Способ получения производных дезоксирибофуранозы






Формула / Реферат
1. Способ получения соединения формулы 11

где R2 означает -CF3, -CH3 или -C6H4CH3,
включающий:
(i) окисление соединения 6

гипохлоритом натрия в присутствии TEMPO (2,2,6,6-тетраметилпиперидина-1-оксил) и ацетата натрия в двухфазной системе с изопропилацетатом с образованием соединения 8

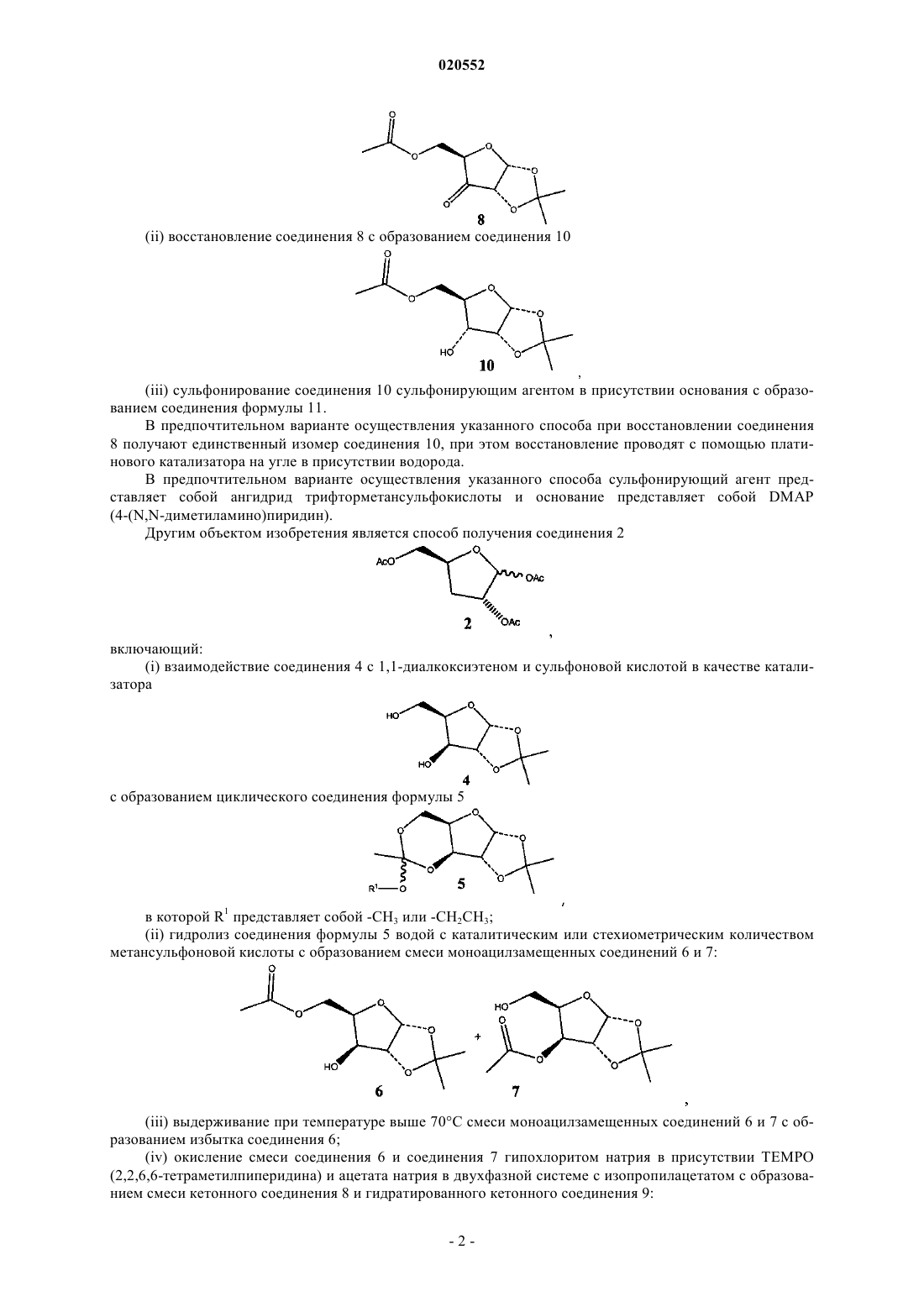
(ii) восстановление соединения 8 с образованием соединения 10

(iii) сульфонирование соединения 10 сульфонирующим агентом в присутствии основания с образованием соединения формулы 11.
2. Способ по п.1, в котором при восстановлении соединения 8 получают единственный изомер соединения 10 и в котором восстановление проводят с помощью платинового катализатора на угле в присутствии водорода.
3. Способ по п.1, в котором сульфонирующий агент представляет собой ангидрид трифторметансульфокислоты и основание представляет собой DMAP (4-(N,N-диметиламино)пиридин).
4. Способ получения соединения 2

включающий:
(i) взаимодействие соединения 4 с 1,1-диалкоксиэтеном и сульфоновой кислотой в качестве катализатора

с образованием циклического соединения формулы 5

в которой R1 представляет собой -CH3 или -CH2CH3:
(ii) гидролиз соединения формулы 5 водой с каталитическим или стехиометрическим количеством метансульфоновой кислоты с образованием смеси моноацилзамещенных соединений 6 и 7:

(iii) выдерживание при температуре выше 70°C смеси моноацилзамещенных соединений 6 и 7 с образованием избытка соединения 6;
(iv) окисление смеси соединения 6 и соединения 7 гипохлоритом натрия в присутствии TEMPO (2,2,6,6-тетраметилпиперидина-1-оксил) и ацетата натрия в двухфазной системе с изопропилацетатом с образованием смеси кетонного соединения 8 и гидратированного кетонного соединения 9:

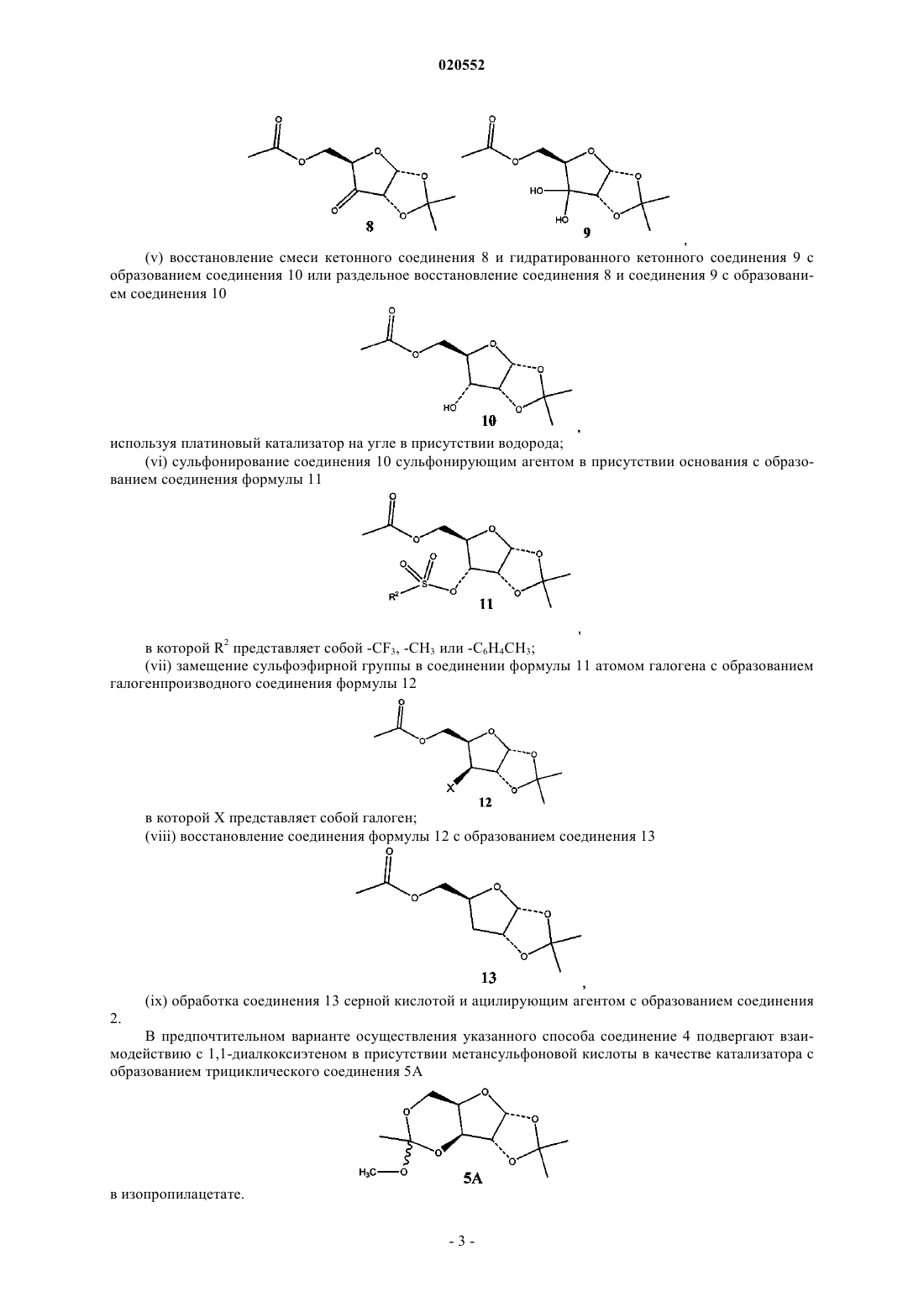
(v) восстановление смеси кетонного соединения 8 и гидратированного кетонного соединения 9 с образованием соединения 10 или раздельное восстановление соединения 8 и соединения 9 с образованием соединения 10

используя платиновый катализатор на угле в присутствии водорода;
(vi) сульфонирование соединения 10 сульфонирующим агентом в присутствии основания с образованием соединения формулы 11

в которой R2 представляет собой -CF3, -CH3 или -C6H4CH3;
(vii) замещение сульфоэфирной группы в соединении формулы 11 атомом галогена с образованием галогенпроизводного соединения формулы 12

в которой X представляет собой галоген;
(viii) восстановление соединения формулы 12 с образованием соединения 13

(ix) обработка соединения 13 серной кислотой и ацилирующим агентом с образованием соединения 2.
5. Способ по п.4, где соединение 4 подвергают взаимодействию с 1,1-диалкоксиэтеном в присутствии метансульфоновой кислоты в качестве катализатора с образованием трициклического соединения 5А

в изопропилацетате.
6. Способ по п.4, где соединение 10 получают как единственный изомер с помощью триацетоксиборгидрида натрия.
7. Способ по п.4, в котором соединение 8 выделяют из смеси соединений 8 и 9, и способ дополнительно включает восстановление соединения 8 с образованием соединения 10 как единственного изомера, где восстановление проводят с помощью платинового катализатора на угле в присутствии водорода.
8. Способ по п.4, где основание представляет собой DMAP (4-(N,N-диметиламино)пиридин).
9. Способ по п.4, где при восстановлении соединения формулы 12 используют гидроксид палладия на угле в присутствии водорода.
10. Соединение, выбранное из:

Текст
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ДЕЗОКСИРИБОФУРАНОЗЫ Изобретение относится к способам получения соединений дезоксирибофуранозы, в том числе соединения (2), которые представляют собой промежуточные соединения для синтеза используемых в фармацевтике соединений, таких как 5-амино-3-(2'-O-ацетил-3'-дезоксиDрибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он и аналогичные. Область техники, к которой относится изобретение Настоящее изобретение относится к способам получения производных дезоксирибофуранозы, которые представляют собой промежуточные соединения для синтеза используемых в фармацевтике соединений,таких как 5-амино-3-(2'-О-ацетил-3'-дезоксиD-рибофуранозил)-3H-тиазоло[4,5d]пиримидин-2-он и аналогичные. Уровень техники Аналоги нуклеозидов составляют важный класс соединений, которые полезны в лечении заболеваний. Например, аналоги нуклеозидов применяли в лечении раковых заболеваний и вирусных инфекций. После введения в клетку аналоги нуклеозидов часто фосфорилируются по механизмам реутилизации нуклеозидов, в которых данные аналоги фосфорилируются в соответствующие моно-, ди- и трифосфаты. Помимо других внутриклеточных функций, трифосфорилированные аналоги нуклеозидов часто служат субстратами для ДНК- или РНК-полимераз и встраиваются в ДНК и/или РНК. Так как трифосфорилированные аналоги нуклеозидов являются сильными ингибиторами полимеразы, они могут индуцировать преждевременный распад возникающей молекулы нуклеиновой кислоты. Когда трифосфорилированные аналоги нуклеозидов встраиваются в репликаты или транскрипты нуклеиновых кислот, это может приводить к экспрессии генов или нарушению их функционирования. Некоторые аналоги нуклеозидов могут быть эффективными вследствие своей способности ингибировать аденозинкиназу. Аденозинкиназа катализирует фосфорилирование аденозина в аденозин-5'монофосфат (AMP). Ингибирование аденозинкиназы может эффективно увеличивать внеклеточный уровень аденозина в организме человека и, таким образом, служить лечению ишемических состояний,включая инсульт, воспаление, артрит, пароксизмы и эпилепсию. В последние несколько десятилетий были приложены значительные усилия по исследованию терапевтических приложений аналогов нуклеозидов. Например, определенные пиримидо[4,5d]пиримидиннуклеозиды описаны в патенте США 5041542 (Robins et al.) как эффективные средства в лечении лимфолейкоза L1210 у гибридных мышей BDF1. Кроме того, 3D-рибофуранозилтиазоло[4,5d]пиримидины, проявляющие значительную иммуноактивность, включая пролиферацию мышечных клеток селезенки, и активность in vivo против вируса леса Семлики, описаны в патентах США 5041426 и 4880784 (Robins et al.). В ряде публикаций также описаны негликозильные производные, содержащие фрагмент тиазоло[4,5-d]пиримидина. См., например, патенты США 5994321 и 5446045 и Revankar etal. (J. Het. Chem., 30, 1341-49 (1993 и Lewis et al. (J. Het. Chem., 32, 547-56 (1995. Показано, что 3,5-двухзамещенные соединения 3H-тиазоло[4,5-d]пиримидин-2-она имеют иммуномодуляторную активность. Получение и применение этого класса соединений описаны в опубликованных патентной заявке СШАUS2006/0160830 (патентная заявка США 11/304691) и в патентной заявке США 11/873202, обе из которых во всей своей полноте включены в настоящий документ посредством ссылки. Сущность изобретения Настоящее изобретение относится к способу получения соединения формулы 11(iii) сульфонирование соединения 10 сульфонирующим агентом в присутствии основания с образованием соединения формулы 11. В предпочтительном варианте осуществления указанного способа при восстановлении соединения 8 получают единственный изомер соединения 10, при этом восстановление проводят с помощью платинового катализатора на угле в присутствии водорода. В предпочтительном варианте осуществления указанного способа сульфонирующий агент представляет собой ангидрид трифторметансульфокислоты и основание представляет собой DMAP(4-(N,N-диметиламино)пиридин). Другим объектом изобретения является способ получения соединения 2 с образованием циклического соединения формулы 5(ii) гидролиз соединения формулы 5 водой с каталитическим или стехиометрическим количеством метансульфоновой кислоты с образованием смеси моноацилзамещенных соединений 6 и 7:(iii) выдерживание при температуре выше 70C смеси моноацилзамещенных соединений 6 и 7 с образованием избытка соединения 6;(iv) окисление смеси соединения 6 и соединения 7 гипохлоритом натрия в присутствии TEMPO(2,2,6,6-тетраметилпиперидина) и ацетата натрия в двухфазной системе с изопропилацетатом с образованием смеси кетонного соединения 8 и гидратированного кетонного соединения 9:(v) восстановление смеси кетонного соединения 8 и гидратированного кетонного соединения 9 с образованием соединения 10 или раздельное восстановление соединения 8 и соединения 9 с образованием соединения 10 используя платиновый катализатор на угле в присутствии водорода;(vi) сульфонирование соединения 10 сульфонирующим агентом в присутствии основания с образованием соединения формулы 11(vii) замещение сульфоэфирной группы в соединении формулы 11 атомом галогена с образованием галогенпроизводного соединения формулы 12(viii) восстановление соединения формулы 12 с образованием соединения 13(ix) обработка соединения 13 серной кислотой и ацилирующим агентом с образованием соединения 2. В предпочтительном варианте осуществления указанного способа соединение 4 подвергают взаимодействию с 1,1-диалкоксиэтеном в присутствии метансульфоновой кислоты в качестве катализатора с образованием трициклического соединения 5 А В другом предпочтительном варианте осуществления указанного способа соединение 10 получают как единственный изомер с помощью триацетоксиборгидрида натрия. В другом предпочтительном варианте осуществления указанного способа соединение 8 выделяют из смеси соединений 8 и 9, и способ дополнительно включает восстановление соединения 8 с образованием соединения 10 как единственного изомера, причем восстановление проводят с помощью платинового катализатора на угле в присутствии водорода. В другом предпочтительном варианте осуществления указанного способа основание представляет собой DMAP (4-(N,N-диметиламино)пиридин). В другом предпочтительном варианте осуществления указанного способа при восстановлении соединения формулы 12 используют гидроксид палладия на угле в присутствии водорода. Ещ одним объектом настоящего изобретения является соединение, выбранное из: Способы по настоящему изобретению особенно пригодны для крупномасштабного промышленного производства описанных в нем соединений. Данные способы просты в исполнении, надежны и эффективны. В частности, данные способы особенно полезны для крупномасштабного производства дезоксисахаров. Кроме того, данные способы рентабельны и показывают высокую производительность и значительно более высокий общий выход по сравнению со способами синтеза, используемыми в прототипах. Подробное описание изобретения Термин "содержащий" (и его грамматические формы) при использовании в настоящем документе применяется в смысле "имеющий" или "включающий", а не в исключительном смысле "состоящий только из". При использовании в настоящем документе термин "галогенид" или "галогенированный" означает фторид, хлорид, бромид и йодид. Термин "галоген" означает фтор, хлор, бром и йод. Термин "Ас" означает ацетил. Термин "алкилкетенацеталь" означает 1,1-диалкоксиэтен. Термин "каталитический" означает содержащий катализатор или действующий как катализатор. Термин "стехиометрический" означает эквивалентное количество. Соединения в настоящем описании могут существовать как индивидуальные стереоизомеры, рацематы и/или различные смеси энантиомеров и/или диастереомеров. Все указанные индивидуальные стереоизомеры, рацематы и/или различные смеси энантиомеров и/или диастереомеров считаются включенными в предмет настоящего описания. При использовании в настоящем документе термин "окислитель" означает вещество или частицы,которые принимают электроны в химической реакции и термин "восстановитель" означает вещество,которое теряет электроны в химической реакции. Термин "иммуномодулятор" означает натуральные или синтетические продукты, способные модифицировать нормальную или отклоняющуюся от нормы иммунную систему посредством стимуляции или подавления.R и S показывают определенную стереохимическую конфигурацию заместителя при асимметричном атоме углерода в рассматриваемой химической структуре. Соединения по настоящему изобретению могут демонстрировать явление таутомерии. Если формулы, приведенные в настоящем документе, не могут определенно показать все возможные таутомерические формы, следует понимать, что формулы, приведенные в настоящем документе,предназначены для представления любой таутомерической формы рассматриваемого соединения, а не просто ограничены определенной формой соединения, изображенной на структурных формулах. Как понятно всем специалистам в данной области, оптически чистое соединение, имеющее один хиральный центр (т.е. один асимметричный атом углерода), представляет собой соединение, которое содержит, главным образом, один из двух возможных энантиомеров (т.е. является энантиомерно чистым), и оптически чистое соединение, имеющее более одного хирального центра, представляет собой соединение, которое является чистым как диастереомерно, так и энантиомерно. Предпочтительно использовать соединения по настоящему изобретению в такой форме, которая по меньшей мере на 90% свободна от других энантиомеров или диастереомеров данного соединения, т.е. чтобы они содержали индивидуальный изомер в количестве не менее 90% (80%-ный энантиомерный избыток EE или диастереомерный избыток DE), предпочтительнее не менее 95% (90%-ный EE или DE), еще предпочтительнее не менее 97,5% (95%-ный EE или DE) и наиболее предпочтительно не менее 99% (98%-ный EE или DE). Соединение 2 полезно как промежуточное соединение для синтеза используемых в фармацевтике соединений,включая 5-амино-3-(2'-О-ацетил-3'-дезоксиD-рибофуранозил)-3H-тиазоло[4,5d]пиримидин-2-он 3 и его фармацевтически приемлемые соли. Как описано в патентной заявке США 11/873202, соединение дезоксирибофуранозы 2 сочетается с 5-амино-3H-тиазоло[4,5-d]пиримидин-2 оном 1 с образованием соединения 3: Соединение 3 используют в способах лечения или предотвращения заболеваний. Например, соединение 3 используют в способах лечения или предотвращения проявления и/или прогрессирования опухолей или раковых заболеваний. Также описаны способы лечения или предотвращения инфекции, вызванной патогенами, например вирусами, включая вирус гепатита B или вирус гепатита C. Соединение 3 также используют в способах модуляции иммунной цитокинной активности. Примеры Следующие примеры приведены только в иллюстративных целях и не предназначены для ограничения объема формулы изобретения. В описанных ниже схемах синтеза, если не определено иное, все температуры приведены в градусах Цельсия и все доли и процентные соотношения приведены по массе. Реагенты приобретали у промышленных поставщиков, включая Aldrich Chemical Company илиLancaster Synthesis Ltd., и использовали без дополнительной очистки, если не определено иное. Все растворители приобретали у промышленных поставщиков, включая Aldrich, EMD Chemicals или Fisher, и использовали в полученном виде. Приведенные ниже реакции проводили, как правило, при положительном давлении азота или аргона и при температуре окружающей среды (если не определено иное) в безводных растворителях, и реакционные колбы снабжали резиновыми мембранами для введения веществ и реагентов с помощью шприца или воронкой для добавления жидкостей или порошковой воронкой для твердых веществ. Реакции исследовали с помощью тонкослойной хроматографии (ТСХ) и/или анализировали с помощью жидкостной хроматомасс-спектрометрии (ЖХМС) и прекращали на основании расходования исходного материала. Аналитическую ТСХ проводили на стеклянных пластинках, предварительно покрытых пластинками силикагеля 60 F254 толщиной 0,25 мм (EMD Chemicals), с визуализацией в УФ-свете(254 нм) и/или с помощью йода на силикагеле и/или нагреванием пятен ТСХ при проявлении в этанольном растворе фосфомолибденовой кислоты, содержащем кислоту растворе параанизальдегида, растворе нингидрина, растворе перманганата калия или растворе сульфата церия(IV). Препаративную ТСХ проводили на стеклянных пластинках, предварительно покрытых пластинками силикагеля 60 F254 толщиной 0,5 мм (2020 см, от Thomson Instrument Company), с визуализацией в УФ-свете (254 нм). Спектры ЯМР 1 Н и спектры 13 С записывали на спектрометре Varian Mercury-VX400 с рабочей частотой 400 МГц. Спектры ЯМР снимали с образцов, растворенных в CDCl3 (шкалам.д.), используя в качестве стандарта хлороформ (7,27 м.д. для 1 Н и 77,00 м.д. для 13 С), CD3OD (3,4 и 4,8 м.д. для 1 Н и 49,3 м.д. для 13 С), DMSO-d6 (2,49 м.д. для 1 Н) или в качестве внутреннего стандарта тетраметилсилан(0,00 м.д.), насколько это было удобно. Другие растворители для ЯМР использовали по мере необходимости. При описании мультиплетности сигналов использовали следующие сокращения: s (синглет),d (дублет), t (триплет), q (квадруплет), m (мультиплет), br (уширенный), bs (широкий синглет), dd (дублет дублетов), dt (дублет триплетов). Константы спин-спинового взаимодействия, если они приведены, выражены в герцах (Гц). Инфракрасные (ИК) спектры записывали на Фурье-спектрометре ATR FT-IR, используя образцы в чистом масле или твердом виде, и выражали приведенные данные в волновых числах (см-1). Приведенные масс-спектры представляют собой эмиссионные масс-спектры (+)-ES или спектры химической ионизации при атмосферном давлении в режиме жидкостной хроматомасс-спектрометрии APCI(+) LC/MS,которые сняты в отделении аналитической химии Anadys Pharmaceuticals, Inc. Элементные анализы проведены в Atlantic Microlab, Inc. (Норкросс, штат Джорджия, США). Температуры плавления (т.пл.) определяли в устройстве с открытым капилляром и приводили без поправки. В описанных способах синтеза и экспериментальных методиках могут использоваться многочисленные общепринятые химические сокращения, в том числеAcN - ацетонитрил,Bn - бензил,Boc - трет-бутоксикарбонил,Bz - бензоил,DBU - 1,8-диазабицикло[5,4,0]ундец-7-ен,DCC - N,N'-дициклогексилкарбодиимид,DCE - 1,2-дихлорэтан,DCM - дихлорметан,DEAD - диэтилазодикарбоксилат,DIEA - диизопропилэтиламин,DMA - N,N-диметилацетамид,DMAP - 4-(N,N-диметиламино)пиридин,DMF - N,N-диметилформамид,DMSO - диметилсульфоксид,EDC - 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид,Et - этил,EtOAc - этилацетат,EtOH - этанол,HATU - О-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат,HBTU - О-бензотриазол-1-ил-N,N,N',N'-тетраметилуронийгексафторфосфат,HF - фтороводород,НОВТ - 1-гидроксибензотриазолгидрат,ЖХВД - жидкостная хроматография высокого давления,IPA - изопропиловый спирт,KOtBu - трет-бутоксид калия,LDA - диизопропиламид лития,МСРВА - 3-хлорпероксибензойная кислота,Me - метил,MeCN - ацетонитрил,МеОН - метанол,NaH - гидрид натрия,NaOAc - ацетат натрия,NaOEt - этоксид натрия,Phe - фенилаланин,PPTS - п-толуолсульфонат пиридиния,PS - полимер на подложке,Py - пиридин,руВОР - бензотриазол-1-илокси)трипирролидинфосфонийгексафторфосфат,TEA - триэтиламин,TFA - трифторуксусная кислота,TFAA - трифторуксусный ангидрид,THF - тетрагидрофуран,ТСХ - тонкослойная хроматография,Tol - толуоил,Val - валин и аналогичные. Пример 1. Получение соединения 6 (основное) и соединения 7 (второстепенное).(а) Стадия 1. Получение трициклического соединения 5 А В 4-литровую четырехгорлую колбу, снабженную патрубком для впуска азота, воронкой для добавления жидкостей, термометром и механической мешалкой, помещали моноацетонксилозу (152,16 г,800 ммоль) и изопропилацетат (1200 мл) и перемешивали до полного растворения твердой фазы с получением слегка мутного раствора. Добавляли кетендиметилацеталь (3,36 мл, 35,5 ммоль) и охлаждали реакционную смесь до 3C с помощью ледяной бани. После добавления метансульфокислоты (0,52 мл,8 ммоль) добавляли по каплям кетендиметилацеталь (80 мл, 844,5 ммоль) в течение 45 мин. В процессе добавления температура реакционной смеси достигала 10C. После окончания добавления анализ мето-6 020552 дом ТСХ с использованием 80% МТВЕ в гексане показал полную чистую конверсию в трициклическое соединение 5 А, которое имеет значительно большую хроматографическую подвижность. Ледяную баню убрали. К вышеуказанной реакционной смеси добавили воду (72 мл, 4000 ммоль) одной порцией и смесь перемешивали при температуре окружающей среды в течение 90 мин. Анализ методом ТСХ реакционной смеси с использованием 80% МТВЕ в гексане показал образование двух новых среднеполярных продуктов, из которых основной продукт имел меньшую хроматографическую подвижность. Реакционную смесь переносили в 2-литровую делительную воронку и встряхивали с 120 мл водного раствора (60 мл 1,0 М NaHCO3, 60 мл 30% NaCl), фазы разделяли, органическую фазу переносили в круглодонную колбу и летучие вещества отгоняли в вакууме.(с) Стадия 3. Выдерживание с образованием соединения 6. Вещество, выделенное при испарении, разбавляли свежим изопропилацетатом (1200 мл) и водой(72 мл) и нагревали до 77C в течение 12 ч, затем охлаждали до температуры окружающей среды. Анализ методом ТСХ с использованием 80% МТВЕ в гексане показал, что из двух продуктов основной продукт имел большую хроматографическую подвижность, а менее подвижный изомер присутствовал лишь в следовых количествах. Досуха выпаривали 0,2 мл образца реакционной смеси и получали 37 мг твердого вещества. 1 Н ЯМР подтверждает, что целевое ацетатное соединение 6 представляет собой основное соединение. 1 Н ЯМР (400 МГц, CDCl3) : 5,92 (1 Н, д, J=3,3 Гц), 4,51-4,56 (2 Н, м), 4,24-4,28 (1 Н, м), 4,13-4,19 (2 Н,м), 2,98 (1 Н, д, J=4,0 Гц), 2,11 (3H, с), 1,51 (3H, с), 1,33 (3H,с). Пример 2. Получение соединений 8 и 9. 4-литровую колбу, которая уже содержала приблизительно 0,8 моль соединения 6 во влажном изопропилацетате с предыдущей стадии, оборудовали патрубком для впуска азота, термометром, воронкой для добавления жидкостей и механической мешалкой. Добавляли TEMPO (800 мг) и смесь перемешивали и охлаждали на ледяной бане. В отдельной колбе водный раствор, содержащий 64,3 г бромида натрия и 98,4 г ацетата натрия в 320 мл деионизированной воды, охлаждали до 5C. Когда температура реакционной смеси достигала 5C, к ней добавляли предварительно охлажденный водный раствор и получали двухфазную реакционную смесь. К данному холодному раствору добавляли по каплям 735 мл водного раствора гипохлорита натрия (титрование непосредственно перед применением показало содержание 10,15% или 1,36 М, 1,002 моль, 1,25 экв.) в течение 2 ч, поддерживая экзотермическую реакцию при температуре не выше 7C. Когда добавление было завершено, перемешивание продолжали еще 30 мин, и анализ методом ТСХ (80% МТВЕ в гексане) показал полную конверсию в кетон, который имел меньшую хроматографическую подвижность. Реакционную смесь переносили в 4-литровую делительную воронку и разделяли фазы. Темную органическую порцию промывали однократно 160 мл водного раствора, содержащего 2,5% тиосульфата натрия. Полученную бледно-желтую органическую порцию промывали 160 мл раствора, содержащего 30% хлорида натрия. Водные фазы объединяли, добавляли 44,1 г твердого хлорида натрия и перемешивали до полного растворения соли. Полученный водный раствор экстрагировали двумя порциями по 400 мл изопропилацетата, органические экстракты объединяли и промывали однократно 50 мл раствора,содержащего 30% хлорида натрия. Все органические порции объединяли и получали слегка мутный раствор. Выпаривали порцию, содержащую 0,25 мл данного раствора и получали 14 мг твердого вещества. 1 Н ЯМР подтверждает присутствие смеси кетона и его гидрата в приблизительном соотношении 1:1. 1 Н ЯМР (400 МГц, CDCl3) : 6,09 (1 Н, д, J=4,4 Гц, соединение 8), 5,84 (1 Н, д, J=3,9 Гц, соединение 9), 4,61 (1 Н, дд, J=11,7 Гц, J2=6,3 Гц, соединение 9), 4,56 (1 Н, т, J=3,3 Гц, соединение 8), 4,36-4,42 (2 Н, м,соединения 8 и 9), 4,20-4,24 (2 Н, м, соединения 8 и 9), 4,06-4,15 (2 Н, м, соединения 8 и 9), 2,11 (3H, с,соединение 9), 2,05 (3H, с, соединение 8), 1,58 (3H, с, соединение 9), 1,50 (3H, с, соединение 8), 1,43 (3H,с, соединение 8), 1,36 (3H, с, соединение 9). В 4-литровую четырехгорлую колбу, снабженную патрубком для впуска азота, порошковой воронкой, термометром и механической мешалкой, помещали мутный органический раствор кетона 8 и его гидрата 9. Его охлаждали при перемешивании до 4C с помощью ледяной бани. К данному холодному раствору добавляли четыре порции по 42,4 г твердого триацетоксиборгидрида натрия с интервалами по 15 мин. После добавления последней порции реакционную смесь перемешивали при 5C в течение 60 мин. При перемешивании при 5C быстро добавляли 1,0 М водного раствора карбоната натрия (800 мл). Температура реакционной смеси увеличивалась до 12C, и выделялось небольшое количество газа. Смесь становилась существенно более густой. После перемешивания в течение 15 мин реакционную смесь переносили в 4-литровую делительную воронку и разделяли фазы, причем водная порция содержала немного твердого вещества. Органическую порцию перемешивали с 2,0 М водным раствором карбоната натрия (400 мл) в течение 10 мин, разделяли фазы и объединяли обе водные фазы. Твердый осадок в водной фазе отфильтровывали и затем растворяли в воде (600 мл) и снова добавляли к полученной гомогенной водной фазе. Водную фазу экстрагировали двумя порциями по 200 мл изопропилацетата и объединяли органические порции. Полная масса органической фазы составила 2370,5 г. Пятиграммовую порцию органической фазы выпаривали и получали 243 мг масла, которое кристаллизовалось в вакууме. Вычисленный выход соединения 10: 2370,5 г раствора 0,243 г продукта/5 г раствора=115,2 г (496,15 ммоль, 62%). 1 Н ЯМР показывает очень высокую чистоту образца. 1(3H, с), 1,38 (3H, с). В 4-литровую четырехгорлую колбу, снабженную коротким дефлегматором, датчиком температуры и механической мешалкой, помещали 2370,5 г органической фазы. Ее нагревали для отгонки 2400 мл дистиллята при атмосферном давлении. В колбу добавляли свежий изопропилацетат (1500 мл) и отгоняли 1500 мл жидкости. Затем реакционную смесь разбавляли 920 мл изопропилацетата и получали слегка мутный раствор. Теперь данный раствор был готов для переноса на следующую стадию. Пример 4. Альтернативное получение соединений 8 и 10.(а) Стадия 1. Получение соединения 8. Колбу, которая содержала соединение 6 (приблизительно 0,2 моль) во влажном изопропилацетате из примера 1, снабжали патрубком для впуска азота, термометром, воронкой для добавления жидкостей и магнитной мешалкой. Добавляли TEMPO (200 мг) и смесь перемешивали и охлаждали при 0C на ледяной бане. В отдельной колбе водный раствор, содержащий бромид натрия (16,08 г) и ацетат натрия(24,6 г) в деионизированной воде (80 мл), охлаждали до 5C. Когда температура реакционной смеси достигала 5C, к ней добавляли предварительно охлажденный водный раствор и получали двухфазную реакционную смесь. К данной холодной смеси добавляли по каплям водный раствор гипохлорита натрия (концентрация 10-15%; 180 мл) в течение 1 ч, поддерживая экзотермическую реакцию при температуре не выше 7C. Когда добавление было завершено, анализ методом ТСХ (80% МТВЕ в гексане) показал полную конверсию в кетон, имеющий меньшую хроматографическую подвижность Rf. Охлаждающую баню убирали и добавляли твердый NaCl (25 г). После перемешивания в течение 30 мин смесь переносили в 1-литровую делительную воронку и затем разделяли фазы. Темную органическую порцию встряхивали с 1,0 МNaHCO3 (25 мл) и затем добавляли 2,0 М Na2SO3 (30 мл) и продолжали встряхивание до полного обесцвечивания (происходило некоторое газовыделение). Полученную прозрачную органическую порцию однократно промывали 15% водным растворомNaCl (20 мл). Прозрачную органическую фазу переносили в 1-литровую колбу, снабженную датчиком температуры, дефлегматором и магнитной мешалкой. Температуру устанавливали на 85C для отгонки растворителя. После остановки отгонки температуру повышали до 105C для завершения дистилляции. Перегонную колбу охлаждали до температуры окружающей среды и разбавляли смесь изопропилацетатом (100 мл). Добавляли активированный уголь (Darco G60; 5 г) и перемешивали смесь при температуре окружающей среды в течение 90 мин. Данную смесь фильтровали через целит и промывали твердую фазу изопропилацетатом (230 мл). Масса бледно-желтого фильтрата составляла 220,5 г. Из этого раствора выпаривали 2,0 мл (масса 1,826 г) и получали 0,189 г бледно-желтого масла. Вычисление показало, что концентрация раствора соединения 8 составляла 0,41 М, полный выход 22,86 г (49,6% в расчете на моноацетонксилозу). 1H ЯМР (400 МГц, CDCl3) : 1,43 (3H, с), 1,50 (3H, с), 2,05 (3H, с), 4,21 (1 Н, дд, J1=11,9 Гц,J2=3,9 Гц), 4,37 (1 Н, д, J=4,7 Гц), 4,40 (1 Н, дд, J1=12,5 Гц, J2=3,2 Гц), 4,56 (1 Н, т, J=3,1 Гц), 6,09 (1 Н, д,J=3,8 Гц). 1 Н ЯМР показал, что присутствовало только соединение 8 (соединение 9 отсутствовало).Matthey, тип В 101018-3, партия С-9264, 58,25% воды). Температуру приводили в равновесие на уровне 26C, смесь дегазировали вакуумной установкой для собственных нужд и три раза продували газообразным водородом, затем смесь интенсивно перемешивали в атмосфере водорода в течение 16 ч. Анализ методом газовой хроматографии (ГХ) показал полную конверсию в соединение 10. Раствор фильтровали через целитовый фильтр, твердую фазу промывали изопропилацетатом (230 мл) и затем прозрачный бесцветный фильтрат выпаривали, получая 5,74 г масла, которое закристаллизовалось. 1H ЯМР подтвердил наличие соединения 10 как единственного продукта. Пример 5. Получение соединения 11 А Четырехлитровую колбу, которая уже содержала приблизительно 496,15 ммоль соединения 10 в сухом изопропилацетате из примера 3, снабжали патрубком для впуска азота, термометром, резиновой мембраной и механической мешалкой. В отдельной колбе DMAP (90,92 г, 744,23 ммоль, 1,5 экв.) растворяли в 255 мл горячего DME. Горячий раствор добавляли в реакционную колбу и реакционную смесь охлаждали на ледяной бане до 5C. Ангидрид трифторметансульфокислоты (104,34 мл, 620,19 ммоль,1,25 экв.) добавляли со скоростью 1,17 мл/мин с помощью шприц-насоса. Максимальная температура,достигнутая в процессе добавления, составляла 7C. Когда добавление заканчивали и температура реакционной смеси возвращалась на уровень 5C, анализ методом ТСХ (20% EtOAc в толуоле) показывал полную чистую конверсию в трифлат, имеющий большую хроматографическую подвижность. К реакционной смеси при 5C добавляли 1,0 М HCl (745 мл), экзотермическая реакция вызывала увеличение температуры до 9C. После перемешивания в течение 5 мин реакционную смесь переносили в делительную воронку и фазы разделяли. Органическую фазу промывали двумя порциями 1,0 М HCl(300 мл) и однократно 240 мл водного раствора (120 мл 1,0 М NaHCO3, 120 мл 30% хлорида натрия). Все водные фазы объединяли и однократно экстрагировали 500 мл изопропилацетата. Экстракт промывали двумя порциями по 100 мл 1,0 М HCl и однократно 80 мл водного раствора (40 мл 1,0 М NaHCO3, 40 мл 30% хлорида натрия). Все органические фазы объединяли и получали слегка мутный раствор трифлата 11 А. Порцию, содержащую 0,25 мл данного раствора, выпаривали и получали 22 мг масла. 1 Н ЯМР показывает очень высокую чистоту образца трифлата, содержащего небольшое количество остаточного изопропилацетата. 1 В 4-литровую четырехгорлую колбу, снабженную патрубком для впуска азота, температурным датчиком, холодильником и механической мешалкой, помещали раствор трифлата в изопропилацетате(предположительно 496,15 ммоль) и 255 мл DME. Добавляли твердый йодид натрия (111,55 г,744,23 ммоль, 1,5 экв.) и смесь перемешивали при 55C в течение 17 ч. Анализ методом ТСХ (10% EtOAc в толуоле) показал полную конверсию в йодид. Добавляли воду (400 мл) и интенсивно перемешивали смесь в течение 5 мин. Смесь переносили в делительную воронку и разделяли фазы. Органическую фазу промывали однократно 400 мл водного раствора (200 мл 1,0 М NaHCO3 и 200 мл 30% NaCl). Водные фазы объединяли и однократно экстрагировали изопропилацетатом (400 мл). Экстракт промывали однократно водой (100 мл) и однократно 100 мл водного раствора (50 мл 1,0 М NaHCO3 и 50 мл 30% NaCl). Все органические фазы объединяли. Раствор соединения 12 А переносили в 3-литровую круглодонную колбу, снабженную коротким дефлегматором. 2 л растворителя отгоняли простой дистилляцией. Смесь охлаждали до температуры окружающей среды и определяли остаточный объем как 500 мл. К нему добавляли 183 мл изопропилацетата и 208 мл абсолютного этанола и получали 0,5 М раствор соединения 12 А в смеси этанола (20%) и изопропилацетата. Аликвоту 0,2 мл отбирали и выпаривали с получением 42 мг масла. 1 Н ЯМР показывает очень высокую чистоту образца соединения 12 А. 1 В 3-литровую колбу, снабженную большой магнитной мешалкой, помещали раствор соединения 12 А (предположительно 496,15 ммоль в виде 0,5 М раствора в смеси этанола (20%) и изопропилацетата),диизопропилэтиламин (112,34 мл, 644,8 ммоль, 1,3 экв.) и 20,37 г 20% Pd(OH)2 на угле (катализатор Перлмана). При интенсивном перемешивании реакционную смесь дегазировали в легком вакууме и затем трехкратно заполняли газообразным водородом. После этого реакционную смесь перемешивали в атмосфере водорода в течение 18 ч. Анализ методом ТСХ (10% EtOAc в толуоле) показал чистую полную конверсию в продукт гидрирования с меньшей хроматографической подвижностью. Реакционную смесь фильтровали через целит и промывали темную твердую фазу двумя порциями по 200 мл изопропилацетата. Фильтрат переносили в 4-литровую делительную воронку и промывали однократно 1,0 М HCl (645 мл), однократно 200 мл водного раствора (100 мл 2,5% тиосульфата натрия,100 мл 1,0 М NaHCO3) и однократно 200 мл 30% NaCl. Все водные фазы объединяли и экстрагировали двумя порциями по 200 мл изопропилацетата. Экстракты объединяли и промывали однократно 80 мл водного раствора (40 мл 2,5% тиосульфата натрия, 40 мл 1,0 М NaHCO3) и однократно 80 мл 30% NaCl. Органические порции объединяли, переносили в 3-литровую круглодонную колбу и отгоняли 1,5 л растворителя дистилляцией при атмосферном давлении. Охлажденный остаток имел объем 450 мл. Добавляли 50 мл изопропилацетата, получали раствор, имеющий концентрацию около 1,0 М, добавляли 10 г активированного угля Norit и перемешивали смесь в течение 2 ч при температуре окружающей среды. Затем ее фильтровали через целит и получали прозрачный фильтрат золотистого цвета. Фильтрат концентрировали в вакууме и получали 103,47 г (478,52 ммоль) прозрачного масла золотистого цвета. 1 Н ЯМР показывает очень высокую чистоту соединения 13. 1 Н ЯМР (400 МГц, CDCl3) : 5,83 (1 Н, д, J=3,7 Гц), 4,74 (1 Н, т, J=4,2 Гц), 4,39-4,45 (1 Н, м), 4,28 (1 Н,дд, J1=11,8 Гц, J2=3,1 Гц), 4,08 (1 Н, дд, J1=12,5 Гц, J2=6,2 Гц), 2,07-2,12 (4 Н, м), 1,62-1,69 (1 Н, м), 1,52 (3H,с), 1,33 (3H, с). Соединение 13 можно дополнительно очищать перегонкой в вакууме, если это потребуется. Температура кипения составляет 70C при давлении 0,025 мм рт. ст. Пример 8. Получение соединения 2 В 25-мл круглодонную колбу, оборудованную магнитной мешалкой и резиновой мембраной, помещали соединение 13 (640 мг, 2,96 ммоль) и 5 мл уксусной кислоты. В отдельной колбе уксусный ангидрид (0,562 мл, 6 ммоль, 2 экв.) разбавляли уксусной кислотой до общего объема 2,0 мл и добавляли 0,1 мл данного раствора уксусного ангидрида к реакционной смеси. Раствор серной кислоты (0,15 мл 1,0 М раствора в уксусной кислоте, 0,15 ммоль, 0,05 экв.) добавляли к реакционной смеси и затем остаток раствора уксусного ангидрида (1,9 мл) добавляли в течение 12 ч с помощью шприц-насоса. Анализ методом ТСХ (30% EtOAc в гексане) показал очень чистую конверсию в целевое соединение 2. Реакционную смесь разбавляли толуолом и выпаривали в вакууме. Остаток растворяли в МТВЕ,перемешивали с 10% раствором карбоната натрия в течение 15 мин и разделяли фазы. Органическую порцию сушили (MgSO4), фильтровали через тонкую подложку силикагеля, выпаривали и получали 680 мг (2,61 ммоль) прозрачного масла. 1 Н ЯМР показывает, что это чистая смесь обоих аномеров. Важно отметить, что толкование и расположение способов и стадий, указанных в примерных вариантах осуществления, является лишь иллюстративным. Хотя подробно описаны только несколько вариантов осуществления настоящего описания, специалисты в данной области легко оценят, что возможны многочисленные модификации без существенного отклонения от новизны и преимуществ предмета, указанного в формуле изобретения. Соответственно, все указанные модификации предназначены для включения в объем настоящего описания, как определено в прилагаемой формуле изобретения. Порядок или последовательность любых способов или стадий способа можно изменить или пересмотреть согласно альтернативным вариантам осуществления. Можно производить другие замещения, модификации, изменения и исключения в плане, условиях работы и расположении вариантов осуществления без отклонения от духа настоящего описания, как описано в прилагаемой формуле изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы 11(iii) сульфонирование соединения 10 сульфонирующим агентом в присутствии основания с образованием соединения формулы 11. 2. Способ по п.1, в котором при восстановлении соединения 8 получают единственный изомер соединения 10 и в котором восстановление проводят с помощью платинового катализатора на угле в присутствии водорода. 3. Способ по п.1, в котором сульфонирующий агент представляет собой ангидрид трифторметансульфокислоты и основание представляет собой DMAP (4-(N,N-диметиламино)пиридин). 4. Способ получения соединения 2 с образованием циклического соединения формулы 5(ii) гидролиз соединения формулы 5 водой с каталитическим или стехиометрическим количеством метансульфоновой кислоты с образованием смеси моноацилзамещенных соединений 6 и 7:(iii) выдерживание при температуре выше 70C смеси моноацилзамещенных соединений 6 и 7 с образованием избытка соединения 6;(iv) окисление смеси соединения 6 и соединения 7 гипохлоритом натрия в присутствии TEMPO(2,2,6,6-тетраметилпиперидина-1-оксил) и ацетата натрия в двухфазной системе с изопропилацетатом с образованием смеси кетонного соединения 8 и гидратированного кетонного соединения 9:(v) восстановление смеси кетонного соединения 8 и гидратированного кетонного соединения 9 с образованием соединения 10 или раздельное восстановление соединения 8 и соединения 9 с образованием соединения 10 используя платиновый катализатор на угле в присутствии водорода;(vi) сульфонирование соединения 10 сульфонирующим агентом в присутствии основания с образованием соединения формулы 11(vii) замещение сульфоэфирной группы в соединении формулы 11 атомом галогена с образованием галогенпроизводного соединения формулы 12(viii) восстановление соединения формулы 12 с образованием соединения 13(ix) обработка соединения 13 серной кислотой и ацилирующим агентом с образованием соединения 2. 5. Способ по п.4, где соединение 4 подвергают взаимодействию с 1,1-диалкоксиэтеном в присутствии метансульфоновой кислоты в качестве катализатора с образованием трициклического соединения 5 А в изопропилацетате. 6. Способ по п.4, где соединение 10 получают как единственный изомер с помощью триацетоксиборгидрида натрия. 7. Способ по п.4, в котором соединение 8 выделяют из смеси соединений 8 и 9, и способ дополнительно включает восстановление соединения 8 с образованием соединения 10 как единственного изомера, где восстановление проводят с помощью платинового катализатора на угле в присутствии водорода. 8. Способ по п.4, где основание представляет собой DMAP (4-(N,N-диметиламино)пиридин). 9. Способ по п.4, где при восстановлении соединения формулы 12 используют гидроксид палладия на угле в присутствии водорода. 10. Соединение, выбранное из:
МПК / Метки
МПК: A01N 43/90
Метки: дезоксирибофуранозы, получения, способ, производных
Код ссылки
<a href="https://eas.patents.su/14-20552-sposob-polucheniya-proizvodnyh-dezoksiribofuranozy.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения производных дезоксирибофуранозы</a>
Способ получения производных 1,3-дигидро-2н-3-бензазепин-2-она

Номер патента: 7744
Опубликовано: 29.12.2006
Авторы: Бриго Даньель, Лерести Жан-Мишель, Лекув Жан-Пьер
МПК: A61P 9/00, C07D 223/16, A61K 31/55...
Метки: способ, 1,3-дигидро-2н-3-бензазепин-2-она, производных, получения
Формула / Реферат:
1. Способ синтеза соединений формулы (I) в которой R1 и R2, которые могут быть одинаковыми или разными, каждый представляет собой линейную или разветвленную (С1-С8)алкоксигруппу или образуют вместе с атомом углерода, к которому они присоединены, 1,3-диоксановое, 1,3-диоксолановое или 1,3-диоксепановое кольцо, в котором соединение формулы (IV) подвергают реакции алкилирования, используя соединение формулы (V) в которой R1 и R2 имеют значения,...
Способ получения оптически чистых производных 2-(4-гидроксифенокси)пропионовой кислоты

Номер патента: 9241
Опубликовано: 28.12.2007
Автор: Глю Эрнст Стивен
МПК: C07D 213/64, C07C 51/367, C07C 59/68...
Метки: способ, кислоты, производных, 2-(4-гидроксифенокси)пропионовой, оптически, чистых, получения
Формула / Реферат:
1. Способ получения R-2-(4-гидроксифенокси)пропановой кислоты или ее соли, включающий взаимодействие гидрохинона или его соли с S-2-галопропановой кислотой или ее солью в присутствии мягкого восстановителя, где мягкий восстановитель представляет собой нейтральный или имеющий низкую степень окисления серосодержащий материал, такой как диоксид серы, сульфит, бисульфит, гидросульфит, метабисульфит, сульфеновая кислота, сульфиновая кислота, например...
Способ получения производных циклогексанола

Номер патента: 7486
Опубликовано: 27.10.2006
Авторы: Парк Дзин-Соо, Ким Кванг-Ил, Ким Кеун-Сик, Ли Сунг-Воо, Чай Ки-Байунг
МПК: C07C 253/30
Метки: способ, производных, циклогексанола, получения
Формула / Реферат:
1. Способ получения производных циклогексанола формулы I в которой R6 и R7 представляют собой орто- или паразаместители, независимо выбранные из группы, состоящей из водорода, гидроксила, C1-С6алкила, C1-С6алкокси, С7-С9аралкокси, С2-С7алканоилокси, C1-С6алкилмеркапто, галогена или трифторметила; R8 представляет водород или C1-С6алкил; р является одним из целых чисел 0, 1, 2, 3 или 4; a R9 представляет водород или C1-С6алкил; включающий...
Новый продукт, способ и промежуточные продукты получения производных азетидина

Номер патента: 11408
Опубликовано: 27.02.2009
Авторы: Лавинь Мишель, Мальпар Жоэль, Крок Вероник, Мютти Стефан, Рике-Цапп Йорг, Грондар Люк
МПК: C07D 401/12
Метки: промежуточные, новый, продукты, продукт, способ, производных, азетидина, получения
Формула / Реферат:
1. Способ синтеза N-{1-[бис-(4-хлорфенил)метил]азетидин-3-ил}-N-(арил или гетероарил)метилсульфонамида, отличающийся тем, что осуществляют реакцию 1-[бис-(4-хлорфенил)метил]азетидин-3-ола гидробромида с N-(арил или гетероарил)метансульфонамидом в присутствии DIAD, трифенилфосфина в толуоле с образованием N-{1-[бис-(4-хлорфенил)метил]азетидин-3-ил}-N-(арил или гетероарил)метилсульфонамида, который выделяют; причем арил представляет собой...
Способ получения эзетимиба и его производных

Номер патента: 17349
Опубликовано: 30.11.2012
Авторы: Гартнер Андрей, Мохар Барбара, Бевц Мойца, Зупет Рок, Стефан Мишель, Плевник Миха, Кросель Весна, Смрколь Матей, Седмак Грегор, Стимац Антон, Бенкиц Примоз, Кляйиц Ален, Кидемет Давор
МПК: A61K 31/397, A61P 3/00, C07D 205/08...
Метки: эзетимиба, получения, способ, производных
Формула / Реферат:
1. Способ получения соединения общей формулыгде R представляет собой атом водорода, защитную группу, выбранную из трехзамещенного силила, арилметила, тетрагидро-2Н-пиранила, моно- или двузамещенного арилметила с заместителями, которые выбраны из группы, состоящей из галогенов, метокси-, нитрогрупп, фенила, нафтила и их любых комбинаций,включающий следующие стадии:а) катализируемая металлами асимметрическая трансферная гидрогенизация...
Предыдущий патент: Оффшорная установка, фундамент оффшорной установки и способ возведения оффшорной установки
Случайный патент: Винт