Меченные радиоактивными изотопами производные хинолина и их применение в качестве лигандов метаботропного глутаматного рецептора
Номер патента: 9334
Опубликовано: 28.12.2007
Авторы: Бисхофф Франсуа Пол, Лаврейсен Хилде, Янссен Корнелус Герардус Мариа, Лесаж Анн Симон Жозефин




























Формула / Реферат
1. Меченное радиоактивным изотопом соединение, представляющее собой соединение (е)

2. Радиоактивная композиция для маркировки и идентификации сайтов метаботропного глутаматного рецептора и для визуализации органа для введения млекопитающим, содержащая терапевтически эффективное количество меченного радиоактивным изотопом соединения по п.1 и фармацевтически приемлемый носитель или разбавитель.
3. Применение меченного радиоактивным изотопом соединения по п.1 в качестве диагностического средства.
4. Применение радиоактивной композиции по п.2 в качестве диагностического средства.
5. Применение меченного радиоактивным изотопом соединения по п.1 при получении диагностического средства для маркировки или идентификации mGlu1-рецептора в биологическом материале.
6. Применение радиоактивной композиции по п.2 в качестве активного компонента при получении диагностического средства для маркировки или идентификации mGlu1-рецептора в биологическом материале.
7. Способ диагностики, включающий маркировку или идентификацию mGlu1-рецептора в биологическом материале меченным изотопом соединением по п.1.
8. Способ диагностики по п.7, где маркировку осуществляют путем введения меченного изотопом соединения по п.1 в биологический материал и идентификацию осуществляют путем обнаружения эмиссии от указанного меченного радиоактивным изотопом соединения.
9. Способ диагностики, включающий маркировку или идентификацию mGlu1-рецептора в биологическом материале радиоактивной композицией по п.2.
10. Способ диагностики по п.9, где маркировку осуществляют путем введения радиоактивной композиции по п.2 в биологический материал и идентификацию путем обнаружения эмиссии от указанного меченного радиоактивным изотопом соединения.
11. Способ диагностики по любому из пп.7-10, включающий оценку того, способно ли тестируемое соединение располагаться на mGlu1-рецепторе или связываться с mGlu1-рецептором в биологическом материале.
12. Способ диагностики путем визуализации органа, заключающийся в том, что в биологический материал в составе соответствующей композиции вводят меченное радиоактивным изотопом соединение по п.1, в количестве, достаточном для связывания указанного меченного радиоактивным изотопом соединения с сайтами mGlu1-рецептора в биологическом материале; и обнаруживают эмиссию от меченного радиоактивным изотопом соединения.
13. Способ диагностики путем визуализации органа, заключающийся в том, что в биологический материал в составе соответствующей композиции вводят радиоактивную композицию по п.2, в количестве, достаточном для связывания указанного меченного радиоактивным изотопом соединения с сайтами mGlu1-рецептора в биологическом материале; и обнаруживают эмиссию от меченного радиоактивным изотопом соединения.
14. Способ диагностики по любому из пп.7-13, где биологический материал выбран из группы, включающей образцы ткани, жидкости плазмы, жидкости организма, части тела и органов, полученных от теплокровных животных и от теплокровных животных как таковых (per se), в частности от человека.
15. Применение меченного радиоактивным изотопом соединение по п.1 для получения диагностического средства для оценки того, обладает ли тестируемое соединения способностью располагаться на mGlu1-рецепторе или связываться с mGlu1-рецептором в биологическом материале.
16. Применение радиоактивной композиции по п.2 для получения диагностического средства для оценки того, обладает ли тестируемое соединение способностью располагаться на mGlu1-рецепторе или связываться с mGlu1-рецептором в биологическом материале.
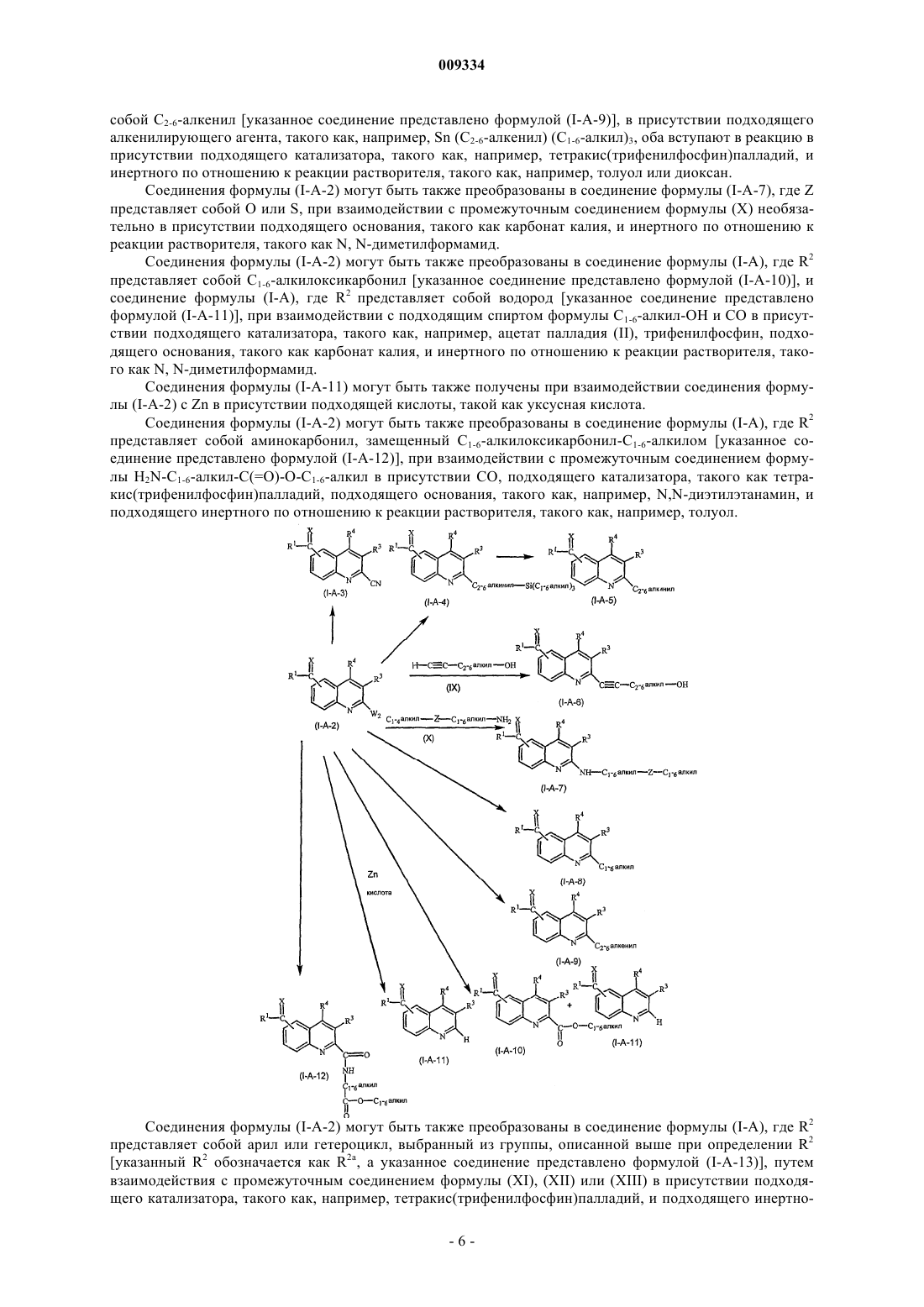
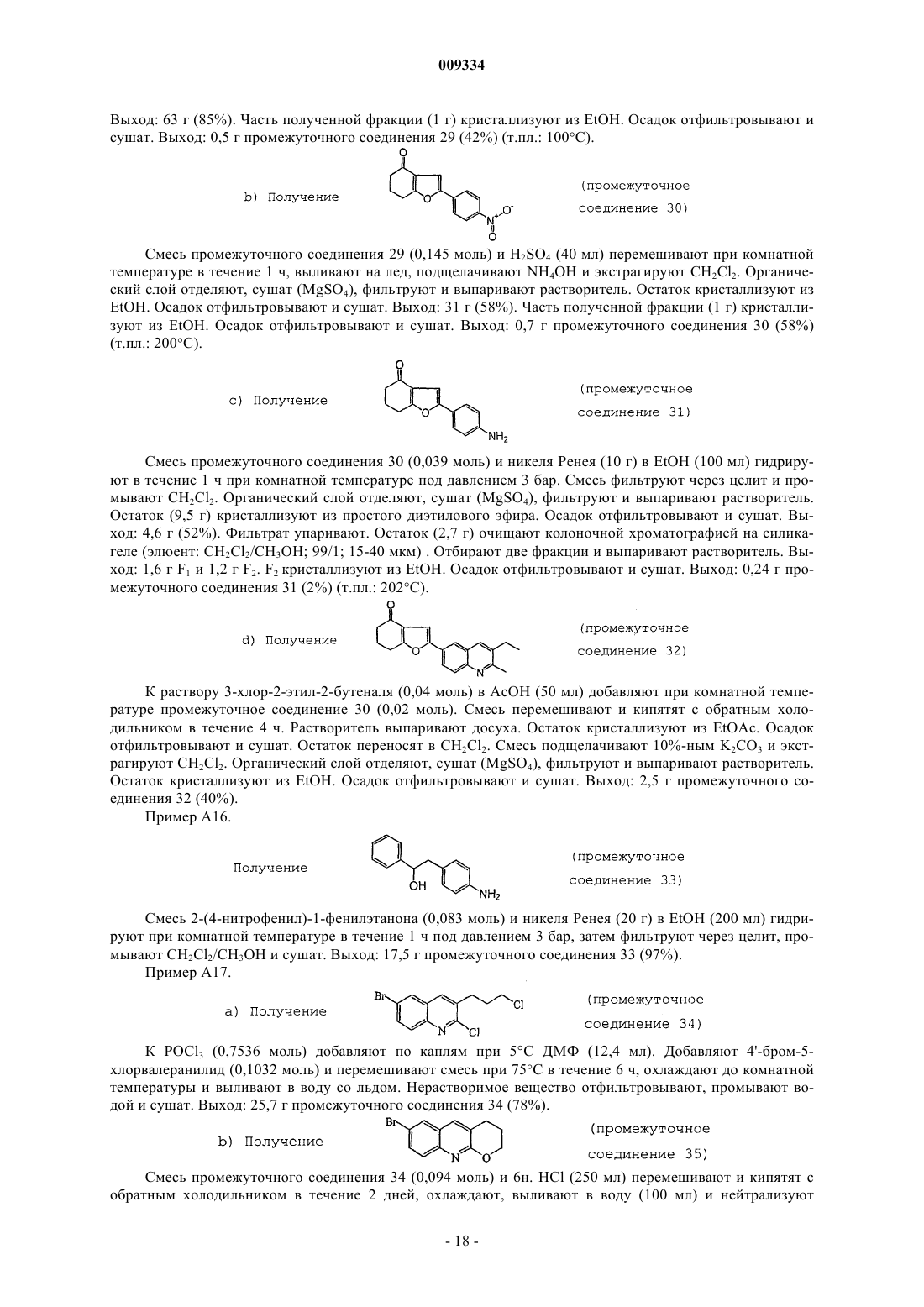
Текст
009334 Настоящее изобретение относится к меченным радиоактивными изотопами производным хинолина,проявляющим антагонистическую активность по отношению к метаботропному глутаматному рецептору, в частности активность по отношению к mGlu1-рецептору, и к получению таких производных; изобретение, кроме того, относится к композициям, содержащим такие производные, а также к их применению для диагностики, в частности для маркировки и идентификации сайтов метаботропного глутаматного рецептора и для визуализации органа. Введение Нейромедиатор глутамат считается главным возбуждающим нейромедиатором в центральной нервной системе млекопитающего. Связывание указанного нейромедиатора с метаботропными глутаматными рецепторами (mGluR), которые представляют собой подсемейство рецепторов, связанных с G-белком, и которые включают 8 отдельных подтипов mGluR, а именно подтипы от mGluR1 до mGluR8, активирует ряд внутриклеточных систем вторичных мессенджеров. Подсемейство mGlu-рецепторов на основе гомологии аминокислотных последовательностей, системы вторичных мессенджеров, используемой рецепторами, и фармакологических характеристик можно разделить на 3 группы. mGlu-рецепторы группы I, которая включает подтипы 1 и 5 mGluR, связываются с фосфолипазой С, и их активация приводит к внутриклеточной активации ионов кальция. mGlu-рецепторы группы II (mGluR2 и 3) и mGlu-рецепторы группы III (mGluR4, 6, 7 и 8) связываются с аденилциклазой, и их активация вызывает снижение (количества) вторичного мессенджера цАМФ, а также торможение нейрональной активности. Как было показано, лечение антагонистами mGluR группы I переводит в парасинапсисе на сниженное высвобождение нейромедиатора глутамата и снижение опосредованного глутаматом нейронального возбуждения через постсинаптические механизмы. Так как различные патофизиологические процессы и болезненные состояния, затрагивающие центральную нервную систему, считаются следствием возбуждения нервных клеток центральной нервной системы, индуцированного избытком глутамата, антагонисты mGluR группы I, в частности антагонисты mGluR1, могли бы быть терапевтически полезны для лечения заболеваний центральной нервной системы, в частности психиатрических и неврологических заболеваний. Однако вплоть до настоящего времени не было специфических mGluR1-лигандов, отсутствие которых значительно затрудняет изучение mGlu1-рецепторов, в частности авторадиографические исследования однозначного распределения и распространенности таких рецепторов в отделах мозга. В группе I пока был доступен только [3H]глутамат, применяемый для крысиных (Thomsen и др., Brain Res. 619: 2228,1993) или человеческих mGlu1a-рецепторов (Kingston и др., Neuropharmacology 37:277-287, 1998). ДляmGlu1a-рецептора и mGlu5-рецептора доступен [3H]квисквалат, однако указанный рецептор не является специфическим для mGlu1-рецептора (он также связывается с АМРА-рецептором) и является конкурентным, то есть вытесняет глутамат (Mutel и др., J.Neurochem. 75: 2590-2601, 2000). Целью настоящего изобретения является получение подходящих специфических, в частности, неконкурентных лигандов mGlu1-рецепторов. В настоящее время авторы обнаружили особую группу соединений, которые в виде меченной радиоактивным изотопом формы обеспечивают подходящие, специфические, в частности, неконкурентные лиганды mGlu1-рецептора, а также способ маркировки и идентификации сайтов метаботропного глутаматного рецептора и визуализации органа. В рамках настоящей заявки термин специфический означает, что лиганд избирательно связывается с сайтом mGlu1-рецептора. Термин неконкурентный означает, что лиганд не вытесняет или только незначительно вытесняет глутамат, связанный с сайтом mGlu1-рецептора. Нерадиоактивные соединения согласно настоящему изобретению описаны в заявке WO 02/28837. В заявке WO 99/26927 описаны антагонисты рецепторов mGluR группы I для лечения неврологических заболеваний и расстройств, основанные - среди прочего - на структуре хинолина. В заявке WO 99/03822 описаны бициклические лиганды метаботропного глутаматного рецептора,ни один из которых не основан на структуре хинолина или хинолинона. В заявке WO 94/27 605 описаны 1,2,3,4-тетрагидрохинолин-2,3,4-трион-3 или 4-оксимы и их применение для лечения и профилактики нейрональной потери, нейродегенеративных заболеваний, неблагоприятных последствий гиперактивности аминокислот, вызывающих возбуждение и тревогу, а также их меченные радиоактивными изотопами соединения. Подробное описание изобретения Настоящее изобретение относится к меченному радиоактивными изотопами соединению формулы Одно из воплощений настоящего изобретения относится к радиоактивной композиции для маркировки и идентификации сайтов метаботропного глутаматного рецептора и для визуализации органа для введения млекопитающим, содержащая терапевтически эффективное количество меченного радиоактив-1 009334 ным изотопом соединения и фармацевтически приемлемый носитель или разбавитель. В другом варианте осуществления настоящего изобретения оно касается применения меченного радиоактивным изотопом соединения в качестве диагностического средства. Вариантом предпочтительного воплощения настоящего изобретения является применение радиоактивной композиции в качестве диагностического средства, в частности для маркировки или идентификации mGlu1-рецептора в биологическом материале. Другим частным воплощением изобретения является способ диагностики, включающий маркировку или идентификацию mGlu1-рецептора в биологическом материале меченным изотопом соединением,причем маркировку осуществляют путем введения меченного изотопом соединения в биологический материал, и идентификацию осуществляют путем обнаружения эмиссии от указанного меченного радиоактивным изотопом соединения. К одному из предпочтительных воплощений настоящего изобретения является способ диагностики,включающий маркировку или идентификацию mGlu1-рецептора в биологическом материале радиоактивной композицией, причем способ диагностики по любому включает оценку того, способно ли тестируемое соединение располагаться на mGlu1-рецепторе или связываться с mGlu1-рецептором в биологическом материале. Другой вариант осуществления изобретения заключается в способе диагностики путем визуализации органа, заключающийся в том, что в биологический материал в составе соответствующей композиции вводят меченное радиоактивным изотопом соединение, в количестве, достаточном для связывания указанного меченного радиоактивным изотопом соединения с сайтами mGlu1-рецептора в биологическом материале; и обнаруживают эмиссию от меченного радиоактивным изотопом соединения. Еще один частный вариант осуществления изобретения относится к способу диагностики, где биологический материал выбран из группы, включающей образцы ткани, жидкости плазмы, жидкости организма, части тела и органов, полученных от теплокровных животных и от теплокровных животных как таковых (per se), в частности от человека. Другое воплощение изобретения относится к применению меченного радиоактивным изотопом соединения для получения диагностического средства для оценки того, обладает ли тестируемое соединение способностью располагаться на mGlu1-рецепторе или связываться с mGlu1-рецептором в биологическом материале. Особый интерес представляют следующие радиоактивные соединения: Все соединения согласно изобретению показывают по отношению к mGluR1 активность от умеренной до сильной. Такая активность, среди прочего, объясняется специфическим связыванием указанного соединения с mGlu1-рецептором, что делает соединения применимыми для диагностики, например для введения меченых атомов и обнаружения сайтов mGlu1-рецепторов. Следовательно, изобретение также относится к меченному радиоактивным изотопом соединению согласно изобретению, применяемому для диагностики. Первое предпочтительное воплощение (радиоактивные изотопы, испускающие гамма-излучение) Согласно первому предпочтительному воплощению меченное радиоактивным изотопом соединение содержит, по меньшей мере, один [3H]-атом или один [125I]-атом. [3H]-атом удобно вводить путем частичного или полного замещения одного или нескольких нерадиоактивных [1H]-атомов водорода в молекуле их радиоактивными изотопами. Выбор того, будет ли применяться [3H]- или [125I]-меченный лиганд,может зависеть частично от доступности жидкостных сцинтилляционных счетчиков (LSC), которые довольно дороги. [125I]-лиганды можно определить количественно с применением либо -счетчика, либоLSC, в то время как для [3H]-лигандов требуется применение LSC. Меченное радиоактивным изотопом соединение, содержащее по меньшей мере один [3H]-атом или один [125I]-атом, успешно применяют в способах, связанных с мечеными лигандами, в частности, при анализах мембранных рецепторов in vitro для маркировки или идентификации mGlu1-рецептора в биологическом материале. Меченные радиоактивным изотопом соединения, содержащие, по меньшей мере, один [3H]-атом или один [125I]-атом, также успешно применяют in vivo в авторадиографии mGlu1-рецепторов мозга, так как соединения согласно изобретению обладают полезной и неожиданной способностью легко пересекать гематоэнцефалический барьер. Следовательно, изобретение также относится к меченному радиоактивным изотопом соединению согласно изобретению, применяемому для диагностики, которая заключается в маркировке или идентификации mGlu1-рецептора в биологическом материале, а также к применению соединений согласно изобретению для производства диагностического средства для маркировки или идентификации mGlu1 рецептора в биологическом материале либо in vivo, либо in vitro.-2 009334 В рамках настоящей заявки термин биологический материал означает любой материал, имеющий биологическое происхождение. В частности, он относится к образцам тканей, жидкостей плазмы, жидкостей организма, частей тела и органов, получаемых от теплокровных животных и от теплокровных животных как таковых (per se), в частности от человека. Основные эксперименты, которые проводили с применением системы мембранного анализа для маркировки или идентификации mGlu1-рецептора в биологическом материале, представляют собой: эксперименты по изучению насыщения, эксперименты по изучению ингибирования, эксперименты по изучению кинетики ассоциации и эксперименты по изучению кинетики диссоциации. Такие методики применимы для большинства рецепторных систем нейромедиаторов и гормонов, включая mGluR1-систему(Methods for Neurotransmitter Receptor Analysis, под редакцией Henry I.Yamamura и др., Raven Press Ltd.,Нью-Йорк, 1990). С этой целью меченное радиоактивным изотопом соединение вводят в биологический материал для маркировки mGlu1-рецепторов и регистрируют эмиссии меченного радиоактивным изотопом соединения, чтобы идентифицировать количество или локализацию mGlu1-рецепторов, например,путем авторадиографии рецептора ex vivo. Меченные радиоактивным изотопом соединения согласно изобретению, содержащие по меньшей мере один [3H]-атом или один [125I]-атом, также применяются в качестве агентов для скрининга, обладает ли тестируемое соединение способностью занимать сайт mGlu1-рецептора или связываться с ним. Степень, с которой тестируемое соединение заместит соединение согласно изобретению на сайте mGlu1 рецептора, покажет способность тестируемого соединения занимать сайт mGlu1-рецептора или связываться с ним и, следовательно, действовать в качестве либо агониста, антагониста, либо смешанного агониста/антагониста mGlu1-рецептора. Меченные радиоактивным изотопом соединения согласно изобретению, содержащие, по меньшей мере, один [3H]-атом или один [125I]- атом преимущественно получают путем замещения атома галогена атомом трития, как описано в экспериментальном разделе. Второе предпочтительное воплощение (радиоактивный изотоп, испускающий позитрон) При втором предпочтительном воплощении меченное радиоактивным изотопом соединение содержит, по меньшей мере, один радиоактивный атом углерода или галогена. В принципе, любое соединение формулы (I), содержащее атом углерода или галогена, подходит для введения радиоизотопного ядра путем замены атома углерода или галогена на подходящий радиоактивный изотоп или путем получения соединения формулы (I) с помощью меченных радиоактивными изотопами реагентов. Подходящие для этой цели радиоактивные изотопы галогенов представляют собой радиоактивный углерод, например,[11C]; радиоактивные йодиды, например [122I], [123I], [131I]; радиоактивные бромиды, например [75Br],[76Br], [77Br] и [82Br]; и радиоактивные фториды, например [18F]. Предпочтительные меченные радиоактивными изотопами соединения представляют собой такие соединения формулы (I-A) и (IB), в которых R1 содержит радиоактивный атом углерода или галогена, в частности [11C], [18F], [123I], [75Br], [76Br] или [77Br]. Получение радиоактивных соединений Введение радиоактивного атома галогена можно проводить с помощью подходящей реакции, такой как приведена ниже в которой все заместители в формуле (I-А-а) и (I-А-а) являются такими же, как в формуле (I-A), а галоген представляет собой атом галогена. Подходящее соединение (I-А-а) вступает в реакцию с H[18F] таким образом, что атом галогена, находящийся на хинолиновом кольце, заменяется путем реакции нуклеофильной замены на радиоактивный [18F]-атом. Для получения меченных радиоактивным изотопом соединений согласно формуле (I-B) введение радиоизотопного ядра можно проводить таким же способом, например, согласно схеме взаимодействия,представленной ниже. Очевидно, что соединения формулы (I-A) также можно получать таким способом, то есть с участием меченого заместителя R1. Другие способы введения меченых атомов трития описаны, например, в публикациях: Peng и др.Compounds and Radiopharmaceuticals 25 (12): 1361-1369 (1988). Введение радиоактивного [11C]-атома можно проводить согласно схеме взаимодействия, приведенной ниже, по которой подходящее соединение (I-А-а) сначала станнилируют (вводят станнильную группу), после чего вводят радиоактивный [11C], применяя, например реакцию конденсации "Stille-типа", катализируемую палладием с применением [11C]метилйодида (Scott W.J.; Crisp G. Т.; Stille J.К., J.Am.Chem. В формуле (I-A-a), (I-A-b) и (I-A-с) все заместители имеют такие же значения, как указано для формулы (I-A), галоген представляет собой атом галогена, и R17 представляет собой метил или бутил. Благодаря неожиданной способности легко проникать через гематоэнцефалический барьер меченные радиоактивным изотопом соединения, содержащие радиоактивный атом галогена, в составе соответствующей композиции применяют преимущественно in vivo для животного, особенно теплокровного животного, и регистрируют локализацию указанных меченных радиоактивным изотопом соединений с помощью методов визуализации, таких как, например, однофотонная эмиссионная компьютерная томография (Single Photon Emission Computered Tomography) (SPECT) или позитронно-эмиссионная томография (Positron Emission Tomography) (PET) и т.п. Таким способом можно регистрировать распределение сайтов mGlu1-рецепторов в организме, и с помощью упомянутых выше способов визуализации можно визуализировать органы, содержащие сайты mGlu1-рецепторов, такие как, например, мозг. Такой способ визуализации органа путем введения меченного радиоактивным изотопом соединения формулы (I-A) или (I-A), которое связывается с сайтами mGlu1-рецепторов, и регистрации эмиссии радиоактивного соединения, также составляет аспект настоящего изобретения. Применение соединений формулы (I-A) и (I-B) в описанных выше способах составляет еще один аспект настоящего изобретения. Изобретение, в частности, относится к применению соединений согласно изобретению для производства диагностического средства для применения в методе PET. Для применения в методе PET наиболее предпочтительными являются меченные радиоактивным изотопом соединения согласно изобретению, в которые входит 18F (патент США 4931270, Horn и др., опубликованный 5 июня 1990). Получение нерадиоактивных соединений Нерадиоактивные соединения согласно изобретению могут быть получены с помощью ряда способов. Чтобы упростить структурное представление некоторых присутствующих соединений и промежуточных соединений, в последующих методиках получения хинолиновая или хинолиноновая группа будут обозначаться далее символом Q. Соединения формулы (I-A) или (I-B), где X представляет собой О, обозначаются формулой (IA/B-а),указанные соединения могут быть получены окислением промежуточного соединения формулы (II) в присутствии подходящего окислителя, такого как перманганат калия, и подходящего катализатора фазового переноса, такого как трис(диокса-3,6-гептил)амин, в подходящем инертном по отношению к реакции растворителе, таком как, например, дихлорметан. Соединения формулы (IA/B-а) могут быть также получены при взаимодействии промежуточного соединения формулы (III) с промежуточным соединением формулы (IV), где W1 представляет собой атом галогена, например бром, в присутствии бутиллития и подходящего инертного по отношению к реакции растворителя, такого как, например, тетрагидрофуран. С другой стороны, соединения формулы (IA/B-а) могут быть также получены при взаимодействии промежуточного соединение формулы (V) с промежуточным соединением формулы (IV) в присутствии бутиллития и подходящего инертного по отношению к реакции растворителя, такого как, например, тетрагидрофуран. Соединения формулы (IA/B-а) , в которых заместитель R1 связан с карбонильной группой через атом кислорода [указанный заместитель R1 обозначается как -O-R1a, а указанные соединения формулой (IA/B-а 1)], могут быть получены при взаимодействии промежуточного соединения формулы (VI) с промежуточным соединением формулы (VII) в присутствии подходящей кислоты, такой как серная кислота. Соединения формулы (I-A), где R2 представляет собой метилкарбонил [указанные соединения обозначаются формулой (I-A-1)], могут быть получены при взаимодействии промежуточного соединения формулы (VIII) в присутствии подходящей кислоты, такой как соляная кислота, и подходящего инертного по отношению к реакции растворителя, такого как, например, тетрагидрофуран. Соединения формулы (I) можно также преобразовывать друг в друга посредством известных в данной области преобразований. Соединения формулы (I-A), где R2 представляет собой атом галогена, такой как хлор, могут быть преобразованы в соединение формулы (I-A), где R2 представляет собой другой атом галогена, такой как фтор или иод, путем взаимодействия с подходящим галогенирующим агентом, таким как, например фторид калия или иодид натрия, в присутствии подходящего инертного по отношению к реакции растворителя, например диметилсульфоксида или ацетонитрила, и, необязательно, в присутствии ацетилхлорида. Соединения формулы (I-A), где R2 представляет собой подходящую удаляемую группу, такую как атом галогена, например хлор, йод [указанная удаляемая группа обозначается как W2, а указанные соединения как (I-A-2)], могут быть преобразованы в соединение формулы (I-А), где R2 представляет собой циано [указанное соединение представлено формулой (I-A-3)], путем взаимодействия с подходящим агентом для ввода цианогруппы, таким как, например, триметилсиланкарбонитрил, в присутствии подходящего основания, такого как N,N-диэтилэтанамин, и подходящего катализатора, такого как, например, тетракис(трифенилфосфин)палладий. Соединения формулы (I-A-2) могут быть также преобразованы в соединения формулы (I-A-4) при взаимодействии с С 2-6-алкинилтри (C1-6-алкил)силаном в присутствии CuI, соответствующего основания,такого как, например, N,N-диэтилэтанамин, и соответствующего катализатора, такого как, например,тетракис(трифенилфосфин)палладий. В свою очередь, соединения формулы (I-A-4) могут быть преобразованы в соединения формулы (I-A-5) при взаимодействии с фторидом калия в присутствии подходящей кислоты, такой как уксусная кислота, или при взаимодействии с подходящим основанием, таким как гидроксид калия, в присутствии подходящего инертного по отношению к реакции растворителя, такого как спирт, например метанол и т.п. Соединения формулы (I-A-2) могут быть также преобразованы в соединения формулы (I-А-6) при взаимодействии с промежуточным соединением формулы (IX) в присутствии CuI, подходящего основания, такого как, например, N,N-диэтилэтанамин, и подходящего катализатора, такого как тетракис(трифенилфосфин)палладий. Соединения формулы (I-A-2) могут быть также преобразованы в соединение, где R2 представляет собой C1-6-алкил [указанное соединение представлено формулой (I-A-8)], в присутствии подходящего алкилирующего агента, такого как, например, Sn (C1-6-алкил)4, или в соединение, где R2 представляет-5 009334 собой С 2-6-алкенил [указанное соединение представлено формулой (I-A-9)], в присутствии подходящего алкенилирующего агента, такого как, например, Sn (С 2-6-алкенил) (С 1-6-алкил)3, оба вступают в реакцию в присутствии подходящего катализатора, такого как, например, тетракис(трифенилфосфин)палладий, и инертного по отношению к реакции растворителя, такого как, например, толуол или диоксан. Соединения формулы (I-A-2) могут быть также преобразованы в соединение формулы (I-A-7), где Z представляет собой О или S, при взаимодействии с промежуточным соединением формулы (X) необязательно в присутствии подходящего основания, такого как карбонат калия, и инертного по отношению к реакции растворителя, такого как N, N-диметилформамид. Соединения формулы (I-A-2) могут быть также преобразованы в соединение формулы (I-A), где R2 представляет собой С 1-6-алкилоксикарбонил [указанное соединение представлено формулой (I-A-10)], и соединение формулы (I-A), где R2 представляет собой водород [указанное соединение представлено формулой (I-A-11)], при взаимодействии с подходящим спиртом формулы С 1-6-алкил-ОН и СО в присутствии подходящего катализатора, такого как, например, ацетат палладия (II), трифенилфосфин, подходящего основания, такого как карбонат калия, и инертного по отношению к реакции растворителя, такого как N, N-диметилформамид. Соединения формулы (I-A-11) могут быть также получены при взаимодействии соединения формулы (I-A-2) с Zn в присутствии подходящей кислоты, такой как уксусная кислота. Соединения формулы (I-A-2) могут быть также преобразованы в соединение формулы (I-A), где R2 представляет собой аминокарбонил, замещенный С 1-6-алкилоксикарбонил-С 1-6-алкилом [указанное соединение представлено формулой (I-A-12)], при взаимодействии с промежуточным соединением формулы H2N-С 1-6-алкил-C(=O)-O-C1-6-алкил в присутствии СО, подходящего катализатора, такого как тетракис(трифенилфосфин)палладий, подходящего основания, такого как, например, N,N-диэтилэтанамин, и подходящего инертного по отношению к реакции растворителя, такого как, например, толуол. Соединения формулы (I-A-2) могут быть также преобразованы в соединение формулы (I-A), где R2 представляет собой арил или гетероцикл, выбранный из группы, описанной выше при определении R2[указанный R2 обозначается как R2a, а указанное соединение представлено формулой (I-A-13)], путем взаимодействия с промежуточным соединением формулы (XI), (XII) или (XIII) в присутствии подходящего катализатора, такого как, например, тетракис(трифенилфосфин)палладий, и подходящего инертно-6 009334 го по отношению к реакции растворителя, такого как, например, диоксан. Соединения формулы (I-A-2) могут быть также преобразованы в соединение формулы (I-В), где Y ИR5 объединены с образованием радикала формулы (b-1) или (b-2) [указанное соединение представлено формулой (I-B-1) или (I-B-2)], путем взаимодействия с гидразинкарбоксальдегидом или азидом натрия в подходящем инертном по отношению к реакции растворителе, таком как спирт, например бутанол, или Соединения формулы (I-A-11) могут быть преобразованы в соответствующий N-оксид, представленный формулой (I-A-14), путем взаимодействия с подходящим пероксидом, таким как 3-хлорбензолнадкарбоновая кислота, в подходящем инертном по отношению к реакции растворителе, таком как, например, метиленхлорид. Указанное соединение формулы (I-A-14) может быть далее преобразовано в соединение формулы (I-B), где R5 представляет собой водород [указанное соединение представлено формулой (I-B-3)], путем взаимодействия с 4-метилбензолсульфонилхлоридом в присутствии подходящего основания, такого как, например, дикарбонат калия, и подходящего инертного по отношению к реакции растворителя, такого как, например, метиленхлорид. Соединения формулы (I-B-3) могут быть также получены из соединения формулы (I-А), где R2 представляет собой С 1-6-алкилокси [указанное соединение представлено формулой (I-A-15)], путем взаимодействия с подходящей кислотой, такой как соляная кислота, в присутствии подходящего инертного по отношению к реакции растворителя, такого как, например,тетрагидрофуран. Соединения формулы (I-B-3) могут быть преобразованы в соединение формулы (I-B), где R5 представляет собой С 1-6-алкил [указанное соединение представлено формулой (I-B-4)], путем взаимодействия с соответствующим алкилирующим агентом, таким как, например промежуточное соединение формулы(XIV), где W3 представляет собой подходящую удаляемую группу, такую как атом галогена, например йод, в присутствии трет-бутилата калия и в присутствии подходящего инертного по отношению к реакции растворителя, такого как, например, тетрагидрофуран.-7 009334 Соединения формулы (I-B-3) могут быть также преобразованы в соединение формулы (I-B), где R5 представляет собой C1-6-алкилоксикарбонил-C1-6-алкил или арил-C1-6-алкил [указанный R5 обозначается как R5a, и указанное соединение представлено формулой (I-B-5)], путем взаимодействия с промежуточным соединением формулы (XV), где W4 представляет собой подходящую удаляемую группу, такую как атом галогена, например бром, хлор и т.п., в присутствии подходящего основания, такого как, например,гидрид натрия, и подходящего инертного по отношению к реакции растворителя, такого как, например,N,N-диметилформамид. Соединения формулы (I-A-2) может быть также преобразовано в соединение формулы (I-B), где R5 представляет собой водород, и Y представляет собой S [указанное соединение представлено формулой(I-B-6)], путем взаимодействия с H2N-C(=S)-NH2 в присутствии подходящего основания, такого как гидроксид калия, и подходящего инертного по отношению к реакции растворителя, такого как спирт, например этанол, или вода. Соединение формулы (I-B-6) может быть далее преобразовано в соединение формулы (I-A), где R2 представляет собой С 1-6-алкилтио [указанное соединение представлено формулой(I-A-16)], путем взаимодействия с подходящим С 1-6-алкилгалогенидом, таким как, например, С 1-6 алкилиодид, в присутствии подходящего основания, такого как карбонат калия, и подходящего растворителя, такого как, например, ацетон. Соединения формулы (IA/B-а) могут быть преобразованы в соединения формулы (I-A) или (I-B), гдеX представляет собой N-R7 [указанное соединение представлено формулой (IА/В-b)], путем взаимодействия с промежуточным соединением формулы (XVI) необязательно в присутствии подходящего основания, такого как, например, N,N-диэтилэтанамин, и в присутствии подходящего инертного по отношению к реакции растворителя, такого как спирт, например этанол. Как уже указывалось в методике получения описанных выше соединений формулы (I-A-13), соединения формулы (I) могут быть также преобразованы в соответствующие N-оксидные формы посредством известных в данной области процедур преобразования трехвалентного азота в его N-оксидную форму. Указанную реакцию N-окисления можно в общем случае проводить путем взаимодействия исходного соединения формулы (I) с соответствующим органическим или неорганическим пероксидом. Соответствующие неорганические пероксиды включают, например, перекись водорода, пероксиды щелочных металлов или щелочно-земельных металлов, например пероксид натрия, пероксид калия; соответствующие органические пероксиды могут включать надкислоты, такие как, например, бензолнадкарбоновая кислота или галогензамещенная бензолнадкарбоновая кислота, например 3-хлорбензолнадкарбоновая кислота,надалкановые кислоты, например надуксусная кислота, алкилгидропероксиды, например третбутилгидропероксид. Подходящими растворителями являются, например, вода, низшие спирты, например этанол и т.п.,углеводороды, например толуол, кетоны, например 2-бутанон, галогенированные углеводороды, например дихлорметан, и смеси таких растворителей. Некоторые из промежуточных соединений и исходных продуктов, применяемых в описанных выше методиках, имеются в продаже или могут быть синтезированы согласно методикам, уже описанным в литературе. Промежуточные соединения формулы (II) могут быть получены путем взаимодействия промежуточного соединения формулы (XVII) с промежуточным соединением формулы (XVIII), где W5 представ-8 009334 ляет собой подходящую удаляемую группу, такую как атом галогена, например хлор, бром и т.п., в присутствии магния, простого диэтилового эфира и подходящего инертного по отношению к реакции растворителя, такого как простой диэтиловый эфир. Промежуточные соединения формулы (XVII) могут быть получены окислением промежуточного соединения формулы (XIX) в присутствии подходящего окислителя, такого как MnО 2, и подходящего инертного по отношению к реакции растворителя, такого как метиленхлорид. Промежуточные соединения формулы (XIX) могут быть получены восстановлением промежуточного соединения формулы (XX) в присутствии подходящего восстановителя, такого как алюмогидрид лития, и подходящего инертного по отношению к реакции растворителя, такого как тетрагидрофуран. Промежуточные соединения формулы (XX), где Q представляет собой хинолиновую группу, необязательно замещенную в положении 3 С 1-6-алкилом, и где карбонильная группа находится в положении 6[указанные промежуточные соединения представлены формулой (XX-а)], могут быть получены путем взаимодействия промежуточного соединения формулы (XXI) с промежуточным соединением формулы(XXII) в присутствии 3-нитробензолсульфоната натрия, подходящей кислоты, такой как серная кислота,и подходящего спирта, например метанола, этанола, пропанола, бутанола и т.п. С другой стороны, промежуточные соединения формулы (II) могут быть также получены путем взаимодействия промежуточного соединения формулы (XXIII) с промежуточным соединением формулы(XXIV), где W6 представляет собой подходящую удаляемую группу, такую как атом галогена, например бром, хлор и т.п., в присутствии подходящего агента, такого как бутиллитий, и подходящего инертного по отношению к реакции растворителя, такого как тетрагидрофуран. Промежуточные соединения формулы (XXIII) могут быть получены окислением промежуточного соединения формулы (XXV) с помощью окисления Пфицнера-Моффата или Сверна (аддукты диметилсульфоксида с дегидратирующими агентами, например, DCC, Ac2O, SO3, P4O10, COCl2 или Cl-CO-COCl) в инертном растворителе, таком как метиленхлорид. Промежуточные соединения формулы (XXV) могут быть получены восстановлением промежуточного соединения формулы (XXVI) в присутствии подходящего восстановителя, такого как, например,алюмогидрид лития, и подходящего инертного по отношению к реакции растворителя, такого как бензол. Промежуточные соединения формулы (XXVI) могут быть получены из промежуточного соединения формулы (XXVII) этерификацией в присутствии подходящего спирта, такого как метанол, этанол,пропанол, бутанол и т.п., и подходящей кислоты, такой как серная кислота. Промежуточные соединения формулы (XXVII), где R1 представляет собой радикал формулы (а-1),где Z1 представляет собой О, Z2 представляет собой CH2, и n равным 1 [указанные промежуточные соединения представлены формулой (XXVII-a)], могут быть получены восстановлением промежуточного соединения формулы (XXVIII) в присутствии подходящего восстановителя, такого как водород, и подходящего катализатора, такого как палладий на активированном угле, и подходящей кислоты, такой как уксусная кислота. Когда R1 промежуточного соединения (XXVII) представляет собой необязательно замещенную фенильную группу, ее можно также преобразовывать в необязательно замещенную циклогексильную группу путем восстановления в присутствии подходящего восстановителя, такого как родий наAl2O3, и подходящего инертного по отношению к реакции растворителя, такого как тетрагидрофуран. Промежуточные соединения формулы (IV), где Q представляет собой хинолиновую группу, замещенную в положении 2 галогеном, например хлором [указанные промежуточные соединения представлены формулой (IV-a)], могут быть получены путем взаимодействия промежуточного соединения формулы (IV), где Q представляет собой хинолиноновую группу, где R5 представляет собой водород [указанное промежуточное соединение представлено формулой (IV-b)], в присутствии POCl3. Промежуточные соединения формулы (IV-a), где R4 представляет собой водород [указанные промежуточные соединения представлены формулой (IV-a-1)], могут быть также получены путем взаимодействия промежуточного соединения формулы (XXIX) с POCl3 в присутствии N, N-диметилформамида Промежуточные соединения формулы (XXIX) могут быть получены путем взаимодействия промежуточного соединения формулы (XXX) с промежуточным соединением формулы (XXXI), где W7 представляет собой подходящую удаляемую группу, такую как атом галогена, например хлор, в присутствии подходящего основания, такого как, например, N,N-диэтилэтанамин, и подходящего инертного по отношению к реакции растворителя, такого как метиленхлорид. Промежуточные соединения формулы (IV-a) могут быть преобразованы в промежуточное соединение формулы (IV-c) путем взаимодействия с промежуточным соединением формулы (XXXII) в присутствии подходящего инертного по отношению к реакции растворителя, такого как спирт, например метанол и т.п. Промежуточные соединения формулы (IV-a) могут быть также преобразованы в промежуточное соединение формулы (IV-d-1) путем взаимодействия с подходящим амином формулы (XXXIII-а), где каждый из Z3 и Z4 независимо представляет собой водород, С 1-6-алкил, С 1-6-алкилокси-С 1-6-алкил, C1-6 алкилтио- C1-6-алкил, или в промежуточное соединение формулы (IV-d-2) путем взаимодействия с подходящим амином формулы (XXXIII-b), где Z3 и Z4 объединены с образованием гетероцикла, как описано выше в определении R2, при условии, что гетероцикл содержит, по меньшей мере, один атом азота, в присутствии подходящего основания, такого как, например, дикарбонат калия, и инертного по отношению к реакции растворителя, такого как N,N-диметилформамид. Промежуточные соединения формулы (IV-a), где R3 представляет собой CH2-CH2-CH2-Cl [указанные промежуточные соединения представлены формулой (IV-a-2)], могут быть также преобразованы в промежуточное соединение формулы (IV), где R2 и R3 объединены с образованием двухвалентного радикала формулы -O-CH2-CH2-CH2-[указанное промежуточное соединение представлено формулой (IV-e1)], путем взаимодействия с подходящей кислотой, такой как соляная кислота и т.п. Промежуточные соединения формулы (IV-a-2) также могут быть преобразованы в промежуточное соединение формулы (IV), где R2 и R3 объединены с образованием двухвалентного радикала формулы -SCH2-CH2-CH2-[указанное промежуточное соединение представлено формулой (IV-e-2)], путем взаимодействия с H2N-C(=S)-NH2 в присутствии подходящего инертного по отношению к реакции растворителя, такого как спирт, например этанол. Промежуточные соединения формулы (V) могут быть получены путем взаимодействия промежуточного соединения формулы (XXVII) с промежуточным соединением формулы CH3-NH-O-CH3 в присутствии 1,1'-карбонилдиимидазола и подходящего инертного по отношению к реакции растворителя,такого как метиленхлорид. Промежуточные соединения формулы (VII), где Q представляет собой хинолиновую группу, в частности, хинолиновую группу, где R2 представляет собой этил, R3 представляет собой метил и R4 представляет собой водород, а карбоксильная группа находится в положении 6 [указанные промежуточные соединения представлены формулой (VII-а)], могут быть получены путем взаимодействия промежуточ- 11009334 ного соединения формулы (XXXIV) в присутствии подходящего альдегида, такого как СН 3-СН 2-СН (=O),(CH2O)n, ZnCl2, FeCl3, и подходящего инертного по отношению к реакции растворителя, такого как спирт, например этанол. Промежуточные соединения формулы (VIII) могут быть получены путем взаимодействия промежуточного соединения формулы (XXXV) с промежуточным соединением формулы (XXXVI) в присутствии подходящего катализатора, такого как, например, тетракис(трифенилфосфин)палладий и подходящего инертного по отношению к реакции растворителя, такого как диоксан. Кроме того, можно разработать некоторые другие способы получения, некоторые из которых описаны далее в примерах настоящей заявки. С помощью известных в данной области методик могут быть получены стереоизомерно чистые формы соединений и промежуточных соединений настоящего изобретения. Диастереомеры можно разделить с помощью физических способов разделения, таких как селективная кристаллизация, и хроматографических методов, например с помощью жидкостной хроматографии с применением хиральных стационарных фаз. Энантиомеры можно отделить друг от друга путем селективной кристаллизации солей их диастереомеров с оптически активными кислотами. С другой стороны, энантиомеры можно разделить хроматографическими методами с применением хиральных стационарных фаз. Указанные стереоизомерно чистые формы могут быть также получены из соответствующих стереоизомерно чистых форм соответствующих исходных продуктов при условии, что реакция происходит стереоселективно или стереоспецифически. Если требуется определенный стереоизомер, предпочтительно синтезировать указанное соединение с помощью стереоселективных или стереоспецифических способов синтеза. При таких способах применяют преимущественно хирально чистые исходные продукты. Очевидно, что стереоизомерные формы соединений формулы (I) включены в объем изобретения. Стереоизомер соединения формулы (I-А) или (I-В), соответствующий цис-форме, может быть преобразован в другой стереоизомер, соответствующий транс-форме, путем взаимодействия соединения с подходящей кислотой, такой как соляная кислота, в присутствии подходящего инертного по отношению к реакции растворителя, такого как, например тетрагидрофуран. Антагонистическую активность настоящих соединений по отношению к mGluR1 можно продемонстрировать при проведении теста по передаче сигнала клонированных крысиных mGluR1 в СНОклетках, и теста на холодовую аллодению (болевого теста) у крыс с лигатурой Беннетта, как описано в дальнейшем. Благодаря их антагонистической активности по отношению к mGluR, более конкретно, их антагонистической активности по отношению к mGluR группы I, и еще более конкретно, их антагонистической активности по отношению к mGluR1, соединения формулы (I-A) или (I-B), их N-оксиды, фармацевтически приемлемые аддитивные соли, четвертичные амины и стереохимически изомерные формы применимы для лечения или профилактики индуцированных глутаматом заболеваний центральной нервной системы. Заболевания, в которых установлена роль глутамата, включают лекарственное привыкание (наркоманию) или абстиненцию (зависимость, толерантность к опиоидам, воздержание от опиоидов), гипоксические, аноксические и ишемические повреждения (ишемический удар, остановку сердечной деятельности), боль (невропатическую боль, боль при воспалении, гипералгезию), гипогликемию, заболевания,связанные с неврональным повреждением, травму мозга, травму головы, повреждение спинного мозга,миелопатию, деменцию, тревогу, шизофрению, депрессию, ослабление когнитивной деятельности, амнезию, биполярные расстройства, расстройства поведения, болезнь Альцгеймера, сосудистую деменцию,смешанную (Альцгеймера и сосудистую) деменцию, заболевание тельца Леви, бредовое состояние или спутанность сознания, болезнь Паркинсона, болезнь Хантингтона, синдром Дауна, эпилепсию, старение,амиотрофический латеральный склероз, рассеянный склероз, СПИД (синдром приобретенного иммунодефицита) и СПИД-ассоциированный комплекс (ARC). Настоящее изобретение также относится к композициям, содержащим терапевтически эффективное количество меченого изотопом соединения формулы (I-A) или (I-B) и фармацевтически приемлемый носитель или разбавитель, для введения животным, в частности, человеку, в частности, в связи с диагно- 12009334 стикой, более конкретно, для визуализации органа. Следовательно, соединения по настоящему изобретению или любой их подгруппы для введения можно получать в различных фармацевтических формах. В качестве соответствующих композиций можно упомянуть все композиции, обычно используемые для системного введения лекарственных средств. Для получения фармацевтических композиций согласно настоящему изобретению терапевтически эффективное количество конкретного соединения, в форме основания или аддитивной соли, в виде активного ингредиента объединяют при тщательном перемешивании с фармацевтически приемлемым носителем, который можно выбрать в зависимости от формы препарата, необходимого для введения, из большого разнообразия форм. Желательно, чтобы такие фармацевтические композиции находились в виде стандартной лекарственной формы, предпочтительно для перорального, ректального, местного введения,через кожу или с помощью парентеральной инъекции. Например, в случае жидких препаратов для орального введения, таких как суспензии, сиропы, эмульсии, эликсиры и растворы, для получения композиции в дозированной форме для перорального введения можно использовать любую обычно применяемую фармацевтическую среду, такую как, например, вода, гликоли, масла, спирты и т.п.; или в случае порошков, пилюль, капсул и таблеток можно использовать твердые носители, такие как крахмалы, сахара, каолин, смазки, связующие компоненты, дезинтеграторы и т.п. Благодаря простоте введения таблетки и капсулы представляют собой наиболее выгодную стандартную лекарственную форму для перорального введения, в которой, безусловно, используются твердые фармацевтические носители. Носитель для парентеральной композиции обычно будет содержать стерилизованную воду, по меньшей мере, в значительной степени, хотя могут быть включены и другие ингредиенты, например вспомогательное средство для растворимости. Например, могут быть получены растворы для инъекции, в которых носитель содержит физиологический раствор, раствор глюкозы или смесь раствора глюкозы и физиологического раствора. Можно также получить суспензии для инъекций, в которых можно использовать соответствующие жидкие носители, суспендирующие агенты и т.п. Также включены препараты в твердой форме, которые незадолго перед использованием переводятся в препараты в жидкой форме. В качестве соответствующих композиций для местного нанесения можно упомянуть все композиции, обычно используемые для местного введения лекарственных средств, например кремы, гель, повязки, шампуни, тинктуры, пасты, мази,бальзамы, порошки и т.п. В композициях, подходящих для введения через кожу, носитель необязательно содержит агент, усиливающий проникновение, и/или подходящее смачивающее вещество, необязательно объединенное в незначительных пропорциях с подходящими добавками любой природы, которые не оказывают значительного вредного воздействия на кожу. Указанные добавки могут способствовать введению через кожу и/или могут быть полезны для получения требуемых композиций. Такие композиции можно вводить различными способами, например в виде трансдермального пластыря, в виде точечных мазков, в виде мази. Для простоты введения и равномерности дозировки особенно удобно получать вышеупомянутые фармацевтические композиции в виде стандартной лекарственной формы. Применяемая здесь в описании и формуле изобретения стандартная лекарственная форма относится к физически дискретным формам, подходящим в качестве стандартных лекарственных форм, каждая единица которых содержит определенное количество активного ингредиента, рассчитанного для получения требуемого терапевтического эффекта, совместно с необходимым фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (включая таблетки с шероховатой поверхностью или таблетки,покрытые оболочкой), капсулы, пилюли, суппозитории, упаковки порошков, облатки, растворы или суспензии для инъекции, дозировки в виде чайной ложки лекарства, столовой ложки и т.п. и сегрегированных кратных количеств. Как известно специалисту в данной области, эффективная диагностическая доза или частота введения зависят от конкретного применяемого соединения формулы (I-A) или (I-B) и конкретного состояния животного, которое подвергается лечению. Следующие примеры предназначены для иллюстрации и не ограничивают объем настоящего изобретения. Экспериментальная часть В дальнейшем N,N-диметилформамид обозначается как ДМФ, простой диизопропиловый эфир обозначается как DIPE, диметилсульфоксид обозначается как ДМСО, 2, 6-бис-(1,1-диметилэтил)-4 метилфенол обозначается как BHT и тетрагидрофуран обозначается как ТГФ. Получение промежуточных соединений Пример A1. Смесь 4-(1-метилэтокси)бензойной кислоты (0,083 моль) и Rh/Al2O3 5% (10 г) в ТГФ(220 мл) подвергают гидрированию при 50C (при давлении H2 3 бар) в течение 1 ночи. Смесь фильтруют через целит, промывают ТГФ и упаривают. Выход: 16 г промежуточного соединения 1 (100%).- 13009334 Пример А 2. Получение 2-этил-3-метил-6-хинолинкарбоновой кислоты (промежуточное соединение 2). Смесь 4-аминобензойной кислоты (0,299 моль) в этаноле (250 мл) перемешивают при комнатной температуре. Добавляют ZnCl2 (0,0367 моль) и (CH2O)n (10 г). По частям добавляют FeCl3 6H2O (0,5 моль) и повышают температуру до 60-65C. По каплям в течение более 2 ч добавляют пропаналь (30 мл). Смесь кипятят с обратным холодильником в течение 2 ч и сохраняют при комнатной температуре в течение 12 ч. Смесь выливают в воду и фильтруют через целит. Фильтрат подкисляют до рН=7 с помощью 6 н HCl и смесь упаривают досуха. Остаток применяют без дальнейшей очистки. Выход: 56,1 г 2-этил-3-метил-6 хинолинкарбоновой кислоты (промежуточное соединение 2). Пример A3. К смеси 4-бромбензоламина (0,232 моль) и N,N-диэтилэтанамина (0,2784 моль) в CH2Cl2 (400 мл) при 5 С добавляют пентаноилхлорид (0,27 84 моль). Смесь перемешивают при комнатной температуре в течение ночи, выливают в воду и экстрагируют CH2Cl2. Органический слой отделяют, промывают концентрированным раствором NH4OH и водой, сушат (MgSO4) , фильтруют и выпаривают растворитель. Остаток (60 г) кристаллизуют из диэтилового эфира. Осадок отфильтровывают и сушат. Остаток (35 г,63%) переносят в CH2Cl2. Органический слой отделяют, промывают 10%-ным раствором К 2 СО 3, промывают водой, сушат (MgSO4) , фильтруют и выпаривают растворитель. Выход: 30 г промежуточного соединения (3) (54%). Пример А 4. Смесь 6-бром-2(1H)-хинолинона (0,089 моль) в POCl3 (55 мл) перемешивают при 60C в течение ночи,затем при 100C в течение 3 ч и выпаривают растворитель. Остаток переносят в CH2Cl2, выливают в воду со льдом, подщелачивают концентрированным NH4OH, фильтруют через целит и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Выход: 14,5 г промежуточного соединения (4) (67%). Пример А 5. К POCl3 (108 мл) при 10C в токе N2 добавляют по каплям ДМФ (37 мл) . После завершения добавления смеси дают нагреться до комнатной температуры. По частям добавляют N-(4-бромфенил)бутанамид(0,33 моль). Смесь перемешивают при 85 С в течение ночи, затем дают охладиться до комнатной температуры и выливают на лед (экзотермическая реакция). Осадок отфильтровывают, промывают небольшим количеством воды и сушат (вакуум). Остаток промывают EtOAc/диэтиловым эфиром и сушат. Выход: 44,2 г промежуточного соединения (5) (50%). Смесь промежуточного соединения (5) (0,162 моль) в метаноле (600 мл) и раствора метилата натрия(метанолнатриевой соли) в метаноле при концентрации 35% (154 мл) перемешивают и кипятят с обратным холодильником в течение ночи. Смесь выливают на лед. Осадок отфильтровывают, промывают небольшим количеством воды и переносят в CH2Cl2. Добавляют 10%-ный К 2 СО 3 и смесь экстрагируют К смеси 4-метоксициклогексанкарбоновой кислоты (0,063 моль) в CH2Cl2(200 мл) по частям добавляют 1,1'-карбонил-бис-1H-имидазол (0,07 4 моль). Смесь перемешивают при комнатной температуре в течение 1 ч. Затем добавляют N-метоксиметанамин (0,074 моль). Смесь перемешивают при комнатной температуре в течение ночи, выливают в Н 2 О и экстрагируют CH2Cl2. Органический слой отделяют,промывают несколько раз Н 2 О, сушат (MgSO4), фильтруют и выпаривают растворитель. Выход: 12,6 г промежуточного соединения 7.(400 мл) гидрируют в присутствии Pd/C (3 г) в качестве катализатора. После поглощения Н 2 (3 экв.) катализатор отфильтровывают. Фильтрат упаривают. Остаток перемешивают в петролейном эфире. Осадок отфильтровывают и сушат (вакуум; 70C). После перекристаллизации из CHCl3/СН 3 ОН осадок отфильтровывают и сушат (вакуум; 80C и высокий вакуум; 85 С). Выход: 8,8 г 6-фтор-3,4-дигидро-2H-1-бензопиран-2-карбоновой кислоты (промежуточное соединение 8) (15,0%).b) Смесь промежуточного соединения (8) (0,255 моль) в этаноле (400 мл) и H2SO4 (5 мл) перемешивают и кипятят с обратным холодильником в течение 8 ч. Выпаривают растворитель досуха. Остаток растворяют (разжижают) в CH2Cl2. Органический слой отделяют, промывают 10%-ным K2 СО 3, сушатc) Реакция в атмосфере N2. Смесь 70%-ного по массе раствора бис-(2-метоксиэтокси)алюмогидрида натрия в метилбензоле 3,4 M (0,44 моль) и бензола (150 мл) (кипячение с обратным холодильником) добавляют по каплям в течение 1 часа к кипящей с обратным холодильником смеси промежуточного соединения 9 (0,22 моль) и бензола (600 мл). После перемешивания в течение 2,5 ч при температуре флегмы смесь охлаждают до 15 С. Смесь растворяют (разлагают), добавляя по каплям этанол (30 мл) и воду(10 мл). Полученную смесь выливают в лед/воду и полученную смесь подкисляют концентрированной соляной кислотой. Полученную смесь экстрагируют диэтиловым эфиром (500 мл). Отделенный органический слой промывают водой, сушат, фильтруют и выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (элюент: CHCl3). Отбирают нужную фракцию и выпаривают элюент. Выход: 34 г 6-фтор-3,4-дигидро-2 Н-1-бензопиран-2-метанол (промежуточное соединение 10) (85%).d) Реакция в атмосфере N2. К смеси этандиоилдихлорида (0,1 моль) в CH2Cl2 (350 мл) при перемешивании и охлаждении (-60C; на бане 2-пропанон/CO2) в течение 10 мин добавляют сульфинил-бис[метан] (30 мл). После перемешивания в течение 10 мин добавляют смесь промежуточного соединения 10 в CH2Cl2 (90 мл) в течение 5 мин. После перемешивания в течение 15 мин добавляют N,Nдиэтилэтанамин (125 мл). Когда смесь нагреется до комнатной температуры, ее выливают в воду. Продукт экстрагируют CH2Cl2. Органический слой промывают водой, HCl (1M), водой, NaHCO3(10%) и водой, сушат и упаривают. Остаток разжижают (растворяют) в диэтиловом эфире, промывают водой, сушат, отфильтруют и упаривают. Остаток очищают колоночной хроматографией на силикагеле (элюент:CHCl3). Нужную фракцию отбирают и элюент выпаривают. Выход: 21,6 г 6-фтор-3,4-дигидро-2 Н-1 бензопиран-2-карбоксальдегида (промежуточное соединение 11) (67%). 1,6 M н-бутиллитий (0,056 моль) медленно добавляют при -70C к раствору промежуточного соединения(5) (0,046 моль) в ТГФ (100 мл). Смесь перемешивают при -70C в течение 30 мин. Медленно добавляют суспензию промежуточного соединения 11 (0,056 моль) в ТГФ (100 мл). Смесь перемешивают при -70C в течение 1 ч, затем доводят температуру до комнатной, выливают в H2O и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (21 г) очищают колоночной хроматографией на силикагеле (элюент:циклогексан/EtOAc 80/10; 15-35 мкм). Очищенные фракции отбирают и выпаривают растворитель. Выход: 9,5 г промежуточного соединения 12 (55%). Пример А 8 Смесь промежуточного соединения (5) (0,1127 моль), 2-метоксиэтанамина (0,2254 моль) и K2CO3(0,2254 моль) в ДМФ (500 мл) перемешивают при 120C в течение 15 ч и затем охлаждают. Выпаривают растворитель. Остаток переносят в CH2Cl2 и H2O. Органический слой отделяют, сушат (MgSO4), фильтруют и досуха выпаривают растворитель. Остаток (33,53 г) очищают колоночной хроматографией на силикагеле (элюент: СН 2 С 12/СН 3 ОН 99,5/0,5; 15-40 мкм). Две фракции отбирают и выпаривают из них растворители. Выход: 5,7 г промежуточного соединения 14 (38%) и промежуточного соединения 13- 15009334 Смесь промежуточного соединения (5) (0,0751 моль), тиоморфолина (0,0891 моль) и K2CO3 (0,15 моль) в ДМФ (200 мл) перемешивают при 120C в течение 12 ч. Выпаривают растворитель досуха. Остаток переносят в CH2Cl2 и H2O. Органический слой отделяют, сушат (MgSO4) , фильтруют и выпаривают растворитель. Остаток (26 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 80/20; 20-45 мкм). Две фракции отбирают и выпаривают из них растворители. Две полученные фракции объединяют. Выход: 9,4 г промежуточного соединения 15 (37%); т.пл. 82 С. Пример А 9.(0,118 моль) в 70%-ной H2SO4 (230 мл) и кипятят смесь с обратным холодильником при перемешивании. Добавляют по каплям 2-пропен-1,1-диол-2-метилдиацетат (0,216 моль) и кипятят смесь с обратным холодильником в течение 4 ч. Добавляют этанол (200 мл) и перемешивают смесь при 80C в течение 48 ч. Смесь упаривают, остаток выливают в воду со льдом/NH4 ОН и экстрагируют CH2Cl2. Органический слой сушат (MgSO4) и упаривают. Остаток очищают колоночной хроматографией на силикагеле (элюент:CH2Cl2/2-пропанол 99/1). Очищенные фракции отбирают и упаривают. Выход: 21 г этил-3-метил-6 хинолинкарбоксилата (промежуточное соединение 16) (45%).LiAlH4 (0,098 моль) в ТГФ в атмосфере N2. Когда добавление завершится, добавляют воду (10 мл). Осадок отфильтровывают и промывают CH2Cl2. Органический слой сушат (MgSO4), фильтруют и упаривают. Продукт применяют без дальнейшей очистки. Выход: 16,71 г 3-метил-6-хинолинметанол (промежуточное соединение 17).c) К раствору промежуточного соединения 17 (0,0 96 моль) в CH2Cl2 (200 мл) добавляют MnO2(0,237 моль) и смесь перемешивают при комнатной температуре в течение 12 ч. Смесь фильтруют через целит и фильтрат снова перемешивают с MnO2 (20 г) в течение 12 ч. Снова добавляют MnO2 (10 г) . Смесь перемешивают в течение 12 ч. Смесь фильтруют через целит и упаривают. Продукт применяют без дальнейшей очистки. Выход: 11,71 г 3-метил-6-хинолинкарбоксальдегида (71%) (промежуточное соединение 18).(50 мл) добавляют при 10C к смеси ТГФ (0,14 моль) в 1,1'-окси-бис-этане (10 мл). Осторожно добавляют раствор промежуточного соединения 18 (0,07 моль) в присутствии Mg-стружки (100 мл) при 5 С, смесь выливают в воду со льдом и экстрагируют EtOAc. Выход: 11,34 г циклогексил-3-метил-6 хинолинметанола (63%) (промежуточное соединение 19). Пример A10.(0,000151 моль) в 1,4-диоксане (5 мл) перемешивают при 80C в течение 3 ч. Добавляют воду. Смесь экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Полученный продукт применяют без дальнейшей очистки. Выход: 1,4 г промежуточного соединения 20. Пример A11.(1,7 мл) перемешивают при комнатной температуре в течение ночи, затем выливают в Н 2 О, подкисляют 3 н. HCl и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток переносят в простой диэтиловый эфир. Осадок отфильтровывают и сушат. Выход: 0,26 г промежуточного соединения 21 (56%). (T.пл.: 232 С) Пример A12.- 16009334 Смесь 5-бром-1H-индол-2,3-диона(0,221 моль) в 3 н. NaOH (500 мл) перемешивают при 80C в течение 30 мин, доводят температуру до комнатной и добавляют 2-пентанон (0,221 моль). Смесь перемешивают и кипятят с обратным холодильником в течение 1 ч 30 мин и подкисляют AcOH до рН=5. Осадок отфильтровывают, промывают водой и сушат. Выход: 52,3 г промежуточного соединения 22 и промежуточного соединения 23. (Общий выход: 80%). 1,6 M н-BuLi (0,0816 моль) добавляют по каплям при -78 С к суспензии промежуточного соединения 22 (0,034 моль) и промежуточного соединения 23 (0,034 моль) в ТГФ (300 мл) в токе N2. Смесь перемешивают при -78C в течение 30 мин. Добавляют по каплям 1,6 M H-BuLi (0,0816 моль). Смесь перемешивают в течение 1 ч. Медленно добавляют смесь промежуточного соединения 9 (0,102 моль) в ТГФ(250 мл). Смесь перемешивают в интервале температур от -78 до -20C, выливают в смесь Н 2 О/HCl (3 н) и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель досуха. Выход: 20,89 г промежуточного соединения 2 4 и промежуточного соединения 25 (86%). Пример A13. К раствору 3-хлор-2-этил-2-бутеналя (0,065 моль) в AcOH (100 мл) при комнатной температуре по частям добавляют 4-амино-3-метоксибензойную кислоту (0,054 моль). Смесь перемешивают, кипятят с обратным холодильником в течение 8 часов и упаривают досуха. Остаток переносят в CH2Cl2, добавляют воду и подщелачивают раствор с помощью Et3N. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток кристаллизуют из 2-пропанона. Осадок отфильтровывают и сушат. Выход: 2,5 г промежуточного соединения 2 6 (18%). К раствору промежуточного соединения 26 (0,011 моль) в CH2Cl2 (30 мл) при комнатной температуре добавляют CDI (0,012 моль). Смесь перемешивают при комнатной температуре в течение 1 ч. Добавляют метоксиаминометил (0,012 моль) и перемешивают смесь при комнатной температуре в течение 8 ч. Добавляют H2O. Осадок отфильтровывают. Фильтрат экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4) , фильтруют и выпаривают растворитель. Остаток перекристаллизовывают из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,95 г промежуточного соединения 27 (31%) (т.пл.: 148 С). Пример A14. К раствору 3-хлор-2-этил-2-бутаналя (0,041 моль) в AcOH (60 мл) добавляют при комнатной температуре 4-бромбензоламик (0,034 моль). Смесь перемешивают и кипятят с обратным холодильником в течение 8 ч, доводят до комнатной температуры и упаривают досуха. Продукт кристаллизуют из EtOAc. Осадок отфильтровывают, промывают 10%-ным K2 СО 3 и переносят в CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Выход: 4,6 г промежуточного соединения 28 (54%). Пример A15. К раствору 1,3-циклогександиона (0,2 68 моль) в H2O (150 мл) медленно добавляют при 5 С растворKOH (0,326 моль) в H2O (150 мл). Температура не должна превышать 12C. Добавляют KI (2 г), затем по частям 2-бром-1-(4-нитрофенил)этанон (0,294 моль). Смесь перемешивают при комнатной температуре в течение 48 ч. Осадок отфильтровывают, промывают H2O, затем простым диэтиловым эфиром и сушат.- 17009334 Выход: 63 г (85%). Часть полученной фракции (1 г) кристаллизуют из EtOH. Осадок отфильтровывают и сушат. Выход: 0,5 г промежуточного соединения 29 (42%) (т.пл.: 100C). Смесь промежуточного соединения 29 (0,145 моль) и H2SO4 (40 мл) перемешивают при комнатной температуре в течение 1 ч, выливают на лед, подщелачивают NH4OH и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток кристаллизуют изEtOH. Осадок отфильтровывают и сушат. Выход: 31 г (58%). Часть полученной фракции (1 г) кристаллизуют из EtOH. Осадок отфильтровывают и сушат. Выход: 0,7 г промежуточного соединения 30 (58%) Смесь промежуточного соединения 30 (0,039 моль) и никеля Ренея (10 г) в EtOH (100 мл) гидрируют в течение 1 ч при комнатной температуре под давлением 3 бар. Смесь фильтруют через целит и промывают CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (9,5 г) кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 4,6 г (52%). Фильтрат упаривают. Остаток (2,7 г) очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH; 99/1; 15-40 мкм) . Отбирают две фракции и выпаривают растворитель. Выход: 1,6 г F1 и 1,2 г F2. F2 кристаллизуют из EtOH. Осадок отфильтровывают и сушат. Выход: 0,24 г промежуточного соединения 31 (2%) (т.пл.: 202C). К раствору 3-хлор-2-этил-2-бутеналя (0,04 моль) в AcOH (50 мл) добавляют при комнатной температуре промежуточное соединение 30 (0,02 моль). Смесь перемешивают и кипятят с обратным холодильником в течение 4 ч. Растворитель выпаривают досуха. Остаток кристаллизуют из EtOAc. Осадок отфильтровывают и сушат. Остаток переносят в CH2Cl2. Смесь подщелачивают 10%-ным K2 СО 3 и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток кристаллизуют из EtOH. Осадок отфильтровывают и сушат. Выход: 2,5 г промежуточного соединения 32 (40%). Пример A16. Смесь 2-(4-нитрофенил)-1-фенилэтанона (0,083 моль) и никеля Ренея (20 г) в EtOH (200 мл) гидрируют при комнатной температуре в течение 1 ч под давлением 3 бар, затем фильтруют через целит, промывают CH2Cl2/CH3OH и сушат. Выход: 17,5 г промежуточного соединения 33 (97%). Пример A17. К POCl3 (0,7536 моль) добавляют по каплям при 5 С ДМФ (12,4 мл). Добавляют 4'-бром-5 хлорвалеранилид (0,1032 моль) и перемешивают смесь при 75C в течение 6 ч, охлаждают до комнатной температуры и выливают в воду со льдом. Нерастворимое вещество отфильтровывают, промывают водой и сушат. Выход: 25,7 г промежуточного соединения 34 (78%). Смесь промежуточного соединения 34 (0,094 моль) и 6 н. HCl (250 мл) перемешивают и кипятят с обратным холодильником в течение 2 дней, охлаждают, выливают в воду (100 мл) и нейтрализуютNH4OH (концентрированный). Нерастворимое вещество фильтруют и промывают водой, затем EtOH. Выход: 19 г. Фильтрат упаривают. Остаток (9,4 г) очищают колоночной хроматографией на силикагеле(элюент: CH2Cl2/CH3OH 99,25/0,75; 15-35 мкм). Отбирают одну фракцию и выпаривают растворитель. Выход: 8 г промежуточного соединения 35 (32%). Получение нерадиоактивных соединений Пример B1. К ДМФ (0,024 моль) медленно добавляют POCl3 (0,024 моль) при 5C. Смесь перемешивают при комнатной температуре в течение 30 мин, затем охлаждают до 5C. Медленно добавляют сложный этиловый эфир 3-оксобутановой кислоты (0,024 моль). Смесь перемешивают при 5C в течение 30 мин. По частям добавляют 1-(4-аминофенил)-2-фенилэтанон (0,024 моль). Смесь перемешивают при 90C в течение 3 ч и растворяют в CH2Cl2. Добавляют воду со льдом. Смесь подщелачивают NH4OH и экстрагируютCH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток кристаллизуют из 2-пропанона/диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,9 г соединения 306 (11%) (т.пл.: 136 С). Пример В 2 К раствору (полученному согласно примеру А 7.е) (0,022 моль) в трис(диокса-3,6-гептил)амине (1 мл) и CH2Cl2 (100 мл) при комнатной температуре по частям добавляют KMnO4 (10 г) . Смесь перемешивают при комнатной температуре в течение 8 ч, фильтруют через целит, промывают CH2Cl2 и сушат. Остаток (6 г, 100%) кристаллизуют из диэтилового эфира/петролейного эфира. Осадок отфильтровывают и сушат. Выход: 2 г соединения (2) (33%); т.пл. 82 С. Пример В 3 1,6 M н-BuLi (0,07 моль) медленно добавляют при -70C к раствору промежуточного соединения (5)(0,058 моль) в ТГФ (150 мл). Смесь перемешивают при -70C в течение 30 мин. Медленно добавляют раствор 2,3-дигидро-1H-инден-2-карбонитрила (0,07 моль) в ТГФ (100 мл). Смесь перемешивают при-70C в течение 1 ч, медленно доводят до комнатной температуры, подвергают гидролизу с H2O и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4) , фильтруют и выпаривают растворитель. Остаток (22 г) очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/циклогексан, от 80/20 до 100; 15-35 мкм). Очищенные фракции отбирают и выпаривают растворитель. Вторую фракцию кристаллизуют из 2-пропанона/диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,11 г соединения (3). Фильтрат концентрируют. Выход: 0,55 г соединения (3); т.пл. 145 С.(5) (0,018 моль) в ТГФ (50 мл). Смесь перемешивают при -70C в течение 1 ч, доводят температуру до 40C, затем охлаждают до -70C. Медленно добавляют раствор промежуточного соединения 7 (0,018 моль) в ТГФ (40 мл). Смесь перемешивают при -70C в течение 1 ч, затем доводят температуру до -20C,подвергают гидролизу с H2O и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4),фильтруют и выпаривают растворитель. Остаток (6,5 г) очищают колоночной хроматографией на силикагеле (элюент: толуол/EtOAc 90/10; 15-40 мкм). Отбирают две фракции (F1 и F2) и выпаривают растворитель. F1 (2,4 г) кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 1,8 г соединения (4) (29%); т.пл. 123 С. F2 (0,9 г) кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,2 г соединения (5) (3%); т.пл. 120C. По каплям при -78 С в токе N2 добавляют 1,6 M н-BuLi в оксане (exane) (0,107 моль) к смеси промежуточного соединения (6) (0,089 моль) в ТГФ. Смесь перемешивают при -78 С в течение 1 ч. При-78 С в токе N2 добавляют смесь промежуточного соединения 7 (150 мл). Смесь перемешивают при 78C в течение 2 ч, доводят температуру до 0C, выливают в H2O и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (31 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 85/15; 20-45 мкм). Отбирают две очищенные фракции и выпаривают из них растворители. Выход: 11 г соединения (7) (38%) и 8,2 г соединения (8) (28%). К суспензии Mg (0,0069 моль) в небольшом количестве простого диэтилового эфира медленно добавляют раствор хлорметилбензола (0,0069 моль) в простом диэтиловом эфире (8 мл) . Смесь перемешивают при комнатной температуре в течение 30 мин (распределение Mg), затем охлаждают до 5C. Медленно добавляют раствор промежуточного соединения 27 (0,0027 моль) в ТГФ (8 мл). Смесь перемешивают при 5 С в течение 15 мин, затем при комнатной температуре в течение 2 ч, выливают в H2O и фильтруют через целит. Осадок промывают EtOAc. Фильтрат экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (1 г) очищают колоночной хроматографией на кромасиле (элюент: от 100 CH2Cl2 до 99/1 СН 2Cl2/СН 3 ОН; 15-40 мкм). Очищенные фракции отбирают и выпаривают растворитель. Остаток (0,5 г, 56%) кристаллизуют из диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,14 г соединения 503 (15%). Пример В 4. Примеры модификаций концевых групп(полученную согласно примеру В 3.с) (0,0122 моль) 3 н. HCl (40 мл) и ТГФ (40 мл) перемешивают и кипятят с обратным холодильником в течение ночи, выливают в воду, подщелачивают 10%-ным K2 СО 3 и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 40/60; 15-40 мкм). Очищенные фракции отбирают и выпаривают растворитель. Выход: 2 г соединения (9)(5 мл) перемешивают при 140C в течение 48 ч. Добавляют H2O. Смесь экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (1 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 60/4 0; 15-40 мкм) . Отбирают две фракции и выпаривают растворитель. Обе фракции по отдельности кристаллизуют из пентана. Осадок отфильтровывают и сушат. Выход: 0,05 г соединения (10) (9%; т.пл. 115 С) и 0,057 г соединения (11)(MgSO4), фильтруют и выпаривают растворитель. Остаток (2,2 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 70/30; 15-40 мкм). Отбирают две фракции и выпаривают растворитель. Первую фракцию кристаллизуют из простого диэтилового эфира/петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,08 г соединения (12) (14%); т.пл.120C. Вторую фракцию кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,18 г соединения Смесь соединения (4) (0,001507 моль), этинилтриметилсилана (0,003013 моль), CuI (0,000151 моль) и Pd(PPh3)4 (0,000151 моль) в N,N-диэтилэтанамине (5 мл) перемешивают при 100C в течение 24 ч. Добавляют воду. Смесь фильтруют через целит, промывают EtOAc и фильтрат экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (1,3 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 85/15; 15-40 м:км). Очищенные фракции отбирают и выпаривают растворитель. Остаток (0,3 г) кристаллизуют из пентана. Осадок отфильтровывают и сушат. Выход: 0,11 г соединения (14) (18%); т.пл. 114C. Смесь соединения (14) (0,013 моль) и KF (0,038 моль) в уксусной кислоте (50 мл) перемешивают при комнатной температуре в течение 2 ч. Добавляют Н 2O и экстрагируют смесь простым диэтиловым эфиром. Органический слой отделяют, сушат (MgSO4) , фильтруют и выпаривают растворитель. Остаток(4,4 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 70/30; 15-40 мкм). Отбирают одну фракцию и выпаривают растворитель. Полученную фракцию (3 г, 7 3%) кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 2,45 г соединения- 21009334 полученное согласно примеру В.7.а) (0,0056 моль) в KOH"[1M, H2O] (10 мл) и метаноле (30 мл) перемешивают при комнатной температуре в течение 1 ч, выливают в воду и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (2,2 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc от 85/15 до 70/30; 15-40 мкм). Отбирают две фракции и выпаривают растворитель. Первую фракцию кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,2 г соединения (15) (11%); т.пл. 133 С. Вторую фракцию кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,3 г соединения (17) (16%); т.пл. 128 С.CuI (0,000121 моль) в N,N-диэтилэтанамине (5 мл) перемешивают при 100C в течение 2 ч. Добавляют воду. Смесь фильтруют через целит, промывают EtOAc и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (0,7 г) очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH 98/2; 15-40 мкм). Очищенные фракции отбирают и выпаривают растворитель. Остаток кристаллизуют из петролейного эфира и простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,1 г соединения (18) (23%); т.пл. 113 С. Смесь соединения (4) (0,006027 моль) и KF (0,024108 моль) в ДМСО (20 мл) перемешивают при 140C. Выпаривают растворитель досуха. Остаток подвергают загустению в воде и простом диэтиловом эфире. Смесь экстрагируют простым диэтиловым эфиром. Органический слой отделяют, промывают простым диэтиловым эфиром, промывают насыщенным раствором NaCl, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (1,7 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 85/15; 15-40 мкм). Отбирают три фракции и выпаривают из них растворители. Первую фракцию кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,21 г соединения (19) (11%); т.пл. 92 С. Вторую фракцию кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,33 г соединения (20) (17%); т.пл. 114 С. Смесь соединения (4) (0,003013 моль), ацетилхлорида (0,003315 моль) и йодида натрия (0,006027 моль) в CH3CN (10 мл) перемешивают и кипятят с обратным холодильником в течение 1 часа. Добавляют 10%-ный K2 СО 3. Смесь экстрагируют СН 2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (1 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 80/20; 15-40 мкм). Отбирают две фракции и выпаривают из них растворители. Первую фракцию кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,12 г соединения (21); т.пл. 110C.(0,00009 моль) в N,N-диэтилэтанамине (5 мл) перемешивают при 100C в течение 2 ч. Добавляют воду. Смесь экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (0,4 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 80/20; 15-40 мкм). Очищенные фракции отбирают и выпаривают растворитель. Остаток(0,18 г, 62%) кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,13 г соединения (22) (45%); т.пл. 138 С.(0,012054 моль) в атмосфере СО (газ) и метаноле (40 мл) перемешивают при 90C в течение 8 ч под давлением СО 5 бар. Добавляют H2O. Смесь экстрагируют EtOAc. Органический слой отделяют, сушат(MgSO4), фильтруют и выпаривают растворитель. Остаток (6 г) очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/СН 3 ОН, от 100/0 до 98/2; 15-35 мкм). Отбирают четыре фракции (F1-F4) и выпаривают растворитель. Выход: 0,13 г (цис) F1; 0,02 г F2 (цис, соединение 25); 0,055 г F3 (транс, 3%) и 0,11 г F4 (транс; соединение 26).F1 кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,03 г соединения (23) (1%); т.пл. 91 С.F3 кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,035 г соединения (24) (1%); т.пл. 99 С. Смесь соединения (4) (0,009 моль) и Zn (0,027 моль) в уксусной кислоте (30 мл) перемешивают при 60C в течение 4 ч, фильтруют через целит, промывают CH2Cl2, упаривают досуха, растворяют в CH2Cl2 и промывают 10%-ным K2 СО 3. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (4 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 75/25; 15-40 мкм). Отбирают одну фракцию и выпаривают растворитель. Полученную фракцию (1 г, 37%) кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: соединениеK2CO3. Смесь экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (0,7 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc, 85/15; 15-40 мкм). Отбирают две фракции (F1 и F2) и выпаривают из них растворители. Выход: 0,27 г (F1111, исходный продукт) и 0,14 г (F2). F2 кристаллизуют из пентана и петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,08 г соединения (27) (17%); т.пл. 110C. Смесь соединения (4) (0,001507 моль), трибутилэтенилолова (0,002260 моль) и Pd(PPh3)4 (0,000151 моль) в диоксане (5 мл) перемешивают при 80C в течение 8 ч. Добавляют воду. Смесь фильтруют через целит, промывают EtOAc и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (0,65 г) очищают колоночной хроматографией на силикагеле(элюент: циклогексан/EtOAc, 90/10; 15-40 мкм). Очищенные фракции отбирают и выпаривают растворитель. Остаток кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,07 г соединения (28) (14%); т.пл. 108C.(0,000151 моль) в диоксане (5 мл) перемешивают при 80C в течение 8 ч. Добавляют воду. Смесь экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (1,4 г) очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/EtOAc, 96/4; 15-40 мкм). Очищенные фракции отбирают и выпаривают растворитель. Остаток (0,38 г) кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,16 г соединения (29) (28%); т.пл. 112C.(0,0001507 моль) в диоксане (5 мл) перемешивают при 80C в течение 8 ч. Добавляют 10%-ный K2CO3. Смесь экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (1,7 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc, 85/15; 15-40 мкм). Очищенные фракции отбирают и выпаривают растворитель. Остаток(0,65 г) кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,35 г соединения (30) (61%); т.пл. 142 С. цис Смесь соединения (4) (0,0015 моль), 3-тиенилбороновой кислоты (0,00226 моль), Pd (PPh3) 4(0,00015 моль) и диоксана перемешивают и кипятят с обратным холодильником в течение 24 ч. Добавляют 10%-ный K2 СО 3. Смесь экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4) ,фильтруют и выпаривают растворитель. Остаток (0,8 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc, 80/20; 15-40 мкм) . Очищенные фракции отбирают и выпаривают растворитель. Остаток (0,4 г, 70%) кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,39 г соединения (31) (68%); т.пл. 113 С. Смесь соединения (4) (0,003 моль), моногидрохлорид сложного метилового эфира глицина (0,0066 моль) и Pd(PPh)4 (0,0003 моль) в Et3N (2 мл) и толуоле (10 мл) перемешивают при 100C под давлением СО 5 бар в течение 8 ч, фильтруют через целит, промывают CH2Cl2 и упаривают. Остаток (2 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc, 80/20; 75-35 мкм). Отбирают одну фракцию и выпаривают растворитель. Полученную фракцию (1 г, 80%) кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,46 г соединения (32) (37%).CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH/NH4OH, 95/5/0,1; 15-40 мкм). Отбирают две фракции (F1 и F2) и выпаривают из них растворители. Выход: 0,3 г F1 и 0,3 г F2.(MgSO4) , фильтруют и выпаривают растворитель. Остаток (6 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc, 60/40; 15-40 мкм). Отбирают первую фракцию и выпаривают растворитель. Остаток кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 1,2 6 г соединения (35) (24%); т.пл. 160C. Смесь соединения (4) (0,009 моль) и тиомочевины (0,0099 моль) в этиловом спирте (30 мл) перемешивают и кипятят с обратным холодильником в течение 12 ч, медленно добавляют раствор KOH (0,0149 моль) в Н 2 О (5 мл) . Смесь перемешивают и кипятят с обратным холодильником в течение 1 ч, выливают в воду и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4) , фильтруют и выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (циклогексан/EtOAc, 70/30; 15-40 мкм). Очищенные фракции отбирают и выпаривают растворитель. Выход: 1,1 г F1111 (37%) и 0,4 г К раствору соединения (36) (0,0015 моль), соединения (37) (0,0015 моль) и К 2 СО 3 (0,0034 моль) в ацетоне (15 мл) при комнатной температуре медленно добавляют CH3I (0,0034 моль). Смесь перемешивают при комнатной температуре в течение 8 ч. Добавляют воду и смесь экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4),фильтруют и выпаривают растворитель. Остаток (1,2 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc,85/15; 15-40 мкм). Очищенные фракции отбирают и выпаривают растворитель. Выход: 0,6 г F1 (57%) и 0,18 г F2 (17%). F1 кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,28 г соединения (38) (27%). F2 кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,065 г соединения (39) (6%). Смесь соединения (41), полученного согласно примеру В 3.b (0,0014 моль), в 3 н. HCl (5 мл) и ТГФ Смесь соединения (5) (0,045 моль), ацетамид (0,90013 моль) и K2CO3 (0,225 моль) перемешивают и кипятят с обратным холодильником при 200C в течение 2 ч, охлаждают до комнатной температуры,выливают в H2O/CH2Cl2 и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и досуха выпаривают растворитель. Остаток (14,4 г) кристаллизуют из СН 3 ОН. Осадок отфильтровывают и сушат. Фильтрат упаривают. Остаток (11,27 г) очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH/NH4OH, 96/4/0,1; 15-35 мкм). Очищенные фракции отбирают и выпаривают растворитель. Выход: 4,2 г соединения (188) (65%). Смесь соединения (188) (0,00032 моль), бензойной кислоты (1,5 эквивалента, 0,00048 моль), 1-этил 3-(3'-диметиламинопропил) карбодиимидHCl (1:1) (1,5 эквиЕ; алента, 0,00048 моль), N-гидроксибензотриазола (1,5 эквивалента, 0,00048 моль) и Et3N (1 эквивалент, 0,00032 моль) в CH2Cl2 (2 мл) перемешивают при комнатной температуре в течение 15 ч. Растворитель выпаривают. Остаток очищают ВЭЖХ, фракции продукта отбирают и выпаривают растворитель. Выход: 0,0 66 г соединения (205) Смесь промежуточного соединения 20 (0,001507 моль) в 3 н. HCl (10 мл) и ТГФ (10 мл) перемешивают при комнатной температуре в течение 8 ч, подщелачивают 10%-ным K2 СО 3 и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (1,2 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc, 85/15; 15-40 мкм). Очищенные фракции отбирают и выпаривают растворитель. Остаток (0,4 г) кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,3 г соединения (6) (58%); т.пл. 108C.(0,00916 моль) в CH3OH (25 мл) перемешивают и кипятят с обратным холодильником в течение 15 ч,затем охлаждают до комнатной температуры, выливают в H2O и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4) , фильтруют и досуха выпаривают растворитель. Остаток (1,1 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc; 40/60; 15-40 мкм) . Отбирают две фракции и выпаривают растворитель. Выход: 0,3 г F1 и 0,5 г F2 (50%). F2 кристаллизуют из простого диэтилового эфира/петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,26 г. F1 кристаллизуют из пентана. Осадок отфильтровывают и сушат. Выход: 0,19 г. Данную фракцию очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/СН 3 ОН; 98/2; 15-40 мкм). Очищенные фракции отбирают и выпаривают растворитель. Выход: 0,11 г. Данную фракцию очищают колоночной хроматографией на кромасиле (элюент: СН 3 ОН/Н 2 О; 70/30). Очищенные фракции отбирают и выпаривают растворитель. Выход: 0,09 г (9%). Полученную фракцию кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,08 г соединения 419 (8%). Пример В 5.(30 мл) в токе N2 при 5 С добавляют йодметан (0,00456 моль). Смесь перемешивают при комнатной температуре в течение ночи, выливают в H2O и экстрагируют CH2Cl2. Органический слой отделяют, сушат(MgSO4), фильтруют и выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc, 65/35; 15-40 мкм). Отбирают две фракции и выпаривают растворитель. Выход: 0,35 г соединения (42) (30%; т.пл. 125 С) и 0,35 г соединения (43) (30%; т.пл. 116 С). Пример В 6.(0,01068 моль). Смесь перемешивают в течение 30 мин. При 0C добавляют этилбромацетат (0,01068 моль). Смесь перемешивают при комнатной температуре в течение 1 ч, подвергают гидролизу с водой и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc, 60/40; 15-40 мкм). Требуемые фракции (F1-F4) отбирают и выпаривают растворитель. Выход: 0,11 г F1; 0,13 г F2; 0,75 г F3 и 0,8 г F4. F3 кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: соединение (44); т.пл.152 С.F4 кристаллизуют из диэтилового эфира. Осадок отфильтровывают и сушат. Выход: соединение К раствору соединения (8) и соединения (9) (0,0064 моль) и 60% NaH (0,007 моль) в ДМФ (40 мл) добавляют по каплям при 0C в токе N2 бромметилбензол (0,007 моль). Смесь перемешивают при комнатной температуре в течение 1 ч, подвергают гидролизу с водой и экстрагируют EtOAc. Органический слой отделяют, промывают водой, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc, 70/30; 15-40 мкм). Требуемые фракции (F1-F4) отбирают и выпаривают растворитель. Выход: 0,15 г F1, 0,1 г F2, 0,6 г F3(23%) и 0,8 г F4.F3 кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,13 г соединения (46); т.пл. 137 С.F4 кристаллизуют из DIPE и петролейного эфира. Осадок отфильтровывают и сушат. Выход: соединение (47); т.пл. 130C. Пример В 7. а) К раствору соединения (48) (полученного согласно примеру В 2) (0,044 моль) в CH2Cl2 (200 мл) при 0C добавляют 3-хлорбензолнадкарбоновую кислоту (0,088 моль) и смесь перемешивают при комнатной температуре в течение 12 ч. Смесь промывают 10%-ным K2CO3. Органический слой сушат(MgSO4), фильтруют и упаривают. Остаток перекристаллизовывают из (C2H5)2O. Выход: 8,2 г циклогексил(3-метил-6-хинолинил)метанон-1-оксида (соединение 49) (69%).b) К раствору соединения (49) (0,028 моль) в К 2 СО 3 (400 мл) и CH2Cl2 (400 мл) добавляют 4 метилбензолсульфонилхлорид (0,043 моль) и перемешивают смесь при комнатной температуре в течение 1 ч. Смесь экстрагируют CH2Cl2. Органический слой сушат (MgSO4) , фильтруют и упаривают. Остаток перекристаллизовывают из (C2H5)2O. Выход: 6,64 г 6-(циклогексилкарбонил)-3-метил-2(1H)-хинолинон- 27009334 Смесь соединения (7) (0,0229 моль), гидроксиламина (0,0252 моль) и N, N-диэтилэтанамина (0,0252 моль) в этаноле (100 мл) перемешивают и кипятят с обратным холодильником в течение 6 ч, выливают в воду и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4) , фильтруют и выпаривают растворитель. Остаток кристаллизуют из CH3CN. Осадок отфильтровывают и сушат. Остаток очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/EtOAc, 80/20; 15-40 мкм). Отбирают две фракции и выпаривают растворитель. Выход: 2,8 г соединения (51) (36%; т.пл. 133 С) и 3 г соединения К раствору соединения (7) (0,015 моль) в этаноле (75 мл) добавляют при комнатной температуре гидразин (0,41 моль). Смесь перемешивают и кипятят с обратным холодильником в течение 1 ночи, выливают в воду и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH/NH4OH 98/2/0,1). Очищенные фракции отбирают и выпаривают растворитель. Остаток кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,8 г соединения (53) (15%); т.пл. 110C. Пример В 9. Методика для получения соединений 400, 401, 402 403, 404 и 405. Смесь промежуточного соединения 21 (полученного согласно A11) (0,000269 моль), гидрохлорида амантадина (0,000404 моль; 1,5 эквивалента), гидрохлорида N'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамина (0,000404 моль; 1,5 эквивалента), 1-гидрокси-1H-бензотриазола (0,000404 моль; 1,5 эквивалента) и Et3N (0,000269 моль) вCH2Cl2 (2 мл) перемешивают при комнатной температуре в течение 12 ч. Выпаривают растворитель. Остаток очищают ВЭЖХ. Фракции продукта отбирают и выпаривают растворитель. Выход: 0,063 г соединения 520 (46.37%). Пример B10. Смесь промежуточного соединения 27 (0,0026 моль) и промежуточного соединения 26 (0,0026 моль) в EtOH (380 мл) и концентр. H2SO4 (19 мл) перемешивают и кипятят с обратным холодильником в течение 15 ч, охлаждают до комнатной температуры, выливают в воду со льдом, подщелачивают K2 СО 3 и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (17,9 г) очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc; 80/20; 15-35 мкм). Очищенные фракции отбирают и выпаривают растворитель. Выход: 0,85 г F1,1,1 г F2 и 11,5 г F3. F1 и F2 по отдельности кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,34 г соединения 233. Пример B11.NH4OH и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (1,2 г) очищают колоночной хроматографией на силикагеле (элюент:CH2Cl2/CH3OH/NH4OH; 93/7/0,5; 15-40 мкм). Отбирают две фракции и выпаривают растворитель. Выход: 0,5 г F1 (41%) и 0,4 г F2. F1 кристаллизуют из петролейного эфира. Осадок отфильтровывают и сушат. Выход: 0,17 г соединения 511 (14%).EtOH (15 мл) перемешивают и кипятят с обратным холодильником в течение 24 ч, выливают в H2O и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/циклогексан, 80/20; 15-40 мкм). Отбирают две фракции и выпаривают растворитель. Выход: 0,35 г F1 (64%) и 0,17 г (SM) F1 кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,33 г соединения 514 (60%) (т.пл.:185 С). Пример B13. Смесь промежуточного соединения 28 (0,019 моль), 2-бензофуранилбороновой кислоты (0,028 моль), Pd(РРh3)4 (0,001 моль) и BHT (небольшое количество) в диоксане (25 мл) и Na2CO3[2] (25 мл) перемешивают, кипятят с обратным холодильником в течение 8 ч и экстрагируют EtOAc. Водный слой подщелачивают NH4OH и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (3,6 г) очищают колоночной хроматографией на силикагеле(элюент: CH2Cl2/СН 3 ОН, 99/1; 15-40 мкм). Очищенные фракции отбирают и выпаривают растворитель. Выход: 1,8 г (33%). Полученную фракцию кристаллизуют из 2-пропанона/диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,39 г соединения 515 (7%) (т.пл.: 134 С). Пример B14. К раствору промежуточного соединения 32 (0,004 моль) в CF3COOH (5 мл) и AcOH (10 мл) медленно добавляют при комнатной температуре триэтилсилан (0,0012 моль). По частям в токе N2 добавляютNaBH4 (0,0012 моль). Смесь перемешивают при комнатной температуре в течение 8 ч, выливают на лед,подщелачивают K2 СО 3 и экстрагируют CH2Cl2. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (1,2 г) очищают колоночной хроматографией на силикагеле(элюент: CH2Cl2/СН 3 ОН, 99/1; 15-40 мкм). Отбирают две фракции и выпаривают растворитель. Выход: 0,5 г F1 (43%) и 0,4 г F2. F1 растворяют в iPrOH. Добавляют HCl/iPrOH (1 экв.). Осадок отфильтровывают и сушат. Выход: 0,32 г соединения 526 (т.пл.: 248 С). Пример B15 Смесь промежуточного соединения 33 (0,082 моль) и 3-хлор-2-этил-2-бутеналя (0,098 моль) в AcOH(200 мл) перемешивают и кипятят с обратным холодильником в течение 8 ч. Досуха выпаривают растворитель. Остаток разжижают (растворяют) в СН 2Cl2 и промывают 10%-ным K2CO3. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают растворитель. Остаток (27 г) очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/EtOAc; от 95/5 до 92/8; 15-35 мкм). Отбирают две фракции и выпаривают растворитель. Выход: 0,7 г F1 и 5,3 г F2. F1 кристаллизуют из 2-пропанона/диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 0,25 г соединения 471 (2%) (т.пл.: 140C). Пример B16. К раствору промежуточного соединения 35 (полученного согласно А 17.b) (0,0379 моль) в ТГФ (200 мл) в токе N2 добавляют по каплям н-BuLi (0,0417 моль). Смесь перемешивают в течение 30 мин. При 78C добавляют по каплям раствор 4-бром-N-метокси-N-метилбензолацетамид (0,0568 моль) в ТГФ (100 мл). Температуру смеси поднимают при перемешивании от -78 до 0C, смесь выливают в H2O и экстра- 29009334 гируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и досуха выпаривают растворитель. Остаток (20,9 г) очищают колоночной хроматографией на силикагеле (элюент: толуол/EtOAc, от 60/40 до 50/50; 15-35 мкм). Отбирают две фракции и выпаривают растворитель. Выход: 4 г фракции 1 и 4 г фракции 2 (28%). Фракцию 2 кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат. Выход: 1 г соединения 528 (т.пл. 195 С). В табл. 1-8 перечислены соединения формулы (I-A) и (I-B), которые были получены согласно одному из описанных выше примеров. Таблица 1
МПК / Метки
МПК: C07D 215/00, A61K 101/00, A61K 51/04
Метки: рецептора, изотопами, хинолина, применение, меченные, производные, качестве, метаботропного, лигандов, глутаматного, радиоактивными
Код ссылки
<a href="https://eas.patents.su/30-9334-mechennye-radioaktivnymi-izotopami-proizvodnye-hinolina-i-ih-primenenie-v-kachestve-ligandov-metabotropnogo-glutamatnogo-receptora.html" rel="bookmark" title="База патентов Евразийского Союза">Меченные радиоактивными изотопами производные хинолина и их применение в качестве лигандов метаботропного глутаматного рецептора</a>
Производные хинолина и их применение в качестве микобактериальных ингибиторов

Номер патента: 8937
Опубликовано: 26.10.2007
Авторы: Декран Лоранс Франсуаз Бернадетт, Оддс Френк Кристофер, Чока Имре Кристиан Франсис, Вернье Даниэль Ф.Ж., Ван Гестел Йозеф Франс Элизабета, Вене Марк Гастон, Гийемон Жером Эмиль Жорж, Андрис Конрад Йозеф Лодевийк Марсель
МПК: A61P 31/06, A61K 31/47, C07D 215/22...
Метки: производные, хинолина, качестве, микобактериальных, применение, ингибиторов
Формула / Реферат:
1. Соединение общей формулы (Iа) или общей формулы (Ib) их фармацевтически приемлемые аддитивные соли с кислотами или основаниями, стереохимически изомерные формы, таутомерные формы и N-оксиды, где R1 означает водород, галоген, галогеналкил, циано, гидрокси, Ar, Het, алкил, алкилокси, алкилтио, алкилоксиалкил, алкилтиоалкил, Ar-алкил или ди(Аr)алкил; р означает целое число, равное 0, 1, 2, 3 или 4; R2 означает водород, гидрокси, тио,...
Производные хинолина и изохинолина, способ их получения и их применение в качестве ингибиторов воспаления

Номер патента: 8540
Опубликовано: 29.06.2007
Авторы: Бергер Маркус, Леманн Манфред, Шэке Хайке, Скубалла Вернер, Ярох Штефан, Кроликевич Конрад, Шоттелиус Арндт, Шмеес Норберт, Ревинкель Хартмут
МПК: A61P 29/00, A61K 31/47, C07D 215/40...
Метки: воспаления, применение, способ, хинолина, получения, ингибиторов, качестве, производные, изохинолина
Формула / Реферат:
1. Соединения общей формулы I в которой А обозначает арильную, бензильную или фенетильную группу, каждая из которых необязательно может быть замещена одним либо несколькими остатками из группы, включающей C1-C5алкил, C1-C5алкоксигруппу, C1-C5алкилтиогруппу, C1-C5перфторалкил, галоген, гидроксигруппу, цианогруппу, нитрогруппу, -О-(СН2)n-О-, -О-(СН2)n-СН2-, -О-СН=СН-, -(СН2)n+2-, где n обозначает 1 или 2, а концевые атомы кислорода и/или атомы...
Производные индолин-2-она, их получение и их применение в качестве лигандов рецепторов окситоцина

Номер патента: 5093
Опубликовано: 28.10.2004
Авторы: Валетт Жерар, Фулон Луак, Гарсия Жорж, Серрадейль-Ле Галь Клодин
МПК: C07D 209/34, A61P 5/10, A61K 31/404...
Метки: получение, рецепторов, лигандов, индолин-2-она, окситоцина, применение, качестве, производные
Формула / Реферат:
1. Соединения в форме чистого энантиомера или смеси энантиомеров формулы в которой R0 представляет собой группу, выбранную из 1) в которой Z1 представляет собой атом хлора, брома, йода или фтора или (C1-C4)алкильную, (C1-C4)алкокси или трифторметильную группу; Z2 представляет собой атом водорода, хлора, брома, йода или фтора или (C1-C4)алкильную, (C3-C5)циклоалкильную, (C1-C4)алкокси, (C3-C5)циклоалкокси или полифтор(C1-C4)алкильную...
Производные аминотиазола и их применение в качестве лигандов рецепторов крф

Номер патента: 5159
Опубликовано: 30.12.2004
Авторы: Галли Даниэль, Фонтен Эвелин, Прадине Антуан, Жеслен Мишель, Роже Пьер
МПК: A61P 25/00, C07D 277/42, A61K 31/426...
Метки: аминотиазола, производные, рецепторов, лигандов, крф, применение, качестве
Формула / Реферат:
1. Соединения, в рацемической форме или в форме чистого энантиомера, формулы где R1 и R2, которые могут быть одинаковыми или разными, каждый независимо представляет собой атом галогена; гидрокси(C1-C5)алкил; (C1-C5)алкил; аралкил, в котором арильная часть представляет собой (C6-C8), а алкильная часть представляет собой (C1-C4); (C1-C5)алкокси; трифторметильную группу; нитрогруппу; нитрильную группу; группу -SR, где R представляет собой...
Производные ацилоксипирролидина и их применение в качестве лигандов v1b или v1b и v1a рецепторов

Номер патента: 8444
Опубликовано: 29.06.2007
Авторы: Оломбар Ален, Праден Антуан, Гарсия Жорж, Ваньон Жан, Серрадей-Ле Галь Клодин
МПК: A61K 31/404, A61P 5/00, C07D 403/04...
Метки: лигандов, ацилоксипирролидина, качестве, производные, применение, рецепторов
Формула / Реферат:
1. Соединение формулы (I) в которой R1 представляет собой атом водорода, (C1-C6)алкил, (C3-C6)циклоалкил, группу -СН2СН2СООН или группу -NR2R3; каждый из R2 и R3 независимо представляет собой атом водорода или (C1-C6)алкил; в форме основания или соли присоединения органического или неорганического основания, а также в форме гидрата или сольвата. 2. Соединение по п.1 формулы (I), в которой R1 представляет собой атом водорода, метильный радикал,...
Предыдущий патент: Фармацевтическая композиция алендроновой кислоты, ее солей или сложных эфиров и способ ее получения
Следующий патент: Способ и состав для связывания ацетальдегида в слюне, желудке и толстой кишке
Случайный патент: Влажное гранулирование с использованием вещества, связывающего воду