Способ получения ситаглиптина и его фармацевтически приемлемых солей







Формула / Реферат
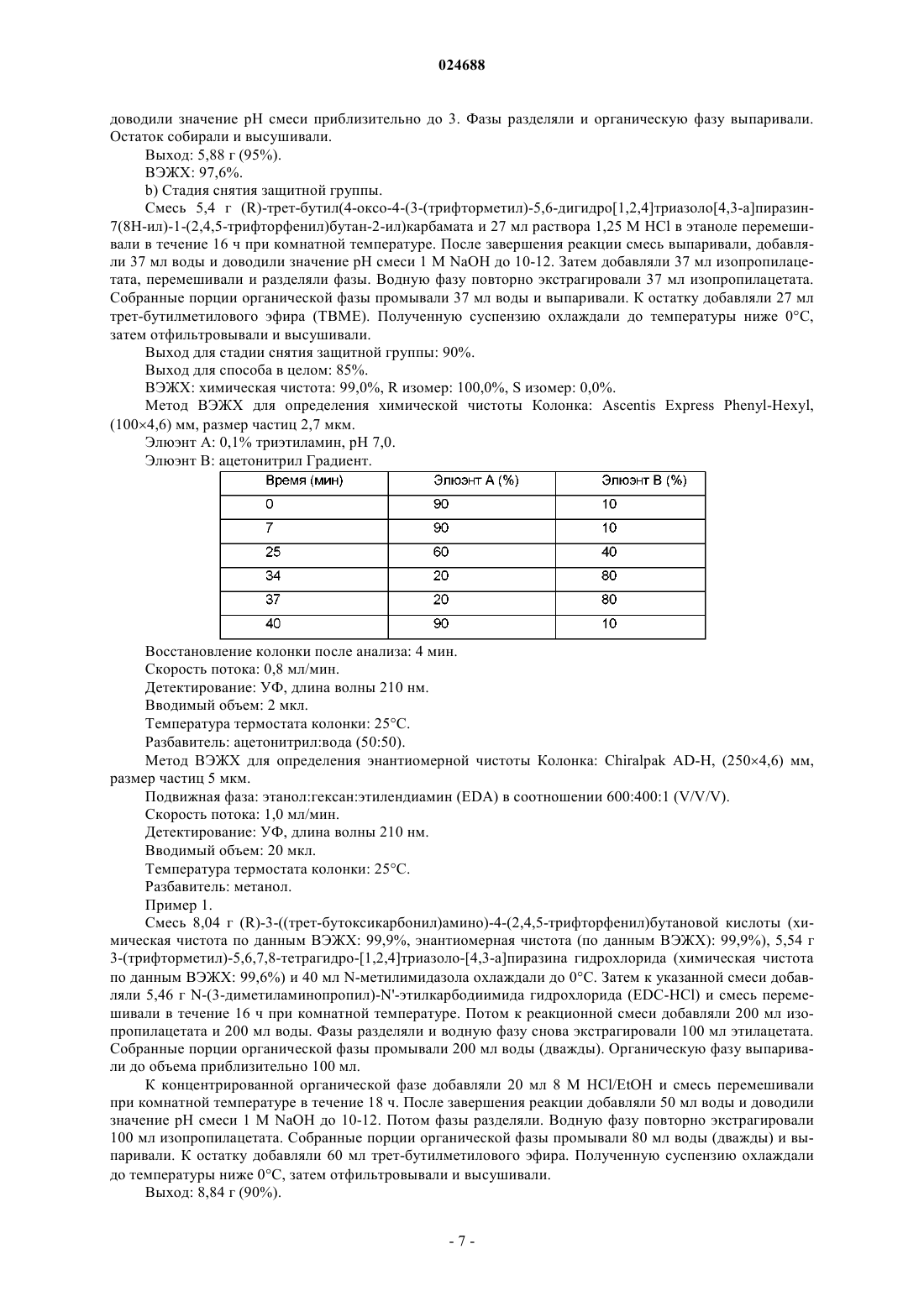
1. Способ получения ситаглиптина или его фармацевтически приемлемых солей формулы 1

включающий стадии:
а) конденсации соединения формулы 2

где R представляет собой аминозащитную группу, которая может быть удалена, с соединением формулы 3

которое может быть использовано в форме свободного основания или в форме соли, в присутствии связывающего агента и основного растворителя, который представляет собой N-метилимидазол, с получением соединения формулы 4

и
b) удаления защитной группы у соединения формулы 4 с получением основания ситаглиптина, которое дополнительно может быть переведено в его фармацевтически приемлемую соль.
2. Способ по п.1, характеризующийся тем, что аминозащитная группа, которая может быть удалена в кислой среде, выбрана из группы, состоящей из карбаматной группы, трет-бутилоксикарбонильной (ВОС) группы, карбобензилокси (Cbz) группы, параметоксибензилкарбонильной (Moz или MeOZ) группы, 9-флуоренилметилоксикарбонильной (FMOC) группы, ацетильной (Ас) группы, бензоильной (Bz) группы, бензильной (Bn) группы, параметоксибензильной (РМВ) группы, 3,4-диметоксибензильной (DMPM) группы, предпочтительно карбаматной группы или ВОС.
3. Способ по любому из пп.1, 2, характеризующийся тем, что связывающий агент выбран из группы, состоящей из фосгена, дифосгена, трифосгена, карбонилдиимидазола, хлортионилимидазола, тионилдиимидазола, дициклогексилкарбодиимида (DCC), N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорида (EDC-HCl), солей тетраметилурония, таких как О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (HBTU), О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (TBTU) или О-(7-аза-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (HATU), предпочтительно N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорида (EDC-HCl).
4. Способ по любому из пп.1-3, характеризующийся тем, что степень химической чистоты полученного ситаглиптина, по данным высокоэффективной жидкостной хроматографии (ВЭЖХ), составляет 99-100%, предпочтительно 99,5-100%, более предпочтительно 99,7-100%.
5. Способ по любому из пп.1-4, характеризующийся тем, что энантиомерная чистота ситаглиптина составляет более чем 99,5% (по данным ВЭЖХ).
6. Способ по любому из пп.1-5, характеризующийся тем, что ситаглиптин дополнительно переводят в одну из его фармацевтически приемлемых солей, полученных из любого неорганического или органического основания или кислоты, таких как гидрохлорид, фосфат, дигидрофосфат, тартрат, бензолсульфонат, сульфат, ацетат, предпочтительно гидрохлорид, фосфат и дигидрофосфат.
7. Применение N-метилимидазола в качестве органического растворителя в реакции конденсации соединения формулы 2

где R представляет собой аминозащитную группу, которая может быть удалена, с соединением формулы 3

которое может быть использовано в форме свободного основания или в форме соли с получением соединения формулы 4

8. Применение по п.7, где N-метилимидазол применяют также в качестве растворителя для удаления защитной группы у соединения формулы 4 с получением основания ситаглиптина.
Текст
СПОСОБ ПОЛУЧЕНИЯ СИТАГЛИПТИНА И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ В настоящем изобретении приведено описание способа получения ситаглиптина или его фармацевтически приемлемых солей формулы (1), включающего стадии: а) конденсации соединения формулы (2), в котором R представляет собой аминозащитную группу с соединением формулы (3), которое может быть использовано в форме свободного основания или в форме соли в присутствии связывающего вещества и основного растворителя, который представляет собой N-метилимидазол, с получением соединения формулы (4), и b) удаления защитной группы у соединения формулы (4) с получением основания ситаглиптина, которое может быть дополнительно переведено в его фармацевтически приемлемую соль. Указанный способ высокорентабелен, экологически безопасен, недорог и легко масштабируем до промышленного уровня. Область изобретения Настоящее изобретение относится к области органической химии. В частности, в настоящем изобретении приведено описание усовершенствованного способа получения ситаглиптина и его фармацевтически приемлемых солей. Указанный способ высокорентабелен, экологически безопасен, недорог и легко масштабируем до промышленного уровня. Предшествующий уровень техники Ситаглиптин,химически обозначаемый как(R)-4-оксо-4-[3-(трифторметил)-5,6 дигидро[1,2,4]триазоло[4,3-а]пиразин-7(8 Н)-ил]-1-(2,4,5-трифторметил)бутан-2-амин, имеет следующую химическую структуру: Ситаглиптин представляет собой эффективное при приеме внутрь антигипергликемическое (противодиабетическое) лекарственное средство из группы ингибиторов дипептидилпептидазы-4 (DPP-4). Он представлен на рынке компанией Merck Co. в виде ситаглиптина фосфата под торговым наименованиемJanuvia. Указанное лекарственное средство ингибитор ферментов применяют или в виде монотерапии,или в комбинации с другими антигипергликемическими средствами для приема внутрь (такими, например, как метформин или тиазолидинедион) для лечения сахарного диабета второго типа. Действие ситаглиптина направлено на конкурентное ингибирование фермента дипептидилпептидазы-4 (DPP-4). Указанный фермент расщепляет инкретины GLP-1 и GIP, гастроинтестинальные гормоны,которые выделяются в ответ на прием пищи. Предотвращая инактивацию GLP-1 и GIP, ситаглиптин способен усиливать секрецию инсулина и подавлять высвобождение глюкагона поджелудочной железой. Подобное действие приближает уровень глюкозы крови к нормальным значениям. Когда уровень глюкозы крови приближается к нормальному, количества высвобожденного инсулина и подавленного глюкагона возвращаются к исходным, способствуя предотвращению "перегрузки" и вызываемому ей низкого содержания сахара крови (гипогликемия), которую наблюдают при приеме некоторых других гипогликемических средств. Описание указанного вещества было впервые приведено в WO 2003/004498 А 1, где было приведено описание общего способа синтеза ситаглиптина путем соединения фрагмента бета-аминокислоты с гетероциклическим фрагментом, как показано на схеме Указанную реакцию проводят при стандартных условиях образования пептидной связи, используя 1-этил-3-3(диметиламинопропил)карбодиимид (EDC), 1-гидроксибензотриазол (НОВТ) и основание,обычно диизопропилэтиламин, в растворителе, таком как N,N-диметилформамид (DMF) или дихлорметан (DCM). Затем трифторуксусной кислотой или раствором хлороводорода в метаноле удаляют защитную группу, если защитной группой является трет-бутоксикарбонильная. Недостатком указанного способа является достаточно низкий выход продукта, полученного после хроматографической очистки, а также использование реагентов подобных НОВТ, который, как сообщалось, взрывоопасен, и поэтому его использования следует избегать. В WO 2005/003135 приведено описание кристаллической дигидрофосфатной соли фосфата ситаглиптина. Описание синтеза ситаглиптина с использованием стереоселективного восстановления описано, например, в WO 2004/085378, WO 2004/085661, WO 2004/085378. В Organic Process ResearchDevelopment 2005, 9, 634-639 раскрыт способ, в котором соединение формулы 2, где R представляет собой OBn (Bn обозначает бензильную группу), соединяют с соединением формулы 3 в присутствии N-метилморфолина (NMM) в качестве основания и EDC-HCl в качестве связывающего агента, позволяя добиться выхода целевого соединения более чем 99%. Недостаток указанного способа заключен в бензильной защитной группе, которая требует для своего удаления каталитического гидрогенирования 10% палладиевый катализатор на углероде. В IPCOM000196065D раскрыт способ получения ситаглиптина, в котором (R)-3-третбутоксикарбонил)амино)-4-(2,4,5-трифторфенил)бутановую кислоту или ее соль реагирует с 3-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло-[4,3-а]пиразином или его солью в присутствии триэтиламина в качестве органического основания и пропилфосфонового ангидрида в качестве связываю-1 024688 щего агента, или N-метилморфолина в качестве органического основания и 1-этил-3-(3 диметиламинопропил)карбодиимида (EDC) или N,N'-дициклогексилкарбодиимида (DCC) в качестве связывающего агента в органическом растворителе, который представляет собой этилацетат. Целевое соединение выделяют в виде масла, которое может быть непосредственно переведено в соль, или выделяют в твердой форме на дополнительных стадиях. Степень химической чистоты, по данным ВЭЖХ, составляет 95,33%, также сообщалось, что у полученного масла наблюдали изменение консистенции с течением времени в сторону воскообразной. В J. Med. Chem. 2005, 48, 141-151 сообщалось о конденсации в присутствии EDC и НОВТ в DMF в качестве растворителя. Недостаток способа все еще оставается и выражается в достаточно низком выходе, который составляет только 71%, и применении для очистки флэш-хроматографии (препаративной хроматографии низкого давления), которая не подходит для промышленных масштабов производства. В Journal of Chemical Research 2010, 230-232 приведено описание указанной реакции конденсации в присутствии EDC-HCI и НОВТ в безводных дихлорметане (DCM) и триэтиламине (Et3N). Полученный продукт перекристаллизовывали из толуола. На стадии удаления защитной группы использовали конц.HCl и метанол. Кроме того, конденсация соединения формулы 2 с соединением формулы 3 в присутствии EDC или EDC-HCl и/или в присутствии НОВТ и в присутствии органического основания в органическом растворителе и последующее выделение промежуточного продукта перед стадией удаления защитной группы раскрыто в ряде других литературных источников, таких, например, какWO 2009064476, WO 2010131025. Тем не менее, все еще существует необходимость в эффективном и экономном синтезе ситаглиптина или его фармацевтически приемлемых солей, который обеспечивает получение высокочистого конечного продукта с высоким выходом. Краткое описание изобретения Согласно первому варианту реализации настоящее изобретение обеспечивает способ, предпочтительно однореакторный способ, получения ситаглиптина или его фармацевтически приемлемых солей формулы 1 включающий стадии: а) конденсации соединения формулы 2 где R представляет собой аминозащитную группу, которая может быть удалена, предпочтительно в кислой среде, и может быть выбрана из группы, состоящей, но не ограниченной только перечисленным,из трет-бутилоксикарбонильной (ВОС) группы, карбобензилокси (Cbz) группы, параметоксибензилкарбонильной (Moz или MeOZ) группы, 9-флуоренилметилоксикарбонильной (FMOC) группы, ацетильной(Ас) группы, бензоильной (Bz) группы, бензильной (Bn) группы, параметоксибензильной (РМВ) группы,3,4-диметоксибензильной (DMPM) группы; предпочтительно карбаматной группы или ВОС, с соединением формулы 3 которое может быть использовано в форме свободного основания или в форме соли, в частности в форме хлороводородной соли, в присутствии связывающего агента, который выбран из группы, состоящей, но не ограниченной только перечисленным, из фосгена, дифосгена, трифосгена, карбонилдиимидазола, хлортионилимидазола, тионилдиимидазола, дициклогексилкарбодиимида (DCC), N-(3 диметиламинопропил)-N'-этилкарбодиимида гидрохлорида (EDC-HCI), солей тетраметилурония, таких как О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (HBTU), О-(бензотриазол-1 ил)-N,N, N',N'-тетраметилурония тетрафторборат (TBTU) или О-(7-аза-бензотриазол-1-ил)-N,N,N',N'тетраметилурония гексафторфосфат (HATU), предпочтительно N-(3-диметиламинопропил)-N'этилкарбодиимида гидрохлорид (EDC-HCI) и основного растворителя, который представляет собой Nметилимидазол, с получением соединения формулы 4b) удаления защитной группы соединения формулы 4 с получением основания ситаглиптина, которое в последующем может быть переведено в фармацевтически приемлемую соль. Согласно еще одному варианту реализации степень химической чистоты полученного ситаглиптина, по данным ВЭЖХ, составляет в пределах 99-100%, предпочтительно 99,5-100%, более предпочтительно 99,7-100%. Согласно еще одному варианту реализации энантиомерная чистота ситаглиптина, полученного по способу согласно настоящему изобретению более чем 99,5%, предпочтительно более чем 99,8%, более предпочтительно более чем 99,9% (по данным ВЭЖХ). Согласно еще одному варианту реализации полученый ситаглиптин переводят в одну из фармацевтически приемлемых солей, приготовленных из любого неорганического или органического основания или кислоты, таких, например, как гидрохлорид, фосфат, дигидрофосфат, тартрат, бензолсульфонат,сульфат, ацетат, предпочтительно гидрохлорид, фосфат и дигидрофосфат. Согласно еще одному варианту реализации настоящее изобретение относится к фармацевтической композиции, включающей ситаглиптин или его фармацевтически приемлемые соли, приготовленные по способу согласно настоящему изобретению и, возможно, по меньшей мере одно другое активное вещество и по меньшей мере один фармацевтически приемлемый эксципиент. Согласно еще одному варианту реализации, настоящее изобретение относится к ситаглиптину или его фармацевтически приемлемым солям, приготовленным по способу согласно настоящему изобретению для использования в лечении, контроле или предотвращении диабета, инсулин-независимого (тип 2) сахарного диабета, гипергликемии, ожирения, резистентности к инсулину, одного или более расстройств липидного обмена, выбранных из группы, состоящей, но не ограниченной только перечисленным, из дислипидемии, гиперлипидемии гипертриглицеридемии, гиперхолестеринемии, низкого содержания липопротеинов высокой плотности (HDL) и высокого содержания липопротеинов промежуточной плотности (IDL), атеросклероза, одного или более расстройств, выбранных из группы, состоящей, но не ограниченной только перечисленным, из нейтропении,нервных расстройств, метастазов новообразований, доброкачественной гипертрофии предстательной железы, гингивита, гипертонии и остеопороза, или одного или более состояний, выбранных из группы, состоящей, но не ограниченной только перечисленным, из поражения желудочно-кишечного тракта(ЖКТ), воспалительного заболевания кишечника, болезни Крона и неспецифического язвенного колита. Подробное описание изобретения Авторы настоящего изобретения неожиданно обнаружили, что использование N-метилимидазола в качестве основного растворителя в реакции конденсации соединения формулы 2 с 3-(трифторметил)5,6,7,8-тетрагидро-[1,2,4]триазоло-[4,3-а]пиразином или его гидрохлоридом, и использованиеN-метилимидазола в последующей реакции удаления защитной группы делает возможным однореакторный синтез ситаглиптина и его фармацевтически приемлемых солей. Кроме того, способ согласно настоящему изобретению обеспечивает получение ситаглиптина и его фармацевтически приемлемых солей с высокой химической и энантиомерной чистотой высокой рентабельными, экологически безопасными, и легко масштабируемыми методами. Согласно первому варианту реализации, настоящее изобретение обеспечивает способ, предпочтительно однореакторный способ, получения ситаглиптина или его фармацевтически приемлемых солей формулы 1 включающий стадии: а) конденсации соединения формулы 2 где R представляет собой аминозащитную группу, которая может быть удалена в кислой среде, с соединением формулы 3 которое может быть использовано в форме свободного основания или в форме соли, в частности в форме гидрохлорида, в присутствии связывающего агента и основного растворителя, который представляет собой N-метилимидазол, с получением соединения формулы 4b) удаления защитной группы соединения формулы 4 с получением основание ситаглиптина, которое дополнительно может быть переведено в фармацевтически приемлемую соль. Аминозащитная группа, которая может быть удалена в кислой среде, выбрана из группы, состоящей, но не ограниченной только перечисленным, из трет-бутилоксикарбонильной (ВОС) группы, карбобензилокси (Cbz) группы, параметоксибензилкарбонильной (Moz или MeOZ) группы,9-флуоренилметилоксикарбонильной (FMOC) группы, ацетильной (Ас) группы, бензоильной (Bz) группы, бензильной (Вп) группы, параметоксибензильной (РМВ) группы, 3,4-диметоксибензилы-юй (DMPM) группы; предпочтительно группы, которые могут быть удалены воздействием кислоты, такие как карбаматная группа, ВОС. Связывающий агент, используемый в способе согласно настоящему изобретению, выбран из группы, состоящей, но не ограниченной только перечисленным, из фосгена, дифосгена, трифосгена, карбонилдиимидазола, хлортионилимидазола, тионилдиимидазола, дициклогексилкарбодиимида (DCC), N-(3 диметиламинопропил)-N'-этилкарбодиимида гидрохлорида (EDC-HCI), солей тетраметилурония, таких как О-(бензотриазол-1-ил)-N, N, N', N'-тетраметилурония гексафторфосфат (HBTU), О-(бензотриазол-1 ил)-N,N,N',N'-тетраметилурония тетрафторборат (TBTU) или О-(7-аза-бензотриазол-1-ил)-N,N,N',N'тетраметилурония гексафторфосфат (HATU), предпочтительно используют N-(3-диметиламинопропил)N'-этилкарбодиимида гидрохлорид (EDC-HCI). Преимущества использования N-метилимидазола в качестве органического растворителя на стадии химического преобразования по способу согласно настоящему изобретению следующие: превосходное химическое преобразование исходных соединений, означающее, что по меньшей мере 95% исходного соединения 2 и исходного соединения 3 переходят в соединение формулы 4; соединение формулы 3 может быть использовано или в форме основания, или в форме кислой соли,что означает отсутствие необходимости добавления дополнительного основания к реакционной смеси; благодаря хорошей растворимости N-метилимидазола в воде легко проводить выделение экстракцией; нет необходимости использовать дополнительное соединение, чтобы избежать рацемизации, как,например, в случае НОВТ, который не ограничен только случаем НОВТ; не нужна очистка полученного продукта методом колоночной хроматографии; из-за хорошей растворимости исходного материала в N-метилимидазоле количество потраченного растворителя может быть меньше, чем при использовании других растворителей, известных из уровня техники, следовательно реакция может быть проведена при высоких концентрациях реагентов;N-метилимидазол не представляет угрозы для окружающей среды и может быть регенерирован. Исходное соединение формулы 2 может быть получено любым известным способом, раскрытым в предшествующем уровне техники, таким, например, как описано в следующих источниках, хотя и не ограничено только перечисленным: IPCOM000176671, Bioorganic and Medicinal Chemistry Letters; т. 17;Chemical Research, Synopses;4; (2010); стр. 230-232, European Journal of Medicinal Chemistry; т. 45;11; (2010); стр. 4953-4962, Tetrahedron: Asymmetry; т. 17;2; (2006); стр. 205-209, EP 2308829,EP 2230241, WO 2010131025, WO 201012278, WO 201078440, WO 200623750, WO 20047468,WO 200382817, US 2008280913. Исходное соединение формулы 3 может быть получено любым известным способом, раскрытым в предшествующем уровне техники, таким, например, как описано в следующих источниках, хотя и не ограничено только перечисленным: IPCOM000176303D, Organic Letters, т. 7;6; (2005); стр. 1039-1042,Journal of Medicinal Chemistry; т. 48;1; (2005); стр. 141-151, Journal of Medicinal Chemistry; т. 48;1;(2005); стр. 141-151, EP 2270009, WO 200985990, WO 200750485, WO 200735198, WO 2006119260,WO 200681151, WO 200633848, WO 200623750, WO 20069886, WO 200597733, WO 200485378,WO 200458266, US 2010331541, US 2008280913. В предпочтительном варианте реализации степень химической чистоты исходного соединения 2 и исходного соединения 3, по данным ВЭЖХ, может быть по меньшей мере 95%, предпочтительно по меньшей мере 96%, более предпочтительно по меньшей мере 97,5%. В предпочтительном варианте реализации энантиомерная чистота соединения формулы 2 в процентном выражении может быть более чем 98,5% (по данным ВЭЖХ). В предпочтительном варианте реализации реакцию конденсации соединения формулы 2 с соединением формулы 3 проводят при температуре от 0 до 60 С, предпочтительно от 10 до 30 С. В дополнительном предпочтительном варианте реализации реакцию конденсации соединения формулы 2 с соединением формулы 3 проводят в течение 2-24 ч., предпочтительно 3-8 ч. Соотношение между любыми реагентами, используемыми в способе согласно настоящему изобретению, эквимолярно. Кроме того, концентрация соединения формулы 3 в реакционном растворителе может быть 50-600 г/л, предпочтительно 100-500 г/л. В предпочтительном варианте реализации удаление защитной группы у in-situ образованного соединения формулы 4 проводят в кислоте, предпочтительно в HCl. HCl может быть использована в виде спиртового раствора или концентрированного водного раствора. Предпочтительно используют концентрированный водный раствор HCl. В предпочтительном варианте реализации удаление защитной группы у in-situ образованного соединения формулы 4 проводят при температуре от 0 до 60 С, предпочтительно от 10 до 30 С. В дополнительном предпочтительном варианте реализации удаление защитной группы у in-situ образованного соединения формулы 4 проводят в течение 2-24 ч., предпочтительно 3-10 ч. Полученный ситаглиптин может быть выделен любым подходящим способом, известным из уровня техники, примерами таких способов являются, но не ограничиваются ими, экстракция, выпаривание,фильтрация, перекристаллизация, перегонка, хроматография и т.п. Согласно дополнительному варианту реализации степень чистоты полученного ситаглиптина, по данным ВЭЖХ, может быть в пределах 99-100%, предпочтительно 99,5-100%, более предпочтительно 99,7-100%. Согласно дополнительному варианту реализации энантиомерная чистота ситаглиптина, полученного по способу согласно настоящему изобретению, более 99,5%, предпочтительно более 99,8%, более предпочтительно более 99,9% (по данным ВЭЖХ). В предпочтительном варианте реализации общий выход в указанном способе получения ситаглиптина согласно настоящему изобретению может быть по меньшей мере 75%, предпочтительно по меньшей мере 80%, более предпочтительно по меньшей мере 85%. В еще одном варианте реализации полученный ситаглиптин переводят в одну из его фармацевтически приемлемых солей, полученных из любого неорганического или органического основания или кислоты, таких, например, как соли, описание которых приведено, но не ограничено только перечисленным, в WO 2003004498, WO 2005003135, WO 2005020920, WO 2005030127, WO 2005072530, WO 2006033848, WO 2006104997, WO 2007035198, KR 20070111099, WO 2009084024, WO 2009070314, WO 2009085990, WO 2009120746, US 2009247532, WO 2010000469, WO 2010012781, WO 2010032264, WO 2010092090, US 2010249140, WO 2010122578, WO 2010131035, WO 2011018494, предпочтительно указанная соль может быть гидрохлоридом, фосфатом, дигидрофосфатом, тартратом, бензолсульфонатом,сульфатом, ацетатом, более предпочтительно гидрохлоридом, фосфатом, дигидрофосфатом. Химическое преобразование может быть проведено согласно любым известным способам, таким как способы, описание которых приведено в вышеуказанных источниках. В предпочтительном варианте реализации полученный ситаглиптин или его фармацевтически приемлемые соли могут быть в аморфной или кристаллической (полиморфной) форме. Кроме того, в контексте настоящего изобретения предполагается получение любой фармацевтически приемлемой гидратной формы ситаглиптина и его фармацевтически приемлемых солей. В еще одном варианте реализации основание ситаглиптина, полученное по способу согласно настоящему изобретению, переводят в гидрохлорид смешиванием основания ситаглиптина с HCl. Полу-5 024688 ченная соль гидрохлорида ситаглиптина может быть выделена или использована непосредственно in-situ для приготовления фармацевтической композиции. Полученная соль гидрохлорида ситаглиптина может быть в аморфной форме, такой, например, как указано в неограниченном описании, приведенном в SIP-201100115, или в кристаллической форме такой, например, как как указано в неограниченном описании, приведенном в WO 2005072530, KR 20070111099, WO 2010000469. В предпочтительном варианте реализации средний размер частиц полученного ситаглиптина или его фармацевтически приемлемых солей может быть 0,1-600 мкм, предпочтительно 1-250 мкм, более предпочтительно 10-50 мкм. Термин "средний размер частиц", использованный в тексте настоящего документа, относится к среднеобъемному диаметру частиц. Среднеобъемный диаметр частиц определяли методом лазерного светорассеяния, используя анализатор Mastersizer MS 2000 компании Malvern, снабженный модулем диспергирования Hydro S. В качестве разбавляющей среды использовали растительное масло. В еще одном варианте реализации настоящее изобретение относится к фармацевтической композиции, включающей ситаглиптин или его фармацевтически приемлемые соли, приготовленные по способу согласно настоящему изобретению и другие фармацевтически приемлемые эксципиенты. В некоторых случаях другие активные вещества могут присутствовать в фармацевтической композиции согласно настоящему изобретению и могут быть введены или по отдельности, или в составе той же самой фармацевтической композиции. Другие активные вещества могут быть выбраны, но не ограничены только перечисленным, из других ингибиторов дипептидилпептидазы-4, бигуанидов, производных фенофибровой кислоты, сульфонилмочевин, глитазонов и любых их смесей. Фармацевтическая композиция согласно настоящему изобретению может быть введена перорально,парентерально, ингаляционно, назально, вагинально, ректально, сублингвально или местно в подходящей лекарственной форме, включающей фармацевтически приемлемые эксципиенты, подходящие для каждого пути введения. Фармацевтически приемлемые эксципиенты могут быть выбраны, но не ограничены только перечисленным, из наполнителей, связующих веществ, разрыхлителей, веществ, способствующих скольжению, красителей, адсорбентов, ПАВ, пленкообразующих веществ, пластификаторов и т.п. Предпочтительно фармацевтическая композиция согласно настоящему изобретению включает 1-200 мг, предпочтительно 10-150 мг, более предпочтительно 25-100 мг ситаглиптина или его фармацевтически приемлемых солей. В еще одном варианте реализации настоящее изобретение относится к ситаглиптину или его фармацевтически приемлемым солям, полученным по способу согласно настоящему изобретению для использования при лечении, контроле или предотвращении диабета, инсулин-независимого (тип 2) сахарного диабета, гипергликемии, ожирения, резистентности к инсулину, одного или более расстройств липидного обмена, выбранных из группы, состоящей, но не ограниченной только перечисленным, из дислипидемии, гиперлипидемии, гипертриглицеридемии, гиперхолестеринемии, низкого содержания липопротеидов высокой плотности (HDL) и высокого содержания липопротеидов промежуточной плотности(IDL), атеросклероза, одного или более расстройств, выбранных из группы, состоящей, но не ограниченной только перечисленным, нейтропению, из нервных расстройств, метастазов новообразований, доброкачественной гипертрофии предстательной железы, гингивита, гипертонии и остеопороза, или одного или более состояний выбранных из группы, состоящей, но не ограниченной только перечисленным, из поражения желудочно-кишечного тракта (ЖКТ), воспалительного заболевания кишечника, болезни Крона и неспецифического язвенного колита. Мы неожиданно обнаружили, что однореакторный способ получения ситаглиптина согласно настоящему изобретению из (R)-3-трет-бутоксикарбонил)амино)-4-(2,4,5-трифторфенил)бутановой кислоты, который проводят в N-метилимидазоле в качестве органического растворителя, приводит к практически полному химическому преобразованию реагирующих веществ, обеспечивая, таким образом, получение высокочистого конечного продукта и почти полное отсутствие каких-либо побочных продуктов. Кроме того, способ согласно настоящему изобретению высокорентабелен, экологически безопасен и легко масштабируем. Настоящее изобретение проиллюстрировано в виде следующих неограниченных Примеров. С помощью указанных примеров мы демонстрируем преимущества способа согласно настоящему изобретению. Примеры Справочный пример 1.a) Стадия конденсации. Смесь 4,02 г Вос(R)-3-амино-(2,4,5-трифторфенил)бутановой кислоты, 2,77 г 3-(трифторметил)5,6,7,8-тетрагидро-[1,2,4]триазоло-[4,3-а]пиразина гидрохлорида и 25 мл пиридина охлаждали до 0 С. Затем добавляли 2,73 г N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорида (EDC) и смесь перемешивали в течение 16 ч при комнатной температуре. Потом к реакционной смеси добавляли 100 мл изопропилацетата и 100 мл воды. Фазы разделяли и водную фазу снова экстрагировали 100 мл этилацетата. Собранные порции органической фазы промывали 100 мл воды, затем добавляли 100 мл воды и доводили значение pH смеси приблизительно до 3. Фазы разделяли и органическую фазу выпаривали. Остаток собирали и высушивали. Выход: 5,88 г (95%). ВЭЖХ: 97,6%.b) Стадия снятия защитной группы. Смесь 5,4 г (R)-трет-бутил(4-оксо-4-(3-(трифторметил)-5,6-дигидро[1,2,4]триазоло[4,3-а]пиразин 7(8 Н-ил)-1-(2,4,5-трифторфенил)бутан-2-ил)карбамата и 27 мл раствора 1,25 М HCl в этаноле перемешивали в течение 16 ч при комнатной температуре. После завершения реакции смесь выпаривали, добавляли 37 мл воды и доводили значение pH смеси 1 М NaOH до 10-12. Затем добавляли 37 мл изопропилацетата, перемешивали и разделяли фазы. Водную фазу повторно экстрагировали 37 мл изопропилацетата. Собранные порции органической фазы промывали 37 мл воды и выпаривали. К остатку добавляли 27 мл трет-бутилметилового эфира (ТВМЕ). Полученную суспензию охлаждали до температуры ниже 0 С,затем отфильтровывали и высушивали. Выход для стадии снятия защитной группы: 90%. Выход для способа в целом: 85%. ВЭЖХ: химическая чистота: 99,0%, R изомер: 100,0%, S изомер: 0,0%. Метод ВЭЖХ для определения химической чистоты Колонка: Ascentis Express Phenyl-Hexyl,(1004,6) мм, размер частиц 2,7 мкм. Элюэнт А: 0,1% триэтиламин, pH 7,0. Элюэнт В: ацетонитрил Градиент. Восстановление колонки после анализа: 4 мин. Скорость потока: 0,8 мл/мин. Детектирование: УФ, длина волны 210 нм. Вводимый объем: 2 мкл. Температура термостата колонки: 25 С. Разбавитель: ацетонитрил:вода (50:50). Метод ВЭЖХ для определения энантиомерной чистоты Колонка: Chiralpak AD-H, (2504,6) мм,размер частиц 5 мкм. Подвижная фаза: этанол:гексан:этилендиамин (EDA) в соотношении 600:400:1 (V/V/V). Скорость потока: 1,0 мл/мин. Детектирование: УФ, длина волны 210 нм. Вводимый объем: 20 мкл. Температура термостата колонки: 25 С. Разбавитель: метанол. Пример 1. Смесь 8,04 г (R)-3-трет-бутоксикарбонил)амино)-4-(2,4,5-трифторфенил)бутановой кислоты (химическая чистота по данным ВЭЖХ: 99,9%, энантиомерная чистота (по данным ВЭЖХ): 99,9%), 5,54 г 3-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло-[4,3-а]пиразина гидрохлорида (химическая чистота по данным ВЭЖХ: 99,6%) и 40 мл N-метилимидазола охлаждали до 0 С. Затем к указанной смеси добавляли 5,46 г N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорида (EDC-HCl) и смесь перемешивали в течение 16 ч при комнатной температуре. Потом к реакционной смеси добавляли 200 мл изопропилацетата и 200 мл воды. Фазы разделяли и водную фазу снова экстрагировали 100 мл этилацетата. Собранные порции органической фазы промывали 200 мл воды (дважды). Органическую фазу выпаривали до объема приблизительно 100 мл. К концентрированной органической фазе добавляли 20 мл 8 М HCl/EtOH и смесь перемешивали при комнатной температуре в течение 18 ч. После завершения реакции добавляли 50 мл воды и доводили значение pH смеси 1 М NaOH до 10-12. Потом фазы разделяли. Водную фазу повторно экстрагировали 100 мл изопропилацетата. Собранные порции органической фазы промывали 80 мл воды (дважды) и выпаривали. К остатку добавляли 60 мл трет-бутилметилового эфира. Полученную суспензию охлаждали до температуры ниже 0 С, затем отфильтровывали и высушивали. Выход: 8,84 г (90%).N-метилимидазола охлаждали до 0 С. Затем добавляли 2,73 г N-(3-диметиламинопропил)-N'этилкарбодиимида гидрохлорида (EDC) и смесь перемешивали в течение 6 ч при комнатной температуре. К реакционной смеси добавляли 100 мл изопропилацетата и 100 мл воды. Фазы разделяли и водную фазу снова экстрагировали 50 мл этилацетата. Собранные порции органической фазы промывали 100 мл воды (дважды). Органическую фазу выпаривали до объема приблизительно 45 мл. К концентрированной органической фазе добавляли 7,5 мл концентрированной HCl и смесь перемешивали при комнатной температуре в течение 18 ч. После завершения реакции добавляли 25 мл воды и доводили значение pH смеси водным раствором NaOH до 10-12. Потом фазы разделяли. Водную фазу повторно экстрагировали 500 мл изопропилацетата. Собранные порции органической фазы промывали 40 мл воды (дважды) и выпаривали. К остатку добавляли 60 мл трет-бутилметилового эфира. Полученную суспензию охлаждали до температуры ниже 0 С, затем отфильтровывали и высушивали. Выход: 4,26 г (86%). ВЭЖХ: химическая чистота: 99,84%, R изомер: 100,0%, S изомер: 0,0%. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения ситаглиптина или его фармацевтически приемлемых солей формулы 1 включающий стадии: а) конденсации соединения формулы 2 где R представляет собой аминозащитную группу, которая может быть удалена, с соединением формулы 3 которое может быть использовано в форме свободного основания или в форме соли, в присутствии связывающего агента и основного растворителя, который представляет собой N-метилимидазол, с получением соединения формулы 4b) удаления защитной группы у соединения формулы 4 с получением основания ситаглиптина, которое дополнительно может быть переведено в его фармацевтически приемлемую соль. 2. Способ по п.1, характеризующийся тем, что аминозащитная группа, которая может быть удалена в кислой среде, выбрана из группы, состоящей из карбаматной группы, трет-бутилоксикарбонильной(DMPM) группы, предпочтительно карбаматной группы или ВОС. 3. Способ по любому из пп.1, 2, характеризующийся тем, что связывающий агент выбран из группы, состоящей из фосгена, дифосгена, трифосгена, карбонилдиимидазола, хлортионилимидазола, тионилдиимидазола, дициклогексилкарбодиимида (DCC), N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорида (EDC-HCl), солей тетраметилурония, таких как О-(бензотриазол-1-ил)-N,N,N',N'тетраметилурония гексафторфосфата (HBTU), О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (TBTU) или О-(7-аза-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата(HATU), предпочтительно N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорида (EDC-HCl). 4. Способ по любому из пп.1-3, характеризующийся тем, что степень химической чистоты полученного ситаглиптина, по данным высокоэффективной жидкостной хроматографии (ВЭЖХ), составляет 99-100%, предпочтительно 99,5-100%, более предпочтительно 99,7-100%. 5. Способ по любому из пп.1-4, характеризующийся тем, что энантиомерная чистота ситаглиптина составляет более чем 99,5% (по данным ВЭЖХ). 6. Способ по любому из пп.1-5, характеризующийся тем, что ситаглиптин дополнительно переводят в одну из его фармацевтически приемлемых солей, полученных из любого неорганического или органического основания или кислоты, таких как гидрохлорид, фосфат, дигидрофосфат, тартрат, бензолсульфонат, сульфат, ацетат, предпочтительно гидрохлорид, фосфат и дигидрофосфат. 7. Применение N-метилимидазола в качестве органического растворителя в реакции конденсации соединения формулы 2 где R представляет собой аминозащитную группу, которая может быть удалена, с соединением формулы 3 которое может быть использовано в форме свободного основания или в форме соли с получением соединения формулы 4 8. Применение по п.7, где N-метилимидазол применяют также в качестве растворителя для удаления защитной группы у соединения формулы 4 с получением основания ситаглиптина.
МПК / Метки
МПК: C07D 487/04
Метки: солей, способ, получения, фармацевтически, приемлемых, ситаглиптина
Код ссылки
<a href="https://eas.patents.su/10-24688-sposob-polucheniya-sitagliptina-i-ego-farmacevticheski-priemlemyh-solejj.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения ситаглиптина и его фармацевтически приемлемых солей</a>
Способ получения (1r, 2s, 4r)-(-)-2-[n, n-(диметиламиноэтокси)]-2-фенил-1,7,7 -триметилбицикло[2,2,1] гептана и его фармацевтически приемлемых кислотных аддитивных солей

Номер патента: 2163
Опубликовано: 24.12.2001
Авторы: Сабо Тибор, Шимиг Дьюла, Надь Калман, Порч-Маккаи Марта, Лукач Дьюла, Краснаи Дьёрдь, Немет Норберт, Будаи Золтан, Суладьи Янош, Мезеи Тибор
МПК: C07C 217/12
Метки: аддитивных, способ, фармацевтически, солей, кислотных, n-(диметиламиноэтокси)]-2-фенил-1,7,7, приемлемых, триметилбицикло[2,2,1, гептана, 4r)-(-)-2-[n, получения
Формула / Реферат:
1. Способ получения (1R,2S,4R)-(-)-2-[N,N-(диметиламиноэтокси)]-2-фенил-1,7,7-триметилбицикло-[2,2,1]гептана высокой степени чистоты по формуле и его фармацевтически приемлемых кислотных аддитивных солей, который включает превращение (+)-1,7,7-триметилбицикло[2,2,1]гептан-2-она {(+)-камфоры} по формуле в (1R, 2S, 4R)-(-)-2-фенил-1,7,7-триметилбицикло[2,2,1]гептан-2-ол по формуле путем реакции соединения по формуле II с...
Способ получения твердых лекарственных форм солифенацина и его фармацевтически приемлемых солей для перорального введения

Номер патента: 23529
Опубликовано: 30.06.2016
Авторы: Турк Урска, Юрсиц Урска, Врбинц Миха, Врецер Франц
МПК: A61K 31/439, A61K 9/28, A61K 9/20...
Метки: получения, лекарственных, солей, введения, форм, твердых, фармацевтически, способ, перорального, солифенацина, приемлемых
Формула / Реферат:
1. Способ получения твердой лекарственной формы для перорального введения, содержащей:а) эффективное количество кристаллического солифенацина или его фармацевтически приемлемой соли,б) фармацевтически приемлемые эксципиенты, подходящие для приготовления твердых лекарственных форм для перорального введения,в отсутствие растворителя,при котором осуществляют стадии:1) прессования гомогенной смеси кристаллического солифенацина или его...
Новый способ синтеза периндоприла и его фармацевтически приемлемых солей

Номер патента: 12034
Опубликовано: 30.06.2009
Авторы: Дюбюффе Тьерри, Лекув Жан-Пьер
МПК: C07K 5/06
Метки: новый, солей, приемлемых, способ, синтеза, фармацевтически, периндоприла
Формула / Реферат:
1. Способ промышленного синтеза периндоприла формулы (I) и его фармацевтически приемлемых солей, отличающийся тем, что сложный бензиловый эфир формулы (IIa) или (IIb) или аддитивная соль сложного эфира формулы (IIa) или (IIb) с минеральной кислотой или органической кислотой взаимодействует с соединением формулы (III) в присутствии конденсирующего агента, который выбирают из следующих реагентов и пар реагентов: гидрохлорид...
Способ стимуляции отказа от курения или его снижения или предупреждения возобновления курения и применение пароксетина или его фармацевтически приемлемых солей или сольватов для получения лекарственного средства

Номер патента: 3584
Опубликовано: 26.06.2003
Автор: Стейнер Мартин Кс.
МПК: A61K 31/445, A61P 25/34
Метки: лекарственного, приемлемых, снижения, возобновления, стимуляции, сольватов, средства, отказа, предупреждения, курения, применение, получения, пароксетина, фармацевтически, способ, солей
Формула / Реферат:
1. Способ стимуляции отказа от курения или его снижения или предупреждения возобновления курения, заключающийся во введении нуждающемуся в этом человеку эффективного нетоксического количества пароксетина или его фармацевтически приемлемой соли или сольвата. 2. Способ по п.1, отличающийся тем, что пароксетин вводится в виде препарата с контролируемым или замедленным высвобождением активного вещества. 3. Способ по п.2, отличающийся тем, что...
Способ синтеза периндоприла и его фармацевтически приемлемых солей

Номер патента: 8644
Опубликовано: 29.06.2007
Авторы: Дюбюффе Тьерри, Ланглуа Паскаль, Фуже Клод
МПК: C07K 5/06, C07D 209/42
Метки: фармацевтически, способ, периндоприла, солей, синтеза, приемлемых
Формула / Реферат:
1. Способ синтеза соединения формулы (I) и его фармацевтически приемлемых солей, который характеризуется тем, что соединение формулы (II) в которой R представляет собой атом водорода или бензил, или линейную, или разветвленную C1-С6-алкильную группу, подвергают реакции с соединением формулы (III), имеющим (R)-конфигурацию в которой G представляет собой атом хлора, брома или йода или гидрокси, n-толуолсульфонилокси, метансульфонилокси- или...
Предыдущий патент: Стержни для применения в курительных изделиях
Следующий патент: Ингибиторы тирозинкиназы брутона