Амидзамещенные индазолы в качестве ингибиторов поли(adp-рибоза)полимеразы (parp)
Номер патента: 16079
Опубликовано: 30.01.2012
Авторы: Шультц-Фадемрехт Карстен, Джоунс Филип, Скарпелли Рита, Онтория Онтория Хесус Мария





























Формула / Реферат
1. Соединение формулы (I)

где R1 представляет водород или фтор;
R2 представляет водород или фтор,
или его фармацевтически приемлемые соли, стереоизомеры или таутомеры.
2. Соединение по п.1 формулы (II)

где R1 и R2 имеют значения, определенные в п.1,
или его фармацевтически приемлемые соли, стереоизомеры или таутомеры.
3. Соединение по п.1 формулы (III)

где R1 и R2 имеют значения, определенные в п.1,
или его фармацевтически приемлемые соли или таутомеры.
4. Соединение по п.1 формулы (IV)

где R1 и R2 имеют значения, определенные в п.1,
или его фармацевтически приемлемые соли или таутомеры.
5. Соединение по любому из предшествующих пунктов, где R1 представляет собой водород и R2 представляет водород или фтор.
6. Соединение по п.1, выбранное из
2-(4-пиперидин-3-илфенил)-2Н-индазол-7-карбоксамида;
2-{4-[(3R)-пиперидин-3-ил]фенил}-2Н-индазол-7-карбоксамида;
2-{4-[(3S)-пиперидин-3-ил]фенил}-2Н-индазол-7-карбоксамида;
5-фтор-2-(4-пиперидин-3-илфенил)-2H-индазол-7-карбоксамида;
5-фтор-2-{4-[(3S)-пиперидин-3-ил]фенил}-2H-индазол-7-карбоксамида;
5-фтор-2-{4-[(3R)-пиперидин-3-ил]фенил}-2H-индазол-7-карбоксамида;
5-фтор-2-(3-фтор-4-пиперидин-3-илфенил)-2H-индазол-7-карбоксамида;
5-фтор-2-{3-фтор-4-[(3R)-пиперидин-3-ил]фенил}-2H-индазол-7-карбоксамида;
5-фтор-2-{3-фтор-4-[(3S)-пиперидин-3-ил]фенил}-2H-индазол-7-карбоксамида,
и его фармацевтически приемлемые соли, таутомеры или стереоизомеры.
7. Соединение по п.6, выбранное из
2-{4-[(3R)-пиперидин-3-ил]фенил}-2H-индазол-7-карбоксамида;
2-{4-[(3S)-пиперидин-3-ил]фенил}-2H-индазол-7-карбоксамида,
и его фармацевтически приемлемые соли или таутомеры.
8. Фармацевтическая композиция, содержащая соединение по любому из предшествующих пунктов или его фармацевтически приемлемую соль, таутомер или стереоизомер в комбинации с фармацевтически приемлемым носителем.
9. Комбинация соединения по любому из пп.1-7 или его фармацевтически приемлемой соли, стереоизомера, таутомера с противораковым агентом для одновременного, раздельного или последовательного введения.
10. Применение соединения по любому из пп.1-7 или его фармацевтически приемлемой соли, стереоизомера или таутомера в терапии.
11. Применение соединения по любому из пп.1-7 или его фармацевтически приемлемой соли, стереоизомера или таутомера для производства лекарственного средства для лечения или предотвращения состояний, которые могут облегчаться путем ингибирования поли(ADP-рибоза)полимеразы (PARP).
12. Применение соединения по любому из пп.1-7 или его фармацевтически приемлемой соли, стереоизомера или таутомера для производства лекарственного средства для лечения или предотвращения рака, воспалительных заболеваний, реперфузионных повреждений, ишемических состояний, инсульта, почечной недостаточности, сердечно-сосудистых заболеваний, сосудистых заболеваний, иных чем сердечно-сосудистые, диабета, нейродегенеративных заболеваний, ретровирусной инфекции, повреждения или старения кожи и УФ-индуцируемого повреждения кожи.
13. Применение соединения по любому из пп.1-7 или его фармацевтически приемлемой соли, стереоизомера или таутомера в качестве химио- и/или радиосенсибилизатора для лечения рака.
14. Способ лечения или предотвращения рака, воспалительных заболеваний, реперфузионных повреждений, ишемических состояний, инсульта, почечной недостаточности, сердечно-сосудистых заболеваний, сосудистых заболеваний, иных чем сердечно-сосудистые, диабета, нейродегенеративных заболеваний, ретровирусной инфекции, повреждения сетчатки или старения кожи и УФ-индуцируемого повреждения кожи, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения по п.1 или композиции, содержащей соединение по п.1.
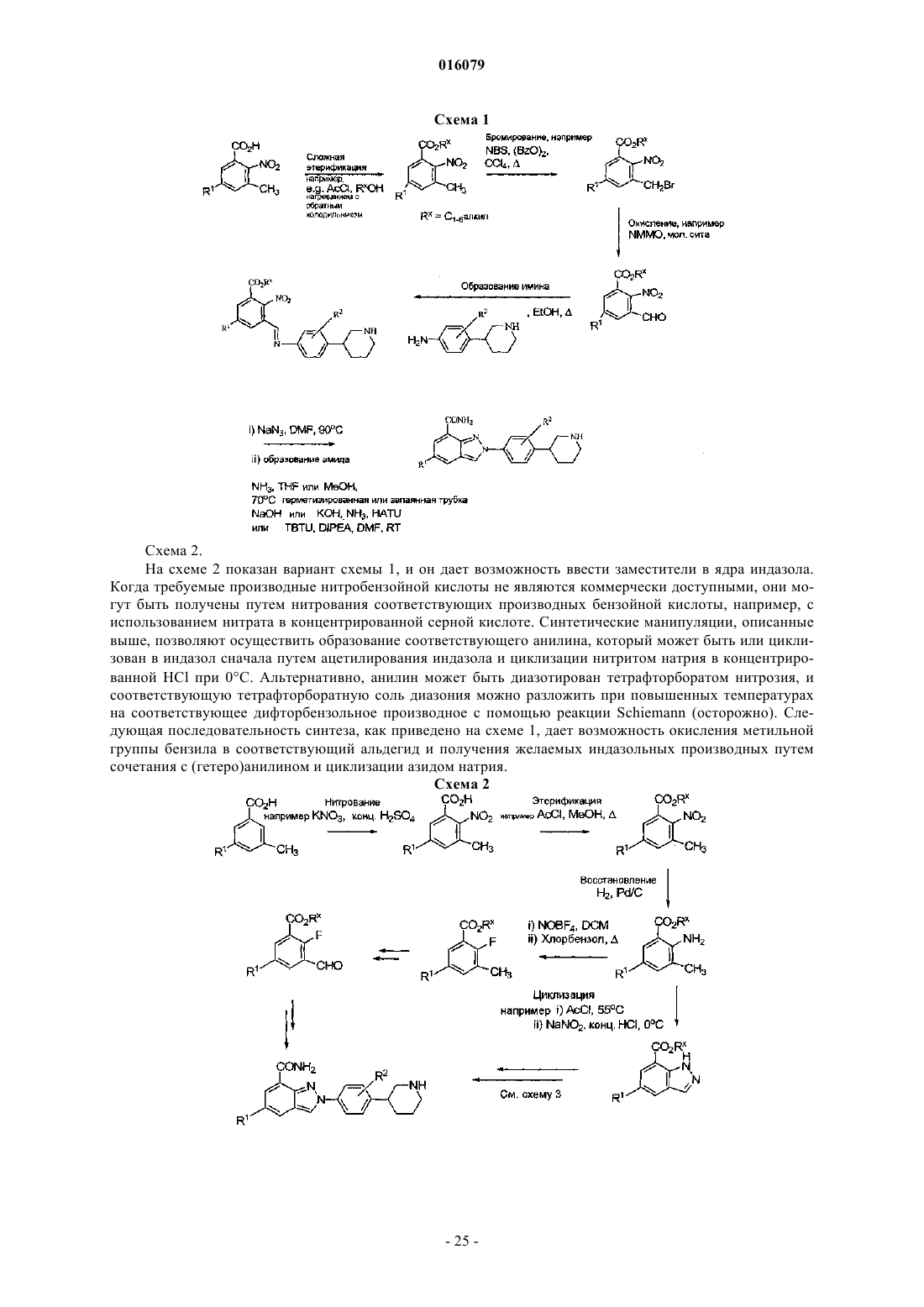
Текст
Дата публикации и выдачи патента Номер заявки Настоящее изобретение относится к соединениям формулы (I) и к их фармацевтически приемлемым солям, стереоизомерам или таутомерам, которые являются ингибиторами поли(ADPрибоза)полимеразы (PARP) и полезны, таким образом, при лечении рака, воспалительных заболеваний, реперфузионных повреждений, ишемических состояний, инсульта, почечной недостаточности, сердечно-сосудистых заболеваний, сосудистых заболеваний, иных чем сердечнососудистые, диабета, нейродегенеративных заболеваний, ретровирусной инфекции, повреждения сетчатки или старения кожи и УФ индуцируемого повреждения кожи, и в качестве химио- и/или радиосенсибилизаторов для лечения рака. Джоунс Филип, Онтория Онтория Хесус Мария, Скарпелли Рита,Шультц-Фадемрехт Карстен (IT) Медведев В.Н. (RU)(71)(73) Заявитель и патентовладелец: ИНСТИТУТО ДИ РИЧЕРКЕ ДИ БИОЛОДЖИА МОЛЕКОЛАРЕ П. АНДЖЕЛЕТТИ СПА (IT) 016079 Настоящее изобретение относится к амидзамещенным индазолам, которые являются ингибиторами фермента поли(ADP-рибоза)полимеразы (PARP), ранее известного как поли(ADP-рибоза)синтаза и поли(ADP-рибозил)трансфераза. Соединения настоящего изобретения полезны в качестве монотерапевтических средств при опухолях со специфическими дефектами в ДНК-репарационных путях и в качестве усилителей некоторых ДНК-повреждающих агентов, таких как противораковые агенты, и при радиотерапии. Далее, соединения настоящего изобретения полезны для снижения некроза клеток (при инсульте и инфаркте миокарда), уменьшения воспаления и повреждения тканей, лечения ретровирусных инфекций и защиты от токсичных эффектов химиотерапии. Поли(ADP-рибоза)полимераза (PARP) составляет сверхсемейство восемнадцати белков, содержащих PARP каталитические домены (Bioessays (2004), 26:1148). Данное семейство белков включаетPARP-1, PARP-2, PARP-3, танкиразу-1, танкиразу-2, vaultPARP и TiPARP. PARP-1, основной член семейства состоит из трех главных доменов: амино (N)-концевого ДНК-связывающего домена (DBD), содержащего два цинковых пальца, автомодификационного домена и карбокси (С)-концевого каталитического домена.PARP являются ядерными и эндоплазматическими ферментами, которые расщепляют NAD+ на никотинамид и ADP-рибозу, образуя длинные и разветвленные ADP-рибозные полимеры в целевых белках,включая топоизомеразы, гистоны и сам PARP (Biochem. Biophys. Res. Commun. (1998), 245:1-10). Поли(ADP-рибозилирование) вовлечено в несколько биологических процессов, включающих репарацию ДНК, генную транскрипцию, развитие клеточного цикла, гибель клеток, хроматиновые функции и геномную стабильность. Было показано, что каталитическая активность PARP-1 и PARP-2 быстро стимулируется разрывами ДНК нити (см. Pharmacological Research (2005), 52:25-33). В ответ на ДНК повреждение PARP-1 связывается с однонитиевым и двунитиевым разрывами ДНК. В нормальных физиологических условиях имеется минимальная активность PARP, однако после повреждения ДНК происходит немедленная активацияPARP вплоть до 500-кратной. Как PARP-1, так и PARP-2 обнаруживают разрывы ДНК нити, действуя как сенсоры разрывов, давая быстрые сигналы остановки транскрипции и задействуя ферменты, требуемые для репарации ДНК в месте повреждения. Поскольку радиотерапия и многие химиотерапевтические подходы к терапии рака основаны на индуцировании повреждения ДНК, ингибиторы PARP являются полезными в качестве химио- и радиосенсибилизаторов для лечения рака. Сообщалось, что ингибиторыPARP являются эффективными в радиосенсибилизации гипоксических опухолевых клеток (патенты США 5032617, 5215738 и 5041653). Большинство биологических эффектов PARP относится к данному процессу поли(ADPрибозилирования), который влияет на свойства и функцию целевых белков; к PAR олигомерам, которые,при отщеплении от поли(ADP-рибозилированных) белков, вызывают различные эффекты в клетках; физической ассоциации PARP с ядерными белками, с образованием функциональных комплексов и к понижению клеточного уровня их субстрата NAD+ (Nature Review (2005), 4:421-440). Помимо того, что она вовлечена в репарацию ДНК, PARP также может действовать как медиатор гибели клеток. Его избыточная активация в таких патологических состояниях, как ишемия и реперфузионное повреждение, может приводить в результате к существенному истощению межклеточного NAD+,что может привести к нарушению нескольких NAD+ зависимых метаболических путей и в результате к гибели клеток (см. Pharmacological Research (2005), 52:44-59). В результате активации PARP уровниNAD+ значительно снижаются. Обширная активация PARP ведет к сильному истощению NAD+ в клетках, страдающих от массивного повреждения ДНК. Короткий период полураспада поли(ADP-рибозы) приводит в результате к быстрой скорости обращения, так как, как только образуется поли(ADP-рибоза),она быстро разлагается под действием конститутивно активной поли(ADP-рибоза)гликогидролазы(PARG). PARP и PARG образуют цикл, который превращает огромное количество NAD+ в ADP-рибозу,вызывая падение уровня NAD+ и АТР до менее чем 20% нормального уровня. Такой сценарий особенно вреден во время ишемии, когда недостаток кислорода уже отрицательно сказывается на выходе клеточной энергии. Предполагается, что последующее продуцирование свободных радикалов во время реперфузии является главной причиной повреждения тканей. Часть падения уровня АТР, которое является типичным во многих органах во время ишемии и реперфузии, может быть связана с истощением уровняNAD+ вследствие оборота поли(ADP-рибозы). Таким образом, ожидается, что ингибирование PARP сохранит уровень клеточной энергии, способствуя, тем самым, выживанию ишемических тканей после инсульта. Следовательно, соединения, которые являются ингибиторами PARP, полезны при лечении состояний, которые являются результатом PARP опосредуемой гибели клеток, включая неврологические состояния, такие как инсульт, травма и болезнь Паркинсона. Было продемонстрировано, что ингибиторы PARP являются полезными для специфичного уничтожения опухолей с дефицитом BRCA-1 и BRCA-2 (Nature (2005), 434:913-916 и 917-921 и Cancer BiologyTherapy (2005), 4:934-936).-1 016079 Было показано, что ингибиторы PARP усиливают эффективность противораковых лекарственных препаратов (Pharmacological Research (2005), 52:25-33), включая соединения платины, такие как цисплатин и карбоплатин (Cancer Chemother Pharmacol (1993), 33:157-162 и Mol. Cancer Ther. (2003), 2:371-382). Было показано, что ингибиторы PARP увеличивают противоопухолевую активность ингибиторов топоизомеразы I, таких как иринотекан и топотекан (Mol. Cancer Ther. (2003), 2:371-382 и Clin. Cancer Res.(2000), 6:2860-2867), и это было продемонстрировано на in vivo моделях (J. Natl. Cancer Inst. (2004),96:56-67). Было показано, что ингибиторы PARP восстанавливают восприимчивость к цитотоксическим антипролиферативным действиям темозоломида (TMZ) (см. Curr. Med. Chem. (2002), 9:1285-1301 и Med.Chem. Rev. Online (2004), 1:144-150). Это было продемонстрировано на ряде in vitro моделей [Br. J.(Me-Lex) (Pharmacological Research (2005), 52:25-33). Было показано, что ингибиторы PARP действуют как сенсибилизаторы радиации. Сообщалось, что ингибиторы PARP являются эффективными в радиосенсибилизации (гипоксических) опухолевых клеток и эффективными в предотвращении выздоровления опухолевых клеток от потенциально летального [Br.(2004), 16(1):29-39) повреждения ДНК после лечения облучением, предположительно под влиянием их способности предотвращать повторное соединение разрыва ДНК нити и повреждения некоторых сигнальных путей повреждения ДНК. Было показано, что ингибиторы PARP являются полезными при лечении острых и хронических заболеваний миокарда (см. Pharmacological Research (2005), 52:34-43). Например, было продемонстрировано, что разовые инъекции ингибиторов PARP снижают размер инфаркта, вызываемого ишемией и реперфузией сердечной или скелетной мышцы у кроликов. В данных исследованиях одна инъекция 3-аминобензамида (10 мг/кг) за 1 мин до окклюзии или за 1 мин до реперфузии вызывает аналогичные снижения размера инфаркта в сердце (32-42%), тогда как 1,5-дигидроксиизохинолин(1 мг/кг), еще один ингибитор PARP, снижает размер инфаркта в сравнимой степени (38-48%). Данные результаты делают обоснованным предположение, что ингибиторы PARP могут заранее не допустить ишемического сердца или реперфузионного повреждения скелетной мышечной ткани (PNAS (1997),94:679-683). Сообщалось также об аналогичных находках на свиньях (Eur. J. Pharmacol. (1998), 359:143150 и Ann. Thorac. Surg. (2002), 73:575-581 и на собаках (Shock. (2004), 21:426-32). Было продемонстрировано, что ингибиторы PARP являются полезными при лечении некоторых сосудистых заболеваний, септического шока, ишемического повреждения и нейротоксичности (Biochim.Biophys. Acta (1989), 1014:1-7; J. Clin. Invest. (1997), 100: 723-735). Кислородное радикальное повреждение ДНК, которое ведет к разрывам нитей в ДНК, которые впоследствии распознаются с помощьюPARP, является главным фактором, способствующим таким болезненным состояниям, как показано исследованиями ингибитора PARP (J. Neuroschi. Res. (1994), 39:38-46 и PNAS (1996), 93:4688-4692). Было продемонстрировано также, что PARP играет роль в патогенезе геморрагического шока (PNAS (2000),97:10203-10208). Было продемонстрировано, что ингибиторы PARP являются полезными при лечении воспалительных заболеваний (см. Pharmacological Research (2005), 52:72-82 и 83-92). Было также продемонстрировано, что эффективная ретровирусная инфекция клеток млекопитающих блокируется ингибированием активности PARP. Было показано, что такое ингибирование инфекций рекомбинантного ретровирусного вектора происходит в многочисленных типах клеток (J. Virology,(1996), 70(6), 3992-4000). Таким образом, ингибиторы PARP были разработаны для применения в антивирусной терапии и при лечении рака (WO 91/18591). Эксперименты in vitro и in vivo продемонстрировали, что ингибиторы PARP могут применяться при лечении или предотвращении аутоиммунных заболеваний, таких как диабет типа I и диабетические осложнения (Pharmacological Research (2005), 52:60-71). Были предположения, что ингибирование PARP задерживает наступление признаков старения в фибробластах человека (Biochem. Biophys. Res. Comm. (1994), 201(2):665-672 и Pharmacological Research(2005), 52:93-99). Это может быть связано с той ролью, которую PARP играет при контроле теломерных функций (Nature Gen. (1999), 23(1):76-80). Подавляющее большинство ингибиторов PARP, известных до настоящего времени, взаимодействуют с никотинамидсвязывающим доменом фермента и ведут себя как конкурентные ингибиторы по отношению к NAD+ (Expert Opin. Ther. Patents (2004), 14:1531-1551). Структурные аналоги никотинамида, такие как бензамид и производные, были среди первых соединений, исследуемых в качестве ингибиторов PARP. Однако данные молекулы обладают слабой ингибирующей активностью и оказывают другие действия, не связанные с ингибированием PARP. Таким образом, существует потребность в разра-2 016079 ботке сильных ингибиторов PARP фермента. Структурно родственные ингибиторы PARP были ранее описаны. В WO 1999/59973 описаны амидзамещенные бензольные кольца, конденсированные с 5-членными гетероароматическими кольцами; вWO 2001/85687 описаны амидзамещенные индолы; в WO 1997/04771, WO 2000/26192, WO 2000/32579,WO 2000/64878, WO 2000/68206, WO 2001/21615, WO 2002/068407, WO 2003/106430 и WO 2004/096793 раскрыты амидзамещенные бензоимидазолы; в WO 2000/29384 раскрыты амидзамещенные бензоимидазолы и индолы и в ЕР 0879820 раскрыты амидзамещенные бензоксазолы. В настоящее время на удивление обнаружено, что амидзамещенные индазолы настоящего изобретения проявляют особенно высокие уровни ингибирования активности поли(ADP-рибоза)полимеразы(PARP). Таким образом, соединения настоящего изобретения являются особенно полезными в качестве ингибиторов PARP-1 и/или PARP-2. Они показывают также особенно хорошие уровни клеточной активности, демонстрируя хорошие антипролиферативные эффекты в клеточных линиях с дефицитом BRCA-1 и BRCA-2. Настоящее изобретение предоставляет соединения формулы (I) где R1 представляет водород или фтор;R2 представляет водород или фтор; или их фармацевтически приемлемые соли, стереоизомеры или таутомеры. В одном варианте осуществления R1 представляет водород. В еще одном варианте осуществления R1 представляет фтор. В еще одном варианте осуществления R2 представляет водород. В еще одном варианте осуществления R2 представляет фтор. В еще одном варианте осуществления R1 представляет водород и R2 представляет водород или фтор. В еще одном варианте осуществления R1 представляет фтор и R2 представляет водород или фтор. В еще одном варианте осуществления R1 представляет водород и R2 представляет водород. В еще одном варианте осуществления R1 представляет водород и R2 представляет фтор. В еще одном варианте осуществления R1 представляет фтор и R2 представляет фтор. В еще одном варианте осуществления R1 представляет водород или фтор и R2 представляет водород. В еще одном варианте осуществления R1 представляет водород или фтор и R2 представляет фтор. Настоящее изобретение также предоставляет соединения формулы (II) где R1 и R2 имеют значения, определенные выше; или их фармацевтически приемлемые соли, стереоизомеры или таутомеры. Настоящее изобретение также предоставляет соединения формулы (III) где R1 и R2 имеют значения, определенные выше; или их фармацевтически приемлемые соли или таутомеры. Настоящее изобретение также предоставляет соединения формулы (IV) где R1 и R2 имеют значения, определенные выше; или их фармацевтически приемлемые соли или таутомеры.-3 016079 Предпочтительные радикалы в отношении формул (II)-(IV) имеют значения, определенные выше для формулы (I) mutatis mutandis. В объем настоящего изобретения также включены N-оксиды соединений формулы (I). Обычно такие N-оксиды могут образовываться на любом доступном атоме азота. N-оксиды могут образовываться общепринятыми средствами, такими как реакция соединения формулы (I) с оксоном в присутствии влажного оксида алюминия. В объем настоящего изобретения также включены пролекарства соединений формулы (I). Обычно такие пролекарства являются функциональными производными соединений формулы (I), которые способны свободно превращаться in vivo в требуемое соединение формулы (I). Общепринятые способы выбора и получения подходящих пролекарственных производных описаны, например, в "Design ofProdrugs", ed. H. Bundgaard, Elsevier, 1985. Пролекарством может быть фармакологически неактивное производное биологически активного вещества ("исходное лекарство" или "исходная молекула"), которое требует преобразования в организме для высвобождения активного лекарственного средства и которое имеет улучшенные свойства доставки по сравнению с исходной молекулой лекарственного средства. Преобразование in vivo может быть, например, результатом некоторого метаболического процесса, такого как химический или ферментный гидролиз карбонового, фосфорного или сульфатного эфира, или восстановление или окисление чувствительной функциональной группы. Настоящее изобретение включает сольваты соединений формулы (I) и их соли, например гидраты. Соединения настоящего изобретения могут иметь асимметрические центры, хиральные оси и хиральные плоскости (как описано у E.L. Eliel and S.H. Wilen, в Stereochemistry of Carbon Compounds, JohnWileySons, New York, 1994, p. 1119-1190) и существуют как рацематы, рацемические смеси и индивидуальные диастереомеры, со всеми возможными изомерами и их смесями, включая оптические изомеры,при этом все такие стереоизомеры включены в объем настоящего изобретения. В дополнение, соединения, описанные в данном описании, могут существовать в виде таутомеров и имеется в виду, что обе таутомерные формы охватываются объемом изобретения, даже если изображена только одна таутомерная структура. Соединения могут существовать в различных изомерных формах, все из которых охватываются настоящим изобретением. Соединения могут существовать в виде ряда различных полиморфных форм. Как использовано в данном описании, С 1-6 алкил представляет разветвленную, линейную и циклическую насыщенную алифатическую углеводородную группу, содержащую 1, 2, 3, 4, 5 или 6 атомов углерода. Например, "C1-6 алкил", в частности, включает метил, этил, н-пропил, изопропил, н-бутил,трет-бутил, изобутил, пентил, гексил, циклопропил, циклобутил, циклопентил и циклогексил и т.д. Предпочтительными алкильными группами являются метил и этил. Конкретными соединениями в объеме настоящего изобретения являются: 3-4-[7-(аминокарбонил)-2H-индазол-2-ил]фенилпиперидинийхлорид; 2-4-[(3R)-пиперидин-3-ил]фенил-2H-индазол-7-карбоксамид; 2-4-[(3S)-пиперидин-3-ил]фенил-2H-индазол-7-карбоксамид; трифторацетат 3-4-[7-(аминокарбонил)-5-фтор-2H-индазол-2-ил]фенилпиперидиния; трифторацетат 5-фтор-2-(3-фтор-4-пиперидин-3-илфенил)-2H-индазол-7-карбоксамида; трифторацетат 3-4-[7-(аминокарбонил)-2H-индазол-2-ил]фенилпиперидиния; 5-фтор-2-(4-пиперидин-3-илфенил)-2 Н-индазол-7-карбоксамид;(S)-5-фтор-2-3-фтор-4-пиперидин-3-илфенил-2H-индазол-7-карбоксамид и их фармацевтически приемлемые соли, свободные основания или таутомеры. Предоставляются также стереоизомеры данных соединений. Конкретным соединением настоящего изобретения является 3-4-[7-(аминокарбонил)-2H-индазол 2-ил]фенилпиперидинийхлорид или его фармацевтически приемлемые свободные основания или таутомеры. Предоставляются также стереоизомеры данного соединения. Конкретным соединением настоящего изобретения является 2-4-[(3R)-пиперидин-3-ил]фенил-2Hиндазол-7-карбоксамид или его фармацевтически приемлемая соль, свободное основание или таутомер. Предоставляются также стереоизомеры данного соединения. Конкретным соединением настоящего изобретения является 2-4-[(3S)-пиперидин-3-ил]фенил-2Hиндазол-7-карбоксамид или его фармацевтически приемлемая соль, свободное основание или таутомер. Предоставляются также стереоизомеры данного соединения.-4 016079 Конкретным соединением настоящего изобретения является трифторацетат 3-4-[7(аминокарбонил)-5-фтор-2H-индазол-2-ил]фенилпиперидиния или его фармацевтически приемлемое свободное основание или таутомер. Предоставляются также стереоизомеры данного соединения. Конкретным соединением настоящего изобретения является трифторацетат 5-фтор-2-(3-фтор-4 пиперидин-3-илфенил)-2H-индазол-7-карбоксамида или его фармацевтически приемлемое свободное основание или таутомер. Предоставляются также стереоизомеры данного соединения. Конкретным соединением настоящего изобретения является 4-метилбензолсульфонат (3S)-3-4-[7(аминокарбонил)-2H-индазол-2-ил]фенилпиперидиния или его фармацевтически приемлемое свободное основание или таутомер. Предоставляются также стереоизомеры данного соединения. В данное изобретение включено свободное основание соединений формулы (I), а также его фармацевтически приемлемые соли и стереоизомеры. Соединения настоящего изобретения могут быть протонированы по атому(атомам) N амина и/или N содержащего гетероциклического фрагмента с образованием соли. Термин "свободное основание" относится к аминным соединениям в несолевой форме. Входящие в объем изобретения фармацевтически приемлемые соли включают не только соли, приведенные в качестве примеров конкретных соединений, описанных в данном описании, но и все типичные фармацевтически приемлемые соли свободной формы соединений формулы (I). Свободная форма конкретной соли соединений, описанных в данном описании, может быть выделена с использованием известных в данной области методов. Например, свободная форма может быть регенерирована путем обработки соли разбавленным водным раствором подходящего основания, таким как разбавленный водный NaOH, карбонат калия, аммиак и бикарбонат натрия. Свободные формы могут несколько отличаться от их соответствующих солевых форм по их некоторым физическим свойствам, таким как растворимость в полярных растворителях, но для целей изобретения соли кислот и оснований в иных отношениях фармацевтически эквивалентны их соответствующим свободным формам. Фармацевтически приемлемые соли данных соединений могут быть синтезированы из соединений данного изобретения, которые содержат основный или кислотный фрагмент, с помощью общепринятых химических способов. Обычно соли основных соединений получают или путем ионообменной хроматографии, или взаимодействия свободного основания со стехиометрическими количествами или с избытком желаемой солеобразующей неорганической или органической кислоты в подходящем растворителе или различных комбинациях растворителей. Аналогичным образом, соли кислотных соединений могут быть образованы взаимодействием с соответствующим неорганическим или органическим основанием. Таким образом, фармацевтически приемлемые соли соединений данного изобретения включают нетоксичные соли соединений данного изобретения, полученные путем взаимодействия основного рассматриваемого соединения с неорганической, органической кислотой или полимерной кислотой. Например, общепринятые нетоксичные соли включают соли, полученные из неорганических кислот, таких как хлористо-водородная, бромисто-водородная, йодисто-водородная, серная, сернистая, сульфаминовая,фосфорная, фосфористая, азотная и аналогичные, а также соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная,салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая,этандисульфоновая, щавелевая, изетионовая, пальмитиновая, глюконовая, аскорбиновая, фенилуксусная,аспарагиновая, коричная, пировиноградная, этансульфоновая, этандисульфоновая, валериановая,трифторуксусная и аналогичные. Примеры подходящих полимерных солей включают соли, полученные из полимерных кислот, таких как дубильная кислота, карбоксиметилцеллюлоза. Предпочтительно фармацевтически приемлемая соль данного изобретения содержит 1 экв. соединения формулы (I) и 1, 2 или 3 экв. неорганической или органической кислоты. В одном варианте осуществления фармацевтически приемлемая соль данного изобретения содержит 2 экв. соединения формулы (I) и 1 экв. неорганической или органической кислоты. Более конкретно, фармацевтически приемлемыми солями данного изобретения являются трифторацетатная, хлоридная или тозилатная соли. Более конкретно, фармацевтически приемлемыми солями данного изобретения являются трифторацетат или хлорид. В одном варианте осуществления солью является трифторацетат. В еще одном варианте осуществления солью является хлорид. В еще одном варианте осуществления солью является тозилат. Термин "толуолсульфоновая кислота" может использоваться взаимозаменяемо с 4-метилбензолсульфоновой кислотой, и толуолсульфонаты также могут называться тозилатами. Когда соединение настоящего изобретения является кислотным, подходящие "фармацевтически приемлемые соли" относятся к солям, полученным из фармацевтически приемлемых нетоксичных оснований, включая неорганические и органические основания. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа [3], железа [2], лития,магния, марганцовистые соли, соли марганца, калия, натрия, цинка и аналогичные. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклические амины и основные ионообменные смолы, такие как соли аргинина, лизина, бетаина, кофеина, холина,-5 016079N,N1-дибензилэтилендиамина, этиламина, диэтиламина, 2-диэтиламиноэтанола, 2-диметиламиноэтанола,этаноламина, диэтаноламина, этилендиамина, N-этилморфолина, N-этилпиперидина, глюкамина, глюкозамина, гистидина, гидрабамина, изопропиламина, лизина, метилглюкамина, морфолина, пиперазина,пиперидина, полиаминовых смол, прокаина, пуринов, теобромина, триэтиламина, триметиламина, трипропиламина, трометамина, дициклогексиламина, бутиламина, бензиламина, фенилбензиламина, трометамина и аналогичные. Получение фармацевтически приемлемых солей, описанных выше, и других типичных фармацевтически приемлемых солей более подробно описано Berg et al. (1977) в J. Pharm. Sci., "PharmaceuticalSalts", 66:1-19. Также следует отметить, что соединения настоящего изобретения являются потенциально внутренними солями или цвиттерионами, поскольку в физиологических условиях депротонируемый кислотный фрагмент в соединении, такой как карбоксильная группа, может быть анионным, и данный электронный заряд может затем уравновешиваться изнутри против катионного заряда протонированного или алкилированного основного фрагмента, такого как четвертичный атом азота. Соединения изобретения могут использоваться в способе лечения людей или животных с помощью терапии. Изобретение предоставляет соединения для применения при лечении или предотвращении состояний, которые могут облегчаться ингибированием поли(ADP-рибоза)полимеразы (PARP) (см., например,Nature Review Drug Discovery (2005), 4:421-440). Таким образом, настоящее изобретение предоставляет соединение формулы (I) для применения в производстве лекарственных средств для лечения или предотвращения состояний, которые могут облегчаться за счет ингибирования поли(ADP-рибоза)полимеразы (PARP). Настоящее изобретение также предоставляет способ лечения или предотвращения состояний, которые могут облегчаться ингибированием поли(ADP-рибоза)полимеразы (PARP), который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). Ингибиторы (PARP) настоящего изобретения полезны при лечении заболеваний, конкретно указанных в WO 2005/082368. Соединения изобретения полезны при лечении воспалительных заболеваний, включающих состояния, являющиеся результатом отторжения органа-трансплантата, таких как хронические воспалительные заболевания суставов, включая артрит, ревматоидный артрит, остеоартрит и заболевания костей, связанные с повышенной костной резорбцией; воспалительные заболевания кишечника, такие как илеит, язвенный колит, синдром Баррета и болезнь Крона: воспалительные заболевания легких, такие как астма,синдром респираторного дистресса взрослых и хроническая обструктивная болезнь дыхательных путей; воспалительные заболевания глаз, включая дистрофию роговицы, трахому, онхоцеркоз, увеит, симпатикофтальмит и эндофтальмит; хронические воспалительные заболевания десны, включающие гингивит и периодонтит; туберкулез; лепра; воспалительные заболевания почек, включающие уремические осложнения, гломерулонефрит и нефроз; воспалительные заболевания кожи, включающие склеродерматит,псориаз и экзему; воспалительные заболевания центральной нервной системы, включающие хронические демиелинирующие заболевания нервной системы, рассеянный склероз, связанную со СПИД нейродегенерацию и болезнь Альцгеймера, инфекционный менингит, энцефаломиелит, болезнь Паркинсона, болезнь Хангтингтона, амиотрофический боковой склероз и вирусный или аутоиммунный энцефалит; диабетические осложнения, включающие, но не ограниченные ими, иммунный сложный васкулит, системную красную волчанку (SLE); воспалительные заболевания сердца, такие как кардиомиопатия, ишемическая болезнь сердца, гиперхолестеринемия и атеросклероз; а также многочисленные другие заболевания,которые могут иметь значительные воспалительные компоненты, включая преэклампсию, хроническую печеночную недостаточность, травму головного и спинного мозга и синдром множественной дисфункции органов (MODS) (множественная недостаточность органов (MOF. Воспалительным заболеванием может быть также системное воспаление организма, примером которого является грамположительный или грамотрицательный шок, геморрагический или анафилактический удар, или шок, вызываемый химиотерапией рака в ответ на провоспалительные цитокины, например шок, связанный с провоспалительными цитокинами. Такой шок может быть вызван, например, химиотерапевтическим агентом, который вводится в качестве лечебного средства от рака. Таким образом, настоящее изобретение предоставляет соединение формулы (I) для применения в производстве лекарственных средств для лечения или предотвращения воспалительных заболеваний. Настоящее изобретение также предоставляет способ лечения или предотвращения воспалительных заболеваний, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). Соединения данного изобретения могут быть также полезны при лечении и предотвращении реперфузионных повреждений, являющихся результатом естественно случающихся эпизодов или событий,и во время хирургических процедур, таких как реперфузионное повреждение кишечного тракта; миокардиальное реперфузионное повреждение; реперфузионное повреждение, являющееся результатом кар-6 016079 диопульмонарной операции с искусственным кровообращением, восстановительной операции аневризмы аорты, операции каротидной эндартерэктомии или геморрагического шока; и реоксогенационное повреждение, являющееся результатом трансплантации органов, таких как сердце, легкие, печень, почки,поджелудочная железа, кишечник и роговица. Таким образом, настоящее изобретение представляет соединения формулы (I) для применения в производстве лекарственного средства для лечения или предотвращения реперфузионных повреждений. Настоящее изобретение также предоставляет способ лечения или предотвращения реперфузионных повреждений, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). Соединения данного изобретения также могут быть полезны при лечении или предотвращении ишемических состояний, включающих состояния, являющиеся результатом трансплантации органов,таких как стабильная ангина, нестабильная ангина, ишемия миокарда, гепатическая ишемия, мезентериальная артериальная ишемия, ишемия кишечного тракта, критическая ишемия конечностей, хроническая критическая ишемия конечностей, церебральная ишемия, острая сердечная ишемия, ишемическая болезнь почек, ишемическая болезнь печени, ишемическое расстройство сетчатки, септический шок и ишемическая болезнь центральной нервной системы, такая как тромбоз или кровоизлияние (или эмболия) или церебральная ишемия. Таким образом, настоящее изобретение предоставляет соединение формулы (I) для применения в производстве лекарственного средства для лечения или предотвращения ишемических состояний. Настоящее изобретение также предоставляет способ лечения или предотвращения ишемических состояний, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). Настоящее изобретение предоставляет соединение формулы (I) для применения в производстве лекарственного средства для лечения или предотвращения удара. Настоящее изобретение также предоставляет способ лечения или предотвращения удара, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). Соединения данного изобретения могут быть также полезны при лечении или предотвращении хронической или острой почечной недостаточности. Таким образом, настоящее изобретение предоставляет соединение формулы (I) для применения в производстве лекарственного средства для лечения или предотвращения почечной недостаточности. Настоящее изобретение также предоставляет способ лечения или предотвращения почечной недостаточности, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). Соединения данного изобретения могут быть также полезны при лечении или предотвращении сосудистых заболеваний, иных чем сердечно-сосудистые заболевания, таких как закупорка периферических артерий, облитерирующий тромбоангит, болезнь и феномен Рейнода, акроцианоз, эритромелалгия,венозный тромбоз, варикозные вены, артериовенозная фистула, лимфодема и жировой отек. Таким образом, настоящее изобретение предоставляет соединение формулы (I) для применения в производстве лекарственного средства для лечения или предотвращения сосудистых заболеваний, иных чем сердечно-сосудистые заболевания. Настоящее изобретение предоставляет также способ лечения или предотвращения сосудистых заболеваний, иных чем сердечно-сосудистые заболевания, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). Соединения данного изобретения могут быть также полезны при лечении или предотвращении сердечно-сосудистых заболеваний, таких как хроническая сердечная недостаточность, атеросклероз, застойная сердечная недостаточность, циркуляторный шок, кардиомиопатия, пересадка сердца, инфаркт миокарда и сердечная аритмия, такая как атриальная фибрилляция, суправентрикулярная тахикардия, частые регулярные сокращения предсердий и пароксизмальная атриальная тахикардия. Таким образом, настоящее изобретение предоставляет соединения формулы (I) для применения в производстве лекарственного средства для лечения или предотвращения сердечно-сосудистых заболеваний. Настоящее изобретение также предоставляет способ лечения или предотвращения сердечнососудистых заболеваний, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). Соединения данного изобретения также могут быть полезны при лечении и предотвращении сахарного диабета, включая диабет типа I (инсулинзависимый сахарный диабет), диабет типа II (инсулиннезависимый сахарный диабет), диабет, связанный с беременностью, аутоиммунный диабет, инсулинопатию,диабет вследствие болезни поджелудочной железы, диабет, связанный с другими болезнями эндокринной системы (такими как синдром Кашинга (Cushing), акромегалия, феохромоцитома, глюкагонома, первичный альдостеронизм или соматостатинома), синдром инсулинорезистентности типа А, синдром инсу-7 016079 линорезистентности типа В, липатрофический диабет и диабет, вызываемый 3-клеточными токсинами. Соединения данного изобретения могут быть также полезны при лечении или предотвращении диабетических осложнений, таких как диабетическая катаракта, глаукома, ретинопатия, нефропатия (такая как микроалюминурия и прогрессивная диабетическая нефропатия), полиневропатия, гангрена ног, атеросклеротическая болезнь коронарной артерии, заболевание периферической артерии, некетотическая гипергликемическо-гиперосмолярная кома, мононейропатии, автономная нейропатия, язвы стопы, проблемы суставов и осложнения кожи или слизистой мембраны (такие как инфекция, пятнистость кожи, грибковая инфекция или липофильный некробиоз), гиперлипидемия, гипертензия, синдром инсулинорезистентности, болезнь коронарной артерии, ретинопатия, диабетическая нейропатия, полинейропатия, мононейропатия, автономная нейроптия, грибковые инфекции, бактериальные инфекции и кардиомиопатия. Таким образом, настоящее изобретение предоставляет соединения формулы (I) для применения в производстве лекарственного средства для лечения или предотвращения диабета. Настоящее изобретение также предоставляет способ лечения или предотвращения диабета, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). Соединения данного изобретения могут быть также полезны при лечении и предотвращении рака,включая солидные опухоли, такие как фибросаркома, миксосаркома, липосаркома, хондросаркома, остеогенная саркома, хордома, ангиосаркома, эндотелиосаркома, лимфангиосаркома, димфангиоэндотелиосаркома, синовиома, мезотелиома, опухоль Эвинга, лейомиосаркома, рабдомиосаркома, рак ободочной кишки, колоректальный рак, рак почек, рак поджелудочной железы, рак костей, рак груди, рак яичников, рак предстательный железы, эзофагеальный рак, рак желудка, рак ротовой полости, рак носовой полости, рак горла, сквамозная клеточная карцинома, базальная клеточная карцинома, аденокарцинома,карцинома потовых желез, карцинома сальных желез, сосочковая карцинома, сосочковая аденокарцинома, цистаденокарцинома, медуллярная карцинома, бронхогенная карцинома, ренальная клеточная карцинома, гепатома, карцинома желчного протока, хориокарцинома, семинома, эмбриональная карцинома,опухоль Уилмса, цервикальный рак, маточный рак, тестикулярный рак, мелкоклеточная легочная карцинома, карцинома мочевого пузыря, рак легких, эпителиальная карцинома, рак кожи, меланома, нейробластома и ретинобластома; виды рака крови, такие как острая лимфобластная лейкемия ("ALL"), острая лимфобластная В-клеточная лейкемия, острая лимфобластная Т-клеточная лейкемия, острая миелобластная лейкемия ("AML"), острая промиелоцитная лейкемия ("APL"), острая монобластная лейкемия, острая эритролейкемическая лейкемия, острая мегакариобластная лейкемия, острая миеломоноцитная лейкемия, острая нелимфоцитная лейкемия, острая недифференцируемая лейкемия, хроническая миелоцитная лейкемия ("CML"), хроническая лимфоцитная лейкемия ("CLL"), лейкемия волосяных клеток и множественная миелома; острые и хронические лейкемии, такие как лимфобластная, миелогенная, лимфоцитная, миелоцитная лейкемии; лимфомы, такие как болезнь Ходжкина, неходжкинская лимфома, множественная миелома, макроглобулинемия Вальденстрома, заболевание тяжелой цепи (полипептидной цепи большой молекулярной массы) и красная или истинная полицитемия; виды рака ЦНС и головного мозга, такие как глиома, пилоцитная астроцитома, астроцитома, анапластическая астроцитома, глиобластома мультиформная, медуллобластома, краниофарингиома, эпендимома, пинеалома, гемангиобластома, акустическая нейрома, олигодендроглиома, менингиома, вестибуляная шваннома, аденома, метастатическая опухоль головного мозга, спинальная опухоль и медуллобластома. Таким образом, настоящее изобретение предоставляет соединения формулы (I) для применения в производстве лекарственного средства для лечения или предотвращения рака. Настоящее изобретение также предоставляет способ лечения или предотвращения рака, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). Соединения настоящего изобретения также могут применяться при лечении рака, который испытывает дефицит зависимой от гомологичной рекомбинации (HR) ДНК DSB восстановительной активностиHR зависимый ДНК DSB восстановительный путь восстанавливает двунитевые разрывы (DSB) ДНК через гомологичные механизмы, реформируя непрерывную ДНК спираль (Nat. Genet. (2001),27(3):247-254). Компоненты HR зависимого ДНК DSB восстановительного пути включают, но не ограничиваются ими, ATM (NM-000051), RAD51 (NM-002875), RAD51LI (NM-002877), RAD51 С(NM-000059), RAD5O (NM-005732), MREI 1A (NM-005590), NBS1 (NM-002485), ADPRT (PARP-1),ADPRTL2, (PARP02) CTPS, RPA, RPA1, RPA2, RPA3, XPD, ERCC1, XPF, MMS19, RAD51, RAD51p,RAD51C, RAD51D, DMC1, XRCCR, XRCC3, BRCA-1, BRCA-2, RAD52, RAD54, RAD50, MRE11, NB51,WRN, BLMKU70, RU80, ATM, ATRCHK1, CHK2, FANCA, FANCB, FANCC, FANCD1, FANCD2,FANCE, FANCF, FANCG, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, RAD1 и RAD9. Другие белки, вовлеченные в HR зависимый ДНК DSB восстановительный путь, включают регуляторные факто-8 016079 ры, такие как EMSY (Cell (2003), 115:523-535). Рак, который испытывает дефицит HR зависимого ДНК DSB восстановления, может включать или состоять из одной или нескольких раковых клеток, которые имеют пониженную или лишены способности восстанавливать DSB по этому пути по сравнению с нормальными клетками, т.е. активность HR зависимого ДНК DSB восстановительного пути может быть пониженной или разрушенной в одной или нескольких раковых клетках. Активность одного или более компонентов HR зависимого ДНК DSB восстановительного пути может разрушаться в одной или нескольких раковых клетках индивидуума, у которого имеется рак с дефицитом HR зависимого ДНК DSB восстановления. Компоненты HR зависимого ДНК DSB восстановительного пути вполне охарактеризованы в литературе (см., например, Science (2001), 291:1284-1289) и включают компоненты, перечисленные выше. Настоящее изобретение предоставляет соединение формулы (I) для применения в производстве лекарственного средства для лечения или предотвращения рака с дефицитом HR зависимой ДНК DSB восстановительной активности. Настоящее изобретение также предоставляет способ лечения или предотвращения рака с дефицитом HR зависимой ДНК DSB восстановительной активности, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). В одном варианте осуществления раковые клетки испытывают дефицит HR зависимой ДНК DSB восстановительной активности одного или более фенотипов, выбранных из ATM (NM-000051), RAD51(NM-005590), NBS1 (NM-002485), ADPRT (PARP-1), ADPRTL2, (PARP02) CTPS, RPA, RPA1, RPA2,RPA3, XPD, ERCC1, XPF, MMS19, RAD51, RAD51p, RAD51C, RAD51D, DMC1, XRCCR, XRCC3,BRCA-1, BRCA-2, RAD52, RAD54, RAD50, MRE11, NB51, WRN, BLMKU70, RU80, ATM, ATRCHK1,CHK2, FANCA, FANCB, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, FANCC, FANCD1,FANCD2, FANCE, FANCF, FANCG, RAD1 и RAD9. В соответствии с еще одним вариантом осуществления раковые клетки испытывают дефицитBRCA-1 и/или BRCA-2 фенотипа. Раковые клетки с данным фенотипом могут испытывать дефицитBRCA-1 и/или BRCA-2, т.е. экспрессия и/или активность BRCA-1 и/или BRCA-2 могут понижаться или исчезать в раковых клетках, например, под действием мутации или полиморфизма в кодирующей нуклеиновой кислоте или под действием амплификации, мутации или полиморфизма в гене, кодирующем регуляторный фактор, например, EMSY гене, который кодирует BRCA-2 регуляторный фактор (CellBRCA-1 и/или BRCA-2 являются известными супрессорами опухолей, чьи аллели дикого типа часто отсутствуют в опухолях гетерозиготных носителей (Oncogen (2002), 21(58):8981-93; Trends Mol. Med.(2002), 8(12):571-6). Хорошо охарактеризована ассоциация BRCA-1 и/или BRCA-2 мутаций с раком груди (Ехр. Clin. Cancer Res. (2002. Известно, что амплификация EMSY гена, который кодирует BRCA-2 связующий фактор, связана с раком груди и яичников. Носители мутаций BRCA-1 и/или BRCA-2 также испытывают повышенный риск рака яичников, простаты и поджелудочной железы. В данной области хорошо известна детекция вариаций в BRCA-1 и/или BRCA-2 и описана, например, в ЕР 699754,ЕР 705903, Genet. Test (1992), 1:75-83; Cancer Treat Res (2002), 107:29-59; Neoplasm (2003), 50(4):246-50;Ceska Gynecol (2003), 68(1):11-16). Определение амплификации BRCA-2 связующего фактора EMSY описано в Cell 115:523-535. Ингибиторы PARP были продемонстрированы как являющиеся полезными для специфичного уничтожения опухолей с дефицитом BRCA-1 и BRCA-2 (Nature (2005), 434:913-916 и 917-920). Таким образом, настоящее изобретение предоставляет соединение формулы (I) для применения в производстве лекарственного средства для лечения или предотвращения опухолей с дефицитом BRCA-1 или BRCA-2. Настоящее изобретение также предоставляет способ лечения или предотвращения опухолей с дефицитом BRCA-1 или BRCA-2, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). В одном варианте осуществления ингибиторы PARP настоящего изобретения могут применяться в профилактической терапии для устранения клеток с BRCA-2-дефицитом (см. Cancer Res. (2005),65:10145). Соединения данного изобретения также могут быть полезны при лечении или предотвращении нейродегенеративных заболеваний, включая связанную с расширением полиглутамина нейродегенерацию, болезнь Хангтингтона, болезнь Кеннеди, спиноцеребральную атаксию, дентато-рубро-паллидольюисову атрофию (DRPLA), связанную с агрегацией белка нейродегенерацию, болезнь МакадоДжозефа (Machado-Joseph), болезнь Альцгеймера, болезнь Паркинсона, амиотрофический боковой склероз, спонгиформную энцефалопатию, связанное с прионом заболевание и рассеянный склероз (MS).-9 016079 Таким образом, настоящее изобретение предоставляет соединение формулы (I) для применения в производстве лекарственного средства для лечения или предотвращения нейродегенеративных заболеваний. Настоящее изобретение представляет также способ лечения или предотвращения нейродегенеративных заболеваний, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I). Соединения настоящего изобретения также могут быть полезны при лечении или предотвращении ретровирусной инфекции (патент США 5652260), ретинальных повреждений (Curr. Eye Res. (2004),29:403), старения кожи и УФ индуцируемого повреждения кожи (патент США 5589483 и Biochem.Pharmacol (2002), 63:921). Соединения изобретения также полезны при лечении или предотвращении преждевременного старения и отсрочки наступления возрастной клеточной дисфункции (Pharmacological Research (2005),52:93-99). Соединения данного изобретения могут вводиться млекопитающим, предпочтительно людям, или как таковое, или в комбинации с фармацевтически приемлемыми носителями, эксципиентами, разбавителями, адъювантами, наполнителями, буферами, стабилизаторами, консервантами, лубрикантами, в фармацевтической композиции согласно стандартной фармацевтической практике. Соединения данного изобретения могут вводиться субъекту любым удобным способом введения,будь то системно/периферически или местно в зависимости от желаемого действия, включающим, но не ограничивающимся ими, оральный (например, с перевариванием); топический или местный (включая,например, трансдермальный, интраназальный, окулярный, защечный и подъязычный); легочный (например, путем ингаляционной или инсуффляционной терапии с использованием, например, аэрозоля, например через рот или нос); ректальный; вагинальный; парентеральный (например, инъекцией, включая подкожную, индрадермальную, внутримышечную, внутривенную, внутриартериальную, внутрисердечную, интратекальную (внутрь синовиального влагалища сухожилия), интраспинальную, интракапсулярную, субкапсулярную, внутриглазную, интраперитонеальную, внутритрахейную, субкутикулярную,внутрисуставную, субарахноидальную и интрастернальную); и с помощью имплантирования базового трансплантата (например, подкожно или внутримышечно). Субъектом может быть межрасовый гибрид, животное, позвоночное животное, млекопитающее,грызуны (например, морские свинки, хомяки, крысы, мыши), мышиные (например, мышь), собачьи (например, собака), кошачьи (например, кошки), лошадиные (например, лошадь), приматы, обезьяноподобные (например, обезьяны), обезьяны (например, мармозетки, бабуины), человекообразные обезьяны (например, гориллы, шимпанзе, орангутанги, гиббоны) или человек. Изобретение также предоставляет фармацевтические композиции, содержащие одно или более соединений данного изобретения и фармацевтически приемлемый носитель. Фармацевтические композиции, содержащие активный ингредиент, могут быть в форме, подходящей для орального применения, например в виде таблеток, пастилок, водных или масляных суспензий,диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул или сиропов или эликсиров. Композиции, предназначенные для орального применения, могут быть получены в соответствии с любым методом, известным в области производства фармацевтических композиций, и такие композиции могут содержать один или более агентов, выбранных из группы, состоящей из подслащивающих, вкусовых или ароматизирующих агентов, окрашивающих агентов и консервантов, для того, чтобы получить фармацевтически мягкие и приятные на вкус препараты. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые являются подходящими для получения таблеток. Данными эксципиентами могут быть, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие или дезинтегрирующие агенты, например микрокристаллическая целлюлоза, натрийкроскармелоза, кукурузный крахмал или альгиновая кислота; связующие агенты, например крахмал, желатин, поливинилпирролидон или камедь акации, и лубриканты, например стеарат магния, стеариновая кислота или тальк. Таблетки могут быть непокрытыми или могут быть покрыты известными способами для маскировки неприятного вкуса или для задержки дезинтеграции и абсорбции в желудочно-кишечном тракте, тем самым обеспечивая замедленное действие на протяжении более длительного периода. Например, можно использовать растворимый в воде маскирующий вкус материал, такой как гидроксипропилметилцеллюлоза или гидроксипропилцеллюлоза, или задерживающий время действия материал, такой как этилцеллюлоза,ацетатбутират целлюлозы. Рецептуры для орального применения могут быть также представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный ингредиент смешан с растворимым в воде носителем, таким как полиэтиленгликоль или масляная среда, например арахисовое масло, жидкий парафин или оливковое масло.- 10016079 Водные суспензии содержат активный ингредиент в смеси с эксципиентами, подходящими для получения водных суспензий. Такими эксципиентами являются суспендирующие агенты, например натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, камедь трагаканта и камедь акации; диспергирующими или смачивающими агентами могут быть природные фосфатиды, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например стеарат полиоксиэтилена, или продукты конденсации этиленоксида с алифатическими спиртами с длинной цепью, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с частичными эфирами, полученными из жирных кислот и гексита, такие как моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с частичными эфирами,полученными из жирных кислот и ангидридов гексита, например полиэтиленсорбитанмоноолеат. Водные суспензии также могут содержать один или более консервантов, например этил или н-пропил п-гидроксибензоат, один или более окрашивающих агентов, один или более вкусовых агентов и один или более подслащивающих агентов, таких как сахароза, сахарин или аспартам. Масляные суспензии могут быть сформированы путем суспендирования активного ингредиента в растительном масле, например арахисовом масле, оливковом масле, кунжутном или кокосовом масле,или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загущающий агент, например пчелиный воск, твердый парафин или цетиловый спирт. Могут добавляться подслащивающие агенты, такие как агенты, представленные выше, и вкусовые или ароматизирующие агенты с обеспечением приятного на вкус орального препарата. Данные композиции могут быть консервированы путем добавления антиоксиданта, такого как бутилированный гидроксианизол или альфатокоферол. Диспергируемые порошки и гранулы, подходящие для получения водной суспензии путем добавления воды, предоставляют активный ингредиент в смеси с диспергирующим или смачивающим агентом,суспендирующим агентом и одним или более консервантами. Подходящие диспергирующие или смачивающие агенты и суспендирующие агенты представлены примерами агентов, приведенных выше. Также могут присутствовать дополнительные эксципиенты, например подслащивающие, вкусовые и окрашивающие агенты. Данные композиции могут быть консервированы путем добавления антиоксиданта, такого как аскорбиновая кислота. Фармацевтические композиции изобретения могут быть также в форме эмульсий "масло-в-воде". Масляной фазой может быть растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например жидкий парафин или их смеси. Подходящими эмульгирующими агентами могут быть природные фосфатиды, например соевый лецитин, и сложные эфиры или частичные эфиры,полученные из жирных кислот и ангидридов гексита, например сорбитанмоноолеат, и продукты конденсации названных частичных эфиров с этиленоксидом, например полиоксиэтиленсорбитанмоноолеат. Эмульсии могут содержать также подслащивающие, вкусовые агенты, консерванты и антиоксиданты. Сиропы и эликсиры могут быть составлены в композицию с подслащивающими агентами, например глицерином, пропиленгликолем, сорбитом или сахарозой. Такие рецептуры могут содержать также смягчающее или успокаивающее средство, консервант, придающий вкус или аромат и окрашивающий агенты и антиоксидант. Фармацевтические композиции могут быть в форме стерильных инъецируемых водных растворов. Среди приемлемых носителей и растворителей, которые могут быть использованы, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Стерильным инъецируемым препаратом может быть также стерильная инъецируемая микроэмульсия "масло-в-воде", в которой активный ингредиент растворен в масляной фазе. Например, активный ингредиент может быть сначала растворен в смеси соевого масла и лецитина. Масляный раствор затем вводят в смесь воды и глицерина и перерабатывают с образованием микроэмульсии. Инъецируемые растворы или микроэмульсии могут вводиться в кровоток пациента путем инъекции местного болюса. Альтернативно, может быть благоприятным вводить раствор или микроэмульсию таким образом, чтобы поддерживать постоянную циркулирующую концентрацию рассматриваемого соединения. Для того чтобы поддерживать такую постоянную концентрацию, можно использовать устройство непрерывной внутривенной доставки. Примером такого устройства является внутривенный насосDeltec CADD-PLUS модель 5400. Фармацевтические композиции могут быть в форме стерильной инъецируемой водной или маслянистой суспензии для внутримышечного и подкожного введения. Данная суспензия может быть составлена в композицию в соответствии с известными в данной области методами с использованием таких подходящих диспергирующих или смачивающих агентов и суспендирующих агентов, которые приведены выше. Стерильным инъецируемым препаратом может быть также стерильный инъецируемый раствор или суспензия в нетоксичном парентерально приемлемом разбавителе или растворителе, например в виде раствора в 1,3-бутандиоле. В дополнение, в качестве растворителя или суспендирующей среды обычно используют стерильные фиксированные масла. Для данной цели можно использовать любое мягкое фиксированное масло, включая синтетические моно- или диглицериды. В дополнение, при получении инъецируемых препаратов находят применение жирные кислоты, такие как олеиновая кислота.- 11016079 Соединения формулы (I) также могут вводиться в форме суппозиториев для ректального введения. Данные композиции могут быть получены путем смешения лекарственного средства с подходящим нераздражающим эксципиентом, который является твердым при обычных температурах, но жидким при температуре прямой кишки, и, следовательно, будет расплавляться в прямой кишке, высвобождая лекарственное средство. Такие материалы включают масло какао, глицеринированный желатин, гидрированные растительные масла, смеси полиэтиленгликолей с различными молекулярными массами и полиэтиленгликолевых эфиров жирных кислот. Для местного применения используют кремы, мази, желе, растворы или суспензии и др., содержащие соединение формулы (I). (Для целей данного изобретения местное применение предусматривает полоскание рта и горла). Соединения настоящего изобретения могут вводиться в виде интраназальной формы с помощью местного применения подходящих интраназальных носителей и доставочных средств или трансдермальными путями с использованием форм трансдермальных кожных пластырей, хорошо известных специалистам в данной области. Чтобы быть введенным в форме трансдермальной системы доставки, введение дозы будет скорее непрерывным, чем не прерывистым, во всем режиме дозировки. Соединения настоящего изобретения также могут доставляться в виде суппозиториев с использованием основ, таких как масло какао, глицеринированный желатин, гидрированные растительные масла, смеси полиэтиленгликолей с различными молекулярными массами и полиэтиленгликолевых эфиров жирных кислот. Когда соединение согласно данному изобретению вводят субъекту, выбираемый уровень доз будет зависеть от множества факторов, включающих, но не ограниченных ими, активность конкретного соединения, тяжесть симптомов у индивидуумов, путь введения, время введения, скорость выведения соединения, длительность лечения, другие лекарственные средства, соединения и/или материалы, используемые в комбинации, и возраст, пол, масса, состояние, общее состояние здоровья и предшествующая медицинская история болезни пациента. Количество соединения и путь введения в конечном итоге отдаются на усмотрение лечащего врача, хотя обычно доза должна быть достаточной для достижения локальных концентраций в участке действия, которые обеспечивают достижение желаемого эффекта, не вызывая существенных пагубных или вредных побочных эффектов. Введение in vivo можно проводить в виде одной дозы непрерывно или прерывисто (например, в виде разделенных доз с соответствующими интервалами) на протяжении курса лечения. Методы определения наиболее эффективных средств и дозы введения хорошо известны специалистам в данной области и будут варьировать в зависимости от рецептуры, используемой для терапии, цели терапии, клеток мишени, подвергаемых лечению, и подвергаемого лечению субъекта. Можно осуществлять одноразовое или множественное введение с учетом уровня и характера дозы, выбираемых лечащим врачом. Обычно подходящая доза активного соединения находится в интервале примерно от 100 мкг до примерно 250 мг на 1 кг массы тела субъекта в день. Когда активным соединением является соль, сложный эфир, пролекарство или аналогичные, вводимое количество вычисляется на основе исходного соединения, и поэтому фактически используемая масса увеличивается пропорционально. Данные соединения полезны также в комбинации с противораковыми агентами или химиотерапевтическими агентами. Соединения данного изобретения могут быть также полезны в качестве химио- и радиосенсибилизаторов для лечения рака. Они полезны при лечении млекопитающих, которые ранее подвергались или подвергаются в настоящее время лечению от рака. Такие предшествующие лечения включают предшествующую химиотерапию, радиационную терапию, хирургию или иммунотерапию, такую как вакцины от рака. Таким образом, настоящее изобретение предоставляет комбинацию соединения формулы (I) и противоракового агента для одновременного, раздельного или последовательного введения. Настоящее изобретение также предоставляет комбинацию соединения формулы (I), радиационной терапии и еще одного химиотерапевтического агента для одновременного, раздельного или последовательного введения. Настоящее изобретение также предоставляет соединение формулы (I) для применения в производстве лекарственного средства для применения в качестве дополнения в терапии рака или для потенцирования опухолевых клеток путем комбинации с ионизирующей радиацией или химиотерапевтическими агентами. Настоящее изобретение также предоставляет применение соединения формулы (I) в производстве лекарственного средства для применения в качестве дополнения в терапии рака или для потенцирования опухолевых клеток путем комбинации с ионизирующей радиацией и другими химиотерапевтическими агентами. Соединения также могут применяться в комбинации с ионизирующей радиацией и другими химиотерапевтическими агентами. Настоящее изобретение также предоставляет способ химиотерапии или радиотерапии, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или композиции, содержащей соединение формулы (I), в комбинации с ионизирующей радиацией или- 12016079 химиотерапевтическими агентами. Соединения также могут вводиться в комбинации с ионизирующей радиацией и другими химиотерапевтическими агентами. В комбинационной терапии соединения данного изобретения могут вводиться до (например, за 5 мин, 15 мин, 30 мин, 45 мин, 1 ч, 2 ч, 4 ч, 6 ч, 12 ч, 24 ч, 48 ч, 72 ч, 96 ч, 1 неделю, 2 недели, 3 недели,4 недели, 5 недель, 6 недель, 8 недель или 12 недель), параллельно с или последовательно после (например, спустя 5 мин, 15 мин, 30 мин, 45 мин, 1 ч, 2 ч, 4 ч, 6 ч, 12 ч, 24 ч, 48 ч, 72 ч, 96 ч, 1 неделю, 2 недели,3 недели, 4 недели, 5 недель, 6 недель, 8 недель или 12 недель) введения другого противоракового агента субъекту, нуждающемуся в этом. В различных вариантах осуществления рассматриваемые соединения и еще один противораковый агент вводят с промежутком в 1 мин, 10 мин, 30 мин, менее чем 1 ч, 1-2 ч,2-3 ч, 3-4 ч, 4-5 ч, 5-6 ч, 6-7 ч, 7-8 ч, 8-9 ч, 9-10 ч, 10-11 ч, 11-12 ч, не более чем 24 ч или не более чем 48 ч. Соединения данного изобретения и другой противораковый агент могут действовать дополнительно или синергически. Синергическая комбинация настоящих соединений и еще одного противоракового агента может дать возможность применения более низких доз одного или обоих данных агентов и/или менее частых доз одного или обоих рассматриваемых соединений и других противораковых агентов и/или введения агентов менее часто, что может снизить любую токсичность, связанную с введением агентов субъекту, не снижая при этом эффективность агентов при лечении рака. В дополнение, синергический эффект может приводить в результате к улучшенной эффективности данных агентов при лечении рака и/или к снижению любых пагубных или нежелательных побочных эффектов, связанных с использованием каждого агента по отдельности. Примеры противораковых агентов или химиотерапевтических агентов для применения в комбинации с соединениями настоящего изобретения можно найти в Cancer Principles and Practice of Oncology byPublishers. Специалист в данной области на основании конкретных характеристик лекарственных средств и ракового заболевания, о которых идет речь, сможет различить, какие сочетания или комбинации агентов будут полезны. Такие противораковые агенты включают, но не ограничиваются ими, следующие: ингибиторы HDAC, модуляторы эстрогеновых рецепторов, модуляторы андрогеновых рецепторов, модуляторы ретиноидных рецепторов, цитотоксические/цитостатические агенты, антипролиферативные агенты, ингибиторы пренил-протеин-трансферазы, ингибиторы HMG-СоА редуктазы, ингибиторы ВИЧ протеазы, ингибиторы обратной транскриптазы и другие ингибиторы ангиогенезиса, ингибиторы клеточной пролиферации и сигнализирующие выживание, индуцирующие апоптоз агенты и агенты, которые мешают контрольным точкам клеточного цикла. Данные соединения особенно полезны, когда вводятся совместно с радиационной терапией. Примеры "ингибиторов HDAC" включают субероиланилидгидроксамовую кислоту (SAHA),LAQ824, LBH589, PXD101, MS275, FK228, вальпроевую кислоту, масляную кислоту и CI-994."Модуляторы эстрогеновых рецепторов" относятся к соединениям, которые препятствуют или ингибируют связывание эстрогена с рецептором независимо от механизма. Примеры модуляторов эстрогеновых рецепторов включают, но не ограничиваются ими, тамоксифен, ралоксифен, идоксифен,LY353381, LY117081, торемифен, фулвестрант, 4-[7-2,2-диметил-1-оксопропокси-4-метил-2-[4-[2-(1 пиперидинил)этокси]фенил]-2 Н-1-бензопиран-3-ил]фенил-2,2-диметилпропаноат, 4,4'-дигидроксибензофенон-2,4-динитрофенилгидразон и SH646."Модуляторы андрогеновых рецепторов" относятся к соединениям, которые препятствуют или ингибируют связывание андрогенов с рецептором, независимо от механизма. Примеры модуляторов андрогеновых рецепторов включают финастерид и другие ингибиторы 5-редуктазы, нилутамид, флутамид,бикалютамиид, лиарозол и абиратеронацетат."Модуляторы ретиноидных рецепторов" относятся к соединениям, которые препятствуют или ингибируют связывание ретиноидов с рецептором, независимо от механизма. Примеры таких модуляторов ретиноидных рецепторов включают бексаротен, третиноин, 13-цис-ретиноевую кислоту,9-цис-ретиноевую кислоту, -дифторметилорнитин, ILX23-7553, транс-N-(4'-гидроксифенил)ретинамид и N-4-карбоксифенилретинамид."Цитотоксические/цитостатические агенты" относятся к соединениям, которые вызывают гибель клеток или ингибируют клеточную пролиферацию, прежде всего путем непосредственного вмешательства в функционирование клеток, или ингибируют или препятствуют митозу клеток, включающим алкилирующие агенты, факторы некроза опухоли, интеркаляторы, активируемые гипоксией соединения, ингибиторы микротрубочек/микротрубочки-стабилизирующие агенты, ингибиторы митотических кинезинов,ингибиторы киназ, вовлеченных в митотическое прогрессирование, антиметаболиты, модификаторы биологической ответной реакции; гормональные/антигормональные терапевтические агенты, гематопоэтические факторы роста, нацеленные на моноклональные антитела терапевтические агенты, ингибиторы топоизомеразы, ингибиторы протеасом и ингибиторы убиквитинлигазы. Примеры цитотоксических агентов включают, но не ограничиваются ими, циклофосфамид, хлорамбуцилкармустин (BCNU), ломустин(CCNU), бусульфан, треосульфан, сертенеф, кахектин, ифосфамид, тасонермин, лонидамин, карбопла- 13016079 тин, алтретамин, преднимустин, дибромдульцитол, ранимустин, фотемустин, недаплатин, ароплатин,оксалиплатин, темозоломид, метилметансульфонат, прокарбазин, дакарбазин, гептаплатин, эстрамустин,импросульфантозилат, трофосфамид, нимустин, диброспидийхлорид, пумитепу, лобаплатин,сатраплатин, профиромицин, цисплатин, ирофульвен, дексифосфамид, цис-аминдихлор(2-метилпиридин)платину, бензилгуанин, глуфосфамид, GPX100, тетрахлорид (транс,транс,транс)-бис-му(гексан-1,6-диамин)-му-[диаминплатина(II)]-бис-[диамин(хлор)платины(II)], диаризидинилспермин, триоксид мышьяка, 1-(11-додециламино-10-гидроксиундецил)-3,7-диметилксантин, зорубицин, идарубицин,даунорубицин, бисантрен, митоксантрон, пирарубицин, пинафид, валрубицин, амрубицин, доксорубицин,эпирубицин,пирарубицин,антинеопластон,3'-дезамино-3'-морфолино-13-дезоксо-10 гидроксикарминомицин, аннамицин, галарубицин, элинафид, MEN10755 и 4-дезметокси-3-дезамино-3 азиридинил-4-мептилсульфонилдаунорубицин (см. WO 00/50032). Дополнительные примеры включают ингибиторы Raf киназы (такие как Вау 43-9006) и ингибиторы mTOR (такие как Wyeth's CCI-779 и AriadAP23573). Дополнительными примерами являются ингибиторы PI3K (например, LY294002). В одном варианте осуществления соединения данного изобретения могут применяться в комбинации с алкилирующими агентами. Примеры алкилирующих агентов включают, но не ограничиваются ими, азотные горчицы: циклофосфамид, ифосфаид, трофорфамид и хлорамбуцил; нитрозомочевины: кармустин (BCNU) и ломустин (CCNU); алкилсульфонаты: бусульфан и треосульфан; триазены: дакарбазин, прокарбазин и темозоломид; содержащие платину комплексы: цисплатин, карбоплатин, ароплатин и оксалиплатин. В одном из вариантов осуществления алкилирующим агентом является дакарбазин. Дакарбазин может вводиться субъекту в дозах в интервале примерно от 150 мг/м 2 (площади поверхности тела субъекта) до примерно 250 мг/м 2. В еще одном варианте осуществления дакарбазин вводится внутривенно субъекту один раз в день в течение пяти последовательных дней в дозе в интервале от примерно 150 до примерно 250 мг/м 2. В одном варианте осуществления алкилирующим агентом является прокарбазин. Прокарбазин может вводиться субъекту в дозах в интервале примерно от 50 мг/м 2 (площади поверхности тела субъекта) до примерно 100 мг/м 2. В еще одном варианте осуществления прокарбазин вводится внутривенно субъекту один раз в день в течение пяти последовательных дней в дозе в интервале примерно от 50 до примерно 100 мг/м 2. В еще одном из вариантов осуществления алкилирующим агентом является темозолоамид. Темозолоамид может вводиться субъекту в дозах в интервале примерно от 150 мг/м 2 (площади поверхности тела субъекта) до примерно 200 мг/м 2. В еще одном варианте осуществления темозолоамид вводится животному орально один раз в день в течение пяти последовательных дней в дозе в интервале примерно от 150 до примерно 200 мг/м 2. Примеры антимитотических агентов включают аллоколхицин, халихондрин В, колхицин, производные колхицина, долстастин 10, мэйтанзин, ризоксин, тиоколхицин и тритилцистеин. Примером активируемого гипоксией соединением является тирапазамин. Примеры ингибиторов протеамосома включают, но не ограничиваются ими, лактацистин, бортезомиб, эпоксомицин и пептидные альдегиды, такие как MG132, MG115 и PSI. Примеры ингибиторов микротрубочек/микротрубочки-стабилизирующих агентов включают паклитаксел, сульфат виндезина, винкристин, винбластин, винорелбин, 3',4'-дидегидро-4'-дезокси-8'норвинкалеукобластин, доцетаксол, ризоксин, доластатин, изетионат мивобулина, ауристатин,цемадотин, RPR109881, BMS184476, винфлунин, криптофицин, 2,3,4,5,6-пентафтор-N-(3-фтор-4 метоксифенил)бензолсульфонамид,ангидровинбластин,N,N-диметил-L-валил-L-валил-N-метил-Lвалил-L-пролил-L-пролин-т-бутиламид, TDX258, эпотилоны (см., например, патенты США 6284781 и 6288237) и BMS188797. Некоторыми примерами ингибиторов топоизомеразы являются топотекан, гикаптамин, иринотекан,лубитекан, эксатекан, гиметекан, дифломотекан, силилкамптотецины, 9-аминокамптотецин,камптотецин, криснатол, митомицин С, 6-этоксипропионил-3',4'-О-экзобензилиденхартреузин,9-метокап-N,N-диметил-5-нитропиразоло[3,4,5-k1]акридин-2-(6H)пропанамин, 1-амино-9-этил-5-фтор 2,3-дигидро-9-гидрокси-4-метил-1H,12H-бензо[de]пирано[3',4':b,7]индолизино[1,2b]хинолин-10,13(9H,15H)дион, луртотекан, 7-[2-(N-изопропиламино)этил]-(20S)камптотецин, BNP1350, BNP11100,BN80915, BN80942, этопозидфосфат, тенипозид, собузоксан, 2'-диметиламино-2'-дезоксиэтопозид,GL331,N-[2-(диметиламино)этил]-9-гидрокси-5,6-диметил-6H-пиридо[4,3-b]карбазол-1-карбоксамид,азулакрин,(5 а,5 аВ,8 аа,9b)-9-[2-[N-[2-(диметиламино)этил]-N-метиламино]этил]-5-[4-гидрокси-3,5 диметоксифенил]-5,5 а,6,8,8 а,9-гексогидрофуро(3',4':6,7)нафто(2,3-d)-1,3-диоксол-6-он, 2,3-(метилендиоксит)-5-метил-7-гидрокси-8-метоксибензо[с]фенантридиний, 6,9-бис-[(2-аминоэтил)амино]бензо[g]изохинолин-5,10-дион, 5-(3-аминопропиламино)-7,10-дигидрокси-2-(2-гидроксиэтиламинометил)-6H-пиразоло[4,5,1-de]акридин-6-он, N-[1-[2-(диэтиламино)этиламино]-7-метокси-9-оксо-9 Н-тиоксантен-4-илметил]формамид, N-(2-(диметиламино)этил)акридин-4-карбоксамид, 6-2-(диметиламино)этил]амино]-3 гидрокси-7 Н-индено[2,1-с]хинолин-7-он и димесна; некамптотециновые ингибиторы топоизомеразы,такие как индолокарбазолы; и двойные ингибиторы топоизомеразы-I и II, такие как бензофеназины,- 14016079XR20 115761MLN 576 и бензопиридоиндолы. В одном варианте осуществления ингибитором топоизомеразы является иринотекан. Иринотекан может вводиться субъекту в дозах в интервале примерно от 50 мг/м 2 (площади поверхности тела субъекта) до примерно 150 мг/м 2. В еще одном варианте осуществления иринотекан вводится внутривенно субъекту один раз в день в течение пяти последовательных дней в дозе в интервале примерно от 50 до примерно 150 мг/м 2 в 1-5 дни,затем снова внутривенно один раз в день в течение пяти последовательных дней в 28-32 дни в дозе в интервале примерно от 50 до около 150 мг/м 2, затем снова внутривенно один раз в день в течение 5 последовательных дней в 55-59 дни в дозе в интервале примерно от 50 до около 150 мг/м 2. Примеры ингибиторов митотических кинезинов и, в частности, митотического кинезина KSP человека, описаны в публикациях заявок РСТ WO 01/30768, WO 01/98278, WO 02/056880, WO 03/050064,WO 03/050122, WO 03/049527, WO 03/049679, WO 03/049678, WO 03/039460, WO 03/079973,WO 03/099211, WO 2004/039774, WO 03/105855, WO 03/106417, WO 2004/087050, WO 2004/058700,WO 2004/058148 и WO 2004/037171 и заявках США 2004/132830 и 2004/132719. В одном варианте осуществления ингибиторы митотических кинозинов включают, но не ограничиваются ими, ингибиторы"Ингибиторы киназы, вовлеченные в митотическое прогрессирование", включают, но не ограничиваются ими, ингибиторы авроракиназы, ингибиторы Polo-подобных киназ (PLK) (в частности, ингибиторы PLK-1), ингибиторы bub-1 и ингибиторы bub-R1."Антипролиферативные агенты" включают антисмысловые РНК и ДНК олигонуклеотиды,такие как G3139, ODN698, RVASKRAS, GEM231 и INX3001, и антиметаболиты, такие как эноцитабин,кармофур, тегафур, пентостатин, доксифлуридин, триметрексат, флударабин, капецитабин, галоцитабин,цитарабинокфосфат, гидрат фостеавиннатрия, ралтитрексед, палтитрексид, эмитефур, тиазофурин,децитабин, нолатрексед, пеметрексед, нелзарабин, 2'-дезокси-2'-метиоиденцитидин, 2'-фторметилен-2'дезоксицитидин,N-[5-(2,3-дигидробензофурил)сульфонил]-N'-(3,4-дихлорфенил)мочевину,N6-[4 дезокси-4-[N2-[2(Е),4(Е)-тетрадекадиеноил]глициламино]-L-глицеро-B-L-манногептопиранозил]аденин,аплидин, эктеинасцидин, троксацитабин, 4-[2-амино-4-оксо-4,6,7,8-тетрагидро-3H-примидино[5,4b][1,4]тиазин-6-(S)-этил]-2,5-тиеноил-L-глутаминовую кислоту, аминоптерин, 5-фторурацил, аланозин,11-ацетил-8-(карбамоилоксиметил)-4-формил-6-метокси-14-окса-1,11-диазатетрацикло(7.4.1.0.0)тетрадека-2,4,6-триен-9-иловый эфир уксусной кислоты, швайнсонин, ломотрексол, дексразоксан, метиониназу,2'-циано-2'-дезокси-N4-пальмитоил-1-В-D-арабинофукранозилцитозин и 3-аминопиридин-2 карбоксиальдегидтиосемикарбазон. Примеры терапевтических агентов, нацеленных на моноклональные антитела, включают такие терапевтические агенты, которые являются цитотоксическими агентами или радоизотопами, присоединяемыми к специфичным раковым клеткам или специфичным моноклональным антителам клеток мишени. Примеры включают Bexxar."Ингибиторы HMG-CoA редуктазы" относятся к ингибиторам 3-гидрокси-3-метилглутарил-СоА редуктазы. Примеры нгибиторов HMG-СоА редуктазы, которые могут использоваться, включают, но не ограничиваются ими, ловастатин (MEVACOR; см. патенты США 4231938, 4294926 и 4319039), симвастатин (ZOCOR; см. патенты США 4444784, 4820850 и 4916239), правастатин (PRAVACHOL; см. патенты США 4346227, 4537859, 4410629, 5030447 и 5180589), флувастатин (LESCOL; см. патенты США 5354772, 4911165, 4929437, 5189164, 5118853, 5290946 и 5356896) и аторвастатин(LIPITOR; см. патенты США 5273995, 4681893, 5489691 и 5342952). Структурные формулы данных и дополнительных ингибиторов HMG-CoA редуктазы, которые могут использоваться в данных способах,описаны на с. 87 публикации M. Yalpani, "Cholesterol Lowing Drugs", ChemistryIndustry, p. 85-89(5 февраля 1996 г. и в патентах США 4782084 и 4885314. Термин ингибитор HMG-CoA редуктазы, используемый в данном описании, включает все фармацевтически приемлемые формы лактонов и открытых кислот (т.е. в которых лактоновое кольцо раскрывается с образованием свободной кислоты), а также солевые и эфирные формы соединений, которые обладают ингибирующей HMG-CoA редуктазу активностью, и, следовательно, применение таких солей,сложных эфиров, открытых-кислотных и лактоновых форм охватывается объемом данного изобретения."Ингибитор пренил-протеин-трансферазы" относится к соединению, которое ингибирует любой один фермент или любое сочетание ферментов пренил-протеин-трансферазы, включая фарнезилпротеин-трансферазу (FPT-аза), геранилгеранил-протеин-трансферазу типа I (GGPT-аза-I) и геранилгеранил-протеин-трансферазу типа II (GGPT-аза-II, называемая также Rab GGPT-азой).- 15016079 Примеры ингибиторов пренил-протеин-трансферазы можно найти в следующих публикациях и патентах: WO 96/30343, WO 97/18813, WO 97/21701, WO 97/23478, WO 97/38665, WO 98/28980,WO 98/29119, WO 95/32987, патенты США 5420245, 5523430, 5532359, 5510510, 5589485, 5602098,европейские патентные публикации 0618221, 0675112, 0604181, 0696593, WO 94/19357, WO 95/08542,WO 95/11917, WO 95/12612, WO 95/12572, WO 95/10514, патент США 5661152, WO 95/10515,WO 95/10516, WO 95/24612, WO 95/34535, WO 95/25086, WO 96/05529, WO 96/06138, WO 96/06193,WO 96/16443, WO 96/21701, WO 96/21456, WO 96/22278, WO 96/24611, WO 96/24612, WO 96/05168,WO 96/05169, WO 96/00736, патент США 5571792, WO 96/17861, WO 96/33159, WO 96/34850,WO 96/34851, WO 96/30017, WO 96/30018, WO 96/30362, WO 96/30363, WO 96/31111, WO 96/31477,WO 96/31478, WO 96/31501, WO 97/00252, WO 97/03047, WO 97/03050, WO 97/04785, WO 97/02920,WO 97/17070, WO 97/23478, WO 97/26246, WO 97/30053, WO 97/44350, WO 98/02436 и патенте США 5532359. Например, что касается роли ингибитора пренил-протеин-трансферазы на ангиогенез, см.European J. Of Cancer (1999), 35(9), 1394-1401. Термин "ингибиторы ангиогенеза" относится к соединениям, которые ингибируют образование новых кровеносных сосудов независимо от механизма. Примеры ингибиторов ангиогенеза включают, но не ограничиваются ими, ингибиторы тирозинкиназы, такие как ингибиторы рецепторов тирозинкиназы Flt-1(VEGFR1) и Flk-1/KDR (VEGFR2), ингибиторы факторов роста эпидермального происхождения, фибробластного или тромбоцитного происхождения, ингибиторы ММР (ингибиторы матриксной металлопротеазы), блокаторы интегрина, интерферон-, интерлейкин-12, пентозанполисульфат, ингибиторы циклооксигеназы, включая нестероидные противовоспалительные средства (NSAID), такие как аспирин и ибупрофен, а также селективные ингибиторы циклооксигеназы-2, такие как целекоксиб и рофекоксибPharmacol. (1997), 75:105; Cancer Res. (1997), 57:1625 (1997); Cell (1998), 93:705; Intl. Jj. Mol. Med. (1998),2:715; J. Biol. Chem. (1999), 274:9116, стероидные противовоспалительные средства (такие как кортикостероиды, минералокортикоиды, дексаметазон, преднизон, преднизолон, метилпред, бетаметазон), карбоксиамидотриазол, комбрестатин а-4, скваламин, 6-O-хлорацетилкарбонил)фумагиллол, талидомид,ангиостатин, тропонин-1, антагонисты ангиотензина II (см. J. Lab. Clin. Med. (1985), 105:141-145) и антитела к VEGF (см. Nature Biothechnology (1999), 17:963-968; Kim et al. (1993), Nature. 362:841-844WO 00/44777; и WO 00/61186). Другие терапевтические агенты, которые модулируют или ингибируют ангиогенез и также могут применяться в комбинации с соединениями данного изобретения, включают агенты, которые модулируют или ингибируют коагуляцию и системы фибринолиза (см. обзор в Clin. Chem. La. Med. (2000), 38:679692). Примеры таких агентов, которые модулируют или ингибируют коагуляцию и пути фибринолиза,включают, но не ограничиваются ими, гепарин (см. Thromb. Haemost. (1998), 80:10-23), низкомолекулярные гепарины и ингибиторы карбоксипептидазы U (известные также как ингибиторы активируемого активным тромбином ингибитора фибринолиза [TAFIa]) (см. Thrombosis Res. (2001), 101:329-354). Ингибиторы TAFIa описаны в публикации РСТ WO 03/013526 и заявке США 60/349925 (поданной 18 января 2002 г.)."Агенты, которые мешают или препятствуют контрольным точкам клеточного цикла", относятся к соединениям, которые ингибируют протеинкиназы, которые преобразовывают сигналы контрольной точки клеточного цикла, сенсибилизируя, тем самым, раковые клетки к ДНК повреждающим агентам. Такие агенты включают ингибиторы ATR, ATM, Chkl и Chk2 киназы и ингибиторы cdk и cdc киназы, и конкретными примерами их являются 7-гидроксистауроспорин, стауроспорин, флавопиридол, CYC202"Ингибиторы клеточной пролицерации и сигнального пути выживания" относятся к фармацевтическим агентам, которые ингибируют рецепторы клеточной поверхности и каскады сигнальной трансдукции вниз по потоку рецепторов поверхности. Такие агенты включают ингибиторы ингибиторов EGFR(например, гефитиниб и эрлотиниб), ингибиторы ERB-2 (например, трастузумаб), ингибиторы IGFR (например, ингибиторы, описанные в WO 03/059951), ингибиторы цитокиновых рецепторов, ингибиторы МЕТ, ингибиторы P13K (например, LY294002), серин/треонинкиназы (включая, но не ограничиваясь ими, ингибиторы Akt, такие как описано в WO 03/086404, WO 03/086403, WO 03/086394, WO 03/086279,WO 02/083675, WO 02/083139, WO 02/083140 и WO 02/083138), ингибиторы Raf киназы (например,BAY-43-9006), ингибиторы MEK (например, CI-1040 и PD-098059) и ингибиторы mTOR (например,Wyeth CCI-779 и Ariad AP23573). Такие агенты включают ингибирующие соединения с малой молекулой и антагонисты антител."Индуцирующие апоптоз агенты" включают активаторы членов семейства TNF рецепторов (включая TRAIL рецепторы). В одном варианте осуществления соединения настоящего изобретения полезны при лечении рака в комбинации с одним или более, в частности одним, двумя или тремя, агентами, выбранными из темозоломида, цисплатина, карбоплатина, оксалиплатина, иринотекано и топотекана.- 16016079 Соединения данного изобретения могут быть полезны при лечении рака в комбинации с любым одним или несколькими следующими терапевтическими агентами: абареликс (Plenaxis depot); альдеслейкин (Prokine); альдеслейкин (Proleukin); алемтозумаб (Campath); алитретиноин (Panretin); аллопуринол (Zyloprim); альтретамин (Hexalen); амифостин (Ethyol); анастрозол (Arimidex); триоксид мышьяка (Trisenox); аспарагиназа (Elspar); азацитидин (Vidaza); бевакузимаб (Avastin); бексаротен в капсулах (Targretin); бексаротен гель (Targretin); блеомицин (Blenoxame); бортезомиб(Tasigna) и дасатиниб (Sprycel). Изобретение охватывает также комбинации с NSAID, которые являются селективными ингибиторами СОХ-2. Для целей данного изобретения NSAID, которые являются селективными ингибиторами СОХ-2, определяются как NSAID, которые обладают специфичностью в отношении ингибирования СОХ-2 по сравнению с СОХ-1 по меньшей мере 100-кратной, измеренной по степени IC50 в отношении СОХ-2 по сравнению с IC50 в отношении СОХ-1, оцененной с помощью клеточного или микросомного анализа. Такие соединения включают, но не ограничиваются ими, соединения, раскрытые в патентах США 5474995, 5861419, 6001843, 6020343, 5409944, 5436265, 5536752, 5550142, 5604260, 5698584,5710140, WO 94/15932, 5344991, 5134142, 5380738, 5393790, 5466823, 5633272 и 5932598, все из которых включены в данное описание посредством ссылки. Ингибиторами СОХ-2, которые особенно полезны в данном способе лечения, являются 5-хлор-3-(4 метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридин или его фармацевтически приемлемая соль. Соединения, которые описаны в качестве специфичных ингибиторов СОХ-2 и, следовательно, полезны в настоящем изобретении, включают, но не ограничиваются ими, парекоксиб, CELEBREX иBEXTRA или его фармацевтически приемлемую соль.- 17016079 Другие примеры ингибиторов ангиогенеэа включают, но не ограничиваются ими, эндостатин,украин, ранпимазу, IM862, 5-метокси-4-[2-метил-3-(3-метил-2-бутенил)оксиранил]-1-оксаспиро[2,5]окт 6-ил(хлорацетил)карбамат, ацетилдинаналин, 5-амино-1-3,5-дихлор-4-(4-хлорбензоил)фенил]метил]1H-1,2,3-триазол-4-карбоксамид, СМ 101, скваламин, комбретастатин, RPI4610, NX31838, сульфатированный маннопентаозфосфат, 7,7-(карбонил-бис-[имино-N-метил-4,2-пирролокарбонилимино[N-метил 4,2-пиррол]карбонилимино]-бис-(1,3-нафталиндиеульфонат) и 3-[(2,4-диметилпиррол-5-ил)метилен]-2 индолинон (SU5416). Используемый выше термин "блокаторы интегрина" относится к соединениям, которые селективно антагонизируют, ингибируют или противодействуют связыванию физиологического лиганда с v3 интегрином, с соединениями, которые селективно антагонизируют, ингибируют или противодействуют связыванию физиологического лиганда с v5 интегрином, к соединениям, которые селективно антагонизируют, ингибируют или противодействуют связыванию физиологического лиганда как с v3, так и сv5 интегрином, и к соединениям, которые антагонизируют, ингибируют или противодействуют активности конкретного интегрина(интегринов), экспрессируемых в капиллярных эндотелиальных клетках. Данный термин относится также к антагонистам v6, v8, 11, 21, 51, 61 и 64 интегринов. Данный термин относится также к антагонистам любого сочетания, v3, v5, v6, v8, 11, 21, 51,61 и 64 интегринов. Некоторые конкретные примеры ингибиторов тирозинкиназы включаютEMD121974. В одном из вариантов осуществления соединения настоящего изобретения являются полезными при лечении или предотвращении появления некроза,индуцируемого селективнымиN3-аденинметилирующими агентами, такими как MeOSO2(CH2)-лекситропсин (Me-Lex). Комбинации с соединениями, иными чем противораковые соединения, также охватываются данными способами. Например, комбинации соединений настоящего изобретения с PPAR- (т.е. PPAR-гамма) агонистами и PPAR- (т.е. PPAR-дельта) агонистами полезны при лечении некоторых злокачественных опухолей. PPAR- и PPAR- являются пероксисом-пролифератор-активируемыми рецепторамии . Об экспрессии PPAR- в эндотелиальных клетках и его вовлечении в ангиогенез сообщалось в литературеVis. Sci. (2000), 41:2309-2317). Несколько позднее было показано, что PPAR- ингибирует ангиогенную ответную реакцию на VEGF in vitro и троглитазон, и росиглитазонмалеат ингибирует развитие ретинальной неоваскуляризации у мышей. (Ach. Ophthalmol. (2001), 119:709-717). Примеры PPAR- агонистов иPPAR-/ агонистов включают, но не ограничиваются ими, тиазолидиндионы (такие как DRF2725,CS-011, троглитазон, росиглитазон и пиоглитазон), фенофибрат, гемфиброзил, клофибрат, GW2570,SB219994, AR-Н 039242, JTT-501, МСС-555, GW2331, GW409544, NN2344, KRP297, NP0110, DRF4158,NN622, GI262570, PNU182716, DRF552926, 2-[(5,7-дипропил-3-трифторметил-1,2-бензизоксазол-6 ил)окси]-2-метилпропионовую кислоту (описана в USSN 09/782856) и 2(R)-7-(3-(2-хлор-4-(4 фторфенокси)фенокси)пропокси)-2-этилхроман-2-карбоновую кислоту (описана в USSN 60/235708 и 60/244697). Еще одним вариантом осуществления данного изобретения является применение описанных в настоящем изобретении соединений в комбинации с противовирусными агентами (такими как аналоги нуклеозида, включающие ганцикловир, для лечения рака. См. WO 98/04290. Еще одним вариантом осуществления данного изобретения является применение описанных в настоящем изобретении соединений в комбинации с генной терапией для лечения рака. Что касается обзора генетических методов подхода к лечению рака, см. Hall et al. (Am. J. Hum. Genet. (1997), 61:787-789) иKufe et al. (Cancer Medicine, 5th Ed., p. 876-889, ВС Decker, Hamilton 2000). Генная терапия может использоваться для доставки любого подавляющего опухоль гена. Примеры таких генов включают, но не ограничиваются ими, р 53, который может доставляться с помощью переноса гена, опосредуемого рекомбинантным вирусом (см., например, патент США 6069134), uPA/uPAR антагонист ("Adenovirus-MediatedDelivery of a uPA/uPAR Antagonist Angiogenesis-Dependent Tumor Growth and Dissemination in Mice",Gene Therapy, August (1998), 5(8):1105-13), и интерферон гамма (J. Immunol. (2000), 164:217-222). Соединения данного изобретения также могут вводиться в комбинации с ингибитором врожденной или свойственной множественной лекарственной устойчивости (MDR), в частности MDR, ассоциированного с высокими уровнями экспрессии белков переносчиков. Такие MDR ингибиторы включают ингибиторы р-гликопротеина (Р-gp), такие как LY335979, XR9576, ОС 144-093, R101922, VX853, верапамил и PSC833 (валсподар). Соединение настоящего изобретения может применяться в комбинации с противорвотными агентами для лечения тошноты или рвоты, включая острую задержанную, в поздней фазе и упреждающую рвоту, которая может быть результатом применения соединения настоящего изобретения, как такового или с радиационной терапией. Для предотвращения или лечения рвоты соединение настоящего изобретения может применяться в комбинации с другими противорвотными агентами, особенно антагонистами рецептора нейрокинина-1, антагонистами рецептора 5 НТ 3, такими как онданосетрон, гранисетрон, трописетрон и затисетрон, GABAB, агонистами рецепторов, такими как баклофен, кортикостериодами, такими как декадрон (дексаметазон), кеналог, аристокорт, назалид, преферид, бенекортен или другие, такие как раскрыто в патентах США 2789118, 2990401, 3048581, 3126375, 3929768, 3996359, 3928326 и 3749712,антидопаминергическим агентом, таким как фенотиазины (например, прохлорперазин, флуфеназин, тиоридазин и мезоридазин), метоклопрамидом или дронабинолом. В одном из вариантов осуществления противорвотный агент, выбранный из антагониста рецептора нейрокинина-1, антагониста рецептора 5 НТ 3 и кортикостероида, вводят в качестве адъюванта для лечения или предотвращения рвоты, которая может быть результатом введения рассматриваемых соединений. Антагонисты рецептора нейрокинина-1 для применения в комбинации с соединениями настоящего изобретения полностью описаны, например, в патентах США 5162339, 5232929, 5942930, 5373003,5387595, 5459270, 5494926, 5496833, 5637699, 5719147; европейских патентных публикациях ЕР 0360390, 0394989, 0428434, 0429366, 0430771, 0436334, 0443132, 0482539, 0498069, 0499313, 0512901,0512902, 0514273, 0514274, 0514275, 0514276, 0515681, 0517589, 0520555, 0522808, 0528495, 0532456,0533280, 0536817, 0545478, 0558156, 0577394, 0585913,0590152, 0599538, 0610793, 0634402, 0686629,0693489, 0694535, 0699655, 0699674, 0707006, 0708101, 0709375, 0709376, 0714891, 0723959, 0733632 и 0776893; международных патентных публикациях РСТ WO 90/05525, 90/05729, 91/09844, 91/18899,92/01688, 92/06079, 92/12151, 92/15585, 92/17449, 92/20661, 92/20676, 92/21677, 92/22569, 93/00330,93/00331, 93/01159, 93/01165, 93/01169, 93/01170, 93/06099, 93/09116, 93/10073, 93/14084, 93/14113,93/18023, 93/19064, 93/21155, 93/21181, 93/23380, 93/24465, 94/00440, 94/01402, 94/02461, 94/02595,94/03429, 94/03445, 94/04494, 94/04496, 94/05625, 94/07843, 94/08997, 94/10165, 94/10167, 94/10168,94/10170, 94/11368, 94/13639, 94/13663, 94/14767, 94/15903, 94/19320, 94/19323, 94/20500, 94/26735,94/26740, 94/29309, 95/02595, 95/04040, 95/04042, 95/06645, 95/07886, 95/07908, 95/08549, 95/11880,95/14017, 95/15311, 95/16679, 95/17382, 95/18124, 95/18129, 95/19344, 95/20575, 95/21819, 95/22525,95/23798, 95/26338, 95/28418, 95/30674, 95/30687, 95/33744, 96/05181, 96/05193, 96/05203, 96/06094,96/07649, 96/10562, 96/16939, 96/18643, 96/20197, 96/21661, 96/29304, 96/29317, 96/29326, 96/29328,96/31214, 96/32385, 96/37489, 97/01553, 97/01554, 97/03066, 97/08144, 97/14671, 97/17362, 97/18206,97/19084, 97/19942 и 97/21702 и в британских патентных публикациях 2266529, 2268931, 2269170,2269590, 2271774, 2292144, 2293168, 2293169 и 2302689. Получение таких соединений полностью описано в приведенных выше патентах и публикациях, которые включены в данное описание посредством ссылки. В одном из вариантов осуществления антагонист рецептора нейрокинина-1 для применения в комбинации с соединениями настоящего изобретения выбирают из 2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-4-фтофенил)-4-(3-(5-оксо-1 Н,4H-1,2,4-триазоло)метил)морфолина или его фармацевтически приемлемой соли, который описан в патенте США 5719147. Соединение данного изобретения также может вводиться с агентом, применяемым при лечении анемии. Таким агентом лечения анемии является, например, активатор рецептора непрерывного эйтропоэза (такой как эпоетин альфа). Соединение данного изобретения также может вводиться с агентом, применяемым при лечении нейтропении. Таким агентом лечения нейтропении является, например, гематопоэтический фактор роста,который регулирует продуцирование и функцию нейтрофилов, такой как фактор, стимулирующий гранулоцитную колонию человека, (G-CSF). Примеры G-CSF включают филграстим. Соединение данного изобретения также может вводиться с усиливающим иммунологию лекарственным средством, таким как левамизол, изопринозин и задаксин. Соединение данного изобретения также может быть полезным при лечении или предотвращении рака, включая рак костей, в комбинации с бисфосфонатами (следует понимать, что они включают бисфосфонаты, дифосфонаты, бисфосфоновые кислоты и дифосфоновые кислоты). Примеры бисфосфонатов включают, но не ограничиваются ими, этидронат (дидронел), памидронат (аредия), алендронат (фосамакс), ризедронат (актонель), золедронат (зомета), ибандронат (бонива), инкадронат или цимадронат,клодронат, ЕВ-1053, минодронат, неридронат, пиридронат и тилудронат, включая любые и все их фар- 19016079 мацевтически приемлемые соли, производные, гидраты и их смеси. Таким образом, объем данного изобретения охватывает применение соединений настоящего изобретения в комбинации с ионизирующими радиацию и/или в комбинации с еще одним соединением, выбранным из ингибиторов HDAC, модуляторов эстрогенового рецептора, модулятора андрогенового рецептора, модулятора ретиноидного рецептора, циктотоксического/цитостатического агента, антипролиферативного агента, ингибитора пренил-протеин-трансферазы, ингибитора HMG-СоА редуктазы, ингибитора ангиогенеза, агониста PPAR-, агониста PPAR- и противовирусного агента, ингибитора свойственной устойчивости к множественному лекарственному средству, противорвотного агента, агента, применяемого при лечении анемии, агента, применяемого при лечении нейтропении, усиливающего иммунологию лекарственного средства, ингибитора клеточной пролиферации и сигнализации выживания,агента, который препятствует контрольной точке клеточного цикла, индуцирующего апоптоз агента и бисфосфоната. Термин "введение" и его варианты (например, "введение" соединения) применительно к соединению изобретения означает введение соединения или пролекарства данного соединения в систему животного, нуждающегося в таком лечении. Когда соединение изобретения или его пролекарство предоставляется в комбинации с одним или несколькими другими активными агентами (например, цитотоксическим агентом и пр.), следует понимать, что каждый из терминов "введение" и его варианты включают параллельное или последовательное введение соединения или его пролекарства и других агентов. Имеется в виду, что используемый в данном описании термин "композиция" охватывает продукт,содержащий конкретно указанные ингредиенты в оговоренных количествах, а также любой продукт, который является результатом, прямо или косвенно, комбинации указанных ингредиентов в указанных количествах. Термин "терапевтически эффективное количество", используемый в данном описании, означает,что количество активного соединения или фармацевтического агента, которое вызывает биологическую или медицинскую ответную реакцию в ткани, системе, животном или человеке, которое подобрано исследователем, ветеринаром, медицинским врачом или другим клиницистом. Термин "лечение" относится к лечению млекопитающего, страдающего от патологического состояния, и относится к действию, которое облегчает данное состояние путем уничтожения раковых клеток, а также действие, которое приводит в результате к ингибированию прогрессирования данного состояния, и включает снижение скорости прогресса, остановку прогрессирования, улучшение состояния и излечение от патологического состояния. В изобретение также включено лечение в качестве профилактической меры (т.е. профилактика). Термин "фармацевтически приемлемый", используемый в данном описании, касается соединений,материалов, композиций и/или дозированных форм, которые являются с учетом суждений медиков подходящими для применения в контакте с тканями субъекта (например, человека) без избыточной токсичности, раздражения, аллергической ответной реакции или других проблем или осложнений, соразмеримыми с разумным соотношением польза/риск. Каждый носитель, эксципиенты и пр. также должны быть"приемлемыми" в смысле совместимости с другими ингредиентами рецептурной формы. Термин "дополнительный или дополнение" относится к применению соединений в комбинации с известными терапевтическими средствами. Такие средства включают цитотоксические режимы лекарственных средств и/или ионизирующую радиацию, используемые при лечении различных типов рака. В частности, известно, что активные соединения потенцируют действия ряда химиотерапевтических средств для лечения рака, которые включают класс ядов топоизомеразы (например, топотекан, иринотекан, рубитекан), большинство известных алкилирующих агентов (например, DTIC, темозоламид) и лекарственные средства на основе платины (например, карбоплатин, цисплатин), используемых в лечении рака. В объем пунктов формулы изобретения также включен способ лечения рака, который включает введение терапевтически эффективного количества соединения формулы (I) в комбинации с радиационной терапией и/или в комбинации с соединением, выбранным из ингибиторов HDAC, модулятора эстрогенового рецептора, модулятора андрогенового рецептора, модулятора ретиноидного рецептора, цитотоксического/цитостатического агента, антипролиферативного агента, ингибитора пренил-протеинтрансферазы, ингибитора HMG-CoA редуктазы, ингибитора ангиогенеза, агониста PPAR-, агонистаPPAR-, противовирусного агента, ингибитора свойственной устойчивости к множественному лекарственному средству, противорвотного агента, агента, полезного при лечении анемии, агента, полезного при лечении нейтропении, усиливающего иммунологию лекарственного средства, ингибитора клеточной пролиферации и сигнализации выживания, агента, который препятствует контрольной точке клеточного цикла, индуцирующего апоптоз агента и бисфосфоната. Данные и другие аспекты изобретения будут очевидны из описания, представленного ниже.- 20016079 Сокращения, используемые в химической части описания и в примерах, являются следующими:IST ISOLUTE SPE колонка SCX - Международная сорбентная технология ISOLUTE катионообменная смола экстракционной колонки в твердой фазе;CDCl3 - дейтерированный хлороформ; ТСХ - тонкослойная хроматография и ТФУК - трифторуксусная кислота. Соединения формулы (I) могут быть получены путем взаимодействия соединения формулы (IA) с аммиакомRx представляет C1-6 алкил, такой как метил. Реакцию обычно осуществляют с использованием водного раствора NH3 в растворителе, таком как ТГФ, примерно при 70 С, в герметизированном реакционном сосуде (с осторожностью). Альтернативно, может добавляться основание, такое как NaOH или KOH, для гидролиза сложного эфира в соответствующую карбоновую кислоту (Rx представляет водород), с последующим добавлениемNH3 в присутствии конденсирующих агентов, таких как HATU или TBTU и DIPEA в растворителе, таком как ДМФА, причем реакцию осуществляют примерно при комнатной температуре. Альтернативно, карбоновая кислота может быть активирована с образованием смешанного ангидрида, например с использованием Вос 2 О и последующим взаимодействием с бикарбонатом аммония, обычно в растворителе, таком как пиридин. Альтернативно, сложный эфир может быть преобразован в соединения формулы (IA) с использованием аммиака в растворителе, таком как МеОН, примерно при 120 С, например, при MW. Атом азота в пиперидиновом кольце соединений формулы (IA) может быть защищен во время описанного выше синтеза, например, группой Boc.- 21016079 Соединения формулы (IA) могут быть получены путем взаимодействия соединения формулы (IB) с азидом где R1, R2 и Rx имеют значения, определенные выше. Азид, такой как NaN3, обычно можно использовать в растворителе, таком как ДМФА, примерно от 90 до 140 С. Можно использовать также добавку, такую как 2,6-лутидин. Реакцию можно осуществлять в атмосфере азота. Соединения формулы (IB) могут быть получены путем конденсации соединения формулы (IC) с соединением формулы (ID)L1 представляет уходящую группу, такую как нитро или галоген, например фтор. Методики включают конденсацию в присутствии дегидратирующего агента, такого как MgSO4 или молекулярные сита, или при нагревании в спиртовом растворителе, таком как этанол, при кипячении с обратным холодильником. Реакцию можно осуществлять в атмосфере азота. Соединение формулы (IC) может быть получено окислением соединения формулы (IE) окисляющим агентом, таким как NMMOL2 представляет уходящую группу, такую как галоген, например бром,обычно в растворителе, таком как MeCN, примерно при комнатной температуре. Реакцию можно осуществлять в атмосфере азота. Соединения формулы (IE), где L2 представляет бром, могут быть получены окислением соединения формулы (IF) бромирующим агентом, таким как NBS, в присутствии инициатора радикалов, такого как бензоилпероксид где R1, Rx и L1 имеют значения, определенные выше,обычно в растворителе, таком как CCl4, при кипячении с обратным холодильником. Реакцию можно осуществлять в атмосфере азота. Соединения формулы (IF), где L1 представляет фтор, могут быть получены путем диазотирования соединения формулы (IG) где R1 и Rx имеют значения, определенные выше,с последующим разложением промежуточной диазониевой соли.- 22016079 Например, диазотирование можно осуществлять с использованием тетрафторбората нитрозония в растворителе, таком как DCM, примерно при 0 С. Соответствующую тетрафторборатную соль диазония можно затем выделить с последующим разложением при повышенных температурах на соответствующее производное фторбензола (осторожно), таким образом, как нагревание до 160 С в растворителе, таком как дихлорбензол. Соединение формулы (IF), где L1 представляет нитро, может быть получено нитрованием соединения формулы (IH) где R1 имеет значения, определенные выше,с последующей сложной этерификацией. Реакцию нитрования можно осуществлять в присутствии нитрата, такого как нитрат калия, и кислоты, такой как серная кислота, примерно при комнатной температуре. Стадию этерификации можно осуществлять в стандартных условиях, таких как взаимодействие с алкилгалогенидом формулы Rx-X, где X представляет галоген, такой как йод, в присутствии основания, такого как карбонат цезия, и в растворителе, таком как ДМФА, примерно при комнатной температуре. Спирт формулы Rx-OH можно также использовать вместе с кислотным катализатором, таким как HCl, полученным на месте из AcCl/МеОН при нагревании с обратным холодильником. Желаемое соединение формулы (IF) может быть затем получено гидрированием нитросоединения в соответствующий анилин с использованием водорода и катализатора,такого как палладий на углероде, обычно в спиртовом растворителе, таком как МеОН. Альтернативно, соединения формулы (I) могут быть получены восстановлением соединения формулы (IJ) где R1 и R2 имеют значения, определенные выше. Восстановление можно осуществлять по реакции Fowler с использованием ацилхлорида, такого какCBz-Cg, и восстанавливающего агента, такого как NaBH4. Гидрирование над палладием на углероде завершает реакцию и удаляет CBz-защитную группу. Соединения формулы (IJ) могут быть получены кросссочетанием соединения формулы (IK) с 3-пиридинилбороновой кислотой формулы (IL) где R1, R2 и L2 имеют значения, определенные выше. Реакцию обычно осуществляют в условиях реакции сочетания Сузуки, таких как использование катализаторов, таких как Pd2(dba) и три(трет-бутил)фосфина, вместе с основанием, таким как карбонат натрия, и растворителей, таких как ДМФА и вода, примерно при 90 С. Соединения формулы (IK) могут быть получены путем конденсации соединения формулы (IM) с соединением формулы (IN)L3 представляет уходящую группу, такую как галоген, например фтор,обычно в растворителе, таком как ДМФА, примерно при 180 С при MW. Также может быть добавлено основание, такое как K2CO3.- 23016079 Соединения формулы (IM) могут быть получены путем взаимодействия соединения формулы (IO) где R1 и Rx имеют значения, определенные выше,с основанием, таким как KOH или NaOH, примерно при комнатной температуре с гидролизом сложного эфира в соответствующую карбоновую кислоту (Rx представляет водород), с последующим добавлениемNH3 в присутствии агентов сочетания, таких как HATU, DIPEA и TBTU, в растворителе, таком как ДМФА, при этом реакцию осуществляют примерно при комнатной температуре. Соединения формулы (IO) могут быть получены из соединения формулы (IG) путем ацетилирования анилиновой группы такими реагентами, как ацетилхлорид, в растворителе, таком как 1,2-DCE, примерно при 55 С. Затем можно проводить циклизацию в желаемый индазол путем обработки нитритом натрия в кислоте, например концентрированной хлористо-водородной кислоте, обычно в присутствии сорастворителя, такого как толуол и вода, примерно при 0 С. Когда синтез промежуточных соединений и исходных веществ не описан, данные соединения являются коммерчески доступными или могут быть получены из коммерчески доступных соединений с помощью стандартных способов или путем использования описанных в данном описании синтезов, схем и примеров. Соединения формулы (I) могут быть преобразованы в другие соединения формулы (I) с помощью известных способов или с помощью способов, описанных выше в данном описании синтезах, схемах и примерах. Во время любых последовательностей синтеза, описанных в данном описании, может быть необходимо и/или желательно защитить восприимчивые или реакционноспособные группы в каких-либо молекулах, о которых идет речь. Это можно осуществить с помощью общепринятых защитных групп, таких как группы, описанные в Protecting Groups in Organic Synthesis, 3rd Edition, Greene, T.W. and Wuts, P.G.M.;Wiley Interscience. 1999 и Kocienski, P. J. Prortecting Groups, Thieme, 1994. Защитные группы могут быть удалены на удобной последующей стадии с использованием известных в данной области методов. Например, когда присутствует Boc (трет-бутоксикарбонильная) или бензилкарбонильная защитная группа,она может быть удалена путем добавления растворителей, таких как ТФУК, DCM и/или MeCN, примерно при комнатной температуре. Соединение также можно гидрировать с использованием стандартных методов, таких как обработка катализатором, таким как Pd/C, в растворителе, таком как метанол, в атмосфере водорода. Также можно добавлять EtOAc в присутствии HCl и 1,4-диоксан для удаления Вос или бензилкарбонильной защитной группы примерно при комнатной температуре. Соединения данного изобретения получали в соответствии со следующими схемами. Все переменные в формулах имеют значения, определенные выше. Когда соединения настоящего изобретения имеют хиральные центры, энантиомеры можно выделять из рацемических смесей стандартными методами разделения, такими как использование SFC, хиральной ВЭЖХ или расщепления хиральными кислотами. Разделение можно осуществлять на любой стадии процесса получения соединений формулы (I). Так, разделение можно осуществлять на конечной стадии или, альтернативно, можно разделить промежуточные соединения и затем отдельные конкретные энантиомеры можно использовать на последующих стадиях с получением желаемых продуктов. Схема 1. Методика синтеза производных соединений данного изобретения показана на схеме 1, в соответствии с которой замещенные 2H-индазолы получают с использованием способа синтеза, аналогично описанному в WO 2005/066136. Следующее далее первоначальное преобразование производного 2-нитрол-3 метилбензойной кислоты в соответствующий эфир, радикальное бромирование метильной группы с использованием реагентов, таких как N-бромсукцинимид и бензоилпероксид, дает ключевое производное бензилбромида. Окисление данного бензилбромида в соответствующий бензальдегид можно осуществлять, например, с использованием N-метилморфолино-N-оксида и молекулярных сит. Последующая конденсация альдегида с амином, замыкание кольца можно осуществлять путем обработки ключевого промежуточного соединения азидом натрия при повышенной температуре для введения концевого атома азота и последующего вытеснения азота, с получением индазольного кольца. В данную реакцию можно добавлять основание, такое как лутидин. Конечное преобразование эфира в первичный амид дает желаемые производные. Это можно осуществить или путем нагревания эфира в растворе аммиака, или преобразования в соответствующую карбоновую кислоту, с последующим присоединением амида. Схема 2. На схеме 2 показан вариант схемы 1, и он дает возможность ввести заместители в ядра индазола. Когда требуемые производные нитробензойной кислоты не являются коммерчески доступными, они могут быть получены путем нитрования соответствующих производных бензойной кислоты, например, с использованием нитрата в концентрированной серной кислоте. Синтетические манипуляции, описанные выше, позволяют осуществить образование соответствующего анилина, который может быть или циклизован в индазол сначала путем ацетилирования индазола и циклизации нитритом натрия в концентрированной HCl при 0 С. Альтернативно, анилин может быть диазотирован тетрафторборатом нитрозия, и соответствующую тетрафторборатную соль диазония можно разложить при повышенных температурах на соответствующее дифторбензольное производное с помощью реакции Schiemann (осторожно). Следующая последовательность синтеза, как приведено на схеме 1, дает возможность окисления метильной группы бензила в соответствующий альдегид и получения желаемых индазольных производных путем сочетания с (гетеро)анилином и циклизации азидом натрия. Схема 2- 25016079 Схема 3. Альтернативная методика включает функционализацию индазола на последней стадии, как показано на схеме 3. В данном случае сложный эфир индазола сначала преобразовывают в соответствующий карбоксамид и подвергают нуклеофильному ароматическому замещению соответствующего фтор(гетеро)ароматического бромида. Это дает возможность получить бромидное производное, которое может быть поперечно сшито в условиях сочетания Сузуки, например, с использованием три(третбутил)фосфина и Pd2(dba)3 в качестве катализаторов в присутствии основания, такого как карбонат натрия. Преобразование в желаемый пиперидиновый фрагмент затем осуществляют путем реакции Fowler с использованием ацилхлорида, такого как CBz-Cl, и восстанавливающего агента, такого как боргидрид натрия. Конечная реакция гидрирования может давать соответствующие пиперидиновые производные. Схема 3PARP-1 SPA анализ. Приведенные в качестве примеров соединения, описанные в данном описании, испытывали в данном анализе, и было обнаружено, что они имеют показатель IC50 менее 5 мкМ, в частности менее 50 нМ. Используемые реагенты. Буфер для анализа: 100 мМ Трис рН 8, 4 мМ MgCl2, 4 мМ спермина, 200 мМ KCl, 0,04% нонидет Р-40. Ферментная смесь: буфер для анализа (12,5 мкл), 100 мМ DTT (0,5 мкл), PARP-1 (5 нМ, Тревиген 4668-500-01), вода (до 35 мкл). Никотинамид-аденин динуклеотид (NAD)/ДНК смесь: [3H-NAD] (250 мкКю/мл, 0,4 мкл,Perkin-Elmer NET-443H), NAD (1,5 мМ, 0,05 мкл, SIGMA N-1511), Биотинилированный-NAD (250 мкМ,0,03 мкл, Тревиген 4670-500-01), Активированный тимус теленка (1 мг/мл, 0,05 мкл, AmershamBiosciences 27-4575), вода (до 10 мкл). Проявляющая смесь: стрептавидин SPA гранулы (5 мг/мл, Amersham Biosciences RPNQ 0007), растворенные в 500 мМ EDTA. Экспериментальная методика. Реакцию проводят на 96-луночном микропланшете с конечным объемом 50 мкл/лунка. Добавляют 5 мкл 5% раствора ДМСО/соединения, добавляют ферментную смесь (35 мкл), начинают реакцию добавлением NAD/ДНК смеси (10 мкл) и инкубируют в течение 2 ч при к.т. Останавливают реакцию добавлением проявляющей смеси (25 мкл) и инкубируют 15 мин при к.т. Измерения проводят с использованием прибора Паккард TOP COUNT. Анализ пролиферации на BRCA-1 приводимых в латентное состояние HeLa клетках. Сокращения:FCS - сыворотка плодного теленка иDMEM - модифицированная Дульбекко среда Игла. Соединения настоящего изобретения испытывали также в антипролиферативном анализе на подобранной паре BRCA-1 масс. и BRCA-1-(shPHK) HeLa клетках. Анализ показывает, что ингибиторы PARP способны проявлять селективность в отношении ингибирования роста BRCA дефицитных клеток. Соединения показали величины СС 50 менее 5 мкМ на BRCA-1 дефицитных клетках и более чем 10-кратную селективность в отношении BRCA профицитных клеток.- 26016079 Анализ основывается на способности живых клеток превращать редокс краситель (резазурин) во флуоресцентный конечный продукт (резофурин). Количество продуцируемого резофурина прямо пропорционально числу клеток. Клеточные линии.HeLa shBRCA-1-GFP - являются HeLa клетками, трансдуцированными при MOI 100 лентивирусом,содержащим shPHK против BRCA-1 и экспрессионную кассету для GFP. Латентный BRCA-1 составляет более 80% по данным оценки анализа Taqman, и клетки стабильно экспрессируют GFP.HeLa THM-GFP - являются HeLa клетками, трансдуцированными при MOI 100 с контрольным вектором, не экспрессирующим какую-либо shPHK. Протокол. Засеивают 300 клеток/лунка на 96-луночный видимый на просвет черный планшет в 90 мкл культуральной среды. Добавляют 10 мкл/лунка 10 соединения (5% ДМСО в воде). Инкубируют в течение 168 ч при 37 С, 5% СО 2. Добавляют 10 мкл раствора Celltiter Blue (Promega G8081), предварительно разведенного 1:1 вPBS1. Инкубируют смесь в течение 45 мин при 37 С, 5% СО 2. Инкубируют 15 мин при к.т. в темноте. Производят считывание планшетов с использованием флуориметра экс.: 550 нм; эм.: 590 нм. Культуральная среда: DMEM (GIBCO, 41966-029), 10% FGS (GIBCO, 10106-169), 0,1 мг/мл пенициллин-стрептомицин (GIBCO, 15140-114), 2 мМ L-глютамин (GIBCO, 3042190). Анализ пролиферации на природных BRCA дефицитных клеточных линиях. Было продемонстрировано также, что соединения настоящего изобретения ингибируют пролиферацию природных BRCA-1 (MDA-MB-436) и BRCA-2 (CAPAN-1) дефицитных клеточных линий с показателями СС 50 менее 5 мМ. Анализ пролиферации. Клетки высевают на 96-луночный планшет при 700 клеток/лунка в 100 мкл соответствующей среды на лунку. На следующий день добавляют серийно разведенное соединение в конечном объеме 200 мкл/лунка. Каждое разведение анализируют в трех повторах. Спустя 6 дней оценивают жизнеспособность клеток с использованием анализа жизнеспособности клеток Celltiter Blue в соответствии с инструкциями производителя (Promega). Считывают пластины с использованием микропланшет-ридераFusion Alfa (Packard Bioscience). Для низкопролиферирующих клеточных линий (т.е. CAPAN-1) пролиферацию анализируют через 14 дней после добавления соединений и замены среды один раз на 7 день (170 мкл среды на лунку аспирируют и заменяют 170 мкл свежей среды, содержащей соединения). Культуральная среда:CAPAN-1: IMDM (GIBCO), 20% FBS (5% СО 2). Соединения, испытанные на онкологических моделях in vivo, показали значительный уровень активности. Препаративный пример. Пример А. 2-Фенил-2H-индазол-7-карбоксамид (А 6). Стадия 1. Метил 3-метил-2-нитробензоат (А 1). К суспензии 3-метил-2-нитро-бензойной кислоты (1,0 экв,) в МеОН (0,4 М) при 0 С по каплям добавляли AcCl (3,0 экв.). Реакционную смесь перемешивали в течение 20 ч при температуре кипения с обратным холодильником. Содержание растворителя понижали в вакууме, остаток растворяли в EtOAc и несколько раз промывали насыщенным водным раствором NaHCO3, насыщенным раствором соли и сушили (Na2SO4). Выпаривание растворителя давало соединение (А 1) в виде белого твердого вещества,которое на следующей стадии использовали без дальнейшей очистки. 1 Н ЯМР (400 МГц, CDCl3, 300 К)7,86 (1 Н, д, J=7,5 Гц), 7,53-7,42 (2 Н, м), 3,89 (3 Н, с), 2,36 (3 Н, с). МС (ES) C9H9NO4 вычислено: 195, найдено: 218 (M+Na)+. Стадия 2. Метил 3-(бромметил)-2-нитробензоат (А 2). Смесь соединения (А 1) (1,0 экв.), (BzO)2 (0,4 M) и NBS (1,18 экв.) в CCl4 (0,2 М, что касается А 1) нагревали при температуре кипения с обратным холодильником в атмосфере N2 в течение 12 ч. Смесь охлаждали до комнатной температуры, разбавляли DCM, концентрировали при пониженном давлении при сухой загрузке на SiO2. Остаток очищали колоночной флэш-хроматографией на SiO2 с использованием смеси 10:90 EtOAc/петролейный эфир, с получением соединения (А 2) в виде белого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3, 300 К)7,93 (1 Н, д, J=7,7 Гц), 7,72 (1 Н, д, J=7,7 Гц), 7,57 (1H, т,J=7,7 Гц), 4,43 (2H, с), 3,88 (3 Н, с). МС (ES) C9H8BrNO4 вычислено: 273:275, найдено: 242:244 (М-МеО)+, 227:229 (M-NO2)+.- 27016079 Стадия 3. Метил 3-формил-2-нитробензоат (A3). К смеси соединения (А 2) (1,0 экв.) и 4 молекулярных сит (15 г) в MeCN (0,2 M) при комнатной температуре добавляли NMMO (2 экв.) и реакционную смесь перемешивали в течение 1,5 ч в атмосфереN2. Затем смесь разбавляли EtOAc, фильтровали и фильтрат промывали Н 2 О, 1 н. HCl, насыщенным раствором соли и сушили (Na2SO4). Выпаривание растворителя давало соединение (A3) в виде белого твердого вещества, которое использовали на следующей стадии без дальнейшей очистки. 1(1 Н, т, J=7,9 Гц), 3,93 (3 Н, с). МС (ES) C9H7NO5 вычислено: 209, найдено: 208 (М-Н)-. Стадия 4. Метил 2-нитро-3-[(фенилимино)метил]бензоат (А 4). Смесь соединения (A3) и анилина (1,05 экв.) в EtOH (0,2 M) перемешивали при температуре кипения с обратным холодильником в атмосфере N2 в течение 2 ч, до тех пор пока ТСХ не показывала завершение реакции (гексан/EtOAc=75:25). Выпаривание растворителя давало соединение (А 4) в виде белого твердого вещества, которое использовали на следующей стадии без дальнейшей очистки. 1(1 Н, т, J=7,8 Гц), 7,43 (2 Н, т, J=7,8 Гц), 7,31 (1 Н, т, J=7,3 Гц), 7,16 (2 Н, д, J=7,8 Гц), 3, 94 (3 Н, с). Стадия 5. Метил 2-фенил-2 Н-индазол-7-карбоксилат (А 5). Смесь соединения (А 4) (1,0 экв.) и NaN3 (1,05 экв.) в сухом ДМФА (0,3 М) перемешивали при 90 С в течение ночи в атмосфере N2. Неочищенную смесь уменьшали в объеме в вакууме и остаток очищали колоночной флэш-хроматографией на силикагеле с использованием градиента смеси EtOAc/петролейный эфир от 10:90 до 40:60 с получением желаемого соединения (А 5) в виде коричневого масла. 1H ЯМР (400 МГц, CDCl3, 300 К)8,50 (1 Н, с), 8,12 (1 Н, д, J=7,0 Гц), 7,96-7,90 (3 Н, м), 7,49 (2 Н, т,J=7,6 Гц), 7,38 (1 Н, т, J=7,4 Гц), 7,15 (1 Н, т, J=7,4 Гц), 4,03 (3 Н, с). МС (ES) C15H12N2O2 вычислено: 252, найдено: 253 (М+Н)+. Стадия 6. 2-Фенил-2H-индазол-7-карбоксилат (А 6). Сложный эфир (А 5) нагревали в смеси ТГФ и 32% водного раствора NH3 при 70 С в течение ночи в закупоренной трубке. Растворитель уменьшали в объеме в вакууме и остаток очищали колоночной флэш-хроматографией на силикагеле с использованием градиента смеси EtOAc/петролейный эфир от 30:70 до 50:50 с получением желаемого соединения (А 6) в виде белого твердого вещества. 1 Н ЯМР (400 МГц, ДМСО, 300 К)9,33 (1 Н, с), 8,56 (1 Н, уш.), 8,16 (2 Н, д, J=7,9 Гц), 8,08-8,00 (2 Н,м), 7,88 (1 Н, уш.), 7,63 (2 Н, т, J=7,7 Гц) 7,50 (1 Н, т, 7,4 Гц), 7,27 (1 Н, т, J=7,9 Гц). МС (ES) C14H11N3O вычислено: 237, найдено: 238 (М+Н)+. Пример 1. 3-4-[7-Аминокарбонил)-2H-индазол-2-ил]фенилпиперидийхлорид (В 4). Стадия 1. трет-Бутил 3-[4-(-[3-(метоксикарбонил)-2-нитрофенил]метиленамино)фенил]пиперидин-1-карбоксилат (В 1). Соединение (В 1) получали, следуя общей методике, представленной для стадии 4 препаративного примера А, с использованием соединения A3 и трет-бутил 3-(4-аминофенил)пиперидин-1-карбоксилата,до тех пор пока ТСХ не показывала завершение реакции (петролейный эфир:EtOAc=4:1) и использовали на следующей стадии без дальнейшей очистки. Стадия 2. Метил 2-4-[1-(трет-бутоксикарбонил)пиперидин-3-ил]фенил-2H-индазол-7-карбоксилат(В 2). Соединение (В 2) получали, следуя общей методике, представленной для стадии 5 препаративного примера А, и неочищенное вещество очищали колоночной флэш-хроматографией на силикагеле с использованием градиента смеси 20-40% EtOAc/петролейный эфир, с получением желаемого соединения(В 2) в виде желтого твердого вещества. 1(В 3). Соединение (В 2) нагревали в 7 н. NH3 в МеОН (0,1 М) в закупоренной трубке в течение 2 дней при 60 С. Растворители удаляли в вакууме и неочищенный продукт очищали растиранием с Et2O с получением желаемого соединения (В 3) в виде желтого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3, 300 К)9,04 (1 Н, уш. с), 8,51 (1 Н, с), 8,31 (1 Н, д, J=6,8 Гц), 7,91 (1 Н, д,J=8,3 Гц), 7,84 (2 Н, д, J=8,2 Гц), 7,42 (2 Н, д, J=8,2 Гц), 7,31-7,22 (1 Н, перекрыто на CDCl3 сигнал), 5,95- 28016079 Стадия 4. 3-4-[7-(Аминокарбонил)-2H-индазол-2-ил]фенилпиперидинийхлорид (В 4). К перемешиваемому раствору соединения (В 3) (1,0 экв.) в EtOAc (0,2 М) добавляли 4 н. растворHCl/1,4-диоксана (10,0 экв.) и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Растворитель выпаривали при пониженном давлении и неочищенный продукт очищали растиранием с Et2O с получением желаемого соединения (В 4) в виде желтого твердого вещества. 1 Н ЯМР (400 МГц, ДМСО-d6, 300 К)9,32 (1 Н, с), 9,12 (1 Н, уш. с), 8,87 (1 Н, уш. с), 8,55 (1 Н, уш. с),8,13 (2 Н, д, J=8,6 Гц), 8,06 (1 Н, J=7,0 Гц), 8,02 (1 Н, д, J=8,4 Гц), 7,89 (1 Н, уш. с), 7,55 (2 Н, д, J=8,6 Гц),7,27 (1 Н, дд, J=8,4, 7,0 Гц), 3,43-3,27 (2 Н, м), 3,17-3,03 (2 Н, м), 3,00-2,85 (1 Н, м), 2,00-1,70 (4 Н, м). МС (ES) C19H21ClN4O вычислено: 320, найдено: 321 (М+Н)+. Пример 2. 2-4[(3R)-Пиперидин-3-ил]фенил-2H-индазол-7-карбоксамид (С 1) и 2-4[(3S)-пиперидин-3 ил]фенил-2H-индазол-7-карбоксамид (С 2). Соединение В 4 примера 1 разделяли хиральной SFC (колонка: Chiralpak AS-H, 125 мм,поток: 10 мл/мин, Тсо 1: 35 С, Pcol: 100 бар, модификатор: 55% (iPrOH+4% Et2NH при использовании CO2 в качестве сверхкритического элюента с получением обоих чистых энантиомеров. Первый элюированный энантиомер (С 1), время удерживания (SFC): 4,80 мин, получали в виде белого порошка. 1H ЯМР (400 МГц, ДМСО-d6, 300 К)9,28 (с, 1H), 8,57 (уш. с, 1 Н), 8,06 (д, 2H, J=7,2 Гц), 8,04 (д,2H, J=8,4 Гц), 7,88 (уш. с, 1 Н), 7,49 (д, 2H, J=8,4 Гц), 7,27 (дд, 1H, J=8,4, 7,2 Гц), 3,08-2,94 (м, 2H), 2,772,67 (м, 1 Н), 2,64-2,52 (м, 1 Н), 1,98-1,90 (м, 1 Н), 1,75-1,47 (м, 4 Н). МС (ES) C19H20N4O вычислено: 320, найдено: 321 (М+Н)+. Свободное основание преобразовывали в(3R)-3-4-[7-(аминокарбонил)-2 Н-индазол-2 ил]фенилпиперидинийхлорид и измеряли вращение плоскости поляризации: []20D=+133,3 (с 0,15,МеОН). Второй элюированный энантиомер (С 2), время удерживания (SFC): 6,51 мин, получали в виде белого порошка. 1 Н ЯМР (400 МГц, ДМСО-d6, 300 К)9,28 (с, 1 Н), 8,57 (уш. с, 1 Н), 8,06 (д, 2 Н, J=7,2 Гц), 8,04 (д,2 Н, J=8,4 Гц), 7,88 (уш. с, 1 Н), 7,49 (д, 2 Н, J=8,4 Гц), 7,27 (дд, 1 Н, J=8,4, 7,2 Гц), 3,08-2,94 (м, 2 Н), 2,772,67 (м, 1 Н), 2,64-2,52 (м, 1 Н), 1,98-1,90 (м, 1 Н), 1,75-1,47 (м, 4 Н). МС (ES) C19H20N4O вычислено: 320, найдено: 321 (М+Н)+. Свободное основание преобразовывали в(3S)-3-4-[7-(аминокарбонил)-2 Н-индазол-2 ил]фенилпиперидинийхлорид и измеряли вращение плоскости поляризации: []20D=-137,9 (с 0,145, МеОН). Пример 3. Трифторацетат 3-4-[7-(аминокарбонил)-5-фтор-2H-индазол-2-ил]фенилпиперидиния (D4). Стадия 1. Метил 5-фтор-1H-индазол-7-карбоксилат (D1). К раствору соединения примера 4, Е 3 (1,0 экв.) в 1,2-дихлорэтане (0,1 М) добавляли AcCl (5 экв.) и нагревали при 55 С в течение 2 ч. Затем растворитель удаляли при пониженном давлении. Белое твердое вещество растворяли в смеси толуол/вода (5/1, 0,1 М). Раствор охлаждали до 0 С и добавляли HCl (10 экв., 37%). Затем медленно порциями добавляли NaNO2 (10 экв.) и смесь перемешивали в течение 3 ч при 0 С. Органическую фазу промывали водой (3), сушили над MgSO4 и растворитель удаляли при пониженном давлении. Желтый раствор в толуоле (0,1 M) нагревали затем в течение 2 ч при 90 С. Выпаривание толуола давало желаемый продукт в виде красного твердого вещества. 1 Н ЯМР (400 МГц, ДМСО, 300 К)13,37 (1 Н, с), 8,23 (1 Н, с), 7,63 (1 Н, дд, J=8,6, J=2,5 Гц), 7,48(1 Н, дд, J=8,6, J=2,5 Гц), 3,66 (3 Н, с). МС (ES+) C9R7FN2O2 вычислено: 194, найдено: 195 (М+Н)+. Стадия 2. 5-Фтор-1H-индазол-7-карбоксамид (D2). Соединение D1 растворяли в смеси диоксан/вода (1/1, 0,1 М) и добавляли KOH (1,5 экв.). После перемешивания в течение 12 ч при комнатной температуре растворители удаляли при пониженном давлении. Белое твердое вещество использовали в последующем сочетании без дальнейшей очистки. Карбоновую кислоту растворяли в ДМФА (0,1 М) и при 0 С добавляли TBTU (1,5 экв.). Через 15 мин добавляли DIPEA (2,0 экв.) и аммиак (3,0 экв., 0,5 М в диоксане) и смесь перемешивали 36 ч при комнатной температуре. Добавляли EtOAc и органическую фазу промывали насыщенным водным раствором NaHCO3 (3) и насыщенным раствором соли (2). Органическую фазу сушили и выпаривали при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией с использованием 1-20%MeOH/DCM с получением соединения (D2) в виде белого твердого вещества.
МПК / Метки
МПК: A61K 31/4439, C07D 401/10, A61P 35/00, A61P 29/00
Метки: амидзамещенные, parp, индазолы, ингибиторов, качестве, поли(adp-рибоза)полимеразы
Код ссылки
<a href="https://eas.patents.su/30-16079-amidzameshhennye-indazoly-v-kachestve-ingibitorov-poliadp-ribozapolimerazy-parp.html" rel="bookmark" title="База патентов Евразийского Союза">Амидзамещенные индазолы в качестве ингибиторов поли(adp-рибоза)полимеразы (parp)</a>
7-фенилалкилзамещённые 2-хинолиноны и 2-хиноксалиноны в качестве ингибиторов поли(адф-рибоза)полимеразы

Номер патента: 11211
Опубликовано: 27.02.2009
Авторы: Гийемон Жером Эмиль Жорж, Мабир Доминик Жан-Пьер, Ваутерс Вальтер Баудевейн Леопольд, Ван Дюн Якобус Альфонсус Йозефус, Сомерс Мария Викторина Франциска
МПК: A61P 43/00, A61K 31/498, C07D 241/44...
Метки: 7-фенилалкилзамещённые, качестве, поли(адф-рибоза)полимеразы, 2-хиноксалиноны, 2-хинолиноны, ингибиторов
Формула / Реферат:
1. Соединение формулы (I) его N-оксидные формы, аддитивные соли и стереохимически изомерные формы, где n равно 0, 1 или 2; X означает N или CR7, где R7 означает водород; R1 означает C1-6-алкил; R2 означает водород; R3 означает радикал, выбираемый из группы, включающей -(CH2)s-NR8R9 (а-1), -O-Н (а-2), где s означает 0 или 2; R8 означает C1-6-алкил или арилС1-6-алкил(C1-6-алкил)аминоС1-6-алкил; R9 означает водород или С1-6-алкил; либо R3 означает...
6-алкенил и 6-фенилалкил замещённые 2-хинолиноны и 2-хиноксалиноны в качестве ингибиторов поли(адф-рибоза) полимеразы

Номер патента: 9875
Опубликовано: 28.04.2008
Авторы: Гийемон Жером Эмиль Жорж, Сомерс Мария Викторина Франциска, Мабир Доминик Жан-Пьер, Ваутерс Вальтер Баудевейн Леопольд, Ван Дюн Якобус Альфонсус Йозефус
МПК: A61K 31/498, A61K 31/4704, A61P 1/04...
Метки: качестве, поли(адф-рибоза, 6-алкенил, замещённые, 2-хинолиноны, полимеразы, 6-фенилалкил, 2-хиноксалиноны, ингибиторов
Формула / Реферат:
1. Соединение формулы (I) его N-оксидные формы, соли присоединения и стереохимически изомерные формы, где n равно 0, 1 или 2; X является N или CR7, где R7 является водородом или взятый вместе с R1 может образовывать двухвалентный радикал формулы -СН=СН-СН=СН-; R1 является C1-6алкилом или тиофенилом; R2 является водородом, гидрокси, C1-6алкилом, С3-6алкинилом или взятый вместе с R3 может образовывать =O; R3 является радикалом, выбранным из...
6-замещённые циклогексилалкил 2-замещённые хинолиноны и 2-хиноксалиноны в качестве ингибиторов поли (adp-рибоза) полимеразы

Номер патента: 10592
Опубликовано: 30.10.2008
Авторы: Ван Дюн Якобус Альфонсус Йозефус, Сомерс Мария Викторина Франциска, Ваутерс Вальтер Баудевейн Леопольд, Мабир Доминик Жан-Пьер
МПК: C07D 241/44, C07D 401/06
Метки: ингибиторов, циклогексилалкил, поли, adp-рибоза, 6-замещённые, хинолиноны, полимеразы, 2-хиноксалиноны, 2-замещённые, качестве
Формула / Реферат:
1. Соединение формулы (I) его N-оксидные формы, аддитивные соли и стереохимические изомерные формы, в котором n представляет собой 0 или 1; s представляет собой 0 или 1; X представляет собой -N= или -СН=; Y представляет собой -N< или -СН<; Q представляет собой -NH-, -O-, -С(О)-, -СН2-СН2- или -CHR5-, где R5 является водородом, гидрокси, C1-6алкилом, арилС1-6алкилом, C1-6алкилоксикарбонилом, C1-6 алкилоксиC1-6алкиламино или...
6-замещённые 2-хинолиноны и 2-хиноксалиноны как ингибиторы поли(адф-рибоза)полимеразы

Номер патента: 10488
Опубликовано: 30.10.2008
Авторы: Ваутерс Вальтер Баудевейн Леопольд, Гийемон Жером Эмиль Жорж, Сомерс Мария Викторина Франциска, Ван Дюн Якобус Альфонсус Йозефус, Мабир Доминик Жан-Пьер
МПК: A61K 31/498, C07D 241/44, A61P 43/00...
Метки: 2-хиноксалиноны, ингибиторы, поли(адф-рибоза)полимеразы, 6-замещённые, 2-хинолиноны
Формула / Реферат:
1. Соединения формулы (I) их N-оксидные формы, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где n равно 0 или 1; X представляет собой N или CR5, где R5 означает водород; R1 представляет собой C1-6-алкил; R2 представляет собой водород или гидроксигруппу или, взятый вместе с R3 или R4, может образовывать =0; R3 представляет собой радикал, выбранный из (а-1), (а-2) или (а-3) или группу формулы (b-1), то есть -Z-;...
Производные хиназолинона в качестве ингибиторов parp

Номер патента: 12837
Опубликовано: 30.12.2009
Авторы: Ван Дюн Якобус Альфонсус Йозефус, Сомерс Мария Викторина Франциска, Мертенс Йозефус Каролус, Ваутерс Вальтер Баудевейн Леопольд, Гийемон Жером Эмиль Жорж, Кенни Людо Эдмон Жозефин
МПК: A61K 31/517, A61P 25/00, C07D 239/91...
Метки: качестве, производные, ингибиторов, хиназолинона
Формула / Реферат:
1. Соединение формулы (I) его N-оксиды, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где пунктирные линии означают необязательные связи; X является >N- или >СН-; является -N-C(O)- или -N=CR4-, где R4 является гидрокси; L является прямой связью или двухвалентным радикалом, выбранным из -С(О)-, -C(O)-NH-, -NH-, -С(О)-O-С1-6алкандиила- или -C1-6алкандиила-; R1 является водородом, галогеном,...
Предыдущий патент: Осветительное оборудование на светоизлучающих диодах
Следующий патент: Способ получения муки амарантовой сортовой экструдированной
Случайный патент: Матричная таблетка, позволяющая пролонгировать выход гликлазида после орального приема