Ингибиторы с-fms киназы
Номер патента: 18936
Опубликовано: 29.11.2013
Авторы: Уолл Марк Дж., Мигалла Санатх К., Чэнь Цзиньшэн, Иллиг Карл Р.




























Формула / Реферат
1. Соединение формулы I

или его сольват, гидрат, таутомер или фармацевтически приемлемая соль, где
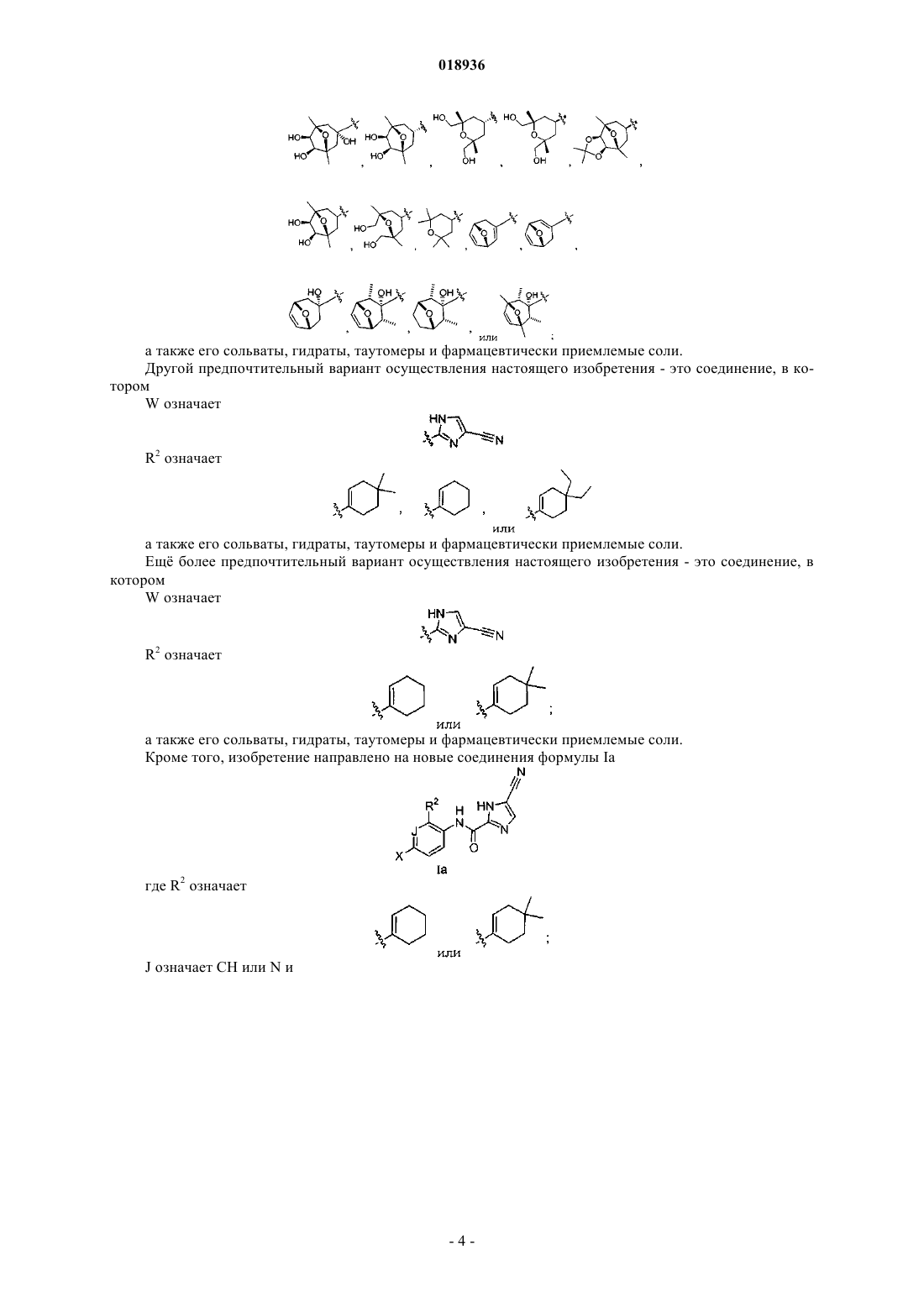
W означает

R2 означает (С3-С8)циклоалкил или (С3-С8)циклоалкенил, любой из которых может быть независимо замещен одним или двумя заместителями, выбранными из С(1-4)алкила;
Z означает Н, F, Cl или СН3;
J означает СН или N;
X означает


где Rw означает Н, -С(1-4)алкил, -СО2С(1-4)алкил, -CONH2, -CONHC(1-4)алкил, -CON(С(1-4)алкил)2 или
-COC(1-4)алкил.
2. Соединение по п.1, в котором
W означает

R2 означает

Z означает Н;
X означает


а также его сольваты, гидраты, таутомеры и фармацевтически приемлемые соли.
3. Соединение по п.2, в котором
W означает

R2 означает

а также его сольваты, гидраты, таутомеры и фармацевтически приемлемые соли.
4. Соединение по п.3, в котором
W означает

R2 означает

а также его сольваты, гидраты, таутомеры и фармацевтически приемлемые соли.
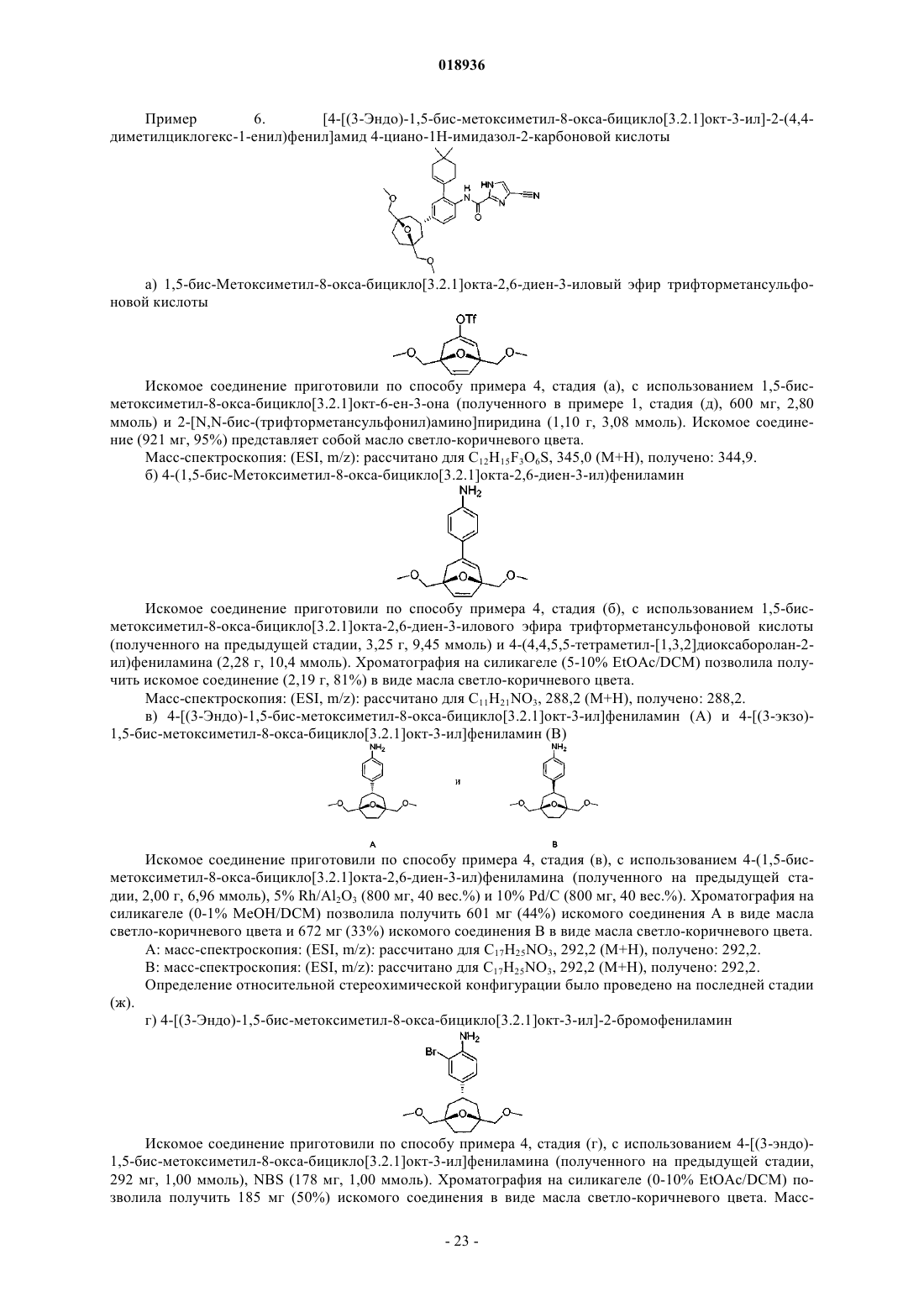
5. Соединение формулы Ia

в котором R2 означает

J означает СН или N и
X означает


где Rw означает Н, -С(1-4)алкил, -СО2С(1-4)алкил, -CONH2, -CONHC(1-4)алкил, -CON(С(1-4)алкил)2 или
-COC(1-4)алкил;
а также его сольваты, гидраты, таутомеры и фармацевтически приемлемые соли.
6. Соединение по п.5, в котором X означает

а также его сольваты, гидраты, таутомеры и фармацевтически приемлемые соли.
7. Соединение формулы

а также его сольваты, гидраты, таутомеры и фармацевтически приемлемые соли.
8. Фармацевтическая композиция, включающая соединение по п.1 и фармацевтически приемлемый носитель.
9. Применение по крайней мере одного из соединений по п.1 для лечения заболевания, выбранного из группы, содержащей остеопороз, болезнь Педжета, ревматоидный артрит и другие формы воспалительного артрита, остеоартрит, отторжение протеза, остеолитическую саркому, миелому и метастазы опухоли в костную ткань.
Текст
Изобретение направлено на соединения формулы I Изобретение относится к новым соединениям, которые действуют как ингибиторы протеинтирозинкиназы. В частности, изобретение относится к новым соединениям, которые действуют как ингибиторы c-fms киназы. Предпосылки создания изобретения Протеинкиназы - это ферменты, которые играют ключевую роль в трансдукции сигналов, осуществляемой путем катализа переноса конечной фосфатной группы от аденозин-5'-трифосфата (АТФ) на гидроксильную группу остатков протеинов тирозина, серина и треонина. Таким образом, ингибиторы и субстраты протеинкиназы являются чрезвычайно полезными средствами для оценки физиологических последствий активации протеинкиназы. Было показано, что сверхсинтез или неадекватное выражение нормальных или мутантных протеинкиназ у млекопитающих играют существенную роль в развитии многих заболеваний, включая рак и диабеты. Протеинкиназы можно разделить на два класса: те, которые фосфорилируют преимущественно остатки тирозина (протеин-тирозинкиназы), и те, которые фосфорилируют преимущественно остатки серина и/или треонина (протеин-серин/треонинкиназы). Протеин-тирозинкиназы выполняют различные функции, от стимулирования роста и дифференциации клеток до остановки пролиферации клеток. Их можно подразделить на рецепторные протеин-тирозинкиназы и внутриклеточные протеинтирозинкиназы. Рецепторные протеин-тирозинкиназы, которые имеют внеклеточный лигандсвязывающий домен и внутриклеточный каталитическим домен с внутренней тирозинкиназной активностью, распределяются на 20 подсемейств. Рецепторные тирозинкиназы семейства эпидермального фактора роста (EGF), к которым относятся рецепторы HER-1, HER-2/neu и HER-3, содержат внеклеточный домен связывания, трансмембранный домен и внутриклеточный цитоплазматический каталитический домен. Связывание рецепторов инициирует многочисленные зависящие от внутриклеточной тирозинкиназы процессы фосфорилирования, которые, в конечном итоге, приводят к онкогенной транскрипции. Была установлена связь рака груди, простаты и колоректального рака с этим семейством рецепторов. Рецептор инсулина (IR) и рецептор инсулиноподобного фактора роста I (IGF-1R) структурно и функционально родственны, однако имеют различный биологический эффект. Была установлена обусловленность рака груди сверхсинтезом IGF-1R. Рецепторы тромбоцитарного фактора роста (PDGF) служат промежуточным звеном клеточных реакций, таких как пролиферация, миграция и выживание, и включают PDGFR, рецептор фактора стволовой клетки (c-kit) и c-fms. Была установлена связь этих рецепторов с такими заболеваниями, как атеросклероз, фиброз и пролиферативная витреоретинопатия. В число рецепторов фактора роста фибробластов (FGR) входят четыре рецептора, которые отвечают за производство кровеносных сосудов, рост конечностей, а также рост и дифференциацию разнообразных типов клеток. Фактор роста эндотелия сосудов (VEGF), мощный митоген эндотелиальных клеток, производится в возрастающих количествах многими опухолями, в том числе карциномой яичника. Известные рецепторы для VEGF обозначаются как VEGFR-1 (Flt-1), VEGFR-2 (KDR), VEGFR-3 (Flt-4). В клетках эндотелия сосудов и в кроветворных клетках были идентифицированы соответствующие группы тирозинкиназных рецепторов-tie-1 и tie-2. Была установлена связь рецепторов VEGF с васкуло- и ангиогенезом. Внутриклеточная протеин-тирозинкиназа известна так же, как нерецепторная протеинтирозинкиназа. Идентифицировано более 24 таких киназ. Они разделены на 11 подсемейств. Серин/треонин-протеинкиназы, подобно клеточным протеин-тирозинкиназам, являются, главным образом,внутриклеточными. Установлена связь аномальной активности протеин-тирозинкиназы с такими патогенными состояниями, как диабет, ангиогенез, псориаз, рестеноз, глазные болезни, шизофрения, ревматоидный артрит,сердечно-сосудистые заболевания и рак. Следовательно, существует необходимость в селективных и сильных ингибиторах протеин-тирозинкиназы с малым размером молекулы. Патенты США 6383790; 6346625; 6235746; 6100254 и Международные заявки РСТ WO 01/47897, WO 00/27820 и WO 02/068406 свидетельствуют о попытках синтеза таких ингибиторов, предпринимаемых в последнее время. Краткое описание изобретения Изобретение направлено на удовлетворение существующей необходимости в селективных и сильных ингибиторах протеин-тирозинкиназы посредством сильных ингибиторов с-fms киназы. Изобретение направлено на новые соединения формулы I или сольват, гидрат, таутомер или фармацевтически приемлемую соль указанных соединений,-1 018936R2 означает (С 3-С 8)циклоалкил или (С 3-С 8)циклоалкенил, любой из которых может быть независимо замещен одним или двумя заместителями, выбранными из C(1-4)алкила;J означает СН или N;-COC(1-4)алкил. Краткое описание чертежей Фиг. 1 - порошковая рентгенограмма соединения примера 31 в зависимости от угла отражения 2; фиг. 2 - порошковая рентгенограмма соединения примера 32 в зависимости от угла отражения 2; фиг. 3 - порошковая рентгенограмма соединения примера 33 в зависимости от угла отражения 2; на фиг. 4 показано воздействие соединения 4 на опухание голеностопного сустава и лапы у крыс в модели артрита, индуцированного стрептококковой клеточной оболочкой (СКО). Подробное описание изобретения Изобретение направлено на удовлетворение существующей необходимости в селективных и сильных ингибиторах протеин-тирозинкиназы посредством сильных ингибиторов c-fms киназы. Изобретение направлено на новые соединения формулы I или сольват, гидрат, таутомер или фармацевтически приемлемую соль указанных соединений, гдеR2 означает (С 3-С 8)циклоалкил или (С 3-С 8)циклоалкенил, любой из которых может быть независимо замещен одним или двумя заместителями, выбранными из C(1-4)алкила;J означает СН или N;-COC(1-4)алкил. Предпочтительный вариант осуществления настоящего изобретения - это соединение, в котором а также его сольваты, гидраты, таутомеры и фармацевтически приемлемые соли. Другой предпочтительный вариант осуществления настоящего изобретения - это соединение, в котором а также его сольваты, гидраты, таутомеры и фармацевтически приемлемые соли. Ещ более предпочтительный вариант осуществления настоящего изобретения - это соединение, в котором а также его сольваты, гидраты, таутомеры и фармацевтически приемлемые соли. Кроме того, изобретение направлено на новые соединения формулы IaJ означает СН или N и-COC(1-4)алкил; а также сольваты, гидраты, таутомеры и фармацевтически приемлемые соли указанных соединений. Предпочтительный вариант осуществления настоящего изобретения - это соединение, в котором а также сольваты, гидраты, таутомеры и фармацевтически приемлемые соли указанных соединений. Ещ один вариант осуществления настоящего изобретения - это соединение формулы Настоящее изобетение также относится к фармацевтической композици, включающей соединение по п.1 и фармацевтически приемлемый носитель. Кроме того, настоящее изобретения относится к применению по крайней мере одного из соединений по п.1 для лечения заболевания, выбранного из группы, содержащей остеопороз, болезнь Педжета,ревматоидный артрит и другие формы воспалительного артрита, остеоартрит, отторжение протеза, остеолитическую саркому, миелому и метастазы опухоли в костную ткань.I. Определения. Термин "алкил" относится к радикалам как с прямой, так и с разветвленной цепочкой, содержащей до 12 атомов углерода, предпочтительно до 6 атомов углерода, если не указано иное, и включает, помимо прочего, следующие радикалы: метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил,пентил, изопентил, гексил, изогексил, гептил, октил, 2,2,4-триметилфенил, нонил, децил, ундецил и додецил. Термин "циклоалкил" относится к насыщенному или частично ненасыщенному кольцу, составленному из 3-8 атомов углерода. На кольце могут произвольно присутствовать до четырех алкильных заместителей. Примеры включают циклопропил, 1,1-диметилциклобутил, 1,2,3-триметилциклопентил, циклогексил, циклопентенил, циклогексенил и 4,4-диметилциклогексенил.II. Терапевтическое применение. Соединения формулы I представляют собой новые мощные ингибиторы протеин-тирозинкиназ, таких как c-fms, и могут быть полезны для профилактики и лечения расстройств, вызванных действием этих киназ. Изобретение также предусматривает способы ингибирования протеин-тирозинкиназы, которые состоят в контактировании протеин-тирозинкиназы с эффективно ингибирующим количеством по крайней мере одного из соединений формулы I. Предпочтительной тирозинкиназой является c-fms. Соединения настоящего изобретения являются также ингибиторами активности тирозинкиназы FLT3. В одном примере ингибирования протеин-тирозинкиназы по крайней мере одно из соединений формулы I сочетается с известным ингибитором тирозинкиназы. В различных вариантах осуществления данного изобретения протеин-тирозинкиназа, ингибируемая соединениями формулы I, располагается в клетках организмов млекопитающих или in vitro. В случае млекопитающих, в том числе людей, назначается терапевтически эффективное количество фармацевтически приемлемой формы по крайней мере одного из соединений формулы I. Изобретение также предусматривает способы лечения рака у млекопитающих, в том числе людей,путем назначения терапевтически эффективного количества фармацевтически приемлемой композиции по крайней мере одного соединения формулы I. Примеры форм рака включают, помимо прочих, следующие заболевания: острый миелолейкоз, острый лимфоцитарный лейкоз, рак яичника, рак матки, рак простаты, рак легких, рак груди, рак толстой кишки, рак желудка и лейкоз ворсистых клеток. Изобретение также предусматривает способы лечения определенных предраковых состояний, включая миелофиброз. В одном примере осуществления данного изобретения эффективное количество по крайней мере одного соединения формулы I назначается в сочетании с эффективным количеством химиотерапевтического препарата. Изобретение также предусматривает способы лечения и профилактики метастазов, являющихся следствием раковых заболеваний, в том числе, помимо прочих, следующих заболеваний: рак яичника,рак матки, рак простаты, рак легких, рак груди, рак толстой кишки, рак желудка и лейкоз ворсистых клеток. Изобретение также предусматривает способы лечения остеопороза, болезни Педжета и других заболеваний, развивающихся при резорбций кости, включая ревматоидный артрит и иные формы воспалительного артрита, остеоартрит, отторжение протеза, остеосаркому, миелому и опухолевые метастазы кости, которые часто наблюдаются при раковых заболеваниях, в том числе, помимо прочих, рак груди,рак простаты и рак прямой кишки. Изобретение также предусматривает способы лечения болевого синдрома, в частности костной боли, вызванной опухолевыми метастазами или остеоартритом, а также висцеральной, воспалительной или нейрогенной боли. Изобретение также предусматривает способы лечения сердечно-сосудистых, воспалительных и аутоиммунных заболеваний у млекопитающих, в том числе людей, путем назначения терапевтически эффективного количества фармацевтически приемлемой формы по крайней мере одного из соединений формулы I. Примеры заболеваний с воспалительным компонентом включают гломерулонефрит, воспалительную болезнь кишечника, отторжение протеза, саркоидоз, острую обструктивную пневмонию,идиопатический легочный фиброз, астму, панкреатит, ВИЧ, псориаз, диабет, патоморфологический ангиогенез, возрастную макулярную дистрофию, диабетическую ретинопатию, рестеноз, шизофрению или болезнь Альцгеймера. Эти заболевания можно эффективно лечить соединениями данного изобретения. Другие заболевания, которые можно эффективно лечить, включают, помимо прочих, атеросклероз и гипертрофию сердца. Аутоиммунные заболевания, такие как системная красная волчанка, ревматоидный артрит и иные формы воспалительного артрита, псориаз, ксеродерматоз, рассеянный склероз или увеит, также можно лечить соединениями настоящего изобретения. Термин "терапевтически эффективное количество", применяемый в данном контексте, означает, что количество активного соединения или фармацевтического средства, которое оказывает биологическое или лечебное воздействие на систему тканей животного или человека, находящегося под наблюдением исследователя, ветеринара, терапевта или другого клинициста; такое воздействие заключается в облегчении, профилактике, лечении или отсрочке начала или развития симптомов заболевания или расстройства,которое подвергается лечению. Когда соединения данного изобретения применяются в качестве ингибиторов протеинтирозинкиназы, их можно назначать в эффективном количестве в пределах дозировки от примерно 0,5 мг до примерно 10 г, предпочтительно от примерно 0,5 мг до примерно 5 г, в виде однократных или разделенных суточных доз. Назначаемая дозировка будет зависеть от таких факторов, как способ приема, состояние здоровья, вес и возраст реципиента, частота лечения и наличие параллельного или несовместимого лечения. Для специалиста в данной области также очевидно, что терапевтически эффективная дозировка для соединений нестоящего изобретения или их фармацевтической композиции будет варьировать в зависимости от желаемого эффекта. Следовательно, оптимальные дозировки, которые должны назначаться,могут быть легко определены соответствующим специалистом и будут варьировать в зависимости от конкретного используемого соединения, способа приема, концентрации препарата и развития болезненного состояния. Кроме того, необходимость регулирования дозировки до приемлемого терапевтического уровня будет обусловлена факторами, связанными с конкретным субъектом лечения, включая его возраст, вес, диету и время назначения препарата. Таким образом, приведенные выше дозировки являются примером для усредненного случая. Разумеется, возможны и отдельные случаи, когда необходимо использовать более низкую или более высокую дозировку, и такие случаи также лежат в области применения данного изобретения. Соединения формулы I можно составлять в фармацевтические композиции, включающие любой из известных фармацевтически приемлемых носителей. Примеры носителей включают, помимо прочих,любые подходящие растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые средства и изотонические вещества. Примеры вспомогательных веществ, которые также могут входить в состав рецептуры, включают наполнители, связующие вещества, вещества для повышения распадаемости и смазывающие вещества. Фармацевтически приемлемые соли соединений формулы I включают распространенные нетоксичные соли или четвертичные аммониевые соли, полученные из неорганических или органических кислот или оснований. К примерам таких кислотно-аддитивных солей относятся ацетат, адипат, бензоат, бензосульфонат, цитрат, камфорат, додецилсульфат, гидрохлорид, гидробромид, лактат, малеат, метансульфонат, нитрат, оксалат, пивалат, пропионат, сукцинат, сульфат и тартрат. Соли оснований включают соли аммония, соли щелочных металлов, такие как соли натрия и калия, соли щелочно-земельных металлов,такие как соли кальция и магния, соли с органическими основаниями, такие как дициклогекиламиновые соли, и соли с аминокислотами, такие как аргинин. Кроме того, основные азотсодержащие группы могут быть кватернизованы, например, алкилгалогенидами. Фармацевтические композиции изобретения можно вводить любыми способами, которые обеспечивают выполнение их назначения. Примеры включают введение парентальным, подкожным, внутривенным, внутримышечным, интраперитонеальным, трансдермальным, внутриротовым или глазным способами. Альтернативно или параллельно возможно пероральное введение. Подходящие рецептуры для парентального введения включают водные растворы активных соединений в водорастворимой форме, например водорастворимые соли, кислые растворы, щелочные растворы, декстрозно-водные растворы,изотонические углеводные растворы и аддукты циклодекстрина. Настоящее изобретение также охватывает способ получения фармацевтической композиции на основе смешивания фармацевтически приемлемого носителя с любым из соединений данного изобретения. Кроме того, настоящее изобретение включает фармацевтические композиции, изготовленные путем смешивания фармацевтически приемлемого носителя с любым соединением настоящего изобретения. Термин "композиция" в данном контексте обозначает продукт, в состав которого входят указанные ингредиенты в указанных количествах, а также любой продукт, который является прямым или косвенным результатом комбинирования указанных ингредиентов в указанных количествах. Полиморфы и сольваты. Кроме того, соединения настоящего изобретения могут иметь одну или более полиморфных или аморфных кристаллических форм, и в качестве таковых попадают в объем притязаний изобретения. В дополнение к этому, соединения могут образовывать сольваты, например, с водой (т.е. гидраты) или обыкновенными органическими растворителями. Термин "сольват" в данном контексте означает физическое связывание соединений настоящего изобретения с одной или несколькими молекулами растворителя. Это физическое связывание включает различные степени ионного или ковалентного связывания, в том числе образование водородных связей. В некоторых случаях сольват будет способен к изоляции,например, когда одна или несколько молекул растворителя включены в кристаллическую решетку кристаллического твердого тела. Термин "сольват" относится как к сольватам в фазе раствора, так и сольватам, способным к изоляции. Примеры подходящих сольватов не ограничиваются следующими: этанолаты, метанолаты и тому подобное. Предполагается, что в объем притязаний настоящего изобретения входят сольваты соединений настоящего изобретения. Следовательно, в способах лечения с использованием настоящего изобретения термин "назначение" должен относиться к средствам для лечения, облегчения или предотвращения описанных в данном документе синдромов, расстройств или заболеваний, содержащим соединения настоящего изобретения или их сольваты, которые, очевидно, должны быть включены в объем притязаний настоящего изобретения, несмотря на отсутствие конкретных указаний на них. В другом примере осуществления данного изобретения предусматривается использование соединения, как описано в примерах, формуле I или формуле Ia, в качестве лекарственного средства. В другом примере осуществления данного изобретения предусматривается использование соединения, как описано в примерах, формуле I или формуле Ia, для приготовления лекарственного средства для лечения заболевания, связанного с повышенным уровнем выработки c-FMS. В другом примере осуществления данного изобретения предусматривается использование соединения в соответствии с примерами, формулой I или формулой Ia для приготовления лекарственного средства для лечения одного из следующих заболеваний: остеопороз, болезнь Педжета, ревматоидный артрит и иные формы воспалительного артрита, остеоартрит, отторжение протеза, остеосаркома, миелома и опухолевые метастазы кости. В другом примере осуществления данного изобретения предусматривается использование соединения в соответствии с примерами, формулой I или формулой Ia для приготовления лекарственного средства для лечения одного из следующих аутоиммунных заболеваний: системная красная волчанка, ревматоидный артрит и иные формы воспалительного артрита, псориаз, ксеродерматоз, рассеянный склероз или увеит. В другом примере осуществления данного изобретения предусматривается использование соединения в соответствии с примерами, формулой I или формулой Ia для приготовления лекарственного средства для лечения одного из следующих заболеваний: гломерулонефрит, воспалительная болезнь кишечника, отторжение протеза, саркоидоз, острая обструктивная пневмония, идиопатический лгочный фиброз, астма, панкреатит, ВИЧ, псориаз, диабет, патоморфологический ангиогенез, возрастная макулярная дистрофия, диабетическая ретинопатия, рестеноз, шизофрения или болезнь Альцгеймера. В другом примере осуществления данного изобретения предусматривается использование соединения в соответствии с примерами, формулой I или формулой Ia для приготовления лекарственного средства для лечения болевого синдрома, в том числе костной боли, вызванной опухолевыми метастазами или остеоартритом, или висцеральной, воспалительной или нейрогенной боли у млекопитающих. В другом примере осуществления данного изобретения предусматривается использование соединения в соответствии с примерами, формулой I или формулой Ia для приготовления лекарственного средства для лечения следующих заболеваний: рак яичника, рак матки, рак груди, рак простаты, рак легких,рак толстой кишки, рак желудка или лейкоз ворсистых клеток. Способы приготовления Схема 1 На схеме 1 показан общий способ приготовления соединений формулы I. Для иллюстрации способа, показанного на данной схеме, определены реагенты и условия для соединений, в которых буквой J обозначена группа CH. Для специалистов в данной области будет ясно, что если J обозначает N, то могут потребоваться лишь небольшие изменения условий реакции и целесообразных реагентов или вообще не потребоваться. Соединения формулы 1-3 можно получить путем катализированной металлом реакции сочетания нитросоединений формулы 1-1, где L - это уходящая или реакционно-способная группа, такая как галоген, триалкилолово, дигидроксибор, диалкоксибор или полифторированный алкилсульфонилокси, со связующим агентом, подходящим для ввода X. Подходящими связующими агентами являются полифторированные алкилсульфонатные эфиры енолов, когда L - это триалкилолово, дигидроксибор или диалкоксибор, и циклоалкенил боронатные эфиры и бороновые кислоты, когда L - это бромо, йодо или полифторированный алкилсульфонилокси. Предпочтительным способом сочетания является реакция кросс-сочетания Сузуки-Мияура (для справки см. N. Miyaura and A. Suzuki, Chem. Rev., 95:2457 (1995);(1988 соединений формулы 1-1, где L - это бромо или йодо. Предпочтительными условиями для реакции Сузуки-Мияура являются палладиевый катализатор,например тетракис(трифенилфосфин)палладий(0) (Pd(PPh3)4), водная основа, такая как водный раствор Na2CO3, и подходящий растворитель, например толуол, этанол, 1,4-диоксан, диметоксиэтан (ДМЭ) или ДМФ (диметилформамид). Синтез связующих агентов описан на приведенных далее схемах. Амины формулы 1-4 могут быть получены по реакции восстановления из нитросоединений формулы 1-3 с использованием стандартных способов синтеза (см. Reductions in Organic Chemistry, M. Hudlicky, Wiley, New York, 1984). Предпочтительными условиями является каталитическое гидрирование с использованием палладиевого катализатора в подходящем растворителе, например в метаноле или этаноле. В случае, когда X содержит алкен, он будет восстановлен до алкана. Для соединений, где X содержит алкен, который должен остаться в конечном соединении, восстановление нитрогрупп можно провести избирательно с использованием железа или цинка в подходящем растворителе, например в уксусной кислоте, или с использованием железа и хлорида аммония в этаноле и воде. С другой стороны, соединения формулы 1-4 могут быть получены из аминов формулы 1-2 с помощью реакций замещения L на X, как описано выше. В соединениях формулы 1-4, где X содержит алкен,в случае необходимости, он может быть восстановлен в алкан описанными выше способами. Соединения формулы 1-2, которых нет в продаже, могут быть получены из соединений формулы 1-1 путем восстановления нитрогруппы с использованием железа или цинка в подходящем растворителе, таком как уксусная кислота, или с использованием железа и хлорида аммония в этаноле и воде. Соединения формулы 1-5 могут быть получены путем ортогалогенирования, предпочтительно бромирования, аминосоединений формулы 1-4 и последующих катализированных металлами реакций сочетания с бороновыми кислотами или бороновыми эфирами (реакции Сузуки-Мияура, где R2M означаетR2B(OH)2 или бороновый эфир, см. ссылку выше) или с реагентами, содержащими олово (реакция Стилла, где R2M означает R2Sn(алкил)3, см. J.K. Stille, Angew. Chem., Int. Ed. Engl., 25: 508-524 (1986, на промежуточном галогенсодержащем соединении. Предпочтительными условиями бромирования соединений 1-4 являются N-бромосукцинимид (NBS) в подходящем растворителе, например N,Nдиметилформамид (DMF), четыреххлористый углерод или предпочтительно дихлорметан (DCM) или ацетонитрил. Затем в соответствии со стандартными способами, описанными в вышеупомянутых литературных источниках, могут быть проведены реакции сочетания, катализированные металлами, предпочтительно реакции Сузуки-Мияура. Соединения формулы 1-6 могут быть получены из соединений формулы 1-5 по реакции аминогруппы с гетероциклической кислотой P1-WCOOH (или ее соответствующей солью P1-WCOOM2, где М 2 означает Li, Na или K), где Р 1 - это необязательная защитная группа (например, 2(триметилсилил)этоксиметил (SEM), причем W означает имидазол, триазол, пиррол или бензимидазол),или где Р 1 отсутствует, как в том случае, когда W означает фуран (перечень подходящих защитных групп для W, см. Theodora W. Greene and Peter G.M. Wuts, Protective Groups in Organic Synthesis, John Wiley andSons, Inc., NY (1991. Сочетание с целью получения соединений формулы 1-6 может проводиться в соответствии со стандартными способами для образования амидных связей (подробнее см. М. Bodanskyand A. Bodansky, The Practice of Peptide Synthesis, Springer-Verlag, NY (1984 или по реакции с хлорангидридами P1-WCOCl либо активированными эфирами Р 1-WCO2Rq (где R4 - это уходящая группа, например пентафторфенил или N-сукцинимид). Предпочтительными условиями реакций сочетания с участиемP1-WCOOH или P1-WCOOM2 являются в случае, когда W означает фуран (необязательная защитная группа Р 1 отсутствует), оксалилхлорид в дихлорметане (DCM) с DMF в качестве катализатора для образования хлорангидрида WCOCl и последующее сочетание в присутствии триалкиламина, например N,Nдиизопропилэтиламин (DIEA); когда W означает пиррол (необязательная защитная группа Р 1 отсутствует), 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (EDCI) и 1-гидроксибензотриазол(HOBt); а если W означает имидазол, триазол, пиррол или бензимидазол (необязательная защитная группа Р 1 присутствует), предпочтительными условиями являются гексафторфосфат бромотрипирролидинфосфония (PyBroP) или DIEA в растворителе, таком как DCM или DMF. Если W в соединениях формулы 1-6 содержат необязательную защитную группу Р 1, как отмечено выше, на этом этапе она может быть удалена и будут получены соединения формулы I. Например, когдаW означает имидазол, защищенный на азоте группой SEM, группу SEM можно удалить либо кислыми реагентами, такими как трифторуксусная кислота (TFA), либо реагентами - источниками фтора, такими как фторид тетрабутиламмония (TBAF) (см. Greene and Wuts, выше). Если соединения формулы 1-6 не содержат защитную группу, они также являются соединениями формулы I. Наконец, понятно, что далее можно получить производные соединений формулы I. Примеры производных, помимо прочего, включают следующие: если соединения формулы I содержат цианогруппу,эту группу можно гидролизовать до амидов или кислот в кислых или основных условиях; если соединения формулы I содержат эфир, последний можно гидролизовать до кислоты, а кислоту можно превратить в амиды способами, описанными выше для образования амидной связи. Кислоты могут быть восстановлены до спиртов. Предпочтительные условия для восстановления карбоновых кислот в присутствии цианогрупп включают борогидрид натрия и этилхлорформиат в тетрагидрофуране (THF). Олефины могут быть восстановлены каталитическим гидрированием. Олефины также можно дигидроксилировать до диолов с использованием ряда способов, включая реакцию с N-метилморфолин N-оксидом, катализированную тетроксидом осмия (подробнее см. Sundermeier, U., Doebler, С. and Beller, M., Modern OxidationA. (Eds.), 2, 1009-1024, VCH, Weinheim, Germany (1996. Если соединения формулы I содержат ациклический или циклический сульфид, последний может быть окислен далее до соответствующих сульфоксидов или сульфонов. Сульфоксиды можно получить путем окисления с использованием подходящего окислителя, такого как один эквивалент мета-хлорпербензойной кислоты (МСРВА) или путем обработкиNaIO4 (см., например, J. Med. Chem., 46: 4676-86 (2003, а сульфоны можно получить с использованием двух эквивалентов МСРВА или путем обработки 4-метилморфолин N-оксидом с тетроксидом осмия в качестве катализатора (см., например, Международный патент WO 01/47919). К тому же, как сульфоксиды, так и сульфоны могут быть приготовлены с использованием одного эквивалента или двух эквивалентов Н 2 О 2 соответственно в присутствии изопропоксида титана(IV) (см., например, J. Chem. Soc, Perkin Схема 2 описывает синтез соединений формулы I, когда X означает Для иллюстрации этого способа реагенты и условия определены в данной схеме для субстратов, гдеX означает Е означает О или NCO2Rw; Rx означает Н или Me; Ry означает Н или CH2Rv, где Rv означает Н, ОМе или OPG, где PG - это подходящая защитная группа, которая устойчива к условиям трансформации данной схемы и может быть удалена в дальнейшем для снятия защиты Rv означает ОН; aRz означает Н или ОН, где две ОН-группы могут быть защищены соответствующим образом с помощью подходящих кетальных или силильных защитных групп, которые в конечных продуктах могут быть либо удалены, либо оставлены. Специалистам в данной области будет ясно, что химические процессы актуальны для всех X, Rx, Ry и Rz, упомянутых выше, и могут быть реализованы с небольшими изменениями реагентов и условий. Исходный материал 2-1 преобразуют в йодированное соединение 2-2 по реакции с I2 или NIS или,что предпочтительнее, с I2/Ag2SO4 в подходящем растворителе, таком как метиловый спирт, изопропиловый спирт или, лучше всего, этиловый спирт. Соединения формулы 2-3, где R2 означает циклоалкенил и циклоалкил, могут быть получены из 2-2 по селективным реакциям сочетания, катализированным металлами, с бороновыми кислотами или бороновыми эфирами, как описано в схеме 1. Аминогруппу соединений формулы 2-3 затем можно присоединить к гетероциклической кислоте P1-WCOOH с образованием соединений формулы 2-4, как описано в схеме 1. Если W в соединениях формулы 2-4 содержит необязательную защитную группу Р 1, она может быть удалена на этой стадии путем, описанным в схеме 1, с образованием соединения 2-5. И наконец, бромзамещенное соединение 2-5 преобразуется в спирт 2-6; при этом вначале проводят депротонирование кислоты с помощью подходящего основания, например хлорида изопропилмагния (i-PrMgCl) в растворителе, таком как этиловый эфир, диметиловый эфир или лучше всего тетрагидрофуран; затем проводят обменную реакцию галоген-литий с подходящим литийсодержащим реагентом, таким как n-бутиллитий, втор-бутиллитий или предпочтительно трет-бутиллитий при температуре от -100 до -40 С, предпочтительно -78 С, а затем перехват литийсодержащего органического промежуточного соединения с помощью подходящего кетона 2-7. Синтез кетонов формулы 2-7 описан в схемах 6 и 7. Для специалистов в данной области будет очевидно, что соединения настоящего изобретения на этом этапе могут быть модифицированы и далее. Например, если соединение 2-6 имеет кислотную группу на W, то эта кислотная группа может быть этерифицирована; также и амид на W может быть дегидрирован с образованием нитрила. Схема 3 На схеме 3 показан общий способ приготовления гетероциклических кетонов формулы 3-2, где Е означает О, S, SO, или SO2 и Ry=Rz=СН 3. Эти кетоны полезны для приготовления соединений формулы I,где X означает Указанные гетероциклические кетоны можно получить по реакциям двойного присоединения по Михаэлю подходящих нуклеофильных реагентов с диенонами формулы 3-1, катализируемым кислотой или основанием при температурах от 0 до 100 С. Если в качестве нуклеофильного реагента используется вода (ЕН 2 означает ОН 2), предпочтительные условия для этого превращения включают реакцию диенонов формулы 3-1 при, например, 40-50 С в течение 4 суток с избытком 1-4 N водного раствора HCl для получения соединения формулы 3-2, где Е означает О (WO 2005012220). Аналогично, когда в качестве нуклеофильного реагента используется H2S, соединения формулы 3-2, где Е означает S, могут быть получены в присутствии неорганических оснований, таких как KOH, при наличии или в отсутствие каталитического количества органического амина, такого как пиперидин, в протоносодержащих растворителях,таких как EtOH, при организованном обратном потоке и непрерывном медленном барботировании H2S(Journal of Industrial and Engineering Chemistry (Washington, D.C.) (1952), 44, 1659-62). Для специалистов в данной области должно быть ясно, что серосодержащие кетоны формулы 3-2, где Е означает S, могут быть окислены одним или двумя эквивалентами подходящего окислителя, такого как mхлорпербензойная кислота, для получения соединений формулы 3-2, где Е означает SO или SO2 соответственно. Схема 4 Два других способа синтеза для приготовления соединений формулы 3-2 показаны на схеме 4, где Е означает S и Ry означает Н, Me и CH2Rv, где Rv означает Н, ОМе или OPG, где PG - это подходящая защитная группа, которая устойчива к условиям трансформации данной схемы и может быть удалена в дальнейшем для снятия защиты, или Rv означает ОН. Ненасыщенные аминокетоны формулы 4-1 и четвертичные аммониевые соли пиперидонов формулы 4-3 (предпочтительно замещенные N,Nдиметилпиперидон галоиды (Rs означает Me), полученные по реакции соответствующим образом замещенного пиперидона с галоидным производным метана, таким как йодометан (L означает I, могут быть преобразованы в соединения формулы 3-2, где Е означает S, под действием H2S или сульфидов металлов(M2S), лучше сульфидов щелочных металлов, таких как Na2S, соответственно (Химия гетероциклических соединений: Сборник, 1970, (2), 174-80 и Известия Академии наук Казахской ССР, Серия химическая,1986, (3), 92-3 соответственно). Схема 5 На схеме 5 показан еще один подход к синтезу гетероциклических кетонов формулы 3-2, где Е означает О, S, Ry означает Me и CH2Rv, где Rv означает Н, ОМе или OPG, где PG - это подходящая защитная группа, которая устойчива к условиям трансформации данной схемы и может быть удалена в дальнейшем для снятия защиты, или Rv означает ОН и Rz означает Me или оба Rz, взятые вместе, означают СН 2-СН 2 или СН означает СН так, что получающийся кетон 3-2 является бициклическим. Это можно осуществить с помощью реакции внутримолекулярной циклизации по Дикману с использованием подходящих прекурсоров формулы 5-1 (Rb означает Me или Et) в кислых или основных условиях и последующим удалением -алкоксикарбонильного заместителя CO2Rt, как показано на схеме 5. Предпочтительный способ для последовательности этого синтеза предусматривает индуцированную основанием циклизацию диэфиров формулы 5-1 при температурах от -78 С до комнатной температуры с целью получения -кетоэфиров формулы 5-2 и последующие катализируемые кислотой гидролиз и декарбоксилирование при температурах от 20 до 200 С. Понятно, что после гидролиза эфира можно провести декарбоксилирование промежуточного соединения 5-2 с выделением или без выделения соответствующей карбоновой кислоты с целью получения соединений формулы 3-2. Предпочтительные основания для первой стадии включают, помимо прочих, такие сильные основания, как алкоксиды и гидроксиды щелочных металлов, например метоксид натрия, этоксид натрия, трет-бутоксид калия и гидроксид лития,а также вторичные органические амины солей щелочных металлов, такие как диизопропиламид лития и гексаметилдисилазид лития. Предпочтительные условия гидролиза и декарбоксилирования включают, помимо прочих, нагревание соединений формулы 5-2 с разбавленными неорганическими кислотами, такими как 1 М водный раствор HCl с подходящим растворителем, таким как THF (тетрагидрофуран), или без него. Гидролиз эфира формулы 5-2 также можно провести путем обработки водным раствором основания, например гидроксида натрия, гидроксида калия или карбоната калия, в подходящей смеси растворителей, например воды и органического растворителя, такого как THF, метанол, этанол или изопропанол. При использовании данного способа гидролиза, катализируемого основанием, полученную соль карбоновой кислоты можно затем обработать неорганической кислотой, такой как 0,01-12 М водный раствор HCl или H2SO4 с подходящим растворителем, таким как THF (тетрагидрофуран) или диоксан, или без него, для получения соответствующей карбоновой кислоты. Для специалистов в данной области ясно, что соответствующие карбоновые кислоты соединений формулы 5-2, полученные либо путем гидролиза, катализированного кислотой, либо путем гидролиза, катализированного основанием, и последующего окисления могут спонтанно декарбоксилировать в присутствии или в отсутствие любого постороннего кислотного или основного реагента, с нагреванием или без нагревания. Кроме того, понятно, что соединения формулы 5-1 можно приготовить с применением известных способов или простого изменения или расширения известных способов (для примеров диэфиров формулы 5-1 и соответствующих двухосновных кислот см. На схеме 6 показан синтез гетеро-бициклических кетонов формулы 6-2, которые используются в качестве промежуточных соединений в реакциях сочетания на схеме 2. Общепринятый способ синтеза состоит в [4+3] циклоприсоединении полученного тут же оксиаллильного катиона формулы 6-1 с подходящим диеном и, при необходимости, последующем дегалогенировании полученного продукта. Предпочтительные прекурсоры для получения оксиаллильных катионов включают полигалокетоны, 2 кислородзамещенные аллиловые эфиры и акролеины, которые могут быть превращены в оксиаллильные катионы и тут же захвачены с помощью подходящего диена в восстановительных основных условиях или в кислоте Льюиса. Необходимый оксиаллильный катион можно также получить путем дисротаторного раскрытия кольца циклопропанонов или путем конротаторной изомеризации оксидов алленов (J.Am. Chem. Soc. (1998), 120, 12310). Дегалогенирование поли -галогенокетонов может быть проведено с помощью таких реагентов, как Cu/ NaI (M означает Na), Zn/Cu ИЛИ Zn/Ag (M означает Zn), Zn/Cu/TMSClFe) (обзор см. Org. React., 1983, 29, 163, J. Org. Chem. (1999), 64, 3398 для получения оксиаллильных катионов. Основные реагенты для галогенирования -галогенокетонов с целью получения оксиаллильных катионов формулы 6-1 включают такие реагенты, как Et3N/CF3CH2OH, алкоксиды натрия 2,2,3,3 тетрафторпропанол и 2,2,2-трифторэтанол (J. Chem. Res., Synop, (1986), 424. J. Chem. Res., Synop. (1981),246, J. Chem. Res., Synop. (1983), 166) и LiClO4/Et3N (J. Org. Chem. (1999), 64, 3398). Кислоты Льюиса,такие как AgO2CCF3, можно использовать для дегалогенирования с целью получения оксиаллильных катионов из 2-метоксиаллил галидов (J. Am. Chem. Soc. (1973), 95, 1338), в то время как AgBF4 можно использовать для 2-аминозамещенных оксиаллильных катионов (Helv. Chim. Acta. (1974), 57, 1883). Другие кислоты Льюиса, такие как SnCl4, Sc(OTf)2 и TiCl4, можно использовать для получения оксиаллильных катионов из 2-O-силилокси-акролеинов (Tett. Lett. (1982), 23, 1693; Org. Lett. (2000), 2, 2703). Одним из способов получения оксиаллильных катионов является обработка -галогенокетонов, например тетрабромацетона, парой Zn/Cu в подходящем органическом растворителе, таком как THF. Вторым способом получения оксиаллильных катионов является обработка -галогенокетонов, например трихлорацетона или пентахлорацетона, натрия 2,2,2-трифторэтоксидом или триэтиламмония 2,2,2-трифторэтоксидом в 2,2,2-трифторэтаноле в качестве растворителя (Lee, K. and Cha, J.K., J. Am. Chem. Soc. (2001), 123, 559091 и Sendelbach, et al., Journal of Organic Chemistry (1999), 64(10), 3398-3408). Кроме того, для получения оксиаллильных катионов из дивинилкетонов можно использовать фотохимические условия реакции (J.Org. Chem (1993), 58, 6795 и J. Am. Chem. Soc. (1968), 90, 6251). Реагентами для захвата диенов являются ароматические гетероциклы, такие как подходящим образом замещенные пирролы и фураны, которые либо имеются в продаже, либо могут быть приготовлены по опубликованным в литературе способам. Исходный продукт [4+3] циклоприсоединения, полученный этим способом, может быть дегалогенирован известными способами, лучше всего восстановительным дегалогенированием с использованием Zn или пары Zn/Cu. Схема 7 Понятно, что двойная связь в оксабицикло аддуктах формулы 6-2 может быть функционализирована далее с использованием соответствующих условий реакций. Как показано на схеме 7, например, соединения формулы 6-2 могут быть бис-гидроксилированы с использованием известных из литературы схем (перечень реагентов и ссылок см. Larock, R.C. Comprehensive Organic Transformations, 2nd Ed.,Wiley-VCH, NY, (1999), p. 996-1003) для получения цис-диолов формулы 7-1, которые могут быть затем защищены для получения соединений формул 7-2 и 7-3. Предпочтительные условия бисгидроксилирования включают, помимо прочих, обработку соединений формулы 6-2 каталитическим количеством OsO4 и трет-BuOOH в качестве реоксиданта в присутствии Et4NOH (Bulletin of the ChemicalSociety of Japan (1984), 57(9), 2515-25). Диолы формулы 7-1 могут быть защищены с целью получения соединений формул 7-2 и 7-3. Примеры подходящих групп для защиты диолов можно найти в публикации Protective Groups in Organic Synthesis, Theodora W. Greene and Peter G.M. Wuts, John WileySons.Inc., NY (1999). Предпочтительными защитными группами являются изопропилидин кеталь (Bulletin ofthe Chemical Society of Japan (1984), 57(9), 2515-25) и ди-трет-бутилсилилен с использованием (третBu)2SiCl2 в качестве силилирующего реагента в хлорированных растворителях, таких как дихлорметан(DCM) или дихлорэтан (DCE) и имидазол, при температурах от -78 С до комнатной температуры, предпочтительно при 0 С. Олефиновые функциональные группы соединений формулы 6-2 также могут быть насыщены для получения соединений формулы 7-4. Предпочтительными условиями для этого превращения является каталитическое гидрирование (например, см. Journal of Organic Chemistry (1999), 64(10),3398-3408). Схема 8 Схема 8 иллюстрирует использование гетероциклических кетонов формул 3-2, 6-2, 7-2, 7-3 и 7-4,которые на схеме 8 представлены формулой 8-1. Эти кетоны формулы 8-1 могут быть переведены в со- 13018936 ответствующие енол-полифторированные алкилсульфонатные эфиры, предпочтительно енолтрифторметансульфонаты и енол-нонафторбутансульфонаты, описанными в литературе способами (примеры см. BioorganicMedicinal Chemistry (2002), 10 (11) и 3583-3591, Chem. Eur. J., 2007, 13, 2410 соответственно). Предпочтительные условия для этого превращения включают, помимо прочих, обработку гетероциклических кетонов формулы 8-1 сильными основаниями, такими как диизопропиламид лития или гексаметилдисилазид лития, при температурах от -78 С до комнатной температуры, предпочтительно -78 С, и последующее добавление фторсульфонил реагентов, таких как нонафторбутансульфонил фторид,2-[N,N-бис-(трифторметансульфонил)амино]пиридин илиN-фенил-бис-(трифторметансульфонимид). Соединения формулы 8-2 могут непосредственно использоваться в катализированных металлами реакциях сочетания, описанных в схеме 1. Кроме того, соединения формулы 8-2 могут быть переведены в соответствующие боронатные эфиры формулы 8-3 перед использованием в реакциях кросс-сочетания по Сузуки-Мияура, описанных в схеме 1 (стандартные способы см. Eastwood, P., Tetrahedron Lett. (2000), 41, 3705-8 и Takahashi, K., et al., Chem. Lett. (2000), 126-7.) И наконец, если соединение формулы 8-2 или 8-3 содержит защитную группу, она может быть удалена на промежуточной или заключительной стадии с использованием соответствующих условий. Например, при наличии цис-диола,защищенного как изопропилидин кеталь, защитная группа может быть удалена в водном кислом растворе при повышенной температуре, предпочтительно при 100 С, а если он защищен как ди-третбутилсилиленовый эфир, защитная группа может быть удалена в кислой среде или предпочтительнее всего источниками фтора, такими как TBAF (фторид тетрабутиламмония) (см. Protective Groups in Organic Synthesis, Theodora W. Greene and Peter G.M. Wuts, John WileySons. Inc, NY, (1999). Схема 9 На схеме 9 показан способ приготовления 2-имидазолкарбоксилатов формулы 9-5, где Ra означает Н или С(1-4)алкил и Rq означает Н, алкил, -CN или -CONH2, которые используются в качестве промежуточных соединений при синтезе соединений формулы I, где W означает имидазол. Имидазолы формулы 9-1, где Ra означает Н или С(1-4)алкил и Rc означает Н, С(1-4)алкил или -CN либо доступны на рынке, либо в случае, если Rc означает -CN, могут быть легко получены из имеющихся на рынке альдегидов (9-1 где Rc означает СНО) по реакции с гидроксиламинами и последующим дегидрированием с использованием подходящего реагента, например оксихлорид фосфора или уксусный ангидрид (Synthesis (Синтез), (2003), 677). Имидазолы формулы 9-1 могут быть защищены подходящей группой (Р 1), такой как метоксиметиламин (MOM), или предпочтительнее всего группой SEM с образованием соединений формулы 9-2 (см. Theodora W. Greene and Peter G.M. Wuts, Protective Groups in Organic Synthesis (Защитные группы в органическом синтезе), John Wiley and Sons, Inc., NY (1991. Имидазолы формулы 9-2, где Rc означает -CN, могут быть галогенированы с помощью подходящего реагента, например N-бромсукцинимида или N-йодосукцинимида, либо в электрофильных условиях в растворителе, таком как DCM или CH3CN, либо в условиях существования радикалов, в присутствии инициатора, такого как азо-бис-(азобутиронитрил) (AIBN) в растворителе, таком как CCl4, с образованием соединений формулы 9-3, где L - это уходящая группа (предпочтительно бромо или йодо). Галогенмагниевый обмен на соединениях формулы 9-3 может дать магнийорганические молекулы, которые затем могут вступать в реакцию с подходящими электрофильными реагентами, с образованием соединений формулы 9-4. Предпочтительными условиями для галоген-магниевого обмена являются использование алкилмагниевого реагента, предпочтительно изопропилмагний хлорида в подходящем растворителе, таком как THF при температурах от -78 до 0 С. Предпочтительными электрофильными реагентами являются этилхлороформат или этилцианоформат (примеры галоген-магниевого обмена на цианоимидазолах см. J. Org. Chem. (2000), 65, 4618). Что касается имидазолов формулы 9-2, где Rc - это не -CN, они могут быть преобразованы непосредственно в имидазолы формулы 9-4 путем депротонирования с использованием подходящего основания, такого как алкиллитий, и последующей реакции с электрофильным реагентом, как описано выше для магнийорганических соединений. Предпочтительными условиями являются обработка имидазола в растворе н-бутиллития в THF при -78 С и гашение образующихся литийорганических соединений с помощью этилхлороформата (примеры см. Tetrahedron Lett. (1988), 29, 3411-3414). Эфиры формулы 9-4 затем могут быть гидролизованы до карбоновых кислот (М означает Н) или солей карбоксилатов (М означает Li, Na или K,) формулы 9-5 с использованием одного эквивалента водного раствора гидроксида металла (MOH), предпочтительно гидроксида калия в подходящем растворителе, таком как этанол или метанол. Синтез соединений формулы 9-5, где R4 означает -CONH2, проводят вначале обработкой соединений формулы 9-4, где Rc означает -CN, соответствующим алкоксидом, таким как этоксид калия, для преобразования цианогруппы в имидат группу (реакция Пиннера), а затем гидролизом как эфиров, так и имидат групп двумя эквивалентами водного раствора гидроксида металла. Схема 10 Схема 10 иллюстрирует способ синтеза 2-имидазолкарбоксилатов формулы 10-3 или 10-5, где Rr это хлоро или бромо, а М означает Н, Li, K или Na, которые используются как промежуточные соединения в синтезе соединений формулы I, где W означает имидазол. Соединения формулы 10-1 можно вначале получить путем защиты имеющегося в продаже этилимидазолкарбоксилата в соответствии со способами, описанными в схеме 9, предпочтительно с группойSEM. Соединения формулы 10-2 могут быть приготовлены по реакции соединений формулы 10-1 с одним эквивалентом соответствующего галогенирующего реагента, таким как NBS или NCS, в подходящем растворителе, например CH3CN, DCM или DMF при 25 С. Соединения формулы 10-4 можно приготовить по реакции соединений формулы 10-1 с двумя эквивалентами соответствующего галогенирующего реагента, такими как NBS или NCS в подходящем растворителе, например CH3CN или DMF, при температурах от 30 до 80 С. Имидазолы формул 10-3 и 10-5 затем можно получить из соответствующих эфиров путем гидролиза, как описано в схеме 9. Схема 11 Схема 11 иллюстрирует способ приготовления имидазолов формулы 11-3, где Rf означает -SCH3,-SOCH3 или -SO2CH3, M означает Н, Li, K или Na, которые используются как промежуточные соединения в синтезе соединений формулы I, где W означает имидазол. Имидазол 11-1 (WO 1996011932) защищен в соответствии со способами, описанными в схеме 9,предпочтительно защитной SEM группой для получения соединений формулы 11-2. Гидролиз эфиров в соответствии со способом схемы 9 дает соединения формулы 11-3, где Rf означает -SCH3. Окисление 2 метилтиоимидазолов формулы 11-2 одним эквивалентом подходящего окислителя и последующий гидролиз эфиров в соответствии со способом схемы 9 дает соединения формулы 11-3, где Rf означает-SOCH3. Окисление двумя эквивалентами подходящего окислителя и последующий гидролиз эфиров в соответствии со способом схемы 9 дает соединения формулы 11-3, где Rf означает -SO2CH3. Предпочтительным реагентом для окисления является МСРВА (мета-хлорпербензойная кислота) в DCM (дихлорметан): рекомендации по превращению сульфидов в сульфоксиды и сульфоны даны на схеме 1. Примеры Пример 1. [2-(4,4-Диметилциклогекс-1-енил)-4-[(3-экзо)-3-гидрокси-1,5-бис-метоксиметил-8-оксабицикло[3.2.1]окт-6-ен-3-ил]фенил]амид 4-циано-1 Н-имидазол-2-карбоновой кислоты Смесь 4-бром-2-йодофениламина (873 мг, 2,93 ммоль), 4,4-диметилциклогексен-1-ил бороновой кислоты (496 мг, 3,22 ммоль), Pd(PPh3)4 (169 мг, 0,147 ммоль) и 2,0 М водного Na2CO3 (11,7 мл, 23, 4 ммоль) в 20 мл 1,4-диоксана перемешивали в атмосфере аргона при 80 С в течение 12 ч. После охлаждения до комнатной температуры реакционную смесь обработали EtOAc (50 мл) и промыли Н 2 О (25 мл) и рассолом (20 мл). Органическую фазу высушили (Na2SO4) и сконцентрировали в вакууме. Полученный остаток очистили флэш-хроматографией на силикагеле (5% EtOAc/гексан), что дало 770 мг (91%) искомого соединения в виде бесцветного масла. Масс-спектроскопия (ESI, m/z): рассчитано для C14H18BrN, 280,1 (М+Н), получено: 280,1. б) [4-Бромо-2-(4,4-диметилциклогекс-1-енил)фенил]амид 4-циано-1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-карбоновой кислоты К смеси 4-бромо-2-(4,4-диметилциклогекс-1-енил)фениламина (полученного на предыдущей стадии, 770 мг, 2,75 ммоль), калиевой соли 4-циано-1-(2-триметилсиланилэтоксиметил)-1H-имидазол-2 карбоновой кислоты (патентная заявка США 2006189623 А 1, 840 мг, 2,75 ммоль) и PyBroP (1,28 г, 2,75 ммоль) в 20 мл DMF добавили DIEA (N,N-диизопропилэтиламин) (1,44 мл, 8,25 ммоль). Полученную смесь перемешивали при комнатой температуре в течение 16 ч в атмосфере аргона. Смесь обработали 80 мл EtOAc, а затем промыли Н 2 О (220 мл), рассолом (20 мл) и высушили (Na2SO4). После отгонки растворителя при пониженном давлении и последующей флэш-хроматографии остатка на силикагеле (510% EtOAc/гексан) было получено 1,28 г (88%) искомое соединение в виде твердого порошка белого цвета. Масс-спектроскопия (ESI, m/z): рассчитано для С 25 Н 33 BrN4O2Si, 529,2 (М+Н), получено: 528,9. в) [4-Бромо-2-(4,4-диметилциклогекс-1-енил)фенил]амид 4-циано-1 Н-имидазол-2-карбоновой кислоты К раствору [4-бромо-2-(4,4-диметилциклогекс-1-енил)фенил]амида 4-циано-1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-карбоновой кислоты (полученного на предыдущей стадии, 350 мг, 0,661 ммоль) в 5 мл DCM (CH2Cl2) добавили 0,15 мл EtOH, а затем 2,5 мл TFA. После перемешивания при комнатной температуре в течение 3 ч смесь обработали 10 мл н-пропанола и сконцентрировали в вакууме. Остаток растерли в порошок с DCM; в результате получили 253 мг (96%) искомого соединения в виде твердого вещества белого цвета. 1 К суспензии гидрида натрия (сухой, 314 мг, 13,1 ммоль) в 2 мл безводного THF в атмосфере аргона осторожно добавили раствор 2,5-бис-гидроксиметилфурана (Международная патентная заявка WO 2006122772 А 1) в 10 мл безводного THF. После перемешивания при комнатной температуре в течение 20 мин добавили метилйодид (672 мкл, 10,8 ммоль) и перемешивали смесь еще 14 ч. Затем очень осторожно добавили воду (15 мл) и сконцентрировали смесь в вакууме для удаления THF. Оставшуюся водную смесь насытили твердым NaCl и экстрагировали Et2O (515 мл). Объединенные органические фазы вы- 16018936 сушили над Na2SO4 и сконцентрировали в вакууме до масла желтого цвета, которое было очищено путем хроматографии на силикагеле (5-30% EtOAc/гексан); в результате было получено искомое соединение К суспензии цинка (нанопорошок, Aldrich Chemical Co., 602 мг, 9,20 ммоль) в растворе 2,5-бисметоксиметилфурана (полученного на предыдущей стадии, 958 мг, 6,13 ммоль) в 1,0 мл безводного THF в атмосфере аргона добавили раствор 1,1,3,3-тетрабромоацетона (3,44 г, 9,20 ммоль) и триэтилбората(2,20 мл, 12,9 ммоль) в 2,8 мл THF по каплям в течение 15 мин. Колбу закрыли алюминиевой фольгой для предотвращения воздействия света и перемешивали смесь при комнатной температуре в течение 18 ч. К смеси добавили воду (10 мл) и перемешивали в течение 15 мин, а затем отфильтровали (целит) и промыли EtOAc (210 мл). Отделили фазы, водную фазу экстрагировали EtOAc (325 мл), а объединенные органические фазы промыли водой (50 мл), высушили (Na2SO4) и сконцентрировали в темное масло. Полученный остаток, растворенный в 5 мл МеОН, по каплям добавили к суспензии цинковой пыли (10 мкм, 2,09 г, 31,9 ммоль), хлорида меди(I) (316 мг, 3,19 ммоль) и хлорида аммония (2,29 г, 42,9 ммоль) в 5 мл МеОН, после чего смесь перемешивали при комнатной температуре в течение 16 ч. Смесь отфильтровали (целит), промыли МеОН (10 мл) и EtOAc (10 мл) и фильтрат сконцентрировали в темное масло. Остаток фракционировали между Et2O-гексаном (3:1, 50 мл) и водой (25 мл). Осажденные твердые вещества растворили путем добавления 1 М HCl (примерно 10 мл), затем экстрагировали водную фазу Et2Oгексаном (3:1, 350 мл). Объединенные органические фазы промыли насыщенным водным раствором(29%) непрореагировавшего 2,5-бис-метоксиметилфурана. Последующим элюированием 2-15% EtOAcDCM было получено искомое соединение (667 мг, 51%, 72% по отношению к восстановленному исходному материалу) в виде бесцветного масла. 1THF при -78 С в атмосфере аргона добавили раствор хлорида изопропилмагния (2,0 М в THF, 321 мкл,0,641 ммоль): реакционную смесь нагрели до комнатной температуры и перемешивали в течение 75 мин,а затем вновь охладили до -78 С. К смеси добавили раствор трет-бутиллития (1,7 М в пентане, 900 мкл,1,53 ммоль) и после перемешивания в течение 20 мин добавили раствор 1,5-бис-метоксиметил-8-оксабицикло[3.2.1]окт-б-ен-3-она (полученного на предыдущей стадии, 141 мг, 0,664 ммоль) в 3,5 мл THF в течение 1,5 мин. Смесь перемешивали при -78 С в течение 30 мин, а затем при комнатной температуре в течение 16 ч. Реакционную смесь погасили 4 мл насыщенного водного раствора NH4Cl, прилили к EtOAc(50 мл), промыли водой (10 мл) и рассолом (10 мл), высушили (Na2SO4) и сконцентрировали, в результате чего было получено 292 мг твердого вещества. Приготовили взвесь полученного осадка в 4 мл MeCN,отфильтровали, промыли MeCN (21 мл), а фильтрат сконцентрировали, что позволило получить 230 мг твердого вещества. Хроматография на 20-г силикагелевой SPE колонке (10-60% EtOAc-DCM) дала стеклообразное вещество, которое после концентрирования от EtOAc-гексана (1:1) позволило получить искомое соединение (32,4 мг, 12%) в виде твердого вещества белого цвета. 1H-ЯМР (CDCl3; 400 МГц)9,62 (с, 1 Н), 8,37 (д, 1 Н, J=8,6 Гц), 7,69 (с, 1 Н), 7,53 (дд, 1 Н, J=8,6, 2,3 Гц), 7,35 (д, 1 Н, J=2,3 Гц), 6,43 (с, 2 Н), 5,74-5,78 (м, 1 Н), 3,59 (с, 4 Н), 3,40 (с, 6 Н), 2,40 (д, 2 Н, J=14,7 Гц),2,25-2,33 (м, 2 Н), 2,08-2,11 (м, 2 Н), 1,96 (д, 2 Н, J=14,7 Гц), 1,58 (т, 2 Н, J=6,2 Гц), 1,10 (с, 6 Н). Масс-спектрометрия: (ESI, m/z): рассчитано для C30H36N4O5, 515,3 (M-Н 2 О+Н), получено 515,0. Определение относительной стереохимической конфигурации было проведено на основе спектров Искомое соединение приготовили по способу примера 1, стадия (е), с использованием [4-бромо-2(4,4-диметилциклогекс-1-енил)фенил]амида 4-циано-1 Н-имидазол-2-карбоновой кислоты (полученного в примере 1, стадия (в), 299 мг, 0,749 ммоль) и 1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-оксабицикло [3.2.1] окт-6-ен-3-она (Lee, K. and Cha, J.K., J. Amer. Chem. Soc, 123: 5590-5591 (2001), 309 мг,0,749 ммоль). Хроматография на силикагеле (1-3% EtOAc/DCM) позволила получить искомое соединение (154 мг, 28%) в виде твердого вещества белого цвета. Масс-спектрометрия: (ESI, m/z): рассчитано для C40H60N4O5Si2, 715,4 (М-Н 2 О+Н), получено: 715,0. б)[4-[(3-эндо)-3-гидрокси-1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-оксабицикло[3.2.1]окт-6-ен-3-ил]-2-(4,4-диметилциклогекс-1-енил)фенил]амида 4-циано-1 Н-имидазол-2 карбоновой кислоты (полученного на предыдущей стадии, 125 мг, 0,171 ммоль) и тетрабутиламмония фторида моногидрата (TBAFH2O) (357 мг, 1,36 ммоль) в 3 мл THF перемешивали при 60 С в течение 1 ч. После охлаждения до комнатной температуры смесь обработали EtOAc (50 мл) и промыли Н 2 О (10 мл), насыщенным водным раствором NH4Cl (210 мл) и рассолом (10 мл). Органическую фазу осушили над Na2SO4 и сконцентрировали в вакууме. Полученный остаток растерли в порошок с DCM и в результате получили искомое соединение (72 мг, 84%) в виде твердого вещества белого цвета. 1(д, 2 Н, J=14,7 Гц), 1,59 (т, 2 Н, J=6,3 Гц), 1,09 (с, 6 Н). Определение относительной стереохимической конфигурации было проведено по аналогии с [2(4,4-диметилциклогекс-1-енил)-4-[(3-экзо)-3-гидрокси-1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-6 ен-3-ил]фенил]амидом 4-циано-1 Н-имидазол-2-карбоновой кислоты (полученным в примере 1, стадия К смеси [2-(4,4-диметилциклогекс-1-енил)-4-[(3-эндо)-3-гидрокси-1,5-бис-гидроксиметил-8-оксабицикло[3.2.1]окт-6-ен-3-ил]фенил]амида 4-циано-1 Н-имидазол-2-карбоновой кислоты (полученного в примере 2, стадия (б), 40,0 мг, 0,0793 ммоль) в 1 мл DCM при 0 С по каплям добавили трифторуксусную кислоту (TFA) (50 мкл). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель отогнали в вакууме, а остаток очистили посредством хроматографии на силикагеле (2-5%MeOH/DCM), что дало искомое соединение (37 мг, 95%) в виде твердого вещества белого цвета. 1 Н-ЯМР (CD3OD; 400 МГц)8,18 (д, 1 Н, J=8,6 Гц), 7,98 (с, 1 Н), 7,32 (дд, 1 Н, J=8,6, 2,3 Гц), 7,21 (д,1 Н, J=2,3 Гц), 6,51 (уш с, 1 Н), 6,41 (д, 1 Н, J=5,8 Гц), 5,96 (д, 1 Н, J=5,8 Гц), 5,73 (м, 1 Н), 3,77-3,88 (м, 4 Н),2,69 (дд, 1 Н, J=17,7, 2,0 Гц), 2,30 (м, 2 Н), 2,17 (дд, 2 Н, J=17,7, 1,7 Гц), 2,07 (м, 2 Н), 1,59 (т, 2 Н, J=6,3 Гц),- 18018936 Раствор 1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окт-6-ен-3-она (Lee,K. and Cha, J.K., J. Amer. Chem. Soc, 123: 5590-5591 (2001), 929 мг, 2,25 ммоль) в 10 мл THF добавили к раствору LHMDS (гексаметилдисилазид лития) (1,0 M в THF, 2,48 мл, 2,48 ммоль) в 20 мл THF при -78 С в атмосфере аргона. Смесь нагрели до комнатной температуры и перемешивали в течение 0,5 ч, затем вновь охладили до -78 С. Затем добавили раствор 2-[N,N-бис-(трифторметансульфонил)амино]пиридина(888 мг, 2,48 ммоль) в 10 мл THF. Полученную смесь нагрели до комнатной температуры и перемешивали в течение 2 ч в атмосфере аргона. После обработки 10 мл насыщенного водного раствора NH4Cl, а затем 100 мл EtOAc, смесь промыли насыщенным водным раствором лимонной кислоты (320 мл), Н 2 О(20 мл), рассолом (10 мл) и высушили (Na2SO4): растворитель отогнали при пониженном давлении и получили 1,22 г искомого соединения в виде масла светло-желтого цвета. 1H-ЯМР (CDCl3; 400 МГц)6,42 (д, 1H, J=5,8 Гц), 6,29 (уш с, 1H), 5,91 (д, 1H, J=5,8 Гц), 3,80 (с,1H), 2,81 (дд, 2 Н, J=17,7, 1,9 Гц), 2,13 (дд, 1 Н, J=17,7, 1,3 Гц), 0,91 (с, 18 Н), 0,08 (с, 12 Н). Продукт использовали на следующей стадии без дополнительной очистки. б) 4-[1,5-бис-(трет-Бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окта-2,6-диен-3 ил]фениламин К смеси 1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окта-2,6-диен-3 илового эфира трифторметансульфоновой кислоты (полученного на предыдущей стадии, 1,22 г, 2,24 ммоль), Pd(PPh3)4 (259 мг, 0,224 ммоль) и 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фениламина(540 мг, 2,46 ммоль) в 20 мл 1,4-диоксана добавили 2,0 М водный раствор Na2CO3 (9,0 мл, 18 ммоль). Полученную смесь перемешивали при 80 С в течение 2 ч, а затем охладили до комнатной температуры. После обработки 100 мл EtOAc смесь промыли H2O (320 мл), рассолом (20 мл) и высушили (Na2SO4): растворитель отогнали при пониженном давлении, после чего провели флэш-хроматографию остатка на силикагеле (1:1 гексан/DCM-DCM) и получили 802 мг (73% для двух стадий) искомого соединения в виде масла светло-коричневого цвета. Масс-спектрометрия: (ESI, m/z): рассчитано для C27H45NO3Si2, 488,3 (М+Н), получено: 488,4. в) 4-[(3-Экзо)-1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окт-3 ил]фениламин Раствор 4-[1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окта-2,6-диен-3 ил]фениламина (полученного на предыдущей стадии, 500 мг, 1,03 ммоль) и 5% Rh/Al2O3 (250 мг, 50 вес.%) в 20 мл МеОН перемешивали при комнатной температуре в присутствии Н 2 (давление камеры) в течение 2 ч. Родиевый (Rh) катализатор выделили фильтрованием на целите (Celite), а фильтрат сконцен- 19018936 трировали в вакууме, что дало 500 мг 4-[1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-оксабицикло[3.2.1]окт-2-ен-3-ил]фениламина в виде масла светло-коричневого цвета. Масс-спектрометрия: (ESI, m/z): рассчитано для C27H47NO3Si2, 490,3 (М+Н), получено: 490,1. Смесь 4-[1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окт-2-ен-3 ил]фениламина (полученного на предыдущей стадии, 500 мг, 1,02 ммоль) и 10% Pd/C (250 мг, 50 вес.%) в 25 мл МеОН перемешивали при комнатной температуре в присутствии Н 2 (344,7 кПа (50 фунт/кв.дюйм в течение 1 ч. Палладиевый катализатор выделили фильтрованием на целите (Celite), а фильтрат сконцентрировали; в результате получили 492 мг (98%) искомого соединения - смесь 2:1 (А:В) в виде масла светло-коричневого цвета. Масс-спектрометрия: (ESI, m/z): рассчитано для C27H49NO3Si2, 492,3(М+Н), получено: 492,4. Определение относительной стереохимической конфигурации было проведено на последней стадии К раствору 4-[(3-экзо)-1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окт-3 ил]фениламина и 4-[(3-эндо)-1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окт-3 ил]фениламина (полученного на предыдущей стадии, 492 мг, 1,00 ммоль) в 10 мл 3:1 DCM/MeCN при 0 С добавили N-бромосукцинимид (NBS) (178 мг, 1,00 ммоль) тремя порциями в течение 5 мин. Смесь нагрели до комнатной температуры и перемешивали в течение 1 ч в атмосфере аргона. Растворитель выпарили в вакууме, а остаток очистили флэш-хроматографией на силикагеле (1:1 гекан/DCM) и получили 345 мг (60%) искомого соединения А в виде масла светло-коричневого цвета и 172 мг (30%) искомого соединения В в виде масла светло-коричневого цвета. А: масс-спектрометрия: (ESI, m/z): рассчитано для C27H48BrNO3Si2, 570,2 (M+Н), получено: 570,1. В: масс-спектрометрия: (ESI, m/z): рассчитано для C27H48BrNO3Si2, 570,2 (М+Н), получено: 570,0. Определение относительной стереохимической конфигурации было проведено на последней стадии К смеси 4-[(3-экзо)-1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окт-3-ил]2-бромофениламина (полученного на предыдущей стадии, 343 мг, 0,600 ммоль), 4,4 диметилциклогексен-1-ил бороновой кислоты (156 мг, 0,660 ммоль) и Pd(PPh3)4 (69 мг, 0,060 ммоль) в 5 мл 1,4-диоксана добавили 2,0 М водного раствора Na2CO3 (2,4 мл, 4,8 ммоль). Полученную смесь перемешивали при 80 С в течение 16 ч в атмосфере аргона, а затем охладили до комнатной температуры. Смесь, обработанную 50 мл EtOAc, промыли Н 2 О (210 мл), рассолом (10 мл) и высушили (Na2SO4): растворитель отогнали при пониженном давлении, затем очистили смесь посредством флэш-хроматографии на силикагеле (DCM) и получили 317 мг (88%) искомого соединения в виде твердого вещества светлокоричневого цвета. Масс-спектрометрия: (ESI, m/z): рассчитано для C35H61NO3Si2, 600,4 (M+Н), получено: 600,5. Определение относительной стереохимической конфигурации было проведено на последней стадии К смеси калиевой соли 4-циано-1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-карбоновой кислоты (полученной согласно патентной заявке США 2006189623 А 1, 192 мг, 0,630 ммоль) и пиридина(51,0 мкл, 0,630 ммоль) в 3 мл DCM при 0 С добавили SOCl2 (46,0 мкл, 0,630 ммоль). После перемешивания при 0 С в течение 0,5 ч в атмосфере аргона полученную смесь нагрели до комнатной температуры и добавили к раствору 4-[(3-экзо)-1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-оксабицикло[3.2.1]окт-3-ил]-2-(4,4-диметилциклогекс-1-енил)фениламина (полученного на предыдущей стадии, 315 г, 0,525 ммоль) в 2 мл DCM при 0C. После перемешивания при 0 С в течение 2 ч в атмосфере аргона реакционную смесь нагрели до комнатной температуры. Полученную смесь обработали 50 млEtOAc, а затем промыли Н 2 О (10 мл), 10% водным раствором лимонной кислоты (10 мл), водным насыщенным раствором NaHCO3 (10 мл) и рассолом (20 мл). Органическую фазу высушили над Na2SO4 и сконцентрировали в вакууме. Полученный остаток очистили хроматографией на силикагеле (2-5%EtOAc/гексан), что позволило получить искомое соединение (401 мг, 90%) в виде масла светлокоричневого цвета. 1 Н-ЯМР (CDCl3; 400 МГц)9,72 (с, 1 Н), 8,29 (д, 1 Н, J=8,6 Гц), 7,76 (с, 1 Н), 7,18 (дд, 1 Н, J=8,6, 2,3 Гц), 7,07 (д, 1 Н, J=2,3 Гц), 5,96 (с, 2 Н), 5,76 (м, 1 Н), 3,59-3,68 (м, 6 Н), 3,02 (м, 1 Н), 2,29 (м, 2 Н), 2,09 (м,2 Н), 1,76-1,88 (м, 6 Н), 1,66 (т, 2 Н, J=12,7 Гц), 1,59 (т, 2 Н, J=6,3 Гц), 1,11 (с, 6 Н), 0,97 (т, 2 Н, J=8,3 Гц),0,89 (с, 18 Н), 0,05 (с, 12 Н), 0,01 (с, 9 Н). Определение относительной стереохимической конфигурации было проведено на последней стадии(ж). ж) [4-3-Экзо)-1,5-бис-Гидроксиметил-8-окса-бицикло[3.2.1]окт-3-ил)-2-(4,4-диметилциклогекс-1 енил)фенил]амид 4-циано-1 Н-имидазол-2-карбоновой кислоты. Смесь [4-[(3-экзо)-1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окт-3-ил]2-(4,4-диметилциклогекс-1-енил)фенил]амида 4-циано-1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол 2-карбоновой кислоты (полученного на предыдущей стадии, 400 мг, 0,471 ммоль) и моногидрата фторида тетрабутиламмония (739 мг, 2,83 ммоль) в 5 мл THF перемешивали при 50 С в течение 16 ч. После охлаждения до комнатной температуры смесь обработали EtOAc (50 мл) и промыли Н 2 О (10 мл), насыщенным водным раствором NH4Cl (210 мл) и рассолом (10 мл). Органическую фазу осушили надNa2SO4 и сконцентрировали в вакууме. Остаток перемололи с DCM и получили искомое соединение (203 мг, 88%) в виде твердого вещества белого цвета. 1(м, 2 Н), 1,84-1,97 (м, 4 Н), 1,63-1,71 (м, 4 Н), 1,59 (т, 2 Н, J=6,3 Гц), 1,08 (с, 6 Н). Масс-спектрометрия: (ESI, m/z): рассчитано для C28H34N4O4, 491,3 (М+Н), получено: 491,1. Определение относительной стереохимической конфигурации было проведено на основе спектров Искомое соединение приготовили по способу примера 4, стадия (д), с использованием 4-[(3-эндо)1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окт-3-ил]-2-бромофениламина (В)(полученного в примере 4, стадия (г), 171 мг, 0,300 ммоль) и 4,4-диметилциклогексен-1-ил бороновой кислоты (77,9 мг, 0,330 ммоль). Хроматография на силикагеле (DCM) позволила получить искомое соединение (165 мг, 92%) в виде масла светло-коричневого цвета. Масс-спектрометрия: (ESI, m/z): рассчитано для C35H61NO3Si2, 600,4 (М+Н), получено: 600,5. Определение относительной стереохимической конфигурации было проведено на последней стадии Искомое соединение приготовили по способу примера 4, стадия (е), с использованием 4-[(3-эндо)1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окт-3-ил]-2-(4,4-диметилциклогекс 1-енил)фениламина (полученного на предыдущей стадии, 150 мг, 0,250 ммоль) и калиевой соли 4-циано 1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-карбоновой кислоты (полученной согласно Патентной заявке США 2006189623 А 1, 92 мг, 0,300 ммоль). Хроматография на силикагеле (2-5% EtOAc/гексан) позволила получить искомое соединение (187 мг, 88%) в виде масла светло-коричневого цвета. 1(м, 2 Н), 2,14 (дд, 1 Н, J=13,8, 6,7 Гц), 2,09 (м, 2 Н), 1,77-1,83 (м, 2 Н), 1,56-1,67 (м, 6 Н), 1,11 (с, 6 Н), 0,97 (т,2 Н, J=8,3 Гц), 0,90 (с, 18 Н), 0,07 (с, 6 Н), 0,06 (с, 6 Н), 0,005 (с, 9 Н). Определение относительной стереохимической конфигурации было проведено на последней стадии(в). в) [4-3-Эндо)-1,5-бис-гидроксиметил-8-окса-бицикло[3.2.1]окт-3-ил)-2-(4,4-диметилциклогекс-1 енил)фенил]амид 4-циано-1 Н-имидазол-2-карбоновой кислоты. Искомое соединение приготовили по способу примера 4, стадия (ж), с использованием [4-[(3-эндо)1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]окт-3-ил]-2-(4,4-диметилциклогекс 1-енил)фенил]амида 4-циано-1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-карбоновой кислоты(342 мг, 1,31 ммоль). Хроматография на силикагеле (1-5% MeOH/DCM) позволила получить искомое соединение (100 мг,93%) в виде масла светло-коричневого цвета. 1H-ЯМР (CD3OD; 400 МГц)8,10 (д, 1 Н, J=8,6 Гц), 7,98 (с, 1 Н), 7,21 (дд, 1 Н, J=8,6, 2,0 Гц), 7,10 (д,1 Н, J=2,0 Гц), 5,72 (м, 1 Н), 3,63 (д, 2 Н, J=11,6 Гц), 3,50 (д, 2 Н, J=11,6 Гц), 2,91 (м, 1 Н), 2,30 (м, 2 Н), 1,932,09 (м, 6 Н), 1,55-1,73 (м, 6 Н), 1,08 (с, 6 Н). Масс-спектрометрия: (ESI, m/z): рассчитано для C28H34N4O4, 491,3 (М+Н), получено: 491,1. Определение относительной стереохимической конфигурации было проведено на основе спектров Искомое соединение приготовили по способу примера 4, стадия (а), с использованием 1,5-бисметоксиметил-8-окса-бицикло[3.2.1]окт-6-ен-3-она (полученного в примере 1, стадия (д), 600 мг, 2,80 ммоль) и 2-[N,N-бис-(трифторметансульфонил)амино]пиридина (1,10 г, 3,08 ммоль). Искомое соединение (921 мг, 95%) представляет собой масло светло-коричневого цвета. Масс-спектроскопия: (ESI, m/z): рассчитано для C12H15F3O6S, 345,0 (М+Н), получено: 344,9. б) 4-(1,5-бис-Метоксиметил-8-окса-бицикло[3.2.1]окта-2,6-диен-3-ил)фениламин Искомое соединение приготовили по способу примера 4, стадия (б), с использованием 1,5-бисметоксиметил-8-окса-бицикло[3.2.1]окта-2,6-диен-3-илового эфира трифторметансульфоновой кислоты(полученного на предыдущей стадии, 3,25 г, 9,45 ммоль) и 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2 ил)фениламина (2,28 г, 10,4 ммоль). Хроматография на силикагеле (5-10% EtOAc/DCM) позволила получить искомое соединение (2,19 г, 81%) в виде масла светло-коричневого цвета. Масс-спектроскопия: (ESI, m/z): рассчитано для C11H21NO3, 288,2 (М+Н), получено: 288,2. в) 4-[(3-Эндо)-1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил]фениламин (А) и 4-[(3-экзо)1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил]фениламин (В) Искомое соединение приготовили по способу примера 4, стадия (в), с использованием 4-(1,5-бисметоксиметил-8-окса-бицикло[3.2.1]окта-2,6-диен-3-ил)фениламина (полученного на предыдущей стадии, 2,00 г, 6,96 ммоль), 5% Rh/Al2O3 (800 мг, 40 вес.%) и 10% Pd/C (800 мг, 40 вес.%). Хроматография на силикагеле (0-1% MeOH/DCM) позволила получить 601 мг (44%) искомого соединения А в виде масла светло-коричневого цвета и 672 мг (33%) искомого соединения В в виде масла светло-коричневого цвета. А: масс-спектроскопия: (ESI, m/z): рассчитано для C17H25NO3, 292,2 (М+Н), получено: 292,2. В: масс-спектроскопия: (ESI, m/z): рассчитано для C17H25NO3, 292,2 (М+Н), получено: 292,2. Определение относительной стереохимической конфигурации было проведено на последней стадии Искомое соединение приготовили по способу примера 4, стадия (г), с использованием 4-[(3-эндо)1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил]фениламина (полученного на предыдущей стадии,292 мг, 1,00 ммоль), NBS (178 мг, 1,00 ммоль). Хроматография на силикагеле (0-10% EtOAc/DCM) позволила получить 185 мг (50%) искомого соединения в виде масла светло-коричневого цвета. Масс- 23018936 спектроскопия: (ESI, m/z): рассчитано для C17H24BrNO3, 370,1 (М+Н), получено: 370,1. Определение относительной стереохимической конфигурации было проведено на последней стадии Искомое соединение приготовили по способу примера 4, стадия (д), с использованием 4-[(3-эндо)1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил]-2-бромофениламина (полученного на предыдущей стадии, 185 мг, 0,500 ммоль), 4,4-диметилциклогексен-1-ил бороновой кислоты (130 мг, 0,550 ммоль). Хроматография на силикагеле (0-15% EtOAc/DCM) позволила получить 150 мг (75%) искомого соединения в виде масла светло-желтого цвета. Масс-спектроскопия: (ESI, m/z): рассчитано для C25H37NO3, 400,3 (М+Н), получено: 400,4. Определение относительной стереохимической конфигурации было проведено на последней стадии Искомое соединение приготовили по способу примера 4, стадия (е), с использованием 4-[(3-эндо)1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил]-2-(4,4-диметилциклогекс-1-енил)фениламина (полученного на предыдущей стадии, 140 мг, 0,350 ммоль) и калиевой соли 4-циано-1-(2 триметилсиланилэтоксиметил)-1 Н-имидазол-2-карбоновой кислоты (полученной согласно патентной заявке США 2006189623 А 1, 128 мг, 0,420 ммоль). Хроматография на силикагеле (5% EtOAc/DCM) позволила получить 164 мг (72%) искомого соединения в виде масла светло-желтого цвета. Масс-спектроскопия: (ESI, m/z): рассчитано для C36H52N4O5Si, 649,4 (М+Н), получено: 649,1. Определение относительной стереохимической конфигурации было проведено на последней стадии(ж). ж) [4-3-Эндо)-1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил)-2-(4,4-диметилциклогекс-1 енил)фенил]амид 4-циано-1 Н-имидазол-2-карбоновой кислоты. Искомое соединение приготовили по способу примера 4, стадия (ж), с использованием [4-[(3-эндо)1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил]-2-(4,4-диметилциклогекс-1-енил)фенил]амида 4 циано-1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-карбоновой кислоты (полученного на предыдущей стадии, 150 мг, 0,231 ммоль) и моногидрата фторида тетрабутиламмония (181 мг, 0,693 ммоль). Хроматография на силикагеле (25% EtOAc/DCM) позволила получить искомое соединение (102 мг, 85%) в виде твердого вещества белого цвета. Масс-спектроскопия: (ESI, m/z): рассчитано для C12H13BrN2O, 281,0 (М+Н), получено: 281,2. 1(м, 2 Н), 2,17 (дд, 1 Н, J=13,5, 6,7 Гц), 2,06 (м, 2 Н), 1,80-1,89 (м, 2 Н), 1,67-1,76 (м, 2 Н), 1,58-1,64 (м, 2 Н),1,58 (т, 2 Н, J=6,3 Гц), 1,08 (с, 6 Н). Масс-спектроскопия: (ESI, m/z): рассчитано для C30H38N4O4, 519,3 (М+Н), получено: 519,0. Определение структуры было проведено по аналогии с [4-[(3-эндо)-1,5-бис-гидроксиметил-8-оксабицикло[3.2.1]окт-3-ил]-2-(4,4-диметилциклогекс-1-енил)фенил]амидом 4-циано-1 Н-имидазол-2 карбоновой кислоты (полученным в примере 5, стадия (в. Искомое соединение получили по способу примера 4, стадия (г), с использованием 4-3-экзо)-1,5 бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил)фениламина (полученного в примере 6, стадия (в), 292 мг, 1,00 ммоль), NBS (178 мг, 1,00 ммоль). Хроматография на силикагеле (0-10% EtOAc/DCM) позволила получить 185 мг (50%) искомого соединения в виде масла светло-коричневого цвета. Масс-спектроскопия (ESI, m/z): рассчитано для C17H24BrNO3, 370,1 (М+Н), получено 370,2. Определение относительной стереохимической конфигурации проводили на последней стадии (г). б) 4-[(3-Экзо)-1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил]-2-(4,4-диметилциклогекс-1 енил)фениламин Искомое соединение получили по способу примера 4, стадия (д), с использованием 4-[(3-экзо)-1,5 бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил]-2-бромфениламина (полученного на предыдущей стадии, 185 мг, 0,500 ммоль), 4,4-диметилциклогексен-1-ил-бороновой кислоты (130 мг, 0,550 ммоль). Хроматографией на силикагеле (0-15% EtOAc/DCM) выделили 156 мг (78%) искомого соединения в виде масла светло-желтого цвета. Масс-спектроскопия (ESI, m/z): рассчитано для C25H37NO3, 400,3 (М+Н), получено 400,3. Определение относительной стереохимической конфигурации проводили на последней стадии (г). в) Искомое соединение получили по способу примера 4, стадия (е), с использованием 4-[(3-экзо)-1,5 бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил]-2-(4,4-диметилциклогекс-1-енил)фениламина (полученного на предыдущей стадии, 140 мг, 0,350 ммоль) и калиевой соли 4-циано-1-(2 триметилсиланилэтоксиметил)-1 Н-имидазол-2-карбоновой кислоты (полученной согласно Патентной заявке США 2006189623 А 1, 128 мг, 0,420 ммоль). Хроматографией на силикагеле (5% EtOAc/DCM) выделили 166 мг (73%) искомого соединения в виде масла светло-желтого цвета. Масс-спектроскопия (ESI, m/z): рассчитано для C36H52N4O5Si, 649,4 (М+Н), получено 649,1. Определение относительной стереохимической конфигурации проводили на последней стадии (г). г)[4-3-Экзо)-1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил)-2-(4,4-диметилциклогекс-1 енил)фенил]амид 4-циано-1 Н-имидазол-2-карбоновой кислоты. Искомое соединение получили по способу примера 4, стадия (ж), с использованием [4-3-экзо)-1,5 бис-метоксиметил-8-окса-бицикло[3.2.1]окт-3-ил)-2-(4,4-диметилциклогекс-1-енил)фенил]амида 4- 25018936 циано-1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-карбоновой кислоты (полученного на предыдущей стадии, 150 мг, 0,231 ммоль) и моногидрата фторида тетрабутиламмония (181 мг, 0,693 ммоль). Хроматографией на силикагеле (25% EtOAc/DCM) выделили искомое соединение (86,4 мг, 72%) в виде твердого вещества белого цвета. 1(м, 2 Н), 2,07 (м, 2 Н), 1,88-1,96 (м, 2 Н), 1,76-1,85 (м, 2 Н), 1,65-1,76 (м, 4 Н), 1,59 (т, 2 Н, J=6,3 Гц), 1,08 (с,6 Н). Масс-спектроскопия (ESI, m/z): рассчитано для C30H38N4O4, 519,3 (М+Н), получено 519,1. Определение структуры проводили по аналогии с [4-[(3-экзо)-1,5-бис-гидроксиметил-8-оксабицикло[3.2.1]окт-3-ил]-2-(4,4-диметилциклогекс-1-енил)фенил]амидом 4-циано-1 Н-имидазол-2 карбоновой кислоты (полученным в примере 4, стадия (ж. Пример 8. [2-(4,4-Диметилциклогекс-1-енил)-4-[(3-экзо)-3-гидрокси-1,5-бис-гидроксиметил-8-оксабицикло[3.2.1]окт-3-ил]фенил]амид 4-циано-1 Н-имидазол-2-карбоновой кислоты Смесь 1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-окса-бицикло[3.2.1]-окт-6-ен-3-она (200 мг,4,84 ммоль) (Lee, K. and Cha, J.K., J. Amer. Chem. Soc, 123:5590-5591 (2001 и 5% Pd/C (30 мг) в 10 мл МеОН перемешивали при комнатной температуре в присутствии H2 (давление камеры) в течение 8 ч. Палладиевый катализатор отделили фильтрацией на целите, фильтрат сконцентрировали и получили 200 мг (100%) искомого соединения в виде бесцветного масла. Масс-спектроскопия (ESI, m/z): расчет для C21H42O4Si2, 415,2 (M+Н), обнаружено 415,1. б) К суспензии [4-бром-2-(4,4-диметилциклогекс-1-енил)фенил]амида 4-циано-1 Н-имидазол-2 карбоновой кислоты (125 мг, 0,314 ммоль) (полученного в примере 1, стадия (в в 2 мл THF при температуре -40 С добавили раствор изо-PrMgCl (2 М THF, 0,392 мл, 0,785 ммоль) и дали смеси нагреться до комнатной температуры. Через 10 мин прозрачный раствор охладили до температуры -78 С и добавили раствор трет-BuLi (1,7 M в пентане, 0,554 мл, 0,942 ммоль). После выдержки в течение 15 мин при температуре-78 С добавили раствор 1,5-бис-(трет-бутилдиметилсиланилоксиметил)-8-оксабицикло[3.2.1]октан-3-она (200 мг, 0,482 ммоль) (полученного на предыдущей стадии) в THF (2 мл), полученную смесь перемешивали в течение 30 мин при температуре -78 С, затем дали смеси нагреться до комнатной температуры и перемешивали в течение еще 5 мин. Реакционную смесь погасили насыщенным раствором NH4Cl (10 мл) и экстрагировали EtOAc (310 мл). Органическую фазу высушили(Na2SO4) и сконцентрировали, искомое соединение очистили на силикагеле, используя в качестве элюента 10% EtOAc/DCM, и получили 116 мг (51%) твердого вещества белого цвета. Масс-спектроскопия (ESI, m/z): рассчитано для C40H62N4O5Si2, 717,4 (М+Н-Н 2 О), получено 717,1. Определение относительной стереохимической конфигурации проводили на последней стадии (в). в)TBAFН 2 О (150 мг, 5,75 ммоль) и полученную смесь перемешивали при температуре 60 С в течение 8 ч. Затем реакционную смесь разбавили EtOAc (10 мл), промыли Н 2 О (210 мл) и рассолом (10 мл), высушили над Na2SO4 и сконцентрировали. Полученное твердое вещество растерли с Et2O и отфильтровали,получив 50 мг (69%) белого твердого вещества. 1(с, 6 Н). Масс-спектроскопия (ESI, m/z): рассчитано для C28H34N4O5, 507,2 (М+Н), получено 507,1. Определение относительной стереохимической конфигурации проводили по аналогии с [2-(4,4 диметилциклогекс-1-енил)-4-[(3-экзо)-3-гидрокси-1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-6-ен 3-ил]фенил]амидом 4-циано-1 Н-имидазол-2-карбоновой кислоты (полученным в примере 1, стадия (е. Пример 9. [2-(4,4-Диметилциклогекс-1-енил)-4-3-экзо)-3-гидрокси-1,5-диметил-8-окса-бицикло Искомое соединение получили по способу примера 1, стадия (е), с использованием [4-бром-2-(4,4 диметилциклогекс-1-енил)фенил]амида 4-циано-1 Н-имидазол-2-карбоновой кислоты (полученного в примере 1, стадия (в и 1,5-диметил-8-окса-бицикло[3.2.1]окт-6-ен-3-она (Chemistry-A European Journal(дд, 1 Н, J=8,5, 2,3 Гц), 7,33 (д, 1 Н, J=2,3 Гц), 6,28 (с, 2 Н), 5,77 (уш с, 1 Н), 3,27 (с, 1 Н), 2,22-2,32 (м, 4 Н),1,94-2,10 (м, 4 Н), 1,45 (с, 6 Н), 1,29 (м, 2 Н), 1,10 (с, 6 Н). Масс-спектроскопия (ESI, m/z): рассчитано для C28H32N4O3, 473,2 (М+Н), получено 473,1. Определение относительной стереохимической конфигурации проводили по аналогии с [2-(4,4 диметилциклогекс-1-енил)-4-[(3-экзо)-3-гидрокси-1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-6-ен 3-ил]фенил]амидом 4-циано-1 Н-имидазол-2-карбоновой кислоты (полученным в примере 1, стадия (е. Пример 10. Метиловый эфир 3-(3-экзо)-[4-[(4-циано-1 Н-имидазол-2-карбонил)амино]-3-(4,4 диметилциклогекс-1-енил)фенил]-(3-экзо)-3-гидрокси-1,5-диметил-8-аза-бицикло[3.2.1]окт-6-ен-8-карбоновой кислоты Искомое соединение получили из метилового эфира 2,5-диметилпиррол-1-карбоновой кислоты (патент США 4551540) и 1,1,3,3-тетрабромацетона, используя реакцию [4+3] циклоприсоединения Кима и Хоффмана (European Journal of Organic Chemistry (2000), (12), 2195-2201). Масс-спектроскопия (ESI, m/z): рассчитано для C11H15NO3, 210,1 (M+H), получено 210,0. б) Метиловый эфир 3-(3-экзо)-[4-[(4-циано-1 Н-имидазол-2-карбонил)амино]-3-(4,4 диметилциклогекс-1-енил)фенил]-(3-экзо)-3-гидрокси-1,5-диметил-8-аза-бицикло[3.2.1]окт-6-ен-8-карбоновой кислоты. Искомое соединение получили по способу примера 1, стадия (е), с использованием [4-бром-2-(4,4 диметилциклогекс-1-енил)фенил]амида 4-циано-1 Н-имидазол-2-карбоновой кислоты (полученного в примере 1, стадия (в и метилового эфира 1,5-диметил-3-оксо-8-аза-бицикло[3.2.1]окт-6-ен-8 карбоновой кислоты (полученного на предыдущей стадии). Н-ЯМР (CDCl3; 400 МГц)9,60 (с, 1 Н), 8,16 (д, 1 Н, J=8,6 Гц), 7,63 (с, 1 Н), 7,17 (дд, 1 Н, J=8,6, 2,0 Гц), 7,12 (д, 1 Н, J=2,0 Гц), 5,98 (с, 2 Н), 5,68 (уш с, 1 Н), 3,69 (с, 3 Н), 3,24 (уш с, 1 Н), 2,45 (д, 2 Н, J=15,2 Гц), 2,20 (м, 2 Н), 2,00 (м, 2 Н), 1,81 (д, 2 Н, J=15,2 Гц), 1,59 (с, 6 Н), 1,48 (м, 2 Н), 1,03 (с, 6 Н). Масс-спектроскопия (ESI, m/z): рассчитано для C30H35N5O4, 530,2 (М+Н), получено 530,2. Определение относительной стереохимической конфигурации проводили по аналогии с [2-(4,4 диметилциклогекс-1-енил)-4-[(3-экзо)-3-гидрокси-1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-6-ен 3-ил]фенил]амидом 4-циано-1 Н-имидазол-2-карбоновой кислоты (полученным в примере 1, стадия (е. Пример 11. [2-(4,4-Диметилциклогекс-1-енил)-4-[(3-экзо)-3-гидрокси-1,5-диметил-6-экзо,7-экзо(диметилметилендиокси)бицикло[3.2.1]окт-3-ил]фенил]амид 4-циано-1 Н-имидазол-2-карбоновой кислоты Искомое соединение получили по способу примера 1, стадия (е), с использованием [4-бром-2-(4,4 диметилциклогекс-1-енил)фенил]амида 4-циано-1 Н-имидазол-2-карбоновой кислоты (полученного в примере, стадия (в и 1,4,4,7-тетраметил-3,5,11-триокса-трицикло [5.3.1.02,6] ундекан-9-она (полученного по схеме, описанной в Bulletin of the Chemical Society of Japan (1983), 56 (9), 2680-99). 1H-ЯМР (CDCl3; 400 МГц)12,65 (с, 1 Н), 9,53 (с, 1 Н), 8,43 (д, 1H, J=8,8 Гц), 7,61 (дд, 1H, J=8,8, 2,3 Гц), 7,59 (д, 1 Н, J=2,5 Гц), 7,24 (д, 1 Н, J=2,3 Гц), 5,70 (уш с, 1 Н), 4,93 (с, 2 Н), 2,22 (м, 2 Н), 2,03 (м, 2 Н),1,82 (д, 2 Н, J=15,1 Гц), 1,18-1,53 (м, 17 Н), 1,03 (с, 6 Н). Масс-спектроскопия (ESI, m/z): рассчитано для C31H38N4O5, 547,2 (М+Н), получено 547,1. Определение относительной стереохимической конфигурации проводили по аналогии с [2-(4,4 диметилциклогекс-1-енил)-4-[(3-экзо)-3-гидрокси-1,5-бис-метоксиметил-8-окса-бицикло[3.2.1]окт-6-ен 3-ил]фенил]амидом 4-циано-1 Н-имидазол-2-карбоновой кислоты (полученным в примере 1, стадия (е. Пример 12. [2-(4,4-Диметилциклогекс-1-енил)-4-(3-экзо)-3,6-экзо,7-экзо-тригидрокси-1,5-диметил 8-окса-бицикло[3.2.1]окт-3-ил)фенил]амид 4-циано-1 Н-имидазол-2-карбоновой кислотыSociety of Japan (1983), 56(9), 2680-99, 550 мг, 2,95 ммоль) в DCE (20 мл) добавили имидазол (2,0 г, 29 ммоль) и ди-трет-бутилдихлорсилан (1,2 мл, 5,9 ммоль). Полученную смесь перемешивали в атмосфере аргона в течение 48 ч. Затем реакционную смесь обработали насыщенным раствором NaHCO3 (20 мл),органическую фазу (DCE) отделили, высушили (Na2SO4) и сконцентрировали, полученный остаток очистили на силикагеле (5-20% EtOAc/гексан) и выделили искомое соединение (869 мг, 90%). 1 Раствор 4-бром-2-(4,4-диметилциклогекс-1-енил)фениламина (полученного в примере 1, стадия (а),3,0 г, 10 ммоль) в N,N-диметилформамида диметилацетале (20 мл) кипятили с обратным холодильником в атмосфере аргона в течение 48 ч. Растворитель отогнали при пониженном давлении, полученный оста- 28018936 ток очистили на силикагеле (5-20% EtOAc/гексан) и выделили искомое соединение (2,6 г, 73%). 1 Н-ЯМР (CDCl3; 400 МГц)7,36 (с, 1 Н), 7,23-7,21 (м, 2 Н), 6,65 (д, 1 Н, J=8,0 Гц), 5,64 (м, 1 Н), 2,97 К раствору N'-[4-бром-2-(4,4-диметилциклогекс-1-енил)фенил]-N,N-диметилформамидина (полученного на предыдущей стадии, 335 мг, 1,00 ммоль) в THF по каплям добавили BuLi (0,68 мл 1,6 М раствора в гексане, 1,1 ммоль) при температуре -78 С. Полученную смесь перемешивали при температуре -78 С в течение 45 мин, затем в нее по каплям добавили раствор 4,4-ди-трет-бутил-1,7-диметил-3,5,11-триокса-4-сила-трицикло[5.3.1.02,6]ундекан-9-она(полученного в данном примере на стадии (а), 358 мг, 1,1 ммоль) в THF (5 мл). Полученной смеси дали нагреться до комнатной температуры и затем перемешивали в течение 1 ч. Затем реакционную смесь обработали насыщенным раствором NH4Cl и экстрагировали продукт EtOAc (310 мл). Полученные органические фазы (EtOAc) объединили, высушили (Na2SO4) и сконцентрировали. Полученный остаток очистили на силикагеле (20% EtOAc/DCM-100% EtOAc) и выделили искомое соединение (197 мг, 34%). Масс-спектроскопия (ESI, m/z): рассчитано для C34H54N2O4Si, 583,4 (М+Н), получено 583,5. Определение относительной стереохимической конфигурации проводили на последней стадии (е). г) (9-Экзо)-[4-амино-3-(4,4-диметилциклогекс-1-енил)фенил]-4,4-ди-трет-бутил-1,7-диметил-3,5,11 триокса-4-сила-трицикло[5.3.1.02,6]ундекан-9-олN'-[4-(9-экзо)-[4,4-ди-трет-бутил-9-гидрокси-1,7-диметил-3,5,11-триокса-4-силатрицикло[5.3.1.02,6]ундец-9-ил]-2-(4,4-диметилциклогекс-1-енил)фенил]-N,N-диметилформамидина (полученного на предыдущей стадии, 180 мг, 0,309 ммоль) в изопропаноле (0,5 мл) добавили безводный гидразин (0,3 мл). Полученную смесь перемешивали при температуре 40 С в атмосфере аргона в течение ночи. Реакционную смесь сконцентрировали в вакууме, а полученный остаток обработали насыщенным рассолом (10 мл) и экстрагировали продукт EtOAc (310 мл). Полученные органические фазы (EtOAc) объединили, высушили (Na2SO4) и сконцентрировали, полученный остаток очистили на силикагеле (50%EtOAc/гексан-100% EtOAc) и выделили искомое соединение (110 мг, 67%). Масс-спектроскопия (ESI, m/z): рассчитано для C31H49NO4Si, 528,3 (М+Н), получено 528,2. Определение относительной стереохимической конфигурации проводили на последней стадии (е). д)(9-Экзо)-[4-амино-3-(4,4-диметилциклогекс-1-енил)фенил]-4,4-ди-трет-бутил-1,7-диметил-3,5,11 триокса-4-сила-трицикло[5.3.1.02,6]ундекан-9-ол (полученный на предыдущей стадии, 148 мг, 0,28 ммоль) ввели в реакцию кросс-сочетания с калиевой солью 4-циано-1-(2-триметилсиланилэтоксиметил)1 Н-имидазол-2-карбоновой кислоты по способу примера 1, стадия (б), и получили искомое соединение(205 мг, выход 94%). Масс-спектроскопия (ESI, m/z): рассчитано для C42H64N4O6Si2, 777,4 (М+Н), получено 777,8. Определение относительной стереохимической конфигурации проводили на последней стадии (е). е)
МПК / Метки
МПК: C07D 409/14, C07D 471/08, C07D 401/14, A61P 19/02, A61K 31/33, C07D 493/08, C07D 405/14, C07D 405/12
Метки: с-fms, киназы, ингибиторы
Код ссылки
<a href="https://eas.patents.su/30-18936-ingibitory-s-fms-kinazy.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы с-fms киназы</a>
Ингибиторы p70 s6 киназы

Номер патента: 16445
Опубликовано: 30.05.2012
Авторы: Хольст Кристиан Л., Чжуан Цзяньпин, Джозеф Саджан, Шеферд Тимоти Алан, Дэлли Роберт Дин
МПК: A61K 31/437, C07D 487/04, A61P 35/00...
Метки: киназы, ингибиторы
Формула / Реферат:
1. Соединение формулыгде Y представляет собой CH;Z1 и Z2 представляют собой независимо CR3 или N при условии, что Z1 и Z2, оба, не являются N;R1 представляет собой H или CH3;R2 представляет собой фенил, содержащий первый заместитель, выбранный из галогена и трифторметила, и необязательно дополнительно содержащий второй заместитель, при этом указанный второй заместитель представляет собой галоген;R3 представляет собой водород, галоген,...
Ингибиторы киназы

Номер патента: 15189
Опубликовано: 30.06.2011
Авторы: Ли Течао, Побанс Марк Эндрю, Лопес Де Уралде-Гармениа Беатрис, Бастиан Джолие Анне, Чжун Боюй, Майерс Майкл Рэй, Мэйдер Мэри Маргарет, Худзиак Кевин Джон, Де Дьес Альфонсо, Ших Чуан, Блас Де Блас Хесус Андрес
МПК: C07D 401/14, A61K 31/44, A61K 31/497...
Метки: киназы, ингибиторы
Формула / Реферат:
1. Соединение формулы Iгде Z выбран из группы, которая включаетX выбран из группы, которая включаетR1 представляет собой С1-С7алкил, необязательно замещенный от одного до шести заместителями, выбранными из группы, которая включает галоген и C1-C4алкилгалоген; С3-С6циклоалкил, необязательно замещенный одним или двумя заместителями, выбранными из группы, которая включает C1-C4алкил и трифторметил; или триметилсилил;R2 представляет собой фенил,...
Ингибиторы c – fms киназы

Номер патента: 13250
Опубликовано: 30.04.2010
Авторы: Баллентайн Шелли К., Иллиг Карл Р., Менти Карл Л., Уолл Марк Дж., Дежарле Рене, Чэнь Цзиньшэн, Флорес Кристофер, Моллой Кристофер Дж., Мигалла Санатх, Рудольф М.Джонатан, Уилсон Кен
МПК: A61K 31/4439
Метки: киназы, ингибиторы
Формула / Реферат:
1. Соединения формулы Iили их сольват, гидрат, таутомер или фармацевтически приемлемая соль, гдеА представляет собой фенил или пиридил, каждый из которых может быть замещен одним из хлора, фтора, метила, -N3, -NH2, -NH(C1-6алкил), -N(C1-6алкил)2, -S(С1-6алкил), -О(С1-6алкил) или 4-аминофенила;W представляет собой пирролил, имидазолил, изоксазолил, оксазолил, 1,2,4-триазолил или фуранил, каждый из которых может быть соединен через любой атом...
Сульфоксиминные производные как ингибиторы p38 мар киназы

Номер патента: 15497
Опубликовано: 31.08.2011
Авторы: Пател Панкадж Раманбхай, Пател Гаутам Д., Чакрабарти Ганес, Чаттерджи Абхиджит, Лохрай Брадж Бхушан, Шетти Шанкар Джайрам, Джаин Мукул Р., Лохрай Видья Бхушан
МПК: A61P 29/00, A61K 31/427, C07D 417/04...
Метки: сульфоксиминные, ингибиторы, мар, киназы, производные
Формула / Реферат:
1. Соединения общей формулы (I)или их фармацевтически приемлемые соли,где R1, R2 могут быть одинаковыми или различными и независимо представляют собой атом водорода, необязательно замещенные группы, выбранные из линейного или разветвленного (C1-С6)алкила, (С3-С7)циклоалкила, фенила или нафтила;R3 и R4могут быть одинаковыми или различными и независимо могут быть выбраны из необязательно замещенного линейного или разветвленного (C1-С6)алкила,...
Замещенные пиразолы, как ингибиторы p 38 киназы

Номер патента: 3925
Опубликовано: 30.10.2003
Авторы: Крич Джойс Зову, Уэйер Ричард М., Коллинз Пол В., Селнесс Шон Радж, Дженг Лифенг, Ханна Иш К., Партис Ричард А., Ляо Шуюань, Саут Майкл С., Флинн Даньел Л., Стили Майкл А., Рао Шашидхар Н., Ю И, Козик Фрэнсис Дж., Хэнсон Гуннар Дж., Клэр Майкл, Ксу Ксянгдонг, Анантанараян Ашок, Деврадж Раджеш
МПК: A61P 19/00, A61K 31/4152, C07D 401/04...
Метки: киназы, пиразолы, замещенные, ингибиторы
Формула / Реферат:
1. Замещенное пиразольное соединение, таутомер этого соединения или фармацевтически приемлемая соль этого соединения или таутомера, причем это соединение соответствует по структуре формуле I где R1 выбирают из группы, включающей гидрид, алкил, циклоалкил, алкенил, циклоалкенил, алкинил, циклоалкилалкилен, циклоалкенилалкилен, гетероциклилалкилен, галогеналкил, галогеналкенил, галогеналкинил, гидроксиалкил, гидроксиалкенил, гидроксиалкинил,...
Предыдущий патент: Нуклеозидный ингибитор для вгс (hcv)
Следующий патент: Способ ухода за волосами
Случайный патент: Присадка в качестве компонента композиции минерального нефтетоплива