Соединения и композиции как ингибиторы киназы с-kit и pdgfr
Номер патента: 19524
Опубликовано: 30.04.2014
Авторы: Лю Сяодон, Брайтенштайн Вернер, Рамси Тимоти, Набакка Джульет, Мольтени Валентина, Чянелли Донателла, Лорен Джон, Ли Сяолинь
























Формула / Реферат
1. Соединение формулы I

в которой L выбран из -NHC(O)- и -C(O)NH-;
R2a и R2b, каждый независимо, представляют собой водород;
R1 выбран из группы, включающей водород, гидроксил, 3-8-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, О и S, C1-C4-алкил, C1-C4-алкоксигруппу, галогензамещенную C1-C4-алкоксигруппу, галогензамещенный C1-C4-алкил, -OX1R8, где X1 обозначает C1-C4-алкилен и R8 обозначает C3-C12-циклоалкил; или
R1 и R2a вместе с атомами углерода, к которым R1 и R2a присоединены, образуют фенил;
R3 и R7 независимо выбраны из водорода и галогена;
R4 и R6 независимо выбраны из группы, включающей водород, C1-C6-алкил, C1-C6-алкоксигруппу, галогензамещенную C1-C6-алкоксигруппу, -OX2R9 и -S(O)2R9, где Х2 обозначает C1-C4-алкилен;
R5 выбран из группы, включающей водород, галоген, C1-C6-алкил, C1-C6-алкоксигруппу, 3-8-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, О и S, и -S(O)2R9;
каждый R9 независимо выбран из группы, включающей C1-C6-алкил, C3-C12-циклоалкил и 3-8-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, О и S, где указанный циклоалкил или гетероциклоалкил в качестве R9 необязательно может быть замещен 1-3 радикалами, которые независимо представляют собой C1-C6-алкил,
при условии, что по меньшей мере один из R4, R5 или R6 обозначает -S(O)2R9, непосредственно связанный с фенильным кольцом,
и его фармацевтически приемлемые соли.
2. Соединение по п.1, в котором
L выбран из -NHC(O)- и -C(O)NH-;
R2a и R2b, каждый независимо, представляют собой водород;
R1 выбран из группы, включающей водород, гидроксил, 5-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N и О, C1-C2-алкил, C1-C2-алкоксигруппу, галогензамещенную C1-C2-алкоксигруппу, галогензамещенный C1-C2-алкил, -OX1R8, где X1 обозначает C1-C4-алкилен и R8 обозначает С3-циклоалкил; или
R1 и R2a вместе с атомами углерода, к которым R1 и R2a присоединены, образуют фенил;
R3 и R7 независимо выбраны из водорода и галогена;
R4 и R6 независимо выбраны из группы, включающей водород, C1-C6-алкил, C1-C6-алкоксигруппу, галогензамещенную C1-C6-алкоксигруппу и -S(O)2R9; и
R5 выбран из группы, включающей водород, галоген, C1-C6-алкил, C1-C6-алкоксигруппу, 5-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N и О, и -S(O)2R9;
каждый R9 независимо выбран из группы, включающей C1-C6-алкил, C3-C12-циклоалкил и 5-6-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, О и S, где указанный циклоалкил или гетероциклоалкил в качестве R9 необязательно может быть замещен 1-3 радикалами, которые независимо представляют собой C1-C6-алкил,
при условии, что по меньшей мере один из R4, R5 или R6 обозначает -S(O)2R9, непосредственно связанный с фенильным кольцом.
3. Соединение по п.2, в котором R1 выбран из группы, включающей водород, гидроксил, пирролидинил, морфолиновую группу, метоксигруппу, дифторметоксигруппу, 2-фторэтоксигруппу и метил; или R1 и R2a вместе с атомами углерода, к которым R1 и R2a присоединены, образуют фенил.
4. Соединение по п.3, в котором R4 и R6 независимо выбраны из группы, включающей водород, метилсульфонил, метил, 2-фторэтоксигруппу, метилпиперазинилсульфонил, пропоксигруппу, изобутоксигруппу, 2,2,2-трифторэтоксигруппу, 2,3-дифтор-2-(фторметил)пропоксигруппу, бутоксигруппу и циклопропилметоксигруппу; и R5 выбран из группы, включающей водород, галоген, метилсульфонил, метил, метоксигруппу, этоксигруппу и морфолиновую группу.
5. Соединение по п.1, выбранное из группы, включающей
N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-3-(метилсульфонил)бензамид;
N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)бензамид;
4-хлор-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-3-(метилсульфонил)бензамид;
2-хлор-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)бензамид;
3-метил-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)бензамид;
4-метил-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-3-(метилсульфонил)бензамид;
4-этокси-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-3-(метилсульфонил)бензамид;
N-(4-метил-3-(4-(5-морфолинопиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)бензамид;
N-(4-метил-3-(4-(5-(пирролидин-1-ил)пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)бензамид;
N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-3-(метилсульфонил)-4-морфолинобензамид;
N-(3-(4-(5-метоксипиридин-3-ил)пиримидин-2-иламино)-4-метилфенил)-4-(метилсульфонил)бензамид;
N-(3-(4-(5-(2-фторэтокси)пиридин-3-ил)пиримидин-2-иламино)-4-метилфенил)-4-(метилсульфонил)бензамид;
N-(3-(4-(5-(циклопропилметокси)пиридин-3-ил)пиримидин-2-иламино)-4-метилфенил)-4-(метилсульфонил)бензамид;
N-(4-метил-3-(4-(5-метилпиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)бензамид;
3-(2-фторэтокси)-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)бензамид;
N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)-3-пропоксибензамид;
3-метокси-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)бензамид;
3-бутокси-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)бензамид;
3-(циклопропилметокси)-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)бензамид;
N-(3-(4-(изохинолин-4-ил)пиримидин-2-иламино)-4-метилфенил)-3-(метилсульфонил)бензамид;
4-метокси-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-3-(метилсульфонил)бензамид;
N-(3-(4-(5-(дифторметокси)пиридин-3-ил)пиримидин-2-иламино)-4-метилфенил)-4-(метилсульфонил)бензамид;
N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-3-(4-метилпиперазин-1-илсульфонил)бензамид;
N-(3-(4-(5-гидроксипиридин-3-ил)пиримидин-2-иламино)-4-метилфенил)-4-(метилсульфонил)бензамид;
3-изобутокси-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)бензамид;
N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)-3-(2,2,2-трифторэтокси)бензамид и
3-(2,3-дифтор-2-(фторметил)пропокси)-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)бензамид.
6. Фармацевтическая композиция, включающая терапевтически эффективное количество соединения по п.1 в комбинации с фармацевтически приемлемым инертным наполнителем.
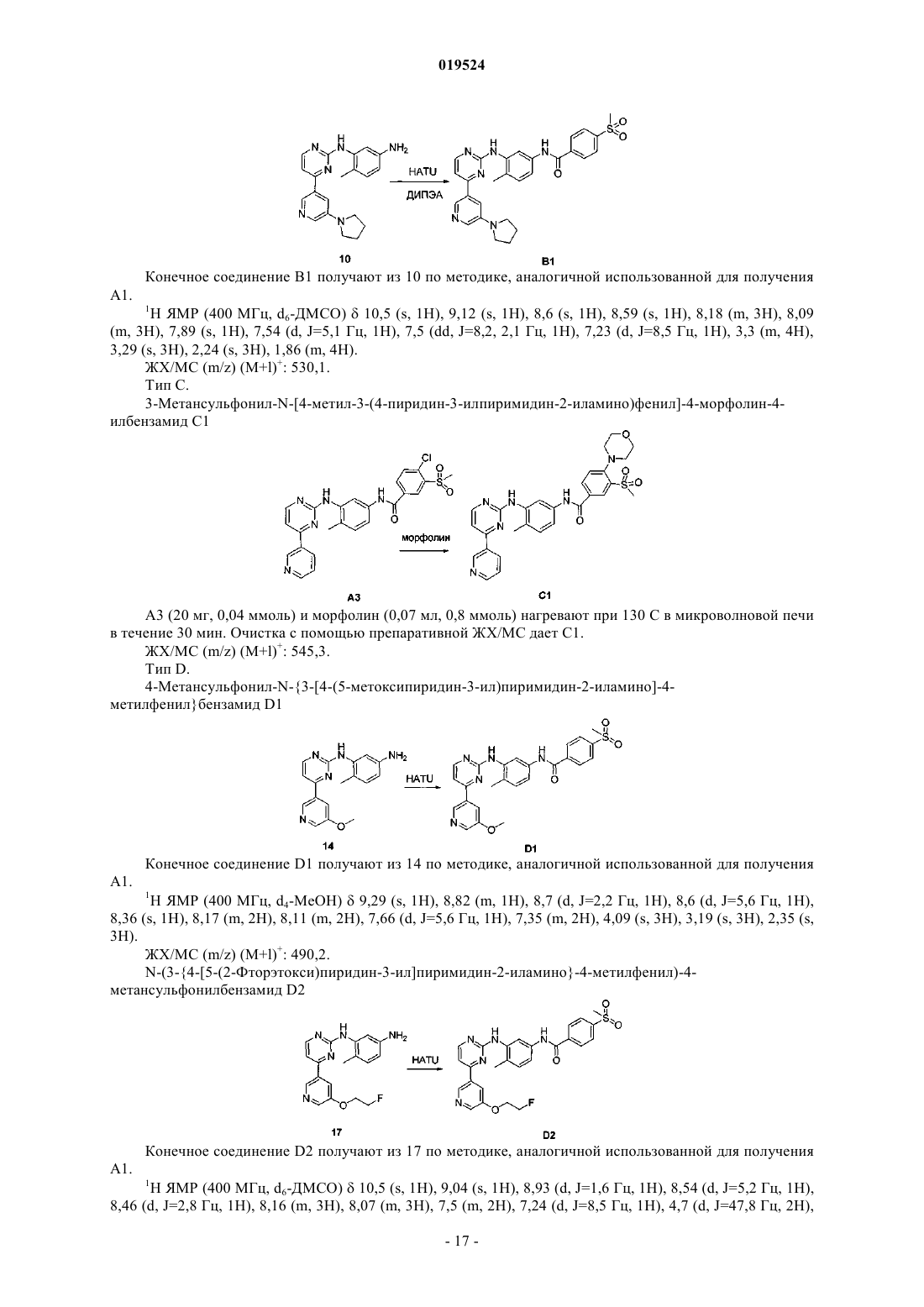
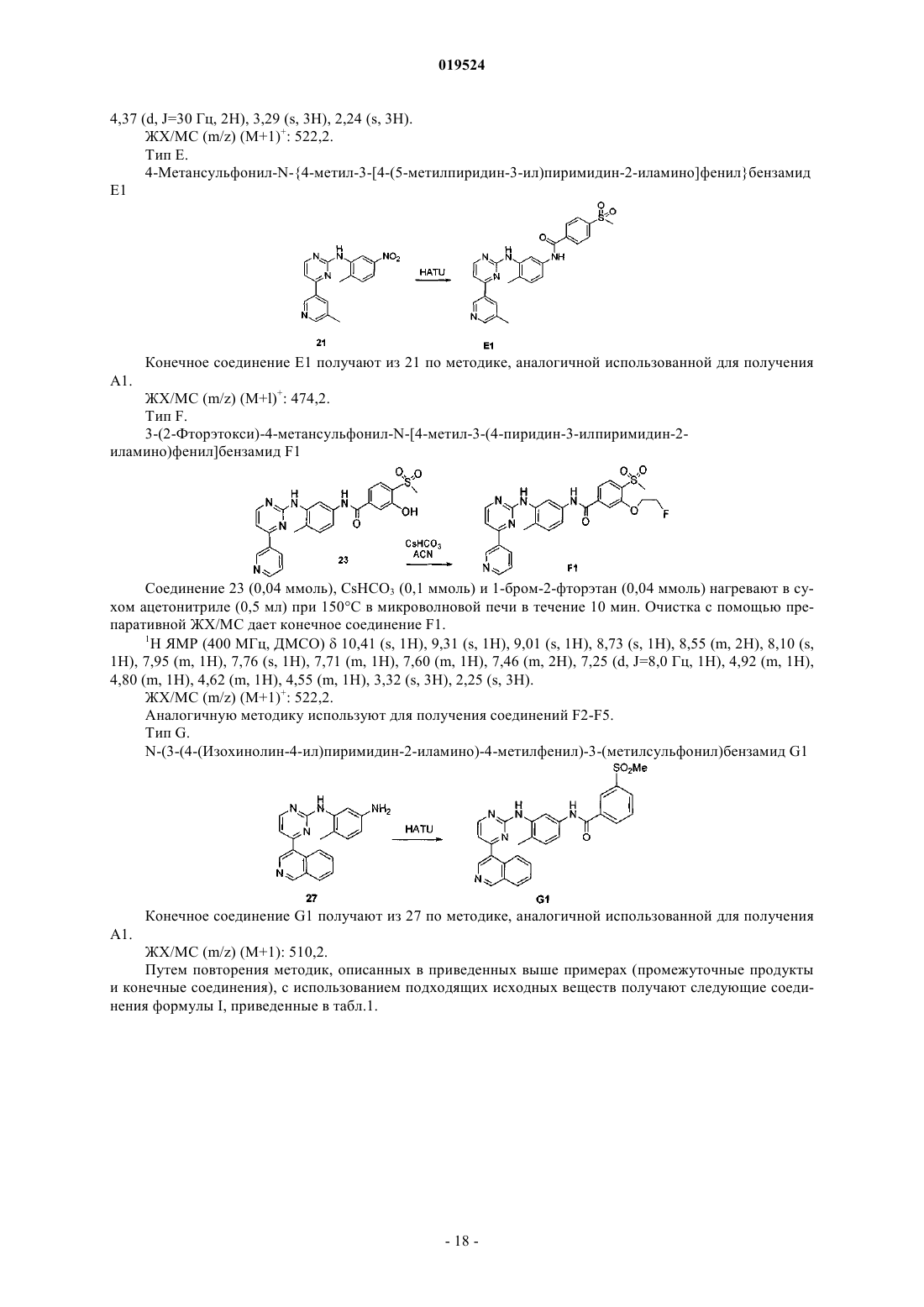
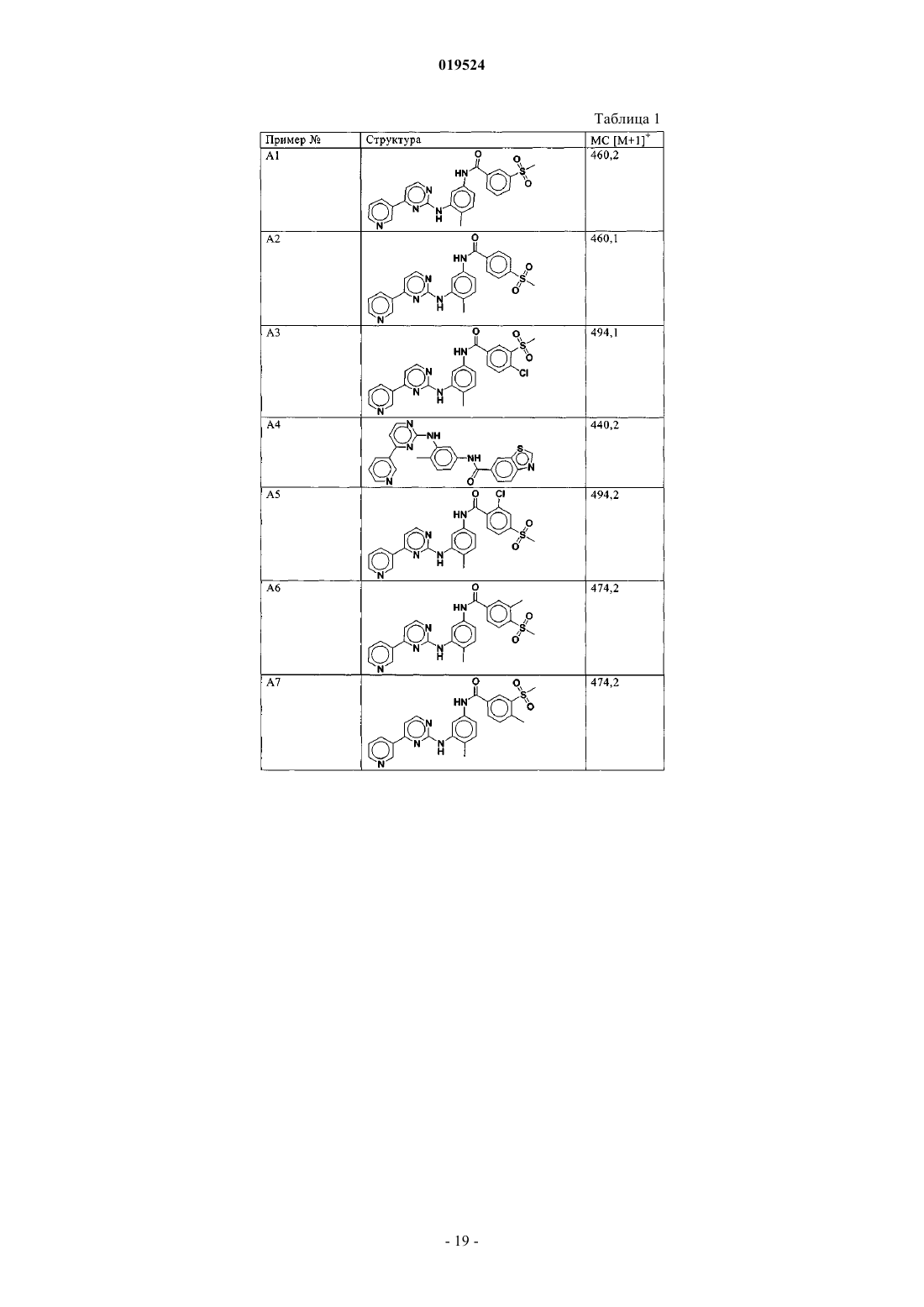
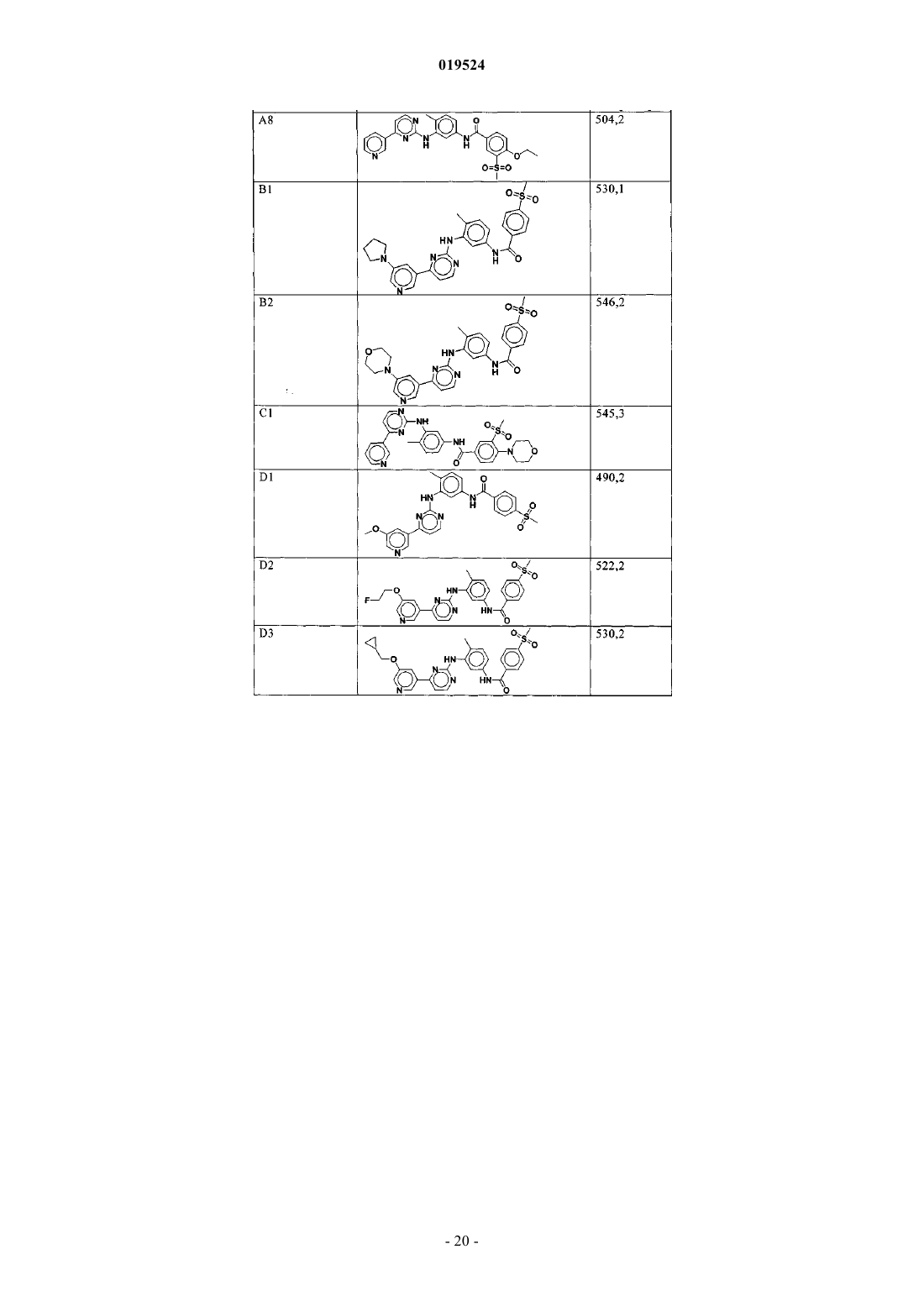
Текст
СОЕДИНЕНИЯ И КОМПОЗИЦИИ КАК ИНГИБИТОРЫ КИНАЗЫ с-kit И PDGFR В изобретении приведено описание соединений формулы I, фармацевтических композиций,включающих такие соединения, и способов применения таких соединений для лечения или предупреждения заболеваний или нарушений, связанных с аномальной или характеризующейся нарушенной регуляцией активностью киназы, в особенности заболеваний или нарушений, которые включают аномальную активацию киназ c-kit, ТРФРР и ТРФРР. Перекрестная ссылка на родственные заявки По заявке на данное изобретение испрашивается приоритет по предварительной заявкеUS60/916051, поданной 4 мая 2007 г. Полное раскрытие этой заявки во всей ее полноте включено в настоящее изобретение в качестве ссылки для всех объектов. Предпосылки создания изобретения Область техники, к которой относится изобретение Настоящее изобретение относится к новому классу соединений, фармацевтическим композициям,включающим такие соединения, и к способам применения таких соединений для лечения или предупреждения заболеваний или нарушений, связанных с аномальной или характеризующейся нарушенной регуляцией активностью киназы, в особенности заболеваний или нарушений, которые включают аномальную активацию киназ c-kit, ТРФРР и ТРФРР. Уровень техники Протеинкиназы представляют собой большую группу белков, которые играют центральную роль в регуляции большого количества клеточных процессов и обеспечивают регулирование клеточных функций. Частичный, неограничивающий перечень этих киназ включает рецепторные тирозинкиназы, такие как киназа рецептора тромбоцитарного фактора роста (ТРФРР), рецептора фактора роста нервов, trkB и рецептора фактора роста фибробластов, FGFR3; нерецепторные тирозинкиназы, такие как Abl, и киназы слияния BCR-Abl, Lck, Bmx и c-src и серин/треонинкиназы, такие как киназы, c-RAF, sgk, MAP (например, MKK4, MKK6 и т.п.) и SAPK2 и SAPK2. Аберрантная активность киназ обнаружена при многих патологических состояниях, включая доброкачественные и злокачественные пролиферативные заболевания, а также заболевания, обусловленные неадекватной активацией иммунной и нервной систем. Новые соединения, предлагаемые в настоящем изобретении, ингибируют активность одной или большего количества протеинкиназ и поэтому предположительно применимы для лечения связанных с киназами заболеваний. Краткое изложение сущности изобретения Первым объектом настоящего изобретения является соединение формулы IR1 выбран из группы, включающей водород, гидроксил, 3-8-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, О и S, C1-C4-алкил, C1-C4-алкоксигруппу, галогензамещенную C1-C4-алкоксигруппу, галогензамещенный C1-C4-алкил, -OX1R8, где X1 обозначаетR3 и R7 независимо выбраны из водорода и галогена;R4 и R6 независимо выбраны из группы, включающей водород, C1-C6-алкил, C1-C6-алкоксигруппу,галогензамещенную C1-C6-алкоксигруппу, -OX2R9 и -S(O)2R9, где Х 2 обозначает C1-C4-алкилен;R5 выбран из группы, включающей водород, галоген, C1-C6-алкил, C1-C6-алкоксигруппу,3-8-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, О и S, и-S(O)2R9; каждый R9 независимо выбран из группы, включающей C1-C6-алкил, C3-C12-циклоалкил и 3-8-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, О и S, где указанный циклоалкил или гетероциклоалкил в качестве R9 необязательно может быть замещен 1-3 радикалами, которые независимо представляют собой C1-C6-алкил; при условии, что по меньшей мере один из R4, R5 или R6 обозначает -S(O)2R9, непосредственно связанный с фенильным кольцом,и его фармацевтически приемлемые соли. Вторым объектом настоящего изобретения является фармацевтическая композиция, которая содержит соединение формулы I или его фармацевтически приемлемую соль в смеси с одним или большим количеством подходящих инертных наполнителей. Подробное описание изобретения Определения."Алкил" в качестве группы или структурного элемента других групп, например галогензамещенного алкила и алкоксигруппы, может обладать линейной или разветвленной цепью. C1-C4-Алкоксигруппы включают метоксигруппу, этоксигруппу и т.п. Галогензамещенный алкил включает трифторметил, пентафторэтил и т.п."Арил" означает моноциклическую или конденсированную бициклическую ароматическую кольцевую систему, содержащую 6-10 кольцевых атомов углерода. Например, C6-C10-арил при использовании в настоящем описании включает, но не ограничивается только ими, фенил или нафтил, предпочтительно фенил. "Арилен" означает двухвалентный радикал, образованный из арильной группы."Гетероарил" означает 5-15-членную ненасыщенную кольцевую систему, содержащую от 1 до 3 гетероатомов, независимо выбранных из группы, включающей -О-, -N=, -NR-, -С(О)-, -S-, -S(O)- и -S(O)2-,где R обозначает водород, C1-C4-алкил или защитную группу атома азота. Например, C1-C10-гетероарил("C1-C10-" указывает, что в кольцевой системе содержатся от 1 до 10 атомов углерода) при использовании в настоящем описании включает, но не ограничивается только ими, пиразолил, пиридинил, индолил,тиазолил, 3-оксо-3,4-дигидро-2 Н-бензо[b][1,4]оксазин-6-ил, фуранил, бензо[b]фуранил, пирролил,1 Н-индазолил, имидазо[1,2-а]пиридин-3-ил, оксазолил, бензо[d]тиазол-6-ил, 1 Н-бензо[d][1,2,3]триазол-5 ил, хинолинил, 1H-индолил, 3,4-дигидро-2 Н-пирано[2,3-b]пиридинил и 2,3-дигидрофуро[2,3b]пиридинил, 3-оксо-3,4-дигидро-2 Н-бензо[b][1,4]оксазин-7-ил и т.п."Циклоалкил" означает насыщенную или частично ненасыщенную моноциклическую, конденсированную бициклическую или мостиковую полициклическую кольцевую систему, содержащую указанное количество кольцевых атомов. Например, C3-C10-циклоалкил включает циклопропил, циклобутил, циклопентил, циклогексил и т.п."Гетероциклоалкил" означает 3-8-членную насыщенную или частично ненасыщенную кольцевую систему, содержащую от 1 до 3 гетероатомов, независимо выбранных из группы, включающей -О-, -N=,-NR-, -С(О)-, -S-, -S(O)- и -S(O)2-, где R обозначает водород, C1-C4-алкил или защитную группу атома азота. Например, C3-C8-гетероциклоалкил при использовании в настоящем описании изобретении для описания соединений, предлагаемых в настоящем изобретении, включает, но не ограничивается только ими, морфолиновую группу, пирролидинил, азепанил, пиперидинил, изохинолинил, тетрагидрофуранил,пирролидинил, пирролидинил-2-он, пиперазинил, пиперидинилон, 1,4-диокса-8-азаспиро[4,5]дец-8-ил и т.п."Галоген" предпочтительно означает хлор или фтор, но также может означать бром или йод."Набор киназ" представляет собой перечень киназ, включающий Abl (человека), Abl (T315I), JAK2,JAK3, ALK, JNK11, ALK4, KDR, Aurora-A, Lck, Blk, MAPK1, Bmx, MAPKAP-K2, BRK, MEK1, CaMKII(крысы), LIMK1, Rsk2, Axl, LKB1, SAPK2, BrSK2, Lyn (h), SAPK3, BTK, МАРКАР-K3, SAPK4, CaMKIV, MARK1, Snk, CDK2/циклин A, MINK, SRPK1, CDK3/циклин Е, MKK4(m), TAK1, CDK5/p25,MKK6(h), TBK1, CDK6/циклин D3, MLCK, TrkA, CDK7/циклин H/MAT1, MRCK, TSSK1, CHK1,MSK1, Yes, CK1d, MST2, ZIPK, c-kit (D816V), MuSK, DAPK2, NEK2, DDR2, NEK6, DMPK, PAK4,DRAK1, PAR-1B, EphAl, PDGFR, EphA2, Pim-1, EphA5, PKB, EphB2, PKCI, EphB4, PKC, FGFR1,PKC, FGFR2, PKC, FGFR4, PKD2, Fgr, PKG1, Flt1, PRK2, Hck, PYK2, HIPK2, Ret, IKK, RIPK2, IRR,ROCK-II (человека), JNK22, Rse, JNK3, Rsk1(h), PI3 K, PI3 K и PI3 K. Для соединений, предлагаемых в настоящем изобретении, проведен скрининг воздействия на этот набор киназ (дикого типа и/или их мутаций) и установлено, что они ингибируют активность по меньшей мере одного представителя указанного набора киназ."Лечение" относится к способу облегчения или смягчения заболевания и/или сопутствующих ему симптомов. Описание предпочтительных вариантов осуществления Ген c-kit кодирует рецепторную тирозинкиназу и лиганд рецептора c-kit называется фактором стволовых клеток (ФСК), который является главным фактором роста, обеспечивающим жизнеспособность мастоцитов. Активность с-kit рецепторной протеинтирозинкиназы в нормальных клетках регулируется, и нормальная функциональная активность продукта гена c-kit важна для поддержания нормального гематопоэза, меланогенеза, генетогенеза и роста и дифференциации мастоцитов. Мутации, которые вызывают конститутивную активацию киназной активности c-kit при отсутствии связывания с ФСК, участвуют в различных заболеваниях в диапазоне от мастоцитоза до раковых заболеваний человека. В одном варианте осуществления соединений формулы I L выбран из -NHC(O)- и -C(O)NH-; R2a иR2b, каждый независимо, представляют собой водород; R1 выбран из группы, включающей водород, гидроксил, 5-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N и О,C1-C2-алкил, C1-C2-алкоксигруппу, галогензамещенную C1-C2-алкоксигруппу, галогензамещенныйC1-C2-алкил, -OX1R8, где X1 обозначает C1-C4-алкилен и R8 обозначает С 3-циклоалкил; или R1 и R2a вместе с атомами углерода, к которым R1 и R2a присоединены, образуют фенил; R3 и R7 независимо выбраны из водорода и галогена; R4 и R6 независимо выбраны из группы, включающей водород, C1-C6-алкил,C1-C6-алкоксигруппу, галогензамещенную C1-C6-алкоксигруппу и -S(O)2R9; и R5 выбран из группы,включающей водород, галоген, C1-C6-алкил, C1-C6-алкоксигруппу, 5-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N и О, и -S(O)2R9; каждый R9 независимо выбран из группы, включающей C1-C6-алкил, C3-C12-циклоалкил и 5-6-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, О и S, где указанный циклоалкил или гетероциклоалкил в качестве R9 необязательно может быть замещен 1-3 радикалами, которые независимо представляют собой C1-C6-алкил; при условии, что по меньшей мере один из R4, R5 или R6 обозначает -S(O)2R9, непосредственно связанный с фенильным кольцом. В другом варианте осуществления R1 выбран из группы, включающей водород, гидроксил, пирролидинил, морфолиновую группу, метоксигруппу, дифторметоксигруппу, 2-фторэтоксигруппу и метил; или R1 и R2a вместе с атомами углерода, к которым R1 и R2a присоединены, образуют фенил. В другом варианте осуществления R4 и R6 независимо выбраны из группы, включающей водород,метилсульфонил, метил, 2-фторэтоксигруппу, метилпиперазинилсульфонил, пропоксигруппу, изобутоксигруппу, 2,2,2-трифторэтоксигруппу, 2,3-дифтор-2-(фторметил)пропоксигруппу, бутоксигруппу и циклопропилметоксигруппу; и R5 выбран из группы, включающей водород, галоген, метилсульфонил, метил, метоксигруппу, этоксигруппу и морфолиновую группу. Другой вариант осуществления относится к соединениям, выбранным из группы, включающей:N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)-3-(2,2,2 трифторэтокси)бензамид и 3-(2,3-дифтор-2-(фторметил)пропокси)-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2 иламино)фенил)-4-(метилсульфонил)бензамид. В одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей терапевтически эффективное количество соединения формулы I, в комбинации с фармацевтически приемлемым инертным наполнителем. Фармакология и применение. Соединения, предлагаемые в настоящем изобретении, модулируют активность киназ и, как таковые,применимы для лечения заболеваний или нарушений, при которых киназы способствуют патологии и/или симптоматике этого заболевания. Примеры киназ, которые ингибируются соединениями и композициями, описанными в настоящем изобретении, и по отношению к которым применимы способы, описанные в настоящем изобретении, включают, но не ограничиваются только ими, киназы c-kit, Abl, Lyn,MAPK14 (р 38-дельта), PDGFR, PDGFR, ARG, BCR-Abl, BRK, EphB, Fms, Fyn, KDR, LCK, b-Raf,c-Raf, SAPK2, Src, Tie2 и TrkB. Мастоциты (МЦ) являются тканевыми элементами, образованными из особой подгруппы гематопоэтических стволовых клеток, которые продуцируют много разных медиаторов, большинство из которых обладает высокой провоспалительной активностью. Поскольку МЦ распределены почти по всем участкам организма, гиперсекреция медиаторов активированными элементами может привести к поражениям множества органов. Поэтому мастоциты являются главными элементами, участвующими во многих заболеваниях. Настоящее изобретение относится к способу лечения связанных с мастоцитами заболеваний,включающему введение человеку, нуждающемуся в таком лечении, соединения, способного уменьшать количество мастоцитов, или соединения, подавляющего дегрануляцию мастоцитов. Такие соединения можно выбрать из числа ингибиторов c-kit и более предпочтительно - из числа не токсичных, селективных и активных ингибиторов c-kit. Предпочтительно, если указанные ингибиторы неспособны стимулировать гибель зависимых от IL-3 клеток, выращенных в присутствии IL-3. Связанные с мастоцитами заболевания включают, но не ограничиваются только ими, акне и акне,вызыванные Propionibacterium (акне включает все формы хронического воспаления кожи, включая вызванные акне, вызванные Propionibacterium); чрезвычайно редкое и приводящее к потере трудоспособности генетическое заболевание соединительной ткани, известное как прогрессирующая оссифицирующая фибродисплазия (ПОФ); вредные последствия воспаления и разрушения ткани, вызванного воздействием химического или бактериологического оружия (такого как сибирская язва, сернистый иприт и т.п.); муковисцидоз (генетическое заболевание легких, пищеварительной и репродуктивной системы); заболевание почек, такое как синдром острого нефрита, гломерулонефрит, почечный амилоидоз, почечный интерстициальный фиброз (конечное общее направление, приводящее к терминальной стадии заболевания почек при различных нефропатиях); воспалительные мышечные нарушения, включая миозит и мышечную дистрофию; ВИЧ (например, уменьшение количества инфицированных посредством ВИЧ мастоцитов может представлять собой новый путь лечения инфицирования посредством ВИЧ и родственных заболеваний); лечения диабета типа II, ожирения и родственных нарушений (мастоциты регулируют целый ряд процессов, которые способствуют развитию атеросклероза, включая гипергликемию, гиперхолестеринемию, гипертензию, эндотелиальную дисфункцию, резистентность к инсулину и ремоделирование сосудов); ишемию головного мозга; мастоцитоз (очень неоднородная группа нарушений, характеризующаяся аномальным накоплением мастоцитов в различных тканях, преимущественно в коже и костном мозге, а также в селезенке, печени, лимфатических узлах и желудочно-кишечном тракте); токсикоманию и симптомы отмены (в особенности привыкание к чрезмерному употреблению лекарственного средства или наркотика, злоупотребление лекарственным средством или наркотиком, привыкание к лекарственному средству или наркотику, зависимость от лекарственного средства или наркотика, синдром отмены и передозировку); нарушения ЦНС (в особенности депрессию, шизофрению, тревожное состояние, мигрень, потерю памяти, боль и нейродегенеративные заболевания); стимулирование роста волос (включая предупреждение или сведение к минимуму потери волос); бактериальные инфекции (в особенности инфекции, вызванные бактериями, экспрессирующими FimH); интерстициальный цистит (хроническое воспаление стенки мочевого пузыря, приводящее к повреждению ткани, в особенности в интерстициальном пространстве между клетками в выстилке мочевого пузыря); воспалительные заболевания кишечника (обычно это относится к четырем заболеваниям кишечника, а именно болезни Крона, язвенному колиту, колиту неопределенной этиологии и инфекционному колиту); ангиогенез опухоли; аутоиммунные заболевания (в особенности рассеянный склероз, язвенный колит, болезнь Крона, ревматоидный артрит и полиартрит, склеродермия, красная волчанка, дерматомиозит, пемфигус, полимиозит, васкулит и заболе-4 019524 вания "трансплантат против хозяина"); воспалительные заболевания, такие как ревматоидный артрит(РА); рассеянный склероз (PC); аллергические нарушения (в особенности аллергический ринит, аллергический синусит, анафилактический синдром, уртикария, ангионевротический отек, атопический дерматит, аллергический контактный дерматит, нодозная эритема, полиморфная эритема, кожный некротический венулит и кожное воспаление вследствие укуса насекомого, бронхиальная астма); полипоз носа и потерю костной массы. ТРФР (тромбоцитарный фактор роста) является широко распространенным фактором роста, который играет важную роль и при нормальном росте, и при патологической пролиферации клеток, такой как наблюдающаяся при канцерогенезе и заболеваниях гладкомышечных клеток кровеносных сосудов, например при атеросклерозе и тромбозе. Соединения, предлагаемые в настоящем изобретении, могут ингибировать активность рецептора ТРФР (ТРФРР) и поэтому пригодны для лечения опухолевых заболеваний, таких как глиомы, саркомы, опухоли предстательной железы и опухоли толстой кишки, молочной железы и яичников, гиперэозинофилии, фиброза, такого как фиброз легких, легочной гипертензии и сердечно-сосудистых заболеваний. Путь передачи сигнала Ras-Raf-MEK-ERK опосредует клеточный ответ на сигналы роста. Ras мутирует в онкогенную форму в 15% случаях рака человека. Группа Raf относится к серин/треонинпротеинкиназам и включает трех представителей, A-Raf, B-Raf и c-Raf (или Raf-1). Воздействие на Raf, как на мишень лекарственного средства, направлено на роль Raf, как расположенного в прямом направлении эффектора Ras. Однако последние данные показывают, что B-Raf может играть заметную роль в образовании некоторых опухолей без необходимости наличия активированного Ras аллеля (Nature, 417, 949-954 (1 Jul. 2002). В частности, мутации B-Raf обнаружены в значительной части злокачественных меланом. Имеющиеся медицинские средства лечения меланомы обладают ограниченной эффективностью, в особенности на поздних стадиях меланом. Соединения, предлагаемые в настоящем изобретении, также подавляют клеточные процессы, включающие b-Raf киназу, и предоставляют новые возможности для лечения раковых заболеваний людей, в особенности меланомы. Соединения, предлагаемые в настоящем изобретении, также подавляют клеточные процессы, включающие c-Raf киназу. c-Raf активируется онкогеном ras, который мутрован при большом количестве раковых заболеваний людей. Поэтому ингибирование киназной активности c-Raf может предоставить средство для предупреждения роста опухоли, опосредуемого ras [Campbell, S.L., Oncogene, 17, 1395(1998)]. Соединения, предлагаемые в настоящем изобретении, можно применять и для лечения незлокачественных пролиферативных нарушений, таких как атеросклероз, тромбоз, псориаз, склеродермия и фиброз,а также для защиты стволовых клеток, например для противодействия гемотоксическому воздействию химиотерапевтических средств, таких как 5-фторурацил, и при астме. Соединения, предлагаемые в настоящем изобретении, оказывают полезное воздействие при лечении нарушений, возникающих вследствие трансплантации, например аллогенной трансплантации, в особенности отторжения тканей, в особенности такого как облитерирующий бронхиолит (ОБ), т.е. хроническое отторжение аллогенных трансплантатов легких. В отличие от пациентов, не страдающих ОБ, у страдающих ОБ часто обнаруживается повышенная концентрация ТРФР в жидких бронхоальвеолярных выделениях. Соединения, предлагаемые в настоящем изобретении, также эффективны при заболеваниях, связанных с миграцией и пролиферацией гладкомышечных клеток сосудов (при которых часто играют роль ТРФР и ТРФРР), таких как рестеноз и атеросклероз. Эти эффекты и их последствия для пролиферации или миграции гладкомышечных клеток сосудов in vitro и in vivo можно продемонстрировать путем введения соединений, предлагаемых в настоящем изобретении, а также путем исследования их влияния на утолщение интимы сосудов in vivo после механического повреждения. Группа trk нейротропиновых рецепторов (trkA, trkB, trkC) стимулирует жизнеспособность, рост и дифференциацию нейронных и не нейронных тканей. Белок TrkB экспрессируется в клетках нейроэндокринного типа в тонком кишечнике и толстой кишке, в альфа-клетках поджелудочной железы, в моноцитах и макрофагах лимфатических узлов и селезенки и в зернистом слое эпидермиса (Shibayama andKoizumi, 1996). Экспрессирование белка TrkB связывали с неблагоприятным прогрессированием опухолей Вилма и нейробластомами. Кроме того, TkrB экспрессируется в раковых клетках предстательной железы, но не в нормальных клетках. Путь передачи сигналов в прямом направлении от рецепторов trk включает каскад активации АМПК с помощью Shc, активированный Ras, гены ERK-1 и ERK-2 и путь трансдукции PLC-гамма (Sugimoto et al., 2001). Киназа c-src передает онкогенные сигналы многих рецепторов. Например, сверхэкспрессированиеEGFR или HER2/neu в опухолях приводит к конститутивной активации c-src, характерной для злокачественной клетки, но отсутствующей в нормальной клетке. С другой стороны, мыши, у которых отсутствует экспрессирование c-src, обладают остеопетротическим фенотипом, что указывает о ключевой роли c-src в функции остеокластов и о возможном участии в связанных с этим нарушениях. Киназа семейства Tec, Bmx, нерецепторная протеинтирозинкиназа, регулирует пролиферацию раковых клеток эпителия млекопитающих. Показано, что рецептор 3 фактора роста фибробластов оказывает негативное регуляторное влияние на рост костей и ингибирование пролиферации хондроцитов. Летальная дисплазия вызывается различными мутациями рецептора 3 фактора роста фибробластов, и одна мутация, TDII FGFR3, обладает конститутивной тирозинкиназной активностью, что активирует фактор транскрипции Stat1, что приводит к экспрессированию ингибитора клеточного цикла, остановке роста и аномальному развитию костей (Su etal., Nature, 1997, 386, 288-292). FGFR3 также часто экспрессируется при раковых заболеваниях типа множественной миеломы. Ингибиторы активности FGFR3 применимы для лечения опосредуемых Т-клетками воспалительных или аутоиммунных заболеваний, включая, но не ограничиваясь только ими,ревматоидный артрит (РА), коллагеновый II артрит, рассеянный склероз (PC), системную красную волчанку (СКВ), псориаз, юношеский диабет, болезнь Шегрена, заболевание щитовидной железы, саркоидоз, аутоиммунный увеит, воспалительную болезнь кишечника (болезнь Крона и язвенный колит), глютеиновую болезнь и миастению gravis. Активность киназы, экспрессируемой под действием сыворотки или глюкокортикоидов (СГК), коррелирует с измененными активностями ионных каналов, в частности натриевых и/или калиевых каналов,и соединения, предлагаемые в настоящем изобретении, можно использовать для лечения гипертензии.Lin et al. (1997), J. Clin. Invest. 100, 8:2072-2078 и Р. Lin (1998), PNAS, 95, 8829-8834 с помощью моделей ксенотрансплататов опухоли молочной железы и меланомы показали, что происходит подавление роста опухоли и васкуляризации, а также уменьшение метастазирования в легкие при аденовирусных инфекциях или при инъекции внутриклеточного домена Tie-2 (Tek). Ингибиторы Tie2 можно использовать в случаях, когда происходит неприемлемая неоваскуляризация (т.е. при диабетической ретинопатии,хроническом воспалении, псориазе, саркоме Капоши, хронической неоваскуляризации вследствие дегенерации желтого пятна, ревматоидном артрите, младенческой гемангиоме и раковых заболеваниях).Lck участвует в Т-клеточной сигнализации. Мыши, лишенные гена Lck, обладают плохой способностью вырабатывать тимоциты. Функция Lck, как положительного активатора Т-клеточной сигнализации, показывает, что ингибиторы Lck могут быть полезными для лечения аутоиммунного заболевания,такого как ревматоидный артрит. Разные JNK, как и другие АМПК, участвуют в опосредовании клеточного ответа на рак, индуцируемой тромбином агрегации тромбоцитов, иммунодефицитных состояниях, аутоиммунных заболеваниях, гибели клеток, аллергиях, остеопорозе и заболевании сердца. Объекты лечебного воздействия, связанные с активацией пути JNK, включают хронический миелолейкоз (ХМЛ), ревматоидный артрит, астму, остеоартрит, ишемию, рак и нейродегенеративные заболевания. Вследствие важности активацииJNK, связанной с заболеванием печени или приступами печеночной ишемии, соединения, предлагаемые в настоящем изобретении, также могут быть применимы для лечения различных заболеваний печени. Также описана роль JNK в сердечно-сосудистом заболевании, таком как инфаркт миокарда или застойная сердечная недостаточность, и было показано, что JNK опосредует гипертрофический ответ на различные формы сердечного стресса. Показано, что каскад JNK также участвует в активации Т-клеток,включая активацию промотора IL-2, таким образом, ингибиторы JNK могут быть полезны для применения при измененных патологических иммунных ответах. Также выявлена роль активации JNK при различных раковых заболеваниях, что свидетельствует о возможности применения ингибиторов JNK при раке. Например, конститутивно активированный JNK связан с опосредуемым HTLV-1 онкогенезом[Oncogene. 13:135-42 (1996)]. JNK может играть роль при саркоме Капоши (СК). Другие пролиферативные эффекты других цитокинов, участвующих в пролиферации при СК, таких как сосудистый эндотелиальный фактор роста (СЭФР), IL-6 и TNF, также могут опосредоваться с помощью JNK. Кроме того,регуляция гена c-jun в р 210 BCR-ABL трансформированных клетках соответствует активности JNK и свидетельствует о роли ингибиторов JNK при лечении хронического миелолейкоза (ХМЛ) [Blood. 92:2450-60 (1998)]. Предполагается, что некоторые аномальные пролиферативные патологические состояния связаны с экспрессированием raf и поэтому предположительно чувствительны к подавлению экспрессирования raf. Аномально высокие уровни экспрессирования белка raf также связаны с трансформацией и аномальной пролиферацией клеток. Также предполагается, что эти аномальные пролиферативные патологические состояния чувствительны к ингибированию экспрессирования raf. Например, экспрессирование белка craf предположительно играет роль при аномальной пролиферации клеток, поскольку сообщали, что 60% всех линий клеток карциномы легких экспрессируют необычно высокие уровни мРНК и белка c-raf. Другими примерами аномальных пролиферативных патологических состояний являются гиперпролиферативные нарушения, такие как раковые заболевания, опухоли, гиперплазия, фиброз легких, ангиогенез,псориаз, атеросклероз и пролиферация гладкомышечных клеток кровеносных сосудов, такие как стеноз или рестеноз после ангиопластики. Клеточные сигнальные пути, частью которых является raf, также участвуют в воспалительных нарушениях, характеризующихся пролиферацией Т-клеток (активация и рост Т-клеток), таких как, например, отторжение тканевого лоскута, эндотоксиновый шок и гломерулонефрит. Активированные стрессом протеинкиназы (АСПК) представляют собой семейство протеинкиназ,-6 019524 которые характеризуют предпоследнюю стадию путей передачи сигналов, которые приводят к активации фактора транскрипции c-jun и экспрессированию генов, регулируемых с помощью c-jun. В частности, cjun участвует в транскрипции генов, кодирующих белки, участвующие в репарации ДНК, которая повреждена вследствие генотоксичных инсультов. Поэтому средства, которые ингибируют активность АСПК в клетке, предотвращают репарацию ДНК и сенсибилизируют клетку по отношению к средствам,которые индуцируют повреждение ДНК или ингибируют синтез ДНК и индуцируют апоптоз клетки, или по отношению к средствам, которые подавляют пролиферацию клеток. Активированные митогеном протеинкиназы (АМПК) являются представителями консервативных путей передачи сигналов, которые активируют факторы транскрипции, факторы трансляции и другие целевые молекулы в ответ на различные внеклеточные сигналы. АМПК активируются фосфорилированием по двойному мотиву фосфорилирования, обладающему последовательностью Thr-X-Tyr, с помощью активированных митогеном протеинкиназкиназ (МКК). У высших эукариотов физиологическая роль передачи сигналов с помощью АМПК была скоррелирована с такими клеточными проявлениями,как пролиферация, онкогенез, развитие и дифференциация. В соответствии с этим способность регулировать трансдукцию сигналов по этим путям (в особенности через МКК 4 и МКК 6) может привести к разработке средств лечения и профилактики заболеваний, связанных с передачей сигналов с помощью АМПК, таких как воспалительные заболевания, аутоиммунные заболевания и рак. Группа рибосомных протеинкиназ S6 человека включает по меньшей мере 8 представителей (RSK1,RSK2, RSK3, RSK4, MSK1, MSK2, p70S6K и p70S6 Kb). Протеинкиназы рибосомного белка S6 выполняют важные плейотропные функции, в частности играют ключевую роль в регуляции трансляции мРНК при биосинтезе белка (Eur. J. Biochem. 2000 November; 267(21):6321-30, Exp. Cell Res. Nov. 25, 1999; 253(1):100-9, Mol. Cell Endocrinol. May 25, 1999; 151(1-2):65-77). Также показано, что фосфорилирование рибосомного белка S6 посредством p70S6 участвует в регуляции клеточной подвижности (Immunol. CellBiol. 2000 August; 78(4):447-51) и росте клеток (Prog. Nucleic Acid Res. Mol. Biol., 2000; 65:101-27) и поэтому может быть важным для метастазирования опухоли, иммунного ответа и восстановления ткани, а также для других патологических состояний. АСПК (также называющиеся "jun N-концевые киназы" или "JNK's") представляют собой семейство протеинкиназ, которые характеризуют предпоследнюю стадию путей передачи сигналов, которые приводят к активации фактора транскрипции c-jun и экспрессированию генов, регулируемых с помощью cjun. В частности, c-jun участвует в транскрипции генов, кодирующих белки, участвующие в репарации ДНК, которая повреждена вследствие генотоксичных инсультов. Средства, которые ингибируют активность АСПК в клетке, предотвращают репарацию ДНК и сенсибилизируют клетку по отношению к средствам лечения рака, которые действуют путем индуцирования повреждения ДНК. ВТК играет роль в аутоиммунном и/или воспалительном заболевании, таком как системная красная волчанка (СКВ), ревматоидный артрит, множественные васкулиты, идиопатическая тромбоцитопеническая пурпура (ИТП), миастения gravis и астма. Вследствие участия ВТК в активации В-клеток ингибиторы ВТК применимы в качестве ингибиторов опосредуемой В-клетками патогенной активности, такой как продуцирование аутоантител, и применимы для лечения В-клеточной лимфомы и лейкоза.CHK2 является представителем семейства checkpoint киназ серин/треонинпротеинкиназ и участвует в механизме, использующемся для контроля повреждения ДНК, такого как повреждение, вызванное мутагенами окружающей среды и эндогенными реакционноспособными кислородсодержащими соединениями. В результате она связана с подавлением опухоли и является объектом противораковой терапии.CSK влияет на метастатическую способность раковых клеток, в особенности при колоректальном раке.Fes является нерецепторной протеинтирозинкиназой, которая участвует в различных путях передачи сигналов цитокинов, а также в дифференциации миелоидных клеток. Fes также является основным компонентом дифференциации гранулоцитов. Активность Flt3 рецепторной тирозинкиназы связана с лейкозами и миелодиспластическим синдромом. Примерно в 25% случаев ОМЛ лейкозные клетки экспрессируют конститутивно активную форму аутофосфорилированной (р) FLT3 тирозинкиназы на поверхности клеток. Активность p-FLT3 способствует росту и жизнеспособности лейкозных клеток. Для пациентов, страдающих острым лейкозом, лейкозные клетки которых экспрессируют активную киназу p-FLT3, прогноз результатов химиотерапии является плохим. Ингибирование активности киназы p-FLT3 индуцирует апоптоз (запрограммированную гибель клеток) лейкозных клеток. Ингибиторы IKK и IKK (1 и 2) являются средствами лечения заболеваний, которые включают ревматоидный артрит, отторжение трансплантата, воспалительную болезнь кишечника, остеоартрит, астму, хроническое обструктивное заболевание легких, атеросклероз, псориаз, рассеянный склероз, удар,системную красную волчанку, болезнь Альцгеймера, ишемию головного мозга, травматическое поражение головного мозга, болезнь Паркинсона, боковой амиотрофический склероз, субарахноидальное кровоизлияние и другие заболевания или нарушения, связанные с избыточным продуцированием воспалительных медиаторов в головном мозге и центральной нервной системе.Met связана с большинством типов основных раковых заболеваний человека, и ее экспрессия часто коррелирует с плохим прогнозом и метастазами. Ингибиторы Met являются средствами лечения заболеваний, которые включают раковые заболевания, такие как рак легких, НМКРЛ (немелкоклеточный рак легких), рак кости, рак поджелудочной железы, рак кожи, рак головы и шеи, кожную или внутриглазную меланому, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, колоректальный рак, рак молочной железы, гинекологические опухоли (например, саркомы матки, карцинома фаллопиевых труб, карцинома эндометрия, карцинома шейки матки, карцинома влагалища и карцинома вульвы), болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы (например, рак щитовидной железы, паращитовидной железы или надпочечников), саркомы мягких тканей, рак мочеиспускательного канала, рак пениса, рак предстательной железы, хронический или острый лейкоз, солидные опухоли у детей, лимфоцитарные лимфомы, рак мочевого пузыря, рак почек или мочеточника (например, почечно-клеточная карцинома, карцинома почечной лоханки), злокачественные новообразования у детей, неоплазмы центральной нервной системы (например, первичная лимфома ЦНС, опухоли оси позвоночника, глиома ствола головного мозга или аденомы гипофиза), раковые заболевания крови, такие как острый миелолейкоз, хронический миелолейкоз и т.п., язву пищевода Баррета (предраковый синдром), опухолевое кожное заболевание, псориаз, фунгоидную гранулему и доброкачественную гипертрофию предстательной железы, связанные с диабетом заболевания, такие как диабетическая ретинопатия, ретинальная ишемия и ретинальная реваскуляризация, цирроз печени, сердечно-сосудистое заболевание, такое как атеросклероз, иммунологическое заболевание, такое как аутоиммунное заболевание и заболевание почек. Предпочтительным заболеванием является рак, такой как острый миелолейкоз и колоректальный рак. Связанная с Nima киназа 2 (Nek2) является регуляторной протеинкиназой клеточного цикла с максимальной активностью в начале митоза, которая локализуется в центросоме. Исследования ее воздействия показали, что Nek2 участвует в регуляции отделения центросомы и образовании веретена. В линиях клеток, полученных из опухолей человека, включая опухоли шейки матки, яичников, предстательной железы и в особенности молочной железы, белок Nek2 содержит от 2 до 5 складок. Опосредуемые с помощью p70S6K заболевания или патологические состояния включают, но не ограничиваются только ими, пролиферативные нарушения, такие как рак и туберозный склероз. Введение и фармацевтические композиции. Обычно соединения, предлагаемые в настоящем изобретении, вводят в терапевтически эффективных количествах посредством любого из обычных и приемлемых путей, известных в данной области техники, по отдельности или в комбинации с одним или большим количеством терапевтических средств. Терапевтически эффективное количество может меняться в широких пределах в зависимости от тяжести заболевания, возраста и относительного состояния здоровья субъекта, активности применяющегося соединения и других факторов. Имеются указания о том, что обычно удовлетворительные результаты получают при системном воздействии и суточных дозах, равных примерно от 0,03 до 2,5 мг/кг массы тела. Указанная суточная доза для более крупного млекопитающего, например человека, находится в диапазоне от примерно 0,5 до примерно 100 мг, обычно вводится, например, в виде разделенных доз вплоть до 4 раз в сутки или в форме пролонгированного действия. Разовые дозированные формы, подходящие для перорального введения, включают примерно от 1 до 50 мг активного ингредиента. Соединения, предлагаемые в настоящем изобретении, можно вводить в виде фармацевтических композиций любым обычным путем, в частности энтерально, например перорально, например в виде таблеток или капсул, или парентерально, например в виде растворов или суспензий для инъекций, местно, например в виде лосьонов, гелей, мазей или кремов, или в назальной, ингаляционной форме или в форме суппозиториев. Фармацевтические композиции, включающие соединение, предлагаемое в настоящем изобретении, в свободной форме или в форме фармацевтически приемлемой соли совместно по меньшей мере с одним фармацевтически приемлемым носителем или разбавителем можно приготовить обычным путем по методикам смешивания, гранулирования или нанесения покрытий. Например, композиции для перорального введения могут представлять собой таблетки или желатиновые капсулы, включающие активный ингредиент совместно с а) разбавителями, например лактозой, декстрозой, сахарозой,маннитом, сорбитом, целлюлозой и/или глицином; b) смазывающими веществами, например диоксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; для таблеток также со с) связующими, например алюмосиликатом магния, крахмальной пастой, желатином, трагакантовой камедью, метилцеллюлозой, натриевой солью карбоксиметилцеллюлозы и/или поливинилпирролидоном; при необходимости с d) разрыхлителями, например крахмалами, агаром, альгиновой кислотой или ее натриевой солью, или шипучими смесями; и/или е) абсорбентами, красителями,вкусовыми добавками и подсластителями. Композиции для инъекций могут представлять собой водные изотонические растворы или суспензии, и суппозитории можно приготовить из эмульсий или суспензий жирных веществ. Композиции могут быть стерилизованы и/или содержать вспомогательные вещества,такие как консервирующие, стабилизирующие, смачивающие или эмульгирующие агенты, стимуляторы растворения, соли для регулирования осмотического давления и/или буферные вещества. Кроме того,они также могут содержать другие терапевтически полезные вещества. Препараты, подходящие для чрескожного введения, включают эффективное количество соединения, предлагаемого в настоящем изобре-8 019524 тении, вместе с носителем. Носитель может включать впитывающиеся фармакологически приемлемые растворители, содействующие проникновению через кожу реципиента. Например, чрескожные устройства представляют собой повязку, включающую защитный элемент, резервуар, содержащий соединение,необязательно совместно с носителями, необязательно регулирующий скорость барьерный элемент для доставки соединения к коже реципиента с регулируемой и заранее заданной скоростью в течение продолжительного периода времени и средства для закрепления устройства на коже. Также можно использовать матричные препараты для чрескожного введения. Препаратами, подходящими для местного нанесения, например на кожу и в глаза, предпочтительно являются водные растворы, мази, кремы или гели,хорошо известные в данной области техники. Они могут содержать солюбилизаторы, стабилизаторы,усиливающие тоничность агенты, буферные вещества и консерванты. Соединения, предлагаемые в настоящем изобретении, можно вводить в терапевтически эффективных количествах в комбинации с одним или большим количеством терапевтических средств (фармацевтические комбинации). Например, синергетический эффект может проявляться при использовании других средств лечения астмы, например стероидов и антагонистов лейкотриена. Например, синергетический эффект может проявляться при использовании других иммуномодулирующих или противовоспалительных средств, например при использовании в комбинации с циклоспорином, рапамицином или аскомицином или с их иммунодепрессантными аналогами, например циклоспорином A (CsA), циклоспорином G, FK-506, рапамицином или сравнимыми соединениями, кортикостероидами, циклофосфамидом, азатиоприном, метотрексатом, брехинаром, лефлуномидом, мизорибином, микофеноловой кислотой, микофенолятмитефилом, 15-дезоксиспергуалином, иммунодепрессантными антителами, в особенности моноклональными антителами к рецепторам лейкоцитов, например МНС, CD2, CD3, CD4, CD7, CD25, CD28, В 7, CD45, CD58 или их лигандами, или другими иммуномодулирующими соединениями, такими как CTLA41g. Когда соединения, предлагаемые в настоящем изобретении, вводят совместно с другими средствами, разумеется, дозы вводимых совместно соединений будут меняться в зависимости от типа используемого совместно лекарственного средства, конкретного используемого лекарственного средства, подвергающегося лечению патологического состояния и т.п. Настоящее изобретение также относится к фармацевтическим комбинациям, например к набору,включающему а) первое средство, которое является соединением, предлагаемым в настоящем изобретении, описанным в настоящем изобретении, в свободной форме или в форме фармацевтически приемлемой соли, и b) по меньшей мере одно дополнительное средство. Набор может включать инструкции по его введению. Термины "совместное введение" или "комбинированное введение" и т.п. при использовании в настоящем изобретении означают введение выбранных терапевтических средств одному пациенту и включает режимы лечения, при которых средства необязательно вводят по одному и тому же пути введения или в одно и то же время. Термин "фармацевтическая комбинация" при использовании в настоящем изобретении означает препарат, который получен смешиванием или объединением более одного активного ингредиента и включает и фиксированные, и нефиксированные комбинации активных ингредиентов. Термин "фиксированная комбинация" означает, что активные ингредиенты, например соединение формулы I и дополнительное средство, оба, вводят пациенту одновременно в виде одного препарата или дозированной формы. Термин "нефиксированная комбинация" означает, что активные ингредиенты, например соединение формулы I и дополнительное средство, оба, вводят пациенту в виде отдельных препаратов одновременно, по отдельности или последовательно без наложения специальных ограничений по времени, и при таком введении в организме пациента создаются терапевтически эффективные уровни этих 2 соединений. Последнее также относится к смешанному лечению, например с введением 3 или большего количества активных ингредиентов. Способы получения соединений, предлагаемых в настоящем изобретении. Настоящее изобретение также относится к способам получения соединений, предлагаемых в настоящем изобретении. Для описанных реакций может оказаться необходимой защита реакционноспособных функциональных групп, например гидрокси-, амино-, имино-, тио- или карбоксигрупп, если необходимо, чтобы они содержались в конечном продукте, чтобы избежать их нежелательного участия в реакциях. Можно использовать обычные защитные группы в соответствии со стандартной практикой; см., например, T.W. Greene and P.G.M. Wuts in "Protective Groups in Organic Chemistry", John WileySons, 1991. Соединения формулы I, в которой L обозначает -NHC(O)-, можно получить по следующей схеме реакций I. где R1, R2a, R2b, R3, R4, R5, R6 и R7 являются такими, как описано в "Кратком изложении сущности изобретения". Соединение формулы I можно получить по реакции соединения формулы 2 с соединением формулы 3 в присутствии подходящего растворителя (например, ДМФ (диметилформамид) и т.п.), подходящего реагента сочетания (например, HATU и т.п.) и подходящего основания (например, ДИЭА (диизопропилэтиламин) и т.п.). Реакция протекает в температурном диапазоне от примерно 0 до примерно 60 С и до завершения может продолжаться в течение до 24 ч. Соединения формулы I, в которой L обозначает -C(O)NH-, можно получить по следующей схеме реакций II. Схема реакций II где R1, R2, R3, R4, R5, R6 и R7 являются такими, как описано в "Кратком изложении сущности изобретения". Соединение формулы I можно получить по реакции соединения формулы 4 с соединением формулы 5 в присутствии подходящего растворителя (например, ДМФ и т.п.), подходящего реагента сочетания(например, HATU и т.п.) и подходящего основания (например, ДИЭА и т.п.). Реакция протекает в температурном диапазоне от примерно 0 до примерно 60 С и до завершения может продолжаться в течение до 24 ч. Подробное описание синтеза соединение формулы I приведено в представленных ниже примерах. Дополнительные методики синтеза соединений, предлагаемых в настоящем изобретении. Соединение, предлагаемое в настоящем изобретении, можно получить в виде фармацевтически приемлемой соли присоединения с кислотой по реакции соединения в свободной форме с фармацевтически приемлемой неорганической или органической кислотой. Альтернативно, фармацевтически приемлемую соль присоединения соединения, предлагаемого в настоящем изобретении, с основанием можно получить по реакции соединения в форме свободной кислоты с фармацевтически приемлемым неорганическим или органическим основанием. Альтернативно, солевые формы соединений, предлагаемых в настоящем изобретении, можно получить с использованием солей исходных веществ или промежуточных продуктов. Формы свободной кислоты или свободного основания соединений, предлагаемых в настоящем изобретении, можно получить из соответствующих солей присоединения с основаниями или солей присоединения с кислотами соответственно. Например, соединение, предлагаемое в настоящем изобретении, в форме соли присоединения с кислотой можно превратить в соответствующее свободное основание путем обработки подходящим основанием (например, раствором гидроксида аммония, гидроксидом натрия и т.п.). Например, соединение, предлагаемое в настоящем изобретении, в форме соли присоединения с основанием можно превратить в соответствующую свободную кислоту путем обработки подходящей кислотой (например, хлористо-водородной кислотой и т.п.). Соединения, предлагаемые в настоящем изобретении, в неокисленной форме можно получить изN-оксидов соединений, предлагаемых в настоящем изобретении, путем обработки восстановительным реагентом (например, серой, диоксидом серы, трифенилфосфином, борогидридом лития, борогидридом натрия, трихлоридом, трибромидом фосфора и т.п.) в подходящем инертном органическом растворителе(например, ацетонитриле, этаноле, водном растворе диоксана и т.п.) при температуре от 0 до 80 С. Пролекарственные производные соединений, предлагаемых в настоящем изобретении, можно получить по методикам, известным специалистам с общей подготовкой в данной области техники (дополнительные подробности см., например, в публикации Saulnier et al. (1994), Bioorganic and MedicinalChemistry Letters, vol. 4, p. 1985). Например, подходящие пролекарства можно получить по реакции не являющегося производным соединения, предлагаемого в настоящем изобретении, с подходящим карбамоилирующим реагентом (например, 1,1-ацилоксиалкилкарбанохлоридатом, п-нитрофенилкарбонатом и т.п.). Защищенные производные соединений, предлагаемых в настоящем изобретении, можно получить по методикам, известным специалистам с общей подготовкой в данной области техники. Подробное описание методик, применимых для введения защитных групп и их удаления, приведено в публикации Т.W. Greene, "Protecting Groups in Organic Chemistry", 3rd edition, John WileySons, Inc., 1999. Соединения, предлагаемые в настоящем изобретении, можно легко получить или сформировать способом, предлагаемым в настоящем изобретении, в виде сольватов (например, гидратов). Гидраты соединений, предлагаемых в настоящем изобретении, можно легко получить путем перекристаллизации из водно/органических смесей растворителей с использованием органических растворителей, таких как диоксан, тетрагидрофуран или метанол. Соединения, предлагаемые в настоящем изобретении, можно получить в виде отдельных стереоизомеров с помощью реакции рацемической смеси соединения с оптически активным разделяющим реагентом с образованием пары диастереоизомерных соединений, разделения диастереоизомеров и выделения оптически чистых энантиомеров. Хотя разделение энантиомеров можно провести с использованием ковалентных диастереоизомерных производных соединений, предлагаемых в настоящем изобретении,предпочтительны диссоциирующие комплексы (например, кристаллические соли диастереоизомеров). Диастереоизомеры обладают разными физическими характеристиками (например, температурами плавления, температурами кипения, растворимостями, реакционной способностью и т.п.), и их можно легко разделить с использованием этих различий. Диастереоизомеры можно разделить с помощью хроматографии или предпочтительно по методикам разделения, основанным на различиях их растворимостей. Затем оптически чистый энантиомер вместе с разделяющим реагентом извлекают по любой возможной методике, которая не приводит к рацемизации. Более подробное описание методик, пригодных для выделения стереоизомерных соединений из их рацемической смеси, приведено в публикации Jean Jacques,Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates и Resolutions", John WileySons, Inc., 1981. Резюмируя, можно сказать, что соединения формулы I можно получить способом, который включает:(a) проведение реакций по схемам реакций I и II;(c) необязательное превращение солевой формы соединения, предлагаемого в настоящем изобретении, в несолевую форму;(d) необязательное превращение неокисленной формы соединения, предлагаемого в настоящем изобретении, в фармацевтически приемлемый N-оксид;(f) необязательное выделение индивидуального изомера соединения, предлагаемого в настоящем изобретении, из смеси изомеров;(g) необязательное превращение не являющегося производным соединения, предлагаемого в настоящем изобретении, в фармацевтически приемлемое пролекарственное производное;(h) необязательное превращение пролекарственного производного соединения, предлагаемого в настоящем изобретении, в его не являющуюся производным форму. Если получение исходных веществ не описано, то эти соединения являются известными или их можно получить по методикам, аналогичным известным в данной области техники или раскрытым в приведенных ниже примерах. Специалист в данной области техники должен понимать, что описанные выше превращения являются только примерами методик получения соединений, предлагаемых в настоящем изобретении, и что также можно использовать другие хорошо известные методики. Примеры Настоящее изобретение дополнительно описывается, но не ограничивается, приведенными ниже примерами, которые иллюстрируют получение соединений формулы I, предлагаемых в настоящем изобретении. Получение промежуточных продуктов. Синтез 6-метил-N1-(4-(пиридин-3-ил)пиримидин-2-ил)бензол-1,3-диамина 5. К 2-амино-4-нитротолуолу 1 (0,033 моль) в н-бутаноле (29 мл) прибавляют азотную кислоту (2,1 г,65% в воде) и затем раствор цианамида в воде (2 мл, 0,047 ммоль). Полученную смесь кипятят с обратным холодильником в течение 25 ч. После охлаждения до 0 С, фильтрования и промывки смесью 1:1 = этанол:диэтиловый эфир (30 мл) получают 2-метил-5-нитрофенилгуанидиннитрат 2. К 2-метил-5-нитрофенилгуанидину 2 (0,0074 моль) в изопропаноле (15 мл) прибавляют 3(0,0074 моль) и чешуйки гидроксида натрия (0,008 моль). Полученную смесь кипятят с обратным холодильником в течение 12 ч. После охлаждения до 0 С продукт собирают фильтрованием и промывают изопропанолом (6 мл) и метанолом (3 мл) и получают 4. 1 Н ЯМР (400 МГц, d6-ДМСО)9,31 (s, 1 Н), 9,24 (s, 1 Н), 8,78 (m, 1 Н), 8,70 (m, 1 Н), 8,61 (m, 1 Н), 8,47(m, 1H), 7,88 (m, 1 Н), 7,55 (m, 3 Н), 2,39 (s, 3 Н). МС (m/z) (M+l)+: 278,2. Реагент 3 можно получить по следующим методикам. Смесь 3-ацетилпиридина (2,47 моль) и диметилацеталя N,N-диметилформамид (240 мл) кипятят с обратным холодильником в течение 16 ч. Растворитель удаляют в вакууме и гексаны (100 мл) прибавляют к остатку для кристаллизации твердого вещества. Твердое вещество перекристаллизовывают из смеси метилен хлорид-гексаны и получают 3-диметиламино-1-(3-пиридил)-2-пропен-1-он. 1 Н ЯМР (400 МГц, d-хлороформ)9,08 (d, J=2,4 Гц, 1 Н), 8,66 (m, 1 Н), 8,20 (m, 1 Н), 7,87 (m, 1 Н),7,37 (m, 1H), 5,68 (d, J=16,4 Гц, 1H), 3,18 (s, 3 Н), 2,97 (s, 3 Н). В реактор помещают концентрированную хлористо-водородную кислоту (17 мл) и затем безводный хлорид олова(II) (0,03 моль). Смесь перемешивают в течение 10 мин и затем охлаждают до 0-5 С. Затем медленно прибавляют (в течение 3-4 мин) раствор соединения 4 (5,6 ммоль) в этилацетате (3 мл), поддерживая температуру равной 0-5 С. Реакционную смесь нагревают до КТ (комнатная температура) и перемешивают в течение 1,5 ч. К ней прибавляют воду (50 мл) и затем медленно прибавляют 50% раствор гидроксида натрия (40 мл). Полученную смесь экстрагируют хлороформом (225 мл). Органический слой тщательно промывают водой и выпаривают. Остаток растворяют в этилацетате (2 мл), охлаждают до 0-10 С, выдерживают при этой температуре в течение 1 ч. Полученный осадок собирают фильтрованием и промывают этилацетатом (1 мл) и получают 1,0 г 5. 1 Н ЯМР (400 МГц, d-хлороформ)9,26 (d, J=2,0 Гц, 1 Н), 8,71 (m, 1 Н), 8,48 (d, J=6,8 Гц, 1H), 8,34 1-(5-Бромпиридин-3-ил)-3-диметиламинопропенон 7 получают из 1-(5-бромпиридин-3-ил)этанона по методике, аналогичной использованной для синтеза 3. ЖХ/МС (m/z) (M+l)+: 386,1, 388,1. Конденсация с N-(2-метил-5-нитрофенил)гуанидиннитратом по методике, аналогичной использованной для получения 4, дает [4-(5-бромпиридин-3-ил)пиримидин-2-ил]-(2-метил-5-нитрофенил)амин 8. 1 Н ЯМР (400 МГц, d-CDCl3)9,33 (d, J=2,4 Гц, 1 Н), 9,09 (d, J=1,6 Гц, 1 Н), 8,74 (d, J=2,4 Гц, 1 Н),8,61 (t, J=2,0 Гц, 1H), 8,53 (d, J=4,8 Гц, 1H), 8,01 (s, 1H), 7,82 (dd, J=8,4, 2,4 Гц, 1 Н), 7,29 (d, J=8,0 Гц, 1H),7,22 (d, J=4,8 Гц, 1H), 7,10 (s, 1H), 2,41 (s, 3 Н). ЖХ/МС (m/z) (M+l)+: 255,0, 257,0. Синтез 6-метил-N1-(4-(5-(пирролидин-1-ил)пиридин-3-ил)пиримидин-2-ил)бензол-1,3-диамина 10(2-Метил-5-нитрофенил)-[4-(5-пирролидин-1-илпиридин-3-ил)пиримидин-2-ил]амин 9 получают путем смешивания 8 (0,40 г, 1,04 ммоль), пирролидина (3,12 ммоль), K3PO4 (0,44 г, 2,08 ммоль), CuI(38,5 мг, 0,10 ммоль) и аминокислоты L-пролина (48,1 мг, 0,21 ммоль) в ДМСО (8 мл) нагревания при 160 С в микроволновой печи в течение 20 мин. Охлажденную смесь подвергают распределению между водой и этилацетатом. Органический слой отделяют и водный слой экстрагируют этилацетатом. Объединенные органические слои промывают рассолом, сушат над Na2SO4 и концентрируют в вакууме. Остаток растворяют в 4 мл ДМСО и очищают с помощью ВЭЖХ (высокоэффективная жидкостная хроматография) и получают чистый продукт 9 (25%). 1 Н ЯМР (400 МГц, d-CDCl3)9,23 (d, J=2,4 Гц, 1H), 8,63 (d, J=5,2 Гц, 1H), 8,58 (s, 1H), 8,10 (s, 1H),8,04 (s, 1H), 7,90 (dd, J=8,4, 2,4 Гц, 1H), 7,44 (s, 1H), 7,39 (d, J=8,4 Гц, 1H), 7,30 (d, J=5,2 Гц, 1H), 3,43-3,48(m, 4H), 2,49 (s, 3H), 2,12-2,16 (m, 4H). ЖХ/МС (m/z) (M+l)+: 377,2. 4-Метил-N3-[4-(5-пирролидин-1-илпиридин-3-ил)пиримидин-2-ил]бензол-1,3-диамин 10 получают гидрированием 9 в EtOH в присутствии 10% Pd/C при давлении водорода, равном 1 атм., при КТ (30 %). Растворитель удаляют после фильтрования через целит и продукт используют без дополнительной очистки. ЖХ/МС (m/z) (M+l)+: 347,2. Путем замены пирролидина на другие амины (например, морфолин) можно синтезировать различные промежуточные продукты, аналогичные 9 и 10. Синтез N1-(4-(5-метоксипиридин-3-ил)пиримидин-2-ил)-6-метилбензол-1,3-диамина 14 Смесь 3-бром-5-метоксипиридина (3,0 г, 16 ммоль) и Pd(PPh3)4 несколько раз дегазируют и продувают с помощью N2. К смеси прибавляют трибутил(1-этоксивинил)олово (7,04 мл, 20,8 ммоль) и толуол(15 мл). Полученную реакционную смесь нагревают при 150 С в течение 30 мин в микроволновой печи. После завершения реакции реакционную смесь фильтруют через слой целита, промывают с помощью МеОН, и концентрируют и получают остаток. К этому остатку прибавляют HCl (1 н., 25 мл) и ТГФ (тетрагидрофуран) (25 мл) и смесь перемешивают при комнатной температуре в течение 2 ч. Растворитель удаляют в вакууме и остаток растворяют в EtOAc. Органический слой промывают раствором Na2CO3,сушат над Na2SO4, фильтруют, концентрируют и получают остаток, который очищают с помощью колоночной хроматографии на силикагеле (EtOAc:гексаны = 1:1) и получают 11 в виде светло-желтого твердого вещества. 1 Н ЯМР (400 МГц, d6-ДМСО)8,75 (d, J=1,6 Гц, 1 Н), 8,52 (d, J=3,2 Гц, 1 Н), 7,74 (dd, J=1,6, 3,2 Гц,1H), 3,91 (s, 3 Н), 2,64 (s, 3 Н). МС (m/z) (M+l)+: 152,1. 3-(Диметиламино)-1-(5-метоксипиридин-3-ил)проп-2-ен-1-он 12 получают из 11 по реакции с диэтокси-N,N-диметилметанамином по методике, аналогичной использованной для получения 3. 1 Н ЯМР (400 МГц, d6-ДМСО)8,69 (d, J=2,0 Гц, 1H), 8,37 (d, J=2,8 Гц, 1 Н), 7,76 (d, J=12,0 Гц, 1 Н),7,70 (dd, J=1,6, 2,8 Гц, 1H), 5,86 (d, J=12,0 Гц, 1H), 3,89 (s, 3H), 3,17 (s, 3H), 2,95 (s, 3H).MC (m/z) (M+l)+: 207,1. Конденсация 12 с N-(2-метил-5-нитрофенил)гуанидиннитратом 2 по методике, аналогичной использованной для получения 4, дает 4-(5-метоксипиридин-3-ил)-N-(2-метил-5-нитрофенил)пиримидин-2 амин 13. 1 Н ЯМР (400 МГц, d6-ДМСО)9,23 (s, 1 Н), 8,93 (d, J=1,2 Гц, 1H), 8,80 (d, J=2,0 Гц, 1 Н), 8,63 (d,J=5,2 Гц, 1H), 8,44 (d, J=2,8 Гц, 1H), 8,00 (s, 1H), 7,91 (dd, J=2,0, 8,4 Гц, 1 Н), 7,62 (d, J=5,2 Гц, 1H), 7,52 (d,J=8,4 Гц, 1H), 3,90 (s, 3 Н), 2,44 (s, 3 Н).N3-[4-(5-Метоксипиридин-3-ил)пиримидин-2-ил]-4-метилбензол-1,3-диамин 14 получают гидрированием 3 по методике, аналогичной использованной для получения 10. 1 Н ЯМР (400 МГц, d6-ДМСО)8,90 (d, J=1,6 Гц, 1 Н), 8,71 (s, 1H), 8,51 (d, J=5,2 Гц, 1 Н), 8,45 (d,J=2,8 Гц, 1H), 8,01 (dd, J=2,0, 3,2 Гц, 1 Н), 7,43 (d, J=5,2 Гц, 1 Н), 6,91 (d, J=8,0 Гц, 1H), 6,87 (d, J=2,4 Гц,1H), 6,38 (dd, J=2,4, 8,0 Гц, 1H), 4,87 (s, 2H), 3,95 (s, 3H), 2,12 (s, 3H).BBR3 (3,25 ммоль) при -78 С в течение ночи. Смесь разбавляют с помощью ДХМ и промывают 1 н. водным раствором NaOH. Полученную водную фазу подкисляют избытком 1 н. HCl и экстрагируют с помощью ДХМ. Концентрирование дает 15, который используют без дополнительной очистки. ЖХ/МС (m/z) (M+l)+: 324,2. Соединение 15 (87,0 мг, 0,27 ммоль) кипятят с обратным холодильником с CsHCO3 (103,6 мг,0,54 ммоль) и 1-бром-2-фторэтаном (102,0 мг, 0,80 ммоль) в ацетонитриле (2,0 мл) в течение ночи. Смесь очищают с помощью препаративной ЖХ/МС (жидкостная хроматография/масс-спектрометрия) и получают 16. ЖХ/МС (m/z) (M+l)+: 370,1. Соединение 16 (50,0 мг, 0,14 ммоль) кипятят с обратным холодильником с SnCl2H2O (77,2 мг,0,41 ммоль) в EtOH (2,0 мл) в течение 1 ч. Смесь растворяют в NaOH (1 н., 50 мл) и экстрагируют с помощью ДХМ. Органический слой отделяют, сушат над Na2O4, фильтруют, концентрируют и получают неочищенный продукт, который затем используют без очистки. ЖХ/МС (m/z) (М+1)+: 340,1. Путем замены 1-бром-2-фторэтана на другие алкилгалогениды (например, бромметилциклопропан) можно синтезировать различные промежуточные продукты, аналогичные 16 и 17. Синтез N-(2-метил-5-нитрофенил)-4-(5-метилпиридин-3-ил)пиримидин-2-амина 21(20 мл) с MgMeBr (3 М раствор в Et2O, 10 мл, 30 ммоль) в течение 2 ч. После охлаждения медленно прибавляют водный раствор Na2CO3 для остановки реакции. Экстракция с помощью ДХМ дает неочищен- 14019524 ную смесь, которую очищают с помощью хроматографии на силикагеле, и получают 1-(5-метилпиридин 3-ил)этанон 18 (0,7 г, 30 %). ЖХ/МС (m/z) (M+l)+: 136,1. 1-(5-Метилпиридин-3-ил)этанон 19 получают по методике, аналогичной использованной для получения 3, по реакции 18 с диэтокси-N,N-диметилметанамином. 1 Н ЯМР (400 МГц, d6-ДМСО)8,87 (d, J=1,2 Гц, 1H), 8,49 (d, J=1,2 Гц, 1 Н), 8,02-8,04 (m, 1H), 7,75(d, J=12,0 Гц, 1H), 5,85 (d, J=12,0 Гц, 1 Н), 3,16 (s, 3 Н), 2,94 (s, 3 Н), 2,35 (s, 3 Н). ЖХ/МС (m/z) (M+l)+: 191,2. К 2-метил-5-нитрофенилгуанидину 2 (2,0 ммоль) в н-бутаноле (15 мл) прибавляют 19 (2,0 ммоль) и чешуйки гидроксида натрия (2,0 ммоль). Полученную смесь нагревают при 180 С в течение 40 мин в микроволновой печи. После охлаждения до 0 С продукт собирают фильтрованием и промывают эфиром(20 мл) и метанолом (10 мл) и получают 20 (90%). 1 Н ЯМР (400 МГц, d6-ДМСО)9,19 (s, 1 Н), 9,13 (d, J=1,6 Гц, 1H), 8,91 (d, J=2,0 Гц, 1 Н), 8,63 (d,J=5,2 Гц, 1 Н), 8,57 (d, J=1,2 Гц, 1H), 8,37 (s, 1 Н), 7,90 (dd, J=8,0, 2,4 Гц, 1 Н), 7,59 (d, J=5,2 Гц, 1H), 7,52 (d,J=8,4 Гц, 1 Н), 2,45 (s, 3 Н), 2,41 (s, 3 Н). ЖХ/МС (m/z) (М+1)+: 322,2. Соединение 21 получают гидрированием 20 в МеОН в присутствии 10% Pd/C с подачей водорода из баллона при КТ. Растворитель удаляют после фильтрования через целит и продукт используют без дополнительной очистки (95%). ЖХ/МС (m/z) (M+l)+: 292,1. Синтез 3-гидрокси-4-метансульфонил-N-[4-метил-3-(4-пиридин-3-илпиримидин-2 иламино)фенил]бензамида 23(5 мл) при КТ. К раствору по каплям прибавляют диизопропилэтиламин (0,732 мл, 4,2 ммоль). Через 30 мин смесь медленно прибавляют к насыщенному водному раствору NaHCO3. Твердое вещество отфильтровывают, промывают водой и сушат в вакууме в течение ночи и получают продукт 22 в виде желтовато-коричневатого твердого вещества (630 мг, 60%). ЖХ/МС (М+1): 524,1. Соединение 22 (580 мг, 1,11 ммоль), CuI (42 мг, 0,272 ммоль), L-пролин (61 мг, 0,444 ммоль), 1 н.NaOH (0,5 мл) и NaSO2Me (267 мг, 2,22 ммоль) суспендируют в ДМСО (3 мл) и нагревают при 180 С в микроволновой печи в течение 25 мин. Смесь разбавляют концентрированным водным раствором аммиака и промывают с помощью EtOAc. К водной фазе прибавляют 1 н. водный раствор HCl до установления рН 7 и твердый осадок собирают фильтрованием и получают продукт 23 (80%). ЖХ/МС (М+1): 476,1. Синтез конечных соединений. Тип А. 6-Метил-N1-(4-(пиридин-3-ил)пиримидин-2-ил)бензол-1,3-диамин 5 (5 ммоль), 3-(метилсульфонил)бензойную кислоту (6 ммоль) и HATU (6 ммоль) растворяют в сухом ДМФ (5 мл) при КТ. Диизопропилэтиламин (6 ммоль) по каплям прибавляют к раствору. Через 30 мин смесь медленно прибавляют к насыщенному водному раствору NaHCO3. Твердое вещество отфильтровывают, промывают водой и сушат в вакууме в течение ночи и получают продукт А 1 в виде светло-желтого твердого вещества. 1 Н ЯМР (400 МГц, d6-ДМСО)10,5 (s, 1 Н), 9,33 (s, 1 Н), 9,04 (s, 1H), 8,75 (d, J=1,7 Гц, 1H), 8,63 (d,J=7,5 Гц, 1 Н), 8,55 (d, J=5,0 Гц, 1H), 8,47 (s, 1 Н), 8,29 (d, J=8 Гц, 1 Н), 8,13 (d, J=8,6 Гц, 1H), 8,11 (s, Н),7,83 (t, J=8 Гц, 1H), 7,66 (dd, J=7,3, 5,3 Гц, 1H), 7,47 (m, 2H), 7,25 (d, J=8,4 Гц, 1H), 3,29 (s, 3H), 2,55 (s, 6H,- 15019524 2 Ме-НОАс), 2,24 (s, 3 Н). ЖХ/МС (m/z) (М+1)+: 460,2. Аналогичную методику используют для получения соединений А 2-А 5 с применением ЖХ/МС для очистки. Промежуточный продукт 6 получают аналогично получению А 1. Соединение 6 (0,1 ммоль), CuI(0,5 мл) и нагревают при 180 С в микроволновой печи в течение 5 мин. Смесь очищают с помощью препаративной ЖХ/МС и получают А 6. 1 Н ЯМР (400 МГц, ДМСО)10,45 (s, 1 Н), 9,24 (s, 1 Н), 8,89 (s, 1H), 8,67 (d, J=2,0 Гц, 1H), 8,49 (m,2 Н), 8,09 (s, 1H), 8,01 (d, J=8,4, 1H), 7,90-7,93 (т, 2 Н), 7,56 (m, 1 Н), 7,39 (d, J=5,6 Гц, 2 Н), 7,23 (d, J=8,4 Гц,1H), 3,32 (s, 3 Н), 2,70 (s, 3 Н), 2,21 (s, 3 Н). ЖХ/МС (m/z) (M+l)+: 474,2. Аналогичную методику используют для получения соединений А 7, А 8. Конечное соединение В 1 получают из 10 по методике, аналогичной использованной для получения А 1. 1 Конечное соединение D1 получают из 14 по методике, аналогичной использованной для получения А 1. 1 Конечное соединение D2 получают из 17 по методике, аналогичной использованной для получения А 1. 1 Конечное соединение Е 1 получают из 21 по методике, аналогичной использованной для получения А 1. ЖХ/МС (m/z) (M+l)+: 474,2. Тип F. 3-(2-Фторэтокси)-4-метансульфонил-N-[4-метил-3-(4-пиридин-3-илпиримидин-2 иламино)фенил]бензамид F1 Соединение 23 (0,04 ммоль), CsHCO3 (0,1 ммоль) и 1-бром-2-фторэтан (0,04 ммоль) нагревают в сухом ацетонитриле (0,5 мл) при 150 С в микроволновой печи в течение 10 мин. Очистка с помощью препаративной ЖХ/МС дает конечное соединение F1. 1 Н ЯМР (400 МГц, ДМСО)10,41 (s, 1H), 9,31 (s, 1H), 9,01 (s, 1H), 8,73 (s, 1H), 8,55 (m, 2 Н), 8,10 (s,1H), 7,95 (m, 1H), 7,76 (s, 1H), 7,71 (m, 1H), 7,60 (m, 1H), 7,46 (m, 2 Н), 7,25 (d, J=8,0 Гц, 1 Н), 4,92 (m, 1 Н),4,80 (m, 1 Н), 4,62 (m, 1H), 4,55 (m, 1H), 3,32 (s, 3 Н), 2,25 (s, 3 Н). ЖХ/МС (m/z) (М+1)+: 522,2. Аналогичную методику используют для получения соединений F2-F5. Тип G. Конечное соединение G1 получают из 27 по методике, аналогичной использованной для получения А 1. ЖХ/МС (m/z) (M+1): 510,2. Путем повторения методик, описанных в приведенных выше примерах (промежуточные продукты и конечные соединения), с использованием подходящих исходных веществ получают следующие соединения формулы I, приведенные в табл.1. Анализы. Соединения, предлагаемые в настоящем изобретении, исследуют для определения их способности селективно подавлять пролиферацию клеток Ba/F3 дикого типа и клеток Ba/F3, трансформированных с киназой Tel c-kit и со слитыми Tel ТРФРР тирозинкиназами. Кроме того, соединения, предлагаемые в настоящем изобретении, селективно подавляют зависимую от ФСК пролиферацию клеток Мо 7 е. Кроме того, соединения исследуют для определения их способности ингибировать киназы Abl, ARG, BCR-Abl,BRK, EphB, Fms, Fyn, KDR, c-kit, LCK, ТРФРР, b-Raf, c-Raf, SAPK2, Src, Tie2 и TrkB. Исследование пролиферации: библиотека BaF3 - протокол считывания Bright glo. Соединения исследуют для определения их способности подавлять пролиферацию клеток дикого типа Ba/F3 и клеток Ba/F3, трансформированных с помощью слитых с Tel тирозинкиназ. Нетрансформированные клетки Ba/F3 держат в средах, содержащих рекомбинантный IL3. Клетки помещают в 384 луночные планшеты ТС при плотности 5000 клеток/лунка в 50 мкл среды на лунку и прибавляют исследуемое соединение в количестве от 0,06 нМ до 10 мкМ. Затем клетки инкубируют в течение 48 ч при 37 С, 5% СО 2. После в каждую лунку прибавляют 25 мкл BRIGHT GLO (Promega) в соответствии с инструкциями изготовителя и планшеты считывают с использованием Analyst GT - режим люминесценции - 50000 интегрирований с представлением результата в виде интенсивностей люминесценции в относительных единицах. Значения IC50, концентрацию соединения, необходимую для 50% подавления, определяют по зависимости доза-ответ. Исследование Мо 7 е. Соединения, описанные в настоящем изобретении, исследуют для определения их способности подавлять зависимую от ФСК пролиферацию с использованием клеток Мо 7 е, которые эндогенно экспрессируют c-kit, с использованием 96-луночных планшетов. Вкратце, методика заключается в следующем,для двукратных серийных разведений исследуемых соединений (Cmax=10 мкМ) исследуют антипролиферативную активность по отношению к клеткам Мо 7 е, стимулированным с помощью рекомбинантного ФСК человека. После 48 ч инкубации при 37 С жизнеспособность клеток определяют с помощью колориметрического анализа МТТ фирмы Promega. Методика исследования c-kit HTRF. Аликвоту (5 мкл) 2 концентрации смеси для исследования фермента c-kit, содержащей 25 нг c-kit бавляют в каждую лунку 384-луночного планшета proxiplate (Packard). Каждая лунка последнего рядаproxiplate содержит 5 мкл смеси для исследования фермента c-kit, не содержащей c-kit, и предназначена для оценки фона. Соединения, предлагаемые в настоящем изобретении, прибавляют в каждую лунку и планшеты инкубируют в течение 30 мин при комнатной температуре. 2 АТФ (40 мкМ) в киназном буфере (5 мкл) прибавляют в каждую лунку и планшеты инкубируют при комнатной температуре в течение 3 ч. В каждую лунку прибавляют смесь для детектирования (50% KF, 40% киназного буфера, 10% ЭДТК,1:100 разведенный Mab PT66-K (cat 61T66KLB) и 1:100 разведенный стрептавидин-XL(cat 611SAXLB)0 (10 мкл) и планшеты дополнительно инкубируют в течение 1-2 ч при комнатной температуре. Затем сигнал HTRF считывают с детектора. Исследование пролиферации TG-HA-VSMC человека. Клетки TG-HA-VSMC человека (АТСС) выращивают в МИСД, к которой прибавлено 10% ФБС, до слияния 80-90% и затем повторно суспендируют в МИСД, к которой прибавлено 1% ФБС и 30 нг/мл рекомбинантного ТРФР-ВВ человека по 6 е 4 клеток/мл. Затем аликвоты клеток помещают в 384-луночные планшеты по 50 мкл/лунка, инкубируют в течение 20 ч при 37 С, затем обрабатывают с помощью 0,5 мкл 100 соединений в течение 48 ч при 37 С. После обработки в каждую лунку прибавляют 25 мклCellTiter-Glo в течение 15 мин, затем планшеты считывают с использованием CLIPR (Molecular Devices). Методика исследования PDGFR/ Lance. Аликвоту (2,5 мкл) 2 концентрации пептида PDGFRP и смеси, содержащей АТФ (аденозинтрифосфат) (4 мкМ пептида биотин-А-А-А-AEEEEYVFIEAKKK, 20 мкМ АТФ в буфере для исследования (20 мМ Hepes, 54 мМ MgCl2, 0,01% БСА, 0,05% Tween-20, 1 мМ ДТТ, 10% глицерин, 50 мкМNa3VO4 прибавляют в каждую лунку 384-луночного планшета proxiplate (Packard). Планшеты центрифугируют и соединения, предлагаемые в настоящем изобретении, (50 нл) прибавляют в каждую лунку с помощью дозатора pintool. В каждую лунку прибавляют (2,5 мкл) 2 концентрации смеси ферментов(cat PV3591) в буфере для анализа) или только буфер для анализа без фермента PDGFR. Планшеты инкубируют в течение 1,5 ч при комнатной температуре. В каждую лунку прибавляют смесь для детектирования (5 мкл; 50% 1 М KF, 40% киназного буфера, 10% ЭДТК, 1:100 разведенный Mab PT66-K(cat 61T66KLB) и 1:100 разведенный стрептавидин-XL (cat 611SAXLB) и proxiplate инкубируют в течение 1 ч при комнатной температуре, затем сигнал HTRF считывают с детектора. Исследование пролиферации Ba/F3 FL FLT3. Используют линию клеток мышей Ba/F3 pro-B, которая сверхэкспрессирует полноразмерную конструкцию FLT3. Эти клетки держат в RPMI 1640/10% фетальной бычьей сыворотки (RPMI/ФБС), к которой прибавлены пенициллин 50 мкг/мл, стрептомицин 50 мкг/мл и L-глутамин 200 мМ с прибавлением мышиного рекомбинантного IL3. Ba/F3 полноразмерные клетки FLT3 выращивают при недостатке IL3 в течение 16 ч и затем помещают в 384-луночные планшеты ТС при плотности 5000 клеток/лунка в 25 мкл среды на лунку и прибавляют исследуемое соединение в количестве от 0,06 нМ до 10 мкМ. После прибавления соединения прибавляют лиганд FLT3 или IL3 для контроля цитотоксичности в 25 мкл среды/лунка при соответствующих концентрациях. Затем клетки инкубируют в течение 48 ч при 37 С, 5%CO2. После инкубации клеток в каждую лунку прибавляют 25 мкл BRIGHT GLO (Promega) в соответствии с инструкциями изготовителя и планшеты считывают с использованием Analyst GT - режим люминесценции - 50000 интегрирований с представлением результата в виде интенсивностей люминесценции в относительных единицах. Подавление пролиферации клеток, зависимой от BCR-Abl (высокопроизводительная методика). Используют линию клеток мышей, представляющую собой гемопоэтическую родительскую линию клеток 32D, трансформированную с помощью BCR-Abl кДНК (32D-p210). Эти клетки выдерживают в МИСД (модифицированная Иглом среда Дульбекко)/10% фетальная телячья сыворотка (МИСД/ФТС) с прибавлением пенициллина 50 мкг/мл, стрептомицина 50 мкг/мл и L-глутамина 200 мМ. Нетрансформированные клетки 32D аналогичным образом выдерживают с прибавлением 15% кондиционированной среды WEHI (Walter и Eliza Hall Institute of Medical Research) в качестве источника IL3. 50 мкл суспензии клеток 32D или 32D-p210 помещают в 384-луночные микропланшеты Greiner(черные) при плотности 5000 клеток/лунка. В каждую лунку прибавляют 50 нл исследуемого соединения(1 мМ исходного раствора в ДМСО) (STI571 включают в качестве положительного контроля). Клетки инкубируют в течение 72 ч при 37 С, 5% СО 2. В каждую лунку прибавляют 10 мкл 60% раствора Alamar(Molecular Devices). Подавление пролиферации клеток, зависимой от BCR-Abl. Клетки 32D-p210 помещают в 96-луночные планшеты ТС при плотности 15000 клеток/лунка. 50 мкл двукратных серийных разведений исследуемого соединения (Cmax=40 мкМ) прибавляют в каждую лунку (STI571 включают в качестве положительного контроля). После инкубации клетки в течение 48 ч при 37 С, 5% CO2 15 мкл МТТ (Promega) прибавляют в каждую лунку и клетки инкубируют в течение еще 5 ч. Оптическую плотность при 570 нм количественно определяют с помощью спектрофотомерии и значения IC50, концентрацию соединения, необходимую для 50% подавления, определяют по зависимости доза-ответ. Влияние на распределение клеточного цикла. Клетки 32D и 32D-p210 помещают в 96-луночные планшеты ТС по 2,5106 клеток/лунка в 5 мл среды и прибавляют исследуемые соединения в количестве 1 или 10 мкМ (STI571 включают в качестве контроля). Затем клетки инкубируют в течение 24 или 48 ч при 37 С, 5% CO2. 2 мл суспензии клеток промывают с помощью ФБС (фетальная бычья сыворотка), фиксируют в 70% EtOH в течение 1 ч и обрабатывают с помощью ФБС/ЭДТК (этилендиаминтетрауксусная кислота)/RNase А в течение 30 мин. Прибавляют пропидиййодид (Cf=10 мкг/мл) и интенсивность флуоресценции количественно определяют с помощью проточной цитометрии с использованием системы FACScalibur (BD Biosciences). Исследуемые соединения, предлагаемые в настоящем изобретении, обнаруживают апоптозное воздействие в случае клеток 32D-p210, но не вызывают апоптоз родительских клеток 32D. Влияние на клеточное аутофосфорилирование BCR-Abl. Аутофосфорилирование BCR-Abl количественно исследуют с помощью твердофазного иммуноферментного анализа с захватом с использованием специфического по отношению к c-abl антитела для захвата и антитела к антифосфотирозину. Клетки 32D-p210 помещают в 96-луночные планшеты ТС по 2105 клеток/лунка в 50 мкл среды. 50 мкл двукратных серийных разведений исследуемых соединений(Cmax=10 мкМ) прибавляют в каждую лунку (STI571 включают в качестве положительного контроля). Клетки инкубируют в течение 90 мин при 37 С, 5% СО 2. Затем клетки в течение 1 ч на льду обрабатывают с помощью 150 мкл литического буфера (50 мМ Tris-HCl (Tris трис-(гидроксиметил)аминометан),рН 7,4, 150 мМ NaCl, 5 мМ ЭДТК, 1 мМ ЭГТК (этиленгликольтетрауксусная кислота) и 1% NP-40), содержащего ингибиторы протеазы и фосфатазы. 50 мкл лизата клеток помещают в 96-луночные планшеты для оптических исследований, предварительно покрытые специфическими анти-abl антителами, и блокируют. Планшеты инкубируют в течение 4 ч при 4 С. После промывки буфером TBS-Tween-20(TBS - забуференный с помощью Tris физиологический раствор), прибавляют 50 мкл конъюгированных со щелочной фосфатазой антифосфотирозиновых антител и планшеты дополнительно инкубируют в течение ночи при 4 С. После промывки буфером TBS-Tween-20 прибавляют 90 мкл люминесцентного субстрата и люминесценцию количественно исследуют с помощью системы Acquest (Molecular Devices). Исследуемые соединения, предлагаемые в настоящем изобретении, которые ингибируют пролиферацию клеток, экспрессирующих BCR-Abl, ингибируют клеточное аутофосфорилирование BCR-Abl зависимым от дозы образом. Влияние на пролиферацию клеток, экспрессирующих мутантные формы BCR-abl. Соединения, предлагаемые в настоящем изобретении, исследуют для определения их антипролиферативного воздействия на клетки Ba/F3, экспрессирующие форму дикого типа или мутантные формыBCR-Abl (G250E, E255V, Т 3151, F317L, М 351 Т), которым придана резистентность или сниженная чувствительность по отношению к STI571. Антипролиферативное воздействие этих соединений на клетки,экспрессирующие мутантную BCR-abl, и на нетрансформированные клетки исследуют при 10, 3,3, 1,1 и 0,37 мкМ, как это описано выше (в средах без IL3). Значения IC50 для соединений, нетоксичных для нетрансформированных клеток, определяют по зависимостям доза-ответ, полученным, как это описано выше.FGFR3 (ферментный анализ). Исследование активности киназы с использованием очищенного FGFR3 (Upstate) проводят при конечном объеме, равном 10 мкл, содержащем 0,25 мкг/мл фермента в киназном буфере (30 мМ Tris-HClpH 7,5, 15 мМ MgCl2, 4,5 мМ MnCl2, 15 мкМ Na3VO4 и 50 мкг/мл БСА (бычий сывороточный альбумин),и субстраты (5 мкг/мл биотин-поли-EY(GLu, Tyr) (CIS-US, Inc.) и 3 мкМ АТФ). Готовят два раствора: сначала первый раствор объемом 5 мкл, содержащий фермент FGFR3 в киназном буфере, помещают в 384-луночные планшеты ProxiPlate (Perkin-Elmer) и затем прибавляют 50 нл соединений, растворенных в ДМСО, затем в каждую лунку прибавляют 5 мкл второго раствора, содержащего субстрат (поли-EY) и АТФ в киназном буфере. Реакционные смеси инкубируют при комнатной температуре в течение 1 ч, реакции останавливают путем прибавления 10 мкл смеси для детектирования HTRF, которая содержит 30 мМ Tris-HCl рН 7,5, 0,5 М KF, 50 мМ ЭДТК, 0,2 мг/мл БСА, 15 мкг/мл стрептавидин-XL665 (CIS-US,Inc.) и 150 нг/мл конъюгированных с криптатом антифосфотирозиновых антител (CIS-US, Inc.). После инкубации в течение 1 ч при комнатной температуре для проведения взаимодействия стрептавидинбиотин с помощью Analyst GT (Molecular Devices Corp.) считывают сигналы флуоресценции с разрешением по времени. Значения IC50 рассчитывают с помощью линейного регрессионного анализа, выраженного в процентах подавления каждым соединением при 12 концентрациях (разведения 1:3 от 50 мкМ до 0,28 нМ). В этом исследовании соединения, предлагаемые в настоящем изобретении, обладают значениями IC50, находящимися в диапазоне от 10 нМ до 2 мкМ.FGFR3 (исследование в клетках). Соединения, предлагаемые в настоящем изобретении, исследуют для определения их способности подавлять пролиферацию трансформированных Ba/F3-TEL-FGFR3 клеток, которая зависит от активности клеточной киназы FGFR3. Ba/F3-TEL-FGFR3 выращивают вплоть до 800000 клеток/мл в суспензии с МИСД 1640 с прибавлением 10% фетальной бычьей сыворотки в качестве культуральной среды. Клетки помещают в 384-луночный планшет по 5000 клеток/лунка в 50 мкл культуральной среды. Соединения,предлагаемые в настоящем изобретении, растворяют и разводят в диметилсульфоксиде (ДМСО). Готовят 12 серийных разведений 1:3 в ДМСО для получения набора концентраций, обычно находящегося в диапазоне от 10 мМ до 0,05 мкМ. К клеткам прибавляли 50 нл разведенных соединений и инкубируют в течение 48 ч в инкубаторе для культур клеток. AlamarBlue (TREK Diagnostic Systems), который можно использовать для мониторинга восстановительной среды, образованной пролиферирующими клетками,прибавляют к клеткам при конечной концентрации, равной 10%. После еще 4 ч инкубации при 37 С в инкубаторе для культур клеток сигналы флуоресценции восстановленного AlamarBlue (возбуждение при 530 нм, испускание при 580 нм) измеряют с помощью Analyst GT (Molecular Devices Corp.). ЗначенияIC50 рассчитывают с помощью линейного регрессионного анализа, выраженного в процентах подавления каждым соединением при 12 концентрациях.b-Raf-ферментный анализ. Соединения, предлагаемые в настоящем изобретении, исследуют для определения их способности ингибировать активность b-Raf. Исследование проводят в 384-луночных планшетах MaxiSorp (NUNC) с черными стенками и прозрачным дном. Субстрат, IВ разводят в ЗФФД (забуференный фосфатом физиологический раствор Дульбекко) (1:750) и в каждую лунку прибавляют 15 мкл. Планшеты инкубируют при 4 С в течение ночи и 3 раза промывают с помощью TBST (25 мМ Tris, pH 8,0, 150 мМ NaCl и 0,05%Tween-20) с использованием устройства для промывки планшетов EMBLA. Планшеты блокируют с помощью Superblock (15 мкл/лунка) в течение 3 ч при комнатной температуре, 3 раза промывают с помощью TBST и сушат. В каждую лунку прибавляют буфер для анализа, содержащий 20 мкМ АТФ (10 мкл),и затем 100 или 500 нл соединения. b-Raf разводят в буфере для анализа (1 мкл в 25 мкл) и в каждую лунку прибавляют 10 мкл разведенного b-Raf (0,4 мкг/лунка). Планшеты инкубируют при комнатной температуре в течение 2,5 ч. Реакцию киназы останавливают путем проводимой 6 раз промывки планшетов с помощью TBST. Антитела Phosph-IBpSer32/36) разводят в Superblock (1:10,000) и в каждую лунку прибавляют 15 мкл. Планшеты инкубируют при 4 С в течение ночи и 6 раз промывают с помощьюTBST. АР-конъюгированный козий-анти-мышиный IgG разводят в Superblock (1:1,500) и в каждую лунку прибавляют 15 мкл. Планшеты инкубируют при комнатной температуре в течение 1 ч и 6 раз промывают с помощью TBST. В каждую лунку прибавляют 15 мкл флуоресцентного субстрата Attophos АР(Promega) и планшеты инкубируют при комнатной температуре в течение 15 мин. Планшеты считывают с использованием Acquest или Analyst GT с использованием программы определения интенсивности флуоресценции (возбуждение при 455 нм, испускание при 580 нм).b-Raf- исследование в клетках. Соединения, предлагаемые в настоящем изобретении, исследуют в клетках A375 для определения их способности ингибировать фосфорилирование MEK. Линию клеток A375 (АТСС) получают из клеток меланомы человека, и она содержит мутацию V599E в гене b-Raf. Содержание фосфорилированногоMEK повышено вследствие мутации b-Raf. Субконфлюэнтные и конфлюэнтные клетки А 375 инкубируют с соединениями в течение 2 ч при 37 С в бессывороточной среде. Затем клетки один раз промывают холодным ЗФФ (забуференный фосфатом физиологический раствор) и подвергают лизису в литическом буфере, содержащем 1% Triton X100. После центрифугирования надосадочные жидкости подвергают электрофорезу в полиакриламидном геле с использованием додецилсульфата натрия и затем переносят на мембраны из нитроцеллюлозы. Затем мембраны подвергают вестерн-блоттингу с анти-фосфо-MEK антителами (ser217/221) (Cell Signaling). За количеством фосфорилированного МЕК следят по плотности полос фосфо-MEK на мембранах из нитроцеллюлозы.Upstate KinaseProfiler - радиоферментное исследование связывания с фильтром. Соединения, предлагаемые в настоящем изобретении, исследуют для определения их способности ингибировать отдельные представители группы киназ. Соединения исследуют по два раза при конечных концентрациях, равных 10 мкМ, по этому типичному протоколу. Отметим, что составы киназного буфера и субстраты были разными для разных киназ, включенных в группу "Upstate KinaseProfiler". Киназный буфер (2,5 мкл, 10 - при необходимости содержащий MnCl2), активную киназу (0,001-0,01 Ед; 2,5 мкл), специфический или Poly(Glu4-Tyr) пептид (5-500 мкМ или 0,01 мг/мл) в киназном буфере и киназный буфер (50 мкМ; 5 мкл) смешивают в пробирке Эппендорфа на льду. Прибавляют смесь Mg/АТФ(10 мкл; 67,5 (или 33,75) мМ MgCl2, 450 (или 225) мкМ АТФ и 1 мкКи/мкл [-32 Р]-АТФ (3000 Ки/ммоль и реакционную смесь инкубируют примерно при 30 С в течение примерно 10 мин. Реакционную смесь наносят (20 мкл) на квадратики бумаги 22 см Р 81 (фосфоцеллюлоза, для положительно заряженных пептидных субстратов) или Whatman No. 1 (для Poly(Glu4-Tyr) пептидного субстрата). Исследуемые квадратики промывают 4 раза по 5 мин с помощью 0,75% фосфорной кислоты и промывают один раз ацетоном в течение 5 мин. Исследуемые квадратики переносят в сцинтилляционный сосуд, прибавляют 5 мл сцинтилляционной смеси и содержание 32 Р (импульсов/мин) в пептидном субстрате количественно определяют с помощью сцинтилляционного счетчика Beckman. Для каждой реакции рассчитывают ингибирование, выраженное в процентах. Соединения формулы I в свободной форме или в форме фармацевтически приемлемой соли обладают ценными фармакологическими характеристиками, например, как об этом свидетельствуют проведенные in vitro исследования, описанные в настоящем изобретении. Следует понимать, что описанные в настоящем изобретении примеры и варианты осуществления предназначены только для иллюстрации и что в соответствии с сущностью и объемом настоящего изобретения и объемом прилагаемой формулы изобретения специалист в данной области техники может внести в нее различные модификации и изменения. Все публикации, патенты и заявки на патенты, цитированные в настоящем изобретении, включены в настоящее изобретение в качестве ссылки для всех объектов. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы IR1 выбран из группы, включающей водород, гидроксил, 3-8-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, О и S, C1-C4-алкил, C1-C4-алкоксигруппу, галогензамещенную C1-C4-алкоксигруппу, галогензамещенный C1-C4-алкил, -OX1R8, где X1 обозначаетR3 и R7 независимо выбраны из водорода и галогена;R4 и R6 независимо выбраны из группы, включающей водород, C1-C6-алкил, C1-C6-алкоксигруппу,галогензамещенную C1-C6-алкоксигруппу, -OX2R9 и -S(O)2R9, где Х 2 обозначает C1-C4-алкилен;R5 выбран из группы, включающей водород, галоген, C1-C6-алкил, C1-C6-алкоксигруппу,3-8-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, О и S, и-S(O)2R9; каждый R9 независимо выбран из группы, включающей C1-C6-алкил, C3-C12-циклоалкил и 3-8-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, О и S, где указанный циклоалкил или гетероциклоалкил в качестве R9 необязательно может быть замещен 1-3 радикалами, которые независимо представляют собой C1-C6-алкил,при условии, что по меньшей мере один из R4, R5 или R6 обозначает -S(O)2R9, непосредственно связанный с фенильным кольцом,и его фармацевтически приемлемые соли. 2. Соединение по п.1, в которомR1 выбран из группы, включающей водород, гидроксил, 5-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N и О, C1-C2-алкил, C1-C2-алкоксигруппу, галогензамещенную C1-C2-алкоксигруппу, галогензамещенный C1-C2-алкил, -OX1R8, где X1 обозначаетR3 и R7 независимо выбраны из водорода и галогена;R4 и R6 независимо выбраны из группы, включающей водород, C1-C6-алкил, C1-C6-алкоксигруппу,галогензамещенную C1-C6-алкоксигруппу и -S(O)2R9; иR5 выбран из группы, включающей водород, галоген, C1-C6-алкил, C1-C6-алкоксигруппу,5-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N и О, и-S(O)2R9; каждый R9 независимо выбран из группы, включающей C1-C6-алкил, C3-C12-циклоалкил и 5-6-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, О и S, где указанный циклоалкил или гетероциклоалкил в качестве R9 необязательно может быть замещен 1-3 радикалами, которые независимо представляют собой C1-C6-алкил,при условии, что по меньшей мере один из R4, R5 или R6 обозначает -S(O)2R9, непосредственно связанный с фенильным кольцом.R1 и R2a вместе с атомами углерода, к которым R1 и R2a присоединены, образуют фенил. 4. Соединение по п.3, в котором R4 и R6 независимо выбраны из группы, включающей водород, метилсульфонил, метил, 2-фторэтоксигруппу, метилпиперазинилсульфонил, пропоксигруппу, изобутоксигруппу, 2,2,2-трифторэтоксигруппу, 2,3-дифтор-2-(фторметил)пропоксигруппу, бутоксигруппу и циклопропилметоксигруппу; и R5 выбран из группы, включающей водород, галоген, метилсульфонил, метил,метоксигруппу, этоксигруппу и морфолиновую группу. 5. Соединение по п.1, выбранное из группы, включающейN-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-иламино)фенил)-4-(метилсульфонил)-3-(2,2,2 трифторэтокси)бензамид и 3-(2,3-дифтор-2-(фторметил)пропокси)-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2 иламино)фенил)-4-(метилсульфонил)бензамид. 6. Фармацевтическая композиция, включающая терапевтически эффективное количество соединения по п.1 в комбинации с фармацевтически приемлемым инертным наполнителем.
МПК / Метки
МПК: A61K 31/506, C07D 401/04, A61P 37/00, C07D 417/14, A61P 35/00
Метки: pdgfr, киназы, соединения, композиции, с-kit, ингибиторы
Код ссылки
<a href="https://eas.patents.su/28-19524-soedineniya-i-kompozicii-kak-ingibitory-kinazy-s-kit-i-pdgfr.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения и композиции как ингибиторы киназы с-kit и pdgfr</a>
Соединения и композиции, как ингибиторы киназы c-kit и pdgfr

Номер патента: 16329
Опубликовано: 30.04.2012
Авторы: Ли Сяолинь, Мольтени Валентина, Лю Сяодон
МПК: A61P 37/00, A61P 35/00, A61K 31/506...
Метки: соединения, ингибиторы, pdgfr, киназы, c-kit, композиции
Формула / Реферат:
1. Соединение формулы Iв которой R1, R2a и R2b все независимо представляют собой водород;R3 и R4 вместе с атомом углерода, к которому R3 и R4 присоединены, образуют фенил, необязательно замещенный 1-3 радикалами, независимо выбранными из группы, включающей галоген, C1-C6-алкил, галогензамещенный C1-C6-алкил и -X2S(O)0-2R5a, гдеX2 выбран из группы, включающей связь и C1-C4-алкилен;R5a независимо выбран из водорода и C1-C6-алкила;и его...
Производные пиримидина и содержащие их композиции как ингибиторы киназы c-kit и pdgfr

Номер патента: 17269
Опубликовано: 30.11.2012
Авторы: Мольтени Валентина, Лю Сяодон, Ли Сяолинь
МПК: A61P 35/00, A61K 31/444, C07D 401/14...
Метки: содержащие, пиримидина, киназы, pdgfr, c-kit, композиции, производные, ингибиторы
Формула / Реферат:
1. Соединение формулы Iв которой L обозначает 5-членное гетероарильное кольцо, содержащее 3 атома азота, выбранное изR1, R2a и R2b обозначают водород;R3 выбран из C6-C10-арила и 5-15-членного гетероарила, содержащего гетероатом, выбранный из О, N или S; где указанный арил или гетероарил в качестве R3 замещен 1-3 радикалами, независимо выбранными из группы, включающей C1-C6-алкоксигруппу, C1-C6-алкоксигруппу, замещенную галогеном, и -X2OR5a, где...
Гетероциклические соединения и содержащие их композиции в качестве ингибиторов киназ c-kit и pdgfr

Номер патента: 18551
Опубликовано: 30.08.2013
Авторы: Мольтени Валентина, Ли Сяолинь, Лорен Джон, Лю Сяодон, Кьянелли Донателла, Йе Венс, Набакка Джульет
МПК: A61K 31/4375, A61P 25/00, A61P 29/00...
Метки: композиции, киназ, содержащие, c-kit, ингибиторов, pdgfr, гетероциклические, качестве, соединения
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая сольФормула (I)в которойR1 выбран из группы, включающей C1-С6-алкил, С3-С6-циклоалкил, 4-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N и О, фенил и 5-9-членный гетероарил, содержащий 1-2 гетероатома, независимо выбранных из N, О и S; где указанный C1-С6-алкил группы R1 необязательно замещен 1-2 радикалами, независимо выбранными из группы, включающей...
Ингибиторы фосфатидилинозит-3-киназы и содержащие их фармацевтические композиции

Номер патента: 19255
Опубликовано: 28.02.2014
Авторы: Тесфай Зером, Баджджалиех Уильям, Сюй Вэй, Мэрлоу Чарльз К., Насс Джон М., Ванг Йонг, Браун С.Дэвид, Баннен Линн Канне, Мак Моррисон Б., Керни Патрик
МПК: A61K 31/498, A61K 31/4985, A61P 35/00...
Метки: композиции, фармацевтические, содержащие, ингибиторы, фосфатидилинозит-3-киназы
Формула / Реферат:
1. Соединение формулы Iaили его таутомер, индивидуальный стереоизомер, рацемат, смесь энантиомеров и диастереомеров или его геометрический изомер и, необязательно, в виде его фармацевтически приемлемой соли или сольвата, гдеW1, W2, W3 и W4 представляют собой -С(Н)=;R50 представляет собой водород;R51 представляет собой метил;R52 представляет собой водород;R53 представляет собой водород и С1-6алкокси;R54 представляет собой водород, С1-6алкил,...
Соединения и композиции как ингибиторы активирующей канал протеазы

Номер патента: 16199
Опубликовано: 30.03.2012
Авторы: Спраггон Глен, Видаль Аньес, Бурсулая Бадри, Чаттерджи Арнаб К., Талли Дейвид С.
МПК: A61K 31/444, A61K 31/425, A61K 31/401...
Метки: ингибиторы, протеазы, канал, композиции, соединения, активирующей
Формула / Реферат:
1. Соединение формулы (1)или его фармацевтически приемлемые соли,в которой O-(CR2)p-R2 обозначает заместитель в любом положении кольца А;J обозначает, пиридил или пиперидинил;где один или более из Z1, Z2, Z3, Z4, Z5, Z6 и Z7 обозначает гетероатом, выбранный из N, NR, О или S, а другие атомы Z1-Z7 обозначают СН при условии, что J не обозначает триазолил;В обозначаетили (CR2)k-R5;Y обозначает связь, -SO2-, -NHCO- или -O-(CO)-;R1 обозначает...