Замещенные имидазольные производные, содержащие их фармацевтические композиции и их применение в качестве ингибиторов ртразы
Номер патента: 19385
Опубликовано: 31.03.2014
Авторы: Яррагунта Равиндра Р., Субраманиан Говиндан, Се Жунъюань, Мджалли Аднан М.М., Полисетти Дхарма Р., Куада Джеймс К., Эндрюс Роберт С.
























Формула / Реферат
1. Соединение, имеющее формулу
где R23-R27 выбирают, независимо, из группы, состоящей из водорода, пропила, бутила, пентила, 1-этилпропила, 1-пропилбутила, 3,3-диметилбутила, 4-метилпентила, 4,4-диметилпентила, 1-(3,3-диметилбутил)-4,4-диметилпентила, изобутила, изопропила, втор-бутила, трет-бутила, трифторметила, 4,4,4-трифторбутокси, этокси, пропокси, бутокси, изобутокси, трет-бутокси, 2-фенетокси, 2,2-диметилпропокси, 3-метилбутокси, 3,3-диметилбутокси, 2-циклогексилэтансульфонила, 3,3-диметилбутан-1-сульфонила, циклогексансульфонила, циклогексилметансульфонила, 2-циклогексилэтансульфинила, 3,3-диметилбутан-1-сульфинила, циклогексилметилсульфинила, 2-циклогексилэтилсульфанила, 3,3-диметилбутилсульфанила, фенетилсульфанила, циклогексилметилсульфанила, циклопентила, циклогексила, циклогексилокси, циклогексилметила, 2-циклопентилэтила, 2-циклогексилэтила, циклопентилметокси, циклогексилметокси, 2-циклопентилэтокси, 2-циклогексилэтокси, 2-циклогексилвинила, 3-этилциклобутила, хлора, фтора и фенила;
по меньшей мере один из R23-R27 отличен от водорода,
или его фармацевтически приемлемая соль.
2. Соединение по п.1, выбранное из группы, состоящей из
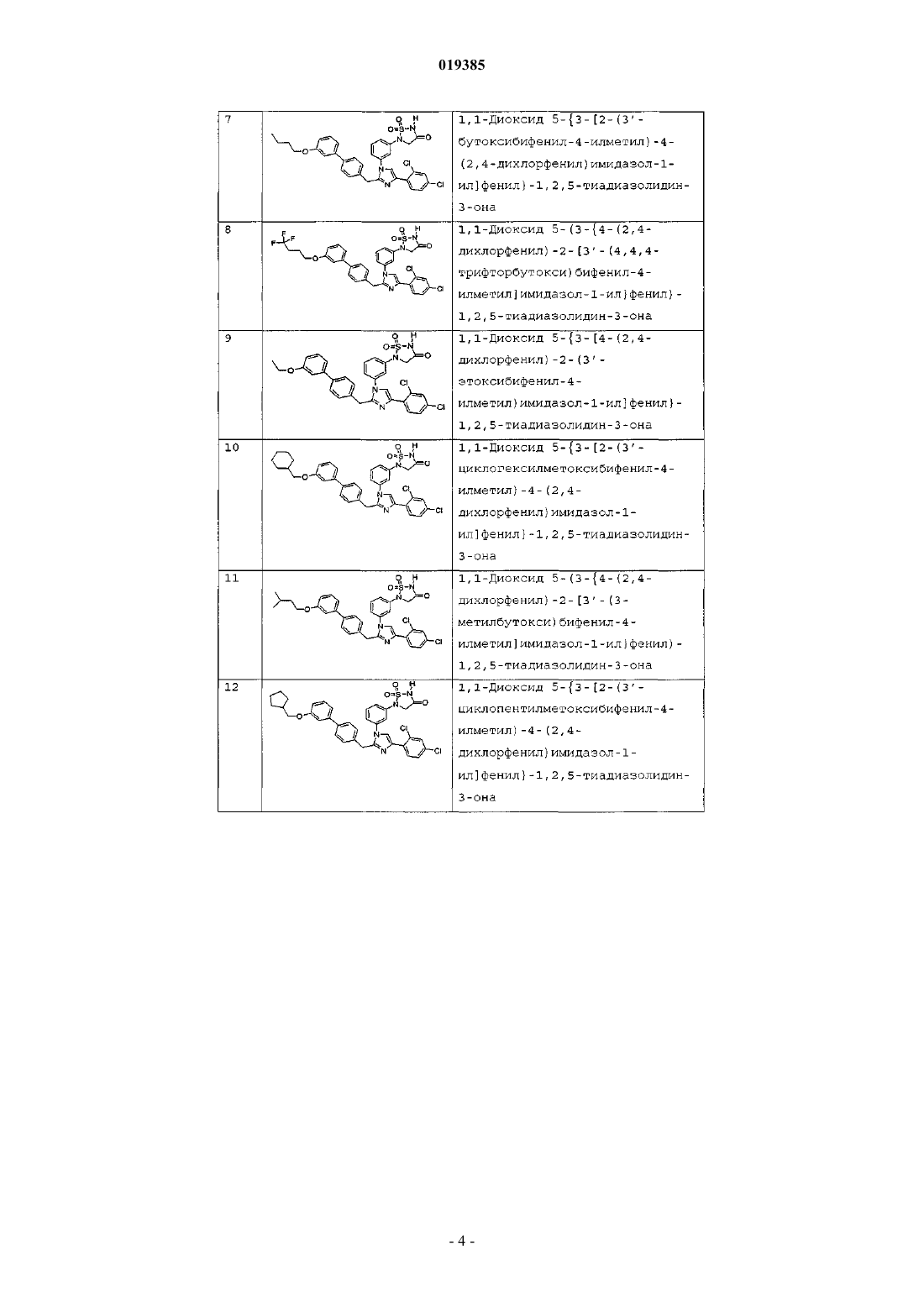
1,1-диоксида 5-{3-[2-(4'-трет-бутилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она;
1,1-диоксида 5-{3-[2-(3'-циклогексилметоксибифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она;
1,1-диоксида 5-{3-[4-(2,4-дихлорфенил)-2-(3'-фенетилоксибифенил-4-илметил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она;
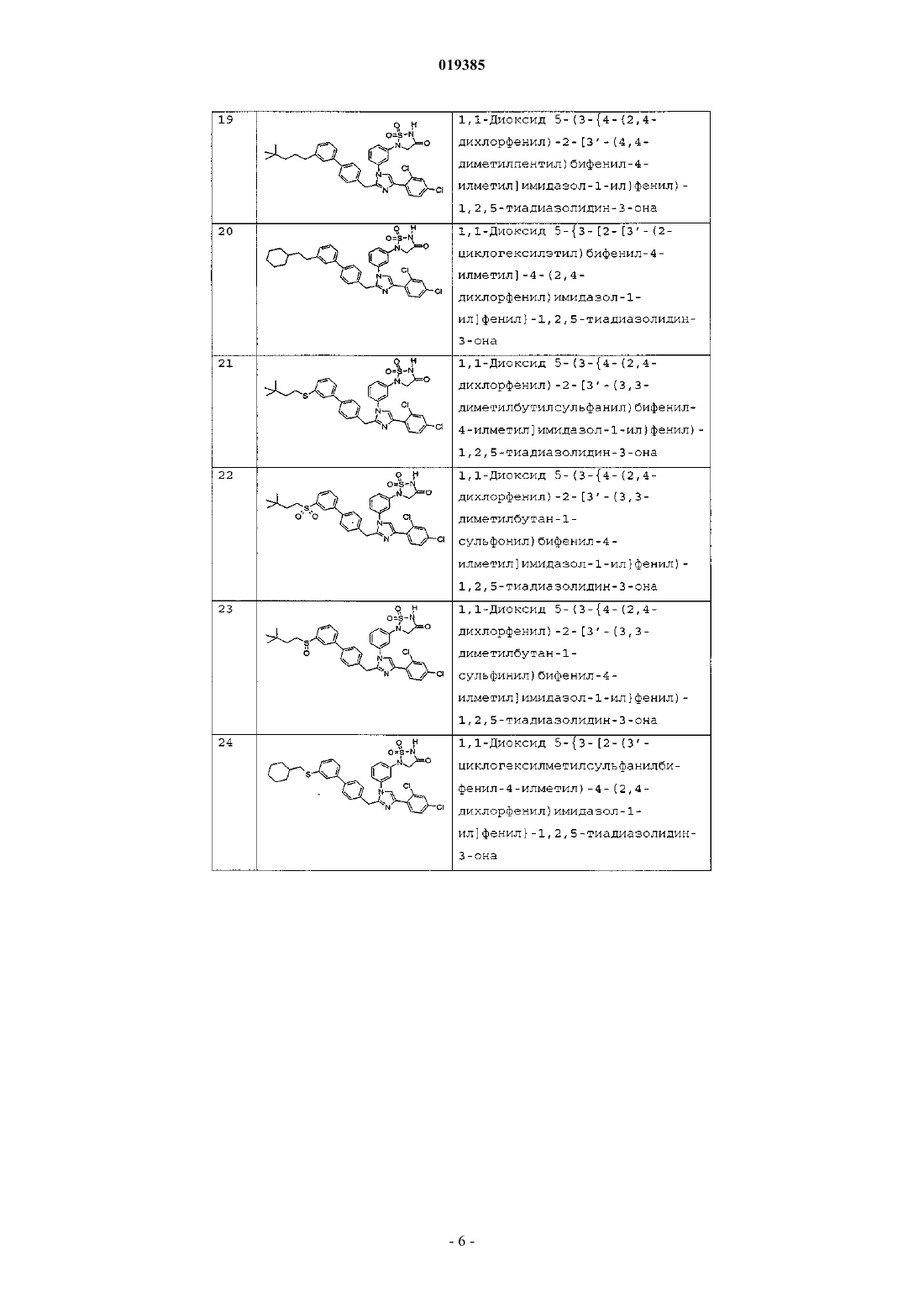
1,1-диоксида 5-(3-{4-(2,4-дихлорфенил)-2-[3'-(3,3-диметилбутилсульфанил)бифенил-4-илметил]имидазол-1-ил}фенил)-1,2,5-тиадиазолидин-3-она;
1,1-диоксида 5-(3-{4-(2,4-дихлорфенил)-2-[3'-(3,3-диметилбутан-1-сульфонил)бифенил-4-илметил]имидазол-1-ил}фенил)-1,2,5-тиадиазолидин-3 -она;
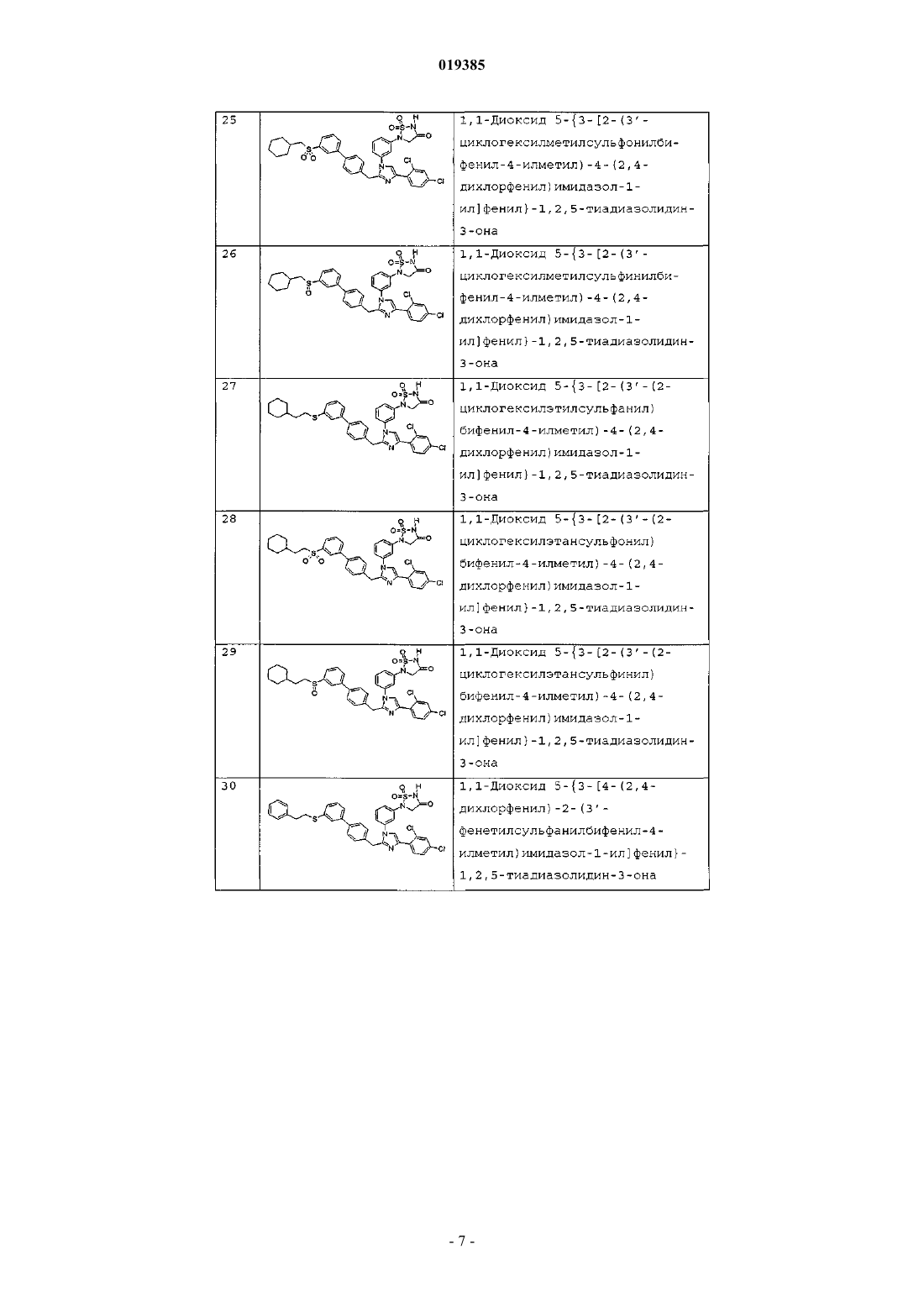
1,1-диоксида 5-{3-[2-(3'-циклогексилметилсульфанилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она;
1,1-диоксида 5-{3-[2-(3'-циклогексилметилсульфонилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она;
1,1-диоксида 5-{3-[2-(3'-(2-циклогексилэтилсульфанил)бифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она;
1,1-диоксида 5-{3-[2-(3'-(2-циклогексилэтансульфонил)бифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она;
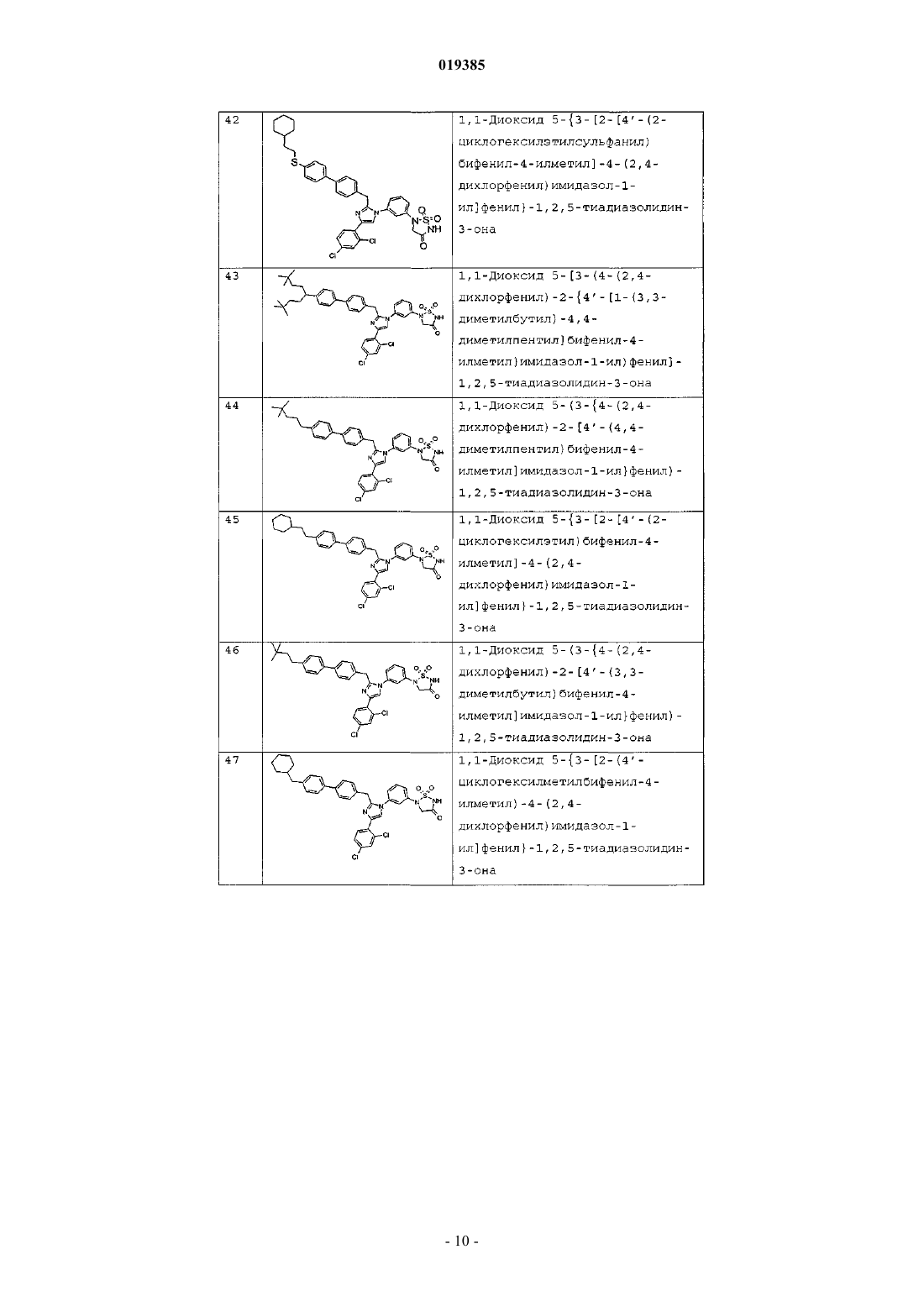
1,1-диоксида 5-(3-{4-(2,4-дихлорфенил)-2-[4'-(3,3-диметилбутокси)бифенил-4-илметил]имидазол-1-ил}фенил)-1,2,5-тиадиазолидин-3-она;
1,1-диоксида 5-{3-[2-(4'-циклогексилметоксибифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она;
1,1-диоксида 5-(3-{4-(2,4-дихлорфенил)-2-[4'-(3,3-диметилбутилсульфанил)бифенил-4-илметил]имидазол-1-ил}фенил)-1,2,5-тиадиазолидин-3-она;
1,1-диоксида 5-{3-[2-(4'-циклогексилметилсульфанилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она;
1,1-диоксида 5-{3-[2-(4'-циклогексилметансульфонилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она;
1,1-диоксида 5-{3-[2-[4'-(2-циклогексилэтокси)бифенил-4-илметил]-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она;
1,1-диоксида 5-{3-[2-[4'-(2-циклогексилэтил)бифенил-4-илметил]-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она
или их фармацевтически приемлемых солей.
3. Соединение по п.1, представляющее собой 1,1-диоксид 5-{3-[2-(4'-циклогексилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она или его фармацевтически приемлемую соль.
4. Соединение по п.3, представляющее собой 1,1-диоксид 5-{3-[2-(4'-циклогексилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она.
5. Соединение по п.3, представляющее собой натриевую или калиевую соль 1,1-диоксида 5-{3-[2-(4'-циклогексилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она.
6. Соединение по п.1, представляющее собой 1,1-диоксид 5-(3-{4-(2,4-дихлорфенил)-2-[3'-(3,3'-диметилбутокси)бифенил-4-илметил]имидазол-1-ил}фенил)-1,2,5-тиадиазолидин-3-она или его фармацевтически приемлемую соль.
7. Соединение по п.6, представляющее собой 1,1-диоксид 5-(3-{4-(2,4-дихлорфенил)-2-[3'-(3,3'-диметилбутокси)бифенил-4-илметил]имидазол-1-ил}фенил)-1,2,5-тиадиазолидин-3-она.
8. Соединение по п.6, представляющее собой натриевую или калиевую соль 1,1-диоксида 5-(3-{4-(2,4-дихлорфенил)-2-[3'-(3,3'-диметилбутокси)бифенил-4-илметил]имидазол-1-ил}фенил)-1,2,5-тиадиазолидин-3-она.
9. Соединение по п.1, представляющее собой 1,1-диоксид 5-{3-[2-(4'-циклогексилметоксибифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она или его фармацевтически приемлемую соль.
10. Соединение по п.9, представляющее собой 1,1-диоксид 5-{3-[2-(4'-циклогексилметоксибифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она.
11. Соединение по п.9, представляющее собой натриевую или калиевую соль 1,1-диоксида 5-{3-[2-(4'-циклогексилметоксибифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил}-1,2,5-тиадиазолидин-3-она.
12. Фармацевтическая композиция, включающая соединение по любому из пп.1-11 или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, разбавитель, эксципиент или их смесь.
13. Фармацевтическая композиция в форме пероральной стандартной лекарственной формы, включающая соединение по любому из пп.1-11 или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, разбавитель, эксципиент или их смесь.
14. Фармацевтическая композиция в форме парентеральной стандартной лекарственной формы, включающая соединение по любому из пп.1-11 или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, разбавитель, эксципиент или их смесь.
15. Применение соединения по любому из пп.1-11 для получения лекарственного средства, предназначенного для лечения диабета типа II.
Текст
ЗАМЕЩЕННЫЕ ИМИДАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РТРазы Настоящее изобретение относится к имидазольным производным, фармацевтическим композициям, содержащим эти соединения, и их применению при лечении расстройств у людей или животных. Соединения по изобретению ингибируют протеинтирозинфосфатазу 1 В и, таким образом, могут применяться для облегчения, лечения, регулирования или вспомогательного лечения заболеваний, опосредуемых активностью РТРазы. Такие заболевания включают диабет типа II.(71)(73) Заявитель и патентовладелец: ТРАНСТЕК ФАРМА ЭлЭлСи (US) Область техники, к которой относится изобретение Настоящее изобретение относится к замещенным имидазольным производным, композициям, способам лечения с использованием соединений и композиций, которые могут применяться для устранения,лечения, регулирования или вспомогательного лечения заболеваний, вызванных активностью протеинфосфатазы. Предпосылки изобретения Фосфорилирование белков в настоящее время признано как главное событие для фундаментальных процессов передачи сигналов в клетках. Поэтому изменение в фосфорилировании белков может составлять или физиологическое или паталогическое изменение в системе in vivo. Дефосфорилирование белков, опосредуемое фосфатазами, также является основным для некоторых процессов передачи сигналов. Двумя основными классами фосфатаз являются (а) протеинсерин/треонинфосфатазы (PSTPаза), которые катализируют дефосфорилирование сериновых и/или треониновых остатков в белках и/или пептидах, и (b) протеинтирозинфосфатазы (РТРазы), которые катализируют дефосфорилирование тирозиновых остатков в белках и/или пептидах. Третьим классом фосфатаз являются специфически двойственные фосфатазы или DSP, которые обладают способностью действовать как РТРазы и как PSTPазы. Среди РТРаз существует два важных семейства - внутриклеточные РТРазы и трансмембранные РТРазы. Внутриклеточные РТРазы включают РТР 1 В, STEP, PTPD1, PTPD2, PTPMEG1, Т-клеточную РТРазу, PTPH1, FAP-1/BAS, PTP1D и РТР 1 С. Трансмембранные РТРазы включают LAR, CD45, РТР,РТР, РТР, РТР, РТР, РТР, РТР, РТР, HePTP, SAP-1 и PTP-U2. Специфически двойственные фосфатазы включают КАР, cdc25, фосфатазу МАРК, РАС-1 и rVH6. РТРазы, в особенности РТР 1 В, органически связаны с невосприимчивостью к инсулину, характерной для диабета типа II (Kennedy В.P., Ramachandran С., Biochem. Pharm., 2000, 60, 877-883). РТРаза, а именно, CD45 и НеРТР, также органически связаны с функционированием иммунной системы, и в частности, Т-клеточной функцией. Некоторые РТРазы, а именно, ТС-РТР, DEP-1, SAP-1 и CDC25, также органически связаны с некоторыми онкозаболеваниями. Некоторые РТРазы, а именно, костная РТРазаOST-PTP, органически связаны с остеопорозом. РТРазы органически связаны с опосредованием действий соматостатина на клетки-мишени, в частности, секрецией гормона и/или секрецией факторов роста. Таким образом, существует потребность в средствах, которые ингибируют действие протеинтирозинфосфатаз. Такие средства могут применяться для лечения непереносимости глюкозы, в том числе диабета типа I и диабета типа II, иммунной дисфункции, в том числе СПИДа, аллергических заболеваний, воспалительных заболеваний и аутоиммунных заболеваний, таких как псориаз, инфекционных заболеваний, ожирения, рака, заболеваний с участием модулированного синтеза гормона роста или модулированного синтеза факторов роста или цитокинов, которые воздействуют на продуцирование гормона роста, или болезни Альцгеймера. Сущность изобретения Настоящее изобретение относится к замещенным имидазольным производным и композициям, которые ингибируют РТР 1 В. В одном варианте осуществления настоящее изобретение относится к соединениям, отображенных ниже. В еще одном варианте осуществления настоящее изобретение относится к применению соединений при лечении расстройств у людей или животных. Соединения по изобретению применимы в качестве ингибиторов протеинтирозинфосфатаз и, таким образом, могут применяться для облегчения, лечения, регулирования и вспомогательного лечения заболевания, опосредуемого активностью РТРазы. Такие заболевания могут включать непереносимость глюкозы, в том числе диабет типа I и диабет типа II, иммунную дисфункцию, в том числе СПИД, аллергические заболевания, воспалительные заболевания и аутоиммунные заболевания, такие как псориаз, инфекционные заболевания, ожирение,рак, заболевания с участием модулированного синтеза гормона роста или модулированного синтеза факторов роста или цитокинов, которые воздействуют на продуцирование гормона роста, или болезнь Альцгеймера. Подробное описание Варианты осуществления настоящего изобретения включают замещенные имидазольные производные, композиции и способы применения. Настоящее изобретение можно осуществить различными путями. В первом аспекте настоящее изобретение относится к имидазольным ингибиторам протеинтирозинфосфатаз (РТРаз), которые применимы для облегчения и лечения заболевания, вызванного РТРазами. В другом аспекте настоящее изобретение относится к соединению формулыR23-R27 выбирают, независимо, из группы, состоящей из водорода, пропила, бутила, пентила, 1 этилпропила, 1-пропилбутила, 3,3-диметилбутила, 4-метилпентила, 4,4-диметилпентила, 1-(3,3 диметилбутил)-4,4-диметилпентила, изобутила, изопропила, втор-бутила, трет-бутила, трифторметила,4,4,4-трифторбутокси, этокси, пропокси, бутокси, изобутокси, трет-бутокси, 2-фенетокси, 2,2 диметилпропокси, 3-метилбутокси, 3,3-диметилбутокси, 2-циклогексилэтансульфонила, 3,3-диметилбутан-1-сульфонила,циклогексансульфонила,циклогексилметансульфонила,2-циклогексилэтансульфинила, 3,3-диметилбутан-1-сульфинила, циклогексилметилсульфинила, 2-циклогексилэтилсульфанила, 3,3-диметилбутилсульфанила, фенетилсульфанила, циклогексилметилсульфанила, циклопентила, циклогексила, циклогексилокси, циклогексилметила, 2-циклопентилэтила, 2-циклогексилэтила, циклопентилметокси, циклогексилметокси, 2-циклопентилэтокси, 2-циклогексилэтокси, 2 циклогексилвинила, 3-этилциклобутила, хлора, фтора и фенила; где по меньшей мере один из R23-R27 отличен от водорода; или его фармацевтически приемлемой соли. Примеры соединений по настоящему изобретению, обладающие потенциально применимой биологической активностью, приводятся с названиями ниже в табл. 1. Способность соединений ингибировать РТР-1 В оценивают с характерными соединениями, перечисленными в табл. 1, с использованием стандартных испытательных процедур первичного/вторичного анализа, в которых измеряют активность ингибирования РТР-1 В. Соединения, которые ингибируют активность РТР-1 В, потенциально применимы при лечении расстройств обмена веществ, родственных инсулинорезистентности или гипергликемии, типично ассоциированных с ожирением или непереносимостью глюкозы. Поэтому соединения по настоящему изобретению особенно применимы при лечении или ингибировании диабета типа II. Соединения по настоящему изобретению также возможно могут применяться при модуляции уровней глюкозы при таких расстройствах, как диабет типа II. Предполагается, что незаполненные валентности в случае гетероатомов, таких как атомы кислорода и азота, в химических структурах, перечисленных в таблице 1, заполнены атомами водорода. В другом аспекте настоящее изобретение относится к фармацевтической композиции, включающей соединение и фармацевтически приемлемый носитель, эксципиент, разбавитель или их смесь. Варианты осуществления настоящего изобретения показывают применимость при ингибировании протеинтирозинфосфатазы РТР 1 В. Обнаружено, что соединения по настоящему изобретению, представленные в примерах, ингибируют протеинтирозинфосфатазу РТР 1 В с ингибирующей эффективностью(IC50) менее примерно 10 мкМ. Вообще, варианты осуществления настоящего изобретения, применимые для фармацевтических применений, могут иметь ингибирующую эффективность (IC50) в отношении белка, представляющего интерес, ниже примерно 100 мкМ. При варианте осуществления настоящего изобретения, применимые для фармацевтических применений, могут иметь ингибирующую эффективность (IC50) в отношении белка, представляющего интерес, ниже примерно 50 мкМ. В случае отдельных применений нужна более низкая ингибирующая эффективность. Так, в другом варианте осуществления соединения по настоящему изобретению могут ингибировать протеинтирозинфосфатазу РТР 1 В с ингибирующей эффективностью(IC50) в интервале от примерно 0,001 до примерно 10 мкМ. В другом варианте осуществления соединения по настоящему изобретению могут ингибировать протеинтирозинфосфатазу РТР 1 В с ингибирующей эффективностью (IC50) в интервале от примерно 0,001 до примерно 3 мкМ. Варианты осуществления соединений настоящего изобретения показывают применимость в качестве ингибиторов протеинтирозинфосфатаз (РТРаз). Варианты осуществления изобретения, описанные в данном описании, относятся, кроме того, к фармацевтическим композициям и способам ингибирования активности РТРазы у млекопитающего, включающим введение млекопитающему, нуждающемуся в ингибировании активности РТРазы, терапевтически определенного количества соединения, приведенных выше, в виде его единственной кристаллической формы или полиморфной кристаллической формы или форм, аморфной формы, отдельного энантиомера, рацемической смеси, отдельного стереизомера, смеси стереоизомеров, отдельного диастереизомера, смеси диастереоизомеров, изотопно обогащенной формы,сольвата, фармацевтически приемлемой соли, сольвата соли, пролекарства, поддающегося биогидролизу эфира или поддающегося биогидролизу амида. Используемые в данном описании термины "фармацевтически приемлемый носитель", "фармацевтически приемлемый разбавитель" и "фармацевтически приемлемый эксципиент" означают, что носитель, разбавитель или эксципиент должен быть совместим с другими ингредиентами препарата и невредным для его реципиента. Используемый в данном описании термин "терапевтически эффективное количество" означает, что количество активного соединения или фармацевтического средства приводит к биологической или лечебной реакции в ткани, системе, организме животного или человека, которой добивается исследователь,ветеринар, лечащий врач или другой врач, которая включает облегчение симптомов заболевания, от которого лечат. Когда активное соединение (т.е., активный ингредиент) вводят в виде соли, ссылки на количество активного ингредиента относятся к форме свободной кислоты или основания соединения. Таким образом, настоящее изобретение относится к способу ингибирования РТРазы, включающему стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по настоящему изобретению. Изобретение также относится к фармацевтической композиции, включающей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по настоящему изобретению, достаточное для ингибирования РТРазы. Количество, ингибирующее РТРазу,может являться количеством, которое снижает или ингибирует активность РТРазы у субъекта. Соединение может включать его единственную кристаллическую форму или полиморфную кристаллическую форму или формы, аморфную форму, отдельный энантиомер, рацемическую смесь, отдельный стереизомер, смесь стереоизомеров, отдельный диастереизомер, смесь диастереоизомеров, изотопно обогащенную форму, сольват, фармацевтически приемлемую соль, сольват соли, пролекарство,поддающийся биогидролизу эфир или поддающийся биогидролизу амид. Кроме того, изобретение относится к фармацевтической композиции, включающей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по настоящему изобретению, достаточное для лечения непереносимости глюкозы. Кроме того, настоящее изобретение относится к фармацевтической композиции, включающей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по настоящему изобретению, достаточное для лечения диабета типа II. Соединения по настоящему изобретению можно вводить субъектам, нуждающимся в ингибировании активности РТРазы. К таким субъектам могут относиться, например, лошади, коровы, овцы, свиньи,мыши, собаки, кошки, приматы, такие как шимпанзе, горилла, макака-резус и люди. В одном варианте осуществления субъектом является человек, нуждающийся в ингибировании активности РТРазы. Фармацевтическая композиция, содержащая соединение по изобретению, может находиться в форме, подходящей для перорального применения, например в виде таблеток, пастилок, лепешек, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул или сиропов или масляных эликсиров. Композиции, предназначенные для перорального применения,можно получить согласно любому известному способу, и такие композиции могут содержать одно или несколько средств, выбранных из группы, состоящей из подслащивающих средств, корригентов, красителей и консервантов, для того, чтобы предоставить фармацевтически элегантные и приятные препараты. Таблетки могут содержать активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые подходят для получения таблеток. Такими эксципиентами могут являться,например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; агенты, способствующие грануляции и рассыпанию, например, кукурузный крахмал или альгиновая кислота; связывающие вещества, например крахмал, желатин или аравийская камедь; и смазывающие вещества, например, стеарат магния, стеариновая кислота или тальк. Такие таблетки могут быть без покрытия, или на них могут быть нанесены покрытия известными методами для замедления рассыпания и абсорбции в желудочно-кишечном тракте и обеспечения посредством этого пролонгированного действия в течение длительного времени. Например, можно использовать такой материал для замедления, как глицерилмоностеарат или глицерилдистеарат. На таблетки также можно нанести покрытие методами, формирующими осмотические лечебные таблетки для регулируемого высвобождения. Препараты для перорального применения также можно предоставить в виде твердых желатиновых капсул, когда активный ингредиент смешивают с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или мягких желатиновых капсул, когда активный ингредиент смешивают с водой или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом. Водные суспензии могут содержать активные соединения в смеси с эксципиентами, подходящими для получения водных суспензий. Такими эксципиентами являются суспендирующие вещества, например натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь; диспергирующие или смачивающие вещества могут представлять собой фосфатиды, встречающиеся в природе, такие как лецитин,или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными эфирами, образованными жирными кислотами и гекситом, такие как моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с неполными эфирами, образованными жирными кислотами и гекситол-ангидридами,например моноолеат полиоксиэтиленсорбитана. Водные суспензии также могут содержать один или несколько красителей, один или несколько корригентов, одно или несколько подслащивающих веществ,таких как сахароза или сахарин. Масляные суспензии можно получить суспендированием активного ингредиента в растительном масле, например арахисовом масле, оливковом масле, сезамовом масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Могут быть добавлены подслащивающие вещества, такие как перечисленные выше, и корригенты для того, чтобы предоставить приятный пероральный препарат. Такие композиции можно сохранить, добавляя антиоксидант, такой как аскорбиновая кислота. Диспергируемые порошки и гранулы, подходящие для получения водной суспензии путем добавления воды, предоставляют активное соединение в смеси с диспергирующим или смачивающим веществом, суспендирующим веществом и одним или несколькими консервантами. Подходящими диспергирующими или смачивающими веществами и суспендирующими веществами являются вещества, примеры которых уже указаны выше. Также могут присутствовать другие эксципиенты, например подслащивающие вещества, корригенты и красители. Фармацевтические композиции по изобретению также могут находиться в форме эмульсий "масло в воде". Масляная фаза может представлять собой растительное масло, например, оливковое масло или арахисовое масло, или минеральное масло, например жидкий парафин, или их смесь. Подходящие эмульгаторы могут представлять собой смолы, встречающиеся в природе, например аравийскую камедь или трагакантовую камедь, встречающиеся в природе фосфатиды, например соевый лецитин, и эфиры или неполные эфиры, полученные из жирных кислот и гекситол-ангидридов, например моноолеат сорбита, и продукты конденсации указанных неполных эфиров с этиленоксидом, например моноолеат полиоксиэтиленсорбита. Эмульсии также могут содержать подслащивающие вещества и корригенты. Сиропы и эликсиры можно получить с подслащивающими веществами, например глицерином,пропиленгликолем, сорбитом или сахарозой. Такие препараты также могут содержать средства, уменьшающие раздражение, консерванты и корригенты и красители. Фармацевтические композиции могут находиться в форме стерильной водной или масляной суспензии для инъекции. Такую суспензию можно получить согласно известным способам с использованием подходящих диспергирующих или смачивающих веществ и суспендирующих веществ, описанных выше. Стерильный препарат для инъекции также может представлять собой стерильный раствор или суспензию для инъекции в нетоксичном парентерально приемлемом разбавителе или растворителе, например раствор в 1,3-бутандиоле. В числе приемлемых сред и растворителей, которые можно использовать, находятся вода, раствор Рингера и изотоничный раствор хлорида натрия. Кроме того, в качестве растворителя или суспензионной среды обычно используют стерильные нелетучие масла. Для такой цели можно использовать любые мягкие нелетучие масла с использованием синтетических моно- и диглицеридов. Кроме того, при получении препаратов для инъекции находят применение жирные кислоты, такие как олеиновая кислота. Композиции также могут находиться в форме суппозиториев для ректального введения соединений по изобретению. Такие композиции можно получить, смешивая лекарственное средство с подходящим невызывающим разражение эксципиентом, который является твердым при обычных температурах, но жидким при ректальной температуре и, таким образом, будет плавиться в прямой кишке, высвобождая лекарственное средство. Такие материалы включают, например, масло какао и полиэтиленгликоли. Для местного применения рассматривают кремы, мази, желе, растворы или суспензии и т.д., содержащие соединения по изобретению. Для целей такого применения местное применение будет включать жидкости для полоскания рта и жидкости для полоскания горла. Соединения по настоящему изобретению также можно вводить в форме липосомных систем доставки, таких как маленькие однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Липосомы можно сформировать из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Настоящее изобретение также относится к пролекарствам по изобретению. Фармацевтически приемлемые соли соединений по настоящему изобретению, в структуре которых присутствует основная или кислотная группа, также входят в объем изобретения. Термин "фармацевтически приемлемые соли" относится к нетоксичным солям соединений по изобретению, которые, как правило, получают взаимодействием свободного основания с подходящей органической или неорганической кислотой или взаимодействием свободной кислоты с подходящим органическим или неорганическим основанием. Характерные соли включают следующие соли: ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат,борат, бромид эдетат кальция, камзилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат,здизилат, эстолат, эзилат, фумарат, глуцептат, глюконат, глутамат, гликолиларсанилат, гексилрезорцинат, соль гидрабамина, гидробромид, гидрохлорид, гидроксинафтоат, иодид, изэтионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, монокалиймалеат, мукат, напсилат, нитрат, соль N-метилглюкамина, оксалат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, калиевую соль, салицилат, натриевую соль, стеарат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид, соль триметиламмония и валерат. Когда присутствует кислотный заместитель, такой как -СООН, для примерения в качестве лекарственной формы можно получить соль аммония, морфолиния, натрия, калия, бария, кальция и т.п. Когда присутствует основная группа, такая как амино, или основной гетероарильный радикал, такой как пиридил, можно получить соль присоединения кислоты, такую как гидрохлорид, гидробромид, фосфат, сульфат, трифторацетат, трихлорацетат, ацетат, оксалат, малеат, пируват, малонат, сукцинат, цитрат, тартрат, фумарат,манделат, бензоат, циннамат, метансульфонат, этансульфонат, пикрат и т.п., и в том числе кислот, соли присоединения которых относятся к фармацевтически приемлемым солям, перечисленным в Journal ofPharmaceutical Science, 66, 2 (1977), p. 1-19. Используемый в данном описании термин "другие фармацевтически приемлемые противоионы" относится к нетоксичным противоионам, таким как, но без ограничения, аммоний, морфолиний, ион натрия, калия, бария, кальция и т.п., и которые можно ввести взаимодействием протона производного тиадиазолидина с соответствующим органическим или неорганическим основанием. Другие соли, которые не являются фармацевтически приемлемыми, могут применяться при получении соединений по изобретению, и они составляют другой аспект изобретения. Кроме того, некоторые соединения по настоящему изобретению могут образовывать сольваты с водой или обычными органическими растворителями. Такие сольваты также охватываются объемом изобретения. Таким образом, в другом варианте осуществления изобретение относится к фармацевтической композиции, включающей соединение по настоящему изобретению или его фармацевтически приемлемую соль, и один или несколько фармацевтически приемлемых носителей, эксципиентов или разбавителей. Соединения по настоящему изобретению могут селективно действовать как ингибиторы одной РТРазы преимущественно перед одной или несколькими другими РТРазами и поэтому могут иметь преимущество при лечении заболевания, опосредованного одной или несколькими РТРазами предпочтительно относительно других. Примеры Общие процедуры, используемые в способах по настоящему изобретению, описаны ниже. Общие экспериментальные положения Данные ЖХ-МС получают с использованием элюирования с градиентом на регуляторе Waters 600,снабженном двухволновым детектором 2487 и автоматическим устройством для ввода проб Leap Technologies HTS PAL Autosampler, с использованием колонки YMC Combiscreen ODS-A, 504,6 мм. При трехминутном градиенте проходят от 25% В (97,5% ацетонитрила, 2,5% воды, 0,05% ТФК) и 75% А(97,5% воды, 2,5% ацетонитрила, 0,05% ТФК) до 100% В. Используемым масс-спектрометром является прибор Micromass ZMD. Все данные получают в положительном варианте, если не указано иное. Даные 1 Н ЯМР получают на спекторометре Varian 400 МГц. Общая процедура В. Арилирование атома азота имидазола с использованием арилфторида/арилбромида К раствору имидазола (1 экв.) в безводном ДМФА (0,1-0,5 М) добавляют соответствующий активированный арилфторид или арилбромид (1,5 экв.), а затем Cs2CO3 (3 экв.). Затем реакционную смесь греют при 120 С в атмосфере азота в течение 2 ч. По завершении периода нагревания реакционную смесь разбавляют смесью вода/EtOAc и слои разделяют. Водный слой повторно экстрагируют EtOAc, и органические слои объединяют и промывают водой и рассолом. Затем органическую фазу сушат над Na2SO4,фильтруют, фильтрат концентрируют и очищают хроматографией на силикагеле и получают арилимидазольное производное. Общая процедура D. Восстановление арилнитрогруппы К суспензии арилнитросоединения (1 экв.) в НОАс (0,1-0,5 M) добавляют железный порошок (-325 меш, 8 экв.) и затем смесь греют при 80 С в атмосфере азота в течение 5-10 мин. Затем реакционную смесь разбавляют водой и EtOAc и оставшийся железный порошок отфильтровывают и промываютEtOAc. Объединенные органические слои промывают водой, насыщенным раствором NaHCO3 и рассолом. Затем органический слой сушат над Na2SO4, фильтруют, фильтрат концентрируют и очищают хроматографией на силикагеле, и получают производное анилина. Общая процедура Е. Алкилирование анилина Способ I) К суспензии производного анилина (1 экв.) в безводном ДМФА (0,1-0,5 М) при комнатной температуре добавляют 1,2 экв. -бромированного эфира, а затем 2,5 экв. DIEA. Затем реакционную смесь перемешивают при комнатной температуре в атмосфере азота в течение 18 ч. Затем реакционную смесь разбавляют водой и EtOAc и слои разделяют. Водный слой повторно экстрагируют EtOAc и органические слои объединяют и промывают водой и рассолом. Затем органическую фазу сушат над Na2SO4,фильтруют, фильтрат концентрируют и очищают хроматографией на силикагеле и получают производное -анилинового эфира. Способ II) К суспензии производного анилина (1 экв.) в безводном ДМФА (0,1-0,5 М) при комнатной температуре добавляют 2 экв. -бромированного эфира, а затем 5 экв. безводного трет-бутоксида калия. Затем реакционную смесь перемешивают при 100 С в атмосфере азота в течение 18 ч. Затем реакционную смесь разбавляют водой и EtOAc и слои разделяют. Водный слой повторно экстрагируютEtOAc и органические слои объединяют и промывают водой и рассолом. Затем органическую фазу сушат над Na2SO4, фильтруют, фильтрат концентрируют и очищают хроматографией на силикагеле, и получают производное -анилинового эфира. Общая процедура F. Получение 1,1-диоксида 1,2,5-тиадиазолидин-3-она К раствору хлорсульфонилизоцианата (1,5 экв.) в безводном 1,2-дихлорэтане (0,1-0,5 М) при 0 С добавляют 1,5 экв. трет-бутанола в виде раствора в безводном 1,2-дихлорэтане (0,5 М). Смеси дают нагреться до комнатной температуры при перемешивании и затем снова охлаждают до 0 С. Суспензию анилинэфира, полученного так, как описано в общей поцедуре Е (1,0 экв.), в 1,2-дихлорэтане (0,3-0,5 M) и 2,5 экв. DIEA охлаждают до 0 С и добавляют по каплям при перемешивании смесь хлорсульфонилизоцианат-трет-бутанол. Смесь перемешивают при комнатной температуре в течение 1 ч, затем разбавляют водой и CH2Cl2, и слои разделяют. Органические слои объединяют и промывают водой и рассолом. Затем органическую фазу сушат над Na2SO4, фильтруют, фильтрат концентрируют и очищают хроматографией на силикагеле, и получают анилин-N-Boc-сульфамидное производное. Содержащий защитную группу Boc сульфамид перемешивают в смеси дихлорметан/трифторуксусная кислота в течение 30 мин. Растворитель удаляют, остаток несколько раз растирают с эфиром, и получают сульфамид без защитной группы. К суспензии производного сульфамида без защитной группы в этаноле (0,1-0,5 М) добавляют 0,5 экв. NaOH в виде 2 М раствора в воде. Смесь перемешивают при комнатной температуре в течение 5-7 мин, затем разбавляют 2% лимонной кислотой и EtOAc и слои разделяют. Органический слой промывают водой и рассолом. Затем органический слой сушат над Na2SO4, фильтруют, фильтрат концентрируют и очищают хроматографией на силикагеле и получают 1,1-диоксид 1,2,5-тиадиазолидин-3-она. Общая процедура G. Сочетание по Судзуки К раствору бромированного соединения (1 экв.) в смеси толуола и этанола 2:1 (0,1-0,5 М) добавляют соответствующую бороновую кислоту (2 экв.) и каталитическое кооличество тетракис(трифенилфосфин)палладия(0) (0,1 экв.), а затем 2 M раствор карбоната натрия в воде (30 экв.). Реакционную смесь перемешивают при 90 С в атмосфере азота в течение 6 ч. После охлаждения реакционную смесь разбавляют водой и экстрагируют этилацетатом. Органический экстракт промывают водой и рассолом и сушат над Na2SO4. После удаления растворителя в вакууме остаток очищают колоночной флэш-хроматографией, и получают нужное соединение. Общая процедура Н. Алкилирование фенольного атома кислорода гидроксифенилбороновой кислоты К раствору гидроксифенилбороновой кислоты (1 экв.) в безводном ДМФА (0,1-0,5 М) добавляют алкилбромид или мезилат (2 экв.), а затем свежеизмельченный K2CO3 (4 экв.) и реакционную смесь греют при 100 С в атмосфере азота в течение 6 ч. Затем смесь разбавляют водой и EtOAc и слои разделяют. Водный слой дополнительно экстрагируют EtOAc и органические слои объединяют и сушат над Na2SO4. Растворитель удаляют в вакууме, остаток очищают хроматографией на силикагеле и получают нужный продукт. Общая процедура M. Получение 1(3-аминофенил)имидазола К смеси 3-аминоацетанилида (1 экв.) и ароматического ацилбромида (2 экв.) в безводном ДМФА(0,1-0,5 М) добавляют NaHCO3 (5 экв.). Реакционную смесь перемешивают при комнатной температуре в атмосфере азота в течение 2 ч. По завершении периода перемешивания реакционную смесь разбавляют водой и EtOAc и слои разделяют. Водный слой экстрагируют EtOAc и органические слои объединяют и промывают водой и рассолом. Затем органический раствор сушат над Na2SO4, фильтруют, фильтрат концентрируют и остаток сушат и используют непосредственно на следующей стадии. Проиводное анилина, полученное выше, растворяют в сухом ДМФА (0,1-0,5 М) и добавляют 5 экв.NaHCO3. При перемешивании в атмосфере азота на льду добавляют по каплям раствор пбромфенилацетилхлорида (1,1 экв.) в бензоле. Затем смесь перемешивают в течение 1 ч и оставляют нагреваться до комнатной температуры. Реакционную смесь разбавляют водой и EtOAc и слои разделяют. Водный слой экстрагируют EtOAc и органические слои объединяют и промывают водой и рассолом. Затем органический раствор сушат над Na2SO4, фильтруют, фильтрат концентрируют и остаток сушат и используют непосредственно на следующей стадии. Третичный анилид, полученный выше, растворяют в ледяной уксусной кислоте (0,1-0,5 М) и добавляют ацетат аммония (10 экв.). Затем смесь греют при 110 С в атмосфере азота в течение 8 ч. По завершении периода нагревания смесь выливают в воду, нейтрализуют насыщенным раствором бикарбоната натрия и экстрагируют этилацетатом. Органический экстракт промывают водой и рассолом и сушат надNa2SO4. После удаления растворителя в вакууме остаток очищают колоночной хроматографией на силикагеле и получают нужное N-(3-ацетамидо)фенильное производное имидазола. Промежуточное соединение, полученное выше, растворяют в смеси 6:1 (об./об.) 4 М HCl/диоксан и воды (0,1-0,5 М) и греют при 70C в течение 3 ч. Затем смесь выливают в воду, нейтрализуют насыщенным раствором бикарбоната натрия и экстрагируют этилацетатом. Органический экстракт промывают водой и рассолом и сушат над Na2SO4. После удаления растворителя в вакууме остаток сушат и используют без дополнительной очистки. Общая процедура R. Сочетание по Ульману К раствору кислород- или азотсодержащего нуклеофила (1 экв.) в безводном NMP (0,1-0,5 М) добавляют соответствующий арилбромид или иодид (1,5 экв.), а затем CuCl (0,2 экв.), 2,2,6,6-тетраметил 3,5-гептандион (0,2 экв.) и Cs2CO3 (3 экв.). Затем реакционную смесь греют при 120 С в атмосфере азота в течение 6-8 ч. По завершении периода нагревания реакционную смесь разбавляют водой и EtOAc и слои разделяют. Водный слой повторно экстрагируют EtOAc и органические слои объединяют и промывают водой и рассолом. Затем органическую фазу сушат над Na2SO4, фильтруют, фильтрат концентрируют и очищают хроматографией на силикагеле и получают нужный продукт (диарилэфирное или заме- 15019385 щенное производное анилина). Общая процедура S. Окисление сульфида с использованием перуксусной кислоты К раствору простого тиоэфира (1 экв.) в смеси DCM/HOAc (1:1, 0,1-0,5 М) добавляют перуксусную кислоту (32 мас.% раствор в уксусной кислоте, 10 экв.) при 0 С и раствор перемешивают при той же температуре в течение получаса. По завершении периода перемешивания реакционную смесь разбавляют водой и EtOAc. Объединенные органические слои промывают водой, насыщенным растворомNaHCO3 и рассолом. Затем органическую фазу сушат над Na2SO4, фильтруют и концентрируют. Остаток растирают несколько раз в гексане и получают нужное сульфоновое производное. Общая процедура Т: окисление сульфида пероксидом водорода К раствору суспензии простого тиоэфира (1 экв.) в метаноле (0,1-0,5 М) добавляют пероксид водорода (50% раствор в воде, 10 экв.) при 0 С и реакционную смесь перемешивают при комнатной температуре в течение часа. По завершении периода перемешивания реакционную смесь разбавляют водой иEtOAc. Объединенные органические слои промывают водой и рассолом. Затем органическую фазу сушат над Na2SO4, фильтруют и концентрируют. Остаток растирают несколько раз в гексане и получают нужное сульфоксидное производное. Общая процедура V. Удаление трет-бутилкарбаматной группы Соединение, содержащее защитную группу Boc, перемешивают в растворе 4 н HCl/диоксан в течение 1 ч. Растворитель удаляют, продукт реакции несколько раз растирают в эфире и получают нужное соединение. Общая процедура Y. Получение бороновой кислоты К суспензии м- или п-бромбензальдегида в ТГФ (0,1-0,5 М) добавляют по каплям алкилмагнийбромид или хлорид в эфире или ТГФ (1,1 экв.) при перемешивании на льду. Смесь оставляют нагреваться до комнатной температуры и перемешивают в течение 30 мин, затем выливают в смесь насыщенный раствор NH4Cl/EtOAc и слои разделяют. Водный слой экстрагируют EtOAc и органические слои объединяют и промывают водой и рассолом. Органическую фазу сушат над Na2SO4, фильтруют и фильтрат концентрируют и остаток сушат и используют непосредственно на следующей стадии. Продукт реакции Гриньяра, полученный выше, растворяют в трифторуксусной кислоте (0,1-0,5 М) в атмосфере азота и охлаждают на ледяной бане с солью при -10 С. К раствору медленно добавляют триэтилсилан (4 экв.) с тем, чтобы температура бани оставалась -10 С. Затем раствор нагревают до комнатной температуры и перемешивают в течение 3 ч. По завершении периода перемешивания смесь выливают в воду и экстрагируют дихлорметаном. Органический экстракт промывают насыщенным водным раствором NaHCO3 и рассолом и сушат над Na2SO4. После удаления растворителя в вакууме остаток очищают колоночной хроматографией на силикагеле и получают нужный продукт м- или пбромфенилалкан. Раствор м- или п-бромфенилалкана в сухом ТГФ (0,1-0,5 M) перемешивают в атмосфере азота на бане с сухим льдом и ацетоном и к смеси на бане с сухим льдом и ацетоном добавляют по каплям вторбутиллитий в циклогексане (2 экв.). Смесь перемешивают 10 мин и затем добавляют по каплям триизопропилборат (3 экв.). Смесь перемешивают еще 30 мин и затем выливают в избыток 2 M HCl при комнатной температуре. Затем добавляют EtOAc и слои разделяют. Водный слой экстрагируют EtOAc и органические слои объединяют, промывают водой и рассолом. Органический раствор сушат над Na2SO4,фильтруют и фильтрат концентрируют. Остаток перекристаллизовывают из EtOAc/гексана, сушат и используют в реакциях сочетания с бороновой кислотой. Общая процедура АА. Восстановление бензилового спирта Спирт, полученный по общей процедуре Z, растворяют в трифторуксусной кислоте (0,1-0,5 M) в атмосфере азота и охлаждают на ледяной бане с солью при -10 С. К раствору медленно добавляют триэтилсилан (4 экв.) с тем, чтобы температура бани оставалась -10 С. Затем раствор нагревают до комнатной температуры и перемешивают в течение 3 ч. По завершении периода перемешивания смесь выливают в воду и экстрагируют дихлорметаном. Органический экстракт промывают насыщенным водным раствором NaHCO3 и рассолом и сушат над Na2SO4. После удаления растворителя в вакууме остаток очищают колоночной хроматографией на силикагеле и получают нужный продукт м- или пбромфенилалкан. Общая процедура AE. Получение 3- или 4-(алкилен)фенилбороновой кислоты К суспензии м- или п-бромбензальдегида в ТГФ (0,1-0,5 М) при перемешивании на льду добавляют по каплям раствор алкилмагнийбромида или хлорида в эфире или ТГФ (1,1 экв.). Смесь нагревают до комнатной температуры и перемешивают в течение 30 мин, затем выливают в смесь насыщенный раствор NH4Cl/EtOAc и слои разделяют. Водный слой экстрагируют EtOAc и органические слои объединяют, промывают водой и рассолом. Затем органический раствор сушат над Na2SO4, фильтруют и фильтрат концентрируют и остаток сушат и используют непосредственно на следующей стадии. Продукт реакции Гриньяра, полученный выше, растворяют в сухом дихлорметане (0,1-0,5 М) и добавляют N,N-диизопропилэтиламин (4 экв.). Смесь охлаждают на льду и добавляют по каплям метансульфонилхлорид (2 экв.). Смесь нагревают до комнатной температуры и перемешивают в течение 18 ч. По завершении периода перемешивания смесь выливают в воду и экстрагируют дихлорметаном. Орга- 16019385 нический экстракт промывают водой и рассолом и сушат над Na2SO4. После удаления растворителя в вакууме остаток очищают колоночной хроматографией на силикагеле и получают нужный продукт мили п-бромфениленалкен. Раствор м- или п-бромфенилалкена в сухом ТГФ (0,1-0,5 М) перемешивают в атмосфере азота на бане с сухим льдом и ацетоном и добавляют по каплям втор-бутиллитий в циклогексане (2 экв.). Смесь перемешивают 10 мин и затем добавляют по каплям изопропилборат (3 экв.). Смесь перемешивают еще 30 мин и затем выливают в избыток 2 M HCl и перемешивают 20 мин. Затем добавляют EtOAc и слои разделяют. Водный слой экстрагируют EtOAc и органические слои объединяют и промывают водой и рассолом. Органический раствор сушат над Na2SO4, фильтруют и фильтрат концентрируют. Остаток перекристаллизовывают из EtOAc/гексана, сушат и используют в реакциях сочетания с бороновой кислотой. Пример 1. 3-[2-(4-Бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фениламин (получен так, как описано в общей процедуре M) (28,4 г, 60 ммоль) обрабатывают так, как описано в общих процедурах E-F, и получают 1,1-диоксид 5-3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 593 (M+Н)+; 1H ЯМР (ДМСО-d6, 400 МГц):4,05 (с, 2 Н), 4,07 (с, 2 Н), 7,15 (д, 2 Н), 7,27 (д, 2 Н), 7,47-7,53 (м, 5 Н),7,64 (д, 1 Н), 7,89 (с, 1 Н), 8,19 (д, 1 Н) м.д. 1,1-Диоксид 5-3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она (592 мг, 1 ммоль) обрабатывают так, как описано в общей процедуре G, с использованием 4 циклогексилфенилбороновой кислоты (408 мг, 2 ммоль), и получают 1,1-диоксид 5-3-[2-(4'циклогексилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 671 (М+Н)+; 1 Н ЯМР (ДМСО-d6, 400 МГц):1,22-1,43 (м, 5 Н), 1,69-1,81 (м, 5 Н), 2,52 (м, 1 Н), 4,05 (с, 2 Н), 4,07 (с,2 Н), 7,15 (д, 2 Н), 7,18 (д, 2 Н), 7,27 (д, 2 Н), 7,37 (д, 2 Н), 7,46-7,54 (м, 5 Н), 7,64 (д, 1 Н), 7,89 (с, 1 Н), 8,19 (д,1 Н) м.д. Названное выше соединение также можно получить с использованием описанной далее процедуры,исходя из 3-нитроанилина. Метилбромацетат (6,77 мл, 73,5 ммоль) добавляют к смеси 3-нитроанилина(9,21 г, 66,7 ммоль), NaHCO3 (14 г, 167 ммоль) и диметилформамида (75 мл). Смесь перемешивают при 70-75 С в течение 3 ч и затем охлаждают до комнатной температуры. К реакционной смеси добавляют воду (600 мл) и продукт реакции собирают фильтрацией и 3 раза промывают водой порциями по 50 мл. Желтый продукт метиловый эфир (3-нитрофениламино)уксусной кислоты сушат при пониженном давлении, и получают 12,4 г (88%) вещества. К раствору трет-бутилового спирта (9,61 мл, 100 ммоль) в дихлорметане (48,2 мл) при 0 С добавляют по каплям хлорсульфонилизоцианат (8,68 мл, 100 ммоль). Смесь нагревают до комнатной температуры и перемешивают в течение 30 мин. Затем 37,5 мл полученного раствора при 0 С добавляют по каплям к смеси метилового эфира (3-нитрофениламино)уксусной кислоты (10,5 г, 50 ммоль), диизопропилэтиламина (26 мл, 150 моль) и дихлорметана (50 мл). По окончании добавления смесь нагревают до комнатной температуры и промывают водой (2500 мл). Экстракт сушат (MgSO4) и фильтруют. Экстракт,содержащий Boc-защищенный сульфамид, разбавляют до общего объема 300 мл, добавляя дихлорметан. К раствору при 0 С добавляют трифторуксусную кислоту, и раствор перемешивают в течение 3 ч или до тех пор, пока ТСХ не покажет отсутствие исходного вещества. Раствор промывают водой (3500 мл), и продукт реакции высаживают, добавляя гексан, и собирают фильтрацией. Продукт промывают 5% раствором дихлорметана в гексане (50 мл) и сушат при пониженном давлении, и получают 10,5 г продукта. К суспензии вещества, из которого удалена защитная группа Boc (10,5 г, 36,3 ммоль), в этаноле (75 мл) при 0C при быстром перемешивании добавляют раствор NaOH (2 М, 2,0 эквивалента) до тех пор,пока не образуется густой осадок (2 мин). Смесь подкисляют до рН 5,0, добавляя 6,0 M соляную кислоту. Продукт реакции 1,1-диоксо-5-(3-нитрофенил) [1,2,5]тиадиазолидин-3-он собирают фильтрацией и промывают диэтиловым эфиром (50 мл). Продукт сушат при пониженном давлении и получают циклизованный продукт (8,5 г, 91%). Раствор нитросоединения (8,5 г) в метаноле (40 мл) обрабатывают 1,7 г катализатора 10% Pd/C при атмосферном давлении водорода в течение 18 ч. Катализатор удаляют фильтрацией. Раствор концентрируют и выкристаллизовывают 5-(3-аминофенил)-1,1-диоксо[1,2,5]тиадиазолидин-3-он, добавляя дихлорметан, собирают продукт фильтрацией и сушат, и получают 6,9 г продукта (выход 92%). К раствору 5-(3-аминофенил)-1,1-диоксо[1,2,5]тиадиазолидин-3-она (5 г, 22 ммоль) в 37,5 мл смеси ацетонитрил-вода (9:1, об./об.) добавляют NaHCO3 (4,62 г, 55 ммоль). Смесь перемешивают при 25 С,добавляя в то же время 2-бром-1-(2,4-дихлорфенил)этанон (7,1 г, 26,4 ммоль). Смесь перемешивают в течение 12-16 ч при 35 С. Смесь охлаждают на ледяной бане для дальнейшего осаждения продукта, который затем собирают фильтрацией. Фильтрат концентрируют при пониженном давлении и собирают вторую порцию сырого продукта. Объединенный сырой продукт реакции промывают один раз 25 мл во- 17019385 ды и сушат, затем промывают один раз 10 мл метил-трет-бутилового эфира и сушат, и получают 6,36 г(выход 70%) 5-3-[2-(2,4-дихлорфенил)-2-оксоэтиламино]фенил-1,1-диоксо-1,2,5-тиадиазолидин-3-она. К смеси фениламина (9,4 г, 23 ммоль) и NaHCO3 (5,04 г, 60 ммоль) в дихлорметане (30 мл) постепенно добавляют раствор в дихлорметане (10 мл) (4'-циклогексилбифенил)ацетилхлорида (34 ммоль),который получают взаимодействием оксалилхлорида с (4'-циклогексилбифенил-4-ил)уксусной кислотой(10 г, 34 ммоль), которую, в свою очередь, получают из (4-циклогексилфенил)бороновой кислоты и 4 бромфенилуксусной кислоты сочетанием по Судзуки. Смесь перемешивают при 25 С в течение 9 ч. Затем смесь разбавляют тетрагидрофураном, промывают разбавленным рассолом, сушат над сульфатом натрия и упаривают. Продукт реакции 2-(4'-циклогексилбифенил-4-ил)-N-[2-(2,4-дихлорфенил)-2 оксоэтил]-N-[3-(1,1,4-триоксо[1,2,5]тиадиазолидин-2-ил)фенил]ацетамид очищают колоночной хроматографией на силикагеле с использованием смесей 1-8% метанола в дихлорметане. К раствору ацетата аммония (223 мг, 2,9 ммоль) в уксусной кислоте (0,5 мл) добавляют полученный выше третичный амид (200 мг, 0,29 ммоль) в диметилформамиде (0,5 мл) и затем смесь перемешивают при 90 С в течение 18 ч, после чего смесь разбавляют тетрагидрофураном и 5 раз промывают разбавленным рассолом, сушат над сульфатом натрия и упаривают досуха. Сырое твердое вещество обильно промывают метанолом и получают 140 мг чистого названного выше продукта. Пример 2. 1,1-Диоксид 5-3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она (59 мг, 0,1 ммоль) обрабатывают так, как описано в общей процедуре G, с использованием 4-трет-бутилфенилбороновой кислоты (36 мг, 0,2 ммоль), и получают 1,1-диоксид 5-3-[2-(4'-третбутилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 645 (М+Н)+; 1 Н ЯМР (ДМСО-d6, 400 МГц):1,30 (с, 9 Н), 4,06 (с, 2 Н), 4,08 (с, 2 Н), 7,15 (д, 2 Н), 7,19 (д, 2 Н), 7,29(д, 2 Н), 7,38 (д, 2 Н), 7,46-7,54 (м, 5 Н), 7,64 (д, 1 Н), 7,90 (с, 1 Н), 8,19 (д, 1 Н) м.д. Пример 3. 1,1-Диоксид 5-3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она (59 мг, 0,1 ммоль) обрабатывают так, как описано в общей процедуре G, с использованием 3-(3,3-диметилбутокси)фенилбороновой кислоты (45 мг, 0,2 ммоль, получена согласно общей процедуре Н), и получают 1,1-диоксид 5-(3-4-(2,4-дихлорфенил)-2-[3'-(3,3'-диметилбутокси)бифенил-4-илметил]имидазол-1-илфенил)-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 689 (M+Н)+; 1H ЯМР (ДМСО-d6, 400 МГц):0,97 (с, 9 Н), 1,67 (т, 2 Н), 4,05 (с, 2 Н), 4,07 (т, 2 Н), 4,08 (с, 2 Н), 7,057,12 (м, 4 Н), 7,29 (д, 2 Н), 7,38 (д, 2 Н), 7,46-7,54 (м, 5 Н), 7,64 (д, 1 Н), 7,90 (с, 1 Н), 8,19 (д, 1 Н) м.д. Способами, аналогичными способам, используемым для получения соединения примера 3, синтезируют соединения, перечисленные далее.(д, 2 Н), 7,21 (д, 2 Н), 7,29-7,41 (м, 4 Н), 7,49-7,58 (м, 5 Н), 7,63 (д, 1 Н), 7,88 (с, 1 Н), 8,18 (д, 1 Н) м.д. Пример 23. 1,1-Диоксид 5-(3-4-(2,4-дихлорфенил)-2-[3'-(3,3-диметилбутилсульфанил)бифенил-4-илметил]имидазол-1-илфенил)-1,2,5-тиадиазолидин-3-она (14 мг, 0,02 ммоль) обрабатывают так, как описано в общей процедуре Т, и получают 1,1-диоксид 5-(3-4-(2,4-дихлорфенил)-2-[3'-(3,3-диметилбутан-1 сульфинил)бифенил-4-илметил]имидазол-1-илфенил)-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 721 (М+Н)+; 1H ЯМР (ДМСО-d6, 400 МГц):0,86 (с, 9 Н), 1,45 (м, 2 Н), 2,98 (м, 2 Н),4,05 (с, 2 Н), 4,08 (с, 2 Н), 7,18 (д, 2 Н), 7,21 (д, 2 Н), 7,26-7,38 (м, 4 Н), 7,47-7,56 (м, 5 Н), 7,63 (д, 1 Н), 7,88 (с,1 Н), 8,18 (д, 1 Н) м.д. Пример 24. 1,1-Диоксид 5-3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она (119 мг, 0,2 ммоль) обрабатывают так, как описано в общей процедуре G, с использованием 3(циклогексилметилсульфанил)фенилбороновой кислоты (100 мг, 0,4 ммоль, получена согласно общей процедуре I), и получают 1,1-диоксид 5-3-[2-(3'-циклогексилметилсульфанилбифенил-4-илметил)-4(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 717 (М+Н)+; 1H ЯМР (CD3OD, 400 МГц):1,00-1,86 (м, 11 Н), 3,03 (м,2 Н), 4,05 (с, 2 Н), 4,08 (с, 2 Н), 7,18 (д, 2 Н),7,21 (д, 2 Н), 7,27-7,39 (м, 4 Н), 7,46-7,58 (м, 5 Н), 7,63 (д, 1 Н), 7,88 (с, 1 Н), 8,18 (д, 1 Н) м.д. Пример 27. 1,1-Диоксид 5-3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она (119 мг, 0,2 ммоль) обрабатывают так, как описано в общей процедуре G, с использованием 3(циклогексилэтилсульфанил)фенилбороновой кислоты (106 мг, 0,4 ммоль, получена согласно общей процедуре I), и получают 1,1-диоксид 5-3-[2-(3'-(2-циклогексилэтилсульфанил)бифенил-4-илметил)-4(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 731 (М+Н)+; 1(м, 4 Н), 7,37-7,47 (м, 4 Н), 7,51-7,59 (м, 5 Н), 7,64 (д, 1 Н), 7,89 (с, 1 Н), 8,19 (д, 1 Н) м.д. Пример 29. 1,1-Диоксид 5-3-[2-(3'-(2-циклогексилэтилсульфанил)бифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она (15 мг, 0,02 ммоль) обрабатывают так, как описано в общей процедуре Т, и получают 1,1-диоксид 5-3-[2-(3'-(2-циклогексилэтансульфинил)бифенил-4 илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 747 (М+Н)+; 1 Н ЯМР (CD3OD, 400 МГц):0,85-1,72 (м, 13 Н), 3,07 (м, 2 Н), 4,05 (с, 2 Н),4,08 (с, 2 Н), 7,21-7,28 (м, 4 Н), 7,37-7,44 (м, 4 Н), 7,49-7,58 (м, 5 Н), 7,64 (д, 1 Н), 7,88 (с, 1 Н), 8,18 (д, 1 Н) м.д. Пример 30. 1,1-Диоксид 5-3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она (119 мг, 0,2 ммоль) обрабатывают так, как описано в общей процедуре G, с использованием 3(фенилэтилсульфанил)фенилбороновой кислоты (104 мг, 0,4 ммоль, получена согласно общей процедуреH ЯМР (ДМСО-d6, 400 МГц):2,89 (т, 2 Н), 3,30 (т, 2 Н), 4,07 (с, 2 Н), 4,09 (с, 2 Н), 7,18-7,22 (м, 5 Н),7,25-7,32 (м, 6 Н), 7,37-7,49 (м, 4 Н), 7,53 (м, 1 Н), 7,57 (д, 2 Н), 7,64 (д, 1 Н), 7,90 (с, 1 Н), 8,20 (д, 1 Н) м.д. Пример 31. 2-(4-Бромбензил)-4-(2,4-дихлорфенил)-1 Н-имидазол (3,9 г, 10 ммоль) обрабатывают так, как описано в общей процедуре В, с использованием 1-иод-3-нитробензола, и получают 2-(4-бромбензил)-1-(3 нитрофенил)-4-(2,4-дихлорфенил)-1 Н-имидазол. 2-(4-Бромбензил)-4-(2,4-дихлорфенил)-1-(3-нитрофенилфенил)-1 Н-имидазол (1,3 г, 2,5 ммоль) восстанавливают до амина и вводят во взаимодействие с метилбромацетатом, следуя общим процедурам D и Е, и получают метиловый эфир 3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты. Метиловый эфир 3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты (55 мг, 0,1 ммоль) вводят в сочетание с 4-н-бутилфенилбороновой кислотой (36 мг, 0,2 ммоль) согласно общей процедуре G, и получают метиловый эфир 3-[2-(4'-бутилбифенил-4-илметил)-4-(2,4 дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты. Метиловый эфир 3-[2-(4'-бутилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты (30 мг, 0,05 ммоль) обрабатывают, следуя общей процедуре F, и получают 1,1 диоксид 5-3-[2-(4'-бутилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 645 (М+Н)+; 1(с, 2 Н), 7,11-7,18 (м, 4 Н), 7,26 (д, 2 Н), 7,37 (д, 2 Н), 7,45-7,53 (м, 5 Н), 7,61 (д, 1 Н), 7,88 (с, 1 Н), 8,19 (д, 1 Н) м.д. Пример 32. Метиловый эфир 3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты (55 мг, 0,1 ммоль) вводят в сочетание с 4-изобутилфенилбороновой кислотой (36 мг, 0,2 ммоль) согласно общей процедуре G и получают метиловый эфир 3-[2-(4'-изобутилбифенил-4-илметил)-4-(2,4 дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты. Метиловый эфир 3-[2-(4'-изобутилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты (20 мг, 0,033 ммоль) обрабатывают, следуя общей процедуре F, и получают 1,1-диоксид 5-3-[4-(2,4-дихлорфенил)-2-(4'-изобутилбифенил-4-илметил)имидазол-1-ил]фенил-1,2,5 тиадиазолидин-3-она. ЖХ-МС: m/z 645 (М+Н)+; 1 Пример 39. Метиловый эфир 3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты (55 мг, 0,1 ммоль) обрабатывают так, как описано в общей процедуре G, с использованием 4(циклогексилметилсульфанил)фенилбороновой кислоты (50 мг, 0,2 ммоль, получена согласно общей процедуре I), и получают метиловый эфир 3-[2-(4'-циклогексилметилсульфанилбифенил-4-илметил)-4(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты. Метиловый эфир 3-[2-(4'-циклогексилметилсульфанилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты (23 мг, 0,033 ммоль) обрабатывают, следуя общей процедуре F, и получают 1,1-диоксид 5-3-[2-(4'-циклогексилметилсульфанилбифенил-4-илметил)-4-(2,4 дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 717 (М+Н)+; 1H ЯМР (ДМСО-d6, 400 МГц):0,99-1,21 (м, 6 Н), 1,47 (м,1 Н), 1,57-1,88H ЯМР (ДМСО-d6, 400 МГц):1,00-1,25 (м, 6 Н), 1,52 (м,1 Н), 1,58-1,87 (м, 4 Н), 3,21 (д, 2 Н), 4,04 (с,2 Н), 4,09 (с, 2 Н), 7,17 (д, 2 Н), 7,21 (д, 2 Н),7,27-7,41 (м, 4 Н), 7,44-7,58 (м, 5 Н), 7,64 (д, 1 Н), 7,87 (с, 1 Н),8,19 (д, 1 Н) м.д. Пример 41. Метиловый эфир 3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты (55 мг, 0,1 ммоль) обрабатывают так, как описано в общей процедуре G, с использованием 3(циклогексилэтокси)фенилбороновой кислоты (50 мг, 0,2 ммоль, получена согласно общей процедуре Н),и получают метиловый эфир 3-[2-[4'-(2-циклогексилэтокси)бифенил-4-илметил]-4-(2,4 дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты. Метиловый эфир 3-[2-[4'-(2-циклогексилэтокси)бифенил-4-илметил]-4-(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты (22 мг, 0,03 ммоль) обрабатывают, следуя общей процедуре(д, 1 Н), 7,87 (с, 1 Н), 8,19 (д, 1 Н) м.д. Пример 42. Метиловый эфир 3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты (55 мг, 0,1 ммоль) обрабатывают так, как описано в общей процедуре G, с использованием 4-(2 циклогексилэтилсульфанил)фенилбороновой кислоты (52 мг, 0,2 ммоль, получена согласно общей процедуре I), и получают метиловый эфир 3-[2-[4'-(2-циклогексилэтилсульфанил)бифенил-4-илметил]-4(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты. Метиловый эфир 3-[2-[4'-(2-циклогексилэтилсульфанил)бифенил-4-илметил]-4-(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты (23 мг, 0,033 ммоль) обрабатывают, следуя общей процедуре F, и получают 1,1-диоксид 5-3-[2-[4'-(2-циклогексилэтилсульфанил)бифенил-4-илметил]-4(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 731 (М+Н)+; 1H ЯМР (ДМСО-d6, 400 МГц):0,86-0,89 (м, 2 Н), 1,14-1,21 (м, 6 Н), 1,45 (м, 1 Н), 1,47-1,80 (м, 4 Н),2,97 (м, 2 Н), 4,01 (с, 2 Н), 4,12 (с, 2 Н), 7,02 (д, 2 Н), 7,04 (д, 2 Н), 7,16 (д, 2 Н), 7,33 (д, 2 Н), 7,42-7,57 (м, 5 Н),7,64 (д, 1 Н), 7,78 (с, 1 Н), 8,17 (д, 1 Н) м.д. Пример 43. 2-(4-Бромбензил)-4-(2,4-дихлорфенил)-1 Н-имидазол (1 г, 2,6 ммоль) обрабатывают трет-бутиловым эфиром (3-иодфенил)карбаминовой кислоты, следуя общей процедуре R, и получают трет-бутиловый эфир 3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенилкарбаминовой кислоты. Содержащий бром и фенил продукт реакции (370 мг, 0,64 ммоль) обрабатывают так, как описано в общей процедуре G, с использованием 4-этоксикарбонилфенилбороновой кислоты, и получают этиловый эфир 4'- [1-(3-трет-бутоксикарбониламинофенил)-4-(2,4-дихлорфенил)-1 Н-имидазол-2-илметил]бифенил 4-карбоновой кислоты. Эфирбензоат, полученный выше (153 мг, 0,24 ммоль), обрабатывают согласно общей процедуре Z с использованием 2 экв. 3,3-диметилбутилмагнийхлорида, с последующим восстановлением продукта реакции Гриньяра согласно общей процедуре АА, и получают трет-бутиловый эфир [3-(4-(2,4 дихлорфенил)-2-4'-[1-(3,3-диметилбутил)-4,4-диметилпентил]бифенил-4-илметилимидазол-1-ил)фенил]карбаминовой кислоты. После удаления группы Boc согласно общей процедуре V полученный анилин последовательно обрабатывают согласно общим процедурам Е и F, и получают 1,1-диоксид 5- [3-(4(2,4-дихлорфенил)-2-4'-[1-(3,3-диметилбутил)-4,4-диметилпентил]бифенил-4-илметилимидазол-1 ил)фенил]-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 771 (М+Н)+. Пример 44. 4-Бромбензальдегид (1 г, 5,4 ммоль) превращают в 4-(4,4-диметилпентил)фенилбороновую кислоту согласно общей процедуре Y с использованием 3,3-диметилбутилмагнийхлорида (1 экв.). 3-[2-(4-Бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фениламин получают из 3-аминоацетанилида согласно общей процедуре М, затем его алкилируют метилбромацетатом согласно общей процедуре Е, и получают сырой метиловый эфир 3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1 ил]фениламиноуксусной кислоты. Бромированное соединение (44 мг, 80 мкмоль) вводят в сочетание с 4-(4,4-диметилпентил)фенил бороновой кислотой так, как описано в общей процедуре G, и получают метиловый эфир 3-(4-(2,4-дихлорфенил)-2-[4'-(4,4-диметилпентил)бифенил-4-илметил]имидазол-1 ил)фениламиноуксусной кислоты. Затем продукт реакции обрабатывают согласно общей процедуре F и получают 1,1-диоксид 5-(3-4-(2,4-дихлорфенил)-2-[4'-(4,4-диметилпентил)бифенил-4-илметил]имидазол-1-илфенил)-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 687 (М+Н)+; 1(д, 1 Н) м.д. Способами, аналогичными способам, используемым для получения соединения примера 44, синтезируют соединения, перечисленные далее. Пример 49. 4-Бромбензальдегид (6,5 г, 35 ммоль) превращают в 4-(2-циклогексилвинил)фенилбороновую кислоту согласно общей процедуре АЕ с использованием циклогексилметилмагнийбромида. 3-[2-(4-Бромбензил)-4-(2,4-дихлорфенил)имидазол-1-ил]фениламин получают из 3-аминоацетанилида согласно общей процедуре М, затем его алкилируют метилбромацетатом согласно общей процедуре Е, и получают сырой метиловый эфир 3-[2-(4-бромбензил)-4-(2,4-дихлорфенил)имидазол-1 ил]фениламиноуксусной кислоты. Бромированное соединение (500 мг, 0,91 ммоль) вводят в сочетание с 4-(2-циклогексилвинил)бороновой кислотой так, как описано в общей процедуре G, и получают метиловый эфир 3-[2-[4'-(2-циклогексилвинил)бифенил-4-илметил]-4-(2,4-дихлорфенил)имидазол-1-ил]фениламиноуксусной кислоты. Затем продукт реакции обрабатывают согласно общей процедуре F и получают 1,1-диоксид 5-3-[2-[4'-(2-циклогексилвинил)бифенил-4-илметил]-4-(2,4-дихлорфенил)имидазол-1 ил]фенил-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 697 (М+Н)+; 1(м, 3 Н), 7,49 (дд, 1 Н), 7,51-7,58 (м, 4 Н), 7,65 (д, 1 Н), 7,94 (с, 1 Н), 8,19 (д, 1 Н) м.д. Пример 50. 1,1-Диоксид 5-3-[2-[4'-(2-циклопентил-1-гидроксиэтил)бифенил-4-илметил]-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она (45 мг, 64 мкмоль) восстанавливают согласно общей процедуре АА и получают 1,1-диоксид 5-3-[2-[4'-(2-циклопентил-этил)бифенил-4-илметил]-4-(2,4 дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. ЖХ-МС: m/z 685 (М+Н)+; 1 Н ЯМР (CD3OD, 400 МГц):1,08-1,20 (м, 2 Н), 1,47-1,57 (м, 2 Н), 1,58-1,68 (м, 4 Н), 1,75-1,86 (м,3 Н), 2,64 (т, 2 Н), 4,17 (с, 2 Н), 4,21 (с, 2 Н), 6,83 (м, 1 Н), 7,07 (м, 2 Н), 7,15 (м, 1 Н), 7,21 (м, 2 Н), 7,28 (м,1 Н), 7,34-7,48 (м, 7 Н), 7,73 (с, 1 Н), 8,01 (д, 1 Н) м.д. Биологический анализ Описанные далее способы анализа используют для идентификации соединений формулы 1, кото- 24019385 рые эффективны при ингибировании активности некоторых фосфатаз, примером которых, используемым в данном описании, является РТР 1 В. Анализ РТР 1 В Анализ ингибирования РТР 1 В основан на детекции комплекса между красителем малахитовым зеленым и свободным фосфатом, высвобожденным из фосфопептидного субстрата действием РТРазы. В каждую лунку аналитического планшета с плоскодонными лунками добавляют 45 мкл буфера для анализа [50 мМ имидазола, рН 7,2, 100 мМ NaCl, 5 мМ DTT и 1 мМ ЭДТК] и 10 мкл пептидного субстрата (тирозинфосфопептид - 1, END(pY)INASL (SEQ ID NO: 1), 80 мкМ FAC, Promega, кат. V256A)] до общего объема 55 мкл. Затем добавляют испытываемое соединение (10 мкл в растворе в ДМСО до 5 0%). Смесь инкубируют в течение 5 мин при 25 С и затем добавляют 10 мкл РТР-1 В (протеинтирозинфосфотаза IB(PTP-1 В); FAC 0,8 нМ; BIOMOL-SE332). Смесь инкубируют при 25 С в течение 30 мин. Затем последовательно добавляют 25 мкл реагента малахитового зеленого (10% (мас./об.) молибдата аммония в воде,Sigma, кат. А-7302, 0,2% (мас./об.) малахитового зеленого в 4 N HCl, Aldrich, кат.21302-0). После инкубации в течение 15 мин при 27 С определяют завершение реакции при 640 нМ. Реагент малахитовый зеленый получают, смешивая один объем 10% раствора молибдата аммония с 3 объемами 0,2% раствора малахитового зеленого, перемешивая смесь при комнатной температуре в течение 30 мин с последующей фильтрацией и сбором фильтрата. Перед применением реагент малахитовый зеленый обрабатывают 10 мкл 5% твина 20 на 990 мкл раствора красителя. Типично испытываемые соединения в вышеуказанном анализе проверяют при восьми концентрациях. Для данного анализа IC50 (мкМ) в анализе ингибирования фермента представляет концентрацию соединения, при которой ингибируется 50% сигнала. Соединения в табл. I ингибируют РТР-1 В с IC50 менее 10 мкМ. Несмотря на то, что изобретение описано и пояснено со ссылкой на определенные его варианты осуществления, специалистам в данной области техники будет понятно, что можно осуществить их различные изменения, модификации и замены без отхода от сущности и объема изобретения. Например,правильными могут быть эффективные дозировки иные, чем дозировки, указанные в данном описании,вследствие изменений в восприимчивости субъекта, которого лечат в случае заболевания(ий), опосредуемого(ых) РТРазой. Подобным образом, наблюдаемые специфические фармакологические реакции могут изменяться согласно и в зависимости от определенного выбранного активного соединения или любого из присутствующих носителей, а также типа препарата и используемого способа введения, и такие предполагаемые изменения или различия в результатах рассматриваются согласно с целями и практикой настоящего изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, имеющее формулу где R23-R27 выбирают, независимо, из группы, состоящей из водорода, пропила, бутила, пентила, 1 этилпропила, 1-пропилбутила, 3,3-диметилбутила, 4-метилпентила, 4,4-диметилпентила, 1-(3,3 диметилбутил)-4,4-диметилпентила, изобутила, изопропила, втор-бутила, трет-бутила, трифторметила,4,4,4-трифторбутокси, этокси, пропокси, бутокси, изобутокси, трет-бутокси, 2-фенетокси, 2,2 диметилпропокси, 3-метилбутокси, 3,3-диметилбутокси, 2-циклогексилэтансульфонила, 3,3-диметилбутан-1-сульфонила, циклогексансульфонила, циклогексилметансульфонила, 2-циклогексилэтансульфинила,3,3-диметилбутан-1-сульфинила,циклогексилметилсульфинила,2-циклогексилэтилсульфанила, 3,3-диметилбутилсульфанила, фенетилсульфанила, циклогексилметилсульфанила, циклопентила, циклогексила, циклогексилокси, циклогексилметила, 2-циклопентилэтила, 2-циклогексилэтила, циклопентилметокси, циклогексилметокси, 2-циклопентилэтокси, 2-циклогексилэтокси, 2 циклогексилвинила, 3-этилциклобутила, хлора, фтора и фенила; по меньшей мере один из R23-R27 отличен от водорода,или его фармацевтически приемлемая соль. 2. Соединение по п.1, выбранное из группы, состоящей из 1,1-диоксида 5-3-[2-(4'-трет-бутилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил 1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-3-[2-(3'-циклогексилметоксибифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1 ил]фенил-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-3-[4-(2,4-дихлорфенил)-2-(3'-фенетилоксибифенил-4-илметил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-(3-4-(2,4-дихлорфенил)-2-[3'-(3,3-диметилбутилсульфанил)бифенил-4-илметил]имидазол-1-илфенил)-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-(3-4-(2,4-дихлорфенил)-2-[3'-(3,3-диметилбутан-1-сульфонил)бифенил-4-илметил]имидазол-1-илфенил)-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-3-[2-(3'-циклогексилметилсульфанилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-3-[2-(3'-циклогексилметилсульфонилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол 1-ил]фенил-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-3-[2-(3'-(2-циклогексилэтилсульфанил)бифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол 1-ил]фенил-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-3-[2-(3'-(2-циклогексилэтансульфонил)бифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол 1-ил]фенил-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-(3-4-(2,4-дихлорфенил)-2-[4'-(3,3-диметилбутокси)бифенил-4-илметил]имидазол-1 илфенил)-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-3-[2-(4'-циклогексилметоксибифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1 ил]фенил-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-(3-4-(2,4-дихлорфенил)-2-[4'-(3,3-диметилбутилсульфанил)бифенил-4-илметил]имидазол 1-илфенил)-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-3-[2-(4'-циклогексилметилсульфанилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол 1-ил]фенил-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-3-[2-(4'-циклогексилметансульфонилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол 1-ил]фенил-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-3-[2-[4'-(2-циклогексилэтокси)бифенил-4-илметил]-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она; 1,1-диоксида 5-3-[2-[4'-(2-циклогексилэтил)бифенил-4-илметил]-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она или их фармацевтически приемлемых солей. 3. Соединение по п.1, представляющее собой 1,1-диоксид 5-3-[2-(4'-циклогексилбифенил-4-илметил)4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она или его фармацевтически приемлемую соль. 4. Соединение по п.3, представляющее собой 1,1-диоксид 5-3-[2-(4'-циклогексилбифенил-4-илметил)4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. 5. Соединение по п.3, представляющее собой натриевую или калиевую соль 1,1-диоксида 5-3-[2(4'-циклогексилбифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3 она. 6. Соединение по п.1, представляющее собой 1,1-диоксид 5-(3-4-(2,4-дихлорфенил)-2-[3'-(3,3'диметилбутокси)бифенил-4-илметил]имидазол-1-илфенил)-1,2,5-тиадиазолидин-3-она или его фармацевтически приемлемую соль. 7. Соединение по п.6, представляющее собой 1,1-диоксид 5-(3-4-(2,4-дихлорфенил)-2-[3'-(3,3'диметилбутокси)бифенил-4-илметил]имидазол-1-илфенил)-1,2,5-тиадиазолидин-3-она. 8. Соединение по п.6, представляющее собой натриевую или калиевую соль 1,1-диоксида 5-(3-4(2,4-дихлорфенил)-2-[3'-(3,3'-диметилбутокси)бифенил-4-илметил]имидазол-1-илфенил)-1,2,5-тиадиазолидин-3-она. 9. Соединение по п.1, представляющее собой 1,1-диоксид 5-3-[2-(4'-циклогексилметоксибифенил 4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она или его фармацевтически приемлемую соль. 10. Соединение по п.9, представляющее собой 1,1-диоксид 5-3-[2-(4'-циклогексилметоксибифенил 4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. 11. Соединение по п.9, представляющее собой натриевую или калиевую соль 1,1-диоксида 5-3-[2(4'-циклогексилметоксибифенил-4-илметил)-4-(2,4-дихлорфенил)имидазол-1-ил]фенил-1,2,5-тиадиазолидин-3-она. 12. Фармацевтическая композиция, включающая соединение по любому из пп.1-11 или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, разбавитель, эксципиент или их смесь. 13. Фармацевтическая композиция в форме пероральной стандартной лекарственной формы, включающая соединение по любому из пп.1-11 или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, разбавитель, эксципиент или их смесь. 14. Фармацевтическая композиция в форме парентеральной стандартной лекарственной формы,- 26019385 включающая соединение по любому из пп.1-11 или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, разбавитель, эксципиент или их смесь. 15. Применение соединения по любому из пп.1-11 для получения лекарственного средства, предназначенного для лечения диабета типа II.
МПК / Метки
МПК: A61K 31/433, C07D 417/14, A61K 31/4178, A61K 31/427, C07D 417/10
Метки: применение, качестве, производные, композиции, содержащие, ртразы, замещенные, ингибиторов, фармацевтические, имидазольные
Код ссылки
<a href="https://eas.patents.su/28-19385-zameshhennye-imidazolnye-proizvodnye-soderzhashhie-ih-farmacevticheskie-kompozicii-i-ih-primenenie-v-kachestve-ingibitorov-rtrazy.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные имидазольные производные, содержащие их фармацевтические композиции и их применение в качестве ингибиторов ртразы</a>
Замещенные пиразоло[4,3-e]диазепины, фармацевтические композиции, содержащие их, и применение в качестве лекарственных продуктов

Номер патента: 5071
Опубликовано: 28.10.2004
Авторы: Наве Мишель, Бюрнуф Катрин, Бересибар Амайа
МПК: A61P 29/00, C07D 487/04, A61K 31/5517...
Метки: их, содержащие, продуктов, лекарственных, качестве, фармацевтические, замещенные, композиции, пиразоло[4,3-e]диазепины, применение
Формула / Реферат:
1. Соединение формулы I ниже где R1 выбран из следующих групп: линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода; циклоалкил, содержащий от 3 до 6 атомов углерода; циклоалкилалкил, включающий в себя алкильную группу, содержащую от 1 до 6 атомов углерода, и циклоалкильную группу, содержащую от 3 до 6 атомов углерода; арил, содержащий от 5 до 10 атомов углерода; арилалкил, содержащий от 6 до 10 атомов углерода; группы...
Новые производные бензимидазолов и бензотиазолов, способ их получения, их применение в качестве лекарственных средств, фармацевтические композиции и новое применение, в частности, в качестве ингибиторов cmet

Номер патента: 14315
Опубликовано: 29.10.2010
Авторы: Клерк Франсуа, Немесек Консепсьон
МПК: C07D 235/32, C07D 235/30, C07D 277/82...
Метки: частности, новое, новые, ингибиторов, средств, фармацевтические, производные, получения, композиции, бензотиазолов, качестве, способ, применение, бензимидазолов, лекарственных
Формула / Реферат:
1. Соединения формулы (I)в которой А обозначает NH или S;R1 и R2, одинаковые или разные, выбирают из атомов водорода, радикала NH2, атомов галогена и алкильных радикалов, необязательно замещенных одним или несколькими атомами галогена,и R3 обозначает атом водорода или выбран из значений R1 и R2, причем по меньшей мере один из R1, R2 и R3 не является водородом,R обозначает циклоалкил или алкил, необязательно замещенный фенилом, гетероарилом,...
Стероиды, замещенные в положении 11, способ их получения, их применение в качестве лекарственного средства и содержащие их фармацевтические композиции

Номер патента: 1868
Опубликовано: 22.10.2001
Авторы: Тетш Жан-Жорж, Ник Франсуа, Буали Иамина, Ван Де Вельд Патрик
МПК: C07J 41/00, A61K 31/565, A61P 19/10...
Метки: качестве, применение, композиции, способ, замещенные, стероиды, лекарственного, средства, фармацевтические, положении, получения, содержащие
Формула / Реферат:
1. Соединения общей формулы в которой n - целое число, равно 2 или 3, либо R1 и R2, идентичные или разные, означают атом водорода или алкил, содержащий от 1 до 4 атомов углерода, либо R1 и R2 образуют вместе с атомом азота, с которым они связаны, моно- или полициклический гетероцикл, имеющий от 5 до 15 звеньев, ароматический или не ароматический, содержащий, при необходимости, от 1 до 3 дополнительных гетероатомов, выбираемых из кислорода,...
Производные бензоксазина в качестве модуляторов 5-нт-6, способ их получения, фармацевтические композиции, содержащие эти производные, и их применение

Номер патента: 9982
Опубликовано: 28.04.2008
Авторы: Чжао Шухай, Кларк Робин Дуглас, Бергер Якоб
МПК: A61K 31/536, C07D 265/36, A61P 25/18...
Метки: качестве, 5-нт-6, эти, способ, бензоксазина, получения, производные, фармацевтические, содержащие, модуляторов, применение, композиции
Формула / Реферат:
1. Соединение формулы его фармацевтически приемлемая соль или пролекарство, где m означает целое число от 0 до 3; n означает 2 или 3; р означает 2; Y означает -S(O2)-; Z1 означает N; R1 и R2 означают водород; R3 означает фенил или нафтил, которые необязательно замещены галогеном, алкилом, алкокси, гидрокси, циано, пиперазинилом, метилсульфониламино, метилсульфонилом, аминокарбонилом, или означает хинолинил, изохинолинил, тиофенил,...
Новое применение гетероциклических соединений, активных в качестве ингибиторов бета-лактамаз, и содержащие их фармацевтические композиции

Номер патента: 7220
Опубликовано: 25.08.2006
Авторы: Лампила Максим, Фроментэн Клод, Асзоди Жозеф, Роулендс Дэвид Ален
МПК: A61K 31/4995, A61K 31/439, A61K 31/55...
Метки: соединений, активных, качестве, новое, ингибиторов, композиции, фармацевтические, бета-лактамаз, применение, содержащие, гетероциклических
Формула / Реферат:
1. Применение соединений общей формулы (I) где R1 означает атом водорода, радикал COOR, CONR6, R7, R выбирают из группы, образованной алкильным радикалом, содержащим от 1 до 6 атомов углерода, не обязательно замещенным пиридильным или карбамоильным радикалом, -СН2-алкенильным радикалом, содержащим в сумме от 3 до 9 атомов углерода, арилом, содержащим от 6 до 10 атомов углерода, или аралкилом, содержащим от 7 до 11 атомов углерода, при этом ядро...
Предыдущий патент: Способ обработки и строительства скважин
Следующий патент: Бензимидазольные модуляторы vr1
Случайный патент: Композиция кондиционирующего шампуня