Окса-и тиадиазолы и их применение в качестве ингибиторов металлопротеиназ
Номер патента: 7609
Опубликовано: 29.12.2006
Авторы: Гийон Жан-Ив, Дэвис Стефен Джон, Пэн Жилль, Эйскаф Эндрю Пол





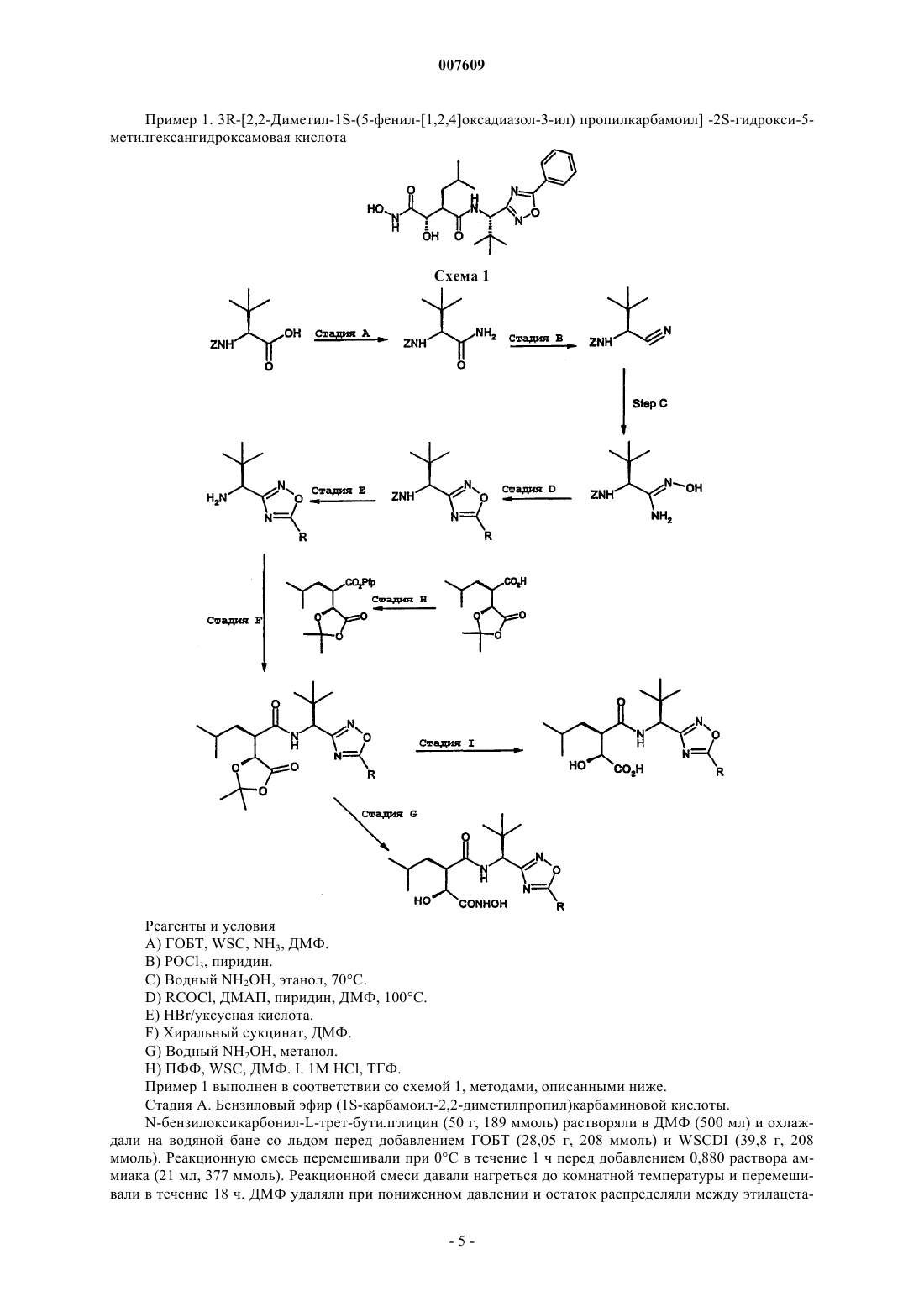


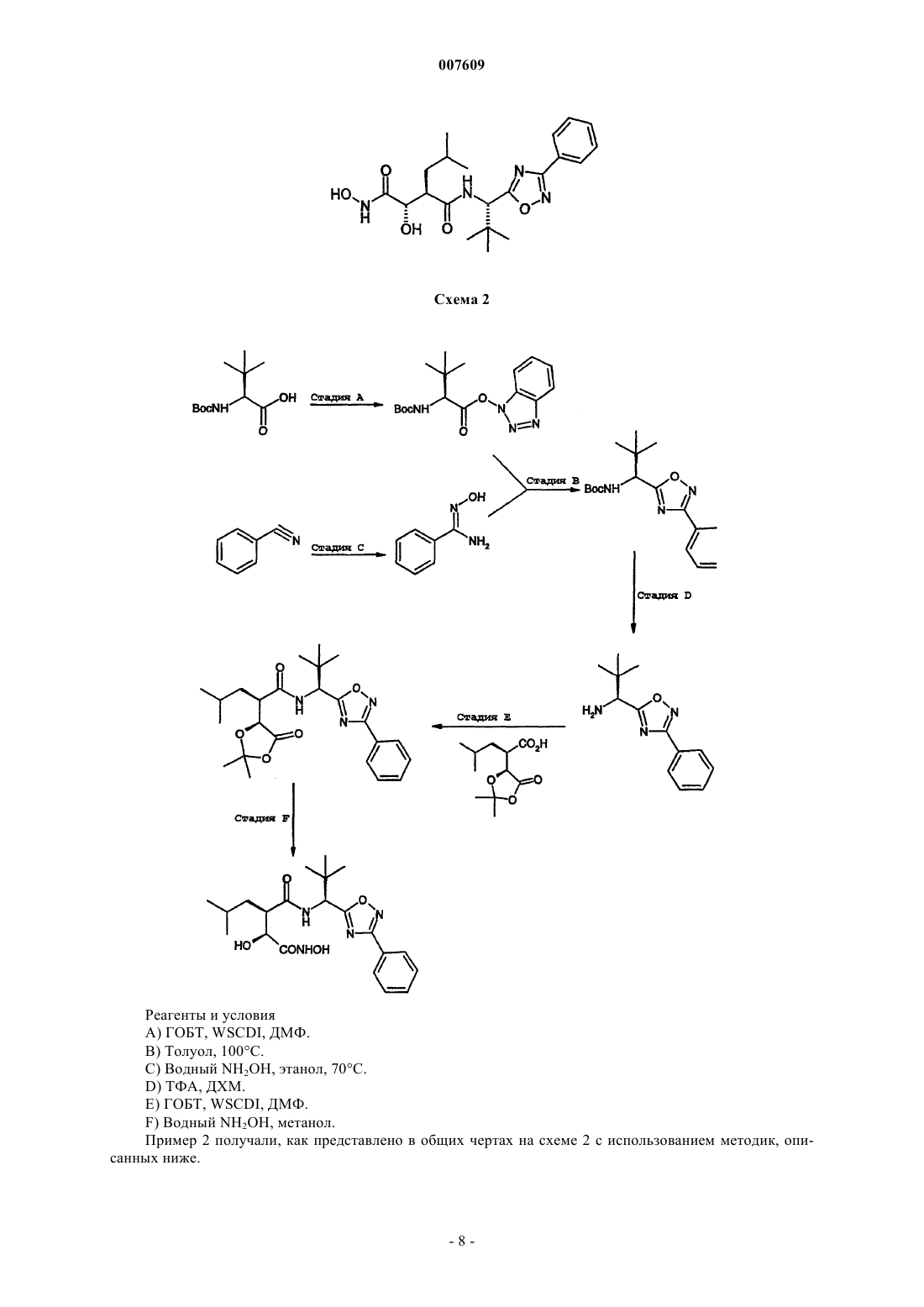



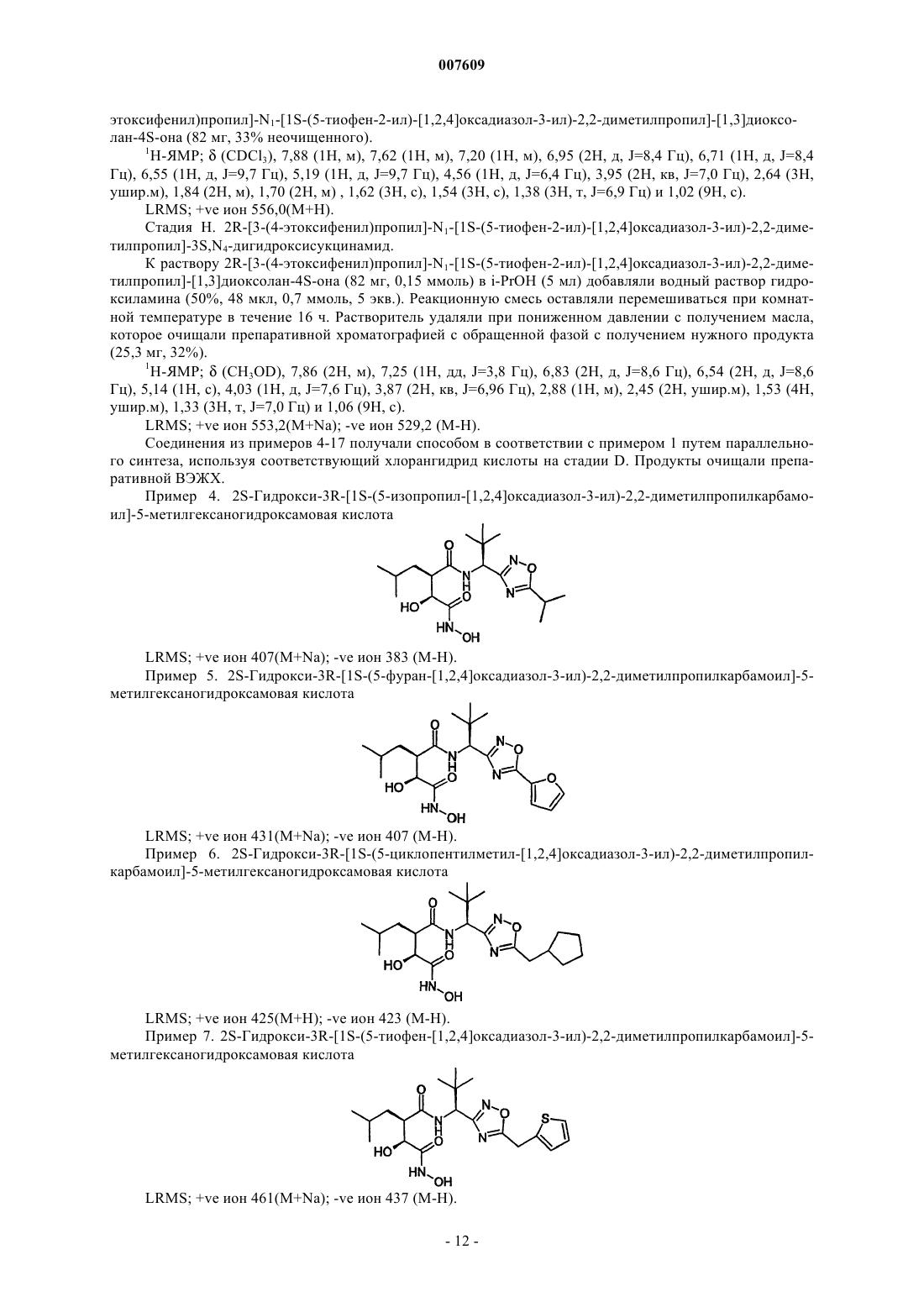
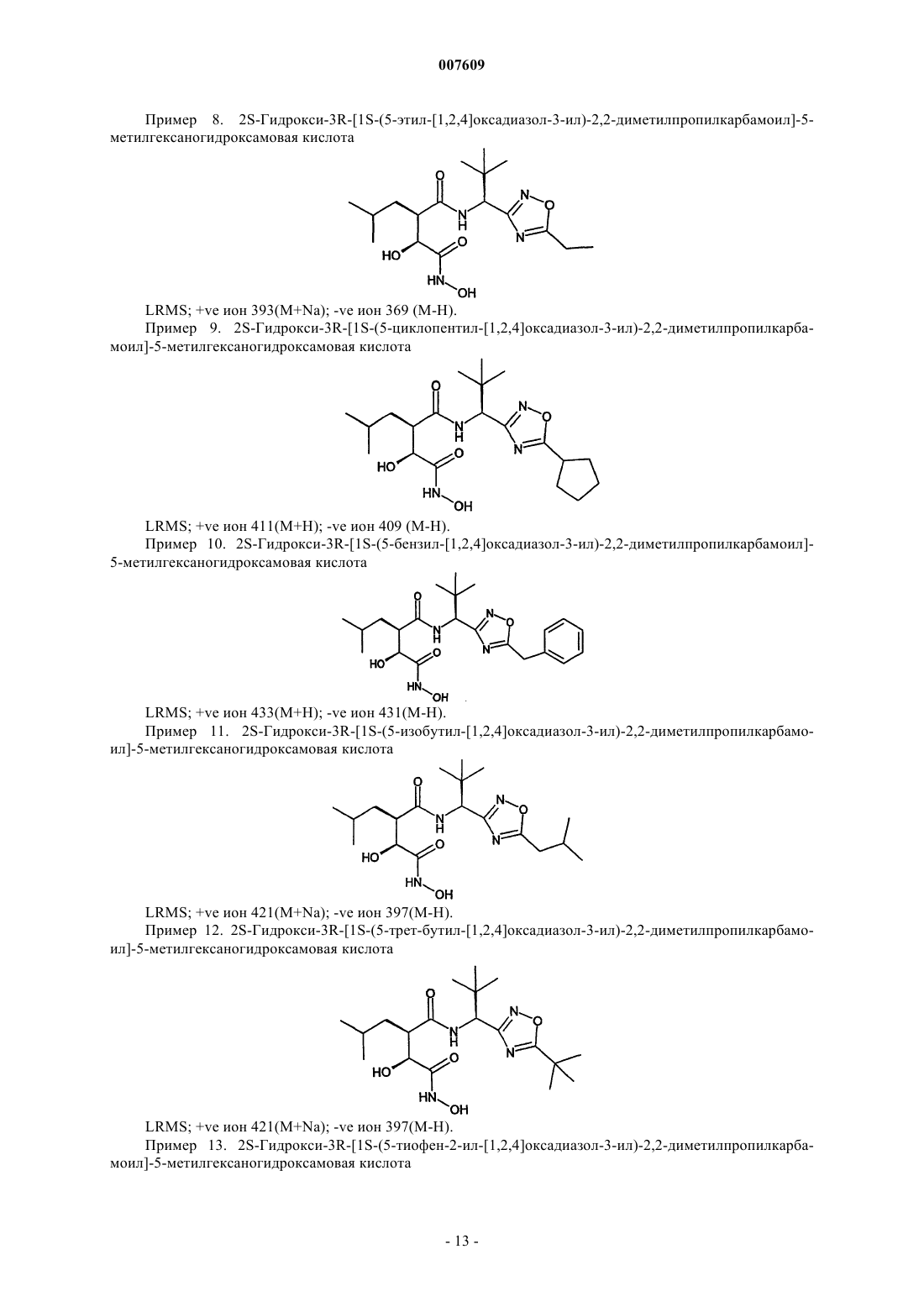

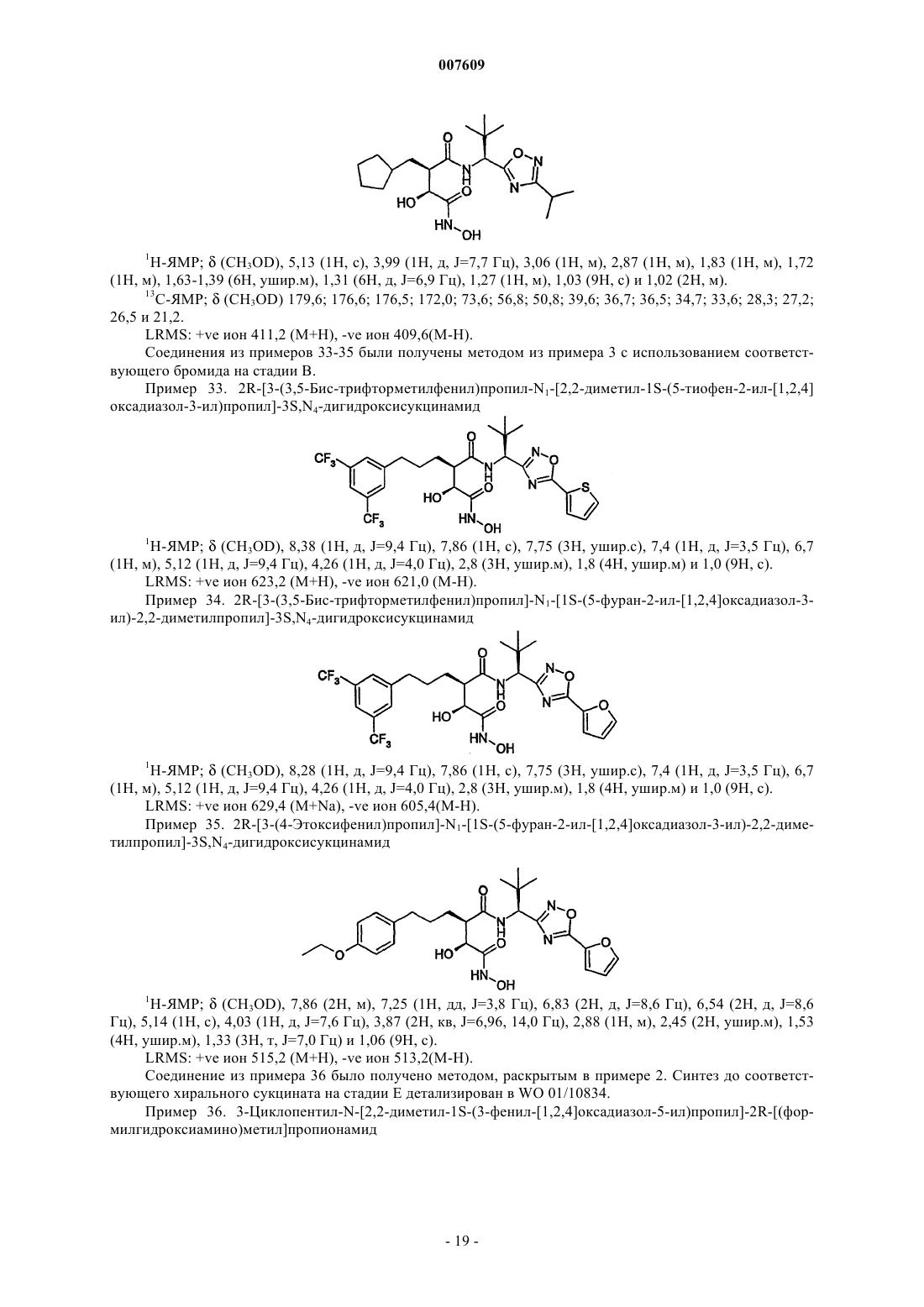






Формула / Реферат
1. Соединение формулы (IA) или (IB)

где W представляет НО(С=O)-, HONH(C=O)- или Н(С=O)N(ОН)-;
X представляет -О- или -S-;
R1 представляет водород, -ОН, метокси, циклопентил, н-пропил или аллил;
R2 представляет (С1-С4)алкил; фенил-(C1-C6алкил)- или фенилаллил, где фенильное кольцо необязательно замещено (C1-C6)алкилом, (C1-С6)алкокси, хлором или трифторметилом; или С3-С6циклоалкил-(C1-С6алкил)-;
R3 представляет (C1-C6алкил), бензил, фенил, циклогексилметил, пиридин-3-илметил, трет-бутоксиметил, изопропил, изобутил, втор-бутил, трет-бутил, 1-бензилтио-1-метилэтил, 1-метилтио-1-метилэтил или 1-меркапто-1-метилэтил;
R4 представляет C1-C6алкил, С3-С6циклоалкил или группу, выбранную из фенила, тиенила, тиенилметила, фуранила, фуранилметила, пирролила, пирролилметила, каждый из которых необязательно замещен в арильном или гетероарильном кольце C1-C6алкилом или галогеном;
и его фармацевтически приемлемые соли, гидраты или сольваты.
2. Соединение по п.1, причем соединение имеет формулу (IA).
3. Соединение по п.1 или 2, где X=-О-.
4. Соединение по любому из предшествующих пунктов, причем W является HONH(C=O)-.
5. Соединение по любому из предшествующих пунктов, где R1 представляет гидроксигруппу.
6. Соединение по любому из пп.1-5, причем R2 представляет изобутил или циклопентилметил.
7. Соединение по любому из пп.1-6, причем R3 представляет трет-бутил.
8. Соединение по любому из предыдущих пунктов, где R4 представляет 2-тиенил, 2-фуранил или 2-пирролил.
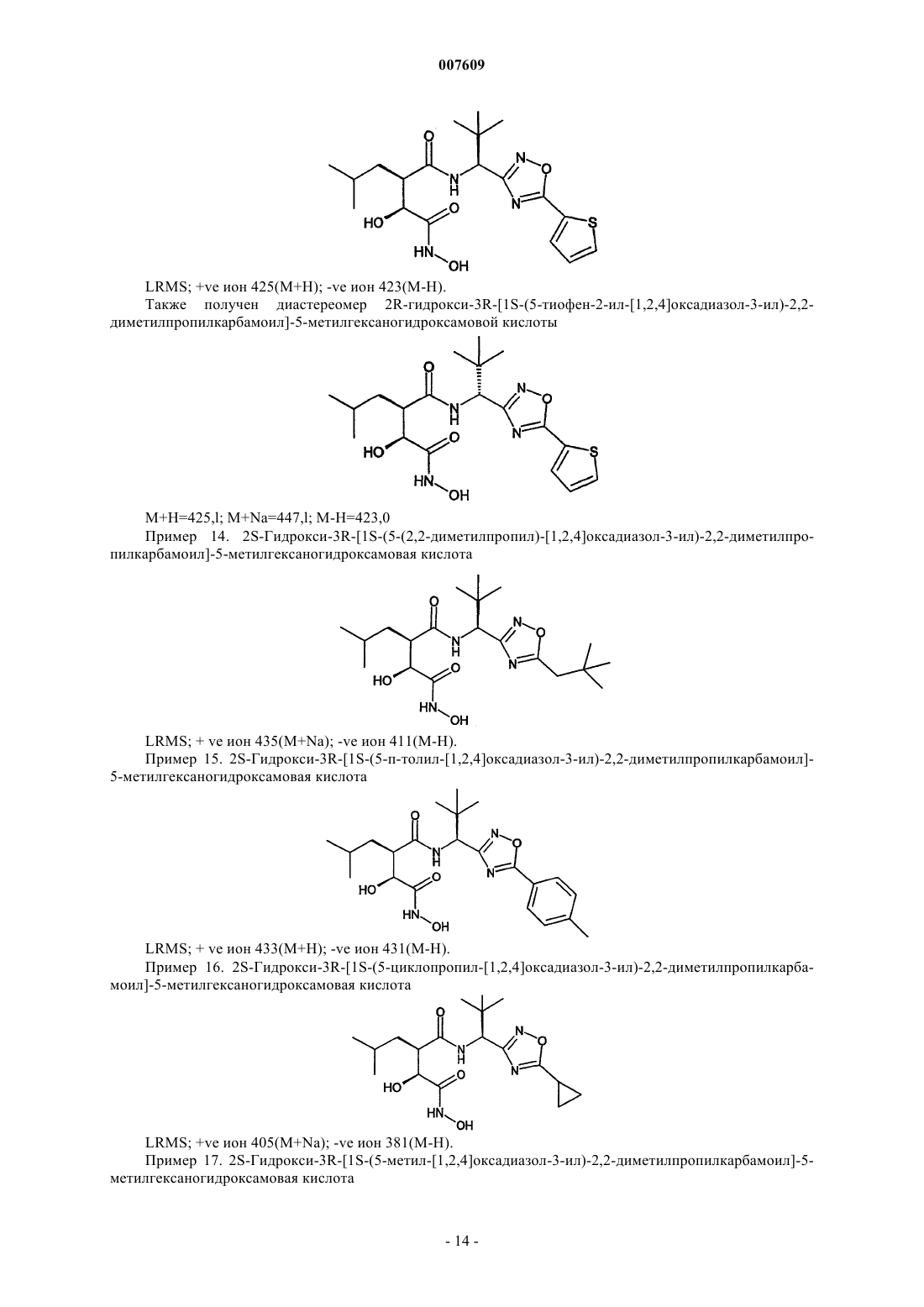
9. Соединение формулы (IА) по п.1, где X=-O-, R1 представляет -ОН; W представляет -C(=O)NHOH, R2 представляет изобутил, R3 представляет трет-бутил и R4 представляет 2-тиенил.
10. Соединение по п.1, выбранное из группы, включающей
3R-[2,2-диметил-1S-(5-фенил-[1,2,4]оксадиазол-3-ил)пропилкарбамоил]-2S-гидрокси-5-метилгексаногидроксамовую кислоту;
3R-[2,2-диметил-1S-(5-фенил-[1,2,4]оксадиазол-3-ил)пропилкарбамоил]-2R-гидpoкcи-5-мeтилгeкcaнoгидpoкcaмoвую кислоту;
3R-[2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропилкарбамоил]-2S-гидрокси-5-метилгексаногидроксамовую кислоту;
2R-[3-(4-этоксифенил)пропил]-N1-[1S-(5-тиофен-2-ил)-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропил]-3S,N4-дигидроксисукцинамид;
2S-гидрокси-3R-[1S-(5-изопропил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(5-фуран-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(5-циклопентилметил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(5-тиофен-2-илметил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(5-этил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(5-циклопентил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(5-бензил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(5-изобутил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(5-трет-бутил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-ме-тилгексаногидроксамовую кислоту;
3R-[1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногид-роксамовую кислоту;
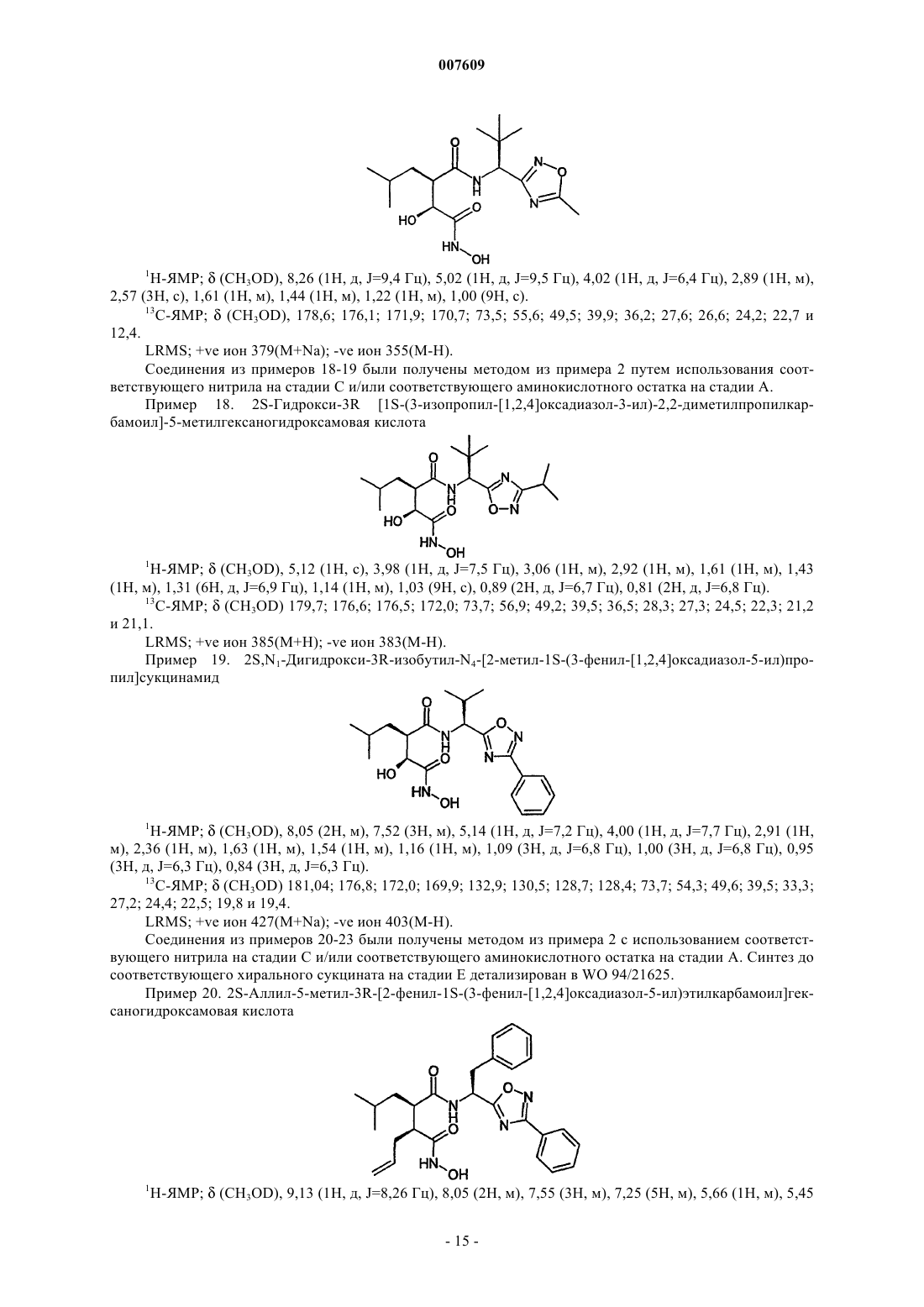
2S-гидрокси-3R[1S-(5-(2,2-диметилпропил)-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(5-п-толил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(5-циклопропил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(5-метил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-гидрокси-3R-[1S-(3-изопропил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S,N1-дигидрокси-3R-изобутил-N4-[2-метил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропил]сукцинамид;
2S-аллил-5-метил-3R-[2-фенил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)этилкарбамоил]гексаногидроксамовую кислоту;
2S-аллил-5-метил-3R-[2-фенил-1S-(3-изопропил-[1,2,4]оксадиазол-5-ил)этилкарбамоил]гексаногид-роксамовую кислоту;
2S-аллил-3R-[2,2-диметил-1S-(3-метил-[1,2,4]оксадиазол-5-ил)пропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
3R-[2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропилкарбамоил]-5-метилгексаногидроксамовую кислоту;
2S-метокси-5-метил-3R-[1S-(3-метил-[1,2,4]оксадиазол-5-ил)-2-фенилэтилкарбамоил]гексаногидроксамовую кислоту;
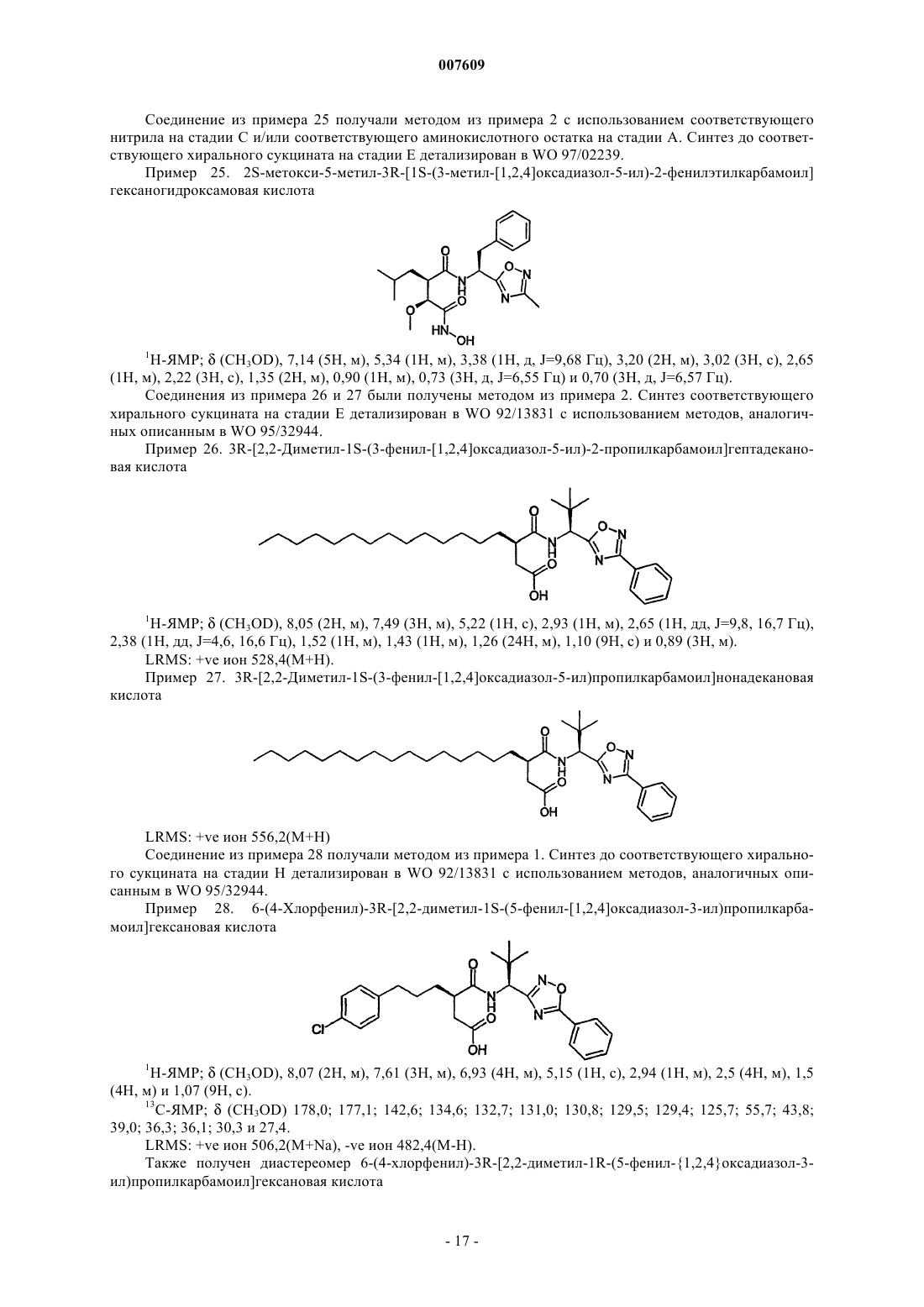
3R-[2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)-2-пропилкарбамоил]гептадекановую кислоту;
3R-[2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропилкарбамоил]нонадекановую кислоту;
6-(4-хлорфенил)-3R-[2,2-диметил-1S-(5-фенил-[1,2,4]оксадиазол-3-ил)пропилкарбамоил]гексановую кислоту;
6-(4-хлорфенил)-3R-[2,2-диметил-1R-(5-фенил-{1,2,4}оксадиазол-3-ил)пропилкарбамоил]гексано-вую кислоту;
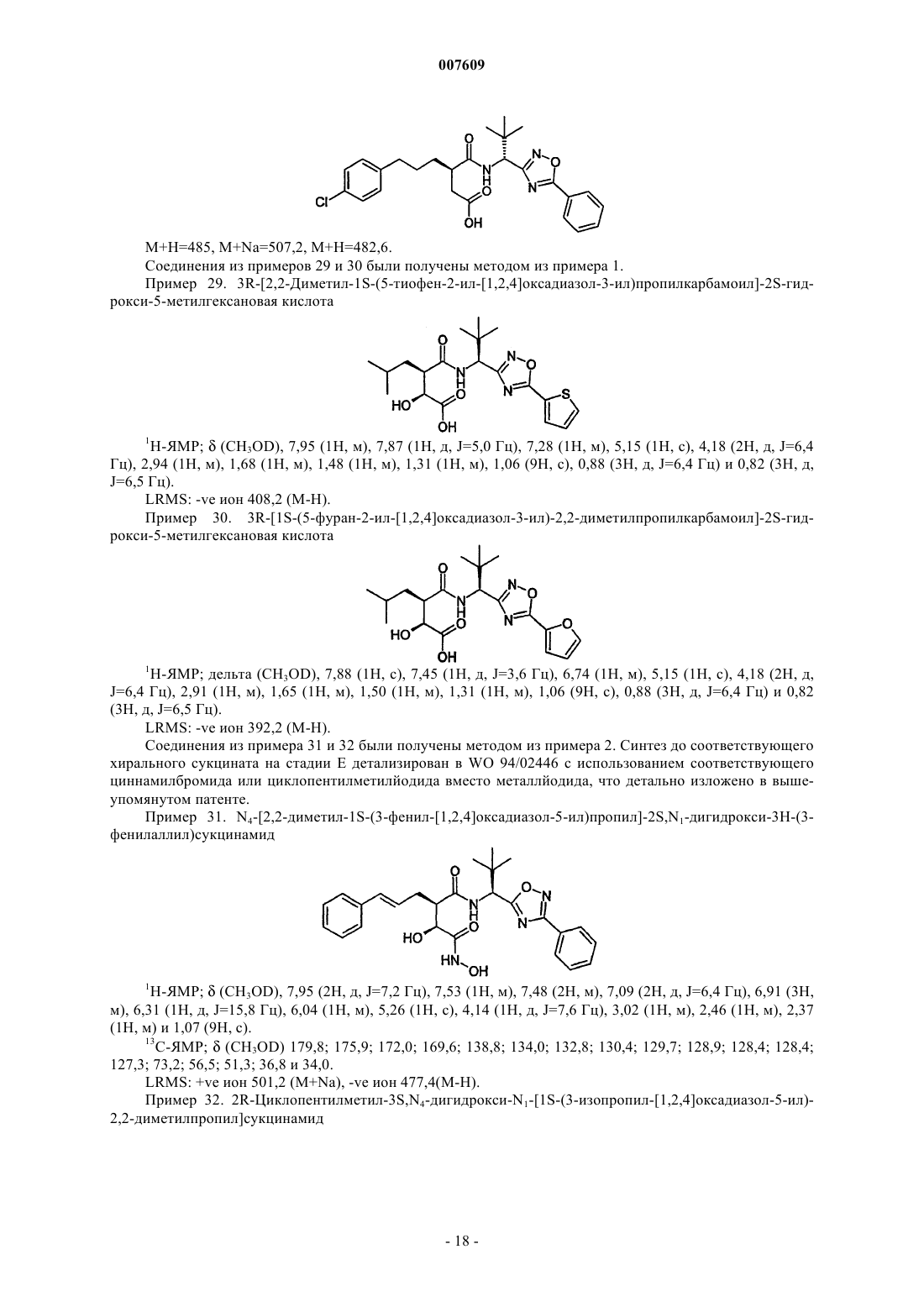
3R-[2,2-диметил-1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)пропилкарбамоил]-2S-гидрокси-5-метилгексановую кислоту;
3R-[1S-(5-фуран-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-2S-гидрокси-5-метилгексановую кислоту;
2R-циклопентилметил-3S,N4-дигидрокси-N1-[1S-(3-изопропил-[1,2,4]оксадиазол-5-ил)-2,2-диметилпропил]сукцинамид;
2R-[3-(3,5-бис-трифторметилфенил)пропил]-N1-[2,2-диметил-1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)пропил]-3S,N4-дигидроксисукцинамид;
2R-[3-(3,5-бис-трифторметилфенил)пропил]-N1-[1S-(5-фуран-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропил]-3S,N4-дигидроксисукцинамид;
2R-[3-(4-этоксифенил)пропил]-N1-[1S-(5-фуран-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропил]-3S,N4-дигидроксисукцинамид;
3-циклопентил-N-[2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропил]-2R-[(формилгидроксиамино)метил]пропионамид;
3-циклопентилметил-N-[2,2-диметил-1S-(5-фенил-[1,2,4]оксадиазол-3-ил)пропил]-2R-[(формилгидроксиамино)метил]пропионамид,
или его фармацевтически приемлемые соли, гидраты или сольваты.
11. Фармацевтическая или ветеринарная композиция, содержащая соединение по любому из предшествующих пунктов.
12. Способ лечения или профилактики заболеваний, опосредованных ММП, у млекопитающих, причем данный способ включает введение млекопитающему эффективного количества 2S-гидрокси-3R-[1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовой кислоты или ее фармацевтически приемлемой соли, гидратов или сольватов.
13. Применение 2S-гидрокси-3R-[1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовой кислоты или ее фармацевтически приемлемой соли, гидратов или сольватов для изготовления лекарственного средства для лечения или профилактики заболеваний, опосредуемых ММП.
14. Способ получения соединения по п.1, где W представляет группу гидроксамовой кислоты
-HONH(C=O)-, включающий взаимодействие кислоты общей формулы (IIА) или (IIB)

или хх активированного производного с гидроксиламином, О-защищенным гидроксиламином или N,O-дизащищенным гидроксиламином или его солью, причем X, R1, R2, R3 и R4 имеют значения, определенные в п.1, за исключением того, что любые заместители в R1, R2, R3 и R4, которые являются потенциально реактивными с гидроксиламином, О-защищенным гидроксиламином, N,О-дизащищенным гидроксиламином или их солями, могут сами быть защищенными от такой реакции, с последующим удалением любых защитных групп у полученной группы гидроксамовой кислоты и у любых защищенных заместителей в R1, R2, R3 и R4.
15. Способ получения соединения по п.1, где W является N-формилгидроксиаминогруппой Н(С=O)NH(ОН)-, включающий N-формилирование соответствующего соединения, в котором W представляет -NH(OP), где Р представляет О-защитную группу, с последующим удалением О-защитной группы Р.
16. Способ получения соединения по п.1, где W является карбоксильной группой -СООН, включающий взаимодействие кислоты формулы (III) или ее активированного производного

с амином формулы (IVA) или (IVB)

где X1, R1, R2, R3 и R4 имеют определения, представленные в п.1, за исключением того, что любые заместители в R1, R2, R3 и R4, которые являются потенциально реактивными в реакции взаимодействия, сами могут быть защищены от такой реакции, и R11 представляет защищающую гидроксил группу, и с последующим удалением защитной группы R11 и любых защитных групп от R1, R2, R3 и R4.
Текст
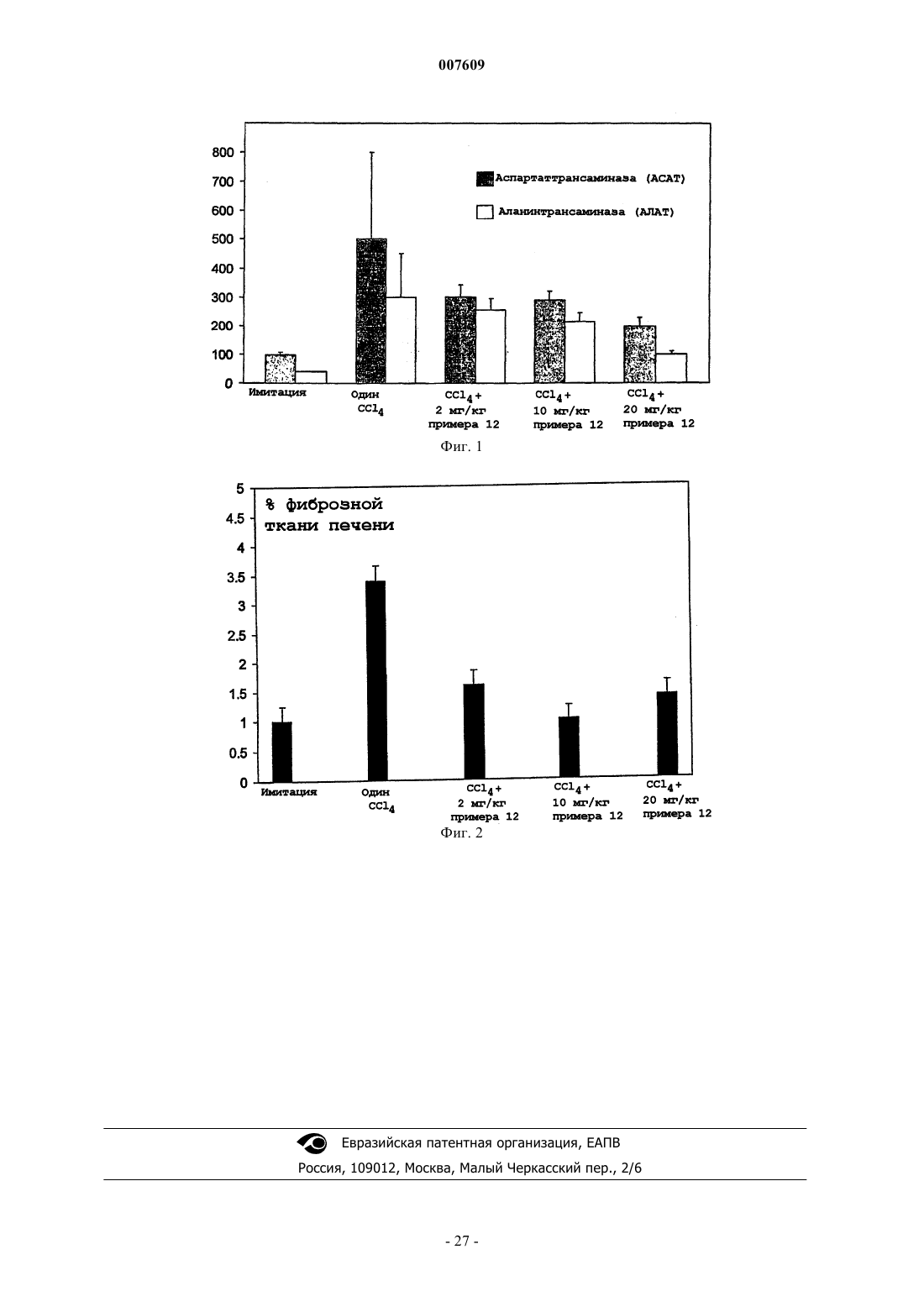
007609 Данное изобретение относится к терапевтически активным производным гидроксамовых и карбоновых кислот, к способам их получения, к содержащим их фармацевтическим композициям и к применению таких соединений в медицине. В частности, данные соединения являются ингибиторами матричных металлопротеиназ. Матричные металлопротеиназы (ММП) представляют собой семейство цинксодержащих эндопептидаз, которые способны к расщеплению больших биомолекул, таких как коллагены, протеогликаны и желатины. Дисбаланс между активными ММП и эндогенными ингибиторами приводит к чрезмерному разрушению тканей. Три главных группы ММП представляют собой коллагеназы, желатиназы и стромелизины. Коллагеназы включают коллагеназу фибробластов (ММП-1), коллагеназу нейтрофилов (ММП 8) и коллагеназу 3 (ММП-13). Желатиназы включают 72 кДа желатиназу (желатиназа А; ММП-2) и 92 кДа желатиназу (желатиназа В; ММП-9) . Стромелизины включают стромелизин 1 (ММП-3), стромелизин 2 (ММП-10) и матрилизин (ММП-7). Однако существуют ММП, которые не точно соответствуют указанным выше группам, например металлоэластаза (ММП-12), ММП мембранного типа (МТ-ММП или ММП-14) и стромелизин 3 (ММП-11). Сверхэкспрессия и активация ММП связаны с широким рядом заболеваний, таких как рак ревматоидный артрит, остеоартрит; хронические воспалительные заболевания, такие как астма, бронхит и эмфизема; сердечно-сосудистые заболевания, такие как атеросклероз; изъязвление роговицы; стоматологические заболевания, такие как гингивит и болезни периодонта; неврологические заболевания, такие как рассеянный склероз и рестеноз. Например, ММП-12 необходима для развития вызываемой курением сигарет эмфиземы у мышей, Science, 277, 2002 (1997). Подавление ММП поэтому является стратегией для лечения таких болезненных состояний. Однако существуют данные о том, что неселективное подавление активности матричных протеиназ может воздействовать на нормальный физиологический процесс,приводящий к ограничивающим дозу побочным эффектам. Селективное подавление ММП-12 и/или ММП-9, как полагают, является особенно значимой стратегией для вмешательства в состояния воспаления. ММП могут гидролизовать связанный с мембраной предшественник провоспалительного цитокинового фактора некроза опухолей(ФНО-). Это расщепление дает зрелый растворимый ФНО-, а ингибиторы ММП могут блокировать продукцию ФНО- как in vitro, так и in vivo. Это фармакологическое действие является вероятным фактором, способствующим противовоспалительному действию этого класса соединений. Обзор, раскрывающий подавление ММП, отраженный в патентной литературе, представлен в Doherty et al. Therapeutic Developments in Matrix Metalloproteinase Inhibition; Expert Opinions on TherapeuticPatients, 2002, 12, 665-707. Краткое описание изобретения Данное изобретение представляет класс соединений, которые являются ингибиторами ММП. Данный класс включает соединения, которые являются селективными ингибиторами ММП-12, относящейся к коллагеназам и стромелизинам. Кроме того, соединения данного изобретения могут проявлять селективную активность в отношении ММП-9. Соединения данного изобретения поэтому показаны в первую очередь для лечения заболеваний, опосредуемых ММП-9 и/или ММП-12, особенно воспалительных патологических состояний, таких как рассеянный склероз и фиброз. Детальное описание изобретения В соответствии с данным изобретением представлено соединение формулы (IA или IB)R2 представляет (С 1-С 4)алкил; фенил-(C1-С 6 алкил)- или фенилаллил, где фенильное кольцо необязательно замещено (C1-С 6)алкилом, (C1-С 6)алкокси, хлором или трифторметилом; или С 3-С 6 циклоалкил(C1-С 6 алкил)-;R4 представляет C1-С 6 алкил, С 3-С 6 циклоалкил, или группу, выбранную из фенила, тиенила, тиенил-1 007609 метила, фуранила, фуранилметила, пирролила, пирролилметила, каждый из которых необязательно замещен в арильном или гетероарильном кольце C1-С 6 алкилом или галогеном; и их фармацевтически приемлемые соли, гидраты или сольваты. В соответствии с описанием термин "(C1-С 6)алкил" означает алкильную группу с прямой или разветвленной цепью, имеющей от 1 до 6 атомов углерода, включая, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, вторичный бутил, трет-бутил, н-пентил и н-гексил. В соответствии с описанием термин "циклоалкил" означает насыщенную алициклическую группу,имеющую 3-8 атомов углерода, и включает, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Существуют по меньшей мере два реальных или потенциальных хиральных центра в соединениях по данному изобретению благодаря наличию асимметричных атомов углерода. Наличие нескольких асимметричных атомов углерода дает увеличение числа диастереоизомеров с R- и S-стереохимией у каждого хирального центра. Данное изобретение включает все такие диастереоизомеры и их смеси. В настоящее время предпочтительной стереоконфигурацией у углеродного атома, несущего R2 группу, является R; предпочтительной стереоконфигурацией у углеродного атома, несущего R1 группу (когда он асимметричен) является R; и предпочтительной стереоконфигурацией у углеродного атома, несущего R3 группу (когда он асимметричен), является S. Группа R1R2 может представлять, например, (С 1-С 4) алкил; фенил-(C1-С 6 алкил)- или фенилаллил, где фенильное кольцо необязательно замещено (C1-С 6)алкилом, (C1-С 6)алкокси, хлором или трифторметилом; или С 3-С 6 циклоалкил-(C1-С 6 алкил)-. Предпочтительные группы R2 включают бензил, н-бутил, изобутил, н-гексил, этоксифенилпропил,предпочтительно, 4-этоксифенилпропил и циклопентилметил. Из этих групп изобутил и этоксифенилпропил, особенно 4-этоксифенилпропил, являются более предпочтительными. Группа R3R3 может представлять, например, C1-C6 алкил, фенил, 2-, 3-или 4-метоксифенил, бензил, 2-, 3- или 4-С 1-С 6 алкоксибензил или бензилокси(С 1-С 6 алкил)-. Примеры конкретных групп R3 включают (С 1-С 6 алкил), бензил, фенил, циклогексилметил, пиридин-3-илметил, трет-бутоксиметил, изопропил, изобутил, втор-бутил, трет-бутил, 1-бензилтио-1 метилэтил, 1-метилтио-1-метилэтил или 1-меркапто-1-метилэтил. Предпочтительные группы R3 включают фенил, бензил, трет-бутоксиметил, изопропил, трет-бутил и изобутил. Из этих групп трет-бутил и бензил являются более предпочтительными. Группа R4R4 может представлять, например, C1-C6 алкил, С 3-С 6 циклоалкил или группу, выбранную из фенила,тиенила, тиенилметила, фуранила, фуранилметила, пирролила, пирролилметила, каждый из которых необязательно замещен в арильном или гетероарильном кольце C1-С 6 алкилом или галогеном. Любая из вышеприведенных групп может быть необязательно замещенной, например, метилом или галогеном. Как упомянуто выше, данные соединения пригодны в медицине и ветеринарии, так как они активны как ингибиторы ММП. Соответственно в другом аспекте данное изобретение относится к:(i) способу лечения (под которым подразумевается лечение или профилактика) заболеваний или патологических состояний, опосредуемых ММП у млекопитающих, в частности у людей, причем способ включает введение млекопитающему эффективного количества соединения, которое охватывается вышеприведенными определениями, или его фармацевтически приемлемыми солями; и(ii) соединению, которое является членом группы, определенной выше, пригодного в медицине и ветеринарии, особенно при лечении (под которым подразумевается лечение и профилактика) заболеваний или патологических состояний, опосредуемых ММП; и(iii) применению соединения, которое является членом группы, определенной выше, для изготовления средства лечения (под которым подразумевается лечение или профилактика) заболеваний или патологических состояний, опосредуемых ММП. Заболевания или патологические состояния, опосредуемые ММП, включают те, которые влекут за собой разрушение ткани, такое как резорбция костей, воспалительные заболевания, дерматологические патологические состояния и рост опухоли или инвазия вторичными метастазами; в частности ревматоидный артрит, остеоартрит, периодонтит, гингивит, изъязвление роговицы, нейровоспалительные заболевания, включающие те, которые влекут за собой разрушение миелина, например рассеянный склероз; рестеноз, эмфизему, бронхит и астму. В дополнительном аспекте данное изобретение относится к фармацевтической или ветеринарной композиции, включающей соединение, которое является членом группы, определенной выше, вместе с приемлемым фармацевтическим или ветеринарным наполнителем или носителем.-2 007609 Будет понятно, что конкретный уровень дозировки для любого конкретного пациента будет зависеть от ряда факторов, включая активность конкретного используемого соединения, возраст, вес тела,общее состояние здоровья, пол, рацион питания, срок введения, путь введения, скорость выведения, сочетание лекарств и тяжесть конкретного заболевания, подвергающегося лечению. Уровни оптимальных доз и частота введения дозы будут определены при клиническом испытании. Соединения настоящего изобретения могут быть приготовлены для введения любым путем, соответствующим их фармакокинетическим свойствам. Композиции для орального введения могут быть в форме таблеток, капсул, порошков, гранул, пастилок, жидких или гелеобразных препаратов, таких как растворы или суспензии для перорального введения, местного применения и стерильные для парентерального введения. Таблетки и капсулы для орального введения могут быть в дозированной форме и могут содержать общепринятые вспомогательные вещества, такие как связывающие вещества, например сироп, камедь акации, желатин, сорбит, камедь трагаканта или поливинилпирролидон; наполнители, например лактозу, сахар, кукурузный крахмал, фосфат кальция, сорбит или глицин; смазывающие вещества для таблетирования, например стеарат магния, тальк, полиэтиленгликоль или двуокись кремния; дезинтеграторы, например картофельный крахмал, или приемлемые улучшающие смачивание вещества,такие как лаурилсульфат натрия. На таблетки может быть нанесено покрытие в соответствии с методиками, хорошо известными в обычной фармацевтической практике. Оральные жидкие препараты могут быть в виде, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров,или могут быть представлены в виде сухого продукта для воспроизведения их с помощью воды или другого подходящего носителя перед приемом. Такие жидкие препараты могут содержать общепринятые добавки, такие как суспендирующие вещества, например сорбит, сироп, метилцеллюлоза, глюкозный сироп, желатин, гидрированные пищевые жиры; эмульгирующие средства, например лецитин, сорбитмоноолеат или камедь акации; неводные носители (которые могут включать пищевые жиры), например миндальное масло, фракционированное кокосовое масло, жирные сложные эфиры, такие как глицерина,пропиленгликоля или этилового спирта; консерванты, например метил- или пропил- п-гидроксибензоат или сорбиновая кислота и, если желательно, общепринятые улучшающие вкус и запах вещества или красители. Для местного применения на коже лекарственное средство может быть изготовлено в виде крема,лосьона или мази. Рецептуры кремов или мазей, которые можно использовать для данного лекарственного препарата, являются обычными рецептурами, хорошо известными специалистам в данной области,например такими, которые описаны в стандартных руководствах для фармацевтов, таких как Британская фармакопея. Для местного применения в глаза данное лекарственное средство может быть изготовлено в виде раствора или суспензии в подходящем стерильном водном растворе или неводном носителе. Могут быть также включены добавки, например буферные вещества, такие как метабисульфит натрия или динатрийэдеат; консерванты, включающие бактерицидные и фунгицидные вещества, такие как фенил ртути ацетат и фенил ртути нитрат, бензалконий хлорид или хлоргексидин, и загустители, такие как гипромеллоза. Активный ингредиент может быть также введен парентерально в стерильной среде. В зависимости от применяемых носителя и концентрации лекарственное вещество может быть или суспендировано или растворено в носителе. Предпочтительно в носителе могут быть растворены вспомогательные вещества,такие как местный анестетик, консервант и буферные средства. Соединения по данному изобретению, в которых W является группой гидроксамовой кислотыHONH(C=O)-, могут быть получены из соответствующих соединений данного изобретения, в которых W является карбоксильной группой -СООН или из соответствующих защищенных производных гидроксамовой кислоты. Другим аспектом данного изобретения является способ, включающий взаимодействие кислоты общей формулы (IIА) или (IIB) или ее активированного производного с гидроксиламином, O-защищенным гидроксиламином илиN,O-дизащищенным гидроксиламином или его солью, причем X, R1, R2, R3 и R4 имеют значения, определенные выше, для общей формулы (IA) или (IB), за исключением того, что любые заместители в R1, R2,R3 и R4, которые являются потенциально реактивными с гидроксиламином, O-защищенным гидроксиламином, N,O-дизащищенным гидроксиламином или их солями, могут сами быть защищенными от такой реакции, с последующим удалением любых защитных групп у полученной группы гидроксамовой кислоты и у любых защищенных заместителей в R1, R2, R3 и R4.-3 007609 Превращение соединений формул (IIА) или (IIВ) в активированное производное, такое как пентафторфениловый, гидроксисукциниловый или гидроксибензотриазолиловый сложный эфир, может быть осуществлено реакцией с соответствующим спиртом в присутствии дегидратирующего средства, такого как дициклогексилдикарбодиимид (DCC; ДЦК), N,N-диметиламинопропил-N-этилкарбодиимид (EDC) или 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (EEDQ; ЭЭДК). Защитные группы, о которых говорилось выше, хорошо известны сами по себе, например, из пептидной химии. Аминогруппы часто могут быть защищены бензилоксикарбонильной, третбутоксикарбонильной или ацетильной группами, или в виде фталимидогруппы. Гидроксильные группы часто могут быть защищены в виде легко расщепляемых простых эфиров, таких как трет-бутиловый или бензиновый простой эфир, или в виде легко расщепляемых сложных эфиров, таких как ацетатный. Карбоксильные группы часто могут быть защищенными в виде легко расщепляемых сложных эфиров, таких как трет-бутиловый или бензиновый сложный эфир. Примеры О-защищенных гидроксиламинов, пригодных в способе (а), выше, включают Обензилгидроксиламин, O-4-метоксибензилгидроксиламин, О-триметилсилилгидроксиламин и O-третбутоксикарбонилгидроксиламин. Примеры O,N-дизащищенных гидроксиламинов, пригодных в способе (а), выше, включают N,Обис(бензил)гидроксиламин, N,O-бис(4-метоксибензил)гидроксиламин, N-трет-бутоксикарбонил-О-третбутилдиметилсилилгидроксиламин, N-трет-бутоксикарбонил-О-тетрагидропиранилгидроксиламин иN,O-бис(трет-бутоксикарбонил)гидроксиламин. Соединения данного изобретения, где W является N-формилгидроксиламиногруппой Н(C=O)NH(ОН)-, могут быть получены N-формилированием соответствующего О-защищенного соединения, в котором W является -NH(OH), и последующее удаление О-защитной группы. Соединения по данному изобретению, в котором W является группой карбоновой кислоты -СООН,т.е. соединения формулы (IIА) или (IIB), выше, могут быть получены способом, включающим взаимодействие кислоты формулы (III) или ее активированного производного где X1, R1, R2, R3 и R4 имеют определения, представленные выше для общей формулы (IA) и (IB), за исключением того, что любые заместители в R1, R2, R3 и R4, которые являются потенциально реактивными в реакции взаимодействия, сами могут быть защищены от такой реакции, и R11 представляет защищающую гидроксил группу, и с последующим удалением защитной группы R11 и любых защитных групп у R1, R2, R3 и R4. Активные производные кислот (III) включают активированные сложные зфиры, такие как пентафторфениловый эфир, ангидриды кислот и галогенангидриды кислот, например хлорангидриды. Подходящие защищающие гидроксил группы могут быть выбраны из известных специалистам в данной области. Соединения формулы (IVA) и (IVB) могут быть получены методами, аналогичными общим методам образования оксадиазольного кольца, проиллюстрированным на схемах 1 и 2 в примерах 1 и 2 ниже. В следующих препаративных примерах описано получение соединений, пригодных в соответствии с данным изобретением. В примерах использованы следующие аббревиатуры: ДХМ - дихлорметан,ДМФ - N,N-диметилформамид,ГОБТ - 1-гидроксибензотриазол,ПФФ - пентафторфенол,WSCDI - N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид,НCl - соляная кислота,ТГФтетрагидрофуран,ТФК - трифторуксусная кислота,Р(O-Тоl)3 - три-О-толилфосфин,AcOEt - этилацетат,CH3CN - ацетонитрил.G) Водный NH2OH, метанол. Н) ПФФ, WSC, ДМФ. I. 1M НСl, ТГФ. Пример 1 выполнен в соответствии со схемой 1, методами, описанными ниже. Стадия А. Бензиловый эфир (1S-карбамоил-2,2-диметилпропил)карбаминовой кислоты.N-бензилоксикарбонил-L-трет-бутилглицин (50 г, 189 ммоль) растворяли в ДМФ (500 мл) и охлаждали на водяной бане со льдом перед добавлением ГОБТ (28,05 г, 208 ммоль) и WSCDI (39,8 г, 208 ммоль). Реакционную смесь перемешивали при 0 С в течение 1 ч перед добавлением 0,880 раствора аммиака (21 мл, 377 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 18 ч. ДМФ удаляли при пониженном давлении и остаток распределяли между этилацета-5 007609 том и 1 М НСl. Органический слой отделяли и промывали 1 М НСl насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли перед сушкой над сульфатом магния, фильтрованием и концентрированием при пониженном давлении с получением бензилового эфира (1S-карбамоил-2,2 диметилпропил)карбаминовой кислоты в виде белого твердого вещества (44,1 г, 89%). 1 Н-ЯМР;(CDCl3), 7,32 (5 Н, м), 6,05 (1 Н, ушир.с), 5,71 (1 Н, ушир.с), 5,60 (1 Н, д, J=6,5 Гц), 5,08LRMS; +ve ион 265 (М+Н), 287 (M+Na). Стадия В. Бензиловый эфир (1S-циано-2,2-диметилпропил)карбаминовой кислоты. Бензиловый эфир (1S-карбамоил-2,2-диметилпропил)карбаминовой кислоты (44,1 г, 167 ммоль) растворяли в безводном пиридине (203 мл, 2,5 ммоль) в атмосфере инертного газа и охлаждали на ледяной бане. Медленно за 15 мин добавляли оксихлорид фосфора и реакционную смесь оставляли перемешиваться на ледяной бане в течение 2 ч перед нагреванием до комнатной температуры и перемешивали в течение 12 ч. Реакционную смесь обрабатывали ледяной водой (400 мл) и экстрагировали этилацетатом(2x300 мл). Органический слой отделяли и промывали 1 М НСl, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли перед сушкой над сульфатом магния, фильтрованием и концентрированием при пониженном давлении. Колоночная хроматография на силикагеле с использованием этилацетата/гексана в качестве элюента приводила к выделению желаемого продукта в виде оранжевого масла (36,72 г, 89%). 1(51 мл, 764 ммоль). Реакционную смесь нагревали с обратным холодильником и перемешивали в течение 3 ч. Реакционную смесь затем охлаждали и концентрировали при пониженном давлении с получением желаемого продукта в виде белой пены/смолы (41,5 г, 97%). 1(0,21 г, 0,75 ммоль) растворяли в ДМФ (5 мл) и обрабатывали пиридином (0,1 мл, 1,28 ммоль), бензоилхлоридом (0,13 мл, 1,1 ммоль) и (DMAP) ДМАФ (каталитическое). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч перед нагреванием до 100 С и перемешиванием в течение 16 ч. Реакционную смесь снова охлаждали до комнатной температуры и концентрировали при пониженном давлении. Реакционную смесь разбавляли этилацетатом и промывали 1 М НСl, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли перед сушкой над сульфатом магния, фильтрованием и концентрированием при пониженном давлении. Желаемый продукт выделяли в виде оранжевого масла (0,22 г, 78%). 1(0,2 г, 0,5 ммоль) обрабатывали 48% гидробромистой кислотой в уксусной кислоте (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении и разделяли между этилацетатом и 1 М Na2CO3. Органический слой дополнительно промывали 1 М Na2CO3 и насыщенным раствором соли перед сушкой над сульфатом магния,фильтрованием и концентрированием при пониженном давлении. Продукт выделяли в виде желтого масла (0,13 г, 98%).(5 мл) и охлаждали на ледяной бане перед добавлением пентафторфенилового эфира 2R-(2,2-диметил 5S-оксо-[1,3]диоксолан-4-ил)-4-метилпентановой кислоты (0,22 г, 0,6 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 15 ч. ДМФ удаляли при пониженном давлении и реакционную смесь разбавляли этилацетатом и промывали 1 М НСl насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли перед сушкой над сульфатом магния,-6 007609 фильтрованием и концентрированием при пониженном давлении. Колоночная хроматография на силикагеле с использованием этилацетата и гексана (1:1) приводила к выделению желаемого продукта в виде белого твердого вещества (0,16 г, 64%). 1 Н-ЯМР;(CDCl3), 8,12 (2 Н, м), 7,55 (3 Н, м), 6,65 (1 Н, д, J=6,4 Гц) 5,25 (1 Н, д, J=6,5 Гц), 4,55 (1 Н,д, J=5,9 Гц), 2,75 (1 Н, м), 1,64 (3 Н, с), 1,55 (3 Н, с), 1,04 (9 Н, с) и 0,88 (6 Н, м).[2,2-Диметил-1S-(5-фенил-[1,2,4]оксадиазол-3-ил)пропил]амид 2R-(2,2-диметил-5S-оксо-[1,3]диоксолан-4-ил)-4-метилпентановой кислоты (0,05 г, 0,11 ммоль) растворяли в метаноле (2 мл) и обрабатывали 50% водным раствором гидроксиламина (0,04 мл, 0,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч перед выпариванием при пониженном давлении. Продукт реакции выделяли препаративной хроматографией с обращенной фазой с получением нужного продукта в виде белого твердого вещества (0,02 г, 44%). 1LRMS; +ve ион 419(M+H); -ve ион 417(М+Н). Стадия Н. Пентафторфениловый эфир 2R-(2,2-диметил-5-оксо-[1,3]диоксолан-4S-ил)-4-метилпентановой кислоты. 2R-(2,2-Диметил-5-оксо-[1,3]диоксолан-4S-ил)-4-метилпентановую кислоту (полученную в соответствии с WO 94/02447) (30 г, 130 ммоль) растворяли в этилацетате (300 мл) и обрабатывали пентафторфенолом (28,8 г, 156 ммоль) и WSCDI (30 г, 156 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 2 ч и затем оставляли перемешиваться при комнатной температуре в течение 12 ч. Реакционную смесь промывали 1 М Na2CO3 и насыщенным раствором соли перед сушкой над сульфатом магния, фильтрованием и концентрированием при пониженном давлении. Данный продукт перекристаллизовывали из этилацетата/гексана с получением желаемого продукта в виде единственного диастереомера (21,2 г, 42%). 1 Н-ЯМР;(CDCl3), 4,55 (1 Н, д, J=6,7 Гц), 3,31 (1 Н, м), 1,85 (3 Н, ушир.м), 1,65 (3 Н, с), 1,58 (3 Н, с),1,05 (3 Н, д, J=6,5 Гц) и 0,99 (3 Н, д, J=6,5 Гц). Также получен диастереомер 3R-[2,2-диметил-1S-(5-фенил-[1,2,4]оксадиазол-3-ил)пропилкарбамоил]-2R-гидрокси-5-метилгексаногидроксамовой кислоты М+Н=420,0; M+Na=441,5; M-H=417,5. Соответствующую карбоновую кислоту получали, как в общих чертах представлено на схеме 1 и по методике, представленной ниже. Стадия I. 3R-[1S-(5-Фуран-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-2S-гидрокси-5-метилгексановая кислота.[2,2-Диметил-1S-(5-фуран-2-ил-[1,2,4]оксадиазол-3-ил)пропил]амид 2R-(2,2-диметил-5S-оксо-[1,3] диоксолан-4-ил)-4-метилпентановой кислоты (0,05 г, 0,12 ммоль) растворяли в тетрагидрофуране (5 мл) и охлаждали до 4 С во время добавления 1 М хлористо-водородной кислоты (5 мл). Раствору давали нагреться до комнатной температуры и затем перемешивали в течение 18 ч. Большую часть растворителя удаляли при пониженном давлении перед сушкой под высоким вакуумом с получением белой пеныF) Водный NH2OH, метанол. Пример 2 получали, как представлено в общих чертах на схеме 2 с использованием методик, описанных ниже.-8 007609 Стадия A. Бензотриазол-1-иловый эфир 2S-трет-бутоксикарбониламино-3,3-диметилмасляной кислоты. Раствор N-трет-бутоксикарбонил-L-трет-бутилглицина (5 г, 21,6 ммоль) в этилацетате (80 мл) охлаждали на ледяной бане. Добавляли ГОБТ (3,22 г, 23,8 ммоль) и WSCDI (4,56 г, 23,8 ммоль) и реакционную смесь оставляли перемешиваться при комнатной температуре на 12 ч. Реакционную смесь промывали 1 М Na2CO3 и насыщенным раствором соли перед сушкой над сульфатом магния, фильтрованием и концентрированием до белой пены (5,74 г, 76%). 1 Н-ЯМР;(CDCl3), 8,05 (1 Н, м), 7,65 (2 Н, м), 7,41 (1 Н, м), 5,10 (1 Н, д, J=6,7 Гц), 4,45 (1 Н, д, J=6,5 Гц), 1,55 (9 Н, с), 1,21 (9 Н, с).LRMS; +ve ион 349(М+Н). Стадия В. трет-Бутиловый эфир [2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропил]карбаминовой кислоты. Бензотриазол-1-иловый эфир 2S-тpeт-бутоксикарбониламино-3,3-диметилмасляной кислоты (3,71 г,10,7 ммоль) растворяли в толуоле (80 мл) и обрабатывали N-гидроксибензамидином (2,9 г, 21,3 ммоль). Реакционную смесь перемешивали при 110 С в течение 18 ч. Раствор концентрировали при пониженном давлении и разделяли между этилацетатом и 1 М Na2CO3. Органический слой дополнительно промывали 1 М Na2CO3 и насыщенным раствором соли перед сушкой над сульфатом магния, фильтрованием и концентрированием при пониженном давлении. Колоночная хроматография на силикагеле с использованием этилацетата и гексана (1:4) приводила к выделению желаемого продукта (2,58 г, 73%). 1LRMS; +ve ион 354,2(M+Na). Стадия С. N-гидроксибензамидин. Бензонитрил (5 г, 48 ммоль) растворяли в этаноле (100 мл) и обрабатывали 50% водным раствором гидроксиламина (16 мл, 242 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 3 ч перед концентрированием при пониженном давлении с получением прозрачной пены (4,5 г, 68%).LRMS; +ve ион 137(М+Н). Стадия D. 2,2-Диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропиламин. Трет-бутиловый эфир [2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропил]карбаминовой кислоты (1 г, 3,0 ммоль) растворяли в ДХМ (5 мл) и обрабатывали ТФА (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении и разделяли между этилацетатом и 1 М Na2CO3. Органический слой дополнительно промывали 1 М Na2CO3 и насыщенным раствором соли перед сушкой над сульфатом магния, фильтрованием и концентрированием при пониженном давлении с получением желаемого продукта (0,65 г, 93%). 1LRMS; +ve ион 232(М+Н). Стадия Е. [2,2-Диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропил]амид 2R-(2,2-диметил-5-оксо[1,3]диоксолан-4S-ил)-4-метилпентановой кислоты. 2R-(2,2-Диметил-5-оксо-[1,3]диоксолан-4S-ил)-4-метилпентановую кислоту (0,27 г, 1,17 ммоль) растворяли в ДМФ (5 мл) и охлаждали на ледяной бане перед добавлением ГОБТ (0,17 г, 1,29 ммоль) иWSCDI (0,25 г, 1,29 ммоль). Реакционную смесь перемешивали при 0 С в течение 1 ч перед добавлением 2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропиламина (0,3 г, 1,29 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 18 ч. ДМФ удаляли при пониженном давлении и остаток разделяли между этилацетатом и 1 М НСl. Органический слой отделяли и промывали 1 М НСl, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли перед сушкой над сульфатом магния, фильтрованием и концентрированием при пониженном давлении. Колоночная хроматография на силикагеле с использованием этилацетата и гексана (1:4) приводила к выделению желаемого продукта (0,26 г, 46%). 1[2,2-Диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропил]амид 2R-(2,2-диметил-5-оксо-[1,3]диоксолан-4S-ил)-4-метилпентановой кислоты (0,26 г, 0,6 ммоль) растворяли в метаноле (5 мл) и обрабатывали 50% водным гидроксиламином (0,2 мл, 2,95 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч перед концентрированием при пониженном давлении. Продукт перекристаллизовывали из этилацетата/гексана с получением желаемого продукта (0,11 г, 41%). 1G) Амин (как детализировано для стадии Е, схема 1), ДМФ. Н) Водный NH2OH, iPrOH. Пример 3 был получен, как представлено в общих чертах на схеме 3, с использованием методик,описанных ниже. Стадия A. Диизопропиловый эфир 2R-аллил-3S-гидроксиянтарной кислоты. К холодному (-78 С) раствору диизопропилового эфира 2S-гидроксиянтарной кислоты (19,70 мл, 95 ммоль) в ТГФ (35 мл) каплями добавляли LiHMDS (200 мл, 0,2 моль, 2,1 экв.). Реакционную смесь перемешивали при -78 С в течение 2 ч, а затем при -30 С в течение 30 мин. Реакционную смесь затем охлаждали до -78 С и каплями добавляли аллилбромид (12,36 мл, 0,14 моль, 1,5 экв.). Реакционной смеси давали нагреться до комнатной температуры в течение ночи. Ее выливали в насыщенный растворNH4 Сl/лед (200 мл). Экстрагирование AcOEt (3x200 мл) с последующим промыванием водой (50 мл) и насыщенным раствором соли (50 мл) давало желтое масло после удаления растворителей в вакууме. Очистка флэш-хроматографией давала диизопропиловый эфир 2R-аллил-3S-гидроксиянтарной кислоты в виде бесцветного масла (7,76 г, de=80%, 40% выход). 1LRMS; +ve ион 281(M+Na). Стадия B. Диизопропиловый эфир 2R-[3-(4-этоксифенил)аллил]-3S-гидроксиянтарной кислоты. К раствору диизопропилового эфира 2R-аллил-3S-гидроксиянтарной кислоты (4,79 г, 18,5 ммоль),4-бромфенетола (3,19 мл, 22,2 ммоль, 1,2 экв) и NEt3 (6,22 мл, 44,6 ммоль, 2,4 экв.) в CH3CN (40 мл) добавляли обработанную ультразвуком (в течение 2 мин) суспензию Р(O-Тоl)3 (0,57 г, 2,22 ммоль, 0,1 экв.)- 10007609 и Pd(OAc)2 (209 мг, 5%) в CH3CN (5 мл). Реакционную смесь нагревали с обратным холодильником в течение 2 ч. CH3CN удаляли в вакууме. Неочищенный продукт экстрагировали AcOEt (3x200 мл), промывали водой (50 мл) и насыщенным раствором соли (50 мл). Очистка флэш-хроматографией давала желаемый диизопропиловый эфир 2R-[3-(4-этоксифенил)аллил]-3S-гидроксиянтарной кислоты (5,92 г,84% выход). 1LRMS; +ve ион 400(M+Na) Стадия C. Диизопропиловый эфир 2R-[3-(4-этоксифенил)пропил]-3S-гидроксиянтарной кислоты. К раствору диизопропилового эфира 2R-[3-(4-этоксифенил)аллил]-3S-гидроксиянтарной кислоты(129 мг, 0,34 ммоль) в МеОН (10 мл) в атмосфере инертного газа добавляли 10% Pd/C (13 мг). Через полученную суспензию пропускали Н 2 в течение 30 мин. Реакционную смесь затем перемешивали при 1 атмосфере Н 2 в течение 16 ч. Pd/C отфильтровывали и растворитель удаляли при пониженном давлении с получением диизопропилового эфира 2R-[3-(4-этоксифенил)пропил]-3S-гидроксиянтарной кислоты (115 мг, 88% выход). 1 Н-ЯМР;(CDCl3), 7,08 (2 Н, д, J=8,6 Гц), 6,81 (2 Н, д, J=8,6 Гц), 4,97-5,14 (2 Н, м), 4,20 (1 Н, дд,J=7,3, 3,5 Гц), 4,01 (2 Н, кв, J=7,0 Гц), 3,18 (1 Н, д, J=7,3 Гц), 2,77-2,83 (1 Н, м), 2,55-2,62 (2 Н, м), 1,45-1,94LRMS; +ve ион 402,0(M+Na). Стадия D. 2R-[3-(4-этоксифенил)пропил]-3S-гидроксиянтарная кислота. К раствору диизопропилового эфира 2R-[3-(4-этоксифенил)пропил]-3S-гидроксиянтарной кислоты(4,78 г, 12,6 ммоль) в ТГФ/воде (3:1, 120 мл) добавляли NaOH (1,66 г, 41,5 ммоль, 5,5 экв.). Реакционную смесь затем перемешивали в течение 16 ч при комнатной температуре. Смесь концентрировали при пониженном давлении и подкисляли до рН=3 добавлением IN HCl. Гидроксидикислоту экстрагировалиAcOEt. Органический слой сушили над MgSO4 и растворитель удаляли при пониженном давлении с получением желаемой 2R-[3-(4-этоксифенил)пропил]-3S-гидроксиянтарной кислоты (3,66 г, 85% выход). 1LRMS; +ve ион 319(M+Na); -ve ион 295(М-Н). Стадия E. 2R- (2,2-диметил-5-оксо-[1,3]диоксолан-4S-ил)-5-(4-этоксифенил)пентановая кислота. К раствору 2R-[3-(4-этоксифенил)пропил]-3S-гидроксиянтарной кислоты (3,66 г, 12,3 ммоль) в ацетоне (50 мл) в инертной атмосфере добавляли диметоксипропан (2,58 мл, 21 ммоль, 1,7 экв.) и хлорид меди (165 мг, 1,2 ммоль, 0,1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель удаляли в вакууме с получением 2R-(2,2-диметил-5-оксо-[1,3]диоксолан-4S-ил)-5(4-этоксифенил)пентановой кислоты (4,03 г, 97% выход). 1LRMS; +ve ион 359(M+Na); -ve ион 335(М-Н). Стадия F. Пентафторфениловый эфир 2R-(2,2-диметил-5-оксо-[1,3]диоксолан-4S-ил)-5-(4-этоксифенил)пентановой кислоты. К холодному (0 С) раствору 2R-(2,2-диметил-5-оксо-[1,3]диоксолан-4S-ил)-5-(4-этоксифенил)пентановой кислоты (4,03 г, 12 ммоль) и пентафторфенола (2,43 г, 13,2 ммоль, 1,1 экв.) в СН 2 Сl2 (50 мл) добавляли WSC (2,54 г, 13,2 ммоль, 1,1 экв.). Реакционной смеси давали нагреться до комнатной температуры в течение ночи. СН 2 Сl2 удаляли в вакууме и полученную неочищенную реакционную смесь растворяли в AcOEt (200 мл). Органический слой промывали водой (50 мл), NaHCO3 нас (20 мл) и наконец насыщенным раствором соли (20 мл). Растворитель удаляли при пониженном давлении с получением масла, которое очищали флэш-хроматографией с получением ожидаемого пентафторфенилового эфира 2R(2,2-диметил-5-оксо-[1,3]диоксолан-4S-ил)-5-(4-этоксифенил)пентановой кислоты (3,94 г, 65% выход). 1G. 2R[3-(4-Этоксифенил)пропил]-N1-[1S-(5-тиофен-2-ил)-[1,2,4]оксадиазол-3-ил)-2,2 диметилпропил] -[1,3]диоксолан-4S-он. К раствору пентафторфенилового эфира 2R-(2,2-диметил-5-оксо-[1,3]диоксолан-4S-ил)-5-(4-этоксифенил)пентановой кислоты (150 мг, 0,30 ммоль) в СН 2 Сl2 (10 мл) добавляли 2,2-диметил-1S-(5-тиофен-2 ил)-[1,2,4]оксадиазол-3-ил)пропиламин (100 мг, 0,42 ммоль, 1,4 экв.). Реакционную смесь перемешивали в течение 16 ч и растворитель удаляли в вакууме. Неочищенный продукт собирали в AcOEt (70 мл) и промывали водой (10 мл), затем Na2CO3 (10 мл) и окончательно насыщенным раствором соли (10 мл). Растворитель осушивали над MgSO4 и удаляли при пониженном давлении с получением 2R-[3-(4- 11007609 этоксифенил)пропил]-N1-[1S-(5-тиофен-2-ил)-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропил]-[1,3]диоксолан-4S-она (82 мг, 33% неочищенного). 1LRMS; +ve ион 556,0(М+Н). Стадия Н. 2R-[3-(4-этоксифенил)пропил]-N1-[1S-(5-тиофен-2-ил)-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропил]-3S,N4-дигидроксисукцинамид. К раствору 2R-[3-(4-этоксифенил)пропил]-N1-[1S-(5-тиофен-2-ил)-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропил]-[1,3]диоксолан-4S-она (82 мг, 0,15 ммоль) в i-PrOH (5 мл) добавляли водный раствор гидроксиламина (50%, 48 мкл, 0,7 ммоль, 5 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 16 ч. Растворитель удаляли при пониженном давлении с получением масла,которое очищали препаративной хроматографией с обращенной фазой с получением нужного продуктаLRMS; +ve ион 553,2(M+Na); -ve ион 529,2 (М-Н). Соединения из примеров 4-17 получали способом в соответствии с примером 1 путем параллельного синтеза, используя соответствующий хлорангидрид кислоты на стадии D. Продукты очищали препаративной ВЭЖХ. Пример 4. 2S-Гидрокси-3R-[1S-(5-изопропил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовая кислотаLRMS; +ve ион 425(М+Н); -ve ион 423(М-Н). Также получен диастереомер 2R-гидрокси-3R-[1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)-2,2 диметилпропилкарбамоил]-5-метилгексаногидроксамовой кислотыLRMS; +ve ион 379(M+Na); -ve ион 355(М-Н). Соединения из примеров 18-19 были получены методом из примера 2 путем использования соответствующего нитрила на стадии С и/или соответствующего аминокислотного остатка на стадии А. Пример 18. 2S-Гидрокси-3R [1S-(3-изопропил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовая кислотаLRMS; +ve ион 427(M+Na); -ve ион 403(М-Н). Соединения из примеров 20-23 были получены методом из примера 2 с использованием соответствующего нитрила на стадии С и/или соответствующего аминокислотного остатка на стадии А. Синтез до соответствующего хирального сукцината на стадии Е детализирован в WO 94/21625. Пример 20. 2S-Аллил-5-метил-3R-[2-фенил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)этилкарбамоил]гексаногидроксамовая кислота(2 Н, м), 2,60 (1 Н, дт, J=11,10, 3,16 Гц), 2,39 (3 Н, с), 1,38 (1 Н, дт, J=13,10, 3,33 Гц), 1,31 (1 Н, м), 1,98 (1 Н,м), 0,98 (9 Н, с), 0,86 (3 Н, д, J=6,6 Гц), 0,84 (3 Н, д, J=6,6 Гц). Соединение из примера 24 было получено методом из примера 2. Синтез до соответствующего хирального сукцината на стадии Е раскрыт в WO 95/19956. Пример 24. 3R-[2,2-Диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропилкарбамоил]-5-метилгексаногидроксамовая кислота- 16007609 Соединение из примера 25 получали методом из примера 2 с использованием соответствующего нитрила на стадии С и/или соответствующего аминокислотного остатка на стадии А. Синтез до соответствующего хирального сукцината на стадии Е детализирован в WO 97/02239. Пример 25. 2S-метокси-5-метил-3R-[1S-(3-метил-[1,2,4]оксадиазол-5-ил)-2-фенилэтилкарбамоил] гексаногидроксамовая кислота(1 Н, м), 2,22 (3 Н, с), 1,35 (2 Н, м), 0,90 (1 Н, м), 0,73 (3 Н, д, J=6,55 Гц) и 0,70 (3 Н, д, J=6,57 Гц). Соединения из примера 26 и 27 были получены методом из примера 2. Синтез соответствующего хирального сукцината на стадии Е детализирован в WO 92/13831 с использованием методов, аналогичных описанным в WO 95/32944. Пример 26. 3R-[2,2-Диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)-2-пропилкарбамоил]гептадекановая кислотаLRMS: +ve ион 556,2(М+Н) Соединение из примера 28 получали методом из примера 1. Синтез до соответствующего хирального сукцината на стадии Н детализирован в WO 92/13831 с использованием методов, аналогичных описанным в WO 95/32944. Пример 28. 6-(4-Хлорфенил)-3R-[2,2-диметил-1S-(5-фенил-[1,2,4]оксадиазол-3-ил)пропилкарбамоил]гексановая кислотаLRMS: +ve ион 506,2(M+Na), -ve ион 482,4(М-Н). Также получен диастереомер 6-(4-хлорфенил)-3R-[2,2-диметил-1R-(5-фенил-1,2,4 оксадиазол-3 ил)пропилкарбамоил]гексановая кислота М+Н=485, M+Na=507,2, М+Н=482,6. Соединения из примеров 29 и 30 были получены методом из примера 1. Пример 29. 3R-[2,2-Диметил-1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)пропилкарбамоил]-2S-гидрокси-5-метилгексановая кислотаLRMS: -ve ион 392,2 (М-Н). Соединения из примера 31 и 32 были получены методом из примера 2. Синтез до соответствующего хирального сукцината на стадии Е детализирован в WO 94/02446 с использованием соответствующего циннамилбромида или циклопентилметилйодида вместо металлйодида, что детально изложено в вышеупомянутом патенте. Пример 31. N4-[2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропил]-2S,N1-дигидрокси-3 Н-(3 фенилаллил)сукцинамидLRMS: +ve ион 411,2 (М+Н), -ve ион 409,6(М-Н). Соединения из примеров 33-35 были получены методом из примера 3 с использованием соответствующего бромида на стадии В. Пример 33. 2R-[3-(3,5-Бис-трифторметилфенил)пропил-N1-[2,2-диметил-1S-(5-тиофен-2-ил-[1,2,4] оксадиазол-3-ил)пропил]-3S,N4-дигидроксисукцинамидLRMS: +ve ион 515,2 (М+Н), -ve ион 513,2(М-Н). Соединение из примера 36 было получено методом, раскрытым в примере 2. Синтез до соответствующего хирального сукцината на стадии Е детализирован в WO 01/10834. Пример 36. 3-Циклопентил-N-[2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропил]-2R-[(формилгидроксиамино)метил]пропионамидLRMS: +ve ион 451 (M+Na), -ve ион 427 (М-Н). Соединение из примера 37 было получено методом в соответствии с примером 1. Синтез до соответствующего хирального сукцината на стадии Е детализирован в WO 01/10834. Пример 37. 3-Циклопентилметил-N-[2,2-диметил-1S-(5-фенил-[1,2,4]оксадиазол-3-ил)пропил]-2R[(формилгидроксиамино)метил]пропионамидLRMS: +ve ион 429(М+Н). Биологические результаты А. Испытание на ингибирование фермента. Соединения данного изобретения испытывали для оценки их активности как ингибиторов ММП 9 и ММП 12. Методика анализа на ММП 9 Испытывали ингибирующее действие соединений в отношении 92 кДа желатиназы (ММП 9) при исследовании с использованием меченного кумарином пептидного субстрата (7-метоксикумарин-4 ил)ацетил-Pro-Leu-Gly-Leu-(3-[2,4-динитрофенил]-L-2,3-диаминопропионил)-Ala-Arg-NH2 (McaPlGLDpaAR) (Knight et al., FEBS Lett. 1992; 263-266). Стандартные исходные растворы готовили следующим образом. Буфер для исследования: 100 мМ Трис-HCl рН 7,6, содержащий 100 мМ NaCl, 10 мМ СаСl2 и 0,05%Brij 35 (полиоксиэтиленовые эфиры). Субстрат: 0,4 мМ McaPlGLDpaAR (от Bachem) (0,437 мг/мл) стандартный раствор в 100%. ДМСО (сохраняемый при -20 С). Разбавить до 8 мкМ буфером для исследования. Фермент: рекомбинантная человеческая 92 кДа желатиназа (ММП 9; АФРА (4-аминофенилртути ацетат), активированный, если необходимо), соответственно разбавленная буфером для исследования. Из испытуемых соединений сначала готовили в виде 10 мМ раствора соединения в 100% ДМСО,разбавляли до 1 мМ 100% ДМСО, затем серийно разбавляли трехкратно 100% ДМСО по колонкам 1-10 96-ячеечной микротитровальной планшеты с интервалом исследуемых концентраций 100 мкМ (колонка 1) до 5,1 нМ (колонка 10). Исследование выполняли в общем объеме, равном 100 мкл на ячейку в 96-ячеечных микротитровальных планшетах. В ячейки добавляли активированный фермент (20 мкл) с последующим добавлением 20 мкл буфера для исследования. Затем добавляли соответствующие концентрации испытуемых соединений, растворенных в 10 мкл ДМСО, с последующим добавлением 50 мкл McaPlGLDpaAR (8 мкМ,приготовленного путем разведения стандартного раствора в ДМСО буфером для исследования). При каждом исследовании оценивали десять концентраций испытуемого соединения в двух повторностях. В контрольных ячейках отсутствовал фермент или отсутствовало испытуемое соединение. Реакционные смеси инкубировали при 37 С в течение 2 ч. Флюоресценцию при 405 нм измеряли сразу же с помощью флюориметра SLT Fluorstar (SLT Labinstruments GmbH, Grdig, Austria), используя возбуждение 320 нм без остановки реакции. Действие испытуемого соединения определяли по кривой зависимости реакции от дозы, получае- 20007609 мой по 10 концентрациям ингибитора в двух повторностях. IС 50 (концентрация соединения, необходимая для получения 50% снижения активности фермента) получали путем подбора данных к равенствуY=a+b-a)/(1+(c/X)d,Y - ингибирование, достигаемое при конкретной дозе;d = угловой коэффициент. Результат округляется до одной значимой цифры. Методика испытания на ММП 12 Испытывали ингибирующую активность соединений в отношении металлоэластазы (ММП 12) при исследовании с использованием меченного кумарином пептидного субстрата (7-метоксикумарин-4-ил) ацетил-Pro-Leu-Gly-Leu-(3-[2,4-динитрофенил]-L-2,3-диаминопропионил)-Ala-Arg-NH2 (McaPLGL DpaAR) (Knight et al., FEBS Lett. 1992, 263-266). Методика для этого исследования описана для испытания с ММП 9, выше. Методика испытания на ММП 1 Испытывали ингибирующее действие соединений в отношении коллагеназы (ММП 1) при испытании с использованием меченного кумарином пептидного субстрата (7-метоксикумарин-4-ил)ацетил-ProLeu-Gly-Leu-(3-[2,4-динитрофенил]-L-2,3-диаминопропионил)-Ala-Arg-NH2 (McaPLGLDpaAR) (Knight etal., FEBS Lett. 1992, 263-266). Методика для этого исследования описана для испытания с ММП 9 выше. Результаты Ключ к биологическим данным Интервал А 100 нМ В 100-1000 нМ С 1000-10000 нМ Эти результаты показывают, что в основном испытуемые соединения были активны в качестве ингибиторов ММП 12, причем некоторые примеры показали селективное подавление как ММП 9, так и 12 по сравнению с ММП 1. В. Модель фиброза печени, индуцированного ССl4. Тетрахлористый углерод (ССl4) вызывает фиброз печени, когда его вводят внутрибрюшинно (Bulbena J., Culat J., Bravo ML., Inflammation 1997, Oct; 21(5):475-88). Соединения данного изобретения можно оценить в отношении их способности предотвращать вызываемое ССl4 образование фиброзной ткани. Животные Самцы крыс Sprague-Dawley, 7-недельного возраста, весом примерно 300 г от Charles River/IffaCredo, St-Germain/l'Arbresle, France. Крысам позволяли акклиматизироваться в течение 5 дней перед началом экспериментов в комнатах с кондиционерами, по 2 животных на клетку, температура: 22 С 2, относительная влажность: 55%10,освещение: 12-часовой цикл (с 7-00 до 19-00), клетка: клетка Macrolon 42,5x26,6x15, на которую помещена крышка-кормушка из нержавеющей стали. Исследование включало следующие группы из 8 животных каждая, как указано ниже. Группа 1. Имитационные животные, получавшие носитель ССl4 (в/б) и один раз в сутки носитель испытуемого вещества (п/к). Группа 2. Группа положительного контроля, получавшая ССl4 (в/б) и один раз в сутки носитель испытуемого вещества (п/к). Группа 3. Экспериментальная группа, получавшая ССl4 (в/б) и один раз в сутки 2 мг/кг п/к соединения примера 13. Группа 4. Экспериментальная группа, получавшая ССl4 (в/б) и один раз в сутки 10 мг/кг п/к соединения примера 13. Группа 5. Экспериментальная группа, получавшая ССl4 (в/б) и один раз в сутки 20 мг/кг п/к соединения примера 13. Крыс метили по их хвостам. Метки проверяли и обновляли, если необходимо, после каждой инъекции ССl4. Методика ССl4 (Prolabo) в оливковом масле вводили каждые 3 дня в течение трех недель путем внутрибрюшинной инъекции (0,25 мл ССl4/кг веса тела, разведенного маслом 1:1 объем/объем до общего объема,равного 0,5 мл/кг). Животных взвешивали ежедневно. Если вес тела снижался на более чем 10% от первоначального веса, животное исключали из исследования. Носители и соединение использовали следующим образом: ССl4 вводили в оливковом масле (prolabo) при разведении 1:1; соединение примера 13 суспендировали в растворе 0,25% твин-80 и 0,25% карбоксиметилцеллюлозы в стерильном 0,9% NaCl. Раствор выдерживали при 4 С в течение всего эксперимента и использовали каждый день для приготовления суспензий. Соединение примера 13 вводили каждый день путем подкожной (п/к) инъекции в объеме введения,равном 5 мл/кг. Группам 1 и 2 п/к вводили дозу 5 мл/кг носителя. Свежеприготовленные растворы использовали в каждый день эксперимента. Введение производили каждый день в одно и то же время. Лечение групп в данном исследовании начинали для каждого животного во время первого введения ССl4 и продолжали в течение 21 последовательного дня. Последнее введение испытуемых веществ или носителя было сделано за день перед забоем животных. Результаты Сообщено о гибели 16 животных. Дата и предполагаемая причина представлены в табл. 1. Уровни фермента в сыворотке Животных забивали через 21 день после первого введения ССl4 путем ингаляции изофурана. Кровь собирали индивидуально во время забоя, т.е. через один день после последнего введения испытуемого вещества или носителя. Кровь центрифугировали при 4 С. Плазму тщательно собирали и делили на али- 22007609 квотные образцы в трех фракциях. Измеряли уровни аспартатаминотрансферазы (АСАТ) и аланинаминотрансферазы (АЛАТ), чтобы определить некроз печени. Повышенные уровни АСАТ и АЛАТ в сыворотке ассоциировали с повреждением печени. Средние уровни АСАТ и АЛАТ для контрольных животных и животных, которых лечили соединением примера 13 в трех разных дозировках, представлены на фиг. 1 (ось У представляет единицы активности фермента на литр крови, МЕ/л). Лечение подкожным введением соединения примера 13 явно снижало уровни АСАТ и АЛАТ по сравнению с животными, которым вводили носитель. Это показывает, что соединение примера 13 обладает защитным действием на печень. Гистологическая оценка фиброза печени Фиброз печени оценивали путем измерения площади фиброза в печени, используя микрохотомию. Результаты представлены в виде процента площади, которая является фиброзной. Печень удаляли, три доли рассекали и отбирали образцы, и/или фиксировали в 10% формальдегиде или замораживали при -80 С. Кусочки печени заключали в парафиновые блоки. Делали срезы и выполняли окрашивание сириусом красным. Количественную оценку фиброза в печени производили по минимум 3 срезам, взятым из разных мест расположения в печени. Количественный анализ выполняли, используя анализатор изображения (Imstar) и программное обеспечение Morphostar. Рассчитывали средний процент площади фиброза в печени животных в разных группах, и результаты показаны на фиг. 2. В. Вызываемое ИЛ-2 перитонеальное скопление лимфоцитов. Введение внутрибрюшинно ИЛ-2 вызывает миграцию лимфоцитов в перитонеальную полость. Это является моделью клеточной миграции, которая происходит во время воспаления. Соединения данного изобретения подавляют (ингибируют) вызываемое ИЛ-2 скопление лимфоцитов. Методика Мышам C3H/HEN (Elevage Janvier, France) внутрибрюшинно вводили ИЛ 2 (Serono PharmaceuticalResearch Institute, 20 мкг/кг, в физиологическом растворе соли). Соединения данного изобретения суспендировали в 0,5% карбоксиметилцеллюлозе (КМЦ)/0,25% твин-20 и вводили п/к (подкожным) или п/о (пероральным) путем (10 мл/кг) за 15 мин перед введением ИЛ-2. Через двадцать четыре часа после введения ИЛ 2 перитонеальные белые клетки крови собирали путем 3 последовательных промываний перитонеальной полости 5 мл фосфатного буферного физиологического раствора (ФБФР)-1 мМ ЭДТА (+4 С). Суспензию центрифугировали (1700 гх 10 мин при +4 С). Полученный осадок суспендировали в 1 мл ФБФР-1 мМ ЭДТА. Лимфоциты идентифицировали и подсчитывали, используя счетчик Beckman/Coulter. План эксперимента Животных делили на 5 групп (6 мышей в каждой группе). Группа 1: (исходный уровень) получала 0,5% КМЦ/0,25% твин-20 (носитель соединения данного изобретения) и физиологический раствор (носитель ИЛ-2). Группа 2: (контроль ИЛ-2) получала 0,5% КМЦ/0,25% твин-20 и инъекцию ИЛ 2. Группа 3: экспериментальная группа (соединение данного изобретения, доза 1) получала соединение данного изобретения и инъекцию ИЛ-2. Группа 4: экспериментальная группа (соединение данного изобретения, доза 2) получала соединение данного изобретения и инъекцию ИЛ-2. Группа 5: экспериментальная группа (соединение данного изобретения, доза 3) получала соединение данного изобретения и инъекцию ИЛ-2. Группа 6: эталонная группа получала эталонное соединение дексаметазон и инъекцию ИЛ-2. Расчет Подавление скопления лейкоцитов рассчитывали следующим образом: где Lyl - число лимфоцитов в группе 1 (Е 3/мкл),Ly2 - число лимфоцитов в группе 2 (Е 3/мкл),Ly X - число лимфоцитов в группе X (3-5) (Е 3/мкл). Дозу соединения данного изобретения, необходимую для подавления скопления лимфоцитов на 50% (ПД 50), рассчитывали, используя обычное вычерчивание кривой по точкам. Результаты представлены в таблице.ID50 для подавления вызываемого ИЛ-2 перитонеального скопления лимфоцитов соединениями данного изобретенияR2 представляет (С 1-С 4)алкил; фенил-(C1-C6 алкил)- или фенилаллил, где фенильное кольцо необязательно замещено (C1-C6)алкилом, (C1-С 6)алкокси, хлором или трифторметилом; или С 3-С 6 циклоалкил(C1-С 6 алкил)-;R4 представляет C1-C6 алкил, С 3-С 6 циклоалкил или группу, выбранную из фенила, тиенила, тиенилметила, фуранила, фуранилметила, пирролила, пирролилметила, каждый из которых необязательно замещен в арильном или гетероарильном кольце C1-C6 алкилом или галогеном; и его фармацевтически приемлемые соли, гидраты или сольваты. 2. Соединение по п.1, причем соединение имеет формулу (IA). 3. Соединение по п.1 или 2, где X=-О-. 4. Соединение по любому из предшествующих пунктов, причем W является HONH(C=O)-. 5. Соединение по любому из предшествующих пунктов, где R1 представляет гидроксигруппу. 6. Соединение по любому из пп.1-5, причем R2 представляет изобутил или циклопентилметил. 7. Соединение по любому из пп.1-6, причем R3 представляет трет-бутил. 8. Соединение по любому из предыдущих пунктов, где R4 представляет 2-тиенил, 2-фуранил или 2 пирролил. 9. Соединение формулы (IА) по п.1, где X=-O-, R1 представляет -ОН; W представляет -C(=O)NHOH,R2 представляет изобутил, R3 представляет трет-бутил и R4 представляет 2-тиенил. 10. Соединение по п.1, выбранное из группы, включающей 3R-[2,2-диметил-1S-(5-фенил-[1,2,4]оксадиазол-3-ил)пропилкарбамоил]-2S-гидрокси-5-метилгексаногидроксамовую кислоту; 3R-[2,2-диметил-1S-(5-фенил-[1,2,4]оксадиазол-3-ил)пропилкарбамоил]-2R-гидpoкcи-5-мeтилгeкcaнoгидpoкcaмoвую кислоту; 3R-[2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропилкарбамоил]-2S-гидрокси-5-метилгексаногидроксамовую кислоту; 2R-[3-(4-этоксифенил)пропил]-N1-[1S-(5-тиофен-2-ил)-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропил]3S,N4-дигидроксисукцинамид; 2S-гидрокси-3R-[1S-(5-изопропил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S-гидрокси-3R-[1S-(5-фуран-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S-гидрокси-3R-[1S-(5-циклопентилметил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5 метилгексаногидроксамовую кислоту; 2S-гидрокси-3R-[1S-(5-тиофен-2-илметил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5 метилгексаногидроксамовую кислоту;- 24007609 2S-гидрокси-3R-[1S-(5-этил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S-гидрокси-3R-[1S-(5-циклопентил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S-гидрокси-3R-[1S-(5-бензил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S-гидрокси-3R-[1S-(5-изобутил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S-гидрокси-3R-[1S-(5-трет-бутил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S-гидрокси-3R-[1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 3R-[1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S-гидрокси-3R[1S-(5-(2,2-диметилпропил)-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]5-метилгексаногидроксамовую кислоту; 2S-гидрокси-3R-[1S-(5-п-толил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S-гидрокси-3R-[1S-(5-циклопропил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S-гидрокси-3R-[1S-(5-метил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S-гидрокси-3R-[1S-(3-изопропил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S,N1-дигидрокси-3R-изобутил-N4-[2-метил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропил]сукцинамид; 2S-аллил-5-метил-3R-[2-фенил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)этилкарбамоил]гексаногидроксамовую кислоту; 2S-аллил-5-метил-3R-[2-фенил-1S-(3-изопропил-[1,2,4]оксадиазол-5-ил)этилкарбамоил]гексаногидроксамовую кислоту; 2S-аллил-3R-[2,2-диметил-1S-(3-метил-[1,2,4]оксадиазол-5-ил)пропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 3R-[2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропилкарбамоил]-5-метилгексаногидроксамовую кислоту; 2S-метокси-5-метил-3R-[1S-(3-метил-[1,2,4]оксадиазол-5-ил)-2-фенилэтилкарбамоил]гексаногидроксамовую кислоту; 3R-[2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)-2-пропилкарбамоил]гептадекановую кислоту; 3R-[2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропилкарбамоил]нонадекановую кислоту; 6-(4-хлорфенил)-3R-[2,2-диметил-1S-(5-фенил-[1,2,4]оксадиазол-3-ил)пропилкарбамоил]гексановую кислоту; 6-(4-хлорфенил)-3R-[2,2-диметил-1R-(5-фенил-1,2,4 оксадиазол-3-ил)пропилкарбамоил]гексановую кислоту; 3R-[2,2-диметил-1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)пропилкарбамоил]-2S-гидрокси-5-метилгексановую кислоту; 3R-[1S-(5-фуран-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-2S-гидрокси-5-метилгексановую кислоту; 2R-циклопентилметил-3S,N4-дигидрокси-N1-[1S-(3-изопропил-[1,2,4]оксадиазол-5-ил)-2,2-диметилпропил]сукцинамид; 2R-[3-(3,5-бис-трифторметилфенил)пропил]-N1-[2,2-диметил-1S-(5-тиофен-2-ил-[1,2,4]оксадиазол 3-ил)пропил]-3S,N4-дигидроксисукцинамид; 2R-[3-(3,5-бис-трифторметилфенил)пропил]-N1-[1S-(5-фуран-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропил]-3S,N4-дигидроксисукцинамид; 2R-[3-(4-этоксифенил)пропил]-N1-[1S-(5-фуран-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропил]3S,N4-дигидроксисукцинамид; 3-циклопентил-N-[2,2-диметил-1S-(3-фенил-[1,2,4]оксадиазол-5-ил)пропил]-2R-[(формилгидроксиамино)метил]пропионамид; 3-циклопентилметил-N-[2,2-диметил-1S-(5-фенил-[1,2,4]оксадиазол-3-ил)пропил]-2R-[(формилгидроксиамино)метил]пропионамид,или его фармацевтически приемлемые соли, гидраты или сольваты. 11. Фармацевтическая или ветеринарная композиция, содержащая соединение по любому из предшествующих пунктов. 12. Способ лечения или профилактики заболеваний, опосредованных ММП, у млекопитающих,- 25007609 причем данный способ включает введение млекопитающему эффективного количества 2S-гидрокси-3R[1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовой кислоты или ее фармацевтически приемлемой соли, гидратов или сольватов. 13. Применение 2S-гидрокси-3R-[1S-(5-тиофен-2-ил-[1,2,4]оксадиазол-3-ил)-2,2-диметилпропилкарбамоил]-5-метилгексаногидроксамовой кислоты или ее фармацевтически приемлемой соли, гидратов или сольватов для изготовления лекарственного средства для лечения или профилактики заболеваний,опосредуемых ММП. 14. Способ получения соединения по п.1, где W представляет группу гидроксамовой кислоты-HONH(C=O)-, включающий взаимодействие кислоты общей формулы (IIА) или (IIB) или ее активированного производного с гидроксиламином, О-защищенным гидроксиламином илиN,O-дизащищенным гидроксиламином или его солью, причем X, R1, R2, R3 и R4 имеют значения, определенные в п.1, за исключением того, что любые заместители в R1, R2, R3 и R4, которые являются потенциально реактивными с гидроксиламином, О-защищенным гидроксиламином, N,О-дизащищенным гидроксиламином или их солями, могут сами быть защищенными от такой реакции, с последующим удалением любых защитных групп у полученной группы гидроксамовой кислоты и у любых защищенных заместителей в R1, R2, R3 и R4. 15. Способ получения соединения по п.1, где W является N-формилгидроксиаминогруппой Н(С=O)NH(ОН)-, включающий N-формилирование соответствующего соединения, в котором W представляет -NH(OP), где Р представляет О-защитную группу, с последующим удалением О-защитной группы Р. 16. Способ получения соединения по п.1, где W является карбоксильной группой -СООН, включающий взаимодействие кислоты формулы (III) или ее активированного производного где X1, R1, R2, R3 и R4 имеют определения, представленные в п.1, за исключением того, что любые заместители в R1, R2, R3 и R4, которые являются потенциально реактивными в реакции взаимодействия,сами могут быть защищены от такой реакции, и R11 представляет защищающую гидроксил группу, и с последующим удалением защитной группы R11 и любых защитных групп от R1, R2, R3 и R4.
МПК / Метки
МПК: C07D 413/04, C07D 285/08, A61P 29/00, A61K 31/4245, C07D 271/06, A61K 31/433, C07D 413/06
Метки: качестве, окса-и, ингибиторов, тиадиазолы, применение, металлопротеиназ
Код ссылки
<a href="https://eas.patents.su/28-7609-oksa-i-tiadiazoly-i-ih-primenenie-v-kachestve-ingibitorov-metalloproteinaz.html" rel="bookmark" title="База патентов Евразийского Союза">Окса-и тиадиазолы и их применение в качестве ингибиторов металлопротеиназ</a>
Тиадиазолы и оксадиазолы и их применение в качестве ингибиторов фосфодиэстеразы-7

Номер патента: 7179
Опубликовано: 25.08.2006
Авторы: Андрианжара Шарль, Вернье Фабрис, Лортиуа Эдвиж, Бернарделли Патрик, Дюкро Пьер
МПК: A61K 31/433, A61K 31/439, A61K 31/4245...
Метки: качестве, применение, фосфодиэстеразы-7, тиадиазолы, ингибиторов, оксадиазолы
Формула / Реферат:
1. Соединение, имеющее следующую формулу (I): в которой Y представляет собой О или S; R1 представляет собой С4-С10алкил, С2-С10алкенил, С2-С10алкинил, циклоалкил, циклоалкенил, гетероцикл, арил или бициклическую группу, каждый(ую) возможно замещенный(ую) одной или несколькими группами X1-R4, одинаковыми или разными, в которых X1 представляет собой одинарную связь, С1-С6алкилен, С2-С6алкенилен, циклоалкилен, арилен или двухвалентный гетероцикл,...
Бетта-сульфонил гидроксамовые кислоты в качестве ингибиторов матричных металлопротеиназ.

Номер патента: 1460
Опубликовано: 23.04.2001
Авторы: Варпехоски Марта А., Харпер Дональд Э.
МПК: A61P 1/04, A61K 31/164, C07C 317/44...
Метки: бетта-сульфонил, металлопротеиназ, ингибиторов, кислоты, качестве, матричных, гидроксамовые
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемые соли, где R1 представляет собой a) С4-12алкил, b) C4-12алкенил, c) C4-12алкинил, d) -(СН2)h-С3-8циклоалкил, e) -(СН2)h-арил, f) -(СН2)h-арил, замещенный C1-4алкилом, C1-4алкокси, галогеном, -NO2, -CF3, -CN, -N(С1-4алкил)2, g) -(СН2)h-гет или h) -(CH2)h-гет, замещенный C1-4алкилом или галогеном; R2 представляет собой a) C1-12алкил, b) C1-12алкил, замещенный от одного до трех...
α-гидрокси, -амино и галоидные производные β-сульфонилгидроксамовых кислот в качестве ингибиторов матриксных металлопротеиназ

Номер патента: 3585
Опубликовано: 26.06.2003
Авторы: Митчелл Марк Аллен, Варпехоски Марта А., Харпер Дональд Э., Маджора Линда Луиза
МПК: A61P 19/04, C07C 317/44, A61K 31/165...
Метки: кислот, beta;-сульфонилгидроксамовых, галоидные, alpha;-гидрокси, амино, ингибиторов, производные, металлопротеиназ, качестве, матриксных
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль, где R1 представляет собой фенил, замещенный C1-4акилом, C1-4 алкокси, галогеном или фенилом; R2 представляет собой фенилсульфонилметил, замещенный C1-4алкилом, C1-4алкокси, галогеном или фенилом; или представляет собой группу -CH2NHC(=O)R3, где R3 представляет собой C5-6циклоалкил или фенил, необязательно замещенный C1-4алкокси; Y представляет собой -OH. 2. Соединение по п.1,...
N-гидрокси-2-(алкил-, арил- или гетероарилсульфанил, -сульфинил или -сульфонил)-3-замещенные алкилов, арилов или гетероариламидов в качестве ингибиторов матриксных металлопротеиназ

Номер патента: 1742
Опубликовано: 27.08.2001
Авторы: Венкатесан Аранапакам Мудумбай, Дэвис Жами Мари, Бэйкер Жанни Леа, Гросу Джордж Теодор
МПК: A61P 5/48, C07D 211/66, A61K 31/164...
Метки: алкилов, арил, сульфонил)-3-замещенные, гетероарилсульфанил, матриксных, качестве, n-гидрокси-2-(алкил, арилов, ингибиторов, сульфинил, металлопротеиназ, гетероариламидов
Формула / Реферат:
1. Соединения формулы I в которой R1 представляет собой алкил из 1-18 углеродных атомов, необязательно замещенный одной или двумя группами, независимо выбранными из R5; алкенил, содержащий 3-18 углеродных атомов с 1-3 двойными связями, необязательно замещенный одной или двумя группами, независимо выбранными из R5; алкинил, содержащий 3-18 углеродных атомов с 1-3 тройными связями, необязательно замещенный одной или двумя группами,...
Простые бисэфиры 1-окса- и 1- азанафталин-2-онов в качестве ингибиторов фосфоламбана

Номер патента: 3084
Опубликовано: 26.12.2002
Авторы: Левийоки Йоуко, Тиайнен Эйя, Хайкала Хеймо, Пюстюнен Ярмо, Карьялайнен Арто, Пархи Сеппо, Норе Пентти, Леннберг Кари
МПК: C07D 407/14, A61P 9/04, A61K 31/4709...
Метки: ингибиторов, фосфоламбана, 1-окса, простые, качестве, бисэфиры, азанафталин-2-онов
Формула / Реферат:
1. Соединение формулы (I) где R1 обозначает водород, C1-7 алкил, С6-10 арил C1-7 алкил, возможно замещенный галогеном или нитро, R2 обозначает водород, R3 обозначает водород, C1-7 алкил, С6-10 арил, А обозначает C1-3 алкилен; R1+R3 являются бутиленом, возможно замещенным С6-10 арилом, Y обозначает О, Х обозначает О, NR11, где R11 обозначает Н, C1-7 алкил или С6-10 арил C1-7 алкил, возможно замещенный галогеном, R4, R5 обозначают независимо одну...
Предыдущий патент: Таблетка с энтеропокрытием
Следующий патент: Таблетка с энтеропокрытием, обеспечивающая отсроченное высвобождение
Случайный патент: Установка для очистки воды