7-дифенил-3,4-дигидро-1,8-нафтиридин-1(2 Н)-ил)гептаноата (стадия 1) (100 мг,0,226 ммоль) и гидроксида лития (37,9 мг, 0,904 ммоль) в ТГФ (2 мл) нагревали при 75 С в течение 6 ч. Реакцию останавливали водой и значение рН доводили до 3-4 путем добавления 1 М НС 1. Смесь подвергали распределению между EtOAc и водой. Органическую порцию отделяли и промывали рассолом, сушили над MgSO4, фильтровали и концентрировали в вакууме. Очистка неочищенного продукта с помощью хроматографии на диоксиде кремния при элюировании смесью 3:2 изогексан/EtOAc давала искомое соединение. ЖХ-МС Rt =1,98 мин; [М+Н]+ 415,5, Методика LowpH30v001. 1 Н ЯМР (400 МГц, ДМСО-d6)12,03 (1 Н, br s), 7,2 (9 Н, m), 7,05 (2 Н, m), 3,6 (2 Н, t), 3,4 (2 Н, m), 2,8(2 Н, m), 2,15 (2 Н, t), 1,9 (2 Н, m), 1,6 (2 Н, m), 1,4 (2 Н, m), 1,3(4 Н,m). Соединения, приведенные в примерах, представленных ниже в таблице (табл. 2), получали по методике, сходной с использованной в примере 1.1, путем замены этил-7-бромгептаноата на соответствующий сложный бромэфир. Таблица 2POCl3 (10 мл, 107 ммоль) при 0 С по каплям добавляли к смеси 6,7-дифенил-1,8-нафтиридин-1 оксида и 2,3-дифенил-1,8-нафтиридин-1-оксида (промежуточные продукты С) (3 г, 10,06 ммоль). Реакционной смеси давали нагреться до комнатной температуры и нагревали при 100 С в течение 2 ч. Смесь осторожно выливали в смесь лед/вода и значение рН доводили до 8-9 путем проводимого порциями добавления Na2CO3 (твердый). Водный слой отделяли и экстрагировали с помощью ДХМ (3150 мл). Органические порции объединяли и промывали рассолом, сушили над MgSO4, фильтровали и концентрировали в вакууме и получали коричневое масло. Очистка неочищенного масла с помощью хроматографии на диоксиде кремния при элюировании с помощью 0-50% EtOAc в изогексане давала 7-хлор-2,3 дифенил-1,8-нафтиридин и 5-хлор-2,3-дифенил-1,8-нафтиридин: 7-Хлор-2,3-дифенил-1,8-нафтиридин: желтое твердое вещество ЖХ-МС Rt = 1,58 мин, [М+Н]+ 317,1, Методика 2minLCv002 1 Н ЯМР (400 МГц, ДМСО-d6)8,63 (1 Н, d), 8,6 (1 Н, s), 7,78 (1 Н d), 7,28-7,7,43 (10 Н, m). 5-Хлор-2,3-дифенил-1,8-нафтиридин: бежевое твердое вещество ЖХ-МС Rt = 1,64 мин, [М+Н]+317,1, Методика 2minLCv002 1 Н ЯМР (400 МГц, ДМСО-d6)9,09 (1 Н, d), 8,52 (1 Н, s), 7,93 (1 Н, d), 7,37-7,5 (10 Н, m). 7-Хлор-2,3-дифенил-1,8-нафтиридин, искомый продукт, использовали на следующей стадии. Стадия 2: Этил-6-(6,7-дифенил-1,8-нафтиридин-2-ил)гексаноат
МПК / Метки
МПК: C07D 471/04, A61P 11/00, A61P 9/00, A61K 31/4375
Метки: гетероциклические, рецептора, агонисты, соединения
Код ссылки
<a href="https://eas.patents.su/30-22046-geterociklicheskie-soedineniya-agonisty-receptora-ip.html" rel="bookmark" title="База патентов Евразийского Союза">Гетероциклические соединения, агонисты рецептора ip</a>
Конденсированные гетероциклические соединения пиримидинона и их применение в качестве агониста ангиотензина ii и агониста γ-рецептора, активирующего пролиферацию пероксисом

Номер патента: 19823
Опубликовано: 30.06.2014
Авторы: Маекава Цуеси, Игава Хидеюки
МПК: C07D 487/04, A61K 31/519, A61P 9/00...
Метки: ангиотензина, агониста, гетероциклические, конденсированные, качестве, соединения, активирующего, пероксисом, пиримидинона, пролиферацию, применение, gamma;-рецептора
Формула / Реферат:
1. Соединение, представленное формулой (I)где R1 представляет собой атом водорода, C1-6алкил, необязательно замещенный 1-5 заместителями, выбранными из указанной ниже группы А заместителей, 3-10-членную неароматическую циклическую группу, необязательно замещенную 1-5 заместителями, выбранными из указанной ниже группы С заместителей, или 5- или 6-членную ароматическую циклическую группу, необязательно замещенную 1-5 заместителями, выбранными из...
Селективные пептидные агонисты рецептора vpac2

Номер патента: 12930
Опубликовано: 26.02.2010
Авторы: Чжан Ляньшань, Алсина-Фернандес Хорхе, Боквист Бенгт Кристер
МПК: A61K 47/48, A61K 38/00, C07K 14/575...
Метки: агонисты, пептидные, рецептора, vpac2, селективные
Формула / Реферат:
1. Пегилированный пептидный агонист рецептора VPAC2, содержащий последовательность, выбранную изи С-концевой удлиняющий сегмент, где N-конец С-концевого удлиняющего сегмента связан с С-концом пептидной последовательности и где С-концевой удлиняющий сегмент содержит последовательность аминокислот формулыгде Xaa1 представляет собой Gly, Cys или отсутствует;Хаа2 представляет собой Gly, Arg или отсутствует;Хаа3 представляет собой Pro, Thr или...
Агонисты β2-адренергического рецептора

Номер патента: 4263
Опубликовано: 26.02.2004
Авторы: Моран Эдмунд Дж., Чои Сеок-Ки
МПК: A61P 11/00, A61K 31/165, A61K 31/135...
Метки: агонисты, рецептора, beta;2-адренергического
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль. 2. Соединение формулы где стереохимия при *C и **C представляет (RS) и (RS); (R) и (R); (R) и (S); (S) и (R) или (S) и (S); или его фармацевтически приемлемая соль. 3. Соединение по п.2, где стереохимия при *C представляет (R) и стереохимия при **C представляет (R). 4. Соединение по п.2, где стереохимия при *C представляет (R) и стереохимия при **C представляет (S). 5....
Агонисты рецептора меланокортина

Номер патента: 19146
Опубликовано: 30.01.2014
Авторы: Ли Хиун Хо, Ли Хиун Мин, Чои Сунг Пил, Ли Коо, Ахн Ин Ае, Шим Донг Суп, Моон Санг Пил, Чунг Соо Йонг, Ли Санг -Дае
МПК: C07D 403/14
Метки: меланокортина, рецептора, агонисты
Формула / Реферат:
1. Соединение следующей формулы 1где R1 представляет собой водород или представляет собой C1-С10алкил, С3-С7циклоалкил, С6-С10арил, гетероцикл или гетероарил, каждый из которых является незамещенным или замещен по меньшей мере одним заместителем, выбранным из группы, состоящей из галогена, амино, С1-С4алкила, трифторметила, С1-С4алкокси, циано и оксо;R2 представляет собой фенил или шестичленный гетероарил, каждый из которых является незамещенным...
Агонисты в2-адренергического рецептора

Номер патента: 3453
Опубликовано: 26.06.2003
Авторы: Моран Эдмунд Дж., Чои Сеок-Ки
МПК: A61K 39/395, A61K 38/00, A61K 39/00...
Метки: рецептора, в2-адренергического, агонисты
Формула / Реферат:
1. Соединение формулы (II) где Ar1 и Ar3 независимо выбирают из группы, включающей арил, гетероарил и гетероциклил; Ar2 выбирают из группы, включающей фенилен, где группы W и X присоединены в 1,2-, 1,3 и 1,4 положениях фениленового кольца; циклогексилен, необязательно замещенный метилом, где группы W и X присоединены в 1,3 и 1,4 положениях циклогексиленового кольца, и пиперазинилен, где группы W и X присоединены в 1,4 положениях...
Предыдущий патент: Способ конверсии синтез-газа в высшие углеводороды (варианты)
Следующий патент: Биоразлагаемая полимерная нить и способ ее получения
Случайный патент: Способ и система для выращивания растений