Комбинация оланзапина и агониста taar1
Номер патента: 24703
Опубликовано: 31.10.2016
Авторы: Зевинг Забине, Рааб Зузанне, Ристеруччи Селин, Хёнер Мариус



















Формула / Реферат
1. Комбинация для лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, включающая оланзапин и агонист TAAR1, где агонист TAAR1 представляет собой
S1 = (S)-4-((S)-2-фенилбутил)-4,5-дигидрооксазол-2-иламин;
S2 = (S)-4-(3-фтор-2-метилфенил)-4,5-дигидрооксазол-2-иламин;
S3 = (S)-4-(4-хлор-2-трифторметилфенил)-4,5-дигидрооксазол-2-иламин;
S4 = (S)-4-[(этилфениламино)метил]-4,5-дигидрооксазол-2-иламин;
S5 = 3-[(S)-1-((S)-2-амино-4,5-дигидрооксазол-4-илметил)пропокси]фенол;
S6 = 5-хлорпиридин-2-карбоновой кислоты (4-пирролидин-3-ил-фенил)амид;
S7 = 4-хлор-N-(4-пирролидин-3-ил-фенил)бензамид;
S8 = 1-(5-хлорпиридин-2-ил)-3-(4-пирролидин-3-ил-фенил)мочевина;
S9 = (S)-4-[(S)-1-(4-фторфенил)этоксиметил]-4,5-дигидрооксазол-2-иламин;
S10 = 5-хлорпиримидин-2-карбоновой кислоты {4-[2-((S)-2-амино-4,5-дигидрооксазол-4-ил)этил]фенил}амид;
S11 = N-{4-[2-((S)-2-амино-4,5-дигидрооксазол-4-ил)этил]фенил}-4-хлорбензамид;
S12 = (R)-2-хлор-6-метил-N-(4-(морфолин-2-ил)фенил)изоникотинамид;
S13 = (S)-N-(4-(морфолин-2-ил)фенил)-6-(2,2,2-трифторэтокси)никотинамид;
S14 = (S)-N-(4-(морфолин-2-ил)фенил)-2-(трифторметил)изоникотинамид;
S15 = (S)-1-(4-фторбензил)-3-(4-(морфолин-2-ил)фенил)мочевина;
S16 = (S)-1-(3-цианофенил)-3-(4-(морфолин-2-ил)фенил)мочевина и
S17 = (S)-6-хлор-N-(4-(морфолин-2-ил)фенил)никотинамид.
2. Соединение S2 = (S)-4-(3-фтор-2-метилфенил)-4,5-дигидрооксазол-2-иламин.
3. Применение комбинации по п.1 для лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, характеризующегося сниженной частотой возникновения метаболического синдрома.
4. Применение комбинации по п.1 для получения лекарственного средства для лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, характеризующегося сниженной частотой возникновения метаболического синдрома.
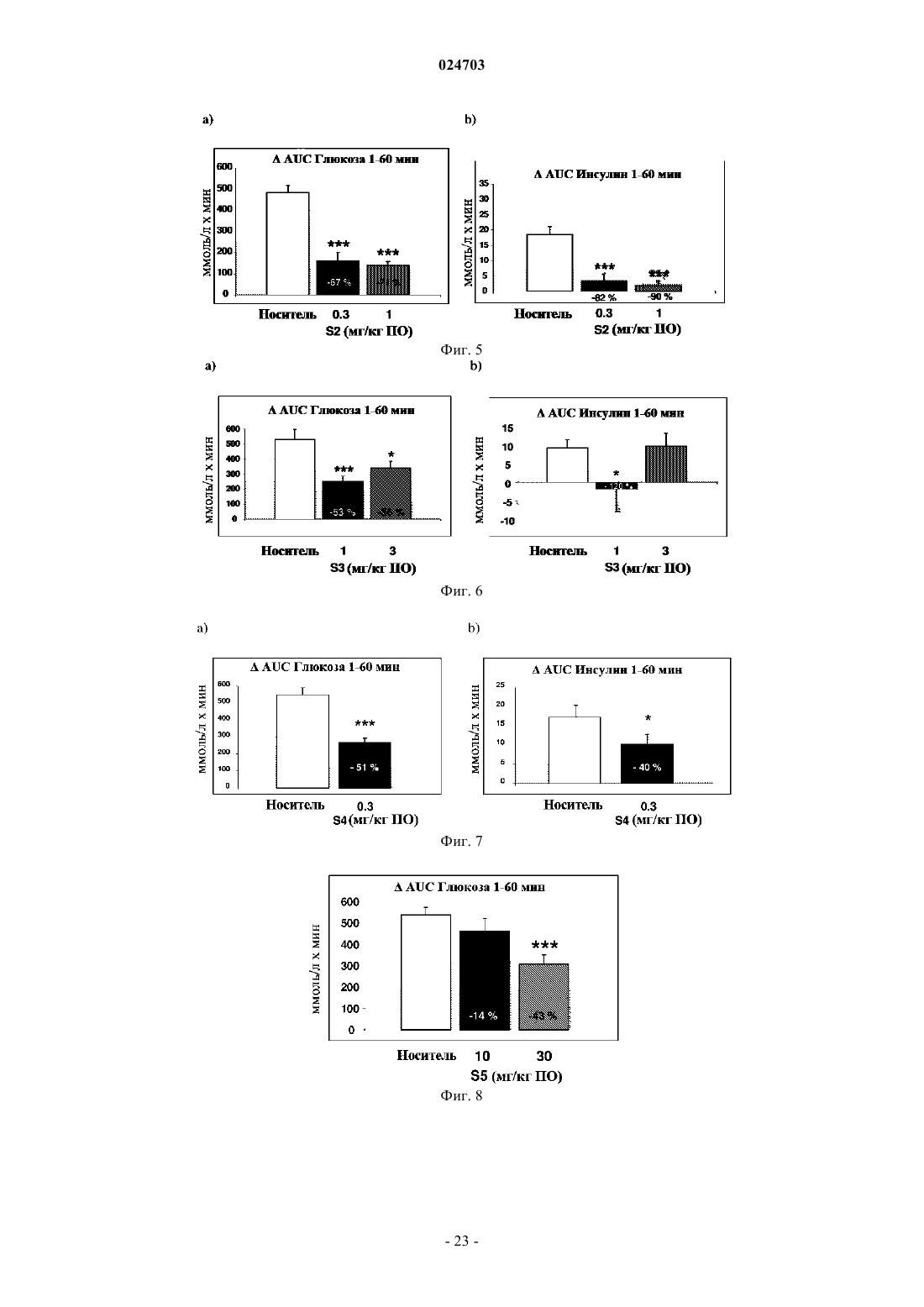
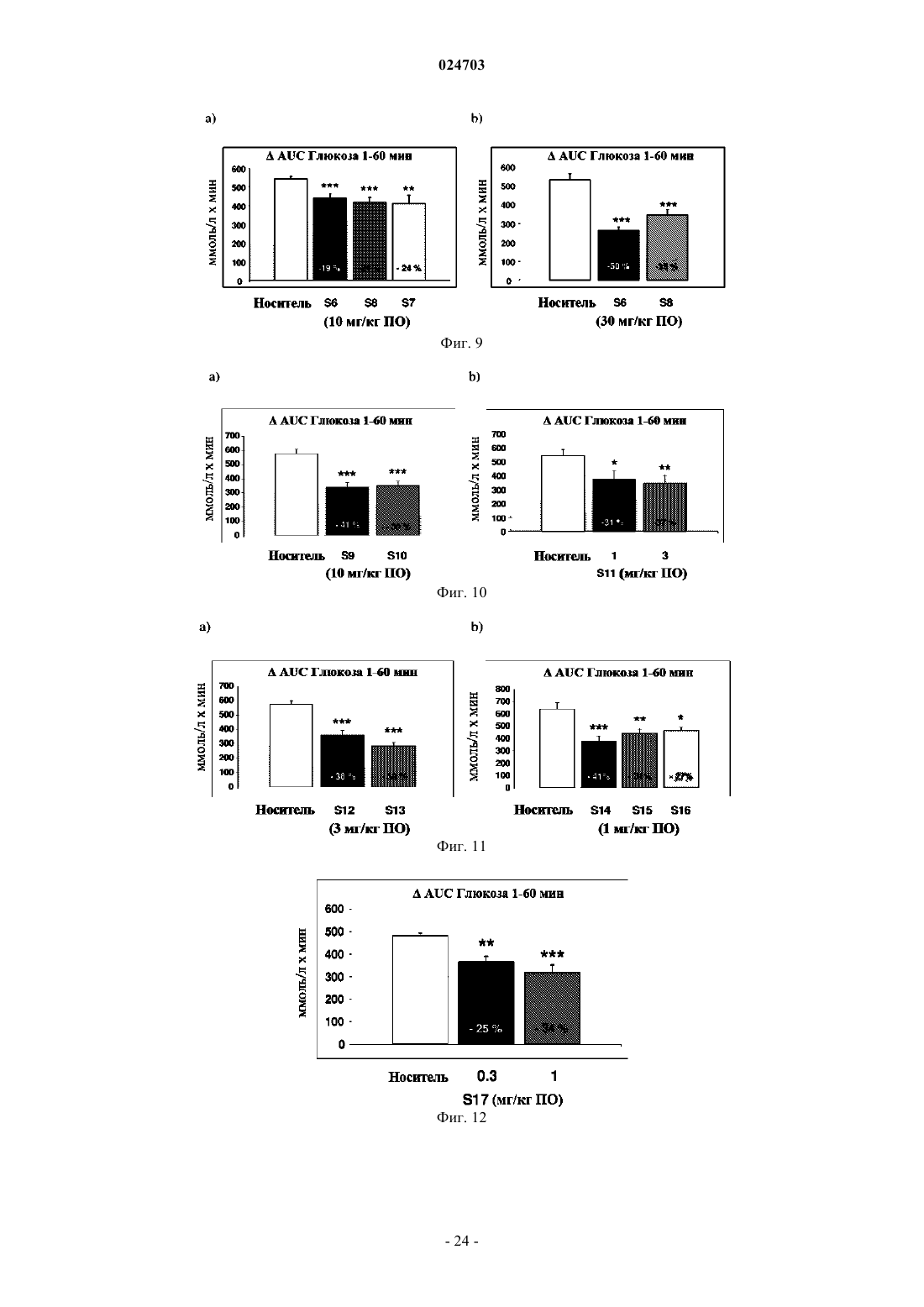
5. Применение по п.4, в котором указанное лекарственное средство обладает противодиабетическим действием.
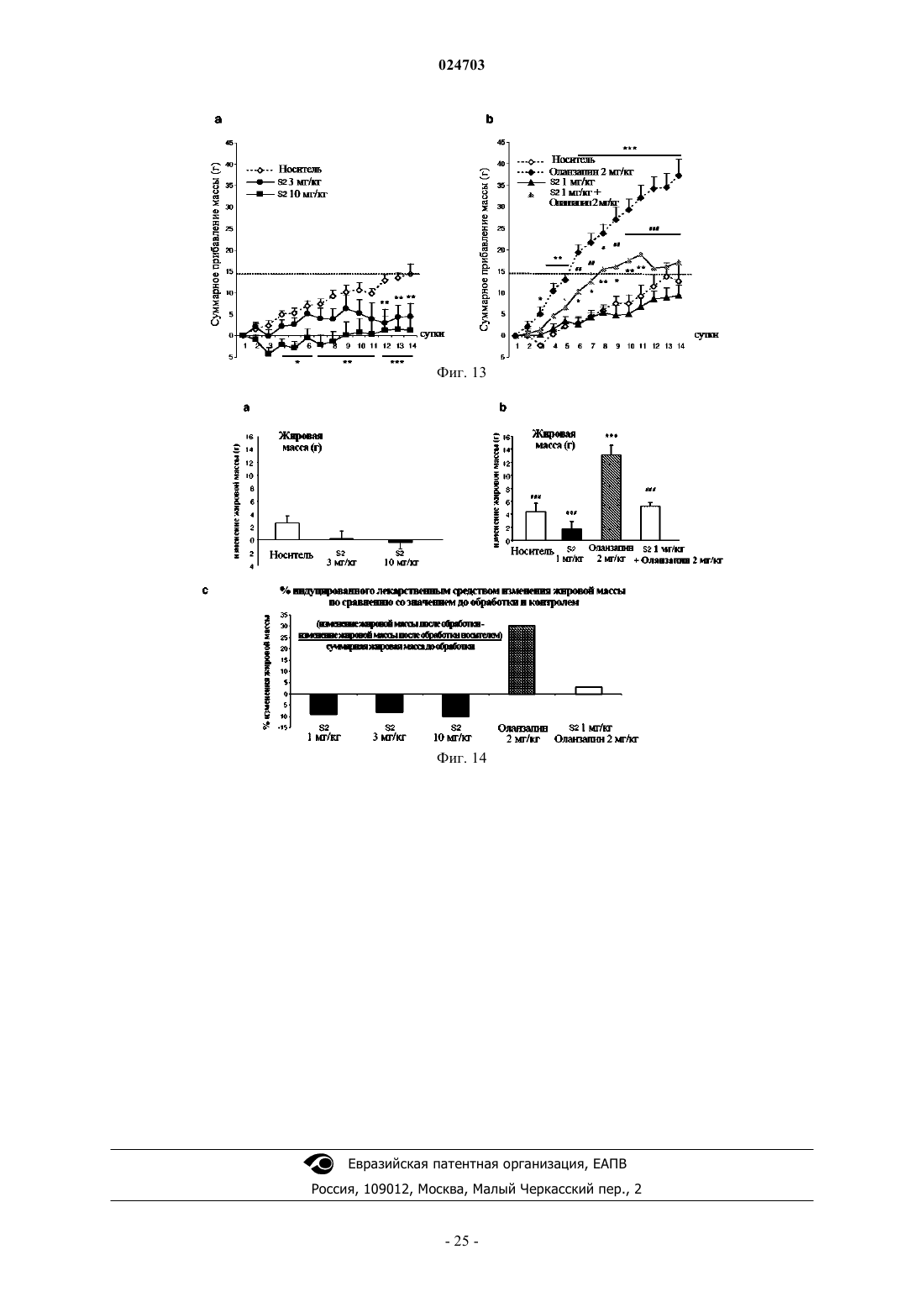
6. Применение по п.5, в котором указанное противодиабетическое действие заключается в уменьшении колебаний концентрации глюкозы в крови.
7. Применение по п.6, в котором указанное противодиабетическое действие заключается в снижении жировой массы и массы тела.
8. Способ лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, характеризующийся сниженной частотой возникновения метаболического синдрома, включающий введение человеку, нуждающемуся в этом, эффективного количества комбинации по п.1.
9. Способ лечения по п.8, где сниженная частота возникновения метаболического синдрома является результатом противодиабетического действия, заключающегося в уменьшении колебаний концентрации глюкозы в крови, снижении жировой массы и массы тела.
10. Фармацевтическая композиция, содержащая комбинацию по п.1 вместе с фармацевтически приемлемыми эксципиентами, для лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, характеризующегося сниженной частотой возникновения метаболического синдрома.
11. Фармацевтическая композиция по п.10, где сниженная частота возникновения метаболического синдрома является результатом противодиабетического действия, заключающегося в уменьшении колебаний концентрации глюкозы в крови, снижении жировой массы и массы тела.
Текст
Изобретение относится к фармацевтической комбинации для лечения шизофрении и острых маниакальных эпизодов, обусловленных биполярными расстройствами, включающей соединение,которое активно в отношении ассоциированного со следовыми аминами рецептора 1 (агонистTAAR1), и оланзапин. Неожиданно обнаружено, что такая комбинация может уменьшить метаболические побочные эффекты, возникающие при применении только одного оланзапина. 024703 Изобретение относится к фармацевтической комбинации для лечения шизофрении и острых маниакальных эпизодов, обусловленных биполярными расстройствами, включающей соединение, которое активно в отношении ассоциированного со следовыми аминами рецептора 1 (агонист TAAR1) и оланзапин. Неожиданно обнаружено, что такая комбинация может уменьшить метаболические побочные эффекты,возникающие при применении только одного оланзапина. Шизофрения является тяжелым и хроническим психическим заболеванием с оценками распространения в диапазоне от 1,4 до 4,6 на 1000 человек населения. Шизофренические расстройства и депрессия вызваны сочетанием генетических факторов и факторов окружающей среды, которые включают, для шизофрении, вероятные аномалии, связанные с неврологическим развитием, в структурах серого и белого вещества. Предполагают, что в основе симптоматических явлений при обоих заболеваниях лежат нарушения моноаминергической нейротрансмиссии (например, серотонина, адреналина и норадреналина). Эти биохимические пути широко представлены в ЦНС и, следовательно, потенциально способны к влиянию на многие зоны, вовлеченные в эмоциональные процессы, познавательную способность и поведение. До недавнего времени гипотеза избытка дофамина была основной патофизиологической теорией шизофрении, в значительной степени основанной на эффективности антагонистов D2 при лечении острых приступов этого заболевания. Симптомы шизофрении, которые типично возникают в пубертатном периоде и в раннем взрослом возрасте, обычно классифицируют как позитивные, негативные или когнитивные. Позитивные симптомы включают галлюцинации, бредовые состояния и тяжелую дезорганизацию мышления. Негативными симптомами является группа дефицитов, включающих уплощение эмоций, апатию, бедность речи, ангедонию и социальное отчуждение. Когнитивные симптомы, такие как дефициты внимания и кратковременной памяти, являются выраженными признаками этого заболевания и идентифицированы как сильные предвестники социального исхода. Современные атипичные антипсихотические средства эффективны, прежде всего, при лечении позитивных симптомов, при этом, обладая минимальными эффектами на негативные симптомы и когнитивную функцию, помимо того, что они ассоциированы со значительными побочными эффектами. Эффективность в отношении когнитивных симптомов и улучшения негативных симптомов является в высшей степени неудовлетворенной потребностью при шизофрении. Лекарственные средства первого поколения эффективны, но ассоциированы со значительной встречаемостью экстрапирамидальных симптомов, тогда как антипсихотические средства второго поколения(атипичные), как оказалось, характеризуются меньшей встречаемостью экстрапирамидальных побочных эффектов и могут быть более эффективными при лечении познавательной способности, но возрастает встречаемость и тяжесть метаболического симптома. Общепринятым антипсихотическим лекарственным средством для лечения шизофрении является оланзапин. Оланзапин (Зипрекса) принадлежит к классу лекарственных средств, известных как атипичные антипсихотические средства. Другие члены этого класса включают клозапин (Клозарил), рисперидон (Рисперидал), арипипразол (Абилифай) и зипразидон (Геодон). Оланзапин связывается с рецепторами альфа-1, дофамина, гистамина, мускариновыми рецепторами и рецепторами серотонина типа 2 (5HT2). Оланзапин одобрен для лечения психотических расстройств, долгосрочного лечения биполярных расстройств и в комбинации с флуоксетином для лечения депрессивных эпизодов, ассоциированных с биполярными расстройствами, а также для лечения устойчивой депрессии. Лечение антипсихотическими лекарственными средствами, такими как оланзапин, может приводить к серьезным побочным эффектам. Управление по надзору за пищевыми продуктами и лекарственными препаратами требует включать во все атипичные антипсихотические средства предупреждение о риске развития гипергликемии и диабета,оба из которых являются факторами при метаболическом синдроме. Эти побочные эффекты могут быть связаны со способностью данного лекарственного средства к индукции прибавления массы тела. Кардиометаболические побочные эффекты, такие как прибавление массы тела, ожирение, гипертензия и липидные и глюкозные аномалии, особенно проблематичны в процессе развития, поскольку они предсказывают ожирение у взрослых, метаболический синдром, сердечно-сосудистую заболеваемость и злокачественные новообразования, особенно при применении у детей и подростков. При применении оланзапина, а также других антипсихотических лекарств данного класса может быть повышен риск повышенных уровней сахара в крови и диабета. Многие педиатрические и подростковые пациенты, которые получали антипсихотические лекарства второго поколения, страдали значительным прибавлением массы параллельно с варьирующими побочными эффектами на уровни холестерина и триглицеридов и другие метаболические показатели, согласно исследованию в выпуске JAMA от 28 октября (JAMA, 2009 Oct.28, 302 (16), 1765-73). Авторы этого исследования пишут, что "Кардиометаболические побочные эффекты антипсихотических лекарств второго поколения вызывают все возрастающее беспокойство. Кардиометаболические побочные эффекты, такие как не соответствующее возрасту прибавление массы, ожирение, гипертензия и липидные и глюкозные аномалии, особенно проблематичны в процессе развития, поскольку они пред-1 024703 сказывают ожирение у взрослых, метаболический синдром, сердечно-сосудистую заболеваемость и злокачественные новообразования". Кардиометаболические эффекты этих лекарств не исследованы в достаточной степени у детей и пациентов-подростков, которые прежде их не получали, в соответствии с информацией предшествующего уровня техники в данной статье.Oaks, и The Feinstein Institute for Medical Research, Manhasset, New York, и сотрудники провели исследование массы и метаболических изменений в группе из 272 педиатрических пациентов (в возрасте от 4 до 19 лет), которые ранее не получали антипсихотические лекарства. Пациенты страдали расстройствами спектра расстройств настроения (47,8%), шизофренического спектра (30,1%) и спектра асоциального поведения и агрессивного поведения (22,1%). Пятнадцать пациентов, которые отказались от участия или были несовместимы с лекарственными средствами, служили в качестве сравнительной группы. Пациентов лечили антипсихотическими лекарствами арипипразолом, оланзапином, кветиапином или рисперидоном в течение 12 недель. После в среднем 10,8 недель лечения масса увеличилась в среднем на 8,5 кг (18,7 фунтов) с оланзапином (n = 45), на 6,1 кг (13,4 фунта) с кветиапином (n = 36), на 5,3 кг (11,7 фунтов) с рисперидоном (n = 135) и на 4,4 кг (9,7 фунтов) с арипипразолом (n = 41) по сравнению с минимальным изменением массы 0,2 кг (0,4 фунта) в не леченной сравнительной группе (n = 15). Авторы пишут, что "каждое антипсихотическое лекарство было ассоциировано со значительно увеличенной жировой массой и окружностью талии". "Всего от 10 до 36% пациентов перешли в состояние избыточной массы или ожирения в течение 11 недель". Исследователи также обнаружили, что побочные изменения в течение периода исследования достигли статистической значимости для оланзапина и кветиапина для суммарного холестерина, триглицеридов, холестерина не липопротеидов высокой плотности (ЛПВП) и отношения триглицеридов к холестерину ЛПВП. "С рисперидоном уровни триглицеридов значимо повысились. Метаболические изменения от базовых до конечных значений не были значимыми с арипипразолом или в не леченной сравнительной группе. Пациенты, получающие кветиапин, обладали умеренно высокими частотами встречаемости гипергликемии и метаболического синдрома, а пациенты, получающие оланзапин, обладали самыми высокими частотами встречаемости". Авторы отметили, что эти результаты вызывают беспокойство, поскольку они включают жировую массу и окружность талии, которые ассоциированы с метаболическим синдромом у взрослых, которых лечили антипсихотическими лекарствами, и сердечными заболеваниями в генеральной совокупности."Кроме того, аномальная масса и метаболический статус в детстве вредно влияют на исходы сердечнососудистых заболеваний у взрослых посредством продолжения существования этих факторов риска, либо независимых или ускоренных механизмов". Авторы сделали вывод, что их "результаты вместе с данными исследований первых эпизодов позволяют предположить, что руководства по воздействию антипсихотических лекарств для восприимчивых педиатрических и подростковых пациентов, не подвергавшихся воздействию антипсихотических лекарств, должны учитывать более частый [например, два раза в год] кардиометаболический мониторинг после первых 3 месяцев лечения. Наконец, в свете неблагоприятных исходов в отношении психического здоровья и субоптимального метаболического мониторинга у тяжелых психических больных, преимущества антипсихотических лекарств второго поколения должно быть сбалансировано против их кардиометаболических рисков посредством осторожной оценки показаний к их применению, учетом альтернатив более низкого риска и упреждающего мониторинга вредных эффектов и их лечения". В сопроводительной редакционной статье Christopher K. Varley, MD, и Jon McClellan, MD, of SeattleChildren's Hospital, Seattle, Washington, пишут, что данные открытия указывают на то, что существуют другие факторы для учета в отношении применения атипичных антипсихотических лекарств у детей и подростков."Эти лекарства могут быть спасительными для молодых людей с серьезными психическими заболеваниями, такими как шизофрения, классически определенное биполярное расстройство или тяжелая агрессия, обусловленная аутизмом. Однако с учетом риска прибавления массы и долгосрочного риска сердечно-сосудистых и метаболических проблем, широко распространенное и возрастающее применение атипичных антипсихотических лекарств у детей и подростков следует пересмотреть". Существует необходимость в новых терапиях с улучшенным профилем безопасности и переносимости по сравнению с современными атипичными антипсихотическими средствами. Например, новые терапии не должны быть ассоциированы с такими побочными эффектами или вредными реакциями, как описано выше. Теперь неожиданно обнаружено, что комбинация антипсихотического лекарственного средства оланзапина с агонистом ассоциированного со следовыми аминами рецептора 1 обладает потенциалом снижения встречаемости метаболического синдрома и позитивных симптомов, как при шизофрении, так и при острых маниакальных эпизодах, ассоциированных с биполярными расстройствами. Метаболический синдром представляет собой комбинацию соматических расстройств, которые повышают риск развития диабета и сердечно-сосудистого заболевания. Факторы риска включают, напри-2 024703 мер, абдоминальное ожирение (избыточную жировую ткань внутри и вокруг брюшной полости), расстройства липидов крови, повышенное кровяное давление, резистентность к инсулину или отсутствие толерантности к глюкозе. Следовые амины (паратирамин, -фенилэтиламин (PEA), октопамин и триптамин) присутствуют во всей ЦНС, в тесной параллели с моноаминергическими биохимическими путями, и на эндогенных уровнях, значительно более низких, чем их нейротрансмиттеры. Их недостаток отчасти является следствием их высокой скорости циркуляции, поскольку они являются хорошими субстратами для МАО А/В. Следовые амины структурно родственны, совместно локализованы и высвобождаются с классическими биогенными аминными нейротрансмиттерами. Предполагают, что они являются нейромодуляторами классических нейротрансмиттеров, таких как дофамин, серотонин и норадреналин, уровни которых являются мишенью всех известных антидепрессантов и большинства антипсихотических средств в настоящее время на рынке или в клинике. Аномалии в физиологии следовых аминов давно связывали с шизофренией и расстройствами настроения. Предполагают, что при шизофрении повышенные уровни PEA (так называемого эндогенного амфетамина) в моче и изменения в метаболизме триптамина и паратирамина включают ферменты, вовлеченные в синтетические и катаболические биохимические пути этих молекул. Поэтому идентификация специфических рецепторов для следовых аминов могла бы привести к разработке специфических лекарственных средств, направленных на данную новую нейромодуляторную систему, с клиническими применениями при расстройствах, таких как шизофрения, биполярное расстройство и депрессия. Недавно идентифицировано семейство рецепторов, сопряженных с G-белком, и названо рецепторами, ассоциированными со следовыми аминами (TAAR, Trace Amine-Associated Receptors), где из этих рецепторов TAAR1 лучше всего охарактеризован и является основной мишенью для эндогенных следовых аминов. TAAR1 экспрессируется в структурах головного мозга, связанных с психиатрическими расстройствами, в частности, в ключевых областях, где происходит модулирование дофамина (вентральная область покрышки) и серотонина (ядро шва), но также в миндалевидном теле, гипоталамусе, прилежащем ядре, обонятельной коре и основании. TAAR1 может быть новой мишенью для антипсихотических лекарственных средств с высоким потенциалом для дифференциации, использования фундаментально нового механизма действия, основанного на модулировании дофаминергической и глутаматергической нейротрансмиссии. Следовательно, даже при отсутствии дефицитов следовых аминов нейромодуляторные эффекты в отношении моноаминергических биохимических путей могли бы предсказуемо привести к улучшению при шизофрении. Кроме того, гены TAAR картированы близко к одному из основных локусов генетической предрасположенности к шизофрении, SCZD5. На основании вышеописанного агонисты TAAR1 должны быть эффективными агентами при лечении психиатрических расстройств, как прямо, так и косвенно, посредством влияния на моноаминергические биохимические пути. Агонисты TAAR1, которые интенсивно профилированы в неклинических экспериментах, показывают антипсихотическую, прокогнитивную, антидепрессантную и антиаддиктивную активность, что приводит к мысли, что они могут составлять абсолютно новый класс лекарственных средств для лечения шизофрении и расстройств настроения. На основании их профиля агонисты TAAR1 могут обладать потенциалом для лечения пациентов с шизофренией с лучшей эффективностью, включающей ослабление негативных и когнитивных симптомов, которые в настоящее время не поддаются лечению существующими терапиями, и возможно снижение злоупотребления химическими веществами у этих пациентов. Наконец, с учетом их благоприятных метаболических, противодиабетических эффектов, эти лекарственные средства могли бы обеспечить значительные преимущества для пациентов с шизофренией, давая возможность контроля позитивных симптомов без возрастания метаболического синдрома. Из группы агонистов TAAR1 выбраны соединения, которые описаны в документах WO 08/092785,WO 08/098857, WO 2010/010014 и PCT/ЕР 2010/070045. Конкретными соединениями, которые использованы в примерах, как описано ниже, являются приведенные ниже:S17 = (S)-6-хлор-N-(4-(морфолин-2-ил)фенил)никотинамид. Неожиданно показано, что комбинация антипсихотического лекарственного средства, в частности,оланзапина, и агониста TAAR1, как упомянуто выше, может уменьшить некоторые нежелательные метаболические побочные эффекты. Объектами настоящего изобретения являются: комбинация атипичного антипсихотического лекарственного средства и агониста TAAR1, где предпочтительным атипичным антипсихотическим лекарственным средством является оланзапин, а предпочтительным агонистом TAAR1 является соединение, выбранное из S1-S17; новое соединение S2, которое представляет собой (S)-4-(3-фтор-2-метил-фенил)-4,5-дигидрооксазол-2-иламин; комбинация, включающая оланзапин и агонист TAAR1, для лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, при сниженной встречаемости метаболического синдрома, где сниженная встречаемость метаболического синдрома является результатом противодиабетической эффективности при снижении изменения величины уровня глюкозы в крови, жировой массы и массы тела; применение комбинации, включающей оланзапин и агонист TAAR1, для лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, при сниженной встречаемости метаболического синдрома, где сниженная встречаемость метаболического синдрома является результатом противодиабетической эффективности при снижении изменения величины уровня глюкозы в крови,жировой массы и массы тела; применение комбинации, включающей оланзапин и агонист TAAR1, для получения лекарственного средства для лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, при сниженной встречаемости метаболического синдрома, где сниженная встречаемость метаболического синдрома является результатом противодиабетической эффективности при снижении изменения величины уровня глюкозы в крови, жировой массы и массы тела; способ лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, при сниженной встречаемости метаболического синдрома, где сниженная встречаемость метаболического синдрома является результатом противодиабетической эффективности при снижении изменения величины уровня глюкозы в крови, жировой массы и окружности талии, включающий введение человеку, нуждающемуся в этом, эффективного количества комбинации, включающей оланзапин и агонистTAAR1; способ лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, при сниженной встречаемости метаболического синдрома, где сниженная встречаемость метаболического синдрома является результатом противодиабетической эффективности при снижении изменения величины уровня глюкозы в крови, жировой массы и окружности талии, где атипичным антипсихотическим лекарственным средством является оланзапин, и агонисты TAAR1 выбраны из соединений S1S17; фармацевтическая композиция, содержащая комбинацию оланзапина и агониста TAAR1, выбранного из соединений S1-S17, вместе с фармацевтически приемлемыми эксципиентами, для лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, при сниженной встречаемости метаболического синдрома, где сниженная встречаемость метаболического синдрома является результатом противодиабетической эффективности при снижении изменения величины уровня глюкозы в крови, жировой массы и массы тела. Агонисты TAAR1 могут быть получены, как описано ниже. Пример S1. (S)-4-S)-2-Фенилбутил)-4,5-дигидрооксазол-2-иламинa) (R)-1-Йодометилпропил)бензол. К раствору трифенилфосфина (15,4 г, 59 ммоль) и имидазола (3,99 г, 59 ммоль) в дихлорметане (150 мл) при комнатной температуре добавляли порциями йод (14,9 г, 50 ммоль) с такой скоростью, чтобы температура реакционной смеси не поднималась выше 30C. Затем к смеси добавляли раствор (R)-2 фенилбутан-1-ола (7,34 г, 41 ммоль, CAS 16460-75-6) в дихлорметане (50 мл), а затем смесь перемешивали при комнатной температуре в течение ночи. Затем смесь концентрировали в вакууме, и остаток ресуспендировали в эфире, и полученные в результате кристаллы собирали фильтрованием. Фильтрат концентрировали в вакууме, и остаток растирали в гептане. Полученные в результате кристаллы удаляли-4 024703 фильтрованием, и фильтрат концентрировали в вакууме. Остаток очищали колоночной хроматографией(SiO2, гептан/EtOAc) с получением бесцветного масла (6,38 г, 60%).b) (2R,5S)-2-Изопропил-3,6-диметокси-5-S)-2-фенилбутил)-2,5-дигидропиразин. Раствор (2R)-(-)-2,5-дигидро-3,6-диметокси-2-изопропилпиразина (4,25 г, 23,1 ммоль) в тетрагидрофуране (30 мл) охлаждали до -78C, затем добавляли н-бутиллитий (1,6 М в гексане, 15,1 мл, 24,2 ммоль), и смесь перемешивали в течение 1 ч. Раствор R)-1-йодометилпропил)бензола (6,30 г, 24,2 ммоль) в тетрагидрофуране (30 мл) добавляли по каплям в течение 30 мин, и смесь перемешивали в течение ночи, давая в это время медленно нагреться с -70C до комнатной температуры. Реакционную смесь гасили добавлением насыщенного водного раствора хлорида аммония, и смесь экстрагировали эфиром. Органический слой отделяли, промывали насыщенным рассолом, затем высушивали над Na2SO4 и концентрировали в вакууме. Остаток очищали колоночной хроматографией (SiO2, гептан/EtOAc) с получением светло-желтого масла (4,69 г, 64%); МС (ISP): 317,0 ([M+H]+).c) (2S,4S)-2-Амино-4-фенилгексановой кислоты метиловый эфир. К раствору трифторуксусной кислоты (3,4 мл) в виде (440 мл) добавляли по каплям в течение 15 мин раствор (2R,5S)-2-изопропил-3,6-диметокси-5-S)-2-фенилбутил)-2,5-дигидропиразина (4,69 г, 14,8 ммоль) в ацетонитриле (75 мл). Смесь перемешивали в течение ночи при комнатной температуре, затем подщелачивали добавлением насыщенного водного раствора карбоната натрия, и смесь экстрагировали этилацетатом. Фазы разделяли, и органическую фазу последовательно промывали водой и насыщенным рассолом, затем высушивали над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией (SiO2, EtOAc/гептан) с получением желтого масла (2,78 г, 85%); МС (ISP): 222,1 ([M+H]+).d) (2S,4S)-2-Амино-4-фенил-гексан-1-ол. К суспензии алюмогидрида лития (121 мг, 3,18 ммоль) в тетрагидрофуране (8 мл) добавляли раствор (2S,4S)-2-амино-4-фенилгексановой кислоты метилового эфира (320 мг, 1,45 ммоль) в тетрагидрофуране (10 мл), и смесь перемешивали в течение 16 ч. Реакционную смесь гасили добавлением по каплям этилацетата, затем подкисляли до pH 5 добавлением соляной кислоты, а затем подщелачивали добавлением насыщенного водного раствора бикарбоната натрия. Смесь растворяли в этилацетате/тетрагидрофуране (1:1), фазы разделяли, и органическую фазу последовательно промывали водой и насыщенным рассолом. Затем органическую фазу высушивали над Na2SO4 и концентрировали в вакууме. Остаток очищали хроматографией (колонка: Isolute Flash-NH2 от фирмы Separtis; элюент: дихлорметан/МеОН) с получением желтого масла, (116 мг, 42%); МС (ISP): 194,4 ([M+H]+).e) (S)-4-S)-2-Фенилбутил)-4,5-дигидро-оксазол-2-иламин. К перемешанному, охлажденному (0C) раствору (2S,4S)-2-амино-4-фенил-гексан-1-ола (270 мг,1,40 ммоль) и ацетата натрия (229 мг, 2,70 ммоль) в метаноле (20 мл) добавляли по каплям раствор цианогенбромида (180 мг, 1,68 ммоль) в метаноле (2 мл) в течение 10 мин. Затем смеси давали нагреться до КТ, и перемешивание продолжали в течение 16 ч. Смесь концентрировали в вакууме, и остаток растворяли в этилацетате и последовательно промывали насыщенным водным раствором бикарбоната натрия и насыщенным рассолом. Органическую фазу высушивали над сульфатом натрия и концентрировали в вакууме. Остаток очищали хроматографией (колонка: Isolute Flash-NH2 от фирмы Separtis; элюент: гептан/EtOAc/МеОН) с получением светло-желтого твердого вещества. МС (ISP): 219,3 ([M+H]+).a) (RS)-Амино-(3-фтор-2-метилфенил)ацетонитрил. К перемешанному раствору 3-фтор-2-метил-бензальдегида (5,0 г) в метаноле (20 мл) последовательно добавляли раствор аммиака (40,5 мл, 7 М раствор в метаноле) и тетраизопропилортотитаната(12,6 мл), и полученную в результате смесь перемешивали при КТ в течение 1 ч. Затем добавляли по каплям триметилсилилцианид (4,69 мл), и перемешивание продолжали при КТ в течение ночи. Реакционную смесь наливали на ледяную воду (400 мл), а затем смесь дважды экстрагировали этилацетатом. Объединенные органические фазы промывали рассолом, а затем высушивали над сульфатом натрия и концентрировали в вакууме с получением (RS)-амино-(3-фтор-2-метил-фенил)-ацетонитрила (5,90 г, количественно) в виде оранжевого твердого вещества. 1H ЯМР(CDCl3, 300 МГц): 7.39 (1H, d, J= 7.3 Гц), 7.31(RS)-Амино-(3-фтор-2-метилфенил)ацетонитрил (5,89 г) суспендировали в 5 н. водной соляной кислоте (40 мл) и смесь нагревали с обратным холодильником в течение 18 ч. Затем смесь экстрагировали этилацетатом и водную фазу концентрировали в вакууме. Остаток ресуспендировали в изопропаноле и снова концентрировали в вакууме. Затем остаток растворяли в воде и нейтрализовали добавлением по-5 024703 каплям 1 н. водного NaOH, в результате чего медленно образовались белые кристаллы. Эти кристаллы собирали фильтрованием и высушивали в вакууме при 50C с получением (RS)-амино-(3-фтор-2-метилфенил)-уксусной кислоты (7,1 г, количественно) в виде беловатого твердого вещества. МС (ISP): 184,1c) (RS)-2-Амино-2-(3-фтор-2-метилфенил)этанол. К перемешанному раствору боргидрида лития в ТГФ (48,9 мл, 2 М раствор) в атмосфере аргона добавляли по каплям хлортриметилсилан (25,0 мл). Полученную в результате суспензию охлаждали до 0C и добавляли порциями (RS)-амино-(3-фтор-2-метилфенил)уксусную кислоту (7,1 г), в результате чего температура реакционной смеси временно поднималась до 45C. Ледяную баню удаляли, а затем перемешивание продолжали при КТ в течение 90 мин. Смесь гасили добавлением по каплям метанола (20 мл), а затем концентрировали в вакууме. Остаток суспендировали в этилацетате и промывали 2 н. воднымNaOH. Фазы разделяли, и водную фазу экстрагировали этилацетатом. Объединенные органические фазы высушивали над сульфатом натрия и концентрировали в вакууме с получением (RS)-2-амино-2-(3-фтор 2-метилфенил)этанола (1,83 г, 28%) в виде светло-желтого твердого вещества. МС (ISP): 170,3 ([M+H]+).(1,82 г) и ацетата натрия (1,72 г) в метаноле (17 мл) добавляли по каплям раствор цианогенбромид (1,18 г) в метаноле (8 мл) в течение 10 мин. Затем смесь перемешивали в течение 1 ч при 0C, затем давали нагреться до КТ, и перемешивание продолжали в течение 2 ч. Смесь концентрировали в вакууме, и остаток ресуспендировали в воде и подщелачивали добавлением 1 М водного раствора гидроксида натрия. Затем смесь дважды экстрагировали дихлорметаном, и объединенные органические фазы высушивали над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией(RS)-4-(3-Фтор-2-метил-фенил)-4,5-дигидрооксазол-2-иламин разделяли с помощью хиральной ВЭЖХ (Chiralpak AD, EtOH/гептан = 10:90) с получением (-)-(R)-4-(3-фтор-2-метил-фенил)-4,5-дигидрооксазол-2-иламина (белое твердое вещество; МС (ISP): 195,3 ([M+H]+ в качестве первой фракции и (+)(S)-4-(3-фтор-2-метилфенил)-4,5-дигидрооксазол-2-иламина (белое твердое вещество; МС (ISP): 195,3a) (RS)-4-(4-Хлор-2-трифторметилфенил)-4,5-дигидрооксазол-2-иламин. Это соединение было получено по аналогии с примером S2 (стадии a-d), начиная с 4-хлор-2 трифторметил-бензальдегида вместо 3-фтор-2-метил-бензальдегида. Белое твердое вещество. МС (ISP): 267,1 ([37ClM+H]+), 265,0 ([35ClM+H]+).(RS)-4-(4-Хлор-2-трифторметилфенил)-4,5-дигидрооксазол-2-иламин разделяли с помощью хиральной ВЭЖХ (Chiralpak AD, EtOH/гептан = 5:95) с получением (+)-(S)-4-(4-хлор-2-трифторметил-фенил)4,5-дигидро-оксазол-2-иламина (белое твердое вещество; МС (ISP): 267,1 ([37ClM+H]+), 265,0([35ClM+H]+ в качестве первой фракции и (-)-(R)-4-(4-хлор-2-трифторметилфенил)-4,5-дигидрооксазол-2-иламина (белое твердое вещество; МС (ISP): 267,1 ([37ClM+H]+), 265,0 ([35ClM+H]+ в качестве второй фракции.(681 мг, CAS 95715-87-0) при КТ в 1,2-дихлорэтане (10 мл) в атмосфере аргона добавляли молекулярные сита 4 (1,5 г) и N-этиланилин (0,25 мл). После перемешивания в течение 15 мин при КТ добавляли триацетоксиборгидрид натрия (1,68 г) одной порцией, а затем уксусную кислоту (5 капель), и перемешивание при КТ продолжали в течение ночи. Смесь гасили осторожным добавлением 10% KHCO3 (15 мл). Двухфазную смесь перемешивали при КТ в течение 20 мин и фильтровали. Водную фазу фильтрата снова экстрагировали CH2Cl2. Объединенные органические вещества промывали Н 2 О и рассолом, высушивали над MgSO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали колоночной хроматографией (SiO2; градиент: циклогексанциклогексан/EtOAc 4:1) с получением (S)-4-[(этил-фениламино)-метил]-2,2-диметил-оксазолидин-3-карбоновой кислоты трет-бутилового эфира (469 мг, 57%) в виде оранжевого вязкого масла. МС (ISP): 335,5 ([M+H]+)b) (S)-2-Амино-3-(этилфениламино)пропан-1-ол. К перемешанному раствору (S)-4-[(этилфениламино)метил]-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутилового эфира (462 мг) при КТ в диоксане (5,85 мл) в атмосфере аргона добавляли раствор HCl (4 M в диоксане; 4,14 мл). Смесь перемешивали при КТ в течение ночи и концентрировали. Остаток растворяли в EtOAc и промывали 10% водным раствором бикарбоната калия. Водный слой снова экстрагировали EtOAc. Объединенные органические вещества промывали водой, а затем рассолом, высушивали над MgSO4 и концентрировали в вакууме. Сырой продукт очищали колоночной хроматографией (колонка Isolute SPE Flash-NH2, аминопропил-функционализированный силикагель; CH2Cl2/ MeOH 9:1) с получением (S)-2-амино-3-(этилфениламино)пропан-1-ола (133 мг, 62%) в виде светлокоричневого вязкого масла. МС (ISP): 195,1 ([M+H]+)c) (S)-4-[(Этилфениламино)метил]-4,5-дигидрооксазол-2-иламин. К перемешанному раствору (S)-2-амино-3-(этилфениламино)пропан-1-ола (128 мг) при КТ в ТГФ (5 мл) в атмосфере аргона добавляли карбонат калия (182 мг) и раствор цианогенбромида (140 мг) в ТГФ (5 мл). Перемешивание при КТ продолжали в течение 21 ч. Смесь (беловатую суспензию) разбавлялиEtOAc и промывали Н 2 О. Водную фазу снова экстрагировали EtOAc. Объединенные органические вещества промывали рассолом, высушивали над MgSO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали колоночной хроматографией (колонка Isolute SPE Flash-NH2, аминопропилфункционализированный силикагель; градиент: CH2Cl2CH2Cl2/MeOH 9:1) с получением (S)-4-[(этилфениламино)метил]-4,5-дигидрооксазол-2-иламина (108 мг, 75%) в виде беловатого твердого вещества. МС (ISP): 220,4 ([M+H]+).a) (S)-4-R)-2-Гидроксибутил)-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутиловый эфир и (S)-4-S)-2-гидроксибутил)-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутиловый эфир. К перемешанному раствору (S)-2,2-диметил-4-(2-оксоэтил)оксазолидин-3-карбоновой кислоты трет-бутилового эфира (15,5 г; CAS 147959-19-1) в сухом диэтиловом эфире (100 мл) в атмосфере аргона при комнатной температуре добавляли по каплям раствор этилмагния бромида в диэтиловом эфире (42,6 мл, 3 М раствор), и перемешивание продолжали в течение 1 ч. Затем реакционную смесь смесь гасили осторожным добавлением воды (10 мл), после чего смесь фильтровали через декалит. Фильтрат последовательно промывали водой и насыщенным рассолом, а затем органическую фазу отделяли, высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией (SiO2; градиент: гептан/EtOAc 100:050:50) с получением (S)-4-R)-2-гидроксибутил)-2,2 диметилоксазолидин-3-карбоновой кислоты трет-бутилового эфира (7,30 г) из фракций, элюировавших первыми, и (S)-4-S)-2-гидроксибутил)-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутилового эфира (6,44 г), из фракций, элюировавших позже, причем оба соединения представляют собой бесцветное масло. (S)-4-R)-2-Гидроксибутил)-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутиловый эфир: 1H ЯМР(CDCl3, 300 МГц): 4.52 (1H, br. D, J = 3.3 Гц), 4.23 (1H, m), 4.00 (1H, dd, J = 8.75.4 Гц),3.66 (1H, d, J = 8.7 Гц), 3.40 (1H, m), 1.79 (1H, td, J = 11.42.1 Гц), 1.60-1.44 (16H, m), 0.95 (3H, t, J = 7.5 Гц). (S)-4-S)-2-Гидрокси-бутил)-2,2-диметил-оксазолидин-3-карбоновой кислоты трет-бутиловый эфир: 1-7 024703 К перемешанному раствору 3-бензилоксифенола (264 мг) в ТГФ (5 мл) последовательно добавляли трифенилфосфин (364 мг) и ди-трет-бутилазодикарбоксилат (309 мг), и полученный в результате желтый раствор перемешивали при комнатной температуре в течение 15 мин. Затем добавляли по каплям раствор(S)-4-R)-2-гидроксибутил)-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутилового эфира (300 мг) в ТГФ (5 мл), и полученную в результате смесь перемешивали при 70C в течение 90 мин. Реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Сырой продукт очищали колоночной хроматографией (SiO2; градиент: гептан/EtOAc 100:070:30) с получением (S)-4-[(S)-2-(3 бензилохуфенокси)бутил]-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутилового эфира (1,35 г,34%) в виде бесцветного вязкого масла. МС (ISP): 456,4 ([M+H]+).c) (2S,4S)-2-Амино-4-(3-бензилоксифенокси)гексан-1-ол. К раствору трифторуксусной кислоты (0,07 мл) в воде (6 мл) добавляли по каплям раствор (S)-4[(S)-2-(3-бензилоксифенокси)бутил]-2,2-диметил-оксазолидин-3-карбоновой кислоты трет-бутилового эфира (135 мг) в ацетонитриле (1 мл). Смесь нагревали в течение 4 ч при 80C при механическом встряхивании. Затем смесь охлаждали до комнатной температуры и разбавляли 1 н. водным раствором гидроксида натрия. Смесь дважды экстрагировали этилацетатом, и объединенные органические фазы высушивали над сульфатом натрия и концентрировали в вакууме с получением (2S,4S)-2-амино-4-(3-бензилоксифенокси)гексан-1-ола (94 мг, количественно) в виде светло-желтого вязкого масла. MC(ISP): 316,1d) (S)-4-[(S)-2-(3-Бензилоксифенокси)бутил]-4,5-дигидрооксазол-2-иламин. К перемешанной смеси (2S,4S)-2-амино-4-(3-бензилоксифенокси)гексан-1-ола (90 мг) и ацетата натрия (46 мг) в метаноле (2 мл) в атмосфере аргона добавляли раствор цианогенбромида (37 мг) в метаноле (1 мл). Смесь перемешивали в течение 18 ч, затем разбавляли 1 н. водным раствором гидроксида натрия. Смесь дважды экстрагировали дихлорметаном, и объединенные органические фазы высушивали над сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали колоночной хроматографией (колонка: Isolute Flash-NH2 от фирмы Separtis; градиент: дихлорметан/ МеОН 100:095:5) с получением (S)-4-[(S)-2-(3-бензилоксифенокси)бутил]-4,5-дигидрооксазол-2-иламина (69 мг, 71%) в виде светло-желтой смолы. МС (ISP): 341,1 ([M+H]+).e) 3-[(S)-1-S)-2-Амино-4,5-дигидрооксазол-4-илметил)пропокси]фенол. К раствору (S)-4-[(S)-2-(3-бензилокси-фенокси)-бутил]-4,5-дигидро-оксазол-2-иламина (60 мг) в метаноле (3 мл) при комнатной температуре добавляли 10% палладий на угле (19 мг). Смесь перемешивали в атмосфере водорода (1 атм) при комнатной температуре в течение 1 ч. Катализатор удаляли фильтрованием через декалит, промывали метанолом и дихлорметаном, и фильтрат концентрировали в вакууме с получением 3-[(S)-1-S)-2-амино-4,5-дигидрооксазол-4-илметил)пропокси]фенола в виде белого твердого вещества (44 мг, количественно); МС (ISP): 251,2 ([M+H]+).a) (RS)-3-[4-(4-Хлорбензоиламино)фенил]пирролидин-1-карбоновой кислоты трет-бутиловый эфир. К перемешанной суспензии (RS)-трет-бутил-3-(4-аминофенил)пирролидин-1-карбоксилата (1,9 г,CAS 908334-28-1) в ТГФ (50 мл) последовательно добавляли триэтиламин (2,0 мл) и 4-хлорбензоилхлорид (0,93 мл), и перемешивание продолжали при комнатной температуре в течение 3 ч. Затем смесь разбавляли этилацетатом. Добавляли воду, и смесь подкисляли до pH 1 добавлением 1 М водной соляной кислоты. Органическую фазу отделяли и последовательно промывали водным раствором гидроксида натрия и насыщенным рассолом. Затем органическую фазу высушивали над сульфатом натрия и концентрировали в вакууме с получением (RS)-3-[4-(4-хлорбензоиламино)фенил]пирролидин-1 карбоновой кислоты трет-бутилового эфира (2,42 г, 83%) в виде белого твердого вещества, который использовали в следующей стадии без дополнительной очистки.b) (RS)-4-Хлор-N-(4-пирролидин-3-ил-фенил)бензамида гидрохлорид. К перемешанному раствору (RS)-3-[4-(4-хлорбензоиламино)фенил]пирролидин-1-карбоновой кислоты трет-бутилового эфира (2,36 г) в ТГФ (30 мл) добавляли по каплям раствор хлорида водорода в диоксане (22 мл, 4 М раствор), и смесь нагревали при 60C в течение ночи. Затем смесь охлаждали до 0C, и полученные в результате кристаллы собирали фильтрованием, промывая диэтиловым эфиром, а затем высушивали в вакууме при 60C с получением (RS)-4-хлор-N-(4-пирролидин-3-ил-фенил)бензамида гидрохлорида (1,65 г, 83%) в виде белого кристаллического твердого вещества. МС (ISP): 303,2 ([37ClM+H]+), 301,3 ([35ClM+H]+).a) (RS)-3-(4-[3-(5-Хлорпиридин-2-ил)уреидо]фенил)пирролидин-1-карбоновой кислоты третбутиловый эфир. К перемешанной суспензии 2-амино-5-хлорпиридина (1,01 г) в дихлорэтане (20 мл) добавляли порциями трифосген (831 мг). Затем добавляли по каплям триэтиламин (2,22 мл), и смесь перемешивали при 50C в течение 1 ч. Затем смесь концентрировали в вакууме с получением бежевого твердого вещества,содержащего смесь триэтиламмония хлорида и 5-хлор-2-изоцианатопиридина. Затем это твердое вещество добавляли к перемешанному раствору (RS)-трет-бутил-3-(4-аминофенил)пирролидин-1-карбоксилата(350 мг, CAS 908334-28-1) и N,N-диизопропилэтиламина (0,68 мл) в дихлорэтане (6 мл), и полученную в результате смесь перемешивали при 60C в течение ночи. Затем смесь разбавляли дихлорметаном и промывали водой. Фазы разделяли, и органическую фазу высушивали над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией (SiO2; градиент: гептан/EtOAc) с получением(RS)-3-4-[3-(5-хлорпиридин-2-ил)уреидо]фенилпирролидин-1-карбоновой кислоты третбутилового эфира (212 мг, 38%) в виде беловатого твердого вещества. МС (ISP): 419,2 ([37ClM+H]+),417,2 ([35ClM+H]+).b (RS)-1-(5-Хлорпиридин-2-ил)-3-(4-пирролидин-3-ил-фенил)мочевины гидрохлорид. К перемешанному раствору (RS)-3-4-[3-(5-хлор-пиридин-2-ил)уреидо]фенилпирролидин-1 карбоновой кислоты трет-бутилового эфира (210 мг) в ТГФ (4 мл) добавляли по каплям раствор хлорида водорода в диоксане (1,89 мл, 4 М раствор), и смесь нагревали при 60C в течение ночи. Затем смесь охлаждали до 0C, и полученные в результате кристаллы собирали фильтрованием, промывая этилацетатом, и высушивали в вакууме при 60C с получением (RS)-1-(5-хлорпиридин-2-ил)-3-(4-пирролидин-3 ил-фенил)-мочевины гидрохлорида (167 мг, 94%) в виде бежевого кристаллического твердого вещества. МС (ISP): 319,1 ([37ClM+H]+), 317,2 ([35ClM+H]+).a) (RS)-3-4-[(5-Хлорпиридин-2-карбонил)амино]фенил)пирролидин-1-карбоновой кислоты третбутиловый эфир. К перемешанной суспензии (RS)-трет-бутил-3-(4-аминофенил)пирролидин-1-карбоксилата (200 мг,CAS 908334-28-1) в ДМФ (10 мл) последовательно добавляли N-метилморфолин (0,22 мл), TBTU (490 мг) и 5-хлор-2-пиридинкарбоновую кислоту (180 мг), и смесь перемешивали при комнатной температуре в течение 90 мин. Затем смесь разбавляли этилацетатом и последовательно промывали 1 М водной соляной кислотой и насыщенным рассолом. Фазы разделяли, и органическую фазу высушивали над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией (SiO2; градиент: гептан/EtOAc) с получением (RS)-3-4-[(5-хлорпиридин-2-карбонил)амино]фенилпирролидин-1 карбоновой кислоты трет-бутилового эфира (310 мг, количественно) в виде белого твердого вещества. МС (ISP): 421,3 ([M+NH4]+), 419,2 ([M+NH4]+).b) (RS)-5-Хлорпиридин-2-карбоновой кислоты (4-пирролидин-3-ил-фенил)амида гидрохлорид. К перемешанному раствору (RS)-3-4-[(5-хлорпиридин-2-карбонил)амино]фенилпирролидин-1 карбоновой кислоты трет-бутилового эфира (310 мг) в ТГФ (6 мл) добавляли по каплям раствор хлорида водорода в диоксане (2,9 мл, 4 М раствор), и смесь нагревали при 60C в течение ночи. Затем смесь охлаждали до 0C, и полученные в результате кристаллы собирали фильтрованием, промывая диэтиловым эфиром, и высушивали в вакууме при 60C с получением (RS)-5-хлор-пиридин-2-карбоновой кислоты (4 пирролидин-3-ил-фенил)-амида гидрохлорида в виде светло-желтого твердого вещества. МС (ISP): 304,2a) 1-S)-1-Аллилокси-этил)-4-фторбензол. К перемешанной суспензии гидрида натрия (3,14 г, 55% дисперсия в масле) в сухом ДМФ (180 мл)-9 024703 в атмосфере аргона добавляли (S)-1-(4-фторфенил)этанол (8,41 г, CAS 101219-73-2). Затем добавляли по каплям аллилбромид (6,6 мл). Реакционную смесь перемешивали в течение 30 мин при комнатной температуре, а затем гасили добавлением воды. Смесь дважды экстрагировали этилацетатом. Объединенные органические слои высушивали (MgSO4) и концентрировали в вакууме. Сырой продукт очищали колоночной хроматографией (SiO2; градиент: гептан/EtOAc) с получением 1-S)-1-аллилоксиэтил)-4-фторбензола (8,66 г, 80%) в виде бесцветной жидкости. 1H ЯМР (300 МГц, CDCl3,млн-1): 1.43 (d, J=6.6 Гц,3H), 3.84 (m, 2H), 4.45 (q, J=6.6 Гц, 1H), 5.15 (dd, J1=10.5 Гц, J2=1.8 Гц, 1H), 5.22 (dd, J1=17.4 Гц, J2=1.8 Гц,1H), 5.89 (m, 1H), 7.03 (m, 2H), 7.29 (m, 2H).AD-MIX-P (62,9 г) перемешивали в t-BuOH/H2O 1:1 (440 мл) в течение 15 мин при комнатной температуре, а затем охлаждали до 0C. К этому раствору добавляли 1-S)-1-аллилоксиэтил)-4-фторбензол(8,00 г). Смесь перемешивали в течение 48 ч при 0C. Реакционную смесь обрабатывали сульфитом натрия и перемешивали в течение 30 мин при 0C и при комнатной температуре в течение 1 суток. Раствор дважды экстрагировали этилацетатом. Объединенные органические слои высушивали (MgSO4) и концентрировали в вакууме. Продукт очищали колоночной хроматографией (SiO2; градиент: гептан/EtOAc 1/1-0/1) с получением смеси 80:20 (S)-3-[(S)-1-(4-фтор-фенил)-этокси]-пропан-1,2-диола и (R)-3-[(S)-1-(4 фторфенил)этокси]пропан-1,2-диола (8,59 г, 90%) в виде светло-желтой жидкости. 1H ЯМР (300 МГц,CDCl3,млн-1): 1.44 (d, J=6.6 Гц, 3H), 2.1 (b, 1H), 2.6 (b, OH), 3.39 (m, 2H), 3.60 (m, 1H), 3.64 (m, 1H), 3.83c) (R)-1-(трет-Бутилдиметилсиланилокси)-3-[(S)-1-(4-фторфенил)этокси]пропан-2-ол. К раствору смеси 80:20 (S)-3-[(S)-1-(4-фторфенил)этокси]пропан-1,2-диола и (R)-3-[(S)-1-(4-фторфенил)этокси]пропан-1,2-диола и (R)-3-[(S)-1-(4-фторфенил)этокси]пропан-1,2-диола (8,40 г) в тетрагидрофуране (84 мл) добавляли триэтиламин (5,74 мл) и 4-диметиламинопиридин (479 мг). Смесь охлаждали до 0C, и раствор трет-бутил(хлор)диметилсилана (6,21 г) в тетрагидрофуране (17 мл) добавляли по каплям. После 2 ч при 0C реакционной смеси давали перемешиваться при комнатной температуре в течение 16 ч. Добавляли воду, и смесь дважды экстрагировали диэтиловым эфиром. Объединенные органические слои высушивали (MgSO4) и концентрировали в вакууме с получением смеси 80:20 (R)-1-(третбутилдиметилсиланилокси)-3-[(S)-1-(4-фторфенил)этокси]пропан-2-ола и (S)-1-(трет-бутилдиметилсиланилокси)-3-[(S)-1-(4-фторфенил)этокси]пропан-2-ола (12,6 г, 98%) в виде желтой жидкости. Сырой продукт использовали в следующей стадии без дополнительной очистки.d) (S)-2-(трет-Бутилдиметилсиланилокси)-1-[(3)-1-(4-фторфенил)этоксиметил]этиламин. К перемешанному раствору смеси 80:20 (R)-1-(трет-бутил-диметил-силанилокси)-3-[(S)-1-(4-фторфенил)этокси]пропан-2-ола и (S)-1-(трет-бутилдиметилсиланилокси)-3-[(S)-1-(4-фторфенил)этокси]пропан-2-ола (12,4 г) и триэтиламина (6,84 мл) в дихлорметане (60 мл) при 0C добавляли по каплям раствор метансульфонилхлорида (3,52 мл) в ТГФ. Смесь перемешивали в течение 1 ч при 0C, а затем добавляли воду и дихлорметан. Водную фазу второй раз экстрагировали дихлорметаном, и объединенные органические слои промывали рассолом и высушивали над MgSO4 Растворитель выпаривали, и продукт высушивали в высоком вакууме. Полученный в результате сырой продукт мезилат (15,5 г) растворяли в ДМФ(100 мл) и добавляли азид натрия (4,94 г). Реакционную смесь перемешивали при 100C в течение 16 ч. Затем реакционную смесь гасили водой, и смесь дважды экстрагировали этилацетатом. Объединенные органические слои высушивали над MgSO4 и концентрировали в вакууме. Сырой азидный продукт (15,6 г) растворяли в метаноле (160 мл) и добавляли 10% палладий на угле (1,6 г). Смесь перемешивали в атмосфере водорода при комнатной температуре в течение 2 ч. Катализатор удаляли фильтрованием через целлит, и фильтрат концентрировали в вакууме с получением смеси 80:20 (S)-2-(трет-бутилдиметилсиланилокси)-1-[(S)-1-(4-фторфенил)этоксиметил]этиламина и (R-2-(трет-бутилдиметилсиланилокси)-1-[(S)1-(4-фторфенил)этоксиметил]этиламина (14,4 г, 100%) в виде желтой жидкости, которую использовали в следующей стадии без дополнительной очистки.e) (R)-2-Амино-3-[(3)-1-(4-фторфенил)этокси]пропан-1-ол. К перемешанному раствору смеси 80:20 (S)-2-(трет-бутилдиметилсиланилокси)-1-[(S)-1-(4-фторфенил)этоксиметил]этиламина и (R)-2-(трет-бутилдиметилсиланилокси)-1-[(S)-1-(4-фторфенил)этоксиметил]-этиламина (14,4 г) в ТГФ (150 мл) при 0C добавляли тетрабутиламмония фторид (22,9 мл, 1 М раствор в ТГФ), и смесь перемешивали при комнатной температуре в течение 18 ч. Растворитель выпаривали, и сырой продукт очищали колоночной хроматографией (колонка: Isolute Flash-NH2 от фирмыf) (R)-2-Амино-3-[(3)-1-(4-фторфенил)этокси]пропан-1-ола (R)-гидроксифенилацетат. К перемешанному раствору смеси 80:20 (R)-2-амино-3-[(S)-1-(4-фторфенил)этокси]пропан-1-ола и- 10024703 в горячем EtOAc (65 мл) при 80C, и смеси давали медленно охладиться. Кристаллы появлялись при достижении температуры 70C, а затем суспензию перемешивали при комнатной температуре в течение 16 ч. Кристаллы собирали фильтрованием и снова растворяли в горячем EtOAc (80 мл) при 80C, и смесь давали медленно охладиться, пока не начиналась кристаллизация, а затем полученную в результате суспензию перемешивали при комнатной температуре в течение 16 ч. Кристаллы собирали фильтрованием с получением чистого (R)-2-амино-3-[(S)-1-(4-фторфенил)этокси]пропан-1-ола (R)-гидроксифенилацетата(3,08 г, 61%) в виде белого твердого вещества. 1H ЯМР (300 МГц, ДМСО,млн-1): 1.34 (d, J=6.3 Гц, 3H),3.13 (m, 1H), 3.23 (m, 1H), 3.37 (m, 4 Н), 3.83 (m, 1H), 4.46 (q, J=6.3 Гц, 1H), 4.54 (s, 1H), 7.20 (m, 5 Н), 7.35EtOAc обрабатывали водным раствором бикарбоната натрия, и смесь перемешивали при комнатной температуре, пока все твердое вещество не растворилось. Фазы разделяли, и органический слой высушивали над MgSO4. Растворитель выпаривали с получением (R)-2-амино-3-[(S)-1-(4-фторфенил)этокси]пропан-1 ола в виде свободного основания. К перемешанному раствору (R)-2-амино-3-[(S)-1-(4-фторфенил)этокси]пропан-1-ола (1,40 г) в ТГФ(80 мл) последовательно добавляли K2CO3 (1,82 г) и цианогенбромид (0,83 г). Смесь перемешивали при комнатной температуре в течение 18 ч, затем добавляли воду. Смесь дважды экстрагировали этилацетатом, и объединенные органические слои высушивали над MgSO4 и выпаривали над силикагелем IsoluteFlash-NH2. В результате хроматографии (колонка: Isolute Flash-NH2 от фирмы Separtis; элюент: гептан/этилацетат = 25:75) получили соединение, указанное в заголовке, в виде белого твердого вещества,(1,15 г, 73% ). МС (ISP): 239,0 ([M+H]+). Соединение, указанное в заголовке, получили по аналогии с примером S3, используя 5 хлорпиримидин-2-карбоновую кислоту (CAS 38275-61-5) вместо 4-хлорбензойной кислоты на стадии с). Белое твердое вещество. МС (ISP): 348,3 ([37ClM+H]+), 346,1 ([35ClM+H]+).(S)-2,2-Диметил-4-[(Е)-2-(4-нитрофенил)винил]оксазолидин-3-карбоновой кислоты третбутиловый эфир. К перемешанному раствору диизопропиламина (1,81 мл) в ТГФ (50 мл), охлажденному до -78C,добавляли по каплям раствор н-бутиллития в гексане (8,05 мл, 1,6 М). Охлаждающую баню удаляли, и реакционной смеси давали нагреться до 10C, после чего повторно охлаждали до -78C. Затем добавляли по каплям раствор (4-нитро-бензил)-фосфоновой кислоты диэтилового эфира (2,71 г, CAS 2609-49-6) в ТГФ (60 мл), и реакционную смесь перемешивали при -78C в течение 1 ч. Затем добавляли по каплям раствор (R)-4-формил-2,2-диметил-оксазолидин-3-карбоновой кислоты трет-бутилового эфира (2,50 г,CAS 95715-87-0) в ТГФ (50 мл) в течение 30 мин, а затем смеси давали нагреться до комнатной температуры в течение 90 мин. Затем смесь разбавляли этилацетатом и подкисляли добавлением 1 н. водной соляной кислоты. Затем смесь последовательно промывали водой и насыщенным рассолом. Органическую фазу отделяли и высушивали над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией (SiO2; градиент: гептан/EtOAc) с получением (S)-2,2-диметил-4-[(Е)-2-(4 нитро-фенил)-винил]-оксазолидин-3-карбоновой кислоты трет-бутилового эфира (2,21 г, 64%) в виде желтого масла. МС (EI): 333 ([М-CH3]+), 292 ([М-С 4 Н 8]+), 277 ([М-СН 3-С 4 Н 8]+), 57 ([С 4 Н 9]+).b (S)-4-[2-(4-Аминофенил)этил]-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутиловый эфир. К перемешанной суспензии (S)-2,2-диметил-4-[(Е)-2-(4-нитрофенил)винил]оксазолидин-3-карбоновой кислоты трет-бутилового эфира (2,08 г) в метаноле (140 мл) добавляли формиат аммония (5,66 г) и палладий на угле (0,51 г, 10% мас./мас.), и смесь нагревали при 60C в течение 90 мин. Затем смесь охлаждали до комнатной температуры, фильтровали через целлит, и фильтрат концентрировали в вакууме. Остаток очищали колоночной хроматографией (SiO2; градиент: гептан/EtOAc) с получением (S)-4-[2-(4 аминофенил)этил]-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутилового эфира (1,58 г, 82%) в виде желтого масла. МС (ISP): 321,4 ([M+H]+).c) (S)-4-2-[4-(4-Хлорбензоиламино)фенил]этил)-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутиловый эфир. К перемешанному раствору (S)-4-[2-(4-аминофенил)этил]-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутилового эфира (20) в ТГФ (4 мл) добавляли 4-хлорбензойную кислоту (147 мг), Nметилморфолин (0,27 мл) и TBTU (401 мг). Реакционную смесь нагревали до 50C и перемешивали в течение 16 ч. Затем реакционную смесь концентрировали в вакууме, и остаток очищали колоночной флэш-хроматографией (SiO2; градиент: EtOAc/гептан) с получением (S)-4-2-[4-(4-хлорбензоиламино) фенил]этил-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутилового эфира в виде белого твердого вещества (254 мг, 89%). МС (ISP): 478,3 ([37ClM+NH4]+), 476,3 ([35ClM+NH4]+), 405,4d) N-[4-S)-3-Амино-4-гидроксибутил)фенил]-4-хлорбензамид. К раствору (S)-4-2-[4-(4-хлор-бензоиламино)фенил]этил-2,2-диметилоксазолидин-3-карбоновой кислоты трет-бутилового эфира (438 мг) в ацетонитриле (5 мл) добавляли воду (4 мл) и трифторуксусную кислоту (0,29 мл). Смесь нагревали при 80C в течение 4,5 ч. Затем смесь охлаждали до комнатной температуры и наливали в 1 М водный NaOH и дважды экстрагировали EtOAc/ТГФ. Объединенные органические слои промывали рассолом, высушивали над Na2SO4, фильтровали и концентрировали в вакууме с получением N-[4-S)-3-амино-4-гидроксибутил)фенил]-4-хлорбензамида (255 мг, 84%) в виде белого твердого вещества. МС (ISP): 321,2 ([37ClM+H]+), 319,2 ([35ClM+H]+).e) N-(4-[2-S)-2-Амино-4,5-дигидрооксазол-4-ил)этил]фенил)-4-хлорбензамид. К перемешанной суспензии N-[4-S)-3-амино-4-гидроксибутил)фенил]-4-хлорбензамида (250 мг) и ацетата натрия (124 мг) в метаноле (10 мл) добавляли по каплям раствор цианогенбромида (100 мг) в метаноле (3 мл). Затем полученный в результате бледно-желтый раствор перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь наливали в 1 н. водный NaOH и дважды экстрагировали дихлорметаном/ТГФ. Объединенные органические слои промывали насыщенным водным NaCl, высушивали над Na2SO4, фильтровали и концентрировали в вакууме. Сырое вещество очищали флэшхроматографией (силикагель; элюент: от 0% до 100% EtOAc в гептане, затем от 0% до 30% MeOH вEtOAc) с получением N-4-[2-S)-2-амино-4,5-дигидрооксазол-4-ил)этил]фенил-4-хлорбензамида (150 мг, 56%) в виде белого твердого вещества. МС (ISP): 346,1 ([37ClM+H]+), 344,2 ([35ClM+H]+). Соединение, указанное в заголовке, получили по аналогии с примером S4, используя (R)-2-(4-бромфенил)морфолин вместо (S)-2-(4-бромфенил)морфолина на стадии b и 2-хлор-6-метилизоникотиновую кислоту (CAS 25462-85-5) вместо 6-(2,2,2-трифторэтокси)никотиновой кислоты на стадии е). Светложелтое твердое вещество. МС (ISP): 334,1 ([37ClM+H]+), 332,1 ([35ClM+H]+).a) (S)-2-(4-Бромфенил)морфолин. Энантиомеры (RS)-2-(4-бромфенил)морфолина (2,27 г, CAS-1131220-82-0) разделяли, используя хиральную ВЭЖХ (колонка: Chiralpak IA, 832 см; элюент: н-гептан/этанол (1:11), содержащий 0,1%(S)-2-(4-Бром-фенил)-морфолин (36,3 г) и N,N-диизопропилэтиламин (31,4 мл) в ТГФ (360 мл) обрабатывали ди-трет-бутилдикарбонатом (39,3 г). Реакционную смесь перемешивали в течение 17 ч при комнатной температуре, затем концентрировали в вакууме, разбавляли этилацетатом, промывали 1 М водной лимонной кислотой (2100 мл), высушивали над сульфатом магния, фильтровали и концентрировали в вакууме. Сырое вещество кристаллизовали из гексана с получением (S)-трет-бутил-2-(4-бромфенил)морфолин-4-карбоксилата (47,1 г, 92%) в виде беловатого твердого вещества. МС (ISP): 344,1 ([M+H]+).(6,41 г) и Pd2(dba)3 (3,14 г) растворяли в атмосфере аргона в сухом и деаэрированном толуоле (940 мл) и обрабатывали трет-бутоксидом натрия (18,5 г). Темно-коричневую смесь перемешивали при 90C в течение 18 ч. Желто-коричневую реакционную смесь разбавляли толуолом (700 мл), охлаждали до комнатной температуры и дважды экстрагировали водой. Органический слой отделяли, высушивали над сульфатом магния и концентрировали в вакууме. Сырой продукт разбавляли 300 мл гексана, перемешивали в течение 1 ч и отфильтровывали с получением оранжевого твердого вещества (68 г), которое очищали колоночной хроматографией (силикагель, 20% этилацетат/гептан). Объединенные и концентрированные фракции суспендировали в гексане, перемешивали в течение 17 ч, отфильтровывали и высушивали в вакууме с получением (S)-трет-бутил 2-(4-(дифенилметиленамино)фенил)морфолин-4-карбоксилата (54,1 г,89%) в виде желтого твердого вещества. МС (ISP): 443,3 ([M+H]+).d) (S)-трет-Бутил 2-(4-аминофенил)морфолин-4-карбоксилат. Суспензию (S)-трет-бутил-2-(4-(дифенилметиленамино)фенил)морфолин-4-карбоксилата (54,1 г),формиата аммония (116 г) и 5% палладия на угле (6,5 г) в метаноле (930 мл) перемешивали при 60C в течение 2 ч. Реакционную смесь фильтровали и концентрировали в вакууме. Остаток растворяли в этилацетате и воде. Органическую фазу дважды экстрагировали 0,5 М водной HCl. Объединенные водные фазы подщелачивали 2 М водным NaOH и дважды экстрагировали дихлорметаном. Органические фазы высушивали над сульфатом магния, фильтровали и высушивали в вакууме с получением (S)-трет-бутил 2-(4-аминофенил)морфолин-4-карбоксилата (31,95 г, 94%) в виде беловатого твердого вещества. МСe) (S)-трет-Бутил-2-(4-(6-(2,2,2-трифторэтокси)никотинамидо)фенил)морфолин-4-карбоксилат. К перемешанной суспензии (S)-трет-бутил-2-(4-аминофенил)морфолин-4-карбоксилата (1,5 г) в ТГФ (75 мл) последовательно добавляли N-метилморфолин (1,78 мл), HBTU (3,07 г) и 6-(2,2,2-трифторэтокси)никотиновую кислоту (1,63 г), и смесь перемешивали при комнатной температуре в течение 17 ч. Суспензию разбавляли EtOAc и последовательно промывали 0,5 М водной HCl, насыщенным воднымNaHCO3 и насыщенным рассолом. Органический слой высушивали над MgSO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали перекристаллизацией из гептана/EtOAc (1:1) с получением (S)-трет-бутил-2-(4-(6-(2,2,2-трифторэтокси)никотинамидо)фенил)морфолин-4-карбоксилата (2,11 г,81%) в виде белого твердого вещества. МС (ISP): 482,1 ([M+H]+).f) (S)-N-(4-(Морфолин-2-ил)фенил)-6-(2,2,2-трифторэтокси)никотинамида гидрохлорид. К перемешанной суспензии (S)-трет-бутил-2-(4-(6-(2,2,2-трифторэтокси)никотинамидо)фенил) морфолин-4-карбоксилата (2,11 г) в диоксане (8 мл) добавляли по каплям раствор хлорида водорода в диоксане (16,7 мл, 4 М раствор), и смесь нагревали при 60C в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли диоксаном, и кристаллический продукт собирали фильтрованием, промывая Et2O. Продукт высушивали в вакууме с получением (S)-N-(4-(морфолин-2-ил)фенил)-6(2,2,2-трифторэтокси)никотинамида гидрохлорида (1,75 г, 94%) в виде светло-желтого твердого вещества. МС (ISP): 382,2 ([M+H]+). Соединение, указанное в заголовке, получили по аналогии с примером S4, используя 2(трифторметил)изоникотиновую кислоту (CAS 131747-41-6) вместо 6-(2,2,2-трифторэтокси)никотиновой кислоты на стадии е). Беловатое твердое вещество. МС (ISP): 352,3 ([M+H]+).a) (S)-трет-Бутил-2-(4-(3-(4-фторбензил)уреидо)фенил)морфолин-4-карбоксилат. К перемешанному раствору (S)-трет-бутил-2-(4-аминофенил)морфолин-4-карбоксилата (100 мг) в ДМФ (3,5 мл) последовательно добавляли триэтиламин (62 мкл) и 1-фтор-4-(изоцианатометил)бензол(58,4 мкл), и смесь перемешивали при 60C в течение 17 ч. Суспензию охлаждали до комнатной температуры, затем разбавляли водой и дважды экстрагировали EtOAc. Объединенные органические фазы последовательно промывали водой и насыщенным рассолом. Органический слой высушивали над MgSO4,фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (силикагель; градиент: EtOAc/гептан) с получением (S)-трет-бутил-2-(4-(3-(4-фторбензил)уреидо)фенил)морфолин-4 карбоксилата (164 мг, количественно) в виде белого твердого вещества. МС (ISP): 374,0 ([M+H-С 4 Н 8]+).b) (S)-1-(4-Фторбензил)-3-(4-(морфолин-2-ил)фенил)мочевины гидрохлорид. К перемешанной суспензии (S)-трет-бутил-2-(4-(3-(4-фторбензил)уреидо)фенил)морфолин-4-карбоксилата (163 мг) в ТГФ (9 мл) добавляли по каплям раствор хлорида водорода в диоксане (1,42 мл, 4 М раствор), и смесь нагревали при 60C в течение 6 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли EtOAc, и кристаллический продукт собирали фильтрованием, промывая Et2O. Продукт высушивали в вакууме с получением (S)-1-(4-фторбензил)-3-(4-(морфолин-2-ил)фенил)мочевины гидрохлорида (101 мг, 73%) в виде белого твердого вещества. МС (ISP): 330,1 ([M+H]+). Соединение, указанное в заголовке, получили по аналогии с примером S7, используя 3 изоцианатобензонитрил (CAS 16413-26-6) вместо 1-фтор-4-(изоцианатометил)бензола на стадии а). Беловатое твердое вещество. МС (ISP): 323,2 ([M+H]+). Соединение, указанное в заголовке, получили по аналогии с примером S4, используя 6 хлорникотиновую кислоту (CAS 5326-23-8) вместо 6-(2,2,2-трифторэтокси)никотиновой кислоты на стадии е). Беловатое твердое вещество. МС (ISP): 320,1 ([37ClM+H]+), 318,1 ([35ClM+H]+). Функциональная активность in vitro агонистов TAAR1 при рецепторе TAAR1 мыши Рекомбинантные клетки HEK293, экспрессирующие TAAR1 мыши, выращивали при 37C и 5%CO2/95% воздуха в 250 мл культуральных флаконах Falcon в 30 мл культуральной среды. Среда для клеточной культуры содержала DMEM с высоким содержанием глюкозы, фетальную сыворотку теленка(10%, инактивирована нагреванием в течение 30 мин при 56C), генетицин G418 (500 мкг/мл) и пенициллин/стрептомицин (1%). Клетки собирали при 80-90% конфлюентности. Затем культуральную среду удаляли из культуральных флаконов, клетки промывали один раз 5 мл ФСБ. После удаления промывочного раствора добавляли 5 мл раствора трипсин/ЭДТА в течение 5 мин при 37C. Затем 45 мл культуральной среды добавляли к 5 мл раствора открепленных клеток, и суммарно 50 мл переносили в пробирку на 50 мл Falcon (ref: 2070), пробирку центрифугировали при 1300 об/мин в течение 3 мин при КТ, и культуральную среду отбрасывали. Осадок клеток ресуспендировали в свежей культуральной среде и доводили до 510 Е 5 клеток на миллилитр. Затем клетки высевали в 96-луночный планшет (BIOCOAT 6640 от фирмы Becton Dickinson) многоканальной пипеткой (100 мкг/лунка, 50000 клеток/лунка) и инкубировали в течение 20 ч при 37C. Анализ цАМФ Среду для клеточной культуры удаляли, и клетки промывали один раз ФСБ. Добавляли 50 мкл ФСБ(AMIMED без эндотоксинов: 8-05F00-1) с 1 мМ IBMX, и клеткам давали инкубироваться в течение 30 мин при 37C и 5% СО 2/95% воздуха. Затем добавляли 50 мкл (например) 20 мкМ раствора соединения или 50 мкл стимулирующей концентрации 50% бета-РЕА в ФСБ (AMIMED без эндотоксинов) с 1 мМIBMX, и клетки снова инкубировали в течение 30 мин при 37C, как указано выше. После инкубации клетки подвергали лизису 50 мкл 3 раствора Detection Mix, содержащего Ru-цАМФ, Alexa700-цАМФAb и и буфер для лизиса, в течение по меньшей мере 60 мин (лучше 2 ч) при КТ при сильном встряхивании. Флуоресценцию измеряли на NanoScan (считывающее устройство IOM) (возбуждение 456 нм, испускание 630 и 700 нм).- 14024703 Соединения проявляют значение EC50 (мкМ) при TAAR1 мыши в диапазоне от менее 0,01 мкМ, как показано в приведенной ниже таблице. Значения эффективности (% эфф.) относятся к фенилэтиламину,обладающему 100% агонистической активностью. Активность, подобная антипсихотической, агонистов TAAR1, аддитивная и синергическая по отношению к имеющемуся в продаже антипсихотическому средству оланзапину в двух животных моделях, показательных для психоза Аддитивный эффект агонистов TAAR1 и оланзапина в тесте индуцированной кокаином локомоции у мышей Было показано, что активация TAAR1 осуществляет понижающую регуляцию дофаминергической нейротрансмиссии, тогда как было показано, что ингибирование TAAR1 усиливает ее (Lindemann et al.,2008; Bradaia et al., 2009). Данные тестов на индуцированную кокаином и L-687414 (бензилоксиамином) повышенную локомоторную активность у мышей указывают на то, что агонисты TAAR1 обладают потенциальной активностью, подобной антипсихотической. При дозах, которые обладали умеренными эффектами на локомоторную активность базового уровня, соединение S2 значимо антагонизировало индуцированную кокаином повышенную локомоторную активность при 1 и 3 мг/кг ПО (перорально) у мышей. Кроме того, частично активные дозы S2 (0,3 мг/кг,ПО) и оланзапина (0,3 мг/кг, ПО), когда их объединяли, полностью вызывали обратное развитие повышенной локомоции, индуцированной кокаином (фиг. 1 а). Это указывает на то, что S2 может обладать аддитивным эффектом на имеющееся в продаже антипсихотическое средство оланзапин. Также наблюдали, что, хотя дозы S1 (0,1 мг/кг, ПО) и оланзапина (0,3 мг/кг, ПО) частично антагонизировали индуцированную кокаином локомоторную активность при тестировании по отдельности,наблюдали полное обратное развитие индуцированной кокаином повышенной локомоторной активности, когда эти соединения комбинировали (фиг. 1b. Это указывает на то, что S1 обладает аддитивным эффектом на имеющееся в продаже антипсихотическое средство оланзапин, подтверждая его потенциал в качестве дополнительной терапии к имеющимся в продаже антипсихотическим средствам. Фиг. 1. Эффекты на индуцированную кокаином локомоцию у мышей.a) Дозы S1 (0,1 мг/кг ПО) и оланзапина (0,3 мг/кг ПО), которые частично антагонизировали индуцированную кокаином повышенную локомоторную активность при тестировании по отдельности, показали эффект нормализации при комбинировании. p0,05, p0,01, p0,002 по сравнению с группойb) Дозы S2 (0,3 мг/кг ПО) и оланзапина (0,3 мг/кг ПО), которые частично антагонизировали индуцированную кокаином повышенную локомоторную активность при тестировании по отдельности, показали эффект нормализации при комбинировании. p0,05, p0,01, p0,002 по сравнению с группой"носитель и кокаин". Синергический эффект агониста TAAR1 и оланзапина в тесте на L-687414-индуцированную локомоцию у мышей Чтобы изучить его потенциальный эффект на глутаматергическую систему, S2 (0,00003-1 мг/кг,ПО) тестировали в остром опыте индуцированной L-687414 (N-гидрокси-3-амино-4-метилпирролидин-2 он, антагонист рецептора NMDA, действующий при глициновом сайте) повышенной локомоции у мышей, где он дозозависимо антагонизировал L-687414, при значимости в диапазоне дозы от 0,003 до 1 мг/кг, ПО (фиг. 2). Фиг. 2. Эффекты агониста TAAR1 на L-687414-индуцированную локомоцию у мышей.L-687414-индуцированная локомоция: S2 (0,00003-1 мг/кг ПО) полностью антагонизировал L- 15024703 687414-индуцированную повышенную локомоторную активность от 0,003 до 1 мг/кг (закрашенные кружки). p0,05, p0,01, p0,001 по сравнению с носителем (пустые кружки). Кроме того, частично активную дозу агониста TAAR1 S2 (0,001 мг/кг, ПО; серый столбик на фиг. 2) добавляли к возрастающим дозам оланзапина (0-0,1 мг/кг, ПО). Как показано на фиг. 3, частично активная доза S2 (0,001 мг/кг, ПО), комбинируемая с неактивными дозами оланзапина (0,02-0,06 мг/кг, ПО) полностью антагонизировала L-687414-индуцированную локомоторную активность, что указывает на то,что агонист TAAR1 и оланзапин проявляют синергические эффекты в данной мышиной модели, показательной для шизофрении. Фиг. 3. Синергизм с оланзапином при L-687414-индуиированной локомоции у мышей.L-687414-индуцированная локомоция: S2 (0,001 мг/кг, ПО), комбинированный с возрастающими дозами оланзапина (0 - 0,1 мг/кг, ПО), полностью антагонизировал L-687414-индуцированную повышенную локомоторную активность при 0,02 и 0,06 мг/кг оланзапина. p0,05, p0,01, p0,001 L687414+S2 по сравнению с группой одного L-687414. Острый эффект агонистов TAAR1 в оральном тесте толерантности к глюкозе (ОГТТ) у самцов обычных мышей C57BI6 Оральный тест толерантности к глюкозе (ОГТТ) проводили у мышей C57BI6, чтобы изучить потенциальные противодиабетические эффекты агонистов TAAR1. Самцов мышей C57BL/6J (Charles RiverLaboratories, Lyon, France) стратифицировали в группы по 8 мышей в соответствии с их массой тела. Накануне вечером животные получали 1 г корма, это соответствовало периоду голодания примерно 10 ч. На сутки эксперимента животных обрабатывали агонистами TAAR1 или плацебо (0,3% Твин 80) за 45 мин до пероральной провокации глюкозой 2 г/кг. Основным регистрируемым значением было содержание глюкозы в крови, измеренное с помощью прибора Accu-Chek Aviva. Параллельно образцы крови брали на определение инсулина.S1 значительно снижал отклонение глюкозы в крови при 0,1 и 0,3 мг/кг, ПО по сравнению с носителем после провокации глюкозой (фиг. 4 а). В то же время, отклонение инсулина было значимо ниже при введении агониста TAAR1 по сравнению с обработкой носителем (фиг. 4b. Никакого эффекта на уровни глюкозы натощак не наблюдали при использовании S1. Поскольку метаболический синдром имеет высокую распространенность при шизофрении, предполагают, что противодиабетический эффект обладает положительным воздействием на пациентов с шизофренией. Фиг. 4. Эффект S1 на AUC глюкозы и инсулина в ОГТТ у мышей.S1 (0,1, 0,3 мг/кг ПО) заметно снижал AUC (0 - 60 мин) (а) глюкозы и (b инсулина во время ОГТТ у мышей. Результаты представлены в виде среднегоSEM. Статистика: Anova, затем апостериорный критерий Даннета,p0,01,p0,001 по сравнению с группой носителя (Veh); n=8/группа. Несколько дополнительных агонистов TAAR1 (S2-8) с различными уровнями эффективности (5190% по сравнению с бета-фенилэтиламином) было протестировано в ОГТТ, и все показали значимый эффект при снижении AUC глюкозы и, если были протестированы, при снижении AUC инсулина (фиг. 59). Фиг. 5. Эффект S2 на AUC глюкозы и инсулина в ОГТТ у мышей.S2 (0,3, 1 мг/кг, ПО) заметно снижал AUC (0-60 мин) (а) глюкозы и (b инсулина во время ОГТТ у мышей. Результаты представлены в виде среднегоSEM. Статистика: Anova, затем апостериорный критерий Даннета,p0,001 по сравнению с группой носителя (Veh); n=8/группа. Фиг. 6. Эффект S3 на AUC глюкозы и инсулина в ОГТТ у мышей.S3 (1, 3 мг/кг ПО) заметно снижал AUC (0-60 мин) (а) глюкозы и (b инсулина во время ОГТТ у мышей. Результаты представлены в виде среднегоSEM. Статистика: Anova, затем апостериорный критерий Даннета,p0,05,p0,001 по сравнению с группой носителя (Veh); n=8/группа. Фиг. 7. Эффект S4 на AUC глюкозы и инсулина в ОГТТ у мышей.S4 (0,3 мг/кг, ПО) заметно снижал AUC (0-60 мин) (а) глюкозы и (b инсулина во время ОГТТ у мышей. Результаты представлены в виде среднегоSEM. Статистика: Anova, затем апостериорный критерий Даннета, p0,05, p0,001 по сравнению с группой носителя (Veh); n=8/группа. Фиг. 8. Эффект S5 на AUC глюкозы в ОГТТ у мышей.S5 (30 мг/кг, ПО) заметно снижал AUC (0-60 мин) глюкозы во время ОГТТ у мышей. Результаты представлены в виде среднегоSEM. Статистика: Anova, затем апостериорный критерий Даннета,p0,001 по сравнению с группой носителя (Veh); n=8/группа. Фиг. 9. Эффекты S6, S7 и S8 (каждый 10 мг/кг, ПО) на AUC глюкозы в ОГТТ у мышей.S6 (10 и 30 мг/кг, ПО), S7 (10 мг/кг, ПО), и S8 (10 и 30 мг/кг, ПО) заметно снижали AUC (0-60 мин) глюкозы во время ОГТТ у мышей. Результаты представлены в виде среднегоSEM. Статистика: Anova,затем апостериорный критерий Даннета, p0,01, p0,001 по сравнению с группой носителя (Veh);a) S9 (10 мг/кг, ПО) и S10 (10 мг/кг, ПО), а также b) S11 (1 и 3 мг/кг, ПО) заметно снижали AUC (0 60 мин) глюкозы во время ОГТТ у мышей. Результаты представлены в виде среднегоSEM. Статистика:Anova, затем апостериорный критерий Даннета, p0,05, p0,01, p0,001 по сравнению с группой носителя (Veh); n=8/группа. Фиг. 11. Эффекты S12, S13, S14, S15 и S16 на AUC глюкозы в ОГТТ у мышей.AUC (0-60 мин) глюкозы во время ОГТТ у мышей. Результаты представлены в виде среднегоSEM. Статистика: Anova, затем апостериорный критерий Даннета, p0,05, p0,01, p0,001 по сравнению с группой носителя (Veh); n=8/группа. Фиг. 12. Эффект S17 (0,3 и 1 мг/кг, ПО) на AUC глюкозы в ОГТТ у мышей.S17 (0,3 и 1 мг/кг, ПО) заметно снижал AUC (0-60 мин) глюкозы во время ОГТТ у мышей. Результаты представлены в виде среднегоSEM. Статистика: Anova, затем апостериорный критерий Даннета,p0,01, p0,001 по сравнению с группой носителя (Veh); n=8/группа. Агонисты TAAR1 снижают увеличение прибавления массы у нормальных крыс и нормализуют увеличение прибавления массы, индуцированное антипсихотическим лекарственным средством оланзапином. Многие антипсихотические лекарственные средства индуцируют прибавление массы у пациентов с шизофренией. Поскольку прибавление массы может привести к серьезным осложнениям состояния здоровья и заболеваниям, таким как диабет, принципиально важно оценить перспективные, более новые соединения у животных на их склонность к изменению массы. У самок крыс линии Спраг-Доули использовали 14-суточный режим обработки S2 и оланзапином,антипсихотическим лекарственным средством, применяемым клинически, которое известно как приводящее к прибавлению массы. Параллельно с измерением массы тела использовали магнитно-резонансную (МР) релаксометрию,чтобы неинвазивным способом определить состав жировой массы у животных. Результаты показывают, что S2 при 3 и 10 мг/кг ПО уменьшал прибавление массы (фиг. 13 а), жировой массы (фиг. 14 а, 14 с) и потребление пищи (табл. 1) по сравнению с животными, обработанными носителем, не индуцируя потерю массы по сравнению со значениями до обработки. Значимый эффект не был измерен при 1 мг/кг ПО (фиг. 13b, 14b, табл. 1). Кроме того, S2 (1 мг/кг ПО) в комбинации с оланзапином (2 мг/кг ПО) снижал увеличение прибавления массы, содержание жировой массы и потребление пищи, измеренные у крыс, обработанных оланзапином (фиг. 13b, 14b, табл. 1). Это указывает на то, что S2 не индуцирует прибавление массы при отдельном введении и может ингибировать прибавление массы, индуцированное имеющимся в продаже антипсихотическим средством оланзапином. Фиг. 13. Эффекты S2 на суммарное прибавление массы у крыс.(a) S2 при 3 и 10 мг/кг, но не при 1 мг/кг ПО уменьшал прибавление массы у самок крыс линии Спраг-Доули (контрасты линейных трендов в группах S2 по сравнению с линейным трендом в группе носителя, p=0,0024 при 3 мг/кг и p=0,012 при 10 мг/кг). (b) S2 (1 мг/кг ПО) в комбинации с оланзапином(2 мг/кг ПО) нормализовал индуцированное оланзапином прибавление массы (линейные тренды оланзапина по сравнению с носителем, p=4,6 Е-10; S2/оланзапин по сравнению с носителем, p=0,41). Межгрупповые сравнения в каждый момент времени: t-критерий, p0,05, p0,01, р 0,001 по сравнению с носителем; p0,05, p0,01, р 0,001 по сравнению с группой оланзапина; n=8/группа. Фиг. 14. Эффекты S2 на содержание жировой массы у крыс.S2 (1 мг/кг ПО) в комбинации с оланзапином (2 мг/кг ПО) нормализовал индуцированное оланзапином увеличение жировой массы у самок крыс линии Спраг-Доули. Значимый эффект одного S2 не был измерен по сравнению с группой носителя. Критерий Даннета p0,05, p0,01, р 0,001 по сравнению с носителем; p0,05, p0,01, р 0,001 по сравнению с группой оланзапина; n=8/группа.- 17024703 Таблица 1. Эффекты S2 на суммарное потребление пищи у крыс.S2 (1 мг/кг ПО) в комбинации с оланзапином (2 мг/кг ПО) нормализовал индуцированное оланзапином увеличение потребления пищи. Значимое снижение потребления пищи было измерено у обработанных S2 крыс при 10 мг/кг; n=8/группа. Оланзапин и соединения S1-S17 и фармацевтически приемлемые соли можно применять в качестве лекарственных средств, например, в форме фармацевтических препаратов. Эти фармацевтические препараты можно вводить перорально, например, в форме таблеток, таблеток с покрытием, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Однако введение можно также осуществлять ректально, например, в форме суппозиториев, или парентерально, например, в форме инъекционных растворов. Соединения, выбранные из S1-S17, можно обрабатывать фармацевтически инертными, неорганическими или органическими носителями для получения фармацевтических препаратов. Лактозу, кукурузный крахмал, целлюлозу или ее производные, тальк, стеариновые кислоты или их соли и тому подобное можно использовать, например, в качестве таких носителей для таблеток, таблеток с покрытием, драже и твердых желатиновых капсул. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и тому подобное. Однако в зависимости от природы активного вещества носители обычно не требуются в случае мягких желатиновых капсул. Подходящими носителями для получения растворов и сиропов являются, например, вода,полиолы, глицерин, растительное масло и тому подобное. Подходящими носителями для суппозиториев являются, например, натуральные или отвержденные масла, воски, жиры, полутвердые или жидкие полиолы и тому подобное. Кроме того, фармацевтические препараты могут содержать консерванты, солюбилизаторы, стабилизаторы, увлажняющие агенты, эмульгаторы, подсластители, красители, корригенты, соли для варьирования осмотического давления, буферы, маскирующие агенты или антиоксиданты. Они могут также содержать еще другие терапевтически ценные вещества. Лекарственные средства, содержащие оланзапин и соединение, выбранное из S1-S17, или его фармацевтически приемлемую соль и терапевтически инертный носитель, также являются объектом настоящего изобретения, как и способ их получения, который включает приведение одного или более чем одного соединения, выбранного из S1-S17, и оланзапина и/или фармацевтически приемлемых солей присоединения кислоты и, если желательно, одного или более чем одного другого терапевтически ценного вещества в форму введения в виде галенова препарата вместе с одним или более чем одним терапевтически инертным носителем. Дозировка может варьировать в широких пределах и должна быть, конечно, отрегулирована в соответствии с индивидуальными потребностями в каждом конкретном случае. В случае перорального введения дозировка для взрослых может варьировать от примерно 0,01 мг до примерно 1000 мг в сутки оланзапина и соединения, выбранного из S1-S17, или соответствующего количества его фармацевтически приемлемой соли. Суточную дозировку можно вводить в виде однократной дозы или в разделенных дозах и, кроме того, верхний предел может быть также превышен, когда это считают показанным. Метод получения: 1. Смешивают пп.1, 2, 3 и 4 и гранулируют с дистиллированной водой. 2. Высушивают гранулы при 50C. 3. Пропускают гранулы через подходящее измельчительное оборудование. 4. Добавляют п.5 и смешивают в течение 3 мин; прессуют на подходящем прессе. Препарат в виде капсулы Метод получения: 1. Смешивают пп.1-5 и гранулируют с дистиллированной водой. 2. Высушивают гранулы при 50C. 3. Пропускают гранулы через подходящее измельчительное оборудование. 4. Добавляют п.6 и смешивают в течение 3 мин; прессуют на подходящем прессе. Комбинированный препарат- 19024703 Метод получения: 1. Смешивают пп.1-6 и гранулируют с дистиллированной водой. 2. Высушивают гранулы при 50C. 3. Пропускают гранулы через подходящее измельчительное оборудование. 4. Добавляют пп.7 и 8 и смешивают в течение трех минут; прессуют на подходящем прессе. Литература- 20024703 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Комбинация для лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, включающая оланзапин и агонист TAAR1, где агонист TAAR1 представляет собойS17 = (S)-6-хлор-N-(4-(морфолин-2-ил)фенил)никотинамид. 2. Соединение S2 = (S)-4-(3-фтор-2-метилфенил)-4,5-дигидрооксазол-2-иламин. 3. Применение комбинации по п.1 для лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, характеризующегося сниженной частотой возникновения метаболического синдрома. 4. Применение комбинации по п.1 для получения лекарственного средства для лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, характеризующегося сниженной частотой возникновения метаболического синдрома. 5. Применение по п.4, в котором указанное лекарственное средство обладает противодиабетическим действием. 6. Применение по п.5, в котором указанное противодиабетическое действие заключается в уменьшении колебаний концентрации глюкозы в крови. 7. Применение по п.6, в котором указанное противодиабетическое действие заключается в снижении жировой массы и массы тела. 8. Способ лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, характеризующийся сниженной частотой возникновения метаболического синдрома, включающий введение человеку, нуждающемуся в этом, эффективного количества комбинации по п.1. 9. Способ лечения по п.8, где сниженная частота возникновения метаболического синдрома является результатом противодиабетического действия, заключающегося в уменьшении колебаний концентрации глюкозы в крови, снижении жировой массы и массы тела. 10. Фармацевтическая композиция, содержащая комбинацию по п.1 вместе с фармацевтически приемлемыми эксципиентами, для лечения шизофрении и маниакальных эпизодов, обусловленных биполярными расстройствами, характеризующегося сниженной частотой возникновения метаболического синдрома. 11. Фармацевтическая композиция по п.10, где сниженная частота возникновения метаболического синдрома является результатом противодиабетического действия, заключающегося в уменьшении колебаний концентрации глюкозы в крови, снижении жировой массы и массы тела.
МПК / Метки
МПК: A61P 25/18, A61K 31/5513, A61K 31/421, A61K 31/422, A61K 31/00
Метки: оланзапина, агониста, taar1, комбинация
Код ссылки
<a href="https://eas.patents.su/26-24703-kombinaciya-olanzapina-i-agonista-taar1.html" rel="bookmark" title="База патентов Евразийского Союза">Комбинация оланзапина и агониста taar1</a>
Комбинация ингибитора обратного захвата серотонина и антагониста, обратного агониста или неполного агониста 5-нт2с-рецептора

Номер патента: 6391
Опубликовано: 29.12.2005
Авторы: Хогг Сандра, Боскер Фокко Ян, Викстрем Хокан Вильхельм, Ден Бур Йохан Антони, Бегесе Клаус Петер, Кремерс Томас Иво Франсискус Хюберт, Мерк Арне, Вестеринк Бернард Хендрик Корнелис
МПК: A61K 31/517, C12Q 1/00, G01N 33/50...
Метки: комбинация, антагониста, агониста, 5-нт2с-рецептора, серотонина, неполного, ингибитора, обратного, захвата
Формула / Реферат:
1. Применение антагониста, обратного агониста или неполного агониста 5-HT2C-рецептора для получения фармацевтической композиции, используемой в комбинации с ингибитором обратного захвата серотонина, при условии что антагонист, обратный агонист или неполный агонист 5-HT2C-рецептора не является миртазепином или миансерином. 2. Применение антагониста, обратного агониста или неполного агониста 5-HT2C-рецептора для получения фармацевтической...
Комбинация агониста бета-2-адренорецепторов и аминосахара и их применение для лечения иммуномодуляторных расстройств

Номер патента: 7906
Опубликовано: 27.02.2007
Автор: Вэйднер Мортэн Слот
МПК: A61K 31/135, A61K 31/726, A61K 31/167...
Метки: применение, лечения, иммуномодуляторных, агониста, комбинация, бета-2-адренорецепторов, расстройств, аминосахара
Формула / Реферат:
1. Химический комплекс, включающий: i) агонист бета-2-адренорецепторов и ii) аминосахар, выбранный из группы, включающей глюкозамин, маннозамин, их производные и соли, при этом их производные выбраны из группы, в которой аминогруппа и/или гидроксигруппа аминосахара алкилирована, арилирована или ацилирована, и где аномерное 2-, 3-, 4- или 6-положение сульфатировано или фосфорилировано. 2. Химический комплекс по п.1, в котором агонист...
Конденсированные гетероциклические соединения пиримидинона и их применение в качестве агониста ангиотензина ii и агониста γ-рецептора, активирующего пролиферацию пероксисом

Номер патента: 19823
Опубликовано: 30.06.2014
Авторы: Игава Хидеюки, Маекава Цуеси
МПК: A61K 31/519, A61P 9/00, C07D 487/04...
Метки: конденсированные, ангиотензина, активирующего, применение, соединения, гетероциклические, gamma;-рецептора, пиримидинона, пролиферацию, пероксисом, агониста, качестве
Формула / Реферат:
1. Соединение, представленное формулой (I)где R1 представляет собой атом водорода, C1-6алкил, необязательно замещенный 1-5 заместителями, выбранными из указанной ниже группы А заместителей, 3-10-членную неароматическую циклическую группу, необязательно замещенную 1-5 заместителями, выбранными из указанной ниже группы С заместителей, или 5- или 6-членную ароматическую циклическую группу, необязательно замещенную 1-5 заместителями, выбранными из...
Форма d дигидрата оланзапина

Номер патента: 1881
Опубликовано: 22.10.2001
Авторы: Рётзель Сюзн М., Стефенсон Грегори А., Ларсен Сэмюель Д., Николс Джон Р.
МПК: C07D 243/10, A61K 31/395
Метки: оланзапина, форма, дигидрата
Формула / Реферат:
1. Полиморфная модификация оланзапина дигидрат D, имеющая типичную порошковую рентгенограмму, представленную следующими межплоскостными расстояниями (d), указанными в табл. 1. Таблица 1 d 9,4511 7,7098 7,4482 6,9807 6,5252 5,7076 5,5539 5,223 4,9803 4,8908 ...
Кристаллические формы оланзапина и способы их получения

Номер патента: 8055
Опубликовано: 27.02.2007
Авторы: Грчман Мария, Котар Йордан Берта, Вречер Франк
МПК: C07D 495/04, A61K 31/551, A61P 25/00...
Метки: способы, кристаллические, формы, оланзапина, получения
Формула / Реферат:
1. Способ получения формы I оланзапина, при котором оланзапин кристаллизуют из смеси растворителей, включающей 2-пропанол. 2. Способ по п.1, в котором смесь растворителей также включает по меньшей мере еще один растворитель, выбираемый из группы, включающей тетрагидрофуран, метиленхлорид, ацетон, толуол, N,N-диметилформамид, диметилсульфоксид и хлороформ. 3. Способ по п.2, в котором смесь растворителей содержит 2-пропанол и по меньшей мере еще...
Предыдущий патент: Новые иммуномодуляторные и противовоспалительные соединения
Следующий патент: Способ очистки воды и аппарат для его осуществления
Случайный патент: Рутениевые комплексы в качестве (пред)катализаторов для реакций метатезиса