Ненуклеозидные ингибиторы обратной транскриптазы






















Формула / Реферат
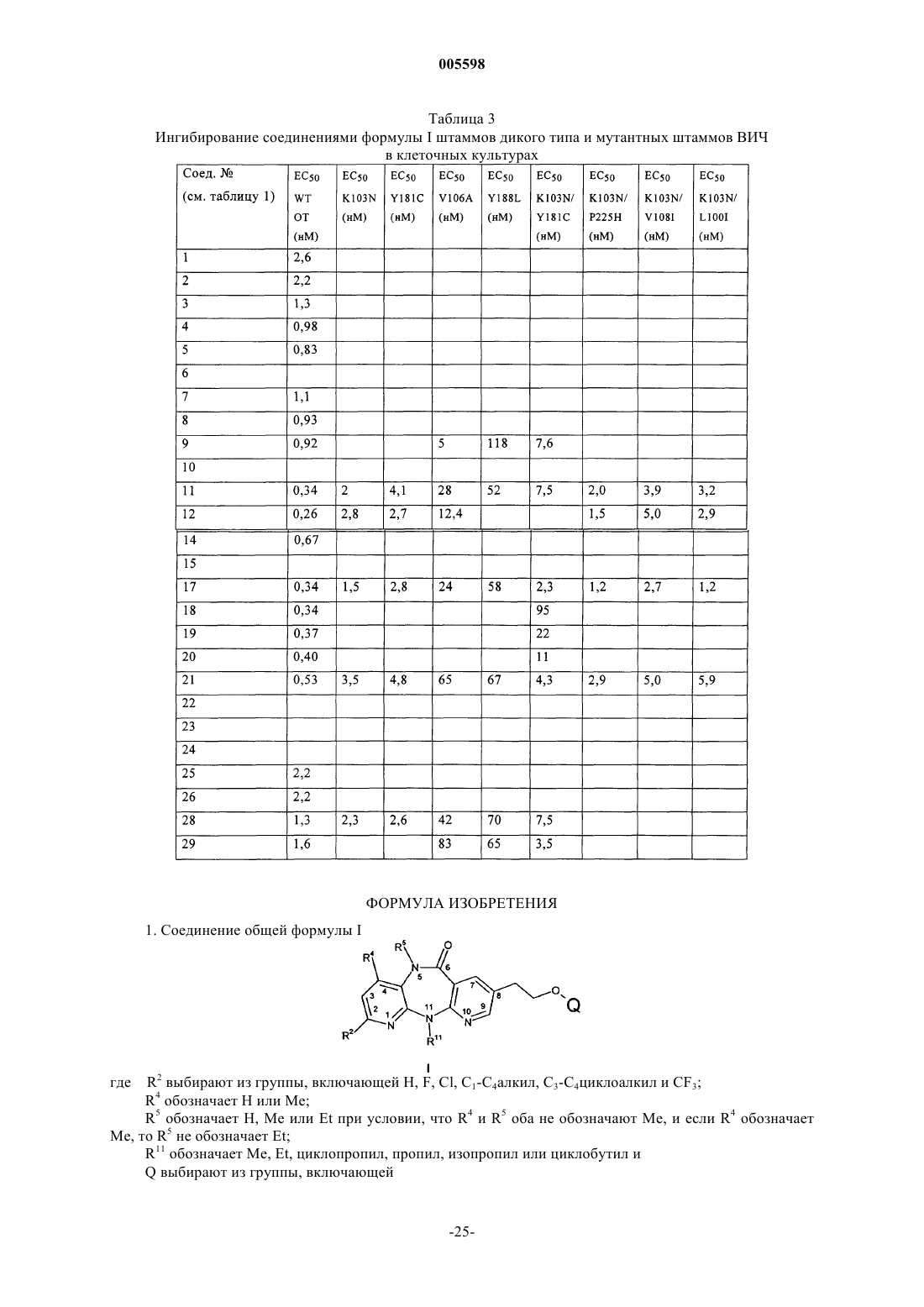
1. Соединение общей формулы I

где R2 выбирают из группы, включающей H, F, Cl, C1-C4алкил, C3-C4циклоалкил и CF3;
R4 обозначает H или Me;
R5 обозначает H, Me или Et при условии, что R4 и R5 оба не обозначают Me, и если R4 обозначает Me, то R5 не обозначает Et;
R11 обозначает Me, Et, циклопропил, пропил, изопропил или циклобутил и
Q выбирают из группы, включающей

или его фармацевтически приемлемая соль.
2. Соединение по п.1, где R11 обозначает Et или циклопропил, или его фармацевтически приемлемая соль.
3. Соединение по п.1, где Q выбирают из группы, включающей

или его фармацевтически приемлемая соль.
4. Соединение по п.1, где Q обозначает

или его фармацевтически приемлемая соль.
5. Соединение по п.1, где R5 обозначает Me, или его фармацевтически приемлемая соль.
6. Соединение по п.1, где R11 обозначает Et, или его фармацевтически приемлемая соль.
7. Соединение

или его фармацевтически приемлемая соль.
8. Соединение

или его фармацевтически приемлемая соль.
9. Соединение

или его фармацевтически приемлемая соль.
10. Применение соединения формулы I по любому из пп.1-9 или его фармацевтически приемлемой соли в качестве ингибитора репликации ВИЧ.
11. Способ лечения или предупреждения ВИЧ-инфекции, заключающийся в том, что пациенту вводят ингибирующее ВИЧ количество соединения формулы I по любому из пп.1-9 или его фармацевтически приемлемой соли.
12. Фармацевтическая композиция, предназначенная для лечения или предупреждения ВИЧ-инфекции, содержащая соединение формулы I по любому из пп.1-9 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Текст
005598 Область техники, к которой относится изобретение Изобретение относится к новым соединениям и их фармацевтически приемлемым солям, их применению либо индивидуально, либо в сочетании с другими терапевтическими агентами для лечения или профилактики ВИЧ-инфекции и к содержащим эти соединения фармацевтическим композициям. Предпосылки создания изобретения Болезнь, известная как синдром приобретенного иммунодефицита (СПИД), вызывается вирусом иммунодефицита человека (ВИЧ), прежде всего штаммом, известным как ВИЧ-1. Для репликации ВИЧ в клетке-хозяине, информация о вирусном геноме должна быть интегрирована в ДНК клетки-хозяина. Однако ВИЧ представляет собой ретровирус, т.е. вирус, у которого генетическая информация считывается с РНК. Вследствие этого в цикле репликации ВИЧ должна присутствовать стадия транскрипции вирусного генома (РНК) с образованием ДНК, что является обратным по отношению к обычному процессу. Транскрипция вирусной РНК с образованием ДНК происходит с помощью фермента, называемого поэтому обратной транскриптазой (ОТ). Вирион ВИЧ наряду с вирусной РНК включает копию ОТ. Для обратной транскриптазы характерны три известные ферментативные функции; она действует как РНК-зависимая ДНК-полимераза, как рибонуклеаза и как ДНК-зависимая ДНК-полимераза. Действуя как РНК-зависимая ДНК-полимераза, ОТ транскрибирует синтез копии одноцепочечной ДНК вирусной РНК. Действуя как рибонуклеаза, ОТ расщепляет исходную вирусную РНК и высвобождает ДНК,непосредственно полученную на матрице исходной РНК. И наконец, действуя как ДНК-зависимая ДНКполимераза, ОТ обеспечивает получение второй комплементарной цепи ДНК, используя первую цепь ДНК в качестве матрицы. Две цепи образуют двухцепочечную ДНК, которая интегрируется в геном клетки-хозяина с помощью другого фермента, называемого интегразой. Соединения, которые ингибируют ферментативные активности обратной транскриптазы ВИЧ-1,могут ингибировать репликацию ВИЧ-1 в зараженных клетках. Такие соединения можно применять для предупреждения или лечения вызываемой ВИЧ-1 инфекции у людей, что продемонстрировано при использовании известных ингибиторов ОТ, таких как 3'-азидо-3'-дезокситимидин (AZT), 2',3'дидезоксиинозин (ddI), 2',3'-дидезоксицитидин (ddC), d4T, 3TC, невирапин, делавирдин, эфавиренз и абакавир, основных лекарственных средств, которые в настоящее время разрешены для применения при лечении СПИДа. Как и в случае любой антивирусной терапии, применение ингибиторов ОТ для лечения СПИДа в конце концов приводит к снижению чувствительности вируса к данному лекарственному средству. Устойчивость (пониженная чувствительность) к указанным лекарственным средствам является результатом мутаций, затрагивающих сегмент обратной транскриптазы гена ро 1. К настоящему времени охарактеризовано несколько мутантных штаммов ВИЧ, для которых установлено, что устойчивость к известным терапевтическим агентам является результатом мутаций в гене ОТ. Примерами некоторых из наиболее распространенных выявленных, связанных с лечением мутаций являются мутация Y181C, включающая замену тирозина (Y) в ко доне 181 на остаток цистеина (С), и K103N, включающая замену лизина (K) в положении 103 на аспарагин (N). Другие мутанты, частота которых возрастает в процессе лечения с использованием известных антивирусных средств, представляют собой мутанты с одной мутацией V106A,G190A, Y188C и P236L; и мутанты с двумя мутациями K103N/Y181C, K103N/P225H, K103N/V108I иK103N/L100I. Поскольку лечение и предупреждение ВИЧ-инфекции с использованием антивирусных средств продолжается, следует ожидать увеличения числа новых устойчивых штаммов. Отсюда ясно, что существует потребность в разработке новых ингибиторов ОТ, отличающихся различным механизмом действия в отношении разных мутантов. Имеющие трициклические структуры соединения, которые являются ингибиторами ВИЧ-1, описаны в патенте US No. 5366972. Другие ингибиторы обратной транскриптазы ВИЧ-1 описаны у Hargrave и др., J. Med Chem., 34, 2231 (1991). В патенте US No. 5705499 в качестве ингибиторов ОТ предложены 8-арилалкил- и 8-арилгетероалкил-5,11-дигидро-6 Н-дипиридо[3,2-В:2',3'-Е][1,4]диазепины. Установлено, что приведенные в качестве примера соединения обладают определенной активностью в отношении ОТ ВИЧ-1 дикого типа и мутантной ОТ, прежде всего Y181C, но при этом они менее эффективны в отношении других несущих одну мутацию мутантов, таких как K103N. Установлено, что соединения по настоящему изобретению являются эффективными ингибиторами мутаций Y181C и K103N, а также широкого спектра других мутантов с одной мутацией и некоторых известных мутантов с двойной мутацией, таких как K103N/Y181C и K103N/P225H. Краткое изложение сущности изобретения Настоящее изобретение позволяет преодолеть сложности и недостатки прототипов с помощью новых соединений, которые являются эффективными ингибиторами штаммов ВИЧ-1, несущих одну и две мутации ОТ. Первым объектом изобретения является соединение общей формулы IR5 обозначает Н, Me или Et при условии, что R4 и R5 оба не обозначают Me, и если R4 обозначает или его фармацевтически приемлемая соль. Вторым объектом изобретения является ингибитор репликации ВИЧ общей формулы I или его фармацевтически приемлемая соль. Третьим объектом изобретения является ингибитор обратной транскриптазы ВИЧ общей формулы I или его фармацевтически приемлемая соль. Четвертым объектом изобретения является способ лечения или предупреждения ВИЧ-инфекции,заключающийся в том, что пациенту вводят ингибирующее ВИЧ количество соединения формулы I или его фармацевтически приемлемой соли. Пятым объектом изобретения является фармацевтическая композиция, предназначенная для лечения или предупреждения ВИЧ-инфекции, которая содержит соединение формулы I или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Шестым объектом изобретения является способ получения соединения формулы I или его фармацевтически приемлемой соли. Подробное описание изобретения Определения В контексте настоящего описания понятие С 1-С 4 алкил обозначает алкильные радикалы с прямой или разветвленной цепью, содержащие от 1 до 4 атомов углерода, и они представляют собой метил, этил,пропил, изопропил, бутил, втор-бутил и трет-бутил. В контексте настоящего описания понятие С 3-С 4 циклоалкил обозначает насыщенные циклические углеводородные радикалы, содержащие 3-4 атома углерода, и они представляют собой циклопропил и циклобутил. Подробное описание предпочтительных вариантов осуществления Предпочтительным вариантом осуществления изобретения являются соединения формулы I, в которых R2 предпочтительно обозначает Cl, F или Н. Более предпочтительно R2 обозначает Cl или Н. Наиболее предпочтительно R2 обозначает Н. Предпочтительным вариантом осуществления изобретения являются соединения формулы I, в которых R4 предпочтительно обозначает Н. Другим вариантом осуществления изобретения являются соединения формулы I, в которых предпочтительно R5 обозначает Me. Предпочтительными соединениями по изобретению являются соединения формулы I, в которых R11 обозначает Et или циклопропил. Более предпочтительно R11 обозначает Et. Предпочтительным вариантом осуществления изобретения являются соединения формулы I, в которых Q предпочтительно выбирают из группы, включающей Другими предпочтительными вариантами осуществления изобретения являются соединения, выбранные из группы, включающей Соединения по изобретению являются эффективными ингибиторами обратной транскриптазы дикого типа, они также обладают способностью ингибировать ферменты, например, несущие одну мутацию, такие как Y181C, K103N, V106A, G190A, Y188C и P236L. Соединения ингибируют также ферменты, несущие две мутации, такие как K103N/Y181C, K103N/P225H, K103N/V108I и K103N/L100I. Соединения формулы I обладают ингибирующей активностью в отношении обратной транскриптазы ВИЧ-1. При введении в виде приемлемых дозируемых форм их можно применять для лечения СПИДа, СПИД-ассоциированного комплекса (ARC) и аналогичных, связанных с заражением ВИЧ-1 заболеваний. Таким образом, еще одним объектом изобретения является способ лечения вызываемой ВИЧ-1 инфекции, заключающийся во введении инфицированному ВИЧ-1 человеку терапевтически эффективного количества описанного выше нового соединения формулы I. Независимо от того, рекомендованы они для лечения или профилактики, соединения можно применять также для предупреждения перинатальной передачи ВИЧ-1 от матери к ребенку, путем их введения матери до рождения ребенка. Соединения формулы I можно вводить в виде однократной или разделенных доз пероральным или парентеральным путем. Приемлемая пероральная доза соединения формулы I составляет примерно от 0,5 мг до 1 г в день. Предпочтительная пероральная доза соединения формулы I составляет примерно 100800 мг в день для пациента весом 70 кг. В композициях для парентерального введения приемлемая стандартная доза может содержать от 0,1 до 250 мг указанных соединений, предпочтительно от 1 до 200 мг. Однако должно быть очевидно, что вводимая доза должна варьироваться от пациента к пациенту и доза,предназначенная для введения любому конкретному пациенту, должна зависеть от рекомендаций лечащего врача, который в качестве критериев для назначения конкретной соответствующей дозы должен использовать габариты и состояние пациента, а также реакцию пациента на лекарственное средство. Когда соединения по настоящему изобретению подлежат введению пероральным путем, их можно вводить в качестве лекарственных средств в форме фармацевтических препаратов, которые содержат их в сочетании с совместимым фармацевтическим носителем. Такой носитель может представлять собой инертный органический или неорганический носитель, пригодный для перорального введения. Примерами таких носителей являются вода, желатин, тальк, крахмал, стеарат магния, гуммиарабик, растительные масла, полиалкиленгликоли, вазелин и т.п. Фармацевтические композиции можно приготавливать общепринятым методом и полученные в результате дозируемые формы могут представлять собой твердые дозируемые формы, например, таблетки,драже, капсулы и т.п., или жидкие дозируемые формы, например растворы, суспензии, эмульсии и т.п. Фармацевтические препараты можно подвергать общепринятым фармацевтическим обработкам, таким как стерилизация. Кроме того, фармацевтические препараты могут содержать обычные адъюванты, такие как консерванты, стабилизаторы, эмульгаторы, корригенты, смачивающие агенты, буферы, соли для изменения осмотического давления и т.п. Твердые носители, которые можно использовать, представляют собой, например, крахмал, лактозу, маннит, метилцелюлозу, микрокристаллическую целлюлозу, тальк,диоксид кремния, вторичный кислый фосфат кальция и высокомолекулярные полимеры (такие, как полиэтиленгликоль). При парентеральном применении соединение формулы I можно вводить в водном или неводном растворе, суспензии или эмульсии в фармацевтически приемлемом масле или смеси жидкостей, которые могут содержать бактериостатические агенты, антиоксиданты, консерванты, буферы или другие ингредиенты, придающие раствору изотоничность с кровью, загустители, суспендирующие агенты или другие фармацевтически приемлемые добавки. Добавки указанного типа включают, например, тартратный, цит-3 005598 ратный и ацетатный буферы, этанол, пропиленгликоль, полиэтиленгликоль, комплексообразователи (такие как ЭДТА), антиоксиданты (такие как бисульфит натрия, метабисульфит натрия и аскорбиновая кислота), высокомолекулярные полимеры (такие как жидкие полиэтиленоксиды) для регуляции вязкости и полиэтиленовые производные ангидридов сорбита. При необходимости можно также добавлять консерванты, такие как бензойная кислота, метил- или пропил парабен, бензалконийхлорид и другие четвертичные аммонийные производные. Соединения по изобретению можно также вводить в виде назальных растворов, и они могут содержать в водном носителе помимо соединений по изобретению приемлемые буферы, регуляторы тоничности, консерванты, препятствующие микробному заражению, антиоксиданты и агенты, повышающие вязкость. Примерами агентов, применяемых для повышения вязкости, являются поливиниловый спирт, производные целлюлозы, поливинилпирролидон, полисорбаты или глицерин. Применяемые консерванты,препятствующие микробному заражению, могут представлять собой бензалконийхлорид, тимеросал,хлорбутанол или фенилэтиловый спирт. Соединения по изобретению можно вводить также с помощью суппозитория. Соединения по изобретению можно получать с помощью известных методов органического синтеза. Примеры реакционных схем приведены на схемах 1-6. Схема 1. Получение промежуточных продуктов, в которых R4 обозначает Me. Последовательность осуществления реакций, представленных на схеме 2, аналогична последовательности, описанной у J.M. Klunder и др.; J. Med. Сhem., 41, 2960-71, 1998, и у C.L. Cywin и др.; J. Med.-5 005598 Схема 6. Альтернативный путь получения соединений, в которых R4 обозначает Me. Как указано выше, соединения по изобретению ингибируют ферментативную активность ОТ ВИЧ 1. Основываясь на описанной ниже оценке биологической активности, установлено, что указанные соединения ингибируют такой вид активности ОТ ВИЧ-1, как активность РНК-зависимой ДНКполимеразы ВИЧ-1. Установлено (данные не представлены), что они ингибируют также такой вид активности ОТ ВИЧ-1, как активность ДНК-зависимой ДНК-полимеразы. Установлено, что с помощью описанного ниже анализа обратной транскриптазы (ОТ), можно тестировать способность соединений ингибировать активность ОТ ВИЧ-1 как РНК-зависимой ДНК-полимеразы. С помощью этого была изучена активность некоторых конкретных соединений, приведенных ниже в примерах. Результаты исследования представлены в табл. 2 в виде значений IC50 (нМ) и в табл. 3 в виде значений ЕС 50 (нМ). Примеры Настоящее изобретение дополнительно проиллюстрировано с помощью примеров, которые не ограничивает его объем. Все реакции осуществляли в атмосфере азота или аргона. Температура дана в градусах Цельсия. Если не указано иное, то процентное содержание или соотношение компонентов в растворе выражено в об.%. Ниже приведены сокращения или символы, применяемые в настоящем описании: ДЭАД: диэтилазодикарбоксилат; ДИАД: диизопропилазодикарбоксилат; ДИЭА: диизопропилэтиламин; ДМАП: 4-(диметиламино)пиридин; ДМСО: диметилсульфоксид; ДМФ: диметилформамид; МС ES: масс-спектрометрия с использованием электронного пучка;Ph: фенил; ТБЭ: трис-борат-ЭДТА; ТБТУ: тетрафторборат 2-(1H-бензотриазол-1-ил)-N,N,N'N'-тетраметилурония; ТФК: трифторуксусная кислота; ТГФ: тетрагидрофуран; МС (ES): масс-спектрометрия с использованием электронного пучка; МС (FAB) или FAB/MC: масс-спектрометрия с использованием бомбардировки быстрыми атомами; ВРМС: масс-спектрометрия высокого разрешения; БОЕ: бляшкообразующие единицы; ДЭПК: диэтилпирокарбонат; ДМСО: диметилсульфоксид; ДТТ: дитиотреитол; ЭДТА: этилендиаминтетраацетат; УМФ: уридин-5'-монофосфат; УТФ: уридин-5'-трифосфат.-6 005598 Синтез В приведенных ниже примерах проиллюстрированы методы получения соединений по изобретению. Пример 1 (номер 24, табл. 1). 5,11-Дигидро-11-этил-5-метил-8-2-(4-хинолинилокси)этил-6Hдипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. а) 2-(Этиламино)-3-нитропиридин. К раствору 2-хлор-3-нитропиридина (51 г, 325 ммолей) в ТГФ (650 мл) добавляли 2 М раствор этиламина ТГФ (365 мл, 731 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь сливали на воду (1,5 л) и образовавшийся твердый продукт фильтровали и сушили при пониженном давлении, получая указанное в заголовке соединение (52 г). б) Амино-2-(этиламино)пиридин. Раствор 2-(этиламино)-3-нитропиридина (52 г) в МеОН (600 мл) перемешивали в течение ночи при комнатной температуре в атмосфере водорода (1 атм) в присутствии 20% Pd(OH)2/C (10,4 г). Катализатор удаляли фильтрацией через диатомовую землю. Фильтрат концентрировали при пониженном давлении,получая указанное в заголовке соединение в виде твердого вещества черного цвета (39 г, общий выход после стадий а) и б): 88%). в) 2-Хлор-N-2-(этиламино)-3-пиридинил-5-бром-3-пиридинкарбоксамид. К охлажденному раствору 3-амино-2-(этиламино)пиридина (30,6 г, 223 ммоля) в MeCN (740 мл) добавляли твердый NaHCO3 (56,3 г, 669 ммолей). Через 5 мин добавляли неочищенный 5-бром-2-хлор-3 пиридинкарбонилхлорид (1 экв 223 ммоля), полученный из 5-бром-2-гидрокси-3-пиридинкарбоновой кислоты и SOСl2 [согласно методу, описанному у Т.W. Gero и др. в: Synth. Commun., 19, 553-559, 1989(публикация включена в настоящее описание в качестве ссылки), но за исключением того, что не проводили обработку водой]. Через 2 ч реакционную смесь сливали на смесь лед/Н 2 О (1,5 л) и образовавшееся твердое вещество фильтровали, промывали Н 2 О, а затем гексаном. После сушки в течение ночи при пониженном давлении получали указанное в заголовке соединение в виде твердого вещества черного цвета указанное в заголовке соединение (54,9 г, выход 69%). г) 8-Бром-5,11-дигидро-11-этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. К раствору 2-хлор-N-2-(этиламино)-3-пиридинил-5-бром-3-пиридинкарбоксамида (54,9 г, 154,4 ммоля) в пиридине (308 мл) при 50 С добавляли по каплям 1 М раствор NаГМДС (гексаметилдисилазид натрия) в ТГФ (355 мл, 355 ммолей). Через 10 мин реакционной смеси давали охладиться до комнатной температуры, а затем сливали на ледяную воду (2 л). Образовавшийся твердый продукт фильтровали,промывали водой, а затем гексаном. Твердый продукт сушили при пониженном давлении, получая указанное в заголовке соединение (36 г, выход 75%) в виде твердого вещества темно-зеленого цвета. д) 8-Бром-5,11-дигидро-11-этил-5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. К раствору 8-бром-5,11-дигидро-11-этил-6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (36,7 г, 115 ммолей) в ДМФ (380 мл) добавляли NaH (3,5 г, 138 ммолей) и смесь нагревали до 50 С в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры и обрабатывали МеI (14,3 мл, 230 ммолей). Через 1,5 ч реакционную смесь сливали на ледяную воду. Твердый продукт фильтровали, промывали водой, а затем гексаном, получая после сушки указанное в заголовке соединение (37,9 г, выход 99%) в виде твердого вещества темно-зеленого цвета. е) 5,11-Дигидро-11-этил-5-метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Аллилтрибутилолово (30,7 мл, 99,0 ммолей) и Рd(Рh3 Р)4 (5,20 г, 4,50 ммоля) добавляли к дегазированному (полученному путем пропускания через раствор N2 в течение 30 мин) раствору 8-бром-5,11 дигидро-11-этил-5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (30,0 г, 90,0 ммолей) в ДМФ (450 мл) при комнатной температуре. Смесь перемешивали при 90 С в течение 1,5 ч, затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали экспрессхроматографией (гексан:ЕtOАс, от 8:2 до 7:3), получая указанное в заголовке соединение (22,19 г, выход 84%). ж) 5,11-Дигидро-11-этил-8-(2-гидроксиэтил)-5-метил-6H-дипиридо[3,2-b:2'.3'-е][1,4]диазепин-6-он. Струeй озонированного кислорода в течение 2,5 ч барботировали холодный (-78 С) раствор 5,11 дигидро-11-этил-5-метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (22,19 г, 75,4 ммоля) в СН 2 Сl2 (150 мл) и МеОН (150 мл). Затем раствор в течение 15 мин барботировали N2, после этого к раствору добавляли твердый NaBH4 (4,99 г, 132 ммоля). Реакционной смеси давали нагреться до комнатной температуры. Через 1 ч добавляли водный насыщенный раствор NH4Cl (200 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Органические растворители удаляли при пониженном давлении. К остатку добавляли воду (300 мл) и СНСl3 (300 мл). Фазы разделяли и водный слой экстрагировали СНСl3 (3 х 300 мл). Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (ЕtOАс:СНСl3,4:1), получая указанное в заголовке соединение (16,1 г, выход 72%) в виде твердого вещества белого цвета. з) 5,11-Дигидро-11-этил-5-метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он.-7 005598 Диэтилазодикарбоксилат (ДЭАД) (12,8 мл, 81,0 ммоль) добавляли при комнатной температуре по каплям к раствору, содержащему 5,11-дигидро-11-этил-8-(2-гидроксиэтил)-5-метил-6H-дипиридо[3,2b:2',3'-е][1,4]диазепин-6-он (16,1 г, 54,0 ммоля), 4-гидроксихинолин (11,6 г, 81,0 ммоль) и Рh3 Р (213 г,81,0 ммоль) в ТГФ (270 мл). Смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (ЕtOАс:МеОН; 95:5), получая указанное в заголовке соединение (17,7 г, выход 77% ) в виде твердого вещества белого цвета: МС (ESI) m/z 426 (МН)+. Пример 2 (номер 2, табл. 1). 2-Хлор-5,11-дигидро-11-этил-5-метил-8-2-(4-хинолинилокси)этил 6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. а) 6-Хлор-2-(этиламино)-3-нитропиридин. Охлажденный на льду раствор EtNH2 (49,8 г, 1,10 моля) в толуоле (200 мл) добавляли в течение 15 мин к охлажденному на льду раствору 2,6-дихлор-3-нитропиридина (100,0 г, 0,52 моля) в толуоле (225 мл). Смесь перемешивали при 0 С в течение 45 мин. Добавляли воду (500 мл) и ЕtOАс (500 мл) и разделяли фазы. Органический слой последовательно промывали водой (200 мл) и соляным раствором (200 мл), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Оставшийся твердый продукт перекристаллизовывали из МеОН, получая указанное в заголовке соединение (83,7 г, выход 80%) в виде игольчатых кристаллов желтого цвета. б) 3-Амино-6-хлор-2-(этиламино)пиридин. Раствор, содержащий SnCl2H2O (616,3 г, 2,73 моля) в водном 12 н. растворе НСl (500 мл), быстро добавляли при комнатной температуре к раствору 6-хлор-2-(этиламино)-3-нитропиридина (169,5 г, 0,84 моля) в АсОН (1,7 л). Через 20 мин смесь охлаждали до 0 С и добавляли воду (250 мл). Затем небольшими порциями добавляли NaOH (240 г). Образовавшуюся суспензию фильтровали, удаляя соли олова. Фильтрат разбавляли водой (3,5 л), раствор подщелачивали, добавляя водный 10 н. NaOH, а затем экстрагировали EtOAc (3 х 1,7 л).Объединенные органические слои промывали соляным раствором (1 л), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспрессхроматографией (гексан:ЕtOАс, 3:2), получая указанное в заголовке соединение (89,9 г, выход 62%) в виде твердого коричневого цвета: МС (ESI) m/z 172/174 (МН)+. в) 5-Бром-2-хлор-N-2-(этиламино)-6-хлор-3-пиридинил-3 пиридинкарбоксамид. Раствор 5-бром-2-хлор-3-пиридинкарбонилхлорида (30,0 г, 97,0 ммоля) в MeCN (100 мл) добавляли при комнатной температуре через канюлю к раствору 3-амино-6-хлор-2-(этиламино)пиридина (16,6 г,97,0 ммоля) в MeCN (180 мл), содержащему твердый NaHCO3 (14,2 г, 169 ммоля). Смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли воду (200 мл) и смесь перемешивали в течение 10 мин. Образовавшуюся суспензию фильтровали. Твердый продукт промывали Et2O (50 мл) и концентрировали из раствора пиридина (3 х 50 мл), получая указанное в заголовке соединение (28,4 г, выход 75%). г) 8-Бром-2-хлор-5,11-дигидро-11-этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. 1 М раствор NаГМДС в ТГФ (167,5 мл, 167,5 моля) медленно добавляли к раствору 5-бpoм-2-xлopN-2-(этилaминo)-6-xлop-3-пиpидинил-3-пиридинкарбоксамида (28,4 г, 72,8 ммоля) в пиридине (146 мл), нагретому до 50 С. Реакционную смесь перемешивали при 50 С в течение 1,5 ч. Затем смесь сливали на смесь вода/лед (1 л) и через 1 ч образовавшуюся суспензию фильтровали. Твердый продукт промывали водой и сушили при пониженном давлении, получая указанное в заголовке соединение (23,4 г, выход 91%). д) 8-Бром-2-хлор-5,11-дигидро-11-этил-5-метил-6H-дипиридо[3,2-b:2',3'-е] [1,4]диазепин-6-он. Твердый NaH (60%-ная масляная дисперсия, 3,46 г, 86,1 ммоля) добавляли в течение 30 мин к раствору 8-бром-2-хлор-5,11-дигидро-11-этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (23,4 г, 66,3 ммоля) в ДМФ (220 мл) при 50 С. Смесь перемешивали при 50 С в течение 1 ч, затем давали охладиться до комнатной температуры. Смесь сливали на воду (1 л) и образовавшуюся суспензию фильтровали. Твердый продукт последовательно промывали водой и гексаном, затем сушили при пониженном давлении, получая указанное в заголовке соединение (23,0 г, выход 94%). е) 2-Хлор-5,11-дигидро-11-этил-5-метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6 он. Аллилтрибутилолово (21,3 мл, 68,7 ммоля) и Рd(Рh3 Р)4 (3,61 г, 3,12 ммоля) добавляли к дегазированному (пропускание N2 через раствор в течение 30 мин) раствору 8-бром-2-хлор-5,11-дигидро-11-этил 5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (23,0 г, 62,5 ммоля) в ДМФ (312 мл). Смесь нагревали до 90 С в течение 2 ч. Смесь концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (гексан:ЕtOАс, 7:3), получая указанное в заголовке соединение (13,4 г, выход 65%). ж) 2-Хлор-5,11-дигидро-11-этил-8-(2-гидроксиэтил)-5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Озонированный кислород вносили в холодный (-78 С) раствор 2-хлор-5,11-дигидро-11-этил-5 метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (13,4 г, 40,7 ммоля) в МеОН (102 мл) и СН 2 Сl2 (102 мл) до полного исчезновения алкена. Раствор барботировали азотом, удаляя избыток О 3. Затем небольшими порциями добавляли твердый NaBH4 (2,69 г, 71,1 ммоля) и смеси давали нагреться до-8 005598 комнатной температуры. Через 1 ч добавляли водный насыщенный раствор NH4Cl (150 мл) и смесь перемешивали в течение 20 мин. Органические растворители удаляли при пониженном давлении. К водному раствору добавляли воду (100 мл). Раствор экстрагировали СНСl3 (3 х 200 мл). Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (ЕtOАс:СНСl3, 4:1), получая указанное в заголовке соединение (10,4 г,выход 77%). з) 2-Хлор-5,11-дигидро-11-этил-5-метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Диэтилазодикарбоксилат (ДЭАД) (4,26 мл, 27,0 ммолей) добавляли при комнатной температуре по каплям к раствору, содержащему 2-хлор-5,11-дигидро-11-этил-8-(2-гидроксиэтил)-5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он (6,00 г, 18,0 ммоля), 4-гидроксихинолин (3,92 г, 27,0 ммолей) и Рh3 Р (7,09 г, 27,0 ммолей) в ТГФ (90 мл). Смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (ЕtOАс:МеОН,95:5), получая указанное в заголовке соединение (6,22 г, выход 75%) в виде твердого вещества белого цвета: МС (ESI) m/z 460/462. Пример 3 (номер 6, табл. 1). 2-Хлор-11-циклопропил-5,11-дигидро-5-метил-8-2-(4-хинолинилокси) этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. а) 6-Хлор-2-(циклопропиламино)-3-нитропиридин. Раствор циклопропиламина (1,25 г, 22,0 ммоля) в толуоле (11 мл) добавляли в течение 10 мин к охлажденному на льду раствору 2,6-дихлор-3-нитропиридина (2,00 г, 10,4 ммоля) в толуоле (10 мл). Смесь перемешивали при 0 С в течение 1 ч при комнатной температуре в течение 2 ч. К смеси добавляли воду(50 мл) и разделяли фазы. Органический слой промывали соляным раствором (25 мл), сушили (MgSO4),фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией(гексан:ЕtOАс, 3:2), получая указанное в заголовке соединение (1,97 г, выход 89%) в виде твердого вещества желтого цвета. б) 2-Хлор-11-циклопропил-5,11-дигидро-8-(2-гидроксиэтил)-5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4] диазепин-6-он. Указанное в заголовке соединение получали из 6-хлор-2-(циклопропиламино)-3-нитропиридина согласно методу, аналогичному описанному в примере 2 для получения 11-этильного аналога. в) 2-Хлор-11-циклопропил-5,11-дигидро-5-метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2b:2',3'-е][1,4]диазепин-6-он. Диэтилазодикарбоксилат (ДЭАД) (85,6 мкл, 0,54 ммоля) добавляли при комнатной температуре по каплям к раствору, содержащему 2-хлор-11-циклопропил-5,11-дигидро-8-(2-гидроксиэтил)-5-метил-6Hдипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он (125 мг, 0,36 ммоля), 4-гидроксихинолин (78,9 мг, 0,54 ммоля) и Рh3 Р (143 мг, 0,54 ммоля) в ТГФ (1, 8 мл). Смесь перемешивали при комнатной температуре в течение 3 ч и затем концентрировали при пониженном давлении. Остаток частично очищали экспрессхроматографией (EtOAc:MeOH; 95:5). Твердый продукт дополнительно очищали с помощью ЖХВР с обращенной фазой (колонка CombiPrep ADS-AQ 50 х 20 мм, 5 мкм, 120 , 5-100% MeCN+0,10% ТФК/вода+0,10% ТФК в течение 25 мин), получая указанное в заголовке соединение в виде трифторацетата (17,7 г, выход 77% ) в виде твердого вещества белого цвета: МС (ESI) m/z 472/474 (МН)+. Пример 4 (номер 4, табл. 1). 5,11-Дигидро-11-этил-2-фтор-5-метил-8-2-(4-хинолинилокси)этил 6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. а) 2,6-Дифтор-3-нитропиридин. К смеси концентрированной серной кислоты (600 мл) и дымящей азотной кислоты (90%, 400 мл) в ледяной бане (внутренняя температура поддерживалась на уровне 5-10 С) добавляли по каплям 2,6 дифторпиридин (200 г, 1,74 моля). Образовавшуюся смесь перемешивали в течение ночи при комнатной температуре. Смесь медленно сливали на 3 кг льда и экстрагировали Et2O (2 х 2 л). Объединенные органические слои промывали водным 1,5 н. раствором NaOH (2 х 1 л), затем водным насыщенным растворомNаНСО 3 (400 мл) до тех пор пока значение рН не достигало примерно 8-9. Органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении до постоянной массы (до удаления не прореагировавшего 2,6-дифторпиридина: 10-12%). В результате получили указанное в заголовке соединение в виде жидкости желтого цвета (207,3 г, выход 74% ). б) 2-(Этиламино)-6-фтор-3-нитропиридин. К раствору 2,6-дифтор-3-нитропиридина (45,7 г, 285 ммолей) в ТГФ (500 мл) при -40 С добавляли по каплям раствор этиламина (25,7 г, 570 ммолей) в ТГФ (250 мл). Через 30 мин реакционную смесь концентрировали при пониженном давлении и остаток растворяли в ЕtOАс. Органическую фазу промывали соляным раствором, сушили (MgSO4), фильтровали и концентрировали. Образовавшийся продукт желтого цвета очищали экспресс-хроматографией (15% EtOAc в гексане), получая указанное в заголовке соединение (43,2 г, выход 82%) в виде твердого вещества желтого цвета. в) 3-Амино-2-(этиламино)-6-фторпиридин. Раствор 2-(этиламино)-6-фтор-3-нитропиридина (43,2 г, 230 ммолей) в ТГФ (1 л) перемешивали в течение ночи при комнатной температуре в атмосфере водорода (1 атм) в присутствии 20% Pd(OH)2/C(4,35 г). Катализатор удаляли фильтрацией через диатомовую землю. Фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (36,3 г, выход 95%) в виде твердого вещества черного цвета. г) 2-Хлор-N-2-(этиламино)-6-фтор-3-пиридинил-5-бром-3-пиридинкарбоксамид. К охлажденному раствору (4 С) 3-амино-2-(этиламино)-6-фторпиридина (31,0 г, 200 ммолей) вMeCN (160 мл) добавляли твердый NaHCO3 (50,4 г, 600 ммолей). Через 15 мин добавляли раствор 5 бром-2-хлор-3-пиридинкарбонилхлорида (1 экв, 200 ммолей) в MeCN (155 мл). После выдерживания в течение 60 мин при комнатной температуре реакционную смесь сливали на воду (1,2 л) и перемешивали в течение 30 мин. Образовавшееся твердое вещество фильтровали, сушили в течение ночи при пониженном давлении при 50 С. В результате получили указанное в заголовке соединение (73,7 г, выход 99%) в виде твердого вещества черного цвета. д) 8-Бром-5,11-дигидро-11-этил-2-фтор-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. К раствору 2-хлор-N-2-(этиламино)-6-фтор-3-пиридинил-5-бром-3-пиридинкарбоксамида (73,5 г,216 молей) в пиридине (435 мл) при 50 С добавляли по каплям 1 М раствор NaГМДC в ТГФ (520 мл, 520 ммолей). Через 10 мин реакционной смеси давали охладиться до комнатной температуры, затем сливали на ледяную воду (2 л). Образовавшийся твердый продукт фильтровали, промывали водой и затем гексаном. Твердый продукт сушили при пониженном давлении, получая указанное в заголовке соединение(50,6 г, выход 69%) в виде твердого вещества темно-зеленого цвета. е) 8-Бром-5,11-дигидро-11-этил-2-фтор-5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. К раствору 8-бром-5,11-дигидро-11-этил-2-фтор-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (44 г, 130,5 ммоля) в ДМФ (520 мл) добавляли NaH (4,28 г, 178 ммолей) и смесь нагревали до 50 С в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры и обрабатывали МеI (24,4 мл, 522 ммоля). Через 1,5 ч реакционную смесь сливали на ледяную воду. Твердый продукт фильтровали, промывали водой и затем гексаном, сушили при пониженном давлении, получая указанное в заголовке соединение (43,2 г, выход 94%) в виде твердого вещества темно-серого цвета. ж) 5,11-Дигидро-11-этил-2-фтор-5-метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6 он. Аллилтрибутилолово (32,0 мл, 103,4 ммоля) и Рd(Рh3 Р)4 (5,43 г, 4,70 ммоля) добавляли к дегазированному (через раствор в течение 45 мин пропускали N2) раствору 8-бром-5,11-дигидро-11-этил-2-фтор 5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (33,0 г, 94,0 ммоля) в ДМФ (470 мл). Для завершения реакции вводили дополнительное количество Рd(Рh3)4 (1,09 г, 0,94 ммоля вносили в реакционную смесь через 1, 2, 3, 4, 5 ч ). Смесь нагревали до 90 С в течение 6 ч. Смесь концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (гексан:ЕtOАс, от 8:2 до 7:3), получая указанное в заголовке соединение (22,4 г, выход 76%). з) 5,11-Дигидро-11-этил-2-фтор-8-(2-гидроксиэтил)-5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Струeй озонированного кислорода в течение 3 ч барботировали холодный (-78 С) раствор 5,11 дигидро-11-этил-2-фтор-5-метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (22,38 г,71,6 ммоля) в СН 2 Сl2 (100 мл) и МеОН (100 мл). Затем раствор барботировали струeй N2 в течение 15 мин и затем к раствору добавляли твердый NaBH4 (5,05 г, 133 ммоля). Реакционной смеси давали нагреться до комнатной температуры. Через 1 ч в реакционную смесь вносили дополнительную порциюNaBH4 (1,62 г, 43,0 моля). После выдерживания в течение еще 1 ч добавляли насыщенный раствор NH4Cl(150 мл) и смесь перемешивали при комнатной температуре в течение 30 мин. Органические растворители удаляли при пониженном давлении. Добавляли воду (200 мл) и смесь экстрагировали СНСl3 (3 х 300 мл). Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (ЕtOАс:СНСl3, 4:1), получая указанное в заголовке соединение (19,7 г, выход 72%) в виде твердого вещества белого цвета. и) 5,11-Дигидро-11-этил-2-фтор-5-метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Диэтилазодикарбоксилат (ДЭАД) (14,3 мл, 91,0 ммоль) добавляли при комнатной температуре по каплям к раствору, содержащему 5,11-дигидро-11-этил-2-фтор-8-(2-гидроксиэтил)-5-метил-6H-дипиридо[3,2-b:2',3'-е] [1,4]диазепин-6-он (19,2 г, 60,7 ммоля), 4-гидроксихинолин (13,2 г, 91,0 ммоль) и Рh3 Р (23,9 г, 91,0 ммоль) в ТГФ (300 мл). Смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (ЕtOАс:МеОН; 95:5), получая указанное в заголовке соединение (17,9 г, выход 67%) в виде твердого продукта белого цвета: МС (ESI) m/z 444 (МН)+. Пример 5 (номер 1, табл. 1). 2-Хлор-5,11-дигидро-11-этил-4-метил-8-2-(4-хинолинилокси)этил 6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. а) 2-Хлор-N-(2,6-дихлор-4-метил-3-пиридинил)-3-пиридинкарбоксамид.NаНСО 3 (21,72 г, 258,6 ммоля) добавляли к раствору 3-амино-2,6-дихлор-4-метилпиридина (41,61 г,235,1 ммоля; полученному согласно методу, описанному у К.G. Grozinger и др. J. Heterocyclic Chem. 32,259-263, 1995) в MeCN (350 мл) и образовавшуюся суспензию перемешивали в течение 15 мин. Затем в-10 005598 течение 30 мин вводили раствор 2-хлорникотинилхлорида (43,44 г, 246,8 ммоля) в MeCN (500 мл). Образовавшуюся суспензию перемешивали при комнатной температуре. Через 24 ч было установлено, что раствор являлся кислым и поэтому в него вводили дополнительное количество NаНСО 3 (3,00 г, 35,7 ммоля). Затем суспензию перемешивали при комнатной температуре в течение еще 2 дней. Затем смесь сливали на смесь воды (2 л) и льда (200 г) и перемешивали в течение 20 мин. Твердый продукт фильтровали и промывали водой (500 мл) и затем образовавшийся твердый продукт сушили над P2O5 при пониженном давлении, получая указанное в заголовке соединение (62,55 г, выход 84%) в виде порошка белого цвета. б) N-(2,6-Дихлор-4-метил-3-пиридинил)-2-(этиламино)-3-пиридинкарбоксамид. Раствор 2-хлор-N-(2,6-дихлор-4-метил-3-пиридинил)-3-пиридинкарбоксамида (63,17 г, 194,0 ммоля) и этиламина (28,0 г, 583 ммоля) в ксилоле (250 мл) перемешивали при температуре 120-125 С в стальном автоклаве в течение 7 ч. Образовавшуюся суспензию сливали на воду (1 л), перемешивали в течение 15 мин и фильтровали. Остаток растворяли в ЕtOАс и промывали водой (трижды), соляным раствором и сушили над MgSO4. Фильтрат экстрагировали EtOAc и объединенные органические экстракты промывали водой и соляным раствором и сушили над MgSO4. Затем две фракции объединяли и избыток ксилола удаляли совместной перегонкой с бензолом, получая указанное в заголовке соединение (61,98 г, твердое вещество розового цвета) в виде основного компонента смеси соединений. Неочищенный продукт использовали непосредственно в следующей реакции. в) 5-Бром-N-(2,6-дихлор-4-метил-3-пиридинил)-2-(этиламино)-3-пиридинкарбоксамид. Твердый КОАс (22,2 г, 229 ммолей) добавляли к неочищенному раствору N-(2,6-дихлор-4-метил-3 пиридинил)-2-(этиламино)-3-пиридинкарбоксамида (61,98 г, 190 ммолй) в АсОН (600 мл). Затем в течение 30 мин добавляли раствор Вr2 (9,7 мл, 191 ммоль). После перемешивания в течение 30 мин вводили дополнительные количества КОАс и Вr2 до тех пор пока по данным ТСХ не оставалось исходного продукта (всего 11,2 г КОАс и 5,91 мл Вr2). Затем раствор сливали на воду (2 л) и перемешивали в течение 20 мин. Твердый продукт удаляли фильтрацией и затем растворяли в Et2O (1 л). Эфирный раствор промывали водой (трижды), водным насыщенным раствором NaHCO3, соляным раствором, затем сушили(MgSO4), получая смесь, содержащую в качестве основного компонента указанное в заголовке соединение (77,7 г, пена желтого цвета). Продукт непосредственно использовали в следующей реакции. г) 8-Бром-2-хлор-5,11-дигидро-11-этил-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Раствор неочищенного 5-бром-N-(2,6-дихлор-4-метил-3-пиридинил)-2-(этиламино)-3-пиридинкарбоксамида (77,7 г, ммолей) в безводном пиридине (1,0 л) нагревали до 50 С. Затем по каплям добавляли 1 М раствор NаГМДС в ТГФ (418 мл, 418 ммолей) и перемешивание продолжали в течение еще 15 мин. После охлаждения до комнатной температуры добавляли воду (50 мл) и смесь концентрировали до 2/3 объема с помощью роторного испарителя. Затем остаток сливали на воду (3 л). После фильтрации получили указанное в заголовке соединение (23,44 г) в виде твердого вещества рыжевато-коричневого цвета. Фильтрат экстрагировали EtOAc (трижды) и объединенные экстракты промывали водой и соляным раствором, затем сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (гексан:ЕtOАс, 9:1 или СНСl3), получая дополнительное количество указанного в заголовке соединения (14,06 г, всего 37,50 г, выход после трех стадий 50%). д) 2-Хлор-5,11-дигидро-11-этил-4-метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6 он. Раствор 8-бром-2-хлор-5,11-дигидро-11-этил-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она(12,6 г, 34,3 ммоля) в ДМФ (25,0 мл) дегазировали при пониженном давлении в течение 20 мин. Добавляли Рd(РРh3)4 (775 мг, 0,7 ммоля), а затем аллилтрибутилолово (12,5 мл, 41,1 ммоля). После дегазации при пониженном давлении в течение 10 мин смесь нагревали до 100 С в течение 7 ч. Затем смесь концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (гексан:ЕtOАс,19:1), получая указанное в заголовке соединение (8,04 г, выход 71% ) в виде твердого вещества желтого цвета. е) 2-Хлор-5,11-дигидро-11-этил-8-(2-гидроксиэтил)-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Струю О 3 пропускали через холодный (-78 С) раствор 2-хлор-5,11-дигидро-11-этил-4-метил-8-(2 пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (8,04 г, 24,4 ммоля) и Судана III в СН 2 Сl2 (120 мл) и МеОН (120 мл). После превращения раствора розового цвета в коричневый, раствор барботировалиO2 в течение 10 мин. Затем добавляли NaBH4 (1,12 г, 29,4 ммоля) и раствору давали нагреться до комнатной температуры. Через 30 мин вносили дополнительное количество NaBH4 (500 мг). Через 1 ч добавляли водный насыщенный раствор NH4Cl и смесь перемешивали в течение 20 мин. Раствор концентрировали при пониженном давлении и экстрагировали CH2Cl2 (трижды). Объединенные органические экстракты промывали соляным раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (СНСl3:ЕtOН, 97:3), получая указанное в заголовке соединение (6,01 г, выход 74%) в виде твердого вещества белого цвета. ж) 2-Хлор-5,11-дигидро-11-этил-4-метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е]-11 005598 Диэтилазодикарбоксилат (ДЭАД) (83 мкл, 0,53 моля) добавляли при комнатной температуре по каплям к раствору, содержащему 2-хлор-5,11-дигидро-11-этил-8-(2-гидроксиэтил)-4-метил-6H-дипиридо(138 мг, 0,53 ммоля) в ТГФ (2,5 мл). Смесь перемешивали при комнатной температуре в течение 3 ч, затем концентрировали при пониженном давлении. Остаток растворяли в ЕtOАс (60 мл) и раствор последовательно промывали 1 н. водным раствором NaOH (3 х 10 мл) и соляным раствором (15 мл), затем сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспрессхроматографией (ЕtOАс:МеОН, 9:1), получая указанное в заголовке соединение (52 мг, выход 30%) в виде твердого вещества белого цвета: МС (ESI) m/z 460/462 (МН)+. Пример 6 (номер 7, табл. 1). 5,11-Дигидро-11-этил-2-фтор-4-метил-8-2-(4-хинолинилокси)этил 6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. а) 8-Бром-5,11-дигидро-11-этил-2-(4-метоксифенил)метиламино-4-метил-6H-дипиридо[3,2b:2',3'-е][1,4]диазепин-6-он. 8-Бром-2-хлор-5,11-дигидро-11-этил-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он (2,50 г,6,80 ммоля) растворяли в диоксане (10 мл) в стеклянном толстостенном сосуде и к раствору добавляли 4 метоксибензиламин (4,0 мл, 30,6 моля). Смесь нагревали до 170 С в течение 5 дней. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток растворяли в ЕtOАс и промывали 10%-ным водным раствором NH4Cl, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении, получая масло коричневого цвета, которое хроматографировали на силикагеле (гексан:ЕtOАС, 4:1). В результате получили указанное в заголовке соединение в виде твердого вещества желтого цвета (1,35 г, выход 42%). б) 2-Амино-8-бром-5,11-дигидро-11-этил-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. 8-Бром-5,11-дигидро-11-этил-2-(4-метоксифенил)метиламино-4-метил-6H-дипиридо[3,2-b:2',3'е][1,4]диазепин-6-он (6,52 г, 18,7 ммоля) растворяли в трифторуксусной кислоте (100 мл) и раствор перемешивали с помощью магнитной мешалки при комнатной температуре в течение 18 ч. Трифторуксусную кислоту выпаривали при пониженном давлении и остаток сливали на смесь, содержащую воду (450 мл), концентрированный NH4OH (50 мл) и ЕtOАс (400 мл). Смесь перемешивали с помощью магнитной мешалки в течение 2 ч при комнатной температуре. Органическую фазу концентрировали при пониженном давлении. Осадок желтого цвета отфильтровывали и промывали водой, сушили при пониженном давлении, получая 7,51 г твердого вещества желтого цвета, которое растирали в Et2O, получая указанное в заголовке соединение (5,12 г, выход 105%) в виде твердого вещества желтого цвета. в) 8-Бром-5,11-дигидро-11-этил-2-фтор-4-метил-6H-дипиридо[3,2-b:2',3'-е] [1,4]диазепин-6-он. В пластмассовый сосуд вносили 2-амино-8-бром-5,11-дигидро-11-этил-4-метил-6H-дипиридо[3,2b:2',3'-е][1,4]диазепин-6-он (5,10 г, 14,6 ммоля). Добавляли HFпиридин (100 г) и образовавшуюся суспензию охлаждали до 0 С. В течение 30 мин добавляли в виде нескольких порций нитрит натрия (1,12 г,16,1 ммоля), получая раствор фиолетового цвета. Затем смесь перемешивали в течение 16 ч при комнатной температуре. Реакционную смесь сливали на смесь льда и 6 н. NaOH. Суспензию бежевого цвета экстрагировали ЕtOАс. Органические экстракты объединяли, сушили над MgSO4, фильтровали и концентрировали. Остаток растирали с Еt2O/гексаном, получая указанное в заголовке соединение (4,30 г, выход 84%). г) 5,11-Дигидро-11-этил-2-фтор-4-метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6 он. Аллилтрибутилолово (7,77 мл, 25,1 ммоля) и Рd(Рh3 Р)4 (1,32 г, 1,14 ммоля) добавляли к дегазированному раствору (через раствор в течение 30 мин пропускали N2) 8-бром-5,11-дигидро-11-этил-2-фтор 4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (8,0 г, 22,8 ммоля) в ДМФ (114 мл). Смесь нагревали до 90 С в течение 3 ч. Смесь сливали на воду (250 мл) и экстрагировали EtOAc (4 х 250 мл). Объединенные органические слои промывали 20%-ным водным раствором NH4OH (250 мл) и соляным раствором (250 мл), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (гексан:ЕtOАс, от 4:1 до 1:1), получая указанное в заголовке соединение (3,16 г, выход 51%). д) 5,11-Дигидро-11-этил-2-фтор-8-(2-гидроксиэтил)-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Струeй озонированного кислорода в течение 45 мин барботировали охлажденный (-78 С) раствор 5,11-дигидро-11-этил-2-фтор-4-метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (3,16 г, 10,1 ммоля) в CH2Cl2 (25 мл) и МеОН (25 мл). После этого раствор барботировали струeй N2 в течение 15 мин и затем к раствору добавляли твердый NaBH4 (669 мг, 17,1 ммоля). Реакционной смеси давали нагреться до комнатной температуры. Через 2 ч добавляли водный насыщенный раствор NH4Cl (50 мл) и смесь перемешивали при комнатной температуре в течение 15 мин. Органические растворители удаляли при пониженном давлении. Оставшийся водный раствор экстрагировали СНСl3 (3 х 100 мл). Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (ЕtOАс:СНСl3, 1:1), получая указанное в заголовке соединение (2,40 г, выход 75%) в виде твердого вещества белого цвета.[1,4]диазепин-6-он. Диизопропилазодикарбоксилат (ДИАД) (117 мкл, 0,59 ммоля) добавляли по каплям к раствору, содержащему 5,11-дигидро-11-этил-2-фтор-8-(2-гидроксиэтил)-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он (125 мг, 0,40 ммоля), 4-гидроксихинолин (86,0 мг, 0,59 ммоля) и Рh3 Р (156 мг, 0,59 ммоля) в ТГФ (1,9 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 3 ч, затем концентрировали при пониженном давлении. Остаток растворяли в EtOAc (40 мл) и раствор последовательно промывали 3 н. водным раствором NaOH (3 х 20 мл), водным насыщенным растворомNH4Cl (15 мл), соляным раствором (15 мл), затем сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток частично очищали экспресс-хроматографией (ЕtOАс:МеОН; 95:5). Твердый продукт дополнительно очищали с помощью ЖХВР с обращенной фазой (колонка CombiPrep ADSAQ 50 х 20 мм, 5 мкм, 120 , 5-100% MeCN+0,10% ТФК/вода+0,10% ТФК в течение 25 мин), получая трифторацетат указанного в заголовке соединения (106 мг, выход 48%) в виде твердого вещества белого цвета: МС (ESI) m/z 444 (МН)+. Пример 7 (номер 5, табл. 1). 11-(Циклопропил)-5,11-дигидро-2-фтор-4-метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. а) 11-Циклопропил-5,11-дигидро-2-фтор-8-(2-гидроксиэтил)-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4] диазепин-6-он. Указанное в заголовке соединение получали из 8-бром-2-хлор-11-циклопропил-5,11-дигидро-4 метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она с помощью метода, аналогичного методу получения соответствующего 11-этильного аналога, который описан в примере 6. б) 11-Циклопропил-5,11-дигидро-2-фтор-4-метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2b:2',3'-е][1,4]диазепин-6-он. Диизопропилазодикарбоксилат (ДИАД) (131 мкл, 0,66 ммоля) добавляли при комнатной температуре по каплям к раствору, содержащему 11-(циклопропил)-5,11-дигидро-2-фтор-8-(2-гидроксиэтил)-4 метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он (140 мг, 0,44 ммоля), 4-гидроксихинолин (96,4 мг,0,66 ммоля) и Рh3 Р (124 мг, 0,66 ммоля) в ТГФ (2,2 мл). Смесь перемешивали при комнатной температуре в течение 3 ч, затем концентрировали при пониженном давлении. Остаток растворяли в EtOAc (60 мл) и раствор последовательно промывали 3 н. водным раствором NaOH (3 х 20 мл), водным насыщенным раствором NH4Cl (20 мл) и соляным раствором (20 мл), затем сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток частично очищали экспресс-хроматографией(ЕtOАс:МеОН; 95:5). Твердый продукт дополнительно очищали с помощью ЖХВР с обращенной фазой(колонка CombiPrep ADS-AQ 50 х 20 мм, 5 мкм, 120 , 5-100% MeCN+0,10% ТФК/водa+0,10% ТФК в течение 25 мин), получая трифторацетат указанного в заголовке соединения (104 мг, выход 43%) в виде твердого вещества белого цвета: МС (ESI) m/z 456 (МН)+. Пример 8 (номер 8, табл. 1). 5,11-Дигидро-11-этил-4-метил-8-2-(4-хинолинилокси)этил-6Hдипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. а) 5,11-Дигидро-11-этил-8-(2-гидроксиэтил)-4-метил-6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Смесь, содержащую 2-хлор-5,11-дигидро-11-этил-8-(2-гидроксиэтил)-4-метил-6H-дипиридо[3,2b:2',3'-е][1,4]диазепин-6-он (168 мг, 0,51 ммоля), формиат аммония (318 мг, 5,1 ммоля) и 10% Pd/C (50 мг) в EtOH (6 мл), перемешивали при комнатной температуре. Через 2 ч вносили дополнительные количества формиата аммония (200 мг, ммоль) и 10% Pd/C (50 мг). После перемешивания в течение еще 24 ч реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в СНСl3 и смесь промывали водным насыщенным раствором NaHCO3, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение (122 мг, выход 81%) в виде масла желтого цвета. б) 5,11-Дигидро-11-этил-4-метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Диизопропилазодикарбоксилат (ДИАД) (42 мкл, 0,21 ммоля) добавляли при комнатной температуре по каплям к раствору, содержащему 5,11-дигидро-11-этил-8-(2-гидроксиэтил)-4-метил-6H-дипиридо[3,2b:2',3'-е][1,4]диазепин-6-он. (42,0 мг, 0,14 ммоля), 4-гидроксихинолин (31 мг, 0,21 ммоля) и Рh3 Р (55,4 мг,0,21 ммоля) в ТГФ (0,7 мл). Смесь перемешивали при комнатной температуре в течение 2 ч, затем концентрировали при пониженном давлении. Остаток растворяли в ЕtOАс (60 мл) и раствор последовательно промывали 3 н. водным раствором NaOH (3 х 20 мл), водным насыщенным раствором NH4Cl (20 мл) и соляным раствором (20 мл), затем сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток частично очищали экспресс-хроматографией (ЕtOАс:МеОН; 95:5). Твердый продукт дополнительно очищали с помощью ЖХВР с обращенной фазой (колонка CombiPrep ADS-AQ 50 х 20 мм,5 мкм, 120 , 5-100% MeCN+0,10% ТФК/вода+0,10% ТФК в течение 25 мин), получая трифторацет указанного в заголовке соединения (14,2 мг, выход 19%) в виде твердого вещества белого цвета: МС (ESI)-13 005598 а) 5-Бром-2-хлор-N-(2,6-дихлор-4-метил-3-пиридинил)-3-пиридинкарбоксамид. 3-Амино-2,6-дихлор-4-метилпиридин (0,51 г, 2,85 ммоля) растворяли в толуоле (35 мл) и добавляли пиридин (0,27 мл, 3,28 ммоля). Затем по каплям в течение 30 мин добавляли 5-бром-2-хлор-3 пиридинкарбонилхлорид (0,80 г, 3,14 ммоля). Образовавшуюся смесь перемешивали при комнатной температуре в течение 1 ч, разбавляли водой и экстрагировали толуолом (дважды). Объединенные органические экстракты сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Образовавшееся вязкое масло растирали с CH2Cl2, и с помощью вакуумной фильтрации собирали твердое вещество белого цвета, получая указанное в заголовке соединение (0,41 г, выход 36%). б) 5-Бром-2-(циклопропиламино)-N-(2,6-дихлор-4-метил-3-пиридинил)-3-пиридинкарбоксамид. Раствор 5-бром-2-хлор-N-(2,6-дихлор-4-метил-3-пиридинил)-3-пиридинкарбоксамида (3,00 г, 7,61 ммоля) и циклопропиламина (2,11 мл, 30,5 ммоля) в ксилоле выдерживали в запечатанной пробирке при 120 С в течение 24 ч. Реакционную смесь охлаждали до 0 С, разбавляли водой и экстрагировали EtOAc(дважды). Объединенные органические экстракты сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Образовавшийся остаток очищали растиранием (Еt2O:гексан, 3:1), получая указанное в заголовке соединение (2,35 г, выход 75%) в виде твердого вещества светло-желтого цвета. в) 8-Бром-2-хлор-11-циклопропил-5,11-дигидро-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6 он. Раствор 5-бром-2-(циклопропиламино)-N-(2,6-дихлор-4-метил-3-пиридинил)-3-пиридинкарбоксамида (105 мг, 0,254 ммоля) в пиридине (3,0 мл) нагревали до 50 С и добавляли 1 М раствор NaГМДC в ТГФ (0,53 мл, 0,53 ммоля). Образовавшийся раствор перемешивали при 50 С в течение 3 ч. Реакцию прекращали, добавляя водный насыщенный раствор NH4Cl, разбавляли водой и экстрагировали EtOAc(трижды). Объединенные органические экстракты промывали соляным раствором, сушили (MgSO4),фильтровали и концентрировали при пониженном давлении. Образовавшийся остаток очищали экспрессхроматографией (гексан: EtOAc, 7:3), получая указанное в заголовке соединение (32 мг, выход 33%). г) 2-Хлор-11-циклопропил-5,11-дигидро-4-метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4] диазепин-6-он. Раствор 8-бром-2-хлор-11-циклопропил-5,11-дигидро-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (0,046 г, 0,122 ммоля) в ДМФ (2,0 мл) дегазировали, барботируя N2 в течение 10 мин. Добавляли Рd(РРh3)4 (2,8 мг, 0,003 ммоля) и аллилтрибутилолово (0,45 мл, 0,146 ммоля) и образовавшийся раствор нагревали до 90 С в течение 2,5 ч. Реакционную смесь разбавляли водой и экстрагировали CH2Cl2(трижды) и EtOAc (дважды). Объединенные органические экстракты сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Образовавшийся остаток очищали экспресс-хроматографией (от гексана до смеси гексан:ЕtOАс, 7:3), получая указанное в заголовке соединение (0,033 г,выход 80%). д) 2-Хлор-11-циклопропил-5,11-дигидро-8-(2-гидроксиэтил)-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4] диазепин-6-он. Холодный (-78 С) раствор 2-хлор-11-циклопропил-5,11-дигидро-4-метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (0,82 г, 2,43 ммоля) в СН 2 Сl2 (7,5 мл) и МеОН (7,5 мл) насыщали О 3 до приобретения раствором светло-голубой окраски. Реакционную смесь выдерживали при -78 С в течение 10 мин, затем добавляли NaBH4 (0,23 г, 9,71 ммоля) и смеси давали медленно нагреться до комнатной температуры в течение 2 ч. Реакцию прекращали, добавляя водный 10%-ный раствор лимонной кислоты и экстрагировали EtOAc (трижды). Органические экстракты объединяли, промывали соляным раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Образовавшийся остаток очищали экспресс-хроматографией (МеОН:СН 2 Сl2, от 2 до 10%), получая указанное в заголовке соединение (0,41 г, выход 60%) в виде твердого вещества белого цвета. е) 11-Циклопропил-5,11-дигидро-8-(2-гидроксиэтил)-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Указанное в заголовке соединение получали с помощью метода, аналогичного методу, описанному в примере 8a для получения 11-этильного аналога. ж) 11-Циклопропил-5,11-дигидро-4-метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Диизопропилазодикарбоксилат (ДИАД) (167 мкл, 0,85 ммоля) добавляли при комнатной температуре по каплям к раствору, содержащему 11-(циклопропил)-5,11-дигидро-8-(2-гидроксиэтил)-4-метил 6H-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-он (175 мг, 0,57 ммоля), 4-гидроксихинолин (123 мг, 0,85 ммоля) и Рh3 Р (223 мг, 0,85 ммоля) в ТГФ (2,8 мл). Смесь перемешивали при комнатной температуре в течение 3 ч, затем концентрировали при пониженном давлении. Остаток растворяли в ЕtOАс (60 мл) и раствор последовательно промывали 3 н. водным раствором NaOH (3x20 мл), водным насыщенным раствором NH4Cl (20 мл) и соляным раствором (20 мл), затем сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (EtOAc:MeOH; 95:5), получая указанное в заголовке соединение (74 мг, выход 30%) в виде твердого вещества белого цвета: МС (ESI)-14 005598 Пример 10 (номер 21, табл. 1). 5,11-Дигидро-11-этил-5-метил-8-2-(1-оксидо-4-хинолинил)окси этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Твердую мХПБК (80-85%, метахлорпербензойная кислота) (2,21 г, 10,2 ммоля) добавляли при комнатной температуре к раствору 5,11-дигидро-11-этил-5-метил-8-2-(4-хинолинилокси)этил-6Hдипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (2,56 г, 6,02 ммоля) в CH2Cl2 (30 мл) и ТГФ (30 мл) (следует отметить, что реакцию можно осуществлять только в CH2Cl2). Смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали при пониженном давлении. Остаток растворяли в СНСl3(400 мл), раствор последовательно промывали водным 10%-ным раствором Nа 2S2 О 3 (3 х 75 мл), водным насыщенным раствором NaHCO3 (3 х 75 мл) и соляным раствором (75 мл), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией(СНСl3:МеОН, 9:1), получая указанное в заголовке соединение (2,01 г, выход 76%) в виде твердого вещества белого цвета: МС (ESI) m/z 442 (МН)+. Пример 11 (номер 17, табл. 1). 2-Хлор-5,11-дигидро-11-этил-5-метил-8-2-(1-оксидо-4-хинолинил) окси-этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. С помощью процесса, идентичного описанному в примере 10, из 2-хлор-5,11-дигидро-11-этил-5 метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-она (2,70 г, 5,87 ммоля) получали указанное в заголовке соединение (2,05 г, выход 73%) в виде твердого вещества белого цвета: МС (ESI) m/z 476/478 (МН)+. Пример 12 (номер 11, табл. 1). 5,11-Дигидро-11-этил-2-фтор-5-метил-8-2-(1-оксидо-4-хинолинил) оксиэтил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Твердую мХПБК (457 мг, 2,12 ммоля) добавляли к раствору 5,11-дигидро-2-фтор-11-этил-5-метил 8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (470 мг, 1,06 ммоля) в СН 2 Сl2 (10 мл). Смесь перемешивали при комнатной температуре в течение 13 ч, затем концентрировали при пониженном давлении. Остаток растворяли в СНСl3 (75 мл), раствор последовательно промывали водным 10%-ным раствором Nа 2S2 О 3 (3x25 мл), водным насыщенным раствором NaHCO3 (3 х 25 мл) и соляным раствором (25 мл), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (СНСl3:МеОН, от 19:1 до 9:1), получая указанное в заголовке соединение (250 мг, выход 51%) в виде твердого вещества белого цвета: МС (ESI) m/z 460(МН)+. Пример 13 (номер 10, табл. 1). 2-Хлор-11-циклопропил-5,11-дигидро-5-метил-8-2-(1-оксидо-4 хинолинил)оксиэтил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Твердую мХПБК (177 мг, 0,82 ммоля) добавляли к раствору 2-хлор-11-циклопропил-5,11-дигидро 5-метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (143 мг, 0,30 ммоля) в СН 2 Сl2 (3 мл) и ТГФ (3 мл). Смесь перемешивали при комнатной температуре в течение 14 ч, затем концентрировали при пониженном давлении. Остаток растворяли в EtOAc (50 мл), раствор последовательно промывали водным раствором 10% Na2S2O3 (3 х 10 мл), водным насыщенным раствором NaHCO3(3 х 10 мл) и соляным раствором (10 мл), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток частично очищали экспресс-хроматографией (от ЕtOАс:МеОН, 95:5 до СН 2 Сl2:МеОН, 9:1). Неочищенный продукт дополнительно очищали с помощью ЖХВР с обращенной фазой (колонка CombiPrep ADS-AQ 50 х 20 мм, 5 мкм, 120 , 5-100% MeCN+0,10% ТФК/вода+0,10% ТФК в течение 25 мин). Очищенные фракции обрабатывали водным насыщенным раствором NaHCO3,экстрагировали EtOAc и раствор сушили (MgSO4), фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение (28,3 мг, выход 19%) в виде твердого вещества белого цвета: МС (ESI) m/z 488/490 (МН)+. Пример 14 (номер 14, табл. 1). 2-Хлор-5,11-дигидро-11-этил-4-метил-8-2-(1-оксидо-4-хинолинил) оксиэтил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Твердую мХПБК (46,9 мг, 0,22 ммоля) добавляли к раствору 2-хлор-5,11-дигидро-11-этил-5-метил 8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (50,0 мг, 0,11 ммоля) в СН 2 Сl2 (2,2 мл). Смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали при пониженном давлении. Остаток растворяли в EtOAc (50 мл), раствор последовательно промывали 10%-ным водным раствором Na2S2O3 (3 х 10 мл), водным насыщенным раствором NаНСО 3 (3 х 10 мл) и соляным раствором (10 мл), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью ЖХВР с обращенной фазой (колонка CombiPrep ADS-AQ 50 х 20 мм, 5 мкм, 120 , 5-100% MeCN+0,10% ТФК/вода+0,10% ТФК в течение 25 мин). Очищенные фракции обрабатывали водным насыщенным раствором NаНСО 3, экстрагировали EtOAc и раствор сушили (MgSO4),фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение(32,5 мг, выход 63%) в виде твердого вещества белого цвета: МС (ESI) m/z 488/490 (МН)+. Пример 15 (номер 9, табл. 1). 5,11-Дигидро-11-этил-2-фтор-4-метил-8-2-(1-оксидо-4-хинолинил) оксиэтил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. С помощью процесса, идентичного описанному в примере 14, из 5,11-дигидро-11-этил-2-фтор-4 метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (125 мг, 0,28 ммоля)-15 005598 получали указанное в заголовке соединение (40 мг, выход 31%) в виде твердого вещества белого цвета: МС (ESI) m/z 460 (МН)+. Пример 16 (номер 12, табл. 1). 5,11-Дигидро-11-этил-4-метил-8-2-(1-оксидо-4-хинолинил)окси этил 6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Твердую мХПБК (163 мг, 0,75 ммоля) добавляли к раствору 5,11-дигидро-11-этил-4-метил-8-2-(4 хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (144 мг, 0,34 ммоля) в CH2Cl2 (7,5 мл). Раствор перемешивали при комнатной температуре в течение 2 ч. Смесь разбавляли СН 2 Сl2 (125 мл),промывали последовательно водным 10%-ным раствором Na2S2O3 (2x25 мл), водным насыщенным раствором NаНСО 3 (2 х 25 мл) и соляным раствором (25 мл), затем сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (СН 2 Сl2:МеОН, 10:1),получая указанное в заголовке соединение (102 мг, выход 68%) в виде твердого вещества белого цвета: МС (ESI) m/z 442 (МН). Пример 17 (номер 15, табл. 1). 11-Циклопропил-5,11-дигидро-4-метил-8-2-(1-оксидо-4-хинолинил)оксиэтил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. С помощью процесса, идентичного описанному в примере 14, из 11-циклопропил-5,11-дигидро-4 метил-8-2-(4-хинолинилокси)этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (37,1 мг, 0,085 ммоля) получали указанное в заголовке соединение (35 мг, выход 91%) в виде твердого вещества белого цвета: МС (ESI) m/z 454 (МН)+. Пример 18 (номер 25, табл. 1). 2-Хлор-5,11-дигидро-11-этил-4-метил-8-2-(5-хинолинилокси)этил 6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Диэтилазодикарбоксилат (ДЭАД) (77 мкл, 0,49 ммоля) добавляли при комнатной температуре по каплям к раствору, содержащему 2-хлор-5,11-дигидро-11-этил-8-(2-гидроксиэтил)-4-метил-6H-дипиридо(128 мг, 0,49 ммоля) в ТГФ (2,5 мл). Через 1,5 ч в смесь вносили дополнительные количества ДЭАД (35 мкл, 0,22 ммоля) и РРh3 (59 мг, 0,22 ммоля). Смесь перемешивали при комнатной температуре в течение 1 ч, затем разбавляли EtOAc (50 мл) и раствор последовательно промывали водным 1 н. раствором NaOH(4 х 10 мл) и соляным раствором (15 мл), затем сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (СН 2 Сl2:ацетон, 85:15). Образовавшийся твердый продукт растирали с Et2O, получая указанное в заголовке соединение (38 мг, выход 22%) в виде твердого вещества белого цвета: МС (ESI) m/z 460/462 (МН)+. Пример 19 (номер 20, табл. 1). 2-Хлор-5,11-дигидро-11-этил-5-метил-8-2-(1-оксидо-5-хинолинил) оксиэтил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Диэтилазодикарбоксилат (ДЭАД) (160 мкл, 1,01 ммоля) добавляли при комнатной температуре по каплям к раствору, содержащему 2-хлор-5,11-дигидро-11-этил-8-(2-гидроксиэтил)-5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он (225 мг, 0,68 ммоля), 5-гидроксихинолин (147 мг, 1,01 ммоля) и Рh3 Р (266 мг, 1,01 ммоля) в ТГФ (3,4 мл). Смесь перемешивали при комнатной температуре в течение 3 ч, затем концентрировали при пониженном давлении. Остаток частично очищали экспресс-хроматографией(ЕtOАс:МеОН; 95:5). Фракции, содержащие 5-хинолинилоксипроизводное, концентрировали, растворяли в СН 2 Сl2 (2 мл) и ТГФ (5 мл) и обрабатывали мХПБК (80%, 248 мг, 1,15 ммоля) при комнатной температуре. Раствор перемешивали при комнатной температуре в течение 2,5 ч, затем концентрировали при пониженном давлении. Остаток растворяли в EtOAc (50 мл), промывали последовательно 10%-ным водным раствором Na2S2O3 (3 х 10 мл), водным насыщенным раствором NаНСО 3 (3 х 10 мл) и соляным раствором (10 мл), затем сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (от ЕtOАс:МеОН, 95:5 до СН 2 Сl2:МеОН, 9:1), получая указанное в заголовке соединение (113 мг, выход после 2 стадий 35% ) в виде твердого вещества белого цвета: МС(ESI) m/z 476/478 (МН)+. Пример 20 (номер 18, табл. 1). 5,11-Дигидро-11-этил-2-фтор-5-метил-8-2-(1-оксидо-5-хинолинил) оксиэтил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. С помощью процесса, идентичного процессу, описанному в примере 19, из 5,11-дигидро-11-этил-2 фтор-8-(2-гидроксиэтил)-5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (267 мг, 0,84 ммоля) получали указанное в заголовке соединение (204 мг, выход после 2 стадий 52%) в виде твердого вещества белого цвета: МС (ESI) m/z 460 (МН)+. Пример 21 (номер 19, табл. 1). 2-Хлор-5,11-дигидро-11-этил-4-метил-8-2-(1-оксидо-5-хинолинил) оксиэтил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. С помощью процесса, идентичного процессу, описанному в примере 19, но используя диизопропилазодикарбоксилат (ДИАД) вместо диэтилазодикарбоксилата, из 2-хлор-5,11-дигидро-11-этил-8-(2 гидроксиэтил)-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (223 мг, 0,67 ммоля) получали указанное в заголовке соединение (43,4 мг, выход 2 стадий 14%) в виде твердого вещества белого цвета: МС-16 005598 С помощью процесса, идентичного процессу, описанному в примере 21, из 5,11-дигидро-11-этил-2 фтор-8-(2-гидроксиэтил)-4-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (250 мг, 0,79 ммоля) получали указанное в заголовке соединение (64,7 мг, выход после 2 стадий 8%) в виде твердого вещества белого цвета: МС (ESI) m/z 460 (МН)+. Пример 23 (номер 22, табл. 1). 2-Хлор-5,11-дигидро-11-этил-8-2-(1-оксидо-4-хинолинил)окси этил-6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. МС (ESI) m/z 462/464 (МН)+. Пример 24 (номер 26, табл. 1). 5,11-Дигидро-5,11-диэтил-8-2-(4-хинолинилокси)этил-6 Ндипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. МС (ESI) m/z 440 (МН)+. Пример 25 (номер 27, табл. 1). 5,11-Дигидро-5,11 -диэтил-8-2-(1-оксидо-4-хинолинил)оксиэтил 6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. МС (ESI) m/z 456 (МН)+. Пример 26 (номер 28, табл. 1). 2,5-Диметил-5,11-дигидро-11-этил-8-2-(1-оксидо-4-хинолинилокси) этил-6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. а) 5-Бром-2-хлор-N-(6-хлор-2-метил-3-пиридинил)-3-пиридинкарбоксамид.NaHCO3 (3,9 г, 46,4 ммоля) добавляли к раствору 3-амино-2-хлор-6-метилпиридина (2,2 г, 15,4 ммоля), полученному согласно методу, описанному у К.G. Grozinger и др. J. Heterocyclic Chem., 32, 259263, 1995, в MeCN (50 мл). Образовавшуюся суспензию перемешивали в течение 15 мин и в течение 30 мин вводили раствор неочищенного 5-бром-2-хлор-3-пиридинкарбонилхлорида (4 г), полученного из 5 бром-2-гидрокси-3-пиридинкарбоновой кислоты и SOCl2 [согласно методу, описанному у T.W. Gero и др. в: Synth. Commun., 19, 553-559, 1989 (публикация включена в качестве ссылки), но за исключением обработки водой] в MeCN (10 мл). Образовавшуюся суспензию перемешивали при комнатной температуре. Через 24 ч смесь сливали на смесь воды (100 мл) и льда (10 г) и перемешивали в течение 20 мин. Суспензию фильтровали и образовавшийся твердый продукт промывали водой (50 мл) и гексаном (25 мл), затем сушили над Р 2 О 5 при пониженном давлении, получая указанное в заголовке соединение (1,8 г, выход 31% ) в виде порошка белого цвета. б) 5-Бром-N-(2-хлор-6-метил-3-пиридинил)-2-(этиламино)-3-пиридинкарбоксамид. Раствор 5-бром-2-хлор-N-(2-хлор-6-метил-3-пиридинил)-3-пиридинкарбоксамида (1,7 г, 4,8 ммоля) и этиламина (2 М в ТГФ, 10,5 ммоля, 5,2 мл) в ТГФ (5 мл) перемешивали при температуре от 90 до 95 С в закрытом автоклаве в течение 16 ч. Образовавшуюся смесь сливали на воду (100 мл), перемешивали в течение 15 мин и фильтровали. Твердый продукт промывали водой, а затем гексаном, получая указанное в заголовке соединение в виде твердого вещества желтого цвета (1,58 г, выход 89%). в) 8-Бром-5,11-дигидро-2-5-диметил-11-этил-6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Раствор неочищенного 5-бром-N-(2-дихлор-6-метил-3-пиридинил)-2-(этиламино)-3-пиридинкарбоксамида (0,7 г, 1,8 ммоля) в безводном пиридине (18 мл) нагревали до 50 С. Затем по каплям добавляли 1 М раствор NаГМДС в ТГФ (7,2 мл, 7,2 ммоля) и перемешивание продолжали в течение еще 3 ч. После охлаждения до комнатной температуры добавляли метилйодид (0,6 мл, 9 ммолей) и реакционную смесь перемешивали в течение ночи. Добавляли воду (25 мл) и смесь экстрагировали EtOAc (трижды) и объединенные экстракты промывали водой и соляным раствором, затем сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (гексан:ЕtOАс,8:2), получая указанное в заголовке соединение (259 мг, выход 41%). г) 5,11-Дигидро-2,5-диметил-11-этил-8-(2-пропенил)-6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Раствор 8-бром-5,11-дигидро-2,5-диметил-11-этил-6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она(258 мг, 0,7 ммоля) в ДМФ (7,4 мл) дегазировали при пониженном давлении в течение 20 мин. Добавляли Рd(РРh3)4 (43 мг, 0,04 ммоля), а затем аллилтрибутилолово (0,3 мл, 0,85 ммоля). После дегазации при пониженном давлении в течение 10 мин смесь нагревали до 100 С в течение 1,5 ч. Затем смесь концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (гексан:ЕtOАс, 8:2),получая указанное в заголовке соединение (184 мг, выход 98% ) в виде твердого вещества желтого цвета. д) 5,11-Дигидро-2,5-диметил-11-этил-8-(2-гидроксиэтил)-6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6 он. Струю О 3 пропускали через холодный (-78 С) раствор, содержащий 5,11-дигидро-2,5-диметил-11 этил-8-(2-пропенил)-6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он (187 мг, 0,6 ммоля) и Судан III в СН 2 Сl2 (3 мл) и МеОН (3 мл). После превращения розового раствора в коричневый, раствор барботировали в течение 10 мин О 2. Затем добавляли NaBH4 (57 мг, 1,52 ммоля) и раствору давали нагреться до комнатной температуры. Через 30 мин вводили дополнительное количество NaBH4 (20 мг). Через 2 ч добавляли водный насыщенный раствор NH4Cl и смесь перемешивали в течение 20 мин. Раствор концентрировали при пониженном давлении и экстрагировали СН 2 Сl2 (трижды). Объединенные органические экстракты промывали соляным раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (ЕtOАс:гексан 6:4), получая указанное в заголовке соединение (111 мг, выход 58%) в виде твердого вещества белого цвета.-17 005598 е) 2,5-Диметил-5,11-дигидро-11-этил-8-2-(4-хинолинилокси)этил-6 Н-дипиридо[3,2-b:2',3'-е][1,4] диазепин-6-он. Диэтилазодикарбоксилат (ДЭАД) (139 мкл, 0,88 ммоля) добавляли по каплям при комнатной температуре к раствору, содержащему 5,11-дигидро-2,5-диметил-11-этил-8-(2-гидроксиэтил)-6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он (110 мг, 0,35 ммоля), 4-гидроксихинолин (128 мг, 0,88 ммоля) и Рh3 Р (231 мг, 0,88 ммоля) в ТГФ (3,5 мл). Смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли ЕtOАс (60 мл) и раствор последовательно промывали 1 н. водным растворомNaOH (3x10 мл) и соляным раствором (15 мл), затем сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (ЕtOАс, затем ЕtOАс:МеОН,9,5:0,5), получая указанное в заголовке соединение (57 мг, выход 37%) в виде твердого вещества белого цвета: МС (ESI) m/z 440 (МН)+. ж) 5,11-Дигидро-2,5-диметил-11-этил-8-2-(1-оксидо-4-хинолинил)оксиэтил-6H-дипиридо[3,2b:2',3'-е][1,4]диазепин-6-он. Твердую мХПБК (80-85%, мета-хлорпербензойная кислота) (60 мг, 0,28 ммоля) добавляли к раствору 5,11-дигидро-2,5-диметил-11-этил-8-2-(4-хинолинилокси)этил-6 Н-дипиридо[3,2-b:2',3'-е] [1,4]диазепин-6-она (55 мг, 0,13 ммоля) в CH2Cl2 (1,3 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 2,5 ч, затем разбавляли СН 2 Сl2. Образовавшийся раствор последовательно промывали 10%-ным водным раствором Nа 2S2 О 3, водным насыщенным раствором NаНСО 3 и соляным раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (MeOH/EtOAc, от 2% до 5%), получая указанное в заголовке соединение (59 мг, выход 99%) в виде твердого вещества белого цвета: МС (ESI) m/z 456 (МН)+. Пример 27 (номер 29, табл. 1). 5,11-Дигидро-11-этил-5-метил-8-2-(1-оксо-4-хинолинилокси)этил 2-(трифторметил)-6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. а) 2-(Этиламино)-3-нитро-6-(трифторметил)пиридин. К раствору аммонийной соли 2-нитроацетамида (4 г, 33 ммоля) в воде (165 мл) добавляли ацетат пиперидиния (33 ммоля) в воде, а затем медленно добавляли 4-этокси-1,1,1-трифтор-3-бутен-2-он (6,1 мл, 43 ммоля) в МеОН (11 мл). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре и кипятили с обратным холодильником в течение 3 ч. Реакционной смеси давали охладиться 45 С и добавляли водный раствор НСl (1 н.) до доведения значения рН до кислого. После выдерживания в течение 1 ч при комнатной температуре реакционную смесь экстрагировали СН 2 Сl2 (трижды). Объединенный органический слой последовательно промывали водой и соляным раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (EtOAc,затем ЕtOАС:МеОН, 9:1), получая 3-нитро-6-(трифторметил)-2(1H)-пиридинон (2,7 г, выход 40%) в виде твердого вещества желтого цвета. К раствору 3-нитро-6-(трифторметил)-2(1H)-пиридинона (2,4 г, 11,5 ммоля) в ДМФ (69 мл) добавляли гидрид натрия (350 мг, 13,8 ммоля). Реакционную смесь перемешивали в течение 30 мин при 45 С,охлаждали до комнатной температуры и добавляли N-фенилтрифторметансульфонимид (4,9 г, 13,8 ммоля). Через 1 ч добавляли 2 М раствор этиламина в ТГФ (13 мл, 25 ммолей). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь сливали на воду (100 мл) и экстрагировали EtOAc (трижды). Объединенный органический слой последовательно промывали водой и соляным раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (гексан: EtOAc, 9:1), получая требуемое соединение (1,1 г, выход 41%) в виде масла желтого цвета. б) 3-Амино-2-(этиламино)-6-(трифторметил)пиридин. Раствор 2-(этиламино)-3-нитро-6-(трифторметил)пиридина (1,1 г, 4,9 ммоля) в МеОН (40 мл) перемешивали в течение ночи при комнатной температуре в атмосфере водород (1 атм) в присутствии 20% Рd(ОН)2/С (100 мг). Катализатор удаляли фильтрацией через диатомовую землю. Фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде масла оранжевого цвета (1 г). в) 2-Xлop-N-2-(этилaминo)-6-(тpифтopмeтил)-3-пиpидинил-5-бpoм-3-пиридинкарбоксамид. К охлажденному раствору 3-амино-2-(этиламино)-6-трифторметилпиридина (1 г, 4,9 ммоля) вMeCN (24 мл) добавляли твердый NаНСО 3 (906 мг, 11 ммолей). Через 5 мин добавляли неочищенный 5 бром-2-хлор-3-пиридинкарбонил хлорид, полученный из 5-бром-2-гидрокси-3-пиридинкарбоновой кислоты и SOСl2 [согласно методу, описанному у T.W. Gero и др. в: Synth. Commun., 19, 553-559, 1989(публикация включена в качестве ссылки), но за исключением обработки водой] (1 экв., 4,9 ммоля). Через 2 ч реакционную смесь сливали на смесь лед/Н 2 О (1,5 л) и образовавшийся твердый продукт фильтровали, промывали Н 2 О и затем гексаном. После сушки при пониженном давлении в течение ночи получали указанное в заголовке соединение в виде твердого вещества светло-желтого цвета (1,6 г, выход 77%). г) 8-Бром-5,11-дигидро-11-этил-2-(трифторметил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. К раствору 2-хлор-N-2-(этиламино)-6-(трифторметил)-3-пиридинил-5-бром-3-пиридинкарбоксамида (766 мг, 1,8 ммоля) в ДМФ (18 мл) добавляли NaH (137 мг, 5,4 ммоля). Реакционную смесь пере-18 005598 мешивали при 80 С. Через 1 ч реакционной смеси давали охладиться до комнатной температуры и затем сливали на воду (20 мл). Образовавшуюся смесь экстрагировали EtOAc (трижды). Объединенный органический слой последовательно промывали водой и соляным раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (гексан:ЕtOАс, 7:3), получая требуемое соединение (200 мг, выход 28%) в виде твердого вещества желтого цвета. д) 8-Бром-5,11-дигидро-11-этил-5-метил-2-(трифторметил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин 6-он. К раствору 8-бром-5,11-дигидро-11-этил-2-(трифторметил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин 6-она (200 мг, 0,52 ммоля) в ДМФ (2,6 мл) добавляли NaH (20 мг, 0,78 ммоля) и смесь нагревали до 50 С в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры и обрабатывали МеI (97 мкл, 1,56 ммоля). Через 16 ч реакционную смесь разбавляли водой. Образовавшуюся смесь экстрагировали EtOAc (трижды). Объединенный органический слой последовательно промывали водой и соляным раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (гексан:ЕtOАс, 8,5:1,5), получая указанное в заголовке соединение (110 мг, выход 53%) в виде пены белого цвета. е) 5,11-Дигидро-11-этил-5-метил-8-(2-пропенил)-2-(трифторметил)-6H-дипиридо[3,2-b:2',3'-е][1,4] диазепин-6-он. Аллилтрибутилолово (98 мкл, 0,32 ммоля) и Рd(Рh3 Р)4 (16 мг, 0,02 ммоля) добавляли при комнатной температуре к дегазированному (пропускали через раствор N2 в течение 30 мин) раствору 8-бром-5,11 дигидро-11-этил-5-метил-2-(трифторметил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (11 мг, 0,27 ммоля) в ДМФ (1,4 мл). Смесь перемешивали при 90 С в течение 1,5 ч, затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией[1,4]диазепин-6-он. Струeй озонированного кислорода барботировали холодный (-78 С) раствор 5,11-дигидро-11-этил 5-метил-8-(2-пропенил)-2-(трифторметил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она (100 мг, 0,27 ммоля) в CH2Cl2 (2,7 мл) и МеОН (2,7 мл) в течение 2,5 ч. Затем раствор барботировали струeй N2 в течение 15 мин и затем к раствору добавляли твердый NaBH4 (52 мг, 1,4 ммоля). Реакционной смеси давали нагреться до комнатной температуры. Через 1 ч добавляли водный насыщенный раствор NH4Cl (5 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли CH2Cl2 и водный слой повторно экстрагировали CH2Cl2. Объединенный органический слой последовательно промывали водой и соляным раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (ЕtOАс:гексан: от 75:25 до EtOAc 100%),получая указанное в заголовке соединение (60 мг, выход 59% ) в виде твердого вещества белого цвета. з) 5,11-Дигидро-11-этил-5-метил-8-2-(4-хинолинилокси)этил-2-(трифторметил)-6H-дипиридо[3,2b:2',3'-е][1,4]диазепин-6-он. Диэтилазодикарбоксилат (ДЭАД) (38 мкл, 0,24 ммоля) при комнатной температуре добавляли по каплям к раствору, содержащему 5,11-дигидро-11-этил-8-(2-гидроксиэтил)-5-метил-2-(трифторметил)6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он (58 мг, 0,16 ммоля), 4-гидроксихинолин (35 мг, 0,24 ммоля) и Рh3 Р (62 г, 0,24 ммоля) в ТГФ (1,6 мл). Смесь перемешивали при комнатной температуре в течение 1 ч,затем концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией(EtOAc:MeOH; 95:5), получая указанное в заголовке соединение (24 мг, выход 31%) в виде твердого вещества белого цвета: МС (ESI) m/z 494, 495 (МН)+. и) 5,11-Дигидро-11-этил-5-метил-8-2-(1-оксидо-4-хинолинил)оксиэтил-2-(трифторметил)-6 Ндипиридо[3,2-b:2',3'-е][1,4]диазепин-6-он. Твердую мХПБК (80-85%, мета-хлорпербензойная кислота) (16 мг, выход 0,08 ммоля) добавляли при комнатной температуре к раствору 5,11-дигидро-11-этил-5-метил-8-2-(4-хинолинилокси)этил-2(трифторметил)-6H-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-она (22 мг, 0,04 ммоля) в СН 2 Сl2 (0,5 мл). Смесь перемешивали при комнатной температуре в течение 2,5 ч, затем разбавляли CH2Cl2. Образовавшийся раствор последовательно промывали 10%-ным водным раствором Na2S2O3, водным насыщенным раствором NаНСО 3 и соляным раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (МеОН:СНСl3, от 5 до 10%), получая указанное в заголовке соединение (11 мг, выход 52%) в виде твердого вещества белого цвета: МС (ESI) m/z 511 (МH)+.-21 005598 Анализ обратной транскриптазы (ОТ) Теоретическая основа анализа Одним из ферментов, которые кодирует вирус иммунодефицита человека (ВИЧ-1), является обратная транскриптаза (1), названная так, потому что она транскрибирует копию ДНК на матрице РНК. Эту активность можно оценить количественно с помощью бесклеточного ферментного анализа и на основе того факта, что обратная транскриптаза обладает способностью использовать для транскрипции радиоактивномеченной цепи ДНК синтетическую матрицу поли-r(С), примированную биотинилированным олиго д(Г), и 3 Н-дГТФ в качестве субстрата. Описанный ниже анализ основан на применении фермента дикого типа (который представляет собой доминирующую форму фермента у пациентов, зараженных ВИЧ 1), однако, в аналогичных условиях анализа можно применять и мутантные ОТ (например, мутантY181C, полученный сайтнаправленным мутагенезом, в котором остаток тирозина в кодоне заменен остатком цистеина). Этот анализ позволяет оценить эффективность соединений в отношении ингибирования мутантных ферментов. Материалы: а) Получение фермента Несколько мутантов клона ОТ ВИЧ-1 IIIB BH10 получали от Др. С.-К. Shih (фирма BoehringerIngelheim Pharmaceuticals Inc., US) в векторе рКК 233-2 (фирма Pharmacia). Клон ОТ ВИЧ pKRT2, содержащий только ОТ гена р 66, регулируемого опероном lac/промотором trc получали от Др. Summers (Йельский университет) (2). В ген ОТ дикого типа интродуцировали определенные аминокислотные замены с помощью сайтнаправленного мутагенеза. Клоны ОТ субклонировали в бактериальном экспрессионном векторе рКК 233-2. Полученные клоны включали клон дикого типа, Val106Ala, Туr181 Суs, Tyr188Cys,Tyr188Leu, Gly190Ala и Pro236Leu. Другие клоны были получены заявителями с помощью сайтнаправленного мутагенеза клонов ОТ рКК 233-2, и они включали Lys103Asn, Lys103Asn/Tyr181Cys,Lys103Asn/Leu100Ile, Lys103Asn/Pro225His и Lys103Asn/Val108Ile. Очистка фермента Очистку рекомбинантной обратной транскриптазы осуществляли с использованием комбинации описанных ранее методов (3). Одну колонию из полученных на свежем планшете трансформированных клеток линии JM109 использовали для инициации роста предварительной культуры, выращенной в течение ночи при 37 С. Этой предварительной культурой инокулировали два литра среды для роста. При значении ОП 600 1,5 (5-6 ч при 37 С) индуцировали экспрессию гена ОТ изопропилтиогалактозидом(ИПТГ) (конечная концентрация 1 мМ) и продолжали ферментацию в течение еще нескольких часов при 37 С. После центрифугирования супернатанты отбрасывали, а клеточный дебрис объединяли и хранили при -80 С до очистки. Клеткам давали оттаять при 4 С в течение ночи и их суспендировали в буфере для лизиса (MES 50 мМ, рН 6, ЭДТА 1 мМ, 10 об.% глицерина, 0,02% (мас./об.) н-октилD-глюкозида,0,02% (маc./об.) азида натрия). Добавляли лизоцим и смесь инкубировали на льду в течение 40 мин. После гомогенизации с помощью Dounce в присутствии лизоцима и обработки ультразвуком клетки центрифугировали в течение 30 мин. Супернатант (S1) собирали и хранили при 4 С. Полученный с помощью центрифугирования дебрис ресуспендировали в буфере для экстракции (MES 50 мМ, рН 6, КРO4 50 мМ, рН 6, КСl 100 мМ, 10 об.% глицерина, 0,02% (мас./об.) н-октилD-глюкозида, 0,02% (мас./об.) азида натрия) и перемешивали в течение 30 мин при 4 С. Эту вторую смесь вновь центрифугировали и собирали супернатант (S2). Вышеописанный процесс повторяли еще дважды, собирая супернатанты S3 иS4, и затем в течение ночи осуществляли одну последнюю экстракцию (S5). Полимин Р (конечная концентрация 0,005%) добавляли к объединенным супернатантам для удаления нуклеиновых кислот. Этот раствор перемешивали в течение 75 мин при 4 С и центрифугировали в течение 1 ч. Супернатант (SS1) осаждали на льду с помощью 20%-ного (мас./об.) сульфата аммония и перемешивали в течение 1 ч при 4 С. Затем смесь центрифугировали и образовавшийся супернатант (SS2) осаждали, добавляя 40%-ный(мас./об.) сульфат аммония (всего 60%), перемешивали в течение 1 ч и вновь центрифугировали. Конечный дебрис (Р 1) хранили в течение ночи при 4 С до его очистки на следующий день. Если не указано иное, все стадии очистки осуществляли при 4 С. Дебрис (Р 1) ресуспендировали в MES 50 мМ, рН 6, КРO4 10 мМ, рН 6, КСl 100 мМ, 10 об.% глицерина, 0,02% (маc./об.) н-октилD-глюкозида, 0,02% (мас./об.) азида натрия. Суспензию подвергали диализу в противотоке этого же буфера в течение ночи, используя диализную трубку типа 12-14 кДаMWCO. Диализат центрифугировали и супернатант фильтровали через фильтры типа Millex-PF, 0,8 мкм. Профильтрованный образец вносили в заполненную гидроксиапатитом колонку (объем слоя 30 мл) и промывали этим же буфером. Связанный фермент элюировали 220 мл линейного градиента от 10 до 300 мМ КРO4 в вышеописанном буфере. Фракции, содержащие гетеродимер р 66/р 51 (что определяли с помощью ДСН-ПААГ, 8% и Вестерн-блоттинга), объединяли для внесения в следующую колонку. Содержащие ОТ фракции разбавляли в 2 раза с помощью смеси, содержащей бис-Трис-пропан 50 мМ, рН 7,0,(NH4)2SO4 1M, 0,02% (мас./об.) OBG, 10 об.% глицерина, 0,02% (мас./об.) NaN3, и вносили в гепаринсефарозную колонку Hi-Trap (объем слоя 5 мл) и промывали этим же буфером. Связанную ОТ затем элюировали с помощью 75 мл линейного градиента от 0 до 1M сульфата аммония в этом же буфере. Со-22 005598 держащие ОТ фракции объединяли на основе данных, полученных с помощью ДСН-ПААГ и Вестернблоттинга. Концентрацию протеина в этом пуле определяли методом Брадфорда (Bradford), используя БСА в качестве стандарта. Конечный препарат фермента подвергали диализу в противотоке смеси, содержащей MES 50 мМ, рН 6, КРO4 300 мМ, рН 6, КСl 175 мМ, 10 об.% глицерина, 0,02% (мас./об.) азида натрия, и разделяли на аликвоты и хранили при -80 С. Процедура анализа: Радиометрический ферментный анализ был адаптирован к формату 96-луночного титрационного микропланшета и основан на применении сцинтилляционных гранул с нанесенным стрептавидином. Анализ в целом описан ниже. Ферменту ОТ ВИЧ-1 давали оттаять и его соответствующим образом разбавляли в Трис/НСl 50 мМ, рН 7,8 содержащем NaCl 60 мМ, гексагидрат MgCl2 2 мМ, ДТТ 6 мМ, восстановленный глутатион 2 мМ и 0,02% (мас./об.) Chaps, до получения 3 нМ фермента. К 30 мкл этого раствора фермента добавляли 10 мкл раствора ингибитора (от 50 мкМ до 2,5 нМ ингибитора в указанном выше буфере, содержащем 5 об.% ДМСО). Планшет предварительно инкубировали в течение 15 мин при комнатной температуре до обработки на следующей стадии. На этой стадии предварительной инкубации наиболее высокая и наиболее низкая концентрации ингибитора составляли 12,5 мкМ и 0,62 нМ соответственно и концентрация ДМСО составляла 3,75 об.%. Затем ферментативную реакцию инициировали,добавляя 10 мкл растворов субстратов. Конечная реакционная смесь содержала Трис/НСд 50 мМ, рН 7,8,NaCl 60 мМ, MgCl26H2O 2 мМ, ДТТ 6 мМ, GSH 2 мМ, Chaps 0,02% (мас./об.), ДМСО 3 об.% , Поли-rС 179 нМ, биотин дГ 15 18 нМ, дГТФ 288 нМ и 3H-дГТФ 71 нМ. На этой стадии инкубации наиболее высокая и наиболее низкая концентрации ингибитора составляли 10 мкМ и 0,5 нМ соответственно. После добавления субстратов планшет заклеивали пластиковым покрытием и инкубировали в течение 1 ч при 37 С в безводном термостате. Затем реакцию прекращали,добавляя 75 мкл 0,5 М раствора ЭДТА, содержащего 5 мг/мл сцинтилляционных гранул с нанесенным стрептавидином. Планшет встряхивали в течение 2 мин при средней скорости и инкубировали в течение 1 ч при комнатной температуре. Затем добавляли 75 мкл 7 М раствора хлорида цезия, планшет встряхивали в течение 2 мин при средней скорости и вновь инкубировали в течение 1 ч при комнатной температуре. Затем планшет заклеивали пластиковым покрытием и подсчитывали радиоактивность с помощью сцинтилляционного и люминсцентного счетчика для микропланшетов типа TopCount-NXT фирмы Packard. Радиоактивность в каждой лунке подсчитывали в течение 60 с. В каждый анализируемый ряд входили чистая и контрольная лунки. Для расчета процента ингибирования использовали следующее уравнение: С помощью указанного выше анализа оценивали способность соединений по изобретению ингибировать ОТ дикого типа (WT) и мутантные ферменты. Результаты приведены в табл. 2 и представлены в виде IС 50 (нМ). Для подтверждения способности указанных соединений ингибировать репликацию ВИЧ их тестировали также с помощью человеческой Т-клеточной культуры (фирма Syncytia) с использованием описанного ниже анализа. Твердофазный иммуноферментый анализ (ELISA) для оценки активности в клеточной культуре Способность соединений по изобретению ингибировать репликацию ВИЧ в клеточной культуре оценивали в 96-луночном планшете. Полную среду RPMI 1640, содержащую RPMI 1640+10% фетальной бычьей сыворотки, 10 мкг/мл гентамицина и 10 мкМ -меркаптоэтанол, использовали для разбавления соединения, а также питательной среды для клеток. Клеточную линию Т-лимфоцитов С 8166 заражали с множественностью заражения 0,001 вирусами, кодирующими обратную траскриптазу дикого типа и мутантную. Затем клетки инкубировали в течение 3 дней в присутствии серийных разведений соединений по изобретению. Объединяли супернатанты 8 клеток-копий и определяли концентрацию внеклеточного р 24 с помощью имеющегося в продаже набора для анализа антигена ВИЧ-1 р 24 (Beckman-Coulter). Уровень ингибирования (% ингибирования) рассчитывали из следующего уравнения: Результаты представлены в табл. 3 и выражены в виде ЕС 50 (нМ). Анализ специфичности Для оценки специфичности ингибирующей активности соединений по изобретению в отношении различных ферментов для нескольких соединений с помощью известных методов анализа оценивали ингибирующую способность в отношении других вирусных полимераз (таких как РНК-зависимые РНКполимеразы вируса гепатита и респираторно-синцитиального вируса), а также полимераз млекопитающих (таких как РНК-зависимая ДНК-полимераза гипофиза теленка и человеческая теломераза). Ни для одного из изученных таким образом соединений не была обнаружена какая-либо заметная ингибирую-23 005598 щая активность в отношении этих ферментов. Эти результаты свидетельствуют о том, что способность соединений по изобретению ингибировать активность ферментов является прежде всего специфичной по отношению к ОТ ВИЧ-1. Литературные источники (включенные в настоящее описание в качестве ссылок): 1. Benn S., и др., Science 230:949, 1985. 2. D'Aquila R.Т. и Summers W.С., J. Acq. Imm. Def. Syn. 2:579, 1989. 3. a) Warren Т.С. и др., Protein ExpressionPurification 3:479, 1992; 4. b) Kohlstaedt L.A., Science 256(5065): 1783, 1992. Таблица 2 Ингибирование соединениями формулы I ОТ дикого типа и мутантных штаммов ОТ-24 005598 Таблица 3 Ингибирование соединениями формулы I штаммов дикого типа и мутантных штаммов ВИЧ в клеточных культурах ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение общей формулы IR5 обозначает Н, Me или Et при условии, что R4 и R5 оба не обозначают Me, и если R4 обозначаетR11 обозначает Me, Et, циклопропил, пропил, изопропил или циклобутил и или его фармацевтически приемлемая соль. 2. Соединение по п.1, где R11 обозначает Et или циклопропил, или его фармацевтически приемлемая соль. 3. Соединение по п.1, где Q выбирают из группы, включающей или его фармацевтически приемлемая соль. 4. Соединение по п.1, где Q обозначает или его фармацевтически приемлемая соль. 5. Соединение по п.1, где R5 обозначает Me, или его фармацевтически приемлемая соль. 6. Соединение по п.1, где R11 обозначает Et, или его фармацевтически приемлемая соль. 7. Соединение или его фармацевтически приемлемая соль. 8. Соединение или его фармацевтически приемлемая соль. 9. Соединение или его фармацевтически приемлемая соль. 10. Применение соединения формулы I по любому из пп.1-9 или его фармацевтически приемлемой соли в качестве ингибитора репликации ВИЧ. 11. Способ лечения или предупреждения ВИЧ-инфекции, заключающийся в том, что пациенту вводят ингибирующее ВИЧ количество соединения формулы I по любому из пп.1-9 или его фармацевтически приемлемой соли. 12. Фармацевтическая композиция, предназначенная для лечения или предупреждения ВИЧинфекции, содержащая соединение формулы I по любому из пп.1-9 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
МПК / Метки
МПК: A61K 31/55, C07D 471/14
Метки: ингибиторы, ненуклеозидные, обратной, транскриптазы
Код ссылки
<a href="https://eas.patents.su/27-5598-nenukleozidnye-ingibitory-obratnojj-transkriptazy.html" rel="bookmark" title="База патентов Евразийского Союза">Ненуклеозидные ингибиторы обратной транскриптазы</a>
4,4-дизамещенные 3,4-дигидро-2(1н)-хиназолиноны, полезные в качестве ингибиторов обратной транскриптазы вич

Номер патента: 1991
Опубликовано: 22.10.2001
Авторы: Корбетт Джеффри В., Ко Соо Сунг
МПК: A61K 31/517, A61P 31/18, C07D 239/80...
Метки: транскриптазы, 3,4-дигидро-2(1н)-хиназолиноны, вич, 4,4-дизамещенные, обратной, ингибиторов, качестве, полезные
Формула / Реферат:
1. Соединение формулы (I) или его стереоизомер, или фармацевтически приемлемая соль, где R1 обозначает C1-3-алкил, замещенный 1-7 атомами галогена; R2 выбран из C1-5-алкила, замещенного 1-2 R4, С2-5-алкенила, замещенного 1-2 R4, и С2-5-алкинила, замещенного одним R4; R3 каждый независимо выбран из C1-4-алкила, ОН, C1-4-алкокси, F, Cl, Br, I, NR5R5a, NO2, CN, C(O)R6, NHC(O)R7 и NHC(О)NR5R5a; альтернативно, если присутствуют два заместителя R3...
Способ кристаллизации ингибитора обратной транскриптазы с применением противорастворителя

Номер патента: 1805
Опубликовано: 27.08.2001
Авторы: Кларк Уилльям, Крокер Луис С., Кукура Джозеф Л.
МПК: C07D 265/18
Метки: способ, транскриптазы, обратной, применением, ингибитора, кристаллизации, противорастворителя
Формула / Реферат:
1. Способ кристаллизации соединения структурной формулы включающий следующие стадии: (1) растворение соединения в растворителе при соотношении от около 3,0 мл до около 10,0 мл растворителя на 1 г соединения; (2) фильтрацию раствора соединения для удаления твердых частиц вещества; (3) добавление к перемешиваемому раствору противорастворителя в течение периода времени от около 30 мин до около 1 ч при комнатной температуре для достижения точки...
Набор для анализа обратной транскриптазы и его применение

Номер патента: 4880
Опубликовано: 26.08.2004
Авторы: Гроновитз Симон, Келландер Клас, Петтерссон Ингвар
МПК: C12Q 1/68
Метки: набор, обратной, транскриптазы, применение, анализа
Формула / Реферат:
1. Набор для анализа обратной транскриптазы (ОТ), включающий в себя одну или несколько упаковок, содержащих связанную(ые) с твердой фазой матрицу (матрицы), представляющую(ие) собой полирибоадениловую кислоту (prA) и/или полидезоксиадениловую кислоту (pdA), приготавливаемую(ые) путем приведения в контакт твердой фазы на основе полистирола со связывающим раствором, содержащим 1-метилимидазол и prA и/или pdA, с последующими инкубацией, промывкой...
Управление мощностью прямой и обратной линии связи с использованием информации о положении и подвижности

Номер патента: 2893
Опубликовано: 31.10.2002
Автор: Солиман Сеймер С.
МПК: H04B 7/005
Метки: прямой, линии, управление, мощностью, обратной, связи, подвижности, использованием, положении, информации
Формула / Реферат:
1. Способ управления мощностью обратной линии связи между мобильной станцией и, по меньшей мере, одной базовой станцией в системе беспроводной связи МДКР, причем мобильная станция содержит контур управления мощностью обратной линии связи, содержащий этапы, на которых идентифицируют мгновенное положение мобильной станции, идентифицируют мгновенную скорость мобильной станции с использованием информации о мгновенном положении и динамически...
Устройство для индикации изменений в сопротивлении живого тела, схема обратной связи и управления для использования в устройстве, способ поддержания постоянной амплитудной характеристики

Номер патента: 1124
Опубликовано: 30.10.2000
Авторы: Хаббард Лафайетт Рональд, Ставропоулос Джеймс, Маккормик Джон, Стиннетт Ричард
МПК: A61B 5/053
Метки: способ, амплитудной, управления, постоянной, обратной, изменений, тела, схема, поддержания, сопротивлении, устройстве, использования, устройство, живого, индикации, характеристики, связи
Формула / Реферат:
1. Устройство для индикации изменений в сопротивлении живого тела, содержащее схему измерения сопротивления, имеющую внешние выводы, схему усиления, подключенную к схеме измерения сопротивления, и индикаторную схему, подключенную к схеме усиления, и схему регулировки чувствительности, подключенную к указанной схеме усиления, отличающееся тем, что схема измерения сопротивления выполнена с возможностью измерения сопротивлений в первом диапазоне...
Предыдущий патент: Пролекарства производных имидазопиридина
Следующий патент: Замещенные аналоги бензопирана
Случайный патент: Составы для ослабления боли, содержащие целекоксиб