Трициклические азотсодержащие соединения в качестве антибактериальных агентов
Номер патента: 15821
Опубликовано: 30.12.2011
Авторы: Хеннесси Алан Джозеф, Джордано Илариа, Дэвис Дэвид Томас, Дэвис Дэвид Эван, Пирсон Нейл Дэвид





























Формула / Реферат
1. Соединение формулы (I) или его фармацевтически приемлемая соль и/или N-оксид

в которой Z1 и Z2независимо выбраны из CH и N;
R1a и R1b независимо выбраны из водорода; галогена; циано; (C1-6)алкила; (C1-6)алкилтио; трифторметила; трифторметокси; карбокси; гидрокси, в случае необходимости замещенного (C1-6)алкилом или (C1-6)алкоксизамещенным (C1-6)алкилом; (C1-6)алкоксизамещенного (C1-6)алкила; гидрокси(C1-6)алкила; аминогруппы, в случае необходимости N-замещенной одним или двумя (C1-6)алкильными, формильными, (C1-6)алкилкарбонильными или (C1-6)алкилсульфонильными группами; и аминокарбонила, в котором аминогруппа в случае необходимости замещена (C1-4)алкилом; при условии, что R1a обозначает Н, когда Z2обозначает N, и R1b обозначает Н, когда Z1 обозначает N;
R2 обозначает водород или (C1-4)алкил или вместе с R6образует Y, как определено ниже;
А обозначает группу (i)

в которой R3 имеет значения, определенные для R1a и R1b, или обозначает оксо;
n=1 или 2; или
А обозначает группу (ii)

в которой W1, W2и W3 обозначают CR4R8или W2 и W3обозначают CR4R8, W1обозначает связь между W3и N;
X обозначает О, CR4R8 или NR6;
один R4имеет значения, определенные для R1a и R1b, a остальные и R8обозначают водород или один R4и R8 вместе обозначают оксо, а остальные обозначают водород;
R6 обозначает водород или (C1-6)алкил или вместе с R2образует Y;
R7 обозначает водород; галоген; гидрокси, в случае необходимости замещенный (C1-6)алкилом; или (C1-6)алкил;
Y обозначает CR4R8CH2; CH2CR4R8; (C=O); CR4R8; CR4R8(C=O) или (C=O)CR4R8или
когда X обозначает CR4R8, R8 и R7вместе обозначают связь;
U выбран из CO и CH2 и
R5 обозначает в случае необходимости замещенную бициклическую карбоциклическую или гетероциклическую кольцевую систему (В)

содержащую до четырех гетероатомов в каждом кольце, в которой по меньшей мере одно из колец (а) и (b) является ароматическим;
X1 обозначает С или N, когда является частью ароматического кольца, или CR14, когда является частью неароматического кольца;
X2 обозначает N, NR13, О, S(O)x, СО или CR14, когда является частью ароматического или неароматического кольца, или может, кроме того, обозначать CR14R15, когда является частью неароматического кольца;
X3 и X5независимо обозначают N или С;
Y1 обозначает линкерную группу из 0-4 атомов, каждый атом которой независимо выбран из N, NR13, О, S(O)x, СО и CR14, когда является частью ароматического или неароматического кольца, или может дополнительно представлять собой CR14R15, когда является частью неароматического кольца;
Y2 обозначает линкерную группу из 2-6 атомов, причем каждый атом Y2 независимо выбран из N, NR13, О, S(O)x, CO, CR14, когда является частью ароматического или неароматического кольца, или может дополнительно представлять собой CR14R15, когда является частью неароматического кольца;
каждый из R14 и R15 независимо выбран из Н; (C1-4)алкилтио; галогена; карбокси(C1-4)алкила; (C1-4)алкила; (C1-4)алкоксикарбонила; (C1-4)алкилкарбонила; (C1-4)алкокси(C1-4)алкила; гидрокси; гидрокси (C1-4)алкила; (C1-4)алкокси; нитро; циано; карбокси; амино или аминокарбонила, в случае необходимости моно- или дизамещенных (C1-4)алкилом; или
R14 и R15 могут вместе обозначать оксо;
каждый R13 независимо обозначает Н; трифторметил; (C1-4)алкил, в случае необходимости замещенный гидрокси, (C1-6)алкокси, (C1-6)алкилтио, галоген или трифторметил; (С2-4)алкенил; (C1-4)алкоксикарбонил; (C1-4)алкилкарбонил; (C1-6)алкилсульфонил; аминокарбонил, в котором аминогруппа в случае необходимости моно- или дизамещена (C1-4)алкилом; и
каждый х независимо означает 0, 1 или 2.
2. Соединение по п.1, в котором:
(i) Z1и Z2, оба, обозначают СН;
(ii) Z1обозначает N и Z2 обозначает СН;
(iii) Z1обозначает СН и Z2 обозначает N.
3. Соединение по любому из пп.1 или 2, в котором R1aобозначает водород и R1b обозначает водород.
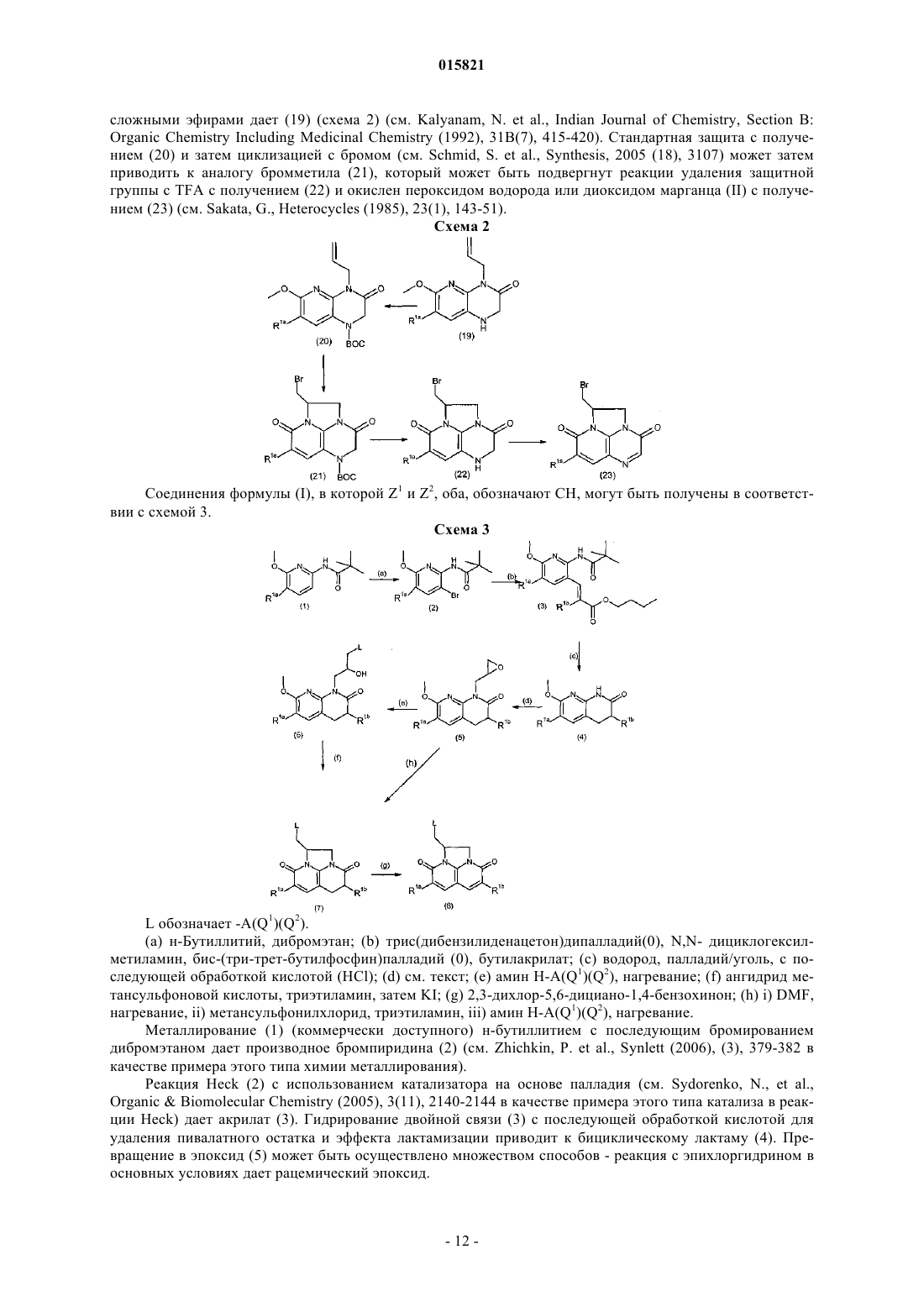
4. Соединение по любому из пп.1-3, в котором А обозначает (ia), n=1 и R3обозначает Н или гидрокси в положении 3; А обозначает (ii); X обозначает CR4R8; где R8 обозначает Н и R4обозначает Н или ОН; или А обозначает (ii); X обозначает О; R7обозначает Н и W1, W2и W3, каждый, обозначают СН2.
5. Соединение по п.4, в котором А обозначает пиперидин-4-ил или пирролидин-4-илметил.
6. Соединение по любому из пп.1-5, в котором U обозначает СН2.
7. Соединение по любому из пп.1-6, в котором R5 обозначает ароматическое гетероциклическое кольцо (В), имеющее 8-11 кольцевых атомов, включая 2-4 гетероатома, из которых по меньшей мере один представляет собой N или NR13; Y2содержит 2-3 гетероатома, один из которых представляет собой S и 1-2 представляют собой N, причем один N связан с X3, или гетероциклическое кольцо (В) имеет кольцо (а) ароматическое, выбранное из, в случае необходимости, замещенного бензо, пиридо, пиридазино и пиримидино, и кольцо (b) неароматическое; и Y2имеет 3-5 атомов, включая по меньшей мере один гетероатом, где О, S, СН2 или NR13 связан с X5, причем R13 является отличным от водорода, и либо NHCO связан через N с X3, либо О, S, СН2 или NH связаны с X3.
8. Соединение по любому из пп.1-6, в котором R5 выбран из следующих соединений:
3-оксо-3,4-дигидро-2Н-пиридо[3,2-b][1,4]оксазин-6-ил;
3-оксо-3,4-дигидро-2Н-пиридо[3,2-b][1,4]тиазин-6-ил;
2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-ил;
[1,3]оксатиоло[5,4-c]пиридин-6-ил;
6-фтор-2,3-дигидро-1,4-бензодиоксин-7-ил;
2,3-дигидро[1,4]оксатиино[2,3-с]пиридин-7-ил;
3,4-дигидро-2Н-пирано[2,3-с]пиридин-6-ил;
5-фтор-2,3-дигидро-1,4-бензодиоксин-7-ил;
5-карбонитро-2,3-дигидро-1,4-бензодиоксин-7-ил;
2,3-дигидробензо[1,4]диоксин-6-ил.
9. Соединение по п.1, выбранное из следующих соединений:
1-({4-[(2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
1-({4-[([1,3]оксатиоло[5,4-с]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
1-({4-[(2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
1-({4-[(7-бром-3-оксо-3,4-дигидро-2Н-пиридо[3,2-b][1,4]тиазин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[([1,3]оксатиоло[5,4-c]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[(2,3-дигидро[1,4]оксатиино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[(5,6,7,8-тетрагидро-3-изохинолинилметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[(6,7-дигидро-5Н-циклопента[с]пиридин-3-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[(1,3-дигидрофуро[3,4-с]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[(3,4-дигидро-2Н-пирано[2,3-с]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
7-[({1-[(4,9-диоксо-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-1-ил)метил]-4-пиперидинил}амино)метил]-2,3-дигидро-1,4-бензодиоксин-5-карбонитрил;
1-[(4-{[(3-оксо-3,4-дигидро-2Н-1,4-бензотиазин-6-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[([1,3]оксатиоло[5,4-c]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-({4-[(2,3-дигидро[1,4]оксатиино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-({4-[(2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(2R)-2-({4-[([1,3]оксатиоло[5,4-c]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(2S)-2-({4-[([1,3]оксатиоло[5,4-c]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
2-({4-[([1,3]оксатиоло[5,4-c]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(2R)-2-({4-[(2,3-дигидро[1,4]оксатиино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
2-({4-[(2,3-дигидро[1,4]оксатиино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(2S)-2-({4-[(2,3-дигидро[1,4]оксатиино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
2-({4-[(2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-({4-[(2,3-дигидро[1,4]оксатиино[2,3-с]пиридин-7-илметил)амино]-4-метил-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-метил-4-[([1,3]оксатиоло[5,4-c]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(2R)-2-({4-[(2,1,3-бензотиадиазол-5-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(2R)-2-[(4-{[(7-фтор-2,3-дигидро-1,4-бензодиоксин-6-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(2R)-2-({4-[(3,4-дигидро-2Н-[1,4]оксатиепино[2,3-с]пиридин-8-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(2R)-2-({4-[([1,3]оксатиоло[4,5-с]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(2R)-2-[(4-{[(3-оксо-3,4-дигидро-2Н-пиридо[3,2-b][1,4]тиазин-6-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-({4-[(2,3-дигидро-1,4-бензодиоксин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[(4-{[(8-фтор-2,3-дигидро-1,4-бензодиоксин-6-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[(4-{[(7-хлор-3-оксо-3,4-дигидро-2Н-пиридо[3,2-b][1,4]оксазин-6-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[(4-{[(4-хлор-7-оксо-6,7-дигидро-1Н-пиримидо[5,4-b][1,4]оксазин-2-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[(4-{[(7-оксо-6,7-дигидро-1Н-пиримидо[5,4-b][1,4]тиазин-2-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[(1,2,3-бензотиадиазол-5-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[(2,3-дигидро-1-бензофуран-5-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[(3,4-дигидро-2Н-пирано[2,3-с]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-({4-[(2,3-дигидрофуро[2,3-с]пиридин-5-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(2R)-2-({4-[(2,3-дигидро-1,4-бензодиоксин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(2R)-2-[(4-{[(8-фтор-2,3-дигидро-1,4-бензодиоксин-6-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
7-{[(1-{[(2R)-3,8-диоксо-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-2-ил]метил}-4-пиперидинил)амино]метил}-2,3-дигидро-1,4-бензодиоксин-5-карбонитрил;
(2R)-2-({4-[(2,3-дигидрофуро[2,3-с]пиридин-5-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-({4-[(2,3-дигидро-1,4-бензодиоксин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-({4-[([1,2,5]тиадиазоло[3,4-b]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[(4-{[(4-фтор-1Н-бензимидазол-2-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[((2S)-2-{[([1,3]оксатиоло[5,4-c]пиридин-6-илметил)амино]метил}-4-морфолинил)метил]-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-{[(2S)-2-({[(7-хлор-3-оксо-3,4-дигидро-2Н-пиридо[3,2-b][1,4]оксазин-6-ил)метил]амино}метил)-4-морфолинил]метил}-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(2R)-2-({4-[([1,2,5]тиадиазоло[3,4-b]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-({4-[(3,4-дигидро-2Н-хромен-7-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[(2,3-дигидро-1-бензофуран-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[(3,4-дигидро-2Н-хромен-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(2R)-2-[(4-{[(5-фтор-2,3-дигидро-1,4-бензодиоксин-6-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-{[(2R)-2-({[(7-фтор-2,3-дигидро-1,4-бензодиоксин-6-ил)метил]амино}метил)-4-морфолинил]метил}-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[((3S)-3-{[([1,3]оксатиоло[5,4-с]пиридин-6-илметил)амино]метил}-1-пирролидинил)метил]-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
7-{[(1-{[(1R)-3,8-диоксо-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-1-ил]метил}-4-пиперидинил)амино]метил}-2,3-дигидро-1,4-бензодиоксин-5-карбонитрил;
(1R)-1-[(4-{[(7-фтор-2,3-дигидро-1,4-бензодиоксин-6-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-[(4-{[(8-фтор-2,3-дигидро-1,4-бензодиоксин-6-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-[(4-{[(2-оксо-2Н-хромен-7-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[(4-{[(7-хлор-3,4-дигидро-2Н-пиридо[3,2-b][1,4]оксазин-6-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[(4-{[(7-хлор-3,4-дигидро-2Н-пиридо[3,2-b][1,4]тиазин-6-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-({4-[(3,4-дигидро-2Н-пиридо[3,2-b][1,4]тиазин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
1-[(4-{[(2,3-дигидро[1,4]оксатиино[2,3-с]пиридин-7-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
1-({4-[([1,3]оксатиоло[5,4-c]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-4Н,9Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
или свободное основание соединения из таблицы



или его фармацевтически приемлемая соль.
10. (2R)-2-({4-[(3,4-Дигидро-2Н-пирано[2,3-с]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион или его фармацевтически приемлемая соль.
11. (2R)-2-({4-[(3,4-Дигидро-2Н-пирано[2,3-с]пиридин-6-илметил)амино]-1-пиперидинил}метил)-1,2-дигидро-3Н,8H-2а,5,8а-триазааценафтилен-3,8-дион гидрохлорид.
12. Применение соединения по любому из пп.1-11 для получения лекарственного средства для применения в лечении бактериальных инфекций у млекопитающих.
13. Применение по п.12 в лечении инфекций верхних и/или нижних дыхательных путей, инфекций кожи и мягких тканей, инфекций мочевых путей и/или туберкулеза.
14. Применение соединения по п.10 или 11 для получения лекарственного средства для применения в лечении бактериальных инфекций у млекопитающих, в котором бактериальная инфекция вызвана грамположительным организмом, выбранным из Staphylococcus aureus, Streptococcus pneumoniae, Streptococcus pyogenes, Enterococcus faecalis и Enterococcus faecium, или грамотрицательным организмом, выбранным из Haemophilus influenzae, Moraxella catarrhalis, Escherichia coli, Pseudomonas aeruginosa, Proteus mirabilis, Enterobacter cloacae, Enterobacter aerogenes, Klebsiella pneumoniae и Stenotrophomonas maltophilia или Mycobacterium tuberculosis.
15. Соединение по любому из пп.1-11 для применения в терапии.
16. Соединение по любому из пп.1-11 для применения в лечении бактериальных инфекций у млекопитающих.
17. Фармацевтическая композиция, содержащая соединение по любому из пп.1-9 и фармацевтически приемлемый носитель.
18. Фармацевтическая композиция, содержащая соединение по п.10 или 11 и фармацевтически приемлемый носитель.
19. Соединение формулы (IIIA)

формулы (IIIB)

или формулы (8)

в которых L обозначает -A-N(R20)R2', где R20 обозначает водород, R2' обозначает R2 или группу, которая может превращаться в нее;
A, R2, R1a и R1b имеют значения, определенные в п.1.
20. (2R)-2-[(4-Амино-1-пиперидинил)метил]-1,2-дигидро-3Н,8Н-2а,5,8а-триазааценафтилен-3,8-дион.
Текст
Дата публикации и выдачи патента Номер заявки ТРИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ АГЕНТОВ Изобретение относится к трициклическим азотсодержащим соединениям формулы (I) Дэвис Дэвид Эван, Дэвис Дэвид Томас, Джордано Илариа, Хеннесси Алан Джозеф (GB), Пирсон Нейл Дэвид (US) Медведев В.Н. (RU) в которой Z1 и Z2 независимо выбраны из СН и N, и их применение в качестве антибактериальных агентов.(71)(73) Заявитель и патентовладелец: ГЛЭКСО ГРУП ЛИМИТЕД (GB) 015821 Это изобретение относится к новым соединениям, содержащим их композициям и их применению в качестве антибактериальных агентов, включая применение в лечении туберкулеза. В WO 02/08224, WO 02/50061, WO 02/56882, WO 02/96907, WO 2003087098, WO 2003010138,WO 2003064421, WO 2003064431, WO 2004002992, WO 2004002490, WO 2004014361, WO 2004041210,WO 2004096982, WO 2002050036, WO 2004058144, WO 2004087145, WO 2006002047, WO 2006014580,WO 2006010040, WO 2006017326, WO 2006012396, WO 2006017468, WO 2006020561, WO 2006081179,WO 2006081264, WO 2006081289, WO 2006081178, WO 2006081182, WO 01/25227, WO 02/40474,WO 02/07572, WO 2004024712, WO 2004024713, WO 2004035569, WO 2004087647, WO 2004089947,WO 2005016916, WO 2005097781, WO 2006010831, WO 2006021448, WO 2006032466, WO 2006038172,WO 2006046552, WO 06099884, WO 06126171, WO 06137485, WO 06105289, WO 06125974,WO 06134378, WO 07016610, WO 07081597, WO 07071936, WO 07115947, WO 07118130, WO 07122258,WO 08006648, WO 08003690 и WO 08009700 раскрыты производные хинолина, нафтиридина, морфолина, циклогексана, пиперидина и пиперазина, имеющие антибактериальную активность. ВWO 2004104000 раскрыты трициклические конденсированные циклические соединения, способные к селективному действию на каннабиноидные рецепторы. В WO 2003048081, WO 2003048158 иUS2003232804 раскрыты глицинамиды как ингибиторы фактора Xa. Это изобретение относится к соединению формулы (I) или к его фармацевтически приемлемой соли и/или N-оксиду в которой Z1 и Z2 независимо выбраны из сН и N;R1a и R1b независимо выбраны из водорода; галогена; циано; (C1-6)алкила; (C1-6)алкилтио; трифторметила; трифторметокси; карбокси; гидрокси, в случае необходимости замещенного (C1-6)алкилом или(C1-6)алкоксизамещенным (C1-6)алкилом; (C1-6)алкоксизамещенного (C1-6)алкила; гидрокси(C1-6)алкила; аминогруппы, в случае необходимости N-замещенной одним или двумя (C1-6)алкильными, формильными, (C1-6)алкилкарбонильными или (C1-6)алкилсульфонильными группами; и аминокарбонила, в котором аминогруппа в случае необходимости замещена (C1-4)алкилом; при условии, что R1a обозначает Н, когда Z2 обозначает N, и R1b обозначает Н, когда Z1 обозначает N;R2 обозначает водород или (C1-4)алкил или вместе с R6 образует Y, как определено ниже; А обозначает группу (i) в которой W1, W2 и W3 обозначают CR4R8 или W2 и W3 обозначают CR4R8 и W1 обозначает связь между W3 и N;X обозначает О, CR4R8 или NR6; один R4 имеет значения, определенные для R1a и R1b, a остальные и R8 обозначают водород или один R4 и R8 вместе обозначают оксо, а остальные обозначают водород;R6 обозначает водород или (C1-6)алкил или вместе с R2 образует Y;R7 обозначает водород; галоген; гидрокси, в случае необходимости замещенный (C1-6)алкилом; илиU выбран из сО и СН 2 иR5 обозначает в случае необходимости замещенную бициклическую карбоциклическую или гетероциклическую кольцевую систему (В) содержащую до четырех гетероатомов в каждом кольце,в которой по меньшей мере одно из колец (а) и (b) является ароматическим;X1 обозначает С или N, когда является частью ароматического кольца, или CR14, когда является частью неароматического кольца;X2 обозначает N, NR13, О, S(O)x, СО или CR14, когда является частью ароматического или неароматического кольца, или может, кроме того, обозначать CR14R15, когда является частью неароматического кольца;Y1 обозначает линкерную группу из 0-4 атомов, каждый атом которой независимо выбран из N,13NR , О, S(O)x, CO и CR14, когда является частью ароматического или неароматического кольца, или может дополнительно представлять собой CR14R15, когда является частью неароматического кольца;Y2 обозначает линкерную группу из 2-6 атомов, причем каждый атом Y2 независимо выбран из N,13NR , О, S(O)x, CO, CR14, когда является частью ароматического или неароматического кольца, или может дополнительно представлять собой CR14R15, когда является частью неароматического кольца; каждый из R14 и R15 независимо выбран из Н; (C1-4)алкилтио; галогена; карбокси(C1-4)алкила;R14 и R15 могут вместе обозначать оксо; каждый R13 независимо обозначает Н; трифторметил; (C1-4)алкил, в случае необходимости замещенный гидрокси, (С 1-6)алкокси, (C1-6)алкилтио, галоген или трифторметил; (С 2-4)алкенил;(C1-4)алкоксикарбонил; (C1-4)алкилкарбонил; (C1-6)алкилсульфонил; аминокарбонил, в котором аминогруппа в случае необходимости моно- или дизамещена (C1-4)алкилом; и каждый х независимо означает 0, 1 или 2. Это изобретение также относится к способу лечения бактериальных инфекций, включая туберкулез,у млекопитающих, особенно у человека, включающему введение млекопитающему эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли и/или N-оксида. Изобретение также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли и/или N-оксида, в получении лекарственного средства для применения в лечении бактериальных инфекций, включая туберкулез, у млекопитающих. Изобретение также относится к фармацевтической композиции, содержащей соединение формулы(I) или его фармацевтически приемлемую соль и/или N-оксид и фармацевтически приемлемый носитель. В одном аспекте один из Z1 и Z2 обозначает СН или N, а другой обозначает СН. В частных аспектах:iii) Z1 обозначает CH, Z2 обозначает N. В частном аспекте R1a и R1b независимо обозначают водород, (C1-4)алкокси, (C1-4)алкилтио,(C1-4)алкил, циано, карбокси, гидроксиметил или галоген; более конкретно, водород, метокси, метил,циано или галоген. В частных вариантах осуществления R1a и R1b обозначают водород. В частном аспекте R2 обозначает водород. Частные примеры R3 включают водород; в случае необходимости замещенный гидрокси; в случае необходимости замещенный амино; галоген; (C1-4)алкил; 1-гидрокси(C1-4)алкил; в случае необходимости замещенный аминокарбонил. Более конкретно, R3 обозначает водород; CONH2; 1-гидроксиалкил, например СН 2 ОН; в случае необходимости замещенный гидрокси, например метокси; в случае необходимости замещенный амино; и галоген, в частности фтор. Наиболее предпочтительно R3 обозначает водород, гидрокси или фтор. В частном аспекте, когда А обозначает (ia), n=1. В другом аспекте R3 находится положении 3 или 4. В более частном аспекте А обозначает (ia), n=1 и R3 находится в положении 3, более конкретно, в цис-положении по отношению к группе NR2. В частных вариантах осуществления А обозначает группу(ia), в которой n=1, и R3 обозначает водород или гидрокси. Более конкретно, если А обозначает 3-гидроксипиперидин-4-ил, конфигурация является конфигурацией (3R,4S) или (3S,4R). Альтернативно и более конкретно, если А обозначает пиперидин-4-ил, конфигурация является конфигурацией (3R,4S). В альтернативном более частном аспекте, когда А обозначает (ia), n=1, R3 находится в положении 4 и обозначает метил.-2 015821 В частном аспекте, когда А обозначает (ii), X обозначает CR4R8, R8 обозначает Н и R4 обозначает Н или ОН и, более конкретно, ОН находится транс-положении по отношению к R7. В другом аспекте W1 обозначает связь. В другом аспекте R7 обозначает Н. В дополнительном аспекте W1 обозначает связь, W2 и W3, оба, обозначают СН 2 и R7 обозначает Н. Если А обозначает 4-гидроксипирролидин-3-илметил, в частном аспекте конфигурация является конфигурацией (3S,4S). Если А обозначает пирролидин-3 илметил, в частном аспекте конфигурация является конфигурацией 3S. В частном аспекте, когда А обозначает (ii), X обозначает О, R7 обозначает Н и W1, W2 и W3, каждый, обозначают СН 2. В некоторых вариантах осуществления U обозначает СН 2. В некоторых вариантах осуществления R5 обозначает ароматическое гетероциклическое кольцо (В),имеющее 8-11 кольцевых атомов, включая 2-4 гетероатома, из которых по меньшей мере один представляет собой N или NR13, в котором, в частных вариантах осуществления, Y2 содержит 2-3 гетероатома,один из которых представляет собой S и 1-2 представляют собой N, причем один N связан с X3. В альтернативных вариантах осуществления гетероциклическое кольцо (В) имеет кольцо (а) ароматическое, выбранное в случае необходимости из замещенного бензо, пиридо, пиридазино и пиримидино,и кольцо (b) неароматическое, и Y2 имеет 3-5 атомов, более конкретно 4 атома, включая по меньшей мере один гетероатом, где О, S, СН 2 или NR13 связан с X5, причем R13 является отличным от водорода и либо NHCO связан через N с X3, либо О, S, СН 2 или NH связаны с X3. В частном аспекте кольцо (а) содержит ароматический азот, более конкретно, кольцо (а) представляет собой пиридин или пиразин. Примеры колец (В) включают в случае необходимости замещенные:-5 015821 В некоторых вариантах осуществления R13 обозначает Н, если находится в кольце (а), или, кроме того, (C1-4)алкил, такой как метил или изопропил, когда находится в кольце (b). Более конкретно, в кольце (b) R13 обозначает Н, когда NR13 связан с X3, и (С 1-4)алкил, когда NR13 связан с X5. В других вариантах осуществления R14 и R15 независимо выбраны из водорода, галогена, гидрокси,(C1-4)алкила, (C1-4)алкокси, нитро и циано. Более конкретно, R15 обозначает водород. Более конкретно, каждый R14 выбран из водорода, хлора, фтора, гидрокси, метила, метокси, нитро и циано. Еще более конкретно, R14 выбран из водорода, фтора или нитро. Наиболее предпочтительно R14 и R15, каждый, обозначают Н. Специфические группы R5 включают:- обозначает точку присоединения. В рамках изобретения термин "алкил" включает группы, имеющие прямые и разветвленные цепи,например метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил и гексил. Термин "алкенил" должен интерпретироваться соответственно.-8 015821 Галоген включает фтор, хлор, бром и йод. Галогеналкил включает 1-3 атома галогена. Соединения в рамках изобретения содержат гетероциклильную группу и могут встречаться в двух или более таутомерных формах в зависимости от природы гетероциклильной группы; все такие таутомерные формы входят в рамки изобретения. Некоторые из соединений по изобретению могут быть кристаллизованы или перекристаллизованы из растворителей, таких как водные и органические растворители. В таких случаях можно образовать сольваты. Это изобретение включает стехиометрические сольваты, включая гидраты, а также соединения, содержащие переменные количества воды, которые могут быть получены такими способами, как лиофилизация. Кроме того, следует понимать, что такие фразы, как "соединение формулы (I), или его фармацевтически приемлемая соль, или N-оксид", охватывают соединение формулы (I), N-оксид формулы (I), фармацевтически приемлемую соль соединения формулы (I), сольват формулы (I) или любую их фармацевтически приемлемую комбинацию. Таким образом, посредством неограничивающего примера, используемого здесь в иллюстративной цели, соединение формулы (I) или его фармацевтически приемлемая соль может включать фармацевтически приемлемую соль соединения формулы (I), которая, кроме того,присутствует в форме сольвата. Так как соединения формулы (I) предназначены для использования в фармацевтических композициях, легко понять, что в частных вариантах осуществления их используют, по существу, в чистой форме, например, с чистотой по меньшей мере 60%, более предпочтительно с чистотой по меньшей мере 75% и особенно по меньшей мере 85%, особенно с чистотой по меньшей мере 98% (% являются весовыми). Неочищенные препараты соединений могут использоваться для получения более чистых форм, используемых в фармацевтических композициях; эти менее чистые препараты соединений должны содержать по меньшей мере 1%, более предпочтительно по меньшей мере 5% и более конкретно от 10% соединения формулы (I) или его фармацевтически приемлемой соли и/или N-оксида. Специфические соединения согласно изобретению включают указанные в примерах и их фармацевтически приемлемые N-оксиды, соли и сольваты. Фармацевтически приемлемые соли вышеуказанных соединений формулы (I) включают соли присоединения с кислотой или соли четвертичного аммониевого основания, например их соли с неорганическими кислотами, например хлористо-водородной, бромисто-водородной, серной, азотной или фосфорной кислотами, или с органическими кислотами, например уксусной, фумаровой, янтарной, малеиновой,лимонной, бензойной, п-толуолсульфоновой, метансульфоновой, нафталинсульфоновой или винной кислотой. Соединения формулы (I) могут также быть получены в форме N-оксида. Изобретение распространяется на все такие производные. Некоторые из соединений формулы (I) могут существовать в форме оптических изомеров, например диастереоизомеров и смесей изомеров во всех отношениях, например рацемических смесей. Изобретение включает все такие формы, в особенности чистые изомерные формы. Например, изобретение включает энантиомеры и диастереоизомеры по месту присоединения NR2 и R3. Различные изомерные формы могут быть отделены одна от другой обычными способами или любой данный изомер может быть получен обычными способами синтеза или стереоспецифическим или асимметрическим синтезом. Некоторые соединения формулы (I) могут также существовать в полиморфных формах, и изобретение включает такие полиморфные формы. В дальнейшем аспекте изобретение относится к способу получения соединений формулы (I), где Z2 обозначает азот, и их фармацевтически приемлемых солей и/или N-оксидов, включающему введение соединения формулы (IIA) в которой L является удаляемой группой или -A(Q1) (Q2), где Q1 и Q2, оба, присоединены к одному и тому же атому углерода на А, Q1 обозначает Н, Q2 обозначает N(R20)R2' или Q1 и Q2 вместе образуют этилендиокси или оксо, R20 обозначает UR5 или группу, способную превращаться в нее, и R2' представляет собой R2 или группу, способную превращаться в нее, A, R1b, R2, U и R5 имеют значения, определенные в формуле (I),в реакцию с (i) этилбромацетатом с последующей циклизацией и окислением или с (ii) этилоксоацетатом с последующей циклизацией, получая соединение формулы (IIIA) и после этого в случае необходимости или по мере необходимости превращение L в -A-NR2-UR5, взаимное превращение любых переменных групп и/или образование фармацевтически приемлемой соли и/илиN-оксида. Вариантом реакции (i) является селективное алкилирование этилбромацетатом в основных условиях (таких как карбонат калия) (см. Yoshizawa, H. et al., Heterocycles (2004), 63(8), 1757-1763 в качестве примера этой селективности в алкилировании 2,3-диаминопиридинов), термическая циклизация в строгих основных условиях (таких как трет-бутоксид калия) и затем окисление диоксидом марганца в обычных условиях (см., например, Smith, М.В.; March, J.M. Advanced Organic Chemistry, Wiley-Interscience,2001). Вариант реакции (ii) может быть осуществлен в толуоле и циклизация может быть осуществлена в сильных основных условиях (таких как трет-бутоксид калия).L может быть гидроксильной группой, которая может быть окислена до альдегида обычными средствами, такими как 1,1,1-трис-(ацетилокси)-1,1-дигидро-1,2-бензйодоксол-3-(1 Н)-он для восстановительного алкилирования с HA-N(R20)R2' в обычных условиях (см., например, Smith, M.B.; March, J.M.Advanced Organic Chemistry, Wiley-Interscience, 2001). Альтернативно, L может быть бромом, который может алкилироваться с HA-N(R20)R2' в обычных условиях. Если Q1 и Q2 вместе образуют этилендиокси, кеталь может быть превращен в кетон (Q1 и Q2 вместе образуют оксо) путем обычного кислотного гидролиза, например, с водным раствором HCl или трифторуксусной кислотой и превращения в NR2UR5 обычным восстановительным алкилированием с аминомNHR2'R20 (см., например, Nudelman, A., et al., Tetrahedron, 60 (2004), 1731-1748) и последующим превращением в нужный замещенный амин или непосредственно с NHR2UR5, таким как триацетоксиборгидрид натрия в смеси дихлорметан/метанол. Предпочтительно один из R20 и R2' обозначает N-защитную группу, такую как третбутоксикарбонил, бензилоксикарбонил или 9-флуоренилметилоксикарбонил. Она может быть удалена несколькими способами, известными специалисту (например, см. "Protective Groups in Organic Synthesis,T.W. Greene and P.G.M. Wuts, Wiley-Interscience, 1999), например обычным кислотным гидролизом, например, с трифторуксусной или соляной кислотой. Изобретение также относится к соединениям формулы (IIIA), в которой L обозначает -A-N (R20) R2' и R20 обозначает водород. Свободный амин формулы (IIIA), в котором R20 обозначает водород, может быть превращен в 2NR UR5 обычными средствами, такими как формирование амида с производным ацила R5COW, для соединений, где U обозначает СО, или, если U обозначает СН 2, алкилированием с алкилгалогенидомR5COW или восстановительным алкилированием с альдегидом R5CHO в обычных условиях (см., например, Smith, M.B.; March, J.M. Advanced Organic Chemistry, Wiley-Interscience, 2001). Подходящие реагенты, содержащие необходимые группы R5, представляют собой известные соединения или могут быть получены аналогично известным соединениям, см., например, WO 02/08224, WO 02/50061, WO 02/56882,WO 02/96907, WO 2003087098, WO 2003010138, WO 2003064421, WO 2003064431, WO 2004002992,WO 2004002490, WO 2004014361, WO 2004041210, WO 2004096982, WO 2002050036, WO 2004058144,WO 2004087145, WO 06002047, WO 06014580, WO 06010040, WO 06017326, WO 06012396,WO 06017468, WO 06020561, WO 2004/035569, WO 2004/089947, WO 2003082835, WO 06002047,WO 06014580, WO 06010040, WO 06017326, WO 06012396, WO 06017468, WO 06020561, WO 06132739,WO 06134378, WO 06137485, WO 06081179, WO 06081264, WO 06081289, WO 06081178, WO 06081182,WO 07016610, WO 07081597, WO 07071936, WO 07115947, WO 07118130, WO 07122258, WO 08006648,WO 08003690, WO 08009700, WO 2007067511 и ЕР 0559285. Если R5 содержит группу NH, она может быть защищена подходящей защитной группой N-группы,такой как трет-бутоксикарбонил, бензилоксикарбонил или 9-флуоренилметилоксикарбонил, в ходе сочетания R5-производного со свободным амином формулы (IIB). Защитная группа может быть удалена обычными способами, такими как обработка трифторуксусной кислотой.- 10015821 В другом аспекте изобретение относится к способу получения соединений формулы (I), где Z1 обозначает азот, и их фармацевтически приемлемых солей и/или N-оксидов, включающему введение соединения формулы (IIB) в котором L является удаляемой группой или -A(Q1)(Q2), где Q1 и Q2 присоединены к одному и тому же атому углерода на A, Q1 обозначает Н, Q2 обозначает N(R20)R2' или Q1 и Q2 вместе образуют этилендиокси или оксо, R20 обозначает UR5 или группу, способную превращаться в нее, и R2' представляет собой R2 или группу, способную превращаться в нее, A, R1a, R2, U и R5 имеют значения, определенные в формуле (I), в реакцию с (i) этилбромацетатом с последующей циклизацией и окислением или с (ii) этилоксоацетатом с последующей циклизацией, получая соединение формулы (IIIB) и после этого в случае необходимости или по мере необходимости превращение L в -A-NR2-UR5, взаимное превращение любых переменных групп и/или образование фармацевтически приемлемой соли и/илиN-оксида. Реакцию и последующие превращения осуществляют, как в случае получения соединений формулы(IIIA). Изобретение также относится к соединениям формулы (IIIB), в которой L обозначает -A-N(R20)R2' и 20R обозначает водород. Соединения формулы (IIB) (L=-A(Q1)(Q2 могут быть получены в соответствии со схемой 1. Схема 1 Хлорпиридин (2) может быть введен в реакцию с аллиламином с получением (3), который может быть затем циклизован с бромобразующим пиридоном (4) после гидролитической обработки (см.Schmid, S. et al., Synthesis, 2005 (18), 3107). Смещение с H-A(Q1XQ2) дает (5), и гидрирование (5) с использованием Pd/C может дать амин (6). Соединения формулы (IIIA) могут быть получены в соответствии со схемой 2 с использованием соединений формулы (IVA) Исходный материал может быть получен реакцией соединения (3) из схемы 1 метилатом натрия и затем восстановлением хлоридом олова (II) или дитионита натрия. Циклизация (IVA) с пропиолатными- 11015821 сложными эфирами дает (19) (схема 2) (см. Kalyanam, N. et al., Indian Journal of Chemistry, Section B:Organic Chemistry Including Medicinal Chemistry (1992), 31B(7), 415-420). Стандартная защита с получением (20) и затем циклизацией с бромом (см. Schmid, S. et al., Synthesis, 2005 (18), 3107) может затем приводить к аналогу бромметила (21), который может быть подвергнут реакции удаления защитной группы с TFA с получением (22) и окислен пероксидом водорода или диоксидом марганца (II) с получением (23) (см. Sakata, G., Heterocycles (1985), 23(1), 143-51). Схема 2 Соединения формулы (I), в которой Z1 и Z2, оба, обозначают СН, могут быть получены в соответствии с схемой 3. Схема 3(а) н-Бутиллитий, дибромэтан; (b) трис(дибензилиденацетон)дипалладий(0), N,N- дициклогексилметиламин, бис-(три-трет-бутилфосфин)палладий (0), бутилакрилат; (с) водород, палладий/уголь, с последующей обработкой кислотой (HCl); (d) см. текст; (е) амин H-A(Q1)(Q2), нагревание; (f) ангидрид метансульфоновой кислоты, триэтиламин, затем KI; (g) 2,3-дихлор-5,6-дициано-1,4-бензохинон; (h) i) DMF,нагревание, ii) метансульфонилхлорид, триэтиламин, iii) амин Н-A(Q1)(Q2), нагревание. Металлирование (1) (коммерчески доступного) н-бутиллитием с последующим бромированием дибромэтаном дает производное бромпиридина (2) (см. Zhichkin, P. et al., Synlett (2006), (3), 379-382 в качестве примера этого типа химии металлирования). Реакция Heck (2) с использованием катализатора на основе палладия (см. Sydorenko, N., et al.,OrganicBiomolecular Chemistry (2005), 3(11), 2140-2144 в качестве примера этого типа катализа в реакции Heck) дает акрилат (3). Гидрирование двойной связи (3) с последующей обработкой кислотой для удаления пивалатного остатка и эффекта лактамизации приводит к бициклическому лактаму (4). Превращение в эпоксид (5) может быть осуществлено множеством способов - реакция с эпихлоргидрином в основных условиях дает рацемический эпоксид.- 12015821 Реакция с (коммерчески доступным) R- или S-глицидилнозилатом 2R)- или (2S)-2 оксиранилметил-3-нитробензолсульфонатом) или (2R)- или (2S)-2-оксиранилметил-4-метилбензолсульфонатом с основанием, например гидридом натрия или трет-бутоксидом калия, дает соответствующие хиральные эпоксиды. Альтернативно, аллилирование аллилбромидом в основных условиях дает соответствующий N-аллил, который может быть эпоксидирован в стандартных ахиральных или хиральных условиях, дает соответствующие ахиральные или хиральные эпоксиды. Эпоксид(ы) (5) может быть раскрыт с амином H-A(Q1)(Q2), таким как 1,1-диметилэтил-4-пиперидинилкарбамат, путем нагревания вDMF с получением (6), который может быть затем циклизован ангидридом метансульфоновой кислоты с получением (7). Альтернативно, эпоксид (5) может быть раскрыт и циклизован непосредственно нагреванием, с получением (7) (L=OH). Окисление до (8) может быть осуществлено путем окисления с 2,3 дихлор-5,6-дициано-1,4-бензохиноном (DDQ). Последующее превращение в соединения формулы (I) может быть выполнено, как в целом описано здесь. В частности, превращение L в A(Q1)(Q2) может быть осуществлено на (7) или (8). Как вариант схемы 3 эпоксид (5) может быть получен из (2) введением сначала подходящей группы-предшественника эпоксидной группы (-СН 2-СНОН-СН 2 ОН, защищенной как циклический эфир) с последующим осуществлением стадий (b) и (с). Изобретение также относится к соединениям формулы (8) из схемы 3, в которой L обозначает-A-N(R20)R2' и R20 обозначает водород. Соединения формулы (I), в которой Z2 обозначает N, могут альтернативно быть получены в соответствии со схемой 4. Схема 4(а) 2-Амино-1,3-пропандиол; (b) диметоксипропан, п-толуолсульфоновая кислота; (с) водород, палладий/уголь; (d) этилбромацетат, карбонат калия; (е) гидрид натрия; (f) бензил хлорформиат; (g) водный раствор кислоты; (h) ангидрид метансульфоновой кислоты; (i) водород, палладий/уголь; (j) MnO2. Реакция нитропиридина (1) с 2-амино-1,3-пропандиолом дает диол (2), который защищен как ацеталь (3). Восстановление нитрогруппы дает амин (4), который алкилируют, получая сложный эфир (5). Циклизация может быть осуществлена с использованием гидрида натрия с получением (6). Его защищают карбоксибензильной (CBz) группой (7), затем расщепляют, получая диол (8). Циклизация с ангидри- 13015821 дом метансульфоновой кислоты дает мезилат (9), затем гидрогенолиз группы CBz (10) и последующее окисление диоксидом марганца (II) дают мезилат ключевого промежуточного диона (11). Порядок стадий может быть изменен таким образом, чтобы пройти через стадию (7 а). Мезилат (11) может быть превращен в соединение формулы (I), как в целом описано здесь. Хиральные соединения формулы (I), в которой Z2 обозначает N, могут альтернативно быть получены в соответствии со схемой 4 а. Схема 4 а(a) EtOH, нагревание с обратным холодильником; (b) TBS-Cl; (с) цинк, уксусная кислота; (d) этилбромацетат, карбонат калия; (е) NaH; (f) водород, палладий/уголь; (g) MnO2; (h) ангидрид метансульфоновой кислоты (i) TFA, (j) ангидрид метансульфоновой кислоты. Реакция нитропиридина (1) с хиральным амином (2) дает промежуточное соединение (3). Защита(3) трет-бутилдиметилсилилхлоридом дает (4). Восстановление нитрогруппы дает амин (5), который алкилируют, получая сложный эфир (6). Циклизация (6) может быть осуществлена с использованием гидрида натрия, и затем обработка водородом с использованием катализатора на основе палладия на угле дает промежуточное соединение (7). Окисление диоксидом марганца (II) и обработка ангидридом метансульфоновой кислоты дают (8). Это промежуточное соединение может быть подвергнуто реакции удаления защитной группы с использованием TFA с получением (9) и введено в реакцию с ангидридом метансульфоновой кислоты с получением (10). Полученный мезилат (10) может затем быть превращен в соединение формулы (I), как в целом описано здесь. Соединения формулы (I), в которой Z1 обозначает N, могут альтернативно быть получены в соответствии со схемой 5.(a) NH3/MeOH; (b) водород, палладий/уголь; (с этилбромацетат, карбонат калия; (d) трет-бутоксид калия; (е) CBzCl; (f) NaH; (S)-глицидил нозилат; (g) DMF, нагревание; (h) метансульфонилхлорид (i) амин H-A(Q1)(Q2), нагревание; (j) водород, палладий/уголь, затем MnO2. Реакция нитропиридина (1) с аммиаком дает нитропиридин (2), который восстанавливают до бисанилина (3). Алкилирование этилбромацетатом с последующей циклизацией с использованием третбутоксида калия дает (5). Его защищают карбоксибензильной группой, получая (6), который может быть затем введен в реакцию с (коммерчески доступным) S-глицидилнозилатом 2S)-2-оксиранилметил-3 нитробензолсульфонатом), получая (7). Циклизация в условиях нагревания дает (8). Мезилирование,смещение подходящим амином, гидрогенолиз группы CBz (10) и последующее окисление диоксидом марганца (II) дает (13). Альтернативно, гидрогенолиз группы CBz (10) и последующее окисление диоксидом марганца (II) с последующим мезилированием и смещение подходящим амином также дает (13). Он может быть превращен в соединение формулы (I), как в целом описано здесь. Соединения формулы (I), в которой Z1 и Z2, оба, обозначают N, могут быть получены в соответствии со схемой 6. Схема 6(4). Альтернативно, (1) может быть превращен в (3) путем сочетания с Вос-глицином с последующим удалением защитной группы с использованием кислоты с получением (4). Соединение (4) может затем быть превращено в соединение формулы (I) по аналогии с превращением соединения (5) из схемы 5. Взаимные превращения R1a, R1b, R2, А и R5 осуществляют обычными способами. В соединениях,которые содержат в случае необходимости защищенную гидроксильную группу, подходящие обычные защитные группы для гидроксигруппы, которые могут быть удалены без разрыва остатка молекулы,включают ацил и алкилсилил; N-защитные группы удаляют обычными способами.- 15015821 Взаимное превращение групп R1a и R1b может быть выполнено обычными способами на соединениях формулы (I). Например, R1a и R1b, представляющие собой метокси, могут быть превращены в R1a и R1b,представляющие собой гидрокси, обработкой литием и дифенилфосфином (общий способ, описанныйIreland et al., J. Amer. Chem. Soc., 1973, 7829) или HBr. Алкилирование гидроксильной группы подходящим алкильным производным, несущим удаляемую группу, такую как галогенид, дает алкокси, замещенный R1a и R1b. R1a или R1b, представляющий собой галоген, такой как бром, может быть превращен в циано обработкой цианидом меди (I) в N,N-диметилформамиде. R1a или R1b, представляющий собой карбокси, может быть получен обычным гидролизом R1a или R1b, представляющего собой циано, и карбокси может быть превращен в гидроксиметил обычным восстановлением. Соединения формулы HA-N(R20)R2' являются известными соединениями или могут быть получены аналогично известным соединениям, см., например, WO 2004/035569, WO 2004/089947, WO 02/08224,WO 02/50061, WO 02/56882, WO 02/96907, WO 2003087098, WO 2003010138, WO 2003064421,WO 2003064431, WO 2004002992, WO 2004002490, WO 2004014361, WO 2004041210, WO 2004096982,WO 2002050036, WO 2004058144, WO 2004087145, WO 2003082835, WO 2002026723, WO 06002047,WO 06014580, WO 06134378, WO 06137485, WO 07016610, WO 07081597, WO 07071936, WO 07115947,WO 07118130, WO 07122258, WO 08006648, WO 08003690 и WO 08009700. Дальнейшие детали получения соединений формулы (I) могут быть найдены в примерах. Антибактериальные соединения согласно изобретению могут быть составлены для введения любым удобным способом для использования у человека или в ветеринарии, по аналогии с другими антибактериальными/противотуберкулезными соединениями. Фармацевтические композиции по изобретению могут быть составлены для введения любым путем и включают композиции в форме, адаптированной для перорального, топического или парентерального использования и могут использоваться для лечения бактериальной инфекции, включая туберкулез, у млекопитающих, включая человека. Композиции могут быть в форме таблеток, капсул, порошков, гранул, таблеток для рассасывания,суппозиториев, кремов или жидких препаратов, таких как пероральные или парентеральные стерильные растворы или суспензии. Топические составы согласно настоящему изобретению могут быть представлены как, например,мази, кремы или лосьоны, глазные мази и глазные или ушные капли, пропитанные повязки и аэрозоли и могут содержать подходящие обычные добавки, такие как консерванты, растворители для облегчения проникновения лекарственного средства и мягчители в мазях и кремах. Составы могут также содержать обычные совместимые носители, такие как основы крема или мази и этанол или олеиловый спирт для лосьонов. Такие носители могут присутствовать в количестве от приблизительно 1 до приблизительно 98% от состава. Чаще они составляют до приблизительно 80% от состава. Таблетки и капсулы для перорального введения могут быть в стандартной лекарственной форме и могут содержать обычные эксципиенты, такие как связующие, например сироп, гуммиарабик, желатин,сорбит, трагакант или поливинилпирролидон; наполнители, например лактозу, сахар, кукурузный крахмал, фосфат кальция, сорбит или глицин; лубриканты для таблетирования, например стеарат магния,тальк, полиэтиленгликоль или диоксид крмния; дезинтеграторы, например картофельный крахмал; или приемлемые смачивающие вещества, такие как лаурилсульфат натрия. Таблетки могут быть покрыты согласно способам, известным в обычной фармацевтической практике. Пероральные жидкие препараты могут быть в форме, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров или могут быть представлены как сухой продукт для восстановления с водой или другим подходящим носителем перед использованием. Такие жидкие препараты могут содержать обычные добавки,такие как суспендирующие агенты, например сорбит, метилцеллюлозу, сироп глюкозы, желатин, гидроксиэтилцеллюлозу, карбоксилметилцеллюлозу, гель стеарата алюминия или гидрированные пищевые жиры, эмульгаторы, например лецитин, сорбитан моноолеат или гуммиарабик; неводные носители (которые могут включать пищевые масла), например миндальное масло, масляные сложные эфиры, такие как глицерин, пропиленгликоль или этанол; консерванты, например метил- или пропил-пгидроксибензоат, или сорбиновую кислоту и, если желательно, обычные ароматизаторы или красители. Суппозитории могут содержать обычные основы суппозитория, например масло какао или другой глицерид. Для парентерального введения жидкие стандартные лекарственные формы получают, используя соединение и стерильный носитель, предпочтительно воду. Соединение в зависимости от используемых носителя и концентрации может быть либо суспендировано, либо растворено в носителе. При получении растворов соединение может быть растворено в воде для инъекции и стерилизовано фильтрацией с последующим заполнением в подходящий пузырек или ампулу и запечатыванием. Предпочтительно в носителе могут быть растворены такие средства, как местный анестетик, консервант и буферизующие агенты. Для усиления стабильности композиция может быть заморожена после заполнения в пузырек и вода может быть удалена в вакууме. Сухой лиофилизированный порошок затем герметизируют в пузырьке и может быть приложен сопутствующий пузырек с водой для инъекции для- 16015821 восстановления жидкости перед использованием. Парентеральные суспензии получают в основном тем же самым образом, за исключением того, что соединение суспендируют в носителе вместо растворения,и стерилизация не может быть осуществлена фильтрацией. Соединение может стерилизоваться действием этиленоксида перед суспендированием в стерильном носителе. Предпочтительно в композицию включают поверхностно-активное вещество или смачивающее вещество, чтобы облегчить однородное распределение соединения. Композиции могут содержать от 0,1 вес.%, предпочтительно 10-60 вес.% активного вещества в зависимости от способа введения. Если композиции включают разовые дозы, каждая доза предпочтительно содержит 50-1000 мг активного ингредиента. Доза при использовании для лечения взрослого человека предпочтительно составляет от 100 до 3000 мг в сутки, например 1500 мг в сутки в зависимости от пути и частоты введения. Такая дозировка соответствует от приблизительно 1,5 до приблизительно 50 мг/кг в сутки. Предпочтительно доза составляет от 5 до 30 мг/кг в сутки. Соединение формулы (I) может быть единственным терапевтическим средством в композициях по изобретению или в комбинации с другими антибактериальными агентами, включая противотуберкулезные соединения. Если другое антибактериальное средство представляет собой -лактам, тогда может быть также использован ингибитор -лактамазы. Соединения формулы (I) могут использоваться в лечении бактериальных инфекций, вызванных разнообразными организмами, включая как грамотрицательные, так и грамположительные организмы,таких как инфекции верхних и/или нижних дыхательных путей, кожи и инфекции мягких тканей и/или инфекции мочевых путей. Соединения формулы (I) могут также использоваться в лечении туберкулеза,вызванного Mycobacterium tuberculosis. Некоторые соединения формулы (I) могут быть активными против более одного организма. Это может быть определено способами, описанными здесь. Следующие примеры иллюстрируют получение некоторых соединений формулы (I) и активность некоторых соединений формулы (I) против различных бактериальных организмов, включаяMycobacterium tuberculosis. Примеры и экспериментальные примеры Общие аббревиатуры в примерах:Rf - фактор задержания. Некоторые реагенты также указаны в сокращении. TFA относится к трифторуксусной кислоте, THF относится к тетрагидрофурану, Pd/C относится к катализатору на основе палладия на угле, DCM относится к дихлорметану, МеОН относится к метанолу, DMF относится к диметилформамиду, EtOAc относится к этилацетату, DDQ относится к 2,3-дихлор-5,6-дициано-1,4-бензохинону, NaBH(OAc)3 относится к триацетоксиборгидриду натрия, Pd2(dba)3 относится к трис-(дибензилиденацетон)дипалладию(0). Спектры протонного ядерного магнитного резонанса (1 Н ЯМР) регистрировали при 400 или 250 МГц, химические сдвиги представлены в частях на миллион (ppm) от внутреннего тетраметилсиланового стандарта (TMS). Аббревиатуры для данных ЯМР являются следующими: s=синглет, d=дублет,t=триплет, q=квартет, m=мультиплет, dd=дублет дублетов, dt=дублет триплетов, td=триплет дублетов,каж.=кажущийся, ушир.=уширенный. J указывает константу сочетания ЯМР, измеренную в герцах.CDCl3 обозначает дейтериохлороформ, CD3OD обозначает тетрадейтериометанол. Масс-спектры получали, используя методики ионизации с электрораспылением (ES). Все температуры приведены в градусах Цельсия. МР-карбонат относится к макропористому триэтиламмонийметилполистиролкарбонату (ArgonautTechnologies). AmberlystA21 представляет собой слабоосновную макропористую смолу с функциональной алкиламиногруппой, зарегистрированный товарный знак изготовителя RohmHaas Co.AD mix alpha получали, смешивая осмиат калия (K2OsO42 Н 2 О) (0,52 г), (3a,9R,3a,4b,9R)-9,9'[1,4-фталазиндиил-бис-(окси)]-бис-[6'-(метилокси)-10,11-дигидроцинхонан] [(DHQ)2PHAL] (5,52 г), затем добавляя феррицианид калия [K3Fe(CN)6] (700 г) и порошок карбоната калия (294 г). Эту смесь перемешивают в мешалке в течение 30 мин. Это обеспечивает приблизительно 1 кг AD mix alpha, который является коммерчески доступным от Aldrich. См. K. Barry Sharpless et al., J. Org. Chem., 1992, 57 (10),2771. AD mix beta представляет собой соответствующую смесь, полученную с (9S,9S)-9,9'-[1,4 фталазиндиил-бис-(окси)]-бис-[6'-(метилокси)-10,11-дигидроцинхонан][(DHQD)2PHAL]. Если указан ADmix alpha/beta, он представляет собой смесь 1:1 альфа и бета.Celite представляет собой добавку для фильтрования, состоящую из кислоты-промытого диатомита и является товарным знаком изготовителя Manville Corp., Денвер, Колорадо. Картридж SCX представляет собой ионообменную колонку, содержащую сильную катионообменную смолу (бензолсульфоновая кислота), поставляется Varian, США.Chiralpak IA и Chiralpak AS-H представляют собой колонки для хиральной ВЭЖХ на полисахаридной основе (Chiral Technologies Inc.). Колонка Chiralpak AS-H включает амилоза-трис-[(S)-альфаметилбензилкарбамат], нанесенную на 5 мкм силикагеля. Колонка Chiralpak IA включает силикагель для препаративной колонки (размер частиц 5 мкм, 21 мм внутреннего диаметра 250 мм длины), иммобилизированный амилоза-трис-(3,5-диметилфенилкарбаматом). Колонки Chiralpak AD и AD-H включают силикагель для препаративных колонок (размер частиц 5 мкм AD-H и размер частиц 10 мкм AD,21 мм внутреннего диаметра 250 мм длины; размер частиц 20 мкм AD, 101 мм внутреннего диаметра 250 мм длины), покрытый амилоза-трис-(3,5-диметилфенилкарбаматом) (Chiral Technologies USA). Измеренные времена удерживания зависят от точных условий хроматографических процедур. Если они указаны ниже в примерах, они указывают порядок элюирования. Колонка Kromasil 5 micron C-18(21250 мм) включает октадецилсилан, химически связанный с пористым силикагелем с размером пор 5 мкм. Как будет понятно для специалиста, указание на получение, осуществляемое подобно или общим способом, другим получениям может охватывать вариации в обычных параметрах, таких как время, температура, условия обработки, незначительные изменения в количествах реагента и т.д. Реакции, включающие гидриды металлов, включая гидрид лития, алюмогидрид лития, диизобутилалюминий гидрид, гидрид натрия, боргидрид натрия и триацетоксиборгидрид натрия, осуществляют в атмосфере аргона или другого инертного газа. Пример 1. 1-(4-[(2,3-Дигидро[1,4]диоксино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинилметил)-1,2-дигидро-3H,8 Н-2a,5,8 а-триазааценафтилен-3,8-дион дигидрохлорид(a) 6-Хлор-3-нитро-N-2-пропен-1-ил-2-пиридинамин. Его получали модификацией способа Schmid, S., et al., Synthesis (2005), (18), 3107-3118. Раствор 2,6-дихлор-3-нитропиридина (8,0 г, 41,45 ммоль) в безводном дихлорметане (180 мл) охлаждали до -15 С в атмосфере аргона. Добавляли триэтиламин (6,0 мл, 43 ммоль) и затем небольшими порциями за 3 ч добавляли аллиламин (3,23 мл, 43 ммоль), поддерживая температуру -15 С. Реакционную смесь перемешивали в течение ночи, в течение которой ее нагревали до температуры окружающей среды. Реакционную смесь промывали 0,2 М водным раствором лимонной кислоты (100 мл), насыщенным водным раствором (100 мл) NaHCO3, пропускали через гидрофобную фритту и упаривали до желтого масла, которое очищали хроматографией на силикагеле с элюированием с использованием от 0 до 50% этилацетата в гексане, получая твердое вещество желтого цвета (7,49 г, 85%).(b) 3-(Бромметил)-8-нитро-2,3-дигидроимидазо[1,2-а]пиридин-5(1 Н)-он. Его получали модификацией способа Schmid, S., et al., Synthesis (2005), (18), 3107-3118. Раствор 6-хлор-3-нитро-N-2-пропен-1-ил-2-пиридинамина (20 г, 93,6 ммоль) в хлорбензоле (500 мл) обрабатывали раствором, содержащим бром (4,75 мл, 92,7 ммоль) в хлорбензоле (100 мл), по каплям за 4,5 ч, поддерживая Т 26 С при охлаждении, если требуется. Густую суспензию перемешивали при температуре окружающей среды в течение 18 ч и разбавляли гексаном (200 мл) и затем реакционную смесь вливали в гексан (1000 мл). Через 15 мин оранжевый осадок собирали фильтрацией и промывали гексаном(250 мл), получая 26,6 г твердого вещества оранжевого цвета (3-(бромметил)-5-хлор-8-нитро-2,3 дигидроимидазо[1,2-]пиридин-1-ий бромид). Это промежуточное соединение добавляли, за 45 мин к быстро перемешиваемой смеси насыщенного водного раствора (1000 мл) NaHCO3 и этилацетата(500 мл). Ярко-красную смесь перемешивали в течение 1 ч, разбавляли этилацетатом (200 мл) и слои разделяли. Водный слой промывали этилацетатом (200 мл) и органические экстракты объединяли, высушивали (безводный сульфат натрия), фильтровали и упаривали, получая продукт в форме твердого вещества коричневого цвета (18,3 г, содержит 40% 6-бром-3-(бромметил)-8-нитро-2,3-дигидроимидазо[1,2]пиридин-5(1 Н)-она).(с) 1,1-Диметилэтил 1-[(8-нитро-5-оксо-1,2,3,5-тетрагидроимидазо[1,2-]пиридин-3-ил)метил]-4 пиперидинилкарбамат. Суспензию смеси 3:2 3-(бромметил)-8-нитро-2,3-дигидроимидазо[1,2-]пиридин-5(1 Н)-она и 6-бром-3-(бромметил)-8-нитро-2,3-дигидроимидазо[1,2-а]пиридин-5(1 Н)-она (18,2 г) обрабатывали 1,1-диметилэтил-4-пиперидинилкарбаматом (26,6 г, 132,8 ммоль) в ацетонитриле (900 мл), затем пиридином (10,7 мл, 132 ммоль). Смесь нагревали при 60 С в атмосфере аргона в течение 17 ч и затем нагревали при 70 С в течение 2 ч, охлаждали и упаривали до приблизительно половины объема. Густой желтый осадок удаляли фильтрацией и хорошо промывали простым диэтиловым эфиром. Фильтрат упаривали досуха и остаток разделяли между хлороформом (500 мл) и водой (200 мл). Нерастворенный материал удаляли фильтрацией и промывали хлороформом (100 мл). Слои в фильтрате разделяли и водный слой промывали хлороформом (200 мл). Объединенные органические экстракты пропускали через гидрофобную фритту и упаривали до темно-желтой смолы, которую хроматографировали, элюируя с использованием от 0 до 100% этилацетата в гексане, затем от 0 до 30% метанола в этилацетате, получая твердое вещество желтого цвета (10,98 г).(d) 1,1-Диметилэтил 1-[(3,8-диоксо-1,2,5 а,8b-тетрагидро-3 Н,8 Н-2 а,5,8 а-триазааценафтилен-1 ил)метил]-4-пиперидинилкарбонат. Суспензию 1,1-диметилэтил 1-[(8-нитро-5-оксо-1,2,3,5-тетрагидроимидазо[1,2-]пиридин-3 ил)метил]-4-пиперидинилкарбамата (2,0 г, 5,08 ммоль) и безводного карбоната калия (700 мг,5,06 ммоль) в абсолютном спирте (150 мл) гидрировали при атмосферном давлении в присутствии 10%Pd на С (1 г) в течение 4 ч. Реакционную смесь фильтровали через Keiselguhr, промывали этанолом(100 мл) и темно-фиолетовую смесь немедленно вводили в реакцию, обрабатывая безводным карбонатом калия (1,4 г, 10 ммоль) и этилбромацетатом (550 мкл, 4,95 ммоль), перемешивали при температуре окружающей среды в течение 20 ч и затем нагревали при 60 С в течение 30 мин. Через 45 мин добавляли дополнительное количество 0,25 мл этилбромацетата и нагревали при 60 С в течение 1,5 ч. Добавляли 0,25 мл этилбромацетата и реакционную смесь снова нагревали при 60 С в течение 1 ч. Реакционную смесь фильтровали через Keiselguhr и упаривали досуха. Смесь подвергали азеотропной обработке с хлороформом и затем хроматографировали с элюированием с использованием от 0 до 100% этилацетата в гексане и затем от 0 до 20% метанола в этилацетате. Второе элюирование с использованием от 0 до 50% метанола в этилацетате дало темную смолу (37 мг, 1,6%).(e) 1-[(4-Амино-1-пиперидинил)метил]-1,2,5 а,8b-тетрагидро-3 Н,8 Н-2 а,5,8a-триазааценафтилен-3,8 дион. Раствор 1,1-диметилэтил 1-[(3,8-диоксо-1,2,5 а,8b-тетрагидро-3 Н,8 Н-2 а,5,8 а-триазааценафтилен-1 ил)метил]-4-пиперидинилкарбамата (37 мг, 0,092 ммоль) в безводном дихлорметане (2 мл) обрабатывали TFA (1 мл) и перемешивали при температуре окружающей среды в течение 1 ч, упаривали досуха,смешивали с безводным дихлорметаном и упаривали до темной смолы. Эту смолу растворяли в смеси 1:1 дихлорметан:метанол (10 мл) и обрабатывали МР-карбонатной смолой (600 мг) и перемешивали в течение 1,5 ч. Реакционную смесь фильтровали, смолу промывали смесью 1:1 дихлорметан:метанол(30 мл) и фильтрат упаривали досуха. Очистка на колонке 5 г SCX с элюированием с градиентом от метанола до 2 н. метанольного аммиака дала продукт в форме смолы. Дальнейшее выпаривание из простого диэтилового эфира дало продукт в форме твердого вещества коричневого цвета (22,8 мг, 82%).(f) Целевое соединение. Раствор 1-[(4-амино-1-пиперидинил)метил]-1,2,5 а,8b-тетрагидро-3 Н,8 Н-2 а,5,8 а-триазааценафтилен 3,8-диона (22,8 мг, 0,0757 ммоль) в безводном дихлорметане (3 мл) и безводном метаноле (0,6 мл) обрабатывали 2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-карбальдегидом (12,5 мг, 0,076 ммоль) (в отношении синтеза см. WO 2004058144, пример 2 (с) или WO 03/087098 пример 19 (d, перемешивали в атмосфере аргона в течение 15 мин и затем обрабатывали триацетоксиборгидридом натрия (48 мг,0,226 ммоль) и перемешивали при температуре окружающей среды в течение 17 ч. Реакционную смесь затем обрабатывали дополнительной частью 2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-карбальдегида(2 мг) и триацетоксиборгидрида натрия (10 мг), смесь перемешивали в течение 4 ч, обрабатывали насыщенным водным раствором NaHCO3 (1 мл) и перемешивали в течение 10 мин. Слои разделяли и водный слой промывали раствором 9:1 дихлорметан:метанол (210 мл). Объединенные органические экстракты пропускали через гидрофобную фритту и упаривали до коричневой смолы, которую хроматографировали, элюируя с использованием от 0 до 30% метанола в дихлорметане, получая свободное основания целевого соединения в форме желтой смолы (20,6 мг, 60%).H ЯМР (250 МГц) (CDCl3)1,38-1,54 (2 Н, м), 1,83-1,93 (2 Н, м), 2,19-2,36 (2 Н, м), 2,54-2,73 (3 Н, м),2,93-2,98 (1 Н, м), 3,09-3,15 (1 Н, м), 3,85 (2 Н, с), 4,26-4,61 (6 Н, м), 4,96-5,05 (1 Н, м), 6,33 (1 Н, d), 6,82 (1 Н,- 19015821 с), 7,76 (1 Н, д), 7,87 (1 Н, с) и 8,10 (1 Н, с). Это свободное основание целевого соединения растворяли в безводном дихлорметане (2 мл) и безводном метаноле (0,5 мл) и обрабатывали 1 М HCl в простом диэтиловом эфире (0,5 мл). Добавляли простой диэтиловый эфир (5 мл) и суспензию охлаждали. После центрифугирования растворитель удаляли и твердое вещество высушивали, получая целевое соединение в форме твердого вещества коричневого цвета (23,5 мг).(a) N-(6-Хлор-2-пиридинил)-2,2-диметилпропанамид. Раствор 6-хлор-2-пиридинамина (13,776 г, 107 ммоль) в толуоле (100 мл) и триэтиламине (16,28 мл,118 ммоль) при 50C в атмосфере аргона обрабатывали 2,2-диметилпропаноилхлоридом (13,81 мл,112 ммоль). Реакционную смесь затем перемешивали при 50C в течение 4 ч и затем при температуре окружающей среды в течение 18 ч. Затем добавляли 2 М HCl (200 мл) и смесь экстрагировали простым диэтиловым эфиром (3500 мл). Органические экстракты высушивали (MgSO4), фильтровали и упаривали, получая продукт в форме твердого вещества коричневого цвета (21,005 г, 92%).-78 С в атмосфере аргона обрабатывали н-бутиллитием (20 мл, 2,5 М в гексанах, 50 ммоль) в течение 10 мин и затем давали нагреться до 0 С, перемешивали при 0 С в течение 3 ч и затем повторно охлаждали до -78 С. Реакционную смесь затем обрабатывали, добавляя по каплям дибромэтан (2,057 мл,23,9 ммоль), реакционной среде давали нагреться до температуры окружающей среды и перемешивали при температуре окружающей среды в течение 0,5 ч. Реакционную смесь затем обрабатывали водой(5 мл), перемешивали при температуре окружающей среды в течение 5 мин, обрабатывали дополнительным количеством воды (500 мл) и экстрагировали простым диэтиловым эфиром (3500 мл). Органические экстракты высушивали (MgSO4), фильтровали, упаривали и остаток хроматографировали (0-25% этилацетат:гексан), получая продукт в форме твердого вещества желтого цвета (3,489 г, 53%).(40 мл) при 0 С в атмосфере аргона обрабатывали гидридом натрия (0,696 г, 17,395 ммоль) и затем давали нагреться до температуры окружающей среды в течение 0,25 ч, перемешивали при температуре окружающей среды в течение 0,25 ч и затем обрабатывали аллилйодидом (1,61 мл, 17,395 ммоль) и перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь затем обрабатывали водой (10 мл), концентрировали до приблизительно 5 мл, обрабатывали большим количеством воды(200 мл) и экстрагировали DCM (3200 мл). Органические экстракты высушивали (MgSO4), фильтровали, упаривали и остаток хроматографировали (0-20% этилацетат:гексан), получая продукт в форме желтого масла, которое затвердевало в твердое вещество грязно-белого цвета (5,324 г, 67%).(d) N-[3-Бром-6-(метилокси)-2-пиридинил]-2,2-диметил-N-2-пропен-1-илпропанамид. Раствор N-(3-бром-6-хлор-2-пиридинил)-2,2-диметил-N-2-пропен-1-илпропанамида (12,388 г,37,370 ммоль) в метаноле (100 мл) при температуре окружающей среды в атмосфере аргона обрабатывали раствором метилата натрия (25% вес./об. в метаноле, 17,76 г, 82,212 ммоль) и затем нагревали с обратным холодильником в течение 42 ч. Реакционную смесь затем охлаждали, обрабатывали водой(500 мл) и экстрагировали простым диэтиловым эфиром (3200 мл). Органические экстракты высушивали (MgSO4) и упаривали, получая продукт (10,918 г, 89%).(e) N-[3-Бром-6-(метилокси)-2-пиридинил]-N-(2,3-дигидроксипропил)-2,2-диметилпропанамид. Раствор N-[3-бром-6-(метилокси)-2-пиридинил]-2,2-диметил-N-2-пропен-1-илпропанамида (1,246 г,3,81 ммоль) в трет-бутаноле (40 мл) при температуре окружающей среды в атмосфере аргона обрабатывали водой (40 мл) и затем AD-mix(2,86 г) и AD-mix(2,86 г) и перемешивали при температуре окружающей среды в течение 18 ч. Реакционную смесь затем обрабатывали насыщенным водным раствором сульфита натрия (40 мл), перемешивали в течение 10 мин, экстрагировали смесью 20% метанол/DCM(3100 мл). Органические экстракты высушивали (MgSO4) и упаривали, получая сырой продукт (1,728 г,126%), содержащий остаточный трет-бутанол.N-[3-бром-6-(метилокси)-2-пиридинил]-N-(2,3-дигидроксипропил)-2,2-диметилпропанамида (7,628 г, 21,130 ммоль) в DCM (100 мл) и пиридине (3,407 мл, 42,26 ммоль) при -78 С в атмосфере аргона обрабатывали раствором трифосгена (6,27 г, 21,130 ммоль) в DCM (20 мл) в течение 5 мин, реакционной смеси затем давали нагреться до температуры окружающей среды и перемешивали при температуре окружающей среды в течение 30 мин. Реакционную смесь затем тщательно обрабатывали насыщенным раствором (200 мл) бикарбоната натрия, экстрагировали DCM (3200 мл). Органические экстракты высушивали (MgSO4), упаривали и хроматографировали (0-50% этилацетат:гексан), получая продукт в форме твердого вещества белого цвета (5,722 г, 70%).(g) (2 Е)-3-[2-(2,2-Диметилпропаноил)[(2-оксо-1,3-диоксолан-4-ил)метил]амино-6-(метилокси)-3 пиридинил]-2-бутилпропеноат. Смесь N-[3-бром-6-(метилокси)-2-пиридинил]-2,2-диметил-N-[(2-оксо-1,3-диоксолан-4-ил)метил]пропанамида (5,722 г, 14,722 ммоль), Pd(PtBu3)2 (151 мг, 0,296 ммоль), Pd2(dba)3 (135 мг, 0,149 ммоль) в 1,4-диоксане (40 мл) обрабатывали N,N'-дициклогексилметиламином (3,48 мл, 16,265 ммоль) и н-бутилакрилатом (2,54 мл, 17,743 ммоль) и смесь затем нагревали при 80 С в течение 1 ч. Реакционную смесь затем охлаждали, обрабатывали водой (200 мл), экстрагировали DCM (3200 мл). Органические экстракты высушивали (MgSO4), упаривали и хроматографировали (0-50% этилацетат:гексан), получая продукт в форме желтого масла (6,156 г, 96%).(2 Е)-3-[2-(2,2-диметилпропаноил)[(2-оксо-1,3-диоксолан-4-ил)метил]амино-6(метилокси)-3-пиридинил]-2-бутилпропеноата (6,156 г, 14,184 ммоль) в этаноле (200 мл) обрабатывали палладием на угле (10% паста, 1,23 г) и смесь затем перемешивали при температуре окружающей среды под 1 атм водорода в течение 18 ч. Реакционную смесь затем фильтровали через тонкий слой целита,элюируя дополнительным количеством этанола (200 мл). Органический фильтрат затем упаривали, получая продукт в форме желтого масла (6,065 г, 98%).(i) 1-(2,3-Дигидроксипропил)-7-(метилокси)-3,4-дигидро-1,8-нафтиридин-2(1 Н)-он. Раствор 3-[2-(2,2-диметилпропаноил)[(2-оксо-1,3-диоксолан-4-ил)метил]амино-6-(метилокси)-3 пиридинил]бутилпропаноата (6,065 г, 13,911 ммоль) в метаноле (100 мл) обрабатывали концентрированным водным раствором HCl (12 М, 50 мл) и затем нагревали с обратным холодильником в течение 48 ч. Реакционную смесь затем концентрировали до приблизительно 50 мл, нейтрализовали карбонатом калия и экстрагировали смесью 20% метанол/DCM (3100 мл). Органические экстракты высушивали (MgSO4) и упаривали, получая сырой продукт в форме желтого масла (2,325 г, 66%).(j) 7-(Метилокси)-1-(2-оксиранилметил)-3,4-дигидро-1,8-нафтиридин-2(1 Н)-он. Раствор 1-(2,3-дигидроксипропил)-7-(метилокси)-3,4-дигидро-1,8-нафтиридин-2(1 Н)-она (2,325 г,9,226 ммоль) в DCM (40 мл) и триэтиламине (1,915 мл, 13,839 ммоль) при 0 С в атмосфере аргона обрабатывали метансульфонилхлоридом (0,714 мл, 9,226 ммоль) и перемешивали при 0 С в течение 0,5 ч. Реакционную смесь затем обрабатывали водой (100 мл), экстрагировали DCM (3100 мл). Органические экстракты высушивали (MgSO4) и упаривали. Остаток затем растворяли в метаноле (50 мл) и обрабатывали карбонатом калия (6,366 г, 46,130 ммоль) и перемешивали при температуре окружающей среды в течение 15 мин. Реакционную смесь затем обрабатывали водой (100 мл), экстрагировали DCM(3200 мл). Органические экстракты высушивали (MgSO4), упаривали и хроматографировали (0-100% этилацетат:гексан), получая продукт в форме желтого масла (428 мг, 20%).(k) 1,1-Диметилэтил(1-2-гидрокси-3-[7-(метилокси)-2-оксо-3,4-дигидро-1,8-нафтиридин-1(2 Н)ил]пропил-4-пиперидинил)карбамат. Раствор 7-(метилокси)-1-(2-оксиранилметил)-3,4-дигидро-1,8-нафтиридин-2(1 Н)-она (428 мг,1,829 ммоль) и 1,1-диметилэтил-4-пиперидинилкарбамата (366 мг, 1,829 ммоль) в DMF (2 мл) в атмосфере аргона нагревали при 120 С в течение 1 ч. Смесь затем упаривали и хроматографировали (0-10% метанол/DCM), получая продукт в форме желтого масла (301 мг, 38%).(0,24 мл, 1,735 ммоль) при температуре окружающей среды в атмосфере аргона обрабатывали ангидридом метансульфоновой кислоты (242 мг, 1,388 ммоль) и нагревали с обратным холодильником в течение 2 ч. Реакционную смесь затем упаривали и растворяли в ацетонитриле (10 мл), обрабатывали йодидом натрия (520 мг, 3,47 ммоль) и нагревали при 80 С в течение 0,25 ч. Смесь затем охлаждали, упаривали,затем обрабатывали водой (200 мл), экстрагировали смесью 20% метанол/DCM (3200 мл). Органические экстракты высушивали (MgSO4), упаривали и хроматографировали (0-10% метанол/DCM), получая продукт в форме оранжевого масла (194 мг, 70%).(m) 1,1-Диметилэтил 1-[(4,9-диоксо-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-1 ил)метил]-4-пиперидинилкарбамат. Раствор 1,1-диметилэтил 1-[(4,9-диоксо-1,2,8,9-тетрагидро-4 Н,7 Н-имидазо[1,2,3-ij]-1,8 нафтиридин-1-ил)метил]-4-пиперидинилкарбамата (194 мг, 0,483 ммоль) (301 мг, 0,694 ммоль) в 1,4-диоксане (5 мл) обрабатывали DDQ (164 мг, 0,724 ммоль) и перемешивали при 60C в течение 24 ч. Добавляли дополнительное количество DDQ (164 мг, 0,724 ммоль) и реакционную смесь перемешивали еще 2 ч. Реакционную смесь затем обрабатывали 5%-ным водным раствором карбоната калия (100 мл),экстрагировали смесью 20% метанол/DCM (3200 мл). Органические экстракты высушивали (MgSO4) и упаривали, получая продукт в форме оранжевого масла (159 мг, 82%).(n) 1-[(4-Амино-1-пиперидинил)метил]-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9 дион дигидрохлорид. Раствор 1,1-диметилэтил 1-[(4,9-диоксо-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-1 ил)метил]-4-пиперидинилкарбамата (159 мг, 0,398 ммоль) в хлороформе (2 мл) и метаноле (2 мл) в атмосфере аргона при температуре окружающей среды обрабатывали 4 М HCl в 1,4-диоксане (2 мл) и перемешивали при температуре окружающей среды в течение 0,5 ч. Реакционную смесь затем высушивали и упаривали, получая продукт в форме твердого вещества желтого цвета (138 мг, 93%).(о) Целевое соединение. Смесь 1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9 диона дигидрохлорида (49 мг, 0,131 ммоль) в DCM (2 мл) и метаноле (0,1 мл) в атмосфере аргона при температуре окружающей среды обрабатывали триэтиламином (58 мкл, 0,419 ммоль) и перемешивали при температуре окружающей среды в течение 0,25 ч, после чего добавляли [1,3]оксатиоло[5,4 с]пиридин-6-карбальдегид (в отношении синтеза см. WO 2004058144, пример 61) (22 мг, 0,131 ммоль). Смесь затем перемешивали при температуре окружающей среды в течение 1 ч, после чего добавлялиNaBH(OAc)3 (56 мг, 0,262 ммоль). Реакционную смесь перемешивали при температуре окружающей среды еще 0,5 ч, после чего добавляли насыщенный водный раствор бикарбоната натрия (20 мл). Смесь экстрагировали смесью 20% метанол/DCM (3100 мл). Органические экстракты высушивали (MgSO4), упаривали и хроматографировали (0-20% метанол/DCM), получая продукт в форме прозрачного маслаMS (ES+) m/z 452 (МН+). 1 Н ЯМР (CDCl3, 400 МГц)1,38-1,48 (2 Н, м), 1,78-1,95 (2 Н, м), 2,15-2,37 (2 Н, м) 2,45-2,60 (1 Н, м),2,61-2,72 (2 Н, м), 2,92-3,02 (1 Н, м), 3,05-3,12 (1 Н, м), 3,83 (2 Н, с), 4,32-4,42 (1H, м), 4,52-4,61 (1 Н, м),4,96-5,05 (1 Н, м), 5,74 (2H, с), 6,22-6,32 (2 Н, м), 7,20 (1 Н, с), 7,45-7,52 (2 Н, м), 7,99 (1 Н, с). Свободное основание целевого соединения в метаноле и хлороформе превращали в гидрохлорид,добавляя эквивалент 4 М хлорида водорода в 1,4-диоксане с последующим упариванием досуха. Пример 3. 1-(4-[(2,3-Дигидро[1,4]диоксино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинилметил)-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион гидрохлорид Способ А. Смесь 1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9 диона дигидрохлорида (36 мг, 0,0965 ммоль) (в отношении получения см. пример 2 (n в DCM (2 мл) и метаноле (0,1 мл) в атмосфере аргона при температуре окружающей среды обрабатывали триэтиламином(43 мкл, 0,309 ммоль) и перемешивали при температуре окружающей среды в течение 0,25 ч, после чего добавляли 2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-карбоксальдегид (в отношении синтеза см.WO 2004058144, пример 2 (с) или WO 03/087098, пример 19 (d (16 мг, 0,0965 ммоль). Смесь затем пе- 22015821 ремешивали при температуре окружающей среды в течение 1 ч, после чего добавляли NaBH(OAc)3(41 мг, 0,193 ммоль). Реакционную смесь перемешивали при температуре окружающей среды еще 0,5 ч,после чего добавляли насыщенный водный раствор бикарбоната натрия (20 мл). Смесь экстрагировали смесью 20% метанол/DCM (3100 мл). Органические экстракты высушивали (MgSO4), упаривали и хроматографировали (0-20% метанол/DCM), получая свободное основание целевого соединения в форме прозрачного масла (24 мг, 55%).H ЯМР (CDCl3, 400 МГц)1,30-1,50 (2 Н, м), 1,80-1,92 (2 Н, м), 2,19-2,35 (2 Н, м) 2,49-2,72 (3 Н, м),2,92-3,02 (1 Н, м), 3,07-3,13 (1 Н, м), 3,81 (2 Н, с), 4,22-4,51 (5 Н, м) 4,52-4,60 (1 Н, м), 4,96-5,04 (1 Н, м), 6,226,32 (2 Н, м), 6,81 (1 Н, с), 7,45-7,53 (2 Н, м), 8,04 (1 Н, с). Свободное основание целевого соединения в метаноле и хлороформе превращали в гидрохлорид,добавляя эквивалент 4 M хлорида водорода в 1,4-диоксане с последующим упариванием досуха. Способ В.(a) 2-Бром-3-[(фенилметил)окси]пропионовая кислота. Рацемический О-(фенилметил)серин (5 г, 25,6 ммоль) и бромид калия (10,7 г, 89,6 ммоль) растворяли в охлажденной льдом H2SO4 (2,5 н.) и обрабатывали раствором нитрита натрия (2,65 г) в воде (30 мл) в течение 50 мин (поддерживая температуру реакции 4 С). Реакционную смесь затем перемешивали при 0 С в течение 45 мин и затем при температуре окружающей среды в течение 1 ч экстрагировали этилацетатом (3100 мл). Объединенные органические экстракты промывали водой, солевым раствором, высушивали (MgSO4), фильтровали и упаривали, получая продукт в форме желтого масла (6 г, 90%).(b) 2-Бром-3-[(фенилметил)окси]метилпропаноат. Раствор 2-бром-3-[(фенилметил)окси]пропионовой кислоты (6 г, 23,2 ммоль) в метаноле (40 мл) при температуре окружающей среды в атмосфере аргона обрабатывали тионилхлоридом (1,7 мл, 23,2 ммоль) и реакционную смесь затем перемешивали при температуре окружающей среды в течение 3 ч и затем упаривали, получая продукт в форме желтого масла (6,3 г, 99%).(50 мл) нагревали при 80 С и перемешивали в атмосфере аргона в течение 2,5 ч. Растворители удаляли при пониженном давлении и остаток обрабатывали насыщенным водным раствором бикарбоната натрия. Водную фазу экстрагировали DCM (5100 мл), высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой продукт хроматографировали, элюируя 0-100% EtOAc/40-60 петролейный эфир. Соответствующие фракции объединяли и упаривали при пониженном давлении. Остаток затем растворяли в DCM (50 мл) и промывали водой (20 мл). Органический слой отделяли, высушивалиMgSO4 и упаривали при пониженном давлении, получая продукт (618 мг, 35%-ный выход).THF (8 мл) при -78 С в атмосфере Ar по каплям добавляли LiAlH4 (1,312 мл, 1,312 ммоль). Реакционной смеси давали нагреться до -10 С за 2 ч. Смесь затем перемешивали при 0 С в течение 2 ч, после чего добавляли воду (0,1 мл), затем гидроксид натрия (0,18 мл, 0,360 ммоль) и затем воду (0,2 мл). Смесь затем перемешивали еще 2 ч при температуре окружающей среды. Полученную смесь фильтровали и промывали THF (100 мл). Объединенный фильтрат и смывы упаривали при пониженном давлении, получая продукт (0,519 г, 89%-ный выход).(0,049 мл, 0,350 ммоль) в DCM (5 мл) при 0 С обрабатывали метансульфонилхлоридом (0,025 мл,0,321 ммоль). Раствору давали нагреться до температуры окружающей среды и перемешивали при этой температуре в течение 1 ч. Добавляли дополнительно 0,2 экв. триэтиламина и 0,4 экв. метансульфонилхлорида и реакционную смесь перемешивали в течение 30 мин. Реакционную смесь разбавляли DCM(20 мл) и обрабатывали водой (2 мл). Водный слой экстрагировали DCM (50 мл). Органические слои объединяли и высушивали MgSO4, фильтровали и упаривали при пониженном давлении до сырого про- 23015821 дукта (101 мг, 65%), который использовали без дальнейшей очистки.(f) 1,1-Диметилэтил(2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-илметил)[1-(2-[7-(метилокси)-2 оксо-3,4-дигидро-1,8-нафтиридин-1(2 Н)-ил]-1-[(фенилметил)окси]метилэтил)-4 пиперидинил]карбамат. Способ 1. Раствор 1,1-диметилэтил[1-(2-хлор-1-[(фенилметил)окси]метилэтил)-4-пиперидинил](2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-илметил)карбамата (101 мг, 0,190 ммоль) в DMF (10 мл) добавляли по каплям к раствору натриевой соли 7-(метилокси)-3,4-дигидро-1,8-нафтиридин-2(1 Н)она(33,8 мг, 0,190 ммоль) в DMF (10 мл) (полученной добавлением гидрида натрия (9,11 мг, 0,228 ммоль) к 7-(метилокси)-3,4-дигидро-1,8-нафтиридин-2(1 Н)-ону (33,8 мг, 0,190 ммоль) (в отношении получения см. пример 5 (е в DMF (10 мл. Раствор перемешивали при температуре окружающей среды в течение ночи в атмосфере Ar. Реакционную смесь затем нагревали до 60 С и перемешивали при этой температуре в атмосфере Ar в течение 1 ч. Реакционную смесь охлаждали до температуры окружающей среды и добавляли дополнительный эквтвалент гидрида натрия (9,11 мг, 0,228 ммоль) с перемешиванием в атмосфере аргона. Реакционную смесь перемешивали при температуре окружающей среды в течение 72 ч. Способ 2. Раствор 1,1-диметилэтил[1-(2-хлор-1-[(фенилметил)окси]метилэтил)-4-пиперидинил](2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-илметил)карбамата (343 мг, 0,645 ммоль) в DMF (10 мл) добавляли по каплям к раствору натриевой соли 7-(метилокси)-3,4-дигидро-1,8-нафтиридин-2(1H)-она(138 мг, 0,774 ммоль) (полученной добавлением гидрида натрия (60%, 38,7 мг, 0,967 ммоль) к 7-(метилокси)-3,4-дигидро-1, 8-нафтиридин-2(1 Н)-ону (138 мг, 0,774 ммоль) (в отношении получения см. пример 5 (е в DMF (10 мл. Раствор перемешивали при температуре окружающей среды в течение ночи в атмосфере аргона. Реакционные смеси способа 1 и способа 2 объединяли и DMF удаляли при пониженном давлении. Остаток обрабатывали насыщенным водным раствором (10 мл) бикарбоната натрия и водой (20 мл) и экстрагировали DCM (3100 мл). Объединенные органические слои высушивали (MgSO4), фильтровали и удаляли при пониженном давлении. Сырой продукт хроматографировали, элюируя 0-100%EtOAc/гексан. Соответствующие фракции объединяли, получая две загрузки продукта (загрузка 1: 167 мг, 38% и (загрузка 2: более низкая чистота, 78 мг, 18%).(g) 1,1-Диметилэтил(2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-илметил)[1-(2-гидрокси-1-[7(метилокси)-2-оксо-3,4-дигидро-1,8-нафтиридин-1(2 Н)-ил]метилэтил)-4-пиперидинил]карбамат. Раствор 1,1-диметилэтил(2,3-дигидро[1,4]диоксино[2,3-с]пиридин-7-илметил)[1-(2-[7-(метилокси)2-оксо-3,4-дигидро-1,8-нафтиридин-1(2 Н)-ил]-1-[(фенилметил)окси]метилэтил)-4-пиперидинил]карбамата (167 мг, 0,248 ммоль) в этаноле (20 мл) гидрировали при давлении 1 атм водорода в течение приблизительно 9 дней. Реакционную смесь фильтровали через целит и промывали этанолом. Объединенный фильтрат и смывы упаривали при пониженном давлении, получая продукт (162 мг, 91%).(162 мг, 0,278 ммоль) в DCM (10 мл) в атмосфере аргона охлаждали до 0 С и обрабатывали триэтиламином (0,046 мл, 0,333 ммоль) и метансульфонилхлоридом (0,026 мл, 0,333 ммоль). Реакционной смеси давали нагреться до температуры окружающей среды и перемешивали при этой температуре в течение 1 ч. Добавляли дополнительно 1,2 экв. триэтиламина (0,046 мл, 0,333 ммоль) и метансульфонилхлорид(0,026 мл, 0,333 ммоль) и раствор перемешивали при температуре окружающей среды в течение ночи. Добавляли дополнительно 1,2 экв. триэтиламина (0,046 мл, 0,333 ммоль) и метансульфонилхлорид(0,026 мл, 0,333 ммоль) и раствор нагревали до 50 С в течение 6 ч. Раствор охлаждали до температуры окружающей среды, добавляли насыщенный водный раствор NaHCO3 (10 мл) и водный слой экстрагировали 20% MeOH/DCM (3100 мл). Органические фазы объединяли, высушивали MgSO4, фильтровали и концентрировали. Сырой продукт затем хроматографировали, элюируя 0-15% MeOH/DCM. Соответствующие фракции объединяли и упаривали при пониженном давлении, получая продукт (80 мг, 48%).(80 мг, 0,145 ммоль) и DDQ (49,4 мг, 0,218 ммоль) в 1,4-диоксане (5 мл) перемешивали при 120 С в течение 2 ч. Добавляли дополнительно 0,5 экв. DDQ (17 мг) и раствор перемешивали еще 2 ч. Смеси давали охладиться до температуры окружающей среды и обрабатывали насыщ. NaHCO3 (10 мл). Водный слой экстрагировали 20% MeOH/DCM (3100 мл). Органические слои объединяли, высушивали MgSO4,- 24015821 фильтровали и концентрировали, получая сырой продукт (64 мг, 83%).(64 мг,0,116 ммоль) в DCM (2 мл) и HCl в 1,4-диоксане (0,291 мл, 1,164 ммоль) перемешивали при температуре окружающей среды в течение 2 ч. Растворители удаляли при пониженном давлении. Сырой продукт добавляли на ионообменную колонку и элюировали МеОН (20 мл) и затем 2 М NH3 в МеОН (15 мл), получая свободное основание целевого соединения (34 мг, 65%). 1 Н ЯМР и LC-MS, идентичные продукту примера 3 А. Свободное основание целевого продукта затем превращали в соль HCl, растворяя в DCM (2 мл) и обрабатывая 1 экв. 1 M HCl в простом эфире. Растворители удаляли при пониженном давлении, получая целевой гидрохлорид. Пример 4. 1-(4-[(7-Бром-3-оксо-3,4-дигидро-2 Н-пиридо[3,2-b][1,4]тиазин-6-илметил)амино]-1 пиперидинилметил)-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион гидрохлорид Смесь 1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9 диона дигидрохлорида (51 мг, 0,136 ммоль) (в отношении получения см. пример 2 (n в DCM (2 мл) и метаноле (0,1 мл) в атмосфере аргона при температуре окружающей среды обрабатывали триэтиламином(60 мкл, 0,438 ммоль) и перемешивали при температуре окружающей среды в течение 0,25 ч, после чего добавляли 7-бром-3-оксо-3,4-дигидро-2 Н-пиридо[3,2-b][1,4]тиазин-6-карбоксальдегид (в отношении синтеза см. WO 2002056882, пример 33 (е (37 мг, 0,136 ммоль). Смесь затем перемешивали при температуре окружающей среды в течение 1 ч, после чего добавляли NaBH(ОАс)3 (86 мг, 0,408 ммоль). Реакционную смесь перемешивали при температуре окружающей среды еще 0,5 ч, после чего добавляли насыщенный водный раствор бикарбоната натрия (20 мл). Смесь экстрагировали смесью 20% метанол/DCM(3100 мл). Органические экстракты высушивали (MgSO4), упаривали и хроматографировали (0-20% метанол/DCM), получая свободное основание целевого соединения в форме прозрачного масла (36 мг,48%).(1,488 г, 1,625 ммоль) и Xantphos (4,5-бис-(дифенилфосфино)-9,9-диметилксантен) (1,880 г, 3,249 ммоль) в сухом дегазированном 1,4-диоксане (800 мл) в атмосфере аргона обрабатывали ультразвуком в течение 0,25 ч и затем обрабатывали 2-хлор-6-(метилокси)пиридином (19,32 мл, 162,494 ммоль). Смесь затем нагревали с обратным холодильником в течение 24 ч. Смесь упаривали, обрабатывали водой (1 л) и экстрагировали 3 DCM (1 л и затем 2500 мл). Органические экстракты высушивали (MgSO4), упаривали и хроматографировали (50-100% DCM/40-60 петролейный эфир, затем 0-5% метанол/DCM), получая целевое соединение в форме твердого вещества желтого цвета (25,191 г, 121,111 ммоль, 75%). Неочищенные фракции повторно хроматографировали (элюируя как выше), получая дополнительное количество продукта (4,990 г, 23,990 ммоль, 15%). Полный выход 90%.(450 мл) в трехгорлой колбе на 1 л с внутренним термометром в атмосфере аргона охлаждали до -78 С и обрабатывали н-бутиллитием (232 мл, 581,847 ммоль) в течение 15 мин и затем давали нагреться до 0 С и перемешивали при 0 С в течение 7 ч. Смесь затем повторно охлаждали до -78 С и обрабатывали 1,2-дибромэтаном (27,3 мл, 317 ммоль) в течение 10 мин и затем раствору давали нагреться до температуры окружающей среды и перемешивали при температуре окружающей среды в течение 30 мин, за которые все образовавшееся твердое вещество снова растворялось. На этой стадии образовывался газ, поэтому на одно из горл колбы помещали газовый барбулятор. Затем за 10 мин аккуратно добавляли воду(100 мл). Затем добавляли дополнительное количество воды (500 мл) и смесь экстрагировали простым диэтиловым эфиром (3500 мл). Объединенные органические растворители затем высушивали (MgSO4),фильтровали, упаривали, получая сырой продукт. Затем его растворяли в теплом этилацетате (100 мл) и оставляли в морозилке в течение ночи. Полученное твердое вещество, которое кристаллизовалось, отфильтровывали, промывали охлажденным льдом простым диэтиловым эфиром (20 мл) и высушивали в вакууме, получая продукт в форме твердого вещества белого цвета (45,660 г, 159,011 ммоль, 60%-ный выход). Фильтрат упаривали и остаток хроматографировали (0-25% этилацетат/40-60 петролейный эфир), получая рекуперируемый исходный материал (7,264 г, 34,9 ммоль) и продукт в форме твердого вещества белого цвета (8,038 г, 27,992 ммоль, 10%-ный выход). Продукты от перекристаллизации и хроматографии на силикагеле были идентичны по-своему ЯМР и LC-MS и поэтому их объединяли.(47,1 мл, 329 ммоль) и дициклогексилметиламином (64,5 мл, 302 ммоль). Реакционную смесь затем нагревали при 80 С в течение 4 ч и затем при 120 С в течение 3 ч. Реакционную смесь затем упаривали,добавляли воду (1000 мл) и смесь экстрагировали простым диэтиловым эфиром (3500 мл). Объединенные органические растворители затем высушивали (MgSO4), фильтровали, упаривали, получая сырой продукт. Затем его растворяли в DCM (300 мл) и хроматографировали (10-30% этилацетат:40-60 петролейный эфир) и затем высушивали в вакууме, получая продукт в форме твердого вещества белого цвета(43,706 г, 131 ммоль) в этаноле (450 мл) в атмосфере аргона при температуре окружающей среды обрабатывали палладием на угле (5,0 г, 47,0 ммоль) и затем перемешивали при температуре окружающей среды под 1 атм водорода в течение 90 ч. Реакционную смесь затем фильтровали через тонкий слой Кизельгура,смывая продукт дополнительным количеством этанола (200 мл). Растворитель затем упаривали, получая продукт в форме желтого масла (43,549, 99%).(e) 7-(Метилокси)-3,4-дигидро-1,8-нафтиридин-2(1 Н)-он. Смесь 3-[2-[(2,2-диметилпропаноил)амино]-6-(метилокси)-3-пиридинил]бутилпропаноата (86,01 г,256 ммоль) в соляной кислоте (500 мл, 3000 ммоль) (6 М водный раствор) нагревали при 80 С в течение 6 ч. Реакционную смесь охлаждали, обрабатывали водой (500 мл), помещали в 5-литровую коническую колбу и тщательно нейтрализовывали твердым карбонатом калия (около 250 г) (наблюдается активное вскипание). Смесь затем экстрагировали смесью 20% MeOH/DCM (3500 мл). Объединенные органические растворители затем высушивали (MgSO4), фильтровали и упаривали, получая сырой продукт в форме твердого вещества желтого цвета (35,84 г, 79%).(f) 7-(Метилокси)-1-[(2R)-2-оксиранилметил]-3,4-дигидро-1,8-нафтиридин-2(1 Н)-он. Раствор 7-(метилокси)-3,4-дигидро-1,8-нафтиридин-2(1 Н)она (4,974 г, 27,9 ммоль) в DMF (100 мл) при 0 С в атмосфере аргона обрабатывали гидридом натрия (60%, 1,340 г, 33,5 ммоль) и перемешивали при 0 С в течение 20 мин. Реакционную смесь затем обрабатывали (2S)-2-оксиранилметил-3 нитробензолсульфонатом (7,60 г, 29,3 ммоль), перемешивали при 0 С и затем давали нагреться до температуры окружающей среды и перемешивали при температуре окружающей среды в течение 1 ч. Затем добавляли воду (5 мл). Реакционную смесь упаривали, затем добавляли насыщенный водный раствор бикарбоната (500 мл) и смесь экстрагировали DCM (3500 мл). Объединенные органические растворители затем высушивали (MgSO4), фильтровали и упаривали, получая сырой продукт.(g) (1S)-1-(Гидроксиметил)-1,2,5,6-тетрагидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион. Раствор 7-(метилокси)-1-[(2R)-2-оксиранилметил]-3,4-дигидро-1,8-нафтиридин-2(1 Н)она (1,167 г,4,98 ммоль) в DMF (20 мл) в атмосфере аргона нагревали при 120 С в течение 6 ч. Реакционную смесь затем упаривали и хроматографировали (0-20% метанол/DCM), получая продукт в форме твердого вещества оранжевого цвета (339 мг, 31%).MS (ES+) m/z 221 (МН+, 100%). Альтернативно, реакционная смесь может быть нагрета в микроволновом реакторе при 160 С в течение 40 мин.(1S)-1-(гидроксиметил)-1,2,5,6-тетрагидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9 диона (1,909 г, 8,67 ммоль) в DCM (100 мл) при 0 С в атмосфере аргона обрабатывали триэтиламином(1,450 мл, 10,40 ммоль) и затем метансульфонилхлоридом (0,743 мл, 9,54 ммоль) и затем давали нагреться до температуры окружающей среды и перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь затем обрабатывали насыщенным водным раствором бикарбоната (100 мл) и смесь экстрагировали DCM (2100 мл). Объединенные органические растворители затем высушивали(MgSO4), фильтровали и упаривали, получая сырое промежуточное соединение [(2S)-4,9-диоксо-1,2,8,9 тетрагидро-4 Н,7 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-2-ил]метил метансульфонат, Его растворяли в сухом ацетонитриле (100 мл) и затем обрабатывали пиридином (1,402 мл, 17,34 ммоль) и 1,1-диметилэтил-4 пиперидинилкарбаматом (3,47 г, 17,34 ммоль) и нагревали при 70 С в течение 20 ч. Через 20 ч добавляли дополнительное количество 1,1-диметилэтил-4-пиперидинилкарбамата (3,47 г, 17,34 ммоль) и пиридина(1,402 мл, 17,34 ммоль), температуру увеличивали для нагревания с обратным холодильником (нагревающий блок 95 С) и реакционную смесь перемешивали при этой температуре еще 4 ч. Реакционную смесь затем упаривали, затем добавляли насыщенный водный раствор NaHCO3 (200 мл) и смесь экстрагировали DCM (3200 мл). Объединенные органические растворители затем высушивали (MgSO4),фильтровали и упаривали, получая сырой продукт в форме твердого вещества коричневого цвета.(i) 1,1-Диметилэтил(1-[(1R)-4,9-диоксо-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-1 ил]метил-4-пиперидинил)карбамат. Раствор 1,1-диметилэтил(1-[(2R)-4,9-диоксо-1,2,8,9-тетрагидро-4 Н,7 Н-имидазо[1,2,3-ij]-1,8 нафтиридин-2-ил]метил-4-пиперидинил)карбамата (5,710 г, 14,19 ммоль) в 1,4-диоксане (50 мл) при температуре окружающей среды обрабатывали DDQ (4,83 г, 21,28 ммоль) и затем нагревали при 120 С в течение 1 ч. Реакционную смесь затем охлаждали до температуры окружающей среды. Реакционную смесь обрабатывали насыщенным водным раствором K2CO3 (5%, 1000 мл) и экстрагировали DCM(3500 мл). Объединенные органические растворители затем высушивали (MgSO4), фильтровали и упаривали, получая сырой продукт в форме твердого вещества коричневого цвета. Реакцию повторяли, используя дополнительную часть карбамата (2,889 г, 7,18 ммоль) в 1,4-диоксане (50 мл) с DDQ (2,444 г,10,77 ммоль). Реакцию осуществляли и обработку проводили, как описано выше, и объединенные остатки хроматографировали (0-100% этилацетат:40-60 петролейный эфир, затем 0-20% метанол:этилацетат),получая продукт в форме твердого вещества коричневого цвета (1,532 г).(j) (1R)-1-[(Амино-1-пиперидинил)метил]-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9 дион дигидрохлорид. Раствор 1,1-диметилэтил(1-[(1R)-4,9-диоксо-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин 1-ил]метил-4-пиперидинил)карбамата (1,532 г, 3,83 ммоль) в хлороформе (20 мл) в атмосфере аргона при температуре окружающей среды обрабатывали 4 М HCl в 1,4-диоксане (10 мл, 40,0 ммоль) и перемешивали при температуре окружающей среды в течение 0,25 ч. Затем добавляли метанол (20 мл) и реакционную смесь перемешивали еще 0,25 ч. Реакционную смесь затем упаривали и растирали с простым диэтиловым эфиром (20 мл). Твердое вещество затем высушивали в вакууме, получая неочищенный продукт в форме твердого вещества коричневого цвета (1,443 г, 101%).MS (ES+) m/z 301 (МН+, 100%). 1-[(4-Амино-1-пиперидинил)метил]-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион дигидрохлорид, полученный этим общим способом (пример 5 (a)-(j, анализировали хиральной ВЭЖХ(Chiralpak AS-H (5 мкм) и было обнаружено, что он является единственным энантиомером, предположительно R.(20 мл) и метаноле (1 мл) при температуре окружающей среды в атмосфере аргона обрабатывали триэтиламином (0,644 мл, 4,62 ммоль) и перемешивали при температуре окружающей среды в течение 0,25 ч. Раствор затем обрабатывали [1,3]оксатиоло[5,4-с]пиридин-6-карбальдегидом (в отношении синте- 27015821 за см. WO 2004058144, пример 61) (258 мг, 1,540 ммоль) и перемешивали еще 0,5 ч. Раствор затем обрабатывали NaBH(OAc)3 (979 мг, 4,62 ммоль) и перемешивали при температуре окружающей среды в течение 0,5 ч. Реакционную смесь затем обрабатывали насыщенным водным раствором NaHCO3 (100 мл) и экстрагировали смесью 20% метанол/DCM (3200 мл). Объединенные органические экстракты высушивали (MgSO4), фильтровали, упаривали и хроматографировали (0-20% метанол/DCM), получая свободное основание целевого соединения в форме твердого вещества светло-коричневого цвета (574 мг,1,273 ммоль, 83%). 1 Н ЯМР (CDCl3, 250 МГц)1,25-1,45 (2 Н, м), 1,75-1,95 (2 Н, м), 2,20-2,45 (2 Н, м), 2,45-2,55 (1 Н, м),2,60-2,75 (2 Н, м), 2,90-3,00 (1 Н, м), 3,05-3,15 (1 Н, дд), 3,85 (2 Н, с), 4,30-4,40 (1 Н, м), 4,55-4,65 (1 Н, м),4,95-5,05 (1 Н, м), 5,75 (2 Н, с), 6,25 (1 Н, м), 6,30 (1 Н, м), 7,20 (1 Н, с), 7,45-7,52 (2 Н, м), 8,00 (1 Н, с).MS (ES+) m/z 452 (МН+). Свободное основание в DCM/MeOH 2:1 (15 мл) обрабатывали 1 М HCl в простом диэтиловом эфире и затем упаривали, получая целевой гидрохлорид. Пример 5 В. (1R)-1-(4-[([1,3]Оксатиоло[5,4-с]пиридин-6-илметил)амино]-1-пиперидинилметил)1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион бензоат.(1R)-1-(4-[([1,3]Оксатиоло[5,4-с]пиридин-6-илметил)амино]-1-пиперидинилметил)-1,2-дигидро 4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион растворяли в метаноле и обрабатывали бензойной кислотой (1 экв.). Концентрация, обработка простым диэтиловым эфиром и выпаривание растворителей при пониженном давлении дали продукт в форме бензоата. Пример 5 С. (1R)-1-(4-[([1,3]Оксатиоло[5,4-с]пиридин-6-илметил)амино]-1-пиперидинилметил)1,2-дигидро-4H,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион дитрифторацетат.(1R)-1-(4-[([1,3]Оксатиоло[5,4-с]пиридин-6-илметил)амино]-1-пиперидинилметил)-1,2-дигидро 3 Н,8 Н-2 а,5,8 а-триазааценафтилен-3,8-дион гидрохлорид в элюенте [10% MeCN в воде (содержание TFA 0,1%)] наносили на колонку для препаративной ВЭЖХ с обратной фазой. Содержащие продукт фракции объединяли, концентрировали и концентрат лиофилизировали. Продукт выделяли в форме липкой белой пены после высушивания (в течение выходных) над Р 2 О 5. Пример 6 А.(1R)-1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8 нафтиридин-4,9-диона дигидрохлорида (511 мг, 1,369 ммоль) (в отношении получения см. пример 5 (j в хлороформе (20 мл) и метаноле (1 мл) при температуре окружающей среды в атмосфере аргона обрабатывали триэтиламином (0,572 мл, 4,11 ммоль) и перемешивали при температуре окружающей среды в течение 0,25 ч. Раствор затем обрабатывали 2,3-дигидро[1,4]оксатиино[2,3-с]пиридин-7-карбальдегидом(в отношении синтеза см. WO 2004058144, пример 60) (248 мг, 1,369 ммоль) и перемешивали еще 0,5 ч. Раствор затем обрабатывали NaBH(OAc)3 (870 мг, 4,11 ммоль) и перемешивали при температуре окружающей среды в течение 0,5 ч. Реакционную смесь затем обрабатывали насыщенным водным растворомNaHCO3 (100 мл) и экстрагировали смесью 20% метанол/DCM (3200 мл). Объединенные органические экстракты высушивали (MgSO4), фильтровали, упаривали и хроматографировали (0-20% метанол/DCM),получая свободное основание целевого соединения в форме твердого вещества светло-коричневого цвета(1 Н, с), 7,42-7,53 (2 Н, м), 8,00 (1 Н, с). Свободное основание в DCM/MeOH 2:1 (15 мл) обрабатывали 1 экв. 1 М HCl в простом диэтиловом эфире и затем упаривали, получая целевой гидрохлорид. Пример 6 В.(1R)-1-(4-[(2,3-Дигидро[1,4]оксатиино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинилметил)1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион растворяли в метаноле и обрабатывали бензойной кислотой (1 экв.). Выпаривание растворителей при пониженном давлении дало продукт в форме бензоата. Пример 6 С. (1R)-1-(4-[(2,3-Дигидро[1,4]оксатиино[2,3-с]пиридин-7-илметил)амино]-1-пиперидинилметил)-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион дитрифторацетат.(1R)-1-(4-[([1,3]Оксатиоло[5,4-c]пиридин-6-илметил)амино]-1-пиперидинилметил)-1,2-дигидро 3 Н,8 Н-2 а,5,8 а-триазааценафтилен-3,8-дион гидрохлорид наносили на колонку для препаративной ВЭЖХ с обратной фазой в смеси 10% MeCN в воде, содержащей 0,1% TFA. Содержащие продукт фракции объ- 28015821 единяли, концентрировали и концентрат лиофилизировали. Продукт (соль бис-TFA) выделяли в форме твердого вещества белого цвета после высушивания над Р 2 О 5. Пример 7. (1R)-1-(4-[(5,6,7,8-Тетрагидро-3-изохинолинилметил)амино]-1-пиперидинилметил)1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион гидрохлорид(a) 5,6,7,8-Тетрагидро-3-изохинолинэтилкарбоксилат. Раствор 1,7-октадиина (4,00 мл, 30,1 ммоль) и этилцианоформиата (2,95 мл, 30,1 ммоль) в сухом дегазированном 1,4-диоксане (500 мл) в атмосфере аргона при температуре окружающей среды обрабатывали циклопентадиенил-кобальт(I)-дикарбонилом (0,814 г, 4,52 ммоль) и затем нагревали с обратным холодильником в течение 18 ч. Реакционную смесь затем упаривали, обрабатывали толуолом (100 мл),вторично упаривали, растворяли в DCM (100 мл), фильтровали через тонкий слой Кизельгура, элюируяDCM, органические экстракты упаривали, хроматографировали (0-100% DCM:40-60 петролейный эфир,затем 0-10% метанол/DCM), получая продукт в форме неочищенного коричневого масла (1,27 г, 21%).-78 С в атмосфере аргона обрабатывали LiAlH4 (1 M раствор в THF, 6,19 мл, 6,19 ммоль) и давали нагреться до температуры окружающей среды. После 10 мин при температуре окружающей среды последовательно добавляли воду (1 мл), 2 М водный раствор NaOH (1 мл) и воду (1 мл) и смесь перемешивали при температуре окружающей среды в течение 0,5 ч. Смесь затем фильтровали через тонкий слой Кизельгура, элюируя THF (50 мл), органические экстракты затем упаривали, хроматографировали (0-20% метанол/DCM), получая продукт в форме оранжевого масла (0,572 г, 57%).(c) 5,6,7,8-Тетрагидро-3-изохинолинкарбальдегид. Раствор 5,6,7,8-тетрагидро-3-изохинолинилметанола (572 мг, 3,50 ммоль) в (DCM) (10 мл) при температуре окружающей среды в атмосфере аргона обрабатывали диоксидом марганца (3,047 г,35,0 ммоль) и затем перемешивали при температуре окружающей среды в течение 2 ч. Реакционную смесь затем фильтровали через тонкий слой Кизельгура, элюируя DCM (50 мл), органические экстракты упаривали, получая сырой продукт в форме коричневого масла (435 мг, 77%).(1R)-1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8 нафтиридин-4,9-диона дигидрохлорида (87 мг, 0,233 ммоль) (в отношении получения см. пример 5 (j в хлороформе (5 мл) и метаноле (0,2 мл) при температуре окружающей среды в атмосфере аргона обрабатывали триэтиламином (97 мкл, 0,699 ммоль) и перемешивали при температуре окружающей среды в течение 0,25 ч. Раствор затем обрабатывали 5,6,7,8-тетрагидро-3-изохинолинкарбальдегидом (37,6 мг,0,233 ммоль) и перемешивали еще 0,5 ч. Раствор затем обрабатывали NaBH(OAc)3 (148 мг, 0,699 ммоль) и перемешивали при температуре окружающей среды в течение 0,5 ч. Реакционную смесь затем обрабатывали насыщенным водным NaHCO3 (100 мл) и экстрагировали смесью 20% метанол/DCM (3200 мл). Объединенные органические экстракты высушивали (MgSO4), фильтровали, упаривали и хроматографировали (0-20% метанол/DCM), получая свободное основание целевого соединения в форме твердого вещества светло-коричневого цвета (66 мг, 64%).(1 Н, с), 7,47-7,59 (2H, м), 8,21 (1 Н, с). Свободное основание в DCM/MeOH 2:1 (15 мл) обрабатывали 1 экв. 1 М HCl в простом диэтиловом эфире и затем упаривали, получая целевой гидрохлорид. Пример 8. (1R)-1-(4-[(6,7-Дигидро-5 Н-циклопента[с]пиридин-3-илметил)амино]-1-пиперидинилметил)-1,2-дигидро-4 Н,9 Н-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион гидрохлорид(a) 6,7-Дигидро-5 Н-циклопента[с]пиридин-3-этилкарбоксилат. Раствор 1,6-гептадиина (1,242 мл, 10,85 ммоль) и этилцианоформиата (1,063 мл, 10,85 ммоль) в сухом дегазированном 1,4-диоксане (100 мл) в атмосфере аргона при температуре окружающей среды обрабатывали циклопентадиенил-кобальт(I)-дикарбонилом (0,293 г, 1,628 ммоль) и затем нагревали с об- 29
МПК / Метки
МПК: C07D 487/16, C07D 471/16, C07D 519/00, A61K 31/4985, A61P 31/00
Метки: агентов, азотсодержащие, качестве, антибактериальных, трициклические, соединения
Код ссылки
<a href="https://eas.patents.su/30-15821-triciklicheskie-azotsoderzhashhie-soedineniya-v-kachestve-antibakterialnyh-agentov.html" rel="bookmark" title="База патентов Евразийского Союза">Трициклические азотсодержащие соединения в качестве антибактериальных агентов</a>
Производные хинолина в качестве антибактериальных агентов

Номер патента: 14431
Опубликовано: 30.12.2010
Авторы: Мотт Магали Мадлен Симон, Андрис Кунрад Йозеф Лодевейк Марсель, Гийемон Жером Эмиль Жорж, Коул Анил, Лансуа Давид Франсис Ален
МПК: A61K 31/4353, C07D 215/227, A61P 31/04...
Метки: качестве, производные, антибактериальных, агентов, хинолина
Формула / Реферат:
1. Применение соединения для получения лекарственного средства для лечения бактериальной инфекции, причем указанное соединение является соединением формулы (Ia)его N-оксидом или его стереохимически изомерной формой, гдеА- обозначает фармацевтически приемлемый противоион;R1 обозначает водород или галоген;р является целым числом, равным 1, 2, 3 или 4;R2 обозначает алкилокси или алкилтио;R3 обозначает Ar, Ar-алкил или Het;q является целым числом,...
Производные хинолина в качестве антибактериальных агентов

Номер патента: 14163
Опубликовано: 29.10.2010
Авторы: Коул Анил, Андрис Кунрад Йозеф Лодевейк Марсель, Мотт Магали Мадлен Симон, Гийемон Жером Эмиль Жорж
МПК: C07D 215/22, A61P 31/04, A61K 31/4353...
Метки: хинолина, агентов, производные, качестве, антибактериальных
Формула / Реферат:
1. Применение соединения для получения лекарственного средства для лечения бактериальной инфекции, причем указанное соединение является соединением формулы (Ia)его фармацевтически приемлемой аддитивной солью кислоты или основания, его стереохимически изомерной формой или его N-оксидной формой,где R1означает водород, галоген, фенил, пиридинил, тиенил, фуранил, алкил или алкилокси;р является целым числом, равным 1, 2, 3 или 4;R2 означает...
Производные гигромицина а в качестве антибактериальных агентов

Номер патента: 4303
Опубликовано: 26.02.2004
Автор: Хейуард Маттью Меррилл
МПК: A61K 31/70, C07H 15/203, A61P 31/04...
Метки: антибактериальных, агентов, качестве, производные, гигромицина
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемые соль, пролекарство или сольват, где каждый X1 представляет собой -CR6R7- и X2 представляет собой O; R1 представляет собой H или OH; R2 представляет собой H или C1-C6алкил; каждый R3 независимо выбран из H, -(CR6R7)t(C6-C10арил), -(CR6R7)t(4-10-членный гетероцикл), -C(O)(CR6R7)t(C6-C10арил) и -C(O)NR6(CR6R7)t(C6-C10арил), где t представляет собой целое число в диапазоне от 0 до 1, и...
Производные хинолина в качестве антибактериальных агентов

Номер патента: 10601
Опубликовано: 30.10.2008
Авторы: Паскье Элизабет Тереза Жанна, Андрис Кунрад Йозеф Лодевейк Марсель, Коул Анил, Гийемон Жером Эмиль Жорж
МПК: C07D 215/26, A61K 31/47, A61K 31/4709...
Метки: хинолина, производные, качестве, антибактериальных, агентов
Формула / Реферат:
1. Применение соединения для изготовления лекарственного средства для лечения бактериальной инфекции, причем соединение является соединением формулы его фармацевтически приемлемой кислотно- или основно-аддитивной солью, его стереохимически изомерной или N-оксидной формой, где R1 представляет собой водород, галоген, полигалоC1-6алкил, C1-6алкил, Ar или Het; р представляет собой целое число, равное 1 или 2; R2 представляет собой C1-6алкилокси,...
Трициклические соединения, представляющие собой ингибиторы протеинкиназ, для увеличения эффективности противоопухолевых агентов и лучевой терапии

Номер патента: 9337
Опубликовано: 28.12.2007
Авторы: Уэнг Йонг, Руй Юджин Йюэнджин, Бенедикт Сюзанн Притчетт, Жу Джинджианг, Нинковиц Саша, Тенг Мин, Уэнг Фен, Беннетт Майкл Джон
МПК: A61P 35/00, A61K 31/55, C07D 487/06...
Метки: противоопухолевых, эффективности, трициклические, ингибиторы, увеличения, соединения, представляющие, протеинкиназ, агентов, терапии, собой, лучевой
Формула / Реферат:
1. Соединение формулы где X представляет собой =O; А представляет собой =CR1-; группа -Y-Z- имеет формулу-О-CH2- или -N=CH-; R1 представляет собой: (а) (C1-C8)алкил; (б) -C(=O)-R5; (в) -C(=O)-NR6R7; или (г) R35 или R36, (С2-С8)алкенил или (С2-С8)алкинил {где каждый из указанных (С2-С8)алкенила или (С2-С8)алкинила не замещен или замещен 1-4 заместителями, независимо выбранными из группы, состоящей из F, Cl, ОН, -NH2, R40 и R42}; R2 представляет...
Предыдущий патент: Производные пиперазинила, предназначенные для лечения заболеваний, опосредованных рецептором gpr38
Следующий патент: Биологически активная добавка
Случайный патент: Пиридинаминосульфонил-замещенные бензамиды в качестве ингибиторов цитохрома р450 3а4 (cyp3a4)