Ингибиторы вируса гепатита с
Номер патента: 21260
Опубликовано: 29.05.2015
Авторы: Белема Маконен, Хаманн Лоренс Г., Чэнь Ци, Лопез Омар Д.







Формула / Реферат
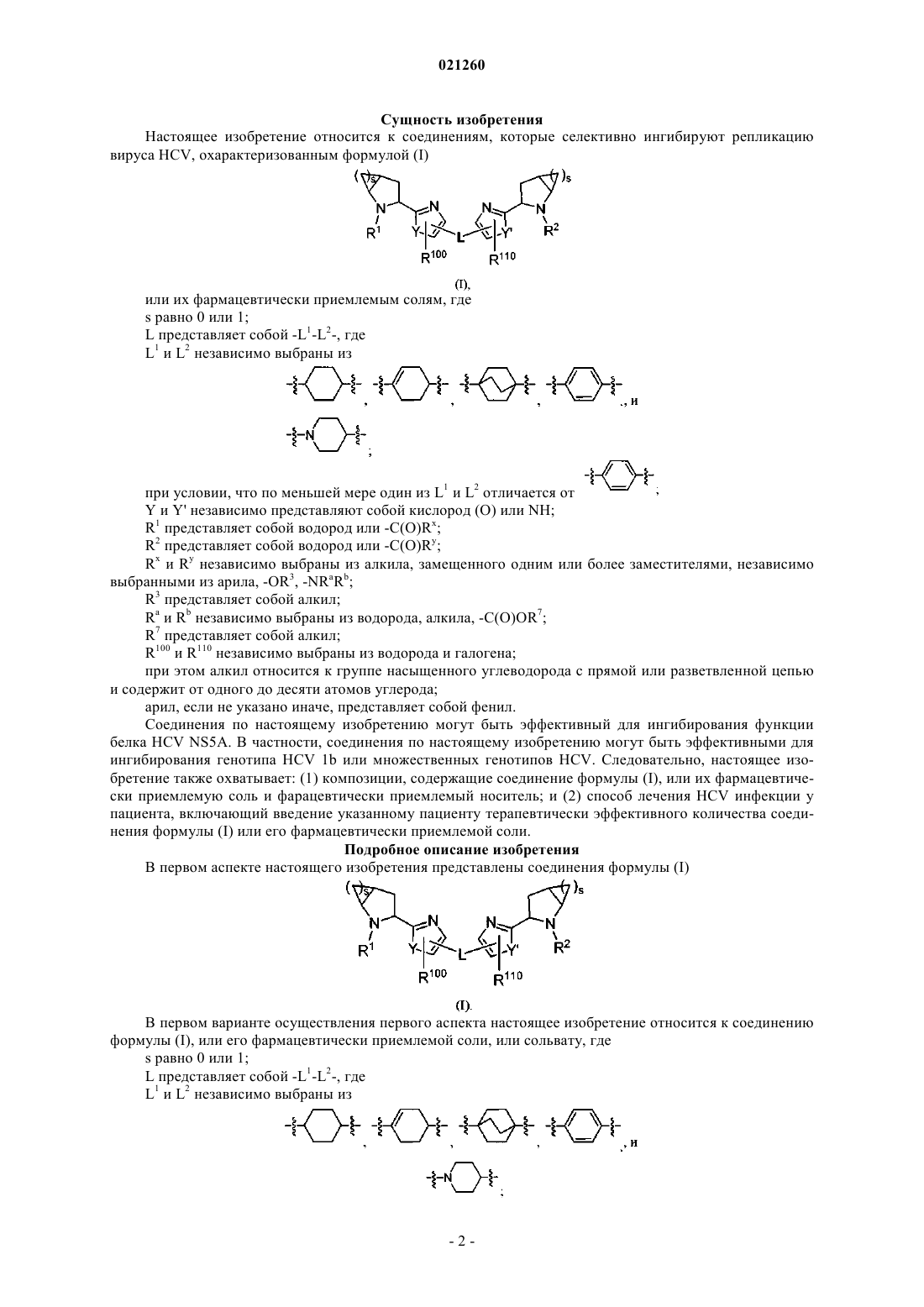
1. Соединение формулы (I)

или его фармацевтически приемлемая соль, где
s равно 0 или 1;
L представляет собой -L1-L2-, где
L1 и L2 независимо выбраны из
 при условии, что по меньшей мере один из L1 и L2 отличается от
при условии, что по меньшей мере один из L1 и L2 отличается от
Y и Y' независимо представляют собой кислород (О) или NH;
R1 представляет собой -C(O)Rx;
R2 представляет собой -C(O)Ry;
Rx и Ry независимо выбраны из алкила, независимо замещенного одним или более заместителями, независимо выбранными из арила, -OR3, -NRaRb;
R3 представляет собой алкил;
Ra и Rb независимо выбраны из водорода, алкила, -C(O)OR7;
R7 представляет собой алкил;
R100 и R110 независимо выбраны из водорода и галогена;
при этом алкил относится к группе насыщенного углеводорода с прямой или разветвленной цепью и содержит от одного до десяти атомов углерода;
арил представляет собой фенил.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где L выбран из


3. Соединение по п.1 или его фармацевтически приемлемая соль, где Y и Y', каждый, представляют собой NH.
4. Соединение по п.1 или его фармацевтически приемлемая соль, где Y представляет собой кислород (О) и Y' представляет собой NH.
5. Соединение по п.1 или его фармацевтически приемлемая соль, где
R1 представляет собой -С(О)Rx;
R2 представляет собой -С(О)Ry;
Rx и Ry независимо представляют собой алкил, замещенный по меньшей мере одним -NRaRb, охарактеризованный формулой (А)

где m равно 0 или 1;
R8 представляет собой водород или алкил;
R9 выбран из алкила, необязательно замещенного заместителем, выбранным из арила, и -OR3;
R100 и R110 независимо выбраны из водорода и галогена;
R3, Ra и Rb определены в п.1.
6. Соединение по п.5 или его фармацевтически приемлемая соль, где
m равно 0;
R8 представляет собой водород или C1-C4-алкил;
R9 выбран из водорода, C1-C6-алкила;
Ra представляет собой водород или C1-C4-алкил;
Rb представляет собой C1-C4-алкил, -C(O)OR7;
R7 представляет собой C1-C4-алкил.
7. Соединение по п.5 или его фармацевтически приемлемая соль, где
m равно 0;
R8 представляет собой водород;
R9 представляет собой фенил.
8. Соединение по п.5 или его фармацевтически приемлемая соль, где
m равно 1;
R8 представляет собой водород;
R9 представляет собой C1-C6-алкил;
Ra представляет собой водород;
Rb представляет собой -C(O)OR4;
R4 представляет собой C1-C6-алкил.
9. Соединение по п.1 или его фармацевтически приемлемая соль, где
R1 представляет собой -C(O)Rx;
R2 представляет собой -C(O)Ry;
Rx и Ry, оба, представляют собой т-бутокси.
10. Соединение формулы (II)

или его фармацевтически приемлемая соль, где
s равно 0 или 1;
L представляет собой -L1-L2-, где
L1 и L2 независимо выбраны из

 при условии, что по меньшей мере один из L1 и L2 отличается от
при условии, что по меньшей мере один из L1 и L2 отличается от
Y и Y' независимо представляют собой кислород (О) или NH;
R1 представляет собой -C(O)Rx;
R2 представляет собой -C(O)Ry;
Rx и Ry независимо выбраны из алкила, независимо замещенного одним или более заместителями, независимо выбранными из арила, OR3, -NRaRb;
R3 представляет собой алкил;
Ra и Rb независимо выбраны из водорода, алкила и -C(O)OR7;
R7 представляет собой алкил;
при этом алкил относится к группе насыщенного углеводорода с прямой или разветвленной цепью и содержит от одного до десяти атомов углерода;
арил представляет собой фенил.
11. Соединение или его фармацевтически приемлемая соль, выбранное из группы, состоящей из



и их соответствующих стереоизомеров.
12. Фармацевтическая композиция для ингибирования функции белка NS5A, содержащая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Текст
ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С Настоящее изобретение относится к новым соединениям формулы (I), как определено в описании, и композициям, содержащим такие новые соединения. Эти соединения являются полезными противовирусными средствами, в особенности при ингибировании функции белкаNS5A, кодируемого вирусом гепатита С (HCV) Область техники Настоящее описание в основном направлено на противовирусные соединения и, более конкретно,направлено на соединения, которые обладают способностью ингибировать функцию белка NS5A, кодируемого вирусом гепатита С (HCV), композиции, содержащие такие соединения, и способы ингибирования функции белка NS5A. Уровень техникиHCV является основным патогеном человека, инфицирующим приблизительно 170 миллионов человек во всем мире - примерно в пять раз больше инфицированных вирусом иммунодефицита 1 типа. У значительной части этих людей, инфицированных HCV, развивается тяжелое прогрессирующее заболевание печени, в том числе цирроз и гепатоклеточная карцинома. В настоящее время стандартное лечение HCV, в котором используется комбинация пэгилированного интерферона и рибавирина, имеет неоптимальный показатель эффективности в достижении устойчивого вирусологического ответа и вызывает многочисленные побочные эффекты. Таким образом, существует очевидная и назревшая потребность в разработке эффективных терапевтических средств, чтобы направить их на эти недостаточно удовлетворенные медицинские нужды.HCV представляет собой вирус с положительной цепью РНК. На основании сравнения расшифрованной аминокислотной последовательности и большого сходства в 5' нетранслируемой области, HCV был классифицирован как отдельный род в семействе флавивирусов (Flaviviridae). Все представители семейства флавивирусов имеют вирионы, заключенные в оболочку, которые содержат геном положительную цепь РНК, кодирующий все известные вирус-специфичные белки посредством трансляции одной непрерывной открытой рамки считывания. Значительная гетерогенность обнаружена в пределах нуклеотидной и кодируемой аминокислотной последовательности по всему геному HCV вследствие высокой частоты появления ошибки кодируемой РНК-зависимой РНК полимеразы, которая лишена способности проверочного считывания. Было охарактеризовано по меньшей мере шесть основных генотипов и более 50 подтипов было описано по всему миру. Клиническая значимость генетической гетерогенности HCV продемонстрировала способность к возникновению мутаций во время монотерапевтического лечения, следовательно, для применения желательны дополнительные варианты лечения. Возможное модулирующее действие генотипов на патогенез и лечение остается неясным. Одноцепочечный РНК геном HCV составляет приблизительно 9500 нуклеотидов в длину и имеет одну открытую рамку считывания (ORF), кодирующую один крупный полипротеин примерно 3000 аминокислот. В инфицированных клетках этот полипротеин расщепляется во множестве сайтов под действием клеточных и вирусных протеаз для образования структурных и неструктурных (NS) белков. В случае HCV образование зрелых неструктурных белков (NS2, NS3, NS4A, NS4B, NS5A и NS5B) осуществляется под действием двух вирусных протеаз. Считается, что первая является металлопротеазой и расщепляет в месте соединения NS2-NS3; вторая представляет собой сериновую протеазу, находящуюся вN-концевой области NS3 (также называемая в настоящем описании NS3 протеазой), и опосредует все последующие расщепления ниже NS3 как в цис, в NS3-NS4A сайте расщепления, так и в транс для оставшихся сайтов NS4A-NS4B, NS4B-NS5A, NS5A-NS5B. Белок NS4A, по-видимому, выполняет множество функций, действуя и как кофактор для NS3 протеазы и способствуя мембранной локализации NS3 и других компонентов вирусной репликазы. Образование комплекса NS3-NS4A необходимо для надлежащей активности протеазы, приводящей в результате к повышенной протеолитической эффективности событий расщепления. Белок NS3 также проявляет нуклеозидтрифосфатазную и РНК геликазную активности. NS5B (в настоящем изобретении также называется HCV полимеразой) представляет собой РНКзависимую РНК полимеразу, которая вовлечена в репликацию HCV с другими белками HCV, в том числеNS5A, в репликазном комплексе. Необходимы соединения, применяемые для лечения HCV-инфицированных пациентов, которые селективно ингибируют репликацию вируса HCV. В частности, необходимы соединения, которые эффективно ингибируют функцию белка NS5A. Белок NS5A HCV описан, например, в следующих ссылках: Сущность изобретения Настоящее изобретение относится к соединениям, которые селективно ингибируют репликацию вируса HCV, охарактеризованным формулой (I) или их фармацевтически приемлемым солям, где при условии, что по меньшей мере один из L1 и L2 отличается отY и Y' независимо представляют собой кислород (О) или NH;R1 представляет собой водород или -C(O)Rx;R2 представляет собой водород или -C(O)Ry;Rx и Ry независимо выбраны из алкила, замещенного одним или более заместителями, независимо выбранными из арила, -OR3, -NRaRb;Ra и Rb независимо выбраны из водорода, алкила, -C(O)OR7;R100 и R110 независимо выбраны из водорода и галогена; при этом алкил относится к группе насыщенного углеводорода с прямой или разветвленной цепью и содержит от одного до десяти атомов углерода; арил, если не указано иначе, представляет собой фенил. Соединения по настоящему изобретению могут быть эффективный для ингибирования функции белка HCV NS5A. В частности, соединения по настоящему изобретению могут быть эффективными для ингибирования генотипа HCV 1b или множественных генотипов HCV. Следовательно, настоящее изобретение также охватывает: (1) композиции, содержащие соединение формулы (I), или их фармацевтически приемлемую соль и фарацевтически приемлемый носитель; и (2) способ лечения HCV инфекции у пациента, включающий введение указанному пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Подробное описание изобретения В первом аспекте настоящего изобретения представлены соединения формулы (I) В первом варианте осуществления первого аспекта настоящее изобретение относится к соединению формулы (I), или его фармацевтически приемлемой соли, или сольвату, где при условии, что по меньшей мере один из L1 и L2 отличается отY и Y' независимо представляют собой кислород (О) или NH;R1 представляет собой водород или -C(O)Rx;R2 представляет собой водород или -C(O)Ry;Rx и Ry независимо выбраны из алкила, независимо замещенного одним или более заместителями,независимо выбранными из арила, -OR3, -NRaRb;Ra и Rb независимо выбраны из водорода, алкила и -C(O)OR7;R100 и R110 независимо выбраны из водорода и галогена; при этом алкил относится к группе насыщенного углеводорода с прямой или разветвленной цепью и содержит от одного до десяти атомов углерода; арил, если не указано иначе, представляет собой фенил. В первом классе соединений первого варианта осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где s равно 0. Во втором классе соединений первого варианта осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где s равно 1. Во втором варианте осуществления первого аспекта настоящее изобретение относится к соединению формулы (I), или его фармацевтически приемлемой соли, или сольвату, где s равно 0 или 1; L выбран из В первом классе соединений во втором варианте осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где L представляет собой Во втором классе соединений второго варианта осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где L представляет собой В третьем классе соединений второго варианта осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где L представляет собой В четвертом классе соединений второго варианта осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где L представляет собой В пятом классе соединений второго варианта осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где L представляет собой В шестом классе соединений второго варианта осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где L представляет собой В седьмом классе соединений второго варианта осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где L представляет собой В восьмом классе соединений второго варианта осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где L представляет собой В девятом классе соединений второго варианта осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где L представляет собой В третьем варианте осуществления первого аспекта настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где Y и Y', каждый, представляют собойNH. В следующем варианте осуществления первого аспекта настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, гдеRx и Ry независимо представляют собой алкил, замещенный по меньшей мере одним -NRaRb, охарактеризованный формулой (А)R8 представляет собой водород или алкил;R100 и R110 независимо выбраны из водорода и галогена. В другом варианте осуществления первого аспекта настоящее изобретение относится к соединению формулы (I), или его фармацевтически приемлемой соли, или сольвату, гдеR8 представляет собой водород или C1-C4-алкил;Ra представляет собой водород или C1-C4-алкил;R7 представляет собой C1-C4-алкил. Еще в одном варианте осуществления первого аспекта настоящее изобретение относится к соединению формулы (I), или его фармацевтически приемлемой соли, или сольвату, гдеRx и Ry, оба, представляют собой т-бутокси. В следующем варианте осуществления первого аспекта настоящее изобретение относится к соединению формулы (II) или его фармацевтически приемлемой соли, где при условии, что по меньшей мере один из L1 и L2 отличается отY и Y' независимо представляют собой кислород (О) или NH;Rx и Ry независимо выбраны из алкила, независимо замещенного одним или более заместителями,независимо выбранными из арила, -OR3, -NRaRb;Ra и Rb независимо выбраны из водорода, алкила и -C(O)OR7;R7 представляет собой алкил,при этом алкил относится к группе насыщенного углеводорода с прямой или разветвленной цепью и содержит от одного до десяти атомов углерода; арил, если не указано иначе, представляет собой фенил. В следующем варианте осуществления первого аспекта настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, выбранному из группы, состоящей из их соответствующих стереоизомеров. Во втором аспекте настоящее изобретение относится к композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Другие аспекты настоящего изобретения могут включать подходящие сочетания вариантов осуществления, описанных в настоящем изобретении. Другие аспекты и варианты осуществления можно найти в представленном здесь описании. Описание настоящего изобретения следует истолковывать в соответствии с законами и принципами образования химических связей. В некоторых случаях может быть необходимо удалить атом водорода для размещения заместителя в любом заданном положении. Некоторые особенности структуры формулы (I) дополнительно проиллюстрированы ниже: В формуле (I), как указано выше, "пирролидиновая группа" с левой стороны "линкера" не зависит от "пирролидиновой группы" с правой стороны, например, применительно к (1) таутомерной форме имидазольного кольца, где Y или Y' представляет собой NH, (2) абсолютной конфигурации стереогенных центров на пирролидиновом кольце и (3) заместителям на атоме азота пирролидина, т.е. R1 и R2 не зависят друг от друга, хотя в некоторых обстоятельствах они предпочтительно являются одинаковыми. Что касается соединения между линкером "L" и пирролидиновыми группами, формула (I) охватывает все следующие возможные комбинации: где Y и Y' независимо представляют собой кислород (О) или NH. В пирролидиновом кольце стереогенный углеродный центр, к которому присоединен пятичленный гетероцикл, может принимать либо (R)-, либо (S)-конфигурацию, как изображено ниже: Когда циклопропильное кольцо конденсировано на пирролидиновом кольце, т.е. когда s равно 1,CH2 группа конденсированного циклопропильного кольца может принимать либо -, либо -положение относительно пирролидинового кольца, как изображено ниже: В формуле (I), когда либо Y, либо Y' представляет собой NH, связь между линкером "L" и полученным в результате имидазольным кольцом может иметь место либо в С-4, либо в С-5 положении (см. ниже) имидазольного кольца. Как было бы понятно специалисту в данной области, вследствие таутомеризации имидазольного кольца, связывание линкера "L" с положением С-4 может быть эквивалентным связыванию линкера с положением С-5, как показано в следующем уравнении: Таким образом, настоящее изобретение охватывает обе возможные связи, даже когда структура изображает только одну из них. В настоящем изобретении плавающая связь или плавающий заместитель (например,-R13) на структуре указывает на то, что эта связь или этот заместитель могут присоединяться к любому доступному положению структуры посредством удаления атома водорода из доступного положения. Должно быть понятно, что в бициклической или полициклической кольцевой структуре, если особым образом не определено иное, положение плавающей связи или плавающего заместителя не ограничивает положение такой связи или заместителя конкретным кольцом. Таким образом, следующие две группы заместителей должны интерпретироваться как эквивалентные: Должно быть понятно, что соединения, охватываемые настоящим изобретением, представляют собой соединения, которые являются достаточно стабильными для применения в качестве фармацевтического средства. Предполагается, что определение любого заместителя или переменной в конкретном положении в молекуле не зависит от его определений в другом месте в этой молекуле. Например, для заместителя (R10)n, когда n равно 2, каждая из двух групп R10 может быть одинаковой или различной. Все патенты, патентные заявки и литературные ссылки, процитированные в описании, включены посредством ссылки в полном объеме. В случае несоответствий настоящее описание, включая определения, будет иметь приоритетное значение. Определения. Определения были представлены выше для каждой из определяемых групп. Кроме того, будут использованы следующие определения. В контексте настоящего изобретения формы единственного числа включают ссылку на формы множественного числа, если из контекста явным образом не следует иное. Если не указано иное, все арильные, циклоалкильные, гетероарильные и гетероциклильные группы в соответствии с настоящим изобретением могут быть замещены, как описано в каждом из соответствующих им определений. Например, арильная часть арилалкильной группы может быть замещена, как описано в определении термина "арил". Термин "ацетил" в контексте настоящего изобретения относится к -С(О)СН 3. Термин "алкенил" в контексте настоящего изобретения относится к моновалентной, прямой или разветвленной углеводородной цепи, имеющей одну или более двойных связей. Двойная связь алкенильной группы может быть неконъюгированной или конъюгированной с другой ненасыщенной группой. Подходящие алкенильные группы включают, но не ограничиваются ими, C2-C10-алкенильные группы,такие как винил, аллил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил, 2 этилгексенил, 2-пропил-2-бутенил, 4-(2-метил-3-бутен)пентенил. Алкенильная группа может быть незамещенной или замещенной одним или двумя подходящими заместителями. Термин "алкокси" в контексте настоящего изобретения относится к алкильной группе, присоединенной к исходной части молекулы через атом кислорода. Характерные примеры алкоксигруппы включают, но не ограничиваются ими, метокси (CH3O-), этокси (СН 3 СН 2 О-) и т-бутокси CH3)3 СО-). Термин "алкил" в контексте настоящего изобретения относится к группе, происходящей из насыщенного углеводорода с прямой или разветвленной цепью, путем удаления атома водорода от одного из насыщенных атомов углерода. Алкильная группа предпочтительно содержит от одного до десяти атомов углерода. Характерные примеры алкильной группы включают, но не ограничиваются ими, метил, этил,изопропил и трет-бутил. Термин "алкилкарбонил" в контексте настоящего изобретения относится к алкильной группе, присоединенной к исходной части молекулы через карбонильную группу. Характерные примеры алкилкарбонильной группы включают, но не ограничиваются ими, ацетил (-С(О)CH3), пропаноил (-C(O)CH2CH3),н-бутирил (-C(O)CH2CH2CH3) и 2,2-диметилпропаноил или пивалоил (-С(О)С(СН 3)3). Термин "аллил" в контексте настоящего изобретения относится к -СН 2 СН=СН 2 группе. Термин "арил" в контексте настоящего изобретения относится к группе, происходящей из ароматического карбоцикла, путем удаления атома водорода из ароматического кольца. Арильная группа может быть моноциклической, бициклической или полициклической, где в бициклической или полициклической арильной группе ароматический карбоцикл может быть конденсирован с другим четырехшестичленным ароматическим или неароматическим карбоциклом. Характерные примеры арильных групп включают, но не ограничиваются ими, фенил, инданил, инденил, нафтил и 1,2,3,4-тетрагидронафт 5-ил. Термин "арилалкил" в контексте настоящего изобретения относится к алкильной группе, замещенной одним, двумя или тремя арильными группами, где арильная часть арилалкильной группы необязательно может быть замещена одним-пятью заместителями, независимо выбранными из C1-C6-алкила, C1C4-галоалкила, C1-C6-алкокси, галогена, циано и нитрогрупп. Характерные примеры арилалкила включают, но не ограничиваются ими, бензил, 2-фенил-1-этил (PhCH2CH2-), (нафт-1-ил)метил и (нафт-2 ил)метил. Термин "бензил" в контексте настоящего изобретения относится к метильной группе, на которой один из атомов водорода замещен фенильной группой, где указанная фенильная группа необязательно может быть замещена одним-пятью заместителями, независимо выбранными из метила, трифторметила(-CF3), метокси (-OCH3), галогена и нитро (-NO2). Характерные примеры бензильной группы включают,но не ограничиваются ими, PhCH2-, 4-МеО-С 6 Н 4 СН 2- и 2,4,6-триметил-С 6 Н 4 СН 2-. Термины "Кэп" и "кэп" в контексте настоящего изобретения относятся к группе, которая расположена на атоме азота пирролидинового кольца в соединениях формулы (I). Должно быть понятно, что"Кэп" или "кэп" также может относиться к реагенту, который является предшественником конечного"кэпа" в соединениях формулы (I), и используется в качестве одного из исходных веществ в реакции,чтобы присоединить группу к атому азота пирролидина, что в результате приводит к получению конечного продукта, соединения, которое содержит функционализированный пирролидин, который будет присутствовать в соединении формулы (I). Термин "карбонил" в контексте настоящего изобретения относится к -С(О)-. Термин "карбоксил" в контексте настоящего изобретения относится к -СО 2 Н. Термин "циано" в контексте настоящего изобретения относится к -CN. Термин "циклоалкил" в контексте настоящего изобретения относится к группе, полученной из насыщенного карбоцикла, имеющей предпочтительно от трех до восьми атомов углерода, путем удаления атома водорода из насыщенного карбоцикла, где насыщенный карбоцикл необязательно может быть конденсирован с одним или двумя другими ароматическими или неароматическими карбоциклами. Характерные примеры циклоалкильных групп включают, но не ограничиваются ими, циклопропил, циклопентил, циклогексил и 1,2,3,4-тетрагидронафт-1-ил. Термин "формил" в контексте настоящего изобретения относится к -СНО. Термины "гало" и "галоген" в контексте настоящего изобретения относятся к F, Cl, Br или I. Термин "галоалкокси" в контексте настоящего изобретения относится к галоалкильной группе, присоединенной к исходной части молекулы через атом кислорода. Термин "галоалкил" в контексте настоящего изобретения относится к алкильной группе, замещенной по меньшей мере одним атомом галогена. Галоалкильная группа может представлять собой алкильную группу, в которой все атомы водорода замещены галогенами. Характерные примеры галоалкила включают, но не ограничиваются ими, трифторметил (CF3-), 1-хлорэтил (ClCH2CH2-) и 2,2,2 трифторэтил (CF3CH2-). Термин "гетероарил" в контексте настоящего изобретения относится к группе, происходящей из моноциклического, бициклического или полициклического соединения, содержащего по меньшей мере одно ароматическое кольцо, содержащее один или более гетероатомов, предпочтительно от одного до трех гетероатомов, независимо выбранных из азота, кислорода и серы, путем удаления атома водорода из их ароматического кольца. Как хорошо известно специалистам в данной области, гетероарильные кольца имеют менее ароматический характер, чем их полностью углеродные эквиваленты. Таким образом, для целей настоящего изобретения гетероарильной группе нужно иметь некоторую степень ароматичности. Иллюстративные примеры гетероарильных групп включают, но не ограничиваются ими, пиридил, пиридазинил, пиримидил, пиразил, триазинил, пирролил, пиразолил, имидазолил, (1,2,3)- и (1,2,4)-триазолил,пиразинил, пиримидинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, изоксазолил, оксазолил,индолил, хинолинил, изохинолинил, бензизоксазолил, бензотиазолил, бензотиенил и пирролопиридинил. Термин "гетероарилалкил" в контексте настоящего изобретения относится к алкильной группе, замещенной одним, двумя или тремя гетероарильными группами. Термин "гетеробициклил" в контексте настоящего изобретения относится к кольцевой структуре,содержащей два конденсированных кольца или кольца с внутренним мостиком, которая включает в себя атом углерода и один или более гетероатомов, независимо выбранных из азота, кислорода и серы в качестве атома (атомов) кольца. Гетеробициклическая кольцевая структура является подгруппой гетероциклического кольца и может быть насыщенной или ненасыщенной. Примеры гетеробициклических кольцевых структур включают тропан, хинуклидин и 7-азабицикло[2.2.1]гептан. Термин "гетероциклил" в контексте настоящего изобретения относится к группе, происходящей из моноциклического, бициклического или полициклического соединения, содержащего по меньшей мере одно неароматическое кольцо, содержащее один или более гетероатомов, предпочтительно от одного до трех гетероатомов, независимо выбранных из азота, кислорода и серы, путем удаления атома водорода из неароматического кольца. Гетероциклильная группа охватывает гетеробициклильную группу. Гетероциклильные группы по настоящему изобретению могут быть присоединены к исходной части молекулы через атом углерода или атом азота в этой группе. Примеры гетероциклильных групп включают, но не ограничиваются ими, морфолинил, оксазолидинил, пиперазинил, пиперидинил, пирролидинил, тетрагидрофурил, тиоморфолинил и индолинил. Термин "гетероциклилалкил" в контексте настоящего изобретения относится к алкильной группе,замещенной одним, двумя или тремя гетероциклильными группами. Термины "гидрокси" или "гидроксил" в контексте настоящего изобретения относятся к -ОН. Термин "нитро" в контексте настоящего изобретения относится к -NO2. Термин "-NRaRb" в контексте настоящего изобретения относится к двум группам, Ra и Rb, которые присоединены к исходной части молекулы через атом азота, или, альтернативно, Ra и Rb, вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членное кольцо или конденсированную или имеющую внутренний мостик бициклическую кольцевую структуру, необязательно содержащую один,два или три дополнительных гетероатома, независимо выбранных из азота, кислорода и серы. Термин"-NRcRd" определяется аналогично. Термин "(NRaRb)алкил" в контексте настоящего изобретения относится к алкильной группе, замещенной одной, двумя или тремя группами -NRaRb. Термин "(NRcRd)алкил" определяется аналогичным образом. Термин "оксо" в контексте настоящего изобретения относится к =О. Термин "сульфонил" в контексте настоящего изобретения относится к -SO2-. В соединениях по настоящему изобретению должно быть понятно, что, когда s равно 0, результатом является соединение формулы (Z), показанное ниже: В соединениях по настоящему изобретению существуют асимметричные центры. Эти центры обозначаются символами "R" или "S", в зависимости от конфигурации заместителей вокруг хирального атома углерода. Должно быть понятно, что настоящее изобретение охватывает все стереохимические изомерные формы или их смеси, которые обладают способностью ингибировать NS5A. Отдельные стереоизомеры соединений могут быть получены синтетическим путем из коммерчески доступных исходных веществ, которые содержат хиральные центры, или путем получения смесей энантиомерных продуктов с последующим их разделением, например превращением в смесь диастереомеров с последующим разделением или рекристаллизацией, методами хроматографии или прямым разделением энантиомеров на хиральных хроматографических колонках. Исходные соединения конкретной стереохимии либо являются коммерчески доступными, либо могут быть получены и разделены способами, известными в данной области. Некоторые соединения по настоящему изобретению также могут существовать в различных стабильных конформационных формах, которые могут быть разделимыми. Торсионная асимметрия вследствие ограниченного вращения около асимметричной одинарной связи, например вследствие стерического несоответствия или напряжения кольца, может дать возможность разделить различные конформационные изомеры. Настоящее изобретение включает каждый конформационный изомер этих соединений и их смеси. Термин "соединения по настоящему изобретению" и эквивалентные выражения охватывают соединения формулы (I) и фармацевтически приемлемые энантиомеры, диастереомеры и их соли. Аналогично,ссылки на производные охватывают их соли, в случае, если это допускается контекстом. Настоящее изобретение включает все изотопы атомов, встречающиеся в соединениях по настояще- 11021260 му изобретению. Изотопы включают те атомы, которые имеют такое же атомное число, но различные массовые числа. В качестве общего примера и без ограничения, изотопы водорода включают дейтерий и тритий. Изотопы углерода включают 13 С и 14 С. Изотопно меченные соединения по изобретению в основном могут быть получены с помощью общепринятых методик, известных специалистам в данной области, или способами, аналогичными тем, которые описаны в настоящем изобретении, используя соответствующий изотопно меченный реагент вместо немеченого реагента, используемого в иных случаях. Такие соединения могут иметь целый ряд потенциальных применений, например, в качестве стандартов и реагентов при определении биологической активности. В случае стабильных изотопов такие соединения могут иметь возможность положительно модифицировать биологические, фармакологические или фармакокинетические свойства. Соединения по настоящему изобретению могут существовать в виде фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль" в контексте настоящего изобретения, представляет соли или цвиттерионные формы соединений по настоящему изобретению, которые являются водорастворимыми или растворимыми в масле или диспергируемыми, которые в объеме тщательной медицинской проверки подходят для применения в контакте с тканями пациентов без чрезмерной токсичности,раздражения, аллергической реакции или других проблем или осложнений, сопоставимых с разумным соотношением польза/риск, и являются эффективными для предназначенного применения. Соли могут быть получены во время конечного выделения и очистки соединений или отдельно путем взаимодействия подходящего атома азота с подходящей кислотой. Характерные кислотно-аддитивные соли включают ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат,камфорсульфонат, диглюконат, глицерофосфат, гемисульфат, гептаноат, гексаноат, формиат, фумарат,гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, мезитиленсульфонат,метансульфонат, нафтиленсульфонат, никотинат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат,персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, трихлорацетат, фосфат,глутамат, бикарбонат, пара-толуолсульфонат и ундеканоат. Примеры кислот, которые могут быть использованы для образования фармацевтически приемлемых кислотно-аддитивных солей, включают неорганические кислоты, такие как соляная, бромисто-водородная, серная и фосфорная, и органические кислоты, такие как щавелевая, малеиновая, янтарная и лимонная. Основные аддитивные соли могут быть получены во время конечного выделения и очистки соединений путем взаимодействия карбоксильной группы с подходящим основанием, таким как гидроксид,карбонат или бикарбонат катиона металла или с аммиаком или органическим первичным, вторичным или третичным амином. Катионы фармацевтически приемлемых солей включают литий, натрий, калий,кальций, магний и алюминий, а также катионы нетоксичных четвертичных аминов, таких как аммоний,тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, диэтиламин, этиламин, трибутиламин, пиридин, N,N-диметиланилин, N-метилпиперидин, N-метилморфолин,дициклогексиламин, прокаин, дибензиламин, N,N-дибензилфенэтиламин и N,N'-дибензилэтилендиамин. Другие характерные органические амины, применяемые для образования основных аддитивных солей,включают этилендиамин, этаноламин, диэтаноламин, пиперидин и пиперазин. В тех случаях, когда это возможно, для использования в терапии, терапевтически эффективные количества соединения формулы (I), а также его фармацевтически приемлемых солей, можно вводить в виде необработанного химического вещества, возможно представить активный ингредиент в виде фармацевтической композиции. Соответственно, настоящее изобретение дополнительно относится к фармацевтическим композициям, которые включают терапевтически эффективные количества соединений формулы (I) или их фармацевтически приемлемых солей, и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов. Термин "терапевтически эффективное количество" в контексте настоящего изобретения относится к общему количеству каждого активного компонента, которое является эффективным для демонстрации значимой пользы для пациента, например устойчивого снижения вирусной нагрузки. При применении к индивидуальном активному ингредиенту, вводимому отдельно, этот термин относится к только к тому ингредиенту. При применении к комбинации этот термин относится к объединенным количествам активных ингредиентов, которые в результате приводят к терапевтическому эффекту, при введении будь то в сочетании, либо последовательно, либо одновременно. Соединения формулы (I) и их фармацевтически приемлемые соли описаны выше. Носитель(и), разбавитель(и) или эксципиент(ы) должны быть приемлемыми в смысле совместимости с другими ингредиентами состава и безвредными для реципиента. В соответствии с другим аспектом настоящего изобретения также представлен способ получения фармацевтической композиции, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли с одним или несколькими фармацевтически приемлемыми носителями, разбавителями или эксципиентами. Термин "фармацевтически приемлемый" в контексте настоящего изобретения относится к тем соединениям, веществам, композициям и/или лекарственным формам, которые в объеме тщательной медицинской проверки подходят для применения в контакте с тканями пациентов без чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, сопоставимой с разумным соотношением польза/риск, и которые являются эффективными для предназначенного использования. Фармацевтические составы могут быть представлены в единичных лекарственных формах, содержащих заранее определенное количество активного ингредиента на единичную дозу. Уровни доз составляют примерно от 0,01 примерно до 250 мг/кг массы тела в день, предпочтительно примерно от 0,05 примерно до 100 мг/кг массы тела в день соединений по настоящему изобретению являются характерными в монотерапии для профилактики и лечения заболевания, опосредованного HCV. Обычно фармацевтические композиции по настоящему изобретению будут вводить примерно от 1 примерно до 5 раз в день или, альтернативно, в виде непрерывной инфузии. Такое введение может быть использовано в качестве длительной или неотложной терапии. Количество активного ингредиента, которое можно объединить с веществами носителями для получения отдельной лекарственной формы, будет варьировать в зависимости от состояния подвергаемого лечению, тяжести состояния, времени введения, пути введения,скорости экскреции используемого соединения, длительности лечения и возраста, пола, массы тела и состояния пациента. Предпочтительными составами единичной дозы являются те, которые содержат суточную дозу или суб-дозу, как указано выше, или их соответствующую фракцию активного ингредиента. В основном лечение начинают с малых доз, существенно ниже оптимальной дозы соединения. После этого дозу повышают небольшим шагом повышения дозы до достижения оптимального эффекта в этих условиях. В основном, соединение наиболее целесообразно вводить с уровнем концентрации, который в основном будет давать эффективные противовирусные результаты, не вызывая никаких опасных или вредных побочных эффектов. В тех случаях, когда композиции по настоящему изобретению содержат комбинацию соединения по настоящему изобретению и одного или более дополнительных терапевтических или профилактических средств, как соединение, так и дополнительное средство обычно присутствует на уровнях доз примерно от 10 до 150% и более предпочтительно примерно от 10 до 80% дозы обычно вводимой в режиме монотерапии. Фармацевтические составы могут быть адаптированы для введения любым подходящим путем, например пероральным (в том числе буккальным или подъязычным), ректальным, назальным, местным (в том числе буккальным, подъязычным или трансдермальным), вагинальным или парентеральным путем(в том числе подкожным, внутрикожным, внутримышечным, внутрь сустава, интрасиновиальным,внутрь грудины, интратекально, внутрь пораженных тканей, внутривенно или внутрикожными инъекциями или инфузиями). Такие составы могут быть получены любым способом, известным в области фармации, например, путем объединения активного ингредиента с носителем (носителями) или эксципиентом (эксципиентами). Пероральное введение или введение инъекционным путем являются предпочтительными. Фармацевтические составы, адаптированные для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; пригодных к употреблению пен или муссов; или жидких эмульсий масло-в-воде или вода-в-масле. Например, для перорального введения в форме таблетки или капсулы, активный лекарственный компонент может быть объединен с пероральным нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и подобное. Порошки получают путем измельчения до подходящего мелкого размера и перемешивания с аналогично измельченным фармацевтическим носителем, таким как пригодный для употребления в пищу углевод, как, например, крахмал или маннит. Также может присутствовать вкусоароматический, консервирующий, диспергирующий и красящий агент. Капсулы получают путем приготовления порошкообразной смеси, как описано выше, и заполнения сформированных желатиновых оболочек. Регуляторы сыпучести и смазывающие вещества, такие как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль,могут быть добавлены к порошкообразной смеси перед операцией заполнения. Вещество для улучшения распадаемости или солюбилизирующий компонент, например агар-агар, карбонат кальция или карбонат натрия, также могут быть добавлены для улучшения доступности лекарственного средства при приеме капсулы внутрь. Кроме того, при желании или необходимости, подходящие связующие, смазывающие вещества,вещества для улучшения распадаемости и красители также могут быть включены в эту смесь. Подходящие связующие включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза,сахаристые вещества из кукурузы, природные и синтетические камеди, такие как акациевая, трагакантовая, или альгинат натрия, карбоксиметилцеллюлоза, полиэтиленгликоль и подобные. Смазывающие вещества, используемые в этих лекарственных формах, включают олеат натрия, хлорид натрия и подобные. Вещества для улучшения распадаемости включают, без ограничения, крахмал, метилцеллюлозу, агар,бетонит, ксантановую камедь и подобное. Таблетки получают, например, путем получения порошкообразной смеси, гранулирования или агрегирования, добавления смазыающего вещества и вещества для улучшения распадаемости и прессования в таблетки. Порошкообразную смесь получают путем перемешивания соединения, измельченного соответствующим образом, с разбавителем или основой, как описано выше, и, необязательно, со связующим, таким как карбоксиметилцеллюлоза, альгинат, желирующее вещество или поливинилпирролидон, вещество, замедляющее растворение, такое как парафин, вещество,- 13021260 ускоряющее всасывание, такое как четвертичная соль и/или абсорбент, например бетонит, каолин или дикальция фосфат. Порошкообразная смесь может быть гранулирована путем увлажнения со связующим, таким как сироп, крахмальная паста, клейкое вещество акадии, или растворами целлюлозных или полимерных материалов и просеивания через сито. В качестве альтернативы гранулированию порошкообразная смесь может быть пропущена через таблетировочную машину и полученные в результате неполностью сформированные крупинки, раздробленные на гранулы. Гранулы могут быть смазаны для предотвращения слипания с фасонной матрицей путем добавления стеариновой кислоты, соли - стеарата,талька или минерального масла. Смазанную смесь затем прессуют в таблетки. Соединения по настоящему изобретению также могут быть объединены с сыпучим инертным носителем и спрессованы в таблетки напрямую без прохождения через стадии гранулирования и агрегации. Может быть представлено прозрачное или непрозрачное защитное покрытие, состоящее из изолирующего слоя шеллака, покрытия сахаром или полимерным материалом и полирующего воскового покрытия. Красящие вещества могут быть добавлены к этим покрытиям для отличия различных единичных доз. Жидкости для перорального приема, такие как раствор, сиропы и эликсиры, могут быть получены в единичной дозированной форме таким образом, чтобы заданное количество содержало заранее определенное количество соединения. Сиропы могут быть получены путем растворения соединения в водном растворе с подходящими вкусоароматическими добавками, тогда как эликсиры получают путем использования нетоксичного носителя. Также могут быть добавлены солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и простые эфиры полиоксиэтиленсорбитола, консерванты,вкусоароматические добавки, такие как масло мяты перечной или натуральные подсластители, или сахарин или другие искусственные подсластители, и подобное. В соответствующих случаях составы единичной дозы для перорального введения могут быть микроинкапсулированы. Состав также может быть получен для пролонгирования или замедления высвобождения, например, путем нанесения покрытия или заключения зернистого материала в полимеры, воск или подобное. Соединения формулы (I) и их фармацевтически приемлемые соли, также можно вводить в форме липосомных систем доставки, таких как небольшие однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из целого ряда фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Соединения формулы (I) и их фармацевтически приемлемые соли также могут быть доставлены путем использования моноклональных антител в качестве индивидуальных носителей, к которым присоединены молекулы соединения. Эти соединения также могут быть присоединены к растворимым полимерам в качестве носителей лекарственных средств направленной доставки. Такие полимеры могут включать поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидефенол или полиэтиленоксидеполилизин, замещенный остатками палитоила. Кроме того, соединения могут быть присоединены к классу биоразлагаемых полимеров, применяемых для достижения контролируемого высвобождения лекарственного средства, например полимолочной кислоте,полэпсилонкапролактону, полигидроксимасляной кислоте, сложным полиортоэфирам, полиацеталям,полидигидропиранам, полицианоакрилатам и поперечно сшитым или амфипатическим блоксополимерам гидрогелей. Фармацевтические композиции, адаптированные для трансдермального введения, могут быть представлены в виде отдельных пластырей, предназначенных для сохранения тесного контакта с эпидермисом реципиента в течение длительного периода времени. Например, активный ингредиент может быть доставлен из пластыря путем ионофореза, как в основном описано в Pharm. Res., 3(6): 318 (1986). Фармацевтические композиции, адаптированные для местного применения, могут быть получены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел. Для лечения глаз или других наружних тканей, например полости рта и кожи, композиции предпочтительно применяют в виде наружной мази или крема. При получении мази активный ингредиент может быть использован либо с парафиновой, либо несмешиваемой с водой мазевой основой. Альтернативно,активный ингредиент может быть составлен в композицию крема с кремовой основой масло-в-воде или основой вода-в-масле. Фармацевтические композиции, адаптированные для наружних применений для глаз, включают глазные капли, где активный ингредиент растворен или суспендирован в подходящем носителе, особенно в водном растворителе. Фармацевтические композиции, адаптированные для наружного применения в полости рта, включают леденцы, пастилки и жидкости для полоскания полости рта. Фармацевтические композиции, адаптированные для ректального введения, могут быть получены в виде суппозиториев или в виде клизм. Фармацевтические композиции, адаптированные для назального введения, в которых носителем является твердое вещество, включают курсовой порошок, имеющий размер частиц, например, в диапазоне от 20 до 500 мкм, который вводят таким же образом, как и нюхательный табак, т.е. быстрым вдыханием через носовой ход из контейнера с порошком близко к носу. Подходящие составы, в которых носитель представляет собой жидкость, для введения в качестве спрея для носа или капель в нос, включают водные или масляные растворы активного ингредиента. Фармацевтические композиции, адаптированные для введения ингаляционным путем, включают тонкодисперсные порошки или аэрозоли, которые могут быть получены посредством различных типов мерных, находящихся под давлением дозирующих аэрозолей, небулайзеров или инжекторов. Фармацевтические композиции, адаптированные для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или спреев. Фармацевтические композиции, адаптированные для парентерального введения, включают водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатики и растворы, которые придают составу изотоничность с кровью определенного реципиента; водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Эти составы могут быть представлены в контейнерах для единичной дозы или в контейнерах для множественных доз, например герметично закрытых ампулах и флаконах, и могут храниться в высушенном замораживанием (лиофилизированном) состоянии, требуя только добавления стерильного жидкого носителя, например воды для инъекций непосредственно перед применением. Приготовленные для немедленного приема инъекционные растворы и суспензии могут быть получены из стерильных порошков,гранул и таблеток. Должно быть понятно, что помимо ингредиентов, в частности упомянутых выше, составы могут включать другие агенты, общепринятые в данной области, имеющие отношение к типу рассматриваемого состава, например, те, которые подходят для перорального введения, могут включать вкусоароматические агенты. Термин "пациент" включает как людей, так и других животных. Термин "лечение" относится к (i) профилактике заболевания, нарушения или патологического состояния, от возникновения у пациента, который может быть предрасположен к этому заболеванию, нарушению и/или патологическому состоянию, но которое у него еще не диагностировано; (ii) ингибированию заболевания, нарушения или патологического состояния, т.е. задержке его развития; и (iii) к облегчению заболевания, нарушения или патологического состояния, т.е. к регрессии заболевания, нарушения и/или патологического состояния. Соединения по настоящему изобретению также можно вводить с циклоспорином, например циклоспорином А. Было показано, что циклоспорин А является активным в отношении HCV в клинических испытаниях (Hepatology 38: 1282 (2003); Biochem. Biophys. Res. Commun. 313: 42; 2004, J. Gastroenterol. 38: 567 (2003. В приведенной ниже табл. 1 приведены некоторые иллюстративные примеры соединений, которые можно вводить с соединениями по настоящему изобретению. Соединения по настоящему изобретению можно вводить с другими соединениями, активность которых направлена против HCV, в комбинированной терапии, либо совместно, либо отдельно, или путем объединения этих соединений в композицию. Соединения по настоящему изобретению также могут быть использованы в качестве лабораторных реагентов. Соединения могут служить средством в обеспечении инструмента исследования для разработки исследований репликации вирусов, определения надежности систем исследования животных и изучений структурной биологии для дальнейшего расширения знаний о механизмах HCV заболевания. Далее, соединения по настоящему изобретению применимы в установлении или определении сайта связывания других противовирусных соединений, например, путем конкурентного ингибирования. Соединения по настоящему изобретению также могут быть использованы для лечения или предотвращения вирусной контаминации материалов и, следовательно, уменьшения риска вирусного инфицирования лаборатории или медицинского персонала или пациентов, которые контактируют с такими материалами, например кровью, тканью, хирургическими инструментами и спецодеждой, лабораторными инструментами и спецодеждой, и материалами и устройствами для сбора крови и трансфузии. Настоящее изобретение охватывает соединения формулы (I), полученные с помощью процессов синтеза или путем метаболических процессов, включая те, которые имеют место в организме человека или животного (in vivo) или процессов, происходящих in vitro. Сокращения, используемые в настоящем изобретении, в том числе, в частности, в иллюстративных примерах, которые следуют далее, хорошо известны специалистам в данной области. Некоторые используемые сокращения представляют собой следующее:Rt или RT - комнатная температура или время удерживания (контекст будет указывать);MCX LP - картридж для экстракции. Соединения и способы по настоящему изобретению будут лучше понятны в сочетании со следующими схемами синтеза, которые иллюстрируют способы, которыми могут быть получены соединения по настоящему изобретению. Исходные материалы могут быть получены из коммерческих источников или получены общепринятыми способами, описанными в литературе, известными специалистам в данной области. Специалисту в данной области будет очевидно, что соединения, определенные выше, могут быть синтезированы путем замены соответствующих реагентов и веществ в синтезе, показанном ниже. Специалисту в данной области также будет очевидно, что стадии селективной защиты и снятия защиты,а также порядок самих стадий может проводиться в различном порядке в зависимости от природы изменяемых компонентов для успешного завершения синтеза, представленного ниже. Изменяемые компоненты определены выше, если иное не определено ниже. Схема 1. Замещенные производные фенилглицина. Замещенные производные фенилглицина могут быть получены целым рядом способов, показанных ниже. Сложный т-бутиловый эфир фенилглицина может быть восстановительно алкилирован (путь А) с использованием соответствующего альдегида и восстановителя, такого как цианоборгидрид натрия в кислой среде. Гидролиз сложного т-бутилового эфира может осуществляться с использованием сильной кислоты, такой как HCl, или трифторуксусной кислоты. Альтернативно, фенилглицин может быть алкилирован алкилгалидом, таким как этилйодид и основанием, таким как бикарбонат натрия или карбонат калия (путь В). Путь С иллюстрирует восстановительно алкилирование фенилглицина как в пути А с последующим вторым восстановительным алкилированием альтернативным альдегидом, таким как формальдегид в присутствии восстановителя и кислоты. Путь D иллюстрирует синтез замещенных фенилглицинов через соотстветствующие аналоги миндальной кислоты. Превращение вторичного спирта в соответствующую требованиям уходящую группу может осуществляться с использованием птолуолсульфонилхлорида. Замещение тозилатной группы соответствующим амином с последующим восстановительным удалением бензилового сложного эфира может предоставить замещенные производные фенилглицина. В пути Е рацемическое замещенное производное фенилглицина может быть выделено путем этерификации с использованием энантиомерно чистой хиральной добавки, такой как, но не только (+)-1-фенилэтанол, (-)-1-фенилэтанол, оксазолидинон Эвана, или энантиомерно чистого пантолактона. Разделение диастереомеров осуществляется путем хроматографии (силикагель, HPLC, кристаллизация и т.д.) с последующим удалением хиральной добавки, получая энантиомерно чистые производные фенилглицина. Путь Н иллюстрирует последовательность синтеза, которая пересекается с путем Е,где указанную выше хиральную добавку помещают перед добавлением амина. Альтернативно, сложный эфир арилуксусной кислоты может быть бромирован с использованием источника иона бромония, такого как бром, N-бромсукцинимид или CBr4. Полученный в результате бензиловый бромид может быть замещен целым рядом моно- или двузамещенных аминов в присутствии третичного аминного основания,такого как триэтиламин или основание Хунига. Гидролиз сложного метилового эфира путем обработки гидроксидом лития при низкой температуре или 6 н. HCl при повышенной температуре обеспечивает получение замещенных производных фенилглицина. Другой способ покаан в пути G. Аналоги глицина могут быть преобразованы в производные с использованием целого ряда арилгалидов в присутствии источника палладия(0), такого как палладий бис(трибутилфосфин) и основания, такого как фосфат калия. Полученный в результате сложный эфир затем может быть гидролизован обработкой основанием или кислотой. Должно быть понятно, что другие хорошо известные способы получения производных фенилглицина существуют в уровне техники, и могут быть изменены для получения желаемых соединений в этом описании. Также должно быть понятно, что конечные производные фенилглицина могут быть очищены до энантиомерной чистоты более 98% ее посредством препаративной HPLC. Схема 2. Ацилированные аминокислотные производные. В другом варианте осуществления настоящего изобретения ацилированные производные фенилглицина могут быть получены, как показано ниже. Производные фенилглицина, где карбоновая кислота защищена как легко удаляемый сложный эфир, могут быть ацилированы с использованием хлорангидрида в присутствии основания, такого как триэтиламин для получения соответствующих амидов (путь А). Путь В иллюстрирует ацилирование исходного производного фенилглицина соответствующим хлорформиатом, тогда как путь С показывает взаимодействие с соответствующим изоцианатом или карбамоилхлоридом. С каждого из трех промежуточных соединений, показанных в путях А-С, может быть снята защита способами, известными специалистам в данной области (т.е. обработкой сложного т-бутилового эфира сильной кислотой, такой как HCl или трифторуксусная кислота). Схема 3. Аминозамещенные фенилуксусные кислоты могут быть получены путем обработки хлорметилфенилуксусной кислоты избытком амина. Синтез основных "кэп"-структур. Условия анализа соединений: оценку чистоты и массовый анализ с низким разрешением проводили на системе Shimadzu LC, соединенной с системой Waters Micromass ZQ MS. Следует отметить, что время удерживания может несколько варьировать между устройствами. Дополнительные условия LC применимы к текущему разделу, если не указано иное. Суспензию 10% Pd/C (2,0 г) в метаноле (10 мл) добавляли к смеси (R)-2-фенилглицина (10 г, 66,2 ммоль), формальдегида (33 мл 37 мас.%, в воде), 1 н. HCl (30 мл) и метанола (30 мл) и подвергали воздействию H2 (60 фунтов/кв.дюйм) в течение 3 ч. Реакционную смесь фильтровали через диатомовую землю(Celite) и фильтрат концентрировали под вакуумом. Полученное в результате сырое вещество перекристаллизовывали из изопропанола с получением HCl соли "Кэп"-1 в виде белых игл (4,0 г). Вращение плоскости поляризации света: -117,1 [с=9,95 мг/мл в Н 2 О; =589 нм]. 1LC (Cond. I): RT=0,25; LC/MS: Аналитический расчет для [М+Н]+ C10H14NO2 180,10; найдено: 180,17; HRMS: Аналитический расчет для [М+Н]+ C10H14NO2 180,1025; найдено: 180,1017.NaBH3CN (6,22 г, 94 ммоль) добавляли частями на протяжении нескольких минут в охлажденную(лед/вода) смесь (R)-2-фенилглицина (6,02 г, 39,8 ммоль) и метанола (100 мл) и перемешивали в течение 5 мин. Ацетальдегид (10 мл) добавляли по каплям на протяжении 10 мин и перемешивание продолжали при той же охлажденной температуре в течение 45 мин и при окружающей температуре в течение 6,5 ч. Реакционную смесь снова охлаждали на бане с ледяной водой, обрабатывали водой (3 мл) и затем гасили путем добавления по каплям концентрированной HCl на протяжении 45 мин, пока рН смеси не составил 1,5-2,0. Охлаждающую баню удаляли и перемешивание продолжали, добавляя концентрированную HCl для поддержания рН смеси около 1,5-2,0. Реакционную смесь перемешивали в течение ночи, фильтровали для удаления белой суспензии и фильтрат концентрировали под вакуумом. Сырое вещество перекристаллизовывали из этанола с получением HCl соли соединения "Кэп"-2 в виде сверкающего твердого вещества белого цвета в двух порциях (порция-1: 4,16 г; порция-2: 2,19 г). 1LC (условие I): RT=0,43 мин; LC/MS: Аналитический расчет для [М+Н]+ C12H18NO2: 208,13; найдено: 208,26.HCl (30 мл) и метанола (40 мл). Охлаждающую баню удаляли и реакционную смесь перемешивали в камере с Н 2 в течение 17 ч. Добавляли дополнительное количество ацетальдегида (10 мл, 178,2 ммоль) и перемешивание продолжали в атмосфере Н 2 в течение 24 ч [Примечание: доставку Н 2 пополняли по мере необходимости на протяжении реакции]. Реакционную смесь фильтровали через диатомовую землю(Celite), фильтрат концентрировали под вакуумом. Полученное в результате сырое вещество перекристаллизовывали из изопропанола с получением HCl соли (R)-2-(этиламино)-2-фенилуксусной кислоты в виде сверкающего твердого вещества белого цвета (2,846 г). 1[М+Н]+ C10H14NO2: 180,10; найдено: 180,18. Суспензию 10% Pd/C (536 мг) в метаноле/Н 2 О (3 мл/1 мл) добавляли к смеси (R)-2-(этиламино)-2 фенилуксусной кислоты/HCl (1,492 г, 6,918 ммоль), формальдегида (20 мл 37 мас.%, в воде), 1 н. HCl (20 мл) и метанола (23 мл). Реакционную смесь перемешивали в камере с Н 2 в течение 72 ч, где доставку H2 восполняли по мере необходимости. Реакционную смесь фильтровали через диатомовую землю (Celite) и фильтрат концентрировали под вакуумом. Полученное в результате сырое вещество перекристаллизовывали из изопропанола (50 мл) с получением HCl соли соединения "Кэп"-3 в виде твердого вещества[М+Н]+ C11H16NO2: 194,12; найдено: 194,18; HRMS: Аналитический расчет для [M+H]+ C11H16NO2: 194,1180; найдено: 194,1181.ClCO2Me (3,2 мл, 41,4 ммоль) добавляли по каплям к охлажденному (лед/вода) THF (410 мл) золю(R)-трет-бутил 2-амино-2-фенилацетат/HCl (9,877 г, 40,52 ммоль) и диизопропилэтиленамина (14,2 мл,81,52 ммоль) на протяжении 6 мин и перемешивали при аналогичной температуре в течение 5,5 ч. Летучий компонент удаляли под вакуумом и остаток разделяли между водой (100 мл) и этилацетатом (200 мл). Органический слой промывали с использованием 1 н. HCl (25 мл) и насыщенного раствора NaHCO3(30 мл), сушили (MgSO4), фильтровали и концентрировали под вакуумом. Полученное в результате бесцветное масло растирали с гексаном, фильтровали и промывали гексаном (100 мл) с получением (R)трет-бутил 2-(метоксикарбониламино)-2-фенилацетата в виде твердого вещества белого цвета (7,7 г). 1TFA (16 мл) добавляли по каплям к охлажденному (лед/вода) CH2Cl2 (160 мл) раствору указанного выше продукта на протяжении 7 мин и охлаждающую баню удаляли и реакционную смесь перемешивали в течение 20 ч. Поскольку снятие защиты все еще не было завершено, добавляли дополнительное количество TFA (1,0 мл) и перемешивание продолжали в течение дополнительных 2 ч. Летучий компонент удаляли под вакуумом и полученный в результате масляный остаток обрабатывали простым диэтиловым эфиром (15 мл) и гексаном (12 мл) с получением осадка. Осадок фильтровали и промывали простым диэтиловым эфиром/гексаном (соотношение 1:3; 30 мл) и сушили под вакуумом с получением "Кэп"-4 в виде рассыпчатого твердого вещества белого цвета (5,57 г). Вращение плоскости поляризации света:[М+Н]+ C10H12NO4 210,08; найдено: 210,17; HRMS: Аналитический расчет для [М+Н]+ C10H12NO4 210,0766; найдено: 210,0756. Смесь (R)-2-фенилглицина (1,0 г, 6,62 ммоль), 1,4-дибромбутана (1,57 г, 7,27 ммоль) и Na2CO3 (2,10 г, 19,8 ммоль) в этаноле (40 мл) нагревали при 100C в течение 21 ч. Реакционную смесь охлаждали до окружающей температуры, фильтровали и фильтрат концентрировали под вакуумом. Остаток растворяли в этаноле и подкисляли 1 н. HCl до рН 3-4 и летучий компонент удаляли под вакуумом. Полученное в результате сырое вещество очищали с помощью HPLC с обращенной фазой (вода/метанол/TFA) с получением TFA соли "Кэп"-5 в виде полувязкой пены белого цвета (1,0 г). 1RT=0,30 мин (условие I); 98% индекс гомогенности; LC/MS: Аналитический расчет для [М+Н]+TFA соль "Кэп"-6 синтезировали из (R)-2-фенилглицина и 1-бром-2-(2-бромэтокси)этана, используя способ получения "Кэп"-5. 1RT=0,32 мин (условие I); 98%; LC/MS: Аналитический расчет для [M+H]+ C12H16NO3: 222,11; найдено: 222,20; HRMS: Аналитический расчет для [М+Н]+ C12H16NO3: 222,1130; найдено: 222,1121.CH2Cl2 (200 мл) раствор п-толуолсульфонилхлорида (8,65 г, 45,4 ммоль) добавляли по каплям к охлажденному (-5C) CH2Cl2 (200 мл) раствору (S)-бензил 2-гидрокси-2-фенилацетата (10,0 г, 41,3 ммоль),триэтиламина (5,75 мл, 41,3 ммоль) и 4-диметиламинопиридина (0,504 г, 4,13 ммоль), поддерживая температуру между -5 и 0C. Реакционную смесь перемешивали при 0C в течение 9 ч и затем хранили в морозильной камере (-25C) в течение 14 ч. Указанную реакционную смесь оставляли оттаивать до окружающей температуры и промывали водой (200 мл), 1 н. HCl (100 мл) и солевым раствором (100 мл),сушили (MgSO4), фильтровали и концентрировали под вакуумом с получением бензил 2-фенил-2(тозилокси)ацетата в виде вязкого масла, которое твердеет при стоянии (16,5 г). Хиральную чистоту продукта не проверяли и этот продукт использовали на следующей стадии без дополнительной очистки. 1RT=3,00 (условие III); 90% индекс гомогенности; LC/MS: Аналитический расчет для [М+Н]+(3,36 мл, 30,3 ммоль) и N,N-диизопропилэтиленамина (13,2 мл, 75,8 ммоль) нагревали при 65C в течение 7 ч. Реакционную смесь оставляли охлаждаться до окружающей температуры и летучий компонент удаляли под вакуумом. Остаток разделяли между этилацетатом и водой и органический слой промывали водой и солевым раствором, сушили (MgSO4), фильтровали и концентрировали под вакуумом. Полученное в результате сырое вещество очищали с помощью флэш-хроматографии (силикагель, этилацетат) с получением бензил 2-(4-метилпиперазин-1-ил)-2-фенилацетата в виде оранжевато-коричневого вязкого масла (4,56 г). Хиральный HPLC анализ (Chiralcel OD-H) показал, что образец представляет собой смесь энантиомеров в соотношении от 38,2 до 58,7. Разделение энантиомеров осуществляли следующим образом: продукт растворяли в 120 мл смеси этанол/гептан (1:1) и вводили (5 мл/введение) в хиральнуюRT=2,10 (условие III); 98% индекс гомогенности; LC/MS: Аналитический расчет для [М+Н]+C20H25N2O2: 325,19; найдено: 325,20. Метанольный (10 мл) раствор любого энантиомера бензил 2-(4-метилпиперазин-1-ил)-2 фенилацетата (1,0 г, 3,1 ммоль) добавляли к суспензии 10% Pd/C (120 мг) в метаноле (5,0 мл). Реакционную смесь помещали в водородную камеру под пристальным мониторингом в течение 50 мин. Сразу же после завершения реакции катализатор фильтровали через диатомовую землю (Celite) и фильтрат концентрировали под вакуумом с получением соединения "Кэп"-7, с загрязнением фенилуксусной кислотой в виде пены желтовато-коричневого цвета (867,6 мг; масса выше теоретического выхода). Этот продукт использовали на следующей стадии без дополнительной очистки. 1(app. br s, 2H), 2,48-2,32 (m, 6H), 2,19 (s, 3H); RT=0,31 (условие II); 90% индекс гомогенности; LC/MS: Аналитический расчет для [М+Н]+ C13H19N2O2: 235,14; найдено: 235,15; HRMS: Аналитический расчет для [М+Н]+ C13H19N2O2: 235,1447; найдено: 235,1440. Синтез "Кэп"-8 и "Кэп"-9 проводили в соответствии с синтезом "Кэп"-7, используя соответствующие амины для стадии замещения SN2 (т.е. 4-гидроксипиперидина для "Кэп"-8 и (S)-3-фторпирролидина для "Кэп"-9) и модифицированные условия для разделения соответствующих стереоизомерных промежуточных соединений, описанные ниже.
МПК / Метки
МПК: A61K 31/4184, A61P 31/00, A61K 31/422, C07D 403/14, C07D 413/14
Метки: вируса, гепатита, ингибиторы
Код ссылки
<a href="https://eas.patents.su/30-21260-ingibitory-virusa-gepatita-s.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы вируса гепатита с</a>
Ингибиторы вируса гепатита с (hcv)

Номер патента: 15415
Опубликовано: 31.08.2011
Авторы: Граупе Михаель, Венкатарамани Чандрэсикар, Линк Джон О.
МПК: A61K 31/74, A61K 31/00, A61K 47/00...
Метки: вируса, гепатита, hcv, ингибиторы
Формула / Реферат:
1. Соединение формулы (I)где Е представляет собой -COCONHR6, где R6представляет собой водород, алкил, циклоалкил, аралкил или гетероаралкил, где ароматическое кольцо необязательно замещено одним или двумя галоидами;R1 представляет собой алкил, циклоалкилалкил, где алифатические или алициклические группы в R1необязательно замещены одним или двумя радикалами Rb, которые независимо выбирают из гидрокси, алкокси, арилокси, гетероарилокси, алкилтио,...
Ингибиторы вируса гепатита с

Номер патента: 15756
Опубликовано: 30.12.2011
Авторы: Рудигер Эдвард Х., Сен-Лоран Денис Р., Нгуен Ван Н., Хаманн Лоуренс Г., Джеймс Клинт А., Гудрич Джейсон, Гуд Эндрю С., Лопез Омар Д., Белема Маконен, Минвелл Николас А., Ромайн Джеффри Ли, Мартель Ален, Деон Дэниел Х., Лавой Рико, Лэнгли Дэвид Р., Бэчэнд Кэрол, Ван Гань, Ян Фукан, Снайдер Лоуренс Б.
МПК: A61P 31/12, A61K 31/4025, A61K 31/4178...
Метки: вируса, ингибиторы, гепатита
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, гдеm и n независимо имеют значения 0, 1 или 2;q и s независимо имеют значения 0, 1, 2, 3 или 4;u и v независимо имеют значения 0, 1, 2 или 3;X выбран из О, S, S(O), SO2, CH2, CHR5 и C(R5)2;при условии, что когда n равен 0, X выбран из СН2, CHR5 и C(R5)2;Y выбран из О, S, S(O), SO2, CH2, CHR6 и C(R6)2;при условии, что когда m равен 0, Y выбран из СН2, CHR6 и C(R6)2;каждый R1и R2...
Ингибиторы вируса гепатита с

Номер патента: 17348
Опубликовано: 30.11.2012
Авторы: Гудрич Джейсон, Хаманн Лоуренс Г., Лавой Рико, Сен-Лоран Денис Р., Гуд Эндрю С., Ромайн Джеффри Ли, Ян Фукан, Белема Маконен, Джеймс Клинт А., Бэчэнд Кэрол, Рудигер Эдвард Х., Снайдер Лоуренс Б., Деон Дэниел Х., Лопез Омар Д., Минвелл Николас А., Нгуен Ван Н., Лэнгли Дэвид Р., Мартель Ален
МПК: A61K 31/4439, A61K 31/444, A61K 31/497...
Метки: вируса, гепатита, ингибиторы
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, гдеm и n равны 1;q и s равны 0;u и v независимо имеют значения 0,1;А и В выбраны из фенила и содержащего шесть членов гетероароматического кольца, включающего один или два атома азота, выбранного из пиридинила, пиримидинила, пиразинила, пиридазинила; при условии, что по крайней мере один из А и В отличен от фенила;X и Y выбраны из СН2;каждый R1 и R2 независимо выбран из...
Ингибиторы вируса гепатита с

Номер патента: 18313
Опубликовано: 30.07.2013
Авторы: Хаманн Лоуренс Г., Лопез Омар Д., Гудрих Джейсон, Мартел Алан, Дэон Даниэль Х., Лэнгли Дэвид Р., Ромин Джеффри Ли, Бачанд Кэрол, Лавуа Рико, Минвелл Николас А., Снайдер Лоуренс Б., Нгуен Ван Н., Рюдигер Эдвард Х., Гуд Эндрю К., Ст.Лоран Денис Р., Джеймс Клинт А., Ванг Гэн, Янг Фуканг, Белема Маконен
МПК: A61K 31/4188, A61K 31/4178, A61K 31/4184...
Метки: вируса, ингибиторы, гепатита
Формула / Реферат:
1. Соединение согласно формуле (II)или его фармацевтически приемлемая соль, в которомR2 выбирается из водорода и галогеналкила, где галогеналкил представляет собой насыщенный углеводород, содержащий от 1 до 6 атомов углерода, замещенный одним, двумя, тремя или четырьмя атомами галогена; аR3 выбирается из водорода и галогена илиR2 и R3 совместно с углеродными атомами, к которым они присоединены, образуют 6-8-членное ароматическое или...
Ингибиторы вируса гепатита с

Номер патента: 18793
Опубликовано: 30.10.2013
Авторы: Белема Маконен, Кэдоу Джон Ф.
МПК: A61K 31/4178, A61K 31/439, A61P 31/14...
Метки: ингибиторы, гепатита, вируса
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, где L выбран из связи,R1 и R2 представляют собойили R1 представляет собойи R2 выбран изгде обозначает точку присоединения к исходной молекуле;R3 и R4 независимо выбраны из водорода и гало;каждый R5 независимо выбран из водорода;каждый R6 независимо выбран из алкила;R6a представляет собой алкил, где алкил необязательно может образовывать конденсированное 3-членное кольцо со смежным...
Предыдущий патент: Моечный узел для мойки по меньшей мере одного участка головки доильного стакана
Следующий патент: Пестицидная композиция, включающая фторпиколид и инсектицидное соединение
Случайный патент: Вакуумная насадка и способ подводного вакуумного оздоровительного массажа