Производные n-(аминогетероарил)-1н-индол-2-карбоксамидов, их получение и их применение в терапии
Номер патента: 17375
Опубликовано: 28.12.2012
Авторы: Жилль Катрин, Дюбуа Лоран, Маланда Андре, Машник Давид, Эванно Янник














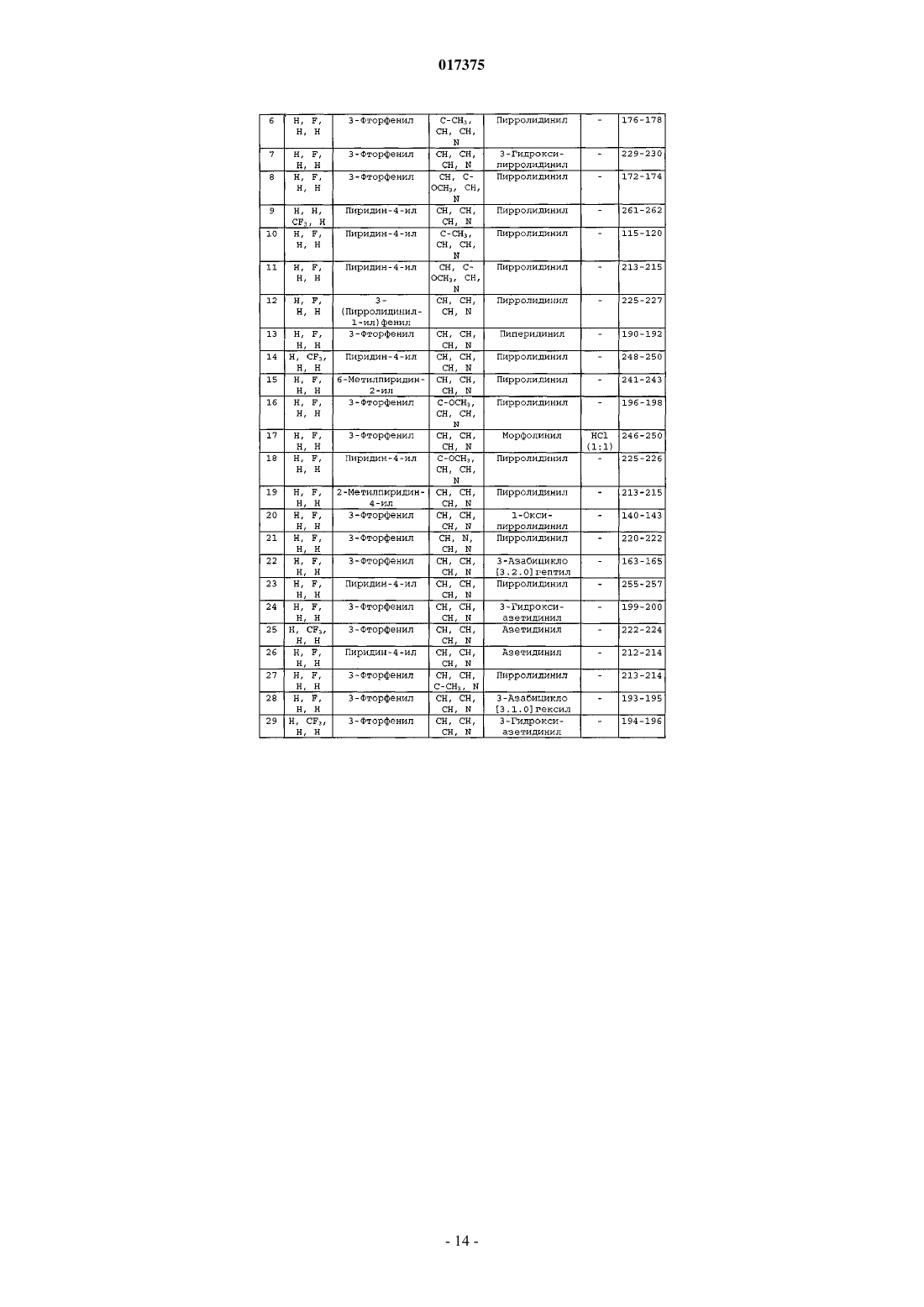





Формула / Реферат
1. Соединение формулы (I)

в которой X1 обозначает атом водорода или галогена либо C1-C6-алкил;
Х2 обозначает атом водорода или галогена, C1-C6-алкил, C1-C6-фторалкил или -S(О)2-C1-C6-алкил;
Х3 и Х4 обозначают независимо друг от друга атом водорода или галогена, C1-C6-алкил, C1-C6-фторалкил, C1-C6-алкоксил, NR1R2 или C1-С6-тиоалкил;
Z1, Z2, Z3 и Z4 обозначают независимо друг от друга атом азота или группу C(R6), при этом по меньшей мере один из Z1, Z2, Z3 и Z4 соответствует атому азота и по меньшей мере один из Z1, Z2, Z3 и Z4 соответствует группе C(R6);
n равно 1;
Y обозначает фенил или пиридинил, необязательно замещенный одной или несколькими группами, выбранными из атома галогена, C1-С6-алкила или NR1R2;
Z обозначает циклический амин, выбранный из азетидина, пирролидина, пиперидина, морфолина, азабицикло[3.1.0]гексана и азабицикло[3.2.0]гептана;
при этом атом азота циклического амина Z может быть в форме N-оксида;
атомы углерода циклического амина Z необязательно замещены группой R12;
R1 и R2 обозначают независимо друг от друга атом водорода или C1-C6-алкил или R1 и R2 образуют вместе с атомом азота, с которым они связаны, пирролидинил;
R3 обозначает атом водорода;
R5 обозначает C1-C6-алкил;
R6 обозначает атом водорода, C1-C6-алкил, C1-C6-фторалкил или C1-C6-алкоксил;
R12 обозначает группу NR1R2, NR3COOR5 или гидроксил;
в виде основания или кислотно-аддитивной соли, а также в виде гидрата или сольвата.
2. Способ получения соединения формулы (I) по п.1, отличающийся тем, что осуществляют взаимодействие соединения общей формулы (IV)

в которой X1, Х2, Х3, Х4, Y и n такие, как определено в общей формуле (I) по п.1, и В обозначает гидроксильную группу с амином общей формулы (V)

в которой Z1, Z2, Z3, Z4 и Z такие, как определено в общей формуле (I) по п.1, в присутствии связующего вещества и основания в растворителе.
3. Способ получения соединения формулы (I) по п.1, отличающийся тем, что осуществляют взаимодействие соединения общей формулы (VI)

в которой X1, Х2, Х3, Х4, Z1, Z2, Z3, Z4, Y и n такие, как определено в общей формуле (I) по п.1, и W обозначает атом галогена, с амином формулы ZH, в которой Z обозначает циклический амин, такой как определен в общей формуле (I) по п.1.
4. Амин общей формулы (V), выбранный из

в виде основания или соли присоединения с кислотой.
5. Фармацевтическая композиция, отличающаяся тем, что она содержит соединение формулы (I) по п.1 или фармацевтически приемлемую соль, гидрат или сольват этого соединения, а также по меньшей мере один фармацевтически приемлемый эксципиент.
Текст
Изобретение относится к соединениям общей формулы (I), в которой Х 1 обозначает атом водорода или галогена либо C1-C6-алкил; Х 2 обозначает атом водорода или галогена, C1-C6-алкил, C1-C6-фторалкил или S(О)2-C1-C6-алкил; Х 3 и Х 4 обозначают независимо друг от друга атом водорода или галогена, C1-C6-алкил,C1-C6-фторалкил, C1-C6-алкоксил, NR1R2 или C1-C6-тиоалкил; Z1, Z2, Z3 и Z4 обозначают независимо друг от друга атом азота или группу С(R6), при этом по меньшей мере один из Z1, Z2, Z3 и Z4 соответствует атому азота и по меньшей мере один из Z1, Z2, Z3 и Z4 соответствует группе C(R6); n равно 1; Y обозначает фенил или пиридинил, необязательно замещенный одной или несколькими группами, выбранными из атома галогена, C1-С 6-алкила или NR1R2; Z обозначает циклический амин, выбранный из азетидина, пирролидина,пиперидина, морфолина, азабицикло[3.1.0]гексана и азабицикло[3.2.0]гептана; при этом атом азота циклического амина Z может быть в форме N-оксида; атомы углерода циклического амина Z необязательно замещены группой R12; R1 и R2 обозначают независимо друг от друга атом водорода или C1-C6-алкил или R1 и R2 образуют вместе с атомом азота, с которым они связаны, пирролидинил; R3 обозначает атом водорода;R12 обозначает группу NR1R2, NR3COOR5 или гидроксил; в виде основания или кислотно-аддитивной соли,а также в виде гидрата или сольвата. Изобретение также относится к способу получения соединения формулы (I), амину общей формулы (V), охарактеризованному в формуле изобретения, и к фармацевтической композиции, содержащей соединение формулы (I). 017375 Объектом изобретения являются производные соединения N-(аминогетероарил)-1 Н-индол-2 карбоксамидов, которые обладают активностью антагонистов in vitro и in vivo в отношении рецепторов типа TRPV1 (или VR1). Первым объектом изобретения являются соединения нижеследующей общей формулы (I). Другим объектом изобретения являются способы получения соединений нижеследующей общей формулы (I). Другим объектом изобретения является применение соединений общей формулы (I), в частности, в составе фармацевтических композиций. Соединения по изобретению соответствуют общей формуле (I) в которой X1 обозначает атом водорода или галогена либо C1-C6-алкил; Х 2 обозначает атом водорода или галогена, C1-C6-алкил, C1-C6-фторалкил или -S(О)2-C1-C6-алкил; Х 3 и Х 4 обозначают независимо друг от друга атом водорода, галогена, C1-C6-алкил, C1-C6 фторалкил, C1-C6-алкоксил, NR1R2 или C1-C6-тиоалкил;Z1, Z2, Z3 и Z4 обозначают независимо друг от друга атом азота или группу C(R6), при этом по меньшей мере один соответствует атому азота и по меньшей мере один соответствует группе С(R6);Y обозначает фенил или пиридинил, необязательно замещенный одной или несколькими группами,выбранными из атома галогена, C1-C6-алкила или NR1R2;Z обозначает циклический амин, выбранный из азетидина, пирролидина, пиперидина, морфолина,азабицикло[3.1.0]гексана и азабицикло[3.2.0]гептана; при этом атом азота циклического амина Z может быть в форме N-оксида; атомы углерода циклического амина Z необязательно замещены группой R12;R1 и R2 обозначают независимо друг от друга атом водорода или C1-C6-алкил или R1 и R2 образуют вместе с атомом азота, с которым они связаны, пирролидинил;R12 обозначает группу NR1R2, NR3COOR5 или гидроксил. В соединениях общей формулы (I) один или несколько атомов азота могут быть в оксидированной форме (N-оксид). Из соединений общей формулы (I), объектов изобретения, первая подгруппа соединений состоит из соединений, в которых Х 1 обозначает атом водорода или галогена либо C1-C6-алкильную группу; Х 2 обозначает атом водорода или галогена либо группу: C1-С 6-алкил, C1-C6-фторалкил или -S(О)2C1-C6-алкил; Х 3 и Х 4 обозначают независимо друг от друга атом водорода или галогена либо группу: C1-C6 алкил, C1-C6-фторалкил, C1-C6-алкоксил, NR1R2 или C1-C6-тиоалкил;R1 и R2 обозначают независимо друг от друга C1-C6-алкильную группу. Из соединений общей формулы (I), объектов изобретения, вторая подгруппа соединений состоит из соединений, в которыхX1 обозначает атом водорода, фтора или хлора либо метильную группу; Х 2 обозначает атом водорода, фтора или хлора либо группу: метильную, изопропильную, третбутильную, трифторметильную или -S(O)2CH3; Х 3 и Х 4 обозначают независимо друг от друга атом водорода, фтора или хлора либо группу: метильную, этильную, изопропильную, трет-бутильную, трифторметильную, -О-изопропильную, NR1R2 или тиометильную;R1 и R2 обозначают независимо друг от друга метильную группу. Из соединений общей формулы (I), объектов изобретения, третья подгруппа соединений состоит из соединений, в которых Z1 и Z3 обозначают группу C(R6) и Z2 и Z4 обозначают атом азота; при этом R6 такой, как определено в общей формуле (I). Из соединений общей формулы (I), объектов изобретения, четвертая подгруппа соединений состоит из соединений, в которых Z1 и Z3 обозначают группу C(R6) и Z2 и Z4 обозначают атом азота; при этом R6 соответствует атому водорода. Из соединений общей формулы (I), объектов изобретения, пятая подгруппа соединений состоит из соединений, в которых Z4 обозначает атом азота и Z1, Z2 и Z3 обозначают независимо друг от друга груп-1 017375 пу C(R6); при этом R6 обозначает атом водорода или группу: C1-C6-алкильную, C1-C6-фторалкильную илиC1-C6-алкоксильную. Из соединений общей формулы (I), объектов изобретения, шестая подгруппа соединений состоит из соединений, в которых Z4 обозначает атом азота и Z1, Z2 и Z3 обозначают независимо друг от друга группу C(R6); при этом R6 обозначает атом водорода или метильную, трифторметильную или метоксигруппу. Из соединений общей формулы (I), объектов изобретения, седьмая подгруппа соединений состоит из соединений, в которых Y обозначает фенил или пиридинил, причем фенил необязательно замещен группой, выбираемой из атома фтора или метильной группы либо NR1R2;R1 и R2 образуют вместе с атомом азота, с которым они связаны, пирролидинильную группу. Из соединений общей формулы (I), объектов изобретения, восьмая подгруппа соединений состоит из соединений, в которыхZ обозначает циклический амин, выбранный из азетидина, пирролидина, пиперидина, морфолина,азабицикло[3.1.0]гексана и азабицикло[3.2.0]гептана; при этом атом азота циклического амина Z может быть в форме N-оксида; атомы углерода циклического амина Z необязательно замещены группой R12;R1 и R2 обозначают независимо друг от друга атом водорода;R5 обозначает C1-C6-алкильную группу. Из соединений общей формулы (I), объектов изобретения, восемнадцатая подгруппа соединений состоит из соединений, в которыхZ обозначает циклический амин, выбранный из азетидина, пирролидина, пиперидина, морфолина,азабицикло[3.1.0]гексана и азабицикло[3.2.0]гептана; при этом атом азота циклического амина Z может быть в форме N-оксида; атомы углерода циклического амина Z необязательно замещены группой R12;R1 и R2 обозначают независимо друг от друга атом водорода;R5 обозначает трет-бутильную группу. Из соединений общей формулы (I), объектов изобретения, девятая подгруппа соединений состоит из соединений, в которыхZ обозначает циклический амин, выбранный из азетидина, пирролидина, пиперидина, морфолина,азабицикло[3.1.0]гексана и азабицикло[3.2.0]гептана; атомы углерода азетидина необязательно замещены гидроксильной группой; атомы углерода пирролидина необязательно замещены группой NR1R2, NR3COOR5, гидроксильной; атом азота пирролидина может быть в форме N-оксида;R1 и R2 обозначают независимо друг от друга атом водорода;R5 обозначает C1-C6-алкильную группу. Из соединений общей формулы (I), объектов изобретения, десятая подгруппа соединений состоит из соединений, в которыхZ обозначает циклический амин, выбранный из азетидина, пирролидина, пиперидина, морфолина,азабицикло[3.1.0]гексана и азабицикло[3.2.0]гептана; при этом атомы углерода азетидина необязательно замещены гидроксильной группой; атомы углерода пирролидина необязательно замещены группой NR1R2, NR3COOR5, гидроксильной; атом азота пирролидина может быть в форме N-оксида;R1 и R2 обозначают независимо друг от друга атом водорода;R5 обозначает трет-бутильную группу. Из соединений общей формулы (I), объектов изобретения, можно назвать следующие соединения: 1) N-6-[R)-3-трет-бутоксикарбониламино)пирролидин-1-ил]пиридин-3-ил-5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоксамид; 2) N-[6-(пирролидин-1-ил)пиридин-3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоксамид; 3) N-6-[(R)-3-аминопирролидин-1-ил]пиридин-3-ил-5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоксамид; 4) N-[6-(азетидин-1-ил)пиридин-3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоксамид; 5)-3 017375 48) N-[6-(азетидин-1-ил)пиридин-3-ил]-5-изопропил-1-(3-фторбензил)-1 Н-индол-2-карбоксамид; 49) N-[6-(азетидин-1-ил)пиридин-3-ил]-5-трет-бутил-1-(3-фторбензил)-1 Н-индол-2-карбоксамид; 50) N-[6-(азетидин-1-ил)пиридин-3-ил]-6-этил-1-(3-фторбензил)-1H-индол-2-карбоксамид; 51) N-[6-(азетидин-1-ил)пиридин-3-ил]-6-изопропил-1-(3-фторбензил)-1 Н-индол-2-карбоксамид; 52) N-[6-(азетидин-1-ил)пиридин-3-ил]-6-трифторметил-1-(3-фторбензил)-1 Н-индол-2-карбоксамид; 53) N-[6-(пирролидин-1-ил)-4-(трифторметил)пиридин-3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол-2 карбоксамид. В рамках настоящего изобретения понимают подCt-Cz, где t и z могут принимать значения от 1 до 7, углеродную цепочку, которая может содержать от t и z атомов углерода, например C1-C3-углеродную цепочку, которая может содержать от 1 до 3 атомов углерода; алкилом: насыщенную алифатическую линейную или разветвленную группу; в качестве примеров можно назвать метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, третбутильную, пентильную группы и т.д.; фторалкилом: алкильную группу, один или несколько атомов водорода которой были замещены атомом фтора; алкоксилом: радикал -O-алкил, где алкильная группа такая, как определено выше; тиоалкилом: радикал -S-алкил, где алкильная группа такая, как определено выше; атомом галогена: фтор, хлор, бром или йод;"тио" означает "=S". Соединения формулы (I) могут содержать один или несколько асимметричных атомов углерода. Таким образом, они могут иметь форму энантиомеров или диастереоизомеров. Эти энантиомеры, диастереоизомеры, а также их смеси, включая рацемические смеси, относятся к изобретению. Соединения формулы (I) могут быть в виде оснований или кислотно-аддитивных солей. Такие аддитивные соли относятся к изобретению. Эти соли преимущественно получают с использованием фармацевтически приемлемых кислот, но соли других пригодных кислот, например, для очистки или выделения соединений формулы (I) также относятся к изобретению. Соединения формулы (I) могут быть в виде гидратов или сольватов, а именно в виде ассоциаций или комбинаций с одной или несколькими молекулами воды или с растворителем. Такие гидраты и сольваты также относятся к изобретению. Далее в тексте под отделяемой группой понимают группу, которая может легко отщепляться от молекулы путем разрыва гетеролитической связи с отделением электронной пары. Такая группа, таким образом, может легко заменяться другой группой, например, в ходе реакции замещения. Такими отделяемыми группами являются, например, галогены или активированная гидроксильная группа, такая как метансульфонат, бензолсульфонат, п-толуолсульфонат, трифлат, ацетат и т.д. Примеры отделяемых групп,а также информация об их получении приведены в "Advances in Organic Chemistry", J. March, 5th Edition,Willey Interscience, 2001. Согласно изобретению соединения общей формулы (I) можно получать способом, который иллюстрирует нижеследующая схема. На схеме соединения общей формулы (IV) можно получать путем взаимодействия соединения общей формулы (II), в которой X1, Х 2, Х 3, Х 4 такие, как определено в общей формуле (I), и В обозначает C1 С 6-алкоксильную группу, с соединением общей формулы (III), в которой Y и n такие, как определено в общей формуле (I), и GP обозначает отделяемую группу, где GP обозначает гидроксильную группу. Соединения общей формулы (I) имеются в продаже или их можно получить несколькими способами, описанными в литературе (D. Knittel Synthesis 1985, 2, 186; Т.М. Williams J. Med. Chem. 1993, 36(9),1291; JP 2001151771A2, например). Если соединение общей формулы (III) определено так, что n равно 1, 2 или 3, и GP обозначает отделяемую группу, такую как атом хлора, брома или йода, взаимодействие можно проводить в присутствии основания, такого как гидрат натрия или карбонат калия, в полярном растворителе, таком как диметилформамид, диметилсульфоксид или ацетон (n=1: Kolasa Т., Bioorg, Med. Chem. 1997, 5(3)507, n=2:Abramovitch R., Synth. Commun., 1995, 25(1), 1). Если соединение общей формулы (III) определено так, что n равно 1, 2 или 3, и GP обозначает гидроксильную группу, соединения общей формулы (IV) можно получать путем взаимодействия соединения общей формулы (II) с соединением общей формулы (III) в присутствии фосфина, такого как, например,трифенилфосфин, и реактива, такого как, например, диэтилазодикарбоксилат, в растворе в растворителе,таком как дихлорметан или тетрагидрофуран (О. Mitsonobu, Synthesis, 1981, 1-28). Если соединение общей формулы (III) определено так, что n равно 0, GP обозначает отделяемую группу, такую как атом хлора, брома или йода, и взаимодействие может происходить при температуре от 80 до 250 С в присутствии катализатора на основе меди, такого как бромид меди или оксид меди, а также основания, такого как карбонат калия (Murakami Y., Chem. Pharm. Bull., 1995, 43(8), 1281). Условия могут также быть более мягкими, как описано в S.L. Buchwald, J. Am. Chem. Soc. 2002, 124, 11684. Соединение общей формулы (IV), в которой В обозначает C1-C6-алкоксильную группу, может превращаться в соединение общей формулы (IV), в которой В обозначает гидроксильную группу, под действием основания, такого как гидроксид натрия или карбонат калия, в растворе в растворителе, таком как этанол. Соединение общей формулы (IV), в которой В обозначает гидроксильную группу, может затем превращаться в соединение общей формулы (IV), в которой В обозначает атом хлора, под действием хлорирующего агента, такого как тионилхлорид, в растворителе, таком как дихлорметан. Соединения общей формулы (I) и (VI) можно затем получить, например, путем взаимодействия соединения общей формулы (IV), в которой В обозначает атом хлора, такого как получено выше, с амином общей формулы (V), в которой Z1, Z2, Z3, Z4 такие, как определено в общей формуле (I), и W обозначает атом галогена, такой как хлор, бром или йодид, или циклическим амином Z, таким как определено в общей формуле (I), в растворителе, таком как дихлорэтан, толуол или тетрагидрофуран. Соединения общей формулы (I) и (VI) можно также получить, например, путем взаимодействия соединения общей формулы (IV), в которой В обозначает гидроксильную группу, такого как получено выше, с амином общей формулы (V), в которой Z1, Z2, Z3, Z4 такие, как определено в общей формуле (I), иW обозначает атом галогена, такой как хлор, бром или йодид, или циклическим амином Z, таким как определено в общей формуле (I), в присутствии связующего вещества, такого как диэтилцианофосфонат, в присутствии основания, такого как триэтиламин, в растворителе, таком как диметилформамид, или в присутствии связующего вещества, такого как N-(3-диметиламинопропил)-N'-этилкарбодиимид, в присутствии N-1-гидроксибензотриазола в растворителе, таком как диметилформамид. В случае если W обозначает атом галогена, получают соединение общей формулы (VI). В случае если W обозначает циклический амин Z, такой как определено в общей формуле (I), получают соединение общей формулы (I). Соединение общей формулы (I) можно также получить взаимодействием соединения общей формулы (VI), где W обозначает атом галогена, в присутствии амина формулы ZH, в которой Z обозначает циклический амин, такой как определено в общей формуле (I), без растворителя или в растворителе, таком как N-метилпирролидинон, или методом катализуемого N-арилирования, такого как описано J.Hartwig (J.F. Hartwig, Angew Chem. Int. Ed. 2005, 44, 1371-1375), предпочтительно в инертной атмосфере в присутствии основания, такого как бис-триметилсилиламидид лития в присутствии источника палладия в каталитическом количестве, такого как диацетат палладия, и каталитического количества лиганда палладия, такого как фосфин, причем все в целом растворено в растворителе, таком как диметоксиэтан(DME). Соединения общих формул (I), (II) и (IV), в которых Х 1, Х 2, Х 3 и/или Х 4 обозначают циано или арильную группу, могут быть получены реакцией сочетания, катализируемой металлом, таким как палладий, проводимой с использованием соединений общих формул (I), (II) и (IV), в которых X1, X2, Х 3-5 017375 и/или Х 4 обозначают отделяемую группу, например бром, методами, описанными в литературе или известными специалисту. Соединения общих формул (I), (II) и (IV), в которых X1, X2, Х 3 и/или Х 4 обозначают группуC(O)NR1R2, можно получить из соответствующих соединений общих формул (I), (II) и (IV), в которыхX1, X2, Х 3 и/или Х 4 обозначают группу циано, методами, описанными в литературе или известными специалисту. Соединения общих формул (I), (II) и (IV), в которых X1, X2, Х 3, Х 4 и/или R6 обозначают -S(О)алкильную или -S(О)2-алкильную группу, можно получить оксидированием соответствующих соединений общих формул (I), (II) и (IV), в которых Х 1, Х 2, Х 3, Х 4 и/или R6 обозначают C1-C6-тиоалкильную группу, методами, описанными в литературе или известными специалисту. Также соединения общих формул (I) и (IV), в которых Y замещен -S(О)-алкильной или -S(О)2 алкильной группой, можно получить оксидированием соответствующих соединений общих формул (I) и(IV), в которых Y замещен C1-C6-тиоалкильной группой, методами, описанными в литературе или известными специалисту. Соединения общей формулы (I), в которых Z1, Z2, Z3 и/или Z4 обозначают группу C-R6, соответствующую группе С-ОН, можно получить из соответствующих соединений общей формулы (I), в которыхZ1, Z2, Z3 и/или Z4 обозначают группу C-R6, соответствующую C1-C6-алкоксильной группе, методами,описанными в литературе или известными специалисту. Соединения общих формул (I), (II) и (IV), в которых Х 1, Х 2, Х 3 и/или Х 4 обозначают группу NR1R2,NR3COR4 или NR3SO2R5, можно получить из соответствующих соединений общих формул (I), (II) и (IV),в которых X1, X2, Х 3 и/или Х 4 обозначают нитрогруппу, например, восстановлением с последующим ацилированием или сульфонилированием методами, описанными в литературе или известными специалисту. Соединения общих формул (I), (II) и (IV), в которых Х 1, Х 2, Х 3 и/или Х 4 обозначают группу NR1R2,NR3COR4 или NR3SO2R5, можно получить из соответствующих соединений общих формул (I), (II) и (IV),в которых X1, X2, Х 3 и/или Х 4 обозначают, например, атом брома, реакцией сочетания соответственно с амином, амидом или сульфонамидом в присутствии основания, фосфина и катализатора на основе палладия методами, описанными в литературе или известными специалисту. Соединения общих формул (I), (II) и (IV), в которых Х 1, Х 2, Х 3 и/или Х 4 обозначают группуSO2NR1R2, можно получить методом, аналогичным описанному в Pharmazie 1990, 45, 346, или методами,описанными в литературе или известными специалисту. Соединения общей формулы (III) имеются в продаже, описаны в литературе (Carling R.W. et al., J.Med. Chem. 2004 (47), 1807-1822 или Russel M.G.N. J. Med. Chem. 2005 (48), 1367-1383) или доступны при использовании методов, известных специалисту. Некоторые соединения общей формулы (IV) описаны в литературе (WO 07/010144, например). Соединения (V) и другие реагенты, если способ их получения не описан, имеются в продаже или описаны в литературе (WO 05028452, WO 02048152, WO 06040522, WO 04052869, Heterocycles 1977, 6(12), 1999-2004, J. Chem. Soc. Perkin trans 1, 1973 (1) 68-69,JP 07/051121, WO 05035526, WO 07/011284, WO 04/062665, GB 870027, US 4104385). В соответствии с другим аспектом объектом изобретения также являются соединения формул (Va),(Vb), (Vc), (Vd), (Ve), (Vf), (Vg). Эти соединения пригодны в качестве полупродуктов синтеза соединений формулы (I). Амины формул (Va)-(Vg) можно получить, например, способом, описанным в примере 5, путем ароматического нуклеофильного замещения предшественника 6-хлорпиридина, необязательно замещенного (Va: 5-метоксигруппой, Vb: 4-метоксигруппой, Vd: 2-метильной группой, Vg: 4-трифторметильной группой), амином, таким как пирролидин, например, в растворителе, таком как этанол. Доступ к аминамVa-Vg может затем потребовать восстановления нитрогруппы, например, каталитической гидрогенизацией в присутствии катализатора, такого как палладий на угле, или любыми другими методами восстановления нитрогруппы до амина, известными специалисту. Амины формул (Va)-(Vg) были получены в виде порошка или масла, в виде основания или кислотно-аддитивной соли. В табл. 1 приведены данные ЯМР 1 Н этих аминов. В следующих примерах описано получение некоторых соединений по изобретению. Эти примеры не ограничивают, а только иллюстрируют изобретение. Номера соединений в примерах соответствуют приведенным в табл. 2. Элементарные микроанализы, анализы ЖХ-МС (жидкостная хроматография в сочетании с масс-спектрометрией), инфракрасные спектры или спектры ЯМР подтверждают структуры полученных соединений. Пример 1 (соединение 1). N-6-[R)-3-трет-Бутоксикарбониламино)пирролидин-1-ил]пиридин-3 ил-5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоксамид. 1.1. 5-Фтор-1-(3-фторбензил)-1 Н-индол-2-карбоновая кислота. Водный раствор гидроксида натрия, полученный из 1,15 г (28,92 ммоль) таблеток гидроксида натрия в 50 мл воды, вводят в раствор 7,6 г (24,10 ммоль) этил-5-фтор-1-(3-фторбензил)-1 Н-индол-2 карбоксилата (WO 2006/024776) в 241 мл этанола. Смесь нагревают в течение 2 ч, затем концентрируют при пониженном давлении. Полученное твердое вещество обрабатывают в 200 мл воды. Раствор промывают два раза по 100 мл простого этилового эфира, подкисляют путем последовательного введения небольших количеств концентрированной соляной кислоты, затем экстрагируют 200 мл этилацетата. Органическую фазу, наконец, промывают два раза по 100 мл воды, один раз 50 мл насыщенного раствора натрия хлорида, сушат над сульфатом магния и концентрируют при пониженном давлении. После сушки при 50 С при пониженном давлении получают 6,4 г целевого продукта в виде твердого вещества, который используют как таковой на следующей стадии. 1.2.N-6-[R)-3-трет-Бутоксикарбониламино)пирролидин-1-ил]пиридин-3-ил-5-фтор-1-(3 фторбензил)-1 Н-индол-2-карбоксамид (соединение 1). Вводят по каплям в атмосфере аргона и при 20 С в раствор 0,47 г (1/64 ммоль) 5-фтор-1-(3 фторбензил)-1 Н-индол-2-карбоновой кислоты (полученной на стадии 1.1), 0,5 г (1,8 ммоль) 3-амино-6[R)-3-трет-бутоксикарбониламино)пирролидин-1-ил]пиридина (JP 2004175739) и 0,85 г (1,64 ммоль) трис-пирролидинофосфония гексафторфосфата (PYBOP) в 20 мл дихлорметана, 0,85 мл (4,91 ммоль) диизопропилэтиламина. Смесь перемешивают в течение 12 ч при комнатной температуре, концентрируют при пониженном давлении, затем обрабатывают в 50 мл этилацетата. Затем этот раствор последовательно промывают три раза по 15 мл насыщенного раствора гидрогенокарбоната натрия и 20 мл насыщенного раствора хлорида натрия, затем сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный продукт очищают хроматографией на колонке с кремнеземом,элюируя смесью дихлорметана и метанола. Полученное твердое вещество измельчают в простом этиловом эфире. Получают 0,788 г твердого вещества, которое сушат при пониженном давлении. Точка плавления: 207-209 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 1,49 (с, 9 Н); 1,85 (секст, 1 Н); 2,1 (секст, 1 Н); 3,2 (м, 4 Н); 4,09 (м, 1 Н); 5,88 (с, 2 Н); 6,41 (д, 1 Н); 7,04 (м, 7 Н); 7,61 (м, 2 Н); 7,8 (дд, 1 Н); 8,31 (д, 1 Н); 10,19 (с, 1 Н).-7 017375 Пример 2 (соединение 2). N-[6-(Пирролидин-1-ил)пиридин-3-ил]-5-фтор-1-(3-фторбензил)-1 Ниндол-2-карбоксамид. В раствор 0,402 г (1,4 ммоль) 5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоновой кислоты (пример 1.1) и 0,274 г (1,68 ммоль) 3-амино-6-(пирролидин-1-ил)пиридина (WO 02/48152) в 10 мл сухого диметилформамида вводят по каплям при 20 С в атмосфере аргона 0,28 мл (1,69 ммоль) диэтилцианофосфоната. Смесь перемешивают в течение 10 мин, затем вводят по каплям 0,43 мл (3,08 ммоль) триэтиламина. Смесь перемешивают в течение 10 ч при комнатной температуре, концентрируют при пониженном давлении, затем обрабатывают 50 мл этилацетата. Затем этот раствор последовательно промывают три раза по 20 мл насыщенного раствора гидрогенокарбоната натрия, 50 мл воды и 20 мл насыщенного раствора хлорида натрия, затем сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученное твердое вещество измельчают в горячем простом изопропиловом эфире. Получают 0,527 г твердого вещества, которое сушат при пониженном давлении. Точка плавления: 199-201 С. ЯМР 1H (ДМСО-d6),(м.д.): 1,9 (м, 4 Н); 3,3 (м, 4 Н); 5,82 (с, 2 Н); 6,4 (д, 1 Н); 7,04 (м, 6 Н); 7,51 (м,2 Н); 7,77 (дд, 1 Н); 8,3 (д, 1 Н); 10,25 (с, 1 Н). Пример 3 (соединение 4). N-[6-(Азетидин-1-ил)пиридин-3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол 2-карбоксамид. 3.1. N-[6-(Хлорпиридин-3-ил)]-5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоксамид. В раствор 4,48 г (34,89 ммоль) 6-хлор-3-аминопиридина в 317 мл толуола вводят при 20 С в условиях перемешивания в атмосфере аргона 23,8 мл (47,57 ммоль) раствора 2 М триметилалюминия в толуоле. Затем раствор нагревают до 120 С и вводят в несколько приемов 10 г (31,71 ммоль) этил-5-фтор 1-(3-фторбензил)-1 Н-индол-2-карбоксилата (WO 2006/024776). Реакционную смесь нагревают в течение 4 ч с обратным холодильником, затем охлаждают до 0 С. В реакционную смесь вводят 15 л воды и затем экстрагируют 2 раза по 200 мл этилацетата. Органические фазы объединяют и промывают два раза по 50 мл воды и один раз 50 мл насыщенного раствора хлорида натрия, затем сушат над сульфатом натрия и концентрируют при пониженном давлении. Полученный остаток измельчают в 100 мл простого этилового эфира, фильтруют и получают 10 г целевого продукта в порошкообразной форме. ЯМР 1 Н (ДМСО-d6),(м.д.): 5,9 (с, 2 Н); 6,9 (м, 2 Н); 7,05 (тд, 1 Н); 7,2 (тд, 1 Н); 7,32 (м, 1 Н); 7,58(м, 4 Н); 8,25 (д, 1 Н); 8,79 (с, 1 Н); 10,79 (с, 1 Н). 3.2 N-[6-(Азетидин-1-ил)пиридин-3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоксамид (соединение 4). В смесь 0,7 г (1,76 ммоль) N-[6-хлорпидин-3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол-2 карбоксамида, полученного на стадии 3.1, 0,123 г (2,11 ммоль) азетидина, 8 мг (0,04 ммоль) палладия диацетата и 19,5 мг (0,04 ммоль) (R)-(-)-1-[(S)-2-(дициклогексилфосфино)ферроценил]этилди-третбутилфосфина в 1,8 мл диметоксиэтана, сухого и дегазированного, вводят при 0 С в условиях перемешивания в атмосфере аргона 4,2 мл (4,2 ммоль) раствора бис-триметилсилиламида лития 1 М в тетрагидрофуране. Реактор закрывают, затем нагревают в течение 16 ч до 100 С. Затем реакционную смесь выливают в 50 л воды. Медленно вводят 5 мл молярного раствора соляной кислоты, затем 50 мл насыщенного раствора гидрогенокарбоната натрия. Смесь экстрагируют два раза по 50 мл этилацетата. Органические фазы объединяют, затем промывают два раза по 50 мл гидрогенокарбоната натрия, сушат над сульфатом магния, затем концентрируют при пониженном давлении. Полученный продукт очищают хроматографией на колонке с кремнеземом, элюируя смесью дихлорметана и ацетона. Полученное твердое вещество затем измельчают в 20 мл горячего изопропанола, извлекают фильтрованием, затем перекристаллизовывают в смеси этанола и метанола. Получают после фильтрования 0,31 г твердого вещества, которое сушат при пониженном давлении. Точка плавления: 227-228 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 2,25 (пент, 2 Н); 3,9 (т, 4 Н); 6,01 (с, 2 Н); 6,35 (д, 1 Н); 6,89 (м, 2 Н); 7,01(тд, 1 Н); 7,12 (тд, 1 Н); 7,19 (кв, 1 Н); 7,34 (с, 1 Н); 7,52 (м, 2 Н); 7,82 (дд, 1 Н); 8,31 (с, 1 Н); 10,28 (с, 1 Н). Пример 4 (соединение 7). N-[6-(3-Гидроксипирролидин-1-ил)пиридин-3-ил]-5-фтор-1-(3 фторбензил)-1 Н-индол-2-карбоксамид. В течение 20 мин нагревают в микроволновой печи при 200 С и 300 Вт смесь 0,4 г (1,01 ммоль) N[6-(хлорпиридин-3-ил)]-5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоксамида, полученного на стадии 3.1,и 0,81 мл (10,06 ммоль) 3-гидроксипирролидина в 1,2 мл N-метилпирролидинона. Реакционную смесь затем выливают в 50 мл воды. Получают после фильтрования твердое вещество, которое очищают хроматографией на колонке с кремнеземом, элюируя смесью дихлорметана и метанола. Полученное твердое вещество затем перекристаллизовывают в смеси метанола и дихлорметана. Получают после фильтрования 0,22 г твердого вещества, которое сушат при пониженном давлении. Точка плавления: 229-230 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 1,9 (м, 1 Н); 2,04 (м, 1 Н); 3,3-3,47 (м, 4 Н); 4,49 (с, 1 Н); 4,92 (с, 1 Н); 5,9-8 017375 7,82 (дд, 1 Н); 8,37 (с, 1 Н); 10,22 (с, 1 Н). Пример 5 (соединение 8). N-[5-Метокси-6-(пирролидин-1-ил)пиридин-3-ил]-5-фтор-1-(3 фторбензил)-1H-индол-2-карбоксамид. 5.1. 3-Метокси-5-нитро-2-(пирролидин-1-ил)пиридин. Нагревают до 100 С в течение 4 ч раствор 3 г (15,91 ммоль) 2-хлор-3-метокси-5-нитропиридина и 2 мл (23,86 ммоль) пирролидина в 30 мл диметилформамида. Затем реакционную смесь концентрируют при пониженном давлении, затем обрабатывают 100 мл воды и экстрагируют три раза по 100 мл этилацетата. Органические фазы объединяют, затем промывают два раза по 50 мл воды, затем один раз 50 мл насыщенного раствора хлорида натрия, сушат над сульфатом натрия, затем концентрируют при пониженном давлении. Таким образом выделяют 3,47 г целевого продукта в виде порошка оранжевого цвета. ЯМР 1 Н (ДМСО-d6),(м.д.): 1,9 (м, 4 Н); 3,79 (м, 4 Н); 3,88 (с, 3H); 7,62 (с, 1 Н); 8,69 (с, 1 Н). 5.2. 3-Метокси-2-(пирролидин-1-ил)-4-аминопиридин (соединение Va). Суспензию 3,4 г (15,54 ммоль) 3-метокси-5-нитро-2-(пирролидин-1-ил)пиридина, полученного на стадии 5.1, и 0,33 г 10%-ного палладия на угле в 40 мл этанола перемешивают в течение 5 ч при 20 С под давлением 5,6 бар водорода. Реакционную смесь затем фильтруют через целитный буфер, концентрируют при пониженном давлении, затем очищают хроматографией на колонке с кремнеземом, элюируя смесью дихлорметана и метанола. Выделяют 0,9 г целевого продукта, который используют как таковой в дальнейшем процессе синтеза. ЯМР 1 Н (CDCl3),(м.д.): 2,5 (м, 4 Н); 3,23 (пик уширенный, 2 Н); 3,49 (М, 4 Н); 3,91 (с, 3H); 5,90 (с,1 Н); 7,71 (с, 1 Н). 5.3N-[5-Метокси-6-(пирролидин-1-ил)пиридин-3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол-2 карбоксамид (соединение 8). Перемешивают в течение 15 мин при 20 С раствор 0,5 г (1,74 ммоль) 5-фтор-1-(3-фторбензил)-1 Ниндол-2-карбоновой кислоты, полученной на стадии 1.1, 0,33 г (1,74 ммоль) N-(3-диметиламинопропил)N'-этилкарбодиимида и 0,23 г (1,74 ммоль) N-1-гидроксибензотриазола в 10 мл диметилформамида. Затем вводят в реакционную среду 0,43 г (2,26 ммоль) соединения (Va), полученного на стадии 5.2. Реакционную среду затем перемешивают в течение 12 ч при 20 С, затем концентрируют при пониженном давлении, выливают на 100 мл воды и экстрагируют два раза по 100 мл этилацетата. Органические фазы объединяют, затем промывают два раза по 50 мл воды, затем один раз 50 мл насыщенного раствора хлорида натрия, сушат над сульфатом натрия, затем концентрируют при пониженном давлении. Полученный продукт очищают хроматографией на колонке с кремнеземом, элюируя смесью дихлорметана и метанола. Таким образом получают 0,3 г целевого продукта. Точка плавления: 172-174 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 1,85 (м, 4 Н); 3,61 (м, 4 Н); 3,78 (с, 3H); 5,91 (с, 2 Н); 6,91 (м, 2 Н); 7,07(тд, 1 Н); 7,15 (тд, 1 Н); 7,31 (кв, 1H); 7,4 (с, 1 Н); 7,58 (м, 3H); 8,08 (с, 1 Н); 10,3 (с, 1 Н). Пример 6 (соединение 16). N-[4-Метокси-6-(пирролидин-1-ил)пиридин-3-ил]-5-фтор-1-(3 фторбензил)-1 Н-индол-2-карбоксамид. 6.1. 4-Метокси-5-нитро-2-(пирролидин-1-ил)пиридин. Получают по методике, описанной на стадии 5.1, из 5,4 г (28,64 ммоль) 2-хлор-4-метокси-5 нитропиридина (WO 03/080610) и 4,53 г (63 ммоль) пирролидина. В этом случае полученный продукт очищают хроматографией на колонке с кремнеземом, элюируя смесью гептана и этилацетата. Таким образом выделяют 3 г целевого продукта. ЯМР 1 Н (ДМСО-d6),(м.д.): 2,17 (м, 4 Н); 3,65 (м, 4 Н); 4,07 (с, 3H); 5,79 (с, 1 Н); 8,98 (с, 1 Н). 6.2. 4-Метокси-2-(пирролидин-1-ил)-5-аминопиридин (соединение Vb). Получают способом, описанным на стадии 5.2, из 1,5 г (6,72 ммоль) 4-метокси-5-нитро-2(пирролидин-1-ил)пиридина, полученного на стадии 6.1, и 0,15 г 10%-ного палладия на угле. Получают 1,25 г целевого продукта. ЯМР 1 Н (ДМСО-d6),(м.д.): 1,81 (м, 4 Н); 3,29 (м, 4 Н); 3,71 (с, 3H); 4,52 (пик уширенный, 2 Н); 6,61N-[4-Метокси-6-(пирролидин-1-ил)пиридин-3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол-2 карбоксамид (соединение 16). Получают способом, описанным в примере 2, из 0,5 г (1,74 ммоль) 5-фтор-1-(3-фторбензил)-1 Ниндол-2-карбоновой кислоты (пример 1.1) и 0,424 г (2,09 ммоль) 4-метокси-2-(пирролидин-1-ил)-5 аминопиридина, полученного на стадии 6.1 (соединение Vb). Таким образом получают 0,59 г целевого продукта. Точка плавления: 196-198 С. ЯМР 1H (CDCl3),(м.д.): 2,01 (м, 4 Н); 3,49 (м, 4 Н); 3,91 (с, 3H); 5,86 (м, 3H); 6,79 (дд, 1 Н); 6,9 (м,2 Н); 7,02 (м, 2 Н); 7,23 (м, 2 Н); 7,34 (дд, 1 Н); 7,9 (м, 1 Н); 8,85 (с, 1 Н). Пример 7 (соединение 20). N-[6-(1-Оксипирролидин-1-ил)пиридин-3-ил]-5-фтор-1-(3-фторбензил)1 Н-индол-2-карбоксамид. Перемешивают в течение 24 ч при 20 С раствор 0,5 г (1,16 ммоль) N-[6-(пирролидин-1-ил)пиридин-9 017375 3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоксамида (соединение 2, полученное способом, описанным в примере 2) и 0,31 г (1,27 ммоль) метахлоропербензойной кислоты (70%) в 20 мл дихлорметана. В реакционную смесь вводят 100 мл дихлорметана, затем ее промывают последовательно 20 мл насыщенного раствора карбоната натрия, затем 3 раза 20 мл воды, сушат над сульфатом магния, затем концентрируют при пониженном давлении. Полученный продукт очищают хроматографией на колонке с кремнеземом, элюируя смесью дихлорметана и метанола. Таким образом получают 0,23 г целевого продукта. Точка плавления: 140-143 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 2,1 (м, 2 Н); 2,35 (м, 2 Н); 3,31 (м, 2 Н); 4,08 (м, 2 Н); 5,91 (с, 2 Н); 6,9 (м,2 Н); 7,04 (тд, 1 Н); 7,2 (тд, 1 Н); 7,32 (м, 1 Н); 7,51 (с, 1 Н); 7,62 (м, 2 Н); 8,41 (м, 2 Н); 8,8 (с, 1 Н); 10,91 (с,1 Н). Пример 8 (соединение 21). N-[2-(Пирролидин-1-ил)пиримидин-5-ил]-5-фтор-1-(3-фторбензил)-1 Ниндол-2-карбоксамид. Получают способом, описанным в примере 2, из 5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоновой кислоты (пример 1.1) и 2-(пирролидин-1-ил)-5-аминопиримидина (US 20060281772). Точка плавления: 220-222 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 1,97 (м, 4 Н); 3,51 (м, 4 Н); 5,89 (с, 2 Н); 6,91 (м, 2 Н); 7,05 (тд, 1 Н); 7,16(8,69 ммоль) пирролидина и 0,8 г (5,79 ммоль) карбоната калия. Затем реакционную смесь концентрируют при пониженном давлении, затем выливают в 100 мл воды. Получают путем фильтрования твердое вещество, которое сушат до получения 0,55 г целевого продукта в виде твердого вещества желтого цвета,которое используют как таковое в дальнейшем процессе синтеза. 9.2. 2-Метил-6-(пирролидин-1-ил)-3-аминопиридин (соединение Vd). Получают способом, описанным на стадии 5.2, из 0,55 г (2,65 ммоль) 2-метил-6-(пирролидин-1-ил)3-нитропиридина, полученного на стадии 9,1, и 0,1 г 10%-ного палладия на угле. Получают 0,34 г целевого продукта, который используют как таковой в дальнейшем процессе синтеза. ЯМР 1 Н (CDCl3),(м.д.): 1,89 (м, 4 Н); 2,23 (с, 3H); 2,98 (пик уширенный, 2 Н); 3,31 (м, 4 Н); 6,03 (д,1 Н); 6,82 (д, 1 Н). 9.3. N-[2-Метил-6-(пирролидин-1-ил)пиридин-3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоксамид (соединение 27). Получают способом, описанным в примере 5.3, из 0,4 г (1,39 ммоль) 5-фтор-1-(3-фторбензил)-1 Ниндол-2-карбоновой кислоты (пример 1.1) и 0,32 г (1,81 ммоль) 2-метил-6-(пирролидин-1-ил)-3 аминопиридина (соединение Vd), описанного на стадии 9.2. Таким образом получают 0,3 г целевого продукта. Точка плавления: 213-214 С. ЯМР 1 Н (ДМСО-d6)(м.д.): 1,92 (м, 4 Н); 2,21 (с, 3H); 3,49 (м, 4 Н); 5,88 (с, 2 Н); 6,29 (д, 1 Н); 6,87 (д,1 Н); 6,92 (д, 1 Н); 7,05 (тд, 1 Н); 7,15 (тд, 1 Н); 7,38 (м, 2 Н); 7,39 (с, 1 Н); 7,52 (д, 1 Н); 7,67 (м, 1 Н); 9,91 (с,1 Н). Пример 10N-[6-(3-Гидроксиазетидин-1-ил)пиридин-3-ил]-5-фтор-1-(3 фторбензил)-1 Н-индол-2-карбоксамид. 10.1. 2-(3-Гидроксиазетидин-1-ил)-5-нитропиридин. Получают способом, описанным на стадии 5.1, из 7 г (42,83 ммоль) 2-хлор-5-нитропиридина и 5,75 г (51,4 ммоль) 3-азетидинолхлоргидрата. В этом случае вводят также 13 г (128,45 ммоль) триэтиламина. Таким образом выделяют 8 г целевого продукта. Точка плавления: 175-176 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 3,85 (м, 2 Н); 4,36 (м, 2 Н); 4,62 (м, 1 Н); 5,88 (д, 1 Н); 6,4 (д, 1 Н); 8,18(дд, 1 Н); 8,92 (с, 1 Н). 10.2. 2-(3-Гидроксиазетидин-1-ил)-5-аминопиридин (соединение Ve). Получают способом, описанным на стадии 5.2, из 5,5 г (28,18 ммоль) 2-(3-гидроксиазетидин-1-ил)5-нитропиридина, полученного на стадии 10.1, и 0,5 г 10%-ного палладия на угле. Получают 4,5 г целевого продукта в виде кристаллов фиолетового цвета. Точка плавления: 148-150 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 3,49 (м, 2 Н); 3,98 (м, 2 Н); 4,48 (пик уширенный, 3H); 5,48 (пик уширенный, 1 Н); 6,22 (д, 1 Н); 6,91 (д, 1 Н); 7,58 (с, 1 Н). 10.3 N-[6-(3-Гидроксиазетидин-1-ил)пиридин-3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоксамид (соединение 24). Получают способом, описанным в примере 2, из 0,5 г (1,74 ммоль) 5-фтор-1-(3-фторбензил)-1 Ниндол-2-карбоновой кислоты (пример 1.1) и 0,35 г (2,09 ммоль) 2-(3-гидроксиазетидин-1-ил)-5- 10017375 аминопиридина, полученного на стадии 10.2 (соединение Ve). Точка плавления: 199-200 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 3,67 (м, 2 Н); 4,13 (м, 2 Н); 4,58 (м, 1 Н); 5,61 (м, 1 Н); 5,91 (с, 2 Н); 6,42(д, 1 Н); 6,91 (м, 2 Н); 7,05 (м, 1 Н); 7,18 (тд, 1 Н); 7,36 (м, 1 Н); 7,42 (с, 1 Н); 7,58 (м, 2 Н); 7,88 (дд, 1 Н); 8,39 (с, 1 Н); 10,35 (с, 1 Н). Пример 11 (соединение 53). N-[6-(Пирролидин-1-ил)-4-(трифторметил)пиридин-3-ил]-5-фтор-1-(3 фторбензил)-1 Н-индол-2-карбоксамид. 11.1. Метил-6-(пирролидин-1-ил)-4-(трифторметил)пиридин-3-карбоксамид. Нагревают до 100 С в течение 3 ч смесь 2,5 г (10,43 ммоль) метил-6-хлор-4 трифторметилникотината, 2,88 г (20,87 ммоль) карбоната калия и 2,61 мл (31,3 ммоль) пирролидина в 90 мл диметилформамида. Затем реакционную смесь концентрируют при пониженном давлении, затем обрабатывают 100 мл воды. Получают путем фильтрования осадок, который промывают 150 мл воды. После сушки при пониженном давлении получают 2,5 г целевого продукта. ЯМР 1H (CDCl3),(м.д.): 2,11 (м, 4 Н); 3,61 (пик уширенный, 4 Н); 3,93 (с, 3H); 6,69 (с, 1 Н); 8,89 (8,1 Н). 11.2. 6-(Пирролидин-1-ил)-4-(трифторметил)пиридин-3-карбоновая кислота. В течение 24 ч перемешивают при 20 С смесь 2,5 г (9,12 ммоль) метил-6-(пирролидин-1-ил)-4(трифторметил)пиридин-3-карбоксилата, полученного на стадии 11.1, и 0,76 г (13,67 ммоль) карбоната калия в 50 мл метанола и 2 мл воды. Затем смесь концентрируют при пониженном давлении. Затем вводят 100 мл воды и раствор промывают 100 мл дихлорметана, после чего подкисляют до рН 4 введением концентрированной соляной кислоты. Получают фильтрованием осадок, который промывают 50 мл воды. После сушки при пониженном давлении получают 2,2 г целевого продукта. ЯМР 1H (ДМСО-d6),(м.д.): 1,99 (с, 4 Н); 3,51 (пик уширенный, 4 Н); 6,71 (с, 1 Н); 8,72 (с, 1 Н). 11.3. 6-(Пирролидин-1-ил)-4-трифторметил-3-(трет-бутоксикарбониламино)пиридин. Нагревают в течение 5 ч до 90 С смесь 2,2 г (8,45 ммоль) 6-(пирролидин-1-ил)-4(трифторметил)пиридин-3-карбоновой кислоты, полученной на стадии 11.2, 2,37 мл (10,99 ммоль) дифенилфосфорилазида и 2,95 мл (21,14 ммоль) триэтиламина в 25 мл трет-бутанола. Затем реакционную смесь концентрируют при пониженном давлении, обрабатывают 50 мл воды, затем экстрагируют 3 раза по 50 мл дихлорметана. Органические фазы объединяют, затем промывают 50 мл воды, сушат над сульфатом натрия, затем концентрируют при пониженном давлении. Полученное масло очищают хроматографией на колонке с кремнеземом, элюируя смесью дихлорметана и метанола. Получают 1,05 г целевого продукта. ЯМР 1 Н (CDCl3),(м.д.): 1,53 (с, 9 Н); 2,09 (м, 4 Н); 3,51 (м, 4 Н); 6,2 (пик уширенный, 1 Н); 6,52 (с,1 Н); 8,39 (пик уширенный, 1 Н). 11.4. 6-(Пирролидин-1-ил)-4-трифторметил-3-аминопиридинхлоргидрат (амин Vg). Перемешивают в течение 5 ч с обратным холодильником раствор 1 г (3,02 ммоль) 6-(пирролидин-1 ил)-4-трифторметил-3-(трет-бутоксикарбониламино)пиридина, полученного на стадии 11.3, в 11 мл 4 N соляной кислоты в диоксане. Затем в охлажденную реакционную смесь вводят 200 мл простого этилового эфира. Получают фильтрованием 0,8 г осадка. Точка плавления: 207-209 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 2 (м, 4 Н); 3,52 (м, 4 Н); 7,1 (с, 1 Н); 7,49 (пик уширенный, 2 Н); 7,92 (с,1 Н). 11.5. N-[6-(Пирролидин-1-ил)-4-трифторметилпиридин-3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол-2 карбоксамид (соединение 53). Получают способом, описанным на стадии 5.3, из 0,3 г (1,04 ммоль) 5-фтор-1-(3-фторбензил)-1 Ниндол-2-карбоновой кислоты (пример 1.1), 0,363 г (1,36 ммоль) хлоргидрата 6-(пирролидин-1-ил)-4 трифторметил-3-аминопиридина, полученного на стадии 11.4, в присутствии 0,22 мл (1,57 ммоль) триэтиламина. Точка плавления: 170-171 С. ЯМР 1H (ДМСО-d6),(м.д.): 1,99 (м, 4 Н); 3,49 (м, 4 Н); 5,87 (с, 2 Н); 6,7 (с, 1 Н); 6,86-7,22 (м, 4 Н); 7,31 (м, 1 Н); 7,4 (с, 1 Н); 7,57 (дд, 1 Н); 7,65 (м, 1 Н); 8,11 (с, 1 Н). Пример 12 (соединение 28). N-[6-(3-Азабицикло[3.1.0]гекс-3-ил)пиридин-3-ил]-5-фтор-1-(3 фторбензил)-1 Н-индол-2-карбоксамид. 12.1. 2-(3-Азабицикло[3.1.0]гекс-3-ил)-5-нитропиридин. Перемешивают при комнатной температуре в течение 36 ч суспензию 0,2 г (1,26 ммоль) 2-хлор-5 нитропиридина, 0,166 г (1,39 ммоль) 3-азабицикло[3.1.0]гексанхлоргидрата (Bioorg.Med. Chera. Lett. 2005, 15(8) 2093) и 0,52 г (3,78 ммоль) карбоната калия в 5 мл диметилформамида. Затем смесь выливают в 5 мл воды, после чего экстрагируют 3 раза по 20 мл этилацетата. Органические фазы объединяют, сушат над сульфатом магния, затем концентрируют при пониженном давлении. Получают 0,22 г целевого продукта, который используют как таковой в последующем процессе синтеза.- 11017375 12.2. 2-(3-Азабицикло[3.1.0]гекс-3-ил)-5-аминопиридин (Vf). Соединение получают тем же способом, что и в примере 5.2, из 0,2 г (0,97 моль) 2-(3 азабицикло[3.1.0]гекс-3-ил)-5-нитропиридина, описанного на стадии 12.1, и из 0,5 г 10%-ного палладия на угле. Таким образом получают 0,1 г целевого продукта, который используют как таковой в последующем процессе синтеза. ЯМР 1 Н (ДМСО-d6)(м.д.): 0,2 (м, 1 Н); 0,66 (м, 1 Н); 1,6 (м, 2 Н); 3,11 (м, 2 Н); 3,51 (д, 2 Н); 4,32 (пик уширенный, 2 Н); 6,25 (д, 1 Н); 6,99 (д, 1 Н); 7,55 (с, 1 Н). 12.3.N-[6-(3-Азабицикло[3.1.0]гекс-3-ил)пиридин-3-ил]-5-фтор-1-(3-фторбензил)-1 Н-индол-2 карбоксамид (соединение 28). Получают способом, описанным в примере 2, из 0,15 г (0,52 ммоль) 5-фтор-1-(3-фторбензил)-1 Ниндол-2-карбоновой кислоты и 0,1 г (0,57 ммоль) 2-(3-азабицикло[3.1.0]гекс-3-ил)-5-аминопиридина,полученного на стадии 12.2. Таким образом получают 0,17 г целевого продукта. Точка плавления: 193-195 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 0,02 (м, 1 Н); 0,51 (м, 1 Н); 1,47 (м, 2 Н); 3,41 (д, 2 Н); 5,67 (с, 2 Н); 6,25 (д,1 Н); 6,69 (м, 2 Н); 6,83 (м, 1 Н); 6,93 (м, 1 Н); 7,1 (м, 1 Н); 7,15 (с, 1 Н); 7,31 (дд, 1 Н); 7,39 (м, 1 Н); 7,6 (м,1 Н); 8,11 (с, 1 Н); 10,03 (с, 1 Н). Пример 13 (соединение 29). N-[6-(3-Гидроксиазетидин-1-ил)пиридин-3-ил]-1-(3-фторбензил)-5 трифторметил-1 Н-индол-2-карбоксамид. Получают способом, описанным в примере 2, из 5-трифторметил-1-(3-фторбензил)-1 Н-индол-2 карбоновой кислоты (WO 2006/072736) и 2-(3-гидроксиазетидин-1-ил)-5-аминопиридина, полученного на стадии 10.2 примера 10 (соединение Ve). Точка плавления: 194-196 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 3,68 (м, 2 Н); 4,17 (м, 2 Н); 4,59 (м, 1 Н); 5,6 (д, 1 Н); 5,93 (с, 2 Н); 6,42 (д,1 Н); 6,91 (м, 2 Н); 7,09 (тд, 1 Н); 7,32 (м, 1 Н);7,55 (м, 2 Н); 7,79 (д, 1 Н); 7,88 (д, 1 Н); 8,2 (с, 1 Н); 8,38 (с,1 Н); 10,41 (с, 1 Н). Пример 14 (соединение 30). N-[6-(3-Гидроксиазетидин-1-ил)пиридин-3-ил]-1-(3-фторбензил)-6 трифторметил-1 Н-индол-2-карбоксамид. Получают способом, описанным в примере 2, из 6-трифторметил-1-(3-фторбензил)-1 Н-индол-2 карбоновой кислоты и 2-(3-гидроксиазетидин-1-ил)-5-аминопиридина, полученного на стадии 10.2 примера 10 (соединение Ve). Точка плавления: 253-255 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 3,67 (м, 2 Н); 4,17 (м, 2 Н); 4,59 (м, 1 Н); 5,6 (д, 1 Н); 6,0 (с, 2 Н); 6,42 (д,1 Н); 6,91 (м, 2 Н); 7,09 (тд, 1 Н); 7,36 (м, 1 Н); 7,47 (д, 1 Н); 7,51 (с, 1 Н); 7,85 (д, 1 Н); 7,99 (д, 1 Н); 8,06 (с,1 Н); 8,33 (с, 1 Н); 10,41 (с, 1 Н). Пример 15 (соединение 23). N-[6-(Пирролидин-1-ил)пиридин-3-ил]-1-[(пиридин-4-ил)метил]-5 фтор-1 Н-индол-2-карбоксамид. 15.1. 5-Фтор-1-[(пиридин-4-ил)метил]-1 Н-индол-2-карбоновая кислота. Нагревают в течение 2 ч с обратным холодильником раствор 2,1 г (7,04 ммоль) этил-5-фтор-1[(пиридин-4-ил)метил]-1 Н-индол-2-карбоксилата (WO 2007/010144) и 1,18 г (21,12 ммоль) карбоната калия в 80 мл этанола и 2 мл воды. Затем реакционную смесь концентрируют при пониженном давлении. Вводят 100 мл воды и доводят рН раствора до 8 добавлением раствора концентрированной соляной кислоты. Путем фильтрования получают осадок, который промывают водой, затем сушат при пониженном давлении. Таким образом получают 1,5 г целевого продукта. Точка плавления: 282-283 С. 15.2. N-[6-(Хлорпиридин-3-ил)]-5-фтор-1-[(пиридин-4-ил)метил]-1 Н-индол-2-карбоксамид. Соединение получают способом, описанным в примере 2, из 0,7 г (2,59 ммоль) 5-фтор-1-[(пиридин 4-ил)метил]-1 Н-индол-2-карбоновой кислоты, полученной на стадии 15.1, и 0,4 г (3,11 ммоль) 2-хлор-5 аминопиридина. Получают 0,41 г целевого продукта. Точка плавления: 244-246 С. 15.3. N-[6-(Пирролидин-1-ил)пиридин-3-ил]-1-[(пиридин-4-ил)метил]-5-фтор-1 Н-индол-2-карбоксамид (соединение 23). Соединение получают способом, описанным в примере 3.2, из 0,4 г (1,05 ммоль) N-[6-хлорпиридин 3-ил]-5-фтор-1-[(пиридин-4-ил)метил]-1 Н-индол-2-карбоксамида, полученного на стадии 15.2, и 2,63 мл(тд, 1 Н); 7,43 (с, 1 Н); 7,57 (м, 2 Н); 7,81 (м, 1 Н); 8,31 (с, 1 Н); 8,48 (д, 2 Н); 10,22 (с, 1 Н). Пример 16 (соединение 14). N-[6-(Пирролидин-1-ил)пиридин-3-ил]-1-[(пиридин-4-ил)метил]-5 трифторметил-1 Н-индол-2-карбоксамид. 16.1. 5-Трифторметил-1-[(пиридин-4-ил)метил]-1 Н-индол-2-карбоновая кислота. Получают способом, описанным в примере 15.1, из этил-5-трифторметил-1-[(пиридин-4-ил)метил]- 12017375 1 Н-индол-2-карбоксилата (WO 07/010144). Полученный продукт используют как таковой в дальнейшем процессе синтеза. 16.2. N-[6-Хлорпиридин-3-ил]-1-[(пиридин-4-ил)метил]-5-трифторметил-1 Н-индол-2-карбоксамид. Получают способом, описанным в примере 3.1, из 0,5 г (1,56 ммоль) 5-трифторметил-1-[(пиридин 4-ил)метил]-1 Н-индол-2-карбоновой кислоты, полученной на стадии 16.1, и 0,22 г (1,72 ммоль) 2-хлор-5 аминопиридина. Этим способом получают 0,5 г целевого продукта, который используют как таковой в дальнейшем процессе синтеза. 16.3. N-[6-(Пирролидин-1-ил)пиридин-3-ил]-1-[(пиридин-4-ил)метил]-5-трифторметил-1 Н-индол-2 карбоксамид (соединение 14). Соединение получают способом, описанным в примере 4, из 0,5 г (1,16 ммоль) N-[6-хлорпиридин-3 ил]-1-[(пиридин-4-ил)метил]-5-трифторметил-1 Н-индол-2-карбоксамида, полученного на стадии 16.2, и 2,91 мл (34,82 ммоль) пирролидина. Смесь помещают в трубку под давлением, которую нагревают в течение 50 мин до 170 С при 200 Вт в микроволновой печи. Затем смесь концентрируют при пониженном давлении, обрабатывают 100 мл этилацетата. Затем органическую фазу два раза промывают два раза по 30 мл воды, затем один раз насыщенным раствором натрия хлорида, сушат над сульфатом магния, затем концентрируют при пониженном давлении. Полученный продукт очищают хроматографией на колонке с кремнеземом, элюируя смесью дихлорметана и метанола. Таким образом получают 0,24 г целевого продукта. Точка плавления: 248-250 С. ЯМР 1 Н (ДМСО-d6),(м.д.): 1,95 (м, 4 Н); 3,41 (м, 4 Н); 5,98 (с, 2 Н); 6,45 (д, 1 Н); 7,03 (д, 2 Н); 7,59 (м,2 Н); 7,73 (д, 1 Н); 7,81 (д, 1 Н); 8,19 (с, 1 Н); 8,32 (с, 1 Н); 8,49 (д, 2 Н). Пример 17 (соединение 22). N-[6-(3-Азабицикло[3.2.0]гепт-3-ил)пиридин-3-ил]-5-фтор-1-(3 фторбензил)-1 Н-индол-2-карбоксамид. Нагревают в течение 90 мин в микроволновой печи при 150 С и 150 Вт смесь 0,2 г (0,5 ммоль) N-[6(хлорпиридин-3-ил)]-5-фтор-1-(3-фторбензил)-1 Н-индол-2-карбоксамида, полученного на стадии 3.1, и 0,0806 г (0,6 ммоль) 3-азабицикло[3.2.0]гептана (J. Med. Chem, 1967, 10(4), 621) и 0,0564 г (1,01 ммоль) карбоната калия в 0,5 мл N-метилпирролидинона. Реакционную смесь затем выливают в 20 мл воды. Смесь экстрагируют три раза по 30 мл этилацетата. Объединенные органические фазы промывают 30 мл воды, сушат над сульфатом магния, затем концентрируют при пониженном давлении. Полученное твердое вещество очищают хроматографией на колонке с кремнеземом, элюируя смесью н-гептана и этилацетата. Таким образом, получают 35 мг целевого продукта. Точка плавления: 163-165 С. ЯМР 1H (ДМСО-d6),(м.д.): 1.71 (м, 2 Н); 2,23 (м, 2 Н); 3,02 (м, 2 Н); 3,2 (м, 2 Н); 3,63 (д, 2 Н); 5,9 (с,2 Н); 6,62 (д, 1 Н); 6,91 (м, 2 Н); 7,05 (тд, 1 Н); 7,16 (тд, 1 Н); 7,33 (м, 1 Н); 7,4 (с, 1 Н); 7,54 (дд, 1 Н); 7,6(м, 1 Н); 7,88 (дд, 1 Н); 8,4 (с, 1 Н); 10,3 (с, 1 Н). Нижеследующая табл. 2 иллюстрирует химические структуры и физические свойства нескольких соединений общей формулы (I) по изобретению. На этой таблице в колонке "PF" указаны предельные точки плавления продуктов в градусах Цельсия (С); в колонке "соль/основание" "-" обозначает соединение в виде свободного основания, тогда как Данные ЯМР некоторых соединений из таблицы описаны ниже в качестве примеров. Соединение 25. ЯМР 1 Н (ДМСО-d6),(м.д.): 2,31 (м, 2 Н); 3,92 (т, 4 Н); 5,92 (с, 2 Н); 6,4 (д, 1 Н); 6,94 (м, 2 Н); 7,09- 15017375 Соединение 49. ЯМР 1 Н (ДМСО-d6),(м.д.): 1,32 (с, 9 Н); 2,3 (м, 2 Н); 3,91 (т, 4 Н); 5,85 (с, 2 Н); 6,38 (д, 1 Н); 6,92 (м,2 Н); 7,03 (тд, 1 Н); 7,35 (м, 4 Н); 7,66 (с, 1 Н); 7,85 (дд, 1 Н); 8,43 (с, 1 Н); 10,19 (с, 1 Н). Соединение 52. ЯМР 1 Н (ДМСО-d6),(м.д.): 2,31 (м, 2 Н); 3,91 (т, 4 Н); 6 (с, 2 Н); 6,39 (д, 1 Н); 6,91 (м, 2 Н); 7,07 (тд,1 Н); 7,32 (м, 1 Н); 7,45 (д, 1 Н); 7,49 (с, 1 Н); 7,82 (дд, 1 Н); 7,92 (д, 1 Н); 8,02 (с, 1 Н); 8,34 (с, 1 Н); 10,41 (с,1 Н). Соединения по изобретению подвергали фармакологическим испытаниям in vitro и in vivo, выявившим интерес, который они представляют в качестве веществ, обладающих терапевтической активностью. Соединения по изобретению обладают также свойством растворяться в воде, которое способствует их высокой активности in vivo. Тест на ингибирование тока, индуцируемого капсаицином, в DRG крысы. Первичная культура клеток узлов задних корешков (DRG) крысы. Нейроны DRG, естественно, выражают рецептор TRPV1. Первичные культуры DRG новорожденных крыс получают из крысят в возрасте одного дня. Коротко, после препарирования узлы трипсинируют и клетки механически диссоциируют путем бережного растирания. Клетки повторно суспендируют в базальной культурной среде Eagle, содержащей 10% эмбриональной бычьей сыворотки, 25 мМ KCl, 2 мМ глютамина, 100 мкм/мл гентамицина и 50 нг/мл NGF, затем накладывают на стеклянные пластинки,покрытые ламинином (0,25106 клеток на пластинку), которые затем помещают в 12-луночные планшеты Corning. Клетки инкубируют при 37 С во влажной атмосфере, содержащей 5% СО 2 и 95% воздуха. Через 48 ч после начала культивирования вводят цитозин -D-арабинозид (1 мкМ) для предупреждения развития не нейронных клеток. Пластинки переносят в экспериментальные камеры для исследованияpatch-clamp через 7-10 дней после начала культивирования. Электрофизиология. Измерительные камеры (объемом 800 мкл), содержащие клеточный препарат, помещают на предметный столик инвертированного микроскопа (Olympus IMT2), снабженный оптикой Hoffman (Modulation Contrast, New York), и наблюдают с увеличением 400 Х. Камеры подвергают непрерывной перфузии самотеком (2,5 мл/мин) при помощи распределителя растворов, имеющего 8 входов, единственный выход которого, состоящий из полиэтиленовой трубки (отверстие 500 мкм), находится в 3 мм от исследуемой клетки. Применяется конфигурация "целая клетка" технологии patch-clamp. Пипетки из боросиликатного стекла (прочность 5-10 MOhms) приближают к клетке при помощи пьезоэлектрического микроманипулятора 3D (Burleigh, PC1000). Общие токи (потенциал мембраны установлен на -60 мВ) регистрируются при помощи усилителя Axopatch ID (Axon Instruments, Foster city, Californie), соединенного с ПК с программным обеспечением Pclamp8 (Axon Instrument). Следы тока регистрируются на бумаге, одновременно оцифровываются (частота эталонирования от 15 до 25 Гц) и записываются на жесткий диск ПК. Использование микромолярного раствора капсаицина вызывает в клетках DRG (напряжение -70 мВ) входящий катионный ток. В целях минимизации десенсибилизации рецепторов между двумя применениями капсаицина соблюдают интервал 1 мин минимум. После контрольного периода (стабилизация ответа только одного капсаицина) тестируемые соединения используют отдельно в заданной концентрации (концентрации 10 и 0,1 нМ) в течение 4-5-минутного периода, в течение которого осуществляют несколько тестов капсицин + соединение (получение максимального ингибирования). Результаты выражают в проценте ингибирования ответа контрольного капсаицина. Процент ингибирования ответа капсаицина (1 мкМ) составляет от 20 до 100% для наиболее активных тестируемых соединений по изобретению в концентрации от 10 до 0,1 нМ (см. пример в табл. 3). Соединения по изобретению, таким образом, являются антагонистами in vitro рецепторов типа Тест на раздражение роговицы мыши. Раздражающее свойство капсаицина можно легко оценить на уровне роговицы, т.к. этот орган является одним из наиболее пронизанных волокнами С. В этом контексте после проведения предварительных экспериментов нанесение очень малого количества капсаицина (2 мкл в концентрации 160 мкМ) на поверхность роговицы животного вызывает несколько стереотипных поведенческих проявлений, связанных с раздражением, которое можно легко описать. Из них отмечают: животное моргает глазом, трет- 16017375 глаз, в который закапали вещество, гомолатеральной передней лапой, трет морду обеими передними лапами, чешет морду гомолатеральной задней лапой. Продолжительность этих проявлений не превышает 2 мин наблюдения, и животное возвращается к нормальной активности. Его внешний вид также является обычным. Мышь не забивается в угол со стоящей дыбом шерстью и не проявляет никаких видимых признаков болезненного состояния. Из этого можно сделать вывод, что продолжительность действия капсаицина в этих дозах меньше 2 мин. Методологический вывод. Принцип серии экспериментов заключается в определении того, могут ли соединения по изобретению оказывать влияние на поведенческий ответ, вызванный данным количеством капсаицина. Первоначально капсаицин разводят до концентрации 25 мМ в ДМСО и для конечного применения разводят в физиологическом растворе 10%-ным Tween 80. Оказывается, что по сравнению с контрольными испытаниями в этих условиях растворитель не оказывает никакого действия. На практике тестируемый продукт, полученный в концентрации 25 мМ в ДМСО и для конечного применения разведенный в физиологическом растворе 10%-ным Tween 80 в более высокой концентрации 500 мкМ, наносят локально на поверхность роговицы в объеме 2 мкл за 10 мин до применения капсаицина. Животному закапывают в глаз 2 мкл раствора капсаицина 160 мкМ, полученного, как указано выше. Во время наблюдений в течение 2 мин после закапывания подсчитывают, сколько раз каждое животное трет глаз после закапывания передней гомолатеральной лапой. В данной группе процент защиты вычисляют следующим образом: Р=100-среднее число царапаний в группе, обработанной соединением/среднее число царапаний в группе, обработанной растворителем)100) Вычисляют средний процент защиты для каждой группы животных (n=число животных, тестируемых с применением соединения по изобретению). В этой модели процент защиты наиболее активными соединениями, используемыми в концентрации 500 мкМ, составляет от 20 до 100% (см. пример в табл. 4). Таблица 4 Результаты этих испытаний показывают, что наиболее активные соединения по изобретению блокируют действие, вызванное стимуляцией рецепторов TRPV1. Таким образом, соединения по изобретению можно использовать для получения лекарственных средств, в частности, для получения лекарственного средства, предназначенного для предупреждения или лечения патологий, в которых участвуют рецепторы типа TRPV1. Таким образом, в соответствии с другим аспектом настоящего изобретения его объектом являются лекарственные средства, содержащие соединение формулы (I) или фармацевтически приемлемую соль,или гидрат, или сольват этого соединения. Эти лекарственные средства находят свое применение в терапии, в частности, для предупреждения и/или лечения боли и воспаления, боли хронической, невропатической (травматической, диабетической,метаболической, инфекционной, токсической, вызванной противораковым лечением или ятрогенным воздействием) (остео-)артритной, ревматической, фибромиалгий, болей в спине, болей, связанных с раком, лицевой невралгии, головных болей, мигрени, зубной боли, ожога, солнечного удара, укусов животных или насекомых, постгерпетической невралгии, мышечной боли, сдавления нервов (центральной и/или периферической системы), травм спинного и/или головного мозга, ишемии (спинного и/или головного мозга), нейродегенерации, нарушения кровообращения (спинного и/или головного мозга), боли после инсульта. Соединения по изобретению могут применяться для получения лекарственного средства, предназначенного для предупреждения и/или лечения урологических заболеваний, таких как гиперактивность мочевого пузыря, гиперрефлекторный мочевой пузырь, нестабильность мочевого пузыря, недержание,императивное мочеиспускание, недержание мочи, цистит, почечные колики, повышенная чувствительность в области таза и боль в области таза. Соединения по изобретению можно применять для получения лекарственного средства, предназначенного для предупреждения и/или лечения гинекологических заболеваний, таких как вульводиния, боли, связанные с сальпингитом, дисменореей. Можно также использовать эти продукты для получения лекарственного средства, предназначенного для предупреждения и/или лечения желудочно-кишечных расстройств, таких как гастроэзофагиальный рефлюкс, язва желудка, язва двенадцатиперстной кишки, функциональная диспепсия, колит, ИБС,болезнь Крона, панкреатит, эзофагит, печеночные колики. Соединения по изобретению можно также применять для получения лекарственного средства,предназначенного для лечения диабета. Также продукты по настоящему изобретению могут быть пригодны для предупреждения и/или лечения дыхательных расстройств, таких как астма, кашель, COPD, бронхостеноз и воспалительные забо- 17017375 левания. Эти продукты можно также применять для предупреждения и/или лечения псориаза, пруриго,раздражения кожи, глаз или слизистых оболочек, герпеса, опоясывающего лишая. Соединения по изобретению могут также применяться для получения лекарственного средства,предназначенного для лечения депрессии. В соответствии с другим аспектом настоящее изобретение относится к фармацевтическим композициям, содержащим в качестве активного вещества соединение по изобретению. Эти фармацевтические композиции содержат эффективную дозу по меньшей мере одного соединения по изобретению или фармацевтически приемлемую соль, гидрат или сольват данного соединения, а также по меньшей мере один фармацевтически приемлемый эксципиент. Указанные эксципиенты выбирают из известных специалисту эксципиентов в зависимости от лекарственной формы и требуемого способа введения. В фармацевтических композициях по настоящему изобретению для перорального, сублингвального, подкожного, внутримышечного, внутривенного, местного, локального, через трахею, через нос, чрескожного или ректального введения активное вещество указанной выше формулы (I), или его возможные соль, сольват или гидрат, может вводиться в форме для разового введения в смеси с традиционными фармацевтическими эксципиентами животным и людям для профилактики или лечения нарушений или заболеваний, указанных выше. Соответствующие разовые формы введения содержат формы для перорального введения, такие как таблетки, мягкие или твердые желатиновые капсулы, порошки, гранулы и питьевые растворы или суспензии, формы для сублингвального, перорального, через трахею, через глаза, через нос, путем ингаляции, формы для введения местного, чрескожного, подкожного, внутримышечного или внутривенного введения, формы для введения ректального и имплантаты. Для местного применения можно использовать соединения по изобретению в составе кремов, гелей, мазей или лосьонов. В качестве примера разовая форма введения соединения по изобретению в форме таблетки может содержать следующие компоненты: Указанные разовые формы имеют такую дозировку, чтобы обеспечить суточную дозу от 0,001 до 30 мг активного вещества на 1 кг веса тела в соответствии с галеновой формой. Возможны частные случаи, когда требуются большие или меньшие дозы, такие дозы не выходят за рамки изобретения. Обычно на практике дозу для каждого пациента определяет врач в зависимости от способа введения, веса и ответа данного пациента. В соответствии с другим аспектом настоящее изобретение относится к способу лечения вышеуказанных патологий, включающему в себя введение пациенту эффективной дозы соединения по изобретению или одной из его фармацевтически приемлемых солей, или гидратов, или сольватов. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) в которой X1 обозначает атом водорода или галогена либо C1-C6-алкил; Х 2 обозначает атом водорода или галогена, C1-C6-алкил, C1-C6-фторалкил или -S(О)2-C1-C6-алкил; Х 3 и Х 4 обозначают независимо друг от друга атом водорода или галогена, C1-C6-алкил, C1-C6 фторалкил, C1-C6-алкоксил, NR1R2 или C1-С 6-тиоалкил;Z1, Z2, Z3 и Z4 обозначают независимо друг от друга атом азота или группу C(R6), при этом по меньшей мере один из Z1, Z2, Z3 и Z4 соответствует атому азота и по меньшей мере один из Z1, Z2, Z3 и Z4 соответствует группе C(R6);Y обозначает фенил или пиридинил, необязательно замещенный одной или несколькими группами,выбранными из атома галогена, C1-С 6-алкила или NR1R2;Z обозначает циклический амин, выбранный из азетидина, пирролидина, пиперидина, морфолина,- 18017375 азабицикло[3.1.0]гексана и азабицикло[3.2.0]гептана; при этом атом азота циклического амина Z может быть в форме N-оксида; атомы углерода циклического амина Z необязательно замещены группой R12;R1 и R2 обозначают независимо друг от друга атом водорода или C1-C6-алкил или R1 и R2 образуют вместе с атомом азота, с которым они связаны, пирролидинил;R12 обозначает группу NR1R2, NR3COOR5 или гидроксил; в виде основания или кислотно-аддитивной соли, а также в виде гидрата или сольвата. 2. Способ получения соединения формулы (I) по п.1, отличающийся тем, что осуществляют взаимодействие соединения общей формулы (IV) в которой X1, Х 2, Х 3, Х 4, Y и n такие, как определено в общей формуле (I) по п.1, и В обозначает гидроксильную группу с амином общей формулы (V) в которой Z1, Z2, Z3, Z4 и Z такие, как определено в общей формуле (I) по п.1, в присутствии связующего вещества и основания в растворителе. 3. Способ получения соединения формулы (I) по п.1, отличающийся тем, что осуществляют взаимодействие соединения общей формулы (VI) в которой X1, Х 2, Х 3, Х 4, Z1, Z2, Z3, Z4, Y и n такие, как определено в общей формуле (I) по п.1, и W обозначает атом галогена, с амином формулы ZH, в которой Z обозначает циклический амин, такой как определен в общей формуле (I) по п.1. 4. Амин общей формулы (V), выбранный из в виде основания или соли присоединения с кислотой. 5. Фармацевтическая композиция, отличающаяся тем, что она содержит соединение формулы (I) по п.1 или фармацевтически приемлемую соль, гидрат или сольват этого соединения, а также по меньшей мере один фармацевтически приемлемый эксципиент.
МПК / Метки
МПК: C07D 403/12, A61P 29/00, C07D 401/12, C07D 401/04, C07D 401/14, A61P 13/00, A61P 3/10, A61P 15/00, A61P 25/24, A61P 17/00, A61P 11/00, C07D 213/74, A61P 25/04, A61K 31/4439
Метки: применение, n-(аминогетероарил)-1н-индол-2-карбоксамидов, производные, терапии, получение
Код ссылки
<a href="https://eas.patents.su/20-17375-proizvodnye-n-aminogeteroaril-1n-indol-2-karboksamidov-ih-poluchenie-i-ih-primenenie-v-terapii.html" rel="bookmark" title="База патентов Евразийского Союза">Производные n-(аминогетероарил)-1н-индол-2-карбоксамидов, их получение и их применение в терапии</a>
Производные n-(аминогетероарил)-1h-индол-2-карбоксамидов, их получение и их применение в терапии
Номер патента: 16829
Опубликовано: 30.07.2012
Авторы: Маланда Андре, Дюбуа Лоран, Эванно Янник
МПК: A61P 13/00, A61P 11/00, A61K 31/435...
Метки: n-(аминогетероарил)-1h-индол-2-карбоксамидов, применение, получение, терапии, производные
Формула / Реферат:
1. Соединение, отвечающее формуле (I)в которой X1 означает атом водорода или галогена или C1-С6-фторалкил;Х2 означает атом водорода или галогена или C1-С6-фторалкил;Х3 и Х4 означают, независимо друг от друга, атом водорода или галогена или C1-С6-фторалкил;Z1, Z2, Z3, Z4 означают, независимо друг от друга, атом азота или группу С(R6), причем по меньшей мере один соответствует атому азота и по меньшей мере один соответствует группе C(R6);n равно...
Производные n-(1h-индолил)-1н-индол-2-карбоксамидов, их получение и их применение в терапии

Номер патента: 11714
Опубликовано: 28.04.2009
Авторы: Эвен Люк, Эванно Янник, Дюбуа Лоран
МПК: A61P 29/00, C07D 209/42, A61K 31/404...
Метки: терапии, получение, применение, n-(1h-индолил)-1н-индол-2-карбоксамидов, производные
Формула / Реферат:
1. Соединение формулы (I) в которой X1, X2, X3, Х4, Z1, Z2, Z3, Z4 и Z5 обозначают независимо друг от друга атом водорода или галогена или C1-С6-алкильную, С3-С7-циклоалкильную, C1-С6-фторалкильную, C1-С6-алкокси, C1-С6-фторалкокси, циано, C(O)NR1R2, нитро, NR1R2, C1-С6-тиоалкильную, -S(О)-C1-С6-алкильную, -S(О)2-C1-С6-алкильную, SO2NR1R2, NR3COR4, NR3SO2R5 или C6-С10-арильную группу; X5 обозначает атом водорода или галогена или...
Производные n-(гетероарил)-1-гетероарилалкил-1н-индол-2-карбоксамидов, их получение и их применение в терапии

Номер патента: 13748
Опубликовано: 30.06.2010
Авторы: Дюбуа Лоран, Эванно Янник, Маланда Андре
МПК: A61K 31/4178, A61K 31/427, A61K 31/4184...
Метки: терапии, n-(гетероарил)-1-гетероарилалкил-1н-индол-2-карбоксамидов, применение, производные, получение
Формула / Реферат:
1. Соединение, отвечающее формуле (I)в которой X1, X2, Х3, Х4 означают, независимо друг от друга, Н, F, CF3, N(CH3)2или tBu;W означает группу, выбранную из бензимидазолила, индолила, хинолила, тетрагидрохинолила, бензотиазолила или бензоксазинила; причем эти группы могут быть замещены 1 или 2 раза группами, выбранными из (C1-С6)алкила, С3-циклоалкила и оксо, которые сами могут быть замещены С3-циклоалкилом;n равно 1, 2 или 3;Y означает группу,...
Производные n-(арилалкил)-1н-пирролопиридин-2-карбоксамидов, их получение и их применение в терапии

Номер патента: 13747
Опубликовано: 30.06.2010
Авторы: Эванно Янник, Дюбуа Лоран, Маланда Андре
МПК: A61P 25/00, A61K 31/437, C07D 471/04...
Метки: терапии, n-(арилалкил)-1н-пирролопиридин-2-карбоксамидов, производные, применение, получение
Формула / Реферат:
1. Соединение, отвечающее формуле (I)в которойn равно 0, 1 или 2;пирролопиридиновое ядро представляет собой пирроло[3,2-b]пиридиновую группу, пирроло[3,2-c]пиридиновую группу, пирроло[2,3-c]пиридиновую группу или пирроло[2,3-b]пиридиновую группу;причем пирролопиридиновое ядро возможно замещено в положении углерода 4, 5, 6 и/или 7 одним или несколькими заместителями X, одинаковыми или отличающимися друг от друга, выбираемыми среди атома галогена...
Производные n-(гетероарил)-1н-индол-2-карбоксамидов и их применение в качестве лигандов ваниллоидного рецептора trpv1

Номер патента: 13264
Опубликовано: 30.04.2010
Авторы: Эванно Янник, Маланда Андре, Дюбуа Лоран
МПК: A61P 29/00, C07D 209/42, A61K 31/33...
Метки: применение, рецептора, ваниллоидного, производные, качестве, n-(гетероарил)-1н-индол-2-карбоксамидов, лигандов, trpv1
Формула / Реферат:
1. Соединение, отвечающее формуле (1)в которой n равно 0, 1;X1, Х2, Х3, Х4, Z1, Z2, Z3, Z4и Z5 обозначают, независимо один от другого, атом водорода или галогена или группу C1-С6-алкил, C1-С6-фторалкил или C1-С6-алкокси,W обозначает конденсированную бициклическую группу формулысвязанную с атомом азота через положения 1, 2, 3 или 4;А обозначает гетероцикл, состоящий из 5-7 звеньев, содержащий от одного до трех гетероатомов, выбранных из О, S или...
Предыдущий патент: Горловинный затвор для бутылки и способ укупоривания бутылок
Следующий патент: Производные тетрагидронафталин-1-карбоновой кислоты, ингибирующие мтр
Случайный патент: Способ получения гликозилированной белковой молекулы, содержащей структуру lewis x, и/или сиалил-lewis x, и/или lacdinac