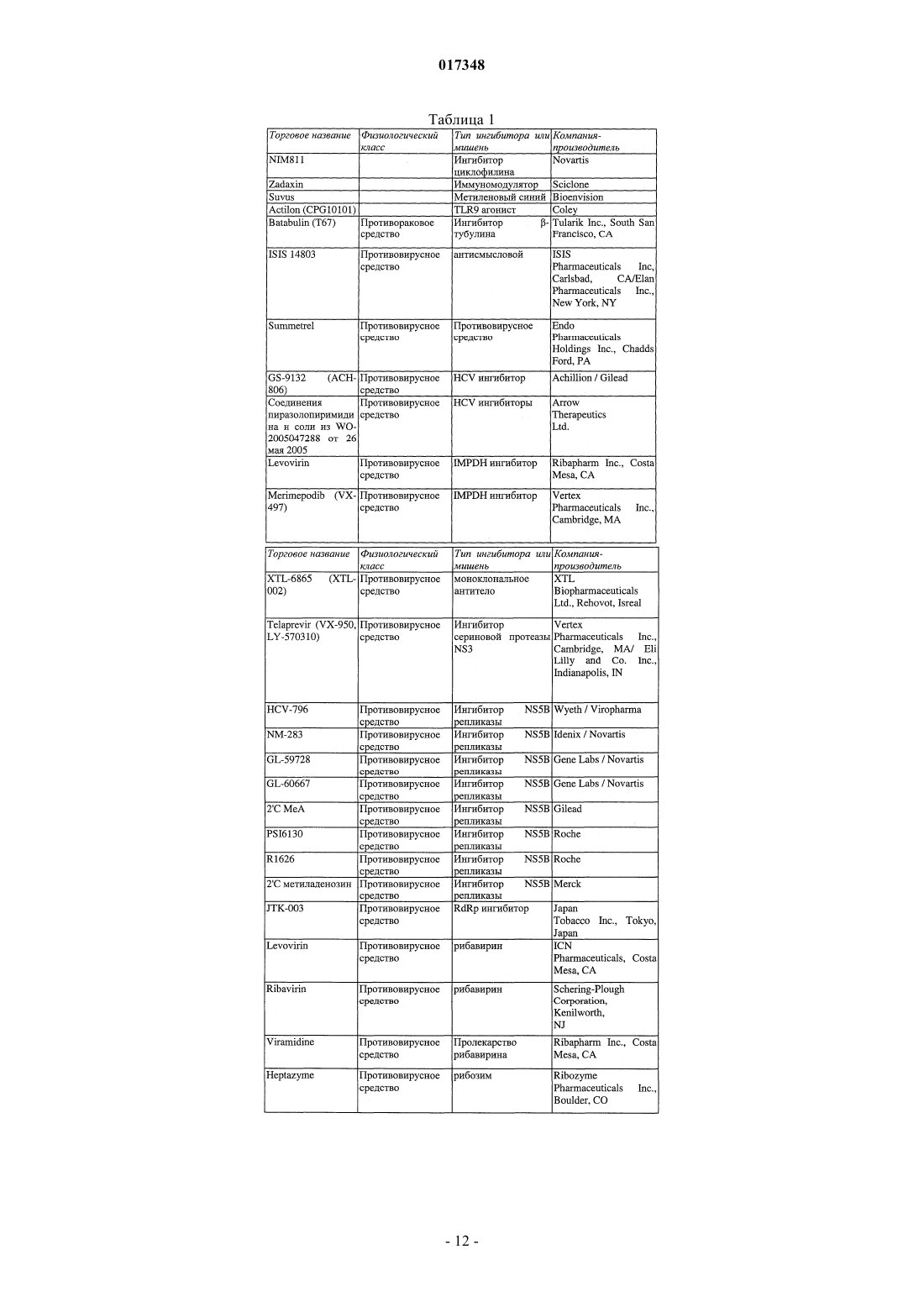
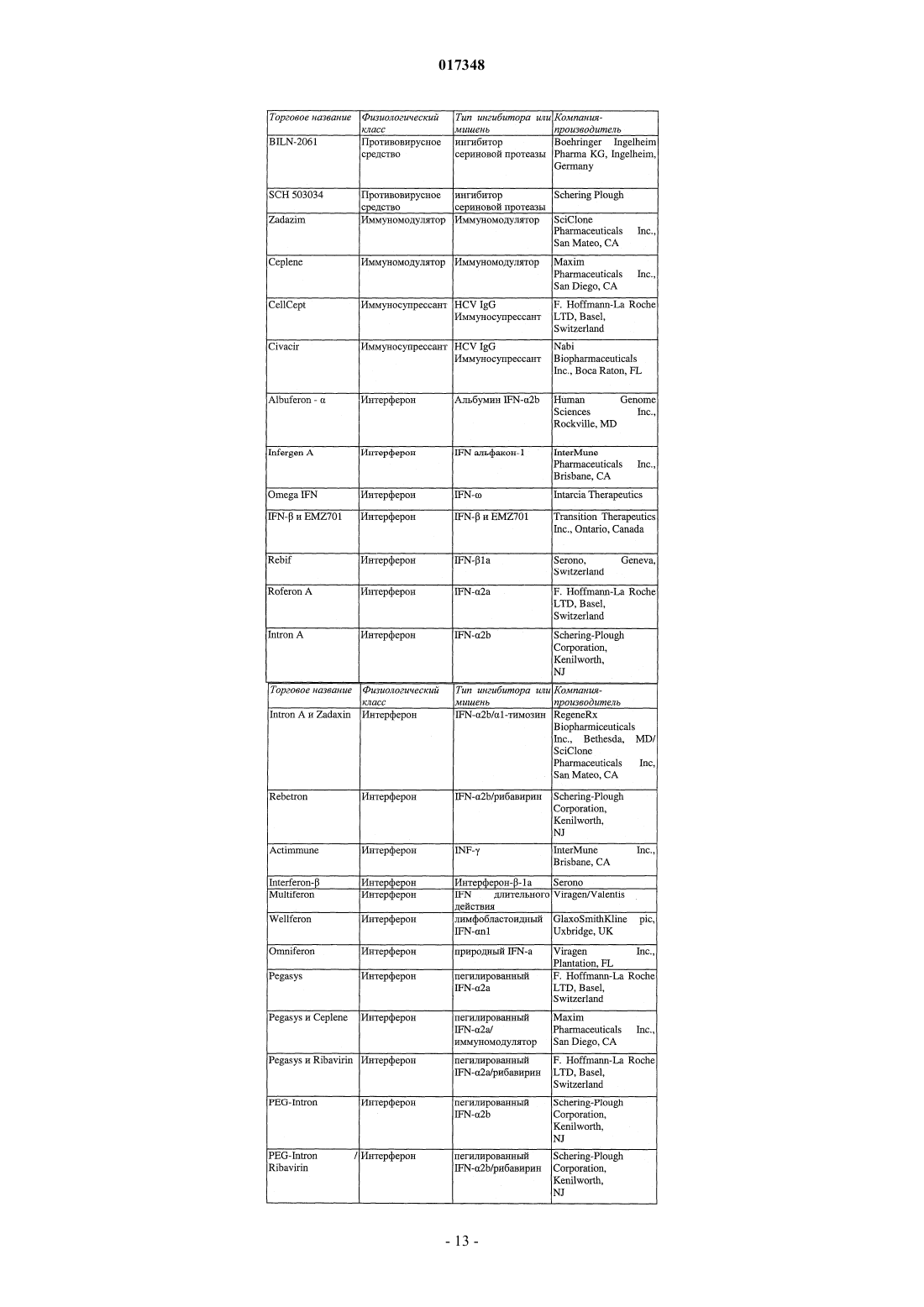
Ингибиторы вируса гепатита с
Номер патента: 17348
Опубликовано: 30.11.2012
Авторы: Сен-Лоран Денис Р., Бэчэнд Кэрол, Белема Маконен, Ромайн Джеффри Ли, Лавой Рико, Гудрич Джейсон, Лопез Омар Д., Ян Фукан, Хаманн Лоуренс Г., Лэнгли Дэвид Р., Снайдер Лоуренс Б., Мартель Ален, Нгуен Ван Н., Рудигер Эдвард Х., Минвелл Николас А., Джеймс Клинт А., Гуд Эндрю С., Деон Дэниел Х.




























Формула / Реферат
1. Соединение формулы (I)

или его фармацевтически приемлемая соль, где
m и n равны 1;
q и s равны 0;
u и v независимо имеют значения 0,1;
А и В выбраны из фенила и содержащего шесть членов гетероароматического кольца, включающего один или два атома азота, выбранного из пиридинила, пиримидинила, пиразинила, пиридазинила; при условии, что по крайней мере один из А и В отличен от фенила;
X и Y выбраны из СН2;
каждый R1 и R2 независимо выбран из С1-С6алкила и галогена;
каждый R3 и R4 независимо выбран из водорода и R9-C(O)-;
каждый R7 и R8 независимо выбран из водорода, галогенС1-С6алкила и триС1-С6алкилсилилС1-С6алкоксиС1-С6алкила;
каждый R9 независимо выбран из C1-С6алкокси, С6-С10арилС1-С6алкокси, NRcRdC1-С6алкила, С6-С10арилС1-С6алкила (где алкильная часть необязательно замещена одной или двумя дополнительными группами, независимо выбранными из C1-С6алкокси, гидрокси, гетероциклила, выбранного из пиперидинила, -NRcRd, где Rc и Rd независимо выбраны из водорода, C1-С6алкоксикарбонила, C1-С6алкила), гетероциклила, выбранного из фуранила, пиперидинила, необязательно замещенного C1-С6алкилом, морфолинила, С3-С7циклоалкилС1-С6алкила (где алкильная часть необязательно замещена NRcRd), C1-С6алкилкарбонила, и гетероциклилС1-С6алкила, где гетероциклил выбран из пиридинила.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где m и n каждый имеет значение 1.
3. Соединение по п.1 или его фармацевтически приемлемая соль, где
u и v каждый независимо имеет значение 0 или 1 и
каждый R1 и R2 независимо выбран из C1-С6алкила и галогена.
4. Соединение по п.1 или его фармацевтически приемлемая соль, где X и Y выбраны из СН2.
5. Соединение по п.1 или его фармацевтически приемлемая соль, где каждый R7 и R8 представляет собой водород.
6. Соединение по п.1 или его фармацевтически приемлемая соль, где каждый R3 и R4 представляет собой R9-C(O)-.
7. Соединение по п.1 или его фармацевтически приемлемая соль, где каждый R9 независимо выбран из C1-С6алкокси, С6-С10арилС1-С6алкила (где алкильная часть необязательно замещена одной или двумя дополнительными группами, независимо выбранными из C1-С6алкокси, гидрокси, гетероциклила, выбранного из пиперидинила, -NRcRd, где Rc и Rd независимо выбраны из водорода, C1-С6алкоксикарбонила, С1-С6алкила), (С3-С7циклоалкил)C1-С6алкила (где алкильная часть необязательно замещена NRcRd), гетероциклила, выбранного из фуранила, пиперидинила, необязательно замещенного C1-С6алкилом, морфолинила, гетероциклилС1-С6алкила, где гетероциклил выбран из пиридинила, и (NRcRd)С1-С6алкила.
8. Соединение формулы (II)

или его фармацевтически приемлемая соль, где
m и n равны 1;
q и s равны 0;
u и v независимо имеют значения 0, 1;
А и В выбраны из фенила и содержащего шесть членов гетероароматического кольца, включающего один или два атома азота, выбранного из пиридинила, пиримидинила, пиразинила, пиридазинила; при условии, что по крайней мере один из А и В отличен от фенила;
X и Y выбраны из CH2;
когда присутствуют, R и/или R представляют собой галоген, где каждый галоген представляет собой фтор;
каждый R3 и R4 представляет собой R9-C(O)-;
каждый R7 и R8 независимо выбран из водорода, галогенС1-С6алкила и триС1-С6алкилсилилС1-С6алкоксиС1-С6алкила; и
каждый R9 независимо выбран из C1-С6алкокси, С6-С10арилС1-С6алкокси, NRcRdC1-С6алкила, С6-С10арилС1-С6алкила (где алкильная часть необязательно замещена одной или двумя дополнительными группами, независимо выбранными из C1-С6алкокси, гидрокси, гетероциклила, выбранного из пиперидинила, -NRcRd, где Rc и Rd независимо выбраны из водорода, C1-С6алкоксикарбонила, C1-С6алкила), гетероциклила, выбранного из фуранила, пиперидинила, необязательно замещенного C1-С6алкилом, морфолинила, С3-С7циклоалкилС1-С6алкила (где алкильная часть необязательно замещена NRcRd), С1-С6алкилкарбонила, и гетероциклилС1-С6алкила, где гетероциклил выбран из пиридинила.
9. Соединение формулы (III)

или его фармацевтически приемлемая соль, где
m и n равны 1;
q и s равны 0;
u и v независимо имеют значения 0, 1;
А и В выбраны из фенила и содержащего шесть членов гетероароматического кольца, включающего один или два атома азота, выбранного из пиридинила, пиримидинила, пиразинила, пиридазинила; при условии, что по крайней мере один из А и В отличен от фенила;
каждый R1 и R2 независимо выбран из C1-С6алкила и галогена;
каждый R3 и R4 независимо выбран из водорода и R9-C(O)-;
каждый R7 и R8 независимо выбран из водорода, галогенС1-С6алкила и триС1-С6алкилсилилС1-С6алкоксиС1-С6алкила; и
каждый R9 независимо выбран из C1-С6алкокси, С6-С10арилС1-С6алкила (где алкильная часть необязательно замещена одной или двумя дополнительными группами, независимо выбранными из C1-С6алкокси, гидрокси, гетероциклила, выбранного из пиперидинила, -NRcRd, где Rc и Rd независимо выбраны из водорода, С1-С6алкоксикарбонила, C1-С6алкила), (С3-С7циклоалкил)C1-С6алкила (где алкильная часть необязательно замещена NRcRd), гетероциклила, выбранного из фуранила, пиперидинила, необязательно замещенного C1-С6алкилом, морфолинила, гетероциклилС1-С6алкила, где гетероциклил выбран из пиридинила, и (NRcRd) С1-С6алкила.
10. Соединение, выбранное из
(1R)-2-((2S)-2-(5-(6-(4-(2-((2S)-1-((2R)-2-(диметиламино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-3-пиридинил)-1Н-имидазол-2-ил)-1-пирролидинил)-N,N-диметил-2-оксо-1-фенилэтанамина;
(1R)-2-((2S)-2-(5-(6-(4-(2-((2S)-1-((2R)-2-гидрокси-2-фенилацетил)-2-пирролидинил)-1H-имидазол-5-ил)фенил)-3-пиридинил)-1H-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтанола;
метил-((1R)-2-((2S)-2-(5-(6-(4-(2-((2S)-1-((2R)-2-((метоксикарбонил)амино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-3-пиридинил)-1H-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтил)карбамата;
5-(2-((2S)-1-((2R)-2-метокси-2-фенилацетил)-2-пирролидинил)-1H-имидазол-5-ил)-2-(4-(2-((2S)-1-((2R)-2-метокси-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)пиридина;
(1R)-2-((2S)-2-(5-(6-(4-(2-((2S)-1-((2R)-2-(диметиламино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)-2-метилфенил)-3-пиридинил)-1H-имидазол-2-ил)-1-пирролидинил)-N,N-диметил-2-оксо-1-фенилэтанамина;
метил-((1R)-2-((2S)-2-(5-(6-(4-(2-((2S)-1-((2R)-2-((метоксикарбонил)амино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)-2-метилфенил)-3-пиридинил)-1Н-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтил)карбамата;
N-((1R)-2-((2S)-2-(5-(6-(4-(2-((2S)-1-((2R)-2-ацетамидо-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-3-пиридинил)-1Н-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтил)ацетамид;
метил-((1R)-2-((2S)-2-(5-(4-(5-(2-((2S)-1-((2R)-2-(диметиламино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)-2-пиридинил)фенил)-1Н-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтил)карбамата;
метил-((1R)-2-оксо-1-фенил-2-((2S)-2-(5-(4-(5-(2-((2S)-1-((2R)-тетрагидро-2-фуранилкарбонил)-2-пирролидинил)-1Н-имидазол-5-ил)-2-пиридинил)фенил)-1H-имидазол-2-ил)-1-пирролидинил)этил)карбамата;
метил-((1R)-2-((2S)-2-(5-(4-(5-(2-((2S)-1-((1-метил-4-пиперидинил)карбонил)-2-пирролидинил)-1H-имидазол-5-ил)-2-пиридинил)фенил)-1H-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтил)карбамата;
метил-((1R)-2-оксо-1-фенил-2-((2S)-2-(5-(4-(5-(2-((2S)-1-(3-пиридинилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)-2-пиридинил)фенил)-1Н-имидазол-2-ил)-1-пирролидинил)этил)карбамата;
метил-((1R))-2-((2S)-2-(5-(4-(5-(2-((2S)-1-(4-морфолинилкарбонил)-2-пирролидинил)-1H-имидазол-5-ил)-2-пиридинил)фенил)-1Н-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтил)карбамата;
диметил-(2,2'-бипиридин-5,5'-диилбис(1H-имидазол-5,2-диил(2S)-2,1-пирролидиндиил((1R)-2-оксо-1-фенил-2,1-этандиил)))бискарбамата;
(1R)-2-((2S)-2-(5-(5-(4-(2-((2S)-1-((2R)-2-(диметиламино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-2-пиридинил)-1Н-имидазол-2-ил)-1-пирролидинил)-N,N-диметил-2-оксо-1-фенилэтанамина;
метил-((1R)-2-((2S)-2-(5-(5-(4-(2-((2S)-1-((2R)-2-((метоксикарбонил)амино)-2-фенилацетил)-2-пирролидинил)-1H-имидазол-5-ил)фенил)-2-пиридинил)-1H-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтил)карбамата;
(S)-2-[5-(2-{4-[2-((S)-1-трет-бутоксикарбонилпирролидин-2-ил)-3Н-имидазол-4-ил]фенил}пиридинин-5-ил)-1-(2-триметилсиланилэтоксиметил)-1H-имидазол-2-ил]-пирролидин-1-карбоновой кислоты трет-бутилового эфира;
(S)-2-(5-{2-[4-((S)-2-пирролидин-2-ил-3Н-имидазол-4-ил)фенил]пиридинин-5-ил}-1Н-имидазол-2-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира;
(1R)-2-((2S)-2-(5-(2-(4-(2-((2S)-1-((2R)-2-(диметиламино)-2-фенилацетил)-2-пирролидинил)-1H-имидазол-5-ил)фенил)-5-пиримидинил)-1H-имидазол-2-ил)-1-пирролидинил)-N,N-диметил-2-оксо-1-фенилэтанамина;
метил-((1R)-2-((2S)-2-(5-(2-(4-(2-((2S)-1-((2R)-2-((метоксикарбонил)амино)-2-фенилацетил)-2-пирролидинил)-1H-имидазол-5-ил)фенил)-5-пиримидинил)-1H-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтил)карбамата;
метил-((1R)-2-оксо-1-фенил-2-((2S)-2-(5-(4-(5-(2-((2S)-1-(3-пиридинилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)-2-пиримидинил)фенил)-1Н-имидазол-2-ил)-1-пирролидинил)этил)карбамата;
метил-((1R)-2-оксо-1-фенил-2-((2S)-2-(5-(2-(4-(2-((2S)-1-(3-пиридинилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-5-пиримидинил)-1Н-имидазол-2-ил)-1-пирролидинил)этил)карбамата;
5-(2-((2S)-1-((2R)-2-фенил-2-(1-пиперидинил)ацетил)-2-пирролидинил)-1H-имидазол-5-ил)-2-(4-(2-((2S)-1-((2R)-2-фенил-2-(1-пиперидинил)ацетил)-2-пирролидинил)-1Н-имидазол-4-ил)фенил)пиридинина;
(1R)-2-((2S)-2-(5-(5-(4-(2-((2S)-1-((2R)-2-(диметиламино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-2-пиразинил)-1Н-имидазол-2-ил)-1-пирролидинил)-N,N-диметил-2-оксо-1-фенилэтанамина;
метил-((1R)-2-((2S)-2-(5-(5-(4-(2-((2S)-1-((2R)-2-((метоксикарбонил)амино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-2-пиразинил)-1Н-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтил)карбамата;
(1R)-2-((2S)-2-(5-(6-(4-(2-((2S)-1-((2R)-2-(диметиламино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-3-пиридазинил)-1Н-имидазол-2-ил)-1-пирролидинил)-N,N-диметил-2-оксо-1-фенилэтанамина;
метил-((1R)-2-((2S)-2-(5-(6-(4-(2-((2S)-1-((2R)-2-((метоксикарбонил)амино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-3-пиридазинил)-1H-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтил)карбамата;
{(R)-2-оксо-1-фенил-2-[(S)-2-(5-{4-[5-((S)-2-пирролидин-2-ил-3Н-имидазол-4-ил)пиридинин-2-ил]фенил}-1Н-имидазол-2-ил)пирролидин-1-ил]этил}карбаминовой кислоты метилового эфира;
метил-((1R)-2-оксо-1-фенил-2-((2S)-2-(4-(4-(5-(2-((2S)-1-((2R)-2-фенил-2-(1-пиперидинил)ацетил)-2-пирролидинил)-1H-имидазол-5-ил)-2-пиримидинил)фенил)-1Н-имидазол-2-ил)-1-пирролидинил)этил) карбамата;
(S)-2-[5-{5'-[2-((S)-1-трет-бутоксикарбонилпирролидин-2-ил)-3-(2-триметилсиланилэтоксиметил)-3Н-имидазол-4-ил]-[2,2']бипиримидинил-5-ил}-1-(2-триметилсиланилэтоксиметил)-1Н-имидазол-2-ил]-пирролидин-1-карбоновой кислоты трет-бутилового эфира;
(1R,1'R)-2,2'-(3,3'-бипиридазин-6,6'-диилбис(1Н-имидазол-5,2-диил(2S)-2,1-пирролидиндиил))бис(N,N-диметил-2-оксо-1-фенилэтанамина);
диметил-(3,3'-бипиридазин-6,6'-диилбис(1H-имидазол-5,2-диил(2S)-2,1-пирролидиндиил((1R)-2-оксо-1-фенил-2,1-этандиил)))бискарбамата;
(1R,1'R)-2,2'-(2,2'-бипиридинин-5,5'-диилбис(1Н-имидазол-5,2-диил(2S)-2,1-пирролидиндиил))бис(N,N-диметил-2-оксо-1-фенилэтанамина);
диметил-(2,2'-бипиридинин-5,5'-диилбис(1Н-имидазол-5,2-диил(2S)-2,1-пирролидиндиил((1R)-2-оксо-1-фенил-2,1-этандиил)))бискарбамата;
(1R,1'R)-2,2'-(2,2'-бипиризин-5,5'-диилбис(1Н-имидазол-5,2-диил(2S)-2,1-пирролидиндиил))бис(N,N-диметил-2-оксо-1-фенилэтанамина);
диметил-(2,2'-бипиризин-5,5'-диилбис(1Н-имидазол-5,2-диил(2S)-2,1-пирролидиндиил((1R)-2-оксо-1-фенил-2,1-этандиил)))бискарбамата;
трет-бутил(2S)-2-(5-(2-(4-(2-((2S)-1-((2R)-2-(диэтиламино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-5-пиримидинил)-1Н-имидазол-2-ил)-1-пирролидинкарбоксилата;
(1R)-N,N-диэтил-2-оксо-1-фенил-2-((2S)-2-(5-(4-(5-(2-((2S)-2-пирролидинил)-1Н-имидазол-5-ил)-2-пиримидинил)фенил)-1Н-имидазол-2-ил)-1-пирролидинил)этанамина;
метил-((1S)-2-((2S)-2-(5-(4-(5-(2-((2S)-1-(N-(метоксикарбонил)-L-аланил)-2-пирролидинил)-1Н-имидазол-5-ил)-2-пиримидинил)фенил)-1Н-имидазол-2-ил)-1-пирролидинил)-1-метил-2-оксоэтил)карбамата;
метил-((1S)-1-(((2S)-2-(5-(2-(4-(2-((2S)-1-((2S)-2-((метоксикарбонил)амино)-3-метилбутаноил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-5-пиримидинил)-1Н-имидазол-2-ил)-1-пирролидинил)карбонил)-2-метилпропил)карбамата;
метил-((1S)-1-циклопропил-2-((2S)-2-(5-(2-(4-(2-((2S)-1-((2S)-2-циклопропил-2-((метоксикарбонил)амино)ацетил)-2-пирролидинил)-1H-имидазол-5-ил)фенил)-5-пиримидинил)-1Н-имидазол-2-ил)-1-пирролидинил)-2-оксоэтил)карбамата;
метил-((1S)-1-(((2S)-2-(5-(2-(4-(2-((2S)-1-((2R)-2-(диэтиламино)-2-фенилацетил)-2-пирролидинил)-1H-имидазол-5-ил)фенил)-5-пиримидинил)-1H-имидазол-2-ил)-1-пирролидинил)карбонил)-2-метилпропил)карбамата;
метил-((1S)-2-((2S)-2-(5-(2-(4-(2-((2S)-1-((2R)-2-(диэтиламино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-5-пиримидинил)-1Н-имидазол-2-ил)-1-пирролидинил)-1-метил-2-оксоэтил)карбамата;
трет-2-бутил(2S)-2-(5-(4-(5-(2-((2S)-1-(трет-бутоксикарбонил)-2-пирролидинил)-1Н-имидазол-5-ил)-2-пиримидинил)фенил)-4-(трифторметил)-1Н-имидазол-2-ил)-1-пирролидинкарбоксилата;
метил-((1R)-2-((2S)-2-(5-(4-(5-(2-((2S)-1-((2R)-2-((метоксикарбонил)амино)-2-фенилацетил)-2-пирролидинил)-1H-имидазол-5-ил)-2-пиримидинил)фенил)-4-(трифторметил)-1H-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтил)карбамата;
(2R)-2-((2S)-2-(5-(4-(5-(2-((2S)-1-((2R)-2-(диэтиламино)-2-фенилацетил)-2-пирролидинил)-1Н-имидазол-5-ил)-2-пиримидинил)фенил)-4-(трифторметил)-1Н-имидазол-2-ил)-1-пирролидинил)-N,N-диэтил-2-оксо-1-фенилэтанамина;
метил-((1S)-1-(((2S)-2-(5-(2-(4-(2-((2S)-1-((2S)-2-((метоксикарбонил)амино)-3-метилбутаноил)-2-пирролидинил)-4-(трифторметил)-1H-имидазол-5-ил)фенил)-5-пиримидинил)-1Н-имидазол-2-ил)-1-пирролидинил)карбонил)-2-метилпропил)карбамата;
метил-((1S)-1-циклопропил-2-((2S)-2-(5-(4-(5-(2-((2S)-1-((2S)-2-циклопропил-2-((метоксикарбонил)амино)ацетил)-2-пирролидинил)-1H-имидазол-5-ил)-2-пиримидинил)фенил)-4-(трифторметил)-1Н-имидазол-2-ил)-1-пирролидинил)-2-оксоэтил)карбамата;
метил-((1S)-2-((2S)-2-(5-(4-(5-(2-((2S)-1-(N-(метоксикарбонил)-L-аланил)-2-пирролидинил)-1H-имидазол-5-ил)-2-пиримидинил)фенил)-4-(трифторметил)-1H-имидазол-2-ил)-1-пирролидинил)-1-метил-2-оксоэтил)карбамата;
(2R)-1-((2S)-2-(5-(4-(5-(2-((2S)-1-((2R)-2-(диэтиламино)пропаноил)-2-пирролидинил)-1H-имидазол-5-ил)-2-пиримидинил)фенил)-4-(трифторметил)-1H-имидазол-2-ил)-1-пирролидинил)-N,N-диэтил-1-оксо-2-пропанамина;
трет-бутил(2S)-2-(5-(2-(4-(2-((2S)-1-(трет-бутоксикарбонил)-2-пирролидинил)-1Н-имидазол-4-ил)-3-фторфенил)-5-пиримидинил)-1Н-имидазол-2-ил)-1-пирролидинкарбоксилата;
2-(3-фтор-4-(2-((2S)-2-пирролидинил)-1Н-имидазол-5-ил)фенил)-5-(2-((2S)-2-пирролидинил)-1Н-имидазол-5-ил)пиридинина;
метил-((1S)-2-((2S)-2-(5-(фтор-4-(5-(2-((2S)-1-(N-(метоксикарбонил)-L-аланил)-2-пирролидинил)-1H-имидазол-5-ил)-2-пиримидинил)фенил)-1H-имидазол-2-ил)-1-пирролидинил)-1-метил-2-оксоэтил)карбамата;
метил-((1S)-1-(((2S)-2-(5-(2-фтор-4-(5-(2-((2S)-1-((2S)-2-((метоксикарбонил)амино)-3-метилбутаноил)-2-пирролидинил)-1Н-имидазол-5-ил)-2-пиримидинил)фенил)-1Н-имидазол-2-ил)-1-пирролидинил)карбонил)-2-метилпропил)карбамата;
метил-((1R)-2-((2S)-2-(5-(2-(3-фтор-4-(2-((2S)-1-((2R)-2-((метоксикарбонил)амино)-2-фенилацетил)-2-пирролидинил)-1H-имидазол-5-ил)фенил)-5-пиримидинил)-1Н-имидазол-2-ил)-1-пирролидинил)-2-оксо-1-фенилэтил)карбамата и
метил-((1S,2R)-1-(((2S)-2-(5-(2-фтор-4-(5-(2-((2S)-1-(N-(метоксикарбонил)-0-метил-L-треонил)-2-пирролидинил)-1Н-имидазол-5-ил)-2-пиримидинил)фенил)-1Н-имидазол-2-ил)-1-пирролидинил)карбонил)-2-метоксипропил)карбамата;
или его фармацевтически приемлемой соли.
11. Соединение

или его фармацевтически приемлемая соль.
12. Фармацевтическая композиция для лечения HCV инфекции, содержащая терапевтически эффективное количество соединения по п.1 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
Текст
(US), Мартель Ален (CA), Минвелл Николас А., Нгуен Ван Н., Ромайн Джеффри Ли (US), Рудигер Эдвард Х. или их фармацевтически приемлемым солям, а также к композициям, содержащим указанные соединения, для лечения вируса гепатита С (HCV инфекция). 017348 Изобретение главным образом направлено на противовирусные соединения и более конкретно, направлено на соединения, которые могут ингибировать функцию белка NS5A, кодируемого вирусом гепатита С (HCV), на композиции, содержащие такие соединения, и способы ингибирования функции белкаNS5A. Вирус гепатита С (HCV) является основным патогеном человека, инфицировавшим предположительно 170 миллионов человек по всему миру - грубо в пять раз больше, чем инфицированных вирусом иммунодефицита человека 1 типа. У значительного числа инфицированных HCV индивидуумов развивается серьезное прогрессирующее заболевание печени, включая цирроз и печеночно-клеточный рак. В настоящее время наиболее эффективная терапия HCV включает комбинацию альфа-интерферона и рибавирина, которые приводят к устойчивому эффекту у 40% пациентов. Последние клинические результаты демонстрируют, что пегилированный альфа-интерферон предпочтителен по сравнению с немодифицированным альфа-интерфероном в виде монотерапии. Тем не менее, даже при экспериментальных терапевтических режимах, включающих комбинации пегилированного альфа-интерферона и рибавирина, существенная часть пациентов не отмечает устойчивого снижения вирусной нагрузки. Таким образом, существует очевидная и важная необходимость развивать эффективные лекарственные средства для лечения HCV инфекции.HCV является положительно закрученным РНК вирусом. Основываясь на сравнении вычисленной последовательности аминокислот и значительном сходстве в 5'-нетранслируемой области, HCV классифицируется как отдельный род в семействе Flaviviridae. Все представители семейства Flaviviridae имеют оболочечные вирионы, которые содержат положительно закрученный РНК геном, кодирующий все известные вирус-специфические белки с помощью трансляции единичной непрерывной открытой рамки считывания. Существенная гетерогенность найдена в нуклеотидной и кодированной аминокислотной последовательности по всему геному HCV. По меньшей мере шесть основных генотипов и более чем 50 подтипов были описаны. Основные генотипы HCV отличаются по распространенности в мире и клиническое значение генетической гетерогенности HCV остается неясным, несмотря на множество исследований возможного действия генотипов на патогенез и терапию. Геном одноцепочечной РНК HCV составляет приблизительно 9500 нуклеотидов в длинну и имеет одну открытую рамку считывания (ORF), кодирующую один большой полипротеин из примерно 3000 аминокислот. В инфицированных клетках этот полипротеин расщепляется по многим сайтам клеточными и вирусными протеазами до получения структурных и неструктурных (NS) белков. В случае HCV поколение зрелых неструктурных белков (NS2, NS3, NS4A, NS4B, NS5A и NS5B) подвергается воздействию двух вирусных протеаз. Первая, как предполагают, является металлопротеазой и расщепляет NS2NS3 связь; вторая является сериновой протеазой, содержащей на N-конце NS3 (также обозначаемая какNS3 протеаза) и опосредует все последующие расщепления ниже по каскаду NS3, как в цис-положении,в сайте расщепления NS3-NS4A, так и в транс-положении, для остальных сайтов NS4A-NS4B, NS4BNS5A, NS5A-NS5B. NS4A белок, как представляется, выполняет множество функций, действуя как кофактор для NS3 протеазы и возможно способствует расположению в мембране NS3 и других компонентов вирусной репликазы. Образование комплекса NS3 белка и NS4A представляется необходимым для этапов процессинга, повышая эффективность протеолиза во всех сайтах. Белок NS3 также проявляет активность нуклеозидной трифосфатазы и РНК хеликазы. NS5B (также обозначаемая как HCV полимераза) является РНК-зависимой РНК полимеразой, которая вовлечена в репликацию HCV. Имеется потребность в соединениях, применимых в лечении HCV-инфицированных пациентов, которые избирательно ингибируют репликацию вируса HCV. В частности, являются востребованными соединения, которые эффективно ингибируют функцию белка NS5A. Белок HCV NS5A описан, например,в Tan, S.-L., Katzel, M.G. Virology 2001, 284, 1-12; and in Park, K.-J.; Choi, S.-H, J. Biological Chemistry 2003. В первом аспекте настоящее изобретение обеспечивает соединение формулы (I) или его фармацевтически приемлемую соль, где m и n равны 1; q и s равны 0; u и v независимо имеют значения 0, 1; А и В выбраны из фенила и содержащего шесть членов гетероароматического кольца, включающего один или два атома азота, выбранного из пиридинила, пиримидинила, пиразинила, пиридазинила; при условии, что по крайней мере один из А и В отличен от фенила;X и Y выбраны из СН 2; каждый R1 и R2 независимо выбран из C1-С 6 алкила и галогена; каждый R3 и R4 независимо выбран-1 017348 из водорода и R9-C(O)-; каждый R7 и R8 независимо выбран из водорода, галогенС 1-С 6 алкила и триС 1-С 6 алкилсилилС 1-С 6 алкоксиС 1-С 6 алкила; каждый R9 независимо выбран из C1-С 6 алкокси, С 6-С 10 арилС 1-С 6 алкокси, NRcRdC1-С 6 алкила, С 6-С 10 арилС 1-С 6 алкила (где алкильная часть необязательно замещена одной или двумя дополнительными группами, независимо выбранными из C1-С 6 алкокси, гидрокси, гетероциклила, выбранного из пиперидинила,-NRcRd, где Rc и Rd независимо выбраны из водорода, C1-С 6 алкоксикарбонила, C1-С 6 алкила), гетероциклила, выбранного из фуранила, пиперидинила, необязательно замещенного C1-С 6 алкилом, морфолинила,С 3-С 7 циклоалкилС 1-С 6 алкила (где алкильная часть необязательно замещена NRcRd), С 1-С 6 алкилкарбонила, и гетероциклилС 1-С 6 алкила, где гетероциклил выбран из пиридинила. В первом воплощении первого аспекта m и n каждый имеет значение 1. Во втором воплощении первого аспекта u и v каждый независимо имеет значение 0 или 1; и каждыйR1 и R2 независимо выбран из С 1-С 6 алкила и галогена. В тертьем воплощений первого аспекта X и Y выбраны из СН 2. В четвертом воплощении первого аспекта каждый R7 и R8 представляет собой водород. В пятом воплощении первого аспекта каждый R3 и R4 представляет собой R9-C(O)-. В шестом воплощении первого аспекта каждый R9 независимо выбран из C1-С 6 алкокси, С 6-С 10 арилС 1-С 6 алкила (где алкильная часть необязательно замещена одной или двумя дополнительными группами, независимо выбранными из C1-С 6 алкокси, гидрокси, гетероциклила, выбранного из пиперидинила,-NRcRd, где Rc и Rd независимо выбраны из водорода, C1-С 6 алкоксикарбонила, C1-С 6 алкила), (С 3 С 7 циклоалкил)C1-С 6 алкила (где алкильная часть необязательно замещена NRcRd), гетероциклила, выбранного из фуранила, пиперидинила, необязательно замещенного C1-С 6 алкилом, морфолинила, гетероциклилС 1-С 6 алкила, где гетероциклил выбран из пиридинила, и (NRcRd)C1-С 6 алкила. Во втором аспекте настоящее изобретение обеспечивает соединение формулы (II) или его фармацевтически приемлемую соль, гдеu и v независимо имеют значения 0,1; А и В выбраны из фенила и содержащего шесть членов гетероароматического кольца, включающего один или два атома азота, выбранного из пиридинила, пиримидинила, пиразинила, пиридазинила; при условии, что по крайней мере один из А и В отличен от фенила;X и Y выбраны из CH2; когда присутствуют, R1 и/или R2 представляют собой галоген, где каждый галоген представляет собой фтор; каждый R3 и R4 представляет собой R9-C(O)-; каждый R7 и R8 независимо выбран из водорода, галогенС 1-С 6 алкила и триС 1-С 6 алкилсилилС 1-С 6 алкоксиС 1-С 6 алкила; и каждый R9 независимо выбран из C1-С 6 алкокси, С 6-С 10 арилС 1-С 6 алкокси,NRcRdC1-С 6 алкила, С 6-С 10 арилС 1-С 6 алкила (где алкильная часть необязательно замещена одной или двумя дополнительными группами, независимо выбранными из C1-С 6 алкокси, гидрокси, гетероциклила, выбранного из пиперидинила, -NRcRd, где Rc и Rd независимо выбраны из водорода, C1-С 6 алкоксикарбонила, C1-С 6 алкила), гетероциклила, выбранного из фуранила, пиперидинила, необязательно замещенногоC1-С 6 алкилом, морфолинила, С 3-С 7 циклоалкилС 1-С 6 алкила (где алкильная часть необязательно замещенаNRcRd), C1-С 6 алкилкарбонила, и гетероциклилС 1-С 6 алкила, где гетероциклил выбран из пиридинила. В тертьем аспекте настоящее изобретение обеспечивает соединение формулы (III) или его фармацевтически приемлемую соль, где m и n равны 1;u и v независимо имеют значения 0,1; А и В выбраны из фенила и содержащего шесть членов гетероароматического кольца, включающего один или два атома азота, выбранного из пиридинила, пиримидинила, пиразинила, пиридазинила; при условии, что по крайней мере один из А и В отличен от фенила; каждый R1 И R2 независимо выбран из C1-С 6 алкила и галогена;-2 017348 каждый R3 и R4 независимо выбран из водорода и R9-C(O)-; каждый R7 и R8 независимо выбран из водорода, галогенС 1-С 6 алкила и триС 1-С 6 алкилсилилС 1-С 6 алкоксиС 1-С 6 алкила; и каждый R9 независимо выбран из C1-С 6 алкокси, С 6-С 10 арилС 1-С 6 алкила (где алкильная часть необязательно замещена одной или двумя дополнительными группами, независимо выбранными из C1-С 6 алкокси, гидрокси, гетероциклила, выбранного из пиперидинила, -NRcRd, где Rc и Rd независимо выбраны из водорода, C1-С 6 алкоксикарбонила, C1-С 6 алкила), (С 3-С 7 циклоалкил)C1-С 6 алкила (где алкильная часть необязательно замещена NRcRd), гетероциклила, выбранного из фуранила, пиперидинила, необязательно замещенного C1-С 6 алкилом, морфолинила, гетероциклилС 1-С 6 алкила, где гетероциклил выбран из пиридинила, и (NRcRd) C1-С 6 алкила. В четвертом аспекте настоящее изобретение обеспечивает композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. В первом воплощении четвертого аспекта композиция, кроме того, включает один или два дополнительных соединения, имеющих противо-HCV активность. Во втором воплощении четвертого аспекта по крайней мере одно из дополнительных соединений представляет собой интерферон или рибавирин. В третьем воплощении четвертого аспекта интерферон является выбранным из интерферона альфа 2 В, пегилированного интерферона альфа, консенсусного интерферона, интерферона альфа 2 А и лимфобластоидного интерферона тау. В четвертом воплощении четвертого аспекта композиция также содержит одно или два дополнительных соединений, обладающих анти-HCV активностью, при этом по меньшей мере одно из этих дополнительных соединений выбрано из следующих: интерлейкин 2, интерлейкин 6, интерлейкин 12, соединение, которое улучшает развитие реакции хелперных Т-клеток 1-го типа, интерферирующая РНК,антисмысловая РНК, имиквимод, рибавирин, ингибитор инозин 5'-монофосфат дегидрогеназы, амантадин и римантадин. В четвертом воплощении четвертого аспекта композиция также содержит одно или два дополнительных соединений, обладающих анти-HCV активностью, при этом по меньшей мере одно из этих дополнительных соединений эффективно ингибирует функцию мишени, выбранной из следующих: HCV метллопротеаза, HCV сериновая протеаза, HCV полимераза, HCV хеликаза, HCV NS4B белок, HCV вход,HCV сборка, HCV выход, HCV NS5A белок и IMPDH для лечения HCV инфекции. В пятом аспекте настоящее изобретение предусматривает способ лечения HCV инфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или фармацевтически приемлемой его соли. В первом воплощении пятого аспекта способ также включает введение одного или двух дополнительных соединений, обладающих анти-HCV активностью, перед, после или одновременно с соединением формулы (I) или фармацевтически приемлемой его соли. Во втором воплощении пятого аспекта по крайней мере одно из дополнительных соединений является интерфероном или рибавирином. В третьем воплощении пятого аспекта интерферон является выбранным из интерферона альфа 2 В,пегилированного интерферона альфа, консенсусного интерферона, интерферона альфа 2 А и лимфобластоидного интерферона тау. В четвертом воплощении пятого аспекта способ также включает введение одного или двух дополнительных соединений, обладающих анти-HCV активностью перед, после или одновременно с соединением формулы (I) или фармацевтически приемлемой его соли, при этом по крайней мере одно из дополнительных соединений выбрано из следующих: интерлейкин 2, интерлейкин 6, интерлейкин 12, соединение, которое улучшает развитие реакции хелперных Т-клеток 1-го типа, интерферирующая РНК, антисмысловая РНК, имиквимод, рибавирин, ингибитор инозин 5'-монофосфат дегидрогеназы, амантадин и римантадин. В пятом воплощении пятого аспекта способ также включает введение одного или двух дополнительных соединений, обладающих анти-HCV активностью перед, после или одновременно с соединением формулы (I) или фармацевтически приемлемой его соли, при этом по крайней мере одно из дополнительных соединений эффективно ингибирует функцию мишени, выбранной из следующих: HCV метллопротеаза, HCV сериновая протеаза, HCV полимераза, HCV хеликаза, HCV NS4B белок, HCV вход, HCV сборка, HCV выход, HCV NS5A белок и IMPDH для лечения HCV инфекции. Другие воплощения по настоящему изобретению могут содержать приемлемые комбинации из двух или большего количества воплощений и/или аспектов, раскрытых в настоящем изобретении. Дополнительные другие воплощения и аспекты изобретения будут очевидны в соответствии с описанием, обеспеченным ниже. Соединения по настоящему изобретению также существуют в виде таутомеров; поэтому настоящее изобретение также охватывает все таутомерные формы. Описание настоящего изобретения должно быть рассмотрено в соответствии с правилами и принципами образования химической связи. В некоторых случаях это может быть необходимо, чтобы удалить атом водорода для размещения заместителя на любом заданном месте. Например, в структуре, показан-3 017348 ной нижеR8 может быть присоединен или к атому углерода в имидазольном кольце или, альтернативно, R8 может быть введен вместо атома водорода на азотном кольце, чтобы образовать N-замещенный имидазол. Должно быть понятно, что соединения, охваченные настоящим изобретением, являются теми, которые обеспечивают подходящую стабильность для применения в качестве фармацевтического агента. Подразумевается, что определение любого заместителя или радикала (например, R1, R2, R5, R6, и так далее) на конкретном месте в молекуле является независимым от определений в другом месте той же молекулы. Например, когда и равно 2, каждая из двух групп R1 может быть одинаковой или различной. Все патенты, патентные заявки и печатные ссылки, процитированные в описании, при этом включены как ссылки в своей полноте. В случае несовместимости будет преобладать настоящее изобретение,включая определения. Как используют в настоящем описании, следующие термины имеют соответствующие значения: Как используют в настоящем изобретении, единственные формы "a", "an" и "the" подразумевают множественные ссылки, за исключением контекста, ясно определяющего иное. За исключением иным образом установленного, все арильные, циклоалкильные и гетероциклильные группы по настоящему изобретению могут быть замещены как описано в каждом из соответствующих определений. Например, арильная часть арилалкильной группы может быть замещена как описано при определении термина "арил". Термин "алкенил,", как используют в настоящем изобретении, относится к прямой или разветвленной цепи группы, содержащей от двух до шести атомов углерода, по крайней мере с одной углеродуглеродной двойной связью. Термин "алкенилокси", как используют в настоящем изобретении, относится к алкенильной группе,присоединенной к остатку исходной молекулы через атом кислорода. Термин "алкенилоксикарбонил", как используют в настоящем изобретении, относится к алкенилоксигруппе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "алкокси", как используют в настоящем изобретении, относится к алкильной группе, присоединенной к остатку исходной молекулы через атом кислорода. Термин "алкоксиалкил", как используют в настоящем изобретении, относится к алкильной группе,замещенной одной, двумя или тремя алкоксигруппами. Термин "алкоксиалкилкарбонил", как используют в настоящем изобретении, относится к алкоксиалкильной группе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "алкоксикарбонил", как используют в настоящем изобретении, относится к алкоксигруппе,присоединенной к остатку исходной молекулы через карбонильную группу. Термин "алкоксикарбонилалкил", как используют в настоящем изобретении, относится к алкильной группе, замещенной одной, двумя или тремя алкоксикарбонильными группами. Термин "алкил", как используют в настоящем изобретении, относится к группе, производной от прямой или разветвленной цепи насыщенного углеводорода, содержащего от одного до шести атомов углерода. В соединениях по настоящему изобретению, когда m и/или n равны 1 или 2; X и/или Y представляет собой CHR5 и/или CHR6, соответственно, и R5 и/или R6 представляет собой алкил, каждый алкил может необязательно образовывать конденсированное, содержащее от трех до шести членов кольцо со смежным атомом углерода для обеспечения одной из структур, представленных ниже: где z имеет значение 1, 2, 3 или 4, w имеет значение 0, 1 или 2 и R50 представляет собой алкил. Когда w имеет значение 2, две R50 алкильные группы могут быть одинаковыми или различными. Термин "алкилкарбонил", как используют в настоящем изобретении, относится к алкильной группе,присоединенной к остатку исходной молекулы через карбонильную группу. Термин "алкилкарбонилалкил", как используют в настоящем изобретении, относится к алкильной группе, замещенной одной, двумя или тремя алкилкарбонильными группами. Термин "алкилкарбонилокси", как используют в настоящем изобретении, относится к алкилкарбонильной группе, присоединенной к остатку исходной молекулы через атом кислорода. Термин "алкилсульфанил", как используют в настоящем изобретении, относится к алкильной груп-4 017348 пе, присоединенной к остатку исходной молекулы через атом серы. Термин "алкилсульфонил", как используют в настоящем изобретении, относится к алкильной группе, присоединенной к остатку исходной молекулы через сульфонильную группу. Термин "арил", как используют в настоящем изобретении, относится к фенильной группе или бициклической конденсированной кольцевой системе, где одно или оба кольца представляют собой фенильную группу. Бициклические конденсированные кольцевые системы состоят из фенильной группы,конденсированной с содержащей от четырех до шести членов ароматическим или неароматическим карбоциклическим кольцом. Арильные группы по настоящему изобретению могут быть присоединены к остатку исходной молекулы через любой замещаемый атом углерода в группе. Типичные примеры арильных групп включают, но без ограничения, инданил, инденил, нафтил, фенил и тетрагидронафтил. Арильные группы по настоящему изобретению необязательно замещены одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из алкокси, алкоксиалкила, алкоксикарбонила, алкила, алкилкарбонила, вторичной арильной группы, арилалкокси, арилалкила, арилкарбонила, циано, галогена, галогеналкокси, галогеналкила, гетероциклила, гетероциклилалкила, гетероциклилкарбонила, гидрокси, гидроксиалкила, нитро, -NRxRy, (NRxRy)алкила, оксо и -P(O)OR2, где каждый R независимо выбран из водорода и алкила; и где алкильная часть арилалкила и гетероциклилалкила является незамещенной и где вторичная арильная группа, арильная часть арилалкила, арильная часть арилкарбонила,гетероциклила и гетероциклильная часть гетероциклилалкила и гетероциклилкарбонила, кроме того, необязательно замещены одной, двумя или тремя заместителями, независимо выбранными из алкокси, алкила, циано, галогена, галогеналкокси, галогеналкила и нитро. Термин "арилалкенил", как используют в настоящем изобретении, относится к алкенильной группе,замещенной одной, двумя или тремя арильными группами. Термин "арилалкокси", как используют в настоящем изобретении, относится к арильной группе,присоединенной к остатку исходной молекулы через алкоксигруппу. Термин "арилалкоксиалкил", как используют в настоящем изобретении, относится к алкильной группе, замещенной одной, двумя или тремя арилалкоксигруппами. Термин "арилалкоксиалкилкарбонил", как используют в настоящем изобретении, относится к арилалкоксиалкильной группе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "арилалкоксикарбонил", как используют в настоящем изобретении, относится к арилалкоксигруппе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "арилалкил", как используют в настоящем изобретении, относится к алкильной группе, замещенной одной, двумя или тремя арильными группами. Алкильная часть арилалкила, кроме того, необязательно замещена одной или двумя дополнительными группами, независимо выбранными из алкокси, алкилкарбонилокси, галогена, галогеналкокси, галогеналкила, гетероциклила, гидрокси и -NRcRd, где гетероциклил, кроме того, необязательно замещен одним или двумя заместителями, независимо выбранными из алкокси, алкила, незамещенного арила, незамещенного арилалкокси, незамещенного арилалкоксикарбонила, галогена, галогеналкокси, галогеналкила, гидрокси и -NRxRy. Термин "арилалкилкарбонил", как используют в настоящем изобретении, относится к арилалкильной группе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "арилкарбонил", как используют в настоящем изобретении, относится к арильной группе,присоединенной к остатку исходной молекулы через карбонильную группу. Термин "арилокси", как используют в настоящем изобретении, относится к арильной группе, присоединенной к остатку исходной молекулы через атом кислорода. Термин "арилоксиалкил", как используют в настоящем изобретении, относится к алкильной группе,замещенной одной, двумя или тремя арилоксигруппами. Термин "арилоксикарбонил", как используют в настоящем изобретении, относится к арилоксигруппе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "арилсульфонил", как используют в настоящем изобретении, относится к арильной группе,присоединенной к остатку исходной молекулы через сульфонильную группу. Термины "Подвеска" и "подвеска", как используют в настоящем изобретении, относится к группе,которая располагается на атоме азота конечного азотсодержащего кольца, то есть, пирролидиновых кольцах соединения 1 е. Должно быть понятно, что термин "Подвеска" или "подвеска" может относится к реагенту, используемому для присоединения группы к конечному азотсодержащему кольцу или к фрагменту в конечном продукте, то есть, "Подвеска-51" или "Фрагмент Подвески-51, найденный в LS-19". Термин "карбонил", как используют в настоящем изобретении, относится к -С(О)- группе. Термин "карбокси", как используют в настоящем изобретении, относится к -СО 2 Н группе. Термин "циано", как используют в настоящем изобретении, относится к -CN группе. Термин "циклоалкил", как используют в настоящем изобретении, относится к насыщенной моноциклической, углеводородной кольцевой системе, имеющей от трех до семи атомов углерода и ноль гетероатомов. Типичные примеры циклоалкильных групп включают, но без ограничения, циклопропил,циклопентил и циклогексил. Циклоалкильные группы по настоящему изобретению необязательно замещены одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из алкокси,-5 017348 алкила, арила, циано, галогена, галогеналкокси, галогеналкила, гетероциклила, гидрокси, гидроксиалкила, нитро и -NRxRy, где арил и гетероциклил, кроме того, необязательно замещены одной, двумя или тремя заместителями, независимо выбранными из алкокси, алкила, циано, галогена, галогеналкокси, галогеналкила, гидрокси и нитро. Термин "(циклоалкил)алкенил", как используют в настоящем изобретении, относится к алкенильной группе, замещенной одной, двумя или тремя циклоалкильными группами. Термин "(циклоалкил)алкил", как используют в настоящем изобретении, относится к алкильной группе, замещенной одной, двумя или тремя циклоалкильными группами. Алкильная часть (циклоалкил)алкила, кроме того, необязательно замещена одной или двумя группами, независимо выбранными из гидрокси и -NRcRd. Термин "циклоалкилокси", как используют в настоящем изобретении, относится к циклоалкильной группе, присоединенной к остатку исходной молекулы через атом кислорода. Термин "циклоалкилоксиалкил", как используют в настоящем изобретении, относится к алкильной группе, замещенной одной, двумя или тремя циклоалкилоксигруппами. Термин "циклоалкилсульфонил", как используют в настоящем изобретении, относится к циклоалкильной группе, присоединенной к остатку исходной молекулы через сульфонильную группу. Термин "формил", как используют в настоящем изобретении, относится к -СНО группе. Термины "гало" и "галоген", как используют в настоящем изобретении, относится к F, Cl, Br или I. Термин "галогеналкокси", как используют в настоящем изобретении, относится к галогеналкильной группе, присоединенной к остатку исходной молекулы через атом кислорода. Термин "галогеналкоксикарбонил", как используют в настоящем изобретении, относится к галогеналкоксигруппе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "галогеналкил", как используют в настоящем изобретении, относится к алкильной группе,замещенной одним, двумя, тремя или четырьмя атомами галогена. Термин "гетероциклил", как используют в настоящем изобретении, относится к содержащему четыре-, пять-, шесть- или семь- членов кольцу, включающему один, два, три или четыре гетероатома, независимо выбранных из азота, кислорода и серы. Содержащее четыре члена кольцо не имеет двойных связей, содержащее пять членов кольцо имеет от нуля до двух двойных связей и содержащие шесть и семь членов кольца имеют от нуля до трех двойных связей. Термин "гетероциклил" также включает бициклические группы, в которых гетероциклильное кольцо является конденсированным с еще моноциклической гетероциклильной группой или содержащим от четырех до шести членов ароматическими или неароматическими карбоциклическими кольцами; так же как мостиковые бициклические группы, такие как 7-азабицикло[2.2.1]гепт-7-ил, 2-азабицикло[2.2.2]ок-2-тил и 2-азабицикло[2.2.2]ок-3-тил. Гетероциклильные группы по настоящему изобретению могут быть присоединены к остатку исходной молекулы через любой атом углерода или атом азота в группе. Примеры гетероциклильных групп включают, но без ограничения, бензотиенил, фурил, имидазолил, индолинил, индолил, изотиазолил, изоксазолил, морфолинил, оксазолил, пиперазинил, пиперидинил, пиразолил, пиридинил, пирролидинил, пирролопиридинил, пирролил, тиазолил, тиенил, тиоморфолинил, 7-азабицикло[2.2.1]гепт-7-ил, 2-азабицикло[2.2.2]ок-2 тил и 2-азабицикло[2.2.2]ок-3-тил. Гетероциклильные группы по настоящему изобретению необязательно замещены одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из алкокси, алкоксиалкила, алкоксикарбонила, алкила, алкилкарбонила, арила, арилалкила, арилкарбонила,циано, галогена, галогеналкокси, галогеналкила, вторичной гетероциклильной группы, гетероциклилалкила, гетероциклилкарбонила, гидрокси, гидроксиалкила, нитро, -NRxRy, (NRxRy)алкила и оксо, где алкильная часть арилалкила и гетероциклилалкила являются незамещенными и где арил, арильная часть арилалкила, арильная часть арилкарбонила, вторичная гетероциклильная группа и гетероциклильная часть гетероциклилалкила и гетероциклилкарбонила, кроме того, необязательно замещены одной, двумя или тремя заместителями, независимо выбранными из алкокси, алкила, циано, галогена, галогеналкокси,галогеналкила и нитро. Термин "гетероциклилалкенил", как используют в настоящем изобретении, относится к алкенильной группе, замещенной одной, двумя или тремя гетероциклильными группами. Термин "гетероциклилалкокси", как используют в настоящем изобретении, относится к гетероциклильной группе, присоединенной к остатку исходной молекулы через алкоксигруппу. Термин "гетероциклилалкоксикарбонил", как используют в настоящем изобретении, относится к гетероциклилалкоксигруппе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "гетероциклилалкил", как используют в настоящем изобретении, относится к алкильной группе, замещенной одной, двумя или тремя гетероциклильными группами. Алкильная часть гетероциклилалкила, кроме того, необязательно замещена одной или двумя дополнительными группами, независимо выбранными из алкокси, алкилкарбонилокси, арила, галогена, галогеналкокси, галогеналкила, гидрокси и -NRcRd, где арил, кроме того, необязательно замещен одним или двумя заместителями, независимо выбранными из алкокси, алкила, незамещенного арила, незамещенного арилалкокси, незамещенного арилалкоксикарбонила, галогена, галогеналкокси, галогеналкила, гидрокси и -NRxRy. Термин "гетероциклилалкилкарбонил", как используют в настоящем изобретении, относится к ге-6 017348 тероциклилалкильной группе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "гетероциклилкарбонил", как используют в настоящем изобретении, относится к гетероциклильной группе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "гетероциклилокси", как используют в настоящем изобретении, относится к гетероциклильной группе, присоединенной к остатку исходной молекулы через атом кислорода. Термин "гетероциклилоксиалкил", как используют в настоящем изобретении, относится к алкильной группе, замещенной одной, двумя или тремя гетероциклилоксигруппами. Термин "гетероциклилоксикарбонил", как используют в настоящем изобретении, относится к гетероциклилоксигруппе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "гидрокси", как используют в настоящем изобретении, относится к -ОН группе. Термин "гидроксиалкил", как используют в настоящем изобретении, относится к алкильной группе,замещенной одной, двумя или тремя гидроксигруппами. Термин "гидроксиалкилкарбонил", как используют в настоящем изобретении, относится к гидроксиалкильной группе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "нитро", как используют в настоящем изобретении, относится к -NO2 группе. Термин "-NRaRb", как используют в настоящем изобретении, относится к двум группам, Ra и Rb которых присоединены к остатку исходной молекулы через атом азота. Радикалы Ra и Rb независимо выбраны из водорода, алкенила и алкила. Термин "(NRaRb)алкил", как используют в настоящем изобретении, относится к алкильной группе,замещенной одной, двумя или тремя -NRaRb группами. Термин "(NRaRb)карбонил", как используют в настоящем изобретении, относится к -NRaRb группе,присоединенной к остатку исходной молекулы через карбонильную группу. Термин "-NRcRd", как используют в настоящем изобретении, относится к двум группам, радикалыR и Rd которых присоединены к остатку исходной молекулы через атом азота. Радикалы Rc и R независимо выбраны из водорода, алкенилоксикарбонила, алкоксиалкилкарбонила, алкоксикарбонила, алкила,алкилкарбонила, алкилсульфонила, арила, арилалкоксикарбонила, арилалкила, арилалкилкарбонила,арилкарбонила, арилоксикарбонила, арилсульфонила, циклоалкила, циклоалкилсульфонила, формила,галогеналкоксикарбонила, гетероциклила, гетероциклилалкоксикарбонила, гетероциклилалкила, гетероциклилалкилкарбонила, гетероциклилкарбонила, гетероциклилоксикарбонила, гидроксиалкилкарбонила,(NReRf)алкила, (NReRf)алкилкарбонила, (NReRf)карбонила, (NReRf)сульфонила, -C(NCN)OR' и-C(NCN)NRxRy, где R' выбран из алкила и незамещенного фенила и где алкильная часть арилалкила, арилалкилкарбонила, гетероциклилалкила и гетероциклилалкилкарбонила, кроме того, необязательно замещена одной -NReRf группой; и где арил, арильная часть арилалкоксикарбонила, арилалкила, арилалкилкарбонила, арилкарбонила, арилоксикарбонила и арилсульфонила, гетероциклила и гетероциклильная часть гетероциклилалкоксикарбонила, гетероциклилалкила, гетероциклилалкилкарбонила, гетероциклилкарбонила и гетероциклилоксикарбонила, кроме того, необязательно замещена одной, двумя или тремя заместителями, независимо выбранными из алкокси, алкила, циано, галогена, галогеналкокси, галогеналкила и нитро. Термин "(NRcRd)алкенил", как используют в настоящем изобретении, относится к алкенильной группе, замещенной одной, двумя или тремя -NRcRd группами. Термин "(NRcRd)алкил", как используют в настоящем изобретении, относится к алкильной группе,замещенной одной, двумя или тремя -NRcRd группами. Алкильная часть (NRcRd)алкила, кроме того, необязательно замещена одной или двумя дополнительными группами, выбранными из алкокси, алкоксиалкилкарбонила, алкоксикарбонила, алкилсульфанила, арилалкоксиалкилкарбонила, карбокси, гетероциклила, гетероциклилкарбонила, гидрокси и (NReRf)карбонил; где гетероциклил, кроме того, необязательно замещен одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из алкокси, алкила, циано, галогена, галогеналкокси, галогеналкила и нитро. Термин "(NRcRd)карбонил", как используют в настоящем изобретении, относится к -NRcRd группе,присоединенной к остатку исходной молекулы через карбонильную группу. Термин "-NReRf", как используют в настоящем изобретении, относится к двум группам, Re и Rf которых присоединены к остатку исходной молекулы через атом азота. Радикалы Re и Rf независимо выбраны из водорода, алкила, незамещенного арила, незамещенного арилалкила, незамещенного циклоалкила, незамещенного (циклоалкил)алкила, незамещенного гетероциклила, незамещенного гетероциклилалкила, (NRxRy)алкила и (NRxRy)карбонила. Термин "(NReRf)алкил", как используют в настоящем изобретении, относится к алкильной группе,замещенной одной, двумя или тремя -NReRf группами. Термин "(NReRf)алкилкарбонил", как используют в настоящем изобретении, относится к(NR R )алкильной группе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин "(NReRf)карбонил", как используют в настоящем изобретении, относится к -NReRf группе,присоединенной к остатку исходной молекулы через карбонильную группу. Термин "(NReRf)сульфонил", как используют в настоящем изобретении, относится к -NReRf группе,-7 017348 присоединенной к остатку исходной молекулы через сульфонильную группу. Термин "-NRxRy", как используют в настоящем изобретении, относится к двум группам, Rx и Ry которых присоединены к остатку исходной молекулы через атом азота. Радикалы Rx и Ry независимо выбраны из водорода, алкоксикарбонила, алкила, алкилкарбонила, незамещенного арила, незамещенного арилалкоксикарбонила, незамещенного арилалкила, незамещенного циклоалкила, незамещенного гетероциклила и (NRx Ry )карбонила, где Rx и Ry независимо выбраны из водорода и алкила. Термин "(NRxRy)алкил", как используют в настоящем изобретении, относится к алкильной группе,замещенной одной, двумя или тремя -NRxRy группами. Термин "оксо", как используют в настоящем изобретении, относится к группе =O. Термин "сульфонил", как используют в настоящем изобретении, относится к -SO2- группе. Термин "триалкилсилил", как используют в настоящем изобретении, относится к -SiR3 группе, гдеR представляет собой алкил. R группы могут быть одинаковыми или различными. Термин "триалкилсилилалкил", как используют в настоящем изобретении, относится к алкильной группе, замещенной одной, двумя или тремя триалкилсилильными группами. Термин "триалкилсилилалкокси", как используют в настоящем изобретении, относится к триалкилсилилалкильной группе, присоединенной к остатку исходной молекулы через атом кислорода. Термин "триалкилсилилалкоксиалкил", как используют в настоящем изобретении, относится к алкильной группе, замещенной одной, двумя или тремя триалкилсилилалкоксигруппами. В соединениях по настоящему изобретению существуют асимметричные центры. Указанные центры обозначают с помощью символов "R" или "S", в зависимости от конфигурации заместителей вокруг хирального атома углерода. Должно быть понятно, что изобретение охватывает все стереохимические изомерные формы или их смеси, которые обладают способностью ингибирования NS5A. Индивидуальные стереоизомеры соединений могут быть получены путем синтеза из коммерчески доступных исходных продуктов, которые содержат хиральные центры или получены из смеси энантиомерных продуктов,с последующим разделением, таким как превращение в смесь диастереомеров, с последующим разделением или перекристаллизацией, хроматографическими спосбами или непосредственным разделением энантиомеров на хиральных хроматографических колонках. Исходные соединения специфической стереохимии являются или коммерчески доступными или могут быть получены и разделены с помощью способов, известных среднему специалисту. Некоторые соединения по настоящему изобретению могут также существовать в различных стабильных конформационных формах, которые могут быть разделимы. Крутящая асимметрия из-за ограниченного вращения около асимметричной простой связи, например, из-за пространственного затруднения или деформации кольца, может допустить разделение различных конформационных вариантов структуры. Настоящее изобретение включает каждый конформационный изомер указанных соединений и их смесей. Термин "соединения по настоящему изобретению" и эквивалентные выражения предназначены,чтобы охватить соединения формулы (I) и их фармацевтически приемлемые энантиомеры, диастереомеры и соли. Подобным образом, ссылки на промежуточные соединения предназначены, чтобы охватить их соли, где так позволяет контекст. Соединения по настоящему изобретению могут существовать в виде фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль", как используют в настоящем изобретении, представляет соли или цвиттерионные формы соединений по настоящему изобретению, которые растворимы в воде или масле, или диспергированы, которые в рамках звучащего медицинского суждения приемлемы для применения в контакте с тканями пациента без излишней токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соразмерных приемлемому соотношению польза/риска и эффективны для их обозначенного применения. Соли могут быть получены в конце при выделении и очистке соединений или раздельно, путем взаимодействия приемлемого атома азота с приемлемыми кислотами. Типичные кислотно-аддитивные соли включают ацетат, адипат, альгинат, цитрат, аспарат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, кафорсульфонат; диглюконат, дигидробромид,дигидрохлорид, дигидроиодид, глицерофосфат, гемисульфат, гептапоат, гексаноат, формиат, фумарат,гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат, мезитиленсульфонат,метансульфонат, нафтиленсульфонат, никотинат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат,персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, трихлорацетат, трифторацетат, фосфат, глютамат, бикарбонат, паратолуолсульфонат и ундеканоат. Примеры кислот, которые могут быть применены, чтобы образовать фармацевтически приемлемые аддитивные соли включают неорганические кислоты, такие как хлористо-водородная, бромисто-водородная, серная и фосфорная и органические кислоты, такие как щавелевая, малеиновая, янтарная и лимонная. Основно-аддитивные соли могут быть получены а конце при выделении и очистке соединений путем взаимодействия карбоксильной группы с приемлемым основанием, таким как гидроксид, карбонат или бикарбонат катиона металла или с аммиаком, или с органическим первичным, вторичным или третичным амином. Катионы фармацевтически приемлемых солей включают литий, натрий, калий, кальций,магний и алюминий, также как катионы нетоксичного четвертичного амина, такого как аммоний, тетра-8 017348 метиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, диэтиламин,этиламин, трибутиламин, пиридин, N,N-диметиланилин, N-метилпиперидин, N-метилморфолин, дициклогексиламин, прокаин, дибензиламин, N,N-дибензилфенэтиламин и N,N'-дибензилэтилендиамин. Другие типичные органические амины, пригодные для образования основно-аддитивных солей включают этилендиамин, этаноламин, диэтаноламин, пиперидин и пиперазин. Когда возможно для использования в терапии, терапевтически эффективные количества соединения формулы (I), также как его фармацевтически приемлемых солей, могут быть введены в виде химического соединения, можно представить активный ингредиент в виде фармацевтической композиции. Таким образом, изобретение, кроме того, обеспечивает фармацевтические композиции, которые включают терапевтически эффективные количества соединений формулы (I) или их фармацевтически приемлемые соли и один или большее количество фармацевтически приемлемых носителей, разбавителей или наполнителей. Термин "терапевтически эффективное количество", как используют в настоящем изобретении, относится к общему количеству каждого активного компонента, которое достаточно, чтобы продемонстрировать значительную пользу для пациента, например снижению вирусного воздействия. Когда используемый индивидуальный активный ингредиент, его вводят сам по себе, термин сам по себе относится к данному ингредиенту. Когда используют комбинацию, термин относится к объединенному количеству активных ингредиентов, которые приводят к терапевтическому эффекту или при введении в комбинации,периодически или одновременно. Соединения формулы (I) и их фармацевтически приемлемые соли являются теми, как описано выше. Носитель(и), разбавитель(и) или наполнитель(и) должны быть совместимы в смысле того, чтобы быть совместимыми с другими ингредиентами состава и не быть опасными для его реципиента. В соответствии с другим аспектом настоящего изобретения также обеспечен способ получения фармацевтического состава, включая смешивание соединения формулы (I) или его фармацевтически приемлемой соли с одним или большим количеством фармацевтически приемлемых носителей,разбавителей или наполнителей. Термин "фармацевтически приемлемый", используемый в данном описании, относится к тем соединениям, веществам, композициям и/или лекарственным формам, которые при взвешенном медицинском решении подходят для применения в контакте с тканями пациентов без избыточной токсичности, раздражения, аллергических реакций или других проблем или осложнений,соответствующих приемлемому отношению польза/риск и являются эффективными в отношении их предполагаемого использования. Фармацевтические препараты могут быть представлены в дозированных лекарственных формах,содержащих заранее определенное количество активного ингредиента на единицу дозирования. Уровни дозировок соединений настоящего изобретения примерно от 0,01 до 250 мг на килограмм ("мг/кг") массы тела в день, предпочтительно примерно от 0,05 до 100 мг/кг массы тела в день являются обычными для монотерапии с целью профилактики и лечения HCV-опосредованного заболевания. Обычно фармацевтические композиции настоящего изобретения вводят примерно от 1 до 5 раз в сутки или альтернативно в виде продолжающейся инфузии. Такое введение может применяться в качестве длительной или срочной терапии. Количество активного ингредиента, которое может быть комбинировано с веществаминосителями, для получения единой лекарственной формы, будет варьироваться в зависимости от патологического состояния, по поводу которого проводится лечение, тяжести состояния, времени введения,пути введения, скорости выведения используемого соединения, продолжительности лечения, возраста,пола, веса и состояния пациента. Предпочтительными дозированными лекарственньми формами являются те, которые содержат суточную дозу или субдозу активного ингредиента, как было выше указано, или их подходящую часть. Лечение может быть начато малыми дозами, существенно меньшими, чем оптимальная доза соединения. После этого дозировку постепенно увеличивают до достижения оптимального эффекта в этих условиях. Вобщем, соединение наиболее желательно вводить при уровне концентрации, который обычно приводит к эффективным противовирусным результатам и не вызывает вредных или опасных побочных эффектов. Когда композиции настоящего изобретения включают комбинацию соединения настоящего изобретения и одного или более дополнительного терапевтического или профилактического агента, и оба соединение и дополнительный агент обычно находятся на уровне дозирования примерно от 10 до 150% и более предпочтительно от около 10 до 80% доз, обычно вводимых в режиме монотерапии. Фармацевтические препараты могут быть адаптированы для введения посредством любого подходящего пути, например пероральным путем (включая защечный подъязычный), ректальным, интраназальным, местным (включая защечный, подъязычный или чрескожный), вагинальным или парентеральным (включая подкожные, внутрикожные, внутримышечные, внутрисуставные, внутрисиновиальные,интрастернальные, интратекальные, внутриочаговые, внутривенные или внутрикожные инъекции или инфузии) путем. Такие препараты могут быть приготовлены с использованием любого из способов, известных в области фармацевтики, например, путем сочетания активного ингредиента с носителем(ями) или наполнителем(ями). Пероральное введение или введение посредством инъекции предпочтительно. Фармацевтические препараты, адаптированные для перорального введения, могут быть приготовлены как дискретные единицы, такие как капсулы или таблетки; порошки или гранулы; растворы или-9 017348 суспензии в водных или неводных жидкостях; пищевые пенки или взбитые массы; или жидкие эмульсии масло-в-воде или эмульсии вода-в-масле. Например, для перорального введения в форме таблетки или капсулы активный лекарственный компонент может быть комбинирован с пероральным нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и т.п. Порошки готовят путем измельчения соединения до подходящего малого размера и смешивания с аналогично измельченным фармацевтическим носителем, таким как пищевой углевод, как, например, крахмал или маннитол. Ароматизатор, консервант, диспергирующий агент или краситель может также присутствовать. Капсулы готовят путем приготовления порошковой смеси, как описано выше, и заполнения ею формованного желатинового футляра. Глиданты и смазывающие вещества, такие как коллоидный кремний, тальк, магния стеарат, кальция стеарат, или твердый полиэтиленгликоль могут быть добавлены к порошковой смеси перед процедурой заполнения. Дезинтегрирующий или солюбилизирующий агент,такой как агар-агар, кальция карбонат или натрия карбонат может быть добавлен для улучшения биодоступности лекарственного средства, в случаях приема капсулы внутрь. Более того, по желанию или по необходимости подходящие связывающие вещества, смазывающие вещества, дезинтегрирующие агенты и красители могут также быть включены в эту смесь. Подходящие смазывающие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или беталактоза, сахаристые вещества кукурузы, природные и синтетические камеди, такие как гуммиарабик,трагакант или натрия альгинат, карбоксиметилцеллюлоза, полиэтиленгликоль и т.п. Смызывающие вещества, используемые в этих лекарственных формах, включают натрия олеат, натрия хлорид и т.п. Дезинтегрирующие вещества включают помимо прочего крахмал, метилцеллюлозу, агар-агар, бетонит,ксантановую камедь и т.п. Таблетки изготавливают, например, путем приготовления порошковой смеси,гранулирования или комкования, добавления смазывающего вещества и дезинтегрирующего вещества и прессования в таблетки. Порошковую смесь готовят путем смешивания соединения, подходящим образом измельченного, с разбавителем или основой, как описано выше, и по необходимости со связывающим веществом, таким как карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, замедлитель растворения, такой как парафин, ускоритель всасывания, такой как четвертичная соль и/или поглощающее вещество, такое как бетонит, каолин или дикальция фосфат. Порошковая смесь может быть приготовлена путем смачивания связующими веществами, такими как сироп, крахмальная паста,смесь гуммиарабика или раствор целлюлозных или полимерных материалов и продавливания через сито. В качестве альтернативы гранулированию порошковая смесь может быть пропущена через таблеточную машину и результатом являются неправильной формы комки, разделенные на гранулы. Гранулы могут быть смазаны для предотвращения прилипания к таблетирующему прессу посредством добавления стеариновой кислоты, стеариновой соли, талька или минерального масла. Смазанную смесь затем прессуют в таблетки. Соединения настоящего изобретения могут быть также комбинированы с сыпучим инертным носителем и прессованы в таблетки напрямую без проведения этапов гранулирования или комкования. Прозрачная или непрозрачная оболочка, состоящая из герметичного слоя шеллака, оболочка из сахара или полимерного материала и гладкая оболочка из воска может быть предусмотрена. Красители могут быть добавлены к этим оболочкам, чтобы различать различные лекарственные формы. Жидкости для перорального применения, такие как раствор, сиропы и эликсиры, могут быть приготовлены в дозированных лекарственных формах, так что заданная доза содержит определенное количество соединения. Сиропы могут быть приготовлены путем растворения соединения в подходящем ароматизированном водном растворе, тогда как эликсиры готовят с помощью нетоксичного носителя. Солюбилизирующие средства и эмульгаторы, такие как этоксилированные изостеариловые спирты и эфиры полиоксиэтиленсорбитола, консерванты, ароматизирующие добавки, такие как перечное масло или природные подсластители, или сахарин, или другие искусственные подсластители и т.п. могут быть добавлены. Если это необходимо, дозированные лекарственные средства для перорального введения могут быть микроинкапсулированными. Препарат также может быть приготовлен для продления или замедления высвобождения, например, с помощью помещения состоящего из частиц материала в оболочку из полимеров, воска или т.п. Соединения формулы (I) и их фармацевтически приемлемые соли могут также быть введены в форме липосомальных систем доставки, таких как малые моноламеллярные везикулы, большие моноламеллярные везикулы и мультиламеллярные везикулы. Липосомы могут быть образованы из множества фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Соединения формулы (I) и их фармацевтически приемлемые соли могут быть также доставлены с помощью моноклональных антител в качестве индивидуальных носителей, к которым молекулы соединения прикреплены. Соединения могут также быть связаны с растворимыми полимерами, такими как носители нацеливаемых лекарств. Такие полимеры могут включать поливинилпирролидон, пиран сополимер, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный остатками пальмитоила. Более того, соединения могут быть связаны с классом биоразлагаемых полимеров, применяемых для достижения контролируемого высвобождения лекар- 10017348 ственного препарата, например полимолочная кислота, полиэпсилонкапролактон, полигидроксимасляная кислота, полиортоэфиры, полиацетали, полигидропираны, полицианоакрилаты и поперечно-сшитые или амфипатические блок-сополимеры гидрогелей. Фармацевтические препараты, адаптированные для чрескожного введения, могут быть представлены в виде отдельных пластырей, предназначенных для пребывания в тесном контакте с эпидермисом реципиента в течение длительного периода времени. Например, активный ингредиент может поступать из пластыря посредством ионофореза, как в общем описано в Pharmaceutical Research 1986, 3(6), 318. Фармацевтические препараты, адаптированные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел. Фармацевтические препараты, адаптированные для ректального введения, могут быть приготовлены в виде суппозиториев или клизм. Фармацевтические препараты, адаптированные для интраназального введения, в которых носитель является твердым веществом, включают сыпучий порошок, имеющий размер частиц, например, в диапазоне от 20 до 500 мкм, который вводится таким же образом, как и нюхательный порошок, т.е. путем быстрого вдыхания через носовой ход порошка из контейнера, который держат близко к носу. Подходящие препараты в случае, когда носитель жидкий для введения в виде интраназального спрея или носовых капель, включают водные или масляные растворы активного ингредиента. Фармацевтические препараты, адаптированные для введения путем ингаляции, включают мелкодисперсные пудры и аэрозоли, которые могут быть получены с помощью различных видов дозирующих аэрозолей, небулайзеров или инсуфляторов. Фармацевтические препараты, адаптированные для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или спреев. Фармацевтические препараты, адаптированные для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буфера, бактериостатические средства, которые делают препарат изотоничным с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Препараты могут быть представлены в однодозных или многодозных контейнерах, например,запаянные ампулы и флаконы, и могут храниться в высушенном сублимацией (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекций,непосредственно перед применением. Растворы для немедленного приготовления препарата для инъекции и суспензии могут быть приготовлены из стерильных порошков, гранул и таблеток. Необходимо понимать, что наряду с ингредиентами, в частности упомянутыми выше, препараты могут включать другие агенты, традиционные в области, имеющей отношение к типу рассматриваемого препарата, например, таковые, подходящие для перорального введения могут включать ароматизаторы. Термин "пациент" включает как людей, так и других млекопитающих. Термин "лечение" относится к: (i) профилактике заболевания, расстройства или патологического состояния, возникающих у пациента, который может быть предрасположен к заболеванию, расстройству и/или патологическому состоянию, но еще не диагностировано их наличие у этого пациента; (ii) подавлению заболевания, расстройства или патологического состояния, т.е. торможение их развития; и (iii) облегчение заболевания, расстройства или патологического состояния, т.е. вызывание регрессии заболевания, расстройства и/или патологического состояния. Соединения настоящего изобретения могут также быть введены с циклоспорином, например, циклоспорином А. Циклоспорин А демонстрирует активность в отношении HCV в клинических исследованиях (Hepatology 2003, 38, 1282; Biochem. Biophys. Res. Commun. 2004, 313,42; J. Gastroenterol. 2003,38,567). В нижеприведенной табл. 1 перечислены некоторые показательные примеры соединений, которые могут быть введены с соединениями настоящего изобретения. Соединения по изобретению могут быть введены с другими обладающими анти-HCV активностью соединениями в комбинированной терапии,либо совместно, либо раздельно, или путем сочетания этих соединений в композиции. Соединения настоящего изобретения могут также быть использованы в качестве лабораторных реактивов. Соединения могут быть средством обеспечения исследовательских инструментов для дизайна исследований вирусной репликации, утверждения животных систем исследования и исследований структурной биологии для дальнейшего углубления знаний механизмов заболевания HCV. Более того, соединения настоящего изобретения пригодны для установления или определения сайта связывания других противовирусных соединений, например, путем конкурентного ингибирования. Соединения по изобретению могут также быть пригодны для лечения или предотвращения вирусной контаминации материалов и, таким образом, снижать риск вирусной инфекции лабораторного или медицинского персонала или пациентов, которые контактируют с такими веществами как, например,кровь, ткань, хирургические инструменты и костюмы, лабораторные инструменты и костюмы, а также аппараты и материалы для забора и переливания крови. Настоящее изобретение предназначено для охватывания соединений, имеющих формулу (I), полученных посредством процессов синтеза или посредством метаболических процессов, включая таковые,происходящие в организме человека или животного (in vivo) или процессов, происходящих in vitro. Сокращения, используемые в настоящем описании, включая в особености включенные в иллюстративные схемы и последующие примеры хорошо известны среднему специалисту. Некоторые из сокращений используют как следующие: HATU для гексафторфосфата O-(7-азабензотриазол-1-ил)-N,N,N',N'тетраметилурония; Boc или ВОС для трет-бутоксикарбонила; NBS для N-бромсукцинимида; tBu или трет-Bu для трет-бутила; SEM для -(триметилсилил)этоксиметила; DMSO для диметилсульфоксида; МеОН для метанола; TFA для трифторуксусной кислоты; RT для комнатной температуры или времени задержки (следуя из контекста); tR для времени задержки; EDCl для гидрохлорида 1-(3 диметиламинопропил)-3-этилкарбодиимида; DMAP для 4-диметиламинопиридина; ТГФ для тетрагидрофурана; DBU для 1,8-диазабицикло[5.4.0]ундек-7-ен; трет-Bu; DEA для диэтиламина; HMDS для гексаметилдисилазида; DMF для N,N-диметилформамида; BzI для бензила; EtOH для этанола; iPrOH или изоPrOH для изопропанола; Me2S для диметилсульфида; Et3N или TEA для триэтиламина; Ph для фенила; ОАс для ацетата; EtOAc для этилацетата; dppf для 1,1'-бис(дифенилфосфино)ферроцена; iPr2EfN илиDIPEA для диизопропилэтиламин; Cbz для карбобензилокси; н-BuLi для н-бутиллитий; ACN для ацетонитрила; h или hr для часов; m или min для минут; s для секунд; LiHMDS для гексаметилдисилазида лития; DIBAL для диизобутил алюминий гидрида; TBDMSCl для трет-бутилдиметилсилилхлорида; Me для метила; са. для приблизительно; ОАс для ацетата; iPr для изопропила; Et для этила; Bn для бензила; иHOAT для 1-гидрокси-7-азабензотриазола. Соединения и процессы по настоящему изобретению будут лучше поняты в совокупности со следующими схемами синтеза, которые демонстрируют способы, с помощью которых могут быть получены соединения по настоящему изобретению. Исходные продукты могут быть получены из коммерческих источников или получены с помощью общепринятых способов из уровня техники, известных среднему специалисту. Среднему специалисту будет очевидно, что соединения, определенные выше, синтезируют путем замещения соответствующих реагентов и агентов и при синтезе, показанным ниже. Среднему специалисту будет также очевидно, что стадии выборочного введения защиты и снятия защиты, так же как порядок проведения указанных стадий, могут быть проведены в различной очередности, в зависимости от природы переменных для успешного завершения синтеза, представленного ниже ниже. Переменные являются теми, как определено выше, если иное ниже не указано. Схема. Симетричные или асимметричные бифенилы. Арилгалоид 1 и бороновый эфир 2 могут быть связаны для получения биарила 3, используя стандартные условия связывания по Сузуки-Миаура (Angew Chem. Int. Ed. Engl 2001, 40, 4544). Следует отметить, что аналог бороновый кислоты 2 может быть использован вместо эфира. Моноснятие защиты с пирролидинового остатка может быть осуществлено, когда R12 и R13 являются различными. Когда R12 = бензил и R13 = трет-бутил обрабатывают в условиях гидрогенолиза, получают соединение 4. Например,может быть использован Pd/C катализатор в присутствии основания, такого как карбонат калия. Ацилирование соединения 4 может быть осуществлено в обычных условиях ацилирования. В этом отношении- 14017348 может быть использован реагент связывания, такой как HATU в комбинации с аминовым основанием,таким как основание Ханига. Альтернативно, соединение 4 может взаимодействовать с изоцианатом или карбамоилхлоридом для обеспечения соединений формулы 5, где R представляет собой амин. Кроме того, снятие защиты с соединения 5 может быть осуществлено путем обработки сильной кислотой, такой как HCl или трифторуксусная кислота. Могут быть использованы стандартные условия, аналогичные тем, которые используют, чтобы превратить соединение 4 в соединение 5 для получения соединения 7 из соединения 6. В другом воплощении, где R12 = R13 = трет-Вu, прямое превращение в соединение 8 может быть осуществлено путем обработки соединения 3 сильной кислотой, такой как HCl или трифторуксусная кислота. Превращение соединения 8 в соединение 7 осуществляют аналогичным образом в соответствии со способами, используемыми для получения соединения 5 из соединения 4 или соединения 7 из соединения 6. В этом случае, тем не менее, подвески в соединении 7 будут идентичны. Схема 2. Асимметрично замещенные бифенилы. Превращение соединения 6 (из схемы 1) в соединение 10 может быть выполнено, используя стандартные условия связывания амида, такое как HATU с основанием амина, таким как основание Ханига. Снятие защиты может быть осуществлено сильной кислотой, такой как HCl или трифторуксусной кислотой, получая на выходе соединение 11. Соединение 11 может затем быть превращено в соединения 12, 13 или 14, используя хлорангидрид кислоты, изоцианат, или карбамоилхлорид, или хлорформиат соответственно.- 15017348 Схема 3. Симметрично преобразованные бифенилы. Соединение 15 (15 = 7 (схема 1), где каждый R9 представляет собой -CH(NHBoc)R18) может быть превращено в соединение 16 путем обработки сильной кислотой, такой как HCl или трифторуксусная кислота. Соединения 17, 18 и 19 могут быть получены из соединения 16 путем обработки соединения 16 соответствующим хлорформиатом, изоцианатом, или карбамоилхлоридом, или хлорангидридом кислоты соответственно. Схема 4. Симметричные бифенилы. Симметричные бифенильные аналоги (соединения формулы 7, где обе части молекулы являются эквивалентными) могут быть синтезированы, исходя из бромкетона 20. Аминирование путем замещения нуклеофилом, таким как азид, фталимид или предпочтительно диформиламид натрия (Yinglin and Hongwen, Synthesis 1990, 122) с последующим снятием защиты дает соединение 21. Конденсация в обычных условиях аминирования, таких как HATU и основание Ханига с соответствующей защищенной аминокислотой обеспечивает соединение 22. Нагревание с ацетатом аммония в термальных условиях или в условиях микроволнового облучения приводит к образованию соединения 3, с которого может быть снята защита сильной кислотой, такой как HCl или трифторуксусная кислота (R12 = R13 = трет-Вu) или путем гидрогенолиза газообразным водородом и катализатором из переходного металла, таким как Pd/C (R12 =R13 = бензил). Ацилирование может быть осуществлено карбоновой кислотой (R9CCН) в соответствии со способом, аналогичным превращению соединения 21 в соединение 22. Образование мочевины может быть осуществлено путем обработки соответствующим изоцианатом (R9 = R24R25N; R25 =Н) или карбамоилхлоридом (R9 = R24 R25 N; R25 отличен от водород). Схема 5. Исходные продукты 25 и 2. Схема 5 описывает получение некоторых из исходных продуктов, неоходимых для синтетических последовательностей, представленных на схемах 1-4. Ключевое промежуточное соединение 25 (аналогично соединению 1 на схеме 1) получают из кето-амида 24 или кето-эфира 27 путем нагревания с ацета- 16017348 том аммония в термальных условиях или в условиях микроволнового облучения. Кето-амид 24 может быть получен из соединения 23 путем конденсации с соответствующими цикличными или ацикличными аминокислотами в обычных условиях образования амида. Бромид 26 может привести к получению соединения 23 путем обработки нуклеофилом, таким как азид, фталимид или диформиламид натрия (Synthesis 1990, 122), с последующим снятием защиты. Бромид 26 может также быть превращен в соединение 27 путем взаимодействия с соответствующими цикличными или ацикличными N-защищенными аминокислотами в присутствии основания, такого как карбонат калия или бикарбонат натрия. Броминирование соединения 28 с источником иона броминирования, таким как бром, NBS или CBr4 приводит к образованию соединения 2d. Бромид 25 может быть превращен в бороновый эфир 2 путем обработки биспинакалотодибором при катализе палладия в соответствии с способом, описанным в Journal of Organic Схема 6. Исходный продукт 31 а. В другом воплощении исходные продукты, такой как 31 а (аналогичный соединению 25 на схеме 5 и соединению 1 на схеме 1), могут быть получены путем взаимодействия бромимидазольного производного 31 в условиях связывания по Сузуки с различными хлорзамещенными арилбороновыми кислотами,которые могут или быть получены с помощью стандартных методик (см., например, Organic Letters 2006,8, 305 и ссылки, приведенные там), или приобретены от коммерческих поставщиков. Бромимидазол 31 может быть получен путем броминирования имидазола 30 источником иона бромирования, таким как бром, CBr4 или N-бромсукцинимид. Имидазол 30 может быть получен из N-защищенных аминокислот,которые являются соответствующим образом замещенными путем взаимодействия с глиоксалем в метанольном растворе гидроксида аммония. Схема 7. Гетероарилы. В еще другом воплощении настоящего изобретения, арилгалоид 32 может быть связан в условиях по Сузуки-Миура катализа палладием, чтобы образовать гетероарильное производное 34. Соединение 34 может быть превращено в соединение 35 путем обработки в условиях гидрогенолиза водородом и катализатором из переходного металла, таким как палладий на угле (R13 = бензил). Ацилирование соединения 35 может быть осуществлено соответствующим хлорангидридом кислоты (R9COCl) в присутствии основания, такого как триэтиламин, с соответствующим образом замещенной карбоновой кислотой (R9CO2H) в присутствии стандартного реагента связывания, такого как HATU или с помощью изоцианата (R27NCO,где R9 = R27R28N-; R28 =Н) или карбамоилхлорида (R27R28NCOCl, где R9 = R27R28N-). Соединение 37 может быть получено из соединения 36 (R12 = трет-Вu) путем обработки сильной кислотой, такой как HCl или трифторуксусная кислота. Ацилирование полученного амина в соединение 37, что дает соединение 38, может быть осуществлено как преобразование соединения 35 в соединение 36. В случаях, где R12 =R13, соединение 34 может быть непосредственно преобразовано в соединение 39 путем обработки сильной кислотой, такой как HCl или трифторуксусная кислота (R12 = R13 = трет-Вu) или путем применения условий гидрогенолиза водородом и катализатора из переходного металла, такого как палладий на угле(R12 = R13 = бензил). Ацилирование соединения 39 может быть осуществлено аналогичным образом, так как описано для преобразования соединения 35 в соединение 36. Схема 8. Гетероарилхлорид 29 может быть превращен в симетричный аналог соединения 40 путем обработки исходным палладием, таким как дихлорбис(бензонитрил)палладия в присутствии тетракис(диметиламино)этилена при повышенной температуре. Удаление SEM эфира и Boc карбаматов, находящихся в соединении 40, может быть осуществлено в одну стадию путем обработки сильной кислотой,такой как HCl или трифторуксусная кислота, обеспечивая соединение 41. Превращение в соединение 42 может быть осуществлено в соответствии с условиями, аналогичными условиям, используемым при превращении соединения 38 в соединение 39 на схеме 7. Схема 9. Симметричная подвеска замещенных гетероарилов. Соединение 43 (аналогичное соединению 42, где R23 = -CH(NHBoc)R24) может быть превращено в соединения 45, 46 и 47 через методики, аналогичные тем, которые описаны на схеме 3. В случаях, где R20= алкоксиметил-(то есть SEM), удаление может быть осуществлено вместе с удалением Boc карбамата(II) для обеспечения соединения 55, которое может быть затем преобразовано в бромкетон 51 путем обработки источником иона бромирования, таким как N-бромсукцинимид, CBr4 или бром. Альтернативно,кето-замещенные гетероарилбромиды 53 могут быть непосредственно превращены в соединение 51 путем обработки источником иона бромирования, таким как бром, CBr4 или N-бромсукцинимид. Бромид 51 может быть превращен в аминокетон 48 путем добавления азида натрия, фталимида калий или диформиламида натрия (Synthesis 1990 122) с последующим снятием защиты. Аминокетон 48 может затем быть связан с соответствующим образом замещенной аминокислотой в обычных условиях образования амида(то есть; реагент связывания, такой как HATU в присутствии мягкого основания, такого как основание Ханига) для обеспечения соединения 49. Соединение 49 может затем быть, кроме того, преобразовано в имидазол 50 путем взаимодействия с ацетатом аммония в термальных условиях или в условиях микроволнового облучения. Альтернативно, соединение 51 может непосредственно взаимодействовать с соответствующим образом замещенной аминокислотой в присутствии основания, такого как бикарбонат натрия или карбонат калия, обеспечивая соединение 52, которое может в свою очередь взаимодействовать с ацетатом аммония в термальных условиях или в условиях микроволнового облучения для обеспечения соединения 50. Имидазол 50 может быть защищенньм алкоксилметильной группой путем обработки соответствующим алкоксиметилгалоидом, таким как 2-(триметилсилил)этоксиметилхлорид после певичного депротонирования сильным основанием, таким как гидрид натрия. Схема 11. Замещенные фенилглициновые производные. Замещенные фенилглициновые производные могут быть получены с помощью ряда способов,представленных ниже. трет-Бутиловый эфир фенилглицина может быть восстановительно алкилирован(направление А) соответствующим альдегидом и восстановителем, таким как цианоборогидрид натрия в кислой среде. Гидролиз трет-бутилового эфира может быть осуществлен сильной кислотой, такой какHCl или трифторуксусная кислота. Альтернативно, фенилглицин может быть алкилирован алкилгалоидом, таким как этилиодид и основанием, таким как бикарбонат натрия или карбонат калия (направление В). Направление С демонстрирует восстановительное алкилирование фенилглицина как в направлении А и последующее вторичное восстановительное алкилирование альтернативным альдегидом, таким как формальдегид в присутствии агента восстановления и кислоты. Направление D демонстрирует синтез замещенных фенилглицинов через соответствующие аналоги миндальной кислоты. Превращение вторичных спиртов в компонент уходящей группы может быть осуществлено с помощью птолуолсульфонилхлорида. Замещение тозилатной группы соответствующим амином с последующим восстановительным удалением бензилового эфира может обеспечить замещенные фенилглициновые производные. В направлении Е рацемическое замещенное фенилглициновое производное разделяют путем эстерификации энантиомерно чистым хиральным вспомогательным веществом, таким как, но без ограничения, (+)-1-фенилэтанол, (-)-1-фенилэтанол, оксазолидон Эванса или энантиомерно чистотый пантолактон. Разделение диастереомеров осуществляют через хроматографию (силикагель, HPLC, кристаллизация и т.д.) с последующим удалением хирального вспомогательного вещества, обеспечивая энантиомерно чистотые фенилглициновые производные. Направление Н демонстрирует синтетическую последовательность, которая пересекается с направлением Е, где вышеупомянутое хиральное вспомогательное вещество вводят до ввода амина. Альтернативно, эфир арилуксусной кислоты может быть броминирован источником иона бромирования, таким как бром, N-бромсукцинимид или CBr4. Полученный бензилированный бромид может быть замещен различными моно- или дизамещенными аминами в присутствии основания третичного амина, такого как триэтиламин или основание Ханига. Гидролиз метилового эфира путем обработки гидроксидом лития при пониженной температуре или 6N раствором HCl- 19017348 при повышенной температуре, обеспечивает замещенные фенилглициновые производные. Другой способ показан в направлении G. Аналоги глицина могут быть произведены различными арилгалоидами в присутствии исходного палладия (0), такого как бис(трибутилфосфин) палладий и основания, такого как фосфат калия. Полученный эфир может затем быть гидролизован путем обработки основанием или кислотой. Должно быть понятно, что в уровне техники существуют другие, хорошо известные способы получения фенилглициновых производных и могут быть улучшены для обеспечения требуемых соединений в настоящем описании. Также должно быть понятно, что конечные фенилглициновые производные могут быть очищены до энантиомерной чистоты, выше чем 98% через препаративную HPLC. Схема 12. Ацилированные аминокислотные производные. В другом воплощении по настоящему изобретению ацилированные фенилглициновые производные могут быть получены как представлено ниже. Фенилглициновые производные, где карбоновую кислоту защищают в виде легко удаляемого эфира, могут быть ацилированы хлорангидридом кислоты в присутствии основания, такого как триэтиламин для обеспечения соответствующих амидов (направление А). Направление В демонстрирует ацилирование исходной фенилглициновой производной соответствующим хлорформиата, в то время как направление С представляет взаимодействие с соответствующим изоцианатом или карбамоилхлоридом. С каждого из трех промежуточных соединений, показанных в направлениях А-С, может быть снята защита с помощью способов, известных среднему специалисту (например, обработка трет-бутилового эфира сильным основанием, таким как HCl или трифторуксусная кислота). Схема 13. Аминозамещенные фенилуксусные кислоты могут быть получены путем обработки хлорметилфенилуксусной кислоты избытком амина. Условия анализа структуры Оценку чистоты и масс-спектра низкого разрешения проводят на Shimadzu LC system, связанной сWaters Micromass ZQ MS system. Следует отметить, что времена задержки могут незначительно меняться между устройствами. LC условия, применяемые для определения времени задержки (RT), следующие:- 20017348 Условие 1. Колонка = Phenomenex-Luna 3.050 мм S10 Исходный %В = 0 Конечный %В = 100 Градиент времени = 2 мин Время окончания = 3 мин Скорость потока = 4 мл/мин Длина волны = 220 нм Растворитель А = 0.1% TFB 10% смесь метанол/90%Н 2 О Растворитель В = 0.1% TFB 90% смесь метанол/10% H2O Условие 2. Колонка = Phenomenex-Luna 4.650 мм S10 Исходный %В = 0 Конечный %В = 100 Градиент времени = 2 мин Время окончания = 3 мин Скорость потока = 5 мл/мин Длина волны = 220 нм Растворитель А = 0.1% TFB 10% смесь метанол/90%Н 2 О Растворитель В = 0.1% TFB 90% смесь метанол/10% H2O Условие 3. Колонка = HPLC XTERRA C18 3.050 мм S7 Исходный %В = 0 Конечный %В = 100 Градиент времени = 3 мин Время окончания = 4 мин Скорость потока = 4 мл/мин Длина волны = 220 нм Растворитель А = 0.1% TFB 10% смесь метанол/90%Н 2 О Растворитель В = 0.1% TFB 90% смесь метанол/10% Н 2 О Условие M1. Колонка: Luna 4.650 мм S10 Исходный %В = 0 Конечный %В = 100 Градиент времени = 3 мин Время окончания = 4 минFlow rate = 4 мл/мин Растворитель А: = 95% Н 2 О: 5% CH3CN, 10 мм ацетат аммония Растворитель В: = 5% Н 2 О : 95% CH3CN; 10 мм ацетат аммония. Синтез общих частей Подвеска-1 Суспензию 10% Pd/C (2.0 г) в метаноле (10 мл) добавляют к смеси (R)-2-фенилглицина (10 г, 66.2 ммоль), формальдегида (33 мл 37 вес.% в воде), 1 N раствора HCl (30 мл) и метанола (30 мл) и подвергают воздействию Н 2 (60 psi) (1 psi = 1 фунт на квадратный дюйм = 0.069 бар) в течение 3 ч. Реакционную смесь фильтруют через диатомит (Целит) и фильтрат концентрируют в вакууме. Полученный сырой продукт перекристаллизовывают из изопропанола для обеспечения HCl соли Подвески-1 в виде игл белого цвета (4.0 г). Оптическое вращение: -117.1 [с = 9.95 мг/мл в Н 2 О;= 589 нм]. 1 Н ЯМР (DMSO-d6,NaBH3CN (6.22 г, 94 ммоль) добавляют порционно в течение более нескольких минут к охлажденной (лед/вода) смеси (R)-2-фенилглицина (6.02 г, 39.8 ммоль) и МеОН (100 мл) и полученную смесь пе- 21017348 ремешивают в течение 5 мин. По каплям добавляют ацетальдегид (10 мл) в течение более 10 мин и перемешивание продолжают при той же самой охлаждающей температуре в течение 45 мин и при температуре окружающей среды в течение 6.5 ч. Реакционную смесь охлаждают снова на бане воды со льдом,обрабатывают водой (3 мл) и затем гасят путем добавления по каплям концентрированной HCl в течение более 45 мин пока значение рН смеси не станет равным 1.5 - 2.0. Охлаждающую баню удаляют и перемешивание продолжают пока происходит добавление концентрированной HCl для того, чтобы поддержать значение рН смеси около 1.5-2.0. Реакционную смесь перемешивают в течение более ночи,фильтруют, чтобы удалить суспензию белого цвета и фильтрат концентрируют в вакууме. Сырой продукт перекристаллизовывают из этанола, что дает HCl соль Подвески-2 в виде блестящего твердого вещества белого цвета в два порции (порция-1: 4.16 г; порция-2: 2.19 г). 1 Н ЯМР (DMSO-d6,= 2.5 ч. на млн, 400 МГц): 10.44 (1.00, широкий с, 1H), 7.66 (м, 2 Н), 7.51 (м, 3 Н), 5.30 (с, 1H), 3.15 (широкий м, 2 Н),2.98 (широкий м, 2 Н), 1.20 (видимый широкий с, 6 Н). Порция-1: []25-102.21 (с = 0.357, H2O); порция-2:N раствора HCl (30 мл) и метанола (40 мл). Охлаждающую баню удаляют и полученную реакционную смесь перемешивают в атмосфере Н 2 из баллона в течение 17 ч. Вводят дополнительное количество ацетальдегида (10 мл, 178.2 ммоль) и перемешивание продолжают в атмосфере Н 2 в течение 24 ч [замечание: подачу Н 2 возобновляют при необходимости в течение реакции]. Реакционную смесь фильтруют через диатомит (Целит) и фильтрат концентрируют в вакууме. Полученный сырой продукт перекристаллизовывают из изопропанола для обеспечения HCl соли (R)-2-(этиламино)-2-фенилуксусной кислоты в виде блестящего твердого вещества белого цвета (2.846 г). 1 Н ЯМР (DMSO-d6,= 2.5 част. на млн,400 МГц):14.15 (широкий с, 1H), 9.55 (широкий с, 2 Н), 7.55-7.48 (м, 5 Н), 2.88 (широкий м, 1H), 2.73(широкий м, 1 Н), 1.20 (видимый т, J = 7.2, 3 Н). LC (состояние 1): tR = 0.39 мин; 95 % показатель гомогенности; LC/MS: вычислено для [М+Н]+C10H14NO2: 180.10; получено 180.18. Суспензию 10% Pd/C (536 мг) в смеси метанол/Н 2 О (3 мл/1 мл) добавляют к смеси (R)-2(этиламино)-2-фенилуксусной кислоты/HCl (1.492 г, 6.918 ммоль), формальдегида (20 мл 37 вес.% в воде), 1 N раствора HCl (20 мл) и метанола (23 мл). Реакционную смесь перемешивают в атмосфере Н 2 из баллона в течение 72 ч, где подачу Н 2 возобновляют при необходимости. Реакционную смесь фильтруют через диатомит (Целит) и фильтрат концентрируют в вакууме. Полученный сырой продукт перекристаллизовывают из изопропанола (50 мл) для обеспечения HCl соли Подвески-3 в виде твердого вещества белого цвета (985 мг). 1 Н ЯМР (DMSO-d6,= 2.5 част. на млн, 400 МГц):10.48 (широкий с, 1H),7.59-7.51 (м, 5 Н), 5.26 (с, 1H), 3.08 (видимый широкий с, 2 Н), 2.65 (широкий с, 3 Н), 1.24 (широкий м, 3 Н).ClCO2Me (3.2 мл, 41.4 ммоль) по каплям добавляют к охлажденному (лед/вода) ТГФ (410 мл) полураствору (R)-трет-бутил-2-амино-2-фенилацетат/HCl (9.877 г, 40.52 ммоль) и диизопропилэтиламину(14.2 мл, 81.52 ммоль) в течение более 6 мин и полученную смесь перемешивают при той же температуре в течение 5.5 ч. Летучий компонент удаляют в вакууме и остаток распределяют между водой (100 мл) и этилацетатом (200 мл). Органический слой промывают 1N раствором HCl (25 мл) и насыщенным раствором NaHCO3 (30 мл), сушат (MgSO4), фильтруют и концентрируют в вакууме. Полученное бесцветное масло растирают в порошок с гексаном, фильтруют и промывают гексаном (100 мл) для обеспечения (R)трет-бутил-2-(метоксикарбониламино)-2-фенилацетата в виде твердого вещества белого цвета (7.7 г). 1 Н ЯМР (DMSO-d6,= 2.5 част. на млн, 400 МГц): 7.98 (д, J = 8.0, 1H), 7.37-7.29 (м, 5 Н), 5.09 (д, J = 8,1H),3.56 (с, 3 Н), 1.33 (с, 9 Н). LC (состояние 1): tR = 1.53 мин; -90 % показатель гомогенности; LC/MS: вычислено для [M+Na]+ C14H19NNaO4: 288.12; получено 288.15. Трифторуксусную кислоту (16 мл) по каплям добавляют к охлажденному (лед/вода) CH2Cl (160 мл)- 22017348 раствору указанного выше продукта в течение более 7 мин, охлаждающую баню удаляют и полученную реакционную смесь перемешивают в течение 20 ч. Так как снятие защиты еще не завершено вводят дополнительное количество трифторуксусной кислоты (1.0 мл) и перемешивание продолжают в течение еще 2 ч. Летучий компонент удаляют в вакууме и полученное масляный остаток обрабатывают диэтиловым эфиром (15 мл) и гексаном (12 мл) для обеспечения осадка. Осадок фильтруют и промывают смесью диэтиловый эфир/гексан (1:3 соотношение; 30 мл) и сушат в вакууме для обеспечения Подвески-4 в виде пушистого твердого вещества белого цвета (5.57 г). Оптическое вращение: -176.9 [с = 3.7 мг/мл в Н 2 О;= 589 нм]. 1 Н ЯМР (DMSO-d6,= 2.5 част. на млн, 400 МГц):12.84 (широкий с, 1H), 7.96 (д, J = 8.3, 1H), 7.41-7.29 (м, 5 Н), 5.14 (д, J = 8.3, 1H), 3.55 (с, 3 Н). LC (состояние 1): tR = 1.01 мин; 95 % показатель гомогенности; LC/MS: вычислено для [М+Н]+ C10H12NO4 210.08; получено 210.17; HRMS: вычислено для [М+Н]+ C10H12NO4 210.0766; получено 210.0756. Подвеска-5 Смесь (R)-2-фенилглицина (1.0 г, 6.62 ммоль), 1,4-дибромбутана (1.57 г, 7.27 ммоль) и Na2CO3 (2.10 г, 19.8 ммоль) в этаноле (40 мл) нагревают при температуре 100 С в течение 2 ч. Реакционную смесь охлаждают до температуры окружающей среды, фильтруют и фильтрат концентрируют в вакууме. Остаток растворяют в этаноле и подкисляют с помощью 1 N раствора HCl до значения рН 3-4 и летучий компонент удаляют в вакууме. Полученный сырой продукт очищают с помощью HPLC с обращенной фазой(смесь вода/метанол/трифторуксусная кислота) для обеспечения соли трифторуксусной кислоты Подвески-5 в виде полувязкой пены белого цвета (1.0 г). 1 Н ЯМР (DMSO-d6,= 2.5, 500 МГц)10.68 (широкий с, 1H), 7.51 (м, 5 Н), 5.23 (с, 1H), 3.34 (видимый широкий с, 2 Н), 3.05 (видимый широкий с, 2 Н), 1.95 (видимый широкий с, 4 Н); tR = 0.30 мин (состояние 1); 98% показатель гомогенности; LC/MS: вычислено для [M+H]+ C12H16NO2: 206.12; получено 206.25. Подвеска-6 Соль трифторуксусной кислоты из Подвески-6 синтезируют из (R)-2-фенилглицина и 1-бром-2-(2 бромэтокси)этана путем использования способ получения соединения, описанного в подвески-5. 1H ЯМРCH2Cl2 (200 мл) раствор п-толуолсульфонилхлорида (8.65 г, 45.4 ммоль) по каплям добавляют к охлажденному (-5 С) CH2Cl2 (200 мл) раствору (S)-бензил-2-гидрокси-2-фенилацетата (10.0 г, 41.3 ммоль),триэтиламину (5.75 мл, 41.3 ммоль) и 4-диметиламинопиридину (0.504 г, 4.13 ммоль), пока температура поддерживается в интервале между -5 и 0 С. Реакционную смесь перемешивают при температуре 0 С в течение 9 ч и затем выдерживают в холодильнике (-25 С) в течение 14 ч. Ей дают возможность нагреться до температуры окружающей среды и промывают водой (200 мл), 1N раствором HCl (100 мл) и рассолом(100 мл), сушат (MgSO4), фильтруют и концентрируют в вакууме для обеспечения бензил-2-фенил-2(тозилокси)ацетата в виде вязкого масла, которое застывает при стоянии (16.5 г). Хиральную чистоту продукта не контролируют и этот продукт используют на следующей стадии без дополнительной очистки. 1 Н ЯМР (DMSO-d6,= 2.5, 500 МГц)7.78 (д, J = 8.6, 2 Н), 7.43-7.29 (м, 10 Н), 7.20 (м, 2 Н), 6.12 (с,1H), 5.16 (д, J = 12.5, 1H), 5.10 (д, J = 12.5, 1H), 2.39 (с, 3 Н). tR = 3.00 (состояние 3); 90% показатель гомогенности; LC/MS: вычислено для [М+Н]+ C22H2ONaO5S: 419.09; получено 419.04. ТГФ (75 мл) раствор бензил-2-фенил-2-(тозилокси)ацетата (6.0 г, 15.1 ммоль), 1-метилпиперазин(3.36 мл, 30.3 ммоль) и N,N-диизопропилэтиламин (13.2 мл, 75.8 ммоль) нагревают при температуре 65 С в течение 7 ч. Реакционной смеси дают возможность остыть до температуры окружающей среды и летучий компонент удаляют в вакууме. Остаток распределяют между этилацетатом и водой и органический слой промывают водой и рассолом, сушат (MgSO4), фильтруют и концентрируют в вакууме. Полу- 23017348 ченный сырой продукт очищают с помощью флеш хроматографии (силикагель, этилацетат) для обеспечения бензил-2-(4-метилпиперазин-1-ил)-2-фенилацетата в виде вязкого масла оранжево-коричневого цвета (4.56 г). Хиральный HPLC анализ (Chiralcel OD-H) показывает, что образец представляет собой смесь энантиомеров в соотношении от 38.2 до 58.7. Разделение энантиомеров осуществляют следующим образом: продукт растворяют в 120 мл смеси этанол/гептан (1:1) и впрыскивают (5 мл/впрыск) на хиральную HPLC колонку (Chiracel OJ, 5 см ID50 см L, 20 мкм), элюируя смесью 85:15 гептан/этанол при скорости потока 75 мл/мин и контролируют при 220 нм. Энантиомер-1 (1.474 г) и энантиомер-2[М+Н]+ C20H25N2O2: 325.19; получено 325.20. Метанольный (10 мл) раствор любого энантиомера бензил-2-(4-метилпиперазин-1-ил)-2 фенилацетата (1.0 г, 3.1 ммоль) добавляют к суспензии 10% Pd/C (120 мг) в метаноле (5.0 мл). Реакционную смесь подвергают воздействию водорода из баллона при тщательном контролировании в течение 50 мин. Сразу после завершения реакции катализатор фильтруют через диатомит (Целит) и фильтрат концентрируют в вакууме для обеспечения Подвески-1, загрязненную фенилуксусной кислотой, в виде пены рыжего цвета (867.6 мг; масса имеет значение выше теоретического выхода). Продукт используют на следующей стадии без дополнительной очистки. 1 Н ЯМР (DMSO-d6,= 2.5, 500 МГц)7.44-7.37 (м,2 Н), 7.37-7.24 (м, 3 Н), 3.92 (с, 1H), 2.63-2.48 (видимый уширенный с, 2 Н), 2.48-2.32 (м, 6 Н), 2.19 (с, 3 Н);tR = 0.31 (состояние 2); 90% показатель гомогенности; LC/MS: вычислено для [М+Н]+ C13H19N2O2: 235.14; получено 235.15; HRMS: вычислено для [М+Н]+ C13H19N2O2: 235.1447; получено 235.1440. Синтез соединений Подвески-8 и Подвески-9 проводят в соответствии с синтезом Подвески-1 путем использования соответствующих аминов для стадии замещения SN2 (то есть, 4-гидроксипиперидин для Подвески-8 и (S)-3-фторпирролидин для Подвески-9) и изменяют условия для разделения соответствующих стереоизомерных промежуточных соединений, как описано ниже. Подвеска-8 Энантиомерное разделение промежуточного бензил-2-(4-гидроксипиперидин-1-ил)-2-фенилацетата осуществляют путем применения следующих условий: соединение (500 мг) растворяют в смеси этанол/гептан (5 мл/45 мл). Полученный раствор впрыскивают (5 мл/впрыск) на хиральную HPLC колонку(Chiracel OJ, 2 см ID25 см L, 10 мкм), элюируя смесью 80:20 гептан/этанол при скорости потока 10 мл/мин, контролируют при 220 нм, для обеспечения 186.3 мг энантиомерп-1 и 209.1 мг энантиомерп-2 в виде вязких масел светло-желтого цвета. Указанный бензиловый эфир гидрогенолизуют в соответствии с методикой получения Подвески-7, что обеспечивает Подвеску-8: 1 Н ЯМР (DMSO-d6,= 2.5, 500 МГц) 7.40 (д, J = 1,2 Н), 7.28-7.20 (м, 3 Н), 3.78 (с 1H), 3.46 (м, 1H), 2.93 (м, 1H), 2.62 (м, 1H), 2.20 (м, 2 Н), 1.70 Диастереомерное разделение промежуточного бензил-2-S)-3-фторпирролидин-1-ил)-2 фенилацетата осуществляют путем применения следующих условий: эфир (220 мг) разделяют на хиральной HPLC колонки (Chiracel OJ-H, 0.46 см ID25 см L, 5 мкм), элюируя смесью 95% СО 2/5% метанол с 0.1% трифторуксусной кислотой, при давлении 10 бар, 70 мл/мин скорости потока и температуре 35 С. HPLC элюат для соответствующих стереоизомеров концентрируют, остаток растворяют в CH2Cl2(20 мл) и промывают в водной среде (10 мл воды + 1 мл насыщенного раствора NaHCO3). Органическую фазу сушат (MgSO4), фильтруют и концентрируют в вакууме для обеспечения 92.5 мг фракции-1 и 59.6 мг фракции-2. Указанные бензиловые эфиры гидрогенолизуют в соответствии с методикой получения Подвески 7, чтобы получить Подвески 9 а и 9b. Подвеска-9 а (дистереомер-1; образец представляет собой соль трифторуксусной кислоты как результат очистки на HPLC с обращенной фазой, используя смесь Н 2 О/метанол/TFA растворитель): 1 Н ЯМР (DMSO-d6,= 2.5, 400 МГц) 7.55-7.48 (м, 5 Н), 5.38 (д из м, J = 53.7, 1H), 5.09 (широкий с, 1H), 3.84-2.82 (широкий м, 4 Н), 2.31-2.09 (м, 2 Н). tR = 0.42 (состояние 1); 95% показатель гомогенности; LC/MS: вычислено для [М+Н]+ C12H15FNO2: 224.11; получено 224.14; К раствору D-пролина (2.0 г, 17 ммоль) и формальдегида (2.0 мл 37 вес.% в H2O) в метаноле (15 мл) добавляют суспензию 10% Pd/C (500 мг) в метаноле (5 мл). Смесь перемешивают в атмосфере водорода из баллона в течение 23 ч. Реакционную смесь фильтруют через диатомит (Целит) и концентрируют в вакууме для обеспечения Подвески-10 в виде твердого вещества грязно-белого цвета (2.15 г). 1 Н ЯМР Смесь (2S,4R)-4-фторпирролидин-2-карбоновой кислоты (0.50 г, 3.8 ммоль), формальдегида (0.5 мл 37 вес.% в H2O), 12 N раствора HCl (0.25 мл) и 10% Pd/C (50 мг) в метаноле (20 мл) перемешивают в атмосфере водорода из баллона в течение 19 ч. Реакционную смесь фильтруют через диатомит (Целит) и фильтрат концентрируют в вакууме. Остаток перекристаллизовывают из изопропанола для обеспеченияHCl соли Подвески-11 в виде твердого вещества белого цвета (337.7 мг). 1 Н ЯМР (DMSO-d6,= 2.5, 500 МГц) 5.39 (д м, J = 53.7, 1H), 4.30 (м, 1H), 3.90 (ддд, J = 31.5, 13.5,4.5, 1H), 3.33 (дд, J = 25.6, 13.4, 1H),2.85 (с, 3 Н), 2.60-2.51 (м, 1H), 2.39-2.26 (м, 1H). tR = 0.28 (состояние 2); 98% показатель гомогенности;L-Аланин (2.0 г, 22.5 ммоль) растворяют в 10% водном растворе карбоната натрия (50 мл) и к нему добавляют ТГФ (50 мл) раствор метилхлорформиата (4.0 мл). Реакционную смесь перемешивают в условиях температуры окружающей среды в течение 4.5 ч и концентрируют в вакууме. Полученное твердое вещество белого цвета растворяют в воде и подкисляют с помощью 1N раствора HCl до значения рН 23. Полученные растворы экстрагируют этилацетатом (3100 мл) и объединенную органическую фазу сушат (Na2SO4), фильтруют и концентрируют в вакууме для обеспечения бесцветного масла (2.58 г). 500 мг указанного продукта очищают с помощью HPLC с обращенной фазой (смесь Н 2 О/метанол/трифторуксусная кислота) для обеспечения 150 мг Подвески-12 в виде бесцветного масла. 1 Н ЯМР (DMSO-d6,Pd/C (500 мг) в метаноле (30 мл) перемешивают в атмосфере водорода (давление 50 psi) в течение 5 ч. Реакционную смесь фильтруют через диатомит (Целит) и фильтрат концентрируют в вакууме для обеспечения HCl соли Подвески-13 в виде масла, которое застывает при стоянии в вакууме (4.4 г; масса имеет значение выше теоретического выхода). Продукт используют без дополнительной очистки. 1H ЯМР(0.773 г, 12.3 ммоль), KOH (0.690 г, 12.3 ммоль) и уксусной кислоты (0.352 мл, 6.15 ммоль) перемешивают в метаноле при температуре 0 С. К полученной смеси добавляют по каплям глутаровый диальдегид(2.23 мл, 12.3 ммоль) в течение более 5 мин. Реакционную смесь перемешивают, чтобы дать ей возможность нагреться до температуры окружающей среды и перемешивание продолжают при той же самой температуре в течение 16 ч. Затем растворитель удаляют и остаток распределяют с 10% водным раствором NaOH и этилацетатом. Органическую фазу отделяют, сушат (MgSO4), фильтруют и концентрируют досуха для обеспечения прозрачного масла. Указанный продукт очищают с помощью препаративнойHPLC с обращенной фазой (Primesphere С-18, 30100 мм; CH3CN-Н 2 О-0.1% трифторуксусная кислота),что дает промежуточный эфир (2.70 г, 56%) в виде прозрачного масла. 1 Н ЯМР (400 МГц, CDCl3)7.537.44 (м, 3 Н), 7.40-7.37 (м, 2 Н), 3.87 (д, J = 10.9 Гц, 1H), 3.59 (д, J = 10.9 Гц, 1H), 2.99 (т, J = 11.2 Гц, 1H),2.59 (т, J = 11.4 Гц, 1H), 2.07-2.02 (м, 2 Н), 1.82 (д, J = 1.82 Гц, 3 Н), 1.40 (с, 9 Н). LC/MS: вычислено дляC17H25NO2: 275; получено: 276 (М+Н)+. Стадия 2. К перемешиваемому раствору промежуточного эфира (1.12 г, 2.88 ммоль) в дихлорметане(10 мл) добавляют трифторуксусную кислоту (3 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 4 ч и затем ее концентрируют досуха, что дает масло светло-желтого цвета. Масло очищают, используя препаративную HPLC с обращенной фазой (Primesphere С-18, 30100 мм; CH3CN-Н 2 О-0.1% трифторуксусная кислота). Соответствующие фракции объединяют и концентрируют досуха в вакууме. Остаток затем растворяют в минимальном количестве метанола и используют МСХ LP экстракционные картриджи (26 г). Картриджи промывают метанолом (40 мл) и затем требуемое соединение элюируют, используя 2 М раствор аммиака в метаноле (50 мл). Продукт, содержащий фракции объединяют и концентрируют и остаток помещают в воду. Лиофилизация указанного раствора обеспечивает указанное в заголовке соединение (0.492 г, 78%) в виде твердого вещества светло-желтого цвета. 1H ЯМР (DMSO-d6)7.50 (с, 5 Н), 5.13 (с, 1H), 3.09 (широкий с, 2 Н), 2.92-2.89 (м, 2 Н), 1.74 (м, 4 Н),1.48 (широкий с, 2 Н). LC/MS: вычислено для C13H17NO2: 219; получено: 220 (М+Н)+. Подвеска-15(100 мл) добавляют твердый EDCl (12.46 г, 0.065 моль), все одновременно. Полученный раствор перемешивают при комнатной температуре в атмосфере Ar в течение 18 ч и затем его разбавляют этилацетатом, промывают (Н 2 О 2, рассол), сушат (Na2SO фильтруют и концентрируют, что дает масло бледножелтого цвета. Флеш хроматография (SiO2/гексан-этилацетат, 4:1) данного масла обеспечивает указанное в заголовке соединение (11.64 г, 73%) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3)7.53-7.17 (м, 10 Н), 5.95 (кв., J = 6.6 Гц, 0.5 Н), 5.94 (кв., J = 6.6 Гц, 0.5 Н), 5.41 (с, 0.5 Н), 5.39 (с, 0.5 Н),1.58 (д, J = 6.6 Гц, 1.5 Н), 1.51 (д, J = 6.6 Гц, 1.5 Н). Стадия 2. (S)-1-Фенилэтил-(R)-2-(4-гидрокси-4-метилпиперидин-1-ил)-2-фенилацетат. К раствору (S)-1-фенилэтил-2-бром-2-фенилацетата (0.464 г, 1.45 ммоль) в ТГФ (8 мл) добавляют триэтиламин (0.61 мл, 4.35 ммоль), а затем иодид тетрабутиламмония (0.215 г, 0.58 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 5 мин и затем добавляют раствор 4-метил-4 гидроксипиперидина (0.251 г, 2.18 ммоль) в ТГФ (2 мл). Смесь перемешивают в течение часа при комнатной температуре и затем ее нагревают при температуре 55-60 С (температура масляной бани) в течение 4 ч. Охлажденную затем реакционную смесь разбавляют этилацетатом (30 мл), промывают (Н 2 О 2,рассол), сушат (MgSO4), фильтруют и концентрируют. Остаток очищают с помощью хроматографии на силикагеле (0-60% смесь этилацетат-гексан) для обеспечения первого (S,R)-изомера, указанного в заголовке соединения (0.306 г, 60%) в виде твердого вещества белого цвета и затем соответствующего (S,S)изомера (0.120 г, 23%), также в виде твердого вещества белого цвета. (S,R)-изомер: 1 Н ЯМР (CD3OD)7.51-7.45 (м, 2 Н), 7.41-7.25 (м, 8 Н), 5.85 (кв., J = 6.6 Гц, 1H), 4.05 (с, 1H), 2.56-2.45 (м, 2 Н), 2.41-2.29 (м,2 Н), 1.71-1.49 (м, 4 Н), 1.38 (д, J = 6.6 Гц, 3 Н), 1.18 (с, 3 Н). LCMS: вычислено для C22H27NO3: 353; получено: 354 (М+Н)+. (S,S)-изомер: 1H ЯМР(CD3OD)7.41-7.30 (м, 5 Н), 7.20-7.14 (м, 3 Н), 7.06-7.00 (м, 2 Н),5.85 (кв., J = 6.6 Гц, 1H), 4.06 (с, 1H), 2.70-2.60 (м, 1H), 2.51 (дт, J = 6.6, 3.3 Гц, 1H), 2.44-2.31 (м, 2 Н),1.75-1.65 (м, 1H), 1.65-1.54 (м, 3 Н), 1.50 (д, J = 6.8 Гц, 3 Н), 1.20 (с, 3 Н). LCMS: вычислено для C22H27NO3: 353; получено: 354 (М+Н)+. Стадия 3. (R)-2-(4-Гидрокси-4-метилпиперидин-1-ил)-2-фенилуксусная кислота. К раствору (S)-1-фенилэтал-(R)-2-(4-гидрокси-4-метилпиперидин-1-ил)-2-фенилацетата (0.185 г,0.52 ммоль) в дихлорметане (3 мл) добавляют трифторуксусную кислоту (1 мл) и полученную смесь перемешивают при комнатной температуре в течение 2 ч. Затем летучие вещества удаляют в вакууме и остаток очищают с помощью препаративной HPLC с обращенной фазой (Primesphere С-18, 20100 мм;CH3CN-H2O-0.1% трифторуксусная кислота), что дает указанное в заголовке соединение (в виде соли- 26017348 трифторуксусной кислоты) в виде твердого вещества бледно-голубоватого цвета (0,128 г, 98%). LCMS: вычислено для C14H19NO3: 249; получено: 250 (М+Н)+. Подвеска-16 Стадия 1. (S)-1-Фенилэтил-2-(2-фторфенил)ацетат. Смесь 2-фторфенилуксусной кислоты (5.45 г, 35.4 ммоль), (S)-1-фенилэтанола (5.62 г, 46.0 ммоль),EDCl (8.82 г, 46.0 ммоль) и DMAP (0.561 г, 4.60 ммоль) в CH2Cl2 (100 мл) перемешивают при комнатной температуре в течение 12 ч. Затем растворитель концентрируют и остаток распределяют в смеси Н 2 Оэтилацетат. Фазы разделяют и водный слой снова экстрагируют этилацетатом (2 х). Объединенные органические фазы промывают (Н 2 О, рассол), сушат (Na2SO4), фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии на силикагеле (Biotage/ 0-20% смесь этилацетат-гексан) для обеспечения указанного в заголовке соединения в виде бесцветного масла (8.38 г, 92%). 1H ЯМР (400 МГц, CD3OD)7.32-7.23 (м, 7 Н), 7.10-7.04 (м, 2), 5.85 (кв., J= 6.5 Гц, 1H), 3.71 (с, 2 Н), 1.48 (д, J = 6.5 Гц,3 Н). Стадия 2. (R)-S)-1-Фенилэтил)-2-(2-фторфенил)-2-(пиперидин-1-ил)ацетат. К раствору (S)-1-фенилэтил-2-(2-фторфенил)ацетата (5.00 г, 19.4 ммоль) в ТГФ (1200 мл) при температуре 0 С добавляют DBU (6.19 г, 40.7 ммоль) и полученному раствору дают возможность нагреться до комнатной температуры при перемешивании в течение 30 мин. Затем раствор охлаждают до температуры -78 С, добавляют раствор CBr4 (13.5 г, 40.7 ммоль) в ТГФ (100 мл), смеси дают возможность нагреться до температуры -10 С и полученную смесь перемешивают при данной температуре в течение 2 ч. Реакцию гасят насыщенным водным раствором NH4Cl и слои разделяют. Водный слой снова экстрагируют этилацетатом (2 х) и объединенные органические фазы промывают (Н 2 О, рассол), сушат (Na2SO4),фильтруют и концентрируют в вакууме. К остатку добавляют пиперидин (5.73 мл, 58.1 ммоль) и полученный раствор перемешивают при комнатной температуре в течение 24 ч. Затем летучие вещества концентрируют в вакууме и остаток очищают с помощью хроматографии на силикагеле (Biotage/ 0-30% смесь диэтиловый эфир-гексан) для обеспечения чистой смеси диастереомеров (2:1 соотношение с помощью 1H ЯМР) в виде масла желтого цвета (2.07 г, 31%), вместе с непрореагировавшим исходным продуктом (2.53 г, 51%). Последующая хроматография диастереомерной смеси (Biotage/ 0-10% смесь диэтиловый эфир-толуол) обеспечивает указанное в заголовке соединение в виде бесцветного масла (0.737 г,11%). 1 Н ЯМР (400 МГц, CD3OD)7.52 (ддд, J = 9.4, 7.6,1.8 Гц, 1H), 7.33-7.40 (м, 1), 7.23-7.23 (м, 4 Н),7.02-7.23 (м, 4 Н), 5.86 (кв., J = 6.6 Гц, 1H), 4.45 (с, 1H), 2.39-2.45 (м, 4 Н), 1.52-1.58 (м, 4 Н), 1.40-1.42 (м,1H), 1.38 (д, J = 6.6 Гц, 3 Н). LCMS: вычислено для C21H24FNO2: 341; получено: 342 (М+Н)+. Стадия 3. (R)-2-(2-Фторфенил)-2-(пиперидин-1-ил)уксусная кислота. Смесь (R)-(S)-1-фенилэтил-2(2-фторфенил)-2-(пиперидин-1-ил)ацетата (0.737 г, 2.16 ммоль) и 20% Pd(OH)2/С (0.070 г) в этаноле (30 мл) гидрируют при комнатной температуре и атмосферном давлении (H2 из баллона) в течение 2 ч. Затем раствор продувают Ar, фильтруют через диатомит (Целит) и концентрируют в вакууме. Указанный обеспечивает указанное в заголовке соединение в виде бесцветного твердого вещества (0.503 г, 98%). 1 Н ЯМР (400 МГц, CD3OD)7.65 (ддд, J = 9.1, 7.6, 1.5 Гц, 1H), 7.47-7.53 (м, 1H), 7.21-7.30 (м, 2 Н), 3.07-3.13 Стадия 1. (S)-1-Фенилэтил-(R)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-2-фенилацетат. К раствору (S)-1-фенилэтил-2-бром-2-фенилацетата (1.50 г, 4.70 ммоль) в ТГФ (25 мл) добавляют триэтиламин (1.31 мл, 9.42 ммоль), а затем иодид тетрабутиламмония (0.347 г, 0.94 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 5 мин и затем добавляют раствор 4-фенил-4 гидроксипиперидина (1.00 г, 5.64 ммоль) в ТГФ (5 мл). Смесь перемешивают в течение 16 ч и затем ее разбавляют этилацетатом (100 мл), промывают (Н 2 О х 2, рассол), сушат (MgSO4), фильтруют и концентрируют. Остаток очищают на колонке с силикагелем (0-60% смесь этилацетат-гексан) для обеспечения приблизительно 2:1 смеси диастереомеров, как оценивают с помощью 1 Н ЯМР. Разделение указанных изомеров проводят, используя сверхкритическую жидкостную хроматографию (Chiralcel OJ-H, 30250 мм; 20% этанол в СО 2 при температуре 35 С), что дает первый (R)-изомер указанного в заголовке соеди- 27017348 нения (0.534 г, 27%) в виде масла желтого цвета и затем соответствующий (S)-изомер (0.271 г, 14%),также в виде масла желтого цвета. (S, R)-изомер: 1 Н ЯМР (400 МГц, CD3OD)7.55-7.47 (м, 4 Н), 7.447.25 (м, 10 Н), 7.25-7.17 (м, 1H), 5.88 (кв., J = 6.6 Гц, 1H), 4.12 (с, 1H), 2.82-2.72 (м, 1H), 2.64 (At, J= 11.1,2.5 Гц, 1H), 2.58-2.52 (м, 1H), 2.40 (дт, J= 11.1,2.5 Гц, 1H), 2.20 (дт, J = 12.1,4.6 Гц, 1H), 2.10 (дт, J = 12.1,4.6 Гц, 1 Н), 1.72-1.57 (м, 2 Н), 1.53 (д, 7 = 6.5 Гц, 3 Н). LCMS: вычислено для C27H29NO3: 415; получено: 416 (М+Н)+; (S,S)-изомер: 1H ЯМР (400 МГц, CD3OD)7.55-7.48 (м, 2 Н), 7.45-7.39 (м, 2 Н), 7.38-7.30LCMS: вычислено для C27H29NO3: 415; получено: 416 (М+Н)+. Следующие эфиры получают аналогичным образом, применяя методику, представленную в стадии 1 при синтезе Подвески-17.- 28017348 Хиральные SFC условия для определения tR для промежуточных соединений 17b-17d Условие 1. Колонка: Chiralpak AD-H Колонка, 4.6250 мм, 5 мкм Растворители: 90% СО 2 - 10% метанол с 0.1%DEA Темп: 35 С Давление: 150 бар Скорость потока: 2.0 мл/мин УФ-контролирование 220 нм Впрыск: 1.0 мг/3 мл метанол Условие 2. Колонка: Chiralcel OD-H Колонка, 4.6250 мм, 5 мкм Растворители: 90% CO2-10% метанол с 0.1%DEA Темп: 35 С Давление: 150 бар Скорость потока: 2.0 мл/мин УФ-контролирование 220 нм Впрыск: 1.0 мг/мл метанол. Подвеска-17. Стадия 2. (R)-2-(4-Гидрокси-4-фенилпиперидин-1-ил)-2-фенилуксусная кислота. К раствору (S)-1-фенилэтил-(R)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-2-фенилацетата (0.350 г,0.84 ммоль) в дихлорметане (5 мл) добавляют трифторуксусную кислоту (1 мл) и полученную смесь перемешивают при комнатной температуре в течение 2 ч. Затем летучие вещества удаляют в вакууме и остаток очищают с помощью препаративной HPLC с обращенной фазой (Primesphere С-18, 20100 мм;CH3CN-Н 2 О-0.1% трифторуксусная кислота), что дает указанное в заголовке соединение (в виде соли трифторуксусной кислоты) в виде твердого вещества белого цвета (0.230 г, 88%). LCMS: вычислено дляC19H21NO3: 311; получено: 312 (М+Н)+. Следующие карбоновые кислоты получают аналогичным образом:LCMS условия для определения времени задержки для Подвесок 17 а-17d Условие 1. Колонка: Phenomenex-Luna 4.650 мм S10 Исходный % В = 0 Конечный% В = 100 Градиент времени = 4 мин Скорость потока = 4 мл/мин
МПК / Метки
МПК: A61K 31/506, A61P 31/12, C07D 401/14, A61K 31/4439, A61K 31/497, A61K 31/501, C07D 403/14, A61K 31/444
Метки: гепатита, ингибиторы, вируса
Код ссылки
<a href="https://eas.patents.su/30-17348-ingibitory-virusa-gepatita-s.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы вируса гепатита с</a>
Ингибиторы вируса гепатита с

Номер патента: 15756
Опубликовано: 30.12.2011
Авторы: Хаманн Лоуренс Г., Белема Маконен, Деон Дэниел Х., Гуд Эндрю С., Джеймс Клинт А., Минвелл Николас А., Снайдер Лоуренс Б., Сен-Лоран Денис Р., Бэчэнд Кэрол, Рудигер Эдвард Х., Мартель Ален, Ван Гань, Лэнгли Дэвид Р., Ромайн Джеффри Ли, Лавой Рико, Гудрич Джейсон, Ян Фукан, Лопез Омар Д., Нгуен Ван Н.
МПК: A61P 31/12, A61K 31/4178, A61K 31/4025...
Метки: ингибиторы, гепатита, вируса
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, гдеm и n независимо имеют значения 0, 1 или 2;q и s независимо имеют значения 0, 1, 2, 3 или 4;u и v независимо имеют значения 0, 1, 2 или 3;X выбран из О, S, S(O), SO2, CH2, CHR5 и C(R5)2;при условии, что когда n равен 0, X выбран из СН2, CHR5 и C(R5)2;Y выбран из О, S, S(O), SO2, CH2, CHR6 и C(R6)2;при условии, что когда m равен 0, Y выбран из СН2, CHR6 и C(R6)2;каждый R1и R2...
Макроциклические ингибиторы вируса гепатита с

Номер патента: 14293
Опубликовано: 29.10.2010
Авторы: Рабуассон Пьер Жан-Мари Бернар, Симмен Кеннет Алан, Антонов Дмитрий, Йенссон Карл Эрик Даниель, Росенквист Оса Анника Кристина, Нильссон Карл Магнус, Айеса Алварес Сусана, Де Кок Херман Аугустинус, Сальвадор Оден Лоурдес, Классон Бьерн Олоф, Самуэльссон Бенгт Бертил
МПК: A61K 31/4523, C07D 245/04, A61P 31/14...
Метки: ингибиторы, макроциклические, гепатита, вируса
Формула / Реферат:
1. Соединение формулыего N-оксид, соль или стереоизомер,где каждая пунктирная линия (представленная как ----) представляет собой необязательную двойную связь;X представляет собой N, СН и, когда X содержит двойную связь, он представляет собой С;R1 представляет собой -OR6, -NH-SO2R7;R2 представляет собой водород и, когда X представляет собой С или СН, R2 также может представлять собой C1-6алкил;R3 представляет собой водород, C1-6алкил,...
Ингибиторы вируса гепатита с (hcv)

Номер патента: 15415
Опубликовано: 31.08.2011
Авторы: Линк Джон О., Венкатарамани Чандрэсикар, Граупе Михаель
МПК: A61K 47/00, A61K 31/74, A61K 31/00...
Метки: гепатита, вируса, hcv, ингибиторы
Формула / Реферат:
1. Соединение формулы (I)где Е представляет собой -COCONHR6, где R6представляет собой водород, алкил, циклоалкил, аралкил или гетероаралкил, где ароматическое кольцо необязательно замещено одним или двумя галоидами;R1 представляет собой алкил, циклоалкилалкил, где алифатические или алициклические группы в R1необязательно замещены одним или двумя радикалами Rb, которые независимо выбирают из гидрокси, алкокси, арилокси, гетероарилокси, алкилтио,...
Ингибиторы вируса гепатита с

Номер патента: 15827
Опубликовано: 30.12.2011
Авторы: Ван Алан Сяндун, Чжао Цянь, Д`андреа Стэнли, Чжэн Барбара Чжичжэнь, Скола Пол Майкл
МПК: A61P 31/00, C07K 5/08, A61K 31/401...
Метки: гепатита, ингибиторы, вируса
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, гдеn равно 1;R1 выбран из гидрокси и -NHSO2R7;R2 представляет собой С2алкил, С2алкенил или дифторметил;R3 представляет собой 4-бифенил-4-ил;R4 представляет собой -OR8;R5 представляет собой С3-4алкил, С7алкенил или 4-трет-бутилбензил;R6 представляет собой трет-бутоксикарбонил, возможно замещенный галогеном, трет-бутиламинокарбонил, адамантан-1-илоксикарбонил,...
Макроциклические ингибиторы вируса гепатита с

Номер патента: 14188
Опубликовано: 29.10.2010
Авторы: Симмен Кеннет Алан, Самуэльссон Бенгт Бертил, Бельфраге Анна Карин Гертруд Линнеа, Линдквист Карин Карлотта, Валльберг Ханс Кристиан, Ху Лили, Рабуассон Пьер Жан-Мари Бернар, Канберг Пиа Сесилия, Классон Бьерн Олоф, Росенквист Оса Анника Кристина, Сальберг Свен Кристер, Вехлинг Хорст Юрген, Нильссон Карл Магнус, Де Кок Херман Аугустинус, Линдстрем Матс Стефан
МПК: A61P 31/12, A61K 31/517, C07D 239/72...
Метки: вируса, гепатита, ингибиторы, макроциклические
Формула / Реферат:
1. Соединение формулы (I)и его N-оксиды, соли и стереоизомеры,где А представляет собой OR1, NHS(=O)pR2; гдеR1 представляет собой водород, C1-C6-алкил;R2 представляет собой C3-C7-циклоалкил, причем указанный C3-C7-циклоалкил необязательно замещен 1-3 заместителями, независимо выбранными из C1-C6-алкила;р независимо равно 2;n равно 3, 4, 5 или 6;---- означает необязательную двойную связь;L представляет собой N или CRz;Rz представляет собой Н;Rq...
Предыдущий патент: Частицы для обработки подложек и способ их приготовления
Следующий патент: Способ получения эзетимиба и его производных
Случайный патент: Способ отделения минерала от тонкоизмельченного зернистого материала