Производные тетрагидронафталин-1-карбоновой кислоты, ингибирующие мтр
Номер патента: 17376
Опубликовано: 28.12.2012
Авторы: Бакс Лео Якобус Йозеф, Бюсхер Гююске Фредерике, Тен Холтэ Петер, Мерпул Ливен







Формула / Реферат
1. Соединение формулы (I)

его фармацевтически приемлемые кислотно-аддитивные соли, его N-оксиды и его стереохимически изомерные формы, в которых А представляет собой -СН2- или -(C=O)-;
X представляет собой

n представляет собой целое число 2 или 3;
R5 представляет собой водород или C1-4алкил;
R6 представляет собой водород;
R1 представляет собой NR7R8 или OR9, в котором каждый R7 и R8 представляет собой независимо выбранный из водорода или С1-8алкила, замещенного C1-4алкилоксикарбонилом; и
R9 представляет собой C1-8алкил или С3-8алкенил;
R2a, R3a, R2b, R3b, R2c и R3c представляют собой водород;
R4 представляет собой фенил; фенил, замещенный C1-4алкилокси; фенил, замещенный фенилом, который замещен полигалогенС1-4алкилом; или пиридинил, замещенный гидрокси или С1-4алкилокси.
2. Соединение по п.1, в котором А представляет собой -(C=O)-.
3. Соединение по п.1, в котором R1 представляет собой NR7R8.
4. Соединение по п.1, в котором R1 представляет собой OR9.
5. Фармацевтическая композиция, включающая фармацевтически приемлемый носитель и терапевтически активное количество соединения по любому из пп.1-4.
6. Способ получения фармацевтической композиции по п.5, в котором терапевтически активное количество соединения по любому из пп.1-4 непосредственно смешивают с фармацевтически приемлемым носителем.
7. Применение соединения по любому из пп.1-4 в качестве лекарственного средства, обладающего апоВ секрецией/МТР ингибирующей активностью.
8. Соединение формулы (II), в котором R1, R2a, R2b, R2c, R3a, R3b, R3c, R5 и R6 определены в п.1.

9. Соединение формулы (XVII), в котором заместители R2a, R2b, R2c, R3a, R3b, R3c, R4, R5, A и X определены в п.1.

10. Способ получения соединения формулы (I), в котором промежуточное соединение формулы (II) взаимодействует с промежуточным соединением формулы (III) в реакционно-инертном растворителе и необязательно в присутствии подходящего связывающего реагента и/или подходящего основания.

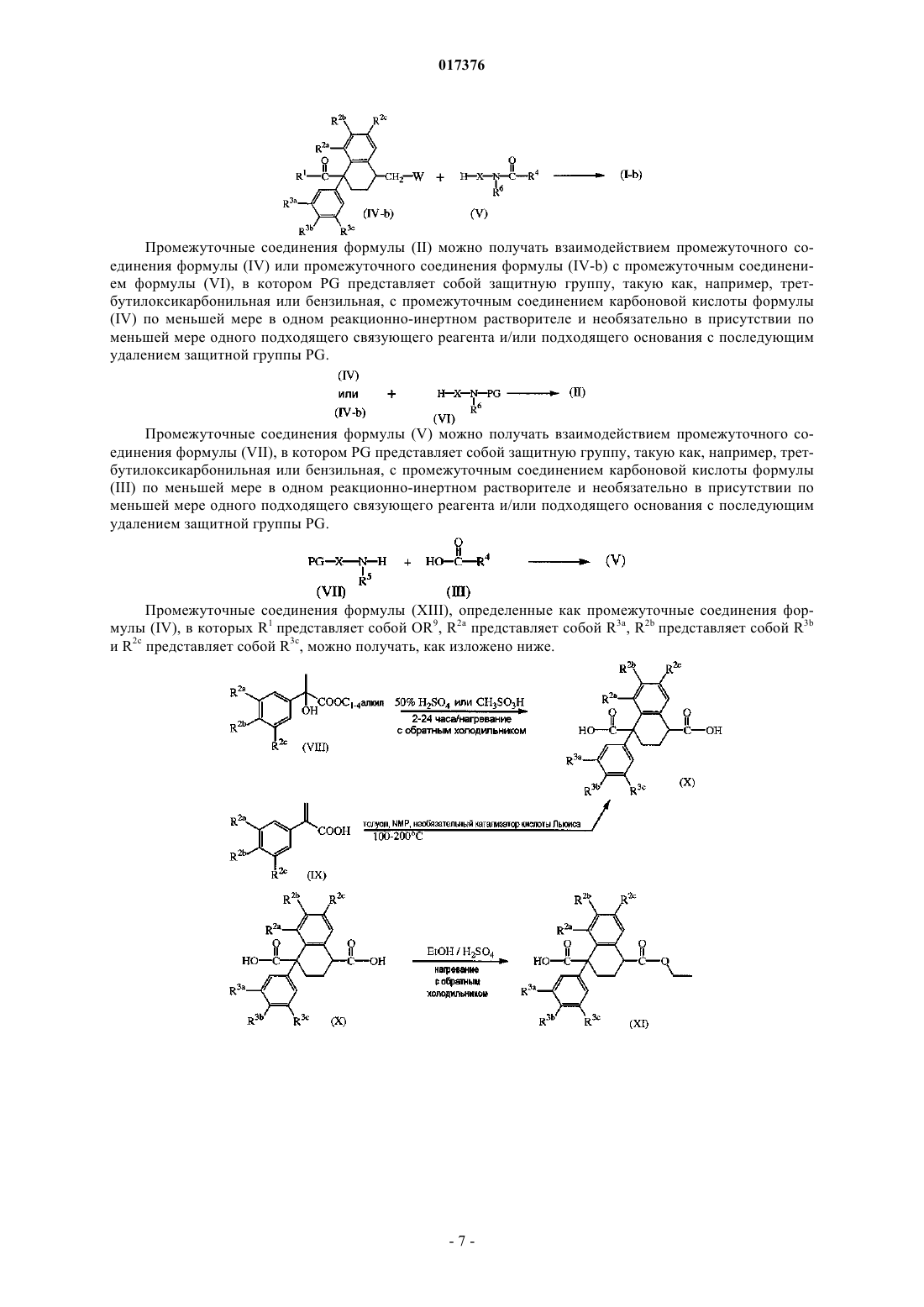
11. Способ получения соединения формулы (I-а), определенного как соединение формулы (I), в котором радикал А представляет собой -(C=O)-, в котором промежуточное соединение формулы (V) взаимодействует с промежуточным соединением формулы (IV) в реакционно-инертном растворителе и необязательно в присутствии подходящего связывающего реагента и/или подходящего основания.

Текст
Настоящее изобретение относится к новым производным тетрагидронафталин-1-карбоновой кислоты, обладающим апоВ секрецией/МТР ингибирующей активностью и сопутствующей гиполипидемической активностью. Изобретение, кроме того, относится к способам получения таких соединений, фармацевтических композиций, включающих указанные соединения, а также к применению указанных соединений в качестве лекарственных средств для лечения атеросклероза,панкреатита, ожирения, гипертриглицеридемии, гиперхолестеролемии, гиперлипидемии, диабета и диабета II типа. Мерпул Ливен, Бакс Лео Якобус Йозеф, Тен Холтэ Петер, Бюсхер Гююске Фредерике (BE) Медведев В.Н. (RU)(71)(73) Заявитель и патентовладелец: ЯНССЕН ФАРМАЦЕВТИКА НВ (BE) 017376 Настоящее изобретение относится к новым производным тетрагидронафталин-1-карбоновой кислоты, обладающим апоВ секрецией/МТР ингибирующей активностью и сопутствующей гиполипидемической активностью. Изобретение, кроме того, относится к способам получения таких соединений, фармацевтическим композициям, включающим указанные соединения, а также к применению указанных соединений в качестве лекарственных средств для лечения атеросклероза, панкреатита, ожирения, гипертриглицеридемии, гиперхолестеролемии, гиперлипидемии, диабета и диабета II типа. Ожирение представляет собой причину бесчисленных серьезных проблем со здоровьем у взрослых людей, таких как диабет и заболевание сердца. Кроме того, потеря веса становится навязчивой идеей среди увеличивающегося числа людей. Обычную взаимосвязь между гиперхолестеролемией, особенно которая связана с увеличивающейся концентрацией плазмы липопротеинов низкой плотности (в дальнейшем называемые ЛПНП), липопротеинов очень низкой плотности (в дальнейшем называемые ЛПОНП) и преждевременным атеросклерозом и/или кардиоваскулярным заболеванием сейчас широко исследуют. Однако ограниченное число лекарственных средств в настоящее время доступно для лечения гиперлипидемии. Лекарственные средства, используемые для лечения гиперлипидемии, включают смолысеквестранты желчных кислот, такие как холестирамин и колестипол, производные фибриновой кислоты, такие как безафибрат, клофибрат, фенофибрат, ципрофибрат и гемфиброзил, ингибиторы синтеза никотиновой кислоты и холестерина, такие как ингибиторы ГМГ-КоА энзим-редуктазы. Все еще остается необходимость в новых гиполипидемических агентах с повышенной эффективностью и/или действием через другие механизмы, чем вышеупомянутые лекарственные средства. Липопротеины плазмы представляют собой водорастворимые комплексы с высокой молекулярной массой, образованные из липидов (холестерин, триглицерид, фосфолипиды) и аполипопротеинов. Пять основных классов липопротеинов, которые отличаются соотношением липидов и типом аполипопротеина, все полученные из печени и/или кишечника, определяли согласно их плотности (как измерено при помощи ультрацентрифугирования). Они включают ЛПНП, ЛПОНП, липопротеины средней плотности(в дальнейшем называемые ЛПСП), липопротеины высокой плотности (в дальнейшем называемые ЛПВП) и хиломикроны. Десять основных аполипопротеинов плазмы человека идентифицировали. ЛПОНП, который выделяется печенью и содержит аполипопротеин В (в дальнейшем называемый АпоВ), подвергается расщеплению до ЛПНП, который транспортирует от 60 до 70% общего сывороточного холестерина. Апо-В также представляет собой главный белковый компонент ЛПНП. Увеличенный ЛПНП-холестерин в сыворотке вследствие чрезмерного синтеза или пониженного метаболизма приводит к атеросклерозу. В отличие от этого липопротеины высокой плотности (в дальнейшем называемые ЛПВП), которые содержат аполипопротеин A1, обладают защитным эффектом и обратно пропорционально коррелируются с риском коронарных сердечных заболеваний. Отношение ЛПВП/ЛПНП таким образом представляет собой удобный способ оценки атерогенного потенциала профиля липидов плазмы человека. Две изоформы аполипопротеинов (апо) В, апо В-48 и апо В-100 представляют собой важные протеины в метаболизме липопротеинов человека. Апо В-48 представляет собой приблизительно 48% размера апо В-100 на гелях додецилсульфат натрия-полиакриламид, синтезируется в кишечнике людей. Апо В-4 8 необходим для скопления хиломикронов и, следовательно, имеет обязательную роль в кишечной абсорбции жиров, входящих в рацион. Апо В-100, который продуцируется в печени человека, требуется для синтеза и секреции ЛПОНП. ЛПНП, которые содержат 2/3 холестерина в плазме человека, представляют собой метаболические продукты ЛПОНП. Апо В-100 в действительности единственный белковый компонент ЛПНП. Повышенные концентрации апо В-100 и холестерина ЛПНП в плазме распознают факторы риска для развития атеросклеротического коронарно-артериального заболевания. Большое число генетических и приобретенных заболеваний может приводить к гиперлипидемии. Они могут быть классифицированы на первичные и вторичные состояния. Наиболее стандартные случаи вторичных гиперлипидемий представляют собой сахарный диабет, злоупотребление алкоголем, лекарственными средствами, гипотиреоз, хроническую почечную недостаточность, нефротический синдром,холестаз и булимию. Первичные гиперлипидемии также были классифицированы на стандартную гиперхолестеролемию, наследственную комбинированную гиперлипидемию, наследственную гиперхолестеринемию, остаточную гиперлипидемию, синдром хиломикронемии и наследственную гипертриглицеридемию. Микросомальный белок переноса триглицерида (в дальнейшем называемый МТР), как известно, катализирует перенос триглицерида, холестерилового эфира и фосфолипидов, таких как фосфатидилхолин. Это указывает, что МТР требуется для синтеза апо В-содержащих липопротеинов, таких как хиломикроны и ЛПОНП, предшественники ЛПНП. Из этого следует, что ингибитор МТР ингибировал бы синтез ЛПОНП и ЛПНП, таким образом понижая уровни ЛПОНП, ЛПНП, холестерина и триглицерида у людей. Соединения, способные к ингибированию МТР, как полагают, являются пригодными для лечения заболеваний, таких как ожирение, гиперлипидемия, гиперхолестеринемия, гипертриглицеридемия, диабет II типа, атеросклероз и для сокращения постпрандиальных уровней триглицеридов плазмы сыворот-1 017376 ки. Настоящее изобретение основано на неожиданном обнаружении, что группа производных тетрагидронафталин-1-карбоновой кислоты обладает апоВ секрецией/МТР ингибирующей активностью. Эти соединения формулы (I) могут действовать систематически и/или в качестве селективных ингибиторов МТР, то есть способны селективно блокировать МТР на уровне стенки кишки у млекопитающих. Настоящее изобретение относится к группе новых соединений формулы (I) его фармацевтически приемлемые кислотно-аддитивные соли, его N-оксиды и его стереохимически изомерные формы, в которых A представляет собой -СН 2- или -(С=O)-;n представляет собой целое число 2 или 3;R5 представляет собой водород или C1-4 алкил;R1 представляет собой NR7R8 или OR9, в котором каждый R7 и R8 представляет собой независимо выбранный из водорода или C1-8 алкила, замещенного C1-4 алкилоксикарбонила; иR4 представляет собой фенил; фенил, замещенный C1-4 алкилокси; фенил, замещенный фенилом, который замещен полигалогенС 1-4 алкилом; или пиридинил, замещенный гидрокси или C1-4 алкилокси. Как используют в вышеуказанных определениях: галоген характерен для фтора, хлора, брома и йода;C1-4 алкил определяет прямые и разветвленные цепи, насыщенные углеводородными радикалами,имеющими от 1 до 4 атомов углерода, такие как, например, метил, этил, пропил, бутил, 1-метилэтил, 2 метилпропил и подобное; С 1-6 алкил, как обозначают, включает C1-4 алкил и его высшие гомологи, имеющие 5 или 6 атомов углерода, такие как, например, 2-метилбутил, пентил, гексил и подобное; С 1-8 алкил, как обозначают, включает С 1-6 алкил и его высшие гомологи, имеющие 7-8 атомов углерода, такие как, например, гептил, этилгексил, октил и подобное; полигалогенС 1-4 алкил определяют как полигалогензамещенный С 1-4 алкил, в частности С 1-4 алкил(как упомянуто выше), замещенный 1-4 атомами галогена, такой как фторметил, дифторметил, трифторметил, трифторэтил и подобное; С 3-8 циклоалкил характерен для циклопропила, циклобутила, циклопентила, циклогексила, циклогептила и циклооктила; С 3-8 циклоалкенил характерен для циклопропенила, циклобутенила, циклопентенила, циклогексенила, циклогептенила и циклооктенила;C3-8 алкенил определяет прямые и разветвленные цепи углеводородных радикалов, содержащие одну двойную связь и имеющие от 3 до 8 атомов углерода, такие как, например, 2-пропенил, 3-бутенил, 2 бутенил, 2-пентенил, 3-пентенил, 3-метил-2-бутенил, 3-гексенил, 2-гексенил, 2-пентенил, 2-октенил и подобное;C3-8 алкинил определяет прямые и разветвленные цепи углеводородных радикалов, содержащие одну тройную связь и имеющие от 3 до 8 атомов углерода, такие как, например, 2-пропинил, 3-бутинил, 2 бутинил, 2-пентенил, 3-пентенил, 3-метил-2-бутинил, 3-гексинил, 2-гексинил, 2-пентинил, 2-октинил и подобное. Фармацевтически приемлемые кислотно-аддитивные соли, как упомянуто выше, как обозначают,включают формы терапевтически активных нетоксичных кислотно-аддитивных солей, которые способ-2 017376 ны образовывать соединения формулы (I). Эти фармацевтически приемлемые кислотно-аддитивные соли могут стандартно быть получены обработкой формы основания такой соответствующей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты,например, хлористо-водородная или бромисто-водородная кислота, серная, азотная, фосфорная и подобные кислоты; или органические кислоты, такие как, например, уксусная, пропановая, гидроксиуксусная,молочная, пировиноградная, щавелевая (т.е. этандиоловая), малоновая, янтарная (т.е. бутандионовая кислота), малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламиновая, салициловая, п-аминосалициловая, памовая и подобные кислоты. Противоположно формы указанных солей могут быть превращены обработкой соответствующим основанием в форму свободного основания. Соединения формулы (I) могут существовать как в несольватированных, так и сольватированных формах. Термин сольват используют здесь для описания молекулярного комплекса, включающего соединение изобретения и одну или более молекул фармацевтически приемлемого растворителя, например этанол. Термин гидрат используют, когда указанный растворитель представляет собой воду. Форма N-оксида соединения согласно формуле (I), как обозначают, включает соединение формулы(I), в которой один или несколько атомов азота окислены до так называемых N-оксидов, в частности, этиN-оксиды, в которых один или более четвертичных азотов (например, пиперазинильный или пиперидинильный радикал) N-окислены. Такие N-оксиды могут быть легко получены специалистом в данной области техники без дополнительных навыков, и они представляют собой очевидную альтернативу для соединения согласно формуле (I), так как эти соединения представляют собой метаболиты, которые образованы при окислении в организме человека при поглощении. Общеизвестно, что окисление представляет собой обычно первую стадию, включенную в метаболизм лекарственных веществ (Textbook of Organic Medicinal and Pharmaceutical Chemistry, 1977, pages 70-75). Также общеизвестно, что форма метаболита соединения может также сама по себе быть введена человеку вместо соединения с почти такими же эффектами. Соединение формулы (I) может быть превращено в соответствующую форму N-оксида с помощью известных в данной области техники методик, превращением трехвалентного азота в его форму Nоксида. Указанную реакцию N-окисления могут обычно проводить взаимодействием соединения формулы (I) с соответствующим органическим или неорганическим пероксидом. Соответствующие неорганические пероксиды включают, например, пероксид водорода, пероксиды щелочных металлов или щелочно-земельных металлов, например пероксид натрия; пероксид калия; соответствующие органические пероксиды могут включать пероксикислоты, такие как, например, бензолкарбопероксидная кислота или галогензамещенная бензолкарбопероксидная кислота, например, 3-хлорбензолкарбопероксидная кислота, пероксоалкановые кислоты, например пероксиуксусная кислота, алкилгидропероксиды, например трет-бутилгидропероксид. Подходящие растворители представляют собой, например, воду, низшие алканолы, например этанол и подобные, углеводороды, например толуол, кетоны, например 2-бутанон, галогенированные углеводороды, например дихлорметан и их смеси растворителей. Термин стереохимически изомерные формы, как используют выше, определяет все возможные формы изомеров, которые соединения формулы (I) могут иметь. До тех пор, пока не определено и не показано иначе, химическая разработка соединений представляет собой все возможные стереохимически изомерные формы, указанные смеси, содержащие все диастереомеры и энантиомеры основных молекулярных структур. В частности, стереогенные центры могут иметь R- или S-конфигурацию; заместители на бивалентных циклических (частично) насыщенных радикалах могут иметь или цис-, или трансконфигурацию. Соединения, содержащие двойную связь, могут иметь Е- или Z-стереохимию при указанной двойной связи. Стереохимически изомерные формы соединений формулы (I), как очевидно предназначают,охватывают масштабы данного изобретения. Абсолютная стереохимическая конфигурация соединений формулы (I) и промежуточных соединений, используемых в их получении, может быть легко определена специалистом в данной области техники при использовании хорошо известных способов, таких как, например, дифракция рентгеновских лучей. Соединения формулы (I) имеют по меньшей мере два ассиметричных атома углерода, как иллюстрировано ниже, в которых асимметричные атомы углерода идентифицированы при помощи а. Вследствие присутствия по меньшей мере двух асимметричных атомов углерода обычно термин соединение формулы (I) охватывает смесь четырех стереоизомеров. Большинство соединений настоящего изобретения получали или с транс-конфигурацией, или с цис-конфигурацией Каждое из вышеобозначенных цис- или транс-соединений состоит из рацемической смеси двух энантиомеров, и жирные связи или прерывистые связи использовали, чтобы показать относительную стереохимическую конфигурацию. В случае цис- или транс-соединение разделяли на его два индивидуальных энантиомера, жирные и прерывистые связи заменяли клиновидными связями, чтобы указать, что соединение представляет собой единственный энантиомер. Если абсолютная стереохимия определенного хирального атома углерода в единственном энантиомере не была известна, его стереохимическую конфигурацию определяли как R или S, указывающую относительную стереохимию. Кроме того, некоторые соединения формулы (I) и некоторые из промежуточных соединений, используемые в их получении, могут показать полиморфизм. Следует понимать, что настоящее изобретение охватывает любые полиморфные формы, обладающие свойствами, пригодными для лечения состояний, отмеченных выще. Некоторые из соединений формулы (I) могут также существовать в их таутомерной форме. Такие формы, хотя не явно обозначенные в вышеупомянутой формуле, как предполагают, включены в объем настоящего изобретения. Например, когда ароматическое гетероциклическое кольцо замещено гидрокси,кето-форма может быть главным образом распространенным таутомером. В основе этой заявки выражение соединение согласно изобретению, как полагают, включает соединение согласно общей формуле (I) и его пролекарство или его изотопный индикатор.-4 017376 Также в объеме изобретения находятся так называемые пролекарства соединений формулы (I). Пролекарства представляют собой определенные производные фармацевтически активных соединений,которые могут обладать небольшой или никакой фармакологической активностью самостоятельно, которые могут быть превращены при введении в или на тело в соединения формулы (I), имеющие желательную фармацевтическую активность, например, гидролитическим расщеплением. Такие производные называются пролекарства. В основе этой заявки соединение согласно изобретению по существу, как предполагают, включает все изотопные комбинации его химических элементов. В основе этой заявки химический элемент, в особенности, когда упомянут относительно соединения согласно формуле (I), включает все изотопы и изотопные смеси этого элемента, или встречающиеся в природе, или искусственно полученные, или с распространенностью в природе, или в изотопно обогащенной форме. В частности, когда упоминают водород, понимают, что относится к 1 Н, 2 Н, 3 Н и их смесям; когда упоминают углерод, понимают, что относится к 11 С, 12 С, 13 С, 14 С и их смесям; когда упоминают азот, понимают, что относится к 13N, 14N, 15N и их смесям; когда упоминают кислород, понимают, что относится к 14O, 15O, 16O, 17O, 18O и их смесям; и когда упоминают фтор, понимают, что относится к 18F, 19F и их смесям. Соединение согласно изобретению, следовательно, по существу включает соединение с одним или более изотопами одного или более элементов и их смеси, включая радиоактивное соединение, также называемое радиоактивномеченным соединением, в котором один или более нерадиоактивных атомов были замещены одним из его радиоактивных изотопов. Под термином радиоактивномеченное соединение понимают любое соединение согласно формуле (I), его фармацевтически приемлемую кислотно- или основно-аддитивную соль, его N-оксидную форму или его четвертичную соль аммония, которые содержат по меньщей мере один радиоактивный атом. Например, соединение может быть мечено позитронили гамма-излучающими радиоактивными изотопами. Для радиолиганд-связывающих методик (анализ мембранных рецепторов), 3 Н-атом или 125I-атом представляет собой атом по выбору, который будет замещен. Для изображения обычно используемые позитронно-активные (ПЭТ) радиоактивные изотопы представляют собой 11C, 18F, 15O и 13N, все из которых произведены ускорителем и имеют периоды полураспада 20, 100, 2 и 10 мин, соответственно. Так как периоды полураспада этих радиоактивных изотопов настолько коротки, их возможно использовать только в учреждениях, которые имеют ускоритель на участке их производства, таким образом ограничивая их использование. Наиболее широко используемые из них представляют собой 18F, 99mTc, 201Tl и 123I. Обработка этих радиоактивных изотопов, их получение,выделение и введение в молекулу известны специалисту в данной области техники. В частности, радиоактивный атом выбирают из группы водорода, углерода, азота, серы, кислорода и галогена. Предпочтительно радиоактивный атом выбирают из группы водорода, углерода и галогена. В частности, радиоактивный изотоп выбирают из группы 3H, 11C, 18F, 122I, 123I, 125I, 131I, 75Br, 76Br,77Br и 82Br. Предпочтительно, радиоактивный изотоп выбирают из группы 3H, 11C и 18F. В варианте осуществления настоящее изобретение относится к тем соединениям формулы (I), в которых А представляет собой -(C=O)-; R1 представляет собой OR9, в котором R9 представляет собой С 1-6 алкил или С 3-8 алкенил; R2a, R3a, R2b, R3b, R2c и R3c представляют собой водород; R4 представляет собой фенил, фенил, замещенный С 1-4 алкилокси, пиридинил, замещенный гидрокси, или пиридинил, замещенный С 1-4 алкилокси; и X представляет собой радикал (a-1), в котором R5 представляет собой водород илиC1-4 алкил и R6 представляет собой водород. Интересные соединения формулы (I) представляют собой те соединения формулы (I), в которых применяют одно или более следующих ограничений:a) X представляет собой радикал (a-1), в котором n представляет собой 2; илиb) X представляет собой радикал (a-1), в котором n представляет собой 3; илиc) X представляет собой радикал (а-2); илиd) X представляет собой радикал (а-4); илиe) X представляет собой радикал (а-5); илиi) R1 представляет собой NR7R8, в котором каждый R7 и R8 независимо выбирают из водорода; С 1-8 алкила; С 1-8 алкила, замещенного С 1-4 алкилоксикарбонила; илиl) R4 представляет собой фенил; фенил, замещенный C1-4 алкилокси; или пиридинил, замещенный гидрокси или C1-4 алкилокси. Обычно соединения формулы (I) могут быть получены N-алкилированием промежуточного соединения формулы (II) с промежуточным соединением карбоновой кислоты формулы (III), по меньшей мере в одном реакционно-инертном растворителе и необязательно в присутствии по меньшей мере одного-5 017376 подходящего связующего реагента и/или подходящего основания, указанный способ, далее необязательно включающий превращение соединения формулы (I) в его аддитивную соль, и/или получение его стереохимически изомерных форм. Может быть удобно активировать карбоновую кислоту формулы (III) добавлением эффективного количества ускорителя реакции. Неограничивающие примеры таких ускорителей реакции включают карбонилдиимидазол,диимиды,такие какN,N'-дициклогексилкарбодиимид или 1-(3 диметиламинопропил)-3-этилкарбодиимид и их функциональные производные. Реакцию могут проводить в дальнейшем в присутствии эффективного количества соединения, такого как гидроксибензотриазол (НОВТ), бензотриазолилокситрис(диметиламино)фосфония гексафторфосфат, тетрапирролидинфосфония гексафторфосфат, бромтрипирролидинфосфония гексафторфосфат или его функциональные производные. Соединения формулы (I-а), определенные как соединения формулы (I), в которых радикал А представляет собой -(C=O)-, можно получать взаимодействием промежуточного соединения формулы (V) с промежуточным соединением формулы (IV), по меньшей мере в одном реакционно-инертном растворителе и необязательно в присутствии по меньшей мере одного подходящего связующего реагента и/или подходящего основания, указанный способ далее необязательно включает превращение соединения формулы (I) в его аддитивную соль и/или получение его стереохимических изомерных форм. Может быть удобно активировать карбоновую кислоту формулы (IV) добавлением эффективного количества ускорителя реакции. Неограничивающие примеры таких ускорителей реакции включают карбонилдиимидазол,диимиды,такие какN,N'-дициклогексилкарбодиимид или 1-(3 диметиламинопропил)-3-этилкарбодиимид и их функциональные производные. В случае, если используют хирально чистый реагент формулы (IV), быструю и без энантиомеризации реакцию промежуточного соединения формулы (IV) с указанным промежуточным соединением (V) могут проводить в дальнейшем в присутствии эффективного количества соединения, такого как гидроксибензотриазол (НОВТ),бензотриазолилокситрис(диметиламино)фосфония гексафторфосфат, тетрапирролидинфосфония гексафторфосфат, бромтрипирролидинфосфония гексафторфосфат или его функциональные производные, такие как раскрытые D. Hudson, J.Org.Chem. (1988), 53:617. Соединения формулы (I-b), определенные как соединения формулы (I), в которых радикал А представляет собой -CH2-, можно получать N-алкилированием промежуточного соединения формулы (V) с промежуточным соединением формулы (IV-b), в котором W представляет собой соответствующую уходящую группу, такую как, например, галоген, например, хлор, бром, йод, или в некоторых случаях W может также быть сульфонилоксигруппой, например метансульфонилокси, бензолсульфонилокси, трифторметансульфонилокси и подобные реактивные уходящие группы. Реакцию могут проводить в реакционно-инертном растворителе, таком как, например, ацетонитрил, 2-пентанол, изобутанол, диметилацетамид или ДМФА, и необязательно в присутствии подходящего основания, такого как, например, карбонат натрия, карбонат калия или триэтиламин. Перемешивание может увеличить скорость реакции. Реакцию могут удобно проводить при температуре реакционной смеси в интервале от комнатной температуры до температуры нагревания с обратным холодильником. Промежуточные соединения формулы (II) можно получать взаимодействием промежуточного соединения формулы (IV) или промежуточного соединения формулы (IV-b) с промежуточным соединением формулы (VI), в котором PG представляет собой защитную группу, такую как, например, третбутилоксикарбонильная или бензильная, с промежуточным соединением карбоновой кислоты формулы(IV) по меньшей мере в одном реакционно-инертном растворителе и необязательно в присутствии по меньшей мере одного подходящего связующего реагента и/или подходящего основания с последующим удалением защитной группы PG. Промежуточные соединения формулы (V) можно получать взаимодействием промежуточного соединения формулы (VII), в котором PG представляет собой защитную группу, такую как, например, третбутилоксикарбонильная или бензильная, с промежуточным соединением карбоновой кислоты формулы(III) по меньшей мере в одном реакционно-инертном растворителе и необязательно в присутствии по меньшей мере одного подходящего связующего реагента и/или подходящего основания с последующим удалением защитной группы PG. Промежуточные соединения формулы (XIII), определенные как промежуточные соединения формулы (IV), в которых R1 представляет собой OR9, R2a представляет собой R3a, R2b представляет собой R3b и R2c представляет собой R3c, можно получать, как изложено ниже. Промежуточные соединения формулы (XV) можно получать, как изложено ниже. Промежуточные соединения формулы (XV) представляют собой промежуточные соединения формулы (IV), в которой R1 представляет собой NR7R8. Промежуточные соединения формулы (IV-b) можно получать, как изложено ниже. Промежуточные соединения формулы (IV-b-1) определяют как промежуточные соединения формулы (IV-b), в которой R1 представляет собой NR7R8, и промежуточные соединения формулы (IV-b-2) определяют как промежуточные соединения формулы (IV-b), в которой R1 представляет собой OR9. Промежуточные соединения формулы (XVII), в которой заместители R2a, R2b, R2c, R3a, R3b, R3c, R4,R , А , А 2 и X представляют собой, как определено для соединений формулы (I), могут быть превращены в соединения формулы (I-c), определенные как соединения формулы (I), в которой R1 представляет собойNR7R8, известными в данной области техники способами N-ацилирования, используя H-NR7R8 в качестве реагента. 5 Соединения формулы (I), как полученные в вышеописанных процессах, могут синтезировать в форме рацемических смесей энантиомеров, которые могут быть отделены друг от друга следующими известными в данной области техники методиками разделения. Те соединения формулы (I), которые получают в рацемической форме, могут быть превращены в формы соответствующих диастереомерных солей взаимодействием с подходящей хиральной кислотой. Указанные формы диастереомерных солей впоследствии разделяют, например, селективной или фракционной кристаллизацией, и энантиомеры освобождают оттуда щелочью. Альтернативный способ разделения энантиомерных форм соединений формулы (I) включает жидкостную хроматографию, используя хиральную неподвижную фазу. Указанные стереохимически чистые изомерные формы могут также быть получены из соответствующих стереохимически чистых изомерных форм соответствующих исходных материалов, при условии, что реакция протекает стереоспецифически. Предпочтительно, если определенный стереоизомер желателен, указанное соединение будут синтезировать стереоспецифическими способами получения. Эти способы будут предпочтительно использовать энантиомерно чистые исходные вещества. Соединения формулы (I), формы N-оксида, фармацевтически приемлемые соли и их стереоизомерные формы обладают благоприятными апоВ секрецией и МТР-ингибирующей активностью и сопутствующей гиполипидемической активностью. Поэтому настоящие соединения формулы (I) полезны в ка-9 017376 честве лекарственных средств, особенно в способе лечения пациентов, страдающих от гиперлипидемии,ожирения, атеросклероза или диабета II типа. Впоследствии настоящие соединения могут использовать для получения лекарственных средств для лечения нарушений, вызванных избытком липопротеинов очень низкой плотности (ЛПОНП) или липопротеинов низкой плотности (ЛПНП), и особенно нарушений, вызванных холестерином, связанным с указанными ЛПОНП и ЛПНП. В частности, настоящие соединения могут использовать для изготовления лекарственных средств для лечения гиперлипидемии,ожирения, атеросклероза или диабета II типа. Принципиальный механизм действия соединений формулы (I), как неожиданно обнаружено, включает ингибирование активности МТР (микросомального белка-переносчика триглицеридов) у гепатоцитов и эпителиальных клеток кишечника, приводя к уменьшенному получению ЛПОНП и хиломикронов,соответственно. Это представляет собой новый и инновационный подход к гиперлипидемии и, как ожидают, понижает ЛПНП-холестерин и триглицериды через уменьшенное получение в печени ЛПОНП и получение в кишечнике хиломикронов. Большое число генетических и приобретенных заболеваний может приводить к гиперлипидемии. Они могут быть классифицированы на первичные и вторичные гиперлипидемические состояния. Большинство общих случаев вторичных гиперлипидемии представляет собой сахарный диабет, злоупотребление алкоголем, лекарственными средствами, гипотиреоз, хроническую почечную недостаточность,нефротический синдром, холестаз и булимию. Первичные гиперлипидемии представляют собой общую гиперхолестеролемию, наследственную комбинированную гиперлипидемию, наследственную гиперхолестеринемию, остаточную гиперлипидемию, синдром хиломикронемии, наследственную гипертриглицеридемию. Настоящие соединения могут также быть использованы для предотвращения заболевания или лечения пациентов, страдающих от ожирения или от атеросклероза, особенно коронарного атеросклероза и более общих нарушений, которые относятся к атеросклерозу, такие как ишемическая болезнь сердца, болезнь периферических сосудов, цереброваскулярная болезнь. Настоящие соединения могут вызывать регрессию атеросклероза и ингибировать клинические последствия атеросклероза, в особенности болезненность и смертность. Ввиду полезности соединений формулы (I) следует, что настоящее изобретение также обеспечивает способ лечения теплокровных животных, включая людей (обычно называемые здесь пациенты), страдающих от нарушений, вызванных избытком липопротеинов очень низкой плотности (ЛПОНП) или липопротеинов низкой плотности (ЛПНП), и особенно нарушений, вызванных холестерином, связанным со указанными ЛПОНП и ЛПНП. Следовательно, способ лечения обеспечивает освобождение пациентов,страдающих от состояний, таких как, например, гиперлипидемия, ожирение, атеросклероз или диабет II типа. Апо В-48, синтезированный кишечником, является необходимым для скопления хиломикронов и поэтому имеет обязательную роль в кишечной абсорбции диетических жиров. Настоящее изобретение обеспечивает соединения, которые действуют в качестве селективных МТР ингибиторов на уровне стенки кишечника. Кроме того, настоящее изобретение обеспечивает фармацевтические композиции, включающие по меньшей мере один фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы (I). Для получения фармацевтических композиций настоящего изобретения эффективное количество определенного соединения в форме основно- или кислотно-аддитивной соли в качестве активного ингредиента объединяют в однородную смесь по меньшей мере с одним фармацевтически приемлемым носителем, носитель могут взять из широкого разнообразия форм в зависимости от формы лекарственного средства, желательной для введения. Эти фармацевтические композиции находятся желательно в форме однократной дозировки, подходящей предпочтительно для перорального введения, ректального введения, подкожного введения или парентерального введения. Например, при получении композиций в виде пероральной дозированной формы можно применять любой из обычных жидких фармацевтических носителей, такой как, например, вода, гликоли, масла,спирты и подобное, в случае пероральных жидких лекарственных средств, таких как суспензии, сиропы,эликсиры и растворы; или твердых фармацевтических носителей, таких как крахмалы, сахара, каолин,смазывающие вещества, связующие вещества, дезинтегрирующие агенты и подобные, в случае порошков, пилюль, капсул и таблеток. Вследствие их легкого введения таблетки и капсулы представляют собой самую предпочтительную форму однократной пероральной дозировки, в случае которых очевидно применяют твердые фармацевтические носители. Для композиций парентерального введения фармацевтический носитель будет главным образом включать стерильную воду, хотя другие ингредиенты могут быть включены для улучшения растворимости активного ингредиента. Инъецируемые растворы можно получать, например, при использовании фармацевтического носителя, включающего солевой раствор, раствор глюкозы или смесь обоих. Вводимые суспензии можно также получать при использовании соответствующих жидких носителей, суспендирующих агентов и подобное. В композициях, подходящих для подкожного введения, фармацевтический носитель может необязательно включать улучшающий проникновение агент и/или подходящий смачивающий агент, необязательно объединенный с незначитель- 10017376 ными количествами подходящих добавок, которые не наносят существенного вреда коже. Указанные добавки могут быть отобраны для облегчения введения активного ингредиента к коже и/или быть полезными для получения желательных композиций. Эти локальные композиции можно вводить различными способами, например в качестве трансдермального пластыря, непосредственно на цель или мази. Аддитивные соли соединений формулы (I), вследствие их увеличенной водной растворимости через соответствующую основную форму, являются, очевидно, более подходящими при получении водных композиций. Особенно предпочтительно получать фармацевтические композиции изобретения в виде единичной дозированной формы для легкости введения и однородности дозированной формы. Единичная дозированная форма, как используется здесь, относится к физически дискретным единицам, подходящим в качестве единичных дозированных форм, каждая единица, содержащая предопределенное количество активного ингредиента, вычисленного для получения желательного терапевтического эффекта совместно с необходимым фармацевтическим носителем. Примеры таких единичных дозированных форм представляют собой таблетки (включая с насечкой или покрытые таблетки), капсулы, пилюли, порошковые пакеты, облатки, инъецируемые растворы или суспензии, чайные ложки, столовые ложки и подобные и их отдельные множества. Для перорального введения фармацевтические композиции настоящего изобретения могут быть в виде твердых дозированных форм, например таблетки (как формы для проглатывания, так и для жевания), капсулы или гелевые капсулы, полученные стандартными средствами с фармацевтически приемлемыми эксципиентами и носителями, такими как связующие агенты (например, желатинированный кукурузный крахмал, поливинилпирролидон, гидроксипропилметилцеллюлоза и подобные), наполнители(например, лактоза, микрокристаллическая целлюлоза, фосфат кальция и подобные), смазывающие вещества (например, стеарат магния, тальк, диоксид кремния и подобные), дезинтегрирующие агенты (например, картофельный крахмал, натрия крахмалгликолят и подобные), смачивающие агенты (например,натрия лаурилсульфат) и подобные. Такие таблетки могут также быть покрыты с помощью способов,известных в данной области техники. Жидкие препараты для перорального введения могут принимать форму, например, растворов, сиропов или суспензий, или их можно получать в качестве сухого продукта для смешивания с водой и/или другим подходящим жидким носителем перед использованием. Такие жидкие препараты могут получать стандартными средствами, необязательно с другими фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сорбитовый сироп, метилцеллюлоза, гидроксипропилметилцеллюлоза или гидрогенизированные пищевые жиры), эмульгирующие агенты (например, лецитин или гуммиарабик), неводные носители (например, миндальное масло, масляные сложные эфиры или этиловый спирт), подслащивающие вещества, ароматизаторы, дезодораторы и консерванты (например, метил или пропил п-гидроксибензоаты или сорбиновая кислота). Фармацевтически приемлемые подслащивающие агенты, пригодные в фармацевтических композициях изобретения, включают предпочтительно по меньшей мере один интенсивный подслащивающий агент, такой как аспартам, ацесульфам калия, цикламат натрия, алитам, сахаристое вещество на основе дигидрохалькона, монеллин, стевиозид сукралозы (4,1',6'-трихлор-4,1',6'-тридезоксигалактосахароза) или предпочтительно сахарин, натрия или кальция сахарин, и необязательно по меньшей мере один сыпучий подслащивающий агент, такой как сорбит, маннит, фруктоза, сахароза, мальтоза, изомальт, глюкоза, гидрогенизированный сироп глюкозы, ксилит, карамель или мед. Интенсивные подслащивающие агенты удобно используют в низких концентрациях. Например, в случае сахарина натрия, указанная концентрация может находиться в интервале от приблизительно 0,04 до 0,1% (мас./об.) окончательного состава. Сыпучий подслащивающий агент могут эффективно использовать в больших концентрациях в пределах от приблизительно 10 до приблизительно 35%, предпочтительно от приблизительно 10 до 15% (мас./об.). Фармацевтически приемлемые ароматизаторы, которые могут маскировать ингредиенты с горьким привкусом в составах низкой дозированной формы, представляют собой предпочтительно ароматизаторы фруктов, таких как вишневые, малиновые, черносмородиновые или клубничные ароматизаторы. Комбинация двух ароматизаторов может привести к очень хорошим результатам. В составах высокой дозированной формы могут требоваться более сильные фармацевтически приемлемые ароматизаторы,такие как Карамель Шоколад, Охлаждающая Мята, Фантазия и подобные. Каждый ароматизатор может присутствовать в окончательной композиции в концентрации в интервале от приблизительно 0,05 до 1%(мас./об.). Предпочтительно используют комбинации указанных сильных ароматизаторов. Предпочтительно используют ароматизатор, который не подвергается никакому изменению или потере вкуса и/или цвета в условиях состава. Соединения формулы (I) можно получать для парентерального введения инъекцией, удобно внутривенной, внутримышечной или подкожной инъекцией, например болюсной инъекцией или непрерывным внутривенным вливанием. Составы для инъекции могут быть представлены в виде единичной дозированной формы, например, в ампулах или контейнерах мультидозы, включая добавленный консервант. Они могут иметь такие формы в качестве суспензий, растворов или эмульсий в масляных или водных- 11017376 наполнителях и могут содержать составляющие агенты, такие как изотонизирующие, суспендирующие,стабилизирующие и/или рассеивающие агенты. Альтернативно, активный ингредиент перед использованием может присутствовать в порошковой форме для смешивания с подходящим наполнителем, например стерильная апирогенная вода. Соединения формулы (I) могут также быть получены в ректальных композициях, таких как суппозитории или удерживающие клизмы, например, содержащих стандартные суппозиторные основы, такие как какао-масло и/или другие глицериды. Соединения формулы (I) могут использовать совместно с другими фармацевтическими агентами, в частности, фармацевтические композиции настоящего изобретения могут, кроме того, включать по меньшей мере один дополнительный гиполипидемический агент, таким образом приводя к так называемой комбинированной гиполипидемической терапии. Указанный дополнительный гиполипидемический агент может быть, например, известным препаратом, стандартно используемым для лечения гиперлипидемии, таким как, например, смолысеквестранты желчных кислот, производные фибриновой кислоты или никотиновой кислоты, как предварительно упомянуто в уровне техники. Подходящие дополнительные гиполипидемические агенты также включают другие ингибиторы биосинтеза холестерина и ингибиторы абсорбции холестерина, особенно ингибиторы ГМГ-КоА редуктазы и ингибиторы ГМГ-КоА синтазы, ингибиторы ГМГ-КоА редуктазы экспрессии генов, ингибиторы СЕТР, ингибиторы АСАТ, ингибиторы сквален-синтетазы, антагонисты СВ-1, ингибиторы абсорбции холестерина, такие как эзетимиб и подобные. Любой ингибитор ГМГ-КоА редуктазы могут использовать в качестве второго соединения в аспекте комбинированной терапии настоящего изобретения. Термин ингибитор ГМГ-КоА редуктазы, как используют здесь, если иначе не заявлено, относится к соединению, которое ингибирует биотрансформацию гидроксиметилглутарилкоэнзима А в мевалоновую кислоту как катализируемую энзимом ГМГКоА редуктазы. Такие ингибиторы ГМГ-КоА редуктазы представляют собой, например, ловастатин,симвастатин, флувастатин, правастатин, ривастатин и аторвастатин. Любой ингибитор ГМГ-КоА синтазы могут использовать в качестве второго соединения в аспекте комбинированной терапии настоящего изобретения. Термин ингибитор ГМГ-КоА синтазы, как используют здесь, если иначе не заявлено, относится к соединению, которое ингибирует биосинтез гидроксиметилглутарилкоэнзима А из ацетилкоэнзима А и ацетоацетилкоэнзима А, катализируемый энзимом ГМГКоА синтазы. Любой ингибитор экспрессии генов ГМГ-КоА редуктазы могут использовать в качестве второго соединения в аспекте комбинированной терапии настоящего изобретения. Эти агенты могут быть ингибиторами транскрипции ГМГ-КоА редуктазы, которые блокируют транскрипцию ДНК, или ингибиторами трансляции, которые предотвращают трансляцию мРНК, кодирующей ГМГ-КоА редуктазу в протеин. Такие ингибиторы могут затронуть или транскрипцию, или трансляцию непосредственно, или могут быть биотрансформированы в соединения, имеющие вышеупомянутые признаки, одним или более энзимами в холестериновый биосинтетический каскад или могут привести к аккумулированию метаболита,имеющего вышеупомянутые активности. Любой ингибитор СЕТР могут использовать в качестве второго соединения в аспекте комбинированной терапии настоящего изобретения. Термин ингибитор СЕТР, как используют здесь, если иначе не заявлено, относится к соединению, которое ингибирует опосредованный транспорт различных холестериловых сложных эфиров и триглицеридов от ЛПВП до ЛПНП и ЛПОНП белком-переносчиком холестериловых эфиров (СЕТР). Любой ингибитор АСАТ могут использовать в качестве второго соединения в аспекте комбинированной терапии настоящего изобретения. Термин ингибитор АСАТ, как используют здесь, если иначе не заявлено, относится к соединению, которое ингибирует внутриклеточную этерификацию диетического холестерина энзимом ацил КоА:холестерин ацилтрансфераза. Любой ингибитор сквален-синтетазы могут использовать в качестве второго соединения в аспекте комбинированной терапии настоящего изобретения. Термин ингибитор сквален-синтетазы, как используют здесь, если иначе не заявлено, относится к соединению, которое ингибирует конденсацию двух молекул фарнесилпирофосфата для получения сквалена, катализируемую энзимом сквален-синтетазы. Специалисты в области лечения гиперлипидемии легко определят терапевтически эффективное количество соединения формулы (I) из результатов теста, представленных в дальнейшем. Обычно полагают, что терапевтически эффективная доза будет от приблизительно 0,001 до приблизительно 50 мг/кг веса тела, более предпочтительно от приблизительно 0,01 до приблизительно 5 мг/кг веса тела пациента,которого лечат. Может быть соответствующим вводить терапевтически эффективные дозы в форме двух или более поддоз через соответствующие интервалы в течение дня. Указанные поддозы могут быть получены в виде единичных дозированных форм, например, каждая содержащая от приблизительно 0,1 до приблизительно 350 мг, более детально от приблизительно 1 до приблизительно 200 мг активного ингредиента в виде единичной дозированной формы. Точная дозированная форма и частота введения зависят от определенного соединения используе- 12017376 мой формулы (I)/определенного состояния, которое лечат, тяжести состояния, которое лечат, возраста,веса и общего физического состояния определенного пациента, так же как других лекарственных средств(включая вышеупомянутые дополнительные гиполипидемические агенты), пациента, который может быть взят, как хорошо известно специалистам в данной области техники. Кроме того, указанное эффективное ежедневное количество может быть понижено или увеличено в зависимости от ответной реакции пациента, которого лечат, и/или в зависимости от оценки врача, предписывающего соединения настоящего изобретения. Эффективные ежедневные диапазоны количества,упомянутые выше, представляют собой поэтому только методические рекомендации. Экспериментальная часть В методиках, описанных в дальнейшем, использовали следующие сокращения: АЦН означает ацетонитрил; ДХМ означает дихлорметан; ДМФА означает N,N-диметилформамид; ТГФ означает тетрагидрофуран и ДИПЭ означает диизопропиловый эфир.Novabiochem 01-64-021; полимерподдерживаемое карбонатное основание [полистирилметил]триметил аммония бикарбонатная смола (5,8 ммоль/г) представляет собой смолу Novabiochem 01-64-041; полистиролкарбодимидная смола (1,90 ммоль/г) представляет собой смолу Novabiochem 01-64-024; полистиролN-метилморфолин HL (3,80 ммоль/г) смола представляет собой смолу Novabiochem 01-64-0211; полистиролбикарбонатная (5,8 ммоль/г) смола представляет собой смолу Novabiochem-01-064-0419. Смолы Novabiochem могут быть получены от Calbiochem-Novabiochem AG, Weidenmattweg 4, CH4448 Laufelfingen, Швейцария. А. Синтез промежуточных соединений. Пример А.1. промежуточного соединения (1).a) Получение Бензолкарбоновую кислоту (0,00012 моль, 1,2 эквивалента) растворяли в ДМФА (0,5 мл) и смешивали с N-циклогексилкарбодиимид N-метилполистирол HL смолой (1,90 ммоль/г) (0,10526 г, 0,0002 моль, 2 эквивалента). Добавляли 1-гидроксибензотриазол (НОВТ) (0,02027 г, 0,00015 моль, 1,5 эквив) в ДМФА (0,5 мл). Смесь перемешивали в течение 15 мин с последующим добавлением N-(третбутоксикарбонил)-1,2-этандиамина (0,0001 моль) в ДХМ (3 мл). После завершения реакции добавляли полимерподдерживаемое карбонатное основание [полистирилметил]триметил аммония бикарбонатную смолу (5,8 ммоль/г) (0,076 г, 0,00045 моль, 4,5 эквивалента) и смесь перемешивали в течение 3 ч. Окончательно смолы удаляли фильтрованием и промывали три раза смесью ДХМ/ДМФА (3/1 об/об, 1,0 мл) с последующим испарением растворителей при пониженном давлении, таким образом получая промежуточное соединение (1) (количественный выход; используемый на следующей стадии реакции, без дальнейшей очистки). промежуточного соединения (2).b) Получение Промежуточное соединение (1) (0,0001 моль) растворяли в смеси 6 н. HCl в изопропаноле (2 мл),перемешивали и нагревали в течение 5 ч при 65C. Реакционную смесь концентрировали при пониженном давлении, таким образом получая промежуточное соединение (2) в виде его аддитивной соли хлористо-водородной кислоты. Аналогичным способом от промежуточного соединения (3) до промежуточного соединения (8) получали в форме их солей хлористо-водородной кислоты. С этой целью на стадии реакции а) бензолкарбоновую кислоту заменяли на 2-метоксибензолкарбоновую кислоту или 4'-(трифторметил)-2 бифенилкарбоновую кислоту; и N-(трет-бутоксикарбонил)-1,2-этандиамин заменяли на N-(третбутоксикарбонил)-1,3-пропандиамин, N-метил-N-(трет-бутоксикарбонил)-1,2-этандиамин или N-метилN-(трет-бутоксикарбонил)-1,3-пропандиамин. Пример А.2. Получение промежуточного соединения (14). Метиловый эфир 2-гидрокси-2-фенилпропионовой кислоты (0,1 моль) добавляли к раствору серной кислоты (300 мл) в воде (250 мл) и реакционную смесь перемешивали при 100 C в течение 20 ч. Осадок отфильтровывали и растворяли в ДХМ (600 мл). Органический слой отделяли, высушивали, отфильтровывали и растворитель испаряли до объема 100 мл. Осадок отфильтровывали и высушивали, получая 9 г промежуточного соединения (14). промежуточного соединения (15) (транс). Пример А.3. Получение Смесь промежуточного соединения (14) (1,327 моль) в сухом этаноле (2360 мл) перемешивали и добавляли концентрированную серную кислоту (4 мл). Реакционную смесь нагревали с обратным холодильником в течение 22 ч в атмосфере азота и затем реакционную смесь охлаждали в течение ночи до комнатной температуры. Результирующий осадок отфильтровывали, промывали сухим этанолом и высушивали, получая 120 г промежуточного соединения (15) (т.пл.: 186-187C). Слои этанола собирали и испаряли и результирующий осадок растворяли в ДХМ (1450 мл), промывали водным раствором NaHCO3 (дважды 500 мл), высушивали и растворитель испаряли. Осадок перемешивали в ДИПЭ (680 мл) при температуре 50-55C, остаточный ДХМ отгоняли и концентрат оставляли отстаиваться в течение более 2 ч при комнатной температуре. Результирующие твердые частицы отфильтровывали, промывали ДИПЭ (120 мл) и пентаном и затем высушивали при 40C, получая другие 103,2 г промежуточного соединения (15) (т.пл.: 187-188C). промежуточного соединения (16) (цис). Получение Предыдущие слои ДИПЭ/пентан испаряли и осадок растворяли в сухом АЦН (200 мл), затем растворитель испаряли снова, получая 166,3 г промежуточного соединения (16) (т.пл.: 75C). Пример А.4. Получение промежуточного соединения (17). Промежуточное соединение (15) (0,03 моль) перемешивали в хлороформе (50 мл). Тионилхлорид(0,06 моль) добавляли и реакционную смесь перемешивали и нагревали с обратным холодильником в течение 4 ч, пока не прекращалось выделение газа. Реакционную смесь концентрировали испарением растворителя. Хлороформ (200 мл) добавляли и растворитель снова испаряли, получая осадок, который медленно добавляли к сухому этанолу (100 мл), который охлаждали на ледяной бане при 5C. Ледяную баню удаляли и реакционную смесь нагревали до комнатной температуры. Реакционную смесь перемешивали в течение 4 ч при комнатной температуре. Растворитель испаряли, получая промежуточное соединение (17) (т.пл.: 78-80C). Промежуточное соединение (18) получали аналогично, но начиная с промежуточного соединения Пример А.5. Получение промежуточного соединения (19). Смесь промежуточного соединения (17) (0,0567 моль) и п-толуолсульфоновой кислоты (1 г) перемешивали и нагревали с обратным холодильником в смеси муравьиной кислоты (500 мл) и концентрированной HCl (125 мл) в течение 3 ч. Реакционную смесь концентрировали испарением растворителя, осадок растворяли в ДХМ, промывали водным раствором NaHCO3 и высушивали. Растворитель испаряли и осадок очищали с помощью колоночной хроматографии на силикагеле (элюент:этилацетат/гексан 1/9),получая промежуточное соединение (19) (т.пл.: 115-118C). Промежуточное соединение (20) (т.пл.: 133-135C) получали аналогично, но начиная с промежуточного соединения (18). а) Получение промежуточного соединения (21). 2-метоксибензойную кислоту (0,028 моль) растворяли в ДХМ (150 мл). Тионилхлорид (8,2 мл) добавляли по каплям к этой смеси и смесь нагревали с обратным холодильником в течение 2 ч и 30 мин. Реакционную смесь охлаждали и растворитель испаряли. Затем добавляли ДХМ (150 мл), и растворитель снова испаряли. Неочищенное соединение растворяли в ДХМ (150 мл). Сначала добавляли 1(фенилметил)-3-пирролидинамин (0,028 моль) и затем добавляли насыщенный водный раствор NaHCO3(75 мл). Смесь взаимодействовала в течение 2 ч. Затем слои отделяли. Отделенный органический слой высушивали (MgSO4), отфильтровывали и растворитель испаряли. Осадок обрабатывали диизопропиловым эфиром и неочищенное соединение очищали с помощью колоночной хроматографии (элюент: от 100% CH2Cl2 до 3% CH3OH/CH2Cl2). Фракции продукта собирали и растворитель испаряли, получая 7,14 г промежуточного соединения (21).b) Получение промежуточного соединения (22). Смесь промежуточного соединения (21) (0,023 моль) в СН 3 ОН (150 мл) гидрогенировали в присутствии палладия на углероде 10% (1 г) в качестве катализатора. После захвата водорода (1 эквивалент) катализатор отфильтровывали и фильтрат испаряли. Осадок очищали с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Shandon Hyperprep C18 BDS (Base Deactivated Silica диоксид кремния, дезактивированный основанием) 8 мкм, 250 г, вн.д. 5 см). Применяли градиент с двумя или тремя подвижными фазами (фаза А: 0,25% раствор NH4HCO3 в воде; фаза В: СН 3 ОН (необязательная); фаза С: CH3CN). Фракции продукта собирали и растворитель испаряли, получая 2,56 г промежуточного соединения (22). Пример А.7.(8,2 мл; 0,112 моль) добавляли по каплям к этой смеси и смесь нагревали с обратным холодильником в течение 2 ч и 30 мин. Растворитель испаряли. Затем добавляли ДХМ (150 мл) и растворитель снова испаряли. Неочищенное соединение растворяли в ДХМ (150 мл). Сначала добавляли 1-(фенилметил)-3 пирролидинамин (0,028 моль) и затем добавляли насыщенный водный раствор NaHCO3 (75 мл). Смесь взаимодействовала в течение 2 ч. Затем слои отделяли. Отделенный органический слой высушивали (MgSO4), отфильтровывали и растворитель испаряли. Осадок очищали с помощью колоночной хроматографии (элюент: от 100% CH2Cl2 до СН 3 ОН/CH2Cl2 1/100). Фракции продукта собирали и растворитель испаряли, получая 7,97 г промежуточного соединения (23).- 15017376 Смесь промежуточного соединения (23) (0,026 моль) в CH3OH (150 мл) гидрогенировали в присутствии палладия на углероде 10% (1 г) в качестве катализатора. После захвата водорода (1 эквивалент) катализатор отфильтровывали и фильтрат испаряли. Осадок очищали с помощью высокоэффективной жидкостной хроматографии с обращенной фазой(Shandon Hyperprep C18 BDS (диоксид кремния, дезактивированный основанием) 8 мкм, 250 г, вн.д. 5 см). Применяли градиент с двумя или тремя подвижными фазами (фаза А: 0,25% раствор NH4HCO3 в воде; фаза В: CH3OH (необязательная); фаза С: CH3CN). Фракции продукта собирали и растворитель испаряли, получая 3,01 г промежуточного соединения (24). Пример А.8. а) Получение промежуточного соединения (25). Раствор 4-этилового эфира транс-1-фенил-1,2,3,4-тетрагидронафталин-1,4-дикарбоновой кислоты(0,15 моль) в NaHCO3 (0,15 М в 200 мл воды) перемешивали и добавляли Aliquat (0,15 моль) (смесь хлоридов три-С 8-10-алкилметил четвертичного аммония) и 3-бром-1-пропен (0,75 моль) в ДХМ (200 мл),затем реакционную смесь перемешивали в течение 4 дней при 20 C и органический слой отделяли. Водный слой экстрагировали ДХМ (300 мл) и объединенные органические слои высушивали (MgSO4). Растворитель испаряли и осадок перемешивали в гексане (500 мл), затем охлаждали до 0C. Результирующий осадок отфильтровывали, промывали гекданом и высушивали в течение ночи при 60C, получая 46 г промежуточного соединения (25).b) Получение промежуточного соединения (26). Концентрированную хлористо-водородную кислоту (28%) (100 мл) и 4-метилбензолсульфоновую кислоту (0,7 г) добавляли к раствору промежуточного соединения (25) (0,13 моль) в муравьиной кислоте(400 мл), затем реакционную смесь перемешивали и нагревали с обратным холодильником в течение 6 ч. Растворитель испаряли и осадок разделяли между ДХМ (300 мл) и насыщенным водным растворомNaHCO3 (200 мл). ДХМ-слой отделяли, высушивали (MgSO4) и растворитель испаряли. Осадок растирали в порошок в эфире с получением твердого вещества (I), и исходные слои концентрировали, затем кристаллизовали из смеси этилацетат/гексан с получением твердого вещества (II). Твердые вещества (I) и(II) объединяли и очищали с помощью колоночной хроматографии (элюент: ДХМ/СН 3 ОН 95/5). Фракции продукта собирали, растворитель испаряли и осадок растирали в порошок в гексане. Этот осадок затем растирали в порошок в эфире и отфильтровали с получением твердого вещества, производя 7 г промежуточного соединения (26) (т.пл.: 138-139C). Пример А.9. а) Получение промежуточного соединения (27). 2-метокси-3-пиридинкарбоновую кислоту (0,028 моль) растворяли в ДХМ (150 мл). Тионилхлорид(8 мл; 0,112 моль) добавляли по каплям к этой смеси и смесь нагревали с обратным холодильником в течение 2 ч и 30 мин. Растворитель испаряли. Затем добавляли ДХМ (150 мл) и растворитель снова испаряли. Неочищенное соединение растворяли в ДХМ (150 мл). Сначала добавляли N-метил-N-(фенилметил)-1,3-пропандиамин (0,028 моль) и затем добавляли насыщенный водный раствор NaHCO3 (75 мл). Смесь взаимодействовала в течение 2 ч. Затем слои отделяли. Отделенный органический слой высушивали (MgSO4), отфильтровывали и растворитель испаряли. Осадок очищали с помощью колоночной хроматографии (элюент: от 100% CH2Cl2 до CH3OH/CH2Cl2 1/100). Фракции продукта собирали и растворитель испаряли, получая 8,71 г промежуточного соединения (27).b) Получение промежуточного соединения (28). Смесь промежуточного соединения (27) (0,028 моль) в CH3OH (150 мл) гидрогенировали в присутствии палладия на углероде 10% (2 г) в качестве катализатора. После захвата водорода (1 эквивалент),катализатор отфильтровывали и фильтрат испаряли. Осадок кристаллизовали из 2-пропанола с добавлением HCl (6 н.) в 2-пропаноле. Осадок очищали с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Shandon Hyperprep С 18 BDS (диоксид кремния, дезактивированный основанием) 8 мкм, 250 г, вн.д. 5 см). Применяли градиент с двумя или тремя подвижными фазами (фаза А: 0,25% раствор NH4HCO3 в воде; фаза В: СН 3 ОН (необязательная); фаза С: CH3CN). Фракции продукта собирали и растворитель испаряли, получая 2,65 г промежуточного соединения (28).(1,4 мл) добавляли по каплям к этой смеси и смесь нагревали с обратным холодильником в течение 2 ч и 30 мин. Растворитель испаряли. Затем добавляли ДХМ (50 мл) и растворитель испаряли снова. Неочищенное соединение растворяли в ДХМ (50 мл). Сначала добавляли 4-(фенилметил)-2 морфолинметанамин (0,00485 моль) и затем добавляли насыщенный водный раствор NaHCO3 (25 мл). Смесь взаимодействовала в течение 2 ч. Затем слои отделяли. Отделенный органический слой высушивали (MgSO4), отфильтровывали и растворитель испаряли. Осадок очищали с помощью колоночной хроматографии (элюент: от 100% CH2Cl2 до СН 3 ОН/CH2Cl2 1/100). Фракции продукта собирали и растворитель испаряли, получая 1,6 г промежуточного соединения (29).b) Получение промежуточного соединения (30). Смесь промежуточного соединения (29) (0,004 9 моль) в СН 3 ОН (50 мл) гидрогенировали в присутствии палладия на углероде (0,4 г) в качестве катализатора. После захвата водорода (1 эквивалент) катализатор отфильтровывали и фильтрат испаряли. Осадок кристаллизовали из 2-пропанола с добавлением а) Получение промежуточного соединения (31). 2-метокси-3-пиридинкарбоновую кислоту (0,0269 моль) растворяли в ДХМ (150 мл). Тионилхлорид(8 мл) добавляли по каплям к этой смеси и смесь нагревали с обратным холодильником в течение 2 ч и 30 мин. Растворитель испаряли. Затем добавляли ДХМ (150 мл) и растворитель испаряли снова. Неочищенное соединение растворяли в ДХМ (150 мл). Сначала добавляли 4-аминогексагидро-1 Н-азепин-1 карбоновую кислоту, сложный этиловый эфир (0,0269 моль) и затем добавляли насыщенный водный раствор NaHCO3 (75 мл). Смесь взаимодействовала в течение 2 ч. Затем слои отделяли. Отделенный органический слой высушивали (MgSO4), отфильтровывали и растворитель испаряли. Осадок очищали с помощью колоночной хроматографии (элюент: от 100% CH2Cl2 до СН 3 ОН/CH2Cl2 1/100). Фракции продукта собирали и растворитель испаряли, получая 8,63 г промежуточного соединения (31).b) Получение Промежуточное соединение (31) (0,02 6 моль) растворяли в CH3OH (60 мл). Гидроксид калия (7 г) добавляли и реакционную смесь нагревали с обратным холодильником в течение 5 ч. ДХМ добавляли к реакционной смеси и органический слой промывали два раза водой. Отделенный органический слой высушивали (MgSO4), отфильтровывали и растворитель испаряли. Осадок очищали с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Shandon Hyperprep C18 BDS (диоксид кремния, дезактивированный основанием) 8 мкм, 250 г, вн.д. 5 см). Применяли градиент с двумя или тремя подвижными фазами (фаза А: 0,25% раствор NH4HCO3 в воде; фаза В: СН 3 ОН (необязательная); фаза С: CH3CN). Фракции продукта собирали и растворитель испаряли, получая 2,01 г промежуточного соединения (32). Пример А.12. промежуточного соединения (33). а) Получение 2-метоксибензойную кислоту (0,02 69 моль) растворяли в ДХМ (150 мл). Тионилхлорид (8 мл) добавляли по каплям к этой смеси и смесь нагревали с обратным холодильником в течение 2 ч и 30 мин. Растворитель испаряли. Затем добавляли ДХМ (150 мл) и растворитель испаряли снова. Неочищенное соединение растворяли в ДХМ (150 мл). Сначала добавляли 4-аминогексагидро-1 Н-азепин-1-карбоновую кислоту, сложный этиловый эфир (0,0269 моль) и затем добавляли насыщенный водный раствор NaHCO3(75 мл). Смесь взаимодействовала в течение 2 ч. Затем слои отделяли. Отделенный органический слой высушивали (MgSO4), отфильтровывали и растворитель испаряли. Осадок очищали с помощью колоночной хроматографии (элюент: от 100% CH2Cl2 до СН 3 ОН/CH2Cl2 1/100). Фракции продукта собирали и растворитель испаряли, получая 8,5 г промежуточного соединения (33).b) Получение Промежуточное соединение (33) (0,0262 моль) растворяли в СН 3 ОН (120 мл) и добавляли воду (1 мл). Затем добавляли гидроксид натрия (7 г) и реакционную смесь нагревали с обратным холодильником в течение 72 ч. Растворители испаряли и добавляли воду и ДХМ. Органический слой промывали водой. Отделенный органический слой высушивали (MgSO4), отфильтровывали и растворитель испаряли. Осадок очищали с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (ShandonHyperprep C18 BDS (диоксид кремния, дезактивированный основанием) 8 мкм, 250 г, вн.д. 5 см). Применяли градиент с двумя или тремя подвижными фазами (фаза А: 0,25% раствор NH4HCO3 в воде; фаза В: СН 3 ОН (необязательная); фаза С: CH3CN). Фракции продукта собирали и растворитель испаряли, получая 4,02 г промежуточного соединения (34). В. Получение конечных соединений. Пример В.1. Смесь промежуточного соединения (2) (0,0001 моль), полистирол-карбодиимидную (1,90 ммоль/г) смолу (0,0002 моль, 0,105 г), полистирол-N-метилморфолиновую HL (3,80 ммоль/г) смолу (0,0005 моль,0,132 г), раствор промежуточного соединения (19) (0,00015 моль) в ДХМ (1 мл) и 1 гидроксибензотриазол (НОВТ) (0,0015 моль, 0,020 г) в ТГФ (1 мл) встряхивали в течение ночи при комнатной температуре. Полистирол-бикарбонатную (5,8 ммоль/г) смолу (0,0005 моль, 0,086 г) добавляли в качестве акцептора для удаления избытка НОВТ. Реакционную смесь встряхивали в течение двух часов,отфильтровывали и фильтрат испаряли, получая соединение (1). Пример В.2. ДМФА (3 капли) добавляли к раствору промежуточного соединения (19) (0,025 моль) в ДХМ (100 мл) и затем добавляли тионилхлорид (0,1 моль). Реакционную смесь перемешивали и нагревали с обратным холодильником в течение 1 ч и затем растворитель испаряли. ДХМ добавляли и растворитель испаряли. Результирующий осадок растворяли в ДХМ (100 мл) и затем добавляли промежуточное соединение(7) (0,025 моль) с последующим водным раствором NaHCO3 (50 мл). Реакционную смесь перемешивали в течение 4 ч при комнатной температуре и слои отделяли. Органический слой высушивали и растворитель испаряли. Осадок разделяли на его энантиомеры с помощью высокоэффективной жидкостной хроматографии (Chiralpak AD) (элюент: гексан/этанол 80/20). Две фракции продукта собирали и растворитель испаряли. Каждый осадок растирали в порошок в ДИПЭ и затем желательные продукты собирали,получая 4,8 4 г соединения (25) и 4,72 г соединения (26). Пример В.3. Раствор промежуточного соединения (19) (0,025 моль) в ДХМ (100 мл) перемешивали и нагревали с обратным холодильником с тионилхлоридом (0,1 моль) в течение 1 ч и затем растворитель испаряли. Свежий ДХМ добавляли и избыток тионилхлорида удаляли испарением. Осадок растворяли в ДХМ (50 мл) и результирующий раствор добавляли к смеси промежуточного соединения (9) (0,025 моль) в ДХМ(50 мл). Водный раствор NaHCO3 (50 мл) добавляли и реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Слои отделяли и органический слой промывали разбавленной HCl, высушивали и растворитель испаряли. Полученный осадок разделяли на его энантиомеры очищением с помощью ВЭЖХ (Chiral phase AD) (элюент: гексан/этанол 60/40), получая 5,02 г соединения (27) и 5,05 г соединения (28). Пример В.4. Промежуточное соединение (26) (1 г; 0,0030 моль) растворяли в ДХМ (15 мл). Тионилхлорид (0,54 мл; 0,0075 моль) добавляли по каплям к этому раствору и добавляли несколько капель ДМФА. Реакцию нагревали с обратным холодильником в течение 1 ч. Растворитель испаряли. ДХМ (15 мл) добавляли к осадку и растворитель испаряли снова. Неочищенную смесь растворяли в ДХМ (15 мл) и добавляли сперва промежуточное соединение (9) (0,003 моль) и затем насыщенный водный раствор NaHCO3 (15 мл). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Слои отделяли. Отделенный органический слой высушивали (MgSO4), отфильтровывали и растворитель испаряли, получая 1,6 г соединения (34). Пример В.5. Соединение (38) (0,00297 моль) растворяли в ТГФ (20 мл). Реакцию барботировали азотом, и затем добавляли тетракис(трифенилфосфин)палладий (0,070 г). Смесь охлаждали до 0 C ледяной баней и затем добавляли боргидрид натрия (0,00297 моль). Охлаждение продолжали в течение 4 ч, и смесь взаимодействовала в течение ночи при комнатной температуре. Затем реакцию подавляли HCl (1 н.) и экстрагировали ДХМ. Отделенный органический слой высушивали (MgSO4), отфильтровывали и растворитель испаряли. Продукт очищали с помощью колоночной хроматографии (элюент: (CH2Cl2/CH3OH) от 99/1 до 90/10). Фракции продукта собирали и растворители испаряли в вакууме. Осадок повторно растворяли вCH2Cl2/CH3OH и обрабатывали активированным углем. Смесь отфильтровывали через декалит и раство- 18017376 ритель испаряли. Осадок очищали с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Shandon Hyperprep C18 BDS (диоксид кремния, дезактивированный основанием) 8 мкм,250 г, вн.д. 5 см). Применяли градиент с двумя или тремя подвижными фазами (фаза А: 0,25% растворNH4HCO3 в воде; фаза В: СН 3 ОН (необязательная); фаза С: CH3CN). Чистые фракции собирали и растворитель испаряли. Осадок повторно растворяли в ДХМ и раствор добавляли к диизопропиловому эфиру. Осадок отфильтровывали и твердое вещество высушивали, получая 0,016 г соединения (29). Пример В.6. а) Соединение (34) (0,0030 моль) растворяли в ТГФ (20 мл). Реакцию барботировали с азотом и затем добавляли тетракис(трифенилфосфин)палладий (0,00006 моль). Смесь охлаждали до 0C ледяной баней и затем добавляли боргидрид натрия (0,003 моль). Охлаждение продолжали в течение 4 ч и смесь взаимодействовала в течение ночи при комнатной температуре. Затем взаимодействие подавляли HCl (1 н.) и экстрагировали ДХМ. Отделенный органический слой высушивали (MgSO4), отфильтровывали и растворитель испаряли. Продукт очищали с помощью колоночной хроматографии (элюент:(ДХМ/МеОН) от 99/1 до 90/10). Фракции продукта собирали и растворители испаряли в вакууме. Осадок повторно растворяли в CH2Cl2/CH3OH и обрабатывали активированным углем. Смесь отфильтровывали через декалит и растворитель испаряли. Осадок очищали с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Shandon Hyperprep C18 BDS (диоксид кремния, дезактивированный основанием) 8 мкм, 250 г, вн.д. 5 см). Применяли градиент с двумя или тремя подвижными фазами(фаза А: 0,25% раствор NH4HCO3 в воде; фаза В: СН 3 ОН (необязательная); фаза С: CH3CN). Фракции продукта собирали и растворитель испаряли. Осадок растворяли в ДХМ и этот раствор добавляли к диизопропиловому эфиру. Осадок отфильтровывали и белое твердое вещество высушивали, получая транс 4-[3-(2-метоксибензоиламино)пропил]метилкарбамоил-1-фенил-1,2,3,4-тетрагидронафталин-1 карбоновую кислоту (промежуточное соединение (35.b) Промежуточное соединение (35) (0,000199 моль, 0,100 г) растворяли в сухом ДХМ (2 мл). Затем к смеси добавляли 1-гидрокси-1 Н-бензотриазол (1,2 экв., 0,032 г), N'-(этилкарбонимидоил)-N,N-диметил 1,3-пропандиамин, моногидрохлорид (1,2 экв, 0,046 г) и гидрохлорид метил 3-аминопропаноата (3 эквивалента, 0,083 г) и N-этил-N-(1-метилэтил)-2-пропанамин (10 экв., 0,329 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли дополнительный гидрохлорид метил 3-аминопропаноата (3 экв., 0,083 г) и смесь промывали 3 раза насыщенным водным раствором NaHCO3. Отделенный органический слой высушивали (MgSO4), отфильтровывали и растворитель испаряли. Осадок затем очищали с помощью колоночной хроматографии (100%CH2Cl2 до 2% CH3OH/CH2Cl2). Желательные фракции собирали и растворитель испаряли, получая соединение (31). Пример В.7. Промежуточное соединение (19) (0,00376 моль, 1,22 г) растворяли в сухом ДХМ (20 мл). Затем к смеси добавляли(0,00451 моль) и DIPEA (0,0376 моль, 7,4 мл). Реакционную смесь перемешивали при комнатной температуре комнаты в течение ночи. Дополнительное промежуточное соединение (24) (0,00451 моль) добавляли и смесь промывали 3 раза насыщенным водным раствором NaHCO3. Отделенный органический слой высушивали, фильтровали и растворитель испаряли. Осадок затем очищали с помощью колоночной флэщ-хроматографии (от 100% CH2Cl2 до CH3OH/CH2Cl2 1/20). Желательную фракцию продукта собирали и растворитель испаряли, получая соединение (45). Табл. F-1 перечисляет соединения, которые получали согласно одному из вышеупомянутых примеров. Стереохимическую конфигурацию для некоторых соединений определяли как R или S, указывая относительную стереохимию, когда абсолютная стереохимия неизвестна, хотя само соединение выделяли в качестве единственного стереоизомера, и поэтому оно представляет собой энантиомерно чистое. Для некоторых соединений была включена точка плавления (т.пл.). Идентификация соединений Общая методика A. Измерение ВЭЖХ проводили, используя систему Alliance HT 27 90 (Waters), включающую насос для нагнетания четырехкомпонентных смесей с дегазатором, автодозатор, колоночный термостат (установка при 40C, если иначе не обозначено), детектор на диодной матрице (DAD) и колонку, как определено в соответствующих способах ниже. Поток от колонки распределяли к масс-спектрометру. Детектор масс-спектрометра конфигурировали с источником электрораспылительной ионизации. Масс-спектры получали сканированием от 100 до 1000 через 1 с, используя время задержки 0,1 с. Напряжение на игле капилляра было 3 кВ, и исходную температуру поддерживали при 140C. Азот использовали в качестве распылительного газа. Регистрацию данных проводили с помощью системы сбора и обработки данныхWaters-Micromass MassLynx-Openlynx. Общая методика В. Измерение ЖХ проводили, используя систему Acquity UPLC (Waters), включающую двойной насос,устройство для проб, колоночный термостат (установка при 55C), детектор на диодной матрице (DAD) и колонку, как определено в соответствующих способах ниже. Поток от колонки распределяли к массспектрометру. Детектор масс-спектрометра конфигурировали с источником электрораспылительной ионизации. Масс-спектры получали сканированием от 100 до 1000 через 0,18 с, используя время задержки 0,02 с. Напряжение на игле капилляра было 3 кВ, и исходную температуру поддерживали при 140C. Азот использовали в качестве распылительного газа. Регистрацию данных проводили с помощью системы сбора и обработки данных Waters-Micromass MassLynx-Openlynx. Способ 1. В дополнение к общей методике А: ВЭЖХ с обращенной фазой проводили на колонке Xterra MS C18 (3,5 мкм, 4,6 100 мм) со скоростью потока 1,6 мл/мин. Три подвижные фазы (подвижная фаза А: 95% 25 мМ ацетата аммония и 5% ацетонитрила; подвижная фаза В: ацетонитрил; подвижная фаза С: метанол) использовали для проведе- 22017376 ния условия градиента от 100% А до 1% А, 49% В и 50% С через 6,5 мин, до 1% А и 99% В через 1 мин и поддерживания этих условий в течение 1 мин и переуравновешивания 100% А в течение 1,5 мин. Использовали инъекцию объемом 10 мкл. Напряжение конуса было 10 В для режима положительной ионизации и 20 В для режима отрицательной ионизации. Способ 2. В дополнение к общей методике В: UPLC с обращенной фазой (ультраэффективную жидкостную хроматографию) проводили на соединенном гибриде этилсилоксан/силикагель (ВЕН) колонка С 18 (1,7 мкм, 2,1 50 мм; Waters Acquity) со скоростью потока 0,8 мл/мин. Две подвижные фазы (подвижная фаза А: 0,1% муравьиная кислота в H2O/метаноле 95/5; подвижная фаза В: метанол), использовали для проведения условия градиента от 95% А и 5% В до 5% А и 95% В через 1,3 мин и поддерживания в течение 0,2 мин. Использовали инъекцию объемом 0,5 мкл. Напряжение конуса было 10 В для режима положительной ионизации и 20 В для режима отрицательной ионизации. Способ 3. В дополнение к общей методике А: ВЭЖХ с обращенной фазой проводили на Chromolith (4,625 мм) со скоростью потока 3 мл/мин. Три подвижные фазы (подвижная фаза А: 95% 25 мМ ацетата аммония и 5% ацетонитрила; подвижная фаза В: ацетонитрил; подвижная фаза С: метанол) использовали для проведения условия градиента от 96% А, 2% В и 2% С, до 49% В и 49% С через 0,9 мин, до 100% В через 0,3 мин и поддерживания в течение 0,2 мин. Использовали инъекцию объемом 2 мкл. Напряжение конуса было 10 В для режима положительной ионизации и 20 В для режима отрицательной ионизации. Таблица. Аналитические данные Когда соединение представляет собой смесь изомеров, которые дают различные пики в методе ЖХМС, только время удерживания главного компонента дано в таблице ЖХМС (Rt: время удерживания в минутах). Оптическое вращение Оптическое вращение измеряли, используя поляриметр. []D20 указывает оптическое вращение, измеренное со светом при длине волны D-линии натрия (589 нм) при температуре 20C. После фактического значения концентрации и растворителя раствора, которое использовали для измерения, упоминали оптическое вращение. С. Фармакологические примеры. С.1. Определение количества секреции апоВ.HepG2 клетки культивировали в 24-луночном планшете в MEM Rega 3, содержащем 10% фетальную телячью сыворотку. При 70% слияния среду изменяли и добавляли тестируемое соединение или носитель (ДМСО, 0,4% окончательная концентрация). После 24 ч инкубирования среду переносили в пробирки Eppendorf и очищали при помощи центрифугирования. Антитела овец, направленные против любого апоВ, добавляли к супернатанту и смесь хранили при 8C в течение 24 ч. Затем добавляли кроличьи антиовечьи антитела и иммунный комплекс осаждали в течение 24 ч при 8C. Иммунопреципитат гранулировали центрифугированием в течение 25 мин при 1320 g и промывали дважды буфером, содержащим 40 мМ Mops, 40 мМ NaH2PO4, 100 мМ NaF, 0,2 мМ DTT, 5 мМ ЭТДА, 5 мМ EGTA, 1% Тритон- 23017376 Х-100, 0,5% дезоксихолат натрия (DOC), 0,1% SDS, 0,2 мкМ лейпептин и 0,2 мкМ ПМСФ. Радиоактивность в грануле определяли количественно жидкостно-сцинтилляционным измерением активности. Значения IC50 обычно превращают в значения pIC50 (равно значению -log IC50) для простоты использования и суммируют в табл. С-1. Таблица С-1. Значения pIC50Zilversmit в Chemistry and Physics of Lipids, 38, 205-222 (1985). Для получения донора и акцептора везикул соответствующие липиды в хлороформе помещали в стеклянную пробирку и сушили в потоке N2. Буфер, содержащий 15 мМ Трис-HCl, рН 7,5, 1 мМ ЭДТА, 40 мМ NaCl, 0,02% NaN3 (аналитический буфер) добавляли к высушенному липиду. Смесь недолго перемешивали и липиды затем гидратировали в течение 20 мин на льду. Везикулы затем готовили при помощи ультразвуковой бани (Branson 2200) при комнатной температуре в течение максимум 15 мин. Бутилированный гидрокситолуол включали во все получения везикул при концентрации 0,1%. Испытательная смесь переноса липидов содержала везикулы донора (40 нмоль фосфатидилхолина, 7,5 моль.% кардиолипина и 0,25 моль.% глицерина три[1-14C]олеата), везикулы акцептора (240 нмоль фосфатидилхолина) и 5 мг БСА в полном объеме 675 мкл в 1,5 мл пробирке микроцентрифуги. Тестируемые соединения добавляли растворенными в ДМСО (0,13% окончательная концентрация). После 5 мин предварительного инкубирования при 37C взаимодействие начинали добавлением МТР в 100 мкл буфера для диализа. Взаимодействие прекращали добавлением 400 мкл целлюлозы DEAE-52, предуравновешенной в 15 мМ Трис-HCl, рН 7,5, 1 мМ ЭДТА, 0,02% NaN3(1:1, об/об). Смесь перемешивали в течение 4 мин и центрифугировали в течение 2 мин при максимальной скорости в центрифуге Eppendorf (4C) для гранулирования DEAE-52-связанных липосом донора. Аликвоту супернатанта, содержащую липосомы акцептора, подсчитывали и [14 С]-подсчеты использовали для вычисления процента переноса триглицерида от донорских к акцепторным везикулам. Таблица С-2. Значения pIC50 его фармацевтически приемлемые кислотно-аддитивные соли, его N-оксиды и его стереохимически изомерные формы, в которых А представляет собой -СН 2- или -(C=O)-;n представляет собой целое число 2 или 3;R5 представляет собой водород или C1-4 алкил;R1 представляет собой NR7R8 или OR9, в котором каждый R7 и R8 представляет собой независимо выбранный из водорода или С 1-8 алкила, замещенного C1-4 алкилоксикарбонилом; иR4 представляет собой фенил; фенил, замещенный C1-4 алкилокси; фенил, замещенный фенилом, который замещен полигалогенС 1-4 алкилом; или пиридинил, замещенный гидрокси или С 1-4 алкилокси. 2. Соединение по п.1, в котором А представляет собой -(C=O)-. 3. Соединение по п.1, в котором R1 представляет собой NR7R8. 4. Соединение по п.1, в котором R1 представляет собой OR9. 5. Фармацевтическая композиция, включающая фармацевтически приемлемый носитель и терапевтически активное количество соединения по любому из пп.1-4. 6. Способ получения фармацевтической композиции по п.5, в котором терапевтически активное количество соединения по любому из пп.1-4 непосредственно смешивают с фармацевтически приемлемым носителем. 7. Применение соединения по любому из пп.1-4 в качестве лекарственного средства, обладающего апоВ секрецией/МТР ингибирующей активностью. 8. Соединение формулы (II), в котором R1, R2a, R2b, R2c, R3a, R3b, R3c, R5 и R6 определены в п.1. 10. Способ получения соединения формулы (I), в котором промежуточное соединение формулы (II) взаимодействует с промежуточным соединением формулы (III) в реакционно-инертном растворителе и необязательно в присутствии подходящего связывающего реагента и/или подходящего основания. 11. Способ получения соединения формулы (I-а), определенного как соединение формулы (I), в котором радикал А представляет собой -(C=O)-, в котором промежуточное соединение формулы (V) взаимодействует с промежуточным соединением формулы (IV) в реакционно-инертном растворителе и необязательно в присутствии подходящего связывающего реагента и/или подходящего основания.
МПК / Метки
МПК: A61K 31/40, C07C 233/57, A61K 31/5377, C07C 235/52, A61K 31/216, C07C 235/54, C07D 207/14, C07C 233/78, C07C 231/02, C07C 231/16, C07D 223/12, A61K 31/55, C07C 235/60, C07D 265/30, A61K 31/4025
Метки: кислоты, мтр, ингибирующие, производные, тетрагидронафталин-1-карбоновой
Код ссылки
<a href="https://eas.patents.su/27-17376-proizvodnye-tetragidronaftalin-1-karbonovojj-kisloty-ingibiruyushhie-mtr.html" rel="bookmark" title="База патентов Евразийского Союза">Производные тетрагидронафталин-1-карбоновой кислоты, ингибирующие мтр</a>
Производные пиперидин- или пиперазинзамещенной тетрагидронафталин 1-карбоновой кислоты, ингибирующие мтр

Номер патента: 16311
Опубликовано: 30.04.2012
Авторы: Яроскова Либуше, Бакс Лео Якобус Йозеф, Бюсхер Гююске Фредерике, Вьейевуа Марсель, Мерпул Ливен, Бертело Дидье Жан-Клод, Линдерс Йоаннес Теодорус Мария
МПК: C07D 241/20, C07D 211/58, C07D 295/10...
Метки: ингибирующие, пиперазинзамещенной, мтр, пиперидин, 1-карбоновой, кислоты, производные, тетрагидронафталин
Формула / Реферат:
1. Соединение формулы (I)его фармацевтически приемлемые соли присоединения кислот, N-оксиды и стереохимически изомерные формы, гдеX представляет собой N или CH; когда X представляет собой CH, тогда A2 представляет собой -NR6-, где R6 представляет собой водород или C1-4алкил, или, когда X представляет собой N, тогда A2 отсутствует;A1 представляет собой -(C=O)- или -CH2- ;R1 представляет собой NR7R8, где каждый R7 и R8 независимо выбраны...
Производные фениламида 4-(бензил)пиперазин-1-карбоновой кислоты и родственные соединения в качестве модуляторов амида жирной кислоты гидролазы для лечения страхов, боли и других состояний

Номер патента: 12589
Опубликовано: 30.10.2009
Авторы: Сяо Вэй, Брайтенбухер Дж.Гай, Паттабираман Канака, Сейерстад Марк, Аподака Ричард
МПК: A61P 25/22, A61P 25/04, A61P 25/28...
Метки: гидролазы, амида, родственные, производные, соединения, боли, других, фениламида, качестве, 4-(бензил)пиперазин-1-карбоновой, страхов, кислоты, лечения, модуляторов, состояний, жирной
Формула / Реферат:
1. Соединение формулы (I) в которой Z означает -N- или >СН; R1 представляет собой -Н или -C1-4алкил; Ar1 означает 2-тиазолил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, каждый из которых является незамещенным или замещенным по атому углерода кольца одной или двумя Ra группами; где каждый остаток Ra независимо выбран из группы, включающей -C1-4алкил, -C2-4алкенил, -ОН, -OC1-4алкил, атом галогена, -CF3,...
Новые производные амида гетероциклической карбоновой кислоты

Номер патента: 11635
Опубликовано: 28.04.2009
Авторы: Хорват Чилла, Надь Йожеф, Саги Каталин, Дьертьян Иштван, Колок Шандор, Борза Иштван, Фаркаш Шандор, Галгоци Корнел
МПК: A61K 31/445, A61P 25/28, C07D 401/06...
Метки: карбоновой, новые, гетероциклической, кислоты, производные, амида
Формула / Реферат:
1. Производные амида гетероциклической карбоновой кислоты формулы (I) в которой значение X соответствует водороду или атому галогена, гидрокси, циано, С1-С4алкилсульфонамидо, факультативно замещенной атомом галогена либо атомами галогена, C1-C4алканоиламидо, факультативно замещенной атомом галогена либо атомами галогена, арилсульфонамидогруппе, Y соответствует -СН=группе либо атому -N=, Z соответствует атому водорода либо атому галогена,...
Сложноэфирные производные декагидроизохинолин-3-карбоновой кислоты в качестве анальгетиков

Номер патента: 9526
Опубликовано: 28.02.2008
Автор: Орнштейн Пол Лесли
МПК: A61P 25/00, A61K 31/4725, A61K 31/5377...
Метки: кислоты, качестве, сложноэфирные, производные, декагидроизохинолин-3-карбоновой, анальгетиков
Формула / Реферат:
1. Соединение формулы где R означает С1-С20алкил, или его фармацевтически приемлемые соли. 2. Соединение по п.1, где R означает C1-С10алкил. 3. Соединение по п.2, где R означает 2-этилбутил, изобутил, 3-метилбутил, децил или этил. 4. Соединение по п.3, где R означает 2-этилбутил. 5. Соединение по п.3, где R означает изобутил. 6. Соединение по п.3, где R означает 3-метилбутил. 7. Соединение по п.3, где R означает децил. 8. Соединение по п.3, где...
Производные октагидро-6, 10-диоксо-6н-пиридазино [1,2-а] [1,2] диазепин-1-карбоновой кислоты, способ их получения и их применение для получения терапевтически активных соединений

Номер патента: 3280
Опубликовано: 24.04.2003
Авторы: Ларкин Джон Патрик, Руссель Патрик, Крок Вероник, Колладан Колетт
МПК: C07D 487/04
Метки: 10-диоксо-6н-пиридазино, активных, кислоты, производные, октагидро-6, способ, 1,2, диазепин-1-карбоновой, терапевтически, получения, соединений, применение, 1,2-а
Формула / Реферат:
1. Соединения общей формулы (I) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS, в формуле которых R представляет собой атом водорода, радикал алкил, содержащий от 1 до 4 атомов углерода, a R1 и R2 означают H или защитную группу аминофункции. 2. Соединения по п.1, в которых аминогруппа защищена в форме фталимида, формулы (IA1) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS. 3. Соединения формулы (I) согласно любому из...
Предыдущий патент: Производные n-(аминогетероарил)-1н-индол-2-карбоксамидов, их получение и их применение в терапии
Следующий патент: Слитый белок эритропоэтина
Случайный патент: Соединения, композиции и способы для лечения заболеваний, опосредованных jak-киназой