Соединения и композиции как ингибиторы протеинкиназы b-raf
Номер патента: 20479
Опубликовано: 28.11.2014
Авторы: Хуан Шэньлинь, Чжан Цион, Цзинь Джефф, Пекки Сабина, Косталес Абран К., Лю Зуошэн, Пун Даниэль, Теллью Джон





























Формула / Реферат
1. Соединение формулы (I) или (II)

в которой
R1 обозначает
(i) Н,
(ii) (С3-С6)циклоалкил, необязательно замещенный цианогруппой;
(iii) (С1-С3)алкил, необязательно замещенный цианогруппой, группой -C(O)NH2 или гидроксигруппой, или
(iv) -X1NHC(O)OR1a или -X1NHC(O)NHR1a, где X1 обозначает (С1-С4)алкилен, необязательно содержащий в качестве заместителей 1-3 группы, каждая из которых независимо выбрана из группы, включающей галоген, (С1-С4)алкил или галогензамещенный (С1-С4)алкил, и
R1а обозначает Н, (С1-С4)алкил или галогензамещенный (С1-С4)алкил;
R1b обозначает Н или метил;
R2 обозначает Н или галоген;
R3 обозначает Н, галоген, (С1-С4)алкоксигруппу, (С1-С4)алкил, галогензамещенную (С1-С4)алкоксигруппу или галогензамещенный (С1-С4)алкил;
R4 обозначает галоген, Н или (С1-С4)алкил;
R5 обозначает (С1-С6)алкил, (С3-С6)циклоалкил, разветвленный (С3-С8)алкил, галогензамещенный (С1-С6)алкил, галогензамещенный разветвленный (С3-С8)алкил, (С3-С6)циклоалкил(С1-С3)алкилен или фенил, где указанный фенил необязательно содержит 1-3 заместителя, каждый из которых независимо выбран из группы, включающей галоген, СН3 или CF3;
R6 обозначает Н, (С1-С4)алкил или галоген и
R7 обозначает Н, (С1-С6)алкил, (С3-С6)циклоалкил, 1-метил-(С3-С6)циклоалкил, разветвленный (С3-С8)алкил или фенил, где указанный фенил необязательно содержит 1-3 заместителя, выбранных из группы, включающей галоген, (C1-С4)алкил или галогензамещенный (С1-С4)алкил;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где указанным соединением является соединение формулы (I) или его фармацевтически приемлемая соль.
3. Соединение по любому из предыдущих пунктов, в котором
R1 обозначает -X1NHC(O)OR1a, где X1 обозначает (С1-С4)алкилен, необязательно содержащий в качестве заместителей 1-3 группы, каждая из которых независимо выбрана из группы, включающей (С1-С4)алкил или галогензамещенный (С1-С4)алкил, и R1a обозначает (С1-С2)алкил или галогензамещенный (С1-С2)алкил;
R2 обозначает Н или F;
R3 обозначает Н, галоген, (С1-С2)алкоксигруппу, (С1-С2)алкил, галогензамещенную (С1-С2)алкоксигруппу или галогензамещенный (С1-С2)алкил;
R4 обозначает Н или метил;
R5 обозначает (С1-С4)алкил, (С3-С6)циклоалкил, разветвленный (С3-С5)алкил, галогензамещенный (С1-С4)алкил, галогензамещенный разветвленный (С3-С6)алкил, (С3-С6)циклоалкил(С1-С3)алкилен или фенил, содержащий 1-3 заместителя, каждый из которых независимо выбран из группы, включающей Cl, F, СН3 или CF3;
R6 обозначает Н, (С1-С2)алкил или галоген и
R7 обозначает (С3-С6)циклоалкил, 1-метил-(С3-С6)циклоалкил или разветвленный (С3-С6)алкил;
или его фармацевтически приемлемая соль.
4. Соединение по любому из предыдущих пунктов, в котором
R1 обозначает -X1NHC(O)OR1a, где X1 обозначает (С1-С4)алкилен, необязательно содержащий в качестве заместителей 1-3 группы, каждая из которых независимо выбрана из группы, включающей (С1-С4)алкил или галогензамещенный (C1-С4)алкил, и R1a обозначает (С1-С2)алкил или галогензамещенный (С1-С2)алкил;
R2 обозначает Н или F;
R3 обозначает Н, галоген, (С1-С2)алкоксигруппу, (С1-С2)алкил, галогензамещенную (С1-С2)алкоксигруппу или галогензамещенный (С1-С2)алкил;
R4 обозначает Н или метил;
R5 обозначает (С1-С4)алкил, (С3-С6)циклоалкил, разветвленный (С3-С5)алкил, галогензамещенный (С1-С4)алкил, галогензамещенный разветвленный (С3-С6)алкил или фенил, замещенный с помощью F, СН3 или CF3;
R6 обозначает Н, (С1-С2)алкил или галоген и
R7 обозначает (С3-С6)циклоалкил, 1-метил-(С3-С6)циклоалкил или разветвленный (С3-С6)алкил;
или его фармацевтически приемлемая соль.
5. Соединение по любому из предыдущих пунктов, в котором
R1 обозначает -X1NHC(O)OR1a, где X1 обозначает (С1-С2)алкилен, замещенный (С1-С2)алкилом, и R1a обозначает (С1-С2)алкил;
R2 обозначает Н;
R3 обозначает Н, Cl, F, метоксигруппу, метил или дифторметоксигруппу;
R4 обозначает Н;
R5 обозначает метил, циклопропил, этил, пропил, изопропил, втор-бутил, изобутил, трифторметил или 3,3,3-трифторпропил;
R6 обозначает Н, метил, F или Сl и
R7 обозначает трет-бутил, циклопропил или 1-метилциклопропил;
или его фармацевтически приемлемая соль.
6. Соединение по любому из предыдущих пунктов, в котором R1 описывается следующей формулой (1а)

или его фармацевтически приемлемая соль.
7. Соединение по п.1, где указанным соединением является соединение формулы (I)

в которой
R1 обозначает
(i) (С1-С3)алкил, необязательно замещенный цианогруппой, группой -C(O)NH2 или гидроксигруппой, или
(ii) -X1NHC(O)OR1a, где X1 обозначает (С1-С4)алкилен, необязательно содержащий в качестве заместителей 1-3 группы, каждая из которых независимо выбрана из группы, включающей галоген, (С1-С4)алкил или галогензамещенный (С1-С4)алкил, R1a обозначает Н, (С1-С4)алкил или галогензамещенный (С1-С4)алкил;
R2 обозначает Н или галоген;
R3 обозначает Н, галоген, (С1-С4)алкоксигруппу, (С1-С4)алкил, галогензамещенную (С1-С4)алкоксигруппу или галогензамещенный (С1-С4)алкил;
R4 обозначает галоген, Н или (С1-С4)алкил;
R5 обозначает (С1-С6)алкил, (С3-С6)циклоалкил, разветвленный (С3-С8)алкил, галогензамещенный (С1-С6)алкил или галогензамещенный разветвленный (С3-С8)алкил;
R6 обозначает Н, (С1-С4)алкил или галоген и
R7 обозначает Н, (С1-С6)алкил, (С3-С6)циклоалкил, 1-метил-(С3-С6)циклоалкил, разветвленный (С3-С8)алкил или фенил, где указанный фенил необязательно содержит 1-3 заместителя, выбранных из группы, включающей галоген, (С1-С4)алкил или галогензамещенный (С1-С4)алкил;
или его фармацевтически приемлемая соль.
8. Соединение по п.1, где указанным соединением является соединение формулы (II) или его фармацевтически приемлемая соль.
9. Соединение по п.8, в котором
R2 обозначает Н или F;
R3 обозначает Н, галоген, (С1-С2)алкоксигруппу, (С1-С2)алкил, галогензамещенную (С1-С2)алкоксигруппу или галогензамещенный (С1-С2)алкил;
R4 обозначает Н или метил;
R5 обозначает (С1-С4)алкил, (С3-С6)циклоалкил, разветвленный (С3-С5)алкил, галогензамещенный (С1-С4)алкил, галогензамещенный разветвленный (С3-С6)алкил или (С3-С6)циклоалкил(С1-С3)алкилен;
R6 обозначает Н, (С1-С2)алкил или галоген и
R7 обозначает (С3-С6)циклоалкил, 1-метил-(С3-С6)циклоалкил или разветвленный (С3-С6)алкил;
или его фармацевтически приемлемая соль.
10. Соединение по п.9, в котором
R2 обозначает Н;
R3 обозначает Н, Cl, F, метоксигруппу, метил или дифторметоксигруппу;
R4 обозначает Н;
R5 обозначает метил, циклопропил, этил, пропил, изопропил, втор-бутил, изобутил, трифторметил или 3,3,3-трифторпропил;
R6 обозначает Н, метил, F или Cl и
R7 обозначает трет-бутил, циклопропил или 1-метилциклопропил;
или его фармацевтически приемлемая соль.
11. Соединение по п.1, выбранное из группы, включающей
(S)-метил-1-(4-(4-(2-хлор-5-фтор-3-(метилсульфонамидо)фенил)-2-циклопропил-1Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(R)-метил-1-(4-(4-(2-хлор-5-фтор-3-(метилсульфонамидо)фенил)-2-циклопропил-1Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(S)-метил-1-(4-(2-циклопропил-4-(2-фтор-5-метил-3-(метилсульфонамидо)фенил)-1Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(S)-метил-1-(4-(2-циклопропил-4-(2,5-дихлор-3-(метилсульфонамидо)фенил)-1Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(S)-метил-1-(4-(4-(2-хлор-3-(метилсульфонамидо)фенил)-2-циклопропил-1Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(S)-метил-1-(4-(2-циклопропил-4-(2-фтор-3-(метилсульфонамидо)фенил)-1Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
метил-(2S)-1-(4-(2-циклопропил-4-(2,5-дифтор-3-(метилсульфонамидо)фенил)-1Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамат и
(S)-метил-1-(4-(4-(2-хлор-5-метил-3-(метилсульфонамидо)фенил)-2-циклопропил-1Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
или его фармацевтически приемлемая соль.
12. Соединение по п.1, выбранное из группы, включающей
(S)-метил-1-(4-(5-(5-хлор-3-(циклопропансульфонамидо)-2-фторфенил)-2-циклопропил-1Н-имидазол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(S)-метил-1-(4-(5-(5-хлор-3-(этилсульфонамидо)-2-фторфенил)-2-циклопропил-1Н-имидазол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(S)-метил-1-(4-(5-(5-хлор-2-фтор-3-(3,3,3-трифторпропилсульфонамидо)фенил)-2-циклопропил-1Н-имидазол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(S)-метил-1-(4-(5-(5-хлор-3-(циклопропилметилсульфонамидо)-2-фторфенил)-2-циклопропил-1Н-имидазол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(S)-метил-1-(4-(4-(2-хлор-3-(этилсульфонамидо)-5-фторфенил)-2-циклопропил-1Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(S)-метил-1-(4-(4-(2-хлор-3-(циклопропансульфонамидо)-5-фторфенил)-2-циклопропил-1Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(S)-метил-1-(4-(5-(5-хлор-2-фтор-3-(метилсульфонамидо)фенил)-2-(1-метилциклопропил)-1Н-имидазол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(S)-метил-1-(4-(5-(2-фтор-5-метил-3-(метилсульфонамидо)фенил)-2-(1-метилциклопропил)-1Н-имидазол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
(S)-метил-1-(4-(5-(5-хлор-2-фтор-3-(4-фторфенилсульфонамидо)фенил)-2-циклопропил-1Н-имидазол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамат и
(S)-метил-1-(4-(5-(5-хлор-2-фтор-3-(3-фторфенилсульфонамидо)фенил)-2-циклопропил-1Н-имидазол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамат;
или его фармацевтически приемлемая соль.
13. Соединение по п.1, выбранное из группы, включающей
N-(2-хлор-3-(2-циклопропил-5-(пиримидин-4-ил)-1Н-имидазол-4-ил)-5-фторфенил)-2,6-дифторбензолсульфонамидо;
N-(2-хлор-3-(2-циклопропил-5-(пиримидин-4-ил)-1Н-имидазол-4-ил)-5-фторфенил)метансульфонамид и
N-(2-хлор-3-(2-циклопропил-5-(пиримидин-4-ил)-1Н-имидазол-4-ил)-5-фторфенил)пропан-1-сульфонамид;
или его фармацевтически приемлемая соль.
14. Фармацевтическая композиция для лечения рака, включающая терапевтически эффективное количество соединения по любому из пп.1-13 и разбавитель, носитель или инертный наполнитель.
15. Фармацевтическая композиция по п.14, дополнительно включающая терапевтическое средство, выбранное из группы, включающей противораковое соединение, анальгетик, противорвотное средство, антидепрессант и противовоспалительное средство.
16. Лекарственное средство, предназначенное для лечения рака, представляющее собой соединение по любому из пп.1-13.
17. Лекарственное средство по п.16, где указанный рак, выбран из группы, включающей карциному легких, карциному поджелудочной железы, карциному мочевого пузыря, карциному толстой кишки, миелоидные нарушения, рак предстательной железы, рак щитовидной железы, меланому и аденомы.
18. Способ лечения рака, включающий введение субъекту, нуждающемуся в таком лечении, соединения по любому из пп.1-13 в фармацевтически эффективном количестве.
19. Способ по п.18, в котором указанный рак выбран из группы, включающей карциному легких, карциному поджелудочной железы, карциному мочевого пузыря, карциному толстой кишки, миелоидные нарушения, меланомы и аденомы.
20. Способ лечения патологического состояния, опосредуемого киназой Raf, включающий введение субъекту, нуждающемуся в таком лечении, соединения по любому из пп.1-13 в эффективном количестве.
21. Способ по п.20, в котором киназой Raf является мутантная киназа B-Raf.
22. Способ по п.21, в котором указанной мутантной киназой B-Raf является B-Raf (V600E).
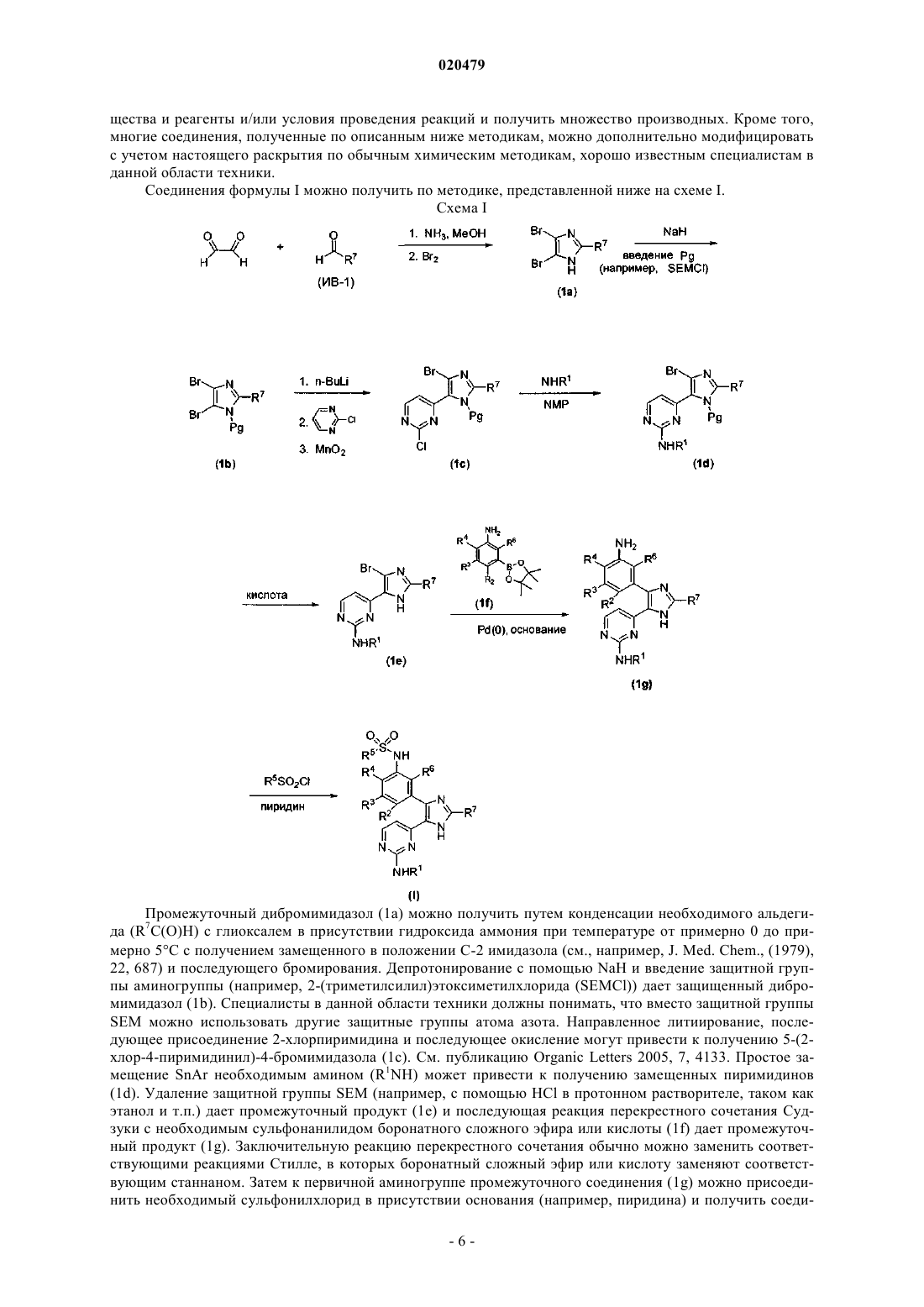
Текст
СОЕДИНЕНИЯ И КОМПОЗИЦИИ КАК ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ B-Raf В изобретении описаны соединения формулы I или II в которых R1, R1b, R2, R3, R4, R5, R6 и R7 являются такими, как определено в настоящем изобретении. Соединения формулы (I) или (II) и включающие их фармацевтические композиции применимы для лечения заболеваний, связанных с киназой B-Raf. Перекрестная ссылка на родственные заявки По настоящей заявке в соответствии с 35 U.S.С. 119(е) испрашивается приоритет по предварительной заявке U.S.61/238083, поданной 28 августа 2009 г., и по предварительной заявке U.S.61/313061, поданной 11 марта 2010 г., которые во всей своей полноте включены в настоящее изобретение в качестве ссылки. Область техники, к которой относится изобретение Настоящее изобретение относится к новому классу соединений, фармацевтическим композициям,включающим такие соединения, и к способам применения таких соединений для лечения или предупреждения заболеваний или нарушений, связанных с аномальной или характеризующейся нарушенной регуляцией активностью киназы, в особенности заболеваний или нарушений, которые включают аномальную активацию B-Raf. Уровень техники Протеинкиназы представляют собой большую группу белков, которые играют центральную роль в регуляции большого количества клеточных процессов и обеспечивают регулирование клеточных функций. Частичный, неограничивающий перечень этих киназ включает рецепторные тирозинкиназы, такие как киназа рецептора тромбоцитарного фактора роста (ТРФРР), рецептора фактора роста нервов, trkB,Met и рецептора фактора роста фибробластов, FGFR3; нерецепторные тирозинкиназы, такие как Abl, и киназы слияния BCR-Abl, Lck, Csk, Fes, Bmx и c-src; и серин/треонинкиназы, такие как киназы, B-Raf,sgk, MAP (например, MKK4, MKK6 и т.п.) и SAPK2, SAPK2 и SAPK3. Аберрантная активность киназ обнаружена при многих патологических состояниях, включая доброкачественные и злокачественные пролиферативные заболевания, а также заболевания, обусловленные неадекватной активацией иммунной и нервной систем. Краткое изложение сущности изобретения В настоящем изобретении описаны приведенные ниже соединения формулы (I) и (II), которые, как показано, обладают активностью по отношению к киназе Raf(iv) -X1NHC(O)OR1a или -X1NHC(O)NHR1a, где X1 обозначает (С 1-С 4)алкилен, необязательно содержащий в качестве заместителей 1-3 группы, каждая из которых независимо выбрана из группы, включающей галоген, (С 1-С 4)алкил или галогензамещенный (С 1-С 4)алкил,R1a обозначает Н, (С 1-С 4)алкил или галогензамещенный (С 1-С 4)алкил;(С 1-С 6)алкил, галогензамещенный разветвленный (С 3-С 8)алкил, (С 3-С 6)циклоалкил(С 1-С 3)алкилен или фенил, где указанный фенил необязательно содержит 1-3 заместителя, каждый из которых независимо выбран из группы, включающей галоген, СН 3 или CF3;R7 обозначает Н, (С 1-С 6)алкил, (С 3-С 6)циклоалкил, 1-метил-(С 3-С 6)циклоалкил, 1-(галогензамещенный метил)-(С 3-С 6)циклоалкил, разветвленный (С 3-С 8)алкил, галогензамещенный (С 1-С 6)алкил,галогензамещенный разветвленный (С 3-С 8)алкил или фенил, где указанный фенил необязательно содержит 1-3 заместителя, выбранных из группы, включающей галоген, (С 1-С 4)алкил или галогензамещенный С 6)циклоалкил, разветвленный (С 3-С 8)алкил или фенил, где указанный фенил необязательно содержит 13 заместителя, выбранных из группы, включающей галоген, (С 1-С 4)алкил или галогензамещенный (С 1 С 4)алкил; или их фармацевтически приемлемые соли. Один вариант осуществления относится к соединениям формулы (I) или к их фармацевтически приемлемым солям. В одном предпочтительном варианте осуществления соединения формулы I или его фармацевтически приемлемой соли(ii) -X1NHC(O)OR1a, где X1 обозначает (С 1-С 4)алкилен, необязательно содержащий в качестве заместителей 1-3 группы, каждая из которых независимо выбрана из группы, включающей галоген, (С 1 С 4)алкил или галогензамещенный (С 1-С 4)алкил, и R1a обозначает Н, (С 1-С 4)алкил или галогензамещенный (С 1-С 4)алкил;(С 1-С 6)алкил или галогензамещенный разветвленный (С 3-С 8)алкил;R7 обозначает Н, (С 1-С 6)алкил, (С 3-С 6)циклоалкил, 1-метил-(С 3-С 6)циклоалкил, 1-(галогензамещенный метил)-(С 3-С 6)циклоалкил, разветвленный (С 3-С 8)алкил, галогензамещенный (С 1-С 6)алкил или галогензамещенный разветвленный (С 3-С 8)алкил или фенил, где указанный фенил необязательно содержит 1-3 заместителя, выбранных из группы, включающей галоген, (С 1-С 4)алкил или галогензамещенный(С 1-С 4)алкил, предпочтительно, если R7 обозначает Н, (C1-С 6)алкил, (С 3-С 6)циклоалкил, 1-метил-(С 3 С 6)циклоалкил или фенил, где указанный фенил необязательно содержит 1-3 заместителя, выбранных из группы, включающей галоген, (С 1-С 4)алкил или галогензамещенный (C1-С 4)алкил. Один предпочтительный вариант осуществления относится к соединениям формулы (1), в которойR1 обозначает -X1NHC(O)OR1a, где X1 обозначает (C1-С 4)алкилен, необязательно содержащий в качестве заместителей 1-3 группы, каждая из которых независимо выбрана из группы, включающей (С 1-С 4)алкил или галогензамещенный (С 1-С 4)алкил, R1a обозначает (С 1-С 2)алкил или галогензамещенный (С 1-С 2)алкил;(С 1-С 4)алкил или галогензамещенный разветвленный (С 3-С 6)алкил;R7 обозначает (С 3-С 6)циклоалкил, 1-метил-(С 3-С 6)циклоалкил или разветвленный (С 3-С 6)алкил; или к их фармацевтически приемлемым солям. Другой предпочтительный вариант осуществления относится к соединениям формулы (I), в которойR1 обозначает -X1NHC(O)OR1a, где X1 обозначает (С 1-С 4)алкилен, необязательно содержащий в качестве заместителей 1-3 группы, каждая из которых независимо выбрана из группы, включающей (С 1-С 4)алкил или галогензамещенный (С 1-С 4)алкил, R1a обозначает (C1-С 2)алкил или галогензамещенный (С 1-С 2)алкил;(С 1-С 4)алкил, галогензамещенный разветвленный (С 3-С 6)алкил, (С 3-С 6)циклоалкил(С 1-С 3)алкилен или фенил, содержащий 1-3 заместителя, каждый из которых независимо выбран из группы, включающей Cl,F, СН 3 или CF3;R7 обозначает (С 3-С 6)циклоалкил, 1-метил-(С 3-С 6)циклоалкил или разветвленный (С 3-С 6)алкил; или к их фармацевтически приемлемым солям. Другой предпочтительный вариант осуществления относится к соединениям формулы (I), в которойR7 обозначает трет-бутил, циклопропил или 1-трифторметилциклопропил; или к их фармацевтически приемлемым солям. Другой предпочтительный вариант осуществления относится к соединениям формулы (I), в которойR1 обозначает -X1NHC(O)OR1a, где X1 обозначает (С 1-С 4)алкилен, необязательно содержащий в качестве заместителей 1-3 группы, каждая из которых независимо выбрана из группы, включающей (С 1-С 4)алкил или галогензамещенный (С 1-С 4)алкил, и R1a обозначает (C1-С 2)алкил или галогензамещенный (С 1 С 2)алкил;R7 обозначает (С 3-С 6)циклоалкил, 1-метил-(С 3-С 6)циклоалкил или разветвленный (С 3-С 6)алкил; или к их фармацевтически приемлемым солям. Еще один предпочтительный вариант осуществления относится к соединениям формулы (I), в которой R1 обозначает -X1NHC(O)OR1a, где X1 обозначает (С 1-С 2)алкилен, замещенный (С 1-С 2)алкилом, и R1a обозначает (C1-С 2)алкил;R7 обозначает трет-бутил, циклопропил или 1-метилциклопропил; или к их фармацевтически приемлемым солям. Один вариант осуществления относится к соединениям формулы (II) или к их фармацевтически приемлемым солям. Предпочтительный вариант осуществления относится к соединениям формулы (II), в которойR7 обозначает (С 3-С 6)циклоалкил, 1-метил-(С 3-С 6)циклоалкил или разветвленный (С 3-С 6)алкил; или к их фармацевтически приемлемым солям. Другой предпочтительный вариант осуществления относится к соединениям формулы (II), в которой R2 обозначает Н; R3 обозначает Н, Cl, F, метоксигруппу, метил или дифторметоксигруппу; R4 обозначает Н; R5 обозначает метил, циклопропил, этил, пропил, изопропил, втор-бутил, изобутил, трифторметил или 3,3,3-трифторпропил; R6 обозначает Н, метил, F или Cl и R7 обозначает трет-бутил, циклопропил или 1-метилциклопропил; или к их фармацевтически приемлемым солям. Один предпочтительный вариант осуществления относится к соединениям формулы (I) или (II), в которой R1 описывается следующей формулой (1 а) или к их фармацевтически приемлемым солям. Предпочтительные соединения формулы (I) включают(S)-метил-1-(4-(4-(2-хлор-5-метил-3-(метилсульфонамидо)фенил)-2-циклопропил-1 Н-имидазол-5 ил)пиримидин-2-иламино)пропан-2-илкарбамат или их фармацевтически приемлемые соли. Наиболее предпочтительным является (S)-метил-1-(4-(4-(2-хлор-5-фтор-3-(метилсульфонамидо) фенил)-2-циклопропил-1 Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамат или его фармацевтически приемлемая соль. Другие предпочтительные соединения формулы (I) включают(S)-метил-1-(4-(5-(5-хлор-2-фтор-3-(3-фторфенилсульфонамидо)фенил)-2-циклопропил-1 Нимидазол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамат или их фармацевтически приемлемые соли. Предпочтительные соединения формулы (II) включают N-(2-хлор-3-(2-циклопропил-5-(пиримидин 4-ил)-1 Н-имидазол-4-ил)-5-фторфенил)-2,6-дифторбензолсульфонамид; N-(2-хлор-3-(2-циклопропил-5(пиримидин-4-ил)-1 Н-имидазол-4-ил)-5-фторфенил)метансульфонамид и N-(2-хлор-3-(2-циклопропил-5(пиримидин-4-ил)-1 Н-имидазол-4-ил)-5-фторфенил)пропан-1-сульфонамид или их фармацевтически приемлемые соли. Соединения, предлагаемые в настоящем изобретении, ингибируют активность B-Raf и поэтому предположительно будут применимы для лечения заболеваний, связанных с B-Raf. Другим объектом настоящего изобретения является фармацевтическая композиция, которая включает соединение формулы (I) или его фармацевтически приемлемую соль и разбавитель, носитель или инертный наполнитель. Фармацевтическая композиция может дополнительно включать дополнительное терапевтическое средство, где дополнительное терапевтическое средство выбрано из группы, включающей противораковое соединение, анальгетик, противорвотное средство, антидепрессант и противовоспалительное средство. Еще одним объектом настоящего изобретения является способ лечения рака, который включает введение субъекту, нуждающемуся в таком лечении, соединения формулы (I) или его фармацевтически приемлемой соли в фармацевтически эффективном количестве или фармацевтической композиции,включающей соединение формулы (I) или его фармацевтически приемлемую соль и разбавитель, носитель или инертный наполнитель. Другим объектом настоящего изобретения является способ лечения патологического состояния,опосредуемого киназой Raf, который включает введение субъекту, нуждающемуся в таком лечении, соединения формулы (I) или его фармацевтически приемлемой соли в эффективном количестве или фармацевтической композиции, включающей соединение формулы (I) или его фармацевтически приемле-4 020479 мую соль и разбавитель, носитель или инертный наполнитель. Предпочтительно, если опосредующей киназой Raf является мутантная киназа b-Raf, более предпочтительно мутантная киназа b-Raf(V600E). Способы могут включать введение дополнительного терапевтического средства. Предпочтительные дополнительные средства включают противораковое лекарственное средство, средство для лечения боли,противорвотное средство, антидепрессант или противовоспалительное средство, более предпочтительно,если дополнительное терапевтическое средство представляет собой другой ингибитор киназы Raf или ингибитор MEK, mTOR, PI3K, CDK9, PAK, протеинкиназы С, киназы MAP, киназы MAPK или ERK. Определения."Алкил" в качестве группы или структурного элемента других групп, например галогензамещенного алкила и алкоксигруппы, может обладать линейной или разветвленной цепью. С 1-С 4-алкоксигруппы включают метоксигруппу, этоксигруппу и т.п."Галогензамещенный алкил" означает алкильную группу (разветвленную или неразветвленную), в которой любой из атомов водорода может быть замещен галогеном. Типичные примеры галогензамещенного С 1-С 4-алкила включают фторметил, дифторметил, трифторметил, хлорфторметил, дифторэтил,пентафторэтил и т.п. Аналогичным образом, гидроксизамещенный C1-С 6-алкил означает алкильную группу (разветвленную или неразветвленную), в которой любой из атомов водорода может быть замещен гидроксигруппой. Например, гидроксизамещенный С 1-С 6-алкил включает 2-гидроксиэтил и т.п. Аналогичным образом, цианозамещенный C1-С 6-алкил означает алкильную группу (разветвленную или неразветвленную), в которой любой из атомов водорода может быть замещен цианогруппой."Арил" означает моноциклическую или конденсированную бициклическую ароматическую кольцевую систему, содержащую 6-10 кольцевых атомов углерода. Например, арил может представлять собой фенил или нафтил, предпочтительно фенил. "Ариден" означает двухвалентный радикал, образованный из арильной группы."Гетероарил" является таким, как выше определено для арила, в котором один или большее количество элементов кольца представляют собой гетероатом. Например, (С 1-С 10)гетероарил включает пиридил,индолил, индазолил, хиноксалинил, хинолинил, бензофуранил, бензопиранил, бензотиопиранил, бензо[1,3]диоксол, имидазолил, бензоимидазолил, пиримидинил, фуранил, оксазолил, изоксазолил, триазолил, тетразолил, пиразолил, тиенил и т.п."Циклоалкил" означает насыщенную или частично ненасыщенную моноциклическую, конденсированную бициклическую или мостиковую полициклическую кольцевую систему, содержащую указанное количество кольцевых атомов. Например, (С 3-С 10)циклоалкил включает циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил и циклодецил. Предпочтительным циклоалкилом является циклопропил."Гетероциклоалкил" означает циклоалкил, в котором один или большее количество кольцевых атомов углерода заменены фрагментом, выбранным из группы, включающей -О-, -N=, -NR-, -С(О)-, -S-,-S(O)- или -S(O)2-, где R обозначает водород, (С 1-С 4)алкил или защитную группу атома азота (-NPg). Типичные примеры (С 3-С 8)гетероциклоалкила включают 2 Н-пиранил, 4 Н-пиранил, пиперидинил, 1,4 диоксан, морфолинил, 1,4-дитианил, тиоморфолиновую группу, имидазолидин-2-он, тетрагидрофуран,пиперазинил, 1,3,5-тритианил, пирролидинил, пирролидинил-2-он, пиперидинон, 1,4-диокса-8 азаспиро[4.5]дец-8-ил и т.п."Лечение", "лечить" относится к способу облегчения или смягчения заболевания и/или сопутствующих ему симптомов. Термин "соединения, предлагаемые в настоящем изобретении" (если специально не указано иное) означает соединения формулы (I) или (II), их пролекарства, фармацевтически приемлемые соли соединений и/или пролекарств и гидраты или сольваты соединений, солей и/или пролекарств, а также все стереоизомеры (включая диастереоизомеры и энантиомеры), таутомеры и изотопно-меченые соединения. Подробное описание изобретения Соединения, предлагаемые в настоящем изобретении, можно синтезировать путями, которые включают методики, аналогичные хорошо известным в химической науке, в частности, с учетом описания,приведенного в настоящем изобретении. Исходные вещества обычно можно приобрести у таких фирм,как Aldrich Chemicals (Milwaukee, Wis.), или легко получить по методикам, хорошо известным специалистам в данной области техники (например, получить по методикам, описанным в публикациях Louis F.Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, New York (1967-1999 ed.) или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая дополнения (также доступные в виде баз данных Beilstein в интернете. Для иллюстративных целей на схемах реакций, представленных ниже, приведены возможные пути синтеза соединений, предлагаемых в настоящем изобретении, а также основных промежуточных продуктов. Более подробное описание отдельных стадий реакций приведено ниже в разделе "Примеры". Специалисты в данной области техники должны понимать, что для синтеза соединений, предлагаемых в настоящем изобретении, можно использовать другие методики синтеза. Хотя на схемах приведены и ниже обсуждены конкретные исходные вещества и реагенты, легко можно использовать другие исходные ве-5 020479 щества и реагенты и/или условия проведения реакций и получить множество производных. Кроме того,многие соединения, полученные по описанным ниже методикам, можно дополнительно модифицировать с учетом настоящего раскрытия по обычным химическим методикам, хорошо известным специалистам в данной области техники. Соединения формулы I можно получить по методике, представленной ниже на схеме I. Схема I Промежуточный дибромимидазол (1a) можно получить путем конденсации необходимого альдегида (R7C(O)H) с глиоксалем в присутствии гидроксида аммония при температуре от примерно 0 до примерно 5 С с получением замещенного в положении С-2 имидазола (см., например, J. Med. Chem., (1979),22, 687) и последующего бромирования. Депротонирование с помощью NaH и введение защитной группы аминогруппы (например, 2-(триметилсилил)этоксиметилхлорида (SEMCl дает защищенный дибромимидазол (1b). Специалисты в данной области техники должны понимать, что вместо защитной группыSEM можно использовать другие защитные группы атома азота. Направленное литиирование, последующее присоединение 2-хлорпиримидина и последующее окисление могут привести к получению 5-(2 хлор-4-пиримидинил)-4-бромимидазола (1 с). См. публикацию Organic Letters 2005, 7, 4133. Простое замещение SnAr необходимым амином (R1NH) может привести к получению замещенных пиримидинов(1d). Удаление защитной группы SEM (например, с помощью HCl в протонном растворителе, таком как этанол и т.п.) дает промежуточный продукт (1 е) и последующая реакция перекрестного сочетания Судзуки с необходимым сульфонанилидом боронатного сложного эфира или кислоты (1f) дает промежуточный продукт (1g). Заключительную реакцию перекрестного сочетания обычно можно заменить соответствующими реакциями Стилле, в которых боронатный сложный эфир или кислоту заменяют соответствующим станнаном. Затем к первичной аминогруппе промежуточного соединения (1g) можно присоединить необходимый сульфонилхлорид в присутствии основания (например, пиридина) и получить соеди-6 020479 нение формулы (I). Соединения формулы I или II также можно получить по методикам, представленным ниже на схеме Изменение порядка проведения реакций перекрестного сочетания и удаления защитной группы,приведенных на схеме I, на обратный приводит к получению тризамещенного имидазола (1g). Удаление защитной группы имидазола также можно провести в конце последовательности реакций синтеза. Обработка необходимым сульфонилхлоридом (R5SO2Cl) в присутствии пиридина при пониженных температурах дает соединение формулы (I). Другой альтернативный путь получения соединений, предлагаемых в настоящем изобретении,представлен ниже на схеме III. Схема III В другом варианте пути, представленного на схеме I, промежуточный бромимидазол (1 с) можно ввести в реакцию перекрестного сочетания с соответствующим боронатным сложным эфиром или соединением (1f) и получить 3-(5-(2-хлорпиримидин-4-ил)-1 Н-имидазол-4-ил)анилин (3 а). Обработка необходимым сульфонилхлоридом дает соответствующий сульфонанилид (3b). Простое замещение SnAr необходимым амином (R1NH) может дать замещенные пиримидины (3 с) и удаление защитной группы имидазола дает соединения формулы (I). Другой вариант схемы I представлен ниже на схеме IV. Защищенный бромимидазол (1d) или соответствующий незащищенный имидазол (1 е), представленный на схеме I, можно ввести в реакцию перекрестного сочетания с сульфонамидом боронатного сложного эфира (4 а) и получить соответствующий замещенный имидазол (4b) или непосредственно соединения формулы (I). Результат пути, описанного на схеме I для синтеза соединений формулы (I), представлен ниже на схеме V. Схема V В трибромимидазол можно ввести защитную группу, как это описано на схеме I, и получить промежуточный продукт (5 а). Селективная реакция перекрестного сочетания Судзуки в положении С-2 с необязательно замещенной фенилбороновой кислотой или эфиром может привести к получению обычного промежуточного продукта (1 а), который затем можно превратить в соединения формулы (I), как это описано на схеме I. См. публикацию Tetrahedron Letters, 1998, 39, 5171. Специалисты в данной области техники должны понимать, что соединения, предлагаемые в настоящем изобретении, можно получить по методикам, аналогичным описанным в представленном ниже разделе "Примеры". Соединения, предлагаемые в настоящем изобретении (включая промежуточные продукты), можно выделить и использовать в качестве самих соединений или в форме их фармацевтически приемлемых солей, сольватов и/или гидратов. Многие из промежуточных продуктов и соединений, описывающихся формулой I, могут образовывать соли присоединения с кислотами, предпочтительно фармацевтически приемлемые соли присоединения с кислотами. Фармацевтически приемлемые соли присоединения с кислотами соединения, предлагаемого в настоящем изобретении, включают соли с неорганическими кислотами, например галогенводородными кислотами, такими как хлористо-водородная кислота, бромисто-водородная кислота или йодисто-водородная кислота, азотная кислота, серная кислота, фосфорная кислота; и органическими кислотами, например алифатическими монокарбоновыми кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота и масляная кислота, алифатическими гидроксикислотами, такими как молочная кислота, лимонная кислота, винная кислота или яблочная кислота, дикарбоновыми кислотами, такими как малеиновая кислота или янтарная кислота, ароматическими карбоновыми кислотами, такими как бензойная кислота, п-хлорбензойная кислота, дифенилуксусная кислота или трифенилуксусная кислота, ароматическими гидроксикислотами, такими как огидроксибензойная кислота, п-гидроксибензойная кислота, 1-гидроксинафталин-2-карбоновая кислота или 3-гидроксинафталин-2-карбоновая кислота, и сульфоновыми кислотами, такими как метансульфоновая кислота или бензолсульфоновая кислота. Эти соли можно получить из соединений формулы I или II по известным методикам получения солей. Соединения, предлагаемые в настоящем изобретении, которые содержат кислотные, например, карбоксигруппы также могут образовывать соли с основаниями, предпочтительно с фармацевтически приемлемыми основаниями, такими как хорошо известные в данной области техники; такие подходящие соли включают соли металла, предпочтительно соли щелочного металла или щелочно-земельного металла, такие как соли натрия, калия, магния или кальция, или соли с аммиаком или фармацевтически приемлемыми органическими аминами или гетероциклическими основаниями, такими как этаноламины, бензиламины или пиридин. Эти соли можно получить из соединений формулы I по известным методикам получения солей. В тех соединениях, в которых имеется асимметрический атом углерода, соединение существует в виде отдельных оптически активных изомерных форм или в виде их смесей, например в виде рацемических смесей или смесей диастереоизомеров. Если не указано иное, настоящее изобретение включает отдельные оптически активные R- и S-изомеры, а также их смеси, например рацемические смеси или смеси диастереоизомеров. Например, если соединение, предлагаемое в настоящем изобретении, содержит двойную связь или конденсированное кольцо, то его цис- и транс-формы, а также их смеси входят в объем настоящего изобретения. Смеси диастереоизомеров можно разделить на отдельные диастереоизомеры на основании различий из физических и химических характеристик по методикам, известным специалистам в данной области техники, например, с помощью хроматографии и/или фракционной кристаллизации. Энантиомеры можно разделить путем превращения смеси энантиомеров в смесь диастереоизомеров путем реакции с подходящим оптически активным соединением (например, хиральным вспомогательным веществом,таким как хиральный спирт или хлорангидрид кислоты Мошера), разделения диастереоизомеров и превращения (например, путем гидролиза) отдельных диастереоизомеров в соответствующие чистые энантиомеры. Кроме того, некоторые из соединений, предлагаемых в настоящем изобретении, могут представлять собой атропоизомеры (например, замещенные биарилы), и они считаются входящими в объем настоящего изобретения. Энантиомеры также можно разделить на имеющейся в продаже хиральной колонке с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). Соединения, предлагаемые в настоящем изобретении, могут существовать в несольватированной, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода,этанол и т.п., и подразумевается, что в объем настоящего изобретения входят сольватированные и несольватированные формы. Для задач настоящего изобретения сольваты (включая гидраты) считаются фармацевтическими композициями, например соединение формулы I или II (или его фармацевтически приемлемая соль) в комбинации с инертным наполнителем, где инертным наполнителем является растворитель. Само соединение, его фармацевтически приемлемая соль или сольват/гидрат может существовать в аморфной или кристаллической форме (например, в полиморфных формах). Кроме того, промежуточные продукты и соединения, предлагаемые в настоящем изобретении, могут существовать в разных таутомерных формах, и все такие формы входят в объем настоящего изобретения. Термин "таутомер" или "таутомерная форма" означает структурные изомеры, обладающие различными энергиями, которые превращаются друг в друга вследствие низкого энергетического барьера. Например, протонные таутомеры (также известные как прототропные таутомеры) включают таутомеры,превращающиеся друг в друга вследствие переноса протона, такие как изомеры кетон-енол и иминенамин. Конкретным примером протонного таутомера является имидазольный фрагмент, в котором протон может перемещаться между двумя кольцевыми атомами азота. Валентные таутомеры включают таутомеры, превращающиеся друг в друга вследствие перегруппировки некоторых связывающих электронов. Настоящее изобретение включает все фармацевтически приемлемые изотопно-меченые соединения формулы (I) или (II), в которой один или большее количество атомов заменены на атомы, обладающие таким же атомным номером, но атомной массой или массовым числом, отличающимся от атомной массы или массового числа, обычно обнаруживающихся в природе. Примеры изотопов, подходящих для включения в соединения, предлагаемые в настоящем изобретении, включают изотопы водорода, такие как 2 Н и 3 Н, углерода, такие как 11 С, 13 С и 14 С, хлора, такой как 36Cl, фтора, такой как 18F, иода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15 О,17 О и 18 О, фосфора, такой как 32 Р, и серы, такой как 35S. Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2 Н, может обеспечить некоторые терапевтические преимущества, обусловленные их более высокой метаболической стабильностью, например увеличенной длительностью полувыведения in vivo или возможностью использования меньших доз, и поэтому при некоторых обстоятельствах они могут быть предпочтительными. Изотопно-меченые соединения, предлагаемые в настоящем изобретении, обычно можно получить по стандартным методикам, известным специалистам в данной области техники, или по методикам, аналогичным описанным в приведенных ниже примерах и в разделе, посвященном синтезу, с использованием подходящего изотопно-меченого реагента вместо использовавшегося ранее не содержащего изотопа реагента. Соединения, предлагаемые в настоящем изобретении, применимы in vitro или in vivo для подавления роста раковых клеток. Следовательно, соединения, предлагаемые в настоящем изобретении (включая композиции и применяющиеся при этом способы), можно использовать для приготовления лекарственного средства, предназначенного для применений в терапии, описанной в настоящем изобретении. Соединения можно применять по отдельности или в композициях вместе с фармацевтически приемлемым носителем, растворителем (включая воду) или инертным наполнителем. Подходящие фармацевтически приемлемые носители, разбавители или инертные наполнители включают, например, технологические добавки и вещества, модифицирующие и улучшающие доставку лекарственного средства, такие как, например, фосфат кальция, стеарат магния, тальк, моносахариды, дисахариды, крахмал, желатин, целлюлоза, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, декстроза, гидроксипропил 9 020479 циклодекстрин, поливинилпирролидинон, низкоплавкие воски, ионообменные смолы и т.п., а также комбинации любых двух или большего количества из них. Другие подходящие фармацевтически приемлемые инертные наполнители описаны в публикации "Remington's Pharmaceutical Sciences", Mack Pub. Co.,New Jersey (1991), которая включена в настоящее изобретение в качестве ссылки. Фармацевтические композиции содержат растворители (включая воду), включенные в кристаллическую матрицу соединения (также называющиеся сольватами и гидратами). Соединения, предлагаемые в настоящем изобретении, модулируют активность киназ и как таковые применимы для лечения заболеваний или нарушений, при которых киназы способствуют патологии и/или симптоматике этого заболевания. Примеры киназ, которые ингибируются соединениями и композициями, описанными в настоящем изобретении, и по отношению к которым применимы способы, описанные в настоящем изобретении, включают, но не ограничиваются только ею, киназу B-Raf. Путь передачи сигнала Ras-Raf-MEK-ERK обеспечивает передачу сигналов от рецепторов клеточной поверхности к ядрам и является необходимым для пролиферации и жизнеспособности клеток. Поскольку 10-20% случаев раковых заболеваний людей включают мутацию Ras в онкогенную форму и многие раковые заболевания людей характеризуются активированными рецепторами фактора роста, этот путь является идеальной мишенью для воздействия. Семейство Raf серин/треонинкиназ включает трех представителей: C-Raf (или Raf-1), B-Raf и ARaf. И важная роль и расположение Raf на многих путях передачи сигнала продемонстрировано с помощью исследований с использованием дерегулированных и преимущественно ингибирующих мутантовRaf в клетках млекопитающих, а также исследований, в которых для моделирования организмов использованы биохимические и генетические методики. Воздействие на Raf, как на мишень противоопухолевого лекарственного средства, ранее было направлено на его действие, как расположенного в прямом направлении эффектора Ras. Однако последние данные показали, что Raf может играть заметную роль в образовании некоторых опухолей без необходимости наличия онкогенного Ras аллеля. В частности, аллели, активирующие B-Raf, обнаружены в 70% случаев меланом, в 40% случаев папиллярной карциномы щитовидной железы, в 30% случаев низкодифференцированной карциномы яичников и в 10% случаев колоректального рака. Показано, что большинство мутаций B-Raf происходит в домене киназы, с одним замещением (V599E), составляющим 80%. Мутантные белки B-Raf активируют путь Raf-MEK-ERK вследствие повышенной активности киназы по отношению к MEK или путем активирования C-Raf. Поэтому разработка ингибиторов киназы B-Raf предоставляет новые возможности для лечения многих типов раковых заболеваний людей, в особенности метастатических меланом, солидных опухолей, опухолей мозга, таких как мультиформная глиобластома (МГБ), острого миелогенного лейкоза(ОМЛ), папиллярной карциномы щитовидной железы, низкодифференцированной карциномы яичников и колоректального рака. Для некоторых ингибиторов киназы Raf с помощью исследований in vitro и/илиin vivo показано, что они эффективно подавляют пролиферацию опухолевых клеток (см., например, патенты U.S.6391636; 6358932; 6037136; 5717100; 6458813; 6204467 и 6268391). В других патентах и заявках на патенты предлагают использовать ингибиторы киназы Raf для лечения лейкоза (см., например, патенты U.S.6268391; 6204467; 6756410 и 6281193 и "брошенные" заявки на патенты U.S.20020137774 и 20010006975) или для лечения рака молочной железы (см., например, патенты U.S.6358932; 5717100; 6458813; 6268391; 6204467 и 6911446). Соединения, предлагаемые в настоящем изобретении, также подавляют клеточные процессы, включающие киназу B-Raf, путем блокирования последовательности сигналов в этих раковых клетках и, в конечном счете, вызывают остановку роста и/или гибель клеток. В соответствии с приведенным выше настоящее изобретение также относится к способу предупреждения или лечения любого из заболеваний или нарушений, описанных выше, у субъекта, нуждающегося в таком лечении, и этот способ включает введение указанному субъекту соединения формулы I или его фармацевтически приемлемой соли в терапевтически эффективном количестве (см. ниже "Введение и фармацевтические композиции"). Для любого из указанных случаев применения необходимая доза меняется в зависимости от пути введения, конкретного подвергающегося лечению патологического состояния и необходимого эффекта. Обычно соединения, предлагаемые в настоящем изобретении, вводят в терапевтически эффективных количествах посредством любого из обычных и приемлемых путей, известных в данной области техники, по отдельности или в комбинации с одним или большим количеством терапевтических средств. Терапевтически эффективное количество может меняться в широких пределах в зависимости от тяжести заболевания, возраста и относительного состояния здоровья субъекта, активности применяющегося соединения и других факторов. Имеются указания о том, что обычно удовлетворительные результаты получают при системном воздействии и суточных дозах, равных от примерно 0,03 до 2,5 мг/(кг массы тела). Указанная суточная доза для более крупного млекопитающего, например человека, находится в диапазоне от примерно 0,5 до примерно 100 мг, обычно вводится, например, в виде разделенных доз вплоть до 4 раз в сутки или в форме пролонгированного действия. Разовые дозированные формы, подходящие для перорального введения, содержат от примерно 1 до 50 мг активного ингредиента. Фармацевтические композиции можно приготовить по обычным методикам растворения и смеши- 10020479 вания. Например, партию лекарственного вещества (т.е. соединения, предлагаемого в настоящем изобретении, или стабилизированной формы соединения (например, комплекс с производным циклодекстрина или другим известным комплексообразующим агентом растворяют в подходящем растворителе в присутствии одного или большего количества инертных наполнителей, описанных выше. Соединение, предлагаемое в настоящем изобретении, обычно готовят в виде фармацевтических дозированных форм, чтобы обеспечить легко регулируемую дозировку лекарственного средства и предоставить пациенту привлекательный и удобный в обращении продукт. Соединения, предлагаемые в настоящем изобретении, можно вводить в виде фармацевтических композиций любым обычным путем, в частности энтерально, например перорально, например в виде таблеток или капсул, или парентерально, например в виде растворов или суспензий для инъекций, местно, например в виде лосьонов, гелей, мазей или кремов, или в назальной форме или в форме суппозиториев. Фармацевтические композиции, включающие соединение, предлагаемое в настоящем изобретении,в свободной форме или в форме фармацевтически приемлемой соли совместно по меньшей мере с одним фармацевтически приемлемым носителем или разбавителем можно приготовить обычным путем по методикам смешивания, гранулирования или нанесения покрытий. Например, композиции для перорального введения могут представлять собой таблетки или желатиновые капсулы, включающие активный ингредиент совместно с а) разбавителями, например лактозой, декстрозой, сахарозой, маннитом, сорбитом,целлюлозой и/или глицином; b) смазывающими веществами, например диоксидом кремния, тальком,стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; для таблеток также со с) связующими, например алюмосиликатом магния, крахмальной пастой, желатином, трагакантовой камедью, метилцеллюлозой, натриевой солью карбоксиметилцеллюлозы и/или поливинилпирролидоном; при необходимости с d) разрыхлителями, например крахмалами, агаром, альгиновой кислотой или ее натриевой солью или шипучими смесями; и/или е) абсорбентами, красителями, вкусовыми добавками и подсластителями. Композиции для перорального введения кроме активного ингредиента могут также включать 20-60% эудрагита ЕРО, гидроксипропилцеллюлозы EF, гидроксипропилметилцеллюлозы или коллидона VA64 и не более 5% плуроника F68, кремофора EL или гелюцира 44/14. Композиции для инъекций могут представлять собой водные изотонические растворы или суспензии, и суппозитории можно приготовить из эмульсий или суспензий жирных веществ. Композиции могут быть стерилизованы и/или содержать вспомогательные вещества, такие как консервирующие, стабилизирующие, смачивающие или эмульгирующие агенты, стимуляторы растворения, соли для регулирования осмотического давления и/или буферные вещества. Кроме того, они также могут содержать другие терапевтически полезные вещества. Препараты, подходящие для чрескожного введения, включают соединение, предлагаемое в настоящем изобретении, в эффективном количестве вместе с носителем. Носитель может включать впитывающиеся фармакологически приемлемые растворители, содействующие проникновению через кожу реципиента. Например, чрескожные устройства представляют собой повязку, включающую защитный элемент, резервуар, содержащий соединение, необязательно совместно с носителями, необязательно регулирующий скорость барьерный элемент для доставки соединения к коже реципиента с регулируемой и заранее заданной скоростью в течение продолжительного периода времени и средства для закрепления устройства на коже. Также можно использовать матричные препараты для чрескожного введения. Препаратами, подходящими для местного нанесения, например на кожу и в глаза, предпочтительно являются водные растворы, мази, кремы или гели, хорошо известные в данной области техники. Они могут содержать солюбилизаторы, стабилизаторы, усиливающие тоничность агенты, буферные вещества и консерванты. В некоторых способах лечения может оказаться предпочтительным введение соединений, предлагаемых в настоящем изобретении, в комбинации с одним или большим количеством терапевтических средств (фармацевтические комбинации). Например, синергетический эффект может проявляться при использовании других противоопухолевых или антипролиферативных средств, например ингибиторов митоза, алкилирующих средств, антиметаболитов, интеркалирующих антибиотиков, ингибиторов фактора роста (например, трастузумаб, панитумумаб, цетуксимаб, гефитиниб, эрлотиниб, лапатиниб, сорафениб и т.п.), ингибиторов клеточного цикла, ферментов, ингибиторов топоизомеразы, модификаторов биологического ответа, антител, цитотоксинов, антигормонов, антиандрогенов, антиангиогенезного средства, ингибитора киназы, ингибитора киназы pan или ингибитора фактора роста. Подходящие терапевтические средства включают эрлотиниб, доцетаксел, гемцитабин, цисплатин, карбоплатин, паклитаксел, бевацизумаб, трастузумаб, пертузумаб, темозоломид, тамоксифен, доксорубицин, рапамицин и лапатиниб. Другие подходящие терапевтические средства перечислены в публикации Physicians Desk Reference. Предпочтительные терапевтические средства, предназначенные для использования в комбинированной терапии, включают ингибиторы MEK (например, сорафениб, AZD6244 (соединение примера 10 вAxon Medchem), XL-765 и XL-147. Когда соединения, предлагаемые в настоящем изобретении, вводят совместно с другими средствами, разумеется, дозы вводимых совместно соединений будут меняться в зависимости от типа используемого совместно лекарственного средства, конкретного используемого лекарственного средства, подвергающегося лечению патологического состояния и т.п. В соответствии со способами, предлагаемыми в настоящем изобретении, соединение, предлагаемое в настоящем изобретении, или комбинацию соединения, предлагаемого в настоящем изобретении, и по меньшей мере одного дополнительного фармацевтического средства вводят субъекту, нуждающемуся в таком лечении, предпочтительно в виде фармацевтической композиции. В комбинированной терапии,предлагаемой в настоящем изобретении, соединение, предлагаемое в настоящем изобретении, и по меньшей мере одно другое фармацевтическое средство (описанное выше) можно вводить по отдельности или в виде фармацевтической композиции, содержащей оба средства. Обычно предпочтительно, если такое введение является пероральным. Однако, если подвергающийся лечению пациент неспособен глотать или пероральное введение приносит вред или является нежелательным в других отношениях, то могут являться подходящими парентеральное или чрескожное введение. В соответствии со способами, предлагаемыми в настоящем изобретении, когда комбинацию соединения, предлагаемого в настоящем изобретении, и по меньшей мере одного другого фармацевтического средства вводят совместно, то такое введение может быть последовательным по времени или одновременным, обычно предпочтительным является способ одновременного введения. В случае последовательного введения соединение, предлагаемое в настоящем изобретении, и дополнительное фармацевтическое средство можно вводить в любом порядке. Обычно предпочтительно, если такое введение является пероральным. Особенно предпочтительно, если такое введение является пероральным и одновременным. Когда соединение, предлагаемое в настоящем изобретении, и дополнительное фармацевтическое средство вводят последовательно, то введение каждого из них можно проводить одинаковыми или разными способами. В зависимости от способа введения лекарственного средства фармацевтическая композиция (или препаративная форма), предназначенная для применения, может быть расфасована различным образом. Обычно изделие, предназначенное для потребителя, включает контейнер с находящейся в нем фармацевтической композицией в соответствующей форме. Подходящие контейнеры хорошо известны специалистам в данной области техники, и они включают такие продукты, как флаконы (пластмассовые и стеклянные), пакеты-саше, ампулы, пластмассовые мешки, металлические цилиндры и т.п. Контейнер также может содержать систему защиты от неумелого обращения, предназначенную для предотвращения неосторожного доступа к содержимому упаковки. Кроме того, на контейнере находится этикетка, на которой описано содержимое контейнера. На этикетке также могут находиться соответствующие предупреждения. Настоящее изобретение также относится к фармацевтическим комбинациям, например к набору,включающему: а) первое средство, которое является соединением, предлагаемым в настоящем изобрете- 12020479 нии, описанным в настоящем изобретении, в свободной форме или в форме фармацевтически приемлемой соли, и b) по меньшей мере одно дополнительное терапевтическое средство. Набор может включать инструкции по его введению. Термины "совместное введение" или "комбинированное введение" и т.п. при использовании в настоящем изобретении означают введение выбранных терапевтических средств одному пациенту и включают режимы лечения, при которых средства необязательно вводят по одному и тому же пути введения или в одно и то же время. Термин "фармацевтическая комбинация" при использовании в настоящем изобретении означает препарат, который получен смешиванием или объединением более одного активного ингредиента, и включает и фиксированные, и нефиксированные комбинации активных ингредиентов. Термин "фиксированная комбинация" означает, что активные ингредиенты, например соединение формулы I и дополнительное средство, оба вводят пациенту одновременно в виде одного препарата или дозированной формы. Термин "нефиксированная комбинация" означает, что активные ингредиенты, например соединение формулы I и дополнительное средство, оба вводят пациенту в виде отдельных препаратов одновременно,по отдельности или последовательно без наложения специальных ограничений по времени, и при таком введении в организме пациента образуются терапевтически эффективные содержания двух соединений. Последнее также относится к смешанному лечению, например, с введением трех или большего количества активных ингредиентов. Примеры Настоящее изобретение дополнительно описано, но без наложения ограничений, с помощью приведенных ниже промежуточных продуктов и примеров соединений, которые иллюстрируют получение соединений, предлагаемых в настоящем изобретении. Препаративные разделения проводили с помощью системы CombiFlash Rf (Teledyne Isco Inc. Lincoln, NE) в комбинации с содержащими диоксид кремния флэш-колонками с нормальной фазой RediSep (4-120 г, размер частиц 35-70 мкм; Teledyne Isco Inc.) или с помощью колоночной флэшхроматографии с использованием силикагеля (230-400 меш) в качестве насадки, или с помощью ВЭЖХ(высокоэффективная жидкостная хроматография) с использованием пробоотборника WATERS 2767, колонки с обращенной фазой С-18, 3050 мм, скорость потока 75 мл/мин. Типичными растворителями,использованными для системы CombiFlash и колоночной флэш-хроматографии, являлись дихлорметан,метанол, этилацетат, гексан, ацетон, водный раствор аммиака (или гидроксид аммония) и триэтиламин. Типичными растворителями, использованными для ВЭЖХ с обращенной фазой, являлись ацетонитрил в разных концентрациях и вода с добавлением 0,1% трифторуксусной кислоты (ТФК). Реакции с обработкой микроволновым излучением проводили в микроволновом реакторе Creator или Initiator (Biotage, Charlottesville, VA). В представленном ниже экспериментальном разделе использованы приведенные ниже аббревиатуры, обладающие указанными значениями. ДЭАД - диэтилазодикарбоксилат,ДИЭА - диизопропилэтиламин,ТГФ - тетрагидрофуран,Et2O - диэтиловый эфир,ДМФ - диметилформамид,NMP - N-метилпирролидинон,ДМЭ - 1,1-диметоксиэтан,ДФФА - дифенилфосфорилазид,EtOAc - этилацетат,ТФК - трифторуксусная кислота,NBS - N-бромсукцинимид,dba - дибензилиденацетон,dppf - бис(дифенилфосфино)ферроцен,ППТС - пиридиний-п-толуолсульфонат. Получение основных исходных веществ и промежуточных продуктов. Получение исходного вещества, (S)-трет-бутил-1-аминопропан-2-илкарбамата (ИВ-1) Стадия 1. Получение (S)-трет-бутил-1-(1,3-диоксоизоиндолин-2-ил)пропан-2-илкарбамата. При перемешивании к раствору (S)-трет-бутил-1-гидроксипропан-2-илкарбамата (7,4 г, 42,2 ммоль) в сухом ТГФ (420 мл) добавляли фталимид (6,83 г, 46,4 ммоль) и PPh3 (12,18 г, 46,4 ммоль). Затем к раствору при перемешивании при комнатной температуре по каплям добавляли ДЭАД (7,3 мл, 46,4 ммоль) и выдерживали в течение 3 ч. Затем реакционную смесь концентрировали и полученный остаток очища- 13020479(жидкостная хроматография-массспектрометрия) (m/z) 205,1 (МН+ - ВОС), tR = 0,86 мин; 1 Н ЯМР (CDCl3, 400 МГц)7,82-7,87 (m, 2 Н), 7,67-7,75 (m, 2 Н), 4,60-4,76 (br d, 1 Н), 4,03-4,20 (br s,1 Н), 3,62-3,72 (m, 2 Н), 1,25 (s, 9 Н), 1,21 (d, J = 6,6 Гц, 3 Н). Стадия 2. Получение (S)-трет-бутил-1-аминопропан-2-илкарбамата (ИВ-1). Гидразинмоногидрат (20 мл, 642,7 ммоль) добавляли к суспензии (S)-трет-бутил-1-(1,3 диоксоизоиндолин-2-ил)пропан-2-илкарбамата (12,5 г, 41,1 ммоль) в сухом метаноле (150 мл) и полученную смесь нагревали при 50 С в течение 1 ч. После охлаждения до комнатной температуры реакционную смесь фильтровали через воронку с фильтром из пористого стекла и фильтрат концентрировали. Полученный остаток суспендировали в диэтиловом эфире (300 мл) и фильтровали, тщательно промывая осадок на фильтре диэтиловым эфиром. Объединенные фильтраты фильтровали и концентрировали и получали 6,3 г (S)-трет-бутил-1-аминопропан-2-илкарбамата; 1 Н ЯМР (CDCl3, 400 МГц)4,44-4,71 (br s, 1 Н), 3,53-3,74 (br m, 1 Н), 2,75 (dd, J = 4,9, 12,9 Гц, 1 Н),2,64 (dd, J = 6,6, 12,9 Гц, 1 Н), 1,45 (s, 9 Н), 1,15-1,34 (br s, 2 Н), 1,12 (d, J = 6,7 Гц, 3H). Получение исходного вещества, (S)-трет-бутил-1-аминопропан-2-илкарбамата (ИВ-2)(R)-трет-бутил-1-аминопропан-2-илкарбамат получали по методике, сходной с описанной выше, с использованием в качестве исходного вещества (R)-трет-бутил-1-гидроксипропан-2-илкарбамата. Получение исходного вещества, 3-амино-2-метилпропаннитрила (ИВ-3) По методике, описанной в патенте US2659739, в стальной автоклав объемом 200 мл помещали 32% водный раствор аммиака (81 мл) и метакрилонитрил (18 г, 23 мл, 270 ммоль). Сосуд для проведения реакции герметизировали и смесь при перемешивании нагревали при 135 С в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и реакционную смесь перегоняли при пониженном давлении (84-85 С, 24 мбар) и получали 12,4 г (147 ммоль, 53%) 3-амино-2-метилпропаннитрила в виде бесцветного масла; 1 Н ЯМР (CDCl3, 400 МГц)2,89 (dd, J = 4,9, 6,5 Гц, 2 Н), 2,63-2,72 (m, 1 Н), 1,37 (2 Н), 1,32 (m, 2 Н),1,31 (d, J = 7,4 Гц, 3 Н). Получение исходного вещества, 3-амино-2-метилпропаннитрила (ИВ-4) Стадия 1. Получение 1-(гидроксиметил)циклопропанкарбонитрила. По методике, описанной в WO 2009/024550, раствор этил-1-цианоциклопропанкарбоксилата (4,0 г,28,7 ммоль) в ДМЭ (80 мл) и метаноле (8 мл) обрабатывали с помощью NaBH4 (8,7 г, 230 ммоль) и перемешивали при комнатной температуре в течение 24 ч. Затем реакцию останавливали насыщенным водным раствором NaHCO3 (100 мл), следя за выделением газа, и затем смесь экстрагировали смесью ДХМ(дихлорметан)-МеОН состава 9:1 (350 мл). Органические слои объединяли, сушили (Na2SO4), концентрировали и получали 1-(гидроксиметил)циклопропанкарбонитрил (2,34 г, 24,1 ммоль, 84%) в виде бесцветного масла, которое использовали без дополнительной очистки; 1 Н ЯМР (300 МГц, CDCl3)3,63 (s, 2 Н), 2,10-2,45 (br s, 1 Н), 1,20-1,35 (m, 2 Н), 0,90-1,05 (m, 2 Н). Стадия 2. Получение 1-1,3-диоксоизоиндолин-2 ил)метил)циклопропанкарбонитрила. Раствор 1-(гидроксиметил)циклопропанкарбонитрила (5,52 г, 56,8 ммоль), фталимида (9,20 г, 62,5 ммоль) и трифенилфосфина (16,4 г, 62,5 ммоль) в ТГФ (550 мл) обрабатывали с помощью ДЭАД (9,90 мл, 62,5 ммоль) и перемешивали при комнатной температуре в течение 17 ч. Затем реакционную смесь концентрировали и полученное твердое вещество растирали с диэтиловым эфиром (150 мл) и собирали фильтрованием. Очистка с помощью флэш-хроматографии (SiO2, 0-20% EtOAc в ДХМ) давала 1-1,3 диоксоизоиндолин-2-ил)метил)циклопропанкарбонитрил (8,8 г, 38,8 ммоль, 68%) в виде белого твердого вещества: ЖХМС (m/z) 227,0 (МН+), tR = 0,66 мин; 1 Н ЯМР (300 МГц, CDCl3)7,90 (dd, J = 5,4, 3,1 Гц, 2 Н), 7,77 (dd, J = 5,4, 3,1 Гц, 2 Н), 3,81 (s, 2 Н),- 14020479 1,30-1,44 (m, 2 Н), 1,28 (d, J = 3,8 Гц, 2 Н). Стадия 3. Получение 1-(аминометил)циклопропанкарбонитрила (ИВ-4). Раствор 1-1,3-диоксоизоиндолин-2-ил)метил)циклопропанкарбонитрила (8,78 г, 38,8 ммоль) и гидразинмоногидрата (9,5 мл, 190 ммоль) в МеОН (150 мл) нагревали при 60 С в течение 3 ч, через 30 мин после начала нагревания выпадал осадок. Затем реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали в вакууме и полученный остаток обрабатывали ультразвуком с использованием Et2O (150 мл). Полученную суспензию фильтровали через воронку с фильтром из пористого стекла и фильтрат концентрировали и сушили в вакууме и получали 1-(аминометил) циклопропанкарбонитрил (3,6 г, 36,3 ммоль, 93%) в виде бесцветного масла; 1 Н ЯМР (300 МГц, CDCl3)2,76 (s, 2 Н), 1,60-2,25 (br s, 2 Н), 1,16-1,35 (m, 2 Н), 0,79-0,98 (m, 2 Н). Получение исходного вещества, 5-хлор-2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)анилина (ИВ-5) Стадия 1. Получение 3-бром-5-хлор-2-фторбензойной кислоты. К охлажденному раствору диизопропиламина (2,4 мл, 17,2 ммоль) в сухом ТГФ (15 мл) в атмосфере аргона при 0 С добавляли н-бутиллитий (7,5 мл, 2,0 М раствор в пентане, 15,0 ммоль). Через 30 мин раствор охлаждали до -78 С и в течение 15 мин добавляли раствор 2-бром-4-хлор-1-фторбензола (3 г, 14,3 ммоль) в сухом ТГФ (15 мл). Через 1 ч этот раствор при -78 С через канюлю в течение 10 мин переносили в смесь твердого диоксида углерода и ТГФ (30 мл). Через 45 мин охлаждающую баню удаляли и избытку диоксида углерода давали испариться при нагревании раствора до комнатной температуры. Затем реакцию останавливали 0,5 М водным раствором HCl (60 мл). После концентрирования оставшуюся водную фазу подщелачивали 0,5 М водным раствором NaOH и промывали этилацетатом (100 мл). Затем водную фазу подкисляли 1,0 М водным раствором HCl и экстрагировали хлороформом (275 мл). Объединенные органические порции промывали рассолом, сушили (Na2SO4), концентрировали и получали 3,34 г полукристаллического твердого вещества, которое представляло собой смесь региоизомеров, в которой искомый изомер содержался в большем количестве: ЖХМС (m/z) не наблюдается (МН+), tR = 0,66 мин; 1 Н ЯМР (400 МГц, CDCl3)7,80 (dd, J = 5,5, 2,7 Гц, 1 Н), 7,95 (dd, J = 5,5, 2,7 Гц, 1 Н). Стадия 2. Получение трет-бутил-3-бром-5-хлор-2-фторфенилкарбамата. Смесь 3-бром-5-хлор-2-фторбензойной кислоты (3,34 г, 13,2 ммоль), ДИЭА (2,04 г, 15,8 ммоль) и дифенилфосфорилазида (ДФФА, 4,53 г, 16,5 ммоль) в смеси сухих трет-бутанола и толуола состава 1:1(35 мл) нагревали и выдерживали при кипячении с обратным холодильником в течение 23 ч. Реакционной смеси давали охладиться до комнатной температуры и затем ее подвергали распределению междуCHCl3 (75 мл) и водой (75 мл). Органический слой отделяли, промывали рассолом, сушили (Na2SO4), затем концентрировали при пониженном давлении и получали маслообразный остаток. Очистка с помощью флэш-хроматографии (SiO2, 0-5% EtOAc в гептане) давала трет-бутил-3-бром-5-хлор-2 фторфенилкарбамат (2,45 г, 7,5 ммоль, 57%): ЖХМС (m/z) не ионизируется (МН+), tR = 1,20 мин; 1 Н ЯМР (400 МГц, CDCl3)8,17 (d, J = 5,1 Гц, 1 Н), 7,16 (m, 1 Н), 6,72 (br s, 1 Н), 1,53 (s, 9 Н). Стадия 3. Получение 3-бром-5-хлор-2-фторанилина. трет-Бутил-3-бром-5-хлор-2-фторфенилкарбамат (1,0 г, 3,1 ммоль) обрабатывали 4,0 М растворомHCl в диоксане (10 мл) и полученную реакционную смесь выдерживали при комнатной температуре в течение 4 ч. Реакционную смесь концентрировали и подвергали распределению между EtOAc (200 мл) и насыщенным водным раствором NaHCO3 (75 мл). Слои разделяли и органическую порцию промывали рассолом (75 мл), сушили (Na2SO4), концентрировали и получали 3-бром-5-хлор-2-фторанилин в виде бесцветного масла, которое использовали на следующей стадии без дополнительной очистки: ЖХМСH ЯМР (400 МГц, CDCl3)6,88 (m, 1 Н), 6,70 (d, J = 7,0 Гц, 1 Н). Стадия 4. Получение 5-хлор-2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ-5). Смесь 3-бром-5-хлор-2-фторанилина (0,69 г, 3,1 ммоль), бис(пинаколято)дибора (0,94 г, 3,7 ммоль) и ацетата калия (0,91 г, 9,2 ммоль) в сухом диоксане (11 мл) продували с помощью N2. Затем добавляли дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладий (0,23 г, 0,31 ммоль) и реакционный сосуд герметизировали. Смесь нагревали на масляной бане при 105 С в течение 3,5 ч. Затем реакционной смеси давали охладиться до комнатной температуры, ее центрифугировали и надосадочную жидкость, которая содержала искомое соединение, 5-хлор-2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин, декантировали и использовали без дополнительной очистки: ЖХМС (m/z) 189,9 (МН+, бороновая кислота), tR = 0,43 мин. Стадия 1. Получение N-(3-бром-5-хлор-2-фторфенил)пропан-1-сульфонамида. К раствору 3-бром-5-хлор-2-фторанилина (ИВ-5, стадия 3, 0,52 г, 2,3 ммоль) в пиридине (2,5 мл),охлажденному в бане из воды со льдом, добавляли пропан-1-сульфонилхлорид (0,3 мл, 2,8 ммоль). Через 4 ч раствор концентрировали и подвергали распределению между EtOAc (75 мл) и 0,1 М водным раствором HCl (30 мл). Слои разделяли и затем органическую фазу промывали рассолом, сушили (Na2SO4) и концентрировали и получали 778 мг N-(3-бром-5-хлор-2-фторфенил)пропан-1-сульфонамида в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки: ЖХМС (m/z) не наблюдается; tR = 0,95 мин. Стадия 2. Получение N-(5-хлор-2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропан-1-сульфонамида (ИВ-6). Это вещество получали по методике, использованной для получения ИВ-5, стадия 4: ЖХМС (m/z) 590,3; (2 МН+), tR = 0,67 мин. Получение исходного вещества,2,5-дифтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)анилина (ИВ-7) Стадия 1. Получение 3-бром-2,5-дифторанилина. Смесь 1,3-дибром-2,5-дифторбензола (4 г, 14,7 ммоль), бензофенонимина (2,6 мл, 15,5 ммоль), третбутоксида натрия (2,1 г, 22,1 ммоль), (S)-бинаф (2,2'-бис(дифенилфосфино)-1,1'-бинафтил) (1,4 г, 2,2 ммоль) в толуоле (15 мл) продували аргоном и добавляли Pd2(dba)3 (0,67 г, 0,74 ммоль). Затем реакционный сосуд герметизировали и смесь облучали в микроволновом реакторе при 100 С в течение 30 мин. Реакционную смесь разбавляли с помощью Et2O и перемешивали в течение 2 ч с поглотителем палладия(Siliabond DMT). Смесь фильтровали через слой целита и собранный фильтрат подвергали распределению между диэтиловым эфиром и водой и полученные слои разделяли. Органическую порцию промывали водой, рассолом, сушили (MgSO4) и концентрировали и получали красно-коричневое твердое вещество. Твердое вещество растворяли в ТГФ (40 мл) и обрабатывали 6,0 н. водным раствором HCl (25 мл, 150 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч и подвергали распределению между водой и Et2O. Слои разделяли и значение рН водной порции 1,0 М водным раствором NaOH доводили до 9. Основный водный слой экстрагировали диэтиловым эфиром (330 мл), объединенные слои, содержащие диэтиловый эфир, промывали 1,0 М водным раствором NaOH, водой, рассолом, сушили (MgSO4), концентрировали. Очистка с помощью флэш-хроматографии (SiO2, 0-10% EtOAc в гептане) давала 3-бром-2,5-дифторанилин (1,7 г, 8,2 ммоль, 56%, следовое количество бензофенона) в виде оранжевого твердого вещества; 1 Н ЯМР (400 МГц, CDCl3)3,93 (br s, 2 Н), 6,43 (m, 1H), 6,62 (m, 1H). Стадия 2. Получение 2,5-дифтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина. К раствору 3-бром-2,5-дифторанилина (2,0 г, 9,4 ммоль), бис(пинаколято)дибора (2,86 г, 11,25 ммоль), трициклогексилфосфина (0,184 г, 0,656 ммоль) и ацетата калия (1,380 г, 14,06 ммоль) в 1,4 диоксане (1,0 мл) добавляли Pd2(dba)3 (0,26 г, 0,28 ммоль) и полученную реакционную смесь облучали в микроволновой печи при 120 С в течение 30 мин. Реакционной смеси давали охладиться до комнатной температуры и ее разбавляли с помощью EtOAc и добавляли поглотитель палладия (Silicycle DMT) и смесь перемешивали в течение 30 мин, затем фильтровали через воронку с фильтром из пористого стекла. Фильтрат промывали водой, рассолом, сушили (MgSO4) и концентрировали и получали отфильтрованный и не содержащий растворителя 2,5-дифтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)анилин (2,4 г, 9,4 ммоль): ЖХМС (m/z) 255,1 (МН+), tR = 0,95 мин; ЖХМС (m/z) 174,0 (МН+, бороновая кислота), tR = 0,3 мин. Получение исходного вещества, 2-хлор-5-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)анилина (ИВ-8) Стадия 1. Получение 3-бром-2-хлор-5-фторанилина. В герметизированную стеклянную пробирку помещали 1,3-дибром-2-хлор-5-фторбензол (12,52 г,43,4 ммоль), бензофенонимин (8,26 г, 45,6 ммоль), трет-бутоксид натрия (6,26 г, 65,1 ммоль) и толуол(100 мл). Полученную смесь тщательно продували аргоном, затем добавляли Pd2(dba)3 (0,398 г, 0,434 ммоль) и (S)-бинаф (0,81 г, 1,3 ммоль) и повторно обрабатывали аргоном. Реакционную пробирку герметизировали и нагревали на масляной бане до 85 С и выдерживали в течение ночи. Реакционной смеси давали охладиться до комнатной температуры и реакцию останавливали водой (20 мл). Полученным слоям давали подвергнуться распределению и их разделяли. Органическую фазу концентрировали и по данным анализа остаток представлял собой смесь моно- и бис-аминированных продуктов (состав 4:1, по данным о площадях пиков, полученным с помощью ВЭЖХ при 220 нм). Остаток растворяли в ТГФ (70 мл), обрабатывали 3,0 М водным раствором HCl (20 мл) при комнатной температуре в течение 1 ч и подщелачивали насыщенным водным раствором Na2CO3 (40 мл). Реакционную смесь подвергали распределению и слои разделяли. Органическую порцию отделяли, промывали рассолом, концентрировали и полученный остаток очищали с помощью флэш-хроматографии (SiO2, 0-15% EtOAc в гептане), получали 3 бром-2-хлор-5-фторанилин в виде светло-желтого твердого вещества (6,82 г, 30,4 ммоль): ЖХМС (m/z) не ионизируется (МН+), tR = 0,95 мин; 1 Н ЯМР (CDCl3, 300 МГц)4,32 (br s, 2 Н), 6,44 (dd, J = 9,8, 2,8 Гц, 1 Н), 6,77 (dd, J = 7,9, 2,6 Гц, 1 Н). Стадия 2. Получение 2-хлор-5-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ-8). В стеклянный сосуд высокого давления добавляли 3-бром-2-хлор-5-фторанилин (10,22 г, 45,5 ммоль), бис(пинаколято)дибор (13,9 г, 54,6 ммоль), трициклогексилфосфин (0,89 г, 3,2 ммоль), ацетат калия (6,70 г, 68,3 ммоль) и Pd(dba)2 (1,31 г, 2,3 ммоль) в 1,4-диоксане (170 мл) и получали красную суспензию, которую продували азотом, и затем сосуд для проведения реакции герметизировали. Реакционную смесь нагревали на масляной бане при 120 С в течение 5 ч и затем ей давали охладиться до комнатной температуры. Добавляли SiliaBond DMT (10 г, выпускающийся фирмой Silicycle) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь разбавляли с помощью EtOAc и фильтровали через нейтральный оксид алюминия с покрытием из силикагеля, тщательно промывая осадок на фильтре с помощью EtOAc. Объединенные фильтраты подвергали распределению с водой и фазы разделяли. Водную фазу экстрагировали с помощью EtOAc. Органические фазы объединяли, промывали водой, рассолом, сушили (Na2SO4), фильтровали, концентрировали и получали желтое масло. Добавляли гептан и смесь кратковременно обрабатывали ультразвуком и получали суспензию, которую концентрировали, и получали 2-хлор-5-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (14,64 г, 35,0 ммоль),чистота 65% по данным 1 Н ЯМР) в виде оранжевого твердого вещества, которое использовали без дополнительной очистки. Для характеризации получали очищенный образец (SiO2, 0-50% EtOAc в гептане): ЖХМС (m/z): 272,0 (МН+), tR = 0,99 мин; 1 Стадия 1. Получение 1-бром-2-хлор-3-нитробензола. В высушенную в сушильном шкафу круглодонную колбу, снабженную стержнем для перемешивания и высушенным в сушильном шкафу холодильником, в атмосфере N2 при комнатной температуре добавляли 2-хлор-3-нитробензойную кислоту (6,0 г, 29,8 ммоль), красный оксид ртути(II) (9,67 г, 44,7 ммоль) и тетрахлорид углерода (200 мл). Реакционную смесь нагревали при 90 С в течение 30 мин при облучении осветительной лампой 150 Вт TYPE A. Затем реакционную смесь охлаждали примерно до 60 С и шприцем по каплям добавляли бром (2,30 мл, 44,7 ммоль) и к патрубку для подачи азота присоединяли баллон с Ar. Реакционную смесь повторно нагревали при 90 С в течение 4 ч при постоянном облучении осветительной лампой. Реакционной смеси давали охладиться до комнатной температуры, реакцию останавливали насыщенным водным раствором NaHCO3 и ДХМ и смесь перемешивали в течение 30 мин. При выдерживании происходило разделение фаз и затем слои разделяли. Водную порцию экстраги- 17020479 ровали с помощью ДХМ. Объединенные органические фазы промывали водой, рассолом, сушили(8,30 г, 127 ммоль), затем NH4Cl (6,79 г, 127 ммоль), что приводило к значительному выделению тепла. Гетерогенную реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь фильтровали через слой целита и концентрировали и получали почти белое твердое вещество. К этому твердому веществу добавляли EtOAc и полученную смесь обрабатывали ультразвуком в течение 10 мин. Смесь фильтровали через целит и промывали с помощью EtOAc. Объединенные фильтраты концентрировали в вакууме и получали 2,17 г (10,5 ммоль, 79%) 3-бром-2-хлоранилина: ЖХМС (m/z) 205,9(МН+); tR = 0,87 мин; 1 Н ЯМР (400 МГц, CDCl3)6,62-6,66 (m, 1 Н), 6,87 (t, J = 8,0 Гц, 1 Н), 6,97 (d, J = 7,8 Гц, 1 Н), 7,22 (d,J = 9,0 Гц, 1 Н). Стадия 3. Получение 2-хлор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ-9). В стеклянный сосуд высокого давления добавляли 3-бром-2-хлоранилин (3,08 г, 14,9 ммоль),бис(пинаколято)дибор (4,55 г, 17,9 ммоль), трициклогексилфосфин (0,29 г, 1,04 ммоль), ацетат калия (2,2 г, 22,4 ммоль) и Pd2dba3 (0,41 г, 0,45 ммоль) в 1,4-диоксане (75 мл) и получали красную суспензию, которую продували азотом, затем сосуд для проведения реакции герметизировали. Реакционную смесь нагревали на масляной бане при 120 С в течение 2 ч. Анализ аликвоты реакционной смеси с помощью ЖХМС указывал на полное превращение исходного вещества в продукт и реакционной смеси давали охладиться до комнатной температуры. Добавляли SiliaBond DMT (4 г, выпускающийся фирмой SiliCycle), полученную смесь перемешивали при комнатной температуре в течение 30 мин, затем фильтровали через слой нейтрального оксида алюминия с покрытием из SiO2. Осадок на фильтре тщательно промывали с помощью EtOAc и объединенные фильтраты подвергали распределению с водой. Фазы разделяли и водную порцию экстрагировали с помощью EtOAc. Объединенные органические порции промывали водой (2), рассолом, сушили (Na2SO4) и концентрировали и получали 2-хлор-3-(4,4,5,5-тетраметил-1,3,2 диоксаборолан-2-ил)анилин (1,6 г) в виде темно-желтого кристаллического твердого вещества, которое использовали без дополнительной очистки: ЖХМС (m/z) 254,0 (МН+); tR = 0,91 мин. Получение исходного вещества,N-(2-хлор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)фенил)пропан-1-сульфонамида (ИВ-10) Стадия 1. Получение N-(3-бром-2-хлорфенил)пропан-1-сульфонамида. Это вещество получали из 3-бром-2-хлоранилина (ИВ-9, стадия 2) по методике, использованной для получения ИВ-6, стадия 1. Очистка с помощью флэш-хроматографии (SiO2; 0-60% EtOAc в гептане) давала N-(3-бром-2-хлорфенил)пропан-1-сульфонамид (51%); 1 Н ЯМР (400 МГц, CDCl3)1,05 (t, J = 7,4 Гц, 3 Н), 1,83-1,95 (m, 2 Н), 3,01-3,15 (m, 2 Н), 4,21 (br s,1 Н), 7,17 (t, J = 8,22 Гц, 1H), 7,44 (d, J = 8,2 Гц, 1H), 7,67 (d, J = 8,22 Гц, 1 Н). Стадия 2. Получение N-(2-хлор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропан-1 сульфонамида (ИВ-10). Это вещество получали с использованием продукта, полученного на предыдущей стадии, и по методике, использованной для получения ИВ-9, стадия 3 (88%): ЖХМС (m/z) 360,1 (МН+); tR = 1,06 мин. Получение 2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ-11) Стадия 1. Получение 3-бром-2-фторанилина. Это вещество получали из 1-бром-2-фтор-3-нитробензола по методике, использованной для получения ИВ-9, стадия 2 (94%): ЖХМС (m/z) 189,9, tR = 0,74 мин; 1 Н ЯМР (300 МГц, CDCl3)3,81 (br s, 2 Н), 6,64-6,75 (m, 1 Н), 6,80 (t, J = 8,2 Гц, 1H), 6,84-6,95 (m,1 Н). Стадия 2. Получение 2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ-11). Это вещество получали по методике, использованной для получения ИВ-9, стадия 3: ЖХМС (m/z) 115,9 (МН+, бороновая кислота); tR = 0,17 мин. Получение исходного вещества,N-(2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)фенил)пропан-1-сульфонамида (ИВ-12) Стадия 1. Получение N-(3-бром-2-фторфенил)пропан-1-сульфонамида. Это вещество получали из 3-бром-2-фторанилина (ИВ-11, стадия 1), по методике, использованной для получения ИВ-6, стадия 1. Очистка с помощью флэш-хроматографии (SiO2, 0-50% EtOAc в гептане) давала N-(3-бром-2-фторфенил)пропан-1-сульфонамид (56%) в виде белого кристаллического твердого вещества; 1H ЯМР (400 МГц, CDCl3)1,05 (t, J = 7,4 Гц, 3 Н), 1,80-1,97 (m, 2H), 2,99-3,20 (m, 2H), 6,60 (br s,1H), 7,04 (t, J = 7,6 Гц, 1H), 7,34 (кажущийся t, J = 6,7 Гц, 1H), 7,56 (кажущийся t, J = 7,6 Гц, 1H). Стадия 2. Получение N-(2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропан-1 сульфонамида (ИВ-12). Это вещество получали по методике, использованной для получения ИВ-9, стадия 3, и использовали на следующих стадиях без дополнительных характеризации и очистки. Получение исходного вещества, 3,3,3-трифтор-N-(2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропан-1-сульфонамида (ИВ-13) Стадия 1. Получение N-(3-бром-2-фторфенил)-3,3,3-трифторпропан-1-сульфонамида. Это вещество получали из 3-бром-2-фторанилина (ИВ-11, стадия 1), в соответствии с методикой,использованной для получения ИВ-6, стадия 1: ЖХМС (m/z) не ионизируется (МН+), tR = 0,92 мин; 1 Н ЯМР (400 МГц, CDCl3)ч./млн 2,62-2,82 (m, 2 Н), 3,30-3,43 (m, 2 Н), 7,05-7,13 (m, 1 Н), 7,43 (t, J = 6,9 Гц, 1 Н), 7,50-7,56 (m, 1 Н). Стадия 2. Получение 3,3,3-трифтор-N-(2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)фенил)пропан-1-сульфонамида (ИВ-13). Это вещество получали в соответствии с методикой, использованной для получения ИВ-9, стадия 3: ЖХМС (m/z) не ионизируется (МН+), tR = 0,62 мин. Получение Стадия 1. Получение бром-2,6-дифторбензойной кислоты. В высушенную в сушильном шкафу 2-горлую круглодонную колбу в атмосфере аргона при комнатной температуре добавляли диизопропиламин (8,1 мл, 57,0 ммоль) и ТГФ (260 мл). Раствор охлаждали до -70 С в бане со смесью твердый диоксид углерода-ацетон. Шприцем по каплям добавляли нбутиллитий (2,0 М раствор в циклогексане, 25,9 мл, 51,8 ммоль) и полученную реакционную смесь кратковременно нагревали до 0 С, затем повторно охлаждали до -70 С. К этому холодному раствору шприцем по каплям добавляли 1-бром-2,4-дифторбензол (5,9 мл, 52,0 ммоль). После добавления реакционную смесь выдерживали при -70 С в течение 1 ч. К раствору добавляли диоксид углерода (кусочки по 5-9 г,предварительно промытые сухим ТГФ). Баллон с Ar удаляли и заменяли на барботер для обеспечения выхода газа. Затем полученной реакционной смеси давали нагреться до комнатной температуры и реакционную смесь нейтрализовывали насыщенным водным раствором NH4Cl до рН 7-8. Водную смесь промывали с помощью EtOAc, подкисляли 6 н. водным раствором HCl до рН 2-3 и экстрагировали с помо- 19020479 щью EtOAc. Органический экстракт промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме и получали 10,9 г (89%) 3-бром-2,6-дифторбензойной кислоты; 1 Н ЯМР (400 МГц, ДМСО (диметилсульфоксид)-d6)7,25 (m, 1 Н), 7,92 (m, 1 Н). Стадия 2. Получение трет-бутил-3-бром-2,6-дифторфенилкарбамата. В круглодонную колбу, снабженную холодильником, содержащую 3-бром-2,6-дифторбензойную кислоту (5 г, 21,1 ммоль), в атмосфере азота добавляли толуол (35 мл) и t-BuOH (35 мл). К этому раствору добавляли ДИЭА (4,4 мл, 25,3 ммоль) и ДФФА (5,7 мл, 26,4 ммоль). Реакционную смесь нагревали на масляной бане при 111 С в течение 48 ч. Реакционной смеси давали охладиться до комнатной температуры и летучие вещества удаляли в вакууме. Полученный остаток суспендировали в воде и экстрагировали с помощью EtOAc. Органическую фазу промывали водой, рассолом, сушили (Na2SO4), фильтровали и концентрировали на диоксиде кремния. Очистка с помощью флэш-хроматографии (SiO2; 0-100%EtOAc в гептане) давала 3,49 г (54%) трет-бутил-3-бром-2,6-дифторфенилкарбамата; 1 Н ЯМР (400 МГц, ДМСО-d6)1,34-1,54 (m, 9 Н), 7,17 (m, 1 Н), 7,63 (ddd, J = 8,9, 7,9, 5,9 Гц, 1 Н). Стадия 3. Получение 3-бром-2,6-дифторанилина. В круглодонную колбу, содержащую трет-бутил-3-бром-2,6-дифторфенилкарбамат (1 г, 3,3 ммоль),добавляли ДХМ (3 мл) и ТФК (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Летучие вещества удаляли в вакууме и полученный остаток нейтрализовывали насыщенным водным раствором NaHCO3 до рН 8. Водную смесь экстрагировали с помощью EtOAc. Органическую фазу промывали водой, рассолом, сушили (Na2SO4), фильтровали и концентрировали на диоксиде кремния. Очистка с помощью флэш-хроматографии (SiO2; 0-50% EtOAc в гептане) давала 425 мг (63%) 3 бром-2,6-дифторанилина: ЖХМС (m/z) 208,0 (МН+); tR = 0,80 мин; 1 Н ЯМР (400 МГц, ДМСО-d6)5,54 (s, 2 Н), 6,78 (ddd, J = 9,0, 7,4, 5,5 Гц, 1 Н), 6,85-6,95 (m, 1H). Стадия 4. Получение N-(3-бром-2,6-дифторфенил)пропан-1-сульфонамида. К раствору 3-бром-2,6-дифторанилина (425 мг, 2,04 ммоль) в сухом пиридине (2,0 мл) добавляли 1 пропансульфонилхлорид (275 мкл, 2,45 ммоль) и полученную реакционную смесь выдерживали при комнатной температуре в течение ночи. Реакционную смесь подвергали распределению между EtOAc и водой и слои разделяли. Водную порцию экстрагировали с помощью EtOAc и объединенные органические порции промывали 10% водным раствором лимонной кислоты, водой, рассолом, сушили (Na2SO4) и концентрировали и получали коричневое вязкое масло, которое представляло собой смесь 3-бром-2,6 дифторанилина и N-(3-бром-2,6-дифторфенил)пропан-1-сульфонамида состава 2:1, которую использовали на следующей стадии без дополнительной очистки, N-(3-бром-2,6-дифторфенил)пропан-1 сульфонамид: ЖХМС (m/z) не ионизируется (МН+); tR = 1,06 мин. Стадия 5. Получение N-(2,6-дифтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропан 1-сульфонамида (ИВ-14). Смесь 3-бром-2,6-дифторанилина и N-(3-бром-2,6-дифторфенил)пропан-1-сульфонамида (560 мг,2,69 ммоль) объединяли с бис(пинаколято)дибором (820 мг, 3,23 ммоль), трициклогексилфосфином (52,8 мг, 0,188 ммоль), ацетатом калия (396 мг, 4,04 ммоль) и Pd2dba3 (74,0 мг, 0,081 ммоль) в 1,4-диоксане (10 мл) и получали желтую суспензию. Реакционную смесь нагревали на масляной бане при 120 С в течение 2 ч, после этого ЖХМС показывала, что превращение завершилось. Реакционной смеси давали охладиться до комнатной температуры и ее подвергали распределению между EtOAc и водой. Слои разделяли и водную порцию экстрагировали с помощью EtOAc. Объединенные органические порции промывали водой (2), рассолом, сушили (Na2SO4) и концентрировали и получали темно-коричневое масло, которое представляло собой смесь боронатных эфиров, которую использовали без дополнительной очистки, N(2,6-дифтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропан-1-сульфонамид: ЖХМС (m/z) 174,0 (МН+, бороновая кислота); tR = 0,33 мин. Получение 3-метокси-2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ-15) Стадия 1. Получение 5-бром-3-метокси-2-метиланилина. Это вещество получали из 5-бром-1-метокси-2-метил-3-нитробензола по методике, использованной для получения ИВ-9, стадия 2: ЖХМС (m/z) 216,0 (МН+); tR = 0,65 мин. Стадия 2. Получение 3-метокси-2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина(ИВ-15). Это вещество получали по методике, использованной для получения ИВ-9, стадия 3: ЖХМС (m/z) 264,3 (МН+); tR = 0,67 мин. Получение 5-хлор-2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ-16) Стадия 1. Получение 3-бром-5-хлор-2-метиланилина. Это вещество получали из 1-бром-5-хлор-2-метил-3-нитробензола по методике, использованной для получения ИВ-9, стадия 3 (96%): ЖХМС (m/z) 219,9 (MH+); tR = 0,99 мин. Стадия 2. Получение 5-хлор-2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ 16). Это вещество получали по методике, использованной для получения ИВ-9, стадия 3: ЖХМС (m/z) 268,1 (МН+); tR = 1,14 мин. Получение исходного вещества, 3-хлор-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина Это вещество получали из 3-бром-5-хлоранилина по методике, сходной с методикой, использованной для получения ИВ-9, стадия 3. Неочищенный продукт очищали с помощью флэш-хроматографии Стадия 1. Получение 1-бром-3-(дифторметокси)-5-нитробензола. К нагретой до 55 С реакционной смеси 3-бром-5-нитрофенола (3,1 г, 14,4 ммоль) и порошкообразного гидроксида натрия (0,63 г, 15,8 ммоль) в ДМФ (14 мл) через каждые 0,5 ч пятью порциями добавляли хлордифторацетат натрия (4,4 г, 28,7 ммоль). Реакционную смесь выдерживали при 55 С в течение 1 дня и затем ей давали охладиться до комнатной температуры. Реакционную смесь подвергали распределению между EtOAc и водой, слои разделяли и водную порцию экстрагировали с помощью EtOAc (225 мл) и объединенные органические слои промывали 1,0 М водным раствором NaOH (325 мл), водой(325 мл) и рассолом (50 мл). Органический слой сушили (MgSO4) и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии (SiO2, 0-50% EtOAc в гексанах) и получали 1-бром-3(дифторметокси)-5-нитробензол (160 мг, 0,6 ммоль). 1 Н ЯМР (400 МГц, CDCl3)6,61 (t, J = 71,6 Гц, 1 Н), 7,65 (s, 1 Н), 7,96 (s, 1 Н), 8,21-8,31 (m, 1H). Стадия 2. Синтез 3-бром-5-(дифторметокси)анилина. Это вещество получали по методике, использованной для получения ИВ-9, стадия 3. Неочищенный продукт очищали с помощью флэш-хроматографии (SiO2, 0-30% EtOAc в гексанах) и получали 3-бром-5(дифторметокси)анилин в виде светло-коричневого масла (выход 41%): ЖХМС (m/z) 237,9 (МН+), tR = 0,81 мин; 1 Н ЯМР (300 МГц, CDCl3)3,83 (br s, 2 Н), 6,45 (t, 1 Н), 6,58-6,79 (m, 1 Н). Стадия 3. Синтез 3-(дифторметокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ 18). Это вещество получали по методике, использованной для получения ИВ-9, стадия 3: ЖХМС (m/z) 204,1 (МН+, бороновая кислота), tR = 0,33 мин.N-(3-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 Стадия 1. Получение N-(3-бром-5-метилфенил)пропан-2-сульфонамида. К раствору 3-бром-5-метиланилина (500 мг, 2,2 ммоль) в ДХМ (5 мл) добавляли изопропилсульфонилхлорид (0,3 мл, 2,7 ммоль), затем добавляли пиридин (0,45 мл, 5,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 25 ч и затем реакцию останавливали водой и смесь экстрагировали с помощью EtOAc. Органический слой промывали рассолом, сушили (MgSO4) и адсорбировали на диоксиде кремния. Очистка с помощью флэш-хроматографии (SiO2, 0-30% EtOAc в гексанах) давала N-(3-бром-5-метилфенил)пропан-2-сульфонамид (558 мг, 1,9 ммоль, 85%) в виде твердого вещества персикового цвета; 1 Н ЯМР (400 МГц, CDCl3)1,41 (d, J = 6,7 Гц, 6 Н), 2,31 (s, 3 Н), 3,33 (m, 1 Н), 6,84 (br s, 1 Н), 6,97 (s,1 Н), 7,10 (s, 1 Н), 7,21 (s, 1 Н). Стадия 2. Получение N-(3-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропан-2 сульфонамида. Это вещество получали по методике, использованной для получения ИВ-9, стадия 3, и использовали на следующей стадии без обработки. Получение 2,5-дихлор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ-20) Стадия 1. Получение 2,5-дихлор-3-нитробензойной кислоты. К раствору 2,5-дихлорбензойной кислоты (3 г, 15,7 ммоль) в H2SO4 (16 мл) при 0 С по каплям добавляли дымящую азотную кислоту (1,4 мл, 31,4 ммоль) и реакционную смесь перемешивали при 0 С в течение 5 мин и затем ей в течение 20 мин давали постепенно нагреваться. Реакционную смесь обрабатывали водой со льдом и экстрагировали с помощью EtOAc. Органический слой промывали водой и рассолом, затем сушили (Na2SO4), фильтровали и концентрировали и получали 3,54 г (48%) 2,5-дихлор-3 нитробензойной кислоты, которая содержала примесь 3,6-дихлор-2-нитробензойной кислоты (40%); 1 Н ЯМР (300 МГц, CD3OD)7,73 (s, 1 Н), 8,06 (d, J = 2,6 Гц, 1 Н), 8,11 (d, J = 2,6 Гц, 1 Н). Стадия 2. Получение 1-бром-2,5-дихлор-3-нитробензола. В высушенную в сушильном шкафу круглодонную колбу, снабженную стержнем для перемешивания и холодильником, в атмосфере азота при комнатной температуре добавляли 2,5-дихлор-3 нитробензойную кислоту (3,54 г, 15,00 ммоль, в виде смеси с 3,6-дихлор-2-нитробензойной кислотой),красный оксид ртути(II) (4,87 г, 22,50 ммоль) и тетрахлорид углерода (100 мл). Реакционную смесь нагревали при 90 С в течение 30 мин при облучении осветительной лампой 150 Вт TYPE А. Затем реакционную смесь охлаждали примерно до 60 С и шприцем по каплям добавляли бром (1,2 мл, 22,5 ммоль). Атмосферу азота заменяли на подающийся из баллона аргон и реакционную смесь повторно нагревали при 90 С в течение 4 ч при постоянном облучении светом. Затем реакционную смесь охлаждали до комнатной температуры, реакцию останавливали насыщенным водным раствором NaHCO3, смесь перемешивали в течение 2 ч, фильтровали через целит и разбавляли с помощью ДХМ. Две фазы разделяли и водную смесь экстрагировали с помощью ДХМ. Органические вещества объединяли, промывали водой,рассолом, сушили (Na2SO4) и концентрировали и получали бледно-желтое кристаллическое твердое вещество, которое очищали с помощью флэш-хроматографии (SiO2, 0-15% EtOAc в гептане) и получали 1,83 г (29%) 1-бром-2,5-дихлор-3-нитробензола; 1 Н ЯМР (300 МГц, CDCl3)7,75 (d, J = 2,3 Гц, 1 Н), 7,87 (d, J = 2,3 Гц, 1 Н). Стадия 3. Получение 3-бром-2,5-дихлоранилина. Это вещество получали в соответствии с методикой, использованной для получения ИВ-9, стадия 2,с использованием 1-бром-2,5-дихлор-3-нитробензола. Очистка с помощью флэш-хроматографии (SiO2, 030% EtOAc в гептане) давала 3-бром-2,5-дихлоранилин (24%): ЖХМС (m/z) 239,9 (МН+), tR = 1,03 мин; Н ЯМР (400 МГц, CDCl3)4,28 (br s, 2 Н), 6,71 (d, J = 2,4 Гц, 1H), 7,02 (d, J = 2,4 Гц, 1 Н). Стадия 4. Получение 2,5-дихлор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ-20). Это вещество получали в соответствии с методикой, использованной для получения ИВ-9, стадия 3. ЖХМС (m/z): 287,9 (МН+); tR = 1,10 мин. Получение 2-хлор-5-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ-21) Стадия 1. Получение 3-бром-2-хлор-5-метиланилина. К продуваемому аргоном раствору 1,3-дибром-2-хлор-5-метилбензола (5 г, 17,6 ммоль), бензофенонимина (3,1 мл, 18,5 ммоль) и трет-бутоксида натрия (2,53 г, 26,4 ммоль) в толуоле добавляли (S)-бинаф(1,6 г, 2,6 ммоль) и Pd2(dba)3 (0,81 г, 0,88 ммоль) и реакционную смесь нагревали на масляной бане. Когда температура достигала 60 С, наблюдали выделение тепла и кипение растворителя. Нагревание продолжали и через 1,5 ч реакция завершалась. Реакционную смесь охлаждали, разбавляли с помощью Et2O и перемешивали с Siliabond DMT (поглотитель Pd) и затем фильтровали через целит. Фильтрат промывали водой, рассолом, сушили (MgSO4), концентрировали и получали липкий коричневый остаток. Остаток растворяли в ТГФ (60 мл) и добавляли 6,0 М водный раствор HCl. Реакционную смесь перемешивали в течение 30 мин, подвергали распределению с Et2O и добавляли 1 М раствор NaOH до установления значения рН водного слоя, равного 9. Слои разделяли и органический слой промывали водой, рассолом, сушили (MgSO4) и концентрировали. Очистка с помощью флэш-хроматографии (SiO2, 0-20% EtOAc в гептане) давало 3-бром-2-хлор-5-метиланилин (1,7 г), содержащий в качестве примеси небольшое количество бензофенона; 1 Н ЯМР (300 МГц, CDCl3)2,21 (s, 3 Н), 4,11 (br s, 2 Н), 6,52 (s, 1H), 6,85 (s, 1H). Стадия 2. Получение 2-хлор-5-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина. Это вещество получали по методике, использованной для получения ИВ-9, стадия 3. 1 Н ЯМР (300 МГц, CDCl3)1,26 (s, 8 Н), 1,36 (s, 10 Н), 2,22 (s, 3 Н), 4,00 (br s, 2 Н), 6,65 (d, J = 2,1 Гц,1H), 6,89 (d, J = 1,8 Гц, 1H). Получение исходного вещества, 2-фтор-5-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)анилина (ИВ-22) Стадия 1. Получение 3-бром-2-фтор-5-метилбензойной кислоты. К раствору диизопропиламина (1,8 мл, 12,7 ммоль) в ТГФ (20 мл) при -10 С добавляли n-BuLi (0,68 г, 10,6 ммоль) и реакционную смесь перемешивали при -10 С в течение 1 ч и затем охлаждали до -78 С. По каплям добавляли раствор 2-бром-1-фтор-4-метилбензола (2,0 г, 10,6 ммоль) в ТГФ (10 мл) и реакционную смесь перемешивали в течение 1 ч, затем добавляли избыток твердого диоксида углерода (4,76 г,106 ммоль). Через 30 мин реакционной смеси давали нагреться до комнатной температуры, давление сбрасывали и реакцию останавливали водой. Полученные слои разделяли и водную порцию экстрагировали с помощью Et2O. Водный слой затем подкисляли 6,0 М водным раствором HCl и полученный белый осадок экстрагировали с помощью Et2O. Объединенные органические порции сушили (MgSO4) и концентрировали в вакууме и получали 3-бром-2-фтор-5-метилбензойную кислоту (2,1 г, 9,0 ммоль, 85%) в виде белого твердого вещества; 1H ЯМР (300 МГц, CDCl3)2,37 (s, 3 Н), 7,60 (dd, J = 5,9, 1,8 Гц, 1 Н), 7,75 (dd, J = 6,2, 1,8 Гц, 1 Н). Стадия 2. Получение трет-бутил-3-бром-2-фтор-5-метилфенилкарбамата. К раствору 3-бром-2-фтор-5-метилбензойной кислоты (2,1 г, 9,01 ммоль) в толуоле (30 мл) и tBuOH (15 мл) добавляли ДИЭА (1,9 мл, 10,8 ммоль) и ДФФА (2,4 мл, 11,3 ммоль) и реакционную смесь нагревали и выдерживали при 120 С в течение 24 ч. Реакционной смеси давали охладиться до комнатной температуры и ее концентрировали и получали коричневое масло. Масло подвергали распределению между диэтиловым эфиром и водой и полученные слои разделяли. Слой, содержащий диэтиловый эфир,промывали водой, рассолом, сушили (MgSO4) и концентрировали. Очистка с помощью флэш- 23020479(985 мг, 3,2 ммоль, 36%) в виде прозрачного бледно-желтого масла: ЖХМС (m/z) 247,9 (МН+ - третбутил); tR = 1,14 мин; 1 Н ЯМР (300 МГц, CDCl3)1,53 (s, 9H), 2,29 (s, 3H), 6,67 (br s, 1H), 6,98 (d, J = 6,2 Гц, 1H), 7,88 (d, J = 6,5 Гц, 1H). Стадия 3. 3-Бром-2-фтор-5-метиланилин. К раствору трет-бутил-3-бром-2-фтор-5-метилфенилкарбамата (985 мг, 3,24 ммоль) в изопропиловом спирте (10 мл) добавляли концентрированный водный раствор HCl (12 М, 2,6 мл, 32,4 ммоль) и реакционную смесь нагревали при 60 С в течение 2 ч, ей давали охладиться до комнатной температуры и затем концентрировали в вакууме и получали белое твердое вещество. Твердое вещество растворяли в воде и полученный водный раствор нейтрализовывали 1,0 М водным раствором NaOH и экстрагировали с помощью Et2O. Органическую фазу промывали рассолом, сушили (MgSO4) и концентрировали и получали 3-бром-2-фтор-5-метиланилин (594 мг, 2,91 ммоль, 90%), который использовали на следующей стадии без дополнительной очистки; 1 Н ЯМР (300 МГц, CDCl3)2,21 (s, 3 Н), 3,71 (br s, 2 Н), 6,51 (d, J = 7,9 Гц, 1 Н), 6,70 (d, J = 4,7 Гц,1 Н). Стадия 4. Получение 2-фтор-5-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (ИВ 22). Это вещество получали по методике, использованной для получения ИВ-9, стадия 3: ЖХМС (m/z) 252,0 (МН+); tR = 0,83 мин. Получение исходного вещества, 2-хлор-4-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)анилина (ИВ-20) Стадия 1. Получение 2-(2-бром-3-хлор-1-фтор-4-нитробензола). 2-Бром-1-хлор-3-фторбензол (3,4 г, 16,2 ммоль) диспергировали в концентрированной серной кислоте (10 мл), затем при перемешивании при 0 С к смеси порциями добавляли NaNO3 (1,52 г, 17,9 ммоль). Затем реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в воду со льдом (60 мл) и выдерживали в течение ночи. Полученное осадившееся белое твердое вещество собирали фильтрованием, промывали водой и растворяли в EtOAc. Органический раствор промывали насыщенным водным раствором Na2CO3, рассолом, сушили (Na2SO4) и концентрировали и получали 3,8 г твердого остатка (смесь двух региоизомеров состава 1:7, искомый изомер содержался в большем количестве). Твердое вещество растворяли в EtOAc (15 мл), охлаждали до 5 С и порциями добавляли порошкообразное железо (2,7 г, 48 ммоль). После добавления реакционную смесь перемешивали при комнатной температуре в течение ночи, разбавляли с помощью EtOAc и затем фильтровали через слой целита. Фильтрат подщелачивали (рН 12) 12 н. водным раствором NaOH и полученную гелеобразную смесь фильтровали через целит. При выдерживании происходило разделение фаз фильтрата и слои разделяли. Водную порцию экстрагировали с помощью EtOAc (2) и объединенные органические порции промывали рассолом, сушили (Na2SO4) и концентрировали. Очистка с помощью флэшхроматографии (SiO2, 0-70% EtOAc в гептане) давала искомый 3-бром-2-хлор-4-фторанилин (2,2 г, 9,0 ммоль, 56%): ЖХМС (m/z) 223,8 (МН+); tR = 0,89 мин; 1 Н ЯМР (400 МГц, CDCl3)6,92-6,88 (m, 1H), 6,71-6,66 (m, 1H), 4,05 (br s 2 Н). Стадия 2. Получение 4-(2-хлор-4-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина(ИВ-23). Это соединение получали по такой же методике, как использованная для получения исходного вещества ИВ-9, стадия 3: ЖХМС (m/z) 272,0 (МН+); tR = 0,95 мин. Получение промежуточного продукта,(S)-трет-бутил-1-(4-(4-бром-2-циклопропил-1-2(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамата (I-1 а) Глиоксаль (40% в воде) (86 мл, 749 ммоль) в воде (200 мл) добавляли к охлажденному (5 С) раствору циклопропанкарбальдегида (50,0 мл, 713 ммоль) в метаноле (100 мл) и получали бесцветный раствор. При 0-5 С в течение 1 ч по каплям добавляли гидроксид аммония (28% водный раствор, 397 мл, 2900 ммоль). Реакционную смесь перемешивали при 0 С в течение 3 ч и затем ей в течение ночи давали нагреться до комнатной температуры. К реакционной смеси добавляли рассол (200 мл) и ее экстрагировали с помощью EtOAc (4400 мл, 4600 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали и получали 2-циклопропил-1 Н-имидазол в виде бежевого твердого вещества (70,1 г, 648 ммоль, 91%). ЖХМС (m/z) 109,0 (МН+), tR = 0,22 мин; 1 Н ЯМР (400 МГц, ДМСО-d6)0,73-0,81 (m, 2 Н), 0,81-0,88 (m, 2 Н), 1,85-1,95 (m, 1 Н), 6,78 (br s, 2 Н),11,65 (br s, 1H). Стадия 2. Получение 4,5-дибром-2-циклопропил-1 Н-имидазола. Бром (61 мл, 1190 ммоль) при 0 С в течение 2 ч по каплям добавляли к охлажденной смеси 2 циклопропил-1 Н-имидазола (70,5 г, 652 ммоль) и KHCO3 (118 г, 1179 ммоль) в ДМФ (360 мл). Добавляли дополнительное количество KHCO3 (20 г, 200 ммоль) и реакционную смесь перемешивали при 0 С в течение еще 45 мин. В течение 45 мин по каплям добавляли воду (1,5 л) и полученную оранжевую взвесь фильтровали в холодном виде. Твердые вещества промывали водой (4150 мл) и сушили в вакуумном сушильном шкафу при 50 С в течение 24 ч и получали 4,5-дибром-2-циклопропил-1 Н-имидазол в виде желтовато-коричневого твердого вещества (122 г, 459 ммоль, 70%). ЖХМС (m/z) 264,8 (МН+), tR = 0,51 мин; 1 Н ЯМР (400 МГц, ДМСО-d6)0,77-0,81 (m, 2 Н), 0,86-0,91 (m, 2 Н), 1,83-1,92 (m, 1 Н), 12,86 (br s,1H). Стадия 3. Получение 4,5-дибром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Нимидазола. Раствор 4,5-дибром-2-циклопропил-1 Н-имидазола (60 г, 226 ммоль) в ТГФ (150 мл) при перемешивании в атмосфере азота при 0 С в течение 50 мин по каплям добавляли к смеси гидрида натрия (95%,6,3 г, 250 ммоль) в сухом ТГФ (150 мл). Реакционную смесь перемешивали в течение 30 мин и при 0 С в течение 50 мин по каплям добавляли 2-(триметилсилил)этоксиметилхлорид (SEMCl, 40 мл, 38 г, 226 ммоль). После перемешивания в течение 1 ч реакцию медленно останавливали водой (20 мл) и к смеси добавляли EtOAc (500 мл). Смесь промывали водой (2750 мл) и объединенные водные порции подвергали обратной экстракции с помощью EtOAc (200 мл). Объединенные органические порции промывали рассолом (1 л), сушили (Na2SO4) и концентрировали. Полученный остаток растворяли в гептане (200 мл) и полученный раствор пропускали через слой силикагеля при элюировании гептаном (4500 мл) и смесью EtOAc-гептан (1:4, 2250 мл) и после концентрирования получали 4,5-дибром-2-циклопропил-1-2(триметилсилил)этокси)метил)-1 Н-имидазол (79 г, 199 ммоль, 88%) в виде бледно-желтого твердого вещества: ЖХМС (m/z) 394,9 (МН+), tR = 1,20 мин,1 Н ЯМР (400 МГц, CDCl3)ч./млн -0,04 - 0,05 (m, 9 Н), 0,87-1,12 (m, 6 Н), 1,88-2,04 (m, 1 Н), 3,553,66 (m, 2 Н), 5,37 (s, 2 Н). Стадия 4. Получение 4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол 5-ил)-2-хлор-4,5-дигидропиримидина. Бутиллитий (2,0 М раствор в пентане, 31 мл, 62 ммоль) при -78 С в течение 40 мин по каплям добавляли к раствору 4,5-дибром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазола (23,4 г, 59,1 ммоль) в ТГФ (175 мл). Реакционную смесь перемешивали при -78 С в течение 10 мин. Затем отбирали аликвоту реакционной смеси и реакцию останавливали водой и завершение литиирования проверяли с помощью ЖХМС. По каплям добавляли раствор 2-хлорпиримидина (8,12 г, 70,9 ммоль) в ТГФ (20 мл) и перемешивали в течение 30 мин. Анализ с помощью ЖХМС указывал на завершение реакции. Реакцию останавливали насыщенным водным раствором NH4Cl (20 мл), смеси давали нагреться до 0 С и ее подвергали распределению между водой (500 мл) и EtOAc (500 мл). Слои разделяли и органическую порцию промывали смесью вода-рассол (500 мл) и рассолом (500 мл), затем сушили (Na2SO4) и концентрировали и получали желтое масло. Неочищенный остаток суспендировали в смеси EtOAc-гексаны (1:5, 50 мл) и гептане (50 мл) и затем обрабатывали ультразвуком в течение 2 мин. Полученной суспензии давали отстояться при 0 С в течение 1 ч. Собранные твердые вещества промывали холодной смесью EtOAc-гептан (1:5, 50 мл) и получали 19,6 г 4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)-2-хлор 4,5-дигидропиримидина в виде белого твердого вещества. Объединенные фильтраты очищали с помощью флэш-хроматографии (SiO2, 20-70% EtOAc в гептане) и получали дополнительно 2,05 г продукта 5-ил)-2-хлорпиримидина. 4-(4-Бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)-2-хлор-4,5 дигидропиримидин (21,65 г, 50,1 ммоль) и диоксид марганца (43,6 г, 501 ммоль) в EtOAc (240 мл) кипятили с обратным холодильником в течение 2,5 ч. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит. Фильтраты концентрировали и остаток очищали с помощью колоночной флэш-хроматографии (10-40% EtOAc в гептане) и получали 4-(4-бром-2-циклопропил-1-2(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)-2-хлорпиримидин в виде желтого масла (17,8 г, 41,4 ммоль, 83%). ЖХМС (m/z) 429,0 (МН+), tR = 1,24 мин; 1 Н ЯМР (400 МГц, CDCl3)-0,06 (s, 9 Н), 0,82-0,90 (m, 2 Н), 1,04-1,11 (m, 2 Н), 1,14-1,20 (m, 2 Н), 2,06(m, 1 Н), 3,55-3,62 (m, 2 Н), 5,92 (s, 2 Н), 7,92 (d, J = 5,5 Гц, 1 Н), 8,62 (d, J = 5,5 Гц, 1 Н). Стадия 6. Получение (S)-трет-бутил-1-(4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамата (I-1 а). Смесь 4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)-2-хлорпиримидина (5,50 г, 12,8 ммоль), (S)-трет-бутил-1-аминопропан-2-илкарбамата (ИВ-1, 2,68 г, 15,4 ммоль), диизопропилэтиламина (2,68 мл, 15,4 ммоль) и карбоната натрия (2,71 г, 25,6 ммоль) в NMP (8 мл) нагревали на масляной бане при 110 С в течение 3,5 ч и анализ аликвоты реакционной смеси с помощью ЖХМС указывал на завершение реакции с получением искомого продукта. Реакционной смеси давали охладиться до температуры окружающей среды, затем ее подвергали распределению междуEtOAc (20 мл) и водой (60 мл). Слой, содержащий EtOAc, промывали водой (60 мл), сушили (Na2SO4) и концентрировали и получали светло-желтое вспененное вещество (6,89 г, 12,1 ммоль). Небольшую порцию вещества дополнительно очищали с помощью флэш-хроматографии (SiO2, EtOAc в гептане): ЖХМС (m/z) 567,3 (МН+), tR = 1,03 мин; 1 Н ЯМР (CDCl3, 300 МГц)-0,09 (s, 9 Н), 0,80 (t, J = 8,2 Гц, 2 Н), 0,98-1,07 (m, 2 Н), 1,10-1,18 (m, 2 Н),1,21 (t, J = 6,5 Гц, 3 Н), 1,40 (s, 9 Н), 1,91-2,08 (m, 1 Н), 3,41 (t, J = 8,2 Гц, 2 Н), 3,44-3,59 (m, 2 Н), 3,82-4,01(S)-трет-бутил-1-(4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Нимидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамата (15,5 г, 27,3 ммоль) в МеОН (60 мл) обрабатывали водным раствором концентрированной HCl (37%, 10 мл, 122 ммоль) при 60 С в течение 3,5 ч. Анализ аликвоты реакционной смеси с помощью ЖХМС указывал на завершение превращения. Реакционную смесь концентрировали в вакууме и получали (S)-N1-(4-(4-бром-2-циклопропил-1 Н-имидазол-5 ил)пиримидин-2-ил)пропан-1,2-диамин в виде соли с HCl (13,3 г): ЖХМС (m/z) 337,1 (МН+), tR = 0,41 мин. Стадия 2. Синтез(13,3 г, 27,3 ммоль, теоретически рассчитанное количество) в смеси ТГФ-вода состава 1:1 (200 мл) охлаждали до 0 С и порциями добавляли твердый NaHCO3 (20,6 г, 245 ммоль). В течение 20 мин по каплям добавляли метилхлорформиат (3,74 мл, 27,3 ммоль) и перемешивали в течение еще 30 мин. анализ аликвоты реакционной смеси с помощью ЖХМС указывал на завершение реакции. Добавляли воду (300 мл) и полученную смесь экстрагировали с помощью EtOAc (3500 мл). Слой, содержащий EtOAc, промывали рассолом (21 л), сушили (Na2SO4) и концентрировали. Полученное твердое вещество суспендировали в смеси EtOAc (7 мл) и этилового эфира (25 мл) и полученной суспензии давали отстояться при 0 С. Твердые вещества собирали и промывали холодным Et2O (20 мл) и получали (S)-метил-1-(4-(4-бром-2 циклопропил-1 Н-имидазол-5-ил)пиримидин-2-иламино)пропан-2-илкарбамат (9,56 г, 24,1 ммоль, 89%) в виде почти белого твердого вещества: ЖХМС (m/z) 395,1 (МН+), tR = 0,61 мин; 1 Н ЯМР (400 МГц, CDCl3)0,91-1,03 (m, 2 Н), 1,08-1,19 (m, 2 Н), 1,22 (d, J = 6,6 Гц, 3 Н), 1,77 (br s,1 Н), 2,13-2,26 (m, 1 Н), 2,70-2,90 (m, 1 Н), 3,70 (s, 3 Н), 3,90-4,03 (m, 1 Н), 4,18-4,32 (m, 1 Н), 4,46-4,86 (m,1 Н), 5,48-5,60 (m, 1 Н), 7,46 (d, J = 5,5 Гц, 1H), 8,24 (d, J = 5,4 Гц, 1H). Получение промежуточного продукта, 3-(4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси) метил)-1 Н-имидазол-5-ил)пиримидин-2-иламино)пропаннитрила (I-1 с) Смесь 4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)-2-хлорпиримидина (1-1 а, стадия 5, 3,50 г, 8,14 ммоль), 3-аминопропионитрила (1,79 мл, 24,4 ммоль), диизопропилэтиламина (2,84 мл, 16,3 ммоль) и Na2CO3 (1,73 г, 16,3 ммоля) в сухом NMP (4 мл) нагревали при 90 С в течение 8 ч. Реакционную смесь охлаждали до комнатной температуры и затем подвергали распределению между EtOAc (100 мл) и водой (100 мл) и слои разделяли. Органическую порцию промывали водой (100 мл), насыщенным водным раствором NaHCO3 (100 мл), рассолом (100 мл), сушили(Na2SO4) и концентрировали. Неочищенный остаток очищали с помощью флэш-хроматографии (SiO2, 050% EtOAc в гептане) и получали 3-(4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Нимидазол-5-ил)пиримидин-2-ил)пропаннитрил в виде бледно-желтого вспененного вещества (2,91 г, 6,28 ммоль). ЖХМС (m/z) 463,1 (MH+), tR = 0,98 мин; 1 Н ЯМР (CDCl3)ч./млн -0,09 (s, 9 Н), 0,73-0,85 (m, 2 Н), 0,99-1,11 (m, 2 Н), 1,12-1,21 (m, 2 Н), 1,902,05 (m, 1 Н), 2,75 (t, J = 6,7 Гц, 2 Н), 3,36-3,48 (m, 2 Н), 3,76 (q, J = 6,5 Гц, 3 Н), 5,50 (br s, 1 Н), 5,85 (s, 2 Н),7,20 (d, J= 5,1 Гц, 1 Н), 8,37 (d, J = 5,1 Гц, 1 Н). Получение промежуточного продукта, 3-(4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси) метил)-1 Н-имидазол-5-ил)пиримидин-2-иламино)-2-метилпропаннитрила (I-1d) Раствор 4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)-2-хлорпиримидина (2,5 г, 5,8 ммоль), ДИЭА (2,0 мл, 11,6 ммоль) и 3-амино-2-метилпропаннитрила (1,49 г, 17,7 ммоль) в NMP (10 мл) обрабатывали с помощью Na2CO3 (1,23 г, 11,6 ммоль) и полученную реакционную смесь нагревали при 90 С в течение ночи. Реакционной смеси давали охладиться до комнатной температуры и ее подвергали распределению между EtOAc (75 мл) и водой (100 мл). Слои разделяли и органическую порцию промывали насыщенным водным раствором NaHCO3 (100 мл), рассолом (150 мл), сушили(0-50% EtOAc-гептан) давала 3-(4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Нимидазол-5-ил)пиримидин-2-иламино)-2-метилпропаннитрил (2,79 г, 5,8 ммоль) в виде белого твердого вещества: ЖХМС (m/z) 477,1 (MH+), tR = 0,98 мин. Получение промежуточного продукта, 1-4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси) метил)-1 Н-имидазол-5-ил)пиримидин-2-иламино)метил)циклопропанкарбонитрила (I-1 е)(I-1e) Смесь 1-(аминометил)циклопропанкарбонитрила (0,67 г, 7,0 ммоль), 4-(4-бром-2-циклопропил-12-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)-2-хлорпиримидина (2,01 г, 4,7 ммоль), ДИЭА (1,64 мл, 9,35 ммоль), Na2CO3 (0,99 г, 9,4 ммоль) и NMP (2 мл) нагревали при 110 С в течение 25 ч. Реакционной смеси давали охладиться до комнатной температуры и ее подвергали распределению между EtOAc(10 мл) и водой (20 мл). Слои разделяли и органическую порцию последовательно промывали водой (20 мл), рассолом (10 мл) и концентрировали. Полученный остаток очищали с помощью флэшхроматографии на силикагеле при элюировании смесью EtOAc-гептан (0-50%) в градиентном режиме и получали 1-4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)пиримидин-2-иламино)метил)циклопропанкарбонитрил (1,50 г, 3,06 ммоль, 66%) в виде белого вспененного вещества. ЖХМС (m/z) 491,1 (МН+), tR = 0,99 мин. Получение промежуточного продукта, 4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)пиримидин-2-амина (I-1g) 4-(4-Бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)-2-хлорпиримидин (1 г, 2,4 ммоль) объединяли с 28% водным раствором NH4OH (8 мл) в 1,4-диоксане (8 мл) и равные части полученной смеси помещали в два реакционных сосуда. Каждый сосуд облучали в микроволновом реакторе при 130 С в течение 40 мин. Анализ с помощью ТСХ (тонкослойная хроматография) указывал на завершение реакции. Затем объединенные реакционные смеси разбавляли водой (100 мл),затем экстрагировали с помощью EtOAc (100 мл). Слой, содержащий EtOAc, промывали рассолом, затем сушили (Na2SO4) и концентрировали. Очистка полученного остатка с помощью флэш-хроматографии на силикагеле в градиентном режиме с использованием смеси EtOAc-гексаны (от 0 до 60%) давала 0,87 г(2,1 ммоль,91%) 4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5 ил)пиримидин-2-амина в виде белого твердого вещества: ЖХМС (m/z): 410,0 (МН+), tR = 0,81 мин. Получение промежуточного продукта, 4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)пиримидина (I-1h) Стадия 1. Получение 4-(2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-4 ил)пиримидина. Раствор 4-(4-бром-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)-2 хлорпиримидина и формиата аммония (12,12 г, 192 ммоль) в МеОН (20 мл) продували аргоном в течение 5 мин. К смеси добавляли Pd/C (200 мг, 4,80 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 5 ч. Анализ аликвоты реакционной смеси с помощью ЖХМС указывал на завершение превращения. Реакционную смесь фильтровали через слой целита и осадок на фильтре тщательно промывали с помощью EtOAc. Объединенные фильтраты концентрировали и полученный остаток очищали с помощью флэш-хроматографии в градиентном режиме с использованием смеси EtOAc-гексаны(0-100%) и получали 4-(2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-4-ил)пиримидин (1,36 г, 4,30 ммоль, 89% выход) в виде липкого светло-желтого масла: ЖХМС (m/z) 317,3 (МН+),tR = 0,71 мин. Стадия 2. Получение 4-(4-бром-2-циклопропил-1-2-(триметилсилилэтокси)метил)-1 Н-имидазол-5 ил)пиримидина (I-1h). При 0 С к охлажденному раствору 4-(2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Нимидазол-4-ил)пиримидина (1,36 г, 4,30 ммоль) в ДХМ добавляли бром, затем насыщенный водный раствор Na2CO3 (16 мл, 4,30 ммоль). Реакционную смесь перемешивали в течение 3 ч при комнатной температуре. После того как анализ с помощью ЖХМС указывал на завершение реакции, реакционную смесь выдерживали и подвергали распределению. Полученные слои разделяли и водный слой экстрагировали с помощью EtOAc (2). Объединенные органические слои промывали рассолом, сушили (Na2SO4) и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии в градиентном режиме с использованием смеси EtOAc-гексаны (20-80%) и получали 4-(5-бром-2-циклопропил-1-2(триметилсилил)этокси)метил)-1 Н-имидазол-4-ил)пиримидин (1,07 г, 2,71 ммоль, 63%) в виде желтого масла: ЖХМС (m/z): 395,0 (МН+), tR = 1,09 мин; 1 Н ЯМР (400 МГц, CDCl3)0,00 (s, 9 Н), 0,80-1,50 (m, 4 Н), 2,06-2,25 (m, 1H), 3,49-3,65 (m, 2 Н), 6,05 В сосуд для микроволновой печи, снабженный стержнем для перемешивания, добавляли 4-(4-бром 2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-5-ил)-2-хлорпиримидин (I-1 а, стадия 5, 0,55 г, 1,3 ммоль), 2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (0,61 г, 2,6 ммоль),2,0 М водный раствор карбоната натрия (3,2 мл, 6,4 ммоль) и ДМЭ (6,4 мл). Полученную смесь продували азотом, затем добавляли аддукт PdCl2(dppf)ДХМ (0,052 г, 0,06 ммоль). Реакционный сосуд герметизировали и облучали в микроволновом реакторе при 120 С в течение 20 мин. Реакционную смесь разбавляли насыщенным водным раствором NH4Cl и экстрагировали с помощью EtOAc. Органическую фазу промывали водой, рассолом, сушили (Na2SO4), фильтровали, концентрировали и адсорбировали на силикагеле. Очистка с помощью флэш-хроматографии (SiO2, 0-100% EtOAc в гептане) давала 3-(5-(2 хлорпиримидин-4-ил)-2-циклопропил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазол-4-ил)-2 фторанилин (223 мг, 0,48 ммоль, 38%) в виде вязкого желтого масла: ЖХМС (m/z) 460,1 (МН+), tR = 0,95 мин. Получение промежуточного продукта, (S)-метил-1-(4-(4-бром-2-трет-бутил-1 Н-имидазол-5 ил)пиримидин-2-иламино)пропан-2-илкарбамата (I-2 а) Стадия 1. Получение 2-трет-бутилимидазола. Раствор глиоксаля (40% в воде, 16,4 г, 113,4 ммоль) в воде (180 мл) добавляли к триметилацетальдегиду (12,4 мл, 112,6 ммоль) и полученный раствор охлаждали до 10 С в бане со смесью лед/вода. К этому раствору при перемешивании добавляли раствор гидроксида аммония (28% в воде, 56 мл). Реакционную смесь перемешивали в течение ночи и полученный осадок отфильтровывали и сушили и получали 12,1 г искомого соединения в виде белого кристаллического твердого вещества. ЖХМС (m/z): 125,10(МН+), tR = 0,26 мин; 1 Н ЯМР (300 МГц, CD3OD)6,86 (2 Н, s), 1,32 (9 Н, s). Стадия 2. Получение 4,5-дибром-2-трет-бутил-1 Н-имидазола. Бром (8,4 г, 52,42 ммоль) по каплям добавляли к смеси 2-трет-бутилимидазола (2,6 г, 20,97 ммоль) и бикарбоната калия (5,4 г, 52,42 ммоль) в сухом ДМФ (25 мл). Затем реакционную смесь перемешивали при 70 С в течение 4 ч. Реакционной смеси давали охладиться до комнатной температуры и затем ее фильтровали через воронку с фильтром из пористого стекла. Собранный фильтрат охлаждали в бане со льдом и при перемешивании разбавляли холодной водой (100 мл). Полученный осадок собирали фильтрованием, промывали холодной водой (3), сушили в вакууме и получали 2,79 г 4,5-дибром-2-третбутил-1 Н-имидазола в виде светло-желтого твердого вещества: ЖХМС (m/z): 281,0 (МН+), tR = 0,63 мин; 1 Н ЯМР (300 МГц, ДМСО-d6)1,23 (9 Н, s). Стадия 3. Получение 4,5-дибром-2-трет-бутил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазола. К охлажденному раствору 2-трет-бутил-4,5-дибромимидазола (1,4 г, 5,0 ммоль; пример 5, стадия 2) в сухом ТГФ (10 мл) при 0 С порциями добавляли гидрид натрия (95%, 0,15 г, 6,0 ммоль). Реакционную смесь перемешивали при 0 С в течение 10 мин, при комнатной температуре в течение 40 мин. Реакционную смесь повторно охлаждали до 0 С и по каплям добавляли SEM-хлорид (0,97 мл, 5,5 ммоль). Реакционную смесь перемешивали в течение ночи, не препятствуя таянию льда в охлаждающей бане, и выливали в смесь воды (30 мл) и EtOAc (50 мл). Полученным слоям давали подвергнуться распределению и их разделяли. Органическую порцию промывали рассолом, затем водой, сушили (Na2SO4) и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии (SiO2, 0-10% EtOAc в гексанах) и получали 2,1 г 4,5-дибром-2-трет-бутил-1-2-(триметилсилил)этокси)метил)-1 Н-имидазола: ЖХМС
МПК / Метки
МПК: A61K 31/506, C07D 403/04, A61P 35/00
Метки: композиции, ингибиторы, соединения, b-raf, протеинкиназы
Код ссылки
<a href="https://eas.patents.su/30-20479-soedineniya-i-kompozicii-kak-ingibitory-proteinkinazy-b-raf.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения и композиции как ингибиторы протеинкиназы b-raf</a>
Соединения и композиции в качестве ингибиторов протеинкиназы

Номер патента: 17405
Опубликовано: 28.12.2012
Авторы: Пэй Вэй, Марсилдж Томас Х., Уно Тецуо, Чэнь Бэй, Мишелли Пьер-Ив, Джин Юнхо, Лу Вэньшо, Цзян Тао
МПК: C07D 239/95, C07D 213/74, C07D 239/48...
Метки: протеинкиназы, ингибиторов, соединения, композиции, качестве
Формула / Реферат:
1. Соединение формулы (2)или его фармацевтически приемлемые соли, гдеR1 означает галоген или С1-6алкил;R2 является Н;R3 является (CR2)0-2SO2R12, (CR2)0-2SO2NRR12, (CR2)0-2C(O)O0-1R12, (CR2)0-2CONRR12, CO2NH2 или циано;R4 является С1-6алкил, С2-6алкенил или С2-6алкинил, OR12, NR(R12), галоген, нитро, SO2R12, (CR2)pR13 или X; или R4 означает Н;R6 является изопропокси или метокси;один из R8 и R9 является (CR2)qY и другой является С1-6алкилом,...
Соединения и композиции в качестве ингибиторов протеинкиназы

Номер патента: 19941
Опубликовано: 30.07.2014
Авторы: Мишелли Пьер-Ив, Марсилдж Томас Х., Джин Юнхо, Уно Тецуо, Цзян Тао, Лу Вэньшо, Чэнь Бэй, Пэй Вэй
МПК: C07D 239/95, C07D 239/48, C07D 213/74...
Метки: ингибиторов, соединения, качестве, протеинкиназы, композиции
Формула / Реферат:
1. Соединение формулы (1)или его фармацевтически приемлемые соли, где W являетсяА1 и А4 независимо означают С;каждый А2 и А3 означает С;R1 и R2 вместе образуют 5-6-членный арил, гетероарил или гетероциклил, содержащий 1-3 атома азота, каждый из которых необязательно может быть замещен C1-6алкокси;R3 является (CR2)0-2SO2R12;R4, R7 и R10 независимо означают Н;R, R5 и R5' независимо означают Н или C1-6алкил;R6 означает галоген или О(C1-6алкил);один...
Соединения и композиции в качестве ингибиторов протеинкиназы

Номер патента: 19966
Опубликовано: 30.07.2014
Авторы: Лу Вэньшо, Чэнь Бэй, Пэй Вэй, Джин Юнхо, Мишелли Пьер-Ив, Цзян Тао, Уно Тецуо, Марсилдж Томас Х.
МПК: C07D 213/74, C07D 239/95, C07D 239/48...
Метки: протеинкиназы, качестве, композиции, соединения, ингибиторов
Формула / Реферат:
1. Соединение формулы (1)или его фармацевтически приемлемые соли, где W являетсяА1 и А4 означают С;каждый А2 и А3 означает С;R1 означает галоген или C1-6алкил;R2 является Н;R3 является SO2(C1-6алкил);R4, R7 и R10 независимо означают Н;R6 означает OR12;R, R5 и R5¢ независимо означают Н или C1-6алкил;один из R8 и R9 означает (CR2)qY, а другой означает C1-6алкил;R12 означает C1-6алкил или 5-7-членное гетероциклическое кольцо, содержащее N, О...
Соединения и композиции в качестве ингибиторов протеинкиназы

Номер патента: 18282
Опубликовано: 28.06.2013
Авторы: Ву Баогэнь, Нгуйен Трук Нгок, Марсилдже Томас Х., Чэнь Бэй, Хуан Чэнь, Чжу Сюфэн, Гао Чжаобо, Пэй Вэй, Цзян Тао, Гэ Йонсюй, Мишелли Пьерр-Ив, Ли Юньчэн
МПК: A61K 31/519, A61K 31/505, A61K 31/506...
Метки: качестве, композиции, ингибиторов, протеинкиназы, соединения
Формула / Реферат:
1. Соединение формулы (1)или его физиологически приемлемая соль,где R1 и R2 представляют собой H;R3 представляет собой галоген;R4 представляет собой H;альтернативно, R3 и R4 вместе с атомами углерода, к которым они присоединены, могут образовывать 5-6-членное кольцо, содержащее 1-3 гетероатома азота и необязательно замещенное 1-2 группами R10, где R10 представляет собой галоген, C1-6алкил, фенил или NR2;R5 представляет собой C1-6алкил или...
Соединения и композиции в качестве ингибиторов протеинкиназы

Номер патента: 18484
Опубликовано: 30.08.2013
Авторы: Мишелли Пьерр-Ив, Цзян Тао, Ву Баогэнь, Нгуйен Трук Нгок, Чжу Сюфэн, Пэй Вэй
МПК: A61K 31/519, A61K 31/53, A61P 31/00...
Метки: композиции, качестве, ингибиторов, протеинкиназы, соединения
Формула / Реферат:
1. Соединение формул (1) или (2)или его физиологически приемлемая соль;где X представляет собой 5-6-членный гетероарил, замещенный C1-6алкилом; 6-членное гетероциклическое кольцо, содержащее NR6 или О; или (CR2)0-4CR(NRR7)(CO2R7);Y представляет собой S(O)0-2R8;Z1 представляет собой N или CH;Ph представляет собой фенил иВ представляет собой 5-6-членное кольцо, необязательно содержащее NR6, О, =O или S;R1, R2, R3 и R4 представляют собой H;R5...
Предыдущий патент: Способ финишной обработки деталей
Следующий патент: Резьбовое соединение для кабеля
Случайный патент: Способ очистки и промывки системы раздачи напитков