Ингибиторы dpiv на основе глутаминила
Номер патента: 7434
Опубликовано: 27.10.2006
Авторы: Хоффманн Торстен, Демут Ханс-Ульрих, Хайзер Ульрих















Формула / Реферат
1. Соединение формулы

где X = СН2 или S, или его соль присоединения кислоты.
2. Соединение по п.1, где соль присоединения кислоты выбрана из группы, состоящей из солей соляной, бромисто-водородной, перхлорной, серной, азотной, фосфорной, уксусной, пропионовой, гликолевой, молочной, янтарной, малеиновой, фумаровой, яблочной, винной, лимонной, бензойной, миндальной, метансульфоновой, гидроксиэтансульфоновой, бензолсульфоновой, щавелевой, памовой, 2-нафталинсульфоновой, паратолуолсульфоновой, циклогексансульфаминовой, салициловой, сахариновой и трифторуксусной кислоты.
3. Соль присоединения кислоты соединения формулы

где X = СН2 или S и где соль присоединения кислоты выбрана из группы, состоящей из солей соляной, бромисто-водородной, перхлорной, азотной, пропионовой, гликолевой, молочной, малеиновой, яблочной, лимонной, бензойной, миндальной, метансульфоновой, гидроксиэтансульфоновой, бензолсульфоновой, щавелевой, памовой, 2-нафталинсульфоновой, паратолуолсульфоновой, циклогексансульфаминовой, салициловой, сахариновой и трифторуксусной кислоты.
4. Соединение по п.3, выбранное из гидрохлорида глутаминилтиазолидина или гидрохлорида глутаминилпирролидина.
5. Гидрохлорид глутаминилтиазолидина.
6. Гидрохлорид глутаминилпирролидина.
7. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и/или разбавитель и соединение по любому из пп.1-6.
8. Способ ингибирования дипептидилпептидазы IV или дипептидилпептидаза IV-подобной ферментной активности для предупреждения или лечения заболеваний или состояний, связанных с дипептидилпептидазой IV или дипептидилпептидаза IV-подобными ферментами, при котором млекопитающему, нуждающемуся в подобном лечении, вводят терапевтически эффективное количество соединения по любому из пп.1-6.
9. Способ по п.8, при котором снижают уровни глюкозы в крови у млекопитающих, являющиеся следствием приема пищи.
10. Способ по п.8 или 9, при котором предупреждают или лечат заболевания или состояния, выбранные из группы, состоящей из инсулиннезависимого сахарного диабета, артрита, ожирения, иммунных и аутоиммунных расстройств, аллотрансплантации, рака, нейрональных расстройств и кожных заболеваний.
11. Способ по п.10, при котором предупреждают или лечат инсулиннезависимый сахарный диабет.
12. Продукт превращения соединения по пп.1, 3 или 5, имеющий формулу

Текст
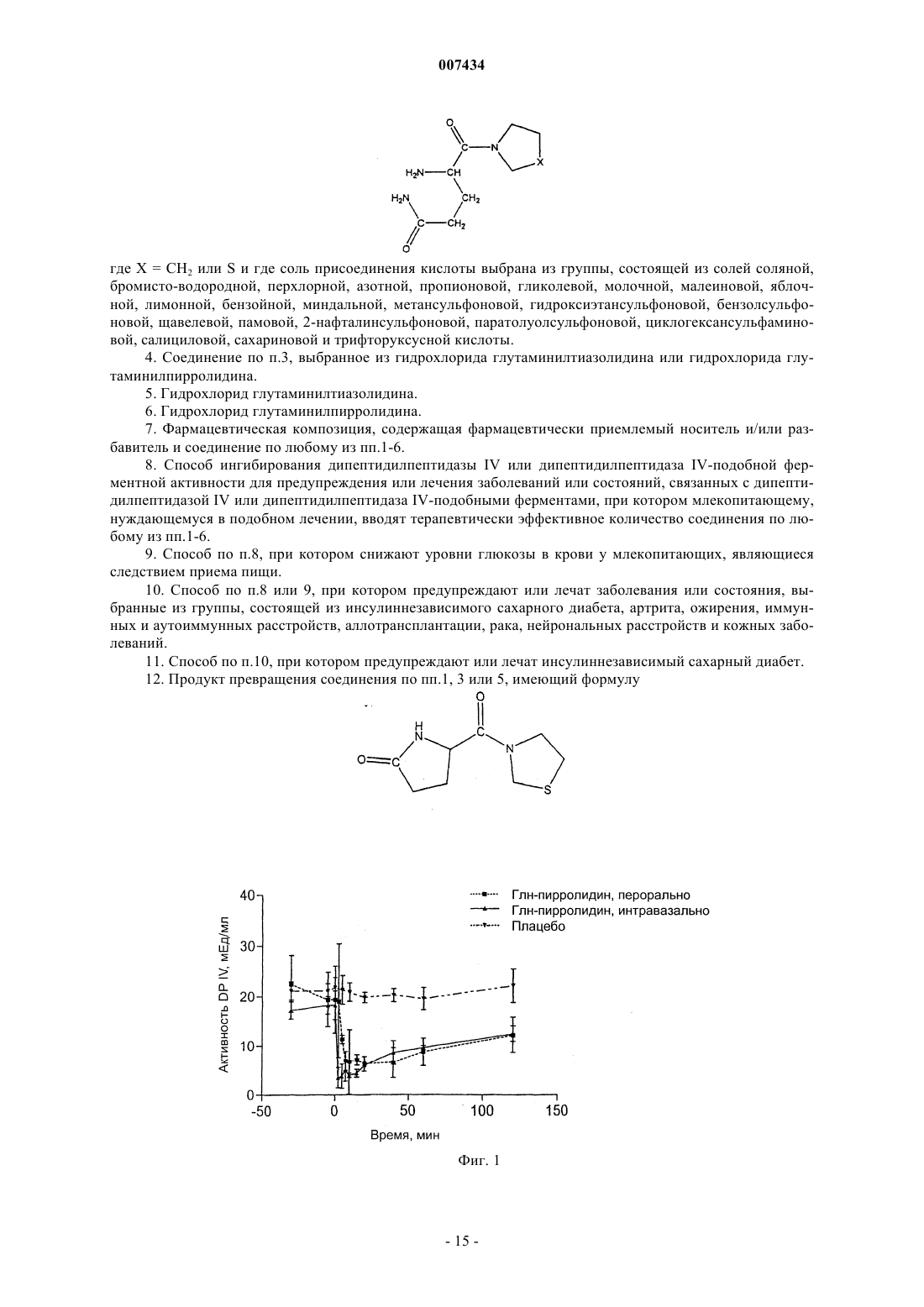
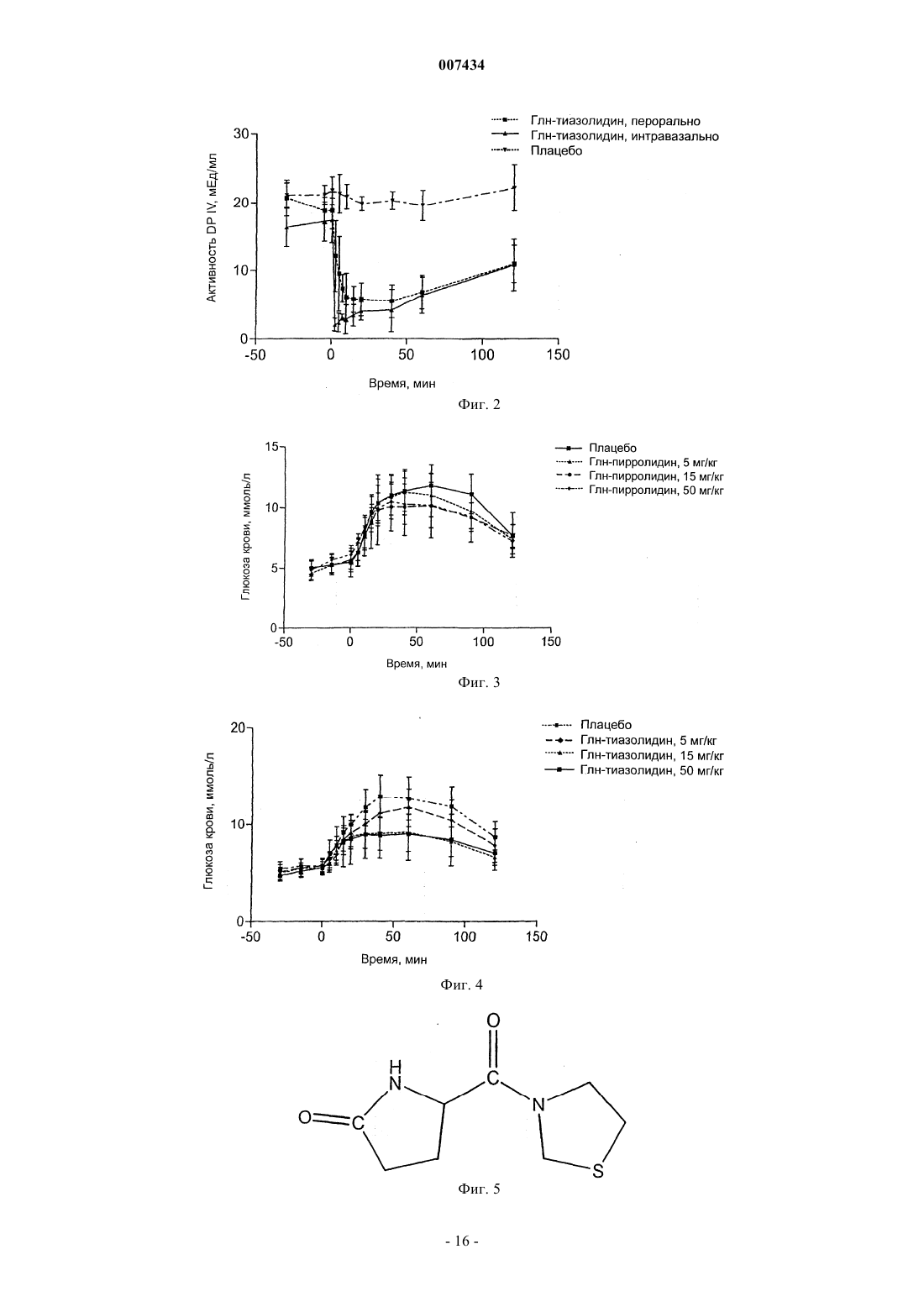
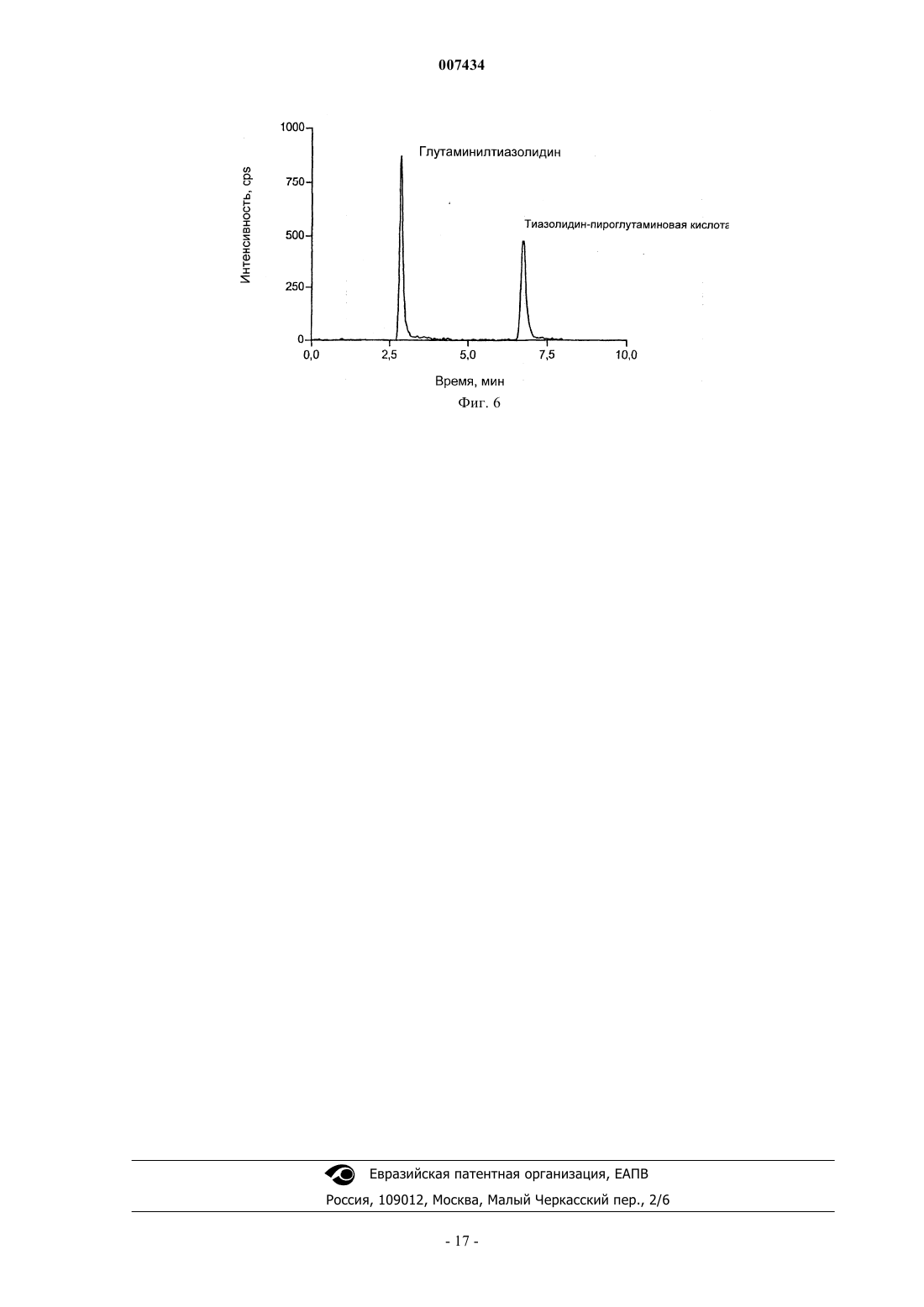
007434 Настоящее изобретение относится к области ингибирования дипептидилпептидазы IV и, более конкретно, относится к глутаминилпирролидину и глутаминилтиазолидину, фармацевтическим композициям, содержащим указанные соединения, и применению указанных соединений при ингибировании дипептидилпептидазы IV и дипептидилпептидаза IV-подобной ферментной активности. Предшествующий уровень техники Дипептидилпептидаза IV (DPIV) представляет собой сериновую протеазу, которая отщепляет Nтерминальные дипептиды от пептидной цепи, содержащей предпочтительно пролиновый остаток в предпоследнем положении. Хотя биологическая роль DRIV в системах млекопитающих до конца не установлена, считают, что она играет важную роль в метаболизме нейропептидов, активации Т-клеток, присоединении раковых клеток к эндотелию и проникновении ВИЧ в лимфоидные клетки. Более того, было обнаружено, что DRIV отвечает за инактивацию глюкагоноподобного пептида-1(GLP-1) и глюкозозависимого инсулинотропного пептида, также известного как желудочный ингибиторный пептид (GIP). Так как GLP-1 является главным стимулятором секреции панкреатического инсулина и оказывает прямое благоприятное воздействие на утилизацию глюкозы, в WO 97/40832 и US 6,303,661 было показано, что ингибирование DPIV и DPIV-подобной ферментной активности представляет собой привлекательный подход, например, к лечению инсулиннезависимого сахарного диабета (NIDDM). Это один аспект настоящего изобретения, в котором предложены новые ингибиторы DPIV, которые эффективны, например, при лечении состояний, опосредованных при помощи DPIV и DPIV-подобных ферментов, фармацевтические композиции, например, полезные при ингибировании DPIV и DPIV-подобных ферментов и способ ингибирования указанной ферментной активности. Другой аспект изобретения относится к способу лечения, в частности, к способу лечения сахарного диабета, особенно инсулиннезависимого сахарного диабета (NIDDM) или диабета 2 типа и состояний,связанных с сахарным диабетом, и к композициям для применения в подобном способе. Дипептидилпептидаза IV (DPIV) представляет собой сериновую протеазу, отщепляющую за остатком пролина (в меньшей степени аланина, серина или глицина), обнаруженную в различных тканях тела,включая почки, печень и кишечник. Известно, что ингибиторы DPIV могут быть полезны при лечении нарушенной толерантности к глюкозе и сахарного диабета (Международная заявка на патент, номер публикации WO 99/61431, Pederson RA и др.,Diabetes. 1998 Aug; 47(8):1253-8 и Pauly RP и др., Metabolism. 1999 Маr; 48(3):385-9). В частности, в WO 99/61431 раскрыты ингибиторы DPIV, содержащие аминокислотный остаток и тиазолидиновую или пирролидиновую группу, и их соли, особенно L-трео-изолейцилтиазолидин, L-алло-изолейцилтиазолидин, L-треоизолейцилпирролидин, L-алло-изолейцилтиазолидин, L-алло-изолейцилпирролидин, и их соли. Дополнительными примерами низкомолекулярных ингибиторов дипептидилпептидазы IV являются агенты, такие как производные тетрагидроизохинолин-3-карбоксамида, N-замещенные 2-цианопирролы и -пирролидины, N-(N'-замещенные глицил)-2-цианопирролидины, (N-замещенные глицил)тиазолидины,N-(замещенный глицил)-4-цианотиазолидины, борониловые ингибиторы и пирролидины, конденсированные с циклопропилом. Ингибиторы дипептидилпептидазы IV описаны в US 6,011,155; US 6,107,317;WO 01/68603; WO 01/40180; WO 01/81337; WO 01/81304; WO 01/55105 и WO 02/02560, содержание которых, касающееся ингибиторов, их получения и их применения, включено сюда полностью посредством этой ссылки. Краткое описание изобретения Согласно настоящему изобретению предложено соединение формулы где X = СН 2 или S, или его фармацевтически приемлемая соль. Такие соединения и формы их соответствующих фармацевтически приемлемых солей присоединения кислоты являются полезными при лечении состояний, опосредованных при помощи DPIV и DPIVподобных ферментов, таких как артрит, ожирение, иммунные и аутоиммунные расстройства, аллотрансплантация, рак, нейрональные расстройства и кожные заболевания. В более предпочтительном воплощении соединения по настоящему изобретению улучшают толерантность к глюкозе при помощи снижения повышенных уровней глюкозы в крови в ответ на пероральное введение глюкозы и поэтому являются полезными при лечении инсулиннезависимого сахарного-1 007434 диабета. Краткое описание чертежей Дальнейшее понимание настоящего изобретения может быть получено при обращении к фигурам,где фиг. 1 показывает активность плазменной DPIV в сыворотке крыс линии Wistar после перорального и интравазального введения 100 мг/кг массы тела глутаминилпирролидина; фиг. 2 - активность плазменной DPIV в сыворотке крыс линии Wistar после перорального и интравазального введения 100 мг/кг массы тела глутаминилтиазолидина; фиг. 3 - дозозависимое снижение уровней глюкозы в крови крыс линии Zucker, больных диабетом после перорального введения 5 мг/кг, 15 мг/кг, 50 мг/кг массы тела глутаминилпирролидина и плацебо,соответственно; фиг. 4 - дозозависимое снижение уровней глюкозы в крови крыс линии Zucker, больных диабетом после перорального введения 5 мг/кг, 15 мг/кг, 50 мг/кг массы тела глутаминилтиазолидина и плацебо,соответственно; фиг. 5 - химическую структуру пироглутамилтиазолидина, продукта превращения, обнаруженного после перорального введения глутаминилтиазолидина крысам линии Wistar, и фиг. 6 показывает хроматограмму экстракта плазмы крови крыс, полученного после перорального введения глутаминилтиазолидина крысам линии Zucker с ожирением. Пик на 2,95 мин соответствует глутаминилтиазолидину, а пик на 6,57 мин соответствует пироглутамилтиазолидину. Подробное описание изобретения Настоящее изобретение относится к области ингибирования дипептидилпептидазы IV (DPIV) и, более конкретно, относится к глутаминилпирролидину и глутаминилтиазолидину, фармацевтическим композициям, содержащим указанные соединения, и применению указанных соединений при ингибировании DPIV и DPIV-подобной ферментной активности. Согласно настоящему изобретению предложены новые ингибиторы DPIV, которые эффективны,например, при лечении состояний, опосредованных при помощи DPIV, фармацевтические композиции,например, полезные при ингибировании DPIV и DPIV-подобной ферментной активности и способ ингибирования DPIV и DPIV-подобной ферментной активности. Согласно настоящему изобретению предложено соединение формулы и особенно соединение формулы (I) или его фармацевтически приемлемая соль. Кроме того, предпочтительным соединением по настоящему изобретению является соединение формулы (II) или его фармацевтически приемлемая соль. Соединения по настоящему изобретению могут быть превращены в соли присоединения кислоты,особенно в фармацевтически приемлемые соли присоединения кислоты. Фармацевтически приемлемая соль обычно принимает форму, в которой основная боковая цепь аминокислот протонирована неорганической или органической кислотой. Типичные органические или неорганические кислоты включают в себя соляную, бромисто-водородную, перхлорную, серную, азотную, фосфорную, уксусную, пропионовую, гликолевую, молочную, янтарную, малеиновую, фумаровую, яблочную, винную, лимонную, бензойную, миндальную, метансульфоновую, гидроксиэтансульфоновую, бензолсульфоновую, щавелевую,памовую, 2-нафталинсульфоновую, пара-толуолсульфоновую, циклогексансульфаминовую, салициловую, сахариновую или трифторуксусную кислоту. Все формы фармацевтически приемлемых солей присоединения кислоты соединений по настоящему изобретению предназначены для включения в объем данного изобретения. Принимая во внимание тесную взаимосвязь между свободными соединениями и соединениями в форме солей, когда бы соединение ни упоминалось в данном контексте, подразумевается также и соответствующая соль, при условии, что такое является возможным или целесообразным при данных обстоятельствах. Далее настоящее изобретение включает в себя в пределах своего объема пролекарства соединений по данному изобретению. Обычно такие пролекарства являются функциональными производными соединений, которые легко превращаются in vivo в требуемое терапевтически активное соединение. Таким образом, в этих случаях для способов лечения по настоящему изобретению, термин "введение" охватывает лечение различных описанных расстройств, пролекарственными вариантами одного или более чем одного заявленного соединения, но которое превращается в вышеуказанное конкретное соединение invivo после введения субъекту. Традиционные методики подбора и получения подходящих пролекарственных производных описаны, например, в "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985 и заявках на патент DE 198 28 113, DE 198 28 114, WO 99/67228 и WO 99/67279, которые включены сюда посредством ссылки. В тех случаях, когда соединения согласно данному изобретению имеют по меньшей мере один хиральный центр, они, соответственно, могут существовать в виде энантиомеров. В тех случаях, когда соединения обладают двумя или более хиральными центрами, они, дополнительно, могут существовать в виде диастереомеров. Следует понимать, что все подобные изомеры и их смеси включены в объем настоящего изобретения. Более того, некоторые из кристаллических форм соединений могут существовать в виде полиморфов и как таковые подразумеваются включенными в настоящее изобретение. Кроме того,некоторые из соединений могут образовывать сольваты с водой (т. е. гидраты) или обычными органическими растворителями, и подразумевается, что такие сольваты также включены в объем данного изобретения. Соединения, включая их соли, также могут быть получены в форме гидратов или включать в себя другие растворители, использованные для их кристаллизации. Как указано выше, соединения по настоящему изобретению, и особенно соединения формул I и II, и формы их соответствующих фармацевтически приемлемых солей присоединения кислоты являются полезными при ингибировании DPIV и DPIV-подобной ферментной активности. Способность соединений по настоящему изобретению и форм их соответствующих фармацевтически приемлемых солей присоединения кислоты ингибировать DPIV и DPIV-подобную ферментную активность можно продемонстрировать, используя анализ активности DPIV для определения значений Ki in vitro и в плазме человека, как описано в примерах 4 и 5. Значения Ki соединений по настоящему изобретению определяли для глутаминилтиазолидина как Ki = 3,12x10-7 М 5,11x10-10 М и для глутаминилпирролидина как Ki = 1,30x10-6 М 8,49x10-8 М в отношении DPIV из почек свиньи. Значения Ki соединений по настоящему изобретению определяли для глутаминилтиазолидина как Ki = 4,03x10-7 М 2,19x10-10 М после 5 мин и 5,13x10-7 М 1,26 х 10-8 М после 22 ч преинкубации, и для глутаминилпирролидина как Ki = 1,30x10-6 М 4,89 х 10-8 М после 5 мин и 1,36 х 10-6 М 3,21x10-8 М после 22 ч преинкубации в плазме человека. Способность соединений по настоящему изобретению и форм их соответствующих фармацевтически приемлемых солей присоединения кислоты ингибировать DPIV in vivo можно продемонстрировать-3 007434 при помощи перорального или интравазального (внутрисосудистого) введения крысам линии Wistar, как описано в примере 9. Соединения по настоящему изобретению ингибируют активность DPIV in vivo как после перорального, так и после интравазального введения крысам линии Wistar.DPIV присутствует в широком ряде органов и тканей млекопитающих, например щеточной каемке кишечника (Gutschmidt S. et al., "In situ" - measurements of protein contents in the brush border region alongmonoclonal antibody detecting dipeptidylpeptidase IV in human tissue. Virchows Arch. A. Pathol. Anat. Histopathol. 1986; 409 (2):263-73), нервных клетках, латеральных мембранах некоторых поверхностных эпителиев, например, фаллопиевых трубах, матке и везикулярных железах, в цитоплазме люминальных клеток, например, эпителия везикулярных желез, и в слизистых клетках бруннеровых желез (Hartel S. et al.,Dipeptidyl peptidase (DPP) IV in rat organs. Comparison of immunohistochemistry and activity histochemistry.Histochemistry 1988; 89 (2): 151-61), половых органах, например хвосте придатка яичка и ампуле, семенных пузырьках и их секретах (AgrawalVanha-Perttula, Dipeptidyl peptidases in bovine reproductive organsand secretions. Int. J. Androl. 1986, 9 (6): 435-52). В сыворотке человека присутствуют две молекулярные формы дипептидилпептидазы (Krepela E. et al., Demonstration of two molecular forms of dipeptidyl peptidase IV in normal human serum. Physiol. Bohemoslov. 1983, 32 (6): 486-96). Высокомолекулярная форма сывороточной DPIV экспрессируется на поверхности активированных Т-клеток (Duke-Cohan J.S. et al.,Serum high molecular weight dipeptidyl peptidase IV (CD26) is similar to a novel antigen DPPT-L releasedfrom activated T cells. J. Immunol. 1996, 156(5): 1714-21). Соединения по настоящему изобретению и формы их соответствующих фармацевтически приемлемых солей присоединения кислоты способны ингибировать DPIV in vivo. В одном воплощении настоящего изобретения все молекулярные формы, гомологи и эпитопы DPIV из всех тканей и органов млекопитающих, а также те, которые еще не открыты, предназначены для включения в объем данного изобретения. Первоначально предполагали, что среди редкой группы пролин-специфичных протеаз DPIV является единственным мембранно-связанным ферментом, специфичным для пролина как предпоследнего остатка в амино (N) конце полипептидной цепи. Однако недавно были установлены другие молекулы даже структурно негомологичные DPIV, но обладающие соответствующей ферментной активностью. DPIVподобными ферментами, которые установлены недавно, являются, например, белок активации фибробластов , дипептидилпептидаза IV , дипептидиламинопептидаза-подобный белок, N-ацетилированная(SedoMalik, Dipeptidyl peptidase IV-like molecules: homologous proteins or homologous activities Biochimica et Biophysica Acta 2001, 36506: 1-10). Кроме того, DPIV-подобные ферменты раскрыты в WO 01/19866, WO 02/04610, WO 02/34900 и WO 02/31134. В WO 01/19866 раскрыта новая дипептидиламинопептидаза (DPP8) человека со структурными и функциональными сходствами с DPIV и белком активации фибробластов (FAP). В WO 02/04610 предложены реагенты, которые регулируют дипептидилпептидаза IV-подобный фермент человека и реагенты, которые связываются с генным продуктом дипептидилпептидаза IV-подобного фермента человека. Эти реагенты могут играть роль в предупреждении, улучшении или корректировке дисфункций или заболеваний, которые включают в себя опухоли и расстройства периферической и центральной нервной системы, включая боль и нейродегенеративные расстройства, но не ограничиваются ими. Дипептидилпептидаза IV-подобный фермент из WO 02/04610 хорошо известен в данной области техники. В базе данных банка генов этот фермент зарегистрирован как KIAA1492 (регистрация в феврале 2001, представлен на рассмотрение 4 апреля 2000 г., АВ 040925). В WO 02/34900 раскрыта дипептидилпептидаза 9(DPP9) со значительной гомологией с аминокислотными последовательностями DPIV и DPP8. В WO 02/31134 раскрыты три DPIV-подобных фермента, DPRP1, DPRP2 и DPRP3. Анализ последовательности показал, что DPRP1 идентичен DPP8, который раскрыт в WO 01/19866, что DPRP2 идентичен DPP9 и чтоDPRP3 идентичен KIAA1492, который раскрыт в WO 02/04610. В другом предпочтительном воплощении настоящего изобретения все молекулярные формы, гомологи и эпитопы протеинов, обладающих DPIV-подобной ферментной активностью, из всех тканей и органов млекопитающих, а также те, которые еще не открыты, предназначены для включения в объем данного изобретения. Способность соединений по настоящему изобретению и форм их соответствующих фармацевтически приемлемых солей присоединения кислоты ингибировать DPIV-подобные ферменты можно продемонстрировать, используя анализ ферментной активности для определения значений Ki in vitro, как описано в примере 6. Значения Ki соединений по настоящему изобретению в отношении свиной дипептидилпептидазы II определяли как Ki = 8,52x10-5 М 6,33x10-6 М для глутаминилпирролидина и Ki = 1,07x10-5 М 3,81x10-7 М для глутаминилтиазолидина. Все соединения ингибируют свиную дипептидилпептидазу II.-4 007434 В другом воплощении настоящего изобретения соединения по настоящему изобретению и формы их соответствующих фармацевтически приемлемых солей присоединения кислоты обладают лишь небольшой, если вообще обладают, ингибиторной активностью в отношении не-DPIV и не-DPIV-подобных пролин-специфичных ферментов. Как описано в примере 7, не обнаружено ингибирования дипептидилпептидазы I и пролилолигопептидазы глутаминилтиазолидином и глутаминилпирролидином. В отношении пролидазы оба соединения проявили значительно более низкую эффективность, чем в отношенииDPIV. Значения IС 50 в отношении пролидазы определяли как IC503 мМ для глутаминилтиазолидина и как IС 50 = 3,4x10-4 М 5,63x10-5 для глутаминилпирролидина. Принимая во внимание способность ингибировать DPIV и DPIV-подобную ферментную активность, соединения по настоящему изобретению, особенно соединения формул I и II, и формы их соответствующих фармацевтически приемлемых солей присоединения кислоты являются полезными при лечении состояний, опосредованных при помощи указанной ферментной активности. Таким образом, соединения, раскрытые здесь, являются полезными при лечении состояний, таких как инсулиннезависимый сахарный диабет, артрит, ожирение, иммунные и аутоиммунные расстройства, аллотрансплантация, рак,нейрональные расстройства подобные рассеянному склерозу и кожные заболевания. В более предпочтительном воплощении настоящего изобретения соединения по настоящему изобретению и формы их соответствующих фармацевтически приемлемых солей присоединения кислоты улучшают толерантность к глюкозе при помощи снижения повышенных уровней глюкозы в крови в ответ на пероральное введение глюкозы и поэтому являются полезными при лечении инсулиннезависимого сахарного диабета. Способность соединений по настоящему изобретению и форм их соответствующих фармацевтически приемлемых солей присоединения кислоты улучшать толерантность к глюкозе в ответ на пероральное введение глюкозы можно оценить на крысах линии Zucker, больных диабетом. Эти способы описаны в примерах 10 и 11. Пероральное введение 5 мг/кг массы тела, 15 и 50 мг/кг массы тела глутаминилтиазолидина или глутаминилпирролидина приводит к дозозависимому снижению повышенных уровней глюкозы в крови и, таким образом, улучшению толерантности к глюкозе у крыс Zucker с диабетом. Соединения по настоящему изобретению, согласно примеру 5, стабильны в плазме человека. Неожиданно, и в качестве дальнейшего предпочтительного воплощения настоящего изобретения, соединения по настоящему изобретению и формы их соответствующих фармацевтически приемлемых солей присоединения кислоты могут претерпевать превращение in vivo после введения млекопитающему. Способность соединений по настоящему изобретению и форм их соответствующих фармацевтически приемлемых солей присоединения кислоты претерпевать превращение in vivo можно продемонстрировать,используя модель крыс линии Wistar и последующий LC/MS анализ (жидкостная хроматография/массспектрометрия). Установлено, что глутаминилтиазолидин превращается после перорального введения крысам линии Wistar в соответствующий пироглутамилтиазолидин. Поэтому, согласно настоящему изобретению предложен способ лечения состояния, опосредованного при помощи модуляции DPIV и DPIV-подобной ферментной активности, у субъекта, которому это необходимо, при котором вводят любое из соединений по настоящему изобретению или их фармацевтические композиции в количестве и режиме дозирования терапевтически эффективном для лечения состояния. Кроме того, настоящее изобретение включает в себя применение соединений по настоящему изобретению и форм их соответствующих фармацевтически приемлемых солей присоединения кислоты для приготовления лекарства для предупреждения или лечения состояния, опосредованного при помощи модуляции активности DPIV, у субъекта. Соединение можно вводить пациенту посредством традиционного пути введения, которые включают в себя внутривенный, пероральный, подкожный, внутримышечный, интрадермальный и парентеральный, но не ограничиваются ими. В дальнейшем воплощении согласно настоящему изобретению предложены прописи для соединений по настоящему изобретению и форм их соответствующих фармацевтически приемлемых солей присоединения кислоты в фармацевтических композициях. Термин "субъект", который использован здесь, относится к животному, предпочтительно млекопитающему, более предпочтительно к человеку, который является объектом лечения, наблюдения или эксперимента. Термин "терапевтически эффективное количество", который использован здесь, означает такое количество активного соединения или фармацевтического агента, которое вызывает биологический или лечебный эффект в тканевой системе, у животного или человека, устанавливаемое исследователем, ветеринаром, врачом или другим клиницистом, которое включает в себя облегчение симптомов заболевания или расстройства, которое лечат. Термин "композиция", который использован здесь, предназначен для описания продукта, содержащего заявленные соединения в терапевтически эффективных количествах, а также любой продукт, который прямо или косвенно является следствием сочетаний заявленных соединений. Для приготовления фармацевтических композиций по данному изобретению, одно или более чем одно соединение по настоящему изобретению, особенно соединение формул I или II, или форму его соответствующей фармацевтически приемлемой соли присоединения кислоты в качестве активного ингре-5 007434 диента тщательно смешивают с фармацевтическим носителем в соответствии с традиционными фармацевтическими методиками приготовления. Такой носитель может принимать большое многообразие форм, в зависимости от формы требуемого для введения препарата, например, пероральную или парентеральную, такую как внутримышечная. При приготовлении композиций в пероральной лекарственной форме могут быть использованы любые обычные фармацевтические среды. Таким образом, для жидких пероральных препаратов, таких как, например, суспензии, эликсиры и растворы, подходящие носители и добавки могут преимущественно включать в себя воду, гликоли, масла, спирты, ароматизаторы, консерванты, красители и тому подобное; для твердых пероральных препаратов, таких как, например, порошки,капсулы, желатиновые капсулы и таблетки, подходящие носители и добавки включают в себя крахмалы,сахара, разбавители, гранулирующие агенты, смазывающие вещества, связующие вещества, разрыхлители и тому подобное. Из-за своего удобства при введении таблетки и капсулы представляют собой наиболее пригодную пероральную стандартную лекарственную форму, в этом случае используются твердые фармацевтические носители. Если требуется, таблетки могут быть покрыты сахаром или энтеросолюбильным покрытием при помощи стандартных методик. Для парентеральных препаратов носитель будет обычно содержать стерильную воду, хотя могут быть включены и другие ингредиенты, например, для целей, таких как вспомогательная растворимость или для консервации. Инъекционные суспензии также могут быть приготовлены, и в этом случае могут быть использованы соответствующие жидкие носители, суспендирующие агенты и тому подобное. Фармацевтические композиции при этом будут содержать на единицу дозировки, например на таблетку, капсулу, порошок,инъекцию, чайную ложку и тому подобное, количество активного ингредиента, необходимое для доставки эффективной дозы, как описано выше. Фармацевтические композиции при этом будут содержать на единицу дозировки, например на таблетку, капсулу, порошок, инъекцию, суппозиторий, чайную ложку и тому подобное, от примерно 0,01 до примерно 1000 мг (предпочтительно от примерно 5 до примерно 500 мг) и могут быть даны в дозировке от примерно 0,1 до примерно 300 мг/кг массы тела в день (предпочтительно от 1 до 50 мг/кг в день). Дозировки, однако, могут варьироваться в зависимости от потребности пациентов, тяжести состояния, которое лечат, и соединения, которое используют. Можно использовать либо суточное введение, либо дозы, повторяемые через определенное время. Типично дозировка будет регулироваться врачом на основании особенностей пациента, его/ее состояния и требуемого терапевтического эффекта. Предпочтительно эти композиции находятся в стандартных лекарственных формах, таких как таблетки, пилюли, капсулы, порошки, гранулы, стерильные парентеральные растворы или суспензии, дозированные аэрозоли или жидкие спреи, капли, ампулы, автоинжекторы или суппозитории, для перорального, парентерального, интраназального, сублингвального или ректального введения или для введения посредством ингаляции или инсуффляции. Альтернативно композиция может быть представлена в форме, подходящей для введения один раз в неделю или один раз в месяц; например, нерастворимая соль активного соединения, такая как деканоат, может быть адаптирована, чтобы обеспечить создание депо для внутримышечной инъекции. Для приготовления твердых композиций, таких как таблетки, основной активный ингредиент идеально смешивают с фармацевтическим носителем, например традиционными таблетирующими ингредиентами, такими как кукурузный крахмал, лактоза, сахароза, сорбит, тальк,стеариновая кислота, стеарат магния, двухкальциевый фосфат или камедь, и другими фармацевтическими разбавителями, например водой, для создания твердой композиции предварительного препарата, содержащей гомогенную смесь соединения по настоящему изобретению или его фармацевтически приемлемой соли. Описывая эти композиции предварительных препаратов как гомогенные, подразумевают,что активный ингредиент идеально диспергирован равномерно по всей композиции, так что композиция может быть легко разделена на равные эффективные лекарственные формы, такие как таблетки, пилюли и капсулы. Эта твердая композиция предварительного препарата может быть затем разделена на стандартные лекарственные формы по типу, описанному выше, содержащие от примерно 0,1 до примерно 1000 мг, предпочтительно от примерно 5 до примерно 500 мг активного ингредиента по настоящему изобретению. Таблетки или пилюли новой композиции предпочтительно могут быть покрыты или иным образом составлены, чтобы обеспечить лекарственную форму, дающую преимущество в виде пролонгированного действия. Например, таблетка или пилюля может содержать внутренний и внешний дозируемый компонент, причем последний находится в виде оболочки над первым. Эти два компонента могут быть разделены энтеросолюбильным слоем, который служит для противодействия разрушению в желудке и позволяет внутреннему компоненту проходить неповрежденным в двенадцатиперстную кишку или высвобождаться замедленно. Ряд веществ может быть использован для таких энтеросолюбильных слоев или покрытий, при этом такие вещества включают в себя ряд полимерных кислот с такими веществами как шеллак, цетиловый спирт и ацетат целлюлозы. Жидкие формы, в которые новые композиции по настоящему изобретению преимущественно могут быть включены для введения перорально или посредством инъекции, включают в себя водные растворы,подходящие ароматизированные сиропы, водные или масляные суспензии и ароматизированные эмульсии с пищевыми маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисо-6 007434 вое масло, а также эликсиры и сходные фармацевтические носители. Подходящие диспергирующие или суспендирующие агенты для водных суспензий включают в себя синтетические или природные камеди,такие как трагакант, гуммиарабик, альгинат, декстран, карбоксиметилцеллюлоза натрия, метилцеллюлоза, поливинилпирролидон или желатин. В тех случаях, когда способы получения соединений согласно изобретению приводят к получению смеси стереоизомеров, эти изомеры могут быть разделены при помощи традиционных методик, таких как препаративная хроматография. Соединения могут быть получены в рацемической форме или отдельные энантиомеры могут быть получены либо при помощи энантиоспецифичного синтеза, либо посредством разделения. Соединения, например, могут быть разделены на их составляющие энантиомеры при помощи стандартных методик, таких как образование диастереомерных пар посредством образования соли с оптически активной кислотой, такой как (-)-ди-паратолуоил-D-винная кислота и/или (+)-ди-паратолуоил-L-винная кислота, с последующей фракционной кристаллизацией и регенерацией свободного основания. Соединения также могут быть разделены при помощи образования диастереомерных эфиров или амидов с последующим хроматографическим разделением и удалением хирального вспомогательного вещества. Альтернативно соединения могут быть разделены при использовании хиральной колонки ВЭЖХ (высокоэффективной жидкостной хроматографии). При любом из способов получения соединений по настоящему изобретению может быть необходимым и/или желательным защищать чувствительные или активные группы в любых из упомянутых молекул. Это может быть достигнуто при помощи традиционных защитных групп, таких как описано в Protective Group in Organic Chemistry, ed. J.F.W. McOmie, Plenum Press, 1973; и T.W. GreeneP.G.M. Wuts,Protective Group in Organic Synthesis, John WileySons, 1991, упомянутые здесь посредством ссылки. Защитные группы могут быть удалены на любой подходящей последующей стадии, используя способы,известные в данной области техники. Способ лечения состояний, модулируемых дипептидилпептидазой IV и DPIV-подобными ферментами, описанный в настоящем изобретении, также может быть осуществлен при использовании фармацевтической композиции, содержащей одно или более чем одно соединение, которое определено здесь, и фармацевтически приемлемый носитель. Фармацевтическая композиция может содержать между примерно 0,01 и 1000 мг, предпочтительно от примерно 5 до примерно 500 мг соединений и может быть составлена в любой форме, подходящей для выбранного способа введения. Носители включают в себя необходимые и инертные фармацевтические эксципиенты, которые включают в себя связующие вещества,суспендирующие агенты, смазывающие вещества, ароматизаторы, подслащивающие вещества, консерванты, красители и покрытия, но не ограничиваются ими. Композиции, подходящие для перорального введения, включают в себя твердые формы, такие как пилюли, таблетки, каплеты, капсулы (каждая включает в себя препараты с немедленным высвобождением, отсроченным высвобождением и продолжительным высвобождением), гранулы и порошки, и жидкие формы, такие как растворы, сиропы, эликсиры, эмульсии и суспензии. Формы, полезные для парентерального введения включают в себя стерильные растворы, эмульсии и суспензии. Преимущественно соединения по настоящему изобретению могут вводиться в однократной суточной дозе, или суммарная суточная доза может вводиться разделенными дозами два, три или четыре раза в день. Кроме того, соединения по настоящему изобретению могут вводиться в интраназальной форме посредством местного применения подходящих интраназальных носителей или посредством трансдермальных кожных пластырей, хорошо известных специалистам в данной области техники. Для введения в форме трансдермальной системы доставки введение дозы будет, конечно, непрерывным, а не дробным на протяжении режима дозирования, и интенсивность дозировки необходимо будет соответственно корректировать, чтобы получить требуемые терапевтические эффекты. Более предпочтительно, для перорального введения в форме таблетки или капсулы, активный компонент лекарственного средства может быть комбинирован с пероральным, нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и тому подобное. Более того,когда требуется или необходимо, подходящие связующие вещества, смазывающие вещества, разрыхлители и красители могут также быть включены в смесь. Подходящие связующие вещества включают в себя, без ограничения, крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, подслащивающие вещества кукурузы, природные и синтетические камеди, такие как гуммиарабик, трагакант, или олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное. Разрыхлители включают в себя, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и тому подобное. Для жидких форм подходят ароматизированные суспендирующие или диспергирующие агенты, такие как синтетические или природные камеди, например, трагакант, гуммиарабик, метилцеллюлоза и тому подобное. Для парентерального введения требуются стерильные суспензии и растворы. Изотонические препараты, которые обычно содержат подходящие консерванты, используются, когда требуется внутривенное введение. Соединение по настоящему изобретению также может быть введено в форме липосомальных систем доставки, таких как однослойные везикулы, большие однослойные везикулы и многослойные вези-7 007434 кулы. Липосомы могут быть образованы из множества фосфолипидов, таких как холестерин, стериламин или фосфатидилхолины, используя способы, хорошо описанные в данной области техники. Соединения по настоящему изобретению также могут быть соединены с растворимыми полимерами в качестве носителей лекарственных средств с целевой доставкой. Такие полимеры могут включать в себя поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидофенол, полигидроксиэтиласпартамидофенол или полиэтиленоксидполилизин, замещенный пальмитоиловым остатком. Кроме того, соединения по настоящему изобретению могут быть соединены с классом биоразрушаемых полимеров, полезных для достижения контролируемого высвобождения лекарственного средства, например, с полиактиковой кислотой, полиэпсилонкапролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианакрилатами и поперечно-сшитыми или амфипатическими блоксополимерами гидрогелей. Соединения по данному изобретению могут быть введены в любых вышеупомянутых композициях и в соответствии с режимами дозирования, установленными в данной области техники, когда бы ни потребовалось лечение расстройств, для терапии которых они предназначены. Суточная дозировка продуктов может варьироваться в широком диапазоне от 0,01 до 1000 мг на взрослого человека в день. Для перорального введения, композиции предпочтительно предлагаются в форме таблеток, содержащих 0,01; 0,05; 0,1; 0,5; 1,0; 2,5; 5,0; 10,0; 15,0; 25,0; 50,0; 100; 150; 200; 250; 500 и 1000 мг активного ингредиента для симптоматического регулирования дозировки для пациента, которого подвергают лечению. Эффективное количество лекарственного средства обычно вводится при уровне дозировки от примерно 0,1 до примерно 300 мг/кг массы тела в день. Предпочтительно, диапазон составляет от примерно 1 до примерно 50 мг/кг массы тела в день. Соединения могут быть введены в режиме от 1 до 4 раз в день. Оптимальные дозировки для введения могут быть легко определены специалистом в данной области техники и будут варьироваться в зависимости от конкретного используемого соединения, способа введения, активности лекарственного препарата, биодоступности, обусловленной способом введения, и развития болезненного состояния. Кроме того, факторы, связанные с конкретным пациентом, которого подвергают лечению, включающие возраст пациента, вес, диету и время введения обычно следует учитывать при подборе дозировок. Соединения или композиции по настоящему изобретению можно принимать перед едой, во время еды или после еды. При приеме перед едой, соединения или композиции по настоящему изобретению можно принимать за 1 ч, предпочтительно за 30 или даже 15 или 5 мин перед едой. При приеме во время еды, соединения или композиции по настоящему изобретению можно смешивать с едой или принимать в виде самостоятельной лекарственной формы, как описано выше. При приеме после еды, соединения или композиции по настоящему изобретению можно принимать через 5, 15 или 30 мин или даже через 1 ч после еды. Примеры Пример 1. Синтез свободного основания глутаминилпирролидина АцилированиеN-Бензилоксикарбонилглутамин (2,02 г; 7,21 ммоль) растворяли в 35 мл ТГФ (тетрагидрофурана) и доводили до -15 С. В эту смесь добавляли CAIBE (изобутилхлорформиат) (0,937 мл; 7,21 ммоль) и 4 метилморфолин (0,795 мл; 7,21 ммоль) и раствор перемешивали в течение 15 мин. Образование смешанного ангидрида проверяли при помощи ТСХ (тонкослойной хроматографии) (элюент: CHCl3/MeOH: 9/1). После нагревания до -10 С добавляли пирролидин (0,596 мл; 7,21 ммоль). Смесь доводили до комнатной температуры и перемешивали в течение ночи. Обработка Образовавшийся осадок отфильтровывали и выпаривали растворитель. Полученное масло собирали в этилацетат (20 мл) и промывали насыщенным раствором гидросульфата натрия, а затем насыщенным раствором гидрокарбоната натрия, водой и рассолом. Органический слой отделяли, сушили и упаривали. Чистоту полученного продукта проверяли при помощи ТСХ (элюент: СНСl3/МеОН: 9/1). Выход: 1,18 г восковидного твердого вещества. Расщепление 1,18 г полученного твердого Z-защищенного соединения растворяли в 40 мл абсолютного этанола. В раствор добавляли приблизительно 20 мг палладия на угле (10%, FLUKA), и суспензию встряхивали в атмосфере водорода в течение 3 ч. За ходом реакции наблюдали при помощи ТСХ (элюент: СНСl3/МеОН: 9/1). После завершения реакции растворитель удаляли с получением свободного основания. Выход: 99%. Чистоту проверяли посредством ТСХ: н-бутанол/АсОН/вода/этилацетат: 1/1/1/1, Rf = 0,4. Идентичность продукта реакции проверяли при помощи анализа ЯМР.-8 007434 Пример 2. Синтез гидрохлорида глутаминилтиазолидина АцилированиеN-трет-Бутилоксикарбонилглутамин (2,0 г; 8,12 ммоль) растворяли в 5 мл ТГФ и доводили до -15 С. В эту смесь добавляли CAIBE (изобутилхлорформиат) (1,06 мл; 8,12 ммоль) и 4-метилморфолин (0,895 мл; 8,12 ммоль), и раствор перемешивали в течение 15 мин. Образование смешанного ангидрида проверяли при помощи ТСХ (элюент: CHCl3/MeOH: 9/1). После нагревания до -10 С добавляли еще один эквивалент 4 метилморфолина (0,895 мл; 8,12 ммоль) и гидрохлорид тиазолидина (1,02 г; 8,12 ммоль). Смесь доводили до комнатной температуры и перемешивали в течение ночи. Обработка Образовавшийся осадок отфильтровывали и выпаривали растворитель. Полученное масло собирали в хлороформ (20 мл) и промывали насыщенным раствором гидросульфата натрия, а затем насыщенным раствором гидрокарбоната натрия, водой и рассолом. Органический слой отделяли, сушили и упаривали. Чистоту полученного продукта проверяли при помощи ТСХ (элюент: СНСl3/МеОН: 9/1). Выход: 1,64 г твердого вещества. Расщепление 640 мг полученного твердого Вос-защищенного соединения растворяли в 3,1 мл ледяной HCl в диоксане (12,98 М; 20 эквивалентов) и оставляли на льду. За ходом реакции наблюдали при помощи ТСХ(элюент: СНСl3/МеОН: 9/1). После завершения реакции растворитель удаляли, и полученное масло собирали в метанол и снова упаривали. После этого полученное масло сушили над оксидом фосфора V и дважды растирали с диэтиловым эфиром. Выход: 0,265 г. Чистоту проверяли при помощи ВЭЖХ. Идентичность продукта реакции проверяли при помощи анализа ЯМР. Пример 3. Синтез гидрохлорида глутаминилпирролидина АцилированиеN-трет-Бутилоксикарбонилглутамин (3,0 г; 12,18 ммоль) растворяли в 7 мл ТГФ и доводили до -15 С. В эту смесь добавляли CAIBE (изобутилхлорформиат) (1,6 мл; 12,18 ммоль) и 4-метилморфолин (1,3 мл; 12,18 ммоль), и раствор перемешивали в течение 15 мин. Образование смешанного ангидрида проверяли при помощи ТСХ (элюент: CHCl3/MeOH: 9/1). После нагревания до -10 С добавляли один эквивалент пирролидина (1,0 мл; 12,18 ммоль). Смесь доводили до комнатной температуры и перемешивали в течение ночи. Обработка Образовавшийся осадок отфильтровывали и выпаривали растворитель. Полученное масло собирали в хлороформ (20 мл) и промывали насыщенным раствором гидросульфата натрия, а затем насыщенным раствором гидрокарбоната натрия, водой и рассолом. Органический слой отделяли, сушили и упаривали. Чистоту полученного продукта проверяли при помощи ТСХ (элюент: CHCl3/MeOH: 9/1). Выход: 2,7 г твердого вещества. Расщепление 2,7 г полученного твердого вещества растворяли в 13 мл ледяной HCl в диоксане (12,98 М; 20 эквивалентов) и оставляли на льду. За ходом реакции наблюдали при помощи ТСХ (элюент: СНCl3/МеОН: 9/1). После завершения реакции растворитель удаляли, и полученное масло собирали в метанол и снова упаривали. После этого полученное масло сушили над оксидом фосфора V и дважды растирали с диэтиловым эфиром. Выход: 980 мг. Чистоту проверяли при помощи ВЭЖХ. Идентичность продукта реакции проверяли при помощи анализа ЯМР. Пример 4. Определение Ki Для определения Ki глутаминилпирролидина и глутаминилтиазолидина использовали дипептидилпептидазу IV из почек свиньи с удельной активностью в отношении глицилпролил-4-нитроанилина 37,5 Ед/мг и концентрацией фермента в исходном растворе 1,41 мг/мл. Анализируемая смесь: Смешивали 100 мкл глутаминилпирролидина или глутаминилтиазолидина с концентрацией в пределах 1x10-5 - 1x10-7 М (глутаминилпирролидин) и 1 х 10-6 - 1 х 10-8 М (глутаминилтиазолидин), соответственно, с 50 мкл глицилпролил-4-нитроанилина в различных концентрациях (0,4 мМ; 0,2 мМ; 0,1 мМ; 0,05 мМ) и 100 мкл HEPES (N-[2-гидроксиэтил]пиперазин-N-[2-этансульфоновой кислоты]) (40 мМ; рН 7,6; ионная сила = 0,125). Анализируемую смесь преинкубировали при 30 С в течение 30 мин. После преинкубации добавляли 20 мкл DPIV (в разведении 1:600) и проводили измерение развития желтого окрашивания из-за высвобождения 4-нитроанилина при 30 С и X = 405 нм в течение 10 мин, используя прибор для считывания планшетов (HTS7000 plus, Applied Biosystems, Weiterstadt, Germany). Значения Ki подсчитывали, используя Graphit 4.0.15 (Erithacus Software, Ltd, UK), на основании конкурентного ингибирования DPIV глутаминилпирролидином или глутаминилтиазолидином. ЗначенияKi определяли для глутаминилтиазолидина как Ki= 3,12x10-7 М 5,11 х 10-10 М и для глутаминилпирролидина как Ki = 1,30x10-6 М 8,49x10-8 М.-9 007434 Пример 5. Определение Ki в плазме человека Плазма человека имеет активность, отщепляющую N-терминальный Хаа-Рrо. Смешивали 70 мкл глутаминилпирролидина или глутаминилтиазолидина с концентрацией в пределах 1x10-5 - 1x10-7 М (глутаминилпирролидин) и 1x10-6 - 1x10-8 М (глутаминилтиазолидин), соответственно, с 50 мкл глицилпролил-4-нитроанилина в различных концентрациях (0,4 мМ; 0,2 мМ; 0,1 мМ; 0,05 мМ) и 100 мкл HEPES (40 мМ; рН 7,6). Анализируемую смесь преинкубировали при 30 С в течение 5 мин и 22 ч соответственно. После преинкубации добавляли 50 мкл плазмы человека и проводили измерение развития желтого окрашивания из-за высвобождения 4-нитроанилина при 30 С и X = 405 нм в течение 10 мин, используя прибор для считывания планшетов (HTS7000 plus, Applied Biosystems, Weiterstadt, Germany). Значения Ki подсчитывали, используя Graphit 4.0.15 (Erithacus Software, Ltd, UK), на основании конкурентного ингибирования DPIV глутаминилпирролидином или глутаминилтиазолидином. ЗначенияKi определяли для глутаминилтиазолидина как Кi = 4,03x10-72,19x10-10 М после 5 мин и 5,13 х 10-71,26 х 10-8 М после 22 ч преинкубации, и для глутаминилпирролидина как Ki = 1,30x10-64,89x10-8 М после 5 мин и 1,36 х 10-63,21 х 10-8 М после 22 ч преинкубации. Пример 6. Ингибирование DPIV-подобных ферментов - дипептидилпептидазы IIDP II (3.4.14.2) отщепляет N-терминальные дипептиды от олигопептидов, если N-конец не протонирован (McDonald, J.К., Ellis, S.Reilly, T.J., 1966, J. Biol. Chem., 241, 1494-1501). Pro и Ala в P1 положении являются предпочтительными остатками. Ферментную активность описывают как DPIVподобную активность, но DP II имеет оптимум рН в кислой области. Используемый фермент выделяли из почек свиньи. Анализ: Смешивали 100 мкл глутаминилпирролидина или глутаминилтиазолидина с концентрацией в пределах 1x10-4 - 5x10-8 М со 100 мкл буферного раствора (40 мМ HEPES; рН 7,6, 0,015% Brij (Бридж - неионный детергент), 1 мМ ДТТ (1,4-дитиотрейтол, 50 мкл раствора лизилаланиламинометилкумарина(5 мМ) и 20 мкл свиной DP II (с 250-кратным разведением в буферном растворе). Проводили измерение флуоресценции при 30 С и возбуждения = 380 нм, эмиссии = 465 нм в течение 25 мин, используя прибор для считывания планшетов (HTS7000 plus, Applied Biosystems, Weiterstadt, Germany). Значения Ki подсчитывали, используя Graphit 4.0.15 (Erithacus Software, Ltd, UK), и определяли какKi = 8,52x10-56,33x10-6 М для глутаминилпирролидина и Ki = 1,07x10-53,81x10-7 М для глутаминилтиазолидина. Пример 7. Перекрестно реагирующие ферменты Глутаминилпирролидин и глутаминилтиазолидин тестировали на способность к перекрестному реагированию с дипептидилпептидазой I, пролилолигопептидазой и пролидазой. Дипептидилпептидаза I (DP I, катепсин С)DP I или катепсин С представляет собой лизосомальную цистеинпротеазу, которая отщепляет дипептиды от N-конца субстратов (Gutman, H.R.Fruton, J.S., 1948, J. Biol. Chem., 174, 851-858). Ее классифицируют как цистеинпротеазу. Используемый фермент закупали у Qiagen (Qiagen GmbH, Hilden, Germany). Для получения в полной мере активного фермента фермент разбавляли в 1000 раз в MES буфере с рН 5,6 (40 мМ MES, 4 мМ ДТТ, 4 мМ KCl, 2 мМ ЭДТА, 0,015% Brij) и преинкубировали в течение 30 мин при 30 С. Анализ: Смешивали 50 мкл глутаминилпирролидина или глутаминилтиазолидина с концентрацией в пределах 1x10-5 - 1x10-7 М со 110 мкл смеси буфера с ферментом. Анализируемую смесь преинкубировали при 30 С в течение 15 мин. После преинкубации добавляли 100 мкл гистидилсерилнитроанилина (2 х 10-5 М) и проводили измерение развития желтого окрашивания из-за высвобождения -нитроанилина при 30 С ивозбуждения = 380 нм,эмиссии = 465 нм в течение 10 мин, используя прибор для считывания планшетов (HTS7000 plus, Applied Biosystems, Weiterstadt, Germany). Значения IC50 подсчитывали, используя Graphit 4.0.15 (Erithacus Software, Ltd, UK). He обнаружено ингибирования ферментной активности DP I глутаминилпирролидином и глутаминилтиазолидином. Пролилолигопептидаза (POP) Пролилолигопептидаза (ЕС 3.4.21.26) представляет собой сериновую эндопротеазу, которая отщепляет пептиды в N-терминальной части связи Хаа-Pro (Walter, R., Shlank, H., Glass, J.D., Schwartz, I.LKerenyi, T.D., 1971, Science, 173, 827-829). Субстратами являются пептиды с молекулярной массой до 3000 Да. Используемый фермент представлял собой рекомбинантную человеческую пролилолигопептидазу. Рекомбинантную экспрессию проводили у E.coli при стандартных условиях, как описано в данном уровне техники. Анализ: Смешивали 100 мкл глутаминилпирролидина или глутаминилтиазолидина с концентрацией в пределах 1x10-4 - 5x10-8 M со 100 мкл буферного раствора (40 мМ HEPES; рН 7,6, 0,015% Brij, 1 мМ ДТТ) и- 10007434 20 мкл раствора POP. Анализируемую смесь преинкубировали при 30 С в течение 15 мин. После преинкубации добавляли 50 мкл раствора глицилпролилпролил-4-нитроанилина (0,29 мМ) и проводили измерение развития желтого окрашивания из-за высвобождения 4-нитроанилина при 30 С и= 405 нм в течение 10 мин, используя прибор для считывания планшетов (Sunrise, Tecan, Crailsheim, Germany). Значения IC50 подсчитывали, используя Graphit 4.0.15 (Erithacus Software, Ltd, UK). He обнаружено ингибирования ферментной активности POP глутаминилпирролидином и глутаминилтиазолидином. Пролидаза (Х-Pro дипептидаза) Пролидазу (ЕС 3.4.13.9) впервые описали BergmannFruton (Bergmann, М.Fruton, JS, 1937, J.Biol. Chem. 189-202). Пролидаза отщепляет N-терминальную аминокислоту от Хаа-Pro дипептидов и имеет оптимум рН между 6 и 9. Пролидазу из почек свиньи (ICN Biomedicals, Eschwege, Germany) растворяли (1 мг/мл) в анализируемом буфере (20 мМ NH4(CH3COO)2, 3 мМ МnСl2, рН 7,6). Для получения в полной мере активного фермента раствор инкубировали в течение 60 мин при комнатной температуре. Анализ: Смешивали 450 мкл глутаминилпирролидина или глутаминилтиазолидина с концентрацией в пределах 5x10-3 - 5x10-7 М с 500 мкл буферного раствора (20 мМ NH4(CH3COO)2; рН 7,6) и 250 мкл IIе-РrоОН (0,5 мМ в анализируемой смеси). Анализируемую смесь преинкубировали при 30 С в течение 5 мин. После преинкубации добавляли 75 мкл пролидазы (с разведением 1:10 в анализируемом буфере) и проводили измерение при 30 С и= 220 нм в течение 20 мин, используя UV/Vis фотометр, UV1 (ThermoSpectronic, Cambridge, UK). Значения IC50 подсчитывали, используя Graphit 4.0.15 (Erithacus Software, Ltd, UK). Их определяли как IС 503 мМ для глутаминилтиазолидина и как IС 50 = 3,4x10-4 М 5,63x10-5 для глутаминилпирролидина. Пример 8. Стабильность в плазме Для исследования стабильности глутаминилпирролидина или глутаминилтиазолидина в плазме человека, активность DPIV в плазме устанавливали в определенное время. Среднюю активность DPIV в плазме человека определяли как 43,69 Ед/мл. В рабочем растворе плазму разводили в 0,9% NaCl для установления уровня активности DPIV 25 Ед/мл. Плазму и глутаминилпирролидин или глутаминилтиазолидин в различных концентрациях (5x10-5; 2,5x10-5; 1,25x10-5 М в плазме) инкубировали при 37 С. В определенное время отбирали образцы, используя автоматическую пипетку (Gilson 215, Liquid handler, Gilson), и переносили в планшет для микротитрования, содержащий 5x10-5 М глицилпролиламинометилкумарина в 0,9% NaCl + 0,15% Brij на ячейку. Через 6 мин реакцию прекращали при помощи добавления изолейцилтиазолидина (5x10-5 М в 0,9% растворе NaCl). Измерение флуоресценции проводили относительно 0,9% NaCl в плазме (стандартный образец), используя прибор для считывания планшетов (HTS7000 plus, Applied Biosystems, Weiterstadt, Germany). Период, за который наблюдалось 50%-ное снижение ингибиторной активности глутаминилпирролидина или глутаминилтиазолидина, подсчитывали при помощи построения графика ферментной активности относительно времени реакции. Для обоих соединений не смогли определить этот полупериод. Считается, что вещество стабильно в плазме человека более 22 ч. Пример 9. Определение ингибиторной активности глутаминилпирролидина или глутаминилтиазолидина в отношении DPIV после интравазального и перорального введения крысам линии Wistar Животные Самцов крыс линии Wistar (Shoe: Wist(Sho с массой тела в пределах между 250 и 350 г закупали уTierzucht Schonwalde (Schonwalde, Germany). Условия содержания Животных содержали в клетках по одному в обычных условиях с контролируемой температурой(222 С) с чередующимся 12/12-часовым циклом свет/темнота (включение света в 6:00). Стандартный гранулированный корм (ssniff Soest, Germany) и подкисленную HCl водопроводную воду давали без ограничений. Введение катетера в сонную артерию Через одну или более чем одну неделю адаптации к условиям содержания в сонную артерию крыс линии Wistar имплантировали катетеры под общим наркозом (интраперитонеальная инъекция 0,25 мл/кг массы тела ромпуна (Rompun) [2%], BayerVital, Germany и 0,5 мл/кг массы тела кетамина 10, AtarostGmbHCo., Twistringen, Germany). Животным давали восстановиться в течение одной недели. Катетеры промывали физиологическим раствором, содержащим гепарин (100 МЕд/мл) три раза в неделю. В случае дисфункции катетера, второй катетер вводили в противоположную сонную артерию соответствующей крысы. Через неделю восстановления после операции это животное повторно включали в исследование. В случае дисфункции второго катетера, животное исключали из исследования. Брали новое животное и продолжали эксперимент в плановом порядке, начиная по меньшей мере через 7 дней после- 11007434 имплантации катетера. Планирование эксперимента Крысам с нормально функционирующим катетером вводили плацебо (1 мл физиологического раствора, 0,154 моль/л) или 100 мг/кг массы тела глутаминилпирролидина или 100 мг/кг массы тела глутаминилтиазолидина посредством перорального и интравазального (внутриартериального) пути введения. После голодания в течение ночи отбирали образцы гепаринизированной артериальной крови по 100 мкл в -30, -5 и 0 мин. Тестируемое вещество растворяли в 1,0 мл физиологического раствора (0,154 моль/л) и сразу же вводили в 0 мин либо перорально при помощи питательной трубки (75 мм; Fine science Tools, Heidelberg, Germany) или посредством интравазального пути введения. В случае перорального введения, в артериальный катетер дополнительно вводили 1 мл физиологического раствора. В случае внутриартериального введения, катетер сразу же промывали 30 мкл физиологического раствора и дополнительно давали 1 мл физиологического раствора перорально при помощи питательной трубки. После применения плацебо или тестируемого вещества отбирали образцы артериальной крови в 2,5; 5; 7,5; 10; 15; 20; 40; 60 и 120 мин из катетера сонной артерии находящихся в сознании нормальных крыс. Все образцы крови отбирали в ледяные пробирки Эппендорфа (Eppendorf-Netheler-Hinz, Hamburg, Germany),наполненные 10 мкл 1 М буферного раствора на основе цитрата натрия (рН 3,0), для измерения активности DPIV в плазме. Пробирки Эппендорфа сразу же центрифугировали (12000 об/мин в течение 2 мин,Hettich Zentrifuge ЕВА 12, Tuttlingen, Germany). Фракции плазмы хранили на льду до анализа или замораживали при -20 С до анализа. Все образцы плазмы помечали следующими данными: кодовый номер,номер животного,дата отбора образца,время отбора образца. Аналитические методы Анализируемая смесь для определения активности DPIV в плазме содержала 80 мкл реагента и 20 мкл образца плазмы. Проводили измерение кинетики образования желтого продукта 4-нитроанилина из субстрата глицилпролил-4-нитроанилина при 390 нм в течение 1 мин при 30 С через 2 мин преинкубации при такой же температуре. Активность DPIV выражали в мЕд/мл. Статистические методы Статистические оценки и построение графиков производили при помощи PRISM 3.02 (GraphPadSoftware, Inc.). Все параметры анализировали описательно, включая среднее значение и среднеквадратичное отклонение. Результаты Соединения глутаминилпирролидин и глутаминилтиазолидин в дозе 100 мг/кг массы тела по сравнению с плацебо ингибировали активность DPIV в плазме после перорального и интравазального введения (см. фиг. 1 и 2). Пример 10. Исследование возрастания доз на крысах линии Zucker с ожирением после перорального введения глутаминилпирролидина Животные 30 самцов крыс линии Zucker (fa/fa) со средним возрастом 11 недель (5-12 недель), средней массой тела 350 г (150-400 г) закупали у Charles River (Sulzfeld, Germany). После доставки их содержали в течение более чем 12 недель до тех пор, пока почти все крысы линии Zucker с ожирением не приобретали признаки явного сахарного диабета. Группу из 8 животных отбирали для тестирования трех возрастающих доз глутаминилпирролидина в сравнении с плацебо (физиологическим раствором). Условия содержания Животных содержали в клетках по одному в обычных условиях с контролируемой температурой(222 С) с чередующимся 12/12-часовым циклом свет/темнота (включение света в 6:00). Стерильный стандартный гранулированный корм (ssniff Soest, Germany) и подкисленную HCl водопроводную воду давали без ограничений. Катетеризация сонной артерии Крыс линии Zucker с ожирением в возрасте 24-31 недели (в среднем: 25 недель), адаптировавшихся к условиям содержания, основательно подготавливали для исследования. В сонную артерию крыс линии Zucker с ожирением имплантировали катетеры под общим наркозом(интраперитонеальная инъекция 0,25 мл/кг массы тела ромпуна [2%], BayerVital, Germany и 0,5 мл/кг массы тела кетамина 10, Atarost GmbHCo., Twistringen, Germany). Животным давали восстановиться в течение одной недели. Катетеры промывали физиологическим раствором, содержащим гепарин (100 МЕд/мл) три раза в неделю. Планирование эксперимента Группам, состоящим из 8 крыс линии Zucker с ожирением, вводили плацебо (1 мл физиологического раствора, 0,154 моль/л) или возрастающие дозы глутаминилпирролидина (5, 15 и 50 мг/кг массы тела). 375 мг глутаминилпирролидина растворяли в 1000 мкл ДМСО (E.Merck, Darmstadt, Germany [диметилсульфоксид р.а.]). Добавляли 10 мл физиологического раствора, и аликвоты по 1 мл, каждая из которых- 12007434 содержала по 34,09 мг глутаминилпирролидина, хранили при -20 С. Для приготовления тестируемого вещества дозозависимые аликвоты разводили в физиологическом растворе. После голодания в течение ночи крысам линии Zucker с ожирением вводили плацебо или тестируемое вещество перорально при помощи питательной трубки (15 G, 75 мм; Fine science Tools, Heidelberg,Germany) в -10 минут. Для постановки теста толерантности к глюкозе (OGTT) вводили 2 г/кг массы тела глюкозы (40%-ный раствор, В. Braun Meisungen, Meisungen, Germany) в 0 мин при помощи второй питательной трубки. Образцы венозной крови из хвостовых вен отбирали в -30 мин, -15 мин,0 мин и в 5,10, 15, 20, 30, 40, 60, 90 и 120 мин в стеклянные капилляры по 20 мкл, которые помещали в стандартные пробирки, наполненные 1 мл раствора для измерения глюкозы в крови. Все образцы плазмы помечали следующими данными: кодовый номер,номер животного,дата отбора образца,время отбора образца. Аналитические методы Уровни глюкозы измеряли, используя глюкозооксидазный метод (анализатор глюкозы Super G; Dr.Muller Geratebau, Freitai, Germany). Статистические методы Статистические оценки и построение графиков производили при помощи PRISM 3.02 (GraphPadSoftware, Inc.). Все параметры анализировали описательно, включая среднее значение и среднеквадратичное отклонение. Воздействие лечения на толерантность к глюкозе Крысы линии Zucker, больные диабетом, которых лечили плацебо, показали сильно возрастающие отклонения уровней глюкозы в крови, указывающие на интолерантность к глюкозе при явном сахарном диабете. Введение 5 мг/кг массы тела глутаминилпирролидина приводило к ограниченному улучшению толерантности к глюкозе у крыс линии Zucker, больных диабетом. Значительное снижение повышенных уровней глюкозы в крови и улучшение толерантности к глюкозе достигали после введения 15 мг/кг и 50 мг/кг массы тела глутаминилпирролидина (см. фиг. 3). Пример 11. Исследование возрастания доз на крысах линии Zucker с ожирением после перорального введения глутаминилтиазолидина Животные 30 самцов крыс линии Zucker (fa/fa) со средним возрастом 11 недель (5-12 недель), средней массой тела 350 г (150-400 г) закупали у Charles River (Sulzfeld, Germany). После доставки их содержали в течение более чем 12 недель до тех пор, пока почти все крысы линии Zucker с ожирением не приобретали признаки явного сахарного диабета. Группу из 8 животных отбирали для тестирования трех возрастающих доз глутаминилтиазолидина в сравнении с плацебо (физиологическим раствором). Условия содержания Животных содержали в клетках по одному в обычных условиях с контролируемой температурой(222 С) с чередующимся 12/12-часовым циклом свет/темнота (включение света в 6:00). Стерильный стандартный гранулированный корм (ssniff Soest, Germany) и подкисленную HCl водопроводную воду давали без ограничений. Катетеризация сонной артерии Крыс линии Zucker с ожирением в возрасте 24-31 недели (в среднем: 25 недель), адаптировавшихся к условиям содержания, основательно подготавливали для исследования. В сонную артерию крыс линии Zucker с ожирением имплантировали катетеры под общим наркозом(интраперитонеальная инъекция 0,25 мл/кг массы тела ромпуна [2%], BayerVital, Germany и 0,5 мл/кг массы тела кетамина 10, Atarost GmbHCo., Twistringen, Germany). Животным давали восстановиться в течение одной недели. Катетеры промывали физиологическим раствором, содержащим гепарин (100 МЕд/мл) три раза в неделю. Планирование эксперимента Группам, состоящим из 8 крыс линии Zucker с ожирением, вводили плацебо (1 мл физиологического раствора, 0,154 моль/л) или возрастающие дозы глутаминилтиазолидина (5, 15 и 50 мг/кг массы тела). Соответствующие количества глутаминилтиазолидина растворяли в 1000 мкл физиологического раствора. После голодания в течение ночи крысам линии Zucker с ожирением вводили плацебо или тестируемое вещество перорально при помощи питательной трубки (15 G, 75 мм; Fine science Tools, Heidelberg,Germany) в -10 мин. Для постановки теста толерантности к глюкозе (OGTT) вводили 2 г/кг массы тела глюкозы (40%-ный раствор, В. Braun Meisungen, Meisungen, Germany) в 0 мин при помощи второй питательной трубки. Образцы венозной крови из хвостовых вен отбирали в -30 мин, -15 мин,0 мин и в 5,10, 15, 20, 30, 40, 60, 90 и 120 мин в стеклянные капилляры по 20 мкл, которые помещали в стандартные пробирки, наполненные 1 мл раствора для измерения глюкозы в крови. Все образцы плазмы помечали следующими данными:- 13007434 кодовый номер,номер животного,дата отбора образца,время отбора образца. Аналитические методы Уровни глюкозы измеряли, используя глюкозооксидазный метод (анализатор глюкозы Super G; Dr.Muller Geratebau, Freital, Germany). Статистические методы Статистические оценки и построение графиков производили при помощи PRISM 3.02 (GraphPadSoftware, Inc.). Все параметры анализировали описательно, включая среднее значение и среднеквадратичное отклонение. Воздействие лечения на толерантность к глюкозе Крысы линии Zucker, больные диабетом, которых лечили плацебо, показали сильно возрастающие отклонения уровней глюкозы в крови, указывающие на интолерантность к глюкозе при явном сахарном диабете. Введение 5 мг/кг массы тела, 15 и 50 мг/кг массы тела глутаминилтиазолидина приводило к дозозависимому снижению повышенных уровней глюкозы в крови и улучшению толерантности к глюкозе у крыс линии Zucker, больных диабетом (см. фиг. 4). Пример 12. Инактивация глутаминилтиазолидина после перорального введения крысам линии Wistar in vivo Животные/Планирование эксперимента Глутаминилтиазолидин вводили перорально крысам линии Wistar, как описано в примере 9. Аналитические методы После применения плацебо или глутаминилтиазолидина отбирали образцы артериальной крови в 2,5; 5; 7,5; 10; 15; 20; 40; 60 и 120 мин из катетера сонной артерии находящихся в сознании нормальных крыс для определения образования продуктов превращения глутаминилтиазолидина. Для анализа использовали простой метод твердофазной экстракции на картриджах С 18 для выделения интересующих соединений из плазмы. Экстракты анализировали, используя жидкостную хроматографию с обращенной фазой на колонке Lichrospher 60 RP Select В, с последующей тандемной массспектрометрией, действующей в режиме регистрации положительных ионов APCI (химическая ионизация при атмосферном давлении). Для определения количества использовали метод внутреннего стандарта. Результаты После перорального введения глутаминилтиазолидина крысам линии Wistar обнаружили превращение соединения. Используя LC/MS, смогли определить продукт превращения как пироглутамилтиазолидин. См. фиг. 5 и 6. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы где X = СН 2 или S, или его соль присоединения кислоты. 2. Соединение по п.1, где соль присоединения кислоты выбрана из группы, состоящей из солей соляной, бромисто-водородной, перхлорной, серной, азотной, фосфорной, уксусной, пропионовой, гликолевой, молочной, янтарной, малеиновой, фумаровой, яблочной, винной, лимонной, бензойной, миндальной, метансульфоновой, гидроксиэтансульфоновой, бензолсульфоновой, щавелевой, памовой, 2 нафталинсульфоновой, паратолуолсульфоновой, циклогексансульфаминовой, салициловой, сахариновой и трифторуксусной кислоты. 3. Соль присоединения кислоты соединения формулы где X = СН 2 или S и где соль присоединения кислоты выбрана из группы, состоящей из солей соляной,бромисто-водородной, перхлорной, азотной, пропионовой, гликолевой, молочной, малеиновой, яблочной, лимонной, бензойной, миндальной, метансульфоновой, гидроксиэтансульфоновой, бензолсульфоновой, щавелевой, памовой, 2-нафталинсульфоновой, паратолуолсульфоновой, циклогексансульфаминовой, салициловой, сахариновой и трифторуксусной кислоты. 4. Соединение по п.3, выбранное из гидрохлорида глутаминилтиазолидина или гидрохлорида глутаминилпирролидина. 5. Гидрохлорид глутаминилтиазолидина. 6. Гидрохлорид глутаминилпирролидина. 7. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и/или разбавитель и соединение по любому из пп.1-6. 8. Способ ингибирования дипептидилпептидазы IV или дипептидилпептидаза IV-подобной ферментной активности для предупреждения или лечения заболеваний или состояний, связанных с дипептидилпептидазой IV или дипептидилпептидаза IV-подобными ферментами, при котором млекопитающему,нуждающемуся в подобном лечении, вводят терапевтически эффективное количество соединения по любому из пп.1-6. 9. Способ по п.8, при котором снижают уровни глюкозы в крови у млекопитающих, являющиеся следствием приема пищи. 10. Способ по п.8 или 9, при котором предупреждают или лечат заболевания или состояния, выбранные из группы, состоящей из инсулиннезависимого сахарного диабета, артрита, ожирения, иммунных и аутоиммунных расстройств, аллотрансплантации, рака, нейрональных расстройств и кожных заболеваний. 11. Способ по п.10, при котором предупреждают или лечат инсулиннезависимый сахарный диабет. 12. Продукт превращения соединения по пп.1, 3 или 5, имеющий формулу
МПК / Метки
МПК: A61P 3/10, A61P 35/00, A61P 19/02, C07D 295/18, A61K 31/41, A61P 25/00, A61P 3/04, C07D 207/04, C07D 277/04, A61P 37/00, A61K 31/395
Метки: основе, глутаминила, ингибиторы
Код ссылки
<a href="https://eas.patents.su/18-7434-ingibitory-dpiv-na-osnove-glutaminila.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы dpiv на основе глутаминила</a>
Ингибиторы герпесвирусов на основе альфа-метилбензилсодержащей тиомочевины, включающие фенилендиаминогруппу

Номер патента: 4205
Опубликовано: 26.02.2004
Авторы: Нортон Эмили Бучер, Росс Эдма Антониа, Блум Джонатан Дэвид, Дашин Расселл Джордж, Лэнг Стэнли Альберт, Дигранди Мартин Джозеф, Каррэн Кевин Джозеф, О'хара Брайан Марк
МПК: A61K 31/17, C07D 335/20, A61P 31/22...
Метки: герпесвирусов, фенилендиаминогруппу, тиомочевины, ингибиторы, включающие, альфа-метилбензилсодержащей, основе
Формула / Реферат:
1. Соединение, имеющее формулу где R1-R5 независимо выбраны из водорода, алкила с 1-6 атомами углерода, алкенила с 2-6 атомами углерода, алкинила с 2-6 атомами углерода, пергалогеналкила с 1-6 атомами углерода, циклоалкила с 3-10 атомами углерода, гетероциклоалкила, арила, гетероарила, галогена, -CN, -NO2, -CO2R6, -COR6, -OR6, -SR6, -SOR6, -SO2R6, -CONR7R8, -NR6N(R7R8), -N(R7R8) или W-Y-(CH2)n-Z, при условии, что по меньшей мере один из...
Ингибиторы протеазы вич широкого спектра действия на основе сульфонамидов 2-(замещенных-амино) бензотиазолов

Номер патента: 6096
Опубликовано: 25.08.2005
Авторы: Вутс Марике Кристиан Йоханна, Де Кок Херман Аугустинус, Вигеринк Пит Том Берт Поль, Де Керпел Ян Октаф Антон, Де Бетюн Мари-Пьер, Гетмен Даниел, Вендевилль Сандрин, Сюрлеро Доминик Луи Нестор Гилейн, Морс Самюэль Лео Кристиан, Версхюэрен Вим Гастон
МПК: A61P 31/18, C07D 277/82, A61K 31/428...
Метки: широкого, спектра, основе, действия, протеазы, вич, бензотиазолов, сульфонамидов, 2-(замещенных-амино, ингибиторы
Формула / Реферат:
1. Соединение, имеющее формулу (I) и его N-оксиды, соли, стереоизомеры, рацемические смеси, пролекарства, эфиры и метаболиты, в которой R1 и R8, каждый независимо, представляет атом водорода, C1-6алкил, C2-6алкенил, арилC1-6алкил, C3-7циклоалкил, C3-7циклоалкилC1-6алкил, арил, Het1, Het1C1-6алкил, Het2 или Het2C1-6алкил; R1 также может быть радикалом формулы в которой R9, R10a и R10b, каждый независимо, представляет атом водорода,...
Ингибиторы агрегации амилоидных белков, их применение (варианты), композиция на их основе и способ визуализации амилоидных отложений

Номер патента: 4632
Опубликовано: 24.06.2004
Авторы: Барвиан Марк Роберт, Кейли Джон Стивен, Сэккэб Аннетт Тереза, Глейз Шелли Энн, Ясунага Томоюки, Суто Марк Джеймс, Оджелли-Зэфрэн Коринн Элизабет, Кимура Такенори, Уокер Лари Кразуэлл, Бигг Кристофер Франклин, Лай Йингджи, Хачийя Шуничиро, Жуанг Ниан
МПК: A61P 25/28, A61K 31/195, C07D 233/54...
Метки: агрегации, амилоидных, белков, визуализации, основе, варианты, отложений, ингибиторы, применение, композиция, способ
Формула / Реферат:
1. Соединение формулы I где Ra представляет собой водород, C1-C6алкил или алкил; n равен числу от 1 до 5 включительно; R1, R2, R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -OH, -NH2, NRbRc, -CO2H, -CO2C1-C6алкил, -NO2, -OC1-C12алкил, C1-C8алкил, -CF3, -CN, -OCH2фенил, -OCH2-замещенный фенил, -(CH2)m-фенил, -O-фенил, -O-замещенный фенил, -CH=CH-фенил, R8 представляет собой COOH, тетразолил, -SO2Rd или -CONHSO2Rd; Rb и...
Способы очистки глинистой корки и гравийной набивки для буровых растворов на масляной основе или водяной основе

Номер патента: 3685
Опубликовано: 28.08.2003
Авторы: Парлар Мехмет, Моррис Элизабет В.А., Прайс-Смит Колин, Тибблз Реймонд Дж., Келкар Шрихари, Брейди Марк Е.
МПК: E21B 37/08
Метки: глинистой, гравийной, основе, буровых, растворов, масляной, корки, водяной, очистки, способы, набивки
Формула / Реферат:
1. Способ гравийной набивки скважины в подземной формации, часть которой, прилегающая к скважине, имеет на себе покрытие глинистой корки, содержащей масляную диспергирующую эмульсию, включающий стадию закачки в скважину состава гравийной набивки, содержащего гравий и жидкость-носитель, содержащую водную фазу; и также стадию разложения или обращения эмульсии глинистой корки. 2. Способ по п.1, в соответствии с которым жидкость-носитель содержит...
Раствор на основе воды для бурения или технического обслуживания нефтяных или газовых скважин (варианты), способ увеличения термической устойчивости и снижения водоотдачи раствора на основе воды

Номер патента: 2815
Опубликовано: 31.10.2002
Автор: Хауз Рой Ф.
МПК: C09K 7/02
Метки: обслуживания, газовых, технического, нефтяных, увеличения, бурения, раствора, устойчивости, способ, термической, снижения, основе, скважин, варианты, водоотдачи, раствор, воды
Формула / Реферат:
1. Раствор на основе воды для бурения или технического обслуживания нефтяных или газовых скважин, отличающийся тем, что он содержит водную жидкость, включающую в себя, по меньшей мере, один полисахарид в количестве, обеспечивающем увеличение вязкости раствора на основе воды при низких скоростях сдвига, олигосахаридную смесь и оксид магния, причем концентрация олигосахаридной смеси является достаточной для увеличения термической устойчивости или...
Предыдущий патент: Макролиды
Следующий патент: Рулон с центральным разматыванием и способ его изготовления
Случайный патент: Способ деаэрации, деаэратор, распылитель для его изготовления (варианты) и применение указанного способа