Ключевые промежуточные соединения для синтеза розувастатина или его фармацевтически приемлемых солей






Формула / Реферат
1. Способ получения соединения формулы II

включающий стадии
получения соединения формулы I

и превращения соединения формулы I путем бромирования в соединение формулы II.
2. Способ по п.1, характеризующийся одной из следующих особенностей (i)-(iv) процесса или их комбинацией:
(i) бромирование осуществляют с помощью N-бромамида в качестве бромирующего агента; и/или
(ii) реакцию бромирования осуществляют в органическом растворителе, выбранном из группы, состоящей из ацетона, этилацетата, углеводородов, ароматических углеводородов и ацетонитрила или их смеси; и/или
(iii) ультрафиолетовое излучение имеет длину волны около 200-400 нм; и/или
(iv) бромирование осуществляют при температуре от около 0 до 90°C.
3. Способ по п.2, в котором в (i) N-бромамид выбран из группы, состоящей из N-бромацетамида, N,N-дибромбензолсульфонамидов, N-бромсукцинимида, N-бромфталимида, N-бромглутаримида, 3-бромгидантоина и 1,3-дибром-5,5-диметилгидантоина.
4. Способ по п.2, в котором начальное количество бромирующего агента составляет от около 1- до около 3-кратного значения молярного стехиометрического количества, основываясь на соединении I.
5. Способ по п.2, в котором в (iii) длина волны составляет около 310 нм.
6. Способ по п.2, в котором в (iv) бромирование осуществляют при температуре от около 15 до 35°C.
7. Способ по п.1 или 2, дополнительно включающий стадию очистки соединения формулы II.
8. Способ по п.7, в котором стадию очистки осуществляют путем кристаллизации.
9. Способ по п.8, в котором кристаллизацию осуществляют с помощью смеси МТВЕ/гексан.
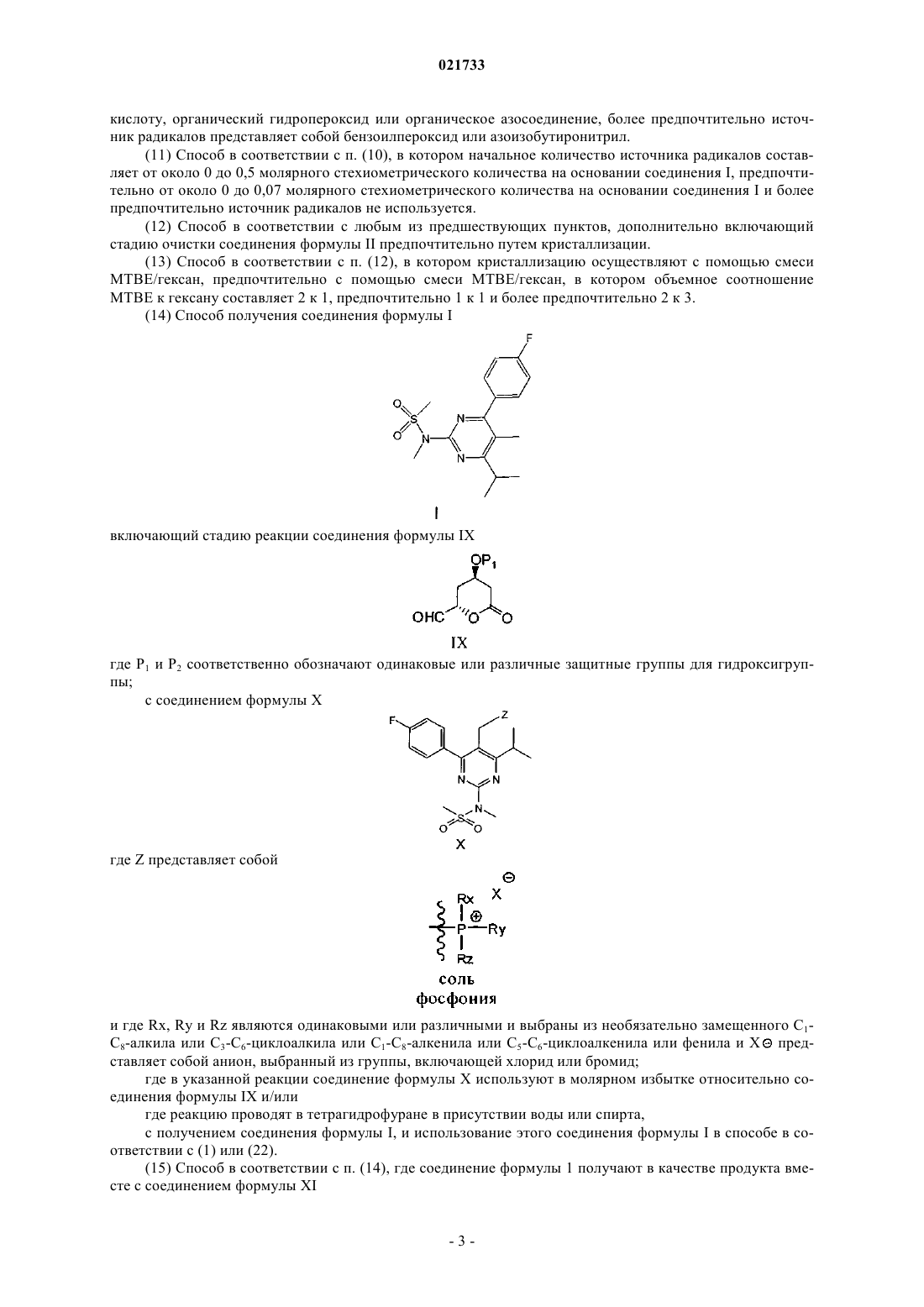
10. Способ получения соединения формулы I

включающий стадию реакции соединения формулы IX

где Р1 соответственно обозначает одинаковые или различные защитные группы для гидроксигруппы;
с соединением формулы X

где Z является

и где Rx, Ry и Rz являются одинаковыми или различными и выбраны из необязательно замещенного C1-C8-алкила или C3-C6-циклоалкила или C1-C8-алкенила или C5-C6-циклоалкенила или фенила, и Xθ представляет собой анион, выбранный из хлорида или бромида;
где в указанной реакции соединение формулы X используют в молярном избытке относительно соединения формулы IX и/или где реакция протекает в тетрагидрофуране в присутствии воды или спирта,
с получением соединения формулы I, и использование этого соединения формулы I в способе по п.1 или 17.
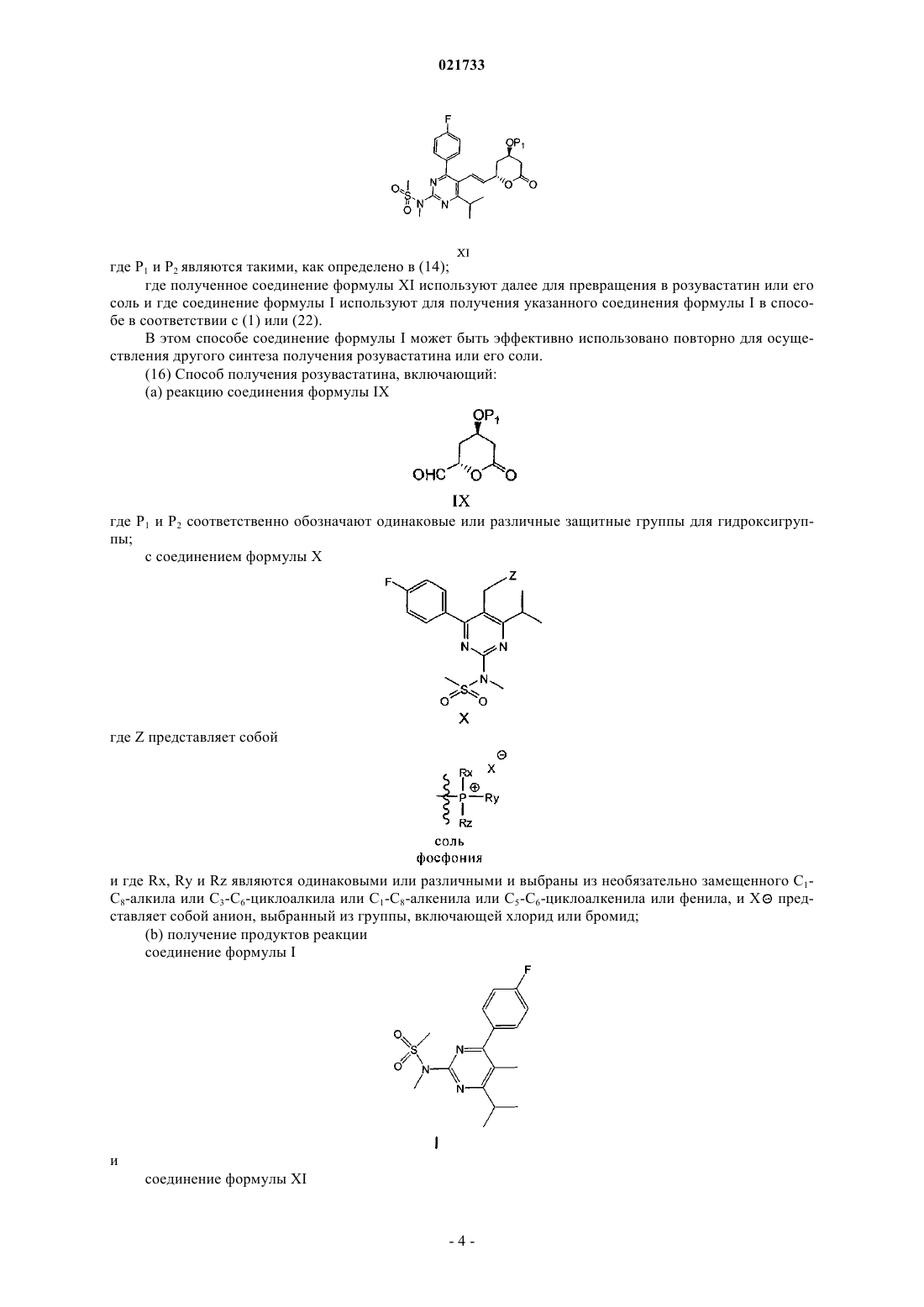
11. Способ по п.10, в котором соединение формулы I получают в виде продукта помимо соединения формулы XI

где Р1 является таким, как определено в п.10;
где полученное соединение формулы XI используют далее для превращения в розувастатин или его соль и где соединение формулы I используют как указанное соединение формулы I в способе по п.1 или 17.
12. Способ получения розувастатина, включающий:
(а) реакцию соединения формулы IX

где Р1 соответственно обозначает одинаковые или различные защитные группы для гидроксигрупп;
с соединением формулы X

где Z является

и где Rx, Ry и Rz являются одинаковыми или различными и выбраны из необязательно замещенного C1-C8-алкила или C3-C6-циклоалкила или C1-C8-алкенила или C5-C6-циклоалкенила или фенила, и Xθ представляет собой анион, выбранный из хлорида или бромида;
(b) получение продуктов реакции
соединения формулы I

и
соединения формулы XI

где P1 является таким, как определено выше;
(c) использование полученного соединения формулы XI для превращения в розувастатин или его соль; и
(d) использование полученного соединения формулы I в качестве указанного соединения формулы I в способе по п.1 или 17 в повторном цикле способа получения розувастатина.
13. Способ по п.12, в котором на стадии (b) полученные продукты реакции соответственно разделяют на соединение формулы I и соединение формулы XI до соответствующего использования на стадии (d).
14. Способ получения соединения формулы III

включающий стадию превращения соединения формулы II

путем гидролиза в соединение формулы III.
15. Способ по п.14, в котором гидролиз осуществляют в присутствии неорганического основания.
16. Способ по п.15, в котором неорганическим основанием является карбонат или гидрокарбонат щелочно-земельного металла.
17. Одностадийный способ получения соединения формулы III

включающий превращение соединения формулы I

реакцией через невыделяемое соединение формулы II

в соединение формулы III.
18. Способ по любому из пп.14-17, дополнительно включающий стадию очистки соединения формулы III.
19. Способ по п.18, в котором стадию очистки осуществляют путем кристаллизации.
20. Способ по п.19, в котором кристаллизацию осуществляют с помощью смеси МТВЕ/гексан.
21. Способ получения розувастатина или фармацевтически приемлемой соли розувастатина, включающий стадии:
a) осуществления способа получения соединения формулы I по п.10, осуществления способа получения соединения формулы II по любому из пп.1-9 или осуществления способа получения соединения формулы III по любому из пп.14-20 и
b) использования соединения формулы I, II или III соответственно на других стадиях синтеза с получением розувастатина или его фармацевтически приемлемых солей.
22. Применение соединения формулы I, полученного согласно способу по любому из пп.10-13 для получения розувастатина или его фармацевтически приемлемых солей.
Текст
КЛЮЧЕВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ СИНТЕЗА РОЗУВАСТАТИНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ Веселицкая И.А., Пивницкая Н.Н.,Кузенкова Н.В., Веселицкий М.Б., Каксис Р.А., Комарова О.М., Белоусов Ю.В. (RU) Настоящее изобретение относится в целом к области органической химии и, в частности, к получению(I),N-(4-(4-фторфенил)-5-(бромметил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамида (II) и N-(4-(4-фторфенил)-5-(гидроксиметил)-6-изопропилпиримидин-2-ил)-Nметилметансульфонамида (III), ключевым промежуточным соединениям для получения розувастатина Область техники, к которой относится изобретение Настоящее изобретение относится к способу получения N-(4-(4-фторфенил)-5-(бромметил)-6 изопропилпиримидин-2-ил)-N-метилметансульфонамида, N-(4-(4-фторфенил)-5-(гидроксиметил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамида и N-(4-(4-фторфенил)-6-изопропил-5-метилпиримидин-2-ил)-N-метилметансульфонамида, полезных в качестве ключевых промежуточных соединений для получения розувастатина или его фармацевтически приемлемых солей. Настоящее изобретение также относится к способу, в котором описанные выше соединения используют в качестве промежуточных соединений. Уровень техники(N-(4-(4-Фторфенил)-5-(бромметил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамид), (N(4-(4-фторфенил)-5-(гидроксиметил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамид) и N-(4(4-фторфенил)-6-изопропил-5-метилпиримидин-2-ил)-N-метилметансульфонамид являются возможными промежуточными соединениями для синтеза розувастатина и его фармацевтически приемлемых солей. Розувастатин кальция, химически обозначаемый как кальциевая соль бис-[(E)-7-[4-(4-фторфенил)-6 изопропил-2-[метил-(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты], представляет собой синтетический липидпонижающий агент, который действует в качестве ингибитора редуктазы 3-гидрокси-3-метилглутарил-коэнзима A (HMG-CoA) (ингибитор редуктазы HMGCoA). Ингибиторы редуктазы HMG-CoA общеизвестны как "статины". Статины являются терапевтически эффективными лекарственными препаратами, используемыми для снижения концентрации частиц липопротеина низкой плотности (LDL) в потоке крови пациентов с риском сердечно-сосудистого заболевания. Следовательно, розувастатин кальция используют для лечения гиперхолестеринемии и смешанной дислипидемии. В патенте EP 521471 A1 описан розувастатин и способ его получения, помимо прочих способ включает стадию получения N-(4-(4-фторфенил)-5-(гидроксиметил)-6-изопропилпиримидин-2-ил)-Nметилметансульфонамида путем восстановления его подходящего сложноэфирного производного диизобутилалюмогидридом (DIBAL-H) в качестве восстанавливающего реагента. Кроме того, в международной заявке на патент WO 2008/059519 А 2 также описано получение розувастатина через N-(4-(4-фторфенил)-5-(гидроксиметил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамид в качестве промежуточного соединения, полученного восстановлением его подходящего сложного эфира с помощью DIBAL-H. В международной заявке на патент WO 2007/017117 А 1 описано получение розувастатина через N(4-(4-фторфенил)-5-(бромметил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамид в качестве промежуточного соединения. Это промежуточное соединение получают нуклеофильным замещением N(4-(4-фторфенил)-5-(гидроксиметил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамида с помощью HBr в качестве источника нуклеофила. Задачей настоящего изобретения является обеспечение улучшенного способа получения N-(4-(4 фторфенил)-5-(бромметил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамида,N-(4-(4-фторфенил)-5-(гидроксиметил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамида и N-(4-(4-фторфенил)-6-изопропил-5-метилпиримидин-2-ил)-N-метилметансульфонамида с получением ценных промежуточных соединений для получения розувастатина и его фармацевтически приемлемых солей. Сущность изобретения Указанная задача решается способами получения N-(4-(4-фторфенил)-5-(бромметил)-6 изопропилпиримидин-2-ил)-N-метилметансульфонамида,N-(4-(4-фторфенил)-5-(гидроксиметил)-6 изопропилпиримидин-2-ил)-N-метилметансульфонамида и N-(4-(4-фторфенил)-6-изопропил-5-метилпиримидин-2-ил)-N-метилметансульфонамида в соответствии с пунктами формулы 1, 10, 14 и 17, способом получения розувастатина или его фармацевтически приемлемых солей в соответствии с пунктами формулы 12 и 21 и применением N-(4-(4-фторфенил)-5-(бромметил)-6-изопропилпиримидин-2-ил)-Nметилметансульфонамида для получения розувастатина или его фармацевтически приемлемых солей в соответствии с пунктом формулы 22. Предпочтительные варианты осуществления приведены далее и в подпунктах формулы изобретения. В соответствии с настоящим изобретением неожиданно было обнаружено, что более эффективный и легкий для осуществления синтез N-(4-(4-фторфенил)-5-(бромметил)-6-изопропилпиримидин-2-ил)-Nметилметансульфонамида и N-(4-(4-фторфенил)-5-(гидроксиметил)-6-изопропилпиримидин-2-ил)-Nметилметансульфонамида соответственно может осуществляться выбором подходящих исходных материалов, которые могут быть превращены в целевой продукт без необходимости агрессивных, трудных для обработки и/или дорогостоящих реагентов. Более того, способ получения является более эффективным, поскольку он включает благоприятные условия реакции, обеспечивая меньшее количество побочных продуктов и, таким образом, более высокую чистоту продуктов и более высокий выход и/или меньшее количество необходимых стадий реакции. Кроме того, способ в соответствии с настоящим изобретением осуществляется с использованием мягких реагентов, также способствуя более легкой обработке в смысле меньшего количества необходимых мер безопасности, касающихся использования и хранения, и меньших мер безопасности, касающихся необходимости специальных условий реакции, таких как атмо-1 021733 сфера защитного газа и/или безводный растворитель. Кроме того, описан эффективный способ извлечения N-(4-(4-фторфенил)-6-изопропил-5-метилпиримидин-2-ил)-N-метилметансульфонамида, который благоприятно влияет на эффективность общего способа синтеза розувастатина. В результате целевые ключевые промежуточные соединения для получения розувастатина или его фармацевтически приемлемых солей обеспечивают существенно улучшенный способ. Различные варианты осуществления, полезные особенности и предпочтительные варианты осуществления настоящего изобретения, которые соответственно отдельно и в комбинации конкретно участвуют в решении задачи изобретения, представлены в следующих пунктах.(1) Способ получения соединения формулы II включающий стадии получения соединения формулы I и превращения соединения формулы I путем бромирования в соединение формулы II.(2) Способ в соответствии с п. (1), в котором бромирование протекает с помощью радикальной реакции.(3) Способ в соответствии с п. (1) или (2), в котором указанное бромирование осуществляют с помощью N-бромамида в качестве бромирующего агента, предпочтительно N-бромамид выбирают из группы, состоящей из N-бромацетамида, N,N-дибромбензолсульфонамидов, N-бромсукцинимида, Nбромфталимида, N-бромглутаримида, 3-бромгидантоина и 1,3-дибром-5,5-диметилгидантоина, более предпочтительно N-бромсукцинимида.(4) Способ в соответствии с п. (2), в котором начальное количество бромирующего агента составляет от около 1- до около 3-кратного значения молярного стехиометрического количества, основываясь на соединении I, предпочтительно от около 1,2- до около 2,5-кратного значения, более предпочтительно от около 1,4- до около 2,2-кратного значения и в частности около 2-кратного значения.(5) Способ в соответствии с любым из предшествующих пунктов, в котором реакцию бромирования осуществляют в органическом растворителе, выбранном из группы, состоящей из ацетона, этилацетата,углеводородов, ароматических углеводородов и ацетонитрила или их смесей, предпочтительно органический растворитель представляет собой ацетонитрил.(6) Способ в соответствии с пп. (1)-(5), в котором бромирование осуществляют путем обработки ультрафиолетовым излучением, в котором указанное ультрафиолетовое излучение имеет длину волны около 200-400 нм, предпочтительно около 310 нм.(7) Способ в соответствии с п. (6), в котором указанное ультрафиолетовое излучение осуществляют в течение от 2 до 10 ч, предпочтительно в течение около 4 ч.(8) Способ в соответствии с любым из пп. (1)-(7), в котором бромирование осуществляют при температуре от около 0 до 90C, предпочтительно от около 10 до 65C, более предпочтительно от около 15 до 35C и в частности от около 19 до 25C.(9) Способ в соответствии с любым из предшествующих пунктов, в котором не используется источник радикалов.(10) Способ в соответствии с любым из пп. (1)-(8), в котором используется источник радикалов, где источник радикалов предпочтительно представляет собой органический пероксид, органическую пер-2 021733 кислоту, органический гидропероксид или органическое азосоединение, более предпочтительно источник радикалов представляет собой бензоилпероксид или азоизобутиронитрил.(11) Способ в соответствии с п. (10), в котором начальное количество источника радикалов составляет от около 0 до 0,5 молярного стехиометрического количества на основании соединения I, предпочтительно от около 0 до 0,07 молярного стехиометрического количества на основании соединения I и более предпочтительно источник радикалов не используется.(12) Способ в соответствии с любым из предшествующих пунктов, дополнительно включающий стадию очистки соединения формулы II предпочтительно путем кристаллизации.(13) Способ в соответствии с п. (12), в котором кристаллизацию осуществляют с помощью смеси МТВЕ/гексан, предпочтительно с помощью смеси МТВЕ/гексан, в котором объемное соотношение МТВЕ к гексану составляет 2 к 1, предпочтительно 1 к 1 и более предпочтительно 2 к 3.(14) Способ получения соединения формулы I включающий стадию реакции соединения формулы IX где Р 1 и Р 2 соответственно обозначают одинаковые или различные защитные группы для гидроксигруппы; с соединением формулы X и где Rx, Ry и Rz являются одинаковыми или различными и выбраны из необязательно замещенного C1C8-алкила или C3-C6-циклоалкила или C1-C8-алкенила или C5-C6-циклоалкенила или фенила и X представляет собой анион, выбранный из группы, включающей хлорид или бромид; где в указанной реакции соединение формулы X используют в молярном избытке относительно соединения формулы IX и/или где реакцию проводят в тетрагидрофуране в присутствии воды или спирта,с получением соединения формулы I, и использование этого соединения формулы I в способе в соответствии с (1) или (22).(15) Способ в соответствии с п. (14), где соединение формулы 1 получают в качестве продукта вместе с соединением формулы XI где P1 и P2 являются такими, как определено в (14); где полученное соединение формулы XI используют далее для превращения в розувастатин или его соль и где соединение формулы I используют для получения указанного соединения формулы I в способе в соответствии с (1) или (22). В этом способе соединение формулы I может быть эффективно использовано повторно для осуществления другого синтеза получения розувастатина или его соли.(а) реакцию соединения формулы IX где P1 и P2 соответственно обозначают одинаковые или различные защитные группы для гидроксигруппы; с соединением формулы X и где Rx, Ry и Rz являются одинаковыми или различными и выбраны из необязательно замещенного C1C8-алкила или C3-C6-циклоалкила или C1-C8-алкенила или C5-C6-циклоалкенила или фенила, и X представляет собой анион, выбранный из группы, включающей хлорид или бромид;(b) получение продуктов реакции соединение формулы I(c) применение полученного соединения формулы XI для превращения в розувастатин или его соль; и(d) применение полученного соединения формулы I для получения указанного соединения формулы I в способе в соответствии с п. (1) или (22) в повторном цикле способа получения розувастатина.(17) Способ в соответствии с п. (16), где на стадии (b) полученные продукты реакции соответственно разделяют на соединение формулы I и соединение формулы XI до соответствующего применения на стадии (d). Методика, описанная в пп. (16) и (17), представляет собой полезный и общепригодный способ с рециркуляцией, который осуществляют для повышения общего выхода розувастатина или его соли.(18) Способ получения соединения формулы III включающий стадию превращения соединения формулы II(19) Способ в соответствии с п. (18), где гидролиз осуществляют в присутствии неорганического основания, предпочтительно карбоната или гидрокарбоната щелочного или щелочно-земельного металла, более предпочтительно NaHCO3.(20) Способ в соответствии с п. (19), где неорганическое основание добавляют к реакционной смеси в форме насыщенного водного раствора.(21) Способ в соответствии с любым из пп. (19)-(20), где начальное количество неорганического основания составляет от около 1- до 10-кратного значения молярного стехиометрического количества на основании соединения II, предпочтительно от около 3- до 7-кратного значения и более предпочтительно от 5- до 6-кратного значения.(22) Одностадийный способ получения соединения формулы III включающий превращение соединения формулы I реакцией через невыделяемое соединение формулы II(23) Способ в соответствии с п. (22), где превращение соединения формулы I в соединение формулы II осуществляют способом по любому из пп. (1)-(11).(24) Способ в соответствии с п. (22) или (23), в котором превращение соединения формулы II в соединение формулы III осуществляют способом по любому из пп. (18)-(21).(25) Способ в соответствии с любым из пп. (22)-(24), в котором реакционную смесь после превращения соединения формулы I в соединение формулы II разбавляют растворителем, как определено в п.(26) Способ в соответствии с любым из пп. (18)-(25), дополнительно включающий стадию очистки соединения формулы III предпочтительно путем кристаллизации.(27) Способ в соответствии с п. (26), в котором кристаллизацию осуществляют с помощью смеси МТВЕ/гексан, предпочтительно с помощью смеси МТВЕ/гексан, где объемное соотношение МТВЕ к гексану составляет 2 к 1, предпочтительно 1 к 1 и более предпочтительно 2 к 3.(28) Способ получения розувастатина или фармацевтически приемлемой соли розувастатина, включающий стадии: а) проведения способа получения соединения формулы I в соответствии с п. (14) и б) подвергания соединения формулы I дальнейшим стадиям синтеза для получения розувастатина или его фармацевтически приемлемых солей.(29) Способ получения розувастатина или фармацевтически приемлемой соли розувастатина, включающий стадии: а) проведения способа получения соединения формулы II в соответствии с любым из пп. (1)-(13) и б) подвергания соединения формулы II дальнейшим стадиям синтеза для получения розувастатина или его фармацевтически приемлемых солей.(30) Способ получения розувастатина или фармацевтически приемлемой соли розувастатина, вклю-6 021733 чающий стадии: а) проведения способа получения соединения формулы III в соответствии с любым из пп. (18)-(27) и б) подвергания соединения формулы III дальнейшим стадиям синтеза для получения розувастатина или его фармацевтически приемлемых солей.(31) Применение соединения формулы I, полученного способом в соответствии с любым из пп.(14)-(15), для получения розувастатина или его фармацевтически приемлемых солей. Подробное описание изобретения Настоящее изобретение далее описано более подробно путем отсылки на другие предпочтительные и также полезные варианты осуществления и примеры, которые, однако, представлены только в целях иллюстрации и не должны пониматься как ограничивающие объем настоящего изобретения. Для улучшения способа получения соединения формулы II (N-(4-(4-фторфенил)-5-(бромметил)-6 изопропилпиримидин-2-ил)-N-метилметансульфонамид) и соединения формулы III (N-(4-(4-фторфенил)5-(гидроксиметил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамид) авторы провели большой ряд испытаний для обнаружения критических факторов, которые особенно подходят для повышения выходов продукта и для снижения побочных продуктов, при существенном упрощении способа благодаря благоприятным условиям реакции и/или меньшему количеству необходимых стадий реакции. Обычно соединение формулы III получают восстановлением подходящего сложноэфирного производного формулы IV (где R предпочтительно обозначает метильный или этильный остаток) с помощью подходящего восстанавливающего агента на поздней или последней стадии многостадийной методики синтеза, как показано на следующей схеме: Однако этот тип восстановления имеет значительные недостатки методики. Во-первых, восстановление осуществляют с помощью диизобутилалюмогидрида (DIBAL-H) в качестве восстанавливающего агента, и, следовательно, восстановление должно осуществляться при температуре около или ниже 0C(предпочтительно вплоть до -70C) в сухих/безводных условиях. Другим недостатком восстановления с помощью DIBAL-H является то, что комплексный гидрид DIBAL-H является дорогостоящим и опасным реагентом. Во-вторых, восстановление осуществляют с помощью KBH4/ZnCl2 в качестве восстанавливающего агента, который также требует наличия сухих/безводных условий. Более того, существует проблема непрореагировавшего исходного материала и образования побочных продуктов, которые сложно удалять в последующих стадиях синтеза розувастатина, если сухие/безводные условия не используются,и реакция не протекает до конца. Как показано на следующей схеме, обычно соединение формулы III затем превращают в соединение формулы II реакцией нуклеофильного замещения с помощью HBr или PBr3 для введения брома Указанная реакция нуклеофильного замещения имеет существенные недостатки, в частности, поскольку HBr является сильно коррозийным и агрессивным реагентом, альтернативный реагент PBr3 является токсичным, выделяет коррозийный HBr и хорошо реагирует с водой и спиртами, что затрудняет его использование. В заключение может быть упомянуто, что описанное выше стандартное получение соединения формулы III или стандартное получение соединения формулы II и соединения формулы IV требует использования реагентов, которые затрудняют использование, являются опасными и/или дорогостоящими. Кроме того, некоторые стадии реакции необходимы для получения соединения формулы II, и стандартные способы имеют недостатки критического получения побочных продуктов, которые влияют далее на синтез розувастатина. В соответствии с одним вариантом осуществления настоящего изобретения соединение формулы II получают превращением соединения формулы I бромированием в соединение формулы II, как показано Поскольку соединение формулы I (N-(4-(4-фторфенил)-5-метил-6-изопропилпиримидин-2-ил)-Nметилметансульфонамид) используется в качестве исходного материала, соединение II (N-(4-(4 фторфенил)-5-(бромметил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамид) может быть получено только в одну стадию бромирования. Реакция может осуществляться наиболее эффективно реакцией радикального бромирования необязательно с помощью УФ-излучения и/или с помощью источников радикалов. Описанное выше бромирование, особенно при проведении радикальной реакции, существенно отличается от введения брома с помощью реакции нуклеофильного замещения (например, где соединение формулы III превращают в соединение формулы II). Реакция нуклеофильного замещения требует наличия уходящей группы, такой как, например, -ОН в соединении формулы III. Наоборот, соединение формулы I не требует наличия такой уходящей группы. В описанной выше реакции бромирования настоящего изобретения предпочтительно используются бромирующие агенты, такие как N-бромамиды. Предпочтительно N-бромамиды используют для постоянной, низкой концентрации брома в реакционной смеси в ходе реакции. Более предпочтительно указанные N-бромамиды выбирают из группы, состоящей из N-бромацетамида, N,N-дибромбензолсульфонамидов; N-бромиминов, таких как N-бромсукцинимид, N-бромфталимид, N-бромглутаримид, 3 бромгидантоин и 1,3-дибром-5,5-диметилгидантоин. N-Бромсукцинимид является наиболее предпочтительным бромирующим агентом, поскольку является легкодоступным коммерческим реагентом и экономичным по цене. Предпочтительно описанные выше бромирующие агенты используют для мягких условий реакции, приводя к меньшему количеству побочных продуктов. Начальное количество указанных бромирующих агентов составляет от около 0,1- до около 3 кратного значения молярного стехиометрического количества на основании соединения I, предпочтительно от около 0,9- до около 2,5-кратного значения, более предпочтительно от около 1,4- до около 2,2 кратного значения и в частности около 2-кратного значения. В этом способе описано эффективное бромирование, приводящее к высоким выходам соединения II, при использовании экономных количеств бромирующего агента. Описанную выше реакцию бромирования обычно осуществляют в органическом растворителе,предпочтительно выбранном из группы, состоящей из ацетона, этилацетата, углеводородов, ароматических углеводородов и ацетонитрила. Наиболее предпочтительно ацетонитрил используют в качестве органического растворителя. Описанные выше органические растворители обеспечивают подходящую растворимость реагентов и улучшенные скорости реакции. Кроме того, эти органические растворители являются гораздо менее токсичными по сравнению с тетрахлоридом углерода или хлорбензолом, которые обычно используются в радикальном бромировании углеводородных боковых цепей ароматических субстратов. Предпочтительно стадию реакции соединения формулы I с бромирующим агентом с получением соединения формулы II осуществляют обработкой ультрафиолетовым излучением, где указанное ультрафиолетовое излучение предпочтительно имеет длину волны от около 200 до 400 нм, более предпочтительно около 310 нм. Указанное ультрафиолетовое излучение предпочтительно осуществляют в течение от 2 до 10 ч, более предпочтительно в течение около 4 ч. В конкретном предпочтительном варианте осуществления по изобретению реакцию бромирования осуществляют при подходящей температуре, предпочтительно при температуре от 0 до 90C, более предпочтительно от 10 до 65C, еще более предпочтительно от 15 до 35C и в частности при комнатной температуре от 19 до 25C. В этом способе могут использоваться благоприятные мягкие условия реакции, которые далее влияют на образование меньшего количества побочных продуктов по сравнению с реакцией нуклеофильного замещения для введения брома, где используются повышенные температуры реакции. Получают более высокие выходы, очистка облегчается, и дальнейшие стадии синтеза для получения розувастатина менее затрагиваются критическими побочными продуктами. Неожиданно, при использовании соединения формулы I в качестве исходного соединения описанное выше радикальное бромирование протекает за относительно короткое время реакции и с высокими выходами, даже если не используется источник радикалов. Отсутствие источника радикалов является выгодным, поскольку реакция становится более безопасной ввиду эксплуатационной безопасности, из-за того что источники радикалов являются довольно реакционоспособными и, следовательно, опасными для использования соединениями. Кроме того, стоимость источника радикалов можно экономить. Следовательно, предпочтительно осуществлять бромирование без источника радикалов. Кроме того, существенно меньшее количество примесей образуется в ходе реакции, если не используется источник радикалов. Тем не менее, если одной целью является дальнейшее ускорение реакции бромирования, источник радикалов может использоваться. Если он используется, источник радикалов предпочтительно представляет собой органический пероксид, органическую перкислоту, органический гидропероксид или органическое азосоединение. Эти источники радикалов подходят для ускорения/поддержания радикальных реакций. Более предпочтительно источник радикалов выбирают из пероксида бензоила или азоизобутиронитрила, поскольку эти источники радикалов являются коммерчески доступными и недорогими. Если источник радикалов используют в реакции бромирования, начальное количество источника радикалов составляет от 0 до 0,5 молярного стехиометрического количества на основании соединения I,предпочтительно от около 0 до 0,07 молярного стехиометрического количества на основании соединенияI и более предпочтительно источник радикалов не используется. Описанные выше количества источника радикалов вводят для полезного ускорения реакции, при этом обеспечивая стабильную и безопасную реакцию. В соответствии с одним вариантом осуществления соединение формулы II выделяют и очищают предпочтительно кристаллизацией. Этим способом осуществляют простой и эффективный способ очистки по сравнению с трудоемкой, затратной по времени и материалам колоночной хроматографией. Поскольку реакцию бромирования осуществляют в мягких условиях, образуется меньшее количество побочных продуктов, и, следовательно, кристаллизации будет достаточно для того, чтобы получить преимущественно чистый продукт. Кроме того, было обнаружено, что кристаллизация, осуществляемая с помощью смеси МТВЕ/гексан и, в частности, с помощью смеси МТВЕ/гексан, где объемное соотношение МТВЕ к гексану составляет 2 к 1, предпочтительно 1 к 1 и более предпочтительно 2 к 3, является особенно предпочтительной. Соединение формулы I может быть получено направленным синтезом. Или в соответствии с предпочтительным вариантом осуществления соединение формулы I получают в виде побочного продукта при получении промежуточных соединений розувастатина, где соединение формулы I образуется реакцией Виттига соли фосфония (соединение формулы X) соответствующего розувастатинового гетероцикла и хиральной статиновой боковой цепи. Иллюстрирующая реакционная система может быть представлена на схеме 1 ниже. На схеме 1 в соединении формулы X Z представляет собой соль фосфония где Rx, Ry, Rz являются одинаковыми или различными и выбраны необязательно из замещенного C1-C8 алкила или C3-C6-циклоалкила или C1-C8-алкенила или C5-C6-циклоалкенила или фенила, и X представляет собой анион, выбранный из группы, включающей хлорид или бромид. Далее на схеме 1 Р 1 и Р 2 независимо обозначают обычные защитные группы для гидроксильной группы. Защитная группа P1 и P2 может быть любой обычно используемой защитной группой для гидроксильных групп, например независимо выбранной из группы, состоящей из алкила, разветвленного алкила, ацила, силила или аналогичной группы, более конкретно выбранной из ацетонида, ацетила (Ас),пивалоила (Piv), n-толуолсульфонила (TOS), -метоксиэтоксиметилового эфира (MEM), метоксиметилового эфира (MOM), n-метоксибензилового эфира (РМВ), метилтиометилового эфира, трет-бутила, тетрагидропиранила (ТНР), бензила (Bn), дифенилметила или трифенилметильной группы, предпочтительно силильной защитной группы, которая может быть представлена формулой SiR1'R2'R3', в которой R1', R2',R3' независимо выбраны из алкила (предпочтительно C1-C6) или арила (предпочтительно С 5-С 10), такого как SiMe3 (TMS), SiMe2 третBu (TBDMS), Si(изо-Pr)3 (TIPS), SiPh2 третBu, SiMe2Ph. Следовательно, как показано на схеме 1, защищенное конечное промежуточное соединение розувастатина может использоваться для проведения последних стадий синтеза получения розувастатина или его солей, тогда как альтернативно или в дополнение может использоваться соединение формулы I с помощью повторного цикла в другом (одинаковом или отличном) способе синтеза розувастатина. До соответствующего дальнейшего применения реакционные продукты, полученные в реакции Виттига, соответственно могут быть разделены подходящими и известными методами на соединение формулы I и соединение, выбранное из формул XI. Преимущественно и неожиданно, соединение формулы I образуется гораздо лучше, когда реакцию Виттига осуществляют с избытком соли фосфония (или ее илида или фосфорана) в качестве реагента Виттига (например, молярный избыток соединения X или X' по отношению к соединению IX или IX' соответственно 5% или более, предпочтительно 10% или более и особенно 15% или более), более эффективно после погашения протонным растворителем, и/или когда реакцию Виттига осуществляют в тетрагидрофуране в присутствии воды или спиртов (например, метанол, этанол, пропанол, изопропанол, бутанол и фенолы) и т.д. Присутствие воды или спиртов может осуществляться добавлением воды или спирта, но альтернативно предпочтительно и достаточно, когда, например, невысушенный, или влажный, или недостаточно высушенный растворитель (растворители) вводят в реакцию Виттига. Исходное соединение формулы IX может быть получено из его гидрата в подходящем растворителе, но без удаления высвобождающихся молекул воды, как показано на следующей схеме реакции: и затем непосредственно (т.е. без удаления воды) введением в реакцию Виттига. Подходящим растворителем для следующей реакции является, например, тетрагидрофуран (ТГФ). Обеспечение и использование соединения формулы I оказывает существенное благоприятное влияние на эффективность общего процесса синтеза розувастатина. Поскольку гетероциклическую часть молекулы получают в несколько трудоемких стадий синтеза, как описано, например, в патенте EP 521471,крайне предпочтительно извлечь ценное соединение формулы I и утилизировать его специфическим превращением в соединения формул II или III, которые, в свою очередь, способны выгодно использоваться далее, например, путем превращения их снова в соль фосфония, обеспечивая дальнейший исходный материал для получения промежуточных соединений розувастатина реакцией Виттига (как показано, например, выше на схеме 1). Соединение формулы II может непосредственно превращаться в производное соли фосфония (см., например, заявку на патент US 2005/0124639). Альтернативно, соединение формулы I может превращаться в соединение формулы III, которое может превращаться в производное соли фосфония (см., например, международную заявку на патент WO 2007/017117). Хотя соединение формулы II может быть получено методиками, известными из уровня техники (см., например, международную заявку на патент WO 2007/017117), эта методика не может использоваться для извлечения соединения I в производное соли фосфония. Аналогично, известные из уровня техники методики получе- 10021733 ния соединения III, описанные в патенте EP 521471, не могут использоваться для извлечения соединения формулы I в производное соли фосфония. Следовательно, обеспечение соединения формулы I, помимо его собственной полезности, может заметно влиять на повышение общего выхода синтеза розувастатина. В соответствии с другим вариантом изобретения соединение формулы III получают способом,включающим стадии превращения соединения формулы II гидролизом в соединение формулы III, как показано на следующей схеме: В соответствии с предпочтительным вариантом осуществления описанное выше превращение осуществляют в присутствии неорганического основания, предпочтительно карбоната или гидрокарбоната щелочного или щелочно-земельного металла, более предпочтительно NaHCO3 используется в качестве неорганического основания. Кроме того, предпочтительно добавлять указанное неорганическое основание к реакционной смеси в форме насыщенного водного раствора. Предпочтительно начальное количество неорганического основания составляет от около 1- до 10 кратного количества от молярного стехиометрического количества, основываясь на соединении II, предпочтительно от около 3- до 7-кратного количества и более предпочтительно от 5- до 6-кратного количества. В соответствии с другим вариантом осуществления настоящего изобретения соединение формулыIII получают одностадийным синтезом превращения соединения формулы I через невыделяемое соединение формулы II в соединение формулы III, как показано на следующей схеме: Было обнаружено, что возможно получить соединение формулы III без выделения и очистки промежуточного соединения формулы II. Следовательно, количество стадий способа может снижаться, что делает общий способ синтеза, по существу, более эффективным. Предпочтительно описанный выше одностадийный синтез осуществляют превращением соединения формулы I в соединение формулы II описанным выше бромированием в соответствии с настоящим изобретением и/или превращением соединения формулы II в соединение формулы III описанным выше гидролизом в соответствии с настоящим изобретением. Кроме того, предпочтительно добавлять растворитель к полученной реакционной партии после осуществления превращения соединения формулы I в соединение формулы II для разбавления реакционной партии. За ходом превращения соединения формулы I в соединение формулы II, например, можно следить по данным тонкослойной хроматографии или высокоэффективной жидкостной хроматографии(ВЭЖХ). Предпочтительно указанный растворитель для разбавления выбран из группы растворителей,описанных выше для реакции бромирования, и более предпочтительно он является тем же растворителем, который используется в реакции бромирования. Таким образом, получают полезную степень растворения соединения формулы II, что в свою очередь обеспечивает мягкий гидролиз с хорошими выходами. В соответствии с другим вариантом осуществления способ получения соединения формулы III также включает стадию очистки соединения формулы III предпочтительно кристаллизацией. В этом способе используют простой и эффективный метод очистки по сравнению с трудоемкой, длительной и требующей дополнительных материалов колоночной хроматографией. Поскольку реакция гидролиза обеспечивает полное превращение соединения формулы II в соеди- 11021733 нение формулы III, кристаллизация является достаточной для обеспечения преимущественно чистого продукта. Кроме того, было обнаружено, что особенно предпочтительной является кристаллизация, проводимая с помощью смеси МТВЕ/гексан, и, в частности, с помощью смеси МТВЕ/гексан, где объемное соотношение МТВЕ к гексану составляет 2 к 1, предпочтительно 1 к 1 и более предпочтительно 2 к 3. Ключевые промежуточные соединения формул II и III затем могут подвергаться дальнейшим стадиям синтеза для получения розувастатина или его фармацевтически приемлемых солей синтетическими методиками, известными или легко определимыми специалистом в данной области техники. Как показано ниже на схеме, могут использоваться следующие синтетические методики: Экспериментальные методики Пример 1. Получение N-(4-(4-фторфенил)-6-изолропил-5-метилпиримидин-2-ил)-N-метилметансульфонамида (1) К холодной (-42C) перемешиваемой суспензии 4-(4-фторфенил)-6-изопропил-2-(N-метилметилсульфонамидо)пиримидин-5-ил)метил)трифенилфосфонийбромида (814 мг, 1,20 ммоль) в тетрагидрофуране (25 мл) добавляли гексаметилдисилазан натрия в ТГФ (1,2 мл 1,0 М, 1,20 ммоль). Реакционную смесь перемешивали в течение 45 мин при -42C, охлаждали до -82C и обрабатывали раствором (2S,4R)4-(трет-бутилдиметилсилилокси)-6-оксотетрагидро-2H-пиран-2-карбальдегида (266 мг, 1,03 ммоль), полученным растворением его гидрата (284 мг, 1,03 ммоль) в 15 мл тетрагидрофурана без удаления высвободившейся воды. Через 30 мин перемешивания раствор нагревали до температуры от -53 до -58C и перемешивали в течение 6 ч. Затем смесь оставляли нагреваться до комнатной температуры в течение 100 мин и обрабатывали насыщенным раствором хлорида аммония (40 мл). После перемешивания в течение 10 мин при 10C водную фазу отрабатывали 20 мл воды и 40 мл насыщенного раствора хлорида натрия. Продукт экстрагировали трет-BuMeO (50 мл + 430 мл). Объединенные органические слои су- 12021733 шили (MgSO4) и концентрировали при пониженном давлении (11 мбар) при 40C с получением белого твердого вещества. Остаток очищали хроматографией на силикагеле (элюируя смесью гексан/AcOEt = 3:1) с получением 170 мг (42%) N-(4-(4-фторфенил)-6-изопропил-5-метилпиримидин-2-ил)-Nметилметансульфонамида (I). Rf (гексан/AcOEt = 3:1) = 0,42. Белое твердое вещество, tпл 113-114C. 1N-4-(4-Фторфенил)-5-метил-6-изопропилпиримидин-2-ил)-N-метилметансульфонамид (112,5 мг,0,33 ммоль, 1 экв.) и N-бромсукцинимид (NBS) (126 мг, 0,72 ммоль, 2,1 экв.) растворяли в 2 мл ацетонитрила. Смесь подвергали облучению светом с длиной волны =310 нм в течение 4 ч при комнатной температуре (около 20C). Затем добавляли воду (10 мл) и смесь экстрагировали CH2Cl2 (310 мл). Объединенные органические фазы промывали 10 мл насыщенного раствора хлорида натрия и полученный раствор сушили Na2SO4. Растворитель удаляли при пониженном давлении с получением 138,6 мг сырогоN-(4-(4-фторфенил)-5-(бромметил)-6-изопропилпиримидин-2-ил)-Nметилметансульфонамида (II), как определено по данным 1 Н-ЯМР интеграла. Этот продукт можно далее очистить кристаллизацией из смеси МТВЕ/гексан с получением чистого материала. Пример 3N-(4-(4-Фторфенил)-5-метил-6-изопропилпиримидин-2-ил)-N-метилметансульфонамид (112,5 мг,0,33 ммоль, 1 экв.) и N-бромсукцинимид (NBS) (118,7 мг, 0,66 ммоль, 2 экв.) растворяли в 2 мл ацетонитрила. Смесь подвергали облучению светом с длиной волны =310 нм в течение 4 ч при комнатной температуре (около 20C). Полученный желтый раствор разбавляли 1 мл ацетонитрила. После добавления 2 мл насыщенного раствора NaHCO3 полученную смесь далее перемешивали при кипении с обратным холодильником в течение 4 ч. Затем смесь охлаждали до комнатной температуры, добавляли воду(10 мл) и смесь экстрагировали CH2Cl2 (310 мл). Объединенные органические фазы промывали 10 мл насыщенного раствора хлорида натрия и полученный раствор сушили Na2SO4. Растворитель удаляли при пониженном давлении с получением 110,8 мг (95%) сырого N-(4-(4-фторфенил)-5-(гидроксиметил)-6 изопропилпиримидин-2-ил)-N-метилметансульфонамида (III), который содержал 77% N-(4-(4 фторфенил)-5-(гидроксиметил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамида (III) по данным 1 Н-ЯМР интеграла. Этот продукт можно далее очистить кристаллизацией из смеси МТВЕ/гексан с получением чистого материала (ВЭЖХ площадь % = 99,6) с Tm = 140-141C. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы II включающий стадии получения соединения формулы I и превращения соединения формулы I путем бромирования в соединение формулы II. 2. Способ по п.1, характеризующийся одной из следующих особенностей (i)-(iv) процесса или их комбинацией:(ii) реакцию бромирования осуществляют в органическом растворителе, выбранном из группы, состоящей из ацетона, этилацетата, углеводородов, ароматических углеводородов и ацетонитрила или их смеси; и/или(iii) ультрафиолетовое излучение имеет длину волны около 200-400 нм; и/или(iv) бромирование осуществляют при температуре от около 0 до 90C. 3. Способ по п.2, в котором в (i) N-бромамид выбран из группы, состоящей из N-бромацетамида,N,N-дибромбензолсульфонамидов, N-бромсукцинимида, N-бромфталимида, N-бромглутаримида, 3 бромгидантоина и 1,3-дибром-5,5-диметилгидантоина. 4. Способ по п.2, в котором начальное количество бромирующего агента составляет от около 1- до около 3-кратного значения молярного стехиометрического количества, основываясь на соединении I. 5. Способ по п.2, в котором в (iii) длина волны составляет около 310 нм. 6. Способ по п.2, в котором в (iv) бромирование осуществляют при температуре от около 15 до 35C. 7. Способ по п.1 или 2, дополнительно включающий стадию очистки соединения формулы II. 8. Способ по п.7, в котором стадию очистки осуществляют путем кристаллизации. 9. Способ по п.8, в котором кристаллизацию осуществляют с помощью смеси МТВЕ/гексан. 10. Способ получения соединения формулы I включающий стадию реакции соединения формулы IX где Р 1 соответственно обозначает одинаковые или различные защитные группы для гидроксигруппы; с соединением формулы X и где Rx, Ry и Rz являются одинаковыми или различными и выбраны из необязательно замещенного C1-C8-алкила или C3-C6-циклоалкила или C1-C8-алкенила или C5-C6-циклоалкенила или фенила, и X представляет собой анион, выбранный из хлорида или бромида; где в указанной реакции соединение формулы X используют в молярном избытке относительно соединения формулы IX и/или где реакция протекает в тетрагидрофуране в присутствии воды или спирта,с получением соединения формулы I, и использование этого соединения формулы I в способе по п.1 или 17. 11. Способ по п.10, в котором соединение формулы I получают в виде продукта помимо соединения формулы XI где Р 1 является таким, как определено в п.10; где полученное соединение формулы XI используют далее для превращения в розувастатин или его соль и где соединение формулы I используют как указанное соединение формулы I в способе по п.1 или 17. 12. Способ получения розувастатина, включающий:(а) реакцию соединения формулы IX где Р 1 соответственно обозначает одинаковые или различные защитные группы для гидроксигрупп; с соединением формулы X и где Rx, Ry и Rz являются одинаковыми или различными и выбраны из необязательно замещенного C1-C8-алкила или C3-C6-циклоалкила или C1-C8-алкенила или C5-C6-циклоалкенила или фенила, и X представляет собой анион, выбранный из хлорида или бромида;(b) получение продуктов реакции соединения формулы I(c) использование полученного соединения формулы XI для превращения в розувастатин или его соль; и(d) использование полученного соединения формулы I в качестве указанного соединения формулы I в способе по п.1 или 17 в повторном цикле способа получения розувастатина. 13. Способ по п.12, в котором на стадии (b) полученные продукты реакции соответственно разделяют на соединение формулы I и соединение формулы XI до соответствующего использования на стадии(d). 14. Способ получения соединения формулы III включающий стадию превращения соединения формулы II путем гидролиза в соединение формулы III. 15. Способ по п.14, в котором гидролиз осуществляют в присутствии неорганического основания. 16. Способ по п.15, в котором неорганическим основанием является карбонат или гидрокарбонат щелочно-земельного металла. 17. Одностадийный способ получения соединения формулы III включающий превращение соединения формулы I реакцией через невыделяемое соединение формулы II в соединение формулы III. 18. Способ по любому из пп.14-17, дополнительно включающий стадию очистки соединения формулы III. 19. Способ по п.18, в котором стадию очистки осуществляют путем кристаллизации. 20. Способ по п.19, в котором кристаллизацию осуществляют с помощью смеси МТВЕ/гексан. 21. Способ получения розувастатина или фармацевтически приемлемой соли розувастатина, включающий стадии:a) осуществления способа получения соединения формулы I по п.10, осуществления способа получения соединения формулы II по любому из пп.1-9 или осуществления способа получения соединения формулы III по любому из пп.14-20 иb) использования соединения формулы I, II или III соответственно на других стадиях синтеза с получением розувастатина или его фармацевтически приемлемых солей. 22. Применение соединения формулы I, полученного согласно способу по любому из пп.10-13 для получения розувастатина или его фармацевтически приемлемых солей.
МПК / Метки
МПК: A61K 31/505, C07D 239/42, A61P 3/06
Метки: приемлемых, промежуточные, фармацевтически, синтеза, ключевые, соединения, розувастатина, солей
Код ссылки
<a href="https://eas.patents.su/19-21733-klyuchevye-promezhutochnye-soedineniya-dlya-sinteza-rozuvastatina-ili-ego-farmacevticheski-priemlemyh-solejj.html" rel="bookmark" title="База патентов Евразийского Союза">Ключевые промежуточные соединения для синтеза розувастатина или его фармацевтически приемлемых солей</a>
Способ синтеза периндоприла и его фармацевтически приемлемых солей

Номер патента: 8644
Опубликовано: 29.06.2007
Авторы: Ланглуа Паскаль, Фуже Клод, Дюбюффе Тьерри
МПК: C07D 209/42, C07K 5/06
Метки: периндоприла, способ, приемлемых, фармацевтически, солей, синтеза
Формула / Реферат:
1. Способ синтеза соединения формулы (I) и его фармацевтически приемлемых солей, который характеризуется тем, что соединение формулы (II) в которой R представляет собой атом водорода или бензил, или линейную, или разветвленную C1-С6-алкильную группу, подвергают реакции с соединением формулы (III), имеющим (R)-конфигурацию в которой G представляет собой атом хлора, брома или йода или гидрокси, n-толуолсульфонилокси, метансульфонилокси- или...
Способ синтеза периндоприла и его фармацевтически приемлемых солей

Номер патента: 8685
Опубликовано: 29.06.2007
Авторы: Фуже Клод, Ланглуа Паскаль, Дюбюффе Тьерри
МПК: C07D 209/42, C07K 5/06
Метки: периндоприла, фармацевтически, солей, способ, синтеза, приемлемых
Формула / Реферат:
1. Способ синтеза соединения формулы (I) и его фармацевтически приемлемых солей, который характеризуется тем, что соединение формулы (II) в которой R представляет собой атом водорода, или бензил, или линейную или разветвленную C1-С6-алкильную группу, подвергают реакции с соединением формулы (III), имеющим (R)-конфигурацию в которой G представляет собой атом хлора, брома или йода или гидрокси-, n-толуолсульфонилокси-, метансульфонилокси- или...
Способ синтеза периндоприла и его фармацевтически приемлемых солей

Номер патента: 5079
Опубликовано: 28.10.2004
Авторы: Тюрб Юг, Ланглуа Паскаль
МПК: C07K 1/02, C07D 209/42
Метки: фармацевтически, солей, периндоприла, способ, приемлемых, синтеза
Формула / Реферат:
1. Способ получения периндоприла формулы (I) а также его фармацевтически приемлемых солей, включающий взаимодействие сложного бензилового эфира формулы (IV) в которой Bn представляет собой бензильную группу, с соединением формулы (V) в этилацетате в присутствии 1-гидроксибензотриазола и дициклогексилкарбодиимида с последующим выделением полученного соединения формулы (VI) в которой Bn представляет собой бензильную группу, и удалением группы,...
Новый способ синтеза периндоприла и его фармацевтически приемлемых солей

Номер патента: 8626
Опубликовано: 29.06.2007
Авторы: Лекув Жан-Пьер, Дюбюффе Тьерри
МПК: C07K 5/02, C07K 5/06, C07D 209/42...
Метки: синтеза, новый, способ, солей, периндоприла, фармацевтически, приемлемых
Формула / Реферат:
1. Способ синтеза соединений формулы (I) и их фармацевтически приемлемых солей, характеризующийся тем, что соединение формулы (II) в которой R1 представляет собой бензил или линейную или разветвленную C1-С6-алькильную группу, подвергают реакции с соединением формулы (III), имеющим S-конфигурацию в которой X представляет собой атом галогена и R2 представляет собой защитную группу для функциональной аминогруппы, в присутствии основания, получая...
Новый способ синтеза периндоприла и его фармацевтически приемлемых солей

Номер патента: 8668
Опубликовано: 29.06.2007
Авторы: Дюбюффе Тьерри, Лекув Жан-Пьер
МПК: C07D 209/42, C07K 5/06, C07K 5/02...
Метки: способ, периндоприла, солей, фармацевтически, приемлемых, новый, синтеза
Формула / Реферат:
1. Способ синтеза соединений формулы (I) и их фармацевтически приемлемых солей, характеризующийся тем, что соединение формулы (II) в которой Вn представляет собой бензильную группу, подвергают реакции с соединением формулы (III), имеющим S-конфигурацию в которой X представляет собой атом галогена и ВОС представляет собой трет-бутоксикарбонильную группу, в присутствии основания, получая после снятия защиты с функциональной аминогруппы...
Предыдущий патент: Матричный материал из графита и неорганических связующих для захоронения радиоактивных отходов, способ его получения, обработки и применения
Следующий патент: Способ получения углеводородов биологического происхождения
Случайный патент: Композиции и способы ингибирования экспрессии гена pcsk9