Способы получения полиморфов бензоатной соли 2-[[6-[(3r)-3-амино-1-пиперидинил]-3,4-дигидро-3-метил-2,4-диоксо-1(2н)-пиримидинил]метил] бензонитрила














Формула / Реферат
1. Способ получения соединения I формулы

где по меньшей мере часть соединения I присутствует в полиморфной форме A, характеризуемой одним или несколькими физическими свойствами, выбранными из группы, состоящей из картины порошковой дифракции рентгеновских лучей с характерными признаками, представляющими собой основные дифракционные линии, которые показаны ниже:
![]()
полученной с использованием Cu-Ka-излучения;
ИК-спектра, содержащего пики поглощения в положениях 1212, 1365, 1447, 1613 и 1697 см-1;
спектра ФП-КР, содержащего пики в положениях 1065, 1103, 1235, 1288, 1337, 1365, 1624, 1689, 2883, 2983 и 3026 см-1; и
спектра дифференциальной сканирующей калориметрии, содержащего эндотермическую область примерно от 173°C до примерно 195°C, при этом способ включает:
(a) кристаллизацию соединения I из любой из следующих систем растворителей, содержащих (i) ацетон, (ii) ацетонитрил; (iii) бутанол, (iv) диметилсульфоксид; (v) диоксан; (vi) этанол; (vii) этанол и изопропиловый спирт; (viii) этанол и воду; (ix) этилацетат; (x) гептан; (xi) изопропанол; (xii) изопропилацетат; (xiii) метанол; (xiv) метилэтилкетон; (xv) метилизобутилкетон; (xvi) 2,2,2-трифторэтанол; (xvii) тетрагидрофуран; (xviii) толуол; (xix) воду и (xx) этанол и гептан; или
(b) превращение аморфной формы 1 соединения I посредством воздействия на аморфную форму 1 нагревания, высокой относительной влажности или органических паров, или посредством мокрого помола аморфной формы 1 в воде.
2. Способ получения соединения I формулы

где по меньшей мере часть соединения I присутствует в аморфной форме 1, при этом способ выбран из группы, состоящей из:
(a) ротационного упаривания из метанола;
(b) быстрого упаривания из воды;
(c) лиофилизации из воды;
(d) кристаллизации соединения I из смеси этилацетата и гексанов; или
из смеси изопропилацетата и гексанов с последующим выделением соединения I из растворителя.
Текст
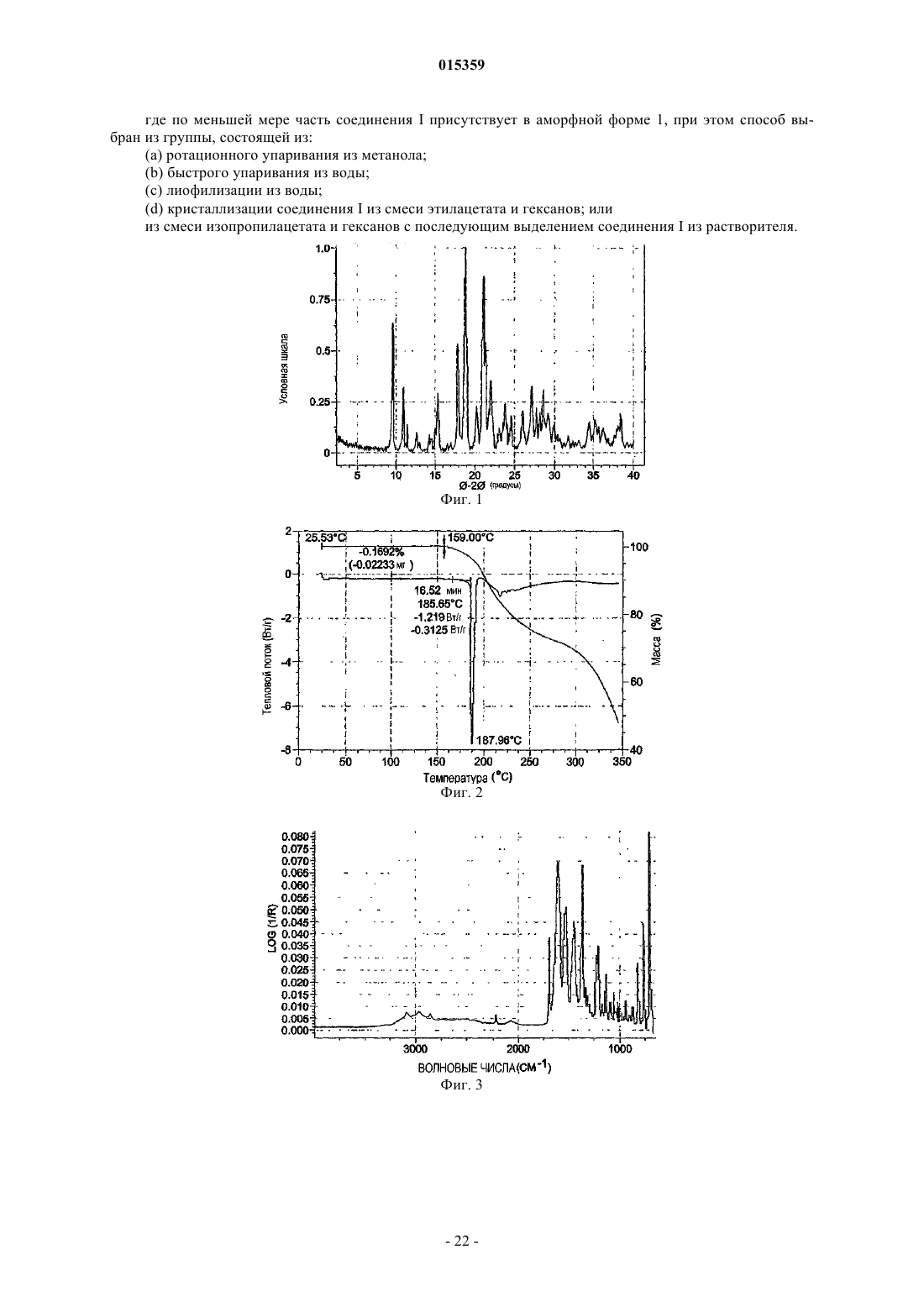
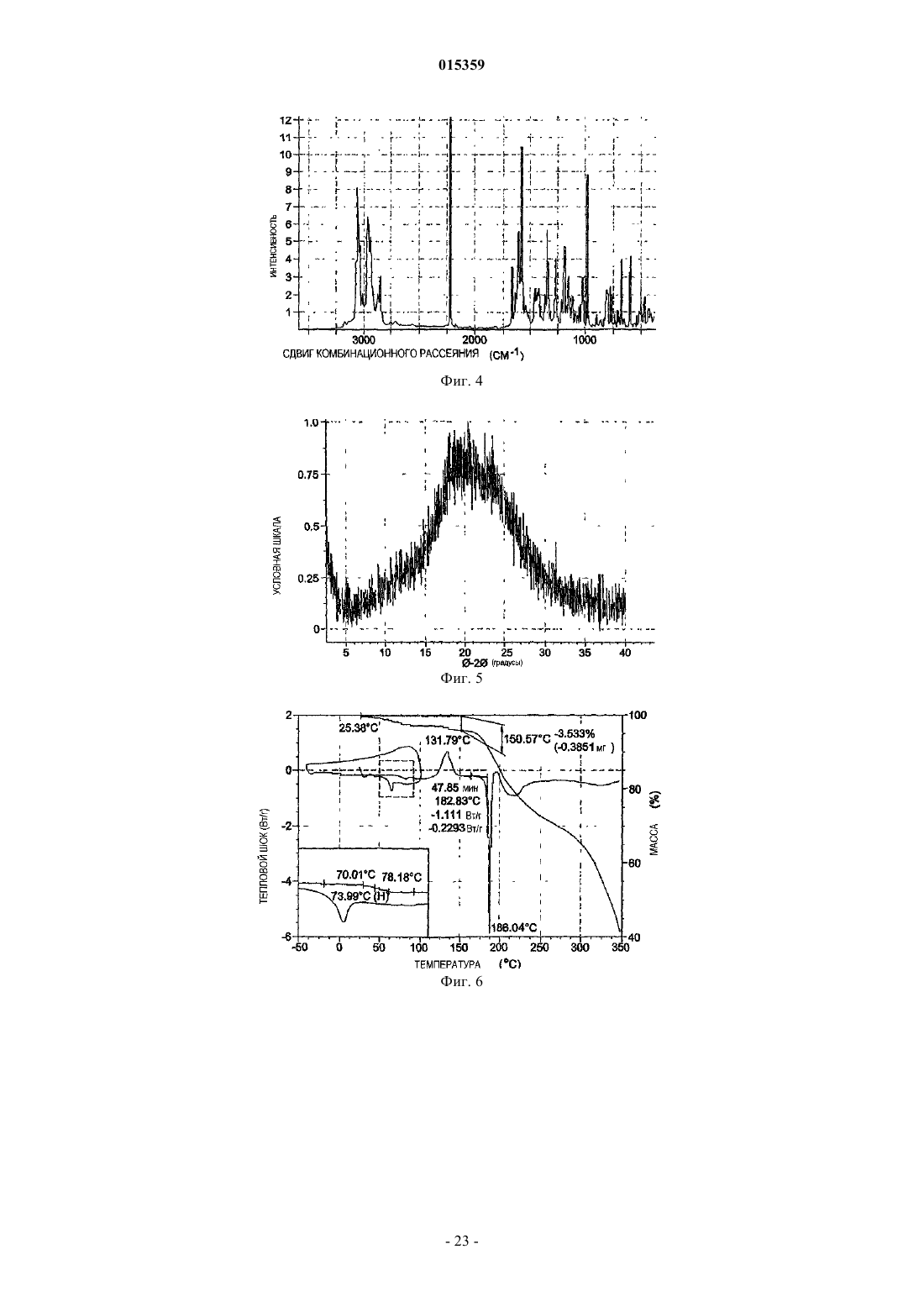
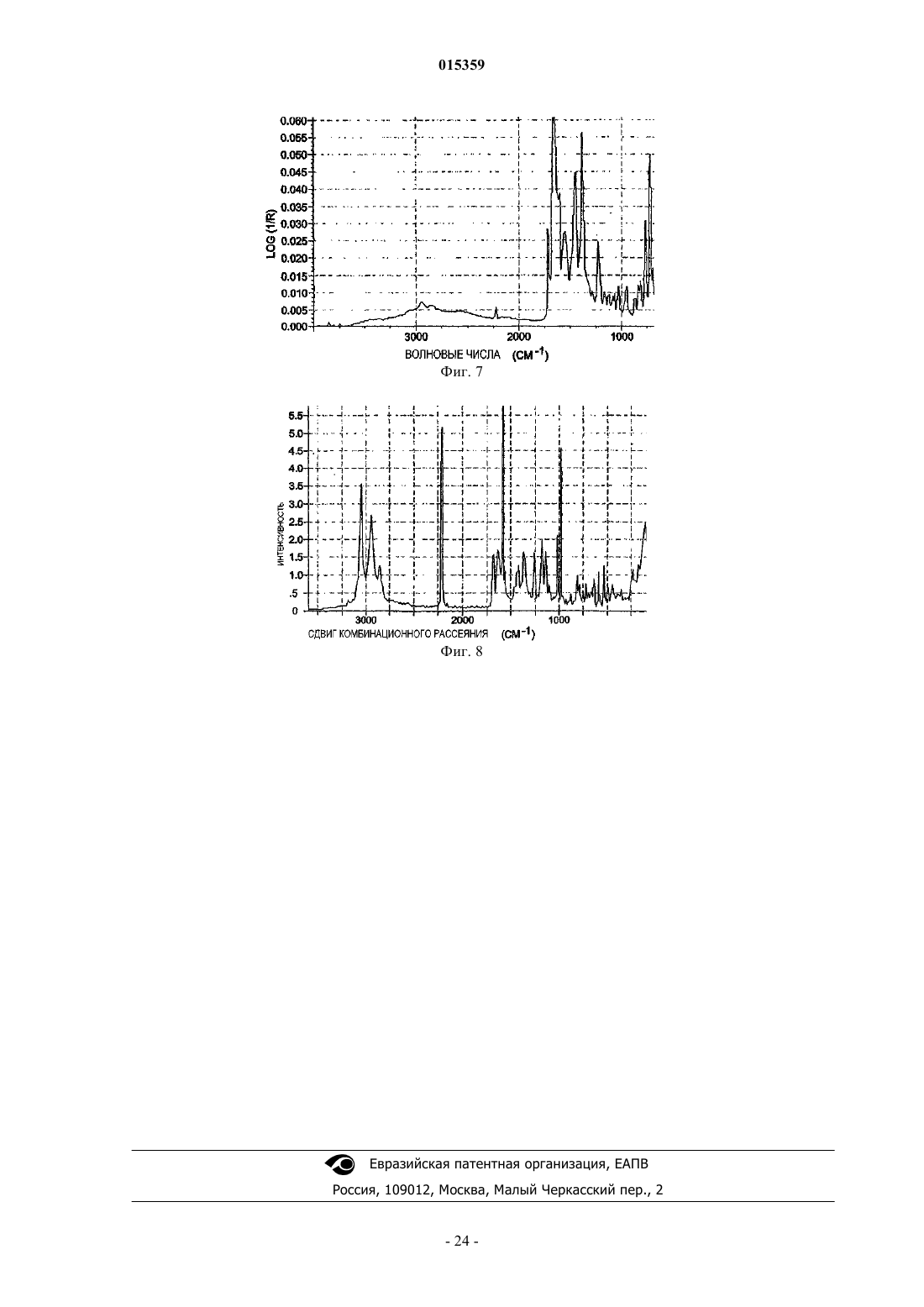
СПОСОБЫ ПОЛУЧЕНИЯ ПОЛИМОРФОВ БЕНЗОАТНОЙ СОЛИ 2-6-[(3R)-3-АМИНО 1-ПИПЕРИДИНИЛ]-3,4-ДИГИДРО-3-МЕТИЛ-2,4-ДИОКСО-1(2 Н)-ПИРИМИДИНИЛ] МЕТИЛ]БЕНЗОНИТРИЛА(71)(73) Заявитель и патентовладелец: ТАКЕДА ФАРМАСЬЮТИКАЛ КОМПАНИ ЛИМИТЕД (JP) Предлагаются способы получения соединения, представленного формулой I в которых часть соединения I присутствует в полиморфной форме A или аморфной форме I. 015359 Изобретение в общем относится к полиморфам бензоатной соли 2-6-[(3R)-3-амино-1 пиперидинил]-3,4-дигидро-3-метил-2,4-диоксо-1(2H)-пиримидинил]метил]бензонитрила (называемого в данном описании соединением I). Дипептидилпептидаза IV (согласно номенклатуре ферментов IUBMB EC.3.4.14.5) является мембранным белком типа II, который в литературе имеет широкое множество названий, включая DPP4, DP4,DAP-IV, FAP, белок 2, образующий комплекс с аденозиндезаминазой, белок, связывающий аденозиндезаминазу (ADAbp), дипептидиламинопептидаза IV; Xaa-Pro-дипептидиламинопептидаза; Gly-Proнафтиламидаза; постпролиндипептидиламинопептидаза IV; лимфоцитарный антиген CD2 6; гликопротеин GP110; дипептидилпептидаза IV; глицилпролинаминопептидаза; глицилпролинаминопептидаза; Xпролилдипептидиламинопептидаза; pep X; лейкоцитарный антиген CD26; глицилпролилдипептидиламинопептидаза; дипептидилпептидгидролаза; глицилпролиламинопептидаза; дипептидиламинопептидазаTp103; X-PDAP. Дипептидилпептидаза IV в данном описании упоминается как "DPP-IV".DPP-IV является неклассической сериновой аминодипептидазой, которая удаляет дипептиды XaaPro с аминоконца (N-конца) полипептидов и белков. Также сообщалось, что зависимое от DPP-IV медленное высвобождение дипептидов типа X-Gly или X-Ser происходит в случае некоторых встречающихся в природе пептидов.DPP-IV конститутивно экспрессируется на эпителиальных и эндотелиальных клетках множества разных тканей (кишечник, печень, легкое, почка и плацента), а также выявляется в жидкостях организма.DPP-IV также экспрессируется на циркулирующих T-лимфоцитах, и было показано, что DPP-IV тождественна антигену клеточной поверхности CD-26. DPP-IV вовлечена в ряд патологических состояний,некоторые из которых обсуждаются ниже.DPP-IV ответственна за метаболическое расщепление некоторых эндогенных пептидов (GLP-1 (736), глюкагон) in vivo и проявляет протеолитическую активность против ряда других пептидов (GHRH,NPY, GLP-2, VIP) in vitro.GLP-1 (7-36) является 29-аминокислотным пептидом, полученным в результате посттрансляционного процессинга проглюкагона в тонком кишечнике. GLP-1 (7-36) оказывает множество воздействий invivo, включая стимуляцию секреции инсулина, ингибирование секреции глюкагона, усиление чувства насыщения и замедление опорожнения желудка. На основании физиологического профиля полагают, что действия GLP-1 (7-36) полезны для профилактики и лечения диабета типа II и, вероятно, для лечения ожирения. Например, обнаружено, что экзогенное введение GLP-1 (7-36) (непрерывная инфузия) пациентам с диабетом является эффективным в данной популяции пациентов. К сожалению, GLP-1 (7-36) быстро распадается in vivo и, как было показано, имеет короткое время полужизни in vivo (t1/2 = 1,5 мин). На основании исследования генетически выведенных нокаутированных по DPP-IV мышей и на основании исследований in vivo/in vitro с использованием избирательных ингибиторов DPP-IV показано,что DPP-IV является основным ферментом, расщепляющим GLP-1 (7-36) in vivo. GLP-1 (7-36) эффективно расщепляется ферментом DPP-IV до GLP-1 (9-36), который, как предполагается, действует в качестве физиологического антагониста GLP-1 (7-36). Поэтому полагают, что ингибирование DPP-IV in vivo применимо для повышения эндогенных уровней GLP-1 (7-36) и уменьшения образования его антагонистаGLP-1 (9-36). Таким образом, считают, что ингибиторы DPP-IV являются средствами, применимыми для профилактики, замедления прогрессирования и/или лечения состояний, опосредованных DPP-IV, в частности диабета, и более конкретно сахарного диабета типа 2, диабетической дислипидемии, состояний нарушения толерантности к глюкозе (IGT), состояний нарушения уровня глюкозы в плазме натощак(IFG), метаболического ацидоза, кетоза, регуляции аппетита и ожирения. Экспрессия DPP-IV возрастает в T-клетках при митогенной или антигенной стимуляции (Mattem, T., et al., Scand. J. Immunol., 1991, 33,737). Сообщалось, что ингибиторы DPP-IV и антитела к DPP-IV подавляют пролиферацию стимулированных митогеном и стимулированных антигеном T-клеток зависимым от дозы образом (Schon, E., et al. ,Biol. Chem. , 1991, 372, 305). Показано, что множество других функций Т-лимфоцитов, таких как продукция цитокинов, опосредованная IL-2 пролиферация клеток и хелперная активность по отношению к В-клеткам, зависят от активности DPP-IV (Schon, E., et al., Scand. J. Immunol., 1989, 29, 127). ИнгибиторыDPP-IV, основанные на борпролине (Flentke, G. R. , et al., Proc. Nat. Acad. Sci. USA, 1991, 88, 1556), хотя и являются нестабильными, были эффективны в ингибировании индуцированной антигеном пролиферации лимфоцитов и продукции IL-2 в T-хелперных клетках CD4+ мышей. Было показано, что такие основанные на бороновой кислоте ингибиторы оказывают действие in vivo, вызывая у мышей подавление продукции антител, индуцированной иммунной стимуляцией (Kubota, T. et al., Clin. Exp. Immun., 1992,89, 192). Роль DPP-IV в регуляции активации T-лимфоцитов также отчасти может быть обусловлена ассоциацией DPP-IV на клеточной поверхности с трансмембранной фосфатазой, CD45. Ингибиторы DPPIV или лиганды неактивного сайта вероятно могут нарушать связь CD45-DPP-IV. Известно, что CD45 является неотъемлемым компонентом аппарата передачи сигнала T-клеток. Сообщалось, что DPP-IV необходима для проникновения и инфекционности вирусов ВИЧ-1 и ВИЧ-2 в T-клетках CD4+ (Wakselman,M., Nguyen, C, Mazaleyrat, J.-P., Callebaut, С, Krust, В., Hovanessian, A. G., Inhibition of HIV-1 infection ofof the 24th European Peptide Symposium 1996). Кроме того, показано, что DPP-IV связывается с ферментом аденозиндезаминазой (ADA) на поверхности Т-клеток (Kameoka, J., et al., Science, 193, 26 466). Дефицит ADA вызывает тяжелый комбинированный иммунодефицитный синдром (SCID) у людей. Указанное взаимодействие ADA-CD26 может быть ключом к патофизиологии SCID. Отсюда следует, что ингибиторы DPP-IV могут быть полезными иммунодепрессантами (или лекарственными средствами,подавляющими высвобождение цитокинов) для лечения среди прочего: отторжения трансплантата органа; аутоиммунных болезней, таких как воспалительное заболевание кишечника, рассеянный склероз и ревматоидный артрит; и для лечения СПИДа. Показано, что DPP-IV в эндотелиальных клетках легких является адгезионной молекулой по отношению к метастазирующим в легкие клеткам карциномы молочной железы и простаты (Johnson, R.С. etal., J. Cell. Biol., 1993, 121, 1423). Известно, что DPP-IV связывается с фибронектином, и известно, что некоторые клетки метастатической опухоли несут большие количества фибронектина на своей поверхности. Сильные ингибиторы DPP-IV могут быть применимы в качестве лекарственных средств для предотвращения метастазов, например опухолей молочной железы и простаты в легкие. Высокие уровни экспрессии DPP-IV также были обнаружены в фибробластах кожи пациентов с псориазом, ревматоидным артритом (RA) и красным плоским лишаем (Raynaud, F., et al., J. Cell. Physiol.,1992, 151, 378). Таким образом, ингибиторы DPP-IV могут быть применимы в качестве средств лечения дерматологических заболеваний, таких как псориаз и красный плоский лишай. Высокая активность DPP-IV обнаружена в гомогенатах тканей, полученных от пациентов с доброкачественной гипертрофией простаты, и в простатосомах. Имеются органеллы, полученные из простаты,которые важны для усиления поступательной подвижности сперматозоидов (Vanhoof, G., et al., Eur. J.Clin. Chem. Clin. Biochem., 1992, 30, 333). Ингибиторы DPP-IV также могут действовать, подавляя подвижность сперматозоидов, и поэтому действуют в качестве средства контрацепции для мужчин. Напротив, ингибиторы DPP-IV рассматриваются как новые средства для лечения бесплодия у человека, и в частности женского бесплодия вследствие синдрома поликистозных яичников (PCOS, синдром ШтейнаЛевенталя), который представляет собой состояние, характеризуемое утолщением капсулы яичника и образованием множества фолликулярных кист. Это приводит к бесплодию и аменорее. Полагают, что DPP-IV играет роль в расщеплении различных цитокинов (стимулирующих гематопоэтические клетки), факторов роста и нейропептидов. Стимулированные гематопоэтические клетки полезны для лечения расстройств, характеризуемых сниженным количеством гематопоэтических клеток или их предшественников in vivo. Такие состояния часто возникают у пациентов с ослабленным иммунитетом, например, вследствие химиотерапии и/или лучевой терапии злокачественной опухоли. Обнаружено, что ингибиторы дипептидилпептидазы типа IV применимы для стимуляции роста и дифференцировки гематопоэтических клеток в отсутствие экзогенно добавляемых цитокинов или других факторов роста или клеток стромы. Указанное открытие противоречит убеждению, существующему в области стимуляции гематопоэтических клеток в том, что добавление цитокинов или клеток, которые продуцируют цитокины (стромальных клеток), является необходимым элементом для поддержания и стимуляции роста и дифференцировки гематопоэтических клеток в культуре. (См., например, международную заявку на выдачу патента PCT No. PCT /US93/017173, опубликованную как WO 94/03055). Показано, что DPP-IV в плазме человека отщепляет N-концевые Tyr-Ala от гормона роста и вызывает инактивацию данного гормона. Поэтому ингибиторы DPP-IV могут быть применимы для лечения низкого роста вследствие недостаточности гормона роста (карликовости) и для стимуляции зависимого от ГР роста или возобновления роста ткани.DPP-IV также может расщеплять нейропептиды и, как было показано, модулирует активность нейроактивных пептидов вещества Р, нейропептида Y и CLIP (Mentlein, R. , Dahms, P., Grandt, D., Kruger, R.,Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV, Regul. Pept., 49, 133,1993; Wetzel, W., Wagner, Т., Vogel, D., Demuth, H.-U., Balschun, D., Effects of the CLIP fragment ACTH 20-24 on the duration of REM sleep episodes, Neuropeptides, 31, 41, 1997). Таким образом, ингибиторы DPPIV также могут быть полезными средствами для регуляции или нормализации состояния при неврологических расстройствах. Все еще существует потребность в ингибиторах DPP-IV, которые обладают полезными с точки зрения активности, стабильности, избирательности, токсичности и/или фармакодинамики свойствами и которые соответственно могут быть эффективно использованы в фармакологических композициях для лечения патологических состояний посредством ингибирования DPP-IV. Сущность изобретения Бензоатная соль 2-6-[(3R)-3-амино-1-пиперидинил]-3,4-дигидро-3-метил-2,4-диоксо-1(2H)-пиримидинил]метил]бензонитрила (называемого в данном описании "соединение I"), которая имеет формулу: является ингибитором DPP-IV, который описан в заявке на выдачу патента США с регистрационным No. 11/080992, поданной 15 марта 2005, которая включена в данное описание в виде ссылки в полном объеме. Настоящее изобретение относится к новому полиморфу соединения I. Для простоты полиморфы,описанные в данной публикации, соответственно названы формой A и аморфной формой 1. 1. Форма A. В одном варианте настоящее изобретение относится к полиморфу соединения I, называемому в данном описании формой A. На основании физических свойств форма A является кристаллической формой. Форму A можно охарактеризовать как форму, имеющую одно или несколько из следующих физических свойств (следует отметить, что композиция не обязательно должна проявлять все указанные свойства, чтобы можно было говорить о присутствии формы A):(a) может быть образована посредством кристаллизации из любой из следующих систем растворителей (i) ацетон, (ii) ацетонитрил; (iii) бутанол, (iv) диметилсульфоксид; (v) диоксан; (vi) этанол; (vii) этанол и изопропиловый спирт; (viii) этанол и вода; (ix) этилацетат; (x) гептан; (xi) изопропанол; (xii) изопропилацетат; (xiii) метанол; (xiv) метилэтилкетон; (xv) метилизобутилкетон; (xvi) 2,2,2 трифторэтанол; (xvii) тетрагидрофуран; (xviii) толуол; (xix) вода и (xx) этанол и гептан.(b) имеет картину порошковой дифракции рентгеновских лучей с характерными признаками, представляющими собой основные дифракционные линии, которые показаны ниже: и, в частности, имеющую следующие характерные пики:(c) имеет ИК-спектр, содержащий пики поглощения 830, 876, 910, 950, 987, 1004, 1026, 1063, 1094,1135, 1173, 1212, 1231, 1284, 1316, 1334, 1365, 1384, 1447, 1458, 1474, 1532, 1592, 1613, 1697, 2082, 2230,2540, 2596, 2743, 2860, 2958, 2979 и 3085 см-1; при этом в ИК-спектре, полученном инфракрасной спектроскопией с Фурье-преобразованием (ФПИК), имеются уникальные пики (пики, в пределах 4 см-1 от которых не наблюдается другого пика, что-3 015359 делает этот набор пиков уникальным) в положениях 1212, 1365, 1447, 1613 и 1697 см-1;(d) имеет в спектрах комбинационного рассеяния с Фурье преобразованием (ФП-КР) пики в положениях 825, 881, 910, 918, 987, 1003, 1027, 1039, 1065, 1084, 1103, 1135, 1157, 1167, 1172, 1184, 1206,1235, 1288, 1337, 1365, 1385, 1417, 1446, 1461, 1474, 1557, 1577, 1597, 1624 1652, 1689, 2230, 2860, 2883,2957, 2970, 2983, 3026, 3053 и 3070 см-1; с уникальными пиками ФП-КР (пиками, в пределах 4 см-1 от которых не наблюдается других пиков, что делает этот набор пиков уникальным) в положениях 1065, 1103, 1235, 1288, 1337, 1365, 1624,1689, 2883, 2983 и 3026 см-1;(e) имеет спектр дифференциальной сканирующей калориметрии, содержащий эндотермическую область в диапазоне примерно от 173C до 195C, необязательно эндотермическую область примерно от 180C до примерно 190C и необязательно эндотермический пик при 186C;(f) по данным термогравиметрического анализа характеризуется потерей 0,2% массы в области 26159C; и/или(g) образуется в результате превращения аморфной формы 1 под воздействием на аморфную форму 1 нагревания, высокой относительной влажности или органических паров или в результате мокрого измельчения аморфной формы 1 в присутствии воды. 2. Амфорная форма 1. Аморфную форму 1 можно охарактеризовать как форму, имеющую одно или несколько из следующих физических свойств (следует отметить, что композиция не обязательно должна проявлять все указанные свойства, чтобы можно было говорить о присутствии аморфной формы 1):(a) может быть образована (i) упариванием в роторном испарителе из метанола; (ii) быстрым упариванием из воды; (iii) лиофилизацией из воды; (iv) кристаллизацией из этилацетата и гексанов и (v) кристаллизацией из изопропилацетата и гексанов;(b) имеет картину порошковой дифракции рентгеновских лучей, которая характеризуется широким гало без конкретных пиков;(c) имеет ИК-спектр, содержащий пики поглощения при 809, 833, 868, 948, 1024, 1068, 1084, 1119,1134, 1172, 1228, 1286, 1375, 1440, 1541, 1599, 1652, 1703, 2136, 2225, 2571, 2861, 2949 и 3062 см-1; при этом в ИК-спектре, полученном инфракрасной спектроскопией с Фурье-преобразованием (ФП-ИК),имеются уникальные пики (пики, в пределах 4 см-1 от которых не наблюдается других пиков, что делает этот набор пиков уникальным) при 809, 868, 1119, 1599 и 1703 см-1;(d) имеет в спектрах ФП-КР пики в положениях 805, 834, 904, 1002, 1024, 1045, 1134, 1168, 1205,1280, 1386, 1443, 1578, 1600, 1654, 1703, 2225, 2864, 2958 и 3065 см-1; с уникальными пиками ФП-КР (пиками, в пределах 4 см-1 от которых не наблюдается других пиков, что делает этот набор пиков уникальным) в положениях 805, 1280 и 1703 см-1;(e) имеет спектр дифференциальной сканирующей калориметрии (циклической ДСК), имеющий Tg= 70C (начало), экзотермический пик при 132C (максимумы), и эндотермический пик при 183C (начальная температура); и/или(f) характеризуется по данным термогравиметрического анализа потерей 4% массы в области от 25 до 151C. Способы, посредством которых осуществляли указанные выше анализы, чтобы идентифицировать указанные физические свойств, описаны в примерах. Краткое описание фигур На фиг. 1 показана картина порошковой дифракции рентгеновских лучей (XRPD) формы A, где"картина XRPD" представляет собой график интенсивности дифракционных линий. Фиг. 2 является графиком данных ТГА и данных ДСК для формы A. Фиг. 3 является графиком, представляющим спектр ИК-поглощения формы A. Фиг. 4 является графиком, представляющим спектр поглощения ФТ-КР формы А. На фиг. 5 показана картина XRPD аморфной формы 1, где "картина XRPD" представляет собой график интенсивности дифракционных линий. Фиг. 6 является графиком данных ТГА и данных ДСК для аморфной формы 1. Фиг. 7 является графиком, представляющим спектр ИК-поглощения аморфной формы 1. Фиг. 8 является графиком, представляющим спектр поглощения ФТ-КР аморфной формы 1. Подробное описание изобретения Настоящее изобретение относится к новым полиморфам соединения I. Предлагаются различные способы, включая способы получения раскрытой формы A и аморфной формы 1. 1. Получение соединения I. Различные способы могут быть использованы для синтеза соединения I. Типичные способы синтеза соединения I приведены в примере 1. Однако следует отметить, что также можно использовать другие способы синтеза, чтобы синтезировать соединение I, включая способы, описанные в заявке на выдачу патента США с регистрационным No. 11/080992, поданной 15 марта 2005, которая включена в данное-4 015359 описание в виде ссылки в полном объеме. 2. Получение полиморфов. В общем, определенный полиморф соединения можно получить прямой кристаллизацией соединения или кристаллизацией соединения с последующим взаимным превращением из другой полиморфной формы или из аморфного состояния. В приведенных ниже примерах описаны способы тестирования растворимости соединения I и способы скрининга условий кристаллизации соединения I. В зависимости от способа, посредством которого кристаллизуют соединение, полученная в результате композиция может содержать разные количества соединения в кристаллической форме по сравнению с аморфным веществом. Также полученная в результате композиция может содержать различные смеси разных полиморфных форм соединения."Кристаллическое" в качестве термина, используемого в данном описании, относится к веществу,которое содержит конкретное соединение, которое может быть гидратировано и/или сольватировано и имеет достаточное содержание кристаллического вещества для проявления различимой картины дифракции при использовании XRPD или другого способа определения дифракции. Часто кристаллическое вещество, которое получают прямой кристаллизацией соединения, растворенного в растворе, или взаимным превращением кристаллов, полученных в разных условиях кристаллизации, будет иметь кристаллы,которые содержат растворитель, используемый для кристаллизации, называемые кристаллосольватами. Также конкретная система растворителей и физический вариант осуществления кристаллизации, вместе называемые условиями кристаллизации, могут приводить к получению кристаллического вещества,имеющего физические и химические свойства, которые являются уникальными для таких условий кристаллизации, обычно вследствие ориентации химических остатков соединения в кристалле относительно друг друга и/или преобладания конкретной полиморфной формы соединения в кристаллическом веществе. В зависимости от полиморфной формы (форм) соединения, которые присутствуют в композиции,также могут присутствовать различные количества соединения в аморфном твердом состоянии, либо в качестве побочного продукта начальной кристаллизации и/либо в качестве продукта разложения кристаллов, содержащих кристаллическое вещество. Таким образом "кристаллическая" в качестве термина,используемого в данном описании, включает случаи, когда композиция может содержать аморфное вещество; при этом присутствие кристаллического вещества среди аморфного вещества можно выявить наряду с другими способами по тому, что композиция имеет характерную картину дифракции. Аморфное содержимое кристаллического вещества может быть увеличено при размалывании или растирании вещества, что подтверждается расширением дифракционных и других спектральных линий по сравнению с кристаллическим веществом до размалывания. Достаточное размалывание и/или растирание может расширять линии по сравнению с линиями кристаллического вещества до размалывания в такой степени, что спектр XRPD или другой конкретный спектр кристаллического вещества становится не различимым, делая вещества по существу аморфными или квазиаморфными."Аморфное", в качестве термина, используемого в данном описании, относится к композиции, содержащей соединение, которое имеет слишком маленькое содержание кристаллического соединения,чтобы получить различимую картину XRPD или исследовать другими способами дифракционного анализа. Стеклообразные вещества являются определенным типом аморфного вещества. Аморфные вещества не имеют правильной кристаллической решетки и, следовательно, являются стеклообразными, а не настоящими твердыми веществами, формально похожими на очень вязкие некристаллические жидкости. Стекла лучше всего могут быть описаны как квазитвердые аморфные вещества, а не настоящие твердые вещества. Таким образом, аморфное вещество относится к квазитвердому стеклообразному веществу. Известно, что при осаждении соединения из раствора, часто осуществляемого быстрым выпариванием растворителя, соединение преимущественно образует аморфное твердое вещество по сравнению с кристаллами. Соединение в аморфном состоянии может быть получено быстрым выпариванием растворителя из сольватированного соединения или измельчением, размалыванием, или иным физическим воздействием под давлением или растиранием соединения в кристаллическом состоянии. Общие способы осаждения и кристаллизации соединения могут быть использованы для получения различных полиморфов,описанных в данной публикации. Такие общие способы известны специалистам в области синтеза органических химических соединений и приготовления фармацевтических препаратов и описаны, например,в J. March, "Advanced Organic Chemistry: Reactions, Mechanisms and Structure", 4th Ed. (New York: WileyInterscience, 1992)."Широкие" или "уширенные" используют в данном описании в качестве термина для описания спектральных линий, включая линии XRPD, ЯМР и ИК-спектроскопии, и термин является относительным термином, который имеет отношение к ширине линии эталонного спектра. Эталонным спектром часто является спектр не подвергнутой обработке кристаллической формы конкретного соединения, которую непосредственно получают при использовании заданного набора физических и химических условий, включая состав растворителей и такие свойства, как температура и давление. Например, термин"уширенные" может быть использован для описания спектральных линий XRPD-спектра размолотого или измельченного вещества, содержащего кристаллическое соединение, по сравнению с веществом до-5 015359 его измельчения. В веществах, в которых составляющие молекулы, ионы или атомы, либо в сольватированном, либо в гидратированном виде, не совершают быстрые ориентационные движения, уширение линий является показателем повышенной хаотичности ориентации химических остатков соединения, и соответственно показателем увеличения содержания аморфной фазы. В случае проведения сравнения между кристаллическими веществами, полученными посредством различных условий кристаллизации,более широкие спектральные линии указывают на то, что вещество, которое дает сравнительно более широкие спектральные линии, имеет более высокий уровень содержания аморфного вещества. Как можно ожидать, длительное измельчение увеличивает содержание аморфного вещества и дополнительно уширяет картину XRPD, и в предельном случае картина XRPD окажется настолько уширенной, что она более не будет отличаться от вышеупомянутого шума. Когда картина XRPD уширена до предела, когда она становится неразличимой, можно полагать, что вещество больше не является кристаллическим веществом, и вместо этого становится полностью аморфным. В случае вещества, имеющего повышенное содержание аморфного вещества, и полностью аморфного вещества невозможно выявить пики, которые могли бы указывать, что измельчение дает другую форму. Композиции, содержащие более высокий процент кристаллического вещества (например, образующего кристаллы с меньшим количеством дефектов решетки и соответственно меньшей долей стеклообразного вещества) обычно получают при использовании условий, которые предпочтительно способствуют замедленному кристаллообразованию, включая условия, которые замедляют испарение растворителя и условия, которые влияют на кинетику. Условия кристаллизации могут быть соответственно скорректированы так, чтобы при необходимости получить кристаллическое вещество более высокого качества. Таким образом, например, если при исходном наборе условий кристаллизации образуются плохие кристаллы, то температуру растворителя можно снизить, а давление окружающей среды над раствором может быть повышено по сравнению с исходным набором условий кристаллизации, чтобы замедлить кристаллизацию. Как будет понятно, в зависимости от того, как была получена композиция, содержащая данное соединение, и затем как после получения хранили и обрабатывали композицию, будет изменяться содержание кристаллического вещества в композиции. Соответственно, в композиции возможно отсутствие содержания кристаллического вещества, или композиция может содержать более высокие концентрации кристаллического вещества. Кроме того, следует отметить, что соединение может присутствовать в данной композиции в виде одной или нескольких разных полиморфных форм, а также необязательно присутствовать в виде аморфного вещества. Указанное может быть результатом (a) физического смешивания двух или более различных полиморфных форм; (b) наличия двух или более разных полиморфных форм, образуемых в результате используемых условий кристаллизации; (c) превращения всей или части данной полиморфной формы в другую полиморфную форму; (d) превращения всей или части соединения в аморфном состоянии в две или более полиморфных форм; а также в силу множества других причин. Как можно видеть, в зависимости от того, как получают композицию, содержащую соединение,процентное содержание по массе данного соединения в конкретной полиморфной форме может варьировать от 0% до 100%. Согласно настоящему изобретению предлагаются композиции, в которых более 1, 5,10, 25, 50, 75, 80, 85, 90, 95, 97 или 99 мас.% или больше соединения I присутствует в композиции в форме A или аморфной форме 1. 3. Полиморфы соединения I. В данной публикации описана форма A и аморфная форма 1 соединения I. Могут быть проведены различные испытания с целью физической характеристики кристаллического состояния соединения I, включая без ограничения рентгеновскую дифракцию порошка ("XRPD"),дифференциальную сканирующую калориметрию ("ДСК"), термогравиметрический анализ ("ТТА"), высокотемпературную микроскопию, инфракрасную спектрометрию ("ИК"), спектрометрию комбинационного рассеяния и анализ по методу Карла Фишера. В приведенных ниже примерах описаны способы,которые использовали для осуществления различных анализов, указанных в данном описании. По возможности результаты каждого испытания для каждого отдельного полиморфа приведены в данном описании. А. Способ получения формы A соединения I. Далее описаны способы, с использованием которых была получена полиморфная формы A соединения I из разных образцов соединения I. Образец No. 1924-73-02: взвесь соединения I в ацетоне фильтровали через нейлоновый шприцевой фильтр 0,2 мкм в чистый флакон. Флакон оставляли открытым в вытяжном шкафу в условиях окружающей среды для быстрого испарения, получая форму A в виде твердого вещества через два дня. Образец No. 1924-73-08: взвесь соединения I в метаноле фильтровали через нейлоновый шприцевой фильтр 0,2 мкм в прозрачный флакон. Флакон закрывали алюминиевой фольгой с проколотыми отверстиями и помещали в вытяжной шкаф для медленного испарения в условиях окружающей среды, получая форму A в виде твердого вещества за два дня. Образец No. 1924-67-05: готовили взвесь соединения I в ацетонитриле на нагревательной плите с-6 015359 установленной температурой 60C и смесь в теплом виде фильтровали через нейлоновый шприцевой фильтр 0,2 мкм в прозрачный теплый флакон. Флакон помещали на нагревательную плиту, которую затем выключали, и давали возможность медленно охладиться до температуры окружающей среды. ФормуA в виде твердого вещества собирали фильтрованием через один день. Образец No. 1994-82-01: соединение I (123 мг) растворяли в смеси 99:1 этанол/изопропилацетат (1 мл) при кипячении с обратным холодильником и затем охлаждали до температуры окружающей среды со скоростью 20C/ч. Твердые вещества преципитировали и полученную в результате взвесь встряхивали в течение 4 ч при температуре окружающей среды. Растворитель сливали и твердые вещества сушили. В эксперименте получали форму A в виде твердого вещества. В. Характеристика формы A соединения I. На фиг. 1 приведена картина XRPD формы A. Наблюдали основные линии дифракции при 2 примерно: 9,44, 10,84, 17,82, 18,75, 25,87 и 28,52. Картина XRPD служит подтверждением того, что вещество представляет собой кристаллическую фазу, которая была названа формой A. Бесцветные пластинки соединения I собирали из раствора этанол-вода. Параметры орторомбической элементарной ячейки и рассчитанный объем ячейки составляют: a= 8,0869(2), b = 9,9030(3), c = 28,5471(10) , V = 2286,18(12) 3. При Z = 4 и массовой формуле 461,53 г рассчитанная плотность составляет 1,34 г/см 3. Полученная модель структуры имеет хорошее качество, на что указывает значение Rфактора 0,068. Обычно значения R-факторов в диапазоне 0,02-0,06 свидетельствуют о наиболее достоверно определенных структурах. ORTEP-изображение соединения I показывает, что кристаллическая структура является такой же, как предполагаемая структура. Отмечено, что асимметричная структурная единица содержит один свободный основной катион и один противоион бензоата. Рассчитанная картина порошковой дифракции, полученная на основе данных для монокристаллического образца очень, сходна с экспериментальной картиной XRPD формы A соединения I, но имеют место небольшие отличия в положении 2 пиков вследствие температурных эффектов. Сбор данных для монокристаллического образца осуществляли при 150 K, тогда как экспериментальную картину XRPD получали при температуре окружающей среды. Различия в интенсивностях вероятно связаны с предпочтительной ориентацией. Термогравиметрические данные и данные ДСК для формы А суммированы ниже в табл. 1A и изображены на фиг. 2. Как следует из фиг. 2, на кривой ДСК наблюдается несколько эндотермических эффектов. Максимум эндотермического пика преобладающего эффекта расположен вблизи 186C. Эксперимент по определению точки плавления подтвердил, что указанный эндотермический эффект связан с плавлением вещества. Серия эндотермических эффектов выше эндотермы плавления далее не была охарактеризована, но вероятно они соответствуют разложению образца. Широкий эндотермический эффект,расположенный ниже эндотермы плавления для образца, также может быть виден на графике ДСК образца формы A. Указанный эффект имеет место в той же самой температурной области что и соответствующая потеря массы, наблюдаемая на графике ТГА для образца формы A, и согласуется с потерей летучих веществ из образца. Таблица 1A фиг. 3. Спектр комбинационного рассеяния для формы A (партияQZ-656-17(1 изображен на фиг. 4. Данные, относящиеся к исследованию свойств сорбции/десорбции влаги формы A, суммированы в табл. 2A ниже. Анализ показал начальное увеличение массы примерно на 0,035% в условиях окружающей среды до 55% RH (относительной влажности) и потерю массы примерно на 0,05% в диапазоне от 9% до 95% RH. Цикл десорбции протекает по несколько отличной траектории по сравнению с циклом сорбции, при этом масса, приобретенная во время цикла сорбции, теряется при десорбции в незначительных количествах в разных интервалах RH. Величину изменения массы в цикле десорбции можно использовать наряду с молекулярной массой соединения I для расчета, согласно которому молекула воды отсутствует в кристалле при 95% RH. Указанные расчеты предполагают, что вода отсутствует в образце в конце анализа при шаге, равном 5% RH. Образец после сорбции/десорбции влаги с помощью XRPD идентифицировали как форму A. В отдельных экспериментах форму A подвергали воздействию различных условий RH при комнатной температуре (табл. 3A). После нескольких недель ни один из указанных-7 015359 образцов не подвергся изменению фазового состояния. Это ясно видно на соответствующих картинахXRPD образцов, которые все соответствуют форме A. Постепенное увеличение/уменьшение массы, наблюдаемое во всем диапазоне уровней RH, исследованных на графике сорбции/десорбции влаги, вместе с отсутствием изменения фазового состояния формы A при воздействии RH, указывает, что рассматриваемая твердая фаза может содержать переменное количество воды, которое будет зависеть от RH окружающей среды. Указанный тип гидратированной фазы называют гидратом переменного состава или нестехиометрическим гидратом. Таблица 2A Таблица 3A Скрининг полиморфов соединения I - эксперименты на основе твердых веществ: исследования воздействий с использованием стеклообразных веществ-8 015359 Таблица 3A Скрининг полиморфов соединения I - эксперименты на основе твердых веществ: исследования воздействий с использованием стеклообразных веществC. Способ получения аморфной формы 1 соединения I. Образец No. 1994-26-01: взвесь соединения I помещали на нагревательную плиту, установленную на 80C. Смесь фильтровали через нейлоновый шприцевой фильтр 0,2 мкм в теплый флакон. Затем флакон помещали на нагревательную плитку, которую затем устанавливали на 40C, и раствору давали возможность охладиться. Добавляли достаточное количество гексанов, что вызывало образование мутной суспензии. Мелкодисперсное твердое вещество собирали фильтрованием и давали возможность высохнуть на воздухе. Эксперименты давали аморфные твердые вещества. Образец No. 1994-07-01: Достаточное количество соединения I добавляли к метанолу так, что оставалось нерастворенное твердое вещество. Полученную в результат взвесь фильтровали через нейлоновый шприцевой фильтр 0,2 мкм в колбу. Раствор упаривали досуха, используя роторный испаритель (Buchi, R-114), при пониженном давлении. Аморфное твердое вещество хранили в эксикаторе.D. Характеристика аморфной формы 1 соединения I. Аморфная полиморфная форма 1. На фиг. 5 приведена картина XRPD аморфной формы 1 (образец 1994-12-01). Данные XRPD показывают низкое отношение сигнала к шуму. Картина XRPD свидетельствует о том, что форма 1 является аморфной формой соединения I. Термогравиметрические данные и данные ДСК для аморфной формы 1 суммированы ниже в табл. 1B и изображены на фиг. 6. Аморфную форму 1 получали несколькими способами, которые указаны выше. Вещество было очень гигроскопичным, его масса увеличивалась на 10% при 85% RH, но происходила потеря массы выше 85% RH, что является показателем кристаллизации. Картина XRPD после установления водного баланса совпадала с формой A. Потеря массы согласно ТГА составила 4% в диапазоне от 25 до 168C, вероятно вследствие потери адсорбированной влаги. Поэтому осуществляли циклический ДСК-эксперимент, чтобы высушить образец и затем определить температуру перехода в стеклообразное состояние Tg, который начинался при 70C. Событие экзотермической перекристаллизации регистрировали при 132C, за которым следует резкий эндотермический пик при 183C (начало пика), который коррелирует с началом плавления (172C), определяемым с помощью высокотемпературной микроскопии. Вышесказанное свидетельствует, что аморфное твердое вещество кристаллизовалось в форму A во время нагревания. Спектры ИК и ФП-КР для аморфной формы 1 (образец 1994-07-01) изображены на фиг. 7 и 8,соответственно. Данные, относящиеся к исследованию свойств сорбции/десорбции влаги аморфной формы 1, суммированы ниже в табл. 2B. Данные исследования сорбции/десорбции влаги показали начальную потерю массы примерно на 1% в диапазоне от условий окружающей среды до 5%RH и затем увеличение массы примерно на 6,5% в диапазоне от 5 до 95% RH. В интервале от 85 до 95% RH образец потерял примерно 5% массы, что свидетельствует о явлении кристаллизации. Образец после проведения эксперимента по исследованию сорбции/десорбции влаги был предположительно идентифицирован как образец, содержащий форму A. Таблица 2B 2-(6-Хлор-2,4-диоксо-3,4-дигидро-2H-пиримидин-1-илметил)бензонитрил (B). К раствору 6 хлорурацила (20 г, 122 ммоль) в смеси ДМФА-ДМСО (6:1, 600 мл) в атмосфере азота при 0C порциями добавляли гидрид натрия (60%, 5,5 г, 137 ммоль). Через 0,5 ч в смесь добавляли бромид лития (8 г, 96 ммоль) и перемешивали в течение 15 мин при 0C. По каплям добавляли раствор -бром-o-толунитрила(25,1 г, 128 ммоль) в ДМФА (30 мл) и перемешивали при такой температуре в течение 1 ч и затем при комнатной температуре в течение ночи. Смесь упаривали и совместно упаривали с водой в вакууме, чтобы удалить большую часть ДМФА, и затем вливали в ледяную воду (1 л) . Осадок собирали фильтрованием. Неочищенный продукт суспендировали в горячей смеси AcOEt-CHCl3 и обрабатывали ультразвуком в течение 5 мин, оставляли стоять при 0C в течение 1 ч и затем фильтровали, получая указанное в заголовке соединение в виде белого твердого вещества (19 г) с выходом 54%. 1 2-(6-Хлор-3-метил-2,4-диоксо-3,4-дигидро-2H-пиримидин-1-илметил)бензонитрил (C). К холодному (0C) раствору бензилированного 6-хлорурацила 2 (10 г, 38 ммоль) в смеси ДМФА-ТГФ (1:1, 300 мл) в атмосфере азота порциями добавляли NaH (60%, 1,6 г, 39,9 ммоль), затем добавляли LiBr (2 г). Смесь перемешивали при комнатной температуре в течение 20 мин. После добавления йодметана (5,4 мл, 76 ммоль) колбу закрывали и перемешивали при указанной температуре в течение 10 мин, при комнатной температуре в течение 2 ч и при 35C в течение ночи и затем концентрировали в вакууме. Остаток растворяли в CHCl3 и промывали водой и насыщенным раствором соли, сушили (Na2SO4) и фильтровали,затем концентрировали в вакууме. Неочищенный продукт кристаллизовали из смеси ТГФ-гексаны, получая 7,6 г (72%) указанного в заголовке соединения 3. 1 2-6-[(3R)-аминопиперидин-1-ил]-3-метил-2,4-диоксо-3,4-дигидро-2H-пиримидин-1-илметилбензонитрил (D). 2-(6-Хлор-3-метил-2,4-диоксо-3,4-дигидро-2H-пиримидин-1-илметил) бензонитрил (330 мг, 1,08 ммоль), дигидрохлорид (R)-3-аминопиперидина (246 мг, 1,4 ммоль) и бикарбонат натрия (500 мг,5,4 ммоль) перемешивали с 200 мг активированных молекулярных сит (4A) в безводном MeOH (5 мл) при 100C в течение 2 ч. Реакционную смесь фильтровали через целит, концентрировали в вакууме и затем разбавляли CHCl3 и промывали водой. Водную фазу экстрагировали CHCl3 и объединенные органические фазы промывали водой, сушили (Na2SO4) и фильтровали. В раствор добавляли ТФУ (1 мл) и затем раствор концентрировали в вакууме. Остаток растворяли в небольшом количестве MeOH и добавляли Et2O, чтобы вызвать преципитацию. Смесь оставляли при комнатной температуре в течение ночи. Растворители сливали и твердое вещество два раза промывали Et2O, получая 270 мг продукта в виде не совсем белого порошка. 1 Соль бензойной кислоты образовывали в результате обработки продукта бензонитрила (D) бензойной кислотой с образованием бензоата 2-[6-(3-аминопиперидин-1-ил)-3-метил-2,4-диоксо-3,4-дигидро 2H-пиримидин-1-илметил]бензонитрила. Получение и выделение бензоатной соли осуществляли обычными способами образования кислотно-аддитивных солей. 1C18H22N5O2, 340,2; найдено 340,2. Пример 2. Характеристика растворимости соединения I в разных растворителях. Следующие эксперименты осуществляли для того, чтобы определить растворимость соединения I в разных растворителях и системах растворителей. Полученную информацию позже использовали для идентификации возможных условий кристаллизации соединения I. Материалы и реагенты Если не оговорено особо, то форму A соединения I и аморфное вещество, полученное из данного образца, использовали в качестве исходных веществ для всех экспериментов по кристаллизации. Растворители и другие реагенты имели чистоту ACS или для ВЭЖХ, и их использовали по мере получения. Оценки растворимости Взвешенный образец соединения I обрабатывали аликвотами тестируемого растворителя при комнатной температуре. Между добавлениями аликвот смесь обрабатывали ультразвуком, чтобы облегчить растворение. Полное растворение тестируемого вещества определяли, используя визуальный контроль. Растворимость оценивали в результате таких экспериментов на основании общего количества растворителя, используемого для достижения полного растворения. Фактическая растворимость может быть выше, чем рассчитанная вследствие использования аликвот растворителя, которые были слишком большими, или вследствие медленной скорости растворения. Растворимость указана с использованием выражения "менее чем", если растворение во время эксперимента не происходило. Если достигали полного растворения в результате добавления только одной аликвоты, то растворимость указывали с использованием выражения "более чем".- 12015359 Таблица 4 Оценки растворимости формы A соединения I Обнаружено, что полиморфная форма A растворима в воде (8 мг/мл), метаноле (13 мг/мл), диметилформамиде (6 мг/мл), метилэтилкетоне (3 мг/мл) и ацетоне, дихлорметане, этаноле, этилацетате и метилизобутилкетоне (во всех 3 мг/мл). Термический анализ показывает, что твердая фаза термически стабильна выше 172C. Анализ ДСК и определения точки плавления показали, что форма A плавится около 172C. Анализ сорбции/десорбции влаги формой A показывает, что данный полиморф является гидратом переменного состава. Таблица 5 Скрининг полиморфов соединения I - эксперименты, основанные на растворении Приготовление образцов Общие способы Медленное упаривание (SE). Форму A добавляли к представляющим интерес растворителям. Использовали обработку ультразвуком, помогающую осуществить растворение. После растворения смеси,судя по визуальному наблюдению, раствор пропускали через нейлоновый фильтр 0,2-мкм в прозрачный флакон, закрывали алюминиевой фольгой и делали отверстия. Раствору давали возможность упариваться в условиях окружающей среды. Быстрое упаривание (FE). Образцы получали согласно способу медленного упаривании за исключением того, что растворы оставляли для упаривания незакрытыми. Медленное охлаждение (SC). Концентрированные растворы формы A получали в разных растворителях при температуре окружающей среды или при повышенной температуре. Концентрированные растворы фильтровали через нейлоновые фильтры 0,2-мкм в прозрачные флаконы. Растворам давали возможность медленно охладиться до комнатной температуры. Дополнительное охлаждение до температур ниже температуры окружающей среды достигали помещением образцов в холодильник или морозильную камеру. Быстрое охлаждение (FC). Концентрированные растворы формы A получали в разных растворителях при повышенных температурах. Концентрированные растворы фильтровали через нейлоновые фильтры 0,2 мкм в прозрачные флаконы. Растворы удаляли от источника нагревания и оставляли при комнатной температуре. Дополнительное охлаждение до температур ниже температуры окружающей среды достигали, помещая образцы в холодильник или морозильную камеру. Ротационное выпаривание (RE). Концентрированные растворы формы A получали в различных тестируемых растворителях. Растворы пропускали через нейлоновые фильтры 0,2 мкм в прозрачные емкости и образцы освобождали от легких компонентов досуха, используя роторный испаритель Bchi R-114. Образцы погружали и вращали в водяной бане при заданной температуре 30 или 40C во время выпари- 17015359 вания. Ускоренная преципитация (CP). Концентрированный раствор формы A получали в разных растворителях при повышенной температуре. Растворы пропускали через нейлоновые фильтры 0,2 мкм в прозрачные флаконы. Затем к растворам образцов добавляли антирастворитель. Образовавшиеся преципитаты собирали вакуумным фильтрованием. Взвеси. Насыщенные растворы формы A, содержащие избыток твердого вещества, готовили в разных растворителях. Образцы помещали на устройство для встряхивания и встряхивали при установленной температуре в течение определенного периода времени. Затем собирали твердые вещества для анализа путем сливания растворов или вакуумным фильтрованием. Диффузия из паровой фазы (VD). Форму A растворяли в тестируемых растворителях. Полученные растворы пропускали через 0,2-мкм фильтр в небольшие флаконы. Небольшие флаконы помещали в открытом виде в большие по размеру флаконы, содержащие смешиваемый антирастворитель. Большие флаконы закрывали крышками, герметизировали парафильмом и хранили в условиях окружающей среды. Образовавшиеся твердые вещества выделяли и анализировали. Эксперименты с воздействием парами (VS). Форму A помещали в небольшой флакон, который в открытом виде помещали в больший по размеру флакон, содержащий диффундирующий растворитель. Большой флакон герметично закрывали и хранили при температуре окружающей среды. Образцы также подвергали воздействию в печи повышенными температурами и разными условиями относительной влажности. Упаривание в капиллярах (CE). Растворами формы A заполняли стеклянные капилляры посредством центрифугирования. Затем образцам в капиллярах обеспечивали возможность упаривания. Ускоренное охлаждение (CC). Готовили образцы формы A в разных растворителях и пропускали через нейлоновые фильтры 0,2 мкм в прозрачные флаконы. Затем флаконы, содержащие растворы, быстро охлаждали погружением в баню со смесью сухой лед/ацетон на несколько секунд. Твердые вещества,которые выпадали в осадок, собирали фильтрованием и сушили. Эксперименты с использованием размалывания. Небольшое количество формы A помещали в патрон для измельчения с мельничным шаром и патрон закрывали. Затем образец размалывали в мельницесмесителе с частотой 30 Гц в течение измеряемого промежутка времени. Твердые вещества исследовали,используя оптическую микроскопию. Несколько образцов подвергали мокрому помолу, добавляя к образцу достаточное количество воды, чтобы смочить его перед размалыванием. Эксперименты с использованием криопомола. Небольшое количество формы A измельчали с частотой 10 Гц в течение 6 минут, используя мельницу с замораживанием Spex Centriprep 6750, заполненную жидким азотом. Образцу давали возможность охладиться в течение одной минуты после каждого двухминутного цикла измельчения. Образец извлекали из мельницы и давали возможность достичь комнатной температуры в большом эксикаторе. Пример 3. Порошковая дифракция рентгеновских лучей. Анализы порошковой дифракции рентгеновских лучей осуществляли, используя рентгеновский порошковый дифрактометр Shimadzu XRD-6000, использующий Cu-K-излучение. Прибор оборудован длинной рентгеновской трубкой с тонкой фокусировкой. Напряжение на трубке и силу тока устанавливали 40 кВ и 40 мА, соответственно. Щели для расходящегося и рассеивающегося пучка устанавливали равными 1 и принимающую щель устанавливали на 0,15 мм. Дифрагированное излучение регистрировали сцинтилляционным датчиком Nal. Использовали непрерывное сканирование 9-29 со скоростью 3/мин (шаг 0,4 с/0,02) в области от 2,5 до 402. Анализировали кремниевый стандарт, чтобы проверить юстировку прибора. Данные собирали и анализировали, используя XRD-6000 v.4.1. Образцы готовили для анализа, помещая их в алюминиевый держатель с кремниевой вставкой. Анализы порошковой дифракции рентгеновских лучей (XRPD) также выполняли, используя дифрактометр Inel XRG-3000, оборудованный CPS (изогнутым позиционно-чувствительным) детектором с 2-диапазоном в 120. Сбор данных осуществляли в режиме реального времени, используя Cu-K излучение, начиная примерно с 42 при разрешении 0,032 . Напряжение на трубке и силу тока устанавливали 40 кВ и 30 мА, соответственно. Щель монохроматора устанавливали на 5 мм на 80 мкм. Картину отображали при 2,5-402. Образцы готовили для анализа, набивая их в тонкостенные стеклянные капилляры. Каждый капилляр устанавливали на головке гониометра, которая является механизированной,чтобы обеспечить возможность вращения капилляра во время сбора данных. Образцы анализировали в течение 5 мин. Калибровку прибора осуществляли, используя кремниевый эталонный стандарт. Сбор данных: бесцветную пластинку C18H22N5O2, C7H5O2, имеющую размеры примерно 0,500,350,28 мм устанавливали на стекловолокно в случайной ориентации. Предварительное исследование и сбор данных осуществляли, используя Mo-K-излучение ( = 0,71073 ) на приборе Nonius KappaCCD, оборудованном графитовым кристаллом в качестве монохроматора падающего луча. Константы элементарной ячейки для сбора данных получали с помощью уточнения по методу наименьших квадратов, используя установленные углы для 13398 отражений в диапазоне 227. Уточненная мозаичность по DENZO/SCALEPACK составила 0,41, что указывает на хорошее качество кристалла. Про- 18015359 странственную группу определяли с помощью программы XPREP. На основании систематического присутствия отражений: h00 h=2n, 0k0 k=2n, 001 l2n и с учетом последующей процедуры уточнения методом наименьших квадратов определили пространственную группу P212121 (no. 19). Сбор данных осуществлен при температуре 423 K. Данные получали до максимального значения по 2, равного 55,1. Обработка данных: было собрано всего 13398 отражений, из которых 4589 были уникальными. Группы данных суммировали с помощью DENZO-SMN. В данные вносили поправки на лоренц-фактор и поляризацию. Линейный коэффициент поглощения равен 0,9/см для Mo-K-излучения. Эмпирическую поправку на поглощение вводили, используя SCALEPACK. Коэффициенты пропускания находились в диапазоне 0,943-0,976. Применяли поправку на вторичную экстинкцию. Интенсивности эквивалентных отражений усредняли. Фактор совпадения для усредненных отражений на основе интенсивности отражений составлял 10,2%. Определение структуры и уточнение: структура решена прямыми способами с использованиемSIR2002. Остальные атомы были локализованы в последующих разностных синтезах Фурье. Атомы водорода были включены в уточнение, но ограничены зависимостью от атома, с которым они были связаны. Структуру уточняли полноматричным методом наименьших квадратов, минимизируя функцию: Весовой множитель w определен в виде 1/[2(Fo2) + (0,1225P)2], где P = (Fo2 +2Fc2)/3. Коэффициенты рассеяния были взяты из "International Tables for Crystallography". Из использованных при уточнении 4589 отражений только 2414 отражений с Fo22(Fo2) использовали при расчете R. Заключительный цикл уточнения включал 321 переменных параметра и получали сходимость (наибольшее изменение параметра составляло менее 0,01 от его установленного стандартного отклонения) с невзвешенными и взвешенными значениями коэффициентов совпадения: Стандартное отклонение при наблюдении за единичной массой составило 1,06. Самый высокий пик в заключительном разностном синтезе Фурье имел высоту 0,34 e/A3. Минимальный отрицательный пик имел высоту, равную -0,45 e/A3. Фактор определения абсолютной структуры уточнен до -1,00. Уточнение осуществляли на LINUX PC, используя SHELX-97. Изображение кристаллической структуры получали,используя программы ORTEP. Вычисление степени разупорядочения и содержания аморфной фазы в измельченных образцах: Массовый процент кристалличности измельченных образцов определяли посредством вычисления с использованием двух пакетов программ. Модуль Шимадзу для определения процента кристалличности,который является частью пакета программ Shimadzu XRD-6000, использовали для образцов с существенным содержанием аморфной фазы. Программу собственной разработки использовали для образцов с большой степенью кристалличности, поскольку компьютерная программа Шимадзу была менее точной при низких концентрациях аморфной фазы. При использовании собственной компьютерной программы сначала производили сглаживание рентгеновских данных для порошков и затем применяли ряд цифровых фильтров, чтобы разделить данные на три компонента: кристаллический, аморфный и разупорядоченный. Также применяли поправки на фон. Затем рассчитывали содержание аморфной фазы в процентах с помощью определения отношения аморфной фазы к общей сумме всех трех компонентов. Оба способа осуществляли в условиях, не соответствующих GMP, и они давали только приблизительные значения. Исследования с измельчением: Сухой помол формы A при температурах окружающей среды и температуре жидкого азота приводил к образованию разупорядоченной формы A. Для картины XRPD сухих измельченных образцов было характерно уширение пика, смещенная базисная линия и пониженная интенсивность. Мокрый помол формы A и аморфного вещества в воде давал форму A в виде кристаллического твердого тела. Четыре измельченных образца анализировали посредством XRPD. Все картины дифракции совпадали с формой A без заметного присутствия аморфной фазы. Расчет содержания аморфной фазы в измельченном образце: Картины XRPD измельченных образцов формы A соединения I имели уширение пика, так же как гало аморфной формы. Это свидетельствует о том, что два процесса вносили вклад в потерю кристалличности: уменьшение размера кристаллита,вызывающее уширение пика, и образование аморфной формы, вызывающее появление смещенной базисной линии. Сухой измельченный образец 1966-04-02 (измельченный в течение 30 мин) выбрали для исследования и получения картины XRPD исходной формы A (партияQZ-656-17 (1, используемой в качестве эталонного стандарта формы A. Вычислен массовый процент содержания аморфного вещества в сухом измельченном образце 1966-04-02, составляющий примерно 58%. Партия 635-181-1 содержала примерно 13 мас.%, аморфного вещества. Значения, определенные при расчетах, являются приблизи- 19015359 тельными, особенно в случае низкого содержания аморфного вещества. Пример 4. Дифференциальная сканирующая калориметрия (ДСК). Дифференциальную сканирующую калориметрию осуществляли, используя дифференциальный сканирующий калориметр ТА Instruments 2920. Образец помещали в алюминиевый тигель для ДСК и точно определяли значение массы. Тигель закрывали крышкой и затем обжимали. Ячейку с образцом уравновешивали при температуре окружающей среды и нагревали в потоке азоте со скоростью 10C/мин до конечной температуры 350C. Металлический индий использовали в качестве калибровочного стандарта. Указанные температуры соответствуют максимальной температуре фазового перехода. Для исследований температуры перехода в стеклообразное состояние (Tg) аморфного вещества,ячейку с образцом уравновешивали при комнатной температуре, затем нагревали в атмосфере азота со скоростью 10C/мин до 100C. Ячейку с образцом затем охлаждали до -40C перед повторным нагреванием со скоростью 10C/мин до конечной температуры 350C. Приведенное значение Tg соответствует точке перегиба при переходе. Циклические ДСК-эксперименты проводили, помещая точно взвешенные образцы в не обжатые тигли. Образцы нагревали в атмосфере азота со скоростью 10C/мин либо до 150, либо до 180C, затем охлаждали до -40C. Указанную процедуру повторяли дважды, затем образец нагревали до 250C. Пример 5. Термогравиметрический анализ (ТГД). Термогравиметрический анализ осуществляли, используя термогравиметрический анализатор TAInstruments 2950. Каждый образец помещали в алюминиевый тигель для образцов и помещали в ТГ-печь. Температуру печи сначала уравновешивали с температурой окружающей среды, затем нагревали в атмосфере азота со скоростью 10C/мин до конечной температуры 350C. Никель и Alumelиспользовались в качестве калибровочных стандартов. Пример 6. Высокотемпературная микроскопия (HSM). Высокотемпературную микроскопию проводили, используя нагревательный столик Linkam, установленный на микроскопе Leica DM LP. Образцы исследовали, используя 20-кратный объектив с лямбда- пластиной со скрещенными поляризаторами. Образцы зажимали между двумя покровными стеклами и исследовали визуально по мере нагревания столика. Изображения фиксировали, используя цветную цифровую камеру SPOT Insight и компьютерную программу SPOT v.3.5.8. Нагревательный столик калибровали, используя в качестве USP-стандартов точки плавления сульфапиридин и ванилин. Пример 7. Инфракрасная спектроскопия с Фурье-преобразованием (ФП-ИК). Инфракрасные спектры получали на инфракрасном спектрофотометре с Фурье-преобразованиемMagna-IR 860 (ФП-ИК) (Thermo Nicolet), снабженном источником ИК-излучения в средней и дальней области спектра Ever-Glo, светоделителем на основе бромида калия (KBr) и детектором на основе дейтерированного триглицинсульфата (DTGS). Дополнительное устройство ATR Thunderdome с невогнутым наконечником использовали для отбора образцов. Подготовка образца заключалась в размещении образца на кристаллическом германии и прессовании вещества на кристалле с использованием плунжера. Каждый спектр представляет собой накопление результатов 256 сканирований, полученных при спектральном разрешении 4 см-1. Получали набор данных для воздушного фона. Спектр в виде зависимости Log 1/R (R = коэффициент отражения) получали, учитывая отношение двух полученных наборов данных друг к другу. Калибровку длин волн осуществляли, используя полистирол. Инфракрасные спектры также получали, используя устройство для измерения диффузного отражения (Collector, Thermo Spectra-Tech). Подготовка образца заключалась в помещении образца в тигель диаметром 13 мм. Получали набор фоновых данных с использованием корректирующего зеркала в рабочем положении. Спектр в виде зависимости Log 1/R (R = коэффициент отражения) получали, учитывая отношение двух полученных наборов данных друг к другу. Калибровку длины волны осуществляли, используя полистирол. Пример 8. Спектроскопия комбинационного рассеяния. Спектры ФП-КР получены на спектрометре FT-Raman 960 (Thermo Nicolet). Указанный спектрометр использует длину волны возбуждения 1064 нм. Для облучения образцов использовали Nd:YVO4 лазер мощностью примерно 0,6-0,8 Вт. Спектры комбинационного рассеяния измеряли с использованием детектора на основе арсенида галлия-индия (InGaAs). Образцы для анализа готовили, помещая вещество в стеклянный капилляр или ЯМР-трубку. Получали результаты 256 сканирований образцов в области 400-3600 см-1 при спектральном разрешении 4 см-1, используя аподизацию Хаппа-Гензеля. Калибровку длины волны осуществляли, используя серу и циклогексан. Пример 9. ЯМР-спектроскопия. Спектры 1H-ЯМР в растворе могут быть получены при температуре окружающей среды на спектрометре Bruker Instruments АМ-250 при напряженности магнитного поля 5,87 тесла (1H ларморовая частота = 250 МГц). Образцы могут быть растворены в ДМСО-d6, имеющем качество очистки для ЯМР. Спектры могут быть получены с длительностью импульса 1H 8,5 мкс (90), временем измерения 2,5 с,временем задержки между сканированиями 5,0 с, шириной спектра 6400,0 Гц с точками данных 32K и числом накоплений результатов сканирования 32. Каждый спад свободной индукции (FID) может быть- 20015359 обработан с помощью компьютерной программы GRAMS/32 AI v. 6.00, использующей число Фурье,равное удвоенному количеству полученных точек [или большее значение, если используют обнуление] с фактором уширения экспоненциальной линии 0,2 Гц для повышения чувствительности. Таблицы пиков могут быть получены с использованием алгоритма поиска пиков с помощью компьютерной программыGRAMS. В случае таких спектров остаточный пик не полностью дейтерированного ДМСО-d6 находится в положении около 2,50 м.д. Пример 10. Анализы сорбции/десорбции влаги. Данные сорбции/десорбции влаги получены на анализаторе сорбции пара VTI SGA-100. Данные сорбции и десорбции получали в диапазоне от 5 до 95% относительной влажности (RH) с 10% интервалами RH в потоке азота. Образцы перед анализом не сушили. Критерием равновесного состояния, используемым для анализа, было изменение массы менее чем на 0,0100% в течение 5 мин при максимальном времени установления равновесия 3 ч, если не удовлетворялся критерий изменения массы. Корректировку данных относительно исходного содержания влаги в образцах не проводили. NaCl и PVP использовали в качестве калибровочных стандартов. Пример 11. Анализ содержания воды способом Карла Фишера. Анализ содержания воды по Карлу Фишеру (титриметрический) можно осуществить согласно U.S.Pharmacopoeia, vol. 24, method 921, U.S.P. Pharmacopeial Convention, Inc, Rockville, Md. Полиморф можно тестировать в отношении содержания воды, проводя титрование по Карлу Фишеру с использованием кулонометра согласно опубликованному способу и инструкциям производителя кулонометра. Хотя настоящее изобретение описано со ссылкой на некоторые варианты и примеры, подробно описанные выше, следует понимать, что такие варианты и примеры являются иллюстративными, а не ограничивающими. По существу, предполагается, что различные модификации и варианты будут очевидны для специалистов в данной области, и подразумевается, что такие модификации и варианты входят в объем изобретения и прилагаемой формулы изобретения. Все патенты, заявки на выдачу патентов,документы и книги, цитированные в данной заявке, включены в данное описание в виде ссылки в полном объеме. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения I формулы где по меньшей мере часть соединения I присутствует в полиморфной форме A, характеризуемой одним или несколькими физическими свойствами, выбранными из группы, состоящей из картины порошковой дифракции рентгеновских лучей с характерными признаками, представляющими собой основные дифракционные линии, которые показаны ниже: полученной с использованием Cu-K-излучения; ИК-спектра, содержащего пики поглощения в положениях 1212, 1365, 1447, 1613 и 1697 см-1; спектра ФП-КР, содержащего пики в положениях 1065, 1103, 1235, 1288, 1337, 1365, 1624, 1689,2883, 2983 и 3026 см-1; и спектра дифференциальной сканирующей калориметрии, содержащего эндотермическую область примерно от 173C до примерно 195C, при этом способ включает:(a) кристаллизацию соединения I из любой из следующих систем растворителей, содержащих (i) ацетон, (ii) ацетонитрил; (iii) бутанол, (iv) диметилсульфоксид; (v) диоксан; (vi) этанол; (vii) этанол и изопропиловый спирт; (viii) этанол и воду; (ix) этилацетат; (x) гептан; (xi) изопропанол; (xii) изопропилацетат; (xiii) метанол; (xiv) метилэтилкетон; (xv) метилизобутилкетон; (xvi) 2,2,2-трифторэтанол; (xvii) тетрагидрофуран; (xviii) толуол; (xix) воду и (xx) этанол и гептан; или(b) превращение аморфной формы 1 соединения I посредством воздействия на аморфную форму 1 нагревания, высокой относительной влажности или органических паров, или посредством мокрого помола аморфной формы 1 в воде. 2. Способ получения соединения I формулы- 21015359 где по меньшей мере часть соединения I присутствует в аморфной форме 1, при этом способ выбран из группы, состоящей из:(a) ротационного упаривания из метанола;(b) быстрого упаривания из воды;(d) кристаллизации соединения I из смеси этилацетата и гексанов; или из смеси изопропилацетата и гексанов с последующим выделением соединения I из растворителя.
МПК / Метки
МПК: C07D 401/04, A61K 31/513, A61P 3/04
Метки: способы, бензонитрила, бензоатной, соли, 2-[[6-[(3r)-3-амино-1-пиперидинил]-3,4-дигидро-3-метил-2,4-диоксо-1(2н)-пиримидинил]метил, полиморфов, получения
Код ссылки
<a href="https://eas.patents.su/25-15359-sposoby-polucheniya-polimorfov-benzoatnojj-soli-2-6-3r-3-amino-1-piperidinil-34-digidro-3-metil-24-diokso-12n-pirimidinilmetil-benzonitrila.html" rel="bookmark" title="База патентов Евразийского Союза">Способы получения полиморфов бензоатной соли 2-[[6-[(3r)-3-амино-1-пиперидинил]-3,4-дигидро-3-метил-2,4-диоксо-1(2н)-пиримидинил]метил] бензонитрила</a>
Способы получения 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила

Номер патента: 7953
Опубликовано: 27.02.2007
Авторы: Лендерс Рубен Герардус Жорж, Виллемс Йоаннес Йозефус Мария, Паскье Элизабет Тереза Жанна, Медар Барт Петрус Анна Мария Йозеф, Хэрес Ян, Жанссен Поль Адриан Ян, Схилс Дидье Филипп Робер, Гийемон Жером Эмиль Жорж
МПК: C07C 253/20, A61K 31/505, C07C 253/30...
Метки: 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, способы, получения
Формула / Реферат:
1. Способ получения 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила формулы (I) его N-оксида, фармацевтически приемлемой кислотно-аддитивной соли, четвертичного амина или стереохимически изомерной формы, который включает осуществление взаимодействия промежуточного соединения формулы (II) его подходящей кислотно-аддитивной соли или стереохимически изомерной формы с промежуточным соединением формулы (III) его...
Гидраты и полиморфы 4-[[(7r)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-n-(1-метил-4-пиперидинил)бензамида, способы их получения и их применение в качестве лекарственных средств

Номер патента: 11407
Опубликовано: 27.02.2009
Авторы: Хертер Рольф, Линц Гюнтер, Ралль Вернер, Хоффманн Маттиас, Зигер Петер, Крэмер Герд Ф., Шмид Рольф
МПК: A61K 31/519, A61P 35/00, C07D 211/58...
Метки: полиморфы, средств, применение, качестве, гидраты, способы, 4-[[(7r)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-n-(1-метил-4-пиперидинил)бензамида, лекарственных, получения
Формула / Реферат:
1. Гидраты соединения формулы (I) 2. Гидрат соединения формулы (I) по п.1, отличающийся тем, что он представляет собой моногидрат соединения (I). 3. Гидрат соединения формулы (I) по п.1, отличающийся тем, что он представляет собой тригидрат соединения (I). 4. Соединение формулы (I) где соединение это ангидрат формы II или формы III. 5. Ангидрат по п.4, отличающийся тем, что он представлен в виде безводной формы II соединения формулы (I),...
Гидрохлорид 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила

Номер патента: 13686
Опубликовано: 30.06.2010
Авторы: Вандекрюйс Рогер Петрус Гереберн, Копманс Алекс Херман, Стапперс Альфред Элизабет, Петерс Йозеф, Стевенс Пол Теодор Агнес
МПК: A61P 31/18, A61K 31/505, C07D 239/02...
Метки: гидрохлорид, 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила
Формула / Реферат:
1. Твердая фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения формулы (I-а)или его N-оксида.2. Фармацевтическая композиция по п.1, в которой соединение формулы (I-а) представляет собой полиморфную форму А, характеризующуюся пиками порошковой рентгеновской дифракции в положениях два тета 9,7±0,2°, 13,5±0,2° и 15,0±0,2°.3. Фармацевтическая...
Фумарат 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила

Номер патента: 11036
Опубликовано: 30.12.2008
Авторы: Стевенс Пол Теодор Агнес, Копманс Алекс Херман, Петерс Йозеф, Вандекрюйс Рогер Петрус Гереберн, Стапперс Альфред Элизабет
МПК: A61K 31/505, A61P 31/18
Метки: 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, фумарат
Формула / Реферат:
1. Соединение формулы (I) его N-оксид или стереохимически изомерная форма. 2. Соединение по п.1, где указанное соединение представляет собой 3. Соединение по п.1 или 2 для использования в качестве лекарственного средства. 4. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и в качестве активного ингредиента - терапевтически эффективное количество соединения по п.1 или 2. 5. Фармацевтическая композиция по п.4, где...
Способ получения 11-амино-3-хлор-6,11-дигидро-5,5-диоксо-6-метилдибензо[c,f][1,2]тиазепина и его применение для синтеза тианептина

Номер патента: 3770
Опубликовано: 28.08.2003
Авторы: Бланшар Жаки, Бриго Даньель, Турбе Юг
МПК: C07D 281/02
Метки: способ, применение, получения, тианептина, 11-амино-3-хлор-6,11-дигидро-5,5-диоксо-6-метилдибензо[c,f][1,2]тиазепина, синтеза
Формула / Реферат:
1. Способ синтеза 11-амино-3-хлор-6,11-дигидро-5,5-диоксо-6-метилдибензо[c,f][1,2]тиазепина формулы (I) и его аддитивных солей, отличающийся тем, что проводят реакцию кетона формулы (III) с боргидридом натрия в двухфазной среде (хлорированный растворитель, такой как, например, хлороформ, дихлорметан или дихлорэтан/водный раствор гидроксида натрия) в присутствии N-додецил-N-метилдиэтаноламмонийбромида с получением спирта формулы (IV) который в...
Предыдущий патент: Хинолины и их терапевтическое применение
Следующий патент: Стерилизуемое паром устройство для разделения крови на компоненты
Случайный патент: Устройство и способ измерения пространственных перемещений производственных конструкций