Новые 1, 2, 3-замещенные производные индолизина, являющиеся ингибиторами факторов роста фибробластов, способ их получения и фармацевтические композиции, содержащие их
Номер патента: 7902
Опубликовано: 27.02.2007
Авторы: Гийо Натали, Борде Мари-Франсуаза, Эрбер Жан-Марк, Бадорк Ален, Боно Франсуаза






Формула / Реферат
1. Соединения формулы I

в которой R1 представляет собой гидроксильный радикал, линейный или разветвленный радикал алкокси с числом атомов углерода от 1 до 5, карбоксильный радикал, алкоксикарбонильный радикал с числом атомов углерода от 2 до 6 или радикал формулы
-NR5R6
-NH-SO2-Alk
-NH-SO2-Ph
-NH-CO-Ph
-N(Alk)-CO-Ph
-NH-CO-NH-Ph
-NH-CO-Alk
-NH-CO2-Alk
-O-(CH2)n-cAlk
-O-Alk-COOR7
-O-Alk-O-R8
-O-Alk-OH
-O-Alk-C(NH2):NOH
-O-Alk-NR5R6
-O-Alk-CN
-O-(CH2)n-Ph
-O-Alk-CO-NR5R6
-CO-NH-(CH2)m-COOR7
-CO-NH-Alk,
где Alk представляет собой алкильный радикал или линейный или разветвленный алкиленовый радикал с числом атомов углерода от 1 до 5,
cAlk представляет собой циклоалкильный радикал с числом атомов углерода от 3 до 6,
n представляет собой целое число от 0 до 5,
m представляет собой целое число от 1 до 5,
R5 и R6, которые являются идентичными или разными, каждый представляет собой атом водорода, линейный или разветвленный алкильный радикал с числом атомов углерода от 1 до 5 или бензильный радикал,
R7 представляет собой атом водорода или алкильный радикал с числом атомов углерода от 1 до 5,
R8 представляет собой алкильный радикал с числом атомов углерода от 1 до 5 или радикал -CO-Alk,
Ph представляет собой фенильный радикал, который возможно замещен одним или более чем одним из галогеновых атомов, одним или более чем одним из радикалов алкокси с числом атомов углерода от 1 до 5, одним или более чем одним из карбоксильных радикалов или одним или более чем одним из алкоксикарбонильных радикалов с числом атомов углерода от 2 до 6,
R2 представляет собой атом водорода, алкильный радикал с числом атомов углерода от 1 до 5, радикал алкилгалогенида с числом атомов углерода от 1 до 5, содержащий от 3 до 5 галогеновых атомов, циклоалкильный радикал с числом атомов углерода от 3 до 6 или фенильный радикал, который возможно замещен одним или более чем одним из галогеновых атомов, одним или более чем одним из радикалов алкокси с числом атомов углерода от 1 до 5, одним или более чем одним из карбоксильных радикалов или одним или более чем одним из алкоксикарбонильных радикалов с числом атомов углерода от 2 до 6,
А представляет собой радикал -СО-, -SO- или -SO2-,
R3 и R4, которые являются идентичными или разными, каждый представляет собой атом водорода, радикал алкокси с числом атомов углерода от 1 до 5, радикал амино, карбоксильный радикал, алкоксикарбонильный радикал с числом атомов углерода от 2 до 6, гидроксильный радикал, радикал нитро, радикал гидроксиамино, радикал формулы
-Alk-COOR7
-NR5R6
-NH-Alk-COOR7
-NH-COO-Alk
-N(R11)-SO2-Alk-NR9R10
-N(R11)-SO2-Alk
-N(R11)-Alk-NR5R6
-N(R11)-CO-Alk-NR9R10
-N(R11)-CO-Alk
-N(R11)-CO-CF3
-NH-Alk-HetN
-O-Alk-NR9R10
-O-Alk-CO-NR5R6
-O-Alk-HetN,
где n, m, Alk, R5, R6 и R7 имеют значения, которые даны выше для R1, и
R9 и R10, которые являются идентичными или разными, каждый представляет собой атом водорода или алкильный радикал с числом атомов углерода от 1 до 5,
R11 представляет собой атом водорода или радикал -Alk-COOR12, где R12 представляет собой атом водорода, алкильный радикал с числом атомов углерода от 1 до 5 или бензильный радикал,
HetN представляет собой 5- или 6-членный гетероцикл, содержащий по меньшей мере один атом азота и возможно другой гетероатом, выбранный из азота и кислорода,
или R3 и R4 совместно образуют 5- или 6-членный ненасыщенный гетероцикл при том условии, однако, что, когда R3 представляет собой радикал алкокси и R4 представляет собой радикал -O-Alk-NR9R10 или гидроксильный радикал, R1 не представляет собой атом водорода или радикал алкокси, возможно в форме одной из их фармацевтически приемлемых солей.
2. Соединения формулы I по п.1, в которой R1 представляет собой гидроксильный радикал, линейный или разветвленный радикал алкокси с числом атомов углерода от 1 до 5, карбоксильный радикал, алкоксикарбонильный радикал с числом атомов углерода от 2 до 6 или радикал формулы
-NR5R6
-NH-SO2-Alk
-NH-SO2-Ph
-NH-CO-Ph
-N(Alk)-CO-Ph
-NH-CO-NH-Ph
-NH-CO-Alk
-NH-CO2-Alk
-O-(CH2)n-cAlk
-O-Alk-COOR7
-O-Alk-O-R8
-O-Alk-OH
-O-Alk-NR5R6
-O-Alk-CN
-O-(CH2)n-Ph
-O-Alk-CO-NR5R6
-CO-NH-(CH2)m-COOR7
-CO-NH-Alk,
где Alk представляет собой алкильный радикал или линейный или разветвленный алкиленовый радикал с числом атомов углерода от 1 до 5,
cAlk представляет собой циклоалкильный радикал с числом атомов углерода от 3 до 6,
n представляет собой целое число от 0 до 5,
m представляет собой целое число от 1 до 5,
R5 и R6, которые являются идентичными или разными, каждый представляет собой атом водорода, линейный или разветвленный алкильный радикал с числом атомов углерода от 1 до 5 или бензильный радикал,
R7 представляет собой атом водорода или алкильный радикал с числом атомов углерода от 1 до 5,
R8 представляет собой алкильный радикал с числом атомов углерода от 1 до 5 или радикал -CO-Alk,
Ph представляет собой фенильный радикал, который, возможно, замещен одним или более чем одним из галогеновых атомов, одним или более чем одним из радикалов алкокси с числом атомов углерода от 1 до 5, одним или более чем одним из карбоксильных радикалов или одним или более чем одним из алкоксикарбонильных радикалов с числом атомов углерода от 2 до 6,
R2 представляет собой алкильный радикал с числом атомов углерода от 1 до 5, трифторметильный радикал, циклоалкильный радикал с числом атомов углерода от 3 до 6 или фенильный радикал, который возможно замещен одним или более чем одним из галогеновых атомов, одним или более чем одним из радикалов алкокси с числом атомов углерода от 1 до 5, одним или более чем одним из карбоксильных радикалов или одним или более чем одним из алкоксикарбонильных радикалов с числом атомов углерода от 2 до 6,
А представляет собой радикал -СО- или -SO2-,
R3 и R4, которые являются идентичными или разными, каждый представляет собой атом водорода, радикал алкокси с числом атомов углерода от 1 до 5, радикал амино, карбоксильный радикал, алкоксикарбонильный радикал с числом атомов углерода от 2 до 6, радикал нитро, радикал гидроксиамино, радикал формулы
-Alk-COOR7
-NR5R6
-NH-Alk-COOR7
-NH-COO-Alk
-N(R11)-SO2-Alk-NR9R10
-N(R11)-SO2-Alk
-N(R11)-Alk-NR5R6
-N(R11)-CO-Alk-NR9R10
-N(R11)-CO-Alk
-N(R11)-CO-CF3
-NH-Alk-HetN,
где n, m, Alk, R5, R6 и R7 имхют значение, данное выше для R1, и
R9 и R10, которые являются идентичными или разными, каждый представляет собой атом водорода или алкильный радикал с числом атомов углерода от 1 до 5,
R11 представляет собой атом водорода или радикал -Alk-COOR12, где R12 представляет собой атом водорода, алкильный радикал с числом атомов углерода от 1 до 5 или бензильный радикал,
HetN представляет собой 5- или 6-членный гетероцикл, содержащий по меньшей мере один атом азота и возможно другой гетероатом, выбранный из азота и кислорода, возможно в форме одной из их фармацевтически приемлемых солей.
3. Соединения формулы I по п.1 или 2, в которой
R1 представляет собой радикал алкокси с числом атомов углерода от 1 до 5, карбоксильный радикал, радикал -O-Alk-COOH, в котором Alk представляет собой алкиленовый радикал с числом атомов углерода от 1 до 5, радикал формулы -O-Alk-Ph, в котором Alk представляет собой алкиленовый радикал с числом атомов углерода от 1 до 5 и Ph представляет собой фенильный радикал, который возможно замещен одним или более чем одним из галогеновых атомов, или одним или более чем одним из радикалов алкокси с числом атомов углерода от 1 до 5, или одним или более чем одним из карбоксильных радикалов, радикал формулы -NH-CO-Ph, радикал формулы -NH-SO2-Ph или радикал формулы -NH-CO-NH-Ph,
R2 представляет собой алкильный радикал с числом атомов углерода от 1 до 5,
А представляет собой радикал -СО-,
R3 и R4, которые являются разными, каждый представляет собой атом водорода, радикал алкокси с числом атомов углерода от 1 до 5, радикал амино, карбоксильный радикал или алкоксикарбонильный радикал с числом атомов углерода от 2 до 6, возможно в форме одной из их фармацевтически приемлемых солей.
4. Соединение формулы I по п.1, которое выбирают из следующих соединений:
(4-амино-3-метоксифенил)-(1-метокси-2-метилиндолизин-3-ил)метанон,
3-(4-амино-3-метоксибензоил)-2-метилиндолизин-1-ил-карбоновая кислота,
2-{[3-(4-амино-3-метоксибензоил)-2-метилиндолизин-1-ил]окси}уксусная кислота,
(4-амино-3-метоксифенил)-{1-[(4-хлорбензил)окси]-2-метилиндолизин-3-ил}метанон,
(4-амино-3-метоксифенил)-{1-[(3-метоксибензил)окси]-2-метилиндолизин-3-ил}метанон,
4-({[3-(4-амино-3-метоксибензоил)-2-метилиндолизин-1-ил]окси}метил)бензойная кислота,
3-(4-карбоксибензоил)-2-метилиндолизин-1-ил-карбоновая кислота,
метил-3-[(1-метокси-2-метилиндолизин-3-ил)карбонил]бензоат,
4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]бензойная кислота,
2-амино-5-[(1-метокси-2-метилиндолизин-3-ил)карбонил]бензойная кислота,
2-амино-5-({1-[(3-метоксибензоил)амино]-2-метилиндолизин-3-ил}карбонил)бензойная кислота,
2-амино-5-({2-метил-1-[(3,4,5-триметоксибензоил)амино]индолизин-3-ил}карбонил)бензойная кислота,
2-амино-5-({1-{[(3-метоксифенил)сульфонил]амино}-2-метилиндолизин-3-ил}карбонил)бензойная кислота,
возможно в форме одной из его фармацевтически приемлемых солей.
5. Способ получения соединений формулы I по пп.1-4, который отличается тем, что производное индолизина формулы II

в которой R1 и R2 имеют значение, которое дано для формулы I, но R2 не представляет собой атом водорода или радикал алкилгалогенида, конденсируют с производным формулы III

в которой X представляет собой атом галогена, a R3 или R4, которые являются идентичными или разными, каждый представляет собой атом водорода, радикал нитро, радикал трифторацетамидо или алкоксикарбонильный радикал с числом атомов углерода от 2 до 6.
6. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R3 и/или R4 представляют собой -NO2, подвергают восстановлению для получения соединений формулы I, в которой R3 и/или R4 представляют собой радикал амино (-NH2).
7. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R3 и/или R4 представляют собой -NH2, подвергают действию алкилгалогенида для получения соединений формулы I, для которых R4 и/или R3 представляют собой радикал -NR5R6 (в котором R5 представляет собой атом водорода, и R6 представляет собой алкильный радикал с числом атомов углерода от 1 до 5) и радикал -NH-Alk-NR5R6 или радикал -NH-Alk-COOR7 (в котором R7 не представляет собой атом водорода).
8. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R3 и/или R4 представляют собой радикал -NH-Alk-COOR7 (в котором R7 не представляет собой атом водорода), подвергают омылению для получения соединений формулы I, в которых R4 и/или R3 представляют собой радикал -NH-Alk-COOH.
9. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R3 и/или R4 представляют собой -NH2, подвергают ацилированию для получения соединений формулы I, для которых R4 и/или R3 представляют собой радикал -NH-CO-Alk или радикал -NH-CO-Alk-NR9R10.
10. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R3 и/или R4 представляют собой -NH2, подвергают сульфонилированию для получения соединений формулы I, для которых R4 и/или R3 представляют собой радикал -NH-SO2-Alk или радикал -NH-SO2-Alk-NR9R10.
11. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения, полученные в пп.9 и 10, подвергают алкилированию для получения соединений, в которых R3 и/или R4 представляют собой радикал -N-(R11)-CO-Alk, или радикал -N-(R11)-CO-Alk-NR9R10, или радикал -N(R11)-SO2-Alk, или радикал -N(R11)-SO2-Alk-NR9R10, где R11 представляет собой радикал -Alk-COOR12, в котором R12 не представляет собой атом водорода.
12. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения, полученные в п.11, подвергают омылению для получения соединений формулы I, для которых R4 и/или R3 представляют собой радикал -N-(R11)-CO-Alk, или радикал -NR9R10-CO-Alk-NR9R10, или радикал -N(R11)-SO2-Alk, или радикал -N(R11)-SO2-Alk-NR9R10, где R11 представляет собой радикал -Alk-COOH.
13. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R3 и/или R4 представляют собой алкоксикарбонильный радикал, подвергают омылению для получения соединений формулы I, в которой R3 и/или R4 представляют собой карбоксильный радикал.
14. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R1 представляет собой радикал бензилокси, a R3 и/или R4 представляют собой -NO2, подвергают действию трифторуксусной кислоты для получения соединений формулы I, в которой R1 представляет собой ОН.
15. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R1 представляет собой радикал бензилокси, a R3 и/шыш R4 представляют собой радикал алкоксикарбоксил, подвергают гидрированию для получения соединений формулы I, в которой R1 представляет собой ОН.
16. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R1 представляет собой ОН, подвергают О-алкилированию для получения соединений формулы I, в которой R1 представляет собой линейный или разветвленный радикал алкокси с числом атомов углерода от 1 до 5, радикал -O-(СН2)n-сАlk, радикал -O-Alk-COOR7, радикал -O-Alk-NR5R6, радикал -O-(CH2)n-Ph или радикал -O-Alk-O-R8.
17. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R1 представляет собой радикал -O-Alk-O-COCH3, подвергают омылению для получения радикала -О-Alk-OH или радикала -O-Alk-CN.
18. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R1 представляет собой радикал -O-Alk-CN, подвергают обработке гидроксиламином для получения радикала -O-Alk-C(NH2)=NOH.
19. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R1 представляет собой радикал -NO2, подвергают омылению для получения соединений формулы I, в которой R1 представляет собой СООН.
20. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения, полученные способом по п.19, подвергают действию производного амина для получения соединений формулы I, в которой R1 представляет собой радикал -CO-NH-Alk.
21. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения, полученные способом по п.19, подвергают действию производного аминокислоты для получения соединений формулы I, в которой R1 представляет собой радикал -CO-NH-(CH2)m-COOR7.
22. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R1 представляет собой радикал -NH-CO2-трет-бутил, a R3 и/или R4 представляют собой -NO2 или -CO2алкил, подвергают алкилированию с последующим снятием защиты и возможным вторым алкилированием для получения соединений формулы I, в которой R1 представляет собой NR5R6.
23. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R1 представляет собой радикал -NH-CO2-трет-бутил, a R3 и/или R4 представляют собой -NO2 или -CO2алкил, подвергают снятию защиты с последующим ацилированием для получения соединений формулы I, в которой R1 представляет собой NH-CO-Alk или NH-CO-Ph.
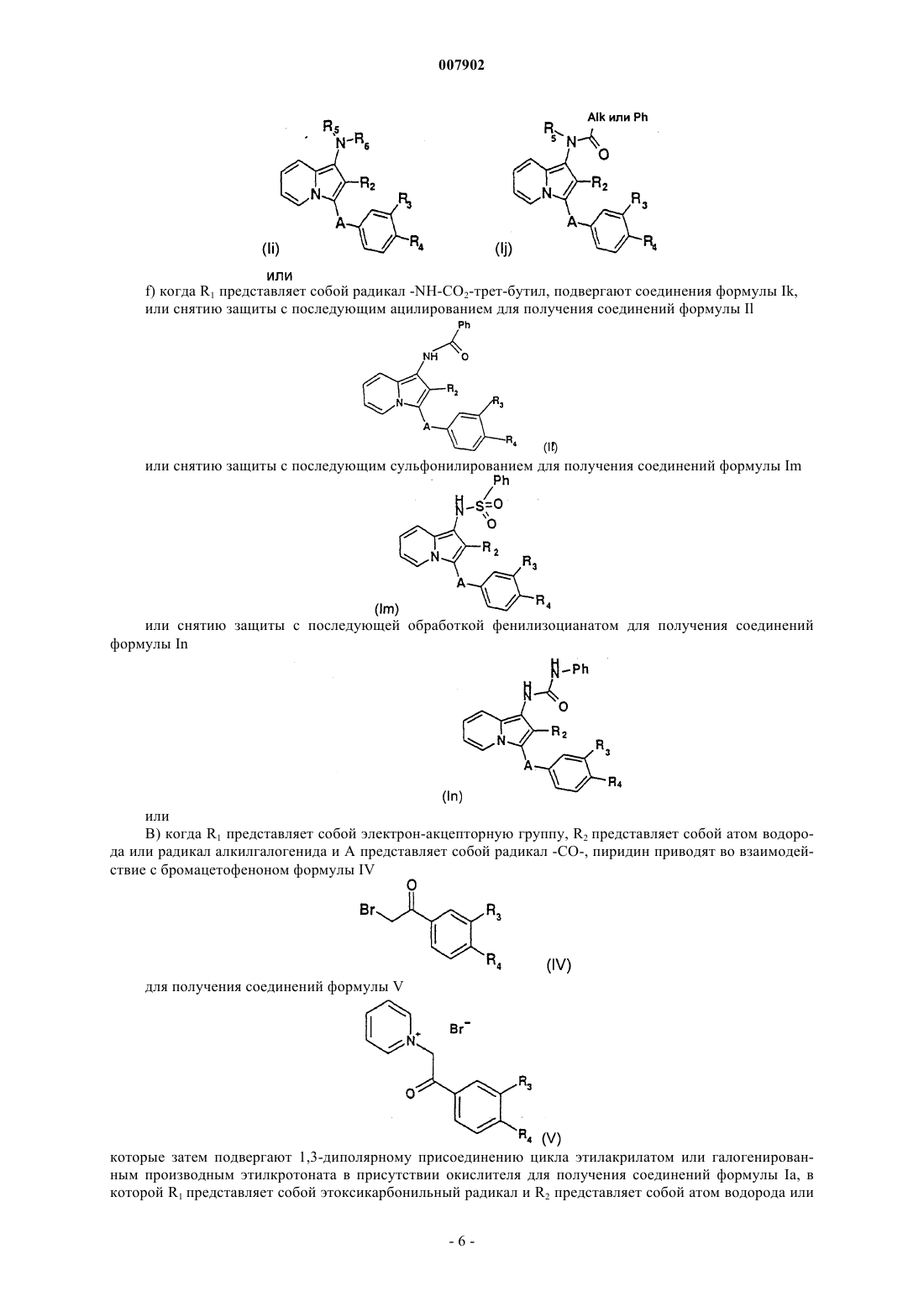
24. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R1 представляет собой NH-CO-Ph, подвергают алкилированию для получения соединений формулы I, в которой R1 представляет собой -N(Alk)-CO-Ph.
25. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R1 представляет собой NH-CO2-трет-бутил, a R3 и/или R4 представляют собой -NH-COCF3, подвергают снятию защиты с последующим ацилированием для получения соединений формулы I, в которой R1 представляет собой NH-CO-Ph.
26. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R1 представляет собой радикал -NH-CO2-трет-бутил, a R3 и/или R4 представляют собой -NH-COCF3, подвергают снятию защиты с последующим сульфонилированием для получения соединений формулы I, в которой R1 представляет собой -NH-SO2-Ph.
27. Способ получения соединений формулы I по пп.1-4, который отличается тем, что соединения формулы I, в которой R1 представляет собой радикал -NH-CO2-трет-бутил, a R3 и/или R4 представляют собой -NH-COCF3, подвергают снятию защиты с последующей обработкой фенилизоцианатом для получения соединений формулы I, в которой R1 представляет собой -NH-CO-NH-Ph.
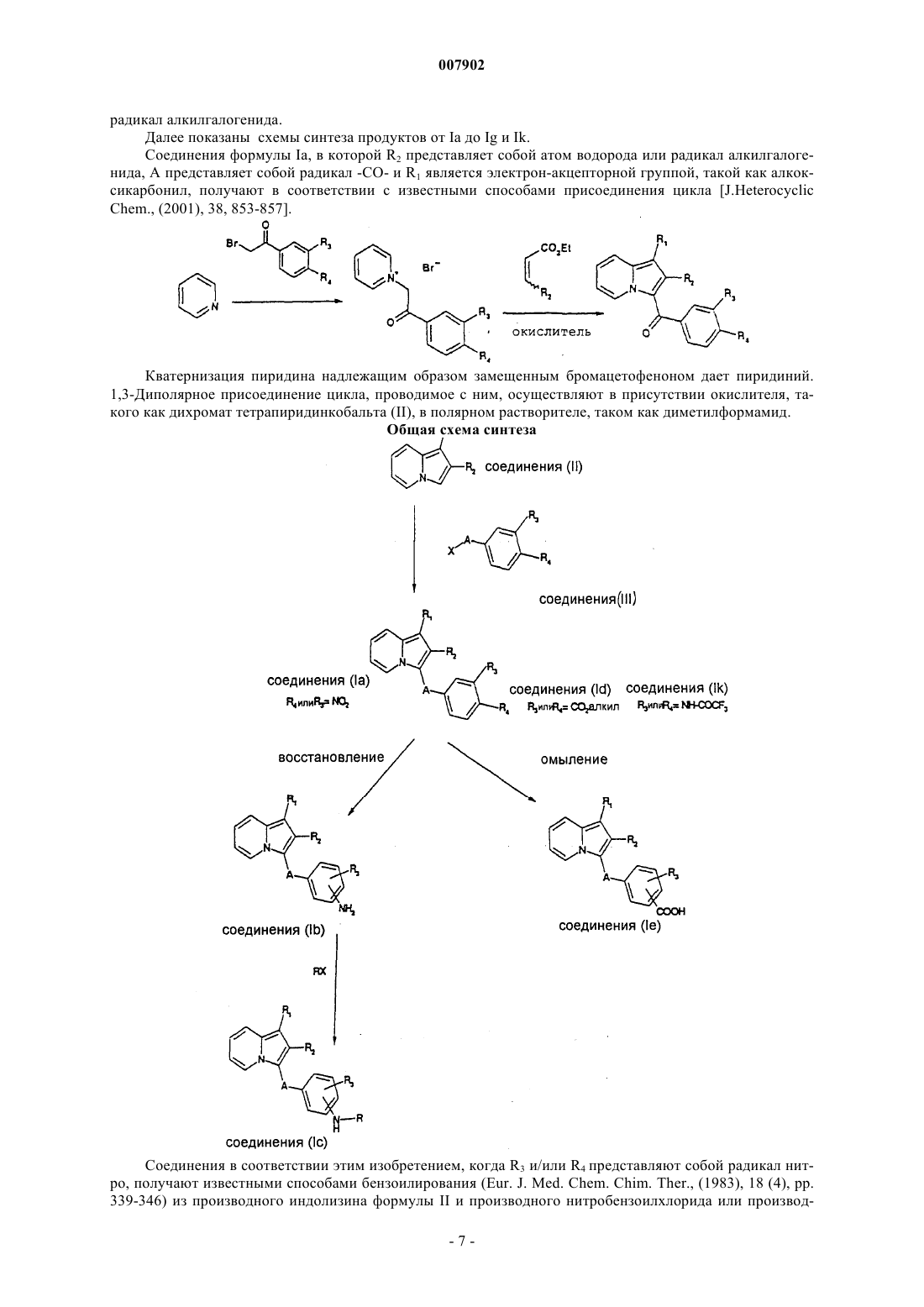
28. Способ получения соединений формулы I по пп.1-4, когда R1 представляет собой электронофильную группу, R2 представляет собой атом водорода или радикал алкилгалогенида и А представляет собой радикал -СО-, который отличается тем, что пиридин приводят во взаимодействие с бромацетофеноном формулы IV

для получения соединений формулы V

которые затем подвергают 1,3-диполярному присоединению цикла этилакрилатом или галогенированным производным этилкротоната в присутствии окислителя для получения соединений формулы Ia, в которой R1 представляет собой этоксикарбонильный радикал и R2 представляет собой атом водорода или радикал алкилгалогенида.
29. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы I по любому из пп.1-4, возможно в комбинации с одним или более чем одним из инертных и подходящих эксципиентов.
30. Фармацевтическая композиция по п.29, которая полезна в лечении заболеваний, требующих модуляции b-FGF.
31. Фармацевтическая композиция по п.29, которая полезна в лечении карцином, имеющих высокую степень васкуляризации, таких как карциномы легких, груди, простаты и пищевода, в лечении раковых заболеваний, которые индуцируют метастазы, таких как рак толстой кишки и рак желудка, и таких как меланомы, глиомы, лимфомы и лейкемии.
32. Фармацевтическая композиция по п.29, которая полезна в лечении сердечно-сосудистых заболеваний, таких как атеросклероз, рестеноз после ангиопластии, заболеваний, связанных с осложнениями, которые появляются вследствие установки внутрисосудистого протеза и/или аорто-коронарного шунтирования или других сосудистых трансплантантов, гипертрофии сердца или сосудистых осложнений при диабете, таких как диабетические ретинопатии.
33. Фармацевтическая композиция по п.29, которая полезна в лечении хронических воспалительных заболеваний, таких как ревматоидный артрит или воспалительные заболевания кишечника.
34. Фармацевтическая композиция по п.29, которая полезна в лечении ахондроплазии (АСН), гипохондроплазии (НСН) и TD (танатофорной дисплазии).
35. Применение соединения формулы I по п.1 для получения фармацевтической композиции, которая полезна в лечении заболеваний, требующих модуляции b-FGF.
Текст
007902 Предметом настоящего изобретения являются новые 1,2,3-замещенные производные индолизина,которые являются ингибиторами факторов роста фибробластов (FGF, basic fibroblast growth factor), способ их получения и фармацевтические композиции, содержащие их. Факторы роста фибробластов представляют собой семейство полипептидов, которых синтезирует большое число клеток во время эмбрионального развития и клетки взрослых тканей в разных патологических состояниях. Известны некоторые производные нафтиридинаминов и соответствующих мочевин, которые являются селективными ингибиторами FGF-1 (Batley В. et al., Life Sciences, (1998), vol. 62 No. 2. pp. 143-150;Thompson A. et al., J. Med. Chem., (2000), vol. 43. pp. 4200-4211). Некоторые производные индолизина раскрыты в заявках на патент и в патентах US 4378362, FR 2341578, GB 2064536, ЕР 0097636, ЕР 302792, ЕР 0382628 и ЕР 0235111. Эти соединения являются полезными в лечении стенокардии и аритмии. Для некоторых из этих соединений описаны свойства ингибирования транслокации кальция. Заявка на патент ЕР 0022762 также раскрывает некоторые производные индолизина, которые обладают активностью ингибирования ксантиноксидазы и аденозиндезаминазы и урикозурической активностью. Эти соединения могут быть полезными в лечении физиологических нарушений, которые происходят вследствие избытка мочевой кислоты и нарушений иммунной системы и паразитических агентов. Было обнаружено, что некоторые соединения, производимые из индолизина, являются мощными антагонистами связывания факторов роста фибробластов с их рецепторами. Соответственно, предметом настоящего изобретения являются новые производные индолизина формулы I в которой R1 представляет собой гидроксильный радикал, линейный или разветвленный радикал алкокси с числом атомов углерода от 1 до 5, карбоксильный радикал, алкоксикарбонильный радикал с числом атомов углерода от 2 до 6 или радикал формулы:-CO-NH-Alk,в которой Alk представляет собой алкильный радикал или линейный или разветвленный алкиленовый радикал с числом атомов углерода от 1 до 5,cAlk представляет собой циклоалкильный радикал с числом атомов углерода от 3 до 6,n представляет собой целое число от 0 до 5,m представляет собой целое число от 1 до 5,R5 и R6, которые являются идентичными или разными, каждый представляет собой атом водорода,линейный или разветвленный алкильный радикал с числом атомов углерода от 1 до 5 или бензильный радикал,R7 представляет собой атом водорода или алкильный радикал с числом атомов углерода от 1 до 5,R8 представляет собой алкильный радикал с числом атомов углерода от 1 до 5 или радикал -CO-Alk,Ph представляет собой фенильный радикал, который возможно замещен одним или более чем одним из галогеновых атомов, одним или более чем одним из радикалов алкокси с числом атомов углерода от 1 до 5, одним или более чем одним из карбоксильных радикалов или одним или более чем одним из-1 007902 алкоксикарбонильных радикалов с числом атомов углерода от 2 до 6,R2 представляет собой атом водорода, алкильный радикал с числом атомов углерода от 1 до 5, радикал алкилгалогенида с числом атомов углерода от 1 до 5, содержащий от 3 до 5 галогеновых атомов,циклоалкильный радикал с числом атомов углерода от 3 до 6 или фенильный радикал, который возможно замещен одним или более чем одним из галогеновых атомов, одним или более чем одним из радикалов алкокси с числом атомов углерода от 1 до 5, одним или более чем одним из карбоксильных радикалов или одним или более чем одним из алкоксикарбонильных радикалов с числом атомов углерода от 2 до 6,А представляет собой радикал -СО-, -SO- или -SO2-, - R3 и R4, которые являются идентичными или разными, каждый представляет собой атом водорода, радикал алкокси с числом атомов углерода от 1 до 5, радикал амино, карбоксильный радикал, алкоксикарбонильный радикал с числом атомов углерода от 2 до 6, гидроксильный радикал, радикал нитро, радикал гидроксиамино, радикал формулы-O-Alk-HetN в которой n, m, Alk, R5, R6 и R7 имеют значение, данное выше для R1, иR9 и R10, которые являются идентичными или разными, каждый представляет собой атом водорода или алкильный радикал с числом атомов углерода от 1 до 5,R11 представляет собой атом водорода или радикал -Alk-COOR12, где R12 представляет собой атом водорода, алкильный радикал с числом атомов углерода от 1 до 5 или бензильный радикал,HetN представляет собой 5- или 6-членный гетероцикл, содержащий по меньшей мере один атом азота и возможно другой гетероатом, выбранный из азота и кислорода, или R3 и R4 совместно образуют 5- или 6-членный ненасыщенный гетероцикл, при том условии, однако, что, когда R3 представляет собой радикал алкокси и R4 представляет собой радикал -O-Alk-NR9R10 или гидроксильный радикал, R1 не представляет собой атом водорода или радикал алкокси, возможно в форме одной из их фармацевтически приемлемых солей. Предпочтительным является соединение формулы I, в котором R1 представляет собой гидроксильный радикал, линейный или разветвленный радикал алкокси с числом атомов углерода от 1 до 5, карбоксильный радикал, алкоксикарбонильный радикал с числом атомов углерода от 2 до 6 или радикал формулыAlk представляет собой алкильный радикал или линейный или разветвленный алкиленовый радикал с числом атомов углерода от 1 до 5,cAlk представляет собой циклоалкильный радикал с числом атомов углерода от 3 до 6,n представляет собой целое число от 0 до 5,-2 007902m представляет собой целое число от 1 до 5,R5 и R6, которые являются идентичными или разными, каждый представляет собой атом водорода,линейный или разветвленный алкильный радикал с числом атомов углерода от 1 до 5 или бензильный радикал,R7 представляет собой атом водорода или алкильный радикал с числом атомов углерода от 1 до 5,R8 представляет собой алкильный радикал с числом атомов углерода от 1 до 5 или радикал -CO-Alk,Ph представляет собой фенильный радикал, который, возможно, замещен одним или более чем одним из галогеновых атомов, одним или более чем одним из радикалов алкокси с числом атомов углерода от 1 до 5, одним или более чем одним из карбоксильных радикалов или одним или более чем одним из алкоксикарбонильных радикалов с числом атомов углерода от 2 до 6,R2 представляет собой алкильный радикал с числом атомов углерода от 1 до 5, трифторметильный радикал, циклоалкильный радикал с числом атомов углерода от 3 до 6 или фенильный радикал, который возможно замещен одним или более чем одним из галогеновых атомов, одним или более чем одним из радикалов алкокси с числом атомов углерода от 1 до 5, одним или более чем одним из карбоксильных радикалов или одним или более чем одним из алкоксикарбонильных радикалов с числом атомов углерода от 2 до 6,А представляет собой радикал -СО- или -SO2-,R3 и R4, которые являются идентичными или разными, каждый представляет собой атом водорода,радикал алкокси с числом атомов углерода от 1 до 5, радикал амино, карбоксильный радикал, алкоксикарбонильный радикал с числом атомов углерода от 2 до 6, радикал нитро, радикал гидроксиамино, радикал формулы-NH-Alk-HetN в которой n, m, Alk, R5, R6 и R7 имеют значение, данное выше для R1, и R9 и R10, которые являются идентичными или разными, каждый представляет собой атом водорода или алкильный радикал с числом атомов углерода от 1 до 5,R11 представляет собой атом водорода или радикал -Alk-COOR12, где R12 представляет собой атом водорода, алкильный радикал с числом атомов углерода от 1 до 5 или бензильный радикал,HetN представляет собой 5- или 6-членный гетероцикл, содержащий по меньшей мере один атом азота и возможно другой гетероатом, выбранный из азота и кислорода, возможно в форме одной из их фармацевтически приемлемых солей. Особенно предпочтительным является соединение формулы I, в котором R1 представляет собой радикал алкокси с числом атомов углерода от 1 до 5, карбоксильный радикал, радикал -O-Alk-COOH, в котором Alk представляет собой линейный или разветвленный алкиленовый радикал с числом атомов углерода от 1 до 5, радикал формулы -O-Alk-Ph, в котором Alk представляет собой алкиленовый радикал с числом атомов углерода от 1 до 5 и Ph представляет собой фенильный радикал, который возможно замещен одним или более чем одним из галогеновых атомов или одним, или более чем одним из радикалов алкокси с числом атомов углерода от 1 до 5, или одним или более чем одним из карбоксильных радикалов, радикал формулы -NH-CO-Ph, радикал формулы -NH-SO2-Ph или радикал формулы -NH-CO-NH-Ph,-R2 представляет собой алкильный радикал с числом атомов углерода от 1 до 5,А представляет собой радикал -СО-,R3 и R4, которые являются разными, каждый представляет собой атом водорода, радикал алкокси с числом атомов углерода от 1 до 5, радикал амино, карбоксильный радикал или алкоксикарбонильный радикал с числом атомов углерода от 2 до 6, возможно в форме одной из его фармацевтически приемлемых солей. Среди соединений по этому изобретению особенно предпочтительными являются следующие:(4-амино-3-метоксифенил)-(1-метокси-2-метилиндолизин-3-ил)метанон,3-(4-амино-3-метоксибензоил)-2-метилиндолизин-1-ил-карбоновая кислота,2-[3-(4-амино-3-метоксибензоил)-2-метилиндолизин-1-ил]оксиуксусная кислота,(4-амино-3-метоксифенил)-1-[(4-хлорбензил)окси]-2-метилиндолизин-3-илметанон,(4-амино-3-метоксифенил)-1-[(3-метоксибензил)окси]-2-метилиндолизин-3-илметанон,4-([3-(4-амино-3-метоксибензоил)-2-метилиндолизин-1-ил]оксиметил)бензойная кислота,3-(4-карбоксибензоил)-2-метилиндолизин-1-ил-карбоновая кислота,метил-3-[(1-метокси-2-метилиндолизин-3-илкарбонил]бензоат,4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]бензойная кислота,-3 007902 2-амино-5-[(1-метокси-2-метилиндолизин-3-ил)карбонил]бензойная кислота,2-амино-5-(1-[(3-метоксибензоил)амино]-2-метилиндолизин-3-илкарбонил)бензойная кислота,2-амино-5-(2-метил-1-[(3,4,5-триметоксибензоил)амино]индолизин-3-илкарбонил)бензойная кислота,2-амино-5-(1-[(3-метоксифенил)сульфонил]амино-2-метилиндолизин-3-илкарбонил)бензойная кислота,возможно в форме одной из его фармацевтически приемлемых солей. Настоящее изобретение также относится к способу получения соединений формулы I, который отличается тем, что А) производное индолизина формулы II где R1 и R2 имеют значение, которое дано для формулы I, но R2 не представляет собой атом водорода или радикал алкилгалогенида, конденсируют с производным формулы III где X представляет собой атом галогена, a R3 или R4, которые являются идентичными или разными,каждый представляет собой атом водорода, радикал нитро, радикал трифторацетамидо или алкоксикарбонильный радикал с числом атомов углерода от 2 до 6, для получения соединений формулы Ia, Id или Ik и затем а) подвергают соединения формулы Iа восстановлению для получения соединений формулы Ib в которой R3 и/или R4 представляют собой радикал амино, причем эти соединения формулы Ib затем подвергают действию алкилгалогенида для получения соединений формулы I, для которых R4 и/или R3 представляют собой радикал -NR5R6 (в котором R5 представляет собой атом водорода и R6 представляет собой алкильный радикал с числом атомов углерода от 1 до 5) и радикал -NH-Alk-NR5R6 или радикал -NH-Alk-COOR7 (в котором R7 не представляет собой атом водорода), из которых последующим омылением получают соединения формулы I,для которых R4 и/или R3 представляют собой радикал -NH-Alk-COOR7, в котором R7 представляет собой атом водорода,или подвергают ацилированию для получения соединений формулы I, для которых R4 и/или R3 представляют собой радикал -NH-CO-Alk или радикал -NH-CO-Alk-NR9R10, которые затем подвергают алкилированию для получения радикала -N-(R11)-CO-Alk или радикала -N-(R11)-CO-Alk-NR9R10, в кото-4 007902 ром R11 представляет собой радикал -Alk-COOR12, в котором R12 не представляет собой атом водорода,причем эти последние соединения затем возможно подвергают омылению для получения соединений формулыI, для которых R4 и/или R3 представляют собой радикал -N-(R11)-CO-Alk или радикал -N-(R11)-CO-Alk-NR9R10,где R11 представляет собой радикал -Alk-COOH, подвергают сульфонилированию для получения соединений формулы I, для которых R4 и/или R3 представляют собой радикал -NH-SO2-Alk или радикал -NH-SO2-AlkNR9R10, которых затем подвергают алкилированию для получения радикала -N(R11)-SO2-Alk или радикала -N(R11)-SO2-Alk-NR9R10, где R11 представляет собой радикал -Alk-COOR12, в котором R12 не представляет собой атом водорода, причем эти последние соединения затем возможно подвергают омылению для получения соединений формулы I, для которых R4 и/или R3 представляют собой радикал -N(R11)-SO2-Alk или радикал, -N(R11)-SO2-Alk-NR9R10, где R11 представляет собой радикал -Alk-COOHb) соединения формулы Id, в которой R3 и/или R4 представляют собой алкоксикарбонильный радикал, подвергают омылению для получения соединений формулы I, в которой R3 и/или R4 представляют собой карбоксильный радикал, илиc) когда R1 представляет собой радикал бензилокси, подвергают соединения формулы Ia действию трифторуксусной кислоты или подвергают соединения формулы Id гидрогенизированию для получения соединений формулы If в которой R3 и/или R4 имеют значения, которые даны выше, и затем соединения формулы If подвергают О-алкилированию для получения соединений формулы Ig в которой R3 и/или R4 имеют значения, которые даны выше, и R1 представляет собой линейный или разветвленный радикал алкокси с числом атомов углерода от 1 до 5, радикал -O-(CH2)n-cAlk, радикал -OAlk-COOR7, радикал -O-Alk-NR5R6, радикал -O-(CH2)n-Ph или радикал -O-Alk-O-R8, который, когда R8 представляет собой радикал -СОСН 3, может дать при последующем омылении радикал -O-Alk-OH- или радикал -O-Alk-CN, который при обработке гидроксиламином дает радикал -O-Alk-C(NH2)=NOH, илиd) когда R1 представляет собой алкоксикарбонильный радикал, подвергают соединения формулы Ia омылению для получения соединений формулы Ih в которой R3 и/или R4 имеют значения, данные выше, которые затем подвергают действию производного амина для получения соединений формулы I, в которой R1 представляет собой радикал -CO-NH-Alk, или действию производного аминокислоты для получения соединений формулы I, в которой R1 представляет собой радикал -CO-NH-(CH2)m-COOR7 или е) когда R1 представляет собой радикал -NH-СО 2-трет-бутил, подвергают соединения формулы Ia или Id или алкилированию с последующим снятием защиты и возможным вторым алкилированием для получения соединений формулы Ii, или снятию защиты с последующим ацилированием для получения соединений формулы Ij, в которой R5 представляет собой атом водорода, с последующим возможным алкилированием для получения соединений формулы Ij, в которой R5 представляет собой алкильный радикалf) когда R1 представляет собой радикал -NH-CO2-трет-бутил, подвергают соединения формулы Ik,или снятию защиты с последующим ацилированием для получения соединений формулы Il или снятию защиты с последующим сульфонилированием для получения соединений формулы Im или снятию защиты с последующей обработкой фенилизоцианатом для получения соединений формулы In или В) когда R1 представляет собой электрон-акцепторную группу, R2 представляет собой атом водорода или радикал алкилгалогенида и А представляет собой радикал -СО-, пиридин приводят во взаимодействие с бромацетофеноном формулы IV для получения соединений формулы V которые затем подвергают 1,3-диполярному присоединению цикла этилакрилатом или галогенированным производным этилкротоната в присутствии окислителя для получения соединений формулы Ia, в которой R1 представляет собой этоксикарбонильный радикал и R2 представляет собой атом водорода или-6 007902 радикал алкилгалогенида. Далее показаны схемы синтеза продуктов от Ia до Ig и Ik. Соединения формулы Ia, в которой R2 представляет собой атом водорода или радикал алкилгалогенида, А представляет собой радикал -СО- и R1 является электрон-акцепторной группой, такой как алкоксикарбонил, получают в соответствии с известными способами присоединения цикла [J.Heterocyclic Кватернизация пиридина надлежащим образом замещенным бромацетофеноном дает пиридиний. 1,3-Диполярное присоединение цикла, проводимое с ним, осуществляют в присутствии окислителя, такого как дихромат тетрапиридинкобальта (II), в полярном растворителе, таком как диметилформамид. Общая схема синтеза Соединения в соответствии этим изобретением, когда R3 и/или R4 представляют собой радикал нитро, получают известными способами бензоилирования (Eur. J. Med. Chem. Chim. Ther., (1983), 18 (4), pp. 339-346) из производного индолизина формулы II и производного нитробензоилхлорида или производ-7 007902 ного нитробензолсульфонилхлорида, причем эти соединения соответствуют соединению формулы III. Так получают соединения формулы Ia. Соединения формулы Ib, в которой R3 и/или R4 представляют собой радикал амино, получают из соединений формулы Ia восстановлением нитрофункциональной группы. Подвергая соединения формулы Ib действию алкилгалогенида, получают соединения формулы Iс, для которых R3 и/или R4 представляют собой радикал -NR5R6 (в котором R5 представляет собой атом водорода и R6 имеет значения, которые даны выше), радикал -NH-Alk-NR5R6 или радикал -NH-Alk-COOR7, в котором R7 не представляет собой атом водорода. Соединения, для которых R7 представляет собой атом водорода, получают из этих последних соединений, подвергая их последующему омылению. Ацилированием соединений формулы Ib получают соединения формулы Ic, для которых R3 и/илиR4 представляют собой радикал -NH-CO-Alk или радикал -NH-CO-Alk-NR9R10. Подвергая эти соединения, для которых R3 и/или R4 представляют собой радикал -NH-CO-Alk или радикал -NH-CO-Alk-NR9R10, алкилированию производным, содержащим алкоксикарбонильный остаток,получают соединения формулы I, для которых R3 и/или R4 представляют собой радикал -N(R11)-CO-Alk или радикал -N(R11)-CO-Alk-NR9R10, где R11 представляет собой радикал -Alk-COOR12, где R12 не представляет собой атом водорода. Подвергая эти последние продукты омылению, получают соединения, для которых R3 и/или R4 представляют собой радикал -N(R11)-CO-Alk или радикал -N(R11)-CO-Alk-NR9R10,где R11 представляет собой радикал -Alk-COOH. Сульфонилированием соединений формулы Ib получают соединения формулы Ic, для которых R3 и/или R4 представляют собой радикал -NH-SO2-Alk или радикал -NH-SO2-Alk-NR9R10. Подвергая эти соединения, для которых R3 и/или R4 представляют собой радикал -NH-SO2-Alk или радикал -NH-SO2-Alk-NR5R10, алкилированию производным, содержащим алкоксикарбонильный остаток, получают соединения формулы I, для которых R3 и/или R4 представляют собой радикал -N(R11)-SO2Alk или радикал -N(R11)-SO2-Alk-NR9R10, где R11 представляет собой радикал -Alk-COOR12, где R12 не представляет собой атом водорода. Подвергая эти последние продукты омылению, получают соединения, для которых R3 и/или R4 представляют собой радикал -N(R11)-SO2-Alk или радикал -N(R11)-SO2-AlkNR9R10, где R11 представляет собой радикал -Alk-COOH. Проведением взаимодействия производного индолизина формулы II с производным алкоксикарбонилбензоилхлорида формулы III получают соединения формулы Id, в которой R3 и/или R4 представляют собой алкоксикарбонильный радикал. Подвергая эти последние соединения омылению, получают соединения формулы Ie, в которой R3 и/или R4 представляют собой карбоксильный радикал. Проведением взаимодействия производного индолизина формулы II с производным трифторацетамидобензоилхлорида формулы III получают соединения формулы Ik, в которой R3 и/или R4 представляют собой трифторацетамидный радикал. Подвергая эти последние соединения щелочному гидролизу,получают соединения формулы Ik, в которой R3 и/или R4 представляют собой карбоксильный радикал и/или радикал амино. Как показывает фиг. 2, начиная с соединений формулы I, в которой R1 представляет собой радикал бензилокси и R3 или R4 представляют собой алкоксикарбонильный радикал, можно получать, подвергая эти соединения гидрогенированию, соединения формулы If. Когда R3 или R4 представляет собой радикал нитро, действием трифторуксусной кислоты получают соединения формулы If. Подвергая соединения формулы If О-алкилированию, получают соединения формулы Ig, в которой R1 представляет собой линейный или разветвленный радикал алкокси с числом атомов углерода от 1 до 5, радикал O-(CH2)n-cAlk, радикал -O-Alk-COOR7, радикал -O-Alk-NR5R6, радикал -O-(CH2)n-Ph, радикал -О-AlkO-R8 (который, когда R8 представляет собой радикал -СОСН 3, может дать при омылении, радикал -OAlk-OH), радикал -O-Alk-CN, который при обработке гидроксиламином может дать радикал -O-AlkC(NH2):NOH. Для получения соединений формулы Ih, в которой R1 представляет собой карбоксильный радикал и А представляет собой радикал -СО- или радикал -SO2, соединения формулы Ia, в которой R1 представляет собой алкоксикарбонильный радикал, подвергают омылению. Полученные таким образом производные индолизин-1-илкарбоновой кислоты формулы Ih можно затем подвергнуть действию амина, чтобы получить соединения формулы Ih, в которой R1 представляет собой радикал -CO-NH-Alk, или действию производного аминокислоты для получения соединений формулы I, в которой R1 представляет собой радикал -CO-NH-(CH2)m-COOR7. Соединения формулы II, когда R1 представляет собой радикал -NH-COO-трет-бутил или радикал -N(CH3)CH2C6H5, получают в соответствии с нижеследующими схемами синтеза при использовании реакции Чичибабина (Synthesis, (1975), р. 209) для получения индолизинов. Соединения формулы II, когда R1 представляет собой радикал -ОСН 3 или радикал -ОСН 2 С 6 Н 5, также получают с использованием реакции Чичибабина в соответствии с нижеследующими схемами синтеза: Соединения формулы I являются мощными антагонистами FGF1 и 2. Их способности ингибировать как образование новых сосудов из дифференцированных эндотелиальных клеток, так и блокировать дифференцировку клеток CD34+ CD133+ костного мозга взрослых в эндотелиальные клетки была продемонстрирована in vitro. Более того, их способность ингибировать патологический ангиогенез была продемонстрирована in vivo. Вообще, факторы роста фибробластов сильно вовлечены посредством аутокринной, паракринной и юкстакринной секреции в феномены нарушенной регуляции стимуляции роста раковых клеток. Более того, факторы роста фибробластов влияют на ангиогенез в опухолях, который играет преобладающую роль как в росте опухолей, так и феномене метастазирования. Ангиогенез является процессом генерирования новых капиллярных сосудов из предсуществующих сосудов или мобилизацией и дифференцировкой клеток костного мозга. Так, в процессах неоваскуляризации опухолей наблюдают как неконтролируемую пролиферацию эндотелиальных клеток, так и мобилизацию ангиобластов из костного мозга. Было показано in vitro и in vivo, что эндотелиальную пролиферацию стимулируют несколько факторов роста и, в частности, FGF1 или a-FGF и FGF2 или b-FGF. Эти два фактора индуцируют пролиферацию, миграцию и продуцирование протеаз эндотелиальными клетками в культуре и неоваскуляризацию in vivo. a-FGF и b-FGF взаимодействуют с эндотелиальными клетками посредством двух классов рецепторов, высокоаффинных рецепторов с тирозинкиназной активностью (FGFR5) и низкоаффинных рецепторов гепаринсульфатпротеогликанового (HSPG) типа, находящихся на поверхности клеток и во внеклеточных матриксах. При том, что паракринная роль этих двух факторов в эндотелиальных клетках описана широко, a-FGF и b-FGF могут действовать на эти клетки в аутокринном процессе. Таким образом, a-FGF и b-FGF и их рецепторы представляют собой весьма подходящие мишени для терапевтических воздействий, направленных на ингибирование процесса ангиогенеза (Keshet E., Ben-Sasson S. A., J. Clin. Invest, (1999), vol. 501, pp. 104-1497; Presta M., Rusnati M.,Dell'Era P., Tanghetti E., Urbinati C., Giuliani R. et al., New York: Plenum Publishers, (2000), pp. 7-34, Billottet C, Janji В., Thiery J.P., Jouanneau J., Oncogene, (2002), vol. 21. pp. 8128-8139). Более того, систематические исследования с целью определения экспрессии, происходящей из-заa-FGF и b-FGF и их рецепторов (FGFR), в разных типах опухолевых клеток показывают, что клеточный ответ на эти два F фактора является функциональным в значительном большинстве исследованных человеческих раковых линий. Эти результаты подтверждают гипотезу, что антагонист a-FGF и b-FGF может также ингибировать пролиферацию опухолевых клеток (Chandler L.A., Sosnowski B.A, Greenlees L, Aukerman S.L., Baird A., Pierce G.F., Int. J. Cancer, (1999), vol. 58. pp. 81-451).a-FGF и b-FGF играют важную роль в росте и поддержании клеток простаты. На животных моделях и людях было показано, что изменение клеточного ответа на эти факторы играет решающую роль в про-9 007902 грессии рака простаты. Действительно, при этих патологиях регистрируют как увеличение продукции aFGF и b-FGF фибробластами и эндотелиальными клетками, присутствующими в опухоли, так и увеличение экспрессии FGFR рецепторов на опухолевых клетках. Таким образом, происходит паракринная стимуляция клеток рака простаты, и этот процесс может быть важным компонентом этой патологии. Соединение, обладающее активностью антагониста FGFR рецепторов, такое как соединения по настоящему изобретению, может представлять собой предпочтительную терапию при этих патологиях (Giri D.,Ropiquet F., Clin. Cancer Res., (1999), vol. 71. pp. 5-1063; Doll J.A., Reiher F.K., Crawford S.E., Pins M.R.,Campbell S.C., Bouck N.P., Prostate, (2001), vol. 305, pp. 49-293). Несколько исследований демонстрируют присутствие a-FGF и b-FGF и их FGFR рецепторов как в линиях клеток опухолей молочной железы человека (в частности MCF7), так и в биопсиях этих опухолей. Эти факторы при этой патологии могут быть причиной появления весьма агрессивного фенотипа,вызывающего сильное метастазирование. Таким образом, соединение, имеющее активность антагониста рецептора FGF, такое как соединения формулы I, может представлять собой предпочтительную терапию при этих патологиях (Vercoutter-Edouart A-S., Czeszak X., Crepin M., Lemoine J., Boilly В., Le Bourhis X. etal., Exp. Cell Res., (2001), vol. 262. pp. 59-68). Злокачественные меланомы являются опухолями, которые с высокой частотой индуцируют метастазы и очень устойчивы к разным видам химиотерапии. Процессы ангиогенеза играют главную роль в прогрессии злокачественной меланомы. Более того, показано, что вероятность появления метастазов очень сильно возрастает при возрастании васкуляризации первичной опухоли. Клетки меланомы продуцируют и секретируют разные ангиогенные факторы, в том числе a-FGF и b-FGF. Более того, показано,что ингибирование клеточных эффектов этих двух факторов растворимыми FGFR1 блокирует пролиферацию и выживание опухолевых клеток меланомы in vitro и прогрессию опухоли in vivo. Таким образом,соединение, имеющее активность антагониста FGFR рецептора, такое как соединения по настоящему изобретению, может представлять собой предпочтительную терапию при этих патологиях (Rofstad E.K.,Halsor E.F., Cancer Res., (2000); Yayon A., Ma Y-S., Safran M., Klagsbrun M., Halaban R., Oncogene, (1997),vol. 14, pp. 2999-3009). Клетки глиомы продуцируют a-FGF и b-FGF in vitro и in vivo и имеют разные FGFR на своей поверхности. Таким образом, из этого следует, что эти два фактора посредством аутокринного и паракринного действия играют центральную роль в прогрессии опухоли этого типа. Более того, как и у большинства солидных опухолей, прогрессия глиом и их способность индуцировать метастазы сильно зависит от ангиогенных процессов в первичной опухоли. Также было показано, что антисмысловые последовательности FGFR1 блокируют пролиферацию человеческих астроцитом. Более того, описаны производные нафталинсульфонатов для ингибирования клеточных эффектов a-FGF и b-FGF in vitro и ангиогенеза, индуцированного этими факторами роста in vivo. Интрацеребральная инъекция этих соединений индуцирует весьма значительное увеличение апоптоза и существенное снижение ангиогенеза, что приводит к значительной регрессии глиом у крыс. Таким образом, соединение, имеющее активность антагониста а-FGF,и/или b-FGF, и/или FGFR рецепторов, такое как соединения по настоящему изобретению, может представлять собой предпочтительную терапию при этих патологиях (Yamada S.M., Yamaguchi F., Brown R.,Berger M.S., Morrison R.S., Glia, (1999), vol. 76 pp. 28-66; Auguste P., Gursel D.B., Lemiere S., Reimers D.,Cuevas P.,Carceller F., et al., Cancer Res., (2001), vol. 26. pp. 61-1717). Совсем недавно была документирована потенциальная роль проангиогенных агентов при лейкемиях и лимфомах. Действительно, было доложено, что вообще клеточные клоны при этих патологиях могут либо быть естественным путем уничтожены иммунной системой, либо могут переключиться на ангиогенный фенотип, который способствует их выживанию и затем их пролиферации. Это изменение фенотипа индуцировано чрезмерной экспрессией ангиогенных факторов, в частности макрофагами, и/или мобилизацией этих факторов из внеклеточного матрикса (Thomas D.A., Giles F.J., Cortes J., Albitar M.,Kantarjian H.M., Acta Haematol, (2001), vol. 207, pp. 106-190). Из числа этих ангиогенных факторов b-FGF найдены во многих линиях клеток лимфобластоидных и гемопоэтических опухолей. FGFR рецепторы также присутствуют у большинства этих линий, откуда следуют возможные аутокринные клеточные эффекты a-FGF и b-FGF, вызывающие пролиферацию этих клеток. Более того, сообщают, что в костном мозге ангиогенез из-за паракринных эффектов коррелирует с прогрессией некоторых из этих патологий. Более конкретно, показано, что b-FGF в клетках хронической лимфоидной лейкемии индуцирует увеличение экспрессии антиапоптозного белка (Вс 12), что приводит к увеличению выживаемости этих клеток и поэтому существенно участвует в их малигнизации. Более того, уровни b-FGF, измеренные в этих клетках, очень хорошо коррелируют с клинической стадией этого заболевания и устойчивостью к химиотерапии, применяемой при этой патологии (флударабин). Таким образом, соединение, имеющее активность антагониста FGFR рецептора, такое как соединения по настоящему изобретению, может представлять собой при этой патологии предпочтительную терапию в комбинации как с флударабином,так и с другими активными продуктами (Thomas D.A., Giles F.J., Cortes J., Albitar M., Kantarjian H.M.,Acta Haematol, (2001), vol. 207, pp. 106-190; Gabrilove J.L. Oncologist, (2001), vol. 6. pp. 4-7). При хронической миелоидной лейкемии существует корреляция между процессом ангиогенеза в костной ткани и экстрамедуллярными заболеваниями. Различные исследования демонстрируют, что ин- 10007902 гибирование ангиогенеза, в частности, соединением, имеющим активность антагониста FGFR рецептора,может представлять собой предпочтительную терапию при этой патологии. Пролиферация и миграция гладкомышечных клеток кровеносных сосудов вносит вклад в гипертрофию интимы артерий и таким образом играет ведущую роль в атеросклерозе ирестенозе после ангиопластии и эндартеректомии. Исследования in vivo показывают происходящую после повреждения сонной артерии введением баллона локальную продукцию a-FGF и b-FGF. В этой же модели нейтрализующие антитела противFGF2 ингибируют пролиферацию гладкомышечных клеток кровеносных сосудов и таким образом снижают гипертрофию интимы. Химерный белок FGF2, связанный с такой молекулой, как сапонин, блокирует связывание b-FGF его FGFR рецепторами, ингибирует пролиферацию гладкомышечных клеток кровеносных сосудов invitro и гипертрофию интимы in vivo (Epstein C.E., Siegall С.В., Biro S., Fu Y.M., FitzGerald D., Circulation,(1991), vol. 87, pp. 84-778; Waltenberger J., Circulation, (1997), pp. 96-4083). Таким образом, антагонисты FGFR рецепторов, такие как соединения по настоящему изобретению,представляют собой предпочтительную терапию в отдельности или в комбинации с соединениями, которые являются антагонистами других факторов роста, участвующих в этих патологиях, таких как фактор роста из тромбоцитов, при лечении патологий, связанных с пролиферацией гладкомышечных клеток кровеносных сосудов, таких как атеросклероз, рестеноз после ангиопластии или после введения внутрисосудистых протезов (расширителей), или во время операции по аорто-коронарному шунтированию. Гипертрофия сердца происходит в ответ на стресс стенки желудочков, вызываемый перегрузкой в смысле давления и объема. Такая перегрузка может быть последствием многочисленных патофизиологических состояний, таких как гипертензия, коарктация аорты, инфаркт миокарда и разные сосудистые нарушения. Последствиями этой патологии являются морфологические, молекулярные и функциональные изменения, такие как гипертрофия миоцитов сердца, накопление белков матрикса и повторная экспрессия эмбриональных генов. В этой патологии участвует b-FGF. Действительно, добавление b-FGF в культуры кардиомиоцитов новорожденных крыс модифицирует профиль соответствующих генов к сократимым белкам, что приводит к генному профилю, который подобен эмбриональному. Кроме того,миоциты взрослой крысы проявляют гипертрофическую реакцию под действием b-FGF, причем эту реакцию блокируют нейтрализующие антитела против b-FGF. Эксперименты, проведенные in vivo на трансгенных мышах, нокаутных по b-FGF, показывают, что b-FGF является главным стимулирующим фактором для гипертрофии миоцитов сердца при этой патологии (Schultz JeJ., Witt S.A., Nieman M.L.,Reiser P.J., Engle S.J., Zhou M. et al., J. Clin. Invest, (1999), vol. 19. pp. 104-709). Соответственно, соединение, такое как соединения по настоящему изобретению, имеющее активность антагониста FGFR рецептора, представляет собой предпочтительную терапию при лечении сердечной недостаточности и другой патологии, связанной с дегенерацией сердечной ткани. Это лечение можно проводить в отдельности или в комбинации с существующими лечебными средствами (бетаблокаторы, диуретики, антагонисты ангиотензина, антиаритмические средства, антагонисты кальция,антитромботические средства и т.п.). Сосудистые нарушения, вызываемые диабетом, характеризуются изменением реактивности сосудов и кровотока, повышенной проницаемостью, чрезмерным пролиферативным ответом и увеличением отложений белков матрикса. Более конкретно, a-FGF и b-FGF присутствуют в преретинальных мембранах пациентов с диабетическими ретинопатиями, в мембранах подлежащих капилляров, и стекловидном теле пациентов, страдающих пролиферативными ретинопатиями. Для сосудистых нарушений, связанных с диабетом, разработан растворимый рецептор для FGF, способный связывать как a-FGF, так и b-FGF(Tilton R.G., Dixon RF., Brock T.A., Exp. Opin. Invest. Drugs, (1997), vol. 84, pp. 6-1671). Таким образом,соединение, такое как соединения формулы I, имеющее активности антагониста FGFR рецептора, представляет собой предпочтительную терапию в отдельности или в комбинации с соединениями, которые являются антагонистами других факторов роста, участвующих в этих патологиях, таких как фактор роста эндотелия сосудов. Ревматоидный артрит (РА) представляет собой хроническое заболевание неизвестной этиологии. При том, что он поражает многие органы, самой тяжелой формой РА является прогрессирующее синовиальное воспаление суставов, приводящее к их разрушению. Оказывается, что на прогрессию этого заболевания сильно влияет ангиогенез. Так, a-FGF и b-FGF были найдены в синовиальной ткани и в жидкости суставов пациента с РА, что указывает на участие этого фактора роста в инициации и/или прогрессии этой патологии. На моделях артрита, вызванного адъювантом у крыс, показано, что чрезмерная экспрессия b-FGF увеличивает тяжесть этого заболевания, тогда как нейтрализующие антитела против b-FGF блокируют прогрессию РА (Yamashita A., Yonemitsu Y., Okano S., Nakagawa К., Nakashima Y., Irisa T. etal., J. Immunol., (2002), vol. 57, pp. 168-450; Manabe N., Oda H., Nakamura K., Kuga Y., Uchida S., Kawaguchi H., Rheumatol, (1999), vol. 20. pp. 38-714). Таким образом, соединения в соответствии с этим изобретением представляют собой предпочтительную терапию при этой патологии. Воспалительные заболевания кишечника (ВЗК) включают в себя две формы хронических воспалительных заболеваний кишечника: язвенный колит и болезнь Крона. ВЗК характеризуются иммунными- 11007902 дисфункциями, которые приводят к неуместной продукции воспалительных цитокинов, индуцирующих возникновение локальной системы микрососудов. Последствием этого ангиогенеза, имеющего воспалительное происхождение, является ишемия кишечника, вызванная сужением сосудов. У пациентов, страдающих этими патологиями, измерены высокие уровни циркулирующего и локального b-FGF (KanazawaS., Tsunoda Т., Onuma E., Majima Т., Kagiyama M., Kkuchi K., American Journal of Gastroenterology, (2001),vol. 28. pp. 96-822; Thorn M., Raab Y., Larsson A., Gerdin В., Hallgren R., Scandinavian Journal of Gastroenterology, (2000), vol. 12, pp. 35-408). Соединения по этому изобретению, которые проявляют высокую антиангиогенную активность в модели воспалительного ангиогенеза, представляют собой предпочтительную терапию при этих патологиях.FGFR 1, 2 и 3 участвуют в процессах хондрогенеза и остеогенеза. Мутации, приводящие к экспрессии перманентно активированных FGFR, были связаны с большим числом наследственных заболеваний человека, которые приводят к порокам развития скелета, таким как синдромы Пфейффера (Pfeiffer), Крузона (Crouzon), Аперта (Apert), Джексона-Вейсса (Jackson-Weiss) и синдром свернутой кожи БираСтивенсона (Beare-Stevenson cutis gyrata). Некоторые из этих мутаций, которые, более конкретно, затрагивают FGFR3, приводят, в частности, к ахондроплазии (АСН), гипохондроплазии (НСН) и танатофорной дисплазии (Thanatophoric dysplasia), причем АСН является наиболее обычной формой карликовости. С биохимической точки зрения, постоянная активация этих рецепторов происходит при димеризации рецептора в отсутствие лиганда (Chen. L, Adar R., Yang X., Monsonego E.G., Li C., Hauschka P.V., Yagon А. и Deng C.X., (1999), The Journal of Clin. Invest, vol. 104, No. 11, pp. 1517-1525). Таким образом, соединения по этому изобретению, которые проявляют активность антагониста связывания b-FGF с FGFR и которые таким образом ингибируют димеризацию этого рецептора, представляют собой предпочтительную терапию при этих патологиях. Благодаря их низкой токсичности и их фармакологическим и биологическим свойствам, соединения по настоящему изобретению находят применение в лечении любой карциномы, имеющей высокий уровень васкуляризации (легких, молочной железы, простаты, пищевода), или индуцирующей метастазы(толстой кишки, желудка, меланомы), или чувствительной к a-FGF или b-FGF аутокринным образом,или, наконец, при патологиях типа лимфомы или лейкемии. Эти соединения представляют собой предпочтительную терапию в отдельности или в комбинации с надлежащей химиотерапией. Соединения в соответствии с этим изобретением также находят применение при лечении сердечно-сосудистых заболеваний, таких как атеросклероз, рестеноз после ангиопластии, при лечении заболеваний, связанных с осложнениями, которые появляются вследствие введения внутрисосудистых протезов и/или аортокоронарного шунтирования, или других трансплантатов сосудов, или гипертрофии сердца, или сосудистых осложнений диабета, таких как диабетические ретинопатии. Соединения в соответствии с этим изобретением также находят применение в лечении хронических воспалительных заболеваний, таких как ревматоидный артрит и ВЗК. Наконец, соединения в соответствии этим изобретением могут быть полезными в лечении ахондроплазии (АСН), гипохондроплазии (НСН) и TD (танатотропной дисплазии). В соответствии с другими из его отличий, предметом настоящего изобретения является поэтому фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы I в соответствии этим изобретением или одну из его фармацевтически приемлемых солей, возможно в комбинации с одним или более чем одним из инертных и подходящих эксципиентов. Выбирают указанные эксципиенты соответственно фармацевтическим стандартным лекарственными формам и желаемым путям введения: перорально, под язык, под кожу, в мышцу, в вену, через кожу,через слизистые, локально или через прямую кишку. Фармацевтические композиции в соответствии с настоящим изобретением предпочтительно вводят пероральным путем. В фармацевтических композициях по настоящему изобретению для перорального введения активные ингредиенты можно вводить в стандартной лекарственной форме для введения в виде смеси с общепринятыми фармацевтическими носителями. Подходящие стандартные лекарственные формы включают в себя, например, таблетки, возможно с насечкой, желатиновые капсулы, порошки, гранулы и пероральные растворы или суспензии. Когда готовят твердую композицию в форме таблеток, главный активный ингредиент смешивают с фармацевтическим носителем, таким как желатин, крахмал, лактоза, стеарат магния, тальк, гуммиарабик и т.п. Можно покрывать таблетки сахарозой или другими подходящими материалами или же можно обрабатывать их так, чтобы они имели продленную или отложенную активность и непрерывно высвобождали предопределенное количество активного ингредиента. Препарат в форме желатиновых капсул получают смешиванием активного ингредиента с разбавителем и вливанием этой полученной смеси в мягкие или твердые желатиновые капсулы. Препарат в форме сиропа или эликсира может содержать активный ингредиент с подсластителем,предпочтительно не содержащим калории, метилпарабеном и пропилпарабеном в качестве антисептиков,усилителем вкуса и подходящим красителем. Диспергируемые в воде порошки или гранулы могут содержать активный ингредиент в виде смеси- 12007902 с диспергирующими агентами, смачивающими агентами или суспендирующими агентами, такими как поливинилпирролидон, и с подсластителями и агентами, корригирующими вкус. Активный ингредиент можно также вводить в стандартную лекарственную форму в виде микрокапсул, возможно с одним или более чем одним из носителей или добавок. В фармацевтических композициях в соответствии с настоящим изобретением активный ингредиент может также быть в форме комплекса включения в циклодекстринах, их простых эфирах или их сложных эфирах. Количество активного ингредиента, подлежащего введению, зависит, как всегда, от степени прогрессии заболевания и от возраста и веса пациента. Композиции в соответствии с этим изобретением для перорального введения поэтому содержат рекомендованные дозы от 0,01 до 700 мг. Нижеследующие примеры, данные без ограничения, иллюстрируют настоящее изобретение. Способы получения Способ получения I. Синтез трет-бутил-2-метилиндолизин-1-илкарбамата К 10 г (48 ммоль) трет-бутил-[(пиридин-2-ил)метил]карбамата в 50 мл ацетонитрила добавляют 11,7 г (62,4 ммоль) карбоната калия и 6,3 г (72 ммоль) бромида лития и далее 5 мл (62,4 ммоль) хлорацетона и эту среду нагревают при дефлегмации в течение ночи. Охлаждают ее и добавляют к ней 40 мл воды и 11,7 г (62,4 ммоль) карбоната калия и нагревают эту среду при 90 С в течение 2 ч 30 мин. Эту реакционную среду охлаждают и экстрагируют этилацетатом. Органическую фазу удаляют после отстаивания, промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют под пониженным давлением. Продукт очищают флэш-хроматографией на колонке с кремнеземом при элюировании смесью толуол/этилацетат(95:5). Собирают 6,27 г белого порошка. Выход: 53% Точка плавления: 111 С Способ получения II. Синтез N-бензил-N-метил-N-(2-метилиндолизин-1-ил)амина Это соединение получают в соответствии с той же процедурой, что и соединение способа получения I, используя реакцию Чичибабина и начиная с 2,47 г N-бензил-N-метил-N-[(пиридин-2 ил)метил]амина и хлорацетона. Получают 970 мг желтого масла. Выход: 34% Масс-спектрометрия (вариант ES+): МН+ = 251 Способ получения III. Синтез 1-метокси-2-метилиндолизина Это соединение получают, начиная с 2-(метоксиметил)пиридина и хлорацетона, используя реакцию Чичибабина. Продукт изолируют в виде желтого масла, которое кристаллизуется в морозильнике. Выход: 77,5% Масс-спектрометрия (вариант ES+): МН+ = 161,8 Способ получения IV. Синтез-бензилокси-2-метилиндолизина Это соединение получают в соответствии с той же процедурой, какая описана в способе полученияI, используя реакцию Чичибабина. Продукт изолируют в форме желтого масла. Выход: 39% Способ получения V. Синтез метил-5-(хлоркарбонил)-2-[(2,2,2-трифторацетил)амино]бензоата Стадия А. Синтез 4-амино-3-(метоксикарбонил)бензойной кислоты 150 мг (1,21 ммоль) 4-диметиламинопиридина добавляют к 2,5 г (12,1 ммоль) 2,4-диоксо-1,4 дигидро-2 Н-3,1-бензоксазин-6-карбоновой кислоты [описана в J. Med. Chem., (1981), 24(6), 735-742] в растворе в 10 мл диметилформамида и 10 мл метанола и эту смесь нагревают при 60 С в течение 3 ч. Эту реакционную среду концентрируют под пониженным давлением. Остаток собирают в воду и экстрагируют этилацетатом. Органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют под пониженным давлением. Полученное твердое вещество собирают в этилацетат, фильтруют и сушат. Получают 1,98 г белого порошка. Выход: 84% Точка плавления: 224,5 С Стадия В. Синтез метил-3-(метоксикарбонил)-4-[(2,2,2-трифторацетил)амино]бензоата 868 мкл (6,15 ммоль) трифторуксусного ангидрида быстро добавляют к 1,0 г (5,12 ммоль) 4-амино 3-(метоксикарбонил)бензойной кислоты в суспензии в 15 мл дихлорметана. Этот раствор перемешивают в течение 30 мин при комнатной температуре. Этот раствор концентрируют досуха и полученное твердое вещество собирают в смесь пентан/этиловый эфир и затем отфильтровывают. После сушки получают 1,48 г белого порошка. Выход: 99% Точка плавления: 239 С Стадия С. 530 мкл (7,27 ммоль) тионилхлорида и 3 капли диметилформамида добавляют к 784 мг (2,69 ммоль) метил 3-(метоксикарбонил)-4-[(2,2,2-трифторацетил)амино]бензоата в растворе в 9 мл дихлорметана и- 13007902 затем эту среду нагревают при дефлегмации в течение 90 мин. Ее упаривают досуха и избыток тионилхлорида удаляют совместным испарением с толуолом. Получают 834 мг хлорангидрида в форме желтого твердого вещества, которое используют как есть, без дальнейшей очистки, в стадиях бензоилирования индолизинов. Выход: количественный. Примеры Пример 1. (1-Метокси-2-метилиндолизин-3-ил)-(3-метокси-4-нитрофенил)метанон 4,21 г (0,0195 моль) 3 метокси-4-нитробензоилхлорида добавляют к 3 г (0,0186 моль) 1-метокси-2 метилиндолизина, способ получения которого описан в способе получения III, растворяют в 50 мл 1,2 дихлорэтана и эту среду перемешивают при комнатной температуре в течение 4 ч. Эту реакционную среду наливают на воду. Органическую фазу отделяют после отстаивания, промывают водным раствором бикарбоната натрия (гидрокарбоната натрия) и затем водой, сушат над сульфатом натрия и концентрируют под вакуумом. Остаток очищают хроматографией на колонке с кремнеземом при элюировании дихлорметаном. После упаривания получают 6,05 г желтого твердого вещества. Выход: 95% Точка плавления: 287 С. Примеры 2-28. Проведением процедуры в соответствии с вышеописанным способом получения синтезируют соединения формулы I, для которых А представляет собой радикал -СО-, описанные в табл. I, бензоилированием по третьей позиции индолизинов, разнообразно замещенных по первой и второй позиции подходящим образом замещенными бензоилхлоридами. Таблица IBOC = трет-бутоксикарбонил Пример 29. (1-Амино-2-метилиндолизин-3-ил)-(3-метокси-4-нитрофенил)метанон 2,32 мл трифторуксусной кислоты добавляют каплями к раствору 643 мг (1,51 ммоль) трет-бутил-3(3-метокси-4-нитробензоил)-2-метилиндолизин-1-илкарбамата в 20 мл дихлорметана, охлажденного до 0 С. Как только добавление завершено, этой среде дают вернуться к комнатной температуре и перемешивают ее в течение 4 ч. Эту реакционную среду наливают на насыщенный водный раствор карбоната- 14007902 калия и экстрагируют этилацетатом. Органическую фазу отделяют после отстаивания, промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют под пониженным давлением. Полученные кристаллы собирают в изопропиловый эфир, отфильтровывают, промывают изопропиловым эфиром и затем сушат. Получают 425 мг коричневого твердого вещества. Выход: 87% Масс-спектрометрия (вариант ES+) МН+ = 326,3 Проведением процедуры в соответствии с вышеописанным способом получения синтезируют соединения формулы I, для которых А представляет собой радикал -СО-, и какие описаны в табл. II, снятием защиты с амина по первой позиции индолизинов с помощью трифторуксусной кислоты. Таблица II Пример 32. N-[3-(3-Метокси-4-нитробензоил)-2-метилиндолизин-1-ил]метансульфонамид 0,292 мл (3,78 ммоль) мезилхлорида добавляют к раствору 350 мг (1,08 ммоль) соединения примера 29 в 3 мл пиридина и эту среду перемешивают при комнатной температуре в течение 4 ч. Эту реакционную среду концентрируют под пониженным давлением. Остаток собирают в 1 н. соляную кислоту и экстрагируют дихлорметаном. Органическую фазу отделяют после отстаивания, промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют под пониженным давлением. Остаток кристаллизуют из этанола. Получают 327 мг желтых кристаллов. Выход: 75% Масс-спектрометрия (вариант ES+) МН+ = 404,3 Пример 33. Метил-5-[(3-[(3-метоксифенил)сульфонил]амино-2-метилиндолизин-3-ил)карбонил]2-[(2,2,2-трифторацетил)амино]бензоат Это соединение получают в соответствии с тем же способом, как в предыдущем примере, сульфонилированием метил-5-[(1-амино-2-метилиндолизин-3-ил)карбонил]-2-[2,2,2-трифторацетил)амино]бензоата 3-метоксибензолсульфонилхлоридом. Получают 466 мг желтого порошка. Выход: 83% Точка плавления: 220,5 С Пример 34. Метил-5-[(1-[(3-метоксианилино)карбониламино-2-метилиндолизин-3-ил)карбонил]2-[(2,2,2-трифторацетил)амино]бензоат 140 мкг (1,05 ммоль) 3-метоксифенилизоцианата добавляют к 400 мг (0,95 ммоль) метил-5-[(1 амино-2-метилиндолизин-3-ил)карбонил]-2-[2,2,2-трифторацетил)амино]бензоата, растворенного в 13 мл тетрагидрофурана. Эту реакционную смесь нагревают при 40 С в течение 20 ч и концентрируют под пониженным давлением. Полученный остаток собирают в ацетон, твердое вещество отфильтровывают и промывают его ацетоном и затем этиловым эфиром. Получают 442 мг желтого порошка. Выход: 82% Точка плавления: 314 С Пример 35. (3-Метокси-4-нитрофенил)-[2-метил-1-(метиламино)индолизин-3-ил]метанон Стадия А. Синтез трет-бутил-3-(3-метокси-4-нитробензоил)-2-метилиндолизин-1-ил-(метил)карбамата 3,05 г (7,2 ммоль) трет-бутил-3-(3-метокси-4-нитробензоил-2-метилиндолизин-1-ил-карбамата в растворе в 50 мл тетрагидрофурана добавляют каплями к 315 мг (7,9 ммоль) гидрида натрия (в виде 60%ой дисперсии в масле) в суспензии в 10 мл тетрагидрофурана, охлажденного до 0 С. После перемешивания в течение 1 ч при 0 С добавляют 0,59 мл (9,5 ммоль) метилйодида, поддерживая эту среду при 0 С. Дают ей вернуться к комнатной температуре и перемешивают ее в течение 1 ч. Эту реакционную среду наливают на насыщенный водный раствор бикарбоната натрия и экстрагируют этилацетатом. Органическую фазу отделяют после отстаивания, промывают насыщенным водным раствором хлорида натрия,сушат над сульфатом натрия и концентрируют под пониженным давлением. Получают 3,47 г пены оранжевого цвета. Выход: 96% Масс-спектрометрия (вариант ES+) МН+ = 440,3 Стадия В. 13 мл трифторуксусной кислоты добавляют каплями к раствору 3,38 г (7,7 ммоль) продукта, полученного в стадии А, в 60 мл дихлорметана, охлажденного до 0 С. Когда это добавление завершено, этой среде дают вернуться к комнатной температуре и перемешивают ее в течение 3 ч. Эту реакционную среду наливают на насыщенный водный раствор бикарбоната натрия и экстрагируют этилацетатом. Органическую фазу отделяют после отстаивания, промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют под пониженным давлением. Остаток очищают флэш-хроматографией на колонке с кремнеземом при элюировании смесью толу- 15007902 ол/этилацетат (9/1). После упаривания получают 2,2 г красного порошка. Выход: 76% Масс-спектрометрия (вариант ES+) МН+ = 340,2 Примеры 36 и 37. Проведением процедуры в соответствии с примером 35, cтадией А, синтезируют соединения формулы I, для которых А представляет собой радикал -СО- и которые описаны в табл. III, алкилированием трет-бутилкарбамата по первой позиции индолизинов 3-метоксибензилхлоридом в присутствии гидрида натрия в растворителе, таком как тетрагидрофуран и диметилформамид. Таблица III Примеры 38 и 39. Проведениемпроцедуры в соответствии примером 35, cтадией В, синтезируют соединения формулы I, для которых А представляет собой радикал -СО- и которые описаны в табл. IV, снятием защиты с амина по первой позиции индолизинов с помощью трифторуксусной кислоты. Таблица IV Пример 40. [1-(Диметиламино)-2-метилиндолизин-3-ил]-(3-метокси-4-нитрофенил)метанон 382 мг (1,1 ммоль) соединения примера 21 в растворе в 10 мл тетрагидрофурана добавляют каплями к 44 мг (1,1 ммоль) гидрида натрия (в виде 60% дисперсии в масле) в суспензии в 5 мл тетрагидрофурана,охлажденного до 0 С. Как только введение завершено, этой среде дают вернуться к комнатной температуре в течение 1 ч и затем добавляют к ней 69 мкл (1,1 ммоль) метилйодида и перемешивают эту среду при комнатной температуре в течение 17 ч. Эту реакционную среду наливают на насыщенный водный раствор хлорида натрия и экстрагируют этилацетатом. Органическую фазу отделяют после отстаивания, промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют под пониженным давлением. Остаток очищают хроматографией на колонке с кремнеземом при элюировании смесью толуол/этилацетат (95/5). Получают 143 мг пены оранжевого цвета. Выход: 37% Пример 41. 1-[(3-Метоксибензил)-(метил)амино]-2-метилиндолизин-3-ил-(3-метокси-4-нитрофенил)метанон 595 мг (1,83 ммоль) карбоната цезия и 83 мкл (1,34 ммоль) метилйодида добавляют к 542 мг (1,22 ммоль) 1-[(3-метоксибензил)амино]-2-метилиндолизин-3-ил-(3-метокси-4-нитрофенилметанона в растворе в 15 мл диметилформамида. Эту реакционную смесь нагревают при 40 С в течение 21 ч. Эту смесь вливают в насыщенный раствор хлорида натрия и экстрагируют этилацетатом. Органическую фазу сушат над сульфатом натрия и концентрируют под пониженным давлением. Продукт очищают хроматографией на силикагеле при элюировании смесью толуол/этилацетат(95/5). Получают красную смолу. Выход: 96% Масс-спектрометрия (вариант ES+) МН+ = 460,3 Пример 42. Метил-2-амино-5-(1-[3-метоксибензил)(метил)амино]-2-метилиндолизин-3-илкарбонил)бензоат Это соединение получают в соответствии с тем же способом, какой описан в примере выше, начиная с 340 мг (0,76 ммоль) метил-2-амино-5-(1-[(3-метоксибензил)амино]-2-метилиндолизин-3-илкарбонил)бензоата. Получают 260 мг твердого вещества оранжевого цвета. Выход: 80% Точка плавления: 60 С Пример 43. Метил-5-(1-[(3-метоксибензоил)амино]-2-метилиндолизин-3-илкарбонил)-2-[2,2,2 трифторацетил)амино]бензоат 3,37 г (7,6 ммоль) бензотриазол-1-илокси-трис(диметиламино)фосфонийгексафторфосфата (ВОР) и- 16007902 2,1 мл триэтиламина добавляют к 1,16 г (7,6 ммоль) 3-метоксибензойной кислоты, растворенной в 30 мл диметилформамида и 60 мл дихлорметана. Эту среду перемешивают при комнатной температуре в течение 15 мин и затем добавляют к ней 3,04 г (7,2 ммоль) метил-5-[(1-амино-2-метилиндолизин-3-ил)-карбонил]-2-[2,2,2-трифторацетил)амино]бензоата. После перемешивания в течение 16 ч при комнатной температуре желтый осадок, полученный в этой реакционной среде, отфильтровывают и промывают его дихлорметаном. Получают 2,38 г желтого порошка. Выход: 60% Точка плавления: 239 С Примеры 44-61. Проведением процедуры в соответствии с вышеописанным способом синтезируют соединения,описанные в табл. V, сочетанием (1-амино-2-метилиндолизин-3-ил)-(3-метокси-4-нитрофенил)метанона или метил-5-[(1-амино-2-метилиндолизин-3-ил)карбонил]-2-[2,2,2-трифторацетил)амино]бензоата с надлежащей карбоновой кислотой в присутствии бензотриазол-1-илокси-трис(диметиламино)фосфонийгексафторфосфата (ВОР). Таблица V Пример 62. N-[3-(3-Метокси-4-нитробензоил)-2-метилиндолизин-1-ил]ацетамид 1,20 мл (12,60 ммоль) уксусного ангидрида добавляют к 410 мг (1,26 ммоль) (1-амино-2 метилиндолизин-3-ил)-(3-метокси-4-нитрофенил)-метанона, растворенного в 10 мл дихлорметана. Эту реакционную смесь перемешивают при комнатной температуре в течение 15 мин. Полученный осадок отфильтровывают и промывают этиловым эфиром и затем сушат с получением 295 мг порошка оранжевого цвета. Выход: 63% Точка плавления: 238 С Пример 63. 3-Метокси-N-[3-(3-метокси-4-нитробензоил)-2-метилиндолизин-1-ил]-N-метилбензамид 51 мг гидрида натрия (60% суспензия в масле) добавляют к 466 мг (1,01 ммоль) 3-метокси-N-[3-(3 метокси-4-нитробензоил)-2-метилиндолизин-1-ил]-бензамида в растворе в 19 мл тетрагидрофурана. Эту среду перемешивают при комнатной температуре в течение 10 мин и затем добавляют к ней 65 мкл метилйодида. После перемешивания в течение 2 ч к этой реакционной среде добавляют воду и затем эту среду экстрагируют этилацетатом. Органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют под пониженным давлением. Продукт очищают хроматографией на силикагеле при элюировании дихлорметаном. Получают 435 мг желтого твердого вещества.- 17007902 Выход: 91% Точка плавления: 190 С Пример 64. Метил-5-(1-[(3-метоксибензоил)-(метил)амино]-2-метилиндолизин-3-илкарбонил)-2 нитробензоат Это соединение получают в соответствии протоколом, описанным в примере выше, метилированием 1,9 г (3,9 ммоль) метил-5-(1-[(3-метоксибензоил)амино]-2-метилиндолизин-3-илкарбонил)-2 нитробензоата метилйодидом. Получают 1,85 г красного твердого вещества. Выход: 84% Точка плавления: 158,5 С Пример 65. Этил-[3-(3-метокси-4-нитробензоил)-2-(трифторметил)индолизин-1-ил]карбоксилат Стадия А. Синтез бромида 1-[2-(3-метокси-4-нитрофенил)-2-оксоэтил]пиридиния 467 мкл (5,78 ммоль) пиридина добавляют к 1,32 г (4,82 ммоль) 2-бром-1-(3-метокси-4 нитрофенил)-1-этанона, описанного в Bull. Soc. Chim. Fr., (1962), 2255-2261, в растворе в 13 мл ацетонитрила и эту среду перемешивают при комнатной температуре в течение 5 ч. Эту реакционную среду преципитируют. Добавляют этиловый эфир, отфильтровывают кристаллы, промывают этиловым эфиром и затем сушат. Получают 1,65 г желтых кристаллов. Выход: 97% Точка плавления: 216 С. Стадия В. 500 мг (1,42 ммоль) бромида 1-[2-(3-метокси-4-нитрофенил)-2-оксоэтил]пиридиния добавляют порциями к 219 мкл (1,56 ммоль) триэтиламина в 4,5 мл диметилформамида с последующим добавлением 1,06 мл (7,08 ммоль) этил-4,4,4-трифторкротоната и 561 мг (0,92 ммоль) дихромата тетрапиридинкобальта(II). Эту реакционную среду нагревают при 90 С в течение 6 ч. Охлаждают эту реакционную среду и затем наливают ее на 1 н соляную кислоту и экстрагируют полученный таким образом продукт этилацетатом. Органическую фазу отделяют после отстаивания, промывают водой и затем насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют под пониженным давлением. Полученный продукт очищают хроматографией на колонке с кремнеземом при элюировании дихлорметаном. Получают 437 мг желтого порошка. Выход: 71% Точка плавления: 63 С Пример 66. Этил-[3-(3-метокси-4-нитробензоил)индолизин-1-ил]карбоксилат Это соединение получают в соответствии с тем же способом, как в предыдущем примере в стадии В,1,3-диполярным присоединением цикла бромидом 1-[2-(3-метокси-4-нитрофенил)-2 оксоэтил]пиридиния (получен в стадии А предыдущего примера) с этилакрилатом. После очистки флэшхроматографией на колонке с кремнеземом при элюировании дихлорметаном получают желтый порошок. Выход: 78% Точка плавления: 168 С Пример 67. (1-Гидрокси-2-метилиндолизин-3-ил)-(3-метокси-4-нитрофенил)метанон Раствор 5 г (12 ммоль) [1-(бензилокси-2-метилиндолизин-3-ил]-(3-метокси-4-нитрофенил)метанона, соединения примера 3, в 30 мл трифторуксусной кислоты нагревают при дефлегмации в течение 2 ч. Эту реакционную среду упаривают под пониженным давлением. Остаток собирают в этилацетат,промывают водным раствором бикарбоната натрия и водой и затем органическую фазу сушат над сульфатом натрия и упаривают под пониженным давлением. Полученный продукт очищают хроматографией на колонке с кремнеземом при элюировании смесью дихлорметан/метанол (99/1). Получают 2,93 г порошка оранжевого цвета. Выход: 75% Точка плавления: 193 С Пример 68. Метил-4-([3-(3-метокси-4-нитробензоил)-2-метилиндолизин-1-ил]оксиметил)бензоат 812 мг (3,37 ммоль) метил-4-(бромметил)-бензоата добавляют к раствору 1 г (3,06 ммоль) (1 гидрокси-2-метил-3-индолизинил)-(3-метокси-4-нитрофенил)метанона в 16 мл диметилформамида в присутствии 508 мг (3,68 ммоль) карбоната калия и эту среду нагревают при 90 С в течение 4 ч. Эту реакционную среду наливают на воду и экстрагируют этилацетатом. Органическую фазу промывают водой, сушат над сульфатом натрия и упаривают досуха. Полученный продукт очищают хроматографией на колонке с кремнеземом при элюировании смесью толуол/этилацетат (9/1). Получают 880 мг желтого порошка. Выход: 60,5% Точка плавления: 154 С- 18007902 Примеры 69-84. Проведением процедуры в соответствии со способом, описанным в примере 68, синтезируют соединения, описанные в табл. VI, алкилированием (1-гидрокси-2-метилиндолизин-3-ил)-(3-метокси-4 нитрофенил)метанона надлежащим образом выбранными галогенированными производными. Для получения соединения примера 80 соединение примера 79 подвергают омылению. Таблица VI Пример 85. Метил 4-[(1-гидрокси-2-метилиндолизин-3-ил)карбонил]бензоат 8,75 мл (86,37 ммоль) циклогексена добавляют к 3,45 г (8,64 ммоль) метил-4-[(1-(бензилокси)-2 метилиндолизин-3-ил)карбонил]бензоата в 40 мл этанола в присутствии 690 мг 10% Pd/C и эту среду нагревают при дефлегмации в течение 1 ч. Эту реакционную среду охлаждают до комнатной температуры, и катализатор удаляют фильтрацией на тальке. Фильтрат под пониженным давлением. Полученный продукт очищают хроматографией на колонке с кремнеземом при элюировании смесью дихлорметан/метанол (98/2). Получают 2,5 г порошка оранжевого цвета. Выход: 93,5% Точка плавления: 192 С Пример 86. Метил-4-[1-(2-этокси-2-оксоэтокси)-2-метилиндолизин-3-ил]карбонилбензоат 202 мкл (1,78 ммоль) этилбромацетата добавляют к 500 мг (1,62 ммоль) метил-4-[(1-гидрокси-2 метилиндолизин-3-ил)карбонил]бензоата, соединения примера 85, в 10 мл диметилформамида в присутствии 268 мг (1,94 ммоль) карбоната калия и эту среду нагревают при 90 С в течение 1 ч. Эту реакционную среду охлаждают, наливают на воду и экстрагируют этилацетатом и затем разделяют после отстаивания. Органическую фазу промывают водой, сушат над сульфатом натрия и упаривают под пониженным давлением. Полученный продукт очищают хроматографией на колонке с кремнеземом при элюировании смесью толуол/этилацетат (9/1). Получают 570 мг желтого порошка.- 19007902 Выход: 89% Точка плавления: 84,5 С Пример 87. Метил-4-(1-[(3-метоксибензил)окси]-2-метилиндолизин-3-илкарбонил)бензоат Это соединение получают в соответствии с той же процедурой, как и в примере 86, О-алкилированием метил-4-[(1-гидрокси-2-метилиндолизин-3-ил)карбонил]бензоата 3-метоксибензилбромидом. Получают желтый порошок, который плавится при 106 С. Выход: 76% Пример 88. 3-(3-Метокси-4-нитробензоил)-2-метилиндолизин-1-илкарбоновая кислота 26,2 мл 1 н. гидроксида натрия добавляют к 5 г (13,1 ммоль) этил-3-(3-метокси-4-нитробензоил)-2 метилиндолизин-1-илкарбоксилата, соединения примера 13, полученного в соответствии с процедурой примера 1 бензоилированием этил-(2-метилиндолизин-1-ил)-карбоксилата, описанного в J. Chem. Soc,(1963), pp. 3277-3280, в суспензии в 50 мл диоксана, и эту среду нагревают при дефлегмации в течение 17 ч. Эту реакционную среду концентрируют под пониженным давлением. Остаток собирают в воду, промывают этиловым эфиром и среду отделяют после отстаивания. Водную фазу подкисляют до рН 6 раствором гидросульфата калия и экстрагируют этилацетатом. Органическую фазу промывают водой, сушат над сульфатом натрия и концентрируют под пониженным давлением. Получают 4,9 г порошка оранжевого цвета. Выход: количественный Точка плавления: 215 С Пример 89. N-Этил-3-(3-метокси-4-нитробензоил)-2-метилиндолизин-1-илкарбоксамид 0,61 мл (4,34 ммоль) триэтиламина добавляют к раствору 750 мг (2,12 ммоль) кислоты примера 88 в 12 мл диметилформамида и затем порциями добавляют 983 мг (2,22 ммоль) гексафторфосфата бензотриазол-1-илокси-трис(диметиламино)фосфония. Эту среду перемешивают в течение 5 мин при комнатной температуре и затем добавляют к ней 182 мг (2,22 ммоль) гидрохлорида этиламина. Эту реакционную среду перемешивают в течение ночи при комнатной температуре, наливают на воду и экстрагируют этилацетатом. Органическую фазу отделяют после отстаивания, промывают водой,сушат над сульфатом натрия и концентрируют под пониженным давлением. Продукт очищают хроматографией на колонке с кремнеземом при элюировании дихлорметаном/метанолом (98/2). Получают 700 мг желтого порошка. Выход: 87% Точка плавления: 188 С Пример 90. Этил-2-([3-(3-метокси-4-нитробензоил)-2-метилиндолизин-1-ил]карбониламино)ацетат Это соединение получают в соответствии с тем же способом, что и предыдущее соединение, сочетанием 3-(3-метокси-4-нитробензоил)-2-метилиндолизин-1-илкарбоновой кислоты с гидрохлоридом этилглицината. Продукт очищают хроматографией на колонке с кремнеземом при элюировании дихлорметаном/метанолом (93/7). Получают желтый порошок. Выход: 86% Точка плавления: 191 С Пример 91. 1-Метокси-2-метил-3-[(4-нитрофенил)сульфонил]индолизин 690 мг (3,1 ммоль) 4-нитробензолсульфонилхлорида в растворе в 4 мл 1,2-дихлорэтана добавляют к 500 мг (3,1 ммоль) 1-метокси-2-метилиндолизина, растворенного в 8 мл 1,2-дихлорэтана, и эту среду перемешивают при комнатной температуре в течение 20 ч. Эту реакционную среду наливают на воду и дихлорметан. Органическую фазу отделяют после отстаивания, промывают водой, сушат над сульфатом натрия и концентрируют под пониженным давлением. Продукт очищают хроматографией на колонке с кремнеземом при элюировании циклогексаном/этилацетатом (9/1). Получают 330 мг желтого масла. Выход: 31% Пример 92. 1-Метокси-2-метил-3-[(3-нитрофенил)сульфонил]индолизин Это соединение получают в соответствии с протоколом, описанным для примера выше, сульфонилированием 1 г (6,2 ммоль) 1-метокси-2-метилиндолизина 3-нитробензолсульфонилхлоридом. Получают 540 мг желтого масла. Выход: 98% Пример 93. Натриевая соль 4-[(1-метокси-2-метилиндолизин-3-ил)сульфонил]бензойной кислоты Это соединение получают в соответствии с той же процедурой, что и соединение примера 91, сульфонилированием 1-метокси-2-метилиндолизина 4-хлорсульфонилбензойной кислотой. Продукт очищают флэш-хроматографией на колонке с кремнеземом при элюировании дихлорметаном/ацетоном (9/1). Получают 120 мг желтого порошка. Выход: 11% Продукт, растворенный в метаноле, переводят в соль добавлением одного эквивалента 1 н. гидроксида натрия. Метанол выпаривают и остаток кристаллизуют из ацетона. Продукт фильтруют, промыва- 20007902 ют ацетоном и затем этиловым эфиром и сушат. Получают 100 мг натриевой соли в виде желтого порошка. Точка плавления: 175 С Пример 94. (4-Амино-3-метоксифенил)-(1-метокси-2-метилиндолизин-3-ил)метанон 700 мг 10% Pd/C добавляют к 6 г (0,0176 моль) (1-метокси-2-метил-3-индолизинил)-(3-метокси-4 нитрофенил)метанона, соединения примера 1, в 100 мл этанола и затем добавляют 35,71 мл (0,352 моль) циклогексена и нагревают эту среду при дефлегмации в течение 2 ч. Эту реакционную среду охлаждают и фильтруют через тальк, а катализатор промывают дихлорметаном. Фильтрат концентрируют под пониженным давлением. Остаток собирают в дихлорметан. Органическую фазу промывают 1 н. гидроксидом натрия и затем водой, сушат над сульфатом натрия и концентрируют под пониженным давлением. Получается 5,05 г желтого порошка. Продукт переводят в соль растворением порошка, полученного выше, в 60 мл дихлорметана плюс 20 мл метанола и затем добавлением 21 мл 1 н. соляной кислоты в этиловом эфире. После добавления этилового эфира полученный осадок отфильтровывают, промывают этиловым эфиром и затем сушат. Получается 5,4 г желтого порошка в форме гидрохлорида. Выход: 88% Точка плавления: 198 С Примеры 95-117. Проведением процедуры в соответствии с вышеописанным способом получения синтезируют соединения, описанные в табл. VII, восстановлением нитрофункциональной группы соединений формулы(5,21 ммоль) гидрата гидразина и перемешивают эту среду при комнатной температуре в течение ночи. Эту реакционную среду фильтруют на тальке и катализатор промывают метанолом. Фильтрат концентрируют под пониженным давлением. Остаток собирают в этилацетат, органическую фазу промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют под пониженным давлением. Получается 460 мг желтого порошка. Этот продукт переводят в соль растворением порошка, полученного выше, в смеси этилацетата и метанола и затем добавляют 1,25 мл (1,2 эквивалента) 1 н. соляной кислоты в этиловом эфире. После добавления этилового эфира полученный осадок фильтруют, промывают этиловым эфиром и затем сушат. Получается 440 мг желтого порошка в форме гидрохлорида с 0,65 Н 2O. Выход: 90% Точка плавления: 177 С Примеры 119-140. Проведением процедуры в соответствии со способом получения, описанным в примере 118, синтезируют соединения, описанные в табл. VIII, восстановлением нитрофункциональной группы соединений формулы Ia гидратом гидразина в присутствии 10% Pd/C в качестве катализатора. Таблица VIII Пример 141. 4-Хлор-N-[3-(4-амино-3-метоксибензоил)-2-метилиндолизин-1-ил]бензамид Смесь 384 мг (0,83 ммоль) 4-хлор-N-[3-(3-метокси-4-нитробензоил)-2-метилиндолизин-1 ил]бензамида и 115 мг оксида платины в 9 мл диметилформамида перемешивают под водородом при давлении 5 бар при комнатной температуре в течение 24 ч и затем фильтруют на тальке. Фильтрат кон- 22007902 центрируют под пониженным давлением. Продукт очищают хроматографией на силикагеле при элюировании толуолом/ацетоном (от 9/1 до 8/2). К полученному желтому порошку, суспендированному в 5 мл дихлорметана и 5 мл метанола, добавляют 1 мл 1 н. соляной кислоты в этиловом эфире. Полученный осадок отфильтровывают, промывают ацетоном и затем растворяют в 2 мл метанола и 40 мл воды. Полученный таким образом гидрохлорид лиофилизируют. Получают 162 мг порошка оранжевого цвета. Выход: 50% Точка плавления: 191 С Пример 142. [1-(2-Гидроксиэтокси)-2-метилиндолизин-3-ил]-(3-метокси-4-нитрофенил)метанон 1,52 мл 1 н. гидроксида натрия добавляют к 420 мг (1,02 ммоль) 2-[3-(3-метокси-4-нитробензоил)2-метилиндолизин-1-ил]оксиэтилацетата, соединения примера 79, растворенного в 6 мл диоксана, и эту среду перемешивают при комнатной температуре в течение 6 ч. Эту реакционную среду наливают на воду и этилацетат. Органическую фазу отделяют после отстаивания, промывают водой, сушат над сульфатом натрия и концентрируют под пониженным давлением. Получают 340 мг порошка оранжевого цвета, который используют без дальнейшей очистки в последующей стадии восстановления нитрогруппы. Выход: 90% Точка плавления: 142 С Пример 143. Натриевая соль 4-[(1-метокси-2-метил-3-индолизинил)карбонил]бензойной кислоты 2,45 мл 1 н. гидроксида натрия добавляют к 720 мг (2,23 ммоль) метил-4-[(1-метокси-2 метилиндолизин-3-ил)карбонил]бензоата, соединения примера 8, в растворе в 15 мл метанола плюс 15 мл диоксана и эту среду перемешивают при комнатной температуре в течение ночи. Эту реакционную среду концентрируют под пониженным давлением. Остаток собирают в воду, промывают этиловым эфиром и разделяют после отстаивания. Водную фазу подкисляют 1 н. соляной кислотой и экстрагируют дихлорметаном. Органическую фазу промывают водой, сушат над сульфатом натрия и концентрируют под пониженным давлением. Получают 700 мг порошка оранжевого цвета, который суспендируют в 20 мл метанола, после чего к нему добавляют один эквивалент 1 н. гидроксида натрия. Полученный раствор концентрируют под пониженным давлением. Остаток кристаллизуют из ацетона. Продукт отфильтровывают,промывают ацетоном и затем этиловым эфиром, сушат и получают 680 мг желтого порошка. Выход (натриевая соль): 92% Точка плавления 400 С Примеры от 144 до 157 Проведением процедуры в соответствии со способом, описанным в примере 143, синтезируют соединения, описанные в табл. IX, омылением эфирной функциональной группы соединений формулы Id. Таблица IX Пример 158. 2-Амино-5-(1-[3-метоксибензоил)амино]-2-метилиндолизин-3-илкарбонил)бензойная кислота 6,6 мл раствора гидроксида натрия (2 н.) добавляют к 3,31 г (6,0 ммоль) метил-5-(1-[(3 метоксибензоил)амино]-2-метилиндолизин-3-илкарбонил)-2-[2,2,2-трифторацетил)амино]бензоата в- 23007902 суспензии в 40 мл диоксана и 20 мл метанола. Эту реакционную среду нагревают при дефлегмации в течение 2,5 ч и затем ей дают вернуться к комнатной температуре и концентрируют ее под пониженным давлением. Полученный остаток собирают в насыщенный водный раствор бикарбоната натрия и экстрагируют этилацетатом. После декантирования водную фазу подкисляют молярным раствором соляной кислоты. Полученный осадок отфильтровывают, тщательно промывают водой и сушат под вакуумом. Получают 2,4 г желтого порошка. Выход: 90% Точка плавления: 290 С Натриевая соль, моногидрат: точка плавления: 265 С Примеры 159-174 Проведением процедуры в соответствии с вышеописанным способом синтезируют соединения формулы I, в которой А представляет собой -СО-, и которые описаны в табл. X, гидролизом метилового эфира и трифторацетамида в R3 и R4 гидроксидом натрия. Таблица X Пример 175. 3-(4-Амино-3-метоксибензоил)-2-метилиндолизин-1-илкарбоновая кислота 30 мл 2 н гидроксида натрия добавляют к 2,1 г (5,96 ммоль) этил 3-(4-амино-3-метоксибензоил)-2 метил-1-индолизинкарбоксилата в растворе в 30 мл диоксана и эту среду нагревают при дефлегмации в течение 20 ч. Эту реакционную среду концентрируют под пониженным давлением. Остаток собирают в воду, промывают этиловым эфиром и отделяют после отстаивания. Водную фазу подкисляют до рН 6,5 10% водным раствором гидросульфата калия и экстрагируют этилацетатом. Органическую фазу промывают водой, сушат над сульфатом натрия и концентрируют под пониженным давлением. Получают 1,8 г желтого порошка. Выход: 93% Затем получают две соли этого соединения: натриевая соль, моногидрат, точка плавления: 224 С; гидрохлорид, точка плавления: 213 С Примеры 176-185 Проведением процедуры в соответствии с вышеописанным способом получения синтезируют соединения, описанные в табл. XI, омылением эфирной функциональной группы, содержащейся в заместителе R1 соединений формулы Id, в которой А представляет собой радикал -СО-, гидроксидом натрия. Пример 186. Двунатриевая соль 3-(4-карбоксибензоил)-2-метилиндолизин-1-илкарбоновой кислоты 7,47 мл 1 н. гидроксида натрия добавляют к 910 мг (2,49 ммоль) этил-3-[4(метоксикарбонил)бензоил]-2-метилиндолизин-1-илкарбоксилата в растворе в 20 мл диоксана плюс 20 мл этанола и эту среду нагревают при дефлегмации в течение 6 ч. Эту реакционную среду концентрируют под пониженным давлением. Остаток собирают в воду, промывают этиловым эфиром и разделяют после отстаивания. Водную фазу подкисляют 1 н. соляной кислотой и экстрагируют этилацетатом. Органическую фазу промывают водой, сушат над сульфатом натрия и концентрируют под пониженным давлением. Получают 650 мг желтого порошка, который суспендируют в 20 мл метанола, и затем к нему добавляют 4,02 мл 1 н. гидроксида натрия (2 экв.). Полученный раствор концентрируют под пониженным давлением. Остаток кристаллизуют из ацетона. Продукт отфильтровывают, промывают ацетоном и затем этиловым эфиром и сушат. Получают 700 мг желтого порошка. Выход двунатриевой соли, дигидрата: 81% Точка плавления: 400 С Примеры 187-191. Проведением процедуры в соответствии с примером 186 синтезируют соединения, описанные в табл. XII, омылением эфирных функциональных групп, содержащихся в заместителях R1 и R4 соединений формулы I, в которой А представляет собой радикал -СО-, 1 н. гидроксидом натрия. Таблица XII Пример 192. (4-[3-(Дибутиламино)пропил]амино-3-метоксифенил)-(1-метокси-2-метилиндолизин-3-ил)метанона гидрохлорид 700 мг (2,25 ммоль) (4-амино-3-метоксифенил)-(1-метокси-2-метилиндолизин-3-ил)метанона, соединения, описанного в примере 94, растворяют в 5 мл тетрагидрофурана, добавляют к 278,4 мг (2,25 ммоль) трет-бутоксида калия в 5 мл тетрагидрофурана и эту среду перемешивают в течение 15 мин при комнатной температуре. Затем добавляют 510,5 мг (2,48 ммоль) хлорида дибутиламинопропила в 5 мл тетрагидрофурана и нагревают эту среду при температуре дефлегмации в течение ночи. Эту реакционную среду охлаждают и наливают на воду и затем экстрагируют этилацетатом. Органическую фазу отделяют после отстаивания, промывают водой, сушат над сульфатом натрия и концентрируют под пониженным давлением. Продукт очищают хроматографией на колонке с кремнеземом при элюировании смесью дихлорметан/ацетон (9/1) и затем (1/1). Получают 700 мг смолы оранжевого цвета, которую переводят в соль в этиловом эфире добавлени- 25007902 ем одного эквивалента 1 н. соляной кислоты в этиловом эфире. Полученные кристаллы отфильтровывают, промывают этиловым эфиром и сушат. Получают оранжевого цвета порошок в форме гидрохлорида с 1,25 Н 2O. Выход: 65% Точка плавления: 51 С Пример 193. [3-Метокси-4-(метиламино)фенил]-(1-метокси-2-метилиндолизин-3-ил)метанона гидрохлорид Это соединение получают в соответствии с той же процедурой, какая описана в примере 192, алкилированием (4-амино-3-метоксифенил)-(1-метокси-2-метилиндолизин-3-ил)метанона метилйодидом. Получают желтый порошок. Выход: 45% Точка плавления: 172 С Пример 194. (4-[3-(Дибутиламино)пропил]амино-3-метоксифенил)-(1-метокси-2-фенилиндолизин-3-ил)метанона дигидрохлорид Получают в соответствии с процедурой, описанной в примере 192, алкилированием (4-амино-3 метоксифенил)-(1-метокси-2-фенилиндолизин-3-ил)метанона, соединения примера 95, хлоридом дибутиламинопропила. Получают оранжевого цвета порошок (дигидрохлорид: 1,3 Н 2O). Выход: 37% Точка плавления: 158 С Пример 195. Этил-2-2-метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]анилиноацетат Это соединение получали в соответствии с процедурой, описанной в примере 192, алкилированием(4-амино-3-метоксифенил)-(1-метокси-2-метилиндолизин-3-ил)метанона, соединения примера 94, этилбромацетатом. Получают желтый порошок. Выход: 60,5% Точка плавления: 125 С Пример 196. 2-2-Метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]анилиноуксусная кислота 3,15 мл 1 н гидроксида натрия добавляют к 1 г (2,52 ммоль) этил-2-2-метокси-4-[(1-метокси-2 метилиндолизин-3-ил)карбонил]анилиноацетата, соединения, полученного в примере 195, в растворе в 10 мл этанола и эту среду перемешивают при комнатной температуре в течение ночи. Эту реакционную среду концентрируют под пониженным давлением. Остаток собирают в воду, промывают этиловым эфиром и разделяют после отстаивания. Водную фазу нейтрализуют 1 н. соляной кислотой. Образовавшийся осадок отфильтровывают, промывают водой, сушат и затем собирают в этиловый эфир, фильтруют и сушат. Получают желтый порошок. Выход: 48,5% Точка плавления: 196 С Пример 197. Этил-2-2-метокси-4-[(1-метокси-2-фенилиндолизин-3-ил)карбонил]анилиноацетат Это соединение получали в соответствии со способом, описанным в примере 192, алкилированием(4-амино-3-метоксифенил)-(1-метокси-2-фенилиндолизин-3-ил)метанона, соединения примера 95, этилбромацетатом. Получают желтый порошок. Выход: 78% Точка плавления: 132 С Пример 198. 2-2-Метокси-4-[(1-метокси-2-фенилиндолизин-3-ил)карбонил]анилиноуксусная кислота Получают в соответствии с той же процедурой, что и соединение примера 196, омылением этил-22-метокси-4-[(1-метокси-2-фенилиндолизин-3-ил)карбонил]анилиноацетата, соединения примера 197,1 н гидроксидом натрия. Получают желтый порошок. Выход: 80% Точка плавления: 206 С Пример 199. 3-(Дибутиламино)-ZN-2-метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]фенилпропанамида гидрохлорид 2,5 мл (17,7 ммоль) триэтиламина добавляют к 2,5 г (8,06 ммоль) (4-амино-3-метоксифенил)-(1 метокси-2-метилиндолизин-3-ил)метанона, соединения примера 94, в 20 мл дихлорметана, охлажденного до 5 С, и далее добавляют 846 мкл (8,86 ммоль) 3-хлорпропионилхлорида в растворе в 10 мл дихлорметана и перемешивают эту среду в течение 3 ч при комнатной температуре. Эту реакционную среду промывают водой и затем насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют под пониженным давлением. Полученный остаток растворяют в 40 мл этанола и к нему добавляют 1,7 г (13,2 ммоль) дибутиламина и затем нагревают эту среду при дефлегмации в течение 7 ч. Эту реакционную среду концентрируют под пониженным давлением. Продукт очищают хроматографией на колонке с кремнеземом при элюировании смесью дихлорметан/метанол (98:2). Получают 2,6 г продукта, который переводят в соль- 26007902 добавлением 1 н. соляной кислоты в этиловом эфире. Получают желтый порошок (гидрохлорид,0,25 Н 2O). Выход: 65% Точка плавления: 82 С Пример 200. 3-(Дибутиламино)-N-2-метокси-4-[(1-метокси-2-фенилиндолизин-3-ил)карбонил]фенилпропанамида гидрохлорид Это соединение получали в соответствии с той же процедурой, какая описана в примере 199, ацилированием (4-амино-3-метоксифенил)-(1-метокси-2-фенилиндолизин-3-ил)метанона 3-хлорпропионилхлоридом с последующим аминированием дибутиламином. Получают желтый порошок (гидрохлорид,гемигидрат). Выход: 52% Точка плавления: 190 С Пример 201. N-(2-Метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]фенилацетамид Это соединение получали в соответствии с процедурой, описанной в примере 199, ацилированием(4-амино-3-метоксифенил)-(1-метокси-2-метилиндолизин-3-ил)метанона, соединения примера 94, ацетилхлоридом. Продукт очищают флэш-хроматографией на кремнеземе при элюировании смесью дихлорметан/метанол (99:1). Получают желтый порошок (0,3 Н 2O). Выход: 73% Точка плавления: 180 С Пример 202. Этил-2-метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]фенилкарбамат Это соединение получали в соответствии с процедурой, описанной в примере 199, ацилированием(4-амино-3-метоксифенил)-(1-метокси-2-метилиндолизин-3-ил)метанона этилхлороформатом. Продукт очищают флэш-хроматографией на кремнеземе при элюировании дихлорметаном. Получают желтый порошок. Выход: 41% Точка плавления: 140 С Пример 203. Этил-2-[3-(дибутиламино)пропаноил]-2-метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]анилиноацетата гидрохлорид Добавляют каплями 1 г (2,03 ммоль) 3-(дибутиламино-N-2-метокси-4-[(1-метокси-2 метилиндолизин-3-ил)карбонил]фенилпропанамида, соединения примера 199, в растворе в 10 мл диметилформамида, к 89,1 мг (2,23 ммоль) гидрида натрия в виде 60% дисперсии в масле в 10 мл диметилформамида и затем перемешивают эту среду при комнатной температуре в течение 1 ч. Затем добавляют 247 мкл (2,23 ммоль) этилбромацетата и перемешивают эту среду при комнатной температуре в течение ночи. Эту реакционную среду наливают на воду и этилацетат. Органическую фазу отделяют после отстаивания, промывают водой, сушат над сульфатом натрия и концентрируют под пониженным давлением. Продукт очищают флэш-хроматографией на колонке с кремнеземом при элюировании смесью дихлорметан/метанол (97:3). Получают 850 мг масла, которое растворяют в этиловом эфире и переводят в соль добавлением одного эквивалента 1 н. соляной кислоты в этиловом эфире. Получают желтые кристаллы (гидрохлорид, гидрат). Выход: 72% Точка плавления: 67 С Пример 204. Гидрохлорид 2-[3-(дибутиламино)пропаноил]-2-метокси-4-[(1-метокси-2 метилиндолизин-3-ил)карбонил]анилиноуксусной кислоты 586 мкл 1 н гидроксида натрия добавляют к 340 мг (0,586 ммоль) этил-2-[3-(дибутиламино)пропаноил]-2-метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]анилиноацетата, полученного в примере 203, в растворе в 5 мл этанола и эту среду перемешивают при комнатной температуре в течение ночи. Эту реакционную среду концентрируют под пониженным давлением. Остаток собирают в воду,промывают этиловым эфиром и разделяют после отстаивания. Водную фазу нейтрализуют 1. соляной кислотой и экстрагируют дихлорметаном. Органическую фазу сушат над сульфатом натрия и концентрируют под пониженным давлением. Продукт очищают флэш-хроматографией на колонке с кремнеземом при элюировании смесью дихлорметан/метанол (8:2). Получают 230 мг масла, которое растворяют в этилацетате и переводят в соль добавлением одного эквивалента 1 н соляной кислоты в этиловом эфире. Получают желтый порошок(гидрохлорид, 0,6 Н 2O) Выход: 71% Точка плавления: 151 С Пример 205. Этил-2-[3-(дибутиламино)пропаноил]-2-метокси-4-[(1-метокси-2-фенил-3 индолизинил)карбонил]анилиноацетата гидрохлорид Получают в соответствии способом, описанным в примере 203, алкилированием 3-(дибутиламино)N-[2-метокси-4-[(1 -метокси-2-фенилиндолизин-3-ил)карбонил]фенилпропанамида, соединения приме- 27007902 ра 200, этилбромацетатом. После перевода в соль 1 н. соляной кислотой в этиловом эфире получают желтый порошок (гидрохлорид). Выход: 55% Точка плавления: 64 С Пример 206. Гидрохлорид 2-[3-(дибутиламино)пропаноил]-2-метокси-4-[(1-метокси-2 фенилиндолизин-3-ил)карбонил]анилиноуксусной кислоты Получают в соответствии с той же процедурой, как в примере 204, омылением этил-2-[3(дибутиламино)пропаноил]-2-метокси-4-[(1-метокси-2-фенилиндолизин-3-ил-карбонил]анилиноацетата,соединения примера 205, 1 н. гидроксидом натрия. После перевода в соль 1 н. соляной кислотой в этиловом эфире получают желтый порошок. Выход: 75% Точка плавления: 113 С Пример 207. 2-(Дибутиламино)-N-2-метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]фенил-1-этансульфонамида гидрохлорид 471,5 мкл (3,38 ммоль) триэтиламина добавляют к 1 г (3,22 ммоль) (4-амино-3-метоксифенил)-(1 метокси-2-метилиндолизин-3-ил)метанона в 15 мл дихлорметана с последующим добавлением 346,9 мкл(3,22 ммоль) 2-хлорэтилсульфонилхлорида в растворе в 5 мл дихлорметана и эту среду перемешивают при комнатной температуре в течение ночи. Эту реакционную среду промывают водой и затем насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют под пониженным давлением. Полученный остаток растворяют в 10 мл этанола. Добавляют 375 мг (2,9 ммоль) дибутиламина и нагревают эту среду при дефлегмации в течение 4 ч. Эту реакционную среду концентрируют под пониженным давлением. Продукт очищают хроматографией на колонке с кремнеземом при элюировании смесью дихлорметан/метанол (98:2). Получают 1,18 г продукта, который переводят в соль добавлением 1 н соляной кислоты в этиловом эфире. Получают желтый порошок (гидрохлорид, гемигидрат). Выход: 69% Точка плавления: 91 С Пример 208. 2-(Дибутиламино)-N-2-метокси-4-[(1-метокси-2-фенилиндолизин-3-ил)карбонил]фенил-1-этансульфонамида гидрохлорид Это соединение получают в соответствии с той же процедурой, что и соединение примера 207,сульфонилированием(4-амино-3-метоксифенил)-(1-метокси-2-фенилиндолизин-3-ил)метанона 2 хлорэтилсульфонилхлоридом с последующим аминированием дибутиламином. Получают желтый порошок (гидрохлорид, гемигидрат). Выход: 63% Точка плавления: 111 С Пример 209. N-2-Метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]фенилметансульфонамид Получают в соответствии с той же процедурой, что и соединение примера 207, сульфонилированием (4-амино-3-метоксифенил)-(1-метокси-2-метилиндолизин-3-ил)метанона метансульфонилхлоридом. Продукт очищают хроматографией на колонке с кремнеземом при элюировании смесью толуол/этилацетат (7:3). Получают желтый порошок. Выход: 67% Точка плавления: 165 С Пример 210. Этил-3-3-метокси-4-[(метилсульфонил)амино]бензоил-2-метилиндолизин-1 илкарбоксилат Это соединение получали в соответствии с той же процедурой, какая описана в примере 207, сульфонилированием этил-3-(4-амино-3-метоксибензоил)-2-метилиндолизин-1-илкарбоксилата, соединения примера 97, метансульфонилхлоридом. Продукт очищают хроматографией на колонке с кремнеземом при элюировании смесью толуол/этилацетат (8:2). Получают желтый порошок. Выход: 57% Точка плавления: 178 С Пример 211. Натриевая соль 3-3-метокси-4-[(метилсульфонил)амино]бензоил-2-метилиндолизин 1-илкарбоновой кислоты 1 мл каустической соды добавляют к 290 мг (0,675 ммоль) этил-3-3-метокси-4[(метилсульфонил)амино]бензоил-2-метилиндолиз-1-илкарбоксилата в 7 мл диоксана плюс 7 мл воды и эту среду нагревают при дефлегмации в течение 6 ч. Эту среду охлаждают, наливают на воду и нейтрализуют водным раствором гидросульфата калия и затем экстрагируют этилацетатом. Органическую фазу промывают водой, сушат над сульфатом натрия и концентрируют под пониженным давлением. Получают 220 мг желтого порошка. Продукт переводят в соль добавлением одного эквивалента 1 н. гидроксида натрия к суспензии продукта в воде и перемешивания при комнатной температуре до достижения растворения.- 28007902 Полученный раствор затем лиофилизируют. Получается желтый лиофилизированный продукт (натриевая соль 1,85 Н 2O). Выход: 81% Масс-спектрометрия (вариант ES+) МН+ = 403,2 Пример 212. Гидрохлорид 2-[2-(дибутиламино)этил]сульфонил-2-метокси-4-[(1-метокси-2 метилиндолизин-3-ил)карбонил]анилиноуксусной кислоты Стадия А. Бензил-2-[2-(дибутиламино)этил]сульфонил-2-метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]анилиноацетата 125 мг (0,906 ммоль) карбоната калия добавляют к 400 мг (0,755 ммоль) 2-(дибутиламино)-N-2 метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]фенил-1-этансульфонамида, соединения примера 207, в растворе в 10 мл диметилформамида с последующим добавлением 142 мкл (0,906 ммоль) бензилбромацетата и эту среду нагревают при 60 С в течение 1 ч. Эту реакционную среду наливают на воду и этилацетат. Органическую фазу отделяют после отстаивания, промывают водой, сушат над сульфатом натрия и концентрируют под пониженным давлением. Продукт очищают флэш-хроматографией на колонке с кремнеземом при элюировании смесью дихлорметан/метанол (98:2). Получают 440 мг масла, которое используют прямо в следующей стадии. Выход: 86% Стадия В. 1,3 мл (12,7 ммоль) циклогексена добавляют к 430 мг (0,634 ммоль) бензил-2-[2(дибутиламино)этил]сульфонил-2-метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]анилиноацетата в 5 мл этанола в присутствии 100 мг 10% Pd/C и эту среду нагревают при дефлегмации в течение 3 ч. Эту реакционную среду охлаждают. Катализатор отфильтровывают и фильтрат концентрируют под пониженным давлением. Продукт очищают флэш-хроматографией на колонке с кремнеземом при элюировании смесью дихлорметан/метанол (9:1). Получают 250 мг масла, которое растворяют в этилацетате и переводят в соль добавлением одного эквивалента 1 н соляной кислоты в этиловом эфире. Получают порошок оранжевого цвета (гидрохлорид, дигидрат). Выход: 67% Точка плавления: 85 С Пример 213. Бензил-2-[2-(дибутиламино)этил]сульфонил-2-метокси-4-[(1-метокси-2 фенилиндолизин-3-ил)карбонил]анилиноацетата гидрохлорид Это соединение получали в соответствии с той же процедурой, как в примере 212, стадии А, алкилированием 2-(дибутиламино)-N-2-метокси-4-[(1-метокси-2-метилиндолизин-3-ил)карбонил]фенил-1 этансульфонамида, бензилбромацетатом. После перевода в соль 1 н соляной кислотой в этиловом эфире получают желтый порошок (гидрохлорид, гемигидрат). Выход: 55% Точка плавления: 95 С Пример 214. Гидрохлорид 2-[2-(дибутиламино)этил]сульфонил-2-метокси-4-[(1-метокси-2 фенилиндолизин-3-ил)карбонил]анилиноуксусной кислоты Это соединение получали в соответствии с той же процедурой, как в примере 212, стадии В, гидрогенированием бензил-2-[2-(дибутиламино)этил]сульфонил-2-метокси-4-[(1-метокси-2-фенилиндолизин-3-ил)карбонил]анилиноацетата, соединения примера 213, циклогексеном в присутствии Pd/C в этаноле. После перевода в соль 1 н соляной кислотой в этиловом эфире получают желтый порошок (гидрохлорид, 1,5 Н 2O). Выход: 75% Точка плавления: 113 С Пример 215. Исследование связывания 125I-b-FGF с очищенным рецептором FGFRIIIc способом проксимальных сцинтилляции На планшеты NBS (белый сплошной планшет NBS с 96 лунками, CORNING 3600) наносят покрытие 0,1% желатином (100 мкл на лунку) в течение 2 ч при 37 С. Под конец этой инкубации средство покрытия удаляют, планшеты ополаскивают и тщательно сушат. В лунки распределяют по 100 мкл буфера для связывания (40 мМ бис-трис-буфер, рН 7,0). Распределяют разведения соединений по этому изобретению в лунки в количестве 10 мкл на лунку. Затем распределяют в лунки b-FGF (AMERSHAM ARM 35050) по 10 мкл на лунку и FGFRIIIc (RDSystems 658 FR) по 10 мкл на лунку. Затем добавляют 125l-b-FGF (Dupont NEN NEX 268, удельная активность 70 мкКи) по 10 мкл на лунку и гранулы SPA (AMERSHAM RPQN 00019) по 50 мкл на лунку. Встряхивают планшет в течение нескольких секунд и инкубируют его в течение 60 мин при 37 С при защите от света. Под конец инкубации просчитывают планшет на счетчике радиоактивности MIBROBETA TR1LUX(WALLAC - PERKINELMER). Соединения по этому изобретению демонстрировали специфическую активность между 10-6 М и 10-9 М. Пример 216. Влияние соединений формулы I на пролиферацию эндотелиальных клеток пуповинной вены человека (HUVEC) против 30 нг/мл b-FGF или 10 нг/мл a-FGF Наносят на 24-луночные планшеты (FALCON PRIMARIA) покрытие раствором фибронектина (50 мкг/мл в забуференном фосфатами физрастворе, PBS) в количестве 200 мкл на лунку. Инокулируют количество клеток, составляющее 30000/мл/лунка, в смеси, представляющей собой среду RPMI 1640+10% фетальной телячьей сыворотки (FCS)+1% глутамина+гепарин-ECGF (НЕ). Инкубируют при 37 С в 5% CO2 в течение времени, нужном клеткам для прикрепления. Растворяют продукты и готовые растворы в диметилсульфоксиде/реакционной среде, имеющей конечную концентрацию 1 мкМ при окончательной 10-7 М. После прикрепления клеток в течение 6 ч при 37 С в присутствии 5% СO2 эту среду заменяют наRPMI 1640 с 0,1% FSC+глутамин+НЕ. Для дериватизации используют в качестве отрицательного контроля 0,1% FCS, в качестве положительного контроля 0% FGS и в качестве контроля 0,1% FCS+30 нг/мл b-FGF или 10 нг/мл a-FGF. Затем проводят инкубирование в течение 24 ч при 37 С в присутствии 5% CO2. На второй день ополаскивают клетки одним миллилитром PBS и трипсином (200 мкл) и затем собирают их в изотоническом растворе. Проводят подсчет (n9 мкм). В этом тесте на пролиферацию эндотелиальных клеток, которую вызывает b-FGF или a-FGF, соединения по этому изобретению показали специфическую активность между 10-5 М и 10-9 М. Пример 217. Модель ангиогенеза in vitro Готовят гели распределением в каждую из лунок слайд-камеры (коллаген типа I из хвоста крыс Biocoat Cellware и 8-луночная слайд-камера: Becton Dickinson 354630) по 160 мкл геля Matrigel, разбавленного 1/6 (Matrigel с пониженным содержанием факторов роста: Becton Dickinson 356230) в коллагене(коллаген типа I из хвоста крыс: Becton Dickinson 354236). Дают гелю образоваться в течение 1 ч при 37 С. Инокулируют эндотелиальные клетки пуповинной вены человека (HUVEC ref: C-015-10C - cascadeBiologies, INC) или эндотелиальные клетки аорты свиньи (РАЕС) в количестве 15103 клеток на лунку в 400 мкл среды ЕВМ (Clonetics C3121)+2% FBS+10 мкг/мл hEGF для HUVECs и DMEM+3% FCS+ 2 мМ глутамин+1 мМ пируват натрия+1% незаменимых аминокислот (GIBCO) для РАЕС. Стимулируют действием 10 нг/мл b-FGF (TEBU/Peprotech) или 10 нг/мл a-FGF (TEBU/Peprotech) в присутствии или в отсутствие продуктов по этому изобретению в течение 24 ч при 37 С в присутствии 5% CO2. После 24 ч фиксируют клетки и окрашивают слайд трихромом Массона перед исследованием под линзами микроскопа при х 4 и анализом изображения (программа BIOСОМ - Visiolab 2000). В тесте ангиогенеза in vitro, который индуцируют b-FGF или a-FGF, соединения по этому изобретению показали специфическую активность между 10-7 М и 10-11 М. Пример 218. Модель воспалительного ангиогенеза у мышей Ангиогенез нужен для развития хронических воспалительных заболеваний, таких как ревматоидный артрит, ВЗК, а также для развития солидных опухолей. Формирование новых сосудов не только создает возможность для перфузии патологических тканей, но также для транспорта цитокинов, являющихся причиной перехода заболевания в хроническую форму. Модель, которую описали Colville-Nash P. et al., (D. JPET., 1995, vol. 274 No. 3, pp. 1463-1472), делает возможным исследование фармакологических агентов, способных модулировать появление ангиогенеза. Животных, неродственных белых мышей весом около 25 г, анестезируют пентобарбиталом натрия(60 мг/кг; Sanofi Nutrition Sante Animate) через внутрибрюшинную трубку. Воздушный карман создают на спине мышей подкожным введением 3 мл воздуха. После возвращения в сознание животные получают воздействие, обычно принудительным кормлением, и получают в карман инъекцию 0,5 мл адъюванта Фрейнда (Sigma) с 0,1% кротоновым маслом(Sigma). Семь дней спустя мышей опять анестезируют и помещают на нагревающую поверхность при 40 С. В хвостовую вену инъецируют 1 мл кармина красного (5% в 10% желатине - Aldrich Chemical). Затем животных помещают в температуру 4 С на 2-3 ч. Затем снимают шкуры и сушат в течение 48 ч в сушильном шкафу при 56 С. Взвешивают сухие ткани и помещают в 1,8 мл буфера для ферментации (2 мМ дитиотрейтол, 2 мМ Na2HPO4, 1 мМ ЭДТА,12 ед./мл папаина) на 24 ч. Затем растворяют краситель в 0,2 мл 5 М NaOH. Центрифугируют шкуры при 2000 g в течение 10 мин. Фильтруют супернатанты на мембранах из ацетата целлюлозы с порами 0,2 мкм. Считывают фильтраты в спектрофотометре при 492 нм против калибровочной серии кармина красного. Исследуют два параметра: сухой вес гранулемы и количество красителя после ферментации этой ткани.
МПК / Метки
МПК: A61K 31/437, C07D 221/00, A61P 9/00, C07D 209/00, C07D 471/04, A61P 35/00, A61P 29/00
Метки: фибробластов, факторов, новые, производные, содержащие, роста, ингибиторами, композиции, фармацевтические, способ, являющиеся, индолизина, получения, 3-замещенные
Код ссылки
<a href="https://eas.patents.su/30-7902-novye-1-2-3-zameshhennye-proizvodnye-indolizina-yavlyayushhiesya-ingibitorami-faktorov-rosta-fibroblastov-sposob-ih-polucheniya-i-farmacevticheskie-kompozicii-soderzhashhie-ih.html" rel="bookmark" title="База патентов Евразийского Союза">Новые 1, 2, 3-замещенные производные индолизина, являющиеся ингибиторами факторов роста фибробластов, способ их получения и фармацевтические композиции, содержащие их</a>
Новые цианоиндольные соединения, являющиеся ингибиторами повторного захвата серотонина, способ их получения и фармацевтические композиции, содержащие их

Номер патента: 3146
Опубликовано: 27.02.2003
Авторы: Миллан Марк, Гобер Ален, Симетьер Бернар, Мюллер Оливье, Лавиелль Жильбер, Риве Жан-Мишель
МПК: C07D 209/04, A61K 31/404
Метки: цианоиндольные, повторного, серотонина, фармацевтические, являющиеся, ингибиторами, получения, способ, композиции, соединения, захвата, содержащие, новые
Формула / Реферат:
1. Соединения формулы (I) в которой R1 и R2, независимо друг от друга, каждый представляет атом водорода или линейную или разветвленную (C1-C6)-алкильную группу; A представляет линейную или разветвленную (C1-C6)-алкиленовую группу, линейную или разветвленную (C2-C6)-алкениленовую группу или линейную или разветвленную (C2-C6)-алкиниленовую группу; G1 представляет группу в которой R3 и R4, независимо друг от друга, каждый представляет атом...
Новые производные полигидроксипиразинов, способ их получения и содержащие их фармацевтические композиции

Номер патента: 3550
Опубликовано: 26.06.2003
Авторы: Бушар Эрве, Коммерсон Алан
МПК: C07D 241/12, A61P 3/10, A61K 31/495...
Метки: композиции, способ, фармацевтические, содержащие, производные, полигидроксипиразинов, новые, получения
Формула / Реферат:
1. Соединения общей формулы в которой R1 обозначает стереоизомерные формы цепи -(CHOH)3-CH2-O-COR (II) и либо R2 обозначает атом водорода, а R3 обозначает стереоизомерные формы цепи -CH2-(CHOH)2-CH2-O-COR (III), либо R2 обозначает стереоиэомерные формы цепей -(CHOH)3-CH2-O-COR (II) или -CH2-(CHOH)2-CH2-COR (III), a R3 обозначает атом водорода, и R обозначает радикал -(Алк)i-(Циклоалк), в котором Алк обозначает алкильный...
Фармацевтические композиции, содержащие производные 3-аминоазетидина, новые производные и способ их получения

Номер патента: 4649
Опубликовано: 24.06.2004
Авторы: Иттэнжер Огюстэн, Букерель Жан, Бушар Эрве, Майерс Майкл, Филош Брюно, Гризони Серж, Ашар Даниель
МПК: C07D 205/04, A61K 31/397, A61P 25/00...
Метки: получения, содержащие, 3-аминоазетидина, композиции, новые, производные, способ, фармацевтические
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы в которой R1 обозначает радикал -NHCOR4 или -N(R5)-Y-R6, Y обозначает CO или SO2, R2 и R3, одинаковые или разные, обозначают либо ароматический радикал, выбранный из фенила, нафтила и инденила, которые могут быть незамещенными или замещенными одним или несколькими заместителями: галоген, алкил, алкокси, формил, гидрокси, трифторметил, трифторметокси,...
Новые замещенные димерные соединения, способ их получения и фармацевтические композиции, содержащие их

Номер патента: 3391
Опубликовано: 24.04.2003
Авторы: Ренар Пьер, Делагранж Филипп, Шев Гвенаэль, Гийоме Жеральд, Беннежан Каролин, Вио Мари-Клод, Декамп-Франсуа Кароль, Ларрайа Карлос, Йус Сэд, Депре Патрик
МПК: A61P 25/00, C07C 233/25, A61K 31/343...
Метки: фармацевтические, димерные, соединения, содержащие, композиции, способ, новые, замещенные, получения
Формула / Реферат:
1. Соединение формулы (I) A-G1-Cy-G2-Cy'-G3-B (I), где A обозначает -NHC(Q)R2, где Q означает O или S, a R2 означает (C1-C6)алкильную группу; B обозначает -NHC(Q)R2, C(Q)OR2, где Q и R2 имеют вышеуказанные значения; G1 и G3 обозначают возможно разветвленную алкиленовую группу, Cy и Cy' обозначает нафтил, 1,4-бензодиоксин, тетрагидронафтил, бензотиенил, дигидробензотиенил, бензофурил, дигидробензофурил, пирроло[3,2-b]пиридинил, индолил,...
Новые производные индолина, способ их получения и фармацевтические композиции, содержащие их

Номер патента: 5620
Опубликовано: 28.04.2005
Авторы: Мюллер Оливье, Лавиелль Жильбер, Миллан Марк, Гобер Ален
МПК: C07D 401/12, A61K 31/435
Метки: индолина, получения, способ, производные, композиции, содержащие, фармацевтические, новые
Формула / Реферат:
1. Соединения формулы (I) в которой R1 и R2 вместе образуют бензокольцо, необязательно замещенное атомом галогена или алкильной группой, алкоксигруппой, цианогруппой, нитрогруппой, гидроксигруппой, аминогруппой, алкиламиногруппой, диалкиламиногруппой или трифторметильной группой, и R3 обозначает атом водорода, или R1 обозначает атом водорода и R2 и R3 вместе образуют бензокольцо, необязательно замещенное атомом галогена или алкильной группой,...
Предыдущий патент: Стабилизированная суспензионная лекарственная форма для перорального применения
Следующий патент: Фармацевтическая композиция, содержащая ингибитор pde4 или рde3/4 и антагонист гистаминового рецептора
Случайный патент: Способ лазерной обработки стекла