Соединения пирроло[2,3-d]пиримидина
Номер патента: 6227
Опубликовано: 27.10.2005
Авторы: Блуменкопф Тодд Эндрю, Манчхоф Майкл Джон, Флэнэган Марк Эдвард






Формула / Реферат
1. Соединение формулы (I)

или его фармацевтически приемлемая соль;
где R1 представляет собой группу формулы

где y равно 0 или 1;
R4 представляет собой (C1-C6)алкил;
R5 представляет собой (C2-C9)гетероциклоалкил, где гетероциклоалкильная группа должна быть замещена в количестве от 1 до 2 (C1-C6)алкилом, (C1-C6)алкокси, (C1-C6)ацилом, (C1-C6)алкиламино-CO-, амино(C1-C6)ацилом, ((C1-C6)алкил)2амино(C1-C6)ацилом, (C1-C6)алкил-S(O)2, R15S(O)2R16N, где R15 и R16, каждый независимо, выбран из водорода или (C1-C6)алкила; или группой формулы

где a равно 0 или 1;
b, c, e, f и g, каждый независимо, представляет собой 0 или 1;
d равно 0, 1, 2 или 3;
X представляет собой S(O)2 или карбонил;
Y представляет собой S(O)2 или карбонил и
Z представляет собой карбонил, C(O)O- или S(O)2;
R6, R7, R8, R9, R10 и R11, каждый независимо, выбран из группы, состоящей из водорода или (C1-C6)алкила;
R12 представляет собой циано, амино, трифторметил, (C1-C6)алкил, трифторметил(C1-C6)алкил, (C1-C6)алкокси, галогено, (C1-C6)ацил, (C1-C6)алкиламино, ((C1-C6)алкил)2амино, амино(C1-C6)алкил, (C1-C6)алкокси-CO-NH, (C1-C6)алкиламино-CO-, (C1-C6)алкокси(C1-C6)алкил, циано(C1-C6)алкил, (C1-C6)ациламино, (C1-C6)ациламино(C1-C6)алкил, (C1-C6)алкокси(C1-C6)ациламино, амино(C1-C6)ацил, ((C1-C6)алкил)2амино(C1-C6)ацил или (C1-C6)алкил-S(O)2;
R2 и R3, каждый независимо, представляет собой водород.
2. Соединение по п.1, где указанное соединение выбрано из группы, состоящей из
метил-[4-метил-1-(пропан-1-сульфонил)пиперидин-3-ил]-(7H-пирроло[2,3-d]пиримидин-4-ил)амина;
3,3,3-трифтор-1-{4-метил-3-[метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}пропан-1-она и
3-{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}-3-оксо-пропионитрила.
3. Фармацевтическая композиция для: а) лечения или предупреждения расстройства или состояния, выбранного из отторжения трансплантата органа, ксенотрансплантации, волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета первого типа и осложнений при диабете, рака, астмы, атопического дерматита, аутоиммунных тиреоидных расстройств, неспецифического язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других аутоиммунных заболеваний, или б) ингибирования протеинкиназ, в частности Janus киназы 3 (JAK3), у млекопитающего, включая человека, содержащая количество соединения по п.1 или его фармацевтически приемлемой соли, одного или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млекопитающих, или с противовоспалительными агентами, эффективное в лечении таких расстройств или состояний, и фармацевтически приемлемый носитель.
4. Способ ингибирования протеинкиназ, в частности Janus киназы 3 (JAK3), у млекопитающего, включая человека, при котором указанному млекопитающему вводят эффективное количество соединения по п.1 или его фармацевтически приемлемой соли, одного или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млекопитающих, или с противовоспалительными агентами.
5. Способ лечения или предупреждения расстройства или состояния, выбранного из отторжения трансплантата органа, ксенотрансплантации, волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета первого типа и осложнений при диабете, рака, астмы, атопического дерматита, аутоиммунных тиреоидных расстройств, неспецифического язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других аутоиммунных заболеваний у млекопитающего, включая человека, при котором указанному млекопитающему вводят количество соединения по п.1 или его фармацевтически приемлемой соли, одного или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млекопитающих, или с противовоспалительными агентами, эффективное в лечении такого состояния.
6. Соединение по п.1, где замещенная (C2-C9)гетероциклоалкильная группа R5 представляет собой пиперидинильную группу.
7. Соединение по п.1, где a равно 0; b равно 1; X является карбонилом; c, e, f и g каждый равен 0 и d равно 1.
8. Соединение по п.2, где соединение представляет собой 3-{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}-3-оксо-пропионитрил.
9. Фармацевтическая композиция по п.3, где указанный агент выбран из группы, состоящей из циклоспорина A, рапамицина, такролимуса, лефлюномида, дезоксипергуалина, микофенолата, азатиоприна, даклизумаба, муромонаб-CD3, антитимоцит глобулина, аспирина, ацетаминофена, ибупрофена, напроксена, пироксикама и противовоспалительного стероида.
10. Способ по п.4, где указанный агент выбран из группы, состоящей из циклоспорина A, рапамицина, такролимуса, лефлюномида, дезоксипергуалина, микофенолата, азатиоприна, даклизумаба, муромонаб-CD3, антитимоцит глобулина, аспирина, ацетаминофена, ибупрофена, напроксена, пироксикама и противовоспалительного стероида.
11. Способ по п.5, где указанный агент выбран из группы, состоящей из циклоспорина A, рапамицина, такролимуса, лефлюномида, дезоксипергуалина, микофенолата, азатиоприна, даклизумаба, муромонаб-CD3, антитимоцит глобулина, аспирина, ацетаминофена, ибупрофена, напроксена, пироксикама и противовоспалительного стероида.
Текст
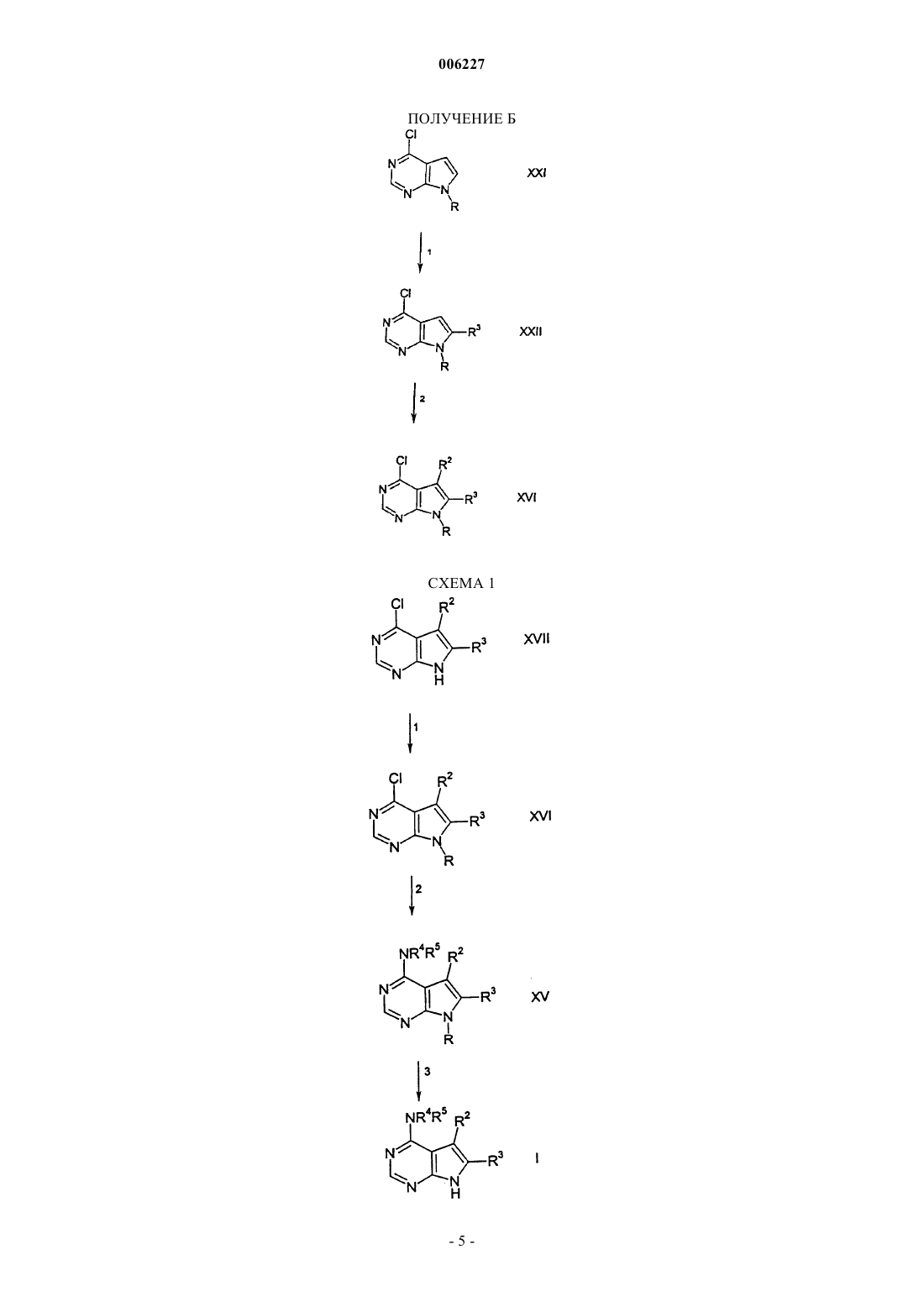
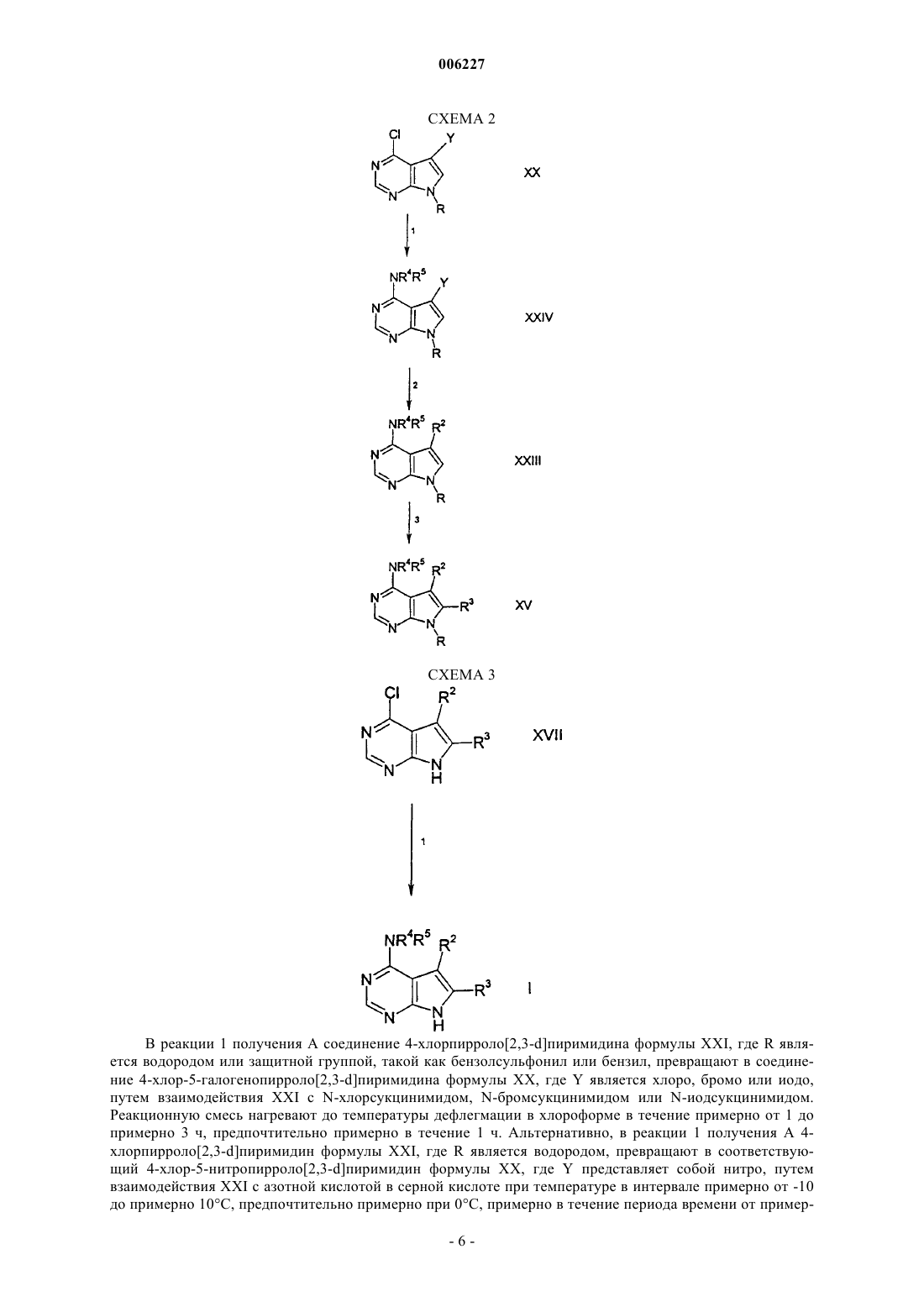
006227 Предпосылки изобретения Настоящее изобретение относится к соединениям пирроло[2,3-d]пиримидина, которые являются ингибиторами протеинкиназ, таких как фермент Janus киназа 3 (в дальнейшем также упоминаемая какJAK3), и как таковые полезны в терапии в качестве иммунодепрессивных агентов при трансплантации органов, ксенотрансплантации, волчанке, рассеянном склерозе, ревматоидном артрите, псориазе, диабете 1 типа и осложнениях при диабете, раке, астме, атопическом дерматите, аутоиммунных тиреоидных расстройствах, неспецифическом язвенном колите, болезни Крона, болезни Альцгеймера, лейкемии и других показаниях, где, вероятно, желательна иммунодепрессия. Это изобретение также относится к способу применения таких соединений в лечении вышеуказанных показаний у млекопитающих, особенно людей, и фармацевтическим композициям, полезным для этого.JAK3 является представителем Janus семейства протеинкиназ. Хотя другие представители этого семейства экспрессируются, по существу, всеми тканями, экспрессия JAK3 ограничивается гемопоэтическими клетками. Это согласуется с ее существенной ролью в передаче сигналов через рецепторы для IL-2(интерлейкин-2), IL-4, IL-7, IL-9 и IL-15 с помощью нековалентной связи JAK3 с гамма-цепью, общей для этих многоцепочечных рецепторов. Были установлены группы XSCID пациентов (Х-сцепленная форма тяжелой комбинированной иммунной недостаточности) с сильно пониженными уровнями белкаJAK3 или с генетическими дефектами на общей гамма-цепи и предположили, что иммуносупрессия является результатом блокирования передачи сигнала через проводящий путь JAK3. Исследования на животных навели на мысль, что JAK3 играет не только существенную роль в созревании лимфоцитов В и Т,но и что JAK3 является существенно необходимой для поддерживания функции Т-клетки. Модуляция иммунной активности по этому новому механизму может пригодиться в лечении Т-клеточных пролиферативных расстройств, таких как отторжение трансплантата, и аутоиммунных заболеваний. Краткое изложение сущности изобретения Настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли; где R1 представляет собой группу формулыR5 представляет собой (С 2-С 9)гетероциклоалкил, где гетероциклоалкильная группа должна быть замещена в количестве от 1 до 2 (С 1-С 6)алкилом, (С 1-С 6)алкокси, (С 1-С 6)ацилом, (С 1-С 6)алкиламино-СО-,амино(С 1-С 6)ацилом, С 1-С 6)алкил)2 амино(С 1-С 6)ацилом, (С 1-С 6)алкил-S(O)2, R15S(O)2R16N, где R15 и R16,каждый независимо, выбран из водорода или (C1-С 6)алкила; или группой формулыR6, R7, R8, R9, R10 и R11, каждый независимо, выбран из группы, состоящей из водорода или (С 1 С 6)алкила;R2 и R3, каждый независимо, представляет собой водород. Предпочтительные соединения формулы I включают в себя соединения, выбранные из группы, состоящей из метил-[4-метил-1-(пропан-1-сульфонил)пиперидин-3-ил]-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амина; 3,3,3-трифтор-1-4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил пропан-1-она и 3-4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил-3-оксопропионитрила. Настоящее изобретение также относится к фармацевтической композиции для (а) лечения или предупреждения расстройства или состояния, выбранного из отторжения трансплантата органа, ксенотрансплантации, волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета первого типа и осложнений при диабете, рака, астмы, атопического дерматита, аутоиммунных тиреоидных расстройств,неспецифического язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других аутоиммунных заболеваний, или (б) ингибирования протеинкиназ, в частности Janus киназы 3 (JAK3), у млекопитающего, включая человека, содержащей количество соединения формулы I или его фармацевтически приемлемой соли, одного или в комбинации с одним или более чем одним дополнительным агентом,который модулирует иммунную систему млекопитающих, или с противовоспалительными агентами,эффективное в лечении таких расстройств или состояний, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к способу ингибирования протеинкиназ, в частности Janus киназы 3 (JAK3), у млекопитающего, включая человека, при котором указанному млекопитающему вводят эффективное количество соединения формулы I или его фармацевтически приемлемой соли, одного или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млекопитающих, или с противовоспалительными агентами. Настоящее изобретение также относится к способу лечения или предупреждения расстройства или состояния, выбранного из отторжения трансплантата органа, ксенотрансплантации, волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета первого типа и осложнений при диабете, рака,астмы, атопического дерматита, аутоиммунных тиреоидных расстройств, неспецифического язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других аутоиммунных заболеваний у млекопитающего, включая человека, при котором указанному млекопитающему вводят количество соединения формулы I или его фармацевтически приемлемой соли, одного или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млекопитающих, или с противовоспалительными агентами, эффективное в лечении такого состояния. Кроме того, предпочтительные соединения формулы I включают в себя те соединения, где замещенная (С 2-С 9)гетероциклоалкильная группа R5 представляет собой пиперидинильную группу. Другие предпочтительные соединения формулы I включают в себя те соединения, где а равно 0;d равно 1. Особо предпочтительное соединение формулы I представляет собой 3-4-метил-3-[метил-(7 Нпирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил-3-оксопропионитрил. Кроме того, настоящее изобретение относится к упомянутой выше фармацевтической композиции для (а) лечения или предупреждения расстройства или состояния, указанного выше, или (б) ингибирования протеинкиназ, в частности Janus киназы 3 (JAK3), в комбинации с одним или более чем одним дополнительным агентом, где указанный агент выбран из группы, состоящей из циклоспорина А, рапамицина, такролимуса, лефлюномида, дезоксипергуалина, микофенолата, азатиоприна, даклизумаба, муромонаб-СD3, антитимоцит глобулина, аспирина, ацетаминофена, ибупрофена, напроксена, пироксикама и противовоспалительного стероида. Кроме того, настоящее изобретение относится к упомянутому выше способу ингибирования протеинкиназ, в частности Janus киназы 3 (JAK3), в комбинации с одним или более чем одним дополнительным агентом, где указанный агент выбран из группы, состоящей из циклоспорина А, рапамицина, такролимуса, лефлюномида, дезоксипергуалина, микофенолата, азатиоприна, даклизумаба, муромонаб-СD3,антитимоцит глобулина, аспирина, ацетаминофена, ибупрофена, напроксена, пироксикама и противовоспалительного стероида. Кроме того, настоящее изобретение относится к упомянутому выше способу лечения или предупреждения расстройства или состояния, указанного выше, в комбинации с одним или более чем одним-2 006227 дополнительным агентом, где указанный агент выбран из группы, состоящей из циклоспорина А, рапамицина, такролимуса, лефлюномида, дезоксипергуалина, микофенолата, азатиоприна, даклизумаба, муромонаб-СD3, антитимоцит глобулина, аспирина, ацетаминофена, ибупрофена, напроксена, пироксикама и противовоспалительного стероида. Термин "алкил", как он использован здесь, если не указано иначе, включает в себя насыщенные моновалентные углеводородные радикалы, имеющие прямые или разветвленные группировки, или их комбинации. Термин "алкокси", как он использован здесь, включает в себя O-алкильные группы, где термин "алкил" определен выше. Термин "галогено", как он использован здесь, если не указано иначе, включает в себя фторо, хлоро,бромо или иодо. Соединения по этому изобретению могут содержать двойные связи. Если такие связи присутствуют, то соединения по изобретению существуют как цис- и транс-конфигурации и как их смеси. Если не указано иначе, алкильная и алкенильная группы, упомянутые здесь, а также алкильные группировки других групп, упомянутые здесь (например, алкокси), могут быть прямыми или разветвленными и они могут быть также циклическими (например, циклопропил, циклобутил, циклопентил,циклогексил или циклогептил) или могут быть прямыми или разветвленными и содержать циклические группировки. Если не указано иначе, галоген включает в себя фтор, хлор, бром и иод.(С 2-С 9)гетероциклоалкил, используемый здесь, относится к пирролидинилу, тетрагидрофуранилу,дигидрофуранилу, тетрагидропиранилу, пиранилу, тиопиранилу, азиридинилу, оксиранилу, метилендиоксилу, хроменилу, изоксазолидинилу, 1,3-оксазолидин-3-илу, изотиазолидинилу, 1,3-тиазолидин-3-илу,1,2-пиразолидин-2-илу, 1,3-пиразолидин-1-илу, пиперидинилу, тиоморфолинилу, 1,2-тетрагидротиазин 2-илу, 1,3-тетрагидротиазин-3-илу, тетрагидротиадиазинилу, морфолинилу, 1,2-тетрагидродиазин-2-илу,1,3-тетрагидродиазин-1-илу, тетрагидроазепинилу, пиперазинилу, хроманилу и так далее. Специалисту в данной области техники будет понятно, что связь вышеупомянутых (С 2-С)гетероциклоалкильных колец осуществляется через атом углерода или находящийся в sp3 гибридизации гетероатом азота.(С 2-С 9)гетероарил, используемый здесь, относится к фурилу, тиенилу, тиазолилу, пиразолилу, изотиазолилу, оксазолилу, изоксазолилу, пирролилу, триазолилу, тетразолилу, имидазолилу, 1,3,5 оксадиазолилу, 1,2,4-оксадиазолилу, 1,2,3-оксадиазолилу, 1,3,5-тиадиазолилу, 1,2,3-тиадиазолилу, 1,2,4 тиадиазолилу, пиридилу, пиримидилу, пиразинилу, пиридазинилу, 1,2,4-триазинилу, 1,2,3-триазинилу,1,3,5-триазинилу, пиразоло[3,4-b]пиридинилу, циннолинилу, птеридинилу, пуринилу, 6,7-дигидро-5 Н[1]пириндинилу, бенз[b]тиофенилу, 5,6,7,8-тетрагидрохинолин-3-илу, бензоксазолилу, бензтиазолилу,бензизотиазолилу, бензизоксазолилу, бензимидазолилу, тианафтенилу, изотианафтенилу, бензофуранилу, изобензофуранилу, изоиндолилу, индолилу, индолизинилу, индазолилу, изохинолилу, хинолилу,фталазинилу, хиноксалинилу, хиназолинилу, бензоксазинилу и так далее. Специалисту в данной области техники будет понятно, что связь вышеупомянутых (С 2-С 9)гетероциклоалкильных колец осуществляется через атом углерода или находящийся в sp3 гибридизации гетероатом азота. Настоящее изобретение также относится к фармацевтически приемлемым солям присоединения кислоты соединений формулы I. Кислоты, которые применяют для получения фармацевтически приемлемых солей присоединения кислоты вышеупомянутых основных соединений по этому изобретению, являются такими, которые образуют нетоксичные соли присоединения кислоты, то есть соли, содержащие фармакологически приемлемые анионы, такие как соли гидрохлорид, гидробромид, гидроиодид, нитрат,сульфат, бисульфат, фосфат, кислый фосфат, ацетат, лактат, цитрат, кислый цитрат, тартрат, битартрат,сукцинат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, паратолуолсульфонат и памоат [то есть 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)]. Изобретение также относится к солям присоединения основания формулы I. Химические основания, которые можно применять в качестве реагентов для получения фармацевтически приемлемых солей присоединения основания соединений формулы I, имеющих кислую природу, являются те основания,которые образуют нетоксичные основные соли с такими соединениями. Такие нетоксичные основные соли включают в себя, но не ограничиваются ими, производные от фармакологически приемлемых катионов, таких как катионы щелочных металлов (например калия и натрия) и катионы щелочноземельных металлов (например кальция и магния), соли аммония или водорастворимые соли присоединения амина, такого как N-метилглюкамин (меглумин), и низшие алканоламмонийные и другие основные соли фармацевтически приемлемых органических аминов. Соединения по этому изобретению включают в себя все конформационные изомеры (например циси транс-изомеры). Соединения по настоящему изобретению имеют асимметрические центры и, следовательно, существуют в различных энантиомерных и диастереомерных формах. Это изобретение относится к применению всех оптических изомеров и стереоизомеров соединений по настоящему изобретению и их смесей и всем фармацевтическим композициям и способам лечения, которые могут применять или содержать их. В этом отношении изобретение включает в себя как Е, так и Z конфигурации. Соединения формулы I могут также существовать как таутомеры. Это изобретение относится к применению всех таких таутомеров и их смесей.-3 006227 Соединения формулы (I) можно вводить в фармацевтически приемлемой форме или одно, или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему у млекопитающих, или с противовоспалительными агентами. Эти агенты могут включать в себя циклоспорин А (например, Sandimmune или Neoral), рапамицин, FK-506 (такролимус), лефлюномид,дезоксипергуалин, микофенолат (например, Cellcept), азатиоприн (например, Imuran), даклизумаб (например, Zenapax), OKT3 (например, Ortoclone), AtGam, аспирин, ацетаминофен, ибупрофен, напроксен, пироксикам и противовоспалительные стероиды (например, преднизолон или дексаметазон), но не ограничиваются ими. Эти агенты можно вводить как часть одной и той же или в разных лекарственных формах тем же самым или другим путем введения и по той же самой или другой схеме введения в соответствии со стандартной фармацевтической практикой. Это изобретение также относится к фармацевтическим композициям, содержащим пролекарства соединений формулы I. Это изобретение также относится к способам лечения или предупреждения расстройств, которые можно лечить или предупреждать путем ингибирования протеинкиназ, таких как фермент Janus киназа 3, при которых вводят пролекарства соединений формулы I. Соединения формулы I,имеющие свободные амино, амидо, гидрокси или карбоксильную группы, можно превратить в пролекарства. Пролекарства включают в себя соединения, в которых аминокислотный остаток или полипептидная цепь, состоящая из двух или более (например, двух, трех или четырех) аминокислотных остатков, ковалентно связана через пептидные связи со свободной амино, гидрокси или карбоксильной группами соединений формулы I. Аминокислотные остатки включают в себя 20 встречающихся в природе аминокислот, обычно обозначаемых трехбуквенными символами, и также включают в себя 4-гидроксипролин,гидроксилизин, десмозин, изодесмозин, 3-метилгистидин, норвалин, бета-аланин, гамма-аминомасляную кислоту, цитруллин, гомоцистеин, гомосерин, орнитин и метионинсульфон. Пролекарства также включают в себя соединения, карбонаты, карбаматы, амиды и алкиловые эфиры которых ковалентно связаны с вышеуказанными заместителями формулы I через атом углерода карбонильной группы, находящейся на боковой цепи пролекарств. Подробное описание изобретения Следующие реакционные схемы иллюстрируют получение соединений по настоящему изобретению. Если не указано иначе, R2, R3, R4 и R5 в последующих реакционных схемах и обсуждении такие, как определено выше. ПОЛУЧЕНИЕ А В реакции 1 получения А соединение 4-хлорпирроло[2,3-d]пиримидина формулы XXI, где R является водородом или защитной группой, такой как бензолсульфонил или бензил, превращают в соединение 4-хлор-5-галогенопирроло[2,3-d]пиримидина формулы XX, где Y является хлоро, бромо или иодо,путем взаимодействия XXI с N-хлорсукцинимидом, N-бромсукцинимидом или N-иодсукцинимидом. Реакционную смесь нагревают до температуры дефлегмации в хлороформе в течение примерно от 1 до примерно 3 ч, предпочтительно примерно в течение 1 ч. Альтернативно, в реакции 1 получения А 4 хлорпирроло[2,3-d]пиримидин формулы XXI, где R является водородом, превращают в соответствующий 4-хлор-5-нитропирроло[2,3-d]пиримидин формулы XX, где Y представляет собой нитро, путем взаимодействия XXI с азотной кислотой в серной кислоте при температуре в интервале примерно от -10 до примерно 10 С, предпочтительно примерно при 0 С, примерно в течение периода времени от пример-6 006227 но 5 до примерно 15 мин, предпочтительно примерно в течение 10 мин. Соединение формулы XXI, где Y представляет собой нитро, превращают в соответствующий 4-хлор-5-аминопирроло[2,3-d]пиримидин формулы XX, где Y представляет собой амино, путем взаимодействия XXI в различных условиях, известных специалисту в данной области техники, таких как гидрогенолиз в присутствии палладия или система хлорид олова (IV) - соляная кислота. В реакции 2 получения А соединение 4-хлор-5-галогенопирроло[2,3-d]пиримидина формулы XX,где R является водородом, превращают в соответствующее соединение формулы XIX, где R2 является-78 С, и полученный таким образом промежуточный дианион подвергают взаимодействию с алкилгалогенидом или бензилгалогенидом при температуре в интервале примерно от -78 С до примерно комнатной температуры, предпочтительно при комнатной температуре. Альтернативно, полученный таким образом дианион подвергают взаимодействию с молекулярным кислородом, чтобы получить соответствующее соединение 4-хлор-5-гидроксипирроло[2,3-d]пиримидина формулы XIX, где R2 представляет собой гидрокси. Соединение формулы XX, где Y является бромом или иодом и R является бензолсульфонатом, превращают в соединение формулы XIX, где R2 является (С 6-С 12)арилом или винилом, путем обработки N-бутиллитием при температуре примерно -78 С с последующим добавлением хлорида цинка при температуре примерно -78 С. Полученное таким образом промежуточное цинкорганическое соединение подвергают взаимодействию с арилиодидом или винилиодидом в присутствии каталитического количества палладия. Реакционную смесь перемешивают при температуре примерно от 50 до примерно 80 С, предпочтительно примерно при 70 С, в течение примерно от 1 до примерно 3 ч, предпочтительно в течение примерно 1 ч. В реакции 3 получения А соединение формулы XIX превращают в соответствующее соединение формулы XVI путем взаимодействия XIX с N-бутиллитием, диизопропиламином лития или гидридом натрия при температуре примерно -78 С в присутствии полярного апротонного растворителя, такого как тетрагидрофуран. Полученное таким образом анионное промежуточное соединение подвергают взаимодействию с (а) алкилгалогенидом или бензилгалогенидом при температуре в интервале примерно от-78 С до примерно комнатной температуры, предпочтительно при -78 С, где R3 является алкилом или бензилом; (б) альдегидом или кетоном при температуре в интервале примерно от -78 С до примерно комнатной температуры, предпочтительно при -78 С, где R3 представляет собой алкокси; и (в) хлоридом цинка при температуре в интервале примерно от -78 С до примерно комнатной температуры, предпочтительно при -78 С, и полученное таким образом соответствующее цинкорганическое промежуточное соединение затем подвергают взаимодействию с арилиодидом или винилиодидом в присутствии каталитического количества палладия. Полученную реакционную смесь перемешивают при температуре в интервале от примерно 50 до примерно 80 С, предпочтительно при 70 С, в течение периода времени примерно от 1 до 3 ч, предпочтительно примерно в течение 1 ч. Альтернативно, полученный таким образом анион подвергают взаимодействию с молекулярным кислородом, чтобы получить соответствующее соединение 4-хлор-6-гидроксипирроло[2,3-d]пиримидина формулы XVI, где R3 представляет собой гидрокси. В реакции 1 получения Б соединение 4-хлорпирроло[2,3-d]пиримидина формулы XXI превращают в соответствующее соединение формулы XXII согласно процедуре, описанной выше в реакции 3 получения А. В реакции 2 получения Б соединение формулы XXII превращают в соответствующее соединение формулы XVI согласно процедурам, описанным выше в реакциях 1 и 2 получения А. В реакции 1 схемы 1 соединение 4-хлорпирроло[2,3-d]пиримидина формулы XVII превращают в соответствующее соединение формулы XVI, где R является бензолсульфонилом или бензилом, путем взаимодействия XVII с бензолсульфонилхлоридом, бензилхлоридом или бензилбромидом в присутствии основания, такого как гидрид натрия или карбонат калия, и полярного апротонного растворителя, такого как диметилформамид или тетрагидрофуран. Реакционную смесь перемешивают при температуре примерно от 0 до 70 С, предпочтительно примерно при 30 С, в течение периода времени примерно от 1 до 3 ч, предпочтительно примерно в течение 2 ч. В реакции 2 схемы 1 соединение 4-хлорпирроло[2,3-d]пиримидина формулы XVI превращают в соответствующее соединение 4-аминопирроло[2,3-d]пиримидина формулы XV путем сочетания XVI с амином формулы HNR4R5. Реакцию проводят в спиртовом растворителе, таком как трет-бутанол, метанол или этанол, или другом высококипящем органическом растворителе, таком как диметилформамид,триэтиламин, 1,4-диоксан или 1,2-дихлорэтан, при температуре от примерно 60 до примерно 120 С,предпочтительно примерно при 80 С. Обычно время реакций составляет от примерно 2 до примерно 48 ч, предпочтительно примерно 16 ч. Если R5 является содержащей азот гетероциклоалкильной группой,то каждый атом азота должен быть защищен защитной группой, такой как бензильная. Удаление защитной группы с R5 проводят при соответствующих для этой определенной защитной группы условиях, если не будет повреждаться защитная группа, находящаяся на R пирроло[2,3-d]пиримидинового кольца. Удаление защитной группы с R5, если она является бензилом, проводят в спиртовом растворителе, таком как этанол, в присутствии водорода и катализатора, такого как гидроксид палладия на углероде. Полученную таким образом содержащую азот гетероциклоалкильную группу R5 можно затем подвергать взаимодей-7 006227 ствию с множеством различных электрофилов формулы II. Для образования производных мочевины электрофилы формулы II, такие как изоцианаты, карбаматы и карбамоилхлориды, подвергают взаимодействию с атомом азота гетероалкильной группы R5 в растворителе, таком как ацетонитрил или диметилформамид, в присутствии основания, такого как карбонат натрия или калия, при температуре примерно от 20 до примерно 100 С в течение периода времени от примерно 24 до примерно 72 ч. Для образования амида и сульфонамида электрофилы формулы II, такие как ацилхлориды и сульфонилхлориды,подвергают взаимодействию с атомом азота гетероалкильной группы R5 в растворителе, таком как метиленхлорид, в присутствии основания, такого как пиридин, при температуре окружающей среды в течение периода времени от примерно 12 до примерно 24 ч. Образование амида можно провести также путем взаимодействия карбоновой кислоты с гетероалкильной группой в присутствии карбодиимида, такого как 1-(3-диметиламинопропил)-3-этилкарбодиимид, в растворителе, таком как метиленхлорид, при температуре окружающей среды в течение 12-24 ч. Для образования алкила электрофилы формулы II, такие как ,-ненасыщенные амиды, кислоты, нитрилы, сложные эфиры и -галогенамиды, подвергают взаимодействию с атомом азота гетероалкильной группы R5 в растворителе, таком как метанол, при температуре окружающей среды в течение периода времени от примерно 12 до примерно 18 ч. Образование алкила можно также провести путем взаимодействия альдегидов с гетероалкильной группой в присутствии восстанавливающего агента, такого как цианоборгидрид натрия, в растворителе, таком как метанол, при температуре окружающей среды в течение периода времени от примерно 12 до примерно 18 ч. В реакции 3 схемы 1 удаление защитной группы с соединения формулы XV, где R является бензолсульфонилом, для получения соответствующего соединения формулы I, проводят путем взаимодействияXV с щелочным основанием, таким как гидроксид натрия или гидроксид калия, в спиртовом растворителе,таком как метанол или этанол, или смеси растворителей, такой как спирт/тетрагидрофуран или спирт/вода. Реакцию проводят при комнатной температуре в течение периода времени от примерно 15 мин до примерно 1 ч, предпочтительно в течение 30 мин. Удаление защитной группы с соединения формулы XV, где R является бензилом, проводят путем взаимодействия XV с раствором натрия в жидком аммиаке при температуре примерно -78 С в течение периода времени от примерно 15 мин до примерно 1 ч. В реакции 1 схемы 2 соединение 4-хлорпирроло[2,3-d]пиримидина формулы XX превращают в соответствующее соединение 4-аминопирроло[2,3-d]пиримидина формулы XXIV согласно процедуре, описанной выше в реакции 2 схемы 1. В реакции 2 схемы 2 соединение 4-амино-5-галогенпирроло[2,3-d]пиримидина формулы XXIV, гдеR является бензолсульфонатом и Z является бромом или иодом, превращают в соответствующее соединение формулы XXIII путем взаимодействия XXIV с (а) арилборной кислотой, где R2 является арилом, в апротонном растворителе, таком как тетрагидрофуран или диоксан, в присутствии каталитического количества палладия (0) при температуре от примерно 50 до примерно 100 С, предпочтительно примерно 70 С, в течение периода времени от примерно 2 до примерно 48 ч, предпочтительно примерно в течение 12 ч; (б) алкинами, где R2 является алкинилом, в присутствии каталитического количества иодида меди(I) и палладия (0) и полярного растворителя, такого как диметилформамид, при комнатной температуре в течение периода времени от примерно 1 до примерно 5 ч, предпочтительно примерно в течение 3 ч; и (в) алкенами или стиролами, где R2 является винилом или стирилом, в присутствии каталитического количества палладия в диметилформамиде, диоксане или тетрагидрофуране при температуре в интервале примерно от 80 до примерно 100 С, предпочтительно примерно 100 С, в течение периода времени от примерно 2 до примерно 48 ч, предпочтительно в течение примерно 48 ч. В реакции 3 схемы 2 соединение формулы XXIII превращают в соответствующее соединение формулы XV согласно процедуре, описанной выше в реакции 3 получения А. В реакции 1 схемы 3 соединение формулы XVII превращают в соответствующее соединение формулы I согласно процедуре, описанной выше в реакции 2 схемы 1. Соединения по настоящему изобретению, основные по своей природе, способны образовывать большое множество различных солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми для введения животным, часто желательно на практике вначале выделить соединение по настоящему изобретению из реакционной смеси в виде фармацевтически неприемлемой соли, а затем превратить последнюю обратно в свободное основание путем взаимодействия со щелочным реагентом с последующим превращением этого основания в фармацевтически приемлемую соль присоединения кислоты. Соли присоединения кислот основных соединений по данному изобретению легко получают путем обработки основного соединения по существу эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как метанол или этанол. При осторожном выпаривании растворителя легко получают желаемую твердую соль. Желаемую соль присоединения кислоты можно также осадить из раствора свободного основания в органическом растворителе путем добавления к раствору соответствующей минеральной или органической кислоты. Те соединения по настоящему изобретению, которые являются кислотными по своей природе, способны образовывать основные соли с различными фармакологически приемлемыми катионами. Примеры таких солей включают в себя соли щелочных или щелочно-земельных металлов и, в частности, соли-8 006227 натрия и калия. Эти соли получают по обычным методикам. Химическими основаниями, которые используют как реагенты для получения фармацевтически приемлемых основных солей этого изобретения,являются те, которые образуют нетоксичные основные соли с кислыми соединениями по данному изобретению. Такие нетоксичные основания соли включают в себя соли, производные от таких фармацевтически приемлемых катионов как натрий, калий, кальций, магний и так далее. Эти соли легко можно получить путем обработки соответствующих кислых соединений водным раствором, содержащим желаемые фармакологически приемлемые катионы, и затем упаривания полученного раствора досуха, предпочтительно при пониженном давлении. Альтернативно, их можно получить путем смешивания вместе растворов кислых соединений в низших спиртах и алкоксида желаемого щелочного металла с последующим упариванием полученного раствора досуха по тому же самому вышеприведенному способу. В каждом случае предпочтительно применяют стехиометрические количества реагентов, чтобы гарантировать завершенность реакции и максимальный выход желаемого конечного продукта. Композиции по настоящему изобретению можно приготовить обычными способами, применяя один или более чем один фармацевтически приемлемый носитель. Так, активные соединения по изобретению можно приготовить в виде препарата для перорального, трансбуккального, интраназального, парентерального (например, внутривенного, внутримышечного или подкожного) или ректального введения или в форме, подходящей для введения путем ингаляции или инсуффляции. Активные соединения по изобретению можно также приготовить в виде препарата для пролонгированной доставки. Для перорального введения фармацевтические композиции могут принимать форму, например,таблеток или капсул, приготовленных обычными способами с фармацевтически приемлемыми эксципиентами, такими как связывающие агенты (например, предварительно желатинизированный кукурузный крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители (например, лактоза,микрокристаллическая целлюлоза или фосфат кальция); смазывающие вещества (например, стеарат магния, тальк или диоксид кремния); разрыхлители (например, картофельный крахмал или крахмал гликолят натрия); или увлажняющие агенты (например, лаурилсульфат натрия). Таблетки могут быть покрыты оболочкой хорошо известными способами. Жидкие препараты для перорального введения могут принимать форму, например, растворов, сиропов или суспензий или они могут быть представлены как сухие препараты для образования состава с водой или другим подходящим носителем перед применением. Такие жидкие препараты можно получить обычными способами с фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сироп сорбита, метилцеллюлоза или гидрированные пищевые жиры); эмульгаторы (например, лецитин или аравийская камедь); неводные носители (например, миндальное масло, жирные эфиры или этиловый спирт); и консерванты (например, метил- или пропилпарагидроксибензоаты или сорбиновая кислота). Для трансбуккального введения композиция может принимать форму таблеток или лепешек, приготовленных в виде препарата обычным способом. Активные соединения по изобретению можно приготовить в виде препарата для парентерального введения путем инъекции, включая применение обычных методик катетеризации или инфузии. Препараты для инъекции могут быть представлены в стандартной лекарственной форме, например в ампулах или многодозовых контейнерах, с добавленным консервантом. Композиции могут принимать такие формы,как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать агенты для приготовления лекарственных препаратов, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный ингредиент может быть в форме порошка для образования состава с подходящим носителем, например стерильной апирогенной водой, перед применением. Активные соединения по изобретению можно также приготовить в виде ректальных композиций,таких как суппозитории или удерживающие клизмы, например, содержащие обычные основы для суппозитория, такие как масло какао или другие глицериды. Для интраназального введения или введения путем ингаляции активные соединения по изобретению обычно доставляют в форме раствора или суспензии из пульверизатора, который сжимается или нагнетается пациентом, или в виде аэрозольного спрея из контейнера, находящегося под давлением, или небулайзера с применением подходящего пропеллента, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае аэрозоля, находящегося под давлением, единица дозировки может быть установлена с помощью клапана для доставки отмеренного количество. Контейнер под давлением или небулайзер может содержать раствор или суспензию активного соединения. Капсулы и картриджи (сделанные, например, из желатина) для применения в ингаляторе или инсуффляторе могут быть приготовлены в виде препарата, содержащего порошковую смесь соединения по изобретению и подходящей порошковой основы, такой как лактоза или крахмал. Рекомендуемая доза активных соединений по изобретению для перорального, парентерального или трансбуккального введения обычному взрослому человеку для лечения вышеупомянутых состояний (например, ревматоидного артрита) составляет от 0,1 до 1000 мг активного ингредиента на стандартную дозу, которую можно вводить, например, от 1 до 4 раз в сутки.-9 006227 Аэрозольные лекарственные препараты для лечения вышеупомянутых состояний (наприме, астмы) у обычного взрослого человека предпочтительно приспособлены так, что каждая отмеренная доза или"пшик" аэрозоля содержит от 20 до 1000 мкг соединения по изобретению. Суммарная суточная доза с аэрозолем будет в пределах интервала 0,1-1000 мг. Вводить можно несколько раз в сутки, например 2, 3,4 или 8 раз, назначая, например, 1, 2 или 3 дозы каждый раз. Соединение формулы (I), вводимое в фармацевтически приемлемой форме само по себе или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему у млекопитающих, или с противовоспалительными агентами, агентами, которые могут включать в себя циклоспорин А (например, Sandimmune или Neoral), рапамицин, FK-506 (такролимус), лефлюномид, дезоксипергуалин, микофенолат (например, Cellcept), азатиоприн (например, Imuran), даклизумаб(например, Zenapax), ОКТЗ (например, Ortoclone), AtGam, аспирин, ацетаминофен, ибупрофен, напроксен, пироксикам и противовоспалительные стероиды (например, преднизолон или дексаметазон), но не ограничиваются ими; и такие агенты можно вводить как часть одной и той же или в разных лекарственных формах тем же самым или другим путем введения и по той же самой или другой схеме введения в соответствии со стандартной фармацевтической практикой.FK-506 (такролимус) назначают перорально по 0,10-0,15 мг/кг массы тела каждые 12 ч в первые 48 ч после операции. Дозировку контролируют по минимальному уровню такролимуса в сыворотке. Циклоспорин A (Sandimmune для перорального или внутривенного введения или Neoral раствор или капсулы для перорального введения) назначают перорально по 5 мг/кг массы тела каждые 12 ч в первые 48 ч после операции. Дозировку контролируют по минимальному уровню циклоспорина А в крови. Активные агенты можно приготовить в виде препаратов для пролонгированной доставки согласно способам, хорошо известным специалисту в данной области техники. Примеры таких препаратов можно найти в патентах США 3538214, 4060598, 4173626, 3119742 и 3492397. Способность соединений формулы I или их фармацевтически приемлемых солей ингибировать Janus киназу 3 и следовательно демонстрировать их эффективность в лечении расстройств или состояний,характеризуемых Janus киназой 3, показаны в следующем тесте in vitro. Биологический тестJAK3 (JH1:GST) ферментативный метод анализа В JAK3 киназном тесте используют белок, экспрессированный в инфицированных бакуловирусом клетках SF9 (гибридный белок GST и каталитического домена человеческой JAK3), очищенный с помощью аффинной хроматографии на глутатион-Sepaharose. Субстратом для реакции является полиглутаминовая кислота-тирозин (ПГТ (4:1), каталог SigmaP0275), нанесенная на планшеты Nunc Maxi Sorp в концентрации 100 мкг/мл в течение ночи при 37 С. Утром планшеты промывают три раза и добавляютJAK3 в лунки, содержащие 100 мкл киназного буфера (50 мМ HEPES (4-(2-гидроксиэтил)-1 пиперазинэтансульфоновая кислота), рН 7,3, 125 мМ NaCl, 24 мМ MgCl2 + 0,2 мкМ АТФ (аденозинтрифосфат) + 1 мМ ортованадата натрия). Реакция продолжается в течение 30 мин при комнатной температуре, и планшеты промывают не менее трех раз. Уровень фосфорилированного тирозина в данной лунке количественно определяют с помощью стандартного теста ELISA, применяя антифосфотирозиновое антитело (ICN PY20, cat. 69-151-1). Ингибирование IL-2 зависимой пролиферации человеческих Т-лимфобластов В этом скрининге определяют ингибирующий эффект соединений на IL-2 зависимую пролиферацию Т-лимфобластов in vitro. Так как для передачи сигнала через IL-2 рецептор необходима JAK-3, то активные клеточные ингибиторы JAK-3 могут ингибировать IL-2 зависимую пролиферацию Тлимфобластов. Клетки для этого теста извлекают из свежей человеческой крови. После выделения мононуклеарных клеток, применяя Accuspin System-Histopaque-1077 (SigmaA7054), первичные Т-клетки человека извлекают с помощью отрицательной селекции, применяя Limpho-Kwik T (One Lamda, Inc., CatLK50T). Т-клетки выращивают при 1-2 х 106/мл в среде (RPMI + 10% инактивированной нагреванием фетальной телячьей сыворотки (Hyclone CatA-1111-L) + 1% пенициллин/стрептомицин (Gibco и вызывают пролиферацию путем добавления 10 мкг/мл ФГА (фитогемагглютинин, Murex Diagnostics, CatНА 16). Через три дня при 37 С в 5% СO2 клетки промывают три раза в среде, ресуспендируют до плотности 1-2 х 106 клеток/мл в среде плюс 100 единиц/мл человеческого рекомбинантного IL-2 (RD Systems, Cat202-IL). Через одну неделю клетки становятся IL-2 зависимыми и могут поддерживаться до трех недель при добавлении два раза в неделю эквивалентных объемов среды + 100 единиц/мл IL-2. Чтобы оценить способность тестируемых соединений ингибировать IL-2 зависимую пролиферацию Т-лимфобластов, IL-2 зависимые клетки промывают три раза, ресуспендируют в среде и затем помещают(50000 клеток/лунка/0,1 мл) в плоскодонный 96-луночный титрационный микропланшет (Falcon353075). Серийные двукратные разведения 10 мМ маточного раствора тестируемого соединения в ДМСО(диметилсульфоксид) добавляют по три раза в лунки, начиная с 10 мкМ. Через один час добавляют к каждой тестовой лунке 10 единиц/мл IL-2. Планшеты затем инкубируют при 37 С в 5% СO2 в течение 72 ч. Планшеты затем импульсно обрабатывают 3H-тимидином (0,5 мкКи/лунка) (NEN CatNET-027A) и до- 10006227 полнительно инкубируют в течение 18 ч. Культуры из планшетов затем собирают в 96-луночный планшет-харвестер, и количество 3H-тимидина, включенное в пролиферативные клетки, определяют с помощью сцинтилляционного счетчика Packard Top Count. Данные анализируют путем построения графика зависимости % ингибирования пролиферации от концентрации тестируемого соединения. Значение IC50(мкМ) определяют из этого графика. Следующие примеры иллюстрируют получение соединений по настоящему изобретению, но не ограничивают его в деталях. Температуры плавления не исправлены. Данные ЯМР представлены в частях на миллион (5) и относятся к сигналу дейтерия растворителя образца (дейтериохлороформ, если не указано иначе). Имеющиеся в продаже реагенты применяли без дополнительной очистки. ТГФ относится к тетрагидрофурану. ДМФ относится к N,N-диметилформамиду. Масс-спектры низкого разрешения(LRMS) записывали или на приборе Hewlett Packard 59809, используя химическую ионизацию (аммиак),или на приборе Fisons (или Micro Mass), платформа с химической ионизацией при атмосферном давлении (ХИАД), применяя в качестве ионизирующего агента смесь ацетонитрил/вода в соотношении 50/50 с 0,1%-ной муравьиной кислотой. Комнатная температура или температура окружающей среды относятся к 20-25 С. Пример 1. 1-4-Метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-илэтанон. Способ А. (1-Бензил-4-метилпиперидин-3-ил)метиламин. К перемешиваемому раствору 1-бензил-4-метилпиперидин-3-она (2,3 г, 11,5 ммоль), полученного согласно способам Iorio, M.A. и Damia, G., Tetrahedron, 26, 5519 (1970) и Grieco et al, Journal of the American Chemical Society, 107, 1768 (1985) (модифицировано применением 5% метанола как сорастворителя),оба способа включены полностью посредством ссылки, растворенному в 23 мл 2 М метиламина в тетрагидрофуране, добавили 1,4 мл (23 ммоль) уксусной кислоты и полученную смесь перемешивали в запечатанной трубке в течение 16 ч при комнатной температуре. Затем добавили триацетоксиборгидрид натрия (4,9 г, 23 ммоль) и новую смесь перемешивали при комнатной температуре в запечатанной трубке в течение 24 ч, после чего реакцию гасили добавлением 1 н раствора гидроксида натрия (50 мл). Реакционную смесь затем экстрагировали эфиром (3 х 80 мл), объединенные эфирные слои сушили над сульфатом натрия (Na2SO4) и концентрировали в вакууме досуха с получением 1,7 г (69%) указанного в заголовке соединения в виде белого твердого вещества. LRMS: 219,1 (М+1). Способ Б. (1-Бензил-4-метилпиперидин-3-ил)метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Раствор 4-хлорпирроло[2,3-d]пиримидина (2,4 г, 15,9 ммоль), полученный согласно способу Davoll,J. Am. Chem. Soc., 82, 131 (1960), который включен полностью посредством ссылки, и продукт, полученный согласно способу А (1,7 г, 7,95 ммоль) и растворенный в двух эквивалентах триэтиламина, нагревали в запечатанной трубке при 100 С в течение 3 дней. После охлаждения до комнатной температуры и концентрирования при пониженном давлении остаток очищали с помощью флэш-хроматографии (силикагель; 3% метанола в дихлорметане) с получением 1,3 г (50%) указанного в заголовке соединения в виде бесцветного масла. LRMS: 336,1 (М+1). Способ В. Метил-(4-метилпиперидин-3-ил)-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. К продукту способа Б (0,7 г, 2,19 ммоль), растворенному в 15 мл этанола, добавили 1,5 мл 2 н соляной кислоты и реакционную смесь дегазировали током азота. К реакционной смеси затем добавили 0,5 г гидроксида палладия на углероде (50% вода) (Aldrich), и полученную смесь перемешивали (Parr-Shaker) в атмосфере водорода при 50 psi (фунт/кв.дюйм, 344,5 кПа) при комнатной температуре в течение 2 дней. Отфильтрованную через целит реакционную смесь концентрировали в вакууме досуха и остаток очищали с помощью флэш-хроматографии (силикагель; 5% метанола в дихлорметане) с получением 0,48 г(90%) указанного в заголовке соединения. LRMS: 246,1 (М+1). Способ Г. 1-4-Метил-3-[метил(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-илэтанон. К перемешиваемому раствору продукта способа В (0,03 г, 0,114 ммоль), растворенного в 5 мл смеси 10:1 дихлорметан/пиридин, добавили ацетилхлорид (0,018 г,0,228 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 18 ч. Затем реакционную смесь распределили между дихлорметаном и насыщенным бикарбонатом натрия (NаНСО 3). Органический слой промыли опять насыщенным NаНСО 3, сушили над сульфатом натрия и концентрировали в вакууме досуха. Остаток очищали с помощью препаративной тонкослойной хроматографии (ТСХ) (силикагель; 4% метанола в дихлорметане) с получением 0,005 мг (15%) указанного в заголовке соединения в виде бесцветного масла. LRMS: 288,1 (М+1). Указанные в заголовках соединения для примеров 2-26 были получены способом, аналогичным описанному в примере 1. Пример 2. [1-(2-Аминоэтансульфонил)-4-метилпиперидин-3-ил]метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин.N-[2-(4-метил-3-метиламинопиперидин-1-ил)-2-оксоэтил]ацетамид. LRMS: 345. Пример 23. 3-Этокси-1-4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1 илпропан-1-он. 3-Этокси-1-(4-метил-3-метиламинопиперидин-1-ил)пропан-1-он. LRMS:346. Пример 24. Метиламид 4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1 карбоновой кислоты Метиламид 4-метил-3-метиламинопиперидин-1-карбоновой кислоты. LRMS: 303. Пример 25. Диэтиламид 4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1 карбоновой кислоты. или его фармацевтически приемлемая соль; где R1 представляет собой группу формулыR5 представляет собой (С 2-С 9)гетероциклоалкил, где гетероциклоалкильная группа должна быть замещена в количестве от 1 до 2 (С 1-С 6)алкилом, (С 1-С 6)алкокси, (С 1-С 6)ацилом, (С 1-С 6)алкиламино-СО-,амино(С 1-С 6)ацилом, С 1-С 6)алкил)2 амино(С 1-С 6)ацилом, (С 1-С 6)алкил-S(O)2, R15S(O)2R16N, где R15 и R16,каждый независимо, выбран из водорода или (C1-С 6)алкила; или группой формулыR6, R7, R8, R9, R10 и R11, каждый независимо, выбран из группы, состоящей из водорода или (С 1 С 6)алкила;R2 и R3, каждый независимо, представляет собой водород. 2. Соединение по п.1, где указанное соединение выбрано из группы, состоящей из метил-[4-метил-1-(пропан-1-сульфонил)пиперидин-3-ил]-(7 Н-пирроло[2,3-d]пиримидин-4 ил)амина; 3,3,3-трифтор-1-4-метил-3-[метил(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1 илпропан-1-она и 3-4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил-3-оксопропионитрила. 3. Фармацевтическая композиция для: а) лечения или предупреждения расстройства или состояния,выбранного из отторжения трансплантата органа, ксенотрансплантации, волчанки, рассеянного склероза,ревматоидного артрита, псориаза, диабета первого типа и осложнений при диабете, рака, астмы, атопического дерматита, аутоиммунных тиреоидных расстройств, неспецифического язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других аутоиммунных заболеваний, или б) ингибирования протеинкиназ, в частности Janus киназы 3 (JAK3), у млекопитающего, включая человека, содержащая количество соединения по п.1 или его фармацевтически приемлемой соли, одного или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млеко- 13006227 питающих, или с противовоспалительными агентами, эффективное в лечении таких расстройств или состояний, и фармацевтически приемлемый носитель. 4. Способ ингибирования протеинкиназ, в частности Janus киназы 3 (JAK3), у млекопитающего,включая человека, при котором указанному млекопитающему вводят эффективное количество соединения по п.1 или его фармацевтически приемлемой соли, одного или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млекопитающих, или с противовоспалительными агентами. 5. Способ лечения или предупреждения расстройства или состояния, выбранного из отторжения трансплантата органа, ксенотрансплантации, волчанки, рассеянного склероза, ревматоидного артрита,псориаза, диабета первого типа и осложнений при диабете, рака, астмы, атопического дерматита, аутоиммунных тиреоидных расстройств, неспецифического язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других аутоиммунных заболеваний у млекопитающего, включая человека, при котором указанному млекопитающему вводят количество соединения по п.1 или его фармацевтически приемлемой соли, одного или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млекопитающих, или с противовоспалительными агентами, эффективное в лечении такого состояния. 6. Соединение по п.1, где замещенная (С 2-С 9)гетероциклоалкильная группа R5 представляет собой пиперидинильную группу. 7. Соединение по п.1, где а равно 0; b равно 1; Х является карбонилом; с, е, f и g каждый равен 0 и d равно 1. 8. Соединение по п.2, где соединение представляет собой 3-4-метил-3-[метил-(7 Н-пирроло[2,3d]пиримидин-4-ил)амино]пиперидин-1-ил-3-оксо-пропионитрил. 9. Фармацевтическая композиция по п.3, где указанный агент выбран из группы, состоящей из циклоспорина А, рапамицина, такролимуса, лефлюномида, дезоксипергуалина, микофенолата, азатиоприна,даклизумаба, муромонаб-СD3, антитимоцит глобулина, аспирина, ацетаминофена, ибупрофена, напроксена, пироксикама и противовоспалительного стероида. 10. Способ по п.4, где указанный агент выбран из группы, состоящей из циклоспорина А, рапамицина, такролимуса, лефлюномида, дезоксипергуалина, микофенолата, азатиоприна, даклизумаба, муромонаб-CD3, антитимоцит глобулина, аспирина, ацетаминофена, ибупрофена, напроксена, пироксикама и противовоспалительного стероида. 11. Способ по п.5, где указанный агент выбран из группы, состоящей из циклоспорина А, рапамицина, такролимуса, лефлюномида, дезоксипергуалина, микофенолата, азатиоприна, даклизумаба, муромонаб-CD3, антитимоцит глобулина, аспирина, ацетаминофена, ибупрофена, напроксена, пироксикама и противовоспалительного стероида.
МПК / Метки
МПК: A61P 37/00, C07D 487/04, A61K 31/519
Метки: соединения, пирроло[2,3-d]пиримидина
Код ссылки
<a href="https://eas.patents.su/15-6227-soedineniya-pirrolo23-dpirimidina.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения пирроло[2,3-d]пиримидина</a>
Соединения пирроло[2,3-d]пиримидина в качестве иммунодепрессантов

Номер патента: 6153
Опубликовано: 27.10.2005
Авторы: Манчхоф Майкл Джон, Флэнэган Марк Эдвард, Блуменкопф Тодд Эндрю
МПК: A61P 17/06, A61K 31/505, C07D 487/04...
Метки: пирроло[2,3-d]пиримидина, качестве, соединения, иммунодепрессантов
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль; где R1 представляет собой группу формулы где R4 представляет собой (C1-C6)алкил; R5 представляет собой пиперидинил, который должен быть замещен группами в количестве от одной до пяти, состоящими из (C1-C6)алкила, циано(C1-C6)алкила, [(циано(C1-C6)алкил)((C1-C6)алкил)амино]ацила, гидрокси(C1-C6)алкилацила, нитро(C1-C6)алкилацила, (C1-C6)алкилSO2HN(C1-C6)алкилацила,...
Пирроло[2,3-d]пиримидиновые соединения

Номер патента: 6034
Опубликовано: 25.08.2005
Авторы: Браун Маттью Фрэнк, Фланаган Марк Эдуард, Чанджелиэн Пол Стивен, Блуменкопф Тодд Эндрю
МПК: C07D 487/04, A61K 31/505, A61P 35/02...
Метки: соединения, пирроло[2,3-d]пиримидиновые
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль, где R1 представляет собой группу формулы где пунктирная линия обозначает возможные двойные связи; m равняется 0, 1 или 2; n равняется 1 или 2, X представляет собой кислород, NR6 или CR7R8; каждый из B и D представляет собой CR7R8; каждый из A и E представляет собой CR7R8; и R6 выбран из группы, состоящей из водорода, (C1-C6)алкила; каждый из R7 и R8 независимо выбран из группы,...
Пирроло[2,3-d]пиримидины

Номер патента: 5852
Опубликовано: 30.06.2005
Авторы: Браун Мэттью Фрэнк, Блюменкопф Тодд Эндрю, Ченджелиан Пол Стивен, Флэнэган Марк Эдвард
МПК: C07D 487/04, A61P 37/06, A61K 31/519...
Метки: пирроло[2,3-d]пиримидины
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль, где R1 представляет группу формулы где y означает 0, 1 или 2; R4 выбирают из группы, состоящей из водорода, (C1-C6)алкила или (C2-C6)алкенила, где алкильные и алкенильные группы необязательно замещены гидроксильной группой, аминогруппой, (C1-C4)алкокси или (C1-C6)ациламиногруппой; или R4 является (C3-C10)циклоалкилом, где циклоалкильная группа необязательно замещена дейтерием,...
Фармкомпозиция, содержащая соединения с анти-ха активностью и соединения антагониста агрегации тромбоцитов, их применение, набор, содержащий эти соединения, способ лечения или профилактики заболеваний, сопутствующих тромбообразованию

Номер патента: 2475
Опубликовано: 27.06.2002
Авторы: Данвидди Кристофер Т., Перроне Марк Х., Кюродо Алан Х., Юзан Андре, Лидли Роберт Дж.
МПК: A61P 7/02, A61K 31/715
Метки: профилактики, содержащий, активностью, тромбоцитов, способ, заболеваний, антагониста, соединения, сопутствующих, агрегации, тромбообразованию, применение, эти, содержащая, набор, лечения, фармкомпозиция, анти-ха
Формула / Реферат:
1. Фармацевтическая композиция, содержащая фармацевтически пригодный носитель и фармацевтически эффективные количества соединения, обладающего анти-Ха активностью, и соединения антагониста агрегации тромбоцитов. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что соединение, обладающее анти-Ха активностью, является низкомолекулярно весовым гепарином. 3. Фармацевтическая композиция по п.2, отличающаяся тем, что низкомолекулярно весовой...
Композиции на основе пирроло [2,3d] пиримидинов и их применение

Номер патента: 3604
Опубликовано: 26.06.2003
Авторы: Уиттер Дэвид Дж., Кастелано Арлиндо Л., Маккиббен Брайан
МПК: A61P 9/00, C07D 487/04, A61K 31/505...
Метки: применение, пирроло, пиримидинов, композиции, основе, 2,3d
Формула / Реферат:
1. Соединение, имеющее формулу I где каждый из R1 и R2 независимо представляет собой атом водорода, замещенный прямой (C1-C30)алкил, замещенный разветвленный (C3-C30)алкил, замещенный (C4-C10)циклоалкил, замещенный циклопропил или замещенный или незамещенный арил; где только один из R1 и R2 может быть водородом; где, когда алкил представляет собой (C1)алкил или (C2)алкил, тогда любой заместитель фенила, если он имеется, замещен; или R1 и R2...
Предыдущий патент: Антинеопластические комбинации
Следующий патент: Улучшенные агенты переноса электронов в композициях для кислотной обработки скважин и способах такой обработки
Случайный патент: Гигиенический набор, включающий банную губку из полимерной сетки с ромбовидными ячейками и жидкое моющее средство с галогенированным биоцидом