Антивирусные соединения
Номер патента: 3327
Опубликовано: 24.04.2003
Авторы: Нореен Рольф, Хегберг Марита, Энгельхардт Пер, Сальберг Кристер

























Формула / Реферат
1. Соединение формулы I

где Rx является цианогруппой или атомом брома;
R1 является атомом галогена;
R2 является C1-C3 алкилом;
R является атомом водорода или группой формулы

где кольцо X представляет собой фенил или пиридил,
R3 и R4 независимо выбраны из H, OH, амино и замещенных аминогрупп;
n равно независимо 0, 1 или 2;
p равно 0 или 1;
и фармацевтически приемлемые соли этих соединений.
2. Соединение по п.1, в котором R1 является атомом фтора.
3. Соединение по п.1, в котором R2 является этилом.
4. Соединение по п.1, представляющее собой, по меньшей мере, на 60%, предпочтительно, по меньшей мере, на 90%, 1S, 2S энантиомерную форму.
5. Соединение по п.1, в котором Rx является цианогруппой.
6. Соединение по п.1, которое представляет собой соединение формулы II

где кольцо X, Rx, R1, R2, R3, R4, n и p определены в п.1, и фармацевтически приемлемые соли этих соединений.
7. Соединение по п.6, в котором кольцо X означает пиридил.
8. Соединение по п.7, в котором кольцо X означает фенил.
9. Соединение по п.6, в котором кольцо X означает пирид-2-ил или предпочтительно пирид-3-ил.
10. Соединение по п.6, в котором R3 означает -NH2-, -NHCH3, -NHCH2CH3 или -N(CH3)2.
11. Соединение по п.6, в котором R3 находится в мета-положении относительно карбонильной группы, особенно в том случае, когда кольцо X является фенилом, либо в котором R3 находится в пара-положении относительно карбонильной группы, особенно в том случае, когда кольцо X является пиридилом.
12. Соединение по п.6, в котором -(CH2)n- и/или -(CH2)p- отсутствуют, то есть p и/или n равно 0.
13. Соединение по п.6, в котором Rx является цианогруппой.
14. Соединение по п.6, в котором R1 является атомом фтора.
15. Соединение по п.6, в котором R2 является этилом.
16. Соединение по п.6, которое, по меньшей мере, на 60%, предпочтительно, по меньшей мере, на 90%, представлено 1S,2S энантиомерной формой.
17. Соединение по п.1, выбранное из следующей группы соединений:
(1S,2S)-N-[цис-2-(6-фтор,2-гидрокси,3-пропионилфенил)циклопропил]-N'-(5-цианопирид-2-ил)мочевина,
(1R,2R)-N-[цис-2-(6-фтор,2-гидрокси,3-пропионилфенил)циклопропил]-N'-(5-цианопирид-2-ил)мочевина,
(1S,2S)-N-[цис-2-(6-фтор,2-гидрокси,3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина и
(1R,2R)-N-[цис-2-(6-фтор,2-гидрокси,3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина
и фармацевтически приемлемые соли этих соединений.
18. Соединение по п.17, представляющее собой (1S,2S)-N-[цис-2-(6-фтор,2-гидрокси,3-пропионилфенил)циклопропил]-N'-(5-цианопирид-2-ил)мочевину или ее фармацевтически приемлемую соль.
19. Соединение по п.1, выбранное из группы, включающей в себя следующие соединения:
(1S,2S)-N-[цис-2-(2-(3-аминофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-цианопирид-2-ил)мочевина,
(1S,2S)-N-[цис-2-(2-(3-этиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-цианопирид-2-ил)мочевина,
(1S,2S)-N-[цис-2-(2-(3-диметиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-цианопирид-2-ил) мочевина,
(1S,2S)-N-{цис-2-[6-фтор-3-пропионил-2-(6-метиламинопирид-3-илкарбонилокси)фенил]циклопропил}-N'-(5-цианопирид-2-ил)мочевина,
(1R,2R)-N-[цис-2-(2-(3-аминофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-цианопирид-2-ил)мочевина,
(1R,2R)-N-[цис-2-(2-(3-этиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-цианопирид-2-ил)мочевина,
(1R,2R)-N-[цис-2-(2-(3-диметиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-цианопирид-2-ил) мочевина,
(1R,2R)-N-{цис-2-[6-фтор-3-пропионил-2-(6-метиламинопирид-3-илкарбонилокси)фенил]циклопропил}-N'-(5-цианопирид-2-ил)-мочевина,
(1S,2S)-N-[цис-2-(2-(3-аминофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина,
(1S,2S)-N-[цис-2-(2-(3-этиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина,
(1S,2S)-N-[цис-2-(2-(3-диметиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина,
(1S,2S)-N-{цис-2-[6-фтор-3-пропионил-2-(6-метиламинопирид-3-илкарбонилокси)фенил]циклопропил}-N'-(5-бромпирид-2-ил)мочевина,
(1R,2R)-N-[цис-2-(2-(3-аминофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина,
(1R,2R)-N-[цис-2-(2-(3-этиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина,
(1R,2R)-N-[цис-2-(2-(3-диметиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина,
(1R,2R)-N-{цис-2-[6-фтор-3-пропионил-2-(6-метиламинопирид-3-илкарбонилокси)фенил]циклопропил}-N'-(5-бромпирид-2-ил)мочевина
и фармацевтически приемлемые соли этих соединений.
20. Соединение по п.19, представляющее (1S,2S)-N-{цис-2-[6-фтор-3-пропионил-2-(6-метиламинопирид-3-илкарбонилокси)фенил]циклопропил}-N'-(5-цианопирид-2-ил)мочевину и ее фармацевтически приемлемую соль.
21. Соединение по п.19, представляющее собой (1S,2S)-N-[цис-2-(2-(3-аминофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевину и ее фармацевтически приемлемую соль.
22. Фармацевтическая композиция, включающая соединение по любому из пп.1-21 и фармацевтически приемлемый носитель или разбавитель.
23. Композиция по п.22, дополнительно включающая от одного до трех дополнительных антиретровирусных средств.
24. Композиция по п.23, в которой дополнительным антиретровирусным средством является средство, выбранное из группы, включающей AZT, ddI, ddC, D4T, 3TC, адефовир, дипивоксил, абакавир, бис-POC-PMPA, фоскарнет, оксимочевину, эфавиренц, тровирдин, невирапин, делавирдин, PFA, H2G, ABT 606, ритонавир, сахинавир, индинавир, ампренавир (Вертекс VX 478), Mitsubishi MKC-442 и нелфинавир.
25. Соединение по любому из пп.1-21 для применения в антивирусной терапии.
26. Применение соединения по любому из пп.1-21 в качестве активного вещества лекарственного средства для лечения или профилактики ВИЧ.
27. Способ подавления или предотвращения ВИЧ инфекции, заключающийся во введении субъекту, который в этом нуждается, эффективного количества соединения по пп.1 или 6.
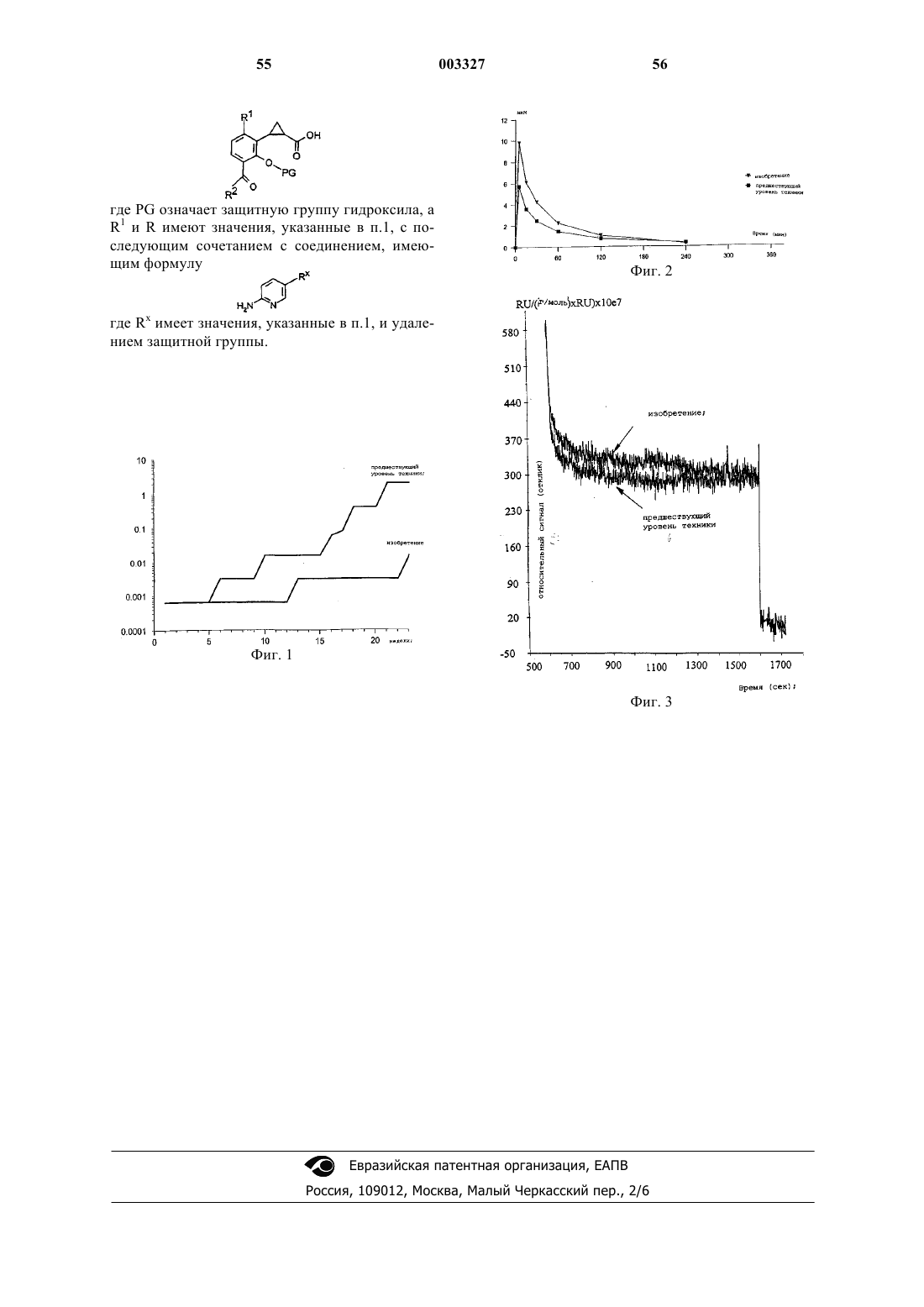
28. Способ получения соединения формулы I по п.1, где R - водород, включающий перегруппировку Курциуса азида кислоты следующей формулы:

где PG означает защитную группу гидроксила, a R1 и R имеют значения, указанные в п.1, с последующим сочетанием с соединением, имеющим формулу

где Rx имеет значения, указанные в п.1, и удалением защитной группы.
Текст
1 Область, к которой относится данное изобретение Это изобретение относится к области антивирусных соединений и, в частности, ингибиторов обратной транскриптазы ВИЧ. В изобретении представлены новые соединения, фармацевтические композиции, включающие в себя эти соединения, и способы ингибирования ВИЧ в результате их применения. Предпосылки изобретения Большинство фармацевтических препаратов, которые проявляют при лечении ВИЧинфекций приемлемую клиническую активность в ингибировании обратной транскриптазы ВИЧ, являются аналогами нуклеозидов, такими как AZT, ddI, ddC и D4T. Эти аналоги нуклеозидов не столь специфичны, как хотелось бы, и поэтому вынужденно используют относительно высокие уровни доз при их введении. При таких уровнях доз аналоги нуклеозидов становятся весьма токсичными, что служит ограничением для их длительного применения. Для того, чтобы преодолеть эти проблемы,связанные со специфичностью и токсичностью,было получено несколько ингибиторов обратной транскриптазы ВИЧ, не относящихся к нуклеозидам. Например, TIBO, обратная транскриптаза производства Janssen, подавляет ВИЧ при наномолярных концентрациях и не проявляет существенной токсичности при клиническом применении. Оба препарата, TIBO и ингибитор обратной транскриптазы не нуклеотидного типа - невирапин, быстро были переведены в фазу II клинических испытаний на пациентах. Однако, вскоре стало очевидным, что эти ненуклеозидные ингибиторы способствуют быстрому отбору in vivo мутантов ВИЧ, которые становятся резистентными к обычно используемым дозам соответствующих ингибиторов. Например, в случае невирапина, вирус, выделенный из сыворотки больных только после четырехнедельной терапии, был в 100 раз менее чувствительным к лекарственному препарату, по сравнению с вирусом, выделенным из сыворотки больных, не подвергавшихся терапии (DragDesign and Discovery 1992, 8, pp. 255-263). Сходная картина наблюдается и для других ненуклеозидных ингибиторов ОТ, с которыми начали клинические испытания, L-697661 Merck и делавирдин (U-87201) Upjohn. А именно, проявляя обнадеживающие результаты при определении активности in vitro, они быстро вызывают появление резистентных мутантов ВИЧ при введении пациентам. Тем не менее, несмотря на этот недостаток, невирапин и делавирдин были недавно зарегистрированы как средства для клинического применения, хотя и ограниченно только при определенных схемах совместного введения с другими препаратами, что является попыткой замедлить развитие резистентности. В международной заявкеWO 95/06034 описана серия новых производных мочевины, 003327 2 которые in vitro проявляют хорошую активность, направленную против обратной транскриптазы ВИЧ, и хорошие результаты по ингибированию репликации ВИЧ в культуре клеток. Однако практическое применение соединений,описанных в заявке WO 95/06034, затруднено из-за их неудовлетворительных фармакокинетических характеристик. Кроме того, как и в случае со многими другими ненуклеозидными ингибиторами обратной транскриптазы, для соединений, описанных в заявке WO 95/06034,сохраняется необходимость улучшить показатель, имеющий ключевое значение, а именно,добиться медленного развития резистентности к этим соединениям и приемлемого проявления активности против мутантов ВИЧ, продуцированных использованием других антивирусных препаратов. В стендовом сообщении berg et al. в 1995 г. ICAR в Санта-Фе, среди других соединений представлено рацемическое соединение,номинально входящее в группу соединений в уже упоминавшейся выше заявке WO 95/06034,и имеющее следующую формулу: При этом в тот момент описанное выше соединение считалось менее интересным, чем варианты тиомочевины, имеющие фенильное кольцо, несущее метокси/ацетильную группу. Однако, авторы данного изобретения обнаружили, что при альтернативном варианте замещения улучшается картина развития резистентности по сравнению с развитием резистентности к соединениям предшествующего уровня техники, в сочетании с хорошими фармакокинетическими характеристиками и пролонгированным периодом развития вирусной резистентности. Таким образом, в данном изобретении представлены ингибиторы, которые сочетают в себе высокую специфичность, присущую ненуклеозидным ингибиторам, с полезными для клинического применения свойствами, которые отсутствуют у всех ингибиторов предшествующего уровня техники. Краткое описание изобретения В соответствии со сказанным, изобретением предложены соединения, имеющие формулу IR1 представляет собой атом галогена;Rx является цианогруппой или атомом брома; 3 и фармацевтически приемлемые соли этих соединений и пролекарства. Далее, в данном изобретении представлены фармацевтические композиции, в состав которых входят соединения, имеющие формулу I,и фармацевтически приемлемые носители или разбавители для этих соединений. Дополнительные аспекты данного изобретения состоят в представлении способов ингибирования ВИЧ,которые заключаются во введении соединения,имеющего формулу I, субъекту, пораженному вирусом ВИЧ. Кроме того, изобретение распространяется на применение соединений, имеющих формулу I, в терапии, а именно для получения лекарственных препаратов для лечения ВИЧ-инфекций. Согласно схеме лечения инфекций, вызванных ВИЧ, соединения, имеющие формулу I,предпочтительно вводятся в таком количестве,чтобы их уровень в плазме достигал примерно от 10 до 1000 нМ и более предпочтительно от 100 до 500 нМ. Это соответствует величине применяемой дозы, в зависимости от биологической доступности композиции, порядка от 0,01 до 10 мг/кг/сутки, предпочтительно от 0,1 до 2 мг/кг/сутки. В обычном случае, для нормального взрослого следует выбирать величину применяемой дозы примерно от 0,05 до 5 г в сутки, предпочтительно от 0,1 до 2 г, например 500-750 мг в сутки, и при этом использовать от одной до четырех стандартных доз. Из соединений по п.1 формулы изобретения предпочтительна, в частности по фармакокинетическим параметрам, группа соединений,имеющих формулу IA где R1 и R2 означают те же группы, что указаны выше, в том числе и фармацевтически приемлемые соли этих соединений и пролекарства. Другая предпочтительная, в частности в отношении простоты образования пролекарства,группа соединений, имеющих формулу I, включает в себя соединения, в которых Rx является атомом брома.R1 является предпочтительно атомом хлора и более предпочтительно - атомом фтора. Подходящими R2 группами являются метил, изопропил, н-пропил и, предпочтительно, этил. Как указано выше, кольцо циклопропила имеет цис-конфигурацию, которая допускает существование двух энантиомеров, 1S,2S и 1R,2R (соответственно и нетрадиционно названных 2R, 1S и 2S,1R в патенте Швеции 980016-7 и патенте Швеции 9800113-4): 4 Каждый из этих энантиомеров является соединением с сильной антиретровирусной активностью, хотя для различных энантиомеров могут быть выявлены некоторые тонкие различия по физиологическим свойствам. Например,энантиомеры 1S,2S и 1R,2R в системе Р 450 могут демонстрировать различные типы метаболизма. В частности, предпочтителен 1S,2S энантиомер соединения, в котором Rx является цианогруппой, поскольку он, по-видимому, является уникальным по своей способности обходить ключевые компоненты системы Р 450. Другие средства против ретровирусов, такие как ингибитор протеазы ВИЧ ритонавир, широко взаимодействуют с системой Р 450, что приводит к появлению ряда нежелательных физиологических ответов, включая значительные изменения в метаболизме других совместно введенных лекарственных средств. Это, в частности, касается фармацевтических препаратов, которые вводятся при хронических инфекциях, в тех случаях, когда предполагается, что пациенты будут принимать большое количество фармацевтических препаратов годами, если не десятилетиями. Пригодные для применения пролекарства на основе соединений, имеющих формулу I,включают в себя соединения, которые можно представить формулой IIR1, R2 и Rx означают те же группы, которые указаны выше;R3 и R4 вместе означают конденсированное 5- или 6-членное кольцо, имеющее 0-2 гетероатома и/или 0-2 ненасыщенных связи и/или 0-2 заместителя;NR R ; Х и окружающее его кольцо означает 5 или 6-членное кольцо, имеющее от 0 до 3 ненасыщенных связей и/или от 0 до 3 любых гетероатомов из следующей группы: S, О и N; 5 и фармацевтически приемлемые соли этих соединений. Соответствующие пролекарства на основе соединений, в которых Rx является атомом хлора, составляют следующий аспект изобретения. Кольцевая структура, внутри которой находится X, упоминаемая в дальнейшем как Хкольцо, может быть насыщенной или иметь 1-3 ненасыщенных связи, включая циклы ароматического типа. Предпочтительно Х-кольцами могут быть кольца циклогексанила или циклогексенила и более предпочтительно фенильное кольцо. Другими предпочтительными Хкольцами являются морфолино или более предпочтительно пиридильное кольцо. В альтернативном случае Х-кольцо может означать пятичленное кольцо, такое как пентенил или пирролил. К приемлемым конденсированным циклическим системам в качестве Х-кольца в случае,когда R3 и R4 объединены с образованием цикла, необязательно содержащего гетероатомы,относятся следующие циклические системы: нафтил, хинолил, тетрагидроизохинолил, индолил или бензимидазол. К замещающим циклам,пригодным в качестве Х-кольца в том случае,когда R4 и R5 объединены с образованием цикла,относятся морфолино- и пиперидиноциклы. Эти конденсированные или замещающие циклы необязательно могут иметь такие заместители как атомы галогенов, галогенметил, аминогруппа,такая как (СНm)nNR5R6, C(=O)NR5R6, гидроксил,гидроксиметил, карбоксил, карбоксиметил, C1-3 алкил, C1-3 алкоксигруппа и им подобные. Х-кольцо может быть отделено от соседнего карбонильного фрагмента группой метилена или этилена, которая необязательно может иметь заместители, такие как атомы галогенов,галогенметил, аминогруппа, аминометил, гидроксильная группа, гидроксиметил, карбоксильная группа, карбоксиметил, C1-3 алкил, C1-3 алкоксигруппа и им подобные. Предпочтительной является структура, в которой Х-кольцо является соседним по отношению к карбонилу. Предпочтительно, чтобы фрагменты, представленные Х-кольцевой системой, R3, R4 и,если имеются R5-R7, имели в некоторой степени основный характер. Это может быть достигнуто выбором подходящего основного гетероцикла,такого как пиридил или бензопиридил, в качестве Х-кольца. Альтернативно или дополнительно один или более радикалов от R3 до R7 могут содержать основные заместители, такие как первичный, вторичный или третичный амины, аминокислота и т.д. Предпочтительно R3 и/или R4 группами являются NH2, N(CH2)2 и NHC1-С 3 алкил, такой как NНСН 3 или NНСН 2 СН 3. ПредпочтительноR3 находится в мета-положении относительно карбонильной группы и ее необязательного спейсера, особенно в том случае, когда Xкольцо является фенилом, или R3 находится в 6 пара-положении, когда Х-кольцом является гетероароматический цикл, такой как пирид-3-ил. В данном случае предпочтительным значением р и/или n является ноль, что означает, что соответствующие группы отсутствуют. К предпочтительным соединениям согласно изобретению относятся следующие соединения:(1S,2S)-N-[цис-2-(2-(3-диметиламинофенилкарбонилокси)-6-фтор-3-ацетилфенил) циклопропил]-N'-(5-цианопирид-2-ил)мочевина; и фармацевтически приемлемые соли этих соединений. Другими предпочтительными соединениями являются следующие соединения:(1S,2S)-N-[цис-2-(2-(6-аминопирид-3 илкарбонилокси)-6-фтор-3-ацетилфенил)циклопропил]-N'-(5-цианопирид-2-ил)мочевина и фармацевтически приемлемые соли этих соединений. Другими приемлемыми соединениями согласно изобретению являются следующие соединения:(1R,2R)-N-[цис-2-(2-(3-диметиламинофенилкарбонилокси)-6-фтор-3-ацетилфенил) циклопропил]-N'-(5-цианопирид-2-ил)мочевина; и фармацевтически приемлемые соли этих соединений. К следующим соединениям, которые могут использоваться, относятся следующие соединения:(1R,2R)-N-[цис-2-(2-(6-аминопирид-3 илкарбонилокси)-6-фтор-3-ацетилфенил)циклопропил]-N'-(5-цианопирид-2-ил)мочевина; и фармацевтически приемлемые соли этих соединений. Предпочтительными соединениями согласно изобретению являются следующие соединения:(1R,2R)-N-[цис-2-(2-(3-диметиламинофенилкарбонилокси)-6-фтор-3-бутирилфенил) циклопропил]-N'-(5-бромпирид-2-ил)мочевина; и фармацевтически приемлемые соли этих соединений. К другим предпочтительным соединениям относятся следующие соединения:(1R,2R)-N-[цис-2-(2-(6-аминопирид-3 илкарбонилокси)-6-фтор-3-ацетилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина; и фармацевтически приемлемые соли этих соединений. 10 Приемлемыми в качестве фармацевтически пригодных солей соединений, имеющих формулу I, являются соли органических карбоновых кислот, таких как уксусная, молочная,глюконовая, лимонная, винная, малеиновая,яблочная, пантотеновая, изэтионовая, щавелевая, лактобионовая и янтарная кислоты, органических сульфоновых кислот, таких как метансульфокислота, этансульфокислота, бензолсульфокислота, п-хлорбензолсульфокислота и п-толуолсульфокислота; и неорганических кислот, таких как хлористо-водородная, иодистоводородная, серная, фосфорная и сульфаминовая кислоты. В соответствии с обычной практикой применения ингибиторов ВИЧ успешным является совместное введение от одного до трех дополнительных антивирусных средств, чтобы обеспечить синергические ответы и обеспечить комплементарную картину резистентности. Такими дополнительными антивирусными средствами могут быть AZT, ddI, ddC, D4T, 3 ТС, абакавир,адефовир,адефовирдипивоксил,бис-РОСРМРА, фоскарнет, оксимочевина, Hoechst-BayerHBY 097, эфавиренц, тровирдин, невирапин,делавиридин, PFA, H2G, АВТ 606, DMP-450,ловирид, ритонавир, сахинавир, индинавир, ампренавир (Vertex VX 478), нелфинавир и подобные им средства, обычно в молярном соотношении, отражающем соответствующие активности и их биологическую доступность. В общем, такое соотношение следует подбирать примерно от 25:1 до 1:25, относительно соединения,имеющего формулу I. Хотя возможно отдельное введение действующего вещества, предпочтительно, чтобы он входил в состав фармацевтической композиции. Такая композиция будет включать в себя указанное выше действующее вещество и один или более приемлемых носителей и необязательно другие терапевтические ингредиенты. Носителем(ями) должен быть приемлемый в смысле совместимости с другими ингредиентами композиции и не вредный для реципиентов носитель(ли). Композиции могут быть пригодны для перорального, ректального, назального, местного(включая трасбуккальное и подъязычное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное и внутрикожное (интрадермальное введения. Удобными являются композиции, представленные в форме стандартных унифицированных доз, например таблеток и капсул замедленного высвобождения, которые могут быть приготовлены способами, хорошо известными в области фармации. К таким способам относится стадия смешения указанного выше действующего вещества и носителя. В общем, композиция готовится путем стандартного и хорошо известного способа соединения действующего вещества и 11 жидких носителей или мелкоизмельченных твердых носителей, или сразу двух видов этих носителей, и затем, если это необходимо, формование продукта. Согласно данному изобретению, композиции, предназначенные для перорального введения, могут представлять собой дискретные единицы, такие как капсулы, крахмальные облатки или таблетки, в каждой из которых содержится заранее определенное количество действующего вещества; порошки или гранулы; растворы или суспензии действующего вещества в водном растворе или неводном растворе; или эмульсии типа масло-в-водном растворе или вода-вмасляном растворе и болюсы и т.д. Что касается композиций, предназначенных для перорального введения (например, таблеток или капсул), то под термином "приемлемый носитель" подразумеваются такие традиционные наполнители как, например, связывающие вещества, например сироп, смола акации, желатин, сорбит, трагакант, поливинил пирролидон (повидон), метилцеллюлоза, этилцеллюлоза, натрий карбоксиметилцеллюлоза,гидроксипропилметилцеллюлоза, сахароза и крахмал; наполнители и носители, например кукурузный крахмал, желатин, лактоза, сахароза, микрокристаллическая целлюлоза, каолин,маннит, двухосновных фосфат кальция, хлорид натрия и альгиновая кислота; и смазывающие вещества, такие как стеарат магния и стеараты других металлов, стеариновая кислота, силиконовая жидкость, тальк, воски, масла и коллоидный оксид кремния. Могут также использоваться отдушки, такие как мята перечная, масло грушанки, вишневая отдушка и им подобные. Добавление отдушек желательно для того, чтобы можно было легко идентифицировать лекарственную форму. Таблетки могут быть также покрыты оболочками с помощью хорошо известных в данной области методов. Подходящими носителями для пероральной лекарственной формы могут быть жидкие композиции в виде растворов, суспензий или эмульсий, необязательно инкапсулированные или представленные в другой форме в виде стандартных доз, которые готовятся традиционным способом. Предпочтительными являются композиции, в состав которых входят следующие компоненты: смола акации/-твин/вода,твин/вода, пропиленгликоль, растительное масло (такое как арахисовое, подсолнечное, оливковое и им подобные) с 10-20% этанола, растительное масло/Capmul MGM, Capmul MCM/пропиленгликоль, метилцеллюлоза/вода, растительное масло/сложный моноэфир стеариновой кислоты и глицерина, растительное масло/ненасыщенные сложные моноэфиры жирной кислоты и глицерина и им подобные. Таблетки могут быть приготовлены путем прессования или формования, необязательно с одним или с большим количеством вспомога 003327 12 тельных ингредиентов. Прессованные таблетки могут быть получены путем прессования в машинах, пригодных для случая, когда действующее вещество находится в свободнотекучей форме, такой как порошок или гранулы, причем оно может быть необязательно смешано со связывающим веществом, смазывающим веществом, инертным разбавителем, консервантом,поверхностно-активным или диспергирующим агентом. Формованные таблетки могут быть получены формованием в машинах, пригодных для смеси порошкообразного соединения, смоченного инертным жидким разбавителем. Таблетки могут быть необязательно покрыты оболочкой или иметь бороздки и могут быть приготовлены так, чтобы обеспечить медленное или контролируемое высвобождение действующего вещества. К композициям, пригодным для локального введения относятся лепешки, состоящие из действующего вещества во вкусовой основе,обычно в сахарозе и смоле акации, или в трагаканте, пастилки, состоящие из действующего вещества в инертной основе, такой как желатин и глицерин, или сахароза и смола акации; или средство для полоскания ротовой полости, состоящее из действующего вещества в подходящем жидком носителе. Композиции, пригодные для локального применения на коже могут быть представлены мазями, кремами, гелями и пастами, состоящими из действующего вещества и фармацевтически активного носителя. Примером системы локальной доставки является пластырь для трансдермального введения, содержащий активное вещество. К другим композициям локального применения относятся антисептические тампоны, из которых действующее вещество высвобождается на кожу и которые применяются перед инвазивными процедурами, такими как инъекция или отбор проб капиллярной крови. Такие тампоны нейтрализуют ВИЧ в крови или сыворотке, вытекающей при инвазивной процедуре, помогая, таким образом,предотвратить перенос вируса ВИЧ к здоровому обслуживающему медицинскому персоналу при повреждениях ткани иглой. Такие тампоны могут представлять собой подушечки из стерильной хирургической марли, пропитанные раствором действующего вещества в летучем растворителе, таком как этанол, и индивидуально упакованные в герметичные пакетики. Композиции для ректального или вагинального введения могут представлять собой суппозиторий или пессарий с пригодной для этой цели основой, включающей в себя, например, масло какао или салицилат. Другие вагинальные препараты могут представлять собой тампоны, кремы, гели, пасты, губки или аэрозольные композиции, содержащие в дополнение к действующему веществу соответствующие носители, которые известны специалистам в данной области. Композиции, пригодные для назального введения с твердым носителем, включают в себя крупнозернистый порошок с размером частиц,например в пределах от 20 до 500 мкм, который вводится путем вдыхания через нос, т.е. путем быстрой ингаляции из контейнера с порошком,который подносится близко к носу. В состав пригодных для введений композиций с жидким носителем, например назального аэрозоля или назальных капель, входят водные или масляные растворы действующего вещества. Композиции, пригодные для парентерального введения, включают в себя водные и неводные стерильные инъекционные растворы,которые могут содержать антиоксиданты, буферные системы, бактериостатические факторы и растворенные вещества, которые делают композицию изотоничной крови реципиента; и водные и неводные стерильные суспензии, которые могут включать в себя суспендирующие средства и загустители. Композиции могут быть представлены в упаковках на один прием или на несколько приемов, например в герметичных ампулах или флаконах, и могут храниться в лиофилизованном состоянии, при котором требуется только добавление стерильного жидкого носителя, например воды для инъекций, непосредственно перед использованием. Инъекционные растворы и суспензии могут быть приготовлены для немедленного приема из стерильных порошков, гранул и таблеток тех видов, которые описаны выше. Следующий аспект изобретения заключается в представлении способов получения соединений, имеющих формулу I, в частности цис-энантиомеров, заключающихся в перегруппировке Курциуса соединения, имеющего следующую формулу 14 в которой R3, R4, Х и n имеют те же значения,которые указаны выше, но группы при этом необязательно защищены, и R8 является водородом или обычной активирующей группой. Альтернативный способ согласно изобретению может далее включать в себя этап алкилирования соединением, имеющим формулу IIIa где n, R3, R4 и Х имеют те же значения, которые указаны выше, но где незащищенные аминогруппа, гидроксил и другие заместители защищены традиционными защитными группами. Энантиомерные соединения, имеющие формулу I, могут быть получены в соответствии со схемой реакций, указанной ниже: Приведенная выше схема является иллюстрацией получения (1S,2S) соединения, представленного в данном изобретении, где Rx является цианогруппой, R1 является F и R2 является этилом, но соответствующая методика применима и при других вариантах Rx, R1 и R2. Хиральный лиганд, указанный на четвертом этапе,может быть представлен, например, соединением, имеющим следующую формулу: и удалением защитных групп, где R , R и R означают те же группы, которые указаны выше,и PG означает защитную группу гидроксила. Способы согласно изобретению могут дополнительно включать в себя этап ацилирования активированным соединением, имеющим формулу III Чтобы получить 1R,2R энантиомер используется зеркально симметричный хиральный лиганд. В альтернативном случае хиральный лиганд можно не включать, чтобы получить рацемат. Пролекарство, имеющее формулу II, в которой р равно 0, синтезируется путем ацилирования соединения, имеющего формулу I, активированным соединением, имеющим формулу где R3, R4, X и n имеют те же значения, которые указаны выше, но группы необязательно защищены, и R8 является водородом или обычно используемой активирующей группой. К активированным соединениям, имеющим формулу III, относятся галогенангидрид,ангидрид карбоновой кислоты, активированный сложный эфир кислоты или кислота в присутствии агента сочетания, такого как дициклогексилкарбодиимид. Типичными активированными производными кислот являются хлорангидрид,смешанные ангидриды муравьиной и уксусной кислот, ангидриды, являющиеся производными алкоксикарбонилгалогенидов, таких как изобутилоксикарбонилхлорида и ему подобных,сложные эфиры - производные N-гидроксисукцинамида, сложные эфиры - производные Nгидроксифталимида, сложные эфиры - производные N-гидрокси-5-норборнен-2,3-дикарбоксамида, сложные эфиры - производные 2,4,5 трихлорфенола и подобных им соединений. Приемлемыми необязательными защитными группами для соединений, имеющих формулуIII, особенно каких-либо входящих в их состав аминов, являются такие группы, которые предназначены для защиты N-концов аминокислоты или пептида, или для защиты аминогруппы от нежелательных реакций в ходе синтеза. Часто используемые N-защитные группы представлены в работе Greene, "Protective Groups in OrganicSynthesis" (John Wiley and Sons, New York,1981), которая здесь включена в качестве ссылки. К N-защитным группам относятся ацильные группы, такие как формил, ацетил, пропионил,пивалоил, трет-бутилацетил, 2-хлорацетил, 2 бромацетил, трифторацетил, трихлорацетил,фталил, о-нитрофеноксиацетил, -хлорбутирил,бензоил, 4-хлорбензоил, 4-бромбензоил, 4 нитробензоил и им подобные; сульфонильные группы, такие как бензолсульфонил, п-толуолсульфонил и им подобные, группы, образующие карбаматы, такие как бензилоксикарбонил, пхлорбензилоксикарбонил,п-метоксибензилоксикарбонил, п-нитробензилоксикарбонил, 2 нитробензилоксикарбонил, п-бромбензилоксикарбонил, 3,4-диметоксибензилоксикарбонил, 4 метоксибензилоксикарбонил, 2-нитро-4,5-диметоксибензилоксикарбонил,3,4,5-триметоксибензилоксикарбонил, 1-(п-бифенилил)-1-метилэтоксикарбонил-диметил-3,5-диметоксибензилоксикарбонил, бензгидрилоксикарбонил,трет-бутоксикарбонил,диизопропилметоксикарбонил, изопропилоксикарбонил, этоксикарбонил, метоксикарбонил, аллилоксикарбонил,2,2,2-трихлорэтоксикарбонил,феноксикарбонил, 4-нитрофеноксикарбонил, флуоренил-9 003327 16 метоксикарбонил, циклопентилоксикарбонил,адамантилоксикарбонил, циклогексилоксикарбонил, фенилтиокарбонил и им подобные; алкильные группы, такие как бензил, трифенилметил, бензилоксиметил и им подобные; и силильные группы, такие как триметилсилил и подобные. Предпочтительными N-защитными группами являются формил, ацетил, бензоил, пивалоил, трет-бутилацетил, фенилсульфонил, бензил, трет-бутоксикарбонил (БОК) и бензилоксикарбонил (Кбз). Ацилирование проводится в обычных для этерификации условиях, таких как в присутствии ДМАП (диметиламинопиридина) и ДЦК(дициклогексилкарбодиимида) в таком растворителе как диметилформамид или пиридин. Необязательные защитные группы могут быть удалены обычными способами, которые всесторонне обсуждались в работе Greene, упоминаемой выше, при таких условиях, как в присутствии ТФУ (трифторуксусной кислоты), НСl(водн.)/диоксана, или гидрированием в присутствии катализатора, чтобы получить соединение, имеющее формулу II. Соединения, имеющие формулу II, в которой р равно 1, могут быть получены в результате реакции соединения, имеющего формулу III,с иодхлорметаном или смесью дихлор/иодхлорметана при обычных условиях алкилирования, чтобы получить соединение, имеющее формулу IIIa: где n, R3, R4 и X имеют те же значения, которые указаны выше, но при этом экспонированные аминогруппа, гидроксильная группа и др. заместители защищены обычными защитными группами. Затем соединение, имеющее формулу IIIa,предпочтительно превращают в соответствующее иодпроизводное в результате реакции с NaI,после чего связывают с соединением, имеющим формулу I, обычно в щелочных условиях, например в органическом растворителе, содержащем гидрид натрия. Детальное описание Аспекты изобретения в этом разделе будут иллюстрироваться только на примерах, со ссылками на следующие, не ограничивающие данное изобретение, примеры и рисунки, на которых на фиг. 1 показана скорость развития резистентности от времени, к соединению данного изобретения по сравнению с соединением предшествующего уровня техники, как описано в биологическом примере 2; на графике, приведенном на фиг. 2, против временных значений показаны значения уровней в плазме соединения данного изобретения или соединения предшествующего уровня тех 17 ники у крыс после перорального введения, как описано в биологическом примере 5; на фиг. 3 приведены кинетические кривые связывания с обратной транскриптазой соединения данного изобретения по сравнению с соединением предшествующего уровня техники,полученные на основании исследования с помощью метода резонанса поверхностных плазмонов, как описано в биологическом примере 10. Получение промежуточных химических соединений Пример 1. 3-[1,1-(Этилендиокси)пропил]6-фтор-2-метоксибензальдегид. К раствору 3-фторфенола (22,4 г, 0,2 моль),пиридина (24 мл, 0,3 моль) и дихлорметана (200 мл) при комнатной температуре добавляли 20 мл (0,225 моль) пропионилхлорида в течение 5 мин. Реакция была экзотермической. Раствор перемешивали в течение следующих 30 мин. После добавления дихлорметана органическую фазу промывали насыщенным растворомNаНСО 3 и водой, высушивали над MgSO4 и концентрировали в вакууме. Было получено 33,8 г (100%) 3-фтор-1-пропионилоксибензола. Это соединение подвергали реакции с 33,3 г(0,25 моль) АlСl3 при 150 С, которая продолжалась в течение 10 мин. После острожного гашения водой проводили трехкратную экстракцию реакционной смеси эфиром. Эфирную фазу высушивали (MgSO4) и выпаривали, чтобы получить 29,5 г (0,176 моль, 88%) продукта перегруппировки. Этот промежуточный продукт растворяли в 200 мл ацетона и добавляли К 2CO2(42 г, 0,3 моль) и МеI (25 мл, 0,4 моль). Реакционную смесь нагревали при 40 С в течение 12 ч. Реакционную смесь фильтровали и выпаривали ацетон. Остаток растворяли в эфире и эфирную фазу промывали 0,5 М раствором NaOH и водой. После высушивания (MgSO4) и выпаривания получали 31,2 г (0,17 моль, выход после трех этапов составил 86%) 4-фтор-2-метоксипропиофенона. К раствору 4-фтор-2-метоксипропиофенона (31,2 г, 0,171 моль), этиленгликоля (10,5 мл, 0,188 моль) в бензоле (300 мл) добавляли 1 г п-толуолсульфокислоты. Реакционную смесь нагревали при кипячении с обратным холодильником в аппарате Dean-Stark примерно в течение 12 ч. После охлаждения органическую фазу несколько раз промывали 1 М раствором NaOH и высушивали (Na2SO4 и К 2 СО 3). Растворитель выпаривали и получали примерно 38 г ацеталя. Чистота в соответствии с данными капиллярной ГХ составляла 88%, и примесь, в основном, была представлена не вступившим в реакцию кетоном. К раствору ацеталя в ТГФ (450 мл) при 65 С в атмосфере азота добавляли по каплям 128 мл (0,32 моль) 2,5 М н-BuLi. При поддержании температуры примерно -65 С добавляли раствор ДМФА (25 мл, 0,32 моль) в ТГФ (50 мл). Реакционной смеси давали возможность 18 медленно нагреться до комнатной температуры,и по данным ГХ примерно через 30 мин исходного материала не оставалось. Спустя еще 1 ч реакционную смесь гасили насыщенным раствором NH4Cl и трижды экстрагировали эфиром. После высушивания (Na2SO4) остаток очищали на колонке с силикагелем (силикагель 60,Merck, размер частиц 0,04-0,063 мм), элюируяEtOAc 1 и гексанами 9, при этом получали 10 г(т, 1 Н), 7,7-7,8 (м, 1 Н), 10,4 (с, 1 Н). Пример 2. 3-[1,1-(Этилендиокси)пропил]6-фтор-2-метоксистирол. К суспензии метилтрифенилфосфоний бромида (14,3 г, 40 ммоль) в ТГФ (250 мл) при комнатной температуре и в атмосфере азота добавляли 16 мл (40 ммоль) 2,5 М н-BuLi. Происходило почти полное растворение, и затем к раствору добавляли 3-[1,1-(этилендиокси)пропил]-6-фтор-2-метоксибензальдегид (10 г, 39,5 ммоль) в ТГФ (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и вливали в смесь гексанов и насыщенного солевого раствора. Органическую фазу промывали дважды насыщенным солевым раствором и один раз водой. После выпаривания растворителя остаток фильтровали через воронку, заполненную глиноземом (оксид алюминия 90 асс. Brockmann, Merk) и элюировали EtOAc 1 и гексанами 9, чтобы удалить образовавшийся оксид трифенилфосфония. Выпаривание органического растворителя приводило к образованию остатка,который окончательно очищали на силикагеле,элюируя EtOAc 1 и гексанами 9, чтобы получить 6,9 г (70%) соединения, приведенного в названии данного примера, чистота которого составляла 94,5%, как было определено с помощью капиллярной ГХ. 1(1S,2R)-цис-2-(6-Фтор-2 метокси-3-пропионилфенил)циклопропилкарбоновая кислота. Сложный этиловый эфир (1S,2R)-цис-2-[3(1,1-этилендиокси)этил]-6-фтор(2-метоксифенил)циклопропилкарбоновой кислоты был получен из 3-[1,1-(этилендиокси)пропил]-6-фтор 2-метоксистирола (19,4 г, 69 ммоль) и этилдиазоацетата (ЭДА) (29 мл, 275 ммоль) с помощью реакции асимметричного циклопропанирования,катализируемой Сu(I)трифлатом (679 мг, 1,35 ммоль), и с использованием хирального лиганда([2,2'-изопропилиденбис-4R)-4-трет-бутил-2 оксазолин)] (794 мг, 2,7 ммоль), что подробно описано Evans et al. в J.Am.Chem.Soc. 1991, 113,726-728. После хроматографии на силикагеле 19 было получено 9,4 г (40,5%) сложного этилового эфира. Избыток энантиомера при ВЭЖХ на хиральной колонке составлял 99%. Сложный эфир растворяли в 150 мл диоксана и добавляли 30 мл 6 М НСl. Реакционную смесь перемешивали в течение ночи и распределяли между эфирной фазой и солевым раствором. Растворитель выпаривали и получали 19 г неочищенного продукта. Продукт растворяли в метаноле (250 мл) и воде (75 мл) и добавляли 6 г (250 ммоль)LiOH. Реакционную смесь нагревали до 90 С и выдерживали при этой температуре в течение 24 ч и большую часть растворителя выпаривали. Оставшуюся смесь подкисляли и трижды экстрагировали дихлорметаном. Выпаривание растворителя позволило получить 11,2 г соединения, приведенного в названии данного примера. 1(м, 1 Н), 11,30 (уширенный с, 1 Н). Пример 4. (1R, 2S)-цис-2-(6-Фтор-2-метокси-3-пропионилфенил)циклопропилкарбоновая кислота. Соединение было получено из 3-[(1,1-этилендиокси)пропил]-6-фтор-2-метоксистирола таким же путем, который описан для получения кислоты в примере 3. В качестве хирального лиганда использовали 2,2'-изопропилиденбисH ЯМР (250 МГц, СDСl3)7,48 (кв, 1 Н),6,84 (т, 1 Н), 3,82 (с, 3H), 2,93 (кв, 2 Н), 2,29 (кв,1 Н), 2,14 (кв, 1 Н), 1,60 (м, 2 Н), 1,16 (т, 3H). Получение соединений, имеющих формулы I и II. Пример 5. N-[цис-2-(2-(6-Фтор-2-гидрокси-3-пропионилфенил)циклопропил]-N'-(5-цианопирид-2-ил)мочевина. Раствор 3-[1,1-(этилендиокси)пропил]-6 фтор-2-метоксистирола (32,4 г, пример 2) и комплекса бромида медиди - метилсульфида(0,30 г) в дихлорэтане (200 мл) нагревали до 80 С в атмосфере азота. Добавляли этилдиазоацетат (54 мл) в дихлорэтане (600 мл) в течение 7 ч. После того как все было добавлено, нагревание прекращали. Через 16 ч растворитель выпаривали и остаток очищали на силикагеле,элюируя этилацетатом и гексанами, при этом получали цис-форму сложного эфира (6,5 г). Сложный эфир в цис-форме (3,7 г, 10,9 ммоль) растворяли в этаноле (20 мл), а КОН (1,8 г, 32,7 ммоль) растворяли в воде (10 мл). Растворы объединяли и нагревали в колбе с обратным холодильником в течение 3 ч. Добавляли воду (30 мл) и раствор дважды промывали гексанами (20 мл). Водную фазу охлаждали на ледяной бане и подкисляли разбавленной НСl. Раствор трижды экстрагировали толуолом. Толуольную фазу высушивали (МgSO4) и выпаривали, при этом получали 1,9 г -цис-2-[3-(1,1 003327 20 этилендиоксипропил)-6-фтор-2-метоксифенил] циклопропилкарбоновой кислоты. К раствору кислоты (120 мг, 0,39 ммоль) в осушенном толуоле добавляли триэтиламин (59 мкл, 0,43 ммоль) и дифенилфосфорилазид (92 мкл, 0,43 ммоль). Раствор перемешивали при комнатной температуре в течение 1 ч и затем нагревали до 120 С. Через 1 ч добавляли 2 амино-5-цианопиридин (51 мг, 0,43 ммоль). Температуру поддерживали еще в течение 3 ч. Через 16 ч растворитель выпаривали, остаток растворяли в дихлорметане (30 мл), промывали разбавленной НСl, сушили (МgSO4) и выпаривали с получением 152 мг. Этот продукт растворяли в диоксане и добавляли НСl (6 н., 1 мл). Спустя 2 ч смесь выпаривали, растворяли в дихлорметане (25 мл), промывали водой (10+10 мл), высушивали (МgSO4) и выпаривали, при этом получали 117 мг. Остаток очищали на силикагеле, элюируя этилацетатом и гексанами,чтобы получить 37 мг промежуточного продукта, представляющего 2-метоксифенил. К раствору промежуточного продукта, содержащего 2-метоксифенил, (37 мг, 0,097 ммоль) в дихлорметане при -60 С добавляли 1 М раствор трибромида бора в дихлорметане (194 мкл; 0,194 ммоль). Через 10 мин убирали охлаждающую баню и продолжали перемешивать в течение 2 ч. Раствор разбавляли дихлорметаном, промывали разбавленным растворомNаНСО 3 и водой, высушивали (MgSO4) и выпаривали. Остаток подвергали перекристаллизации из MeCN, получая при этом 17 мг названного продукта. 1(1,72 г, 6,1 ммоль). Раствор перемешивали при комнатной температуре в атмосфере аргона в течение 30 мин и затем нагревали до 120 С. Через 15 мин добавляли раствор 2-амино-5 цианопиридина (0,99 г, 8,9 ммоль) в ДМФА (3 мл) и продолжали нагревать в течение 4 ч. Толуол выпаривали и смесь разбавляли диэтиловым эфиром (100 мл) и этилацетатом (50 мл) и промывали 1 М НСl, Н 2O и насыщенным солевым раствором. Органический слой высушивали 21 К раствору промежуточного продукта, содержащего 2-метоксифенил, (1,40 г, 3,66 ммоль) в СН 2 Сl2 (80 мл) при -72 С в аргоне добавляли 1 М раствор трихлорида бора в СН 2 Сl2 (11,0 мл,11,0 ммоль). Через 10 мин охлаждающую баню убирали и перемешивание продолжали в течение 1 ч 15 мин. Раствор разбавляли СН 2Cl2 и промывали водным раствором NaHCO3, Н 2 О и насыщенным солевым раствором. Органический слой высушивали (Na2SO4) и концентрировали. Преципитат из смеси ацетонитрила/Н 2O в соотношении 1:1 давал 0,62 г чистого названного продукта. Остаток концентрировали, проводили хроматографию,элюируя этилацетатом/нгексаном в соотношении от 1:10 до 1:1 и этилацетатом и затем проводили кристаллизацию из ацетонитрила, получая при этом 0,2 г названного продукта. Выход составил 0,82 г (61%). Избыток энантиомера при ВЭЖХ на хиральной колонке составил 95%, []D22 -171,2 (с=0,50,СН 2 Сl2). 1(м, 1 Н), 3,01 (кв, 2 Н), 2,03 (м, 1 Н), 1,55 (м, 1 Н),1,29 (м,4 Н). Пример 7. (1R,2R)-N-[цис-2-(2-(3-Аминофенилкарбонилокси)-6-фтор-3-пропионилфенилциклопропил]-N'-(5-цианопирид-2-ил)мочевина. К раствору соединения, описанного в примере 6 (1,64 г, 4,4 ммоль), 3-аминобензойной кислоты, имеющей защитную группу БОК (1,6 г, 6,6 ммоль) и 4-диметиламинопиридина (269 мг, 2,2 ммоль) в 20 мл дихлорметана и 10 мл ДМФА при комнатной температуре и в атмосфере аргона добавляли 1,36 г (6,6 ммоль) ДЦК. Реакционную смесь перемешивали в течение 24 ч. Растворитель осторожно выпаривали и остаток очищали на силикагеле, используя в качестве растворителя смесь гексаны/этилацетат в соотношении 1:1, чтобы получить 2,6 г БОКзащищенного названного в заглавии примера продукта. Этот продукт добавляли к 75 мл трифторуксусной кислоты при 0 С. Затем смесь перемешивали при 0 С в течение 1 ч. Растворитель осторожно удаляли в вакууме. Остаток разделяли между фазами, представленными этилацетатом и насыщенным раствором карбоната калия. Органическую фазу высушивали и выпаривали. Остаток очищали на колонке с силикагелем, используя в качестве элюента смесь этилацетата/гексанов в соотношении 4:1, чтобы получить 1,03 г свободного основания указанного соединения. Этот промежуточный продукт обрабатывали 3 мл 1 М НС 1 в эфире и получали 0,84 г названного соединения. Чистота продукта по данным ВЭЖХ составила примерно 97%. 1H ЯМР свободный амин (250 МГц, СDСl3)1,09 (т, 3H), 1,2-1,3 (м, 1 Н), 1,4-1,5 (м, 1 Н),1,95-2,00 (м, 1 Н), 2,83(кв, 2 Н), 3,15-3,25 (м, 1 Н), 003327(ушир.с, 1 Н). Пример 8. (1S,2S)-N-(цис-2-(6-Фтор-2 гидрокси-3-пропионилфенил)циклопропил)-N'(5-цианопирид-2-ил)мочевина. К раствору кислоты, полученной, как описано в примере 3 (1,2 г, 4,5 ммоль), в осушенном толуоле (10 мл) в атмосфере азота добавляли триэтиламин (670 мкл, 4,8 ммоль) и дифенилфосфорилазид (1,05 мл, 4,9 ммоль). Раствор перемешивали при комнатной температуре 30 мин и затем нагревали до 120 С. Через 15 мин добавляли раствор 2-амино-5-цианопиридина (0,80 г, 6,7 ммоль) в диметилформамиде (1,5 мл) и продолжали нагревать в течение 4 ч. Раствор разбавляли диэтиловым эфиром и промывали 1 М соляной кислотой. Органический слой высушивали (MgSO4) и концентрировали. Остаток очищали флэш-хроматографией на силикагеле(градиентное элюирование начинали смесью нгексана: этилацетата в соотношении 1:1, заканчивали чистым этилацетатом), получая мало очищенное 2-метоксифенильное производное(0,93 г). В результате повторной хроматографии,проводимой так же как описано выше, получали очищенное 2-метоксифенильное производное(0,70 г, 41%). К раствору промежуточного продукта, содержащего 2-метоксифенил (700 мг, 1,8 ммоль), в метиленхлориде при -60 С добавляли 1 М раствор трихлорида бора в метиленхлориде(5,5 мл, 5,5 ммоль). Через 10 мин охлаждающую баню убирали и продолжали перемешивать в течение 2 ч. Раствор разбавляли метиленхлоридом и промывали водным раствором гидрокарбоната натрия. Органический слой высушивали(МgSO4) и концентрировали, остаток очищали флеш-хроматографией на силикагеле (градиент,н-гексан:этилацетат 2:1, 1:1, 1:2, этилацетат: метанол 8:1), получая соединение, приведенное в названии данного примера (500 мг, 74%).(с, 3H), 9,83 (с, 1 Н), 13,22 (с, 1 Н, Ar-OH). Пример 9. (1S,2S)-N-[цис-2-(2-(3-Аминофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил-N'-(5-цианопирид-2-ил) мочевина. Начиная с соединения, описанного в примере 6, и используя способ, описанный в примере 7, получили названный продукт в виде гидрохлорида. 1(3,0 г, 11,3 ммоль), триэтиламин (1,58 мл, 11,3 ммоль) и дифенилфосфорилазид (2,44 мл, 11,3 ммоль) растворяли в осушенном толуоле (8 мл) при комнатной температуре и в атмосфере аргона. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, после чего температуру увеличивали до 120 С и выдерживали при этой температуре еще 15 мин. Затем добавляли 2-амино-5-бромпиридин (2,08 г, 12 ммоль) и реакционную смесь перемешивали при 120 С в течение 2,5 ч. Добавляли бензол и 1 М раствор НСl и органическую фазу выпаривали. Остаток очищали на силикагеле с использованием в качестве элюента смеси гексанов:этилацетата в соотношении 1:1. Собирали соответствующую фракцию и получали 5,0 г(1S,2S)-N-(цис-2-(6-фтор-2-метокси-3-пропионилфенил)циклопропил)-N'-(5-бромпирид-2 ил)мочевины. Это соединение растворяли в дихлорметане (100 мл) и раствор оставляли в атмосфере аргона и охлаждали до -65 С. Добавляли трихлорид бора (30 мл 1 М раствора в дихлорметане, 30 ммоль) и реакционной смеси давали нагреться до комнатной температуры в течение ночи. Добавляли дихлорметан и насыщенный раствор бикарбоната натрия. Органическую фазу выпаривали и остаток очищали на силикагеле с использованием в качестве элюента смеси этилацетата:метанола в соотношении 9:1. Получили 1,96 г (41%) названного соединения. Анализ: расчетные данные: С 51,2; Н 4,1;(ушир.с, 1 Н), 8,53 (ушир.с, 1 Н), 13,32(д, 1 Н). Пример 11. (1R,2R)-N-(цис-2-(6-Фтор-2 гидрокси-3-пропионилфенил)циклопропил)-N'(5-бромпирид-2-ил)мочевина. Реакция асимметричного циклопропанирования соединения, описанного в примере 2, была проведена таким же образом, как описано в примере 3, с использованием хирального лиганда 2,2'-изопропилидинбис(4S)-4-трет-бутил-2 оксазолина (имеющийся в продаже производства Aldrich). Затем полученная (1R,2S)-цис-2-(6 фтор-2-метокси-3-пропионилфенил)циклопропилкарбоновая кислолота была использована для получения соединения, указанного в назва 003327[]D22 - 153,8 (с=0,50, СН 2 Сl2). Пример 12. (1S,2S)-N-[цис-2-(2-(3-Аминофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина. К раствору соединения, полученного в примере 10 (633 мг, 1,5 ммоль), БОК-защищенной 3-аминобензойной кислоты (475 мг, 2 ммоль) и 4-диметиламинопиридина (123 мг, 1 ммоль) в 20 мл смеси дихлорметана:ДМФА 1:1 при комнатной температуре и в атмосфере аргона добавляли 415 мг (2 ммоль) ДЦК. Реакционную смесь перемешивали в течение 36 ч. Растворитель осторожно выпаривали и остаток очищали на силикагеле, используя в качестве растворителя гексаны:этилацетат в соотношении 1:1, чтобы получить 811 мг БОКзащищенного продукта, приведенного в названии примера. Этот продукт растворяли в диоксане (20 мл) и добавляли 10 мл 6 М НСl, смесь перемешивали в течение ночи. Растворитель осторожно удаляли в вакууме. Остаток обрабатывали этанолом и эфиром и получали 255 мг указанного продукта в виде соли НСl. Чистота по данным ВЭЖХ составляла примерно 93%. 1(ушир.д, 1 Н), 8,26 (ушир.с, 1 Н), 8,35-8,37 (м,1 Н), 8,42-8,46 (м, 1 Н). Пример 13. (1S,2S)-N-[цис-2-(2-(3-L-Аланиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2 ил)мочевина. Исходное соединение, БОК-защищенная 3L-аланиламинобензойная кислота, было получено из ТХЭ-защищенной 3-аминобензойной кислоты с использованием стандартной методики способа, см., например, Bodanszky's "TheSpringer. Реакцию этого соединения с соединением, полученным в примере 10, проводили, как описано в примере 12, чтобы получить названный продукт в виде соли НСl. 1 Н ЯМР (250 МГц, свободный амин,CDCl3)1,10 (т, 3H), 1,15-1,25 (м, 1 Н), 1,4-1,5 25 Пример 14. (1S,2S)-N-цис-2-[6-Фтор-3 пропионил-2-(4-пиридилкарбонилокси)фенил] циклопропил-N'-(5-бромпирид-2-ил)мочевина. Конденсацию продукта, полученного в примере 10, с изоникотиновой кислотой провели по способу, аналогичному описанному в примере 12, чтобы получить указанный продукт в виде соли НСl. 1(м, 1 Н), 1,62 (м, 1 Н), 1,38 (м, 1 Н), 1,13 (т, 3H). Пример 15. (1S,2S)-N-цис-2-[2-(3-Диметиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил]циклопропил-N'-(5-бромпирид 2-ил)мочевина. Конденсацию продукта, полученного в примере 10, с 3-диметиламинобензойной кислотой проводили способом, аналогичным способу,описанному в примере 12, чтобы получить названный продукт в виде соли НСl. 1(т, 1 Н), 7,10 (д, 1 Н), 3,48 (с, 6 Н), 3,28 (м, 1 Н),3,00 (м, 2 Н), 2,11 (м, 1 Н), 1,58 (м, 1 Н), 1,38 (м,1 Н), 1,14 (т, 3H). Пример 16. (1S,2S)-N-[цис-2-(2-(3-Аминометилбензоилоксиметилокси)-5-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил) мочевина. 3-трет-Бутоксикарбониламидометилбензойную кислоту обрабатывали раствором гидрохлорида тетрабутиламмония (1 М в МеОН) до рН 9 и выпаривали. Остаток растворяли в дихлорметане и обрабатывали хлориодметаном в течение ночи. Раствор промывали водой и выпаривали для получения неочищенного 3-третбутоксикарбониламидометилбензоилоксиметилхлорида. Этот материал использовался для реакции с натриевой солью, описанной в примере 10 (получена с помощью гидрида натрия в ДМФА), в присутствии небольшого количества иодида натрия в качестве катализатора. Через 2 ч после начала реакции раствор гасили уксусной кислотой и разбавляли дихлорметаном, промывали водой и выпаривали. Сырой продукт очищали на силикагеле, элюируя смесью этилацетата/гексана 1:2 и очищенный материал обрабатывали трифторуксусной кислотой и выпаривали, чтобы получить трифторацетатную соль указанного соединения в виде твердого вещества. 1 26 Конденсацию (1S,2S)-N-(цис-2-(6-фтор-2 гидрокси-3-пропионилфенил)циклопропил)-N'(5-бромпирид-2-ил)мочевины, полученной в примере 10, с 3-трет-бутоксикарбониламидо-4 метилбензойной кислотой проводили в соответствии с описанием процедуры, приведенном в примере 12. Продукт обрабатывали трифторуксусной кислотой и выпаривали, чтобы получить трифторацетат соединения, указанного в названии данного примера, в виде твердого вещества. 1(ушир.с, 1 Н), 7,1 (т, 1 Н), 7,4 (д, 1 Н), 7,8 (м, 1 Н),7,9(м, 2 Н), 8,1 (с, 1 Н), 8,3 (с, 1 Н). Пример 18. (1S,2S)-N-(цис-2-(2-(3-Этиламинобензоилокси)-6-фтор-3-пропионилфенил) циклопропил)-N'-(5-бромпирид-2-ил)мочевина. Конденсацию соединения, полученного в примере 10, с 3-(N-этил-трет-бутоксикарбониламидо)бензойной кислотой проводили в соответствии с процедурой, описанной в примере 12, и полученный продукт обрабатывали трифторуксусной кислотой и выпаривали, чтобы получить трифторацетат соединения, указанного в названии примера, в виде твердого вещества. 1(т, 1 Н), 7,2 (ушир.с, 1 Н), 7,6 (т, 1 Н), 7,7-7,8 (м,2 Н), 7,9 (д, 1 Н), 8,1 (с, 1 Н), 8,2 (д, 1 Н), 8,4 (с,1 Н). Пример 19. (1S, 2S)-N-(цис-2-(2-Хинол-4 илокси-6-фтор-3-пропионилфенил)циклопропил)-N'-(5-бромпирид-2-ил)мочевина. Конденсацию соединения, полученного в примере 10, с 4-хинолиновой кислотой проводили в соответствии с процедурой, описанной в примере 12, продукт растворяли в трифторуксусной кислоте и выпаривали, чтобы получить ацетат соединения, указанного в названии примера, в виде твердого вещества. 1(д, 1 Н), 9,1 (м, 2 Н), 9,2(ушир.с, 1 Н). Пример 20. (1S,2S)-N-(цис-2-(3-Аминометил-2-метилбензоилокси)фтор-3-пропионилфенил)циклопропил)-N'-(5-бромпирид-2-ил)мочевина. Конденсацию соединения, полученного в примере 10, с 3-трет-бутилоксикарбониламидо 2-метилбензойной кислотой проводили в соответствии с процедурой, описанной в примере 12. Продукт обрабатывали трифторуксусной кислотой и выпаривали, чтобы получить соединение, указанное в названии примера, в виде твердого вещества. 1(ушир.с, 1 Н), 4,2 (с, 2 Н), 7,0-7,2 (м, 2 Н), 7,4 (д, 27 1 Н), 7,6-7,7 (м, 2 Н), 7,8-8,0 (м, 2 Н), 8,2 (ушир. с,2 Н). Пример 21. (1S,2S)-N-[цис-2-(6-Фтор-2-(4 аминометилфенилкарбонилокси)-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил) мочевина. 4-(трет-Бутилоксикарбониламидометил) бензойную кислоту получали при добавлении 6,5 г ДЦК к раствору, содержащему 4 г 4 цианобензойной кислоты в 200 мл МеОН. Смесь перемешивали 70 ч при комнатной температуре,фильтровали, чтобы удалить выпавшую в осадок дициклогексилмочевину и фильтрат концентрировали в вакууме, чтобы получить 7 г неочищенного продукта. Сложный метиловый эфир растворяли в 500 мл МеОН и добавляли 9,6 г CoCl26H2O. Смесь обрабатывали добавлением порций NaBH4. Спустя 5 ч реакционную смесь концентрировали и удаляли осадок. Фильтрат подкисляли 150 мл 1 М НСl (водн.) и экстрагировали 2 х 100 мл СН 2 С 12. Кислую водную фазу обрабатывали 100 мл 25% NH3 (водн.),экстрагировали 3 х 100 мл СН 2 Сl2, высушивали с помощью Na2SO4 и концентрировали, чтобы получить 2,64 г коричневатого масла. Масло растворяли в 30 мл смеси диоксана/воды (2:1) и обрабатывали в течение 20 ч 1,5 г NaOH (кристал.). Растворитель удаляли и добавляли 40 мл смеси трет-бутанола/воды (1:1). После добавления 3,7 г ди-трет-бутилдикарбоната раствор перемешивали в течение 24 ч,затем было добавлено больше воды и смесь экстрагировали 2 х 50 мл гексана. Водную фазу подкисляли (рН 1,5-2,0) NaHSO4 и экстрагировали 3 х 75 мл эфира. Объединенные экстракты промывали 50 мл насыщенного солевого раствора,высушивали с помощью Na2SO4 и выпаривали,чтобы получить промежуточный продукт 4(трет-бутилоксикарбониламидометил) бензойную кислоту в виде белого порошка. Проводили конденсацию 4-(трет-бутилоксикарбониламидометил)бензойной кислоты и(1S,2S)-N-(цис-2-(6-фтор-2-гидрокси-3-пропионилфенил)циклопропил)-N'-(5-бромпирид-2-ил) мочевину, полученную в примере 10, и БОКзащитную группу удаляли с помощью способа,описанного в примере 12, чтобы получить продукт, указанный в названии примера, в виде хлористо-водородной соли 1 28 0,1 г индол-5-карбоновой кислоты смешивали с 2 эквивалентами метилтрифторметан сульфоната в 1 мл ДМФА при комнатной температуре. Через 5 ч растворитель выпаривали и регистрировали спектр 1 Н ЯМР. 1ii) Получение указанного в названии примера соединения. Конденсацию N-метилиндол-5-карбоновой кислоты и (1S,2S)-N-(цис-2-(6-фтор-2-гидрокси 3-пропионилфенил)циклопропил)-N'-(5-бромпирид-2-ил)мочевины, полученной в примере 10,проводили способом, описанным в примере 12,чтобы получить указанный продукт в виде хлористоводородной соли. 1(ушир.с, 1 Н), 9,43 (ушир.с, 1 Н). Пример 23. (1S,2S)-N-[цис-2-(6-Фтор-2(индол-4-карбонилокси)-3-пропионилфенил) циклопропил]-N'-(5-бромпирид-2-ил)мочевина. Конденсацию индол-4-карбоновой кислоты и (1S,2S)-N-(цис-2-(6-фтор-2-гидрокси-3 пропионилфенил)циклопропил)-N'-(5-бромпирид-2-ил)мочевины, полученной в примере 10,проводили способом, описанным в примере 12,чтобы получить указанный продукт в виде хлористо-водородной соли. 1(ушир.д, 1 Н), 7,00-7,35 (м, 4 Н), 7,55 (дд, 1 Н),7,60 (д, 1 Н), 7,79 (дд, 1 Н), 7,89 (д, 1 Н), 8,10 (д,1 Н), 9,27 (ушир.д, 2 Н). Пример 24. (1S, 2S)-N-[цис-2-(6-фтор-2-(3 амино-4-хлорфенилкарбонилокси)-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил) мочевина. Конденсацию 3-амино-4-хлорбензойной кислоты и (1S,2S)-N-(цис-2-(6-фтор-2-гидрокси 3-пропионилфенил)циклопропил)-N'-(5-бромпирид-2-ил)мочевины, полученной в примере 10,проводили способом, описанным в примере 12,чтобы получить указанный продукт в виде хлористо-водородной соли. 1 Сухую смесь соединения, полученного в примере 8 (50 г, 0,68 ммоль), N,N'-дициклогексилкарбодиимида (0,168 г, 0,81 ммоль), никотиновой кислоты (0,1 г, 0,81 ммоль) и 4-(диметиламино)пиридина (0,041 г, 0,34 ммоль) растворяли в СН 2 Сl2 (5 мл) и N,N'-диметилформамиде (ДМФА) (2,5 мл). Затем смесь перемешивали при комнатной температуре. Через 20 ч смесь фильтровали и высушивали в вакууме,затем снова растворяли в минимальном количестве дихлорметана и фильтровали. Прозрачный раствор выпаривали на кремнезем и очищали с помощью хроматографии (этилацетат), чтобы получить указанное в названии соединение(0,168 г, 50%). Образцы для анализа были получены перекристаллизацией из хлороформагексана. 1 Сухую смесь соединения, полученного в примере 6 (0,1 г, 0,27 ммоль), N,N'-дициклогексилкарбодиимида (0,067 г, 0,33 ммоль) и никотиновой кислоты (0,037 г, 0,3 ммоль) суспендировали в дихлорметане (2 мл). По каплям добавляли минимальное количество ДМФА, чтобы получить достаточно прозрачный раствор. Добавляли 4-(диметиламино)пиридин (0,016 г,0,14 ммоль). Реакционную смесь перемешивали при комнатной температуре. Через 20 ч растворитель выпаривали в вакууме и неочищенный остаток растворяли в водном растворе соляной кислоты (рН 1-2) и фильтровали. Прозрачный раствор с помощью гидрокарбоната натрия доводили до слабощелочного раствора и отфильтровывали осажденный продукт. В результате хроматографической очистки (дихлорметанметанол, 15:1) получали 0,072 г соединения,указанного в названии примера (56%). 1(0,06 г, 0,5 ммоль) и 3-(N-этил-N-бутоксикарбонил)аминобензойную кислоту (0,320 г, 1,2 ммоль) (полученная в результате гидроаминирования 3-аминобензойной кислоты с последующей защитой аминогруппы) растворяли в дихлорметане (8 мл) и ДМФА (3 мл). Затем смесь перемешивали при комнатной температуре. Через 18 ч растворитель удаляли в условиях вакуума и неочищенный продукт снова растворяли в дихлорметане и фильтровали. Прозрачный раствор выпаривали на кремнезем и очищали с помощью хроматографии (этилацетатгексан, 3:2), чтобы получить достаточно очищенное соединение, указанное в названии примера (0,24 г, 39%). 1 К раствору соединения, полученного в примере 27 (0,120 мг, 0,19 ммоль), в дихлорметане (10 мл) при перемешивании добавляли трифторуксусную кислоту (5 мл). Смесь оставляли при комнатной температуре на 1-2 ч, затем выпаривали досуха. Сырой продукт очищали ВЭЖХ (препаративная колонка С-18, 40% воды в ацетонитриле), чтобы собрать 0,045 г (30%) соединения, указанного в названии примера, в виде соли трифторуксусной кислоты. 1(0,016 г, 0,14 ммоль) и 3-диметиламинобензойную кислоту (0,054 г, 0,39 ммоль) растворяли в дихлорметане (3 мл) и ДМФА (1 мл). Реакцию оставляли при комнатной температуре на 16 ч. Затем растворитель удаляли в вакууме и сухой остаток снова растворяли в дихлорметане и фильтровали. Хроматографическая очистка(колонка С-18, 0,1% ТФУ в ацетонитриле) позволяли получить 0,1 г (58%) соединения, указанного в названии примера, в виде соли трифторуксусной кислоты. 1 Промежуточный продукт, полученный на стадии а) (0,65 мг, 1,8 ммоль) суспендировали в метаноле (6 мл) и воде (2 мл). Добавляли гидроокись лития (0,11 г, 3,9 ммоль) и смесь перемешивали в течение 24 ч при комнатной температуре. Затем добавляли воду (10 мл) и наполовину уменьшали объем. Водный раствор промывали 10-20 мл этилацетата, затем подкисляли соляной кислотой. В результате экстракции этилацетатом (2 х 20 мл), высушивания и выпаривания в вакууме получали 0,524 г чистого промежуточного продукта, указанного выше Это промежуточное соединение было получено аналогично тому, что описано Villaneuve(2,17 г, 10 ммоль) и гексахлорацетона (1,32 г, 5 ммоль) в дихлорметане (20 мл) перемешивали в атмосфере азота и охлаждали вплоть до -78 С. По каплям добавляли трифенилфосфин (2,6 г, 10 ммоль) в дихлорметане (10 мл) и смесь перемешивали в течение 30 мин. Затем по каплям добавляли метил-3-аминобензоат (1,5 г, 10 ммоль) в дихлорметане (10 мл), после чего добавляли триэтиламин (1 г, 10 ммоль) в дихлорметане. Затем реакционной смеси давали нагреться до комнатной температуры, после чего растворитель выпаривали в вакууме. Остаток очищали хроматографией на кремнеземе (гексан-этилацетат, 3:1), с последующей перекристаллизацией из этилацетата-гексана, при этом получали(0,038 г, 0,3 ммоль) и промежуточный продукт,полученный на стадии b) (0,25 г, 0,74 ммоль),растворяли в дихлорметане (9 мл) и ДМФА (3 мл). Реакцию оставляли при комнатной температуре на 19 ч. Затем растворитель удаляли в вакууме и сухой остаток снова растворяли в дихлорметане и фильтровали. После хроматографической очистки (этилацетат-гексан, 1:1),получали 0,029 г (67%) очищенного N-защищенного соединения, указанного в названии примера. 1N-защищенное соединение, полученное на стадии с) (0,16 г, 0,23 ммоль) и тиофенол (0,054 г, 0,46 ммоль) растворяли в дихлорметане (6 мл) и охлаждали до 0 градусов. Добавляли трифторуксусную кислоту (6 мл) и смеси давали возможность нагреться до комнатной температуры и оставляли на 1 ч. В результате выпаривания досуха и последующей хроматографической очистки (дихлорметан-метанол, 10:1,5) получали 0,150 г (90%) соединения, указанного в названии примера, в виде соли ТФУ. 1 а) 6-Этиламиноникотиновая кислота Этот промежуточный продукт получен из 6-хлорникотиновой кислоты и этиламина с помощью такой же процедуры, которая описана в примере 35, стадия а). Вместо этилацетата для экстракции использовали 1-бутанол. В результате перекристаллизации (МеОН-СНСl3) получали 0,53 г (50%). 1 34 растворитель удаляли в вакууме и остаток суспендировали в дихлорметане и фильтровали. Растворитель удаляли и сырой, неочищенный продукт очищали с помощью хроматографии(этилацетат-гексан, 2:1), при этом получали соединение, указанное в названии примера (0,063 г, 45%). 1(0,1 г, 0,27 ммоль), N,N'-дициклогексилкарбодиимид (0,127 г, 0,62 ммоль) и 4 диметиламинопиридин (0,016 г, 0,13 ммоль) растворяли в дихлорметане (4 мл) и оставляли при температуре окружающей среды. Через 19 ч смесь фильтровали и растворитель удаляли в вакууме. Неочищенный продукт очищали с помощью хроматографии (этилацетат-гексан, 1:1),при этом получали соединение, указанное в названии примера (0,040 г, 27%). 1 Соединение, полученное в примере 8 (0,1 г, 0,27 ммоль), 6-этиламиноникотиновую кислоту (0,084 г, 0,54 ммоль), N,N'-дициклогексилкарбодиимид (0,127 г, 0,62 ммоль) и 4 диметиламинопиридин (0,016 г, 0,13 ммоль) растворяли в ДМФА (3 мл) и оставляли при температуре окружающей среды. Через 19 ч 6-Аминоникотиновую кислоту (2 г, 22 ммоль) растворяли в метаноле (10 мл) и серной кислоте (0,5 мл). Раствор нагревали в колбе с обратным холодильником в течение ночи и растворитель выпаривали в вакууме. Неочищенный продукт растворяли в смеси вода-EtOAc и подщелачивали водным раствором гидрокарбоната натрия. В результате экстракции EtOAc получа 35 ли чистый промежуточный продукт, указанный выше (2,3 г, 70%). 1 Промежуточный продукт, полученный на стадии а) (0,75 г, 4,9 ммоль) растворяли ТГФ (5 мл). По каплям добавляли бис-(триметилсилил)амид натрия (5 мл, 2 М в ТГФ). После перемешивания при комнатной температуре в течение 30 мин добавляли ди-трет-бутилдикарбонат (1,1 г, 5 ммоль) в ТГФ (8 мл). Реакционную смесь оставляли на ночь в атмосфере азота. Затем раствор выпаривали в вакууме и растворяли в EtOAc (40 мл) и 0,1 М соляной кислоте (100 мл). Слои разделяли и водную фазу дважды экстрагировали EtOAc (40 мл), затем доводили до слабо щелочной реакции водным раствором гидрокарбоната натрия и еще один раз экстрагировали EtOAc (20 мл). Органические фракции объединяли, высушивали над сульфатом натрия и очищали с помощью хроматографии (EtOAc-гексан, 1:4), при этом получали очищенный промежуточный продукт, указанный выше (0,5 г, 40%). 1 Промежуточный продукт, полученный на стадии с) (0,4 г, 1,6 ммоль) суспендировали в метаноле (4 мл) и воде (1,25 мл). ДобавлялиLiOH (0,1 г, 4 ммоль). Суспензию оставляли при комнатной температуре на 48 ч. Затем прозрачный раствор концентрировали в вакууме и растворяли в воде и подкисляли уксусной кислотой(рН=4-5). В результате экстракции EtOAc получали очищенный промежуточный продукт, указанный выше (0,27 г, 70%). 1(0,150 г, 0,41 ммоль), промежуточный продукт,полученный на стадии с) (0,17 г, 0,49 ммоль),N,N'-дициклогексилкарбодиимид (0,1 г, 0,49 ммоль) и 4-диметиламинопиридин (0,06 г, 0,49 ммоль) растворяли в ДМФА (2 мл) . Смесь перемешивали при комнатной температуре в течение ночи, затем помещали на 50-градусную масляную баню на 2 ч. В результате выпаривания на силикагеле и очистки с помощью хроматографии получали N-защищенное соединение,указанное в названии (0,048 г, 20%). 1 Промежуточный продукт, полученный на стадии d) (0,048 г, 0,08 ммоль) растворяли в дихлорметане (2 мл). Добавляли трифторуксусную кислоту (1 мл) и смесь перемешивали в течение 1 ч. Выпариванием в вакууме получали неочищенное соединение, указанное в названии примера. Этот продукт растворяли в эфире (2 мл) и оставляли на ночь. Образовавшийся белый осадок отфильтровывали, чтобы получить соединение, указанное в названии примера в виде трифторацетатной соли (0,032 г, 65%). 1(0,076 г, 0,49 ммоль), N,N'-дициклогексилкарбодиимид (0,1 г, 0,49 ммоль) и 4-диметиламинопиридин (0,024 г, 0,2 ммоль) растворяли в дихлорметане (4 мл). Смесь оставляли на ночь. В результате выпаривания в вакууме, очистки с помощью хроматографии (EtOAc-гексан,1:2) получали соединение, указанное в названии примера (0,067 г, 32%). 1 а) 6-Диметиламиноникотиновая кислота 6-Хлорникотиновую кислоту (0,5 г, 3,17 ммоль) и диметиламин (10 мл, 40% в воде) нагревали в герметичном автоклаве при 130 С в течение 6 ч. Затем растворитель удаляли и остаток переносили в воду, рН доводили до 4-5. В результате экстракции дихлорметаном получали очищенный промежуточный продукт, указанный выше (0,1 г, 20%). 1b) (1S,2S)-N-цис-2-[6-фтор-3-пропионил 2-(6-диметиламинопирид-3-илкарбонилокси)фенил]циклопропил-N'-(5-цианопирид-2-ил)мочевина. Соединение, полученное в примере 8 (0,13 г, 0,3 ммоль), промежуточный продукт, полученный на стадии а) (0, 05 г, 0,3 ммоль), N,N'дициклогексилкарбодиимид (0, 09 г, 0,4 ммоль) и 4-диметиламинопиридин (0,02 г, 0,18 ммоль) растворяли в дихлорметане (3 мл) и ДМФА (1 мл). Смесь оставляли на ночь. В результате выпаривания в вакууме и очистки с помощью хроматографии (EtOAc-гексан, 2:1) получали соединение, указанное в названии примера (0,06 г,39%). 1(6,9 г, 50 ммоль) в 150 мл ДМФА добавляли трет-бутоксид калия (12,34 г, 110 ммоль) и смесь перемешивали при комнатной температуре в течение одного часа. Добавляли бензилбромид (20,5 г, 120 ммоль) и смесь перемешивали в течение двух дней при комнатной температуре. Смесь выпаривали в условиях пониженного давления и добавляли 100 мл 1,4-диоксана и раствор гидроокиси натрия (6,0 г, 150 ммоль) в 50 мл воды. Смесь нагревали в колбе с обратным холодильником в течение 2 ч, охлаждали и 38 выпаривали при пониженном давлении. Добавляли воду и смесь подкисляли уксусной кислотой. Продукт фильтровали, промывали холодной водой и высушивали. Выход: 10,2 г = 89%.(2,28 г, 10 ммоль) в 20 мл осушенного дихлорметана добавляли пять капель ДМФА и 2,5 мл тионилхлорида. Смесь нагревали в колбе с обратным холодильником в течение трех часов и выпаривали в условиях разрежения. Выход: 2,45 г = 100%.c) (1S,2S)-N-[цис-2-(6-Фтор-2-О-3-пропионилфеил)циклопропил]-N'-[2-(5-цианопирид-2 ил)мочевина-О-4-бензилоксибензоат. К раствору (1S,2S)-N-[цис-2-(6-фтор-2-гидрокси-3-пропионилфенил)циклопропил]-N'-(5 цианопирид-2-ил)мочевины (184 г, 0,5 ммоль) в 3 мл ДМФА добавляли трет-бутоксид калия(78,5 мг, 0,7 ммоль) и смесь перемешивали в течение одного часа при комнатной температуре. Добавляли раствор 4-бензилоксибензоилхлорида (185 мг, 0,75 ммоль) в 1 мл ДМФА и смесь перемешивали в течение ночи при комнатной температуре. Добавляли 40 мл этилацетата и органическую фазу четыре раза промывали водой. Раствор сушили с помощью сульфата натрия и выпаривали в условиях пониженного давления. Продукт выделяли с помощью хроматографии на колонке с силикагелем. Выход: 180 мг = 62%. 1(170 мг, 0,29 ммоль) в 15 мл этилацетата и 15 мл метанола три раза гидрировали в присутствии 10% палладия на угольном носителе (30 мг) при комнатной температуре и нормальном давлении. Катализатор отфильтровывали и промывали этилацетатом и метанолом и раствор выпаривали в условиях пониженного давления. Продукт выделяли хроматографией на колонке с силикагелем. Выход: 100 мг = 70%. 1(6,85 г, 45 ммоль) в 80 мл ДМФА добавляли трет-бутоксид калия (5,6 г, 51 ммоль) и смесь перемешивали при комнатной температуре в течение одного часа. Добавляли 4-метоксибен 39 зилхлорид (8,3 г, 52 ммоль) и смесь перемешивали в течение ночи при комнатной температуре. Смесь выпаривали в условиях пониженного давления и добавляли 200 мл этилацетата. Органическую фазу четыре раза промывали водой, высушивали с помощью сульфата натрия и выпаривали при пониженном давлении. Выход: 12,3 г = 100%. 1b) 4-(4-Метоксибензилокси)бензойная кислота. К раствору метил-4-(4-метоксибензилокси) бензоата (12,2 г, 44,8 ммоль) в 50 мл 1,4-диоксана добавляли раствор гидрооксиси лития(2,15 г, 89,6 ммоль) и смесь перемешивали в течение ночи при 60 С. Смесь выпаривали при пониженном давлении и добавляли 5% уксусную кислоту. Продукт фильтровали, промывали водой и высушивали. Выход: 10,1 г = 87%. 1c) Хлорметил-4-(4-метоксибензилокси) бензоат. К раствору 4-(4-метоксибензилокси) бензойной кислоты (5,16 г, 20 ммоль) в 100 мл 1,4 диоксана добавляли 40% раствор гидроокиси тетрабутиламмония (14,27 г, 22 ммоль) и смесь перемешивали в течение 2 ч при комнатной температуре. Смесь выпаривали при пониженном давлении и дважды выпаривали совместно с 1,4-диоксаном и два раза с толуолом. Высушенный продукт растворяли в 60 мл дихлорметана и добавляли иодхлорметан (35,3 г, 200 ммоль). Раствор перемешивали в течение двух дней при комнатной температуре и выпаривали при пониженном давлении. Добавляли примерно 100 мл этилацетата и органическую фазу дважды промывали водой, высушивали с помощью сульфата натрия и выпаривали в условиях пониженного давления. Продукт выделяли с помощью хроматографии на колонке с силикагелем. Выход: 4,48 г = 73%. 1d) Иодметил-4-(4-метоксибензилокси)бензоат. К раствору хлорметил-4-(4-метоксибензилокси)бензоата (0,77 г, 2,5 ммоль) в 15 мл осушенного ацетона добавляли иодид натрия(1,87 г, 12,5 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Смесь выпаривали при пониженном давлении и экстрагировали этилацетатом/водой. Органическую фазу промывали 5% раствором тиосульфата натрия, сушили с помощью сульфата натрия и выпаривали в условиях пониженного давления. Выход: 0,86 г = 86%.e) (1S,2S)-N-[цис-2-(6-Фтор-2-O-3-пропионилфенил)циклопропил]-N'-[2-(5-цианопиридил)]мочевина-O-метилен-4-(4-метоксибензилокси)бензоат. К раствору (1S,2S)-N-[цис-2-(6-фтор-2 гидрокси-3-пропионилфенил)циклопропил]-N'[2-(5-цианопиридил)]мочевины (368 мг, 1 ммоль) в 5 мл ДМФА добавляли суспензию 60% гидрида натрия в минеральном масле (44 мг, 1,1 ммоль) и смесь перемешивали при комнатной температуре в течение одного часа. Добавляли раствор иодметил-4-(4-метоксибензилокси)бензоата (0,84 г, 2,1 ммоль) в 2 мл ТГФ и смесь перемешивали при комнатной температуре в течение ночи. Добавляли 50 мл этилацетата и органическую фазу промывали четыре раза водой,сушили с помощью сульфата натрия и выпаривали при пониженном давлении. Продукт выделяли с помощью хроматографии на колонке с силикагелем. Выход: 525 мг = 82%. 1f) (1S,2S)-N-[цис-2-(6-Фтор-2-О-3-пропионилфенил)циклопропил]-N'-[2-(5-цианопиридил)]мочевина-O-метилен-4-гидроксибензоат. К раствору (1S,2S)-N-[цис-2-(6-фтор-2-О 3-пропионилфенил)циклопропил]-N'-[2-(5-цианопиридил)]мочевина-О-метилен-4-(4-метоксибензилокси)бензоата (100 мг, 0,156 ммоль) в 4 мл дихлорметана добавляли ТФУ (0,5 мл) и раствор перемешивали в течение одного часа при комнатной температуре. Раствор выпаривали при пониженном давлении и продукт выделяли хроматографией на колонке с силикагелем. Выход: 45 мг = 55%. 1 Это соединение было получено из 6 метиламиноникотиновой кислоты (0,050 г, 0,33 ммоль) и соединения, полученного в примере 8 и непрореагировавшие исходные материалы) очищали с помощью хроматографии (этилацетат), при этом получали 0,030 г (22%) соединения, указанного в заглавии примера. 1 Дикий тип Дикий тип 50% сыворотки В исследование входили многочисленные определения, выполненные с использованием красителя ХТТ на клетках МТ-4 (Weislow et al.,J.Nat.Cancer Inst., 1989, vol.81, no 8, 577 и все последующие), включая определения, выполненные в присутствии 50% сыворотки человека,чтобы показать вклад связывания белков. ЭД 50 представлена в мкг/мл. Также представлены исходные данные относительно рассчитываемого терапевтического индекса (SI-индекса безопасности), который определяется как отношение дозы, вызывающей токсический эффект в 50% соответствующих клеток, не инфицированных ВИЧ, к ЭД 50. Соединение предшествующего уровня техники, предложенное в Санта Фе, 1995ICAR, указано выше. Очевидно, что соединения согласно изобретению, особенно энантиомеры, имеют заметно более низкие значения ЭД 50, чем известные до настоящего времени соединения, включая значения эффективных доз в случае применения против известных проблематичных мутантов K103N и Y181C, так же как L100I и двойного мутанта L100I, Y181C. Более того,терапевтические индексы для энантиомеров в 510 раз выше, чем для соединения предшествующего уровня техники. Эти результаты следует рассматривать в контексте терапии ВИЧ инфекций, когда предполагаться, что пациенты будут принимать лекарственные средства против вирусов ВИЧ, известных своей способностью вырабатывать резистентность, в течение многих лет, если не до конца жизни. Поэтому высокое значение SI необходимо, чтобы избежать кумулятивной токсичности, позволяя в то Характер развития резистентности. Соединения изобретения были тестированы на антивирусную активность против нескольких штаммов ВИЧ, включая вирусы дикого типа и известные мутанты, возникшие в результате применения других ненуклеозидных ингибиторов обратной транскриптазы, которые описаны в обзоре Schinazi et al., InternationalAntiviral News, vol.4 no 6, pp.95-107 (1996). Результаты представлены в таблице 1. Таблица 1 Предшеств. уроПример 6 Пример 8 вень техники 0,0008 +/-0,0004 0,0007 +/-0,0002 0,0056 +/-0,004 0,006+/-0,003 0,007+/-0,001 0,023+/-0,011 0,017+/-0,008 0,037+/-0,007 0,13+/-0,060 0,17+/-0,07 0,39+/-0,31 0,9+/-0,6 0,006 +/-0,002 0,006 +/-0,001 0,13+/-0,02 0,08+/-0,05 0,08+/-0,06 0,13+/-0,07 0,08+/-0,06 0,06+/-0,02 0,17+/-0,03 0,9+/-0,05 1,0+/-0,05 1,9+/-1,5 не опред. 0,34+/-0,06 1,0 не опред. 0,009+/-0,001 0,026+/-0,009 22500 87000 5900 8830 4285 800 же время использовать адекватные дозы, чтобы поддерживать терапевтическое действие на вирусы и предотвратить спонтанное возникновение множества резистентных штаммов ВИЧ. Биологический пример 2. Период развития резистентности. По 2 х 104 клеток МТ 4 помещали в ячейки планшета для микротитрования и инфицировали вирусом ВИЧ-1IIIB в дозе, соответствующей 5-10TCID50. Тестируемые соединения добавляли в концентрациях, соответствующих примерно ЭД 50, по 8 повторов для каждой концентрации. После 6 дней инкубации измеряли активностьRT в 10 мкл надосадочной жидкости. При последующих пассажах культур один раз в неделю придерживались следующей процедуры: вирусы, которые сформировались в присутствии тестируемого соединения в такой концентрации, при которой активность RT составляет 50% от активности в необработанных препаратом инфицированных клетках (НИК,начальная ингибирующая концентрация), пассировали в свежие культуры клеток МТ 4. 15 мкл надосадочной жидкости из каждого из восьми повторов переносили к клеткам без тестируемого соединения (контроль) и к клеткам, к которым добавлено тестируемое соединение в той же самой концентрации и дополнительно в двух других концентрациях, увеличенных, соответственно, в пять раз (см. табл. 2, приведенную ниже). В том случае, когда вирусы продолжали расти при самой высокой нетоксичной концентрации (5-40 мкМ), содержимое 2-4 параллельных ячеек собирали и увеличивали объем, что 43 бы получить материал для анализа последовательностей и перекрестной резистентности. Таблица 2. Рост вирусов возможен. Размножение вирусов ингибировано На фиг. 1 приведен график, на котором против соответствующих временных точек нанесены значения, соответствующие росту вирусной резистентности к соединению изобретения (пример 8). На график также нанесена соответствующая кривая, полученная для наиболее близкого соединения, предложенного в Санта Фе, которое указано выше. Очевидно, что соединения изобретения вызывают развитие резистентности значительно медленнее. Биологический пример 3. Метаболизм в системе Р 450. Метаболизм соединений изобретения основными изоформами системы цитохрома Р 450 человека исследовали в инфицированных бакуловирусом клетках насекомых, трансфицированных кДНК цитохрома Р 450 человека (суперсомы) Gentest Corp. Woburn USA. Тестируемые соединения в концентрациях 0,5, 5 и 50 мкМ инкубировали в повторах в присутствии суперсом, способных к сверхэкспрессии различных изоформ цитохрома Р 450, включая CYP1A2 + Р 450 редуктазу, CYP2A6 + Р 450 редуктазу, CYP2C9-Arg 144 + Р 450 редуктазу,CYP2C19 +Р 450 редуктазу, CYP2D6-Val 374 + Р 450 редуктазу и CYP3A4 + Р 450 редуктазу. Концентрация цитохрома Р 450 в инкубируемых системах была фиксированной (например, 50 пмоль), и эксперименты проводились в течение 1 ч. Участие конкретной изоформы в метаболизме тестируемого соединения определяли с помощью УФ ВЭЖХ, измеряя хроматографическим путем исчезновение исходного соединения. Значения, соответствующие процентному содержанию соединений, оставшихся после 7,5 мин тестирования в трех концентрациях, свидетельствуют о том, что CYP3A4, 1 А 2, 2 С 19 и 2 А 6 вовлечены в метаболизм соединения, полученного в соответствии с примером 7. Сходные сочетания изоформ цитохрома Р 450 участвуют в метаболизме соединений предшествующего уровня техники, содержащих галогенпиридинил, которые были предложены в Санта Фе. Неожиданно для соединения, полученного в соответствии с примером 8, не зарегистрированы значимые метаболические превращения в присутствии некоторых изоформ Р 450, что может являться свидетельством того, что соединение стабильно в условиях in vivo, и что возмож 003327 44 ность нарушения метаболизма совместно вводимых лекарственных средств соответственно низка. Биологический пример 4. Фармакокинетика. Высвобождение соединения, имеющего формулу I, из перорально введенного пролекарства, имеющего формулу II, прослеживали у крыс. Соединение, полученное в соответствии с примером 7, было приготовлено в виде формы,содержащей в качестве носителя пропиленгликоль, и введено перорально паре голодных самцов крыс Sprague Dawley в дозе, соответствующей 0,027 ммоль/кг. В указанные интервалы времени из катетера, имплантированного в canisjugularis, отбирали 0,2 мл крови, центрифугировали и замораживали для последующего анализа. Высвободившееся лекарственное средство,имеющее формулу I (пример 6), исследовали методом ВЭЖХ. Из каждого образца плазмы отбирали пробы объемом 40-100 мкл и смешивали с равным объемом ацетонитрила (10 секунд, Vibrofex). Эти пробы центрифугировали(2 мин, 14000 об/мин) и 30 мкл надосадочной жидкости инъецировали в систему ВЭЖХ, параметры которой указаны ниже. Предварительная колонка Колонка: Подвижная фаза: Скорость потока: Регистрация:YMC основная, 3 мкм,150 х 3 мм 60% ацетонитрил в 3 мМ ацетате аммония, рН 6,4 0,4 мл/мин УФ, 250 нм Таблица 3 Уровень исходного соединения в плазме (мкг/мл) 0,24, 0,35 0,18, 0,28 0,13, 0,17 0,07, 0,12 0,05 0,07 На основании данных табл. 3 очевидно,что при пероральном введении пролекарства,имеющего формулу II, in vivo высвобождаются клинически значимые количества соединения,имеющего формулу I. Биологические примеры 5-8.i) Подготовка. В примерах, посвященных определению фармакокинетических параметров, использовали самцов крыс Sprague-Dawley весом примерно 200-250 г. По меньшей мере 16 ч перед экспериментом крысы голодали, но имели свободный доступ к воде. За день до эксперимента крыс анестезировали, используя смесь Efrane, веселящего газа и кислорода. В vena jugularis вводили катетер. Отмечали вес крыс в день эксперимента. Незадолго до того, как дать крысам дозу препарата перорально или перед в/в инъекцией дозы препарата в тыльную часть шеи, животных анестезировали. Каждое вещество вводили паре крыс. Обезьяны голодали в течение 12 ч перед пероральным введением, но имели свободный доступ к воде. Тестируемое соединение вводили через зонд для назогастрального питания младенцев. Через 6 ч обезьяны получали яблоко.ii) Приготовление доз. Для перорального введения соответствующие количества активных ингредиентов,описанных в последующих примерах, растворяли/суспендировали в растворе пропиленгликоля или 10% смолы акации и 1% твина в воде. Для внутривенного введения соединения растворяли в ДМСО.iii) Взятие образцов крови. Образцы крови (обычно 0,6 мл для крыс, 2 мл для обезьян) брали перед введением и через определенные указанные интервалы времени после введения лекарственного средства. У обезьян образцы откачивали из бедренной вены путем пункции в пробирки, содержащие ЭДТА. Образцы крови центрифугировали, инфекционные агенты нейтрализовали 1% SDS/64/20 мин и плазму хранили при -20 С.iv) Биологические анализы. Образцы плазмы готовили следующим образом: 40-100 мкл плазмы смешивали с равным объемом ацетонитрила (10 с, Vibrofex). Образцы центрифугировали (2 мин, 14000 об/мин) и 30 мкл надосадочной жидкости инъецировали в систему ВЭЖХ, параметры которой указаны далее. Предварительная 46 Соединения, имеющие формулу I, сравнивали по стабильности in vivo и пригодности с близким соединением, предложенным в Санта Фе, имеющим название -N-(цис-2-(6-фтор-2 гидрокси-3-пропионилфенил)циклопропил)-N'(5-хлорпиридил-2-ил)мочевина, для чего соответствующие соединения в наполнителе ДМСО были введены в дозе 0,024 ммоль/кг. На графике, приведенном на фиг. 2, против временных значений отмечены значения уровней в плазме соответствующих соединений (в каждом случаеn=2). Очевидно, что соответствующие кривые имеют сходный характер, но значение параметра AUC (0-4 ч) для соединения изобретения в 1,5 раза выше значения AUC (0-4 ч) для близкого соединения предшествующего уровня техники. Другими словами, соединения изобретения позволяют обеспечить более высокую экспозицию in vivo, на 50% выше, чем описанное ранее производное, хотя еще предстоит выяснить, является ли это следствием низкой скорости выведения из организма соединений изобретения или с большей степенью связывания этих соединений в тканях и т.д. по сравнению с соединениями предшествующего уровня техники. Биологический пример 6. Биоэквивалентность пролекарств и исходного соединения. Крысам вводили различные соединения,имеющие формулу II (которые являются пролекарствами соединений, имеющих формулу I), и в течение определенного времени следили за уровнями в плазме исходного соединения, полученного согласно изобретению (в этом примере использовали соединение, полученное согласно примеру 10). В качестве наполнителя использовали 10% смолу акации и 1% твин в воде или пропиленгликоль Биологический пример 5. Сравнение с близким соединением предшествующего уровня техники. Таблица 4 Соединение Пример 12 Уровень в плазме исходного соединения (мкг/мл) 0,2 0,2 0,3 0,2 0,3 0,3 Очевидно, что пролекарства, имеющие формулу II, in vivo высвобождают в плазму достаточные для клинического действия количества соединений, имеющих формулу I. Абсолютная биологическая доступность при пероральном введении (определенная относительно в/в дозы, как описано в разделе, касающемся подготовки эксперимента) составляла 28-33% для соединения, согласно примеру 37, и 27% для животных, на которых можно было определить доступность соединения, полученного согласно примеру 27. Биологический пример 7. Биологическая доступность у разных видов. Пролекарство, имеющее согласно изобретению формулу II (пример 12), вводили в той же дозе (0,026 ммоль/кг) и с тем же носителем соединения, имеющего формулу I (пример 10) измеряли как функцию времени. Таблица 5 Крыса Уровень в плазме исходного соединения (мкг/мл) 0,09 0,05 0,10 0,07 0,09 0,08 0,08 0,08 0,06 0,05 Очевидно, что пролекарства, имеющие формулу II, in vivo высвобождают достаточные для клинического действия количества соединений, имеющих формулу I. Высвобождение происходит и у грызунов и у приматов, причем значительно более высокий уровень наблюдается в плазме приматов. Соответствующие данные для соединения,согласно примеру 28 (крыса: смола акации/твин; обезьяна: пропиленгликоль), показаны в табл. 5 А. Таблица 5 А Время Уровень в плазме исходВид(мин) ного соединения (мкг/мл) Крыса 30 0,033 0,046 60 0,039 0,084 120 0,066 0,123 240 0,039 0,034 360 0,03 0,03 Обезьяна 30 0,108 0,03 90 0,159 0,098 180 0,062 0,050 240 0,03 0,060 540 0,036 0,070 Биологический пример 8. Антивирусная активность. Соединения, имеющие формулу I, были тестированы на ВИЧ-1 активность против вируса дикого типа ВИЧIIIB и резистентных мутантов в присутствии 50% сыворотки человека или без сыворотки в исследованиях с применением ХТТ-формазана, при которых оценивали ингибирование цитопатогенного действия на клетки МТ 4. В каждом случае значения ЭД 50 указаны в мкМ. Пример 10 ВИЧ Пример 10 50% сывоштамм ротки Дикий тип 0,01 0,06 Таким образом, соединения, имеющие формулу I, обладают высокой активностью против различных штаммов ВИЧ при концентрациях, которые могут быть достигнуты in vivo. Биологический пример 9. Антивирусная активность. Кроме того, соединения, полученные согласно изобретению, сравнивали с близким соединением предшествующего уровня техники,используя известный анализ культуры клеток,при котором Т клетки человека линии МТ 4 выращивали на среде RPMI 1640 с добавлением 10% околоплодной сыворотки теленка, пенициллина и стрептомицина, клетки высевали в микропланшеты на 96 лунок (2 х 104 клеток/лунку) и инфицировали 10-20 TCID50 на лунку ВИЧ-1IIIB (дикий тип) или мутантными 50 вирусами, несущими мутации RT Ilе 100, Cys 181 или Asn 103. Серийно разведенные тестируемые соединения добавляли в соответствующие лунки и культуры инкубировали при 37 С в атмосфере, обогащенной СО 2, и определяли выживаемость клеток на пятый или шестой день с помощью витального красителя ХТТ. Результаты, приведенные ниже, представляют собой средние значения из нескольких определений. Результаты представлены в виде ЭД 50 в мкМ. Таблица 7 Пример Предшествующий уровень техники Санта Фе Пример 10 Пример 11 Пример 8 Пример 6 Соединения изобретения имеют значительно улучшенные характеристики действия против вирусов дикого типа и особенно против важных с клинической точки зрения мутантов,возникающих во время лечения NNRTI (ненуклеозидные ингибиторы обратной транскриптазы). Биологический пример 10. Кинетика связывания. Степень ассоциации и диссоциации NNRTI с ферментом-мишенью может быть непосредственно определена с помощью метода резонанса поверхностных плазмонов, при котором обратная транскриптаза иммобилизуется на поверхности чипа, и связывание или диссоциация предполагаемого ингибитора прослеживаются путем наблюдения за изменениями показателя преломления, вызываемых происходящим при этом увеличением или уменьшением массы чипа. Соединение изобретения (пример 8) сравнивали с ближайшим соединением предшествующего уровня техники из Санта Фе, которое указано выше. Эксперименты проводили на Biacore 2000 (Biacore AB, Uppsala, Sweden), используя компьютерную программу BIAevaluation (версия 3.0) для оценки полученных данных. Связывание небольшого анализируемого вещества(NNRTI) с гораздо более крупным ферментом приводит к ответам, характеризующим связывание, в пределах 10-20 RU. Различия между показателем преломления буфера для промывки и образца усложняет количественную оценку данных при вводе образца. Во время фазы диссоциации изменение суммарного показателя преломления является незначительным, поэтому связывание различных веществ оценивали во время этой фазы. Иммобилизация: фермент и белок-свидетель были иммобилизованы прямым связыванием с первичными аминами на СМ 5 чипе (Mark 51gren et al., 1998). В качестве белка-свидетеля использовали антитела к Fc g (Biacore BR-100057), они были иммобилизованы в соответствии с инструкцией производителя. Обратную транскриптазу ВИЧ (Unge et al., 1990) переносили из 3 М раствора (NН 4)2SO4 в 5 мМ Hepes, pH 1,6,содержащий 4 мМ MgCl2, используя концентрирующую центрифугу Naposept 10K (Pall Filtron,MA, USA). Количество RT, иммобилизованной на сенсорный чип, соответствовало 6800-9700RU. Сенсорную поверхность дезактивировали инъекцией 35 мл 0,5 М Tris pH 7,6; 4 мМ MgCl2 0,5 М КСl. Процедуру иммобилизации проводили при 33 С. Взаимодействие с ингибиторами: исходные растворы ингибиторов (1 мг/мл в ДМСО) разводили в буфере для прокачки RT (10 мМHepes pH 7,6; 4 мМ MgCl2; 0,25 мМ спермин; 40 мМ КСl; 0,5% тритон Х-100; 3% ДМСО; 0,5% околоплодная сыворотка теленка) до концентрации 10 мМ. Связывание вещества с RT анализировали путем инъекции 200 мл разведенного вещества при скорости потока 20 мл/мин и температуре 25 С. После каждой инъекции вещества систему промывали, инъецируя 120 мл 10% ДМСО в буфере для прокачки RT. Результаты показаны на фиг. 3. Очевидно,что существуют различия в кинетике связывания между соединением изобретения и соединением предшествующего уровня техники, причем соединение, полученное согласно изобретению, диссоциирует с меньшей скоростью, что свидетельствует о более эффективном связывании с ферментом. Ссылки:Analytical Biochemistry, 1998, vol.265, in press. Хотя различные аспекты и варианты изобретения были иллюстрированы благодаря ссылкам на конкретные примеры, сравнительные примеры и фигуры, следует понимать, что изобретение не ограничено этими вариантами, а может быть расширено в духе и рамках прилагаемой формулы изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы I где Rx является цианогруппой или атомом брома;R является атомом водорода или группой формулы где кольцо Х представляет собой фенил или пиридил,R3 и R4 независимо выбраны из Н, ОН,амино и замещенных аминогрупп;n равно независимо 0, 1 или 2; р равно 0 или 1; и фармацевтически приемлемые соли этих соединений. 2. Соединение по п.1, в котором R1 является атомом фтора. 3. Соединение по п.1, в котором R2 является этилом. 4. Соединение по п.1, представляющее собой, по меньшей мере, на 60%, предпочтительно, по меньшей мере, на 90%, 1S, 2S энантиомерную форму. 5. Соединение по п.1, в котором Rx является цианогруппой. 6. Соединение по п.1, которое представляет собой соединение формулы II где кольцо X, Rx, R1, R2, R3, R4, n и р определены в п.1, и фармацевтически приемлемые соли этих соединений. 7. Соединение по п.6, в котором кольцо Х означает пиридил. 8. Соединение по п.7, в котором кольцо Х означает фенил. 9. Соединение по п.6, в котором кольцо Х означает пирид-2-ил или предпочтительно пирид-3-ил. 10. Соединение по п.6, в котором R3 означает -NH2-, -NHСН 3, -NНСН 2 СН 3 или -N(СН 3)2. 11. Соединение по п.6, в котором R3 находится в мета-положении относительно карбонильной группы, особенно в том случае, когда кольцо Х является фенилом, либо в котором R3 находится в пара-положении относительно карбонильной группы, особенно в том случае, когда кольцо Х является пиридилом. 12. Соединение по п.6, в котором -(СН 2)nи/или -(СН 2)p- отсутствуют, то есть р и/или n равно 0. 13. Соединение по п.6, в котором Rx является цианогруппой. 14. Соединение по п.6, в котором R1 является атомом фтора. 15. Соединение по п.6, в котором R2 является этилом. 53 16. Соединение по п.6, которое, по меньшей мере, на 60%, предпочтительно, по меньшей мере, на 90%, представлено 1S,2S энантиомерной формой. 17. Соединение по п.1, выбранное из следующей группы соединений:(1R,2R)-N-[цис-2-(6-фтор,2-гидрокси,3 пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина и фармацевтически приемлемые соли этих соединений. 18. Соединение по п.17, представляющее собой (1S,2S)-N-[цис-2-(6-фтор, 2-гидрокси, 3 пропионилфенил)циклопропил]-N'-(5-цианопирид-2-ил)мочевину или ее фармацевтически приемлемую соль. 19. Соединение по п.1, выбранное из группы, включающей в себя следующие соединения:(1S,2S)-N-[цис-2-(2-(3-диметиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил) циклопропил]-N'-(5-бромпирид-2-ил)мочевина,(1S,2S)-N-цис-2-[6-фтор-3-пропионил-2(6-метиламинопирид-3-илкарбонилокси)фенил] циклопропил-N'-(5-бромпирид-2-ил)мочевина,(1R,2R)-N-[цис-2-(2-(3-аминофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина,(1R,2R)-N-[цис-2-(2-(3-этиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевина,(1R,2R)-N-[цис-2-(2-(3-диметиламинофенилкарбонилокси)-6-фтор-3-пропионилфенил) циклопропил]-N'-(5-бромпирид-2-ил)мочевина,(1R,2R)-N-цис-2-[6-фтор-3-пропионил-2(6-метиламинопирид-3-илкарбонилокси)фенил] циклопропил-N'-(5-бромпирид-2-ил)мочевина,и фармацевтически приемлемые соли этих соединений. 20. Соединение по п.19, представляющее(1S,2S)-N-цис-2-[6-фтор-3-пропионил-2-(6-метиламинопирид-3-илкарбонилокси)фенил]циклопропил-N'-(5-цианопирид-2-ил)мочевину и ее фармацевтически приемлемую соль. 21. Соединение по п.19, представляющее собой (1S,2S)-N-[цис-2-(2-(3-аминофенилкарбонилокси)-6-фтор-3-пропионилфенил)циклопропил]-N'-(5-бромпирид-2-ил)мочевину и ее фармацевтически приемлемую соль. 22. Фармацевтическая композиция, включающая соединение по любому из пп.1-21 и фармацевтически приемлемый носитель или разбавитель. 23. Композиция по п.22, дополнительно включающая от одного до трех дополнительных антиретровирусных средств. 24. Композиция по п.23, в которой дополнительным антиретровирусным средством является средство, выбранное из группы, включающей AZT, ddI, ddC, D4T, 3 ТС, адефовир, дипивоксил, абакавир, бис-РОС-РМРА, фоскарнет,оксимочевину, эфавиренц, тровирдин, невирапин, делавирдин, PFA, H2G, АВТ 606, ритонавир, сахинавир, индинавир, ампренавир (Вертекс VX 478), Mitsubishi MKC-442 и нелфинавир. 25. Соединение по любому из пп.1-21 для применения в антивирусной терапии. 26. Применение соединения по любому из пп.1-21 в качестве активного вещества лекарственного средства для лечения или профилактики ВИЧ. 27. Способ подавления или предотвращения ВИЧ-инфекции, заключающийся во введении субъекту, который в этом нуждается, эффективного количества соединения по п.1 или 6. 28. Способ получения соединения формулы I по п.1, где R - водород, включающий перегруппировку Курциуса азида кислоты следующей формулы где PG означает защитную группу гидроксила, a
МПК / Метки
МПК: A61K 31/17, C07D 213/61
Метки: антивирусные, соединения
Код ссылки
<a href="https://eas.patents.su/29-3327-antivirusnye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Антивирусные соединения</a>
Антивирусные соединения

Номер патента: 1389
Опубликовано: 26.02.2001
Авторы: Миллер Шон К., Теббе Марк Дж., Виктор Франц, Спитцер Вейн А., Юнгхейм Луи Н.
МПК: A61K 31/4184, A61P 31/12, C07D 235/30...
Метки: антивирусные, соединения
Формула / Реферат:
1. Соединение формулы I в которой а равно 0, 1, 2 или 3; каждый R независимо представляет водород, галоген, циано, амино, галоген(C1-C4)алкил, ди(С1-С4)алкиламино, азидо, С1-С6-алкил, карбамоил, карбамоилокси, карбамоиламино, C1-C4-алкокси, С1-С4-алкилтио, С1-С4-алкилсульфинил, C1-C4-алкилсульфонил, пирролидино, пиперидино или морфолино; R0 представляет водород, галоген, С1-С4-алкил или C1-C4-алкокси; R1 представляет галоген, циано,...
Фармкомпозиция, содержащая соединения с анти-ха активностью и соединения антагониста агрегации тромбоцитов, их применение, набор, содержащий эти соединения, способ лечения или профилактики заболеваний, сопутствующих тромбообразованию

Номер патента: 2475
Опубликовано: 27.06.2002
Авторы: Данвидди Кристофер Т., Кюродо Алан Х., Юзан Андре, Перроне Марк Х., Лидли Роберт Дж.
МПК: A61P 7/02, A61K 31/715
Метки: способ, содержащая, содержащий, тромбоцитов, анти-ха, антагониста, лечения, агрегации, активностью, профилактики, эти, сопутствующих, заболеваний, тромбообразованию, фармкомпозиция, соединения, применение, набор
Формула / Реферат:
1. Фармацевтическая композиция, содержащая фармацевтически пригодный носитель и фармацевтически эффективные количества соединения, обладающего анти-Ха активностью, и соединения антагониста агрегации тромбоцитов. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что соединение, обладающее анти-Ха активностью, является низкомолекулярно весовым гепарином. 3. Фармацевтическая композиция по п.2, отличающаяся тем, что низкомолекулярно весовой...
Хиральные бифосфиновые соединения, комплексы на их основе и способ получения хирального бифосфинового соединения

Номер патента: 1377
Опубликовано: 26.02.2001
Авторы: Воланте Ральф П., Россен Кай, Пай Филип
МПК: C07F 9/50, C07C 25/22, C07B 53/00...
Метки: хиральные, получения, бифосфиновые, соединения, способ, основе, комплексы, хирального, бифосфинового
Формула / Реферат:
1. Хиральные бифосфины формулы где R представляет C1-4-алкил, С3-6-циклоалкил или арил, незамещенный или замещенный -F, -СН3, -СF3 или СН3О-; и Х1 и Х2 связывают два R2Р-замещенные бензола и независимо образуют 2-4-членную связывающую группу, состоящую из 2-4 атомов углерода и одного незамещенного или замещенного гетероатома, выбранных из О, S, SO, SO2 или 2. Соединение по п.1, в котором число атомов в связи X1 является таким же, как и...
Четырехциклические конденсированные соединения с гетероатомами, замещенные арилом, промежуточные соединения, способы получения, композиции и методы лечения

Номер патента: 1649
Опубликовано: 25.06.2001
Автор: Гриз Тимоти А.
МПК: C07D 471/00, A61K 31/35, A61P 5/30...
Метки: лечения, гетероатомами, соединения, замещенные, получения, методы, конденсированные, способы, композиции, четырехциклические, промежуточные, арилом
Формула / Реферат:
1. Соединение формулы I или II где Y обозначает -О-, -S-, -СН2-, -СН2СН2-, -СН=СН- или -NR4-; В обозначает -СН2- или -СО-; R1 и R2 обозначают каждый независимо -Н, -ОН, -O(C1-C4алкил), -ОСОС6Н5, -ОСО(С1-С6алкил), -ОSО2(С4-С6алкил), OSO2СF3, Сl или F; n равно 1 или 2; W обозначает -СН2- или >С=O; R3 обозначает 1-пиперидинил, 2-оксо-1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 2-оксо-1-пирролидинил,...
Циклоалкильные соединения, лактамы, лактоны и родственные соединения, содержащие их фармацевтические композиции и способы ингибирования высвобождения и/или синтеза β-амилоидного пептида с помощьюуказанных соединений

Номер патента: 2100
Опубликовано: 24.12.2001
Авторы: Плейсс Майкл А., Макданиел Стейси Л., Кви Синтия Л., Торсетт Юджин Д., Рил Джон К., Дрост Джеймс Дж., Фридман Стефен, Бриттон Томас К., Дрессман Брюс А., Скотт Уильям Леонард, Портер Варрен Дж., Ву Джинг, Одия Джеймс Е., Латимер Ли Х., Мабри Томас Э., Ниссен Джеффри С., Танг Джей С., Генри Стивен С., Стаки Расселл Д., Нейц Джеффри, Джон Варгес
МПК: A61P 25/28, A61K 31/55, C07D 243/10...
Метки: соединения, способы, фармацевтические, циклоалкильные, лактоны, помощьюуказанных, ингибирования, лактамы, высвобождения, соединений, пептида, композиции, синтеза, beta;-амилоидного, родственные, содержащие
Формула / Реферат:
1. Способ ингибирования высвобождения и/или синтеза b -амилоидного пептида в клетке, который заключается в том, что в такую клетку вводят соединение или смесь соединений в количестве, эффективном для ингибирования высвобождения и/или синтеза b -амилоидного пептида в клетке, и указанные соединения имеют формулу I в которой R1 выбирают из группы, включающей C1-С10алкил, необязательно замещенный 1-3 заместителями, независимо выбранными из...
Предыдущий патент: Способ лечения рака
Следующий патент: Легочная и назальная доставка ралоксифена
Случайный патент: Шнековый пресс для изготовления брикетов из растительных материалов