Антагонисты рецептора 5-нт7
Номер патента: 16787
Опубликовано: 30.07.2012
Авторы: Ваттон Мария Энн, Бадеску Валентина О., Галлахер Питер Таддеуш, Филла Сандра Энн





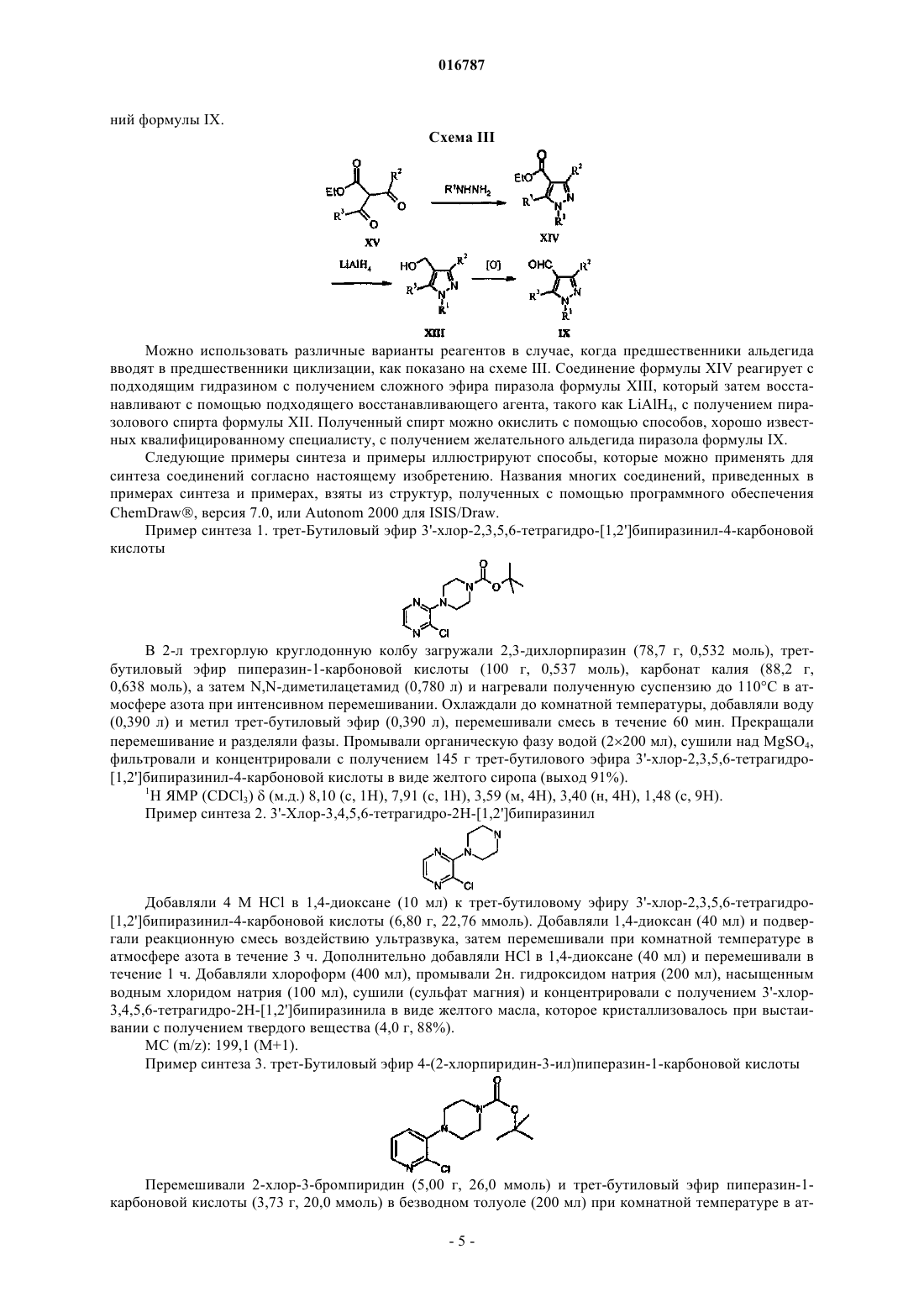






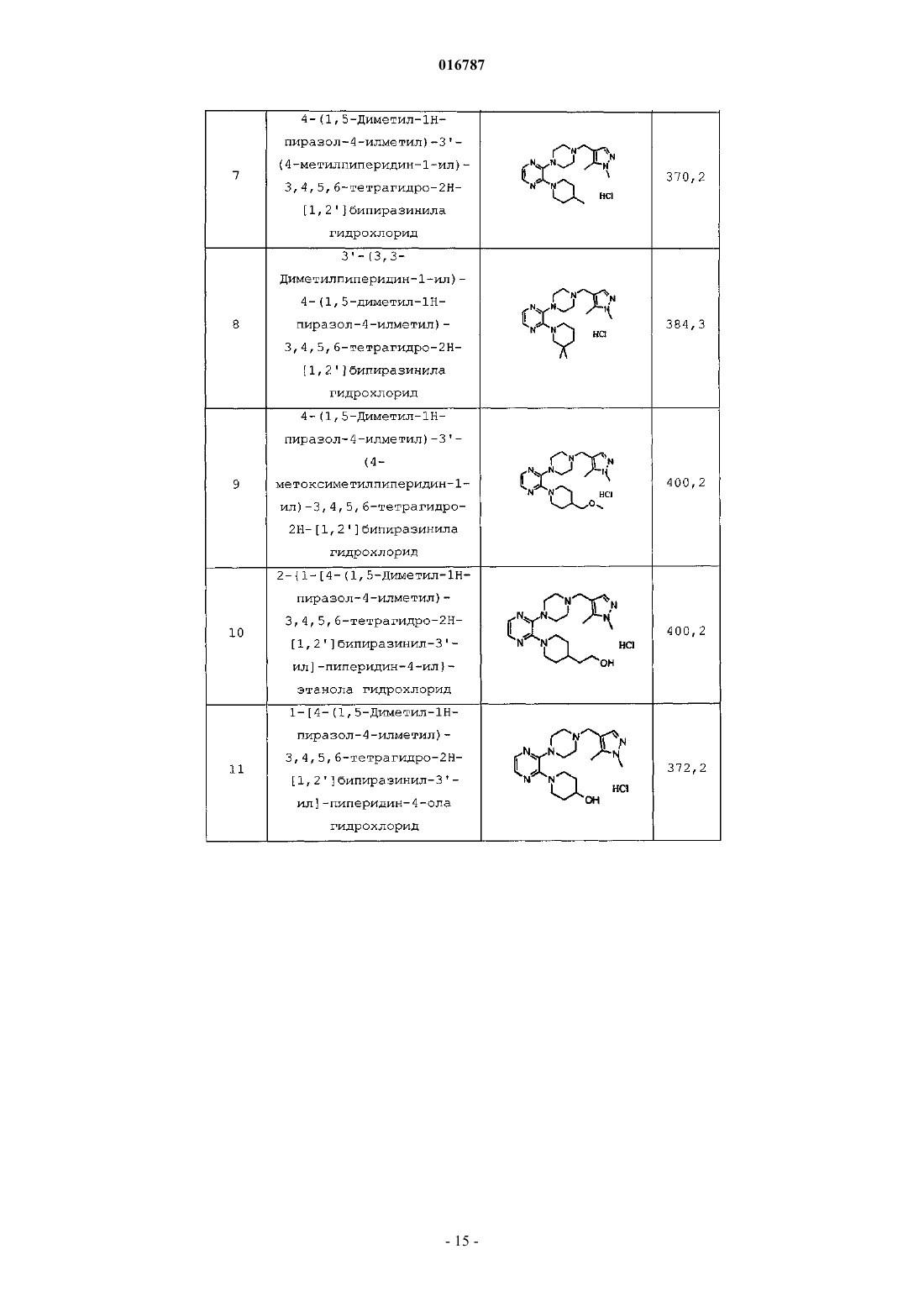
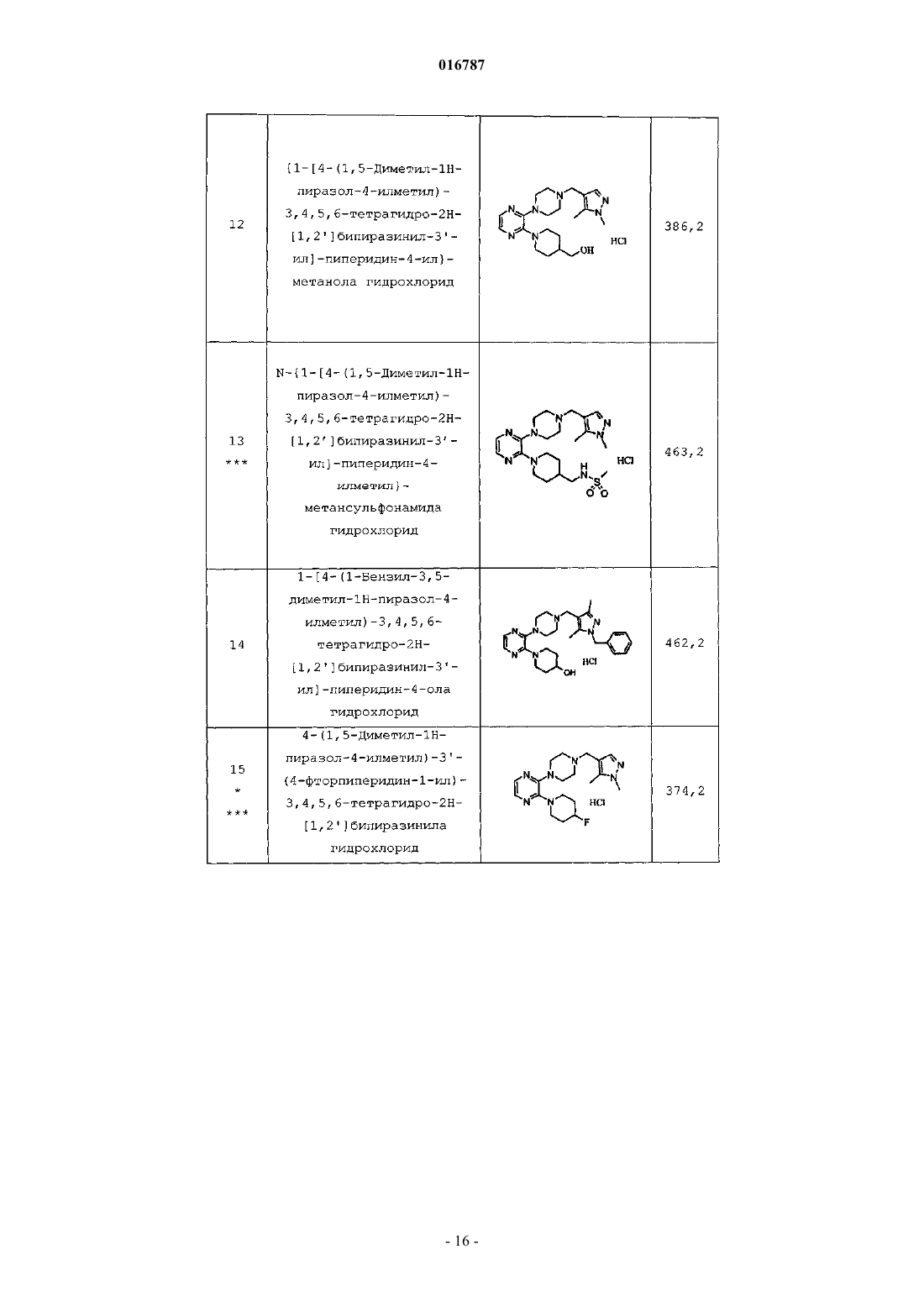
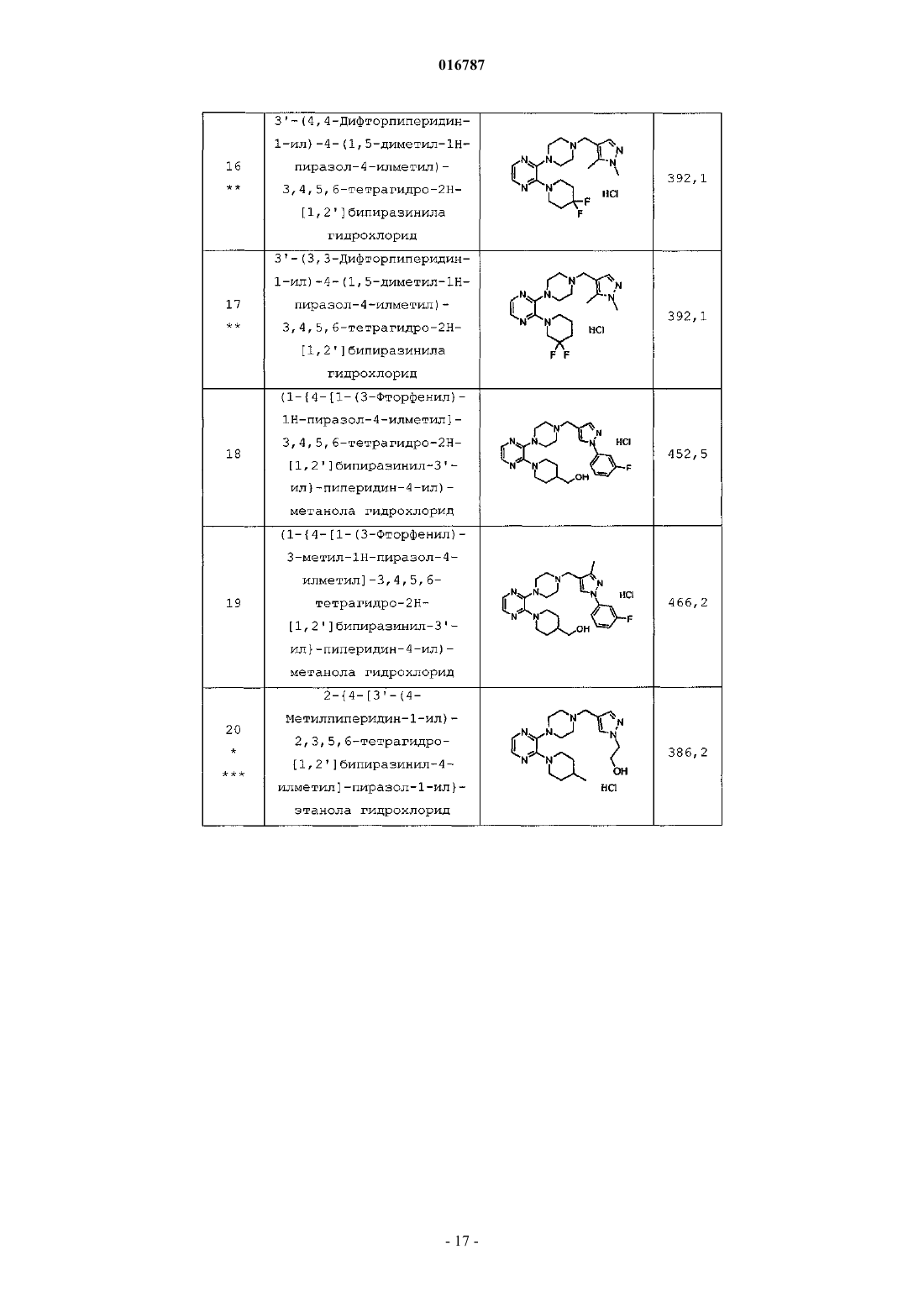

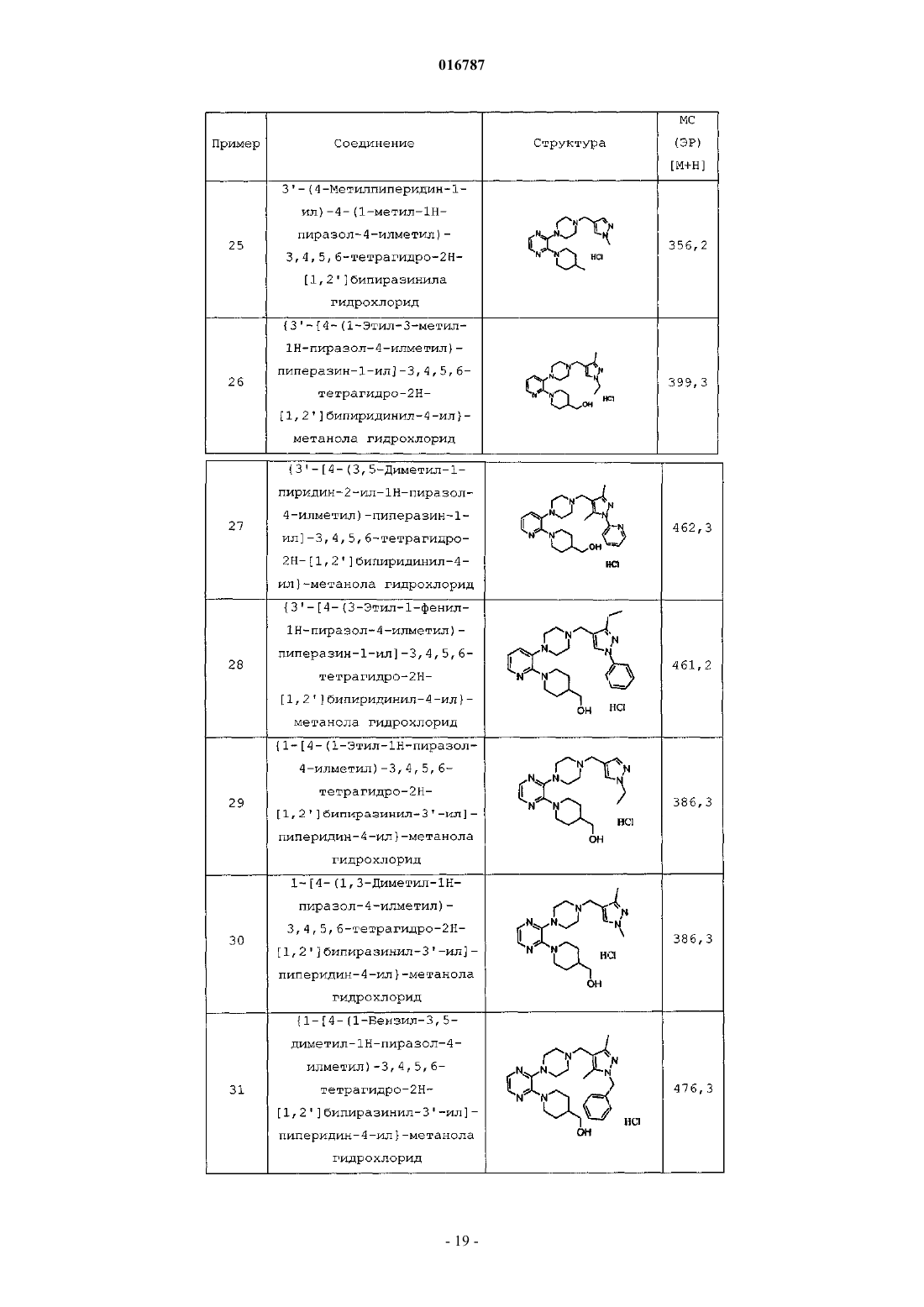
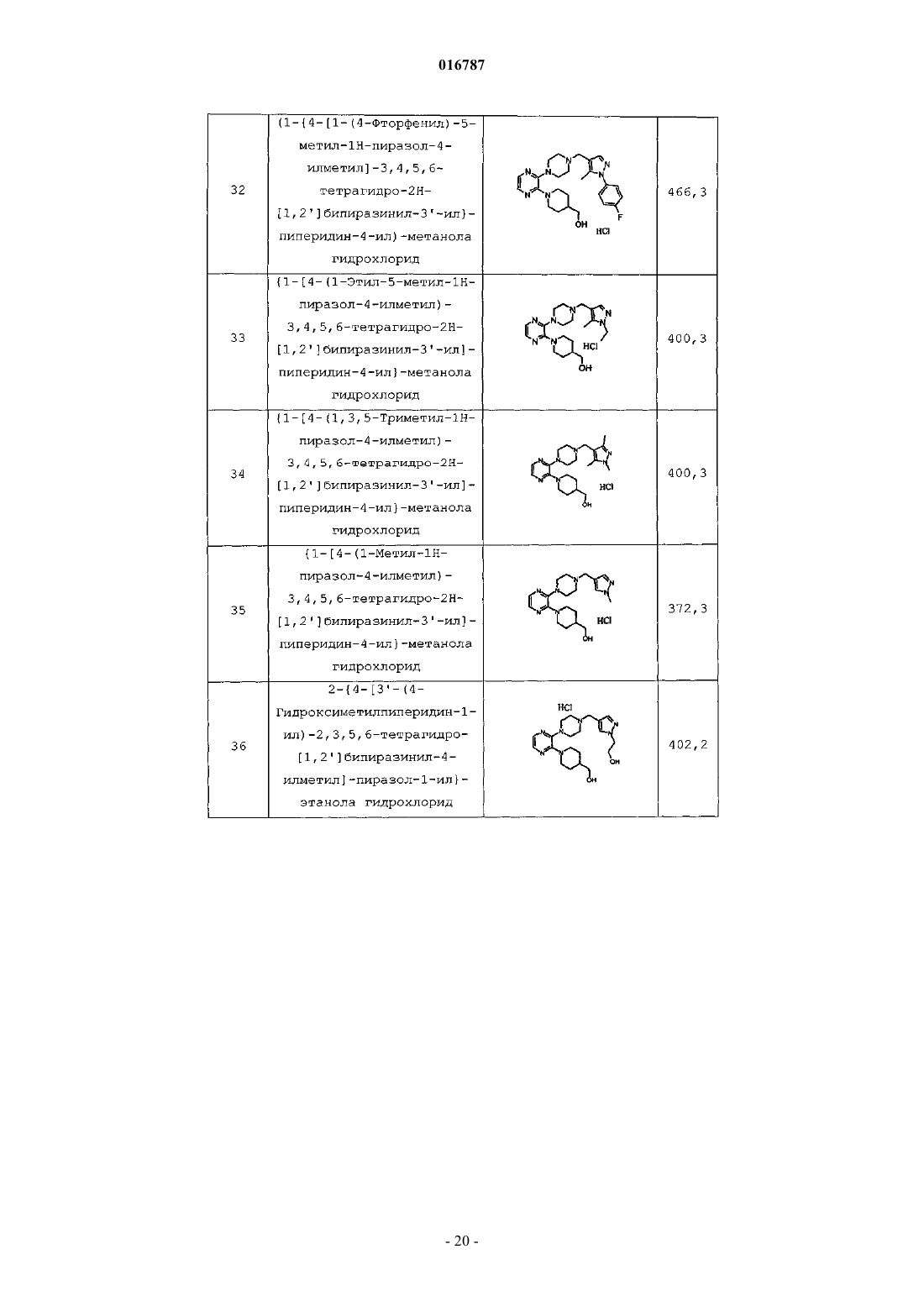
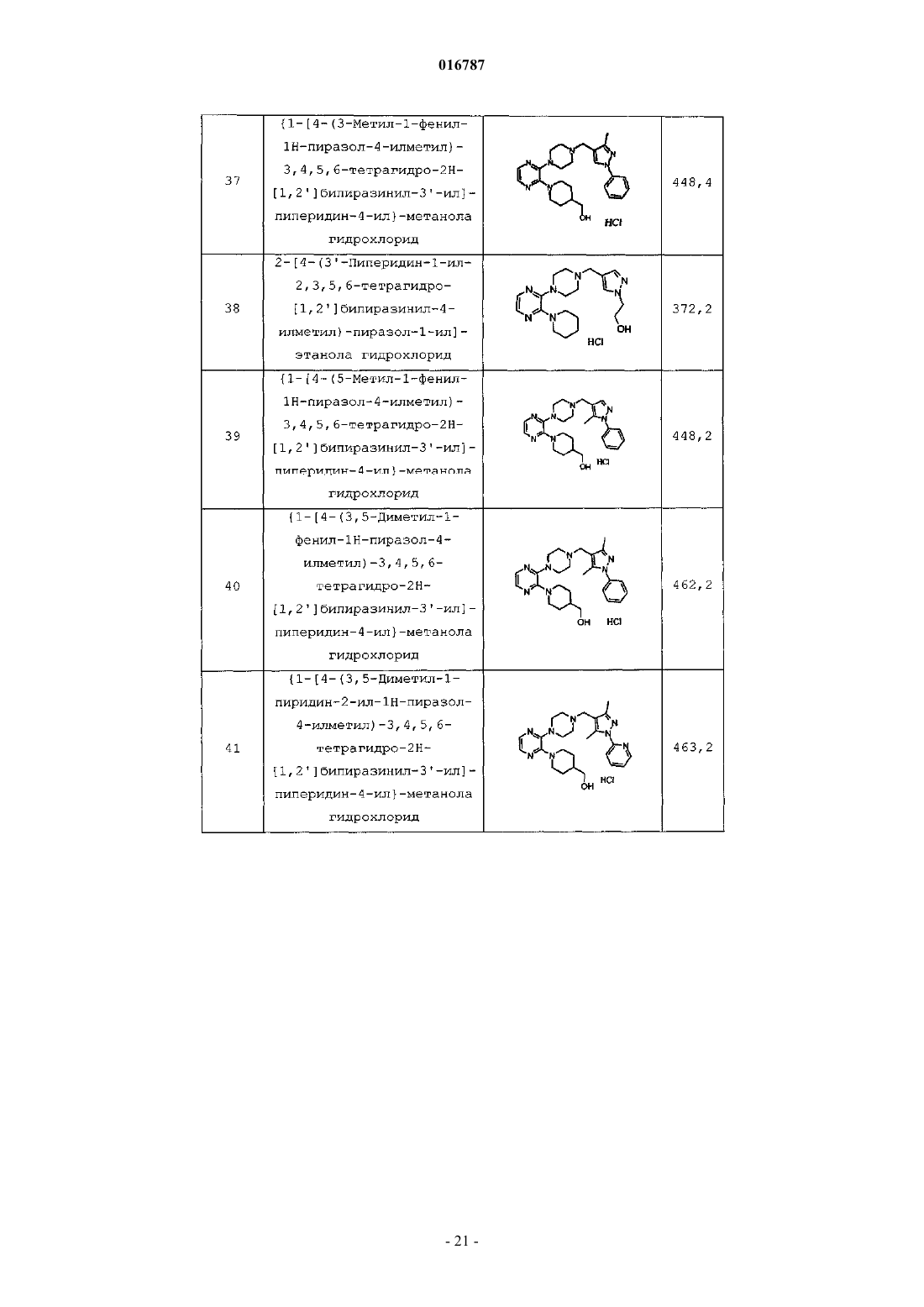








Формула / Реферат
1. Соединение формулы

где А представляет собой -С(Н)= или -N=;
R1 представляет собой заместитель, выбранный из группы, состоящей из i) водорода, ii) метила, iii) этила, iv) гидроксиметила, v) гидроксиэтила, vi) фенила, необязательно содержащего в качестве заместителей от 1 до 3 фторгрупп, vii) бензила, необязательно содержащего в качестве заместителей от 1 до 3 фторгрупп, и viii) пиридила;
R2 представляет собой водород, метил или этил;
R3 представляет собой водород, метил или хлор;
R4 выбран из группы, состоящей из i) водорода, ii) фтора, iii) метила, iv) гидрокси, v) гидроксиметила,
vi) гидроксиэтила, vii) метоксиметила, viii) цианметила и ix) метилсульфониламинометила;
R5 представляет собой водород или фтор при условии, что, если R5 представляет собой фтор, R4 представляет собой фтор;
R6 и R7 одинаковы и вместе выбраны из группы, состоящей из водорода, метила и фтора, при условии, что, если R6 и R7 не являются водородом, R4 и R5, оба, представляют собой водород;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где R1 представляет собой метил, этил или фенил, необязательно содержащий в качестве заместителей от 1 до 2 фторгрупп.
3. Соединение по п.1, где R1 представляет собой метил, этил или фенил, необязательно содержащий в качестве заместителей от 1 до 2 фторгрупп, и R4 представляет собой гидрокси, гидроксиметил или метоксиметил.
4. Соединение по п.1, которое представляет собой 3'-[4-(1-этил-5-метил-1Н-пиразол-4-илметил)пиперазин-1-ил]-3,4,5,6-тетрагидро-2Н-[1,2']бипиридинил-4-ол или его фармацевтически приемлемую соль.
5. Фармацевтическая композиция, содержащая соединение по любому из пп.1-4 в качестве активного ингредиента совместно с фармацевтически приемлемым носителем, разбавителем или эксципиентом.
6. Применение соединения по любому из пп.1-4 при лечении мигрени у людей.
7. Применение соединения по любому из пп.1-4 при профилактическом лечении мигрени у людей.
8. Применение соединения по любому из пп.1-4 в изготовлении лекарственного средства для лечения мигрени.
9. Применение соединения по любому из пп.1-4 в изготовлении лекарственного средства для профилактического лечения мигрени.
Текст
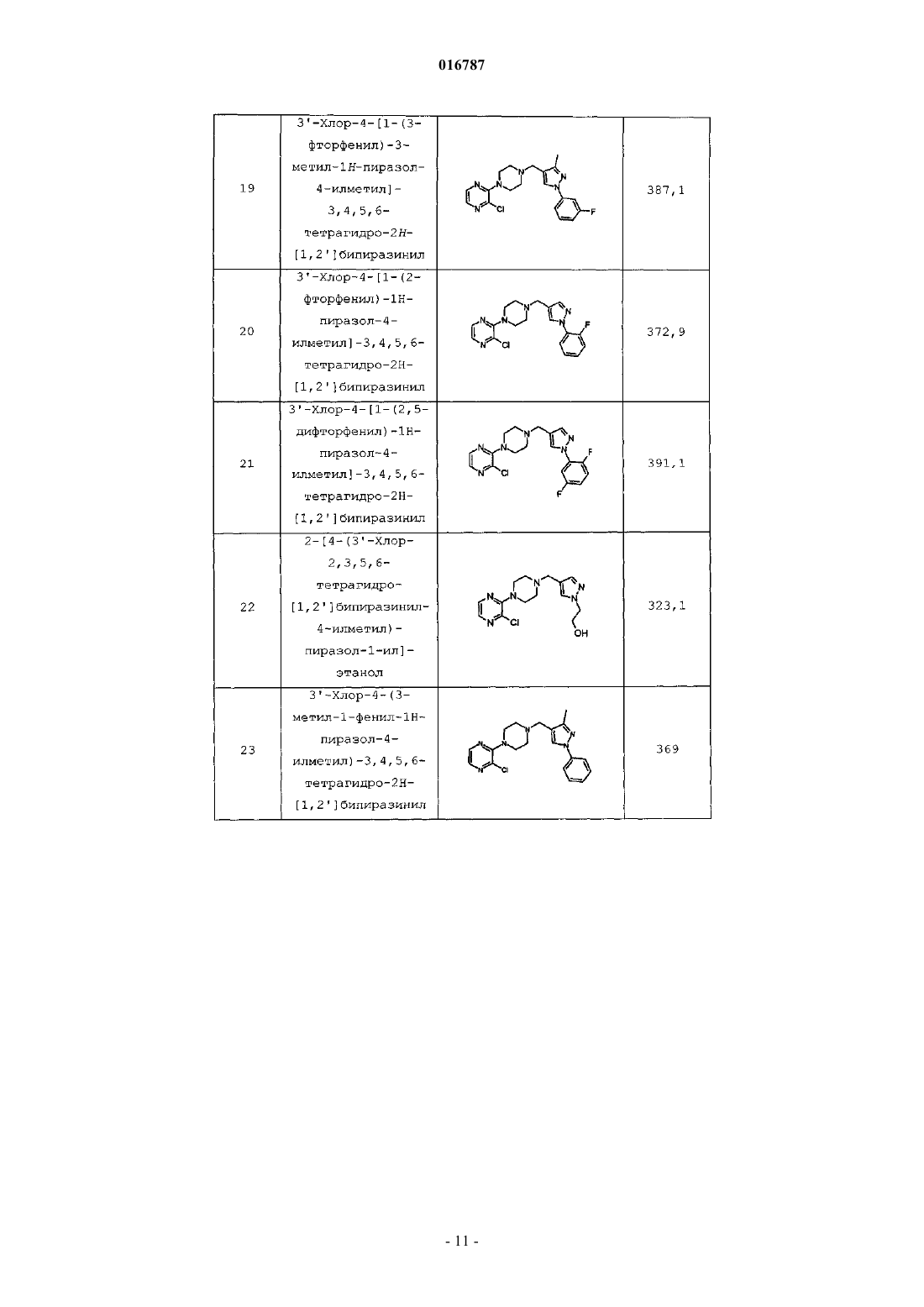
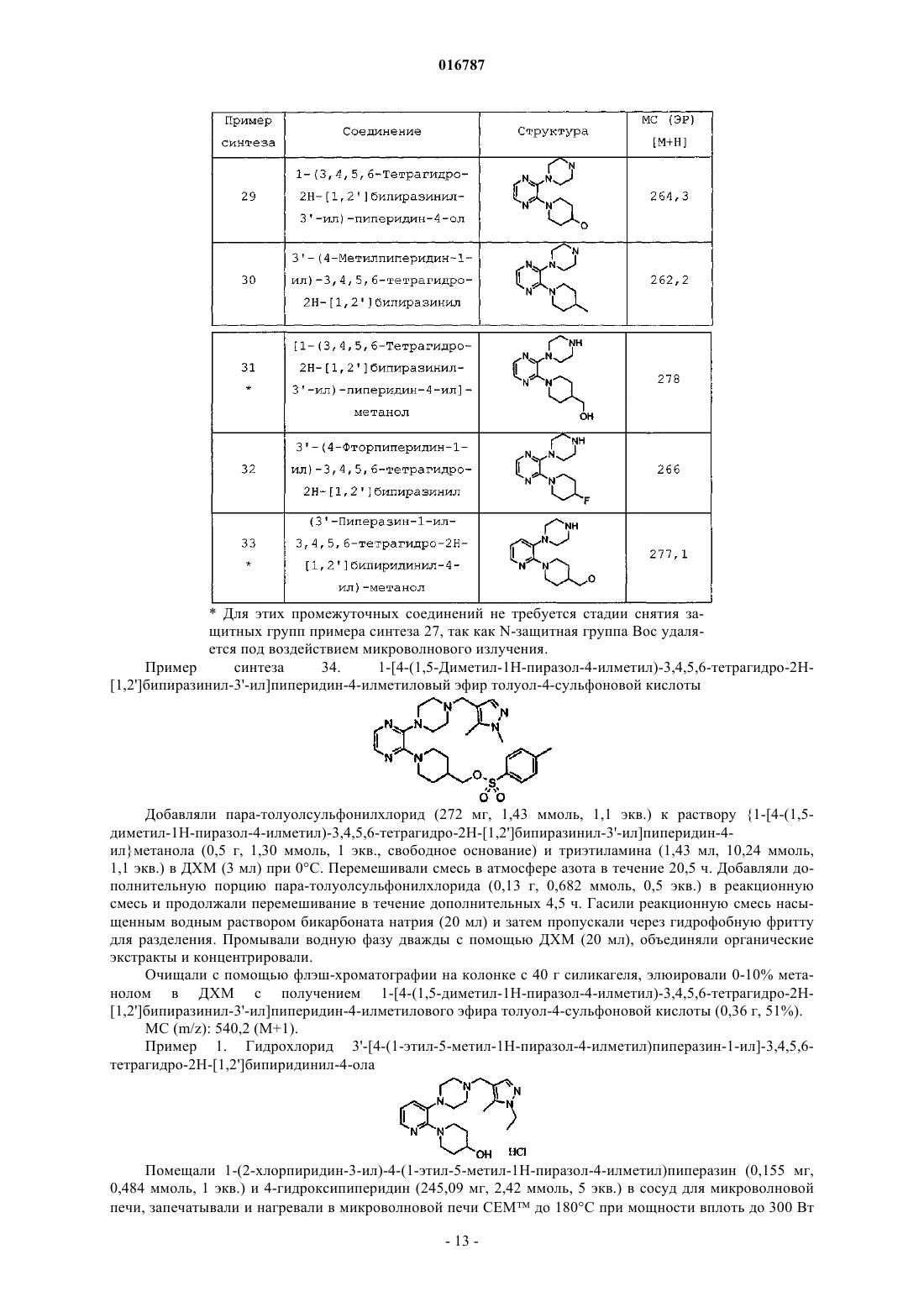
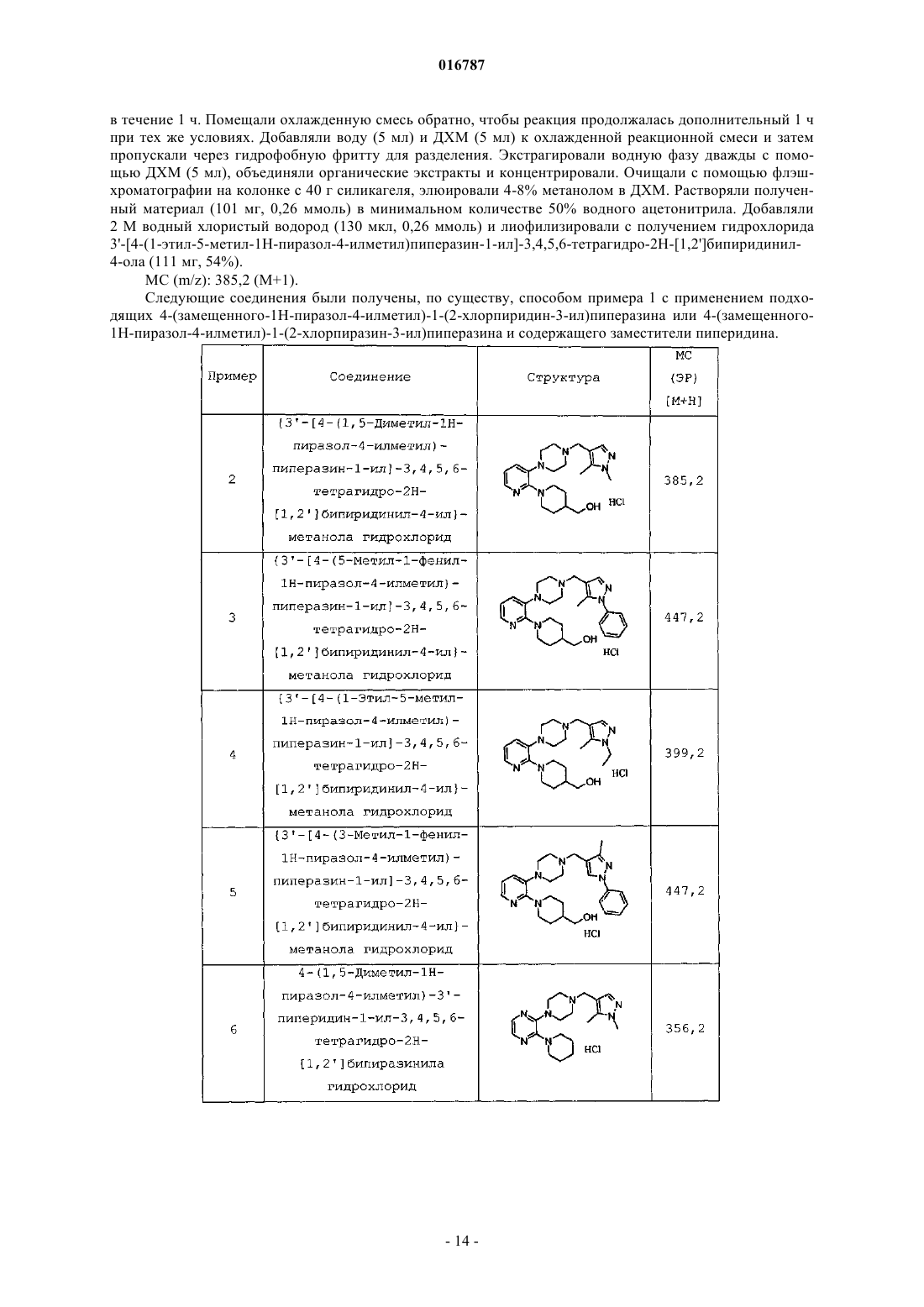
Настоящее изобретение предлагает селективные антагонисты рецептора 5-НТ 7, представляющие собой соединения формулы I, и их применение для лечения мигрени Бадеску Валентина О., Филла Сандра Энн (US), Галлахер Питер Таддеуш,Ваттон Мария Энн (GB) Медведев В.Н. (RU)(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) 016787 Нейромедиатор серотонин (5-гидрокситриптамин, 5-НТ) обладает разнообразными фармакологическими свойствами, обусловленными гетерогенностью популяции, включающей по меньшей мере 14 различных рецепторов. Все рецепторы обладают индивидуальными, хотя зачастую и перекрывающимися областями распределения в организме, и уникальными сайтами связывания серотонина, что обусловливает различия в аффинности к серотонину и различные физиологические ответы на взаимодействие с серотонином. Было показано, что рецептор 5-НТ 7 играет важные функциональные роли в терморегуляции, циркадном ритме, обучении и памяти, передаче сигналов гиппокампа и во сне. Также была установлена связь рецептора 5-НТ 7 с различными неврологическими расстройствами, включая мигрень и тревогу, а также постоянную боль, в частности боль, вызванную воспалением, и невропатическую боль. Высокоаффинные антагонисты рецептора 5-НТ 7 позволяют получить полезные терапевтические средства для лечения упомянутых, связанных с рецептором 5-НТ 7 расстройств, включая мигрень и постоянную боль, в частности воспалительную и невропатическую боль. Высокоаффинные антагонисты рецептора 5-НТ 7, которые также селективны в отношении рецептора 5-НТ 7, обеспечивают такое полезное терапевтическое действие в отсутствие нежелательных побочных явлений, связанных с модуляцией рецепторов других типов, например, относящихся к другим подклассам серотонинергических рецепторов, таким как 5-HT1A, 5-HT1B и 5-HT1D, или альфа-адренергическим рецепторам. Достичь селективности по отношению к рецептору 5-НТ 7, по сравнению с другими подтипами рецептора 5-НТ, при разработке антагонистов 5-НТ 7 оказалось затруднительным. Агонисты рецептора 5-HT1A связаны с серотониновым синдромом. Агонисты рецепторов 5-HT1B и 5-HT1D связаны с нежелательными явлениями, такими как боль в груди. У Leopoldo, M. (2004) "Serotonin(7) receptors (5-HT(7)Rs) and their ligands". Curr. Med. Chem. 11, 629661, описаны различные более ранние подходы к получению лигандов рецептора 5-НТ 7. ВWO 2004/067703 описаны антагонисты 5-НТ 7, включая некоторые 2-(пиперазин-1-ил)-3-фенилпиразины и пиридины. Согласно настоящему изобретению предложены новые эффективные антагонисты рецептора 5-НТ 7. Некоторые соединения согласно настоящему изобретению селективны в отношении рецептора 5-НТ 7 по сравнению с другими рецепторами серотонина. Согласно настоящему изобретению предложены соединения, представляющие собой селективные антагонисты рецептора 5-НТ 7 формулы IR1 представляет собой заместитель, выбранный из группы, состоящей из i) водорода, ii) метила,iii) этила, iv) гидроксиметила, v) гидроксиэтила, vi) фенила, необязательно содержащего в качестве заместителей от 1 до 3 фторгрупп, vii) бензила, необязательно содержащего в качестве заместителей от 1 до 3 фторгрупп, и viii) пиридила;R5 представляет собой водород или фтор, в том случае, когда R5 представляет собой фтор, R4 представляет собой фтор;R6 и R7 одинаковы и вместе выбраны из группы, состоящей из водорода, метила и фтора, при условии, что если R6 и R7 не являются водородом, то R4 и R5, оба, представляют собой водород; или их фармацевтически приемлемая соль. Согласно настоящему изобретению также предложены фармацевтические композиции, содержащие соединение формулы I или его фармацевтически приемлемую соль, совместно с фармацевтически приемлемым носителем, разбавителем или эксципиентом. В другом аспекте настоящего изобретения предложено одно или более соединений формулы I или его фармацевтически приемлемая соль(и) для применения в терапии. Этот аспект включает одно или более соединений формулы I или фармацевтически приемлемую соль такого соединения(и) для применения в качестве фармацевтического препарата. Аналогичным образом, в данном аспекте настоящего изобретения предложены одно или более соединений формулы I или его фармацевтически приемлемая соль(и) для применения при лечении мигрени у млекопитающих, в частности у людей, профилактическом лечении мигрени у млекопитающих, в частности людей, и/или лечении постоянной боли, в частно-1 016787 сти боли, вызванной воспалением, или невропатической боли у млекопитающих, в частности у людей. В одном варианте реализации этого аспекта настоящего изобретения предложен способ лечения мигрени у млекопитающих, включающий введение млекопитающему, нуждающемуся в таком лечении,эффективного количества соединения формулы I или его фармацевтически приемлемой соли. В другом варианте реализации этого аспекта настоящего изобретения предложен способ профилактического лечения мигрени у млекопитающих, включающий введение млекопитающему, нуждающемуся в таком лечении, т.е. млекопитающему, которое подвержено мигрени, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Другой вариант реализации этого аспекта настоящего изобретения обеспечивает способ лечения постоянной боли у млекопитающих, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I, или его фармацевтически приемлемой соли. Конкретные варианты реализации представляют собой лечение боли при воспалении и/или невропатической боли. В другом варианте реализации этого аспекта настоящего изобретения предложен способ лечения тревоги у млекопитающих, включающий введение млекопитающему, нуждающемуся в таком лечении,эффективного количества соединения формулы I, или его фармацевтически приемлемой соли. В предпочтительных вариантах реализации указанных способов лечения с применением соединений формулы I или их фармацевтически приемлемых солей указанное млекопитающее представляет собой человека. В другом аспекте настоящего изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения и/или профилактического лечения мигрени. В другом аспекте настоящего изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения постоянной боли, в частности боли при воспалении и/или невропатической боли. В другом аспекте настоящего изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения тревоги. Дополнительно, согласно настоящему изобретению предложена фармацевтическая композиция для лечения мигрени и/или для профилактического лечения мигрени, содержащая соединение формулы I или его фармацевтически приемлемую соль, совместно с фармацевтически приемлемым носителем, разбавителем или эксципиентом. Аналогичным образом, согласно настоящему изобретению предложена фармацевтическая композиция для лечения постоянной боли, в частности боли при воспалении и/или невропатической боли, содержащая соединение формулы I или его фармацевтически приемлемую соль, совместно с фармацевтически приемлемым носителем, разбавителем или эксципиентом. Дополнительно, согласно настоящему изобретению предложена фармацевтическая композиция для лечения тревоги, содержащая соединение формулы I или его фармацевтически приемлемую соль, совместно с фармацевтически приемлемым носителем, разбавителем или эксципиентом. Общие химические термины, используемые в данном изобретении, имеют обычные значения. Термин "аминозащитная группа" в данном патенте относится к заместителю, обычно используемому для блокировки или защиты функциональной аминогруппы при осуществлении реакции с другими функциональными группами на соединении. Используемые типы аминозащитных групп не являются определяющими при условии, что модифицированная аминогруппа стабильна в условиях последующих реакций в других положениях молекулы и может быть удалена в подходящий момент без разрушения остальной части молекулы. Выбор и использование (присоединение и последующее удаление) аминозащитных групп хорошо известны специалисту в данной области. Дополнительно, примеры групп, описанных указанными терминами, описаны у Т.W. Greene и P.G.M. Wuts, "Protective Groups in Organic Synthesis", 3-е издание, John Wiley и Sons, New York, NY, 1999, глава 7, далее именуемой "Greene". Термин "фармацевтический" или "фармацевтически приемлемый" в случае, когда он используется в данном изобретении как прилагательное, означает, по существу, нетоксичный и, по существу, не наносящий вреда реципиенту. Термин "фармацевтическая композиция" также подразумевает, что указанный носитель, растворитель, эксципиент и/или соль должны быть совместимы с действующим ингредиентом композиции (например, соединением формулы I). Для специалиста в данной области очевидно, что термины "фармацевтический состав" и "фармацевтическая композиция" в целом являются взаимозаменяемыми, поэтому они используются в контексте настоящего патента. Термин "эффективное количество" означает количество соединения формулы I, которое способно вызвать антагонизм рецепторов 5-НТ 7 и/или вызвать данное фармакологическое действие. Термин "подходящий растворитель" относится к любому растворителю или смеси растворителей,которая обеспечивает достаточную степень солюбилизации реагентов для получения среды, в которой осуществляется желательная реакция и которая не препятствует желательной реакции. Соединение, предназначенное для применения в фармацевтической композиции, можно, когда это-2 016787 возможно и желательно, перевести в форму соли, чтобы оптимизировать такие свойства, как удобство обращения, стабильность, фармакокинетика и/или биодоступность и т.д. Для любого соединения невозможно предсказать, какие противоионы будут образовывать солевые формы, такие как, например, кристаллическая солевая форма, имеющая оптимальные комбинации свойств для предполагаемого применения. Способы превращения соединения в солевую форму хорошо известны в данной области (см., например, P. Stahl, и др., Handbook of Pharmaceutical Salts: Properties, Selection and Use, (VCHA/Wiley-VCH,2002); Berge, S.M, Bighley, L.D., и Monkhouse, D.C., J. Pharm. Sci., 66:1, (1977. Такие соли также являются вариантами реализации настоящего изобретения. Хорошо известно, что соли могут быть образованы в различных молярных отношениях с кислотой с получением, например, полукислой, кислой однозамещенной, кислой двузамещенной соли и т.д. В процессе образования соли кислоту добавляют в определенном стехиометрическом отношении, и, если это не подтверждено аналитическими методами, предполагают, хотя это не известно наверняка, что соль образуется в данном молярном отношении. Сокращения, используемые в данном изобретении, имеют определения, указанные далее."Хроматография SCX" означает хроматографию на колонке или картридже SCX."Колонка SCX" или "картридж SCX" в данном патенте относятся к колонке Varian Bond Elute с сильной катионообменной смолой на основе диоксида кремния, или к одноразовому картриджу, или эквиваленту (как, например, картридж SCX-2). Хотя все соединения согласно настоящему изобретению можно применять в качестве антагонистов 5-НТ 7, некоторые группы являются предпочтительными, как, например, соединения, имеющие любые заместители из следующего списка: 1) R1 выбран из группы, состоящей из метила, этила, фенила, необязательно содержащего в качестве заместителей от 1 до 2 фторгрупп, или бензила; 2) R1 выбран из группы, состоящей из метила, этила и фенила, необязательно содержащего в качестве заместителей от 1 до 2 фторгрупп; 3) R1 представляет собой метил или этил; 4) R1 представляет собой фенил; 5) R1 представляет собой фенил, R2 представляет собой водород, R3 представляет собой хлор и R4 представляет собой гидрокси, гидроксиметил или метоксиметил; 6) R4 представляет собой гидрокси, гидроксиметил или метоксиметил; 7) R4 представляет собой гидрокси; 8) R4 представляет собой гидроксиметил; 9) R4 представляет собой метоксиметил; 10) R1 выбран из группы, состоящей из метила, этила и фенила, необязательно содержащего в качестве заместителей от 1 до 2 фторгрупп, и R4 представляет собой гидрокси, гидроксиметил или метоксиметил. В целом, производные пиразинила являются более предпочтительными, чем производные пиридила. Из соединений пиразинила предпочтительны такие, которые имеют заместители, выбранные из списка согласно любому из приведенных выше пп.1-10. Аналогичным образом, из соединений пиридила предпочтительны такие, которые имеют заместители, выбранные из списка согласно любому из приведенных выше абзацев 1-10. Особенно предпочтительные соединения согласно настоящему изобретению представляют собой соединения, описанные в примерах, приведенных в данном патенте, включая их свободные основания и фармацевтически приемлемые соли. Одним особенно предпочтительным соединением является 3'-[4-(1 этил-5-метил-1 Н-пиразол-4-илметил)пиперазин-1-ил]-3,4,5,6-тетрагидро-2 Н-[1,2']бипиридинил-4-ол или его фармацевтически приемлемая соль, как, например, соединение из примера 1. Соединения согласно настоящему изобретению могут быть получены согласно следующим схемам синтеза с помощью способов, хорошо известных и принятых в данной области. Подходящие условия реакции для стадий данных схем хорошо известны в данной области и подходящие замены растворителей и сореагентов известны специалисту в данной области. Аналогичным образом, для специалиста в данной области очевидно, что синтетические промежуточные соединения можно выделять и/или очищать с помощью различных хорошо известных методик при необходимости или желании и что часто может быть возможным применение различных промежуточных соединений непосредственно на следующих стадиях синтеза с незначительной очисткой или вовсе без нее. Более того, для квалифицированного специалиста очевидно, что при некоторых обстоятельствах порядок, в котором молекулы вводят в реакцию, не является принципиально важным. Конкретный порядок стадий, необходимый для получения соединений формулы I, зависит от конкретного синтезируемого соединения, исходного соединения и относительной лабильности содержащей заместители молекулы, что очевидно для опытного химика. Все заместители, если не указано другое, имеют значения, определенные выше, а все реагенты хорошо известны и приняты в данной области.-3 016787 На схеме I ниже показан один общий способ синтеза для получения соединений согласно настоящему изобретению. Схема I На этой схеме для соединений формулы VII, где А представляет собой азот, Hal, как правило, представляет собой хлор. Дигалогенпиперазин подвергают взаимодействию с N-защищенным пиперазином и подходящим основанием, таким как карбонат калия, в подходящем растворителе, таком как N,Nдиметилацетамид, при повышенной температуре с получением соединения формулы VI, где А представляет собой азот. Для соединений формулы VII, где А представляет собой -СН=, Hal обычно представляет собой бром или йод. Дигалогенпиридил сочетают с N-защищенным пиперазином в подходящих условиях каталитического сочетания, хорошо известных в данной области (John P. Wolfe и Stephen L. Buchwald.Organic Syntheses, Coll. том 10, с. 423 (2004); том 78, с. 23 (2002, с получением соединения формулы VI(А представляет собой СН). С соединений формулы VI можно снять защитные группы в условиях, хорошо известных квалифицированному специалисту (например, см. Greene и Wuts, Protective Groups in Organic Synthesis, третье издание, 1999, главы 2 и 7, John Wiley and Sons Inc.), с получением аминов формулы III. Затем можно осуществить реакцию полученных аминов с подходящими альдегидами пиразола в условиях восстановительного аминирования, хорошо известных квалифицированному специалисту (Richard С. Larock, Comprehensive Organic Transformations, второе издание, 1999, с. 835-846, Wiley и Sons Inc.), с получением соединений формулы II. Затем можно осуществить реакцию соединений формулы II с содержащими подходящие заместители пиперидинами, которые можно либо приобрести, либо получить с помощью способов, хорошо известных в данной области, с получением желательных свободных оснований I. При желании указанные свободные основания можно перевести в солевую форму с помощью средств, хорошо известных в данной области, например, путем реакции с фармацевтически приемлемой кислотой. В качестве альтернативы, промежуточные соединения формулы VI можно привести во взаимодействие с пиперидинами VIII при повышенной температуре с получением промежуточных соединений формулы V. С промежуточных соединений V затем снимают защитные группы в условиях, хорошо известных квалифицированному специалисту, с получением соединений формулы IV. Полученные амины затем приводят во взаимодействие с подходящими альдегидами пиразола в условиях восстановительного аминирования, хорошо известных квалифицированному специалисту, с получением соединений формулы I. Схема II Содержащие заместители пиразолы либо доступны для приобретения, либо их можно синтезировать с помощью в целом известных процедур, как, например, процедура, представленная на схеме II, где переменные R1, R2 и R3 такие, как определено ранее. Когда R2 не одинаков с R3, региоизомерные продукты циклизации нужно разделять с помощью обычных хроматографических методик. Если XII представляет собой неустойчивый альдегид, XII обычно присутствует в форме ацеталя. Осуществляют реакцию соединений формулы XII с подходящими гидразинами с получением соединений формулы XI. Затем осуществляют реакцию промежуточных соединений XI с POCl3 в подходящем растворителе, таком как диметилформамид, при повышенной температуре с получением желательных промежуточных соедине-4 016787 ний формулы IX. Схема III Можно использовать различные варианты реагентов в случае, когда предшественники альдегида вводят в предшественники циклизации, как показано на схеме III. Соединение формулы XIV реагирует с подходящим гидразином с получением сложного эфира пиразола формулы XIII, который затем восстанавливают с помощью подходящего восстанавливающего агента, такого как LiAlH4, с получением пиразолового спирта формулы XII. Полученный спирт можно окислить с помощью способов, хорошо известных квалифицированному специалисту, с получением желательного альдегида пиразола формулы IX. Следующие примеры синтеза и примеры иллюстрируют способы, которые можно применять для синтеза соединений согласно настоящему изобретению. Названия многих соединений, приведенных в примерах синтеза и примерах, взяты из структур, полученных с помощью программного обеспечения В 2-л трехгорлую круглодонную колбу загружали 2,3-дихлорпиразин (78,7 г, 0,532 моль), третбутиловый эфир пиперазин-1-карбоновой кислоты (100 г, 0,537 моль), карбонат калия (88,2 г,0,638 моль), а затем N,N-диметилацетамид (0,780 л) и нагревали полученную суспензию до 110 С в атмосфере азота при интенсивном перемешивании. Охлаждали до комнатной температуры, добавляли воду(0,390 л) и метил трет-бутиловый эфир (0,390 л), перемешивали смесь в течение 60 мин. Прекращали перемешивание и разделяли фазы. Промывали органическую фазу водой (2200 мл), сушили над MgSO4,фильтровали и концентрировали с получением 145 г трет-бутилового эфира 3'-хлор-2,3,5,6-тетрагидро[1,2']бипиразинил-4-карбоновой кислоты в виде желтого сиропа (выход 91%). 1 Н ЯМР (CDCl3)(м.д.) 8,10 (с, 1 Н), 7,91 (с, 1 Н), 3,59 (м, 4 Н), 3,40 (н, 4 Н), 1,48 (с, 9 Н). Пример синтеза 2. 3'-Хлор-3,4,5,6-тетрагидро-2 Н-[1,2']бипиразинил Добавляли 4 М HCl в 1,4-диоксане (10 мл) к трет-бутиловому эфиру 3'-хлор-2,3,5,6-тетрагидро[1,2']бипиразинил-4-карбоновой кислоты (6,80 г, 22,76 ммоль). Добавляли 1,4-диоксан (40 мл) и подвергали реакционную смесь воздействию ультразвука, затем перемешивали при комнатной температуре в атмосфере азота в течение 3 ч. Дополнительно добавляли HCl в 1,4-диоксане (40 мл) и перемешивали в течение 1 ч. Добавляли хлороформ (400 мл), промывали 2 н. гидроксидом натрия (200 мл), насыщенным водным хлоридом натрия (100 мл), сушили (сульфат магния) и концентрировали с получением 3'-хлор 3,4,5,6-тетрагидро-2 Н-[1,2']бипиразинила в виде желтого масла, которое кристаллизовалось при выстаивании с получением твердого вещества (4,0 г, 88%). МС (m/z): 199,1 (М+1). Пример синтеза 3. трет-Бутиловый эфир 4-(2-хлорпиридин-3-ил)пиперазин-1-карбоновой кислоты Перемешивали 2-хлор-3-бромпиридин (5,00 г, 26,0 ммоль) и трет-бутиловый эфир пиперазин-1 карбоновой кислоты (3,73 г, 20,0 ммоль) в безводном толуоле (200 мл) при комнатной температуре в ат-5 016787 мосфере азота. Добавляли трет-бутоксид натрия (2,88 г, 30,0 ммоль), трис(дибензилиденацетон)дипалладий(0) (0,366 г, 0,40 ммоль) и 4,5-бис-(дифенилфосфино)-9,9-диметилксантен (0,694 г,1,20 ммоль), дегазировали реакционную смесь и нагревали до 100 С (температура масляной бани) в течение 3 ч. Охлаждали до комнатной температуры, добавляли 100 мл воды, экстрагировали 2200 мл этилацетата. Концентрировали органическую фазу под вакуумом, очищали (хроматография на силикагеле, элюировали смесью 30:70 этилацетат:изогексан) и сушили в вакуумной печи в течение ночи с получением трет-бутилового эфира 4-(2-хлорпиридин-3-ил)пиперазин-1-карбоновой кислоты в виде бежевого порошка (3,01 г, 51%). МС (m/z): 298 (М+1). Пример синтеза 4. 1-(2-Хлорпиридин-3-ил)пиперазин(2,00 г, 6,72 ммоль) в ДХМ (50 мл) при комнатной температуре, затем добавляли трифторуксусную кислоту (5 мл). Перемешивали реакционную смесь в течение 2 ч и удаляли растворители под вакуумом,затем получали свободное основание, применяя хроматографию SCX-2, промывая метанолом, затем элюировали приблизительно 3 М аммиаком в метаноле. Концентрировали под вакуумом с получением 1(2-хлорпиридин-3-ил)пиперазина в виде коричневого масла (1,47 г, выход 110%). МС (m/z): 198 (М+1). Пример синтеза 5. 1-(3-Фторфенил)-3-метил-1H-пиразол Добавляли соляную кислоту (5 М, 12 мл, 60 ммоль) к смеси 4,4-диметоксибута-2-она (6,61 г,6,67 мл, 50 ммоль) и гидрохлорида 3-фторфенилгидразина (8,13 г, 50 ммоль) в этаноле (50 мл). Нагревали и перемешивали при кипении с обратным холодильником в атмосфере азота в течение 7,5 ч, охлаждали до комнатной температуры, оставляли на 60 ч. Выпаривали этанол под вакуумом и проводили хроматографию остатка на диоксиде кремния, элюировали ДХМ. Выпаривали дихлорметан с получением 1-(3 фторфенил)-3-метил-1 Н-пиразола в виде жидкости (4,38 г, 49%). МС (m/z): 171,1 (М+1). Пример синтеза 6. 1-(2,5-Дифторфенил)-1 Н-пиразол(9,022 г, 62,6 ммоль) и соляной кислоты (5 М, 5 мл, 25 ммоль) в этаноле (50 мл) и нагревали и перемешивали при кипении с обратным холодильником в атмосфере азота в течение 17 ч. Охлаждали смесь, выпаривали этанол под вакуумом, суспендировали остаток в ДХМ (80 мл), фильтровали раствор ДХМ и пропускали через колонку SCX-2. Собирали элюент и пропускали через вторую колонку SCX2 и выпаривали элюент с получением 1-(2,5-дифторфенил)-1 Н-пиразола в виде жидкости (8,79 г, 97%). МС (m/z): 181 (М+1). Пример синтеза 7. 1-(3-Фторфенил)-3-метил-1 Н-пиразол-4-карбальдегид Добавляли оксихлорид фосфора (20,8 мл, 34,3 г, 223,7 ммоль) по каплям при помешивании при 95 С в атмосфере азота к 1-(3-фторфенил)-3-метил-1 Н-пиразолу (4,38 г, 24,86 ммоль) в диметилформамиде (19,2 мл, 18,17 г, 248,6 ммоль). Грели при 95 С в течение 15 ч, охлаждали до комнатной температуры, заливали поверх льда и нейтрализовали бикарбонатом натрия. Экстрагировали водный раствор этилацетатом (2150 мл), сушили (сульфат магния), фильтровали и пропускали через колонку SCX-2. Выпа-6 016787 ривали растворитель с получением 1-(3-фторфенил)-3-метил-1 Н-пиразол-4-карбальдегида в виде твердого вещества. (4,22 г, 83%). МС (m/z): 205,1 (М+1). Пример синтеза 8. 1-(2,5-Дифторфенил)-1 Н-пиразол-4-карбальдегид Указанное в заголовке промежуточное соединение получали, применяя способы, аналогичные описанным в примере синтеза 7, с использованием 1-(2,5-дифторфенил)-1H-пиразола. МС (ЭР) [М+Н] 209,1. Пример синтеза 9. 5-Метил-1-пиридин-2-ил-1 Н-пиразол-4-карбальдегид Этиловый эфир 2-диметиламинометилен-3-оксомасляной кислоты. Добавляли этилацетоацетат (15 мл, 0,118 моль) к диметоксиметилдиметиламину (19 мл, 0,142 моль) и кипятили смесь с обратным холодильником в течение 1 ч. Выпаривали смесь с получением этилового эфира 2-диметиламинометилен-3-оксомасляной кислоты (21,7 г, 99%). Этиловый эфир 5-метил-1-пиридин-2-ил-1 Н-пиразол-4-карбоновой кислоты. Растворяли этиловый эфир 2-диметиламинометилен-3-оксомасляной кислоты (0,662 г, 3,57 ммоль) и пиридин-2-илгидразин (0,410 г, 3,75 ммоль) в этаноле (15 мл) и кипятили с обратным холодильником в течение 2 ч. Выпаривали смесь, затем разбавляли насыщенным бикарбонатом натрия и экстрагировали трижды этилацетатом. Сушили раствор (сульфат натрия), фильтровали и концентрировали. Очищали,применяя хроматографию на силикагеле, элюировали смесью 50:50 этилацетат:гексан с получением этилового эфира 5-метил-1-пиридин-2-ил-1 Н-пиразол-4-карбоновой кислоты в виде белого твердого вещества (0,700 г, 85%). МС (m/z): 232 (М+1).(5-Метил-1-пиридин-2-ил-1 Н-пиразол-4-ил)метанол. Добавляли литийалюминий гидрид (0,225 г, 5,92 ммоль) к тетрагидрофурану (15 мл) при 0 С, затем медленно по каплям добавляли этиловый эфир 5-метил-1-пиридин-2-ил-1 Н-пиразол-4-карбоновой кислоты (0,685 г, 2,96 ммоль) в тетрагидрофуране (5 мл). Нагревали смесь до комнатной температуры и перемешивали в течение 2 ч, затем охлаждали раствор до 0 С. Добавляли насыщенный водный сульфат натрия (0,5 мл), нагревали до комнатной температуры, затем перемешивали в течение 2 ч. Отфильтровывали твердые частицы, затем сушили раствор (сульфат натрия), фильтровали и концентрировали с получением (5-метил-1-пиридин-2-ил-1 Н-пиразол-4-ил)метанола в виде белого твердого вещества (0,501 г,89%). 5-Метил-1-пиридин-2-ил-1 Н-пиразол-4-карбальдегид. Растворяли диметилсульфоксид (0,751 мл, 10,6 ммоль) в ДХМ (20 мл) и охлаждали до -78 С. Добавляли оксалилхлорид (0,577 мл, 6,62 ммоль) по каплям в ДХМ (8 мл) и перемешивали в течение 15 мин. Добавляли по каплям (5-метил-1-пиридин-2-ил-1 Н-пиразол-4-ил)метанол (0,501 г, 2,65 ммоль) в ДХМ (20 мл) и перемешивали в течение 1 ч при -78 С. Добавляли триэтиламин (1,85 мл, 13,2 ммоль) и нагревали смесь до комнатной температуры в течение 1 ч. Разбавляли смесь насыщенным бикарбонатом натрия и экстрагировали трижды с помощью ДХМ. Сушили (сульфат натрия) раствор, фильтровали и концентрировали с получением 5-метил-1-пиридин-2-ил-1 Н-пиразол-4-карбальдегида в виде белого твердого вещества (0,496 г, 100%). МС (m/z): 188 (М+1). Пример синтеза 10. 3-Этил-1-фенил-1 Н-пиразол-4-карбальдегид-7 016787 Добавляли уксусную кислоту (1,00 мл, 17,45 ммоль) и фенилгидразин (1,98 мл, 20,00 ммоль) к раствору 2-бутанона (2,15 мл, 24,00 ммоль) в этаноле (90 мл) при комнатной температуре. Перемешивали реакционную смесь в течение 1 ч, затем удаляли растворители под вакуумом с получением N-[1 метилпроп-(Е)-илиден]-N'-фенилгидразина в виде неочищенного оранжевого масла (3,21 г, 99%). МС (m/z): 163 (М+1). 3-Этил-1-фенил-1 Н-пиразол-4-карбальдегид. К ледяному раствору N,N-диметилформамида (4,59 мл, 59,36 ммоль) и фосфорилхлорида (5,52 мл,59,36 ммоль) по каплям добавляли раствор N-[1-метилпроп-(Е)-илиден]-N'-фенилгидразина (3,21 г,19,79 ммоль) в N,N-диметилформамиде (2 мл). Нагревали до комнатной температуры, затем нагревали до 75 С в течение 5 ч. Охлаждали до комнатной температуры и вливали в ледяной раствор насыщенного карбоната калия. Экстрагировали ДХМ (320 мл), пропускали через IST Phase Separator Frit и концентрировали. Очищали (хроматография на силикагеле, элюировали смесью от 0:100 до 20:80 этилацетат:изогексан), с получением 3-этил-1-фенил-1 Н-пиразол-4-карбальдегида в виде коричневого твердого вещества (600 мг, 15%). МС (m/z): 201 (М+1). Пример синтеза 11. 3,5-Диметил-1-пиридин-2-ил-1 Н-пиразол-4-карбальдегид Этиловый эфир 3,5-диметил-1-пиридин-2-ил-1 Н-пиразол-4-карбоновой кислоты. Растворяли этиловый эфир 2-ацетил-3-оксомасляной кислоты (20,74 г, 0,120 моль) и 2 пиридилгидразин (14,5 мл, 0,133 моль) в уксусной кислоте (160 мл) и перемешивали смесь в течение 18 ч. Концентрировали, разбавляли ДХМ, промывали насыщенным бикарбонатом натрия, сушили (сульфат натрия), фильтровали и концентрировали с получением этилового эфира 3,5-диметил-1-пиридин-2 ил-1 Н-пиразол-4-карбоновой кислоты в виде масла (28,6 г, 97%). МС (m/z): 246 (М+1).(3,5-Диметил-1-пиридин-2-ил-1 Н-пиразол-4-ил)метанол. Суспендировали литийалюминийгидрид (0,359 г, 9,46 ммоль) в тетрагидрофуране (25 мл) при -10 С и добавляли по каплям этиловый эфир 3,5-диметил-1-пиридин-2-ил-1 Н-пиразол-4-карбоновой кислоты(1,160 г, 4,73 ммоль) в тетрагидрофуран (5 мл). Позволяли смеси нагреться до 25 С и перемешивали в течение 4 ч. Охлаждали смесь до 0 С, затем осторожно гасили насыщенным раствором сульфата натрия(1 мл). Позволяли смеси перемешиваться при комнатной температуре в течение 2 ч, затем отфильтровывали осадок, сушили раствор и концентрировали с получением (3,5-диметил-1-пиридин-2-ил-1 Нпиразол-4-ил)метанола в виде желтого твердого вещества (0,821 г, 86%). 3,5-Диметил-1-пиридин-2-ил-1 Нпиразол-4-карбальдегид. Растворяли диметилсульфоксид (0,324 мл, 4,56 ммоль) в ДХМ (10 мл) и охлаждали раствор до-78 С. По каплям добавляли к смеси оксалилхлорид (0,239 мл, 2,74 ммоль) и перемешивали при -78 С в течение 20 мин. Добавляли (3,5-диметил-1-пиридин-2-ил-1 Н-пиразол-4-ил)метанол (0,369 г, 1,82 ммоль) в ДХМ (10 мл) и перемешивали смесь при -78 С в течение 1 ч. Добавляли к смеси триэтиламин (1,27 мл,9,12 ммоль) и нагревали до комнатной температуры, затем перемешивали в течение 18 ч. Добавляли насыщенный водный бикарбонат натрия и экстрагировали водную фазу 3 раза с помощью ДХМ, сушили органический раствор, затем фильтровали и концентрировали. Очищали, применяя хроматографию на силикагеле, элюировали смесью 20:80 гексаны:этилацетат с получением 3,5-диметил-1-пиридин-2-ил 1 Н-пиразол-4-карбальдегида в виде желтого твердого вещества (0,358 г, 97%). МС (m/z): 202 (М+1). Пример синтеза 12. 1-(2-Гидроксиэтил)-1 Н-пиразол-4-карбальдегид Объединяли 1 Н-пиразол-4-карбальдегид (0,110 г, 1,14 ммоль), 2-бромэтанол (0,172 г, 1,37 ммоль) и карбонат калия (0,236 г, 1,71 ммоль) в ацетонитриле (2 мл). Нагревали в микроволновой печи при 150 С в течение 20 мин. Охлаждали до комнатной температуры и фильтровали, промывали ацетонитрилом. Концентрировали фильтрат с получением 1-(2-гидроксиэтил)-1 Н-пиразол-4-карбальдегида (0,155 г,97%). ГХ-МС (m/z): 140 (М+). К раствору трет-бутил-4-(аминометил)тетрагидропиридин-1(2 Н)-карбоксилата (1,50 г, 7,0 ммоль,1 экв.) в ДХМ (безводный) (20 мл) добавляли метансульфонилхлорид (569 мкл, 7,35 ммоль, 1,05 экв.). Затем добавляли по каплям триэтиламин (2,05 мл, 14,7 ммоль, 2,1 экв.) в течение 15 мин. Перемешивали при комнатной температуре в течение 3 ч и затем добавляли воду (20 мл) при помешивании. Органическую фазу отделяли, затем промывали 2 М водной соляной кислотой (20 мл) и раствором насыщенного водного бикарбоната натрия (20 мл). Сушили органическую фазу (сульфат магния) и концентрировали с получением трет-бутилового эфира 4-(метансульфониламинометил)пиперидин-1-карбоновой кислоты(2,1 г, 102%). МС (ЭР): m/z=315,1 [M+Na]+. К раствору этого соединения (2,1 г, 7,2 ммоль, 1 экв.) в 1,4-диоксане (25 мл) добавляли 4 М хлористый водород в диоксане (17,95 мл, 72 ммоль, 10 экв.). Перемешивали при комнатной температуре в течение 29 ч, повышали основность с помощью 2 М водного гидроксида натрия, а затем добавляли ДХМ(20 мл). Разделяли фазы и экстрагировали водную фазу дважды с помощью ДХМ (20 мл), сушили объединенные органические фазы над сульфатом магния, фильтровали и концентрировали. Дополнительно экстрагировали водную фазу четыре раза с помощью смеси 3:1 хлороформ:изопропанол (25 мл). Концентрировали водную фазу до менее чем 10 мл по объему и снова экстрагировали четыре раза с помощью смеси 3:1 хлороформ:изопропанол (25 мл). Объединяли со всеми полученными ранее органическими экстрактами с получением N-пиперидин-4-илметилметансульфонамида (703 мг, 50%). МС (m/z): 193 (М+1). Пример синтеза 14. Пиперидин-4-илацетонитрил трет-Бутиловый эфир 4-цианметиленпиперидин-1-карбоновой кислоты. Добавляли диэтилцианметилфосфонат (5,33 г, 4,88 мл, 30,11 ммоль) к карбонату калия (3,47 г,25,09 ммоль) в безводном ТГФ (10 мл) и перемешивали при комнатной температуре в течение 15 мин,затем нагревали при кипении с обратным холодильником в течение 15 мин. К этой смеси добавляли третбутиловый эфир 4-оксопиперидин-1-карбоновой кислоты (5,00, 25,09 ммоль) и грели при кипении с обратным холодильником в атмосфере азота в течение 24 ч, позволяли охладиться до комнатной температуры и выстаивали в течение ночи. Вливали реакционную смесь в водный раствор карбоната калия (10%,80 мл) и экстрагировали полученную смесь этилацетатом (250 мл). Объединяли органические фазы сушили (MgSO4) и выпаривали под вакуумом с получением трет-бутилового эфира 4 цианметиленпиперидин-1-карбоновой кислоты в виде жидкости, которая затвердевала при выстаивании(5,39 г, 96,6%). 1 Н ЯМР (-CDCl3) 1,5 (с, 9 Н), 2,4 (м, 2 Н), 2,6 (м, 2 Н), 3,5 (м, 4 Н), 5,2 (с, 1 Н). трет-Бутиловый эфир 4-цианметилпиперидин-1-карбоновой кислоты. Добавляли трет-бутиловый эфир 4-цианметиленпиперидин-1-карбоновой кислоты (5,39 г,24,25 ммоль) в этаноле (160 мл) в суспензию 5% палладия на древесном угле (0,69 г) в этаноле (20 мл) и гидрировали при комнатной температуре при встряхивании под давлением 60 фунтов на кв.дюйм в течение 6 ч. Фильтровали смесь через целит и выпаривали растворитель под вакуумом с получением третбутилового эфира 4-цианметилпиперидин-1-карбоновой кислоты в виде масла, которое затвердевало при выстаивании с получением твердого вещества (5,43 г, 99,8%). МС (m/z): 247 (M+Na). Пиперидин-4-илацетонитрил. Добавляли трифторуксусную кислоту (23 мл, 34,7 г, 304 ммоль) к трет-бутиловому эфиру 4 цианметилпиперидин-1-карбоновой кислоты (5,43 г, 24,21 ммоль) в ДХМ (25 мл) и перемешивали при комнатной температуре в течение 18 ч. Удаляли растворитель под вакуумом, растворяли в метаноле(50 мл) и вливали на колонку SCX-2. Элюировали 2 М аммиаком в метаноле и выпаривали элюент с получением пиперидин-4-илацетонитрила в виде масла, которое затвердевало при выстаивании (2,78 г,92%). МС (m/z): 125,1 (М+1). 3'-Хлор-4-(1,5-диметил-1 Н-пиразол-4-илметил)-3,4,5,6-тетрагидро-2 Н В 2-л трехгорлую круглодонную колбу загружали 3'-хлор-3,4,5,6-тетрагидро-2 Н-[1,2']бипиразинил(39 г, 0,196 моль), 1,2-дихлорэтан (780 мл), а затем 1,5-диметил-1 Н-пиразол-4-карбальдегид (25,5 г,0,206 моль) и перемешивали в течение 15 мин в атмосфере азота при интенсивном перемешивании. Добавляли триацетоксиборогидрид натрия (45,77 г, 215 ммоль) тремя частями с интервалами 10 мин. Медленно добавляли метанол (100 мл) и перемешивали в течение 20 мин, концентрировали с получением белой пены. Растворяли полученную пену в метиленхлориде и добавляли к 1 кг вставки из диоксида кремния. Элюировали продукт 5-10% изопропиловым спиртом/ДХМ и концентрировали фракции, содержащие продукт, с получением 3'-хлор-4-(1,5-диметил-1 Н-пиразол-4-илметил)-3,4,5,6-тетрагидро-2 Н[1,2']бипиразинила в виде желтого масла (37 г, 60%). МС (m/z): 307 (М+1). Следующие соединения были получены, по существу, с помощью способов примера синтеза 15 с применением подходящих 2-хлор-3-(пиперазин-1-ил)пиразина или 1-(2-хлорпиридин-3-ил)пиперазина и содержащего заместители 1 Н-пиразол-4-карбальдегида. трет-Бутиловый эфир 3'-пиперидин-1-ил-2,3,5,6-тетрагидро-[1,2']бипиразинил-4-карбоновой кислоты. Помещали трет-бутиловый эфир 3'-хлор-2,3,5,6-тетрагидро-[1,2']бипиразинил-4-карбоновой кислоты (0,4 г, 1,34 ммоль, 1 зкв.) и пиперидин (662 мкл, 6,69 ммоль, 5 экв.) в сосуд для микроволновой печи,запечатывали и нагревали в микроволновой печи СЕМ до 180 С при мощности 300 Вт в течение 1 ч(нужно соблюдать осторожность, так как при этом повышается давление). Добавляли 2 М водный раствор гидроксида натрия (5 мл) и ДХМ (5 мл), а затем пропускали через гидрофобную фритту для разделения. Экстрагировали водную фазу дважды ДХМ (5 мл), объединяли органические экстракты и концентрировали с получением трет-бутилового эфира 3'-пиперидин-1-ил-2,3,5,6-тетрагидро-[1,2']бипиразинил 4-карбоновой кислоты (0,46 г, 99%). МС (m/z): 348,3 (М+1). 3'-Пиперидин-1-ил-3,4,5,6-тетрагидро-2 Н-[1,2']бипиразинил. Добавляли трифторуксусную кислоту (1,00 мл, 13,24 ммоль, 10 экв.) к раствору трет-бутилового эфира 3'-пиперидин-1-ил-2,3,5,6-тетрагидро-[1,2']бипиразинил-4-карбоновой кислоты (0,46 г, 1,32 ммоль,1 экв.) в ДХМ (10 мл), затем перемешивали смесь при комнатной температуре в течение 4 ч. Концентрировали реакционную смесь, затем растворяли в метаноле и загружали на 10 г ионообменный картриджSCX-2 (предварительно промытый метанолом). Промывали одним объемом колонки метанола, затем элюировали одним объемом колонки 3,5 М аммиака в метаноле. Концентрировали с получением 3'пиперидин-1-ил-3,4,5,6-тетрагидро-2 Н-[1,2']бипиразинила (0,317 г, 97%). МС (m/z): 248,2 (М+1). Следующие соединения были получены, по существу, способом примера синтеза 28 с применением трет-бутил-4-(2-хлорпиридин-3-ил)пиперазин-1-карбоксилата или трет-бутил-4-(3-хлорпиразин-2 ил)пиперазин-1-карбоксилата и содержащего заместители пиперидина. Для этих промежуточных соединений не требуется стадии снятия защитных групп примера синтеза 27, так как N-защитная группа Boc удаляется под воздействием микроволнового излучения. Пример синтеза 34. 1-[4-(1,5-Диметил-1 Н-пиразол-4-илметил)-3,4,5,6-тетрагидро-2 Н[1,2']бипиразинил-3'-ил]пиперидин-4-илметиловый эфир толуол-4-сульфоновой кислоты Добавляли пара-толуолсульфонилхлорид (272 мг, 1,43 ммоль, 1,1 экв.) к раствору 1-[4-(1,5 диметил-1 Н-пиразол-4-илметил)-3,4,5,6-тетрагидро-2 Н-[1,2']бипиразинил-3'-ил]пиперидин-4 илметанола (0,5 г, 1,30 ммоль, 1 экв., свободное основание) и триэтиламина (1,43 мл, 10,24 ммоль,1,1 экв.) в ДХМ (3 мл) при 0 С. Перемешивали смесь в атмосфере азота в течение 20,5 ч. Добавляли дополнительную порцию пара-толуолсульфонилхлорида (0,13 г, 0,682 ммоль, 0,5 экв.) в реакционную смесь и продолжали перемешивание в течение дополнительных 4,5 ч. Гасили реакционную смесь насыщенным водным раствором бикарбоната натрия (20 мл) и затем пропускали через гидрофобную фритту для разделения. Промывали водную фазу дважды с помощью ДХМ (20 мл), объединяли органические экстракты и концентрировали. Очищали с помощью флэш-хроматографии на колонке с 40 г силикагеля, элюировали 0-10% метанолом в ДХМ с получением 1-[4-(1,5-диметил-1 Н-пиразол-4-илметил)-3,4,5,6-тетрагидро-2 Н[1,2']бипиразинил-3'-ил]пиперидин-4-илметилового эфира толуол-4-сульфоновой кислоты (0,36 г, 51%). МС (m/z): 540,2 (М+1). Пример 1. Гидрохлорид 3'-[4-(1-этил-5-метил-1 Н-пиразол-4-илметил)пиперазин-1-ил]-3,4,5,6 тетрагидро-2 Н-[1,2']бипиридинил-4-ола Помещали 1-(2-хлорпиридин-3-ил)-4-(1-этил-5-метил-1 Н-пиразол-4-илметил)пиперазин (0,155 мг,0,484 ммоль, 1 экв.) и 4-гидроксипиперидин (245,09 мг, 2,42 ммоль, 5 экв.) в сосуд для микроволновой печи, запечатывали и нагревали в микроволновой печи СЕМ до 180 С при мощности вплоть до 300 Вт- 13016787 в течение 1 ч. Помещали охлажденную смесь обратно, чтобы реакция продолжалась дополнительный 1 ч при тех же условиях. Добавляли воду (5 мл) и ДХМ (5 мл) к охлажденной реакционной смеси и затем пропускали через гидрофобную фритту для разделения. Экстрагировали водную фазу дважды с помощью ДХМ (5 мл), объединяли органические экстракты и концентрировали. Очищали с помощью флэшхроматографии на колонке с 40 г силикагеля, элюировали 4-8% метанолом в ДХМ. Растворяли полученный материал (101 мг, 0,26 ммоль) в минимальном количестве 50% водного ацетонитрила. Добавляли 2 М водный хлористый водород (130 мкл, 0,26 ммоль) и лиофилизировали с получением гидрохлорида 3'-[4-(1-этил-5-метил-1 Н-пиразол-4-илметил)пиперазин-1-ил]-3,4,5,6-тетрагидро-2 Н-[1,2']бипиридинил 4-ола (111 мг, 54%). МС (m/z): 385,2 (М+1). Следующие соединения были получены, по существу, способом примера 1 с применением подходящих 4-(замещенного-1 Н-пиразол-4-илметил)-1-(2-хлорпиридин-3-ил)пиперазина или 4-(замещенного 1 Н-пиразол-4-илметил)-1-(2-хлорпиразин-3-ил)пиперазина и содержащего заместители пиперидина. Реакцию вели при обычном нагреве в запечатанной пробирке, а не под действием микроволнового излучения, как в примере 1.Подходящий содержащий заместители пиперидин применяли в виде егоHCl-соли и добавляли диизопропилэтиламин для предотвращения разложения.Применяли подходящий растворитель, такой как 1,4-диоксан или пиридин. Пример 24. 3'-[4-(1-Этил-1 Н-пиразол-4-илметил)пиперазин-1-ил]-3,4,5,6-тетрагидро-2 Н[1,2']бипиридинил-4-илметанола гидрохлорид К раствору (3'-пиперазин-1-ил-3,4,5,6-тетрагидро-2 Н-[1,2']бипиридинил-4-ил)метанола (0,145 г,0,524 ммоль, 1 экв.) и 1-этил-1 Н-пиразол-4-карбальдегида (97,69 мг, 0,787 ммоль, 1,5 экв.) в 1,2-дихлорэтане (10 мл) добавляли сразу весь триацетоксиборогидрид натрия (166,79 мг, 0,787 ммоль,1,5 экв.) в виде твердого вещества. Перемешивали полученную смесь при комнатной температуре в атмосфере азота в течение 20 ч. Добавляли 2 М водный раствор гидроксида натрия (20 мл) и ДХМ (20 мл). Разделяли, применяя разделитель фаз, и экстрагировали водную фазу с помощью ДХМ (10 мл). Концентрировали объединенные органические экстракты и очищали с помощью ВЭЖХ с обращенной фазой при высоком рН. Растворяли полученный материал (120 мг, 0,31 ммоль) в минимальном количестве 50% водного ацетонитрила. Добавляли 2 М водный хлористый водород (155 мкл, 0,31 ммоль) и лиофилизировали с получением указанного в заголовке соединения (127 мг, 58%). МС (m/z): 385,2 (М+1). Следующие соединения были получены, по существу, способом примера 24 с применением подходящих 1-(2-(замещенного-пиперидин-1-ил)пиридин-3-ил)пиперазина или 2-(замещенного-пиперидин-1 ил)-3-(пиперазин-1-ил)пиразина и содержащего заместители 1 Н-пиразол-4-карбальдегида. Добавляли цианид натрия (78,46 мг, 1,60 ммоль, 2,4 экв.) к раствору 1-[4-(1,5-диметил-1 Н-пиразол 4-илметил)-3,4,5,6-тетрагидро-2 Н-[1,2']бипиразинил-3'-ил]пиперидин-4-илметилового эфира толуол-4 сульфоновой кислоты (0,36 г, 0,667 ммоль, 1 экв.) в диметилсульфоксиде (5 мл). Нагревали полученный раствор до 50 С при помешивании в течение 5,75 ч, а затем охлаждали до комнатной температуры. Добавляли воду (20 мл) и экстрагировали водную фазу трижды с помощью ДХМ (20 мл). Объединяли органические экстракты, сушили над сульфатом магния, фильтровали и концентрировали. Очищали с помощью флэш-хроматографии на колонке с 40 г силикагеля, элюировали градиентом 2-10% метанола в ДХМ. Дополнительно очищали с помощью ВЭЖХ с обращенной фазой при высоком рН (управляемой ультрафиолетом). Растворяли полученный материал (148 мг, 0,38 ммоль) в минимальном количестве 50% водного ацетонитрила. Добавляли 2 М водный хлористый водород (190 мкл, 0,38 ммоль) и лиофилизировали с получением гидрохлорида 1-[4-(1,5-диметил-1 Н-пиразол-4-илметил)-3,4,5,6-тетрагидро-2 Н[1,2']бипиразинил-3'-ил]пиперидин-4-илацетонитрила (166 мг, 58%). МС (m/z): 395,2 (М+1). Перемешивали 3-бром-2-хлорпиридин (460 г, 2,39 моль) в толуоле (2,3 л). Добавляли N-третбутоксикарбонилпиперазин (445,2 г, 2,39 моль) и продували азотом в течение 15 мин. Добавляли трис(дибензилиденацетон)дипалладий(0) (43,78 г, 47,8 ммоль) и 4,5-бис-(дифенилфосфино)-9,9 диметилксантен (82,99 г, 143 ммоль) и продували азотом в течение 15 мин. Переносили полученную смесь в автоклав объемом один галлон и держали в атмосфере азота. Добавляли трет-бутоксид натрия(252,69 г, 2,63 моль) (при этом наблюдали небольшой экзотермический эффект). Нагнетали в автоклаве давление с помощью азота до 40 фунтов на кв.дюйм (275,6 кПа) и снимали давление, это делали трижды,а затем нагнетали давление азота до 20-40 фунтов на кв.дюйм (137,8-275,6 кПа) и быстро нагревали смесь до 110 С. Температура поднималась при экзотермической реакции до приблизительно 113 С. Перемешивали реакционную смесь в течение 2,75 ч при 110 С и 20-40 фунтах на кв.дюйм (137,8-275,6 кПа) в атмосфере азота. Охлаждали смесь и проверяли на завершение реакции (анализ с помощью ВЭЖХ). Фильтровали смесь через стекловолокнистую бумагу и промывали толуолом. Переносили фильтрованную смесь в делительный сосуд и экстрагировали водой (2 л). Экстрагировали водную фазу дважды этилацетатом (3 л, затем 2 л). Промывали объединенные органические фазы дважды 15% раствором NaCl (4 л, затем 2 л). Перемешивали органические фазы в течение 30 мин с сульфатом натрия и обесцвечивающим углем (100 г). Фильтровали смесь и выпаривали фильтрат на роторном испарителе с получением темного масла (831 г). Растворяли полученный неочищенный продукт в этилацетате (3 л) и загружали на воронку из спеченного стекла, заполненную силикагелем (6 кг, заполняли, применяя гептан). Промывали колонку смесью 95% гептан:5% этилацетат (8 л), затем элюировали смесью 70% гептан:30% этилацетат, и собирали фракцию, содержащую неочищенный продукт. Дополнительно очищали объединенные фракции, содержащие продукт, с помощью хроматографии на силикагеле с 5% метил-трет-бутиловым эфиром в ДХМ с получением трет-бутилового эфира 4-(2-хлорпиридин-3-ил)пиперазин-1-карбоновой кислоты (331 г,46,5%) в виде желтого твердого вещества. 1 Н ЯМР 500 МГц (CDCl3)8,078 (дд, J=3,2 Гц, 1 Н), 7,30 (дд, J=6,5 Гц, 1 Н), 7,201 (м, 1 Н), 3,616 (м,4 Н), 3,018 (м, 4 Н), 1,485 (с, 9 Н). трет-Бутил-4-(2-(4-гидроксипиперидин-1-ил)пиридин-3 ил)пиперазин-1-карбоксилат Оборудовали 2-л сосуд мешалкой, термопарой и линией для добавления азота под поверхностью и продували азотом в течение 30 мин. Добавляли трет-бутиловый эфир 4-(2-хлорпиридин-3-ил)пиперазин 1-карбоновой кислоты (100 г, 0,336 моль), 4-гидроксипиперидин (37,36 г, 0,369 моль), трет-бутоксид натрия (80,68 г, 0,839 моль) и ацетат(2'-ди-трет-бутилфосфино-1,1' -бифенил-2-ил)палладий(II) (2,33 г,5,04 ммоль). Полученную смесь твердых веществ помещали в атмосферу азота на 15 мин. В отдельном сосуде, азотом барботировали толуол (933 мл) в течение 30 мин. Добавляли толуол в смесь твердых веществ и перемешивали в течение 28 ч, медленно барботируя азотом реакционную смесь и поддерживая температуру на уровне от 16 до 20 С с помощью водяной бани. Добавляли воду (1 л) по каплям, сохраняя температуру на уровне ниже 25 С. Разделяли фазы и экстрагировали водную фазу толуолом (500 мл). Объединяли органические фазы и промывали дважды 15% водным NaCl. Выпаривали органическую фазу на роторном испарителе с получением масла. Добавляли толуол (250 мл) и выпаривали дважды с получением 127,7 г масла. Растворяли полученное масло в этилацетате (255 мл в 2 объемах) и нагревали до 65-70 С. Добавляли гептан (1277 мл в 10 объемах) при 65-70 С. Позволяли раствору охладиться до температуры окружающей среды и выстаивали в течение от 16 до 18 ч. Охлаждали желтую смесь до 0-5 С в- 24016787 течение 1 ч, а затем фильтровали. Промывали твердые вещества раствором 20% этилацетата в гептане при 0-5 С. Сушили твердые вещества при 45-50 С в вакуумной печи с получением трет-бутил-4-(2-(4 гидроксипиперидин-1-ил)пиридин-3 ил)пиперазин-1-карбоксилата (66,6 г, 54,7%). 1 Н ЯМР 500 МГц (CDCl3)7,958 (дд, J=3,3 Гц, 1 Н), 7,10 (д, J=7,1 Гц, 1 Н), 6,834 (м,1 Н), 4,01 (д,J=3,2, 2 Н), 3,865 (м, 1 Н), 3,578 (м, 4 Н), 3,041 (м, 4 Н), 2,945 (м, 2 Н), 1,643 (м, 2 Н), 1,487 (с, 9 Н). Дигидрохлорид 1-(3-(пиперазин-1-ил)пиридин-2-ил)пиперидин-4-ола В охлажденный на ледяной бане 2-л сосуд добавляли газообразный HCl к метанолу (900 мл) с получением 7,31 М раствора, поддерживая температуру на уровне ниже 20 С. Добавляли в 12-литровый сосуд трет-бутил-4-(2-(4-гидроксипиперидин-1-ил)пиридин-3 ил)пиперазин-1-карбоксилат (306,5 г, 0,846 моль), а затем метанол (613 мл) и толуол (3,06 л). Перемешивали смесь с получением раствора, а затем добавляли раствор HCl (579 мл) в метаноле. Нагревали раствор до 35 С в течение 2 ч, а затем держали 4 ч при температуре окружающей среды. Отфильтровывали полученный кристаллический продукт, промывали кристаллы толуолом, а затем сушили в вакуумной печи при 40-45 С с получением дигидрохлорида 1-(3-(пиперазин-1-ил)пиридин-2-ил)пиперидин-4-ола в виде кристаллического твердого вещества (283,5 г, 99,47%). 1 Растворяли дигидрохлорид 1-(3-(пиперазин-1-ил)пиридин-2-ил)пиперидин-4-ола (281,0, 0,838 моль) в насыщенном водном растворе хлорида натрия (2,45 л). Добавляли 2 М NaOH (1 л), чтобы довести рН до 11,3. Экстрагировали смесь трижды с помощью ДХМ (32,04 л). Сушили объединенные органические фазы над сульфатом натрия, фильтровали и выпаривали растворитель на роторном испарителе с отбором азота с получением пены. Когда пена стабилизировалась, дополнительно сушили материал в течение от 2 до 3 ч при 50 С под вакуумом с получением 1-(3-(пиперазин-1-ил)пиридин-2-ил)пиперидин-4-ола Растворяли 1-(3-(пиперазин-1-ил)пиридин-2-ил)пиперидин-4-ол (207 г, 0,789 моль) и 1-этил-5 метил-1 Н-пиразол-4-карбальдегид (130,8 г, 0,947 моль) в дихлорэтане (4,55 л). Охлаждали до -5 С и начинали добавлять по частям триацетоксиборогидрид натрия (334,5 г, 1,578 моль), поддерживая температуру на уровне ниже приблизительно 5 С. Удаляли водяную баню и позволяли реакционной смеси нагреться до 10 С в течение приблизительно 1 ч. Нагревали реакционную смесь до 18-20 С и перемешивали в течение 3 ч. Охлаждали реакционную смесь до 15 С и добавляли 2 н. NaOH (2 л). Разделяли фазы и экстрагировали водные фазы дважды с помощью ДХМ (21,3 л). Фильтровали объединенные органические фазы через стекловолокнистую бумагу. Экстрагировали органические фазы с помощью 1 н. HCl (12,5 л один раз, 21 л). К объединенным водным фазам, которые содержали продукт, добавляли 50% NaOH (400 мл),чтобы довести рН до 11,6. Экстрагировали полученную беловатую водную фазу с помощью ДХМ (13 л,21,5 л). Сушили объединенные органические фазы над сульфатом натрия. Добавляли обесцвечивающий уголь (G-60, 44 г) и перемешивали смесь при температуре окружающей среды в течение 20 мин. Фильтровали через стекловолокнистую бумагу, промывали ДХМ (1 л) и выпаривали растворители с получением 3'-[4-(1-этил-5-метил-1 Н-пиразол-4-илметил)пиперазин-1-ил]-3,4,5,6-тетрагидро-2 Н-[1,2']бипиридинил-4-ола в виде масла (330 г, 109%). Н ЯМР 300 МГц (CDCl3) (Е 29-Н 70357-031)7,87 (дд, J=3,3 Гц, 1 Н), 7,37 (с, 1 Н) 7,05 (дд,J=6,26 Гц, 1 Н), 6,77 (м,1 Н), 4,065 (кв, J=7,35 Гц, 2 Н), 3,99 (уш.м, 2 Н), 3,802 (уш.м, 1 Н), 3,365 (с, 2 Н),2,658 (уш.м, 2 Н), 2,551 (уш.м, 3 Н), 2,236 (с, 3 Н), 1,985 (уш.м, 2 Н), 1,615 (уш.м, 2 Н), 1,381 (т, J=7,26 Гц,3 Н). Дигидрохлорид 3'-[4-(1-этил-5-метил-1 Н-пиразол-4-илметил)пиперазин-1-ил]-3,4,5,6-тетрагидро 2 Н-[1,2']бипиридинил-4-ола. Растворяли 3'-[4-(1-этил-5-метил-1 Н-пиразол-4-илметил)пиперазин-1-ил]-3,4,5,6-тетрагидро-2 Н[1,2']бипиридинил-4-ол (415 г, 1,079 моль) в этаноле (5,5 литра) и метил-трет-бутиловом эфире (6,23 л). Перемешивали раствор в атмосфере азота и нагревали до 50-55 С. Добавляли 2,96 М раствор HCl в этаноле (0,729 л) при 50-55 С в течение 50 мин. Позволяли смеси охладиться до приблизительно 40,1 С в течение 90 мин. Охлаждали смесь до 20 С в течение 20 мин, а затем перемешивали при 20 С в течение 30 мин. Фильтровали смесь и промывали метил-трет-бутиловым эфиром (3500 мл). Сушили твердые осадки в вакуумной печи при 50-55 С под вакуумом с небольшим отбором азота в течение 24 ч с получением дигидрохлорида 3'-[4-(1-этил-5-метил-1 Н-пиразол-4-илметил)пиперазин-1-ил]-3,4,5,6-тетрагидро 2 Н-[1,2']бипиридинил-4-ола (416 г, 84,3%). 1 Н ЯМР 500 МГц (CD3OD)7,89 (дд, J=6,0 Гц, 2 Н), 7,828 (с,1 Н), 7,275 (дд, J=6,0 Гц, 1 Н), 4,86(уш.м, 2 Н), 3,410 (уш.т,2 Н), 3,145 (т, J=8,1, 2H), 2,467 (с, 3 Н), 2,082 (уш.м, 2 Н), 1,716 (уш.м, 2 Н), 1,416 (т,J=7,5 Гц, 3 Н). Анализ хлорида осуществляли с помощью масс-спектрометрии с индуктивно-связанной плазмой(ИСП/МС) (15,6%). Антагонисты рецептора 5-НТ 7 согласно настоящему изобретению относительно селективны по отношению к рецептору 5-НТ 7. Соединения согласно настоящему изобретению особенно относительно селективны по отношению к рецептору 5-НТ 7 по сравнению с другими подтипами рецептора 5-НТ и особенно с рецепторами 5-НТ 1 А, 5-HT1B и 5-HT1D. Эта селективность продемонстрирована в следующих тестах на связывание рецептора и тестах на антагонистическую активность к рецептору. Получение мембран. Мембраны для тестов на аффинность и антагонистическую активность получали, по существу, как описано далее. Клетки AV-12, стабильно экспрессирующие рецептор 5-НТ 7, выращивали в виде монослоя в плоских флаконах 54-150 в среде DMEM/F12 (3:1) с 5% фетальной сыворотки теленка (FBS),20 мМ HEPES, 400 мг/мл генетицина, 50 мг/мл тобрамицина. После того как клетки достигли 90% конфлюентности, среды удаляли и заменяли на среды Hybritech, содержащие 2% лошадиной сыворотки,100 мг/мл сульфата декстрана, 1 мг/мл нуцеллина, 1 мг/мл трансферина человека (частично насыщенного железом), 50 мг/мл тобрамицина, 20 мМ HEPES, 100 мг/мл генетицина, 0,04% плуроник F68 (средыHybritech представляют собой модифицированные среды DMEM/F12 с низким содержанием кальция для поддержания роста клеток в суспензии, имеющие следующий состав: биотин 7,3 мкг/л, ангидрид хлорида кальция 11 мг/л, хлорид холина 8,98 мг/л, сульфат меди 5 Н 2 О 3,75 мкг/л, D-глюкоза (декстроза) 6,00 г/л, DL-липоевая (тиоктовая) кислота 0,21 мг/л, этаноламин-HCl 10 мг/л, нитрат железа 9 Н 2 О 50 мкг/л, сульфат железа 7 Н 2 О 0,42 мг/л, фолиевая кислота 4 мг/л, глицин 30 мг/л, L-инозит 12,6 мг/л,L-аланин 8,9 мг/л, L-аргинин HCL 211 мг/л, L-аспарагин Н 2 О 15 мг/л, L-аспарагиновая кислота 13,3 мг/л,L-цистин 2HCl 62,6 мг/л, L-глутаминовая кислота 7,35 мг/л, L-глутамин 1,46 г/л, L-гистидин HCl Н 2 О 42 мг/л, L-изолейцин, 105 мг/л, L-лейцин 105 мг/л, L-лизин HCl 146 мг/л, L-метионин 30 мг/л,L-фенилаланин 66 мг/л, L-пролин 17,25 мг/л, L-серин 42 мг/л, L-треонин 95 мг/л, L-триптофан 16 мг/л,двунатриевая соль L-тирозина 104 мг/л, L-валин 94 мг/л, безводный хлорид магния 28,64 мг/л, безводный сульфат магния 48,84 мг/л, ниацинамид 4 мг/л, KCl 311,8 мг/л, путресцин 2HCl 0,08 мг/л, пиридоксаль НС 1 4 мг/л, пиридоксин HCl 30 мкг/л, рибофлавин 0,4 мг/л, NaCl 5,50 г/л, гипоксантин натрия 4,77 мг/л,пантотенат натрия 4 мг/л, двухосновный фосфат натрия безводный 71,2 мг/л, одноосновный фосфат натрия 62,5 мг/л, пируват натрия 220 мг/л, селенит натрия 5,00 мкг/л, тиамин HCl 4 мг/л, тимидин 0,73 мг/л, витамин В-120,68 мг/л, сульфат цинка 7H2O 0,43 мг/л.) Клетки растили в течение ночи, чтобы они кондиционировали среды. На следующее утро кондиционированные среды (суммарный объем 150 мл) удаляли и помещали в стерильный контейнер. Клетки трипсинизировали и собирали в кондиционированные среды. Добавляли свежие суспензионные среды, в результате получали общий объем 500 мл и плотность клеток до 5105 клеток/мл. Объем суспензионной культуры несколько раз увеличивали в течение следующих 3 недель до получения желаемого объема и плотности, а затем клетки собрали (целевая плотность клеток приблизительно 3,5-4,0106 клеток в мл). Клетки собирали путем центрифугирования при 1500 g при 4 С в течение 30 мин. Супернатант декантировали и осадки клеток ресуспендировали в ледяном фосфатно-солевом буферном растворе (ФБР). Суспензию клеток делили на аликвоты в 50 мл центрифужные пробирки и центрифугировали при 1500 g при 4 С в течение 15 мин. Супернатант удаляли, осадки взвешивали, а затем замораживали на сухом льду. Для получения мембран полученные осадки ресуспендировали в ледяном буфере Tris (20 мМ TrisHCl, рН 7,4 при 23 С, 5 мМ EDTA) и гомогенизировали с помощью гомогенизатора ткани Wheaton. Ли- 26016787 зат затем центрифугировали при 200g в течение 5 мин при 4 С, что позволяло осадить крупные фрагменты осадка клеток, которые не использовали. Супернатант собирали и центрифугировали при 40000g в течение 60 мин при 4 С. Полученный осадок ресуспендировали в конечном буфере, содержащем 50 мМ Tris HCl и 0,5 мМ EDTA, рН 7,4. Препараты мембран мгновенно замораживали на сухом льду и хранили при -80 С. Концентрации белка определяли методом Брэдфорд. Anal. Biochem., 72:248-254,1976. Для функциональных тестов на циклический аденозинмонофосфат (цАМФ) клетки, экспрессирующие 5-НТ 7, растили в плоских флаконах объемом 150 см 2 и обрабатывали, по существу, как указано далее. Среды извлекали из флаконов и клетки промывали 1 мл ФБР. Клетки снимали с поверхности сосуда,применяя не содержащий ферменты раствор для диссоциации клеток (Specialty media(www.chemicon.com) CATS-004-B), и ресуспендировали в полных средах. Брали образец клеток для подсчета количества клеток, а остальные клетки центрифугировали, как описано выше, в течение 3 мин. Полученный осадок клеток ресуспендировали в ФБР при концентрации 1106 клеток в мл и использовали непосредственно в тесте на цАМФ, описанном далее. Аффинность к рецептору 5-НТ 7: тест на связывание радиолиганда. Связывание [3 Н] 5-НТ осуществляли путем модификации условий анализа, описанного у Kahl и др.(J. Biomol. Screen, 2: 33-40 (1997, по существу, как указано далее. Тесты на связывание радиолиганда проводили в 96-луночных микротитрационных планшетах, в суммарном объеме 125 мкл, содержащих следующий реакционный буфер: 50 мМ Tris, 10 мМ MgCl2, 0,2 мМ EDTA, 10 мМ паргилин, 0,1% аскорбат, рН 7,4, при комнатной температуре. Тест на конкурентное связывание проводили с использованием одиннадцати концентраций тестируемого соединения, лежащих в диапазоне от 0,1 до 10000 нМ, в присутствии 1 нМ [3 Н]5-НТ. Немеченый 5-НТ (10 мкМ) применяли для определения неспецифического связывания. Реакцию связывания инициировали путем добавления 0,15 мкг гомогената мембран(2,31 нг/мкл, 65 мкл на лунку) и 0,5 мг флуоресцентных микросфер для сцинтилляционного анализа сближения. Реакционные смеси инкубировали при комнатной температуре в течение 3 ч, а затем проводили количественный анализ с помощью сцинтилляционного счетчика Trilux Microbeta, что позволяло определить количество связанных с рецептором радиолигандов. Результаты теста на связывание анализировали с помощью автоматизированного анализа с четырехпараметрической аппроксимацией(ID Business Solutions Ltd, Гилфорд, Суррей, Великобритания). Значения IC50 преобразовывали в значения Ki, применяя уравнение Ченга-Прусоффа. Biochem. Pharmacol., 22:3099-3108 (1973). Соединения примеров испытывали, по существу, как описано выше, и найдено, что они имеют значения Ki50 нМ. Соединение примера 1 испытывали, по существу, как описано, и найдено, что оно имеет значение Ki, примерно 16,2 нМ. Аффинность к другим подтипам рецепторов серотонина, а также к адренергическим рецепторам альфа 1 и 2, можно легко определить путем модификации описанного теста на связывание рецептора радиолигандом, с использованием мембран, полученных из клеток, стабильно экспрессирующих желаемый подтип рецептора, включая подтипы 5-HT1A, 5-HT1B и 5-HT1D, а также подтипы рецептора 5-НТ 2 А, 5HT2B, 5-HT2C, 5-НТ 4, 5-НТ 5 и 5-НТ 6. Соотношение селективности Ki-x/Ki-5HT7, где Ki-x представляет собойKi для сравниваемого рецептора, и является показателем относительной аффинности соединений к рецептору 5-НТ 7. Соединения из примеров протестировали и обнаружили, что они имеют соотношения селективности против других серотонинергических рецепторов 4 и против адренергических рецепторов 4. Соединение из примера 1 тестировали, по существу, как описано выше, и обнаружили, что оно имеет следующий профиль селективности. Функциональный тест на антагонистическую активность: измерение образования цАМФ. Рецептор 5-НТ 7 функционально связан с G-белком. Это определили по способности серотонина и серотонинергических лекарственных препаратов стимулировать продукцию цАМФ в клетках СНО,трансфицированных рецептором 5-НТ 7 (Ruat, и др., Proceedings of the National Academy of Sciences(USA), 90:8547-8551, 1993). Следовательно, функциональную активность рецептора можно измерить путем измерения активности аденилатциклазы, с применением коммерчески доступного набора для гомогенного анализа, на основе клеток, который позволяет измерить флуоресценцию с временным разрешением, как, например, набор, производимый Cisbio-US, Inc. (Бедфорд, Массачусетс). Используя, по существу, протокол и реагенты, предоставленные изготовителем, приблизительно 20000 клеток AV-12,экспрессирующих рецептор 5-НТ 7 человека (как описано выше), использовали при концентрациях доз тестируемого соединения, лежащих в диапазоне, описанном для теста на связывание. Параллельно строили кривые доза ЕС-90-ответ для 5-НТ, чтобы продемонстрировать конкурентный антагонизм. Стандартную кривую цАМФ также снимали в каждом эксперименте. После того как планшеты для анализа считывали в измерительном приборе Envision (Perkin-Elmer, Уэллсли, Массачусетс), результаты нормировали по стандартной кривой и преобразовывали в процент ингибирования для анализа результатов, как описано выше для результатов теста на связывание рецептора. Рассчитывали Kb (нМ) как меру антагонистической эффективности соединения. Предпочтительными являлись соединения, имеющие процент ингибирования 75%. К другим предпочтительным соединениям отнесли соединения, имеющиеKb50 нМ. Соединение примера 1 тестировали, по существу, как описано выше, и обнаружили, что оно является полным антагонистом со значением Kb, равным приблизительно 2,97 нМ (ингибирование = приблизительно 108%). Модель на животных экстравазации белка плазмы (РРЕ) в твердую мозговую оболочку. Модель экстравазации белка плазмы в твердую мозговую оболочку представляет собой общепринятую модель мигрени. Способность тестируемого соединения снижать экстравазацию белков плазмы в твердую мозговую оболочку в условиях теста считают свидетельством того, что указанное соединение способно уменьшать или предотвращать воспаление твердой мозговой оболочки, которое считают симптомом мигрени (см. Johnson, K.W., и др., Neuroreport, 8 (1997), 2237-2240). Для тестирования соединений на их способность уменьшать или предотвращать экстравазацию белков плазмы в твердую мозговую оболочку, самцов крыс Spraque-Davwley от Harlan (250-350 г) анестезировали пентобарбиталом натрия (65 мг/кг, интраперитонеально) и помещали в стереотаксическую рамку (David Kopf Instruments), при этом режущую пластинку устанавливали на -2,5 мм. После срединного сагиттального разреза скальпа просверливали 2 пары билатеральных отверстий в черепе (3,2 мм постериально, 1,8 и 3,8 мм латерально, все координаты указаны по отношению к брегме). Пары стимулирующих электродов из нержавеющей стали, изолированные по всей длине, за исключением кончиков(Rhodes Medical Systems, Inc.), погружали в отверстия в оба полушария на глубину 9,2 мм. Тестируемое соединение вводили внутривенно (в.в.) в бедренную вену в объеме дозировки, равном 1 мл/кг. Приблизительно через 8 мин после инъекции животным вводили дозу бычьего сывороточного альбумина, меченного флуоресцеинизотиоцианатом (FITC-BSA) (20 мг/кг, внутривенно). FITC-BSA выполняет функцию маркера экстравазации белка. Через 10 мин после инъекции тестируемого соединения левый ганглий тройничного нерва стимулировали электричеством в течение 5 мин при силе тока, равной 1,0 мА (5 Гц, 5-миллисекундный импульс каждые 200 мс) с помощью Grass Instrument Stimulator моделиS48 с блоком фотоэлектрической развязки PSIU6 (Grass-Telefactor). В качестве альтернативы, крысам, которые в течение ночи были лишены доступа к воде и пище, перорально вводили дозу тестируемого соединения через желудочный зонд в объеме 2 мл/кг. Приблизительно через 50 мин после введения дозы животных анестезировали и помещали в стереотаксическую рамку, как описано выше. Животным вводили дозу FITC-BSA (20 мг/кг, внутривенно) через 58 мин после перорального введения дозы. Через 60 мин после введения дозы соединения животных подвергали стимуляции электричеством, как описано выше. Через 5 мин после окончания стимуляции животных умерщвляли заменой крови на 40 мл физиологического раствора. Верхушку черепа удаляли и брали образцы твердой оболочки из обоих полушарий,ополаскивали водой и распределяли по поверхности предметных стекол. После высыхания ткани помещали на покровные стекла в 70% растворе глицерин/вода. Количество FITC-BSA в каждом образце определяли количественно с помощью флуоресцентного микроскопа (Zeiss), оборудованного монохроматором с дифракционной решеткой, спектрофотометром и платформой, управляемой компьютером. Измерения флуоресценции осуществляли в 25 точках в сетке 55 с шагом 500 мкм на каждом образце твердой мозговой оболочки с возбуждающей длиной волны приблизительно при 490 нм и интенсивность испускания измеряли приблизительно при 535 нм. Определяли среднее и квадратичное отклонение для 25 измерений.- 28016787 Экстравазация, индуцированная электрической стимуляцией ганглия тройничного нерва, представляет собой ипсилатеральный эффект (т.е. происходит лишь на той стороне твердой мозговой оболочки,ганглий тройничного нерва которой был стимулирован). Это позволяет использовать другую (нестимулированную) половину твердой мозговой оболочки в качестве контроля. Вычисляли отношение степени экстравазации в твердую мозговую оболочку для стимулированной стороны к степени экстравазации в нестимулированной половине. Для контрольных животных, которым вводили только физиологический раствор, получали отношение, равное приблизительно 2,0. В отличие от этого соединение, которое эффективно предотвращало экстравазацию в твердую мозговую оболочку для стимулированной стороны,давало отношение, равное приблизительно 1,0. Предпочтительными соединениями являются такие, которые эффективно предотвращают экстравазацию. Соединение примера 1 тестировали, по существу, как описано выше, и обнаружили, что оно имеет ID100, равную 0,1 мг/кг, что дает соотношение, равное приблизительно 1,15. Хотя соединения, используемые в способах согласно настоящему изобретению, можно вводить непосредственно без какого-либо состава, указанные соединения, как правило, вводят в виде фармацевтических композиций, включающих по меньшей мере одно соединение формулы I или его фармацевтически приемлемую соль в качестве активного ингредиента и по меньшей мере один фармацевтически приемлемый носитель, разбавитель и/или эксципиент. Такие композиции можно вводить множеством путей,включая пероральный, сублингвальный, буккальный, интраназальный, трансдермальный, подкожный,внутривенный, внутримышечный и пульмональный. Такие фармацевтические композиции и способы их получения хорошо известны в данной области. См., например, Remington: The Science and Practice ofPharmacy (University of the Sciences in Philadelphia, ред., 21-е изд., Lippincott WilliamsWilkins Co.,2005). Композиции предпочтительно изготавливают в единичной дозированной форме, причем каждая доза содержит от приблизительно 0,1 до приблизительно 200 мг, чаще от приблизительно 1,0 до приблизительно 30 мг активного ингредиента. Термин "единичная дозированная форма" относится к физически дискретным единицам, подходящим для использования в качестве единичных доз для введения человеку и другим млекопитающим; причем каждая доза содержит заранее определенное количество активного вещества, рассчитанное таким образом, чтобы обеспечить необходимый терапевтический эффект совместно по меньшей мере с одним подходящим фармацевтически приемлемым носителем, разбавителем и/или эксципиентом. Соединения, как правило, являются эффективными в широком диапазоне дозировок. Например, дозировки в день обычно будут лежать в диапазоне от приблизительно 0,01 до приблизительно 30 мг/кг,как, например, в диапазоне от приблизительно 0,1 до приблизительно 15 мг/кг/день, в виде одной или разделенных доз. Тем не менее, очевидно, что количество соединения, которое фактически вводят, будет определять лечащий врач с учетом значимых обстоятельств, включая патологическое состояние, от которого лечат, выбранный путь введения, конкретное соединение или соединения, которые вводят, возраст,вес и ответ на лечение конкретного пациента, тяжесть симптомов у данного пациента, и, следовательно,упомянутые выше диапазоны дозировок не предполагают ограничения объема настоящего изобретения каким-либо образом. В некоторых случаях уровни дозировок ниже вышеупомянутого нижнего предела могут оказаться достаточными, тогда как в других случаях можно применять даже еще большие дозировки. Тип композиции, используемый для введения соединений, используемых в способах согласно настоящему изобретению, может определяться конкретным используемым соединением, типом фармакокинетической характеристики, необходимой исходя из выбранного способа введения и состояния пациента.
МПК / Метки
МПК: A61P 25/00, C07D 403/12, C07D 401/14, A61K 31/497
Метки: 5-нт7, рецептора, антагонисты
Код ссылки
<a href="https://eas.patents.su/30-16787-antagonisty-receptora-5-nt7.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонисты рецептора 5-нт7</a>
Антагонисты рецептора гистамина-3

Номер патента: 13602
Опубликовано: 30.06.2010
Авторы: Чандрасекаран Рамалакшми Йегна, Вагер Тревис Т., Батлер Тодд Уилльям
МПК: C07C 237/24, A61K 31/165, C07D 207/04...
Метки: гистамина-3, рецептора, антагонисты
Формула / Реферат:
1. Соединение формулы Iили его фармацевтически приемлемая соль,где R1и R2, каждый независимо, выбран из группы, включающейводород;С1-С8-алкил, необязательно замещенный 1-4 галогенами;С1-С4-алкильную группу, необязательно замещенную заместителем, выбранным из группы, состоящей из ОН, одного до четырех С1-С4-алкила, С3-С7-циклоалкила, С1-С4-диалкиламино, С6-С14-арила, необязательно замещенного галогеном и необязательно замещенного группой...
Пиридин-4-илэтинилимидазолы и -пиразолы, как антагонисты рецептора mglu5

Номер патента: 11722
Опубликовано: 28.04.2009
Авторы: Бюттельманн Бернд, Фиайра Эрик, Чеккарелли Симона-Мария, Йэшке Георг, Кольчевски Забине, Спурр Пол, Портер Ричард Хью Филип
МПК: A61K 31/4439, A61K 31/444, A61P 25/22...
Метки: пиридин-4-илэтинилимидазолы, mglu5, антагонисты, пиразолы, рецептора
Формула / Реферат:
1. Соединение общей формулы в которой один из А и Е обозначает N и другой обозначает C; R1 обозначает галоген или цианогруппу; R2 обозначает С1-С6алкил; R3 обозначает арил, который представляет собой ароматическую карбоциклическую группу, состоящую из одного отдельного кольца, или двух, или большего количества сконденсированных колец, в которой по меньшей мере одно кольцо является ароматическим, или гетероарил, который необязательно замещен с...
Антагонисты рецептора il-8

Номер патента: 1436
Опубликовано: 26.02.2001
Авторы: Вебер Дэниел Франк, Ратледж Мельвин Кларенс Мл., Юревич Энтони Джозеф, Виддаусон Кетрин Луиза, Херцберг Роберт Филип
МПК: A61P 11/06, A61K 31/17
Метки: антагонисты, рецептора
Формула / Реферат:
1. Способ лечения болезненного состояния, опосредованного хемокином, где хемокин связывается у млекопитающих, нуждающихся в таком лечении, с IL-8 а- или b-рецептором, включающий введение млекопитающему эффективного количества соединения формулы где Х является кислородом или серой; R является любой функциональной группой, имеющей ионизируемый водород и рКа, равный 10 или менее; R1 независимо выбирают из водорода; галогена; нитро; циано;...
Бензоксазиноновые антагонисты рецептора допамина d4.

Номер патента: 1486
Опубликовано: 23.04.2001
Авторы: Вайс Лауренс Дейвид, Беллиотти Томес, Вустров Дейвид Юрген
МПК: C07D 265/36, A61K 31/535
Метки: допамина, бензоксазиноновые, антагонисты, рецептора
Формула / Реферат:
1. Соединение формулы (I) где R1 и R2 означают независимо водород или алкил с числом углеродных атомов от 1 до 6, X означает С, N или СН, при этом в случае X=С штрихованная линия означает связь, а в других случаях отсутствует; R3 означает фенил, нафтил, гетероарил, замещенный фенил, замещенный нафтил или замещенный гетероарил, при этом каждый заместитель независимо выбран из группы, включающей галоген, алкоксигруппу с числом углеродных...
Антагонисты рецептора интегрина

Номер патента: 2822
Опубликовано: 31.10.2002
Авторы: Хартман Джордж Д., Аскью Бен С., Мейсснер Роберт С., Коулман Пол Дж., Хант Сесилия, Хатчинсон Джон Х., Смит Гарри Р., Ванг Дзиабинг, Пэйтан Майкл А., Даггэн Марк Е., Халщенко Василь
МПК: C07D 471/04, A61K 31/4375, A61P 19/00...
Метки: интегрина, антагонисты, рецептора
Формула / Реферат:
1. Соединение формулы где Х выбирают из группы, состоящей из Y выбирают из группы, состоящей из (СН2)m, (СН2)m-S-(СН2)n и (СН2)m-NR4-(СН2)n, где каждый атом углерода метиленовой группы (СН2) в группировке Y в отличие от R4 может быть замещен одним или двумя заместителями R3, при условии, что когда Y обозначает -(СН2)m-NR4-(СН2)n- и n=1, то заместитель R3 на атоме углерода метиленовой группы в -(СН2)m-, соседнем с атомом азота, не может...
Предыдущий патент: Способ получения мочевины из аммиака и диоксида углерода
Следующий патент: Противоградовая станция
Случайный патент: Ступень движущейся дорожки или подобного устройства