Кристаллические сольваты производных (1s)-1,5-ангидро-1-с-(3-((фенил)метил)фенил)-d-глюцитола в качестве ингибиторов sglt2 для лечения диабета
Номер патента: 20428
Опубликовано: 28.11.2014
Авторы: Рамакришнан Шривидья, Лай Чиаджень, Гугутас Джек З., Рибель Петер, Сингх Джанак, Нёршль Александра А., Бйн Джеффри Т., Дешпанде Прашант П., Ванг Ченьчи, Лобингер Хильдегард, Гроссо Джон Энтони, Димарко Джон Д.





















Формула / Реферат
1. Кристаллическая форма (R)-PG (форма SD-3) соединения, имеющего формулу Ib

характеризующаяся одной или несколькими следующими характеристиками:
a) рентгенограмма на порошке, включающая значения 2θ (CuKα l = 1.5418 Å), выбранные из группы, состоящей из 3.9 ± 0.1, 8.0 ± 0.1, 8.7 ± 0.1, 15.3 ± 0.1, 15.6 ± 0.1, 17.2 ± 0.1, 19.2 ± 0.1, 19.9 ± 0.1 и 20.3 ± 0.1, при комнатной температуре;
b) спектр ЯМР 13С в твердой фазе, имеющий, по существу, аналогичные положения пиков при 15.8, 17.6, 39.0, 60.9, 63.2, 67.4, 69.7, 77.3, 79.2, 79.8, 113.3, 123.6, 129.0, 130.4, 132.0, 135.6, 139.2 и 157.9 част. на млн, как определено на спектрометре 400 МГц относительно TMS при нуле;
c) термограмма дифференциальной сканирующей калориметрии, имеющая эндотерму в диапазоне от приблизительно 43 до 60°С, или как показано на фиг. 8; или
d) диаграмма термогравиметрического анализа с потерей веса приблизительно 18.7% от приблизительно комнатной температуры до приблизительно 235°С, или как показано на фиг. 6.
2. Кристаллическая форма (R)-PG (форма SD-3) по п.1, характеризующаяся рентгенограммой на порошке, включающей значения 2θ (CuKα l = 1.5418 Å) 3.9 ± 0.1, 8.0 ± 0.1, 8.7 ± 0.1, 15.3 ± 0.1, 15.6 ± 0.1, 17.2 ± 0.1, 19.2 ± 0.1, 19.9 ± 0.1 и 20.3 ± 0.1, при комнатной температуре.
3. Кристаллическая форма EtOH (форма SA-1) соединения, имеющего формулу Ic

характеризующаяся одним или большим количеством параметров элементарной ячейки, по существу, равных следующим:
размеры ячейки

где измерение указанной кристаллической структуры осуществляют при температуре -50°С, и которая характеризуется фракционными атомными координатами, по существу, являющимися такими, как приведено в табл. 6.
4. Кристаллическая форма EG (форма SB-1) соединения, имеющего формулу Id

характеризующаяся 13С-ЯМР спектром в твердой фазе, имеющим, по существу, аналогичные положения пиков при 12.49, 59.16, 60.61, 60.69, 68.10, 72.51, 76.11, 78.51, 79.02, 112.09, 125.16, 126.47, 127.38, 128.61, 129.02, 129.73, 135.62, 137.48 и 154.70 част. на млн, как определено на спектрометре 400 МГц относительно TMS при нуле; или
одним или большим количеством параметров элементарной ячейки, по существу, равных следующим:
размеры ячейки

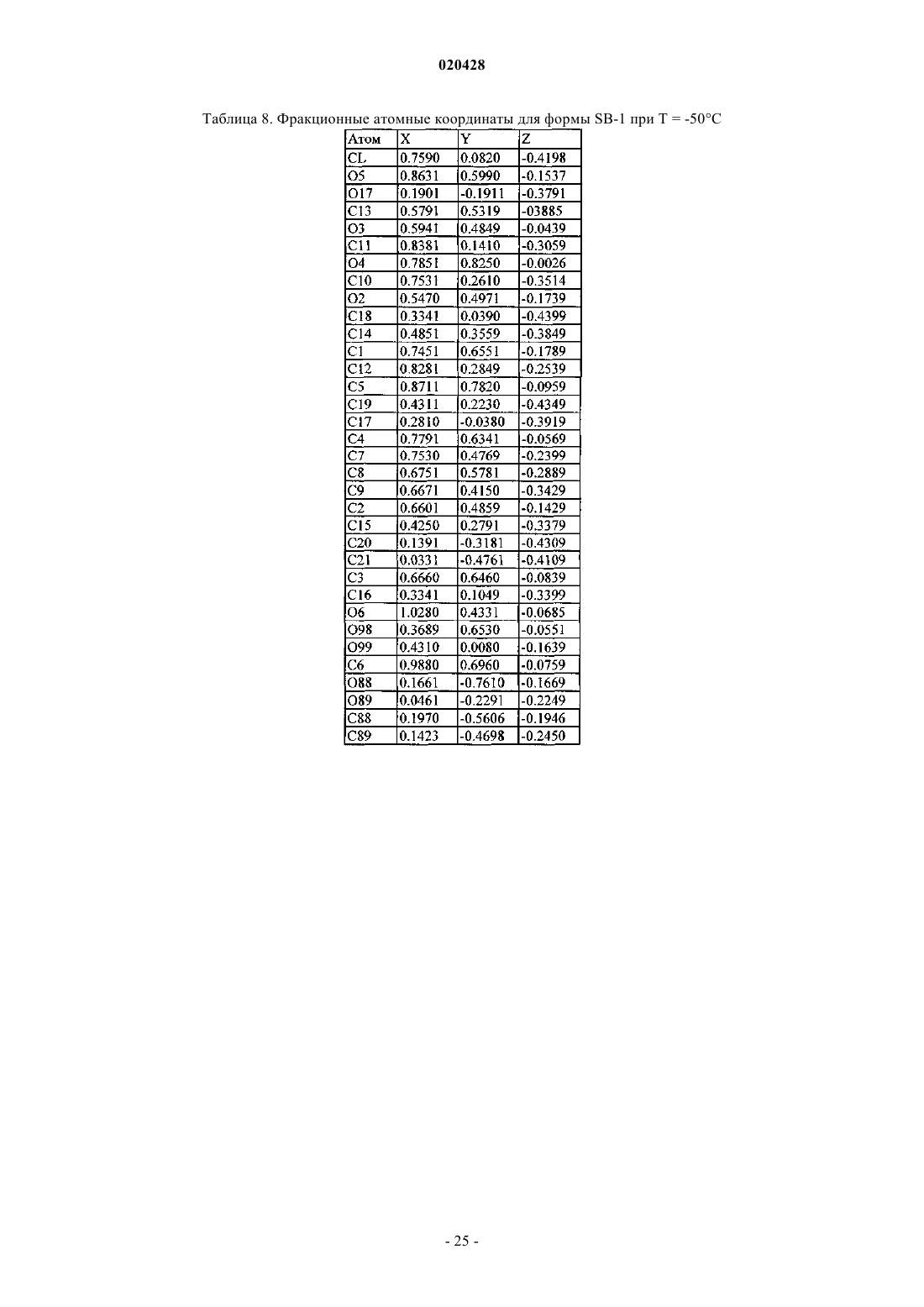
где измерение указанной кристаллической структуры осуществляют при температуре -50°С, и которая характеризуется фракционными атомными координатами, по существу, являющимися такими, как приведено в табл. 8.
5. Кристаллическая форма EG (SB-2) соединения, имеющего формулу Ie

характеризующаяся одним или большим количеством параметров элементарной ячейки, по существу, равных следующим:
размеры ячейки

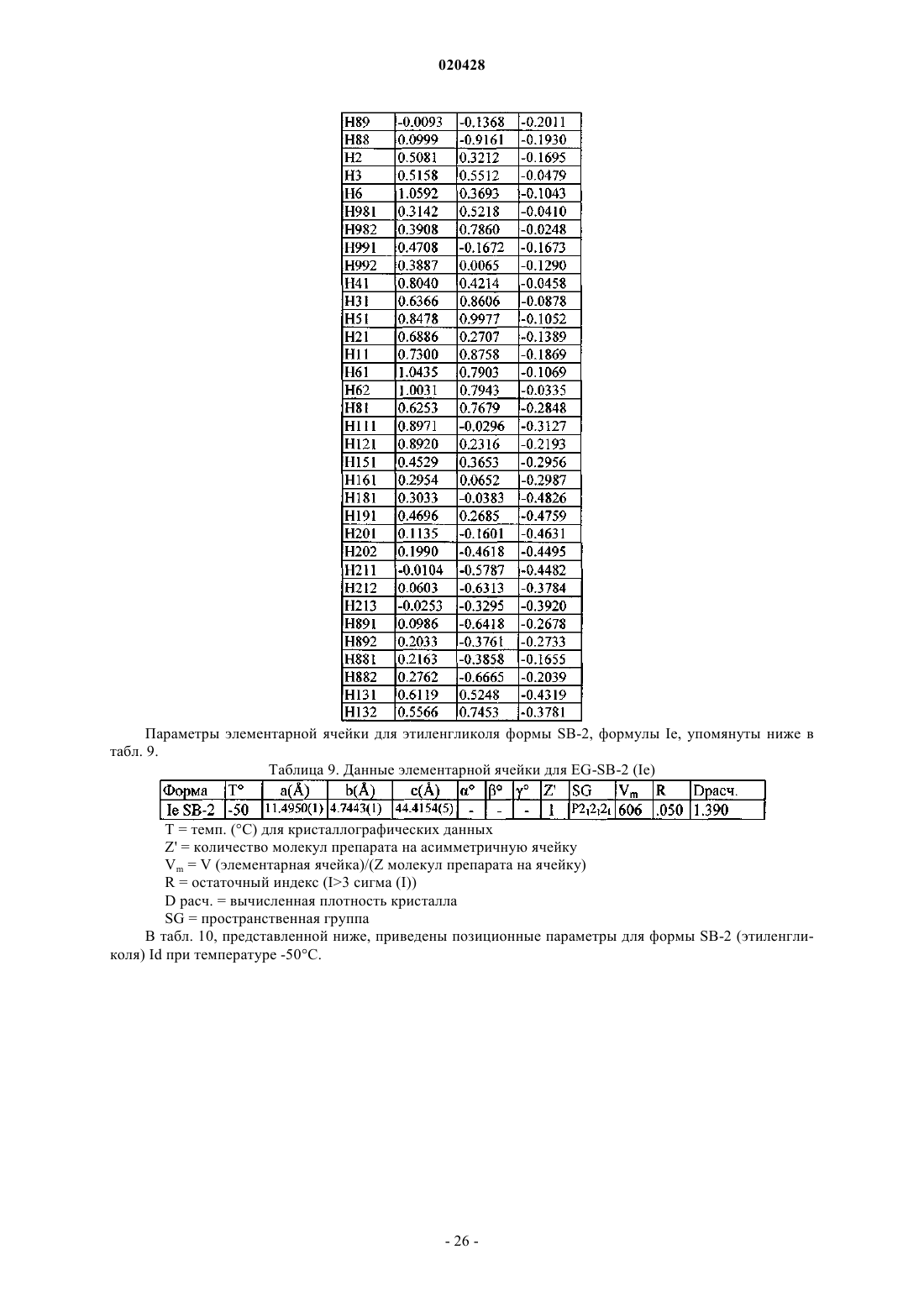
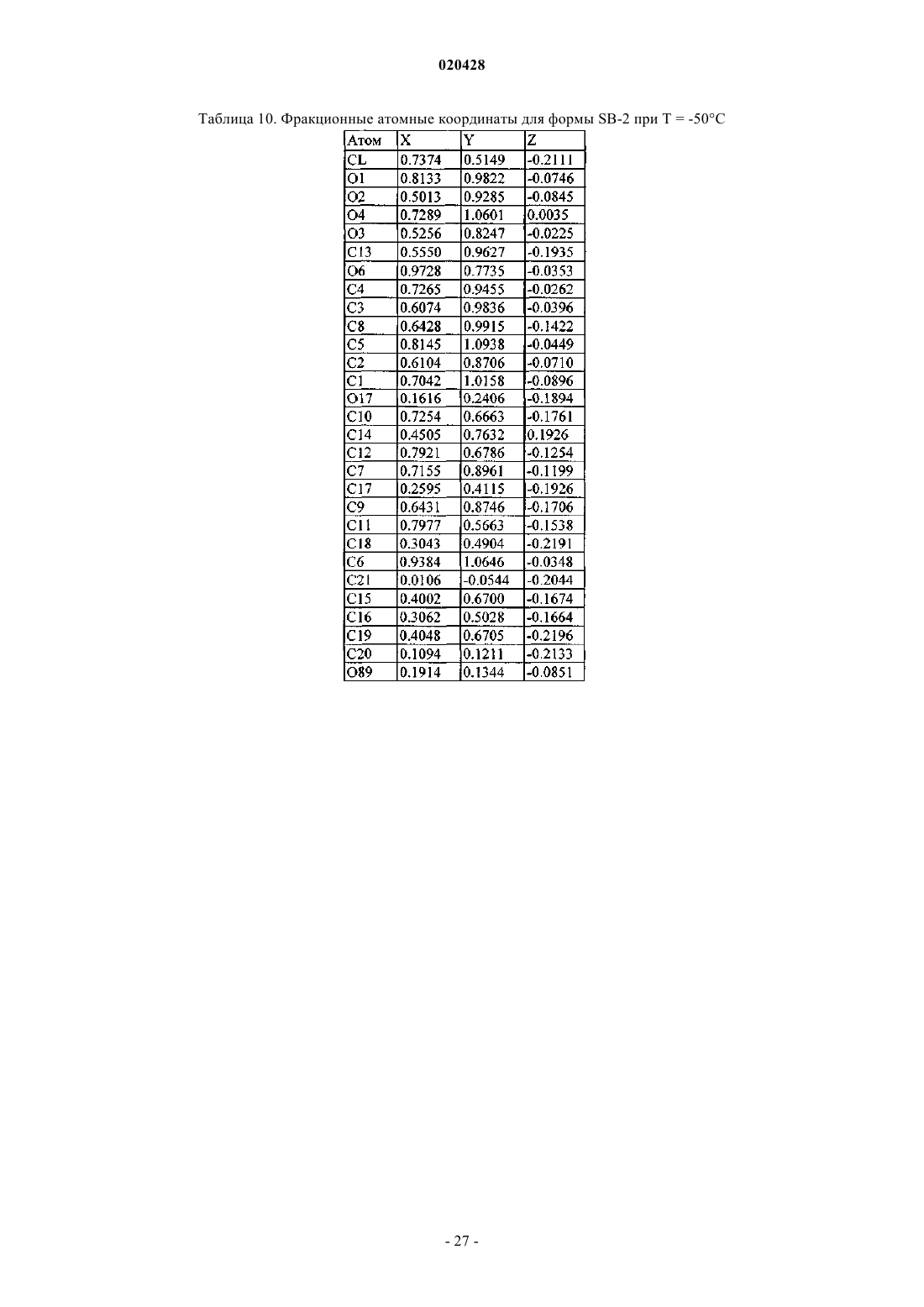
где измерение указанной кристаллической структуры - при комнатной температуре, и которая характеризуется фракционными атомными координатами, по существу, являющимися такими, как приведено в табл. 10.
6. Кристаллическая форма по любому из пп.1-5, находящаяся, по существу, в чистой форме.
7. Фармацевтическая композиция для лечения диабета, диабетической ретинопатии, диабетической невропатии, диабетической нефропатии, замедленного заживления ран, резистентности к инсулину, гипергликемии, гиперинсулинемии, повышенных уровней в крови жирных кислот или глицерина, гиперлипидемии, дислипидемии, ожирения, гипертриглицеридемии, синдрома X, осложнений диабета, атеросклероза или гипертонии или для увеличения уровней липопротеина высокой плотности у млекопитающего, содержащая эффективное количество кристаллической формы по любому из пп.1-5 и фармацевтически приемлемый носитель или разбавитель.
8. Фармацевтическая композиция по п.7, в которой указанная кристаллическая форма находится, по существу, в чистой форме.
9. Фармацевтическая композиция, содержащая эффективное количество кристаллической формы по любому из пп.1-6 в комбинации с одним или большим количеством терапевтических агентов, выбранных из группы, состоящей из антидиабетического агента, агента против ожирения, противогипертонического агента, антиатеросклеротического агента и липидопонижающего агента.
10. Способ лечения диабета, диабетической ретинопатии, диабетической невропатии, диабетической нефропатии, замедленного заживления ран, резистентности к инсулину, гипергликемии, гиперинсулинемии, повышенных уровней в крови жирных кислот или глицерина, гиперлипидемии, дислипидемии, ожирения, гипертриглицеридемии, синдрома X, осложнений диабета, атеросклероза или гипертонии или для увеличения уровней липопротеина высокой плотности у млекопитающего, включающий введение млекопитающему терапевтически эффективного количества кристаллической формы по любому из пп.1-6.
11. Способ по п.10, предназначенный для лечения диабета.
12. Способ получения кристаллической формы соединения, имеющего формулу Ib

по п.1 или 2, включающий реакцию соединения формулы А

в органическом растворителе с основанием и (R)-пропиленгликолем, необязательно с добавлением затравки кристаллической формы (R)-PG соединения, имеющего формулу Ib, с получением кристаллической формы по п.1 или 2.
13. Способ получения кристаллической формы соединения формулы Ic

по п.3, который включает растворение соединения I структуры

в этаноле при охлаждении до температуры в пределах от приблизительно -10 до приблизительно -30°С с образованием кристаллической формы по п.3.
14. Способ по п.13, включающий стадию образования соединения I растворением соединения А структуры

в водном спирте и водном основании нагреванием до кипения и затем нейтрализацией кислотой с образованием кристаллической формы по п.3.
15. Способ получения кристаллической формы соединения формулы Id

по п.4, который включает растворение соединения I структуры

в водном растворе этиленгликоля с получением раствора и добавление затравки кристаллической формы SC-3 соединения формулы Ia (S)-пропиленгликоля

с образованием кристаллической формы по п.4.
16. Способ получения кристаллической формы соединения формулы Ie

по п.5, который включает растворение соединения I структуры

в водном растворе этиленгликоля с получением раствора и добавление затравки кристаллической формы соединения формулы Ic (форма SA-1)

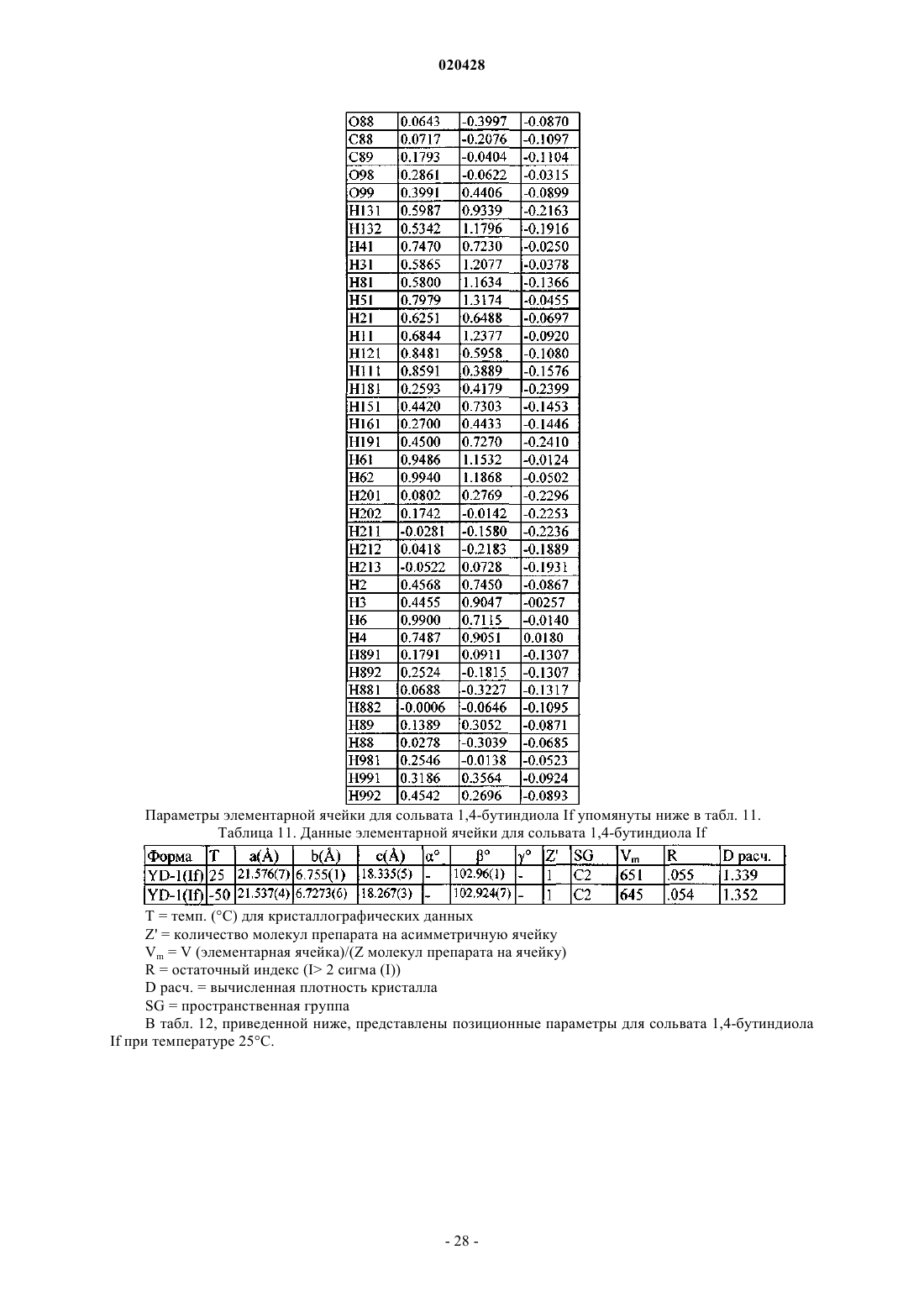
или кристаллической формы SB-1 дигидрата этиленгликоля формулы Id с образованием кристаллической формы по п.5.
Текст
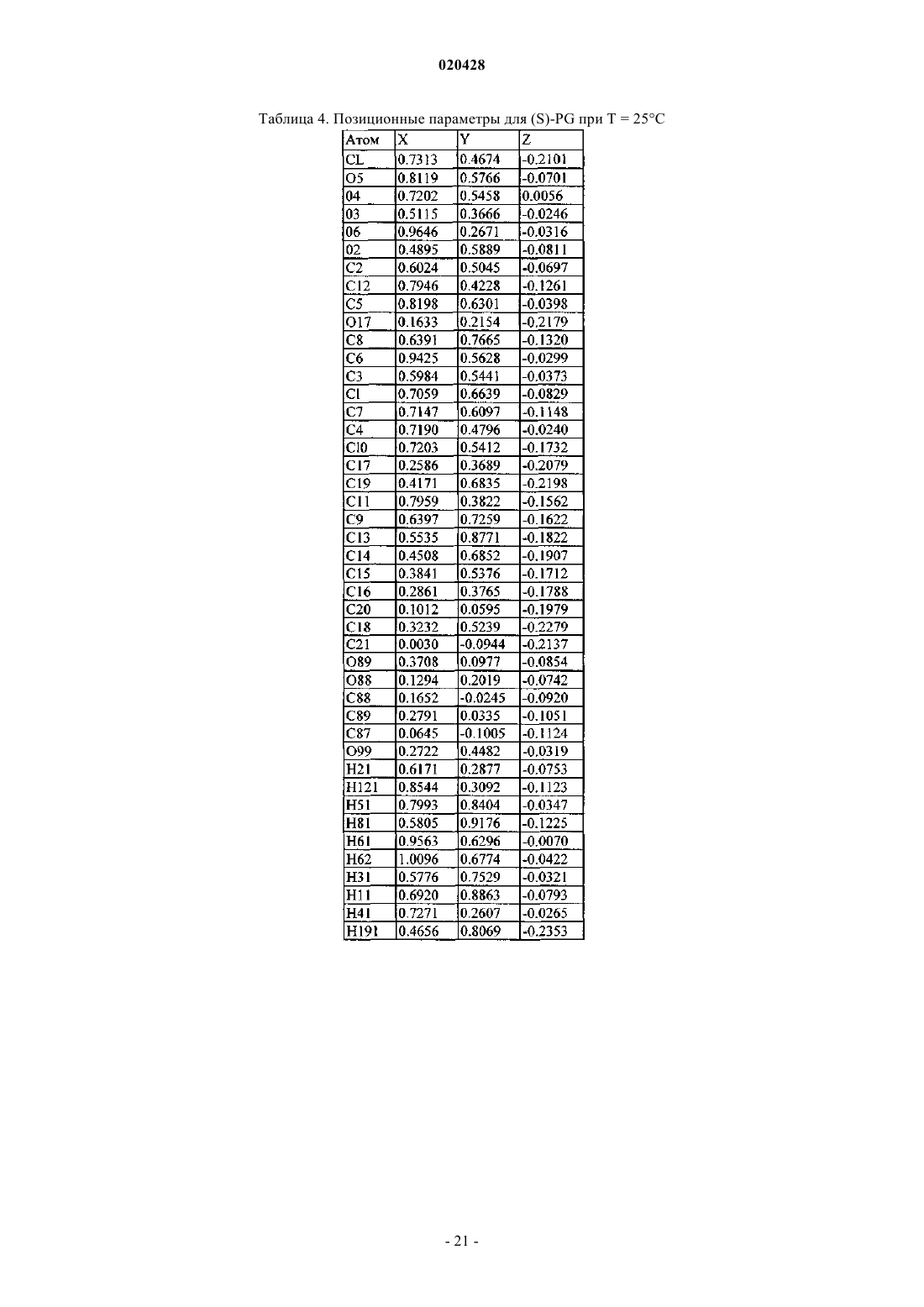
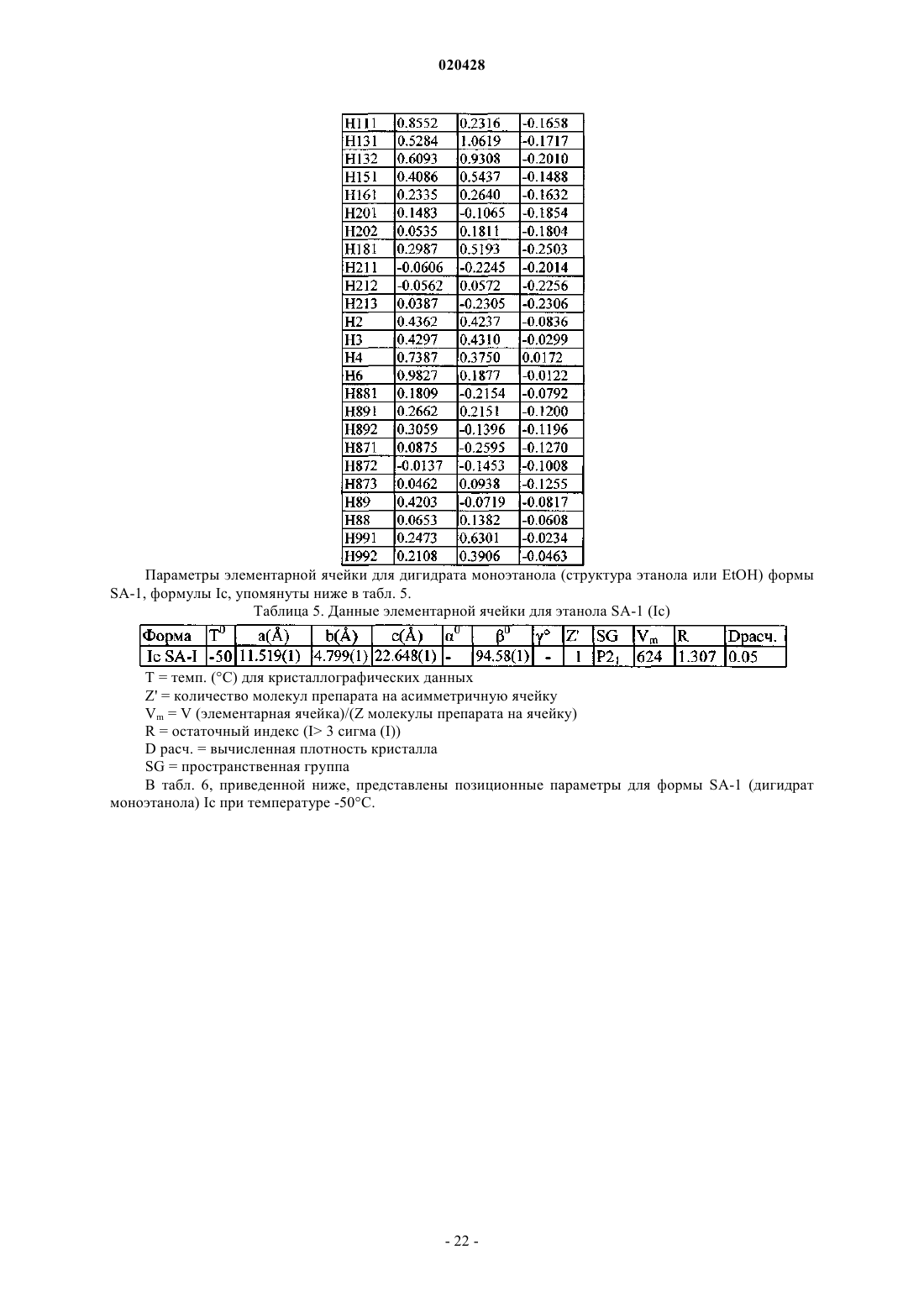
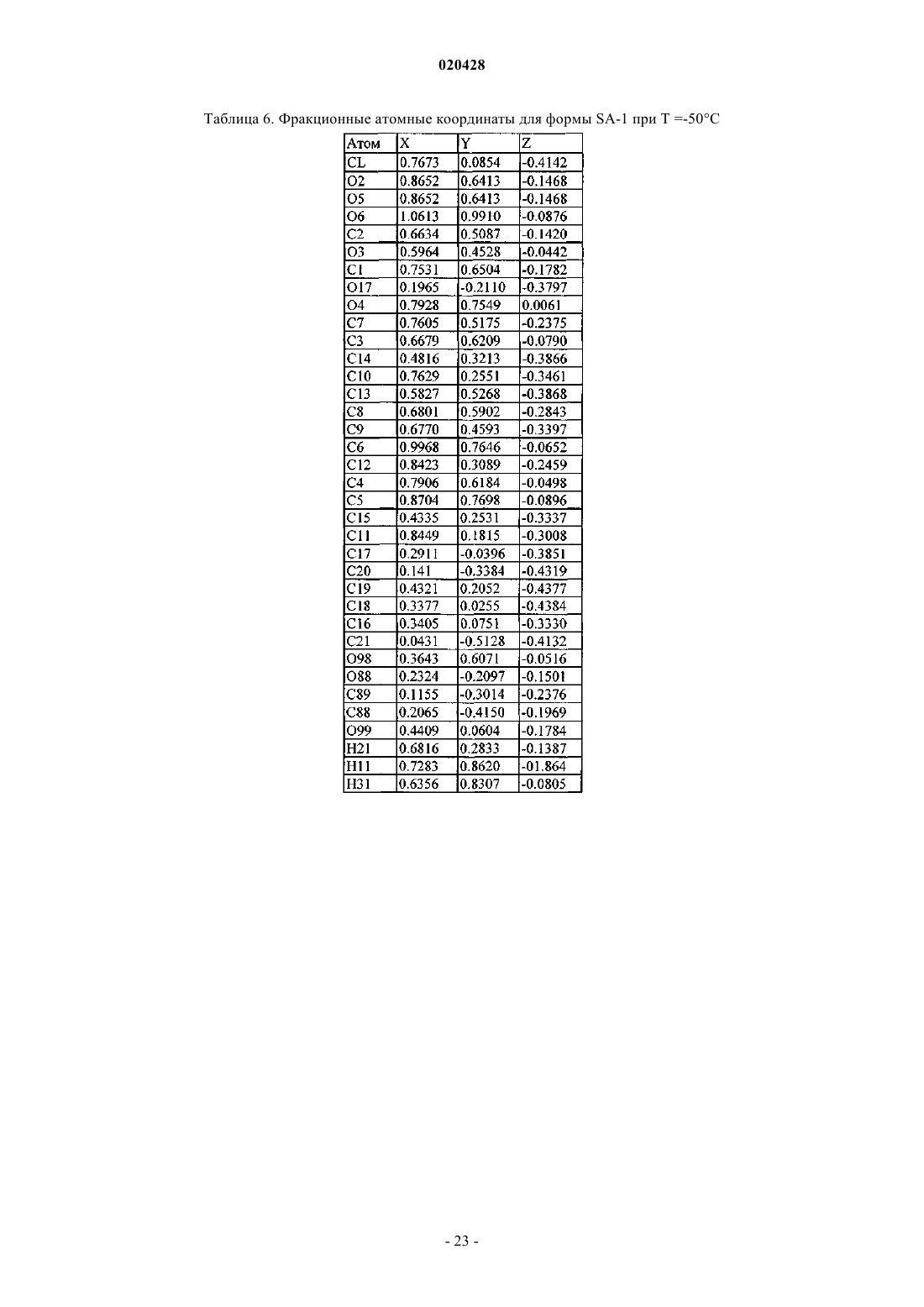
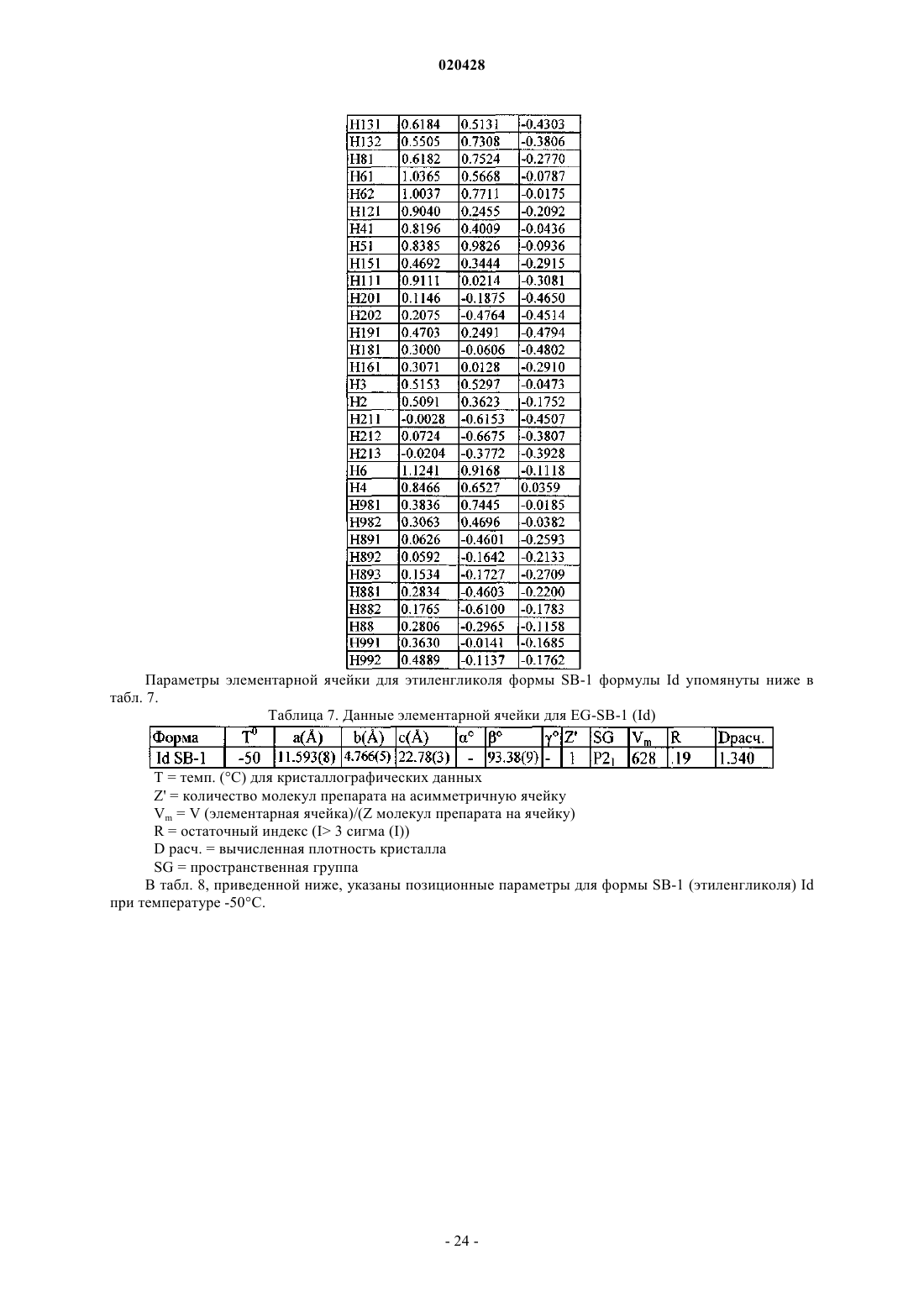
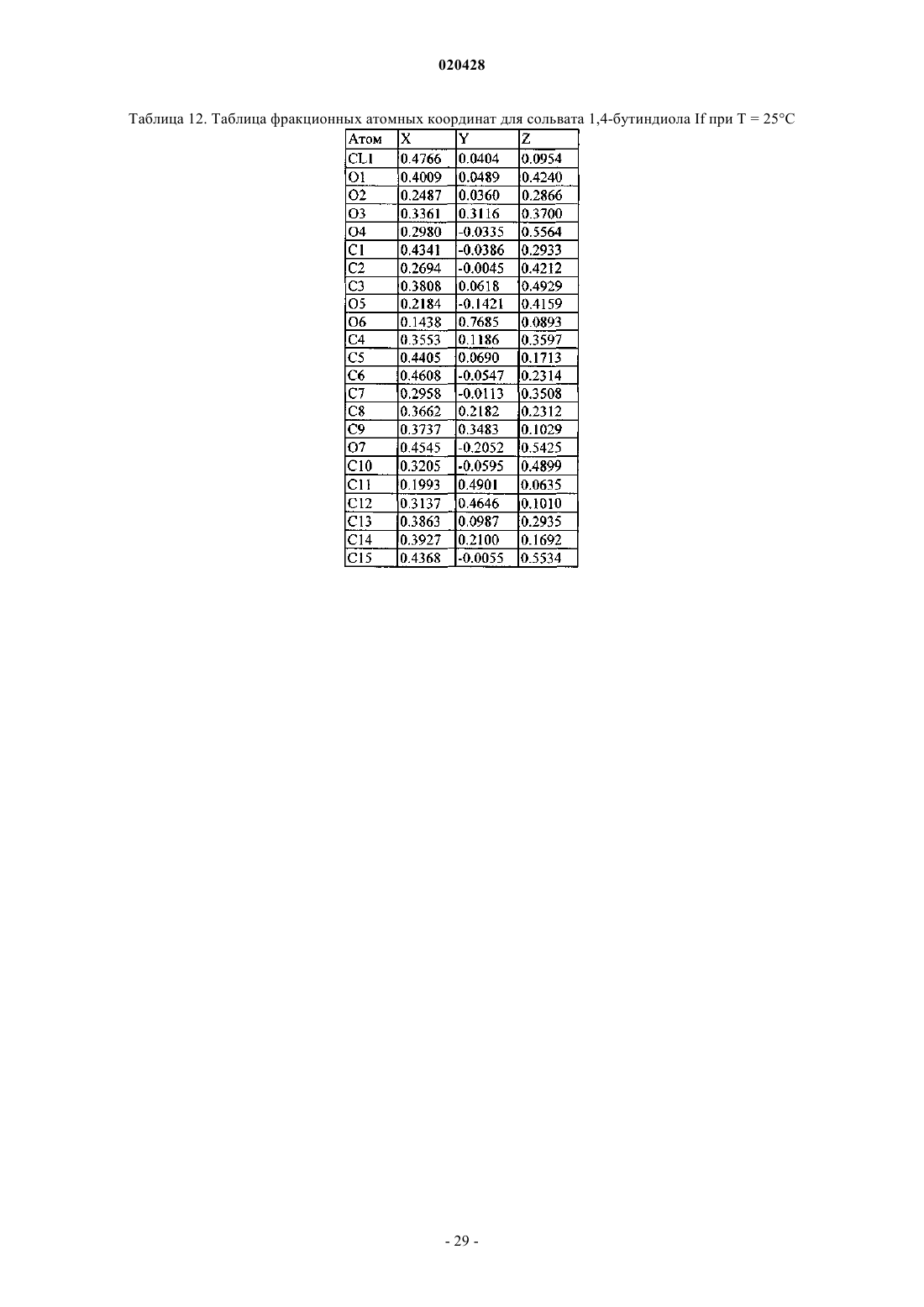
КРИСТАЛЛИЧЕСКИЕ СОЛЬВАТЫ ПРОИЗВОДНЫХ (1S)-1,5-АНГИДРО-1-С-(3 ФЕНИЛ)МЕТИЛ)ФЕНИЛ)-D-ГЛЮЦИТОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ SGLT2 ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА Настоящее изобретение относится к новым кристаллическим формам сольватов соединения формулы I. Также настоящее изобретение относится к способам их получения, к фармацевтическим композициям, содержащим эти кристаллические формы, и к способам лечения диабета,диабетической ретинопатии, диабетической невропатии, диабетической нефропатии, замедленного заживления ран, резистентности к инсулину, гипергликемии, гиперинсулинемии, повышенных уровней в крови жирных кислот или глицерина, гиперлипидемии, дислипидемии, ожирения,гипертриглицеридемии, cиндрома X, осложнений диабета, атеросклероза или гипертонии, или для увеличения уровней липопротеина высокой плотности у млекопитающего с использованием таких кристаллических форм. Область техники, к которой относится изобретение Настоящее изобретение относится к полиморфным кристаллическим структурам свободной кислоты ингибиторов SGLT2, к их фармацевтическим композициям, к способу для получения таких кристаллических структур и также к способам лечения заболеваний, таких как диабет. Сведения о предшествующем уровне техники Приблизительно 100 млн человек во всем мире страдают от диабета типа II (NIDDM), который характеризуется гипергликемией из-за чрезмерной продукции глюкозы в печени и периферической резистентности к инсулину, первопричины для которых являются пока еще неизвестными. Постоянный контроль уровней глюкозы в плазме у пациентов с диабетом может уменьшить развитие диабетических осложнений и декоменсации бета-клеток, отмечаемых в данном заболевании. Глюкоза плазмы обычно фильтруется в клубочках почек и активно реабсорбируется в проксимальном канальце. 90% обратного захвата глюкозы в почке происходит в эпителиальных клетках начального сегмента S1 ренальных кортикальных проксимальных канальцев. SGLT2, 672 аминокислот белка, содержащих 14 перекрывающих мембрану сегментов, которые преобладающе экспрессируются в начальном сегменте S1 ренальных проксимальных канальцев, который вероятно, будет главным транспортером,ответственным за этот обратный захват. Субстратная специфичность, зависимость от натрия и локализация SGLT2 находятся в соответствии со свойствами высокой мкости, низкого сродства, зависимого от натрия переносчика глюкозы, предварительно охарактеризованного в человеческих кортикальных почечных проксимальных канальцах. Кроме того, исследования гибридного истощения выявили SGLT2 в качестве преобладающего сотранспортера Na+/глюкозы в сегменте S1 проксимального канальца, так как фактически вся активность переноса Na-зависимой глюкозы, закодированная в mRNA коры почки крысы, ингибируется антисмысловым олигонуклеотидом, характерным для крысы SGLT2. У людей мутации в SGLT2 были связаны с семейными формами почечной глюкозурии, обеспечивая дальнейшую очевидность первичной роли SGLT2 в почечной реабсорбции глюкозы. У таких пациентов морфология почек и функция почек отлична от нормальной. Ингибирование SGLT2 прогнозирует уменьшение уровней глюкозы в плазме посредством улучшенного выведения глюкозы из организма у пациентов с диабетом. Селективное ингибирование SGLT2 у пациентов с диабетом могло бы нормализовать уровень глюкозы в плазме, увеличивая выделение глюкозы с мочой, таким образом улучшая чувствительность к инсулину и задерживая развитие диабетических осложнений, в отсутствие существенных побочных эффектов со стороны желудочно- кишечного тракта. Сущность изобретения Один из аспектов изобретения относится к кристаллической форме (R)-PG (форма SD-3) соединения структуры Ib Следующий из аспектов изобретения относится к кристаллической форме EtOH (форма SA-1) соединения структуры Ic Следующий из аспектов изобретения относится к кристаллической форме EG (форма SB-1) соединения структуры Id Еще один из аспектов изобретения относится к кристаллической форме EG (форма SB-2) соединения структуры Ie В еще одном из аспектов изобретение относится к фармацевтической композиции для лечения диабета, диабетической ретинопатии, диабетической невропатии, диабетической нефропатии, замедленного заживления ран, резистентности к инсулину, гипергликемии, гиперинсулинемии, повышенных уровней в крови жирных кислот или глицерина, гиперлипидемии, дислипидемии, ожирения, гипертриглицеридемии, синдрома X, осложнений диабета, атеросклероза или гипертонии, или для увеличения уровней липопротеина высокой плотности у млекопитающего, содержащей эффективное количество любой из перечисленных выше кристаллических форм и фармацевтически приемлемый носитель или разбавитель. В своем следующем аспекте изобретение относится к способу лечения диабета, диабетической ретинопатии, диабетической невропатии, диабетической нефропатии, замедленного заживления ран, резистентности к инсулину, гипергликемии, гиперинсулинемии, повышенных уровней в крови жирных кислот или глицерина, гиперлипидемии, дислипидемии, ожирения, гипертриглицеридемии, синдрома X,осложнений диабета, атеросклероза или гипертонии, или для увеличения уровней липопротеина высокой плотности у млекопитающего, включающему введение млекопитающему терапевтически эффективного количества любой из перечисленных выше кристаллических форм. Соединение формулы I в форме некристаллического твердого вещества раскрыто в US 6515117, содержание которого полностью включено в настоящую заявку в качестве ссылки. В другом аспекте настоящего изобретения предлагается способ получения кристаллического соединения (R)-PG структуры Ib (форма SD-3) который включает реакцию соединения формулы А в органическом растворителе с основанием и (R)-пропиленгликолем, необязательно с добавлением затравки кристаллической формы (R)-PG соединения, имеющего формулу Ib. В другом аспекте настоящего изобретения предлагается способ получения формы SA-1, имеющей структуру Ic который включает растворение соединения I структуры в этаноле при охлаждении до температуры в пределах от приблизительно -10 до приблизительно 30 С. Соединение I может быть получено растворением соединения А в этаноле предпочтительно путем нагревания до кипения, чтобы получить продукт в виде масла, который является соединением I. В еще другом воплощении изобретения предлагается способ получения соединения формулы Id который включает растворение соединения I структуры в водном растворе этиленгликоля с получением раствора и добавление затравки кристаллической формы SC-3 соединения формулы Ia (S)-пропиленгликоля В дополнительном воплощении изобретения предлагается способ получения соединения формулы который включает растворение соединения I структуры в водном растворе этиленгликоля с получением раствора и добавление затравки кристаллической формы соединения формулы Ic (форма SA-1), или кристаллической формы SB-1 дигидрата этиленгликоля формулы Id. Краткое описание чертежей Изобретение проиллюстрировано ссылкой на прилагаемые фигуры, описанные ниже. Фиг. 1 иллюстрирует вычисленную (модельную при температуре 25 С) и наблюдаемую (экспериментальную при комнатной температуре) порошковую рентгенограмму (S)-PG кристаллической структуры Ia, форма SC-3. Фиг. 2 иллюстрирует наблюдаемую (экспериментальную при комнатной температуре) порошковую рентгенограмму (R)-PG кристаллической структуры Ib. Фиг. 3 иллюстрирует 13 С ЯМР спектр CPMAS для (S)-PG кристаллической структуры Ia формы SC3. Фиг. 4 иллюстрирует 13 С ЯМР спектр CPMAS для (R)-PG кристаллической структуры Ib. Фиг. 5 иллюстрирует диаграмму термогравиметрического анализа (TGA) (S)-PG кристаллической структуры Ia, формы SC-3. Фиг. 6 иллюстрирует диаграмму термогравиметрического анализа (TGA) (R)-PG кристаллической структуры Ib, формы SD-3. Фиг. 7 иллюстрирует термограмму дифференциальной сканирующей калориметрии (DSC) (S)-PG кристаллической структуры соединения формы Ia, форма SC-3. Фиг. 8 иллюстрирует термограмму дифференциальной сканирующей калориметрии (DSC) (R)-PG кристаллической структуры Ib. Фиг. 9 иллюстрирует наблюдаемую (экспериментальную при комнатной температуре) порошковую рентгенограмму кристаллической структуры сольвата 1,4-бутиндиола If. Фиг. 10 иллюстрирует наблюдаемую (экспериментальную при комнатной температуре) порошковую рентгенограмму кристаллической структуры сольвата диметанола Ig. Фиг. 11 иллюстрирует термограмму дифференциальной сканирующей калориметрии (DSC) кристаллической структуры сольвата 1,4-бутиндиола If. Фиг. 12 иллюстрирует термограмму дифференциальной сканирующей калориметрии (DSC) кристаллической структуры сольвата диметанола Ig. Фиг. 13 является схематическим представлением непрерывного реакционного процесса. Сведения, подтверждающие возможность осуществления изобретения Настоящее изобретение обеспечивает, по крайней мере в части, кристаллические структуры соединения I в качестве новых веществ. Термин "фармацевтически приемлемый", как используется в настоящей заявке, относится к тем соединениям, веществам, композициям и/или дозированным формам, которые находятся в области традиционной медицинской практики, которые являются пригодными для контакта с тканями людей и животных без чрезмерной токсичности, раздражения, аллергической реакции или других осложнений, соразмерных с разумным соотношением польза/риск. В определенных предпочтительных воплощениях кристаллические структуры соединения I изобретения находятся, по существу, в чистой форме. Термин ", по существу, чистый", как используется в настоящей заявке, означает соединение, имеющее чистоту больше чем приблизительно 90%, включая, например, приблизительно 91%, приблизительно 92%, приблизительно 93%, приблизительно 94%, приблизительно 95%, приблизительно 96%, приблизительно 97%,приблизительно 98%, приблизительно 99% и приблизительно 100%. Свойство соединения находится в виде различных кристаллических структур известно как полиморфизм. Как используется в настоящей заявке, термин "полиморф" относится к кристаллическим формам, имеющим тот же самый химический состав, но различные пространственные расположения молекул, атомов и/или ионов, формирующих кристалл. В то время как полиморфы имеют тот же самый химический состав, они отличаются по упаковке и геометрическому расположению и могут показывать различные физические свойства, такие как температура плавления, форма, цвет, плотность, твердость,деформируемость, стабильность, растворимость и т.п. В зависимости от соотношения температурастабильность, два полиморфа могут быть или монотропными, или энантиотропными. Для монотропной системы относительная стабильность между двумя твердыми фазами остается неизменной, когда изменяется температура. Напротив, в энантиотропной системе существует температура перехода, при которой стабильность этих двух фаз полностью изменяется (Theory and Origin of Polymorphism in "Polymorphism in Pharmaceutical Solids" (1999) ISBN: )-8247-0237). Образцам кристаллических структур по изобретению можно обеспечить, по существу, чистую фа-4 020428 зовую однородность, идентифицируемую наличием доминирующего количества единственной кристаллической структуры и, необязательно, незначительных количеств одной или более других кристаллических структур. Присутствие больше чем одной кристаллической структуры по изобретению в образце может быть определено методами, такими как порошковая дифракции рентгеновских лучей (PXRD) или твердофазная ядерная магнитно-резонансная спектроскопия (SSNMR). Например, присутствие экстрапиков при сравнении экспериментально измеренной рентгенограммы PXRD (наблюдаемой) с модельной рентгенограммой PXRD (вычисленной) может указывать на больше чем одну кристаллическую структуру в образце. Модельная рентгенограмма PXRD может быть вычислена на основе данных рентгеновского анализа монокристалла (см. Smith, D.K., "А FORTRAN Program for Calculating X-Ray Powder DiffractionPatterns", Lawrence Radiation Laboratory, Livermore, California, UCRL-7196 (April 1963); см. также Yin. S.,Scaringe, R.P., DiMarco, J., Galella, M. and Gougoutas, J.Z., American Pharmaceutical Review, 2003, 6, 2, 80). Предпочтительно кристаллическая структура имеет, по существу, чистую фазовую однородность, как обозначено посредством меньше чем 10%, предпочтительно меньше чем 5% и более предпочтительно меньше чем 2% общей площади пика в экспериментальной рентгенограмме PXRD, являющейся результатом дополнительных пиков, которые отсутствуют на модельной рентгенограмме PXRD. Наиболее предпочтительна кристаллическая структура по изобретению, имеющая, по существу, чистую фазовую однородность с меньше чем 1% от общей площади пика в экспериментальной рентгенограмме PXRD,являющейся результатом дополнительных пиков, которые отсутствуют на модельной рентгенограммеPXRD. Различные кристаллические структуры по изобретению, описанному в настоящей заявке, могут быть различимы друг от друга с помощью различных аналитических методов, известных специалисту в данной области. Такие методы включают, но не ограничиваясь, твердофазную ядерную магнитнорезонансную спектроскопию (SSNMR), порошковую дифракцию рентгеновских лучей (PXRD), дифференциальную сканирующую калориметрию (DSC) и/или термогравиметрический анализ (TGA). Получение кристаллических структур Кристаллические структуры по изобретению могут быть получены разнообразными методами,включая, например, кристаллизацию или перекристаллизацию из подходящего растворителя, сублимацию, рост из расплава, переход в твердую фазу из другой фазы, кристаллизацию из суперкритической жидкости и реактивное распыление. Методы для кристаллизации или перекристаллизации кристаллических форм из растворяющей смеси включают, например, выпаривание растворителя, уменьшение температуры растворяющей смеси, использование кристаллической затравки в пересыщенной растворяющей смеси молекулы и/или соли, замораживание, высушивающее растворяющую смесь и добавление антирастворителей (противорастворителей) к растворяющей смеси. Могут использоваться высокопроизводительные методы кристаллизации для получения кристаллических форм, включая полиморфы. Кристаллические лекарства, включая полиморфы, способы получения и характеристики кристаллов лекарства обсуждаются в Solid-State Chemistry of Drugs, S.R. Byrn, R.R. Pfeiffer, и J.G. Stowell, 2nd Edition,SSCI, West Lafayette, Indiana, 1999. Кристаллические затравки могут быть добавлены к любой смеси для кристаллизации для ускорения кристаллизации. Как будет понятно квалифицированному специалисту, применение затравки используется для управления ростом конкретной кристаллической структуры или для управления распределением размера частиц кристаллического продукта. Соответственно, вычисление количества необходимых затравок зависит от размера доступной затравки и желательного размера средней частицы продукта, как описано, например, в "Programmed Cooling of Batch Crystallizers", J.W. Mullin и J. Nyvlt, Chemical Engineering Science, 1971, 26, 369-377. Вообще, затравки небольшого размера необходимы для эффективного управления ростом кристаллов в партии. Затравка небольшого размера может быть получена просеиванием, помолом или тонким измельчением больших кристаллов или микрокристаллизацией растворов. Должна быть предпринята осторожность, чтобы размалывание или тонкое измельчение кристаллов не привели ни к какому изменению в кристалличности формы заданной кристаллической структуры (то есть изменения на аморфную форму или на другой полиморф). Как используется в настоящей заявке, термин "комнатная температура" или "RT" обозначает температуру окружающей среды от 20 до 25 С (68-77F). Вообще, в получении кристаллического соединения Ia, как описано ниже, растворитель(и) будет использоваться для образования кристаллического соединения Ia, предпочтительно имеющего объемную плотность, как описано ниже. Кристаллическое соединение структуры Ia (S-PG) SC-3 получено согласно следующей сокращенной реакции, как показано на схеме I. Соединение формулы В (аморфная форма) раскрыто в US 10/745,075, поданной 23 декабря 2003,содержание которой полностью включено в настоящую заявку посредством отсылки. Как отмечено на схеме I, соединение В или If, или Ig (все вместе относятся к соединению В), где соединение В в форме аморфного или кристаллического твердого вещества (If или Ig) обрабатывают агентом восстановления,таким как силилгидрид, предпочтительно алкилсилилгидрид, более предпочтительно триэтилсилан (или триэтилсилилгидрид), в присутствии активирующей группы, которая является кислотой Льюиса, такой как BCl3Me2S, BBr3, BF3OEt2, BCl3 или BF32CH3COOH, предпочтительно BF3OEt2 или BF32 СН 3 СООН,и органического растворителя, такого как CH3CN, CH3CN/толуол или CH3CN/дихлорметан, метиленхлорид или вода, при температуре в пределах диапазона от приблизительно -15 до приблизительно 25 С,предпочтительно от приблизительно 5 до приблизительно 10 С, для восстановления соединения В и образования соответствующего основного соединения I которое отделяют от реакционной смеси и обрабатывают (S)-пропиленгликолем S)-PG) и органическим растворителем, таким как алкилацетат, как изложено выше, предпочтительно изопропилацетат или метилтретбутиловый эфир (МТВЕ), и необязательно, затравкой соединения S)-PG) Ia (молярное отношение затравка Ia:соединение В в пределах диапазона от приблизительно 0.1 до приблизительно 10%, предпочтительно приблизительно от 0.5 до приблизительно 3%), с образованиемкристаллической суспензии соединения S)-PG) Ia, и выделяют кристаллическое соединение S)-PG) Ia из кристаллической суспензии. При выполнении вышеупомянутой сокращенной реакции схемы I, силильный восстанавливающий агент будет использоваться в молярном соотношении к соединению В в пределах диапазона от приблизительно 1.2:1 до приблизительно 4.5:1, предпочтительно от приблизительно 2:1 до приблизительно 4:1,в то время как активирующая группа (кислота Льюиса) будет использоваться в молярном соотношении к силильному восстанавливающему агенту в пределах диапазона от приблизительно 1.2:1 до приблизительно 4.5:1, предпочтительно от приблизительно 2:1 до приблизительно 4:1. (S)-пропиленгликоль S)PG) будет использоваться в молярном соотношении к соединению В в пределах диапазона от приблизительно 0.9:1 до приблизительно 1.5:1, предпочтительно от приблизительно 0.98:1 до приблизительно 1.2:1; вода будет использоваться в молярном соотношении к (S)-PG в пределах диапазона от приблизительно 0.95:1 до приблизительно 5:1, предпочтительно от приблизительно 0.99:1 до приблизительно 2:1. Кристаллическое соединение структуры Ia S)-PG) формы SC-3 может также быть получено согласно реакционной схеме II, изложенной ниже. где соединение А обрабатывают спиртовым растворителем, таким как метанол, этанол или изопропиловый спирт, предпочтительно метанол, водой и водным основанием, таким как гидроксид щелочного металла, таким как NaOH, KOH или LiOH, предпочтительно NaOH, предпочтительно в инертной атмосфере, такой как азот, при повышенной температуре в пределах диапазона от приблизительно 50 до приблизительно 85 С, предпочтительно от приблизительно 60 до приблизительно 80 С, чтобы получить соединение I. Водное основание будет использоваться в молярном соотношении к соединению в пределах диапазона от приблизительно 3.5:1 до приблизительно 5.5:1, предпочтительно от приблизительно 3:1 до приблизительно 5:1. Реакционную смесь, содержащую соединение I, обрабатывают органическим растворителем, таким как метилтретбутиловый эфир (МТВЕ) или алкилацетат, как описано выше, предпочтительно изопропилацетат или МТВЕ, выделяют соединение I, которое обрабатывают с (S)-пропиленгликолем, чтобы получить сгущенную суспензию, содержащую кристаллический продукт Ia (S)-PG, форма SC-3. Необязательно, добавляют затравку соединения S)-PG) Ia к реакционной смеси. Кристаллическое соединение Ia отделяют от суспензии с использованием традиционных процедур, например, суспензию соединения Ia обрабатывают органическим растворителем, таким как циклогексан, изооктан или метилциклогексан,предпочтительно циклогексан, и выделяют кристаллическое соединение Ia. В процессе образования соединения Ia (S)-PG используется в молярном соотношении к соединениюI в диапазоне от приблизительно 0.9:1 до приблизительно 1.5:1, предпочтительно от приблизительно 0.98:1 до приблизительно 1.2:1. Как отмечалось ранее в настоящей заявке, сольват (R)-пропиленгликоля Ib соединения I может быть получен методом, подобным методу получения сольвата(S)-пропиленгликоля Ia, за исключением того, что (R)-пропиленгликоль используется вместо (S)-пропиленгликоля. Способ согласно изобретению для получения моно-EtOH-дигидрата (этанол или EtOH/структура) формы SA-1 (соединение Ic) показан на схеме III, приведенной ниже. где соединение А растворяют в этаноле посредством нагревания до кипения, затем добавляют воду при объемном соотношении к этанолу в пределах диапазона от приблизительно 1:1 до приблизительно 3:1, предпочтительно от приблизительно 1.5:1 до приблизительно 2.5:1. Добавляют этанол и смесь охлаждают до температуры в диапазоне температур от приблизительно -10 до приблизительно -30 С, предпочтительно от приблизительно -15 до приблизительно -25 С. Соединение Ic выделяют в виде кристаллов моно-EtOH-дигидрата. Способ согласно изобретению для получения структур формы SB-1 и формы SB-2 дигидрата этиленгликоля (соединений Id и Ie, соответственно) осуществляют следующим образом. Соединение Id формы SB-1 получают растворением соединения А в водном растворе этиленгликоля (вода:этиленгликоль от приблизительно 1:1 до приблизительно 0.4:1, предпочтительно от приблизительно 0.7:1 до приблизительно 0.5:1) посредством нагревания при температуре в пределах диапазона от приблизительно 35 до приблизительно 55 С, предпочтительно от приблизительно 40 до приблизительно 50 С, в течение приблизительно от 1.5 до приблизительно 2 ч, предпочтительно от приблизительно 0.30 мин приблизительно до 1 ч. Смесь охлаждают до температуры в пределах диапазона от приблизительно 10 до приблизительно 22 С, предпочтительно от приблизительно 14 до приблизительно 16C и добавляют затравку кристаллов Ic моно-EtOH-дигидрата или кристаллов дигидрата этиленгликоля формы SB-1Id в молярном соотношении к соединению А в пределах диапазона от приблизительно 0.1 приблизительно до 10%, предпочтительно от приблизительно 0.5 приблизительно до 3%, для получения кристалла дигидрата этиленгликоля формы SB-1 Id. В соответствии с настоящим изобретением кристалл дигидрата этиленгликоля формы SB-2 Ie образуется посредством растворения соединения А в водном растворе этиленгликоля (вода:этиленгликоль от приблизительно 1:1 до приблизительно 0.4:1, предпочтительно от приблизительно 0.7:1 до приблизительно 0.5:1), нагреванием при температуре в пределах диапазона от приблизительно 35 до приблизительно 55 С, предпочтительно от приблизительно 40 до приблизительно 50 С, в течение приблизительно от 1.5 приблизительно до 2 ч, предпочтительно приблизительно от 0.30 мин приблизительно до 1 ч. Смесь охлаждают до температуры в пределах диапазона от приблизительно 10 С до приблизительно 30 С, предпочтительно от приблизительно 20 до приблизительно 25 С и добавляют затравку кристаллов дигидрата этиленгликоля формы SB-2 Ie при молярном соотношении к соединению А в пределах диапазона от приблизительно 0.1 приблизительно до 10%, предпочтительно от приблизительно 0.5 приблизительно до 3%, для получения кристалла дигидрата этиленгликоля формы SB-2 Ie. Способ согласно изобретению для получения кристаллической формы соединения В, которая является формой If, осуществляют в соответствии со схемой IV, изложенной ниже. Кристаллический сольват 1,4-бутиндиола If по изобретению получают согласно следующей реакционной схеме IV. где некристаллическое соединение В (которое может быть получено, как описано в US10/745075, поданной 23 декабря 2003, или в US 6515117), предпочтительно в, по существу, чистой форме(например, с чистотой от 50 до 100%), смешивают со смесью толуол/алкилацет (такой как этилацетат) и смесь нагревают до температуры в пределах диапазона от приблизительно 50 до приблизительно 70 С,предпочтительно от приблизительно 55 до приблизительно 65 С, добавляют 2-бутин-1,4 диол и нагревают, как указано выше, пока диол не растворится, добавляют затравку соединения If и смесь охлаждают до получения кристаллов соединения If. В альтернативном способе получения кристаллического соединения If соединение В растворяют в алкилацетате (таком как бутилацетат) или в смеси алкилацетат/гептан (от 0.5:1 до 1.5:1), при повышенной температуре в пределах диапазона от приблизительно 50 до приблизительно 70 С, предпочтительно от приблизительно 55 до приблизительно 65 С, добавляют 1,4-бутиндиол и смесь охлаждают до комнатной температуры до образования кристаллов соединения If. В предпочтительном воплощении соединение If кристаллизуют из смеси соединения В и толуол/алкилацета (предпочтительно этилацетата), содержащей объемное отношение толуола к алкилацетату в пределах диапазона от приблизительно 1:1 до приблизительно 19:1, предпочтительно от приблизительно 4:1 до приблизительно 9:1. Смесь толуол/алкилацетат будет включать достаточное количество толуола, чтобы получить молярное соотношению к соединению В в пределах диапазона от приблизительно 40:1 до приблизительно 90:1, предпочтительно от приблизительно 60:1 до приблизительно 80:1, до образования сольвата 1,4-бутиндиола If. Кристаллизация для образования сольвата 1,4-бутиндиола If может быть более легко осуществлена с использованием кристаллов затравки соединения If в количестве от приблизительно 0.1 до приблизительно 10%, предпочтительно от приблизительно 0.5 до приблизительно 3% в расчете на вес исходного соединения В. В другом предпочтительном воплощении соединение If (которое может быть или может не быть очищено) кристаллизуют из смеси соединения В и алкилацетат/гептан (предпочтительно бутилацетат/толуол) необязательно с использованием затравки кристаллического соединения If, использующегося от приблизительно 0.1 до приблизительно 10%, предпочтительно от приблизительно 0.5 до приблизительно 3% затравки If в расчете на вес исходного соединения В. Алкилацетат будет использоваться при объемном соотношении с гептаном в пределах диапазона от приблизительно 0.5:1 до приблизительно 2:1, предпочтительно от приблизительно 1:1 до приблизительно 1:1.5. Кристаллический сольват 1,4-бутиндиола If может также быть получен в непрерывном процессе,как показано на схеме IVA. Синтез сольвата If включает две последовательные стадии с соединением Е и соединением D: (1) литирование соединения Е, чтобы получить литированное промежуточное соединение G, и (2) связывание литированного промежуточного соединения G с соединением D. Со ссылкой на фиг. 13 показана блок-схема процесса (подобного раскрытому в US 7164015, который включен в настоящую заявку в качестве ссылки). В этом воплощении весь способ получения соединения If, как показано на схеме IVA, выполняют при некриогенных условиях. Ароматический реагент Е,имеющий группу, подходящую для лития и галогенового обмена, хранят в первом сосуде 1 при комнатной температуре. Литиевый реактив Q подают во второй сосуд 2 также при комнатной температуре. Ароматический реагент Е и литиевый реактив Q перемещают из сосудов 1 и 2 с помощью насосов 3 и 4,соответственно, к первому накрытому кожухом статическому миксеру 5. Температура реакции для получения литированных анионных частиц регулируется в диапазоне от приблизительно -30 С до приблизительно 20 С, в первом миксере 5 с помощью холодильника 6. Литированные анионные частицы G, таким образом образованные, подаются непосредственно из первого миксера 5 во второй статический миксер 22 по обычной линии подачи 19. Карбонилом замещенный реагент D подается в третий сосуд 20 при комнатной температуре и перемещается насосом 21 через холодильник 26, где он охлаждается до температуры в пределах диапазона от приблизительно -10 до приблизительно -30 С, и затем перемещается во второй покрытый кожухом статический миксер 22. Реакция для получения продукт гликозида Н регулируется во втором миксере 22 с помощью второго холодильника 23. Затем обработка в условиях гликозидирования происходит, где Н подается в обычный реактор 25,где его обрабатывают кислотой в спиртовом растворителе, предпочтительно MSA/MeOH или HCl/МеОН,с образованием Н' (десилилированного гемикеталя), который затем преобразовывают в гликозид В. Затем дополнительное выделение продукта и обратная экстракция и кристаллизация с 2-бутин-1,4-диолом(J) в смеси толуол/EtOAc позволяют получить кристаллический продукт If. Реактор 25 может поддерживаться при комнатной или другой некриогенной температуре в течение любых последующих реакций. Используемый литиевый реагент представляет собой по желанию органолитиевый реагент. Подходящие органолитиевые реагенты включают n-BuLi, S-BuLi и t-BuLi. Другие будут очевидны квалифицированным в данной области специалистам. После завершения реакции желаемый продукт If может быть выделен и очищен согласно методам,широко известным в данной области органической химии (например, осаждением, экстракцией растворителем, перекристаллизацией и хроматографией). Соединение If со снятием защитной группы само по себе может быть полезным в качестве промежуточного продукта или конечного продукта. Соединение If может реагировать далее с получением фармацевтически приемлемых кислотно-аддитивных или основных солей, с использованием методов, которые будут известны квалифицированным в данной области специалистам. Температура и время реакции - два важных параметра в непрерывном способе, показанном на схемеIVA: литированием можно управлять непрерывно от -30 (или ниже) до 20 С (или выше), предпочтительно приблизительно от -17 до приблизительно -10 С, от минут до секунд времени реакции. Для последующей реакции связывания поток литированного производного G затем смешивают с потоком соединения D (третий поток подачи) в миксере. Смешанный поток можно затем направить в проточный реактор, если дополнительное время реакции необходимо для завершения. Реакцией связывания можно управлять непрерывно при более высоких температурах от -30 до -10 С (или выше), предпочтительно приблизительно от -30 до приблизительно -20 С, от минут до секунд времени реакции. Поток реакции связывания затем направляют в реактор периодического действия для дальнейших реакций, как описано в настоящей заявке. При непрерывном процессе и реакция литирования, и реакция связывания могут быть объединены и могут управляться при более высоких температурах, использующих меньшие проточные реакторы с эффективным температурным контролем, по сравнению с криогенными периодическими реакторами в масштабе. Рабочая температура непрерывного литирования в вышеупомянутом процессе может быть до 20 С(не ограничиваясь), предпочтительно от -17 до -10 С, производя 95 RAP желательного литированного промежуточного продукта G. В реакции связывания продукт связывания вышеупомянутого процесса при от -20 до -30 С предпочтительно находится в диапазоне 70-79 RAP. Соединение If может использоваться для получения кристаллического промежуточного продукта,как показано на схеме IVB. Схема IVB Получение промежуточного продукта А Со ссылкой на схему IVB, соединение If, твердый DMAP, жидкий ацетонитрил и жидкий уксусный ангидрид нагревают до температуры в пределах диапазона от приблизительно 70 до приблизительно 85 С и выдерживают до тех пор, пока реакция не завершится. Загрузку охлаждают (например, 5 С). Триэтилсилан и комплекс трифторида бора с уксусной кислотой или другую кислоту Льюиса (как описано в отношении схемы I) добавляют к реакционной смеси. После того как реакция завершена, добавляют ацетон или другой растворитель. Загрузку нагревают (например, от приблизительно 20 до приблизительно 30 С) и выдерживают до тех пор, пока не израсходован триэтилсилан. Добавляют водный раствор NH4OAc и загрузку перемешивают и позволяют ей расслоиться на верхнюю и более нижнюю формы фаз. Восстанавливают загрузочный объем продукта в обогащенной верхней фазе, отгоняя ацетонитрил при минимальном перемешивании. SDA3A этанол добавляют при повышенной температуре ( 60 С). Продукт А кристаллизуют охлаждением или охлаждением с добавлением затравки (5 вес.%, в расчете на соединение If влажного измельчения, измельченного в азотной струе, или предыдущую загрузку). Продукт перекристаллизовывают или в виде влажного, или сухого осадка из этанола SDA3A. Кристаллический сольват диметанола Ig по изобретению получают согласно следующей схеме реакции V. Схема V где некристаллическое соединение В (которое может быть получено, как описано в US 10/745,075, поданной 23 декабря 2003, или в US 6515117), предпочтительно в, по существу, чистой форме (чистота от 50 до 100%), растворяют в метаноле, смеси метанол/толуол или смеси метанол/толуол/гептан, смеси метанол/метилтретбутиловый эфир (МТВЕ)/гептан, или смеси метанол/толуол/этилацетат или другой алкилацетат, при перемешивании, с образованием белой суспензии,содержащей кристаллический сольват диметанола Ig. Кристаллический сольват диметанола Ig можно выделить из суспензии с использованием обычных методик, таких как фильтрация. Вышеупомянутый процесс может быть выполнен при комнатной температуре, хотя могут использоваться повышенные температуры до приблизительно 20-25 С, чтобы усилить кристаллизацию. В предпочтительном воплощении соединение Ig кристаллизуют из смеси метанол/толуол, содержащей объемное соотношение метанола к толуолу в пределах диапазона от приблизительно 6:1 до приблизительно 1:1, предпочтительно от приблизительно 3:1 до приблизительно 5:1. Смесь метанол/толуол будет включать достаточное количество метанола, чтобы получить мольное соотношению к соединению В в пределах диапазона от приблизительно 80:1 до приблизительно 10:1, предпочтительно от приблизительно 40:1 до приблизительно 20:1, чтобы способствовать образованию сольвата диметанола Ig. Кристаллизация для образования сольвата диметанола Ig может быть более легко выполнена с использованием затравочных кристаллов соединения Ig в количестве от приблизительно 0.1 до приблизи- 11020428 тельно 10%, предпочтительно от приблизительно 0.5 до приблизительно 3%, в расчете на вес исходного соединения В. В другом предпочтительном воплощении соединение Ig (которое может быть или, возможно, может не быть очищено) кристаллизуют из смеси метанол/толуол/гептан с добавлением затравки кристаллического соединения Ig, использующего в диапазоне от приблизительно 0.1 до приблизительно 10%, предпочтительно от приблизительно 0.5 до приблизительно 3%, в расчете на вес исходного соединения В. Метанол будет использоваться при объемном соотношении к толуолу в пределах диапазона от приблизительно 1:0.5 до приблизительно 1:6, предпочтительно от приблизительно 1:1.5 до приблизительно 1:2.5 и при объемном соотношении гептан:толуол в пределах диапазона от приблизительно 2:1 до приблизительно 0.5:1, предпочтительно от приблизительно 1.3:1 до приблизительно 0.5:1. Получение соединений формулы I, главным образом, описано в US 6414126 и особенно раскрыто на схеме 1 и в примере 1. US 5515117, 6414126 и 5515117 полностью включены в настоящую заявку в качестве ссылки. Стабильные формы соединений формулы (I) могут быть кристаллизованы в виде сольватов (например, гидратов). Примеры Получение кристаллических структур Пример 1. Получение (S)-пропиленгликоля S)-PG) структуры - формы SC-3 - формулы Ia Соединение А может быть получено, как описано в примере 1, части Е US 6515117. Стеклянный реактор на 10 л, оборудованный термопарой и вводом для азота, промывают МеОН(1.25 л), деминерализованной водой (3.6 л) и затем 50%-ым водным раствором NaOH (205.9 мл, 3.899 моля). Остаточный раствор NaOH в градуированном цилиндре помещают с водой (94 мл) в реактор. Добавляют соединение А (503.11 г, 0.872 моль) и смесь размешивают и нагревают до температуры -68 С в течение более чем 1.5 ч. Через 1 ч температуру бани с циркуляцией понижают от 80 до 70 С; внутренняя температура стала 65 С. После в общей сложности 3 ч ВЭЖХ 1 обозначает завершение реакции, соединение I АР 99.5. После того как смесь была охлаждена до температуры 25 С, добавляют изопропилацетат(2.5 л). Смесь размешивают в течение 10 мин и затем водный слой отделяют (рН 12.5) и органический слой промывают водой (1 л). В течение этой промывки рН двухфазной системы регулируют до 6.0 конц.HCl (5.0 мл) и затем водный слой отделяют 2. Органический слой собирают в отдельном сосуде. Реактор промывают водой (2 л), МеОН (2 л) и продувают азотом. Влажный раствор соединения В добавляют в реактор и вводят (S)-пропиленгликоль S)-PG) (67.03 г, 0.872 моль). Необязательно, на данной стадии могут быть добавлены затравочные кристаллы (S)-PG Ia. Мгновенная кристаллизация позволяет получить сгущенную суспензию. После перемешивания в течение 1 ч быстро добавляют циклогексан (2.5 л) в течение 10 мин и перемешивание продолжают в течение 21 ч. Продукт фильтруют через фильтровальную бумагу (Whatman 5, воронка Бюхнера с диаметром 24"). Фильтрация осуществляется быстро и занимает приблизительно 15 мин. Отфильтрованный осадок промывают смесью (1:1) МТВЕ/циклогексан(21 л) и сушат под вакуумом в течение 0.5 ч. Твердое вещество помещают на тарелку из пирекса и сушат под вакуумом (25 мм рт.ст.) в сушильном шкафу при температуре 25-30 С в течение двух дней, пока водный анализ KF не будет соответствовать моногидрату (3.6 вес.%). Получают продукт (S)-PG Ia (0.425 кг, выход 97%) в виде белоснежного твердого вещества, ВЭЖХ 3 АР 99.7. Затравочные кристаллы могут быть получены растворением соединения I в растворителе, таком как МТВЕ, и обработкой получающегося раствора с (S)-пропиленгликолем и осуществлением процесса, как описано выше, без использования затравки. 1 ВЭЖХ: Колонка: YMC ODS-A (С-18) S3, 4.650 мм. Растворитель А: 0.2% вод. раствор Н 3 РО 4. Растворитель В: 90% CH3CN/10% Н 2 О. Исходное %В = 0, конечное %В = 100. Градиент времени 8 мин; время удерживания 3 мин. Интегрированное время остановки 11.0 мин. Расход 2.5 мл/мин. Длина волны УФ 220 нм. 2 была осуществлена нейтрализация перед фазовым разделением, чтобы предотвратить загрязнение продукта с NaOH. (S)-PG структура, полученная без нейтрализации, была немного основной [рН 8.3 суспензии, диспергированной с помощью ультразвука в воде (20 мг/мл)]. ВЭЖХ метод: Мобильная Фаза А: 0.05% TFA в Н 2 О. Мобильная Фаза В: TFA: 0.05% в CAN. Колонка: YMC Hydrosphere 4.6150 (3 мк). Градиент: 30-90%В в течение 45 мин, удерживание 5 мин; обратно к 30%В и повторное уравновешивание в течение 10 мин. Длина волны: 220 нм. Объем Инъекции: 10 мкл. Температура: Окружающей среды Пример IA. Процедура 20 г Соединения А загружают в реактор при температуре и давлении окружающей среды. В реактор добавляют 30 мл метанола и 49.75 мл 3 Н NaOH и реакционную смесь нагревают до температуры 80 С или кипятят с обратным холодильником и выдерживают приблизительно 2-3 ч для завершения реакции 0.5 АР. Загрузку охлаждают до температуры 20 С и нейтрализуют до рН 6.0-7.5 с использованием конц.HCl или 1 Н уксусной кислоты (требуется 1 мл/гм загрузки). Экстракция Продукт экстрагируют из реакционной смеси 100 мл изопропилацетата, водную фазу отделяют и органическую фазу промывают водой до удельной электропроводности 10 мС ( 4 мл/гм загрузки). Водную фазу отделяют. Кристаллизация 2.8 г (1.05 экв) (S)-(+)-1,2-Пропандиола добавляют к реакционной смеси. В загрузку добавляют затравочные кристаллы 0.1 г соединения I. Добавляют 160 мл циклогексана и загрузку охлаждают от комнатной температуры до температуры 5 С. Загрузку перемешивают от комнатной температуры до температуры 5 С по крайней мере в течение 1 ч до выделения. Выделение и сушка Каждый образец выделенного осадка промывают смесью 50/50 по объему изопропилацетат/циклогексан. Осадок сушат при температуре 30 С в вакуумном сушильном шкафу под глубоким вакуумом. (Осадок высушен, когда KF = 3.6-4.1%). Выход = 84% (без учета погрешностей) Характерная чистота = 99.81 АР Характерное содержание PG = 15.1-15.8% посредством ГХ Пример 2. Получение структуры (R)-пропиленгликоля - Ib Сруктура (R)-пропиленгликоля была получена с использованием аналогичного способа, описанного выше для структуры (S)-пропиленгликоля Ia (пример 1), за исключением того, что (R)-пропиленгликоль используют вместо (S)-пропиленгликоля. Соединение А (1.0 г) растворяют в EtOH (3.0 мл) нагреванием до кипения и раствор разбавляют водой (7 мл). Добавляют EtOH на 1 мл и смесь разделяют на три части для кристаллизации при температурах 20, 5 и -20 С. После охлаждения в интервале температур от -10 до -20 С образуются кристаллы, которые имеют т.пл. 40-41 С. Примеры 4 и 5. Получение структуры этиленгликоля - формы SB-1 и SB-2 - формул Id и Ie, соответственно Чтобы получить полиморфную форму кристалла дигидрата этиленгликоля формы SB-1 Id, соединение А (0.5 грамм) растворяют в водном растворе этиленгликоля (0.3 мл воды: 0.5 мл этиленгликоля) нагреванием при температуре 45 С в течение 30 мин. После охлаждения до комнатной температуры добавляют затравку SB-1 (10 мг). Реакционную смесь размешивают в течение 16 ч, обеспечивая белое кристаллическое твердое вещество. Кристаллы отфильтровывают, промывают водой и сушат. Для получения полиморфной формы затравочных кристаллов дигидрата этиленгликоля SB-1 Id соединение А растворяют в водном растворе этиленгликоля, кристаллическую форму (S)-пропиленгликоля SC-3 Ia добавляют, чтобы получить кристаллическую форму дигидрата этиленгликоля SB-1 Id (пример 4). Указанные кристаллы отфильтровывают и промывают избытком воды. Чтобы получить полиморфную форму дигидрата этиленгликоля кристаллической формы SB-2 Ie(пример 5), соединение А растворяют в водном растворе этиленгликоля нагреванием. После охлаждения добавляют затравку кристаллической формы моно-EtOH-дигидрата SA-1, Ic, чтобы получить кристаллическую форму дигидрата этиленгликоля SB-2 Ie (пример 5). Указанные кристаллы отфильтровывают и промывают избытком воды. 1H ЯМР для форм SB-1 и SB-2: 1H ЯМР (400 МГц, DMSO)1.29 (т, 3 Н, J = 6.98 Гц, -СН 3), 3.15 (м,4 Н,), 3.33 (уш.с, 6 Н, -СН 2), 3.42 (м, 3 Н), 3.6 (уш.дд, J = 11.4 Гц, 1 Н), 3.9 (уш.м, 5 Н, Н-I, -2 СН 2), 4.43 (т,1 Н, J = 7.4 Гц, ОН), 4.86 (д, 1H, J = 2.4, ОН), 4.95 (к, 1 Н, -ОН), 6.82 (д, 2 Н, J = 11.47 Гц, Ar-H), 7.8 (д, 2 Н, J К ацетонитрилу (12 мл) при температуре бани 8-10 С в атмосфере азота загружают диэтилэтерат трифторида бора (2.3 мл, 18.4 ммоль) и воду (0.82 мл, 4.6 ммоль). После выдерживания вышеупомянутой смеси в течение приблизительно 1 ч добавляют триэтилсилан (3 мл, 18.4 ммоль). Полученную смесь выдерживают в течение приблизительно 1 ч и затем добавляют соединение В (полученное, как описано в примере 17) в 10 мл ацетонитрила. Загрузку выдерживают при температуре 5-10 С. При завершении реакции, как определяется ВЭЖХ, реакционную смесь гасят водным ацетатом аммония (24 мл; 85 г) в 200 мл воды. Фазы разделяют и органическую фазу, обогащенную продуктом, сушат над сульфатом натрия. Органическую фазу, обогащенную продуктом, концентрируют при пониженном давлении. Воду (13 мг, 0.7 ммоль, в расчете на 0.3 г сырого соединения В в загрузке), (S)-пропиленгликоль (56 мг, 0.7 ммоль), метил-трет-бутиловый эфир (5 мл, 17 мл/г соединения В в загрузке), затравку соединения Ia (20 мг) смешивают и выдерживают в течение 1 ч до образования суспензии кристаллов. Добавляют циклогексан (10 мл, 33 мл/г соединения В (загрузка. Кристаллический продукт (Ia) выделяют фильтрацией (4-5%) и сушат в вакууме при температуре 20-25 С. Пример 7. Получение кристаллического сольвата МеОН Ig Кристаллы метанольного сольвата Ig получают растворением чистого соединения В в метаноле и перемешиванием при комнатной температуре. Белая суспензия образуется после нескольких дней и, как обнаруживают, является кристаллическим метанольным сольватом Ig. Таким образом полученный кристаллический di-MeOH сольват Ig может использоваться вместо соединения В в получении кристаллического соединения Ia, как описано в примере 6. Пример 8. Получение кристаллического Di-MeOH сольвата Ig из неочищенного соединения В в смеси 80/20 метанол/толуол, используя затравку. 6 г Соединения В (ВЭЖХ АР приблизительно 80%), растворяют в 15 мл смеси 80/20 метанол/толуол. Добавляют затравочные кристаллы (приблизительно 1% от исходного соединения В) соединения Ig и смесь охлаждают, чтобы получить суспензию, содержащую кристаллы. Суспензию размешивают в течение 6 ч перед выделением. Влажный осадок, как было обнаружено, является кристаллическим метанольным сольватом, If, но теряет кристалличность, если оставлен открытым в течение нескольких ч. Пример 9. Получение кристаллического сольвата Di-MeOH Ig из неочищенного соединения В в смеси метанол/толуол/гептан, используя затравку. 2.5 г Соединения В (91.5%), добавляют в сцинтилляционный флакон с магнитной мешалкой. Добавляют 4 мл толуола, чтобы растворить соединение Ia. Добавляют 2 мл метанола. Затем добавляют затравочные кристаллы соединения Ig (приблизительно 1%). Добавляют 4 мл гетана в течение 30 мин и смесь размешивают в течение 12 ч. Влажный осадок выделяют на воронке Бюхнера. Влажный осадок, как обнаруживают, является кристаллическим метанольным сольватом Ig. Его сушат под вакуумом при температуре 30 С. Полученный порошок теряет кристалличность. Выход = 1.7 г = 74.5% (без учета погрешности). Исследование рентгенограммы XRD кристаллов: фиг. 10. Таким образом образованный кристаллический сольват МеОН Ig может использоваться вместо соединения В в получении кристаллического соединения Ia, как описано в примере 6. Пример 10. Получение кристаллического сольвата 1,4-бутиндиола If из соединения В в смеси толуол/этилацетат, используя затравку сольват 1,4-бутиндиола, может быть кристаллизован в алкилацетате(например, этил-, пропил- или бутилацетате), спирте (например, изопропаноле, бутаноле) или даже в воде. Толуол и гептан действуют как антирастворители, когда кристаллизуются в алкилацетате. 50 г (90.3 вес.%) Соединения В растворяют в 675 мл толуола. Раствор нагревают до температуры 60 С и добавляют 75 мл этилацетата. Добавляют 1.5 экв. 2-бутин-1,4-диола (=13.3 г) и смесь выдерживают при температуре 60 С до тех пор, пока бутиндиол не растворится. Раствор охлаждают до температуры 55 С и добавляют 0.1% затравки (50 мг) соединения 1,4-бутиндиола If. Смесь выдерживают в течение 1 ч при температуре 55 С. Соединение If начинает кристаллизоваться. Смесь охлаждают до температуры 25 С в течение 6 ч. Полученную суспензию размешивают в течение 3 ч перед выделением (концентрация маточного раствора составляет 3 мг/мл), отфильтровывают и промывают 180 мл толуола + 20 мл этилацетата и сушат под вакуумом при температуре 45 С, чтобы получить кристаллы сольвата 1,4 бутиндиола If. ВЭЖХ АР = 99.5%. Активность = 80.7 вес.% (рассчитанная активность = 83.6% для сольвата 1:1). Выход = 95%. Пример 11. Получение кристаллического сольвата 1,4-бутиндиола If из соединения В в смеси бутилацетат/гептан 0.5 г Соединения В (91 вес.%) растворяют в 3.5 мл бутилацетата + 3.5 мл гептана при температуре 60 С. Добавляют 1.5 экв. 2-бутин-1,4-диола и смесь охлаждают до комнатной температуры. Полученную суспензию размешивают в течение 12 ч, фильтруют и промывают 1 мл смеси 1:1 бутилацетат:гептан и сушат под вакуумом при температуре 50 С, чтобы получить кристаллы сольвата 1,4-бутиндиола If. Активность = 85.1%. Выход = 90%. Сольват 1,4-бутиндиола If может использоваться вместо соединения В при использовании кислоты Льюиса BF32 СН 3 СООН вместо BF3OEt2, чтобы получить кристаллическое соединение Ia. Пример 16. Получение соединения If с помощью непрерывных реакций литирования и связывания Используется схема реакции, подобная той, которая показана на схеме IVA и фиг. 13. Устанавливают холодильник (-30 С) для реактора литирования 5 (снабженный рубашкой статический смеситель 5). Устанавливают холодильник (-30 С) для реактора связывания 22 (снабженный рубашкой статический смеситель 22) и теплообменник предварительного охлаждения (не показан на фиг. 22) для смеси подачи соединение D/толуол. Непрерывное литирование Два потока подачи смесь E/THF/толуол (2.74 мл/мин) и Q, а именно n-BuLi в гексане (0.41 мл/мин),смешивают и объединяют с помощью снабженного рубашкой статического смесителя 5 (-30 С). Перед перекачкой потока подачи D/толуол, толуол (2.96 мл/мин) направляют в систему в виде свежего потока для поддержания расхода общего потока постоянным при 6.1 мл/мин. Образцы на выходе статического смесителя литирования 5 собирают для анализа ВЭЖХ. Образцы берут прежде (а) начала реакции связывания и (b) после сбора реакционной смеси в реакторе MSAMeOH. Непрерывная реакция связывания Поток подачи D/толуол (2.96 мл/мин) предварительно охлаждают с помощью теплообменника перед смешиванием с потоком литирования. Эти два потока, а именно G и D, смешивают и объединяют в снабженном рубашкой статическом смесителе 22 (между -24 и -30 С). Поток реакции является желоватым по цвету. Образцы собирают на выходе смесителя 22 для анализа ВЭЖХ. Образцы берут до и после сбора в реакторе MSA-MeOH 25. Метилгликозидирование Поток реакции связывания 24 направляют в реактор 25 на 500 мл, содержащий MSA и метанол или смесь HCl/МеОН при температуре -10 С при перемешивании. После окончания сбора реакционную смесь хранят при температуре -10 С при перемешивании в течение следующего ч. Реакционную смесь нагревают до температуры 35 С. Реакцию считают завершенной (приблизительно 6 ч) тогда, когда анализ ВЭЖХ покажет, что RAP десилилированного гемикеталя Н' 0.3%. Реакционную смесь охлаждают до комнатной температуры (20 С) и реакционную смесь выдерживают в течение 16 ч, чтобы получить соединение В. Образование кристаллов If Соединение В кристаллизуют с 2-бутин-1,4-диолом (J) в смеси толуол/EtOAc, чтобы получить кристаллы If. Пример 17. Получение промежуточного продукта А Твердое соединение If (50.0 г), твердый DMAP (1.2 г), жидкий ацетонитрил (450 мл) и жидкий уксусный ангидрид (63 мл) загружают в реактор в виде сосуда на 250 мл. Загрузку (77 С) нагревают и выдерживают до завершения реакции. Загрузку охлаждают (5 С). Триэтилсилан (72 мл) и комплекс уксусной кислоты и трифторида бора (63 мл) загружают в реактор. После завершения реакции добавляют ацетон (36 мл). Загрузку (21 С) нагревают и выдерживают, пока не израсходуется триэтилсилан. Водный раствор NH4OAc (33 вес.%, 450 мл) добавляют и загрузку перемешивают, позволяя расслоиться на верхнюю и нижнюю фазы, которые образуются. Загрузочный объем продукта в богатой верхней фазе восстанавливают отгонкой ацетонитрила при минимальном перемешивании. Этанол SDA3A (1 л) загружают при повышенной температуре ( 60 С). Продукт кристаллизуют охлаждением или охлаждением с добавлением затравки (5 вес.%, в расчете на соединение If влажного измельчения, измельчения в струе азота или предыдущей загрузки). Продукт обычно выделяют с выходом 75% . Продукт перекристаллизовывают или в виде влажного, или сухого осадка из этанола SDA3A. Исследование кристаллических структур Кристаллические структуры, эквивалентные кристаллическим структурам, описанным ниже и заявленным в настоящей заявке, могут демонстрировать подобные, но все же неидентичные, аналитические характеристики в пределах приемлемого диапазона погрешности, в зависимости от испытательных условий, чистоты, оборудования и других обычных переменных, известных квалифицированным в данной области специалистам. Соответственно, будет очевидно квалифицированным в данной технологии специалистам, что могут быть сделаны различные модификации и изменения в настоящем изобретении, не отступая от объема и сущности изобретения. Другие воплощения изобретения будут очевидны квалифицированным в тех- 17020428 нологии специалистам из соображения описания и практики изобретения, раскрытого в настоящей заявке. Указано, что описание и примеры необходимо рассматривать в качестве образца, но не ограничивая объем изобретения. Дифракция рентгеновских лучей на порошке Квалифицированный в данной области специалист оценит, что дифракционная рентгенограмма может быть получена с погрешностью измерения, которая зависит от используемых условий измерения. В частности, общеизвестно, что интенсивность на дифракционной рентгенограмме на порошке может колебаться в зависимости от используемых условий измерения. Должно далее быть понятно, что относительные интенсивности могут также изменяться в зависимости от экспериментальных условий и, соответственно, точный порядок интенсивности не должен быть принят во внимание. Дополнительно, погрешность измерения угла дифракции для обычной дифракционной рентгенограммы на порошке составляет обычно приблизительно 5% или меньше и такая степень погрешности измерения должна быть принята во внимание как имеющая отношение к вышеупомянутым углам дифракции. Следовательно, должно быть понятно, что кристаллические структуры настоящего изобретения не ограничиваются кристаллическими структурами, которые обеспечиваются дифракционными рентгенограммами на порошке,полностью идентичными дифракционным рентгенограммам на порошке, изображенным в приложенных фигурах, раскрытых в настоящей заявке. Любые кристаллические структуры, которые обеспечиваются дифракционными рентгенограммами на порошке, в основном идентичными раскрытым в приложенных фигурах, находятся в объеме настоящего изобретения. Способность устанавливать реальные тождества дифракционных рентгенограмм на порошке находится в пределах навыков квалифицированных в данной области специалистов.(PXRD). Образец был перемещен в ячейку Philips MPD (45 KB, 40 мА, Cu K1). Данные собирают при комнатной температуре при 2-32 2-тета (способ непрерывного сканирования, частота сканирования 0.03 градус/с, щели автодивергенции и антирассеяния, приемная щель: 0.2 мм, вращатель образца: ON). Рентгенограммы на порошке для структур (S)-PG (Ia), (R)-PG (Ib) проиллюстрированы на фиг. 1 и 2,соответственно. Рентгенограммы на порошке для сольвата 1,4-бутиндиола If и сольвата диметанола Ig проиллюстрированы на фиг. 9 и 10, соответственно. Выбранные положения дифракционных пиков (градусы 20.2) для (S)-PG (Ia), (R)-PG (Ib) показаны в табл. 1, приведенной ниже. Характерные положения дифракционного пика (градусы 20.1) при КТ, являются основанными на рентгенограмме высокого качества, снятой с помощью дифрактометра (CuK) с вращающимся капилляром с 2, калиброванным Национальным Институтом Стандартов и методологии Технологии и других пригодных стандартов, известных квалифицированным в данной области специалистам. Относительные интенсивности, однако,могут изменяться в зависимости от размера и морфологии кристалла. Таблица 1. Выбранные пики PXRD (20.2) Ядерный магнитный резонанс в твердой фазе Структуры (S)-PG (Ia), (R)-PG (Ib), сольвата 1,4-бутиндиола If и сольвата диметанола Ig охарактеризованы методами ЯМР в твердой фазе. Все измерения ЯМР С-13 в твердой фазе были сделаны на приборе Bruker DSX-400, спектрометром ЯМР 400 МГц. Спектры с высоким разрешением получают, используя мощное протонное расщепление импульсного режима ТРРМ и поперечную поляризацию линейно нарастающей амплитуды (RAMP-CP) с магическим углом вращения (MAS) при приблизительно 12 кГц (А.Е. Bennett et al., J. Chem. Phys., 1995,103, 6951; G. Metz, X. Wu и S.O. Smith, J. Magn. Reson. A., 1994, 110, 219-227). Приблизительно 70 мг образца, помещенного в контейнер с ротором из двуокиси циркония, использовалось для каждого эксперимента. Химические сдвигибыли отнесены к внешнему адамантану с высокочастотным резонансом,установленным до 38.56 част. на млн (W.L. Earl и D.L. VanderHart, J. Magn. Reson., 1982, 48, 35-54). Полученный I3C ЯМР CPMAS спектр для структуры (S)-PG и (R)-PG показан на фиг. 3 и 4, соответственно. Основные резонансные пики для спектра углерода в твердой фазе (S)-PG и (R)-PG упомянуты ниже в табл. IA и табл. 2 и для сольвата 1,4-бутиндиола If и сольвата диметанола Ig упомянуты ниже в табл. 2 А и 2 В, соответственно. Кристаллические структуры, демонстрирующие в основном аналогичные положения пиков 13 С ЯМР, где "в основном аналогичный" означает от 10 до 15% безразмерного значения,как считается, находятся в рамках настоящего изобретения (то есть эквиваленты к структурам, проиллюстрированным ниже). Таблица 1 А. Положения пика протонного ЯМР для сольвата (S)-пропиленгликоля Ia 1 Указанные данные точно действительны для спектрофотометра на 400 МГц. Таблица 2 А. Положения пика протонного ЯМР для сольвата 1,4-бутиндиола If 1 Н ЯМР (400 М Гц, CDCl3)1.33 (т, 3 Н, J = 7.1 Гц, -СН 3), 2.90 (с, 2 Н, -СН 2), 3.39 (с, 9 Н, -ОСН 3), 3.43.65 (м, 3 Н), 3.81 (уш.м, 2 Н), 3.91 (q, 2 Н, J =7.1 Гц, -СН 2), 3.97 (м, 1H), 6.73 (д, 1H, J=8.6 Гц, Ar-Н), 7.02(д, 2 Н, J = 8.4 Гц, Ar-Н), 7.25 (с, 2 Н, Ar-Н), 7.34 (с, 1 Н, Ar-Н); 13 С (CDCl3)14,78, 38.43, 49.14, 50.57,61.84, 63.34, 69.98, 72.53, 74.63, 100.95, 114.36, (2), 126.64, 129.19, 129.59, 129.71, 131.38, 134.30, 136.61,138.50, 157.27. Т.пл. 103.08 С. Таблица 2 В. Положения пика протонного ЯМР для сольвата диметанола Ig 1= 8.6 Гц, Ar-Н), 7.4 (с, 2 Н, Ar-Н), 7.50 (с, 1 Н, Ar-Н); 13 С (CDCl3)14.69, 48.28, 49.02, 60.81, 62.84, 70.05,74.02, 76.81, 83.97, 100.64, 114.23, 127.40, 128.2, 129.44, 131.2, 131.4, 132.45, 137.38, 138.57, 156.84. Элементный анализ расчетный для C26H33ClO9: Расч. С 59.48, Н 6.34, Cl 6.75; Найдено С 59.35, Н 5.97, Cl 6.19. Термогравиметрический анализ Эксперименты термогравиметрического анализа (TGA) были выполнены на приборе ТА Instruments модели Q500. Образец (приблизительно 10-30 мг) помещают в платиновый резервуар, предварительно измеренный. Вес образца измеряют точно и регистрируют до тысячной миллиграмма с помощью прибора. Печь продувают азотом при 100 мл/мин. Данные собирают между комнатной температурой и 300 С при скорости нагревания 10 С/мин. Диаграммы TGA для структур (S)-PG Ia и (R)-PG Ib показаны на фиг. 5 и 6, соответственно. Потери веса соответствуют одному молю воды и одному молю пропиленгликоля на моль проанализированной структуры. Дифференциальная сканирующая калориметрия Температурный режим твердого состояния структур (S)-PG Ia, (R)-PG Ib, сольвата 1,4-бутиндиолаIf, сольвата диметанола Ig был исследован с помощью дифференциальной сканирующей калориметрии(DSC). Диаграммы DSC для структур (S)-PG Ia и (R)-PG Ib показаны на фиг. 7 и 8, соответственно. Диаграммы DSC для структур сольвата 1,4-бутиндиола If и сольвата диметанола Ig показаны на фиг. 11 и 12,соответственно. Эксперименты дифференциальной сканирующей калориметрии (DSC) были выполнены на приборе ТА Instruments модели Q1000. Образец (приблизительно 2-6 мг) взвешивают в алюминиевом резервуаре и взвешивают точно до одной сотой миллиграмма и направляют на DSC. Прибор продувают азотом при 50 мл/мин. Данные собирают между комнатной температурой и 300 С при скорости нагревания 10 С/мин. График сделан с эндотермическими пиками, направленными вниз. Специалисту, квалифицированному в данной области, будет понятно, что при измерении DSC есть определенная термодинамическая степень изменчивости в фактическом действительном масштабе измерений и пиковых температурах, в зависимости от скорости нагревания, кристаллической формы и чистоты и других параметров измерения. Рентгенографический анализ монокристалла Монокристалл для структуры (S)-PG Ia и для структур сольвата 1,4-бутиндиола If, сольвата диметанола Ig получают и исследуют посредством дифракции рентгеновских лучей. Данные собирают на дифрактометре серии Bruker-Nonius CAD4 (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, WI 53711 USA). Параметры элементарной ячейки получают посредством анализа методом наименьших квадратов экспериментальных значений параметров дифрактометра при 25 отражениях под большими углами. Интенсивности были измерены, используя CuK излучение ( = 1.5418 ) при постоянной температуре с -2 различными методиками сканирования, и были скорректированы только для факторов поляризации Лоренца. Фоновые индексы собирают при крайних наблюдаемых значениях в течение половины времени сканирования. Поочередно, данные монокристалла собирают на приборе Bruker-NoniusKappa CCD 2000 system, используя Cu K излучение ( = 1.5418 ). Индексирование и проведение измерений данных интенсивности были выполнены с программным обеспечением HKL2000 (Otwinowski, Z.Minor, W. (1997) в Macromolecular Crystallography, eds. Carter, W.C. JrSweet, R.M. (Academic, NY),vol. 276, pp. 307-326) с пакетом программ (Collect Data collection and processing user interface: Collect: Datacollection software, R. Hooft, Nonius B.V., 1998). Если указано, то кристаллы охлаждают в холодном потоке криосистемы Oxford cryo system во время сбора данных (Oxford Cryosystems Cryostream cooler: J. Cosier и A.M. Glazer, J. Appl. Cryst, 1986, 19,105). Структуры решены прямыми методами и уточнялись на основе наблюдаемых отражений, используя или пакет программ SDP (SDP, Structure Determination Package, Enraf-Nonius, Bohemia NY 11716 Scattering factors, including/' and/", in the SDP software were taken from the" International Tables for Crystallography", Kynoch Press, Birmingham, England, 1974; vol. IV, Tables 2.2A and 2.3.1) с незначительными изменениями, или пакет программ по кристаллографии MAXUS (maXus solution and refinement software suite: S.the solution and refinement of crystal structures from diffraction data). Полученные атомные параметры (координаты и температурные коэффициенты) уточнены с помощью метода наименьших квадратов с полной матрицей. Функция, минимизированная при уточнении,была w(F0 - FC)2, R определен как F0 - FC/F0, где Rw = [w(F0 - FC)2/wF02]1/2, где w - соответствующая весовая функция, основанная на погрешностях в наблюдаемых интенсивностях. Различные диаграммы исследованы на всех стадиях уточнения. Водороды введены в теоретических положениях с изотропными температурными коэффициентами, но водородные параметры не были изменены. Параметры элементарной ячейки для структуры (S)-PG формы Ia, SC-3 упомянуты ниже в табл. 3. Как используется в настоящей заявке, параметр элементарной ячейки "молекулы/на ячейку" относится к количеству молекул соединения в элементарной ячейке. Таблица 3. Данные элементарной ячейки для (S)-PG (Ia)Z' = количество молекул препарата на асимметричную ячейкуVm = V (элементарная ячейка) / (Z молекул препарата на ячейку)SG = пространственная группа Табл. 4, приведенная ниже, формулирует позиционные параметры для структуры (S)-PG Ia при температуре 25 С. Таблица 4. Позиционные параметры для (S)-PG при Т = 25 С Параметры элементарной ячейки для дигидрата моноэтанола (структура этанола или EtOH) формыSA-1, формулы Ic, упомянуты ниже в табл. 5. Таблица 5. Данные элементарной ячейки для этанола SA-1 (Ic)Z' = количество молекул препарата на асимметричную ячейкуVm = V (элементарная ячейка)/(Z молекулы препарата на ячейку)SG = пространственная группа В табл. 6, приведенной ниже, представлены позиционные параметры для формы SA-1 (дигидрат моноэтанола) Ic при температуре -50 С. Таблица 6. Фракционные атомные координаты для формы SA-1 при Т =-50 С Параметры элементарной ячейки для этиленгликоля формы SB-1 формулы Id упомянуты ниже в табл. 7. Таблица 7. Данные элементарной ячейки для EG-SB-1 (Id)Z' = количество молекул препарата на асимметричную ячейкуVm = V (элементарная ячейка)/(Z молекул препарата на ячейку)SG = пространственная группа В табл. 8, приведенной ниже, указаны позиционные параметры для формы SB-1 (этиленгликоля) Id при температуре -50 С. Таблица 8. Фракционные атомные координаты для формы SB-1 при Т = -50 С Параметры элементарной ячейки для этиленгликоля формы SB-2, формулы Ie, упомянуты ниже в табл. 9. Таблица 9. Данные элементарной ячейки для EG-SB-2 (Ie)Z' = количество молекул препарата на асимметричную ячейкуVm = V (элементарная ячейка)/(Z молекул препарата на ячейку)SG = пространственная группа В табл. 10, представленной ниже, приведены позиционные параметры для формы SB-2 (этиленгликоля) Id при температуре -50 С. Таблица 10. Фракционные атомные координаты для формы SB-2 при Т = -50 С Параметры элементарной ячейки для сольвата 1,4-бутиндиола If упомянуты ниже в табл. 11. Таблица 11. Данные элементарной ячейки для сольвата 1,4-бутиндиола IfZ' = количество молекул препарата на асимметричную ячейкуVm = V (элементарная ячейка)/(Z молекул препарата на ячейку)SG = пространственная группа В табл. 12, приведенной ниже, представлены позиционные параметры для сольвата 1,4-бутиндиола Таблица 12. Таблица фракционных атомных координат для сольвата 1,4-бутиндиола If при Т = 25 С
МПК / Метки
МПК: A61P 3/10, A61K 31/70, C07H 7/04
Метки: сольваты, производных, sglt2, кристаллические, 1s)-1,5-ангидро-1-с-(3-((фенил)метил)фенил)-d-глюцитола, ингибиторов, диабета, лечения, качестве
Код ссылки
<a href="https://eas.patents.su/30-20428-kristallicheskie-solvaty-proizvodnyh-1s-15-angidro-1-s-3-fenilmetilfenil-d-glyucitola-v-kachestve-ingibitorov-sglt2-dlya-lecheniya-diabeta.html" rel="bookmark" title="База патентов Евразийского Союза">Кристаллические сольваты производных (1s)-1,5-ангидро-1-с-(3-((фенил)метил)фенил)-d-глюцитола в качестве ингибиторов sglt2 для лечения диабета</a>
Кристаллические сольваты и комплексы производных (1s)-1,5-ангидро-1-c-(3-((фенил)метил)фенил)-d-глюцитола с аминокислотами в качестве ингибиторов sglt2 для лечения диабета

Номер патента: 18229
Опубликовано: 28.06.2013
Авторы: Димарко Джон Д., Лобингер Хильдегард, Сингх Джанак, Рибель Петер, Нёршль Александра А., Гугутас Джек З., Дешпанде Прашант П., Рамакришнан Шривидья, Бйн Джеффри Т., Гроссо Джон Энтони, Лай Чиаджень, Ванг Ченьчи
МПК: C07H 7/04, A61P 3/10, A61K 31/70...
Метки: 1s)-1,5-ангидро-1-c-(3-((фенил)метил)фенил)-d-глюцитола, сольваты, лечения, качестве, ингибиторов, кристаллические, комплексы, аминокислотами, производных, диабета, sglt2
Формула / Реферат:
1. Кристаллическая форма SC-3 соединения (S)-PG формулы (Ia)охарактеризованная одним или большим количеством следующих характеристик:a) параметры элементарной ячейки:а=11.2688(8) Å,b=4.8093(3) Å,с=46.723(3) Å,пространственная группа = P212121,молекулы/асимметричная ячейка = 1,где измерение указанной кристаллической структуры осуществляют при комнатной температуре и которая охарактеризована фракционными атомными координатами, по...
Аминотетрагидропираны в качестве ингибиторов дипептидилпептидазы-iv для лечения или предупреждения диабета

Номер патента: 18613
Опубликовано: 30.09.2013
Авторы: Кокс Джейсон М., Вебер Энн Э., Чэнь Пин, Бифту Тесфайе
МПК: C07D 487/04, A61K 31/4162, A61P 3/10...
Метки: дипептидилпептидазы-iv, диабета, предупреждения, качестве, аминотетрагидропираны, лечения, ингибиторов
Формула / Реферат:
1. Соединение структурной формулы (I)или его фармацевтически приемлемая соль,где V означает группуAr является фенилом, необязательно замещенным одним-пятью заместителями R1;каждый R1 означает галоген;каждый R2 означает водород;R3a и R3b, каждый независимо, являются водородом или C1-4алкилом, необязательно замещенным 1-5 атомами фтора;R8 выбирают из группы, включающей -SO2C1-6алкил, -SO2C3-6циклоалкил, -SO2-арил, -SO2-гетероарил, где алкил и...
Кристаллические безводные формы лактата n-гидрокси-3-[4-[[[2-(2-метил-1h-индол-3-ил)этил]амино]метил]фенил]-2e-2-акриламида

Номер патента: 17984
Опубликовано: 30.04.2013
Авторы: Штовассер Франк, Карпински Пьотр, Ацемоглы Мурат, Папоутсакис Димитрис, Байва Джоджиндер С., Слейд Джоел
МПК: C07D 209/00, A61P 35/00, A61K 31/404...
Метки: формы, безводные, лактата, кристаллические, n-гидрокси-3-[4-[[[2-(2-метил-1h-индол-3-ил)этил]амино]метил]фенил]-2e-2-акриламида
Формула / Реферат:
1. Кристаллическая безводная форма (форма А) лактата N-гидрокси-3-[4-[[[2-(2-метил-1H-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида, где кристаллическая безводная форма выбрана изDL-лактата, характеризующегося порошковой рентгенограммой, содержащей по меньшей мере два максимума, выбранных из 9,9, 11,4, 13,8, 15,7, 18,2, 19,7, 20,3, 21,5, 25,3, 27,4 и 30,0 (градусов 2θ);L-лактата, характеризующегося порошковой рентгенограммой,...
Соли и кристаллические формы 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)фенил]пропионитрила

Номер патента: 15677
Опубликовано: 31.10.2011
Авторы: Гарад Судхакар Девидасрао, Бэнцигер Маркус, Штовассер Франк
МПК: A61P 35/00, C07D 471/04, A61K 31/4745...
Метки: формы, соли, кристаллические, 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)фенил]пропионитрила
Формула / Реферат:
1. Кристаллическая форма соединения формулы Iили гидрата или сольвата соединения формулы I, или соли соединения формулы I, или гидрата или сольвата соли соединения формулы I.2. Соединение I по п.1 в кристаллической форме A.3. Соединение по п.2, на рентгенограмме которого содержится пик при угле дифракции 2-тэта, равном 8,4±0,3°.4. Соединение I по п.1 в кристаллической форме B.5. Соединение по п.3, на рентгенограмме которого содержится пик при...
Полиморфная форма 2-(r) – (1- (r) – (3,5-бис (трифторметил) фенил) этокси) -3- (s) – (4-фтор) фенил – 4 – (3 – (5-оксо-1h, 4h-1, 2, 4 – триазоло) метил)морфолина в качестве антагониста рецептора тахикинина

Номер патента: 2405
Опубликовано: 25.04.2002
Авторы: Крокер Луис, Макколи Джеймс
МПК: A61K 31/5375, A61P 25/28, C07D 265/32...
Метки: тахикинина, метил)морфолина, 4h-1, фенил, 2-(r, трифторметил, 3,5-бис, качестве, 4-фтор, форма, 5-оксо-1h, антагониста, полиморфная, триазоло, рецептора, этокси
Формула / Реферат:
1. Полиморфная форма соединения 2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фтор)фенил-4-(3-(5-оксо-1Н,4Н-1,2,4-триазоло)метил)морфолина, обозначенная как форма I, по существу, отличающаяся рентгеновской порошковой дифрактограммой с характерными отражениями приблизительно 12,0, 15,3, 16,6, 17,0, 17,6, 19,4, 20,0, 21,9, 23,6, 23,8 и 24,8ш (2 тэта). 2. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и...
Предыдущий патент: Способ предотвращения затопления калийных рудников при прорывах в рудник подземных вод из геологических нарушений
Следующий патент: Способ получения эзомепразола
Случайный патент: Эспандер с упорами