Замещённые тиоацетамиды
Номер патента: 7781
Опубликовано: 27.02.2007
Авторы: Малламо Джон П., Чаттерджи Санкар, Бэйкон Эдвард Р., Трипатхи Рабиндранатх, Данн Дерек, Миллер Мэттью С., Вот Джеффри Л.

























Формула / Реферат
1. Соединение формулы (VI)

где Ar1 и Ar2, каждый независимо, выбраны из фенила и тиенила;
где каждый из Ar1 или Ar2 может быть независимо необязательно замещен 1-3 заместителями, независимо выбранными из Н, F, Cl, Br, I, -ОН, -OR7 или C1-C8алкила;
Q представляет собой C1-С3алкилен;
R2A представляет собой Н,
R4A представляет собой Н или C1-C8алкил,
R7 представляет собой C1-C8алкил,
и его стереоизомерные формы, смеси стереоизомерных форм или фармацевтически приемлемые формы солей и сложных эфиров.
2. Соединение по п.1, где Ar1 и Ar2, каждый независимо, представляют собой незамещенный фенил или тиенил.
3. Соединение по п.2, где Ar1 и Ar2 каждый представляют собой фенил.
4. Соединение по п.1, где Q представляет собой C1 или С2алкилен.
5. Соединение по п.1, где R4A представляет собой Н, незамещенный C1-C6алкил или C1-C6алкил, замещенный OR, где R представляет собой Н или C1-C6алкил.
6. Соединение по п.1, выбранное в соответствии с табл. 2А.
Таблица 2А


7. Соединение формулы (VIII)

где X представляет собой связь или -O-,
каждое из колец А и В вместе с атомами углерода, с которыми они связаны, независимо выбрано из бензо или тиенила;
Q представляет собой C1-С3алкилен;
R2A представляет собой Н;
R4A представляет собой Н, C1-C8алкил, фенил, -ОН или OR7;
R7 представляет собой C1-C8алкил;
и его стереоизомерные формы, смеси стереоизомерных форм или фармацевтически приемлемые формы солей и сложных эфиров.
8. Соединение по п.7, где кольца А и В представляют собой бензо.
9. Соединение по п.7, где Q представляет собой C1- или С2алкилен.
10. Соединение по п.7, где А и В представляют собой бензо; X представляет собой связь; R2A представляет собой Н; Q представляет собой C1- или C2алкилен; R4A представляет собой Н, OR7, фенил или C1-C6алкил, необязательно замещенный OR, где R представляет собой Н или C1-C6алкил.
11. Соединение по п.7, выбранное в соответствии с табл. 2В.
Таблица 2В


12. Соединение по п.7, где X представляет собой связь.
13. Соединение формулы (VII-1)

14. Применение соединения по любому из пп.1, 7 или 13 для получения лекарственного средства для лечения сонливости, усталости, болезни Паркинсона, церебральной ишемии, удара, апноэ по время сна, расстройств питания, расстройства дефицита внимания с гиперактивностью, нарушений познавательной способности или слабости; а также для стимуляции бодрствования, стимуляции аппетита или стимуляции прибавки в весе.
15. Применение по п.14, в котором лекарственное средство вводят для лечения депрессии, шизофрении или синдрома хронической усталости.
16. Фармацевтическая композиция, включающая соединение по пп.1, 7 или 13 в смеси с одним или несколькими фармацевтически приемлемыми эксципиентами для применения в лечении сонливости, усталости, болезни Паркинсона, церебральной ишемии, удара, апноэ во время сна, расстройств питания, расстройства дефицита внимания с гиперактивностью, нарушений познавательной способности или слабости; а также для стимуляции бодрствования, стимуляции аппетита или стимуляции прибавки в весе.
17. Способ лечения заболеваний или расстройств у субъекта, нуждающегося в таком лечении, включающий введение указанному субъекту терапевтически эффективного количества соединения по любому из пп.1, 7 или 13, где указанное соединение вводят для лечения сонливости, усталости, болезни Паркинсона, церебральной ишемии, удара, апноэ во время сна, расстройств питания, расстройства дефицита внимания с гиперактивностью, нарушений познавательной способности или слабости; а также для стимуляции бодрствования, стимуляции аппетита или стимуляции прибавки в весе.
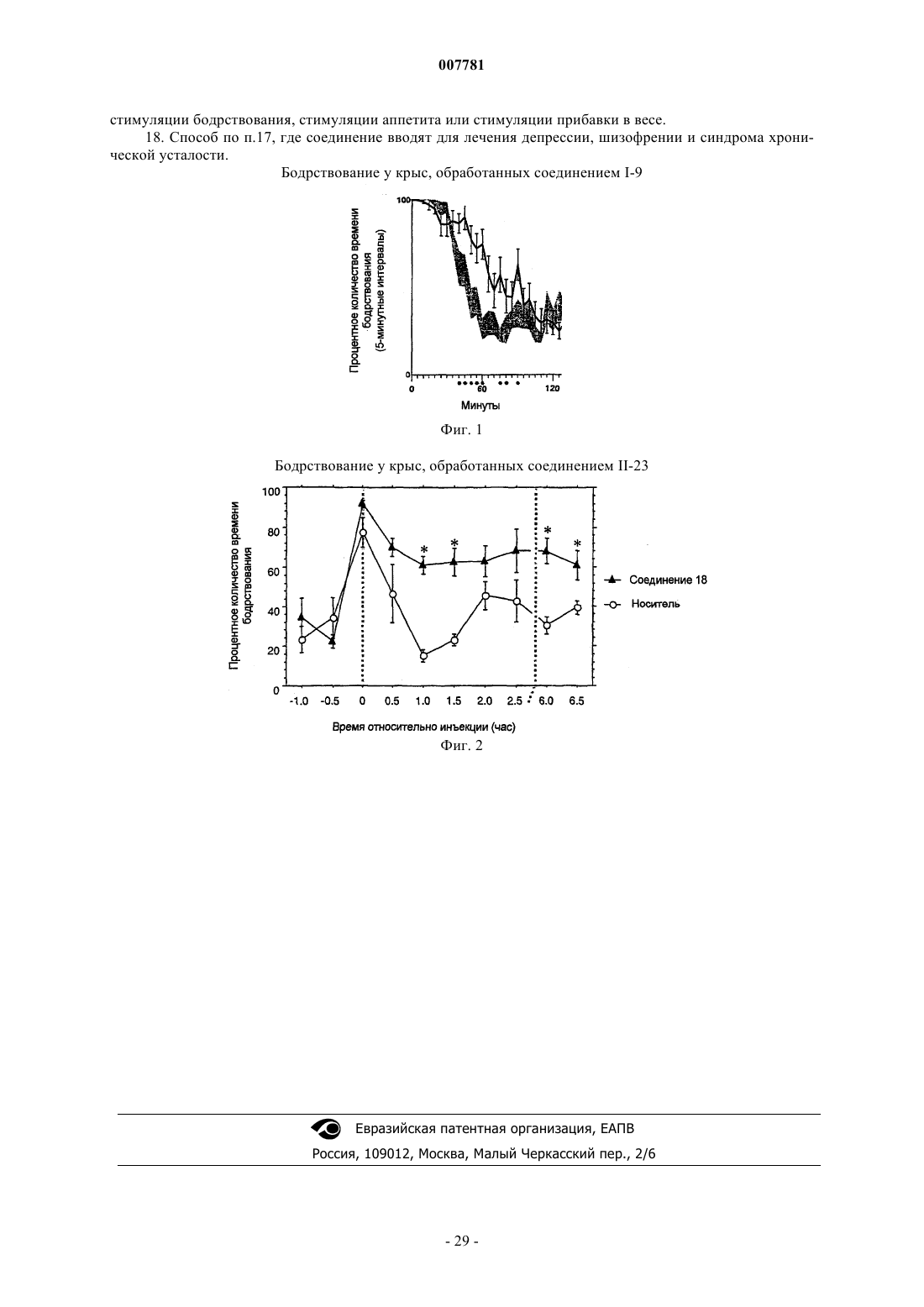
18. Способ по п.17, где соединение вводят для лечения депрессии, шизофрении и синдрома хронической усталости.
Текст
007781 Ссылка на родственные заявки Настоящее изобретение испрашивает приоритет на основании патентной заявки США 10/014645,поданной 26 октября 2001 г. Область изобретения Настоящее изобретение относится к химическим композициям, способам их получения и применению композиций. В частности, настоящее изобретение относится к композициям, которые включают замещенные тиоацетамиды, и к их применению для лечения заболеваний, в том числе для лечения сонливости, стимулирования бодрости, лечения болезни Паркинсона, церебральной ишемии, удара, апноэ во время сна, нарушений питания, стимуляции аппетита и прибавки в весе, лечения расстройства дефицита внимания ("ADHD"), связанного с гиперактивностью, для улучшения дееспособности при заболеваниях,связанных с гиперактивностью коры головного мозга, включая, но не ограничиваясь этим, депрессию,шизофрению, усталость, в частности, усталость, связанную с неврологическими заболеваниями, такими как рассеянный склероз, синдром хронической усталости, и для улучшения при расстройствах познавательной способности. Предпосылки изобретения Соединения, раскрытые в данной заявке, относятся к биологическим и химическим аналогам модафинила. Модафинил, C15H15NO2S, также известный как 2-(бензгидрилсульфинил)ацетамид или 2[(дифенилметил)сульфинил]ацетамид, представляет собой синтетическое производное ацетамида, обладающее активностью по стимуляции бодрости, структура которого была описана в патенте Франции 7805510 и патенте США 4177290 ('290) и который был одобрен Ведомством США по пищевым продуктам и лекарствам для применения в лечении чрезмерной сонливости в дневное время, связанной с нарколепсией. Модафинил был испытан для лечения некоторых поведенческих состояний в сочетании с различными веществами, включая апоморфин, амфетамин, резерпин, оксотреморин, гипнотические средства, йохимбин, 5-гидрокситриптофан и ингибиторы моноаминоксидазы, как описано в ссылочных патентах. Способ получения рацемической смеси описан в патенте '290, а способ получения левовращающего изомера описан в патенте США 4927855 (оба включены в данную заявку в качестве ссылки). Сообщается, что левовращающий изомер является полезным для лечения гиперсомнии, депрессии, болезни Альцгеймера и обладает активностью, направленной на улучшение симптомов деменции и потери памяти, особенно у пожилых людей. Основной фармакологической активностью модафинила является стимулирование бодрости. Модафинил стимулирует бодрость у крыс (Touret et al., 1995; Edgar and Seidel, 1997), кошек (Lin et al., 1992),собак (Shelton et al., 1995) и приматов, отличных от человека (Hernant et al., 1991), а также в моделях,имитирующих клинические ситуации, такие как апноэ во время сна (Модель нарушения дыхания английского бульдога во время сна) (Panckeri et al., 1996) и нарколепсии (нарколептические собаки) (Sheltonet al., 1995). Модафинил также был описан как средство, обладающее активностью в центральной нервной системе, и как полезное средство для лечения болезни Паркинсона (патент США 5180745); в защите церебральной ткани от ишемии (патент США 5391576); при лечении недержания мочи и кала (патент США 5401776); и для лечения апноэ во время сна и расстройств, связанных с центральной нервной системой (патент США 5612379). Патент США 5618845 описывает препараты модафинила с определенным размером частиц, менее 200 мкм. Кроме того, модафинил можно использовать для лечения расстройств питания или для стимулирования прибавки в весе или для стимулирования аппетита у людей и животных (предварительная заявка на патент США 60/150071, включенная в настоящую заявку в качестве ссылки), или для лечения расстройства дефицита внимания (ADHD), связанного с гиперактивностью, или усталости (слабости), особенно усталости, связанной с рассеянным склерозом (предварительная заявка на патент США 60/149612, включенная в настоящую заявку в качестве ссылки). Некоторые опубликованные патентные заявки описывают производные формы модафинила и применение производных модафинила для лечения различных расстройств. Например, публикация РСТ WO 99/25329 описывает аналоги модафинила, в которых фенильные группы замещены F, Cl, Br, CF3, NO2,NH2, С 1-С 4 алкилом, С 1-С 4 алкилокси или метилендиокси, и в которых амид замещен ОН, С 1-С 4 алкилом,С 1-С 4 гидроксиалкилом или C1-C4 углеводородным радикалом. Такие композиции описаны как полезные для лечения вызванной лекарственными средствами сонливости, особенно сонливости, связанной с введением морфина пациентам, больным раком. Аналогично, патент США 4066686 описывает бензгидрилсульфинильные производные, включающие производные модафинила с удлиненной алкильной цепью между сульфинильной и карбонильной группами, и где NR3R4 представляет собой NHOH. Эти соединения описаны как полезные в терапии для лечения расстройств центральной нервной системы. Публикация РСТ WO 95/01333 описывает производные модафинила, полезные для улучшения расстройств питания. Модификации модафинила включают группу хлора в положении 3 одной из фенильных групп, и замещение пиридилом второго фенила, замещение водородов одной или двумя метильными группами в положении 2 углерода, водороды амида могут быть замещены одной или двумя группами,выбранными из Н, пиридилметильной или этильной групп, и, кроме того, сера может быть неокислен-1 007781 ной. Публикация РСТ WO 95/01171 также описывает модифицированные производные модафинила, которые, как указано, полезны для улучшения расстройств питания. Описанные соединения включают 4 фтор-, 3-фтор- и 4-хлорзамещения в первой фенильной группе и 4-фтор- или 3-фторзамещения во второй фенильной группе. Также описаны замещения, когда амид содержит заместители ОН или изопропильную группу. Terauchi, H., et al., описали никотинамидные производные, полезные в качестве ингибиторов АТФ-азы (Terauchi, H, et al., J. Med.Chem., 1997, 40, 313-321). В частности, описаны некоторые N-алкилзамещенные 2-(бензгидрилсульфинил)никотинамиды. Патенты США 4980372 и 4935240 описывают производные бензоиламинофеноксибутановой кислоты. В частности, описаны сульфидные производные модафинила, содержащие фенильную и замещенную фенильную линкерную группу между сульфидом и карбонилом, и замещенный арил в положении концевого амида. Были описаны другие производные модафинила, в которых концевые фенильные группы ограничены связывающей группой. Например, в патенте США 5563169 сообщается о некоторых ксантенильных или тиаксантенильных производных, содержащих замещенный арил в положении концевого амида. Другие ксантенильные или тиаксантенильные производные описаны Annis, I; Barany, G. Pept. Proc.Am. Pept. Symp. 15th (Meeting Date 1997) 343-344, 1999 (получение ксантенильного производного реагента Эллмана (Ellman), полезного в качестве реагента в синтезе пептидов); Han, Y.; Barany, G., J. Org.(тиаксантенольные производные тиогликолевой кислоты). Таким образом, существует потребность в новых классах соединений, обладающих полезными свойствами. Было обнаружено, что класс соединений, который в данной заявке указан как замещенные тиоацетамиды, представляет собой полезные средства для лечения или профилактики заболеваний или расстройств, в том числе для лечения сонливости, стимулирования бодрости, лечения болезни Паркинсона, церебральной ишемии, удара, апноэ во время сна, расстройств питания, стимуляции аппетита и прибавки в весе, расстройства дефицита внимания, связанного с гиперактивностью, улучшения дееспособности при заболеваниях, связанных с гиперактивностью коры головного мозга, включая, но не ограничиваясь этим, депрессию, шизофрению, усталость, в частности, усталость, связанную с неврологическими заболеваниями, такими как рассеянный склероз, синдром хронической усталости, и для улучшения расстройств познавательной способности. Настоящее изобретение направлено на эти, а также другие важные цели. Краткое описание изобретения Настоящее изобретение в одном своем аспекте предлагает, частично, различные новые замещенные тиоацетамиды. Другие аспекты настоящего изобретения также включают их фармацевтические композиции, способы их получения и применение этих соединений для лечения заболеваний. В одном аспекте настоящего изобретения предлагаются соединения формулы (VI) Составляющие группы и предпочтительные воплощения описаны подробно ниже. В другом аспекте настоящего изобретения предлагаются соединения формулы (VIII) Составляющие группы и предпочтительные воплощения описаны подробно ниже. Другой целью настоящего изобретения является создание соединения формулы (VII-1) Другой целью настоящего изобретения является создание способов лечения или профилактики заболеваний или расстройств, в том числе для лечения сонливости, стимулирования бодрости, лечения болезни Паркинсона, церебральной ишемии, удара, апноэ во время сна, расстройств питания, стимуляции аппетита и прибавки в весе, расстройства дефицита внимания, связанного с гиперактивностью,улучшения дееспособности при заболеваниях, связанных с гиперактивностью коры головного мозга,включая, но не ограничиваясь этим, депрессию, шизофрению, усталость, в частности, усталость, связанную с неврологическими заболеваниями, такими как рассеянный склероз, синдром хронической усталости, и для улучшения расстройств познавательной способности. Еще одной целью настоящего изобретения является создание фармацевтических композиций,включающих соединения по настоящему изобретению, где композиции включают один или несколько фармацевтически приемлемых эксципиентов и терапевтически эффективное количество по меньшей мере одного соединения по настоящему изобретению или его фармацевтически приемлемую форму соли или сложного эфира. Эти и другие цели, характерные признаки и преимущества замещенных тиоацетамидов раскрываются ниже в разделе "Подробное описание изобретения". Краткое описание чертежей На фиг. 1 графически представлены данные определенного при помощи ЭЭГ бодрствования у крыс,леченых соединением I-9 (100 мг/кг, в/б, сплошная линия) или метилцеллюлозным носителем (пунктирная линия). Бодрость количественно определяют в 5-минутных бинах. N=13 крыс/группа. р 0,05 против животных, обработанных носителем. На фиг. 2 графически представлены данные определенного при помощи ЭЭГ бодрствования у крыс,леченых соединением II-23 (100 мг/кг, и/п, черные треугольники) или метилцеллюлозным носителем(незаштрихованные круги). Каждая точка представляет средний процент времени бодрствования для следующего получасового периода. р 0,05 против животных, обработанных носителем. Подробное описание изобретения В одном варианте воплощения настоящего изобретения предлагаются новые соединения формулы где Ar1 и Аr2, каждый независимо, выбраны из фенила и тиенила; где каждый из Ar1 или Аr2 может быть независимо необязательно замещен 1-3 заместителями, независимо выбранными из: Н, F, Cl, Br, I, -ОН, -OR7 или C1-C8 алкила;R2A представляет собой Н,R4A представляет собой Н или C1-C8 алкил,R7 представляет собой C1-C8 алкил,и их стереоизомерные формы, смеси стереоизомерных форм или фармацевтически приемлемые формы солей и сложных эфиров. Предпочтительные варианты воплощения соединений формулы (VI) представлены ниже. 1). Соединения формулы (VI), где Ar1 и Аr2, каждый независимо, представляют собой незамещенный фенил или тиенил. 2). Соединения формулы (VI), где Ar1 и Аr2 каждый представляют собой фенил. 3). Соединения формулы (VI), где Q представляет собой C1 или C2 алкилен. 4). Соединения формулы (VI), где R4A представляет собой Н, незамещенный C1-С 6 алкил или C1-C6,замещенный OR, где R представляет собой Н или C1-C6. 5). Соединения формулы (VI), выбранные в соответствии с табл. 2 А. В другом варианте воплощения настоящего изобретения предлагаются новые соединения формулы где X представляет собой связь или -O-,каждое из колец А и В вместе с атомами углерода, с которыми они связаны, независимо выбрано из бензо или тиенила;R7 представляет собой C1-C8 алкил; и его стереоизомерные формы, смеси стереоизомерных форм или фармацевтически приемлемые формы солей и сложных эфиров. Предпочтительные варианты воплощения соединений формулы (VIII) представлены ниже. 1). Соединения формулы (VIII), где кольца А и В представляют собой бензо. 2). Соединения формулы (VIII), где Q представляет собой C1- или С 2 алкилен. 3). Соединения формулы (VIII), где А и В представляют собой бензо; X представляет собой связь; 5). Соединения формулы (VIII), где X представляет собой связь. Еще одно воплощение настоящего изобретения представляет собой соединение формулы (VII-1) Также изобретение относится к применению соединений по изобретению для получения лекарственного средства для лечения сонливости, усталости, болезни Паркинсона, церебральной ишемии, удара,апноэ по время сна, расстройств питания, расстройства дефицита внимания с гиперактивностью, нарушений познавательной способности или слабости; а также для стимуляции бодрствования, стимуляции аппетита или стимуляции прибавки в весе. Предпочтительно лекарственное средство по изобретению вводят для лечения депрессии, шизофрении или синдрома хронической усталости. Еще одно воплощение настоящего изобретения представляет собой фармацевтическую композицию, включающую соединение по изобретению в смеси с одним или несколькими фармацевтически приемлемыми эксципиентами для применения в лечении сонливости, усталости, болезни Паркинсона, церебральной ишемии, удара, апноэ по время сна, расстройств питания, расстройства дефицита внимания с гиперактивностью, нарушений познавательной способности или слабости; а также для стимуляции бодрствования, стимуляции аппетита или стимуляции прибавки в весе. И, наконец, изобретение также относится к способу лечения заболеваний или расстройств у субъекта, нуждающегося в таком лечении, включающему введение указанному субъекту терапевтически эффективного количества соединения формулы (VI), (VIII) или (VII-1), где указанное соединение вводят для лечения сонливости, усталости, болезни Паркинсона, церебральной ишемии, удара, апноэ по время сна, расстройств питания, расстройства дефицита внимания с гиперактивностью, нарушений познавательной способности или слабости; а также для стимуляции бодрствования, стимуляции аппетита или стимуляции прибавки в весе. Предпочтительно соединение по изобретению вводят для лечения депрессии, шизофрении и синдрома хронической усталости. Определения Как используется в данном описании термин "алкил" относится к замещенной или незамещенной,разветвленной или линейной углеводородной цепи из 1-8 атомов углерода, образованной путем удаления одного атома водорода. В некоторых предпочтительных вариантах воплощения, алкильная группа со-5 007781 держит от 1 до 6 атомов углерода. В других предпочтительных вариантах воплощения алкильная группа содержит от 1 до 4 атомов углерода. Обозначение "С 1-С 4 алкил" относится к алкильному радикалу, содержащему от 1 до 4 атомов углерода. Примеры включают метил, этил, н-пропил, изопропил, н-бутил,изобутил, втор-бутил, трет-бутил, пентил, 2-метилпентил, гексил, 2-метилгексил, 2,3-диметилгексил,гептил, октил и т.д. Как использован в данном описании, термин "замещенный" относится к замещению одного или нескольких атомов водорода в указанной группе выбранной группой, называемой в данном описании "заместителем", при условии, что не превышается валентность замещаемого атома и что замещение приводит к стабильному соединению. Замещенная группа имеет 1-5, предпочтительно 1-3 и более предпочтительно 1 независимо выбранных заместителей. Предпочтительные заместители включают, но не ограничиваются этим, F, Cl, Br, I, ОН, OR, NH2, NR2, NHOH, NO2, CN, CF3, CF2CF3, C1-C6, C2-С 6 алкенил, С 2 С 6 алкинил, C1-С 6 алкокси, С 3-С 7 циклоалкил, гетероциклил, C6-С 10 арил, гетероарил, арилалкил, C(=O)R,СООН, CO2R, O-C(=O)R, C(=O)NRR', NRC(=O)R', NRCO2R', OC(=O)NRR', -NRC(=O)NRR',-NRC(=S)NRR' и -SO2NRR', где R и R', каждый независимо, представляет собой водород, C1-C6 или C6 С 10 арил. Как используется в данном описании, термин "алкилен" относится к замещенному или незамещенному углеводороду с разветвленной или прямой цепью, содержащему 1-8 атомов углерода, образованному путем удаления двух атомов водорода. Такое обозначение как "С 1-С 4 алкилен" относится к алкиленовому радикалу, содержащему от 1 до 4 атомов углерода. Примеры включают метилен (-СН 2-), пропилиден (СН 3 СН 2 СН=), 1,2-этандиил (-СН 2 СН 2-) и т.д. Как используется в данном описании, термин "субъект" относится к теплокровному животному, такому как млекопитающее, предпочтительно к человеку или к ребенку, пораженному или который потенциально может быть поражен одним или несколькими заболеваниями и состояниями, описанными в данной заявке. Как используется в данном описании, термин "терапевтически эффективное количество" относится к количеству соединения по настоящему изобретению, которое является эффективным для уменьшения,устранения, лечения или контролирования симптомов описанных в данной заявке заболеваний и состояний. Термин "контролирование" относится ко всем способам, где может иметь место замедление, прерывание, задержка или остановка развития заболеваний и состояний, описанных в данной заявке, но необязательно указывает на полное устранение всех симптомов заболеваний и состояний, и также включает профилактическое лечение. Как используется в данном описании, термин "фармацевтически приемлемый" относится к тем соединениям, веществам, композициям и/или лекарственным формам, которые с точки зрения медицины являются подходящими для контакта с тканями человека и животных, без чрезмерной токсичности, раздражения, аллергических реакций или других проблемных осложнений, в соответствии с разумным соотношением польза/риск. Как используется в данном описании, термин "фармацевтически приемлемые соли" относится к производным раскрываемых в данной заявке соединений, где исходное соединение модифицируют, получая его соль с кислотой или основанием. Фармацевтически приемлемые соли включают обычные нетоксичные соли или четвертичные аммониевые соли исходного соединения, образованные, например, из нетоксичных неорганических или органических кислот. Например, такие обычные нетоксичные соли включают соли, полученные из неорганических кислот, таких как хлористо-водородная, серная, сульфаминовая, фосфорная, азотная и т.д.; и соли, полученные из органических кислот, таких как уксусная,пропионовая, янтарная, винная, лимонная, глутаминовая, бензойная, салициловая, толуолсульфоновая,щавелевая и т.д. Фармацевтически приемлемые соли по настоящему изобретению можно синтезировать из исходного соединения, которое содержит основную или кислотную группу, традиционными химическими способами. Обычно, такие соли можно получить взаимодействием свободной кислоты или основания таких соединений со стехиометрическим количеством подходящего основания или кислоты в воде или в органическом растворителе или в смеси двух таких веществ. Обычно предпочтительной является неводная среда, например эфир, этилацетат, этанол, изопропанол или ацетонитрил. Перечень подходящих солей можно найти в Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, PA, 1985,p. 1418, раскрытие которого включено в данную заявку в качестве ссылки. Как используется в данном описании, термин "пролекарство" предназначен для включения любых ковалентно связанных носителей, которые высвобождают активное исходное лекарственное вещество,соответствующее соединениям по настоящему изобретению, in vivo, когда такое пролекарство вводят млекопитающему субъекту. Поскольку известно, что пролекарства улучшают различные желаемые свойства фармацевтических препаратов (например, растворимость, биодоступность, изготовление и т.д.), соединения по настоящему изобретению могут доставляться в форме пролекарств. Таким образом, настоящее изобретение охватывает пролекарства заявленных соединений, содержащие их композиции и способы их доставки. Пролекарства соединения по настоящему изобретению можно получить путем модификации функциональных групп, присутствующих в соединении, таким образом, чтобы модифицирован-6 007781 ные группы отщеплялись, либо посредством рутинного манипулирования, либо in vivo, до исходного соединения. Соответственно, пролекарства включают, например, соединения по настоящему изобретению, в которых гидрокси, амино или карбоксигруппа связана с любой группой, которая при введении лекарственного средства млекопитающему субъекту отщепляется с образованием свободного гидроксила, свободного амино или карбоновой кислоты соответственно. Примеры включают, но не ограничиваются этим, ацетатные, формиатные и бензоатные производные спиртовых и функциональных групп амина; и алкильные, циклоалкильные, арильные и алкиларильные сложные эфиры, такие как метиловый,этиловый, циклопропиловый, фениловый, бензиловый и фенетиловый сложные эфиры, и т.д. Настоящее изобретение предлагает способ лечения заболеваний и состояний у субъекта, нуждающегося в таком лечении, включающий введение указанному субъекту терапевтически эффективного количества соединения формулы (VI), (VIII) или (VII-1). Например, соединения формулы (VI), (VIII) или(VII-1) можно применять для лечения сонливости, предпочтительно сонливости, связанной с нарколепсией, стимулирования бодрости, лечения болезни Паркинсона, церебральной ишемии, удара, апноэ во время сна, расстройств приема пищи, предпочтительно расстройств приема пищи, связанных с заболеванием, в частности, когда заболевание представляет собой anorexia nervosa, стимуляции аппетита и прибавки в весе, лечения расстройства дефицита внимания, связанного с гиперактивностью, улучшения дееспособности при заболеваниях, связанных с гиперактивностью коры головного мозга, включая, но не ограничиваясь этим, депрессию, шизофрению, усталость, в частности, усталость, связанную с неврологическими заболеваниями, такими как рассеянный склероз, синдром хронической усталости, и для улучшения расстройств познавательной способности. Идентификация субъектов, нуждающихся в лечении указанных в данной заявке заболеваний и состояний находится в пределах компетенции и знаний специалистов. Опытный практикующий врач при помощи клинических тестов, врачебного осмотра и истории болезни/семейного анамнеза может легко определить тех субъектов, которые нуждаются в таком лечении. Терапевтически эффективное количество может легко определить лечащий врач, как специалист в данной области, при помощи обычных методов и путем наблюдения результатов при аналогичных обстоятельствах. При определении терапевтически эффективного количества лечащий врач должен учитывать различные факторы, включая, но не ограничиваясь этим, к какому виду млекопитающих относится субъект; его размер, возраст и общее состояние здоровья; конкретное, подлежащее лечению заболевание; степень поражения или тяжесть заболевания; ответную реакцию конкретного субъекта; конкретное вводимое соединение; способ введения; характеристики биодоступности вводимого препарата; выбранную схему введения; применение сопутствующего лечения; и другие соответствующие обстоятельства. Количество соединения формулы (VI), (VIII) или (VII-1), необходимое для достижения желаемого биологического эффекта, варьирует в зависимости от различных факторов, включая дозу вводимого лекарственного средства, химические характеристики (например, гидрофобность) применяемых соединений, активность соединений, вид заболевания, состояние заболевания у пациента и путь введения. Как правило, соединения по настоящему изобретению могут предоставляться в водном физиологическом буферном растворе, содержащем около 0,1-10% мас./об. соединения для парентерального введения. Типичные дозы находятся в пределах от около 1 мкг/кг до около 1 г/кг веса тела в день; предпочтительные пределы доз составляют от около 0,01 до около 100 мг/кг веса тела в день. Предпочтительная суточная доза для взрослого человека включает около 25, 50, 100 и 200 мг, и эквивалентная доза для ребенка. Предпочтительные дозы подлежащего введению лекарственного средства, по-видимому, зависят от таких переменных, как вид и прогрессирование заболевания, общее состояние здоровья конкретного пациента, относительная биологическая эффективность выбранного соединения и композиция эксципиентов для этого соединения, а также от пути введения. Соединения по настоящему изобретению можно вводить в виде стандартных доз, где термин "стандартная доза" означает отдельную дозу, которую можно вводить пациенту и которой можно легко манипулировать и которую легко можно упаковать, при этом она остается физически и химически стабильной стандартной дозой, включающей либо активное соединение как таковое, либо в виде фармацевтически приемлемой композиции, как описано ниже. Такая доза типично находится в пределах от около 0,1 до 100 мг/кг веса тела. Согласно общему руководству, стандартная доза для взрослых находится в пределах от около 0,1 до около 1000 мг в день. Предпочтительно стандартная доза для взрослых находится в пределах от около 1 до около 500 мг при введении от одного до четырех раз в день и даже более предпочтительно от около 10 мг до около 300 мг два раза в день. Согласно альтернативному способу описания эффективной дозы предпочтительная пероральная стандартная доза представляет собой дозу, необходимую для достижения уровня содержания лекарственного средства в сыворотке крови субъекта от около 0,05 до 20 мкг/мл и более предпочтительно от около 1 до около 20 мкг/мл. Соединения, представленные в настоящей заявке, можно формулировать в фармацевтические композиции смешиванием с одним или несколькими фармацевтически приемлемыми эксципиентами. Такие композиции могут быть приготовлены для применения путем перорального введения, в частности, в форме таблеток или капсул; или для парентерального введения, в частности, в форме жидких растворов,суспензий или эмульсий; или для введения интраназальным путем, в частности, в форме порошков, ка-7 007781 пель в нос или аэрозолей; или для нанесения на кожу, например, местным путем или при помощи чрескожных пластырей. Композиции можно удобным образом вводить в виде стандартных единиц лекарственной формы, и они могут быть получены методами, хорошо известными в области фармации, например, как описано вRemington: The Science and Practice of Pharmacy, 20th ed.; Gennaro, A.R., Ed.; Lippincott Williams and Wilkins: Philadelphia, PA, 2000. В качестве части композиции могут быть включены фармацевтически приемлемые связующие вещества и/или адъюванты. Композиции для перорального применения обычно включают инертный разбавитель или носитель или съедобный носитель. Таблетки, пилюли, порошки, капсулы, драже и т.п. могут содержать один или несколько любых указанных далее ингредиентов, или соединений подобной структуры: связующее, такое как микрокристаллическая целлюлоза или камедь трагаканта; разбавитель, такой как крахмал или лактоза; разрыхлители, такие как крахмал или производные целлюлозы; смазывающее вещество, такое как стеарат магния; агент скольжения, такой как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или отдушку, такую как перечная мята или метилсалицилат. Капсулы могут иметь форму твердых капсул или мягких капсул, которые обычно получают из желатиновых смесей, необязательно смешанных с пластификаторами, а также форму крахмальных капсул. Кроме того, стандартные дозы лекарственной формы могут содержать другие различные вещества, которые модифицируют физическую форму лекарственной формы, например, покрытия из сахара, шеллака или энтеросолюбильных веществ. Другие пероральные лекарственные формы, сироп или эликсир могут содержать подсластители, консерванты, красители, окрашивающие вещества и отдушки. Кроме того, активные соединения можно включать в препараты и композиции быстрого растворения, с измененным или замедленным высвобождением, и где такие композиции замедленного высвобождения предпочтительно являются двухслойными. Предпочтительные композиции включают фармацевтические композиции, в которых соединение по настоящему изобретению сформулировано для перорального или парентерального введения или более предпочтительно композиции, в которых соединение по настоящему изобретению сформулировано в виде таблетки. Предпочтительные таблетки содержат лактозу, кукурузный крахмал, силикат магния, натрийкроскармелозу, повидон, стеарат магния или тальк в любой комбинации. Один аспект настоящего раскрытия также состоит в том, что соединение по настоящему изобретению можно включать в пищевой продукт или жидкость. Жидкие, предназначенные для введения препараты включают стерильные водные или неводные растворы, суспензии и эмульсии. Жидкие композиции также могут включать связующие, буферы, консерванты, хелатообразующие вещества, подсластители, отдушки и красители и т.п. Неводные растворители включают спирты, пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, и органические сложные эфиры, такие как этилолеат. Водные носители включают смеси спиртов и воды, забуференные среды и физиологический раствор. В частности, полезными эксципиентами для контроля высвобождения активных соединений могут быть биосовместимые, биоразлагаемые лактидный полимер, сополимер лактида/гликолида или сополимеры полиоксиэтилена-полиоксипропилена. Внутривенные носители могут включать жидкие и питательные добавки, электролитные добавки, такие как добавки на основе декстрозы Рингера, и т.п. Другие потенциально полезные парентеральные системы доставки для этих активных соединений включают частицы этилен-винилацетатного сополимера, осмотические насосы, имплантируемые системы для инфузий и липосомы. Альтернативные способы введения включают композиции для ингаляций, которые включают такие средства, как сухой порошок, аэрозоль или капли. Они могут представлять собой водные растворы, содержащие, например, полиоксиэтилен-9-лауриловый эфир, гликохолат и деоксихолат, или масляные растворы для введения в форме назальных капель, или гели для интраназального применения. Композиции для буккального применения включают, например, леденцы или пастилки и могут также включать вкусовую основу, такую как сахароза или аравийская камедь, и другие эксципиенты, такие как гликохолат. Композиции, подходящие для ректального введения, предпочтительно представлены в виде суппозиториев, которые включают носитель на твердой основе, такой как масло какао, и могут включать салицилат. Композиции для местного нанесения на кожу предпочтительно имеют форму мази, крема, лосьона,пасты, геля, спрея, аэрозоля или масла. Носители, которые можно использовать, включают вазелин, ланолин, полиэтиленгликоли, спирты или их сочетания. Композиции, подходящие для чрескожного введения, могут быть представлены в виде отдельных пластырей, и могут представлять собой липофильные эмульсии или забуференные водные растворы, растворенные и/или диспергированные в полимере или адгезиве. Соединения по настоящему изобретению можно применять в виде единственного активного ингредиента в фармацевтической композиции. Альтернативно, их можно применять в сочетании или соединенными с другими фармацевтическими средствами для лечения других болезненных состояний. В частности, соединения формулы (VI), (VIII) или (VII-1) можно объединить со средствами, полезными для лечения нарушений познавательной способности, связанных с различными болезненными состояниями,включая, но не ограничиваясь этим, возрастные состояния, травму, стресс или временное ухудшение изза химического дисбаланса или токсичности, гиперсомнию, депрессию, болезнь Альцгеймере, деменции-8 007781 не по типу Альцгеймера, включая деменцию тельца Льюи (Lewy), сосудистую деменцию и деменцию Бинсвангера (Binswanger), шизофрению и т.п. Настоящее изобретение поэтому охватывает комбинации соединений по настоящему изобретению с аналогами эбурнана, гетероциклическими индукторами тирозингидроксилазы, 3,4-дифенилхроманами, такриновыми метаболитами, азациклическими соединениями,полиаминовыми соединениями или тиамином; неантихолинергическими антидепрессантами, такими как бензодиазепины; фенотиазиновыми алифатическими соединениями, такими как хлорпромазин; пиперидинами, такими как тиоридазин; пиперазинами, такими как трифторперазин; флуфеназином и перфеназином; дибензоксазепинами, такими как локсапин; дигидроиндолами, такими как молиндол; тиоксантенами, такими как тиотиксен; бутирофенонами, такими как галоперидол; дифенилбутилпиперидинами,такими как пимозид; дибензодиазепином, таким как рисперидон; тиенобензодиазепином, таким как кветиапин; имидазолидиноном, таким как сертиндол; бензизотиазолилпиперазином, таким как зипразидон,и т.п. Синтез Соединения по настоящему изобретению могут быть получены различными путями, хорошо известными специалистам в данной области. Соединения, например, можно синтезировать способами, описанными ниже, или при помощи вариантов этих способов, как это должно быть понятно специалистам. Подходящие модификации и замещения являются очевидными и хорошо известными, или они доступны для специалистов из научной литературы. Должно быть понятно, что соединения по настоящему изобретению могут содержать один или несколько асимметрично замещенных атомов углерода и могут быть выделены в оптически активной или рацемической форме. Таким образом, охватываются все хиральные, диастереомерные, рацемические формы и все геометрические изомерные формы структуры, если только нет конкретного указания конкретной стереохимии или изомерной формы. Из уровня техники хорошо известно как можно получить и выделить такие оптически активные формы. Например, смеси стереоизомеров можно разделить стандартными методами, включая, но не ограничиваясь этим, разделение рацемических форм, обычную, с обращенной фазой и хиральную хроматографию, избирательное солеобразование, перекристаллизацию и т.п., или путем хирального синтеза либо исходя из хиральных исходных веществ, либо путем целенаправленного синтеза целевых хиральных центров. Как должно быть понятно, функциональные группы, присутствующие в соединениях по настоящему изобретению, могут содержать защитные группы в ходе синтеза. Например, заместители аминокислотной боковой цепи соединений формулы (VI), (VIII) или (VII-1) могут быть замещены защитными группами, такими как бензилоксикарбонильная или трет-бутоксикарбонильная группы. Защитные группы являются известными per se как химические функциональные группы, которые могут быть селективно присоединены к и удалены из функциональных групп, таких как гидроксильные группы и карбоксильные группы. Эти группы присутствуют в химическом соединении для того, чтобы сделать такие функциональные группы инертными к условиям химической реакции, которым подвергают данное соединение. В соответствии с настоящим изобретением можно использовать любую из множества защитных групп. Предпочтительные защитные группы включают бензилоксикарбонильную ("Cbz") группу,трет-бутилоксикарбонильную ("Воc")-группу и тозильную (п-толуолсульфонильную "Tos")-группу. Другие предпочтительные защитные группы, которые могут быть использованы в соответствии с настоящим изобретением, можно найти в Greene, T.W. and Wuts, P.G.M., Protective Groups in Organic Synthesis, 2d.Ed., WileySons, 1991. Примеры Другие характерные признаки настоящего изобретения будут очевидны из представленного ниже описания примеров осуществления изобретения. Эти примеры представлены в целях иллюстрации данного изобретения и не предназначены для его ограничения. Следующие примеры 1-6 были синтезированы в соответствии со схемой 1. Получение соединения С. К интенсивно перемешиваемой смеси тиомочевины (соединение В, 5 г, 0,066 моль), 48% HBr (30 мл) и воды (5 мл) при температуре 70-75 С добавляли 9-гидроксифлуорен (соединение А, 9,28 г, 0,051 моль) небольшими порциями с последующим добавлением дополнительного количества воды (30 мл). Затем реакционную смесь нагревали до температуры 100-105 С (температура бани), поддерживали на бане еще в течение 30 мин и охлаждали до комнатной температуры. Осажденное твердое вещество отфильтровывали, промывали последовательно водой и простым эфиром и сушили в вакууме с получением 14 г соответствующей тиоурониевой соли, которую использовали на следующей стадии без дополнительной очистки. К интенсивно перемешиваемой смеси указанной выше тиоурониевой соли (10,47 г) в 10 н. NaOH(5,24 г, 0,034 моль) в воде (20 мл). Затем реакционную смесь нагревали до температуры 105-110 С (температура бани), поддерживали на бане еще в течение 30 мин, охлаждали до комнатной температуры, разбавляли водой (25 мл) и промывали простым эфиром (350 мл). Щелочной водный слой подкисляли (рН 2-3) концентрированной НСl и экстрагировали в этилацетат (3100 мл). Объединенный органический слой сушили (MgSO4) и концентрировали с получением 7,80 г соединения С, которое использовали на следующей стадии без дополнительной очистки. 1 Н-ЯМР (CDCl3)7,80 (м, 4 Н), 7,30 (м, 4 Н), 4,90 (с, 1 Н),2,10 (м, 4 Н). Получение соединения D. Это соединение получали из соединения А, следуя процедуре, описанной выше для синтеза соединения D, за исключением того, что на стадии алкилирования вместо 3-бромпропионовой кислоты использовали 4-броммасляную кислоту; 1 Н-ЯМР (CDCl3)7,70 (м, 4 Н), 7,40 (м, 4 Н), 4,80 (с, 1 Н), 2,20 (т, 2 Н), 2,00 (т, 2 Н), 1,40 (м, 2 Н). Получение соединения Е. К раствору соединения С (7,8 г, 0,029 моль) в бензоле (40 мл) при кипячении с обратным холодильником медленно добавляли тионилхлорид (5,3 мл). Смесь кипятили с обратным холодильником еще в течение 2 ч, охлаждали, фильтровали и концентрировали при пониженном давлении с получением 8 г соединения Е, которое сразу же использовали на следующей стадии без дополнительной очистки. Получение соединения F. Это соединение получали из соединения D, следуя процедуре, описанной выше для синтеза соединения Е из соединения С. Пример 1. Синтез соединения G. Соединение Е (8 г) с предыдущей стадии растворяли в метиленхлориде (20 мл) и добавляли к интенсивно перемешиваемому, охлажденному (0 С) 28% раствору NH4OH (50 мл). Ледяную баню удаляли и перемешивание продолжали еще в течение часа. Реакционную смесь разбавляли водой (30 мл) и экстрагировали в метиленхлорид (230 мл). Объединенный органический слой промывали водой (230 мл), 3% раствором NaHCO3 (230 мл), насыщенным солевым раствором (130 мл), сушили (Na2SO4) и концентрировали с получением остатка, который растирали с эфиром с получением 6,30 г соединения G;(с, 1 Н), 2,30 (т, 2 Н), 2,10 (т, 2 Н). Пример 2. Синтез соединения Н. Это соединение получали из соединения Е, следуя процедуре, описанной выше для синтеза соединения G, за исключением того, что на стадии аминирования вместо 28% NH4OH использовали диметиламин. 1(м, 4 Н). Пример 3: Синтез соединения I. Это соединение получали из соединения F, следуя процедуре, описанной выше для синтеза соединения G из соединения Е. 1 Н-ЯМР (ДМСО-d6)7,80 (д, 2 Н), 7,60 (д, 2 Н), 7,40 (м, 4 Н), 7,10 (ушир., 1 Н), 6,70 (ушир., 1 Н), 5,10(с, 1 Н), 2,10 (т, 2 Н), 2,00 (т, 2 Н), 1,50 (м, 2 Н). Пример 4. Синтез соединения II-1. К раствору соединения G (5,15 г, 0,019 моль) в ледяной уксусной кислоте (20 мл) при комнатной температуре медленно добавляли 50% Н 2 О 2 (1,2 экв.). Смесь перемешивали в течение 1 ч, выливали в ледяную воду и фильтровали. Осажденное твердое вещество тщательно промывали водой, затем эфиром и сушили в условиях высокого вакуума с получением 4,42 г соединения II-1; белое твердое вещество; т.пл. 163-164 С; Rt=7,57 мин. 1 Н-ЯМР (ДМСО-d6)8,10-7,50 (ряд м, 8 Н), 7,40 (ушир., 1 Н), 6,90 (ушир., 1 Н), 5,70 (с, 1 Н), 2,30 (м,4 Н). Пример 5. Синтез соединения II-2. Это соединение получали из соединения Н, следуя процедуре, описанной выше для синтеза соединения II-1 из соединения G; белое твердое вещество; т.пл. 110-112 С; Rt=8,64 мин. 1 Н-ЯМР (ДМСО-d6)8,00 (т, 2 Н), 7,70 (д, 1 Н), 7,60 (д, 1 Н), 7,50 (м, 2 Н), 7,40 (кв., 2 Н), 5,60 (с, 1 Н),2,80 (с, 3 Н), 2,70 (с, 3 Н), 2,60-2,20 (ряд м, 4 Н). Пример 6. Синтез соединения II-3. Это соединение получали из соединения I, следуя процедуре, описанной выше для синтеза соединения II-1 из соединения G; белое твердое вещество; т.пл. 161-162 С; Rt=7,61 мин. 1 Н-ЯМР (ДМСО-d6)8,20-7,60 (ряд м, 8 Н), 7,40 (ушир., 1 Н), 6,90 (ушир., 1 Н), 5,80 (с, 1 Н), 2,30 (м,4 Н), 1,80 (м, 2 Н). Следующие примеры 7-8 синтезировали в соответствии со схемой 2. Схема 2 Пример 7. Синтез соединения J. К перемешиваемому раствору соединения С (1,9 г, 0,007 моль) в безводном ДМФА (20 мл) при температуре 0 С добавляли N-метилморфолин ("NMM") (1,92 мл) с последующим добавлением тетрафторбората 2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония ("TBTU") (3,38 г, 0,0105 моль). Смесь перемешивали в течение 10 мин и добавляли к ней (L)-аланинамид (в виде гидрохлоридной соли) (1,3 г,0,0105 моль) в безводном ДМФА (5 мл). Охлаждающую баню удаляли и смесь перемешивали еще в течение 2 ч. Затем смесь выливали в холодную воду (25 мл) и экстрагировали в этилацетат (350 мл). Объединенный органический слой промывали последовательно водой, 2% лимонной кислотой, 3% бикарбонатом натрия, водой и насыщенным солевым раствором. В результате сушки (MgSO4) и выпаривания растворителя получали остаток, который при растирании с холодным эфиром давал 1,93 г соединения J; 1 Н-ЯМР (ДМСО-d6)7,70 (м, 3 Н), 7,50 (д, 2 Н), 7,20 (м, 4 Н), 7,10 (ушир., 1 Н), 6,80 (ушир., 1 Н), 5,00- 11007781 Пример 8. Синтез соединения II-4. Это соединение получали из соединения J, следуя процедуре, описанной выше для синтеза соединения II-1 из соединения G (схема 1); белое твердое вещество (смесь диастереомеров); Rt=7,16 мин. 1 Н-ЯМР (ДМСО-d6)8,30 (два перекрывающихся д, 1 Н), 8,20-7,60 (ряд м, 8 Н), 7,50 (д, 1 Н), 7,10 (д,1 Н), 5,80 (с, 1 Н), 4,20 (м, 1 Н), 2,60-2,40 (две группы м, 4 Н), 1,30 (два перекрывающихся д, 3 Н). Следующие примеры 9-18 синтезировали в соответствии со схемой 3. Схема 3 Получение соединения 33. Схема 3, стадия 1. На стадии 1 а 3-бромтиофен (10,22 г) (соединение 31) в безводном эфире при температуре от -70 до 78 С подвергали взаимодействию с н-бутиллитием (25 мл 2,5 М раствора, 1,1 экв.). После перемешивания в течение дополнительного времени для завершения реакции обмена галоген-металл, в реакционную колбу медленно добавляли 3-тиофенкарбоксальдегид (6,39 г) (соединение 32). Перемешивание продолжали еще в течение 2-3 ч при низкой температуре. Охлаждающую баню удаляли и реакционной смеси давали медленно нагреться до комнатной температуры, после чего ее гасили, предпочтительно 50% водным раствором NH4Cl. Смесь экстрагировали в органический растворитель (эфир или этилацетат). Органический слой промывали насыщенным солевым раствором, сушили (MgSO4 или Na2SO4) и концентрировали с получением неочищенного продукта. Очистку можно осуществлять известными методами очистки (предпочтительно колоночной хроматографией и/или перекристаллизацией) с получением чистого соединения 33; 1H-ЯМР (CDCl3)7,40 (д, 2 Н), 7,30 (с, 2 Н), 7,10 (д, 2 Н), 6,00 (д, 1 Н), 2,20 (д, 2 Н). Этот способ представляет собой адаптацию процедуры, ранее описанной Gronowitz, S.; Eriksson, В.Arkiv Kemi 1963, 335, полностью включенной в данную заявку в качестве ссылки. Получение соединения 34. Схема 3, стадия 2. На первой стадии тиомочевину (5 г, 1,3 экв.) поглощали 48% НВr и водой. Смесь нагревали (предпочтительно до 60-70 С) с последующим добавлением соединения 33 (10 г). Температуру реакционной смеси повышали (предпочтительно до 90-95 С) и перемешивание продолжали в течение дополнительного периода времени для завершения реакции. Затем реакционную смесь охлаждали до комнатной температуры (в некоторых случаях необходима ледяная баня) и осажденное твердое вещество фильтровали и тщательно промывали водой. Мокрое твердое вещество затем поглощали дополнительным количеством воды и обрабатывали водным основанием, предпочтительно раствором гидроксида натрия. Смесь нагревали (предпочтительно до 70-80 С, но в некоторых случаях необходима более высокая температура) и к ней добавляли хлоруксусную кислоту (4,8 г, 1,1 экв.) в воде. Реакционную смесь поддерживали при повышенной температуре(предпочтительно 100-110 С) в течение нужного периода времени, охлаждали, поглощали водой и промывали органическим растворителем (предпочтительно эфиром). Водный слой основания подкисляли раствором неорганической кислоты (например, водным раствором НСl). Затем водный (кислотный) раствор экстрагировали несколько раз в органический растворитель (например, эфир или этилацетат). Объединенный органический слой промывали насыщенным солевым раствором, сушили (MgSO4 или Na2SO4) и концентрировали с получением неочищенного продукта 34, который можно использовать непосредственно на следующей стадии. Однако его также можно очистить с использованием известных методов(например, перекристаллизации) с получением чистого соединения 34; 1 Н-ЯМР (CDCl3)7,30 (д, 2 Н), 7,20 (с, 2 Н), 7,10 (д, 2 Н), 5,40 (с, 1 Н), 3,10 (с, 2 Н). Этот способ является адаптацией процедуры, ранее описанной в патенте США 4177290 (выдан 4 декабря 1979 г.), включенном в данную заявку полностью в качестве ссылки. Получение соединения 35. Схема 3, стадия 3. Раствор тиокислоты 34 (9,0 г) в бензоле доводили до температуры кипения с обратным холодильником и к нему медленно добавляли 1,1 экв. тионилхлорида. Смесь кипятили с обратным холодильником до исчезновения исходного вещества (как определяли аналитическими методами), охлаждали и растворитель удаляли с получением неочищенного продукта 35, который можно было использовать непосредственно на следующей стадии. Однако его также можно очистить с использованием известных методов(например, перекристаллизации) с получением чистого соединения 35. Пример 9. Синтез соединения 36. Схема 3, стадия 4. Полученный хлорид тиокислоты 35 (9,5 г) с предыдущей стадии вводили в подходящий органический растворитель (предпочтительно тетрагидрофуран или метиленхлорид) и обрабатывали газообразным аммиаком (или 28% водным раствором). Реакционную смесь затем распределяли между водой и этилацетатом. Отделенный органический слой промывали водой, разбавленной кислотой и насыщенным солевым раствором, сушили над осушителем (например, MgSO4 или Na2SO4) и концентрировали с получением 6,40 г соединения 36. 1 Н-ЯМР (CDCl3)7,40 (д, 2 Н), 7,30 (с, 2 Н), 7,20 (д, 2 Н), 6,40 (ушир., 1 Н), 5,50 (ушир., 1 Н), 5,40 (с,1 Н), 3,10 (с, 2 Н). Пример 10. Синтез соединения 37. Следуя процедуре, аналогичной описанной в примере 9, в результате обработки 2,15 г свежеполученного соединения 35 2,2 г н-пропиламина получали неочищенное вещество, которое очищали колоночной хроматографией (элюент: 30% этилацетат в гексане) с получением 1,71 г соединения 37. Аналитические данные: вязкое масло, Rt=12,30 мин. 1H-ЯМР (ДМСО-d6)7,90 (т, 1 Н), 7,50 (д, 2 Н), 7,40 (с, 2 Н), 7,10 (д, 2 Н), 5,60 (с, 1 Н), 3,30 (д, 1 Н),3,10 (м, 3 Н), 1,30 (м, 2 Н), 0,80 (т, 3 Н). Пример 11. Синтез соединения 38. Следуя процедуре, аналогичной описанной в примере 9, в результате обработки 2,56 г свежеполученного соединения 35 газообразным диметиламином получали неочищенное вещество, которое очищали колоночной флэш-хроматографией (элюент: 30% этилацетат в гексане) с получением 1,96 г соединения 38. Аналитические данные: белое твердое вещество, т.пл. 71-72 С, Rt =11,08 мин. 1H-ЯМР (CDCl3)7,30-7,10 (м, 6 Н), 5,50 (с, 1 Н), 3,20 (с, 2 Н), 3,00 и 2,90 (две группы с, 6 Н). Пример 12. Синтез соединения 39. Следуя процедуре, аналогичной описанной в примере 9, в результате обработки 2,15 г свежеполученного соединения 35 2,74 г диметиламина получали неочищенное вещество, которое очищали колоночной флэш-хроматографией (элюент: 25% этилацетат в гексане) с получением 1,56 г соединения 39. Аналитические данные: белое твердое вещество, т.пл. 83-84 С, Rt=13,37 мин. 1- 13007781 перекрывающихся т, 6 Н). Пример 13. Синтез соединения 40. Следуя процедуре, аналогичной описанной в примере 9, в результате обработки 2,15 г свежеполученного соединения 35 4 г морфолина получали неочищенное вещество, которое очищали колоночной флэш-хроматографией (элюент: 50% этилацетат в гексане) с получением 2,02 г соединения 40. Аналитические данные: белое твердое вещество, т.пл. 75,5-78 С, Rt=11,21 мин. 1 Н-ЯМР (CDCl3)7,40-7,20 (две группы м, 6 Н), 5,50 (с, 1 Н), 3,70 (м, 4 Н), 3,60 (м, 2 Н), 3,40 (м, 2 Н),3,20 (с, 2 Н). Пример 14. Синтез соединения I-9. К охлажденному (-15 до -25 С) раствору соединения 36 (5,50 г) либо в метиленхлориде, либо в хлороформе медленно добавляли 1 экв. окислителя - м-хлорпероксибензойной кислоты (m-СРВА) в этом же растворителе. Перемешивание продолжали при низкой температуре до исчезновения исходного вещества, что определяли различными аналитическими методами. Затем реакционную смесь тщательно промывали водой, насыщенным раствором бикарбоната натрия, водой и насыщенным солевым раствором соответственно и сушили над осушителем (например, MgSO4 или Na2SO4) и концентрировали. Полученное вещество затем очищали колоночной хроматографией и/или перекристаллизацией с получением соединения I-9 (5,50 г). Аналитические данные: белое твердое вещество, т.пл. 131-132 С. 1H-ЯМР (CDCl3)7,40 (м, 4 Н), 7,25 (д, 1 Н), 7,15 (д, 1 Н), 6,90 (ушир., 1 Н), 5,60 (ушир., 1 Н), 5,45 (с,1 Н), 3,45 (д, 1 Н), 3,10 (д, 1 Н). Пример 15. Синтез соединения I-10. Следуя процедуре, аналогичной описанной в примере 14, соединение 37 (1,67 г) окисляли 1 экв. окислителя - м-хлорпероксибензойной кислотой (m-СРВА) и затем очищали с получением соединения I10 (1,40 г). Аналитические данные: полутвердое вещество, Rt=8,95 мин. 1 Н-ЯМР (ДМСО-d6)8,00 (т, 1 Н), 7,40 (м, 4 Н), 7,10 (м, 2 Н), 5,30 (с, 1 Н), 3,20 (д, 1 Н), 3,10 (м, 1 Н),3,00 (д, 1 Н), 2,90 (м, 1 Н), 1,20 (м, 2 Н), 0,80 (т, 3 Н). Пример 16. Синтез соединения I-11. Следуя процедуре, аналогичной описанной в примере 14, соединение 38 (1,91 г) окисляли 1 экв. окислителя - м-хлорпероксибензойной кислотой (m-СРВА) и затем очищали с получением соединения I11 (1,63 г). Аналитические данные: белое твердое вещество, т.пл. 93-96 С, Rt=7,79 мин. 1 Н-ЯМР (CDCl3)7,50-7,30 (м, 6 Н), 5,70 (с, 1 Н), 3,60 (д, 1 Н), 3,40 (д, 1 Н), 3,10 и 2,90 (две группы с,6 Н). Пример 17. Синтез соединения I-12. Следуя процедуре, аналогичной описанной в примере 14, соединение 39 (1,53 г) окисляли 1 экв. окислителя - м-хлорпероксибензойной кислотой (m-СРВА) и затем очищали с получением соединения I12 (1,35 г). Аналитические данные: белое твердое вещество, т.пл. 93-95 С, Rt=9,70 мин. 1 Н-ЯМР (CDCl3)7,40-7,20 (м, 6 Н), 5,70 (с, 1 Н), 3,60 (д, 1 Н), 3,40 (м, 2 Н), 3,30 (д, 1 Н), 3,20 (м, 2 Н),1,20 (т, 3 Н), 1,10 (т, 3 Н). Пример 18. Синтез соединения I-13. Следуя процедуре, аналогичной описанной в примере 14, соединение 40 (2,00 г) окисляли 1 экв. окислителя - м-хлорпероксибензойной кислотой (m-CPBA) и затем очищали с получением соединения I13 (1,60 г). Аналитические данные: белое твердое вещество, т.пл. 59-73 С, Rt=8,03 мин. 1 Н-ЯМР (CDCl3)7,40-7,20 (две группы м, 6 Н), 5,60 (с, 1 Н), 3,80-3,20 (ряд м, 10 Н). Пример 19. Синтез соединения I-22. Соединение 1-22 получали таким же многостадийным способом, как показано на схеме А, используя на стадии 1 3-бромтиофен и бензальдегид. (М+Н)=280. Примеры 20-39. Синтез соединений с I-1 по I-7 и с I-26 по I-38. Соединения I-1 - I-7 и I-26 - I-38 получали таким же многостадийным способом, как показано на схеме А, используя на стадии 3b подходяще замещенный амин NHR3R4. Аналитические данные представлены масс-спектром (М+Н) для каждого соединения, как показано в табл. 3. Следующие примеры 40-41 синтезировали в соответствии со схемой 4. Схема 4 Получение соединения 43. Смесь соединения 41 (0,75 г) (Dondoni, A. et al. J. Org. Chem. 1988, pp. 1748-1761), уксусного ангидрида (3 экв.) и безводного пиридина (2-3 мл/ммоль спирта) перемешивали в течение ночи при комнатной температуре или до завершения реакции, как было показано методом тонкослойной хроматографии. Затем реакционную смесь выливали в холодную воду и экстрагировали в этилацетат (325 мл). Объединенную органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия,водой, насыщенным солевым раствором, сушили (сульфат натрия) и концентрировали с получением же- 15007781 лаемого продукта - соединения 43 (0,84 г). Аналитические данные: Rf=0,6 (2,5% метанол/этилацетат); 1 Н-ЯМР (CDCl3)7,72 (с, 1 Н), 7,47 (м, 1 Н), 7,38-7,22 (м, 5 Н), 7,11 (с, 1 Н), 2,17 (с, 3H). Получение соединения 44. Соединение 42 (0,92 г) подвергали взаимодействию в соответствии со способом, описанным выше для получения соединения 41. Полученный неочищенный сложный эфир очищали флэшхроматографией (элюент: гексан/этилацетат) с получением 0,41 г соединения 44. Аналитические данные:Rf=0,32 (4:1 гексан/этилацетат); 1 Н-ЯМР (CDCl3)7,83 (с, 1 Н), 7,42 (с, 1 Н), 7,36 (м, 1 Н), 7,17 (м, 1 Н), 7,00 (м, 1 Н), 2,19 (с, 3H). Получение соединения 45. К перемешиваемому раствору соединения 43 (0,84 г) и метилтиогликолята (1,2 экв.) в безводном дихлорметане (4-5 мл/ммоль) при 0 С в атмосфере аргона добавляли триметилсилилтрифторметан (TMSтрифлат, 1 экв.), реакционной смеси давали нагреться до комнатной температуры и перемешивали до завершения реакции (2-6 ч). Затем смесь разбавляли дихлорметаном, промывали насыщенным раствором бикарбоната натрия, сушили (сульфат натрия), концентрировали и сушили в условиях высокого вакуума с получением соединения 45 (1,01 г), которое непосредственно использовали на следующей стадии без дополнительной очистки. Аналитические данные: Rf=0,62 (2,5% метанол/этилацетат); 1H-ЯМР (CDCl3)7,75 (с, 1 Н), 7,5 (д, 1 Н), 7,38-7,27 (м, 5 Н), 5,72 (с, 1 Н), 3,69 (с, 3H), 3,25 (кв., 2 Н). Получение соединения 46. Соединение 44 (0,41 г) подвергали взаимодействию в соответствии со способом, описанным выше для получения соединения 45, с получением соединения 46 (0,30 г). Аналитические данные: Rf=0,62(2,5% метанол/этилацетат); 1 Н-ЯМР (CDCl3)7,75 (с, 1 Н), 7,39 (с, 1 Н), 7,36 (м, 1 Н), 7,17 (ушир., 1 Н), 6,94 (м, 1 Н), 6,07 (с, 1 Н),3,72 (с, 3H), 3,30 (кв., 2 Н). Получение соединения 47. В перемешиваемый раствор соединения 45 (1,0 г) в метаноле (10 мл/ммоль) при 0 С в течение 5-10 мин барботировали безводный аммиак. Реакционной смеси давали нагреться до комнатной температуры,перемешивали еще в течение 5-7 ч, концентрировали при пониженном давлении и сушили в вакууме. Неочищенный продукт очищали флэш-хроматографией (элюент: 5% метанол/этилацетат) с получением 0,48 г соединения 47. Аналитические данные: Rf=0,20 (5% метанол/этилацетат); 1 Н-ЯМР (CDCl3)7,77 (с, 1H), 7,47 (д, 1 Н), 7,44-7,27 (м, 5 Н), 5,53 (ушир., 1 Н), 3,22 (кв., 2 Н). Получение соединения 48. Соединение 46 (0,30 г) подвергали взаимодействию в соответствии со способом, описанным выше для получения соединения 47, с получением соединения 48 (0,25 г). Аналитические данные: Rf=0,20 (5% метанол/этилацетат); 1 Н-ЯМР (CDCl3)7,72, (с, 1 Н), 7,31 (с, 1 Н), 7,28 (м, 1 Н), 7,17 (с, 1 Н), 6,97 (м, 1 Н), 6,84 (ушир., 1 Н),6,11 (ушир., 1 Н), 5,86 (с, 1 Н), 3,25(кв., 2 Н). Пример 40. Синтез соединения I-39. К перемешиваемому раствору соединения 47 (0,48) в безводном дихлорметане (10 мл/ммоль) при температуре -78 С добавляли раствор m-CPBA (1,0 экв.) в дихлорметане (5-8 мл/ммоль). После дополнительного перемешивания еще в течение 1 ч реакционной смеси давали нагреться до температуры от -30 до -40 С и гасили 10% водным раствором Na2S2O3. Отделенную органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия, водой и насыщенным солевым раствором, сушили (сульфат натрия) и концентрировали с получением соединения I-37 (0,31 г). Аналитические данные: Rf=0,13 (5% метанол/этилацетат); 1 Н-ЯМР (CDCl3) основной диастереомер:7,92 (с, 1 Н), 7,61 (м, 2 Н), 7,44-7,36 (м, 5 Н), 7,00 (ушир.,1 Н), 5,61 (с, 1 Н), 3,42 (кв., 2 Н); второстепенный диастереомер:7,86 (с, 1 Н), 7,55 (м, 2 Н), 7,44-7,36 (м,5 Н), 6,83 (ушир., 1 Н), 5,55 (с, 1 Н), 3,61 (кв., 2 Н). Пример 41. Синтез соединения I-40. Соединение 48 (0,25 г) подвергали взаимодействию в соответствии со способом, описанным выше для получения соединения 47, с получением соединения I-39 (0,105 г) (смесь диастереомеров). Аналитические данные: 1 Н-ЯМР (ДМСО-d6) основной диастереомер:8,03 (с, 1 Н), 7,92 (с, 1 Н), 7,78(ушир., 1 Н), 7,68 (с, 1 Н), 7,36 (ушир., 1 Н), 7,17 (м, 1 Н), 6,50 (с, 1 Н), 3,47 (кв., 2 Н); второстепенный диастереомер:7,97 (с, 1 Н), 7,86 (с, 1 Н), 7,78 (ушир., 1 Н), 7,72 (с, 1 Н) , 7,36 (ушир., 1 Н), 7,22 (м, 1 Н), 6,39 (с,1 Н), 3,36 (кв., 2 Н). Пример 42. Синтез соединения II-9. Исходя из 9-гидроксифлуорена, это соединение получали в соответствии с многостадийным общим способом, описанным на схеме 3 выше, и используя на стадии аминирования L-аланин-NH2. Аналитические данные: белое твердое вещество (смесь диастереомеров); Rt 7,27 и 7,41 мин. 1 Н-ЯМР (ДМСО-d6)8,40-7,00 (ряд м и д, 11 Н), 5,60 и 5,70 (две группы с, 1 Н), 4,20 (м, 1 Н), 3,20 и 3,00 (две группы дд, 2 Н), 1,20 (два перекрывающихся дуплета, 3H).- 16007781 Пример 43. Синтез соединения II-23. Исходя из 9-гидроксифлуорена, это соединение получали в соответствии с многостадийным общим способом, описанным на схеме 3 выше, и используя на стадии аминирования 28% водный раствор аммиака. Аналитические данные: белое твердое вещество, т.пл. 178,5-180 С; Rt 7,48 мин. 1H-ЯМР (CDCl3)7,90-7,40 (ряд м, 8 Н), 6,60 (ушир., 1 Н), 5,40 (с, 1 Н), 5,30 (ушир., 1 Н), 2,80 (д, 1 Н),2,60 (д, 1 Н). Пример 44. Синтез соединения II-25. Исходя из дибензосуберола, это соединение получали в соответствии с многостадийным общим способом, описанным на схеме 3 выше, и используя на стадии аминирования 28% водный раствор аммиака. Аналитические данные: белое твердое вещество, т.пл. 182-190 С; Rt=8,43 мин. 1 Н-ЯМР (ДМСО-d6)7,80 (д, 1 Н), 7,60 (д, 1 Н), 7,40 (м, 8 Н), 5,50 (с, 1 Н), 3,60 (м, 2 Н), 3,50 (д, 1 Н),3,40 (д, 1 Н), 2,90 (м, 2 Н). Пример 45. Синтез соединения II-26. Исходя из дибензосуберола, это соединение получали в соответствии с многостадийным общим способом, описанным на схеме 3 выше, и используя на стадии аминирования диметиламин. Аналитические данные: белое твердое вещество т.пл. 112,5-115 С; Rt=10,36 мин. 1 Н-ЯМР (ДМСО-d6)7,60 (д, 1 Н), 7,40 (м, 7 Н), 5,50 (с, 1 Н), 4,00 (д, 1 Н), 3,60 (д, 1 Н), 3,50 (м, 2 Н),2,90 (с, 3H), 2,80 (м, 2 Н), 2,70 (с, 3H). Примеры 46-91. Синтез соединений с II-6 по II-8, с II-10 по II-15, II-24, II-27, с II-30 по II-54, с II-56 по II-91. Соединения с II-6 по II-8, с II-10 по II-15, II-24, II-27, с II-30 по II-54, с II-56 по II-91 получали в соответствии с таким же многостадийным общим способом, как показано на схеме В, используя соответствующие реагенты, с получением желаемого продукта. Аналитические данные представлены массспектром (М+Н) для каждого соединения, как показано в табл. 4. Следующий пример 92 синтезировали в соответствии со схемой 5. Схема 5 Получение соединения М. Смесь диметилфталата (соединение K, 10 г, 0,51 моль), 3,4-диметоксиацетофенона (соединение L,9,74 г, 0,054 моль) и порошкообразного метоксида натрия (2,76 г, 0,051 моль) нагревали при температуре кипения с обратным холодильником в течение ночи, охлаждали до комнатной температуры и концентрировали в вакууме. Желтую взвесь суспендировали в воде (100 мл), перемешивали в течение 10 мин,подкисляли 6 н. НСl (рН 1-2) и фильтровали. Остаток помещали в этанол (200 мл), нагревали до температуры кипения с обратным холодильником в течение 30 мин, охлаждали до комнатной температуры и фильтровали. Остаток промывали холодным этанолом и сушили в вакууме с получением соединения M в- 19007781 виде светло-желтого хлопьевидного твердого вещества (4,1 г), которое использовали без дополнительной очистки. Аналитические данные: 1 Н-ЯМР (CDCl3)3,99 (с, 3H), 4,02 (с, 3H), 6,99 (д, 1 Н), 7,68-7,75 (м, 2 Н),7,85 (м, 2 Н), 8,07 (д, 1 Н), 8,09 (с, 1 Н); МС: (М+Н)+=311. Получение соединения N. Смесь соединения М (3,37 г, 0,011 моль), гидразина (0,41 г, 0,013 моль) и этанола (250 мл) в атмосфере азота нагревали до температуры кипения с обратным холодильником в течение 6 ч, охлаждали до комнатной температуры и фильтровали. Остаток промывали этанолом и сушили с получением соединения N в виде желтого твердого вещества (2,0 г). Аналитические данные: 1 Н-ЯМР (CDCl3)3,85 (с, 3H), 3,89 (с, 3H), 7,17 (д, 1 Н), 7,38-7,43 (м, 1 Н),7,55 (м, 2 Н), 7,60 (д, 1 Н), 7,85 (д, 1 Н), 7,95 (с, 1 Н); МС: (М+Н)+=307. Получение соединения О. К перемешиваемому раствору соединения N (0,084 г, 0,2 7 ммоль) в ТГФ/H2O (3:1, 8 мл) при комнатной температуре в атмосфере азота добавляли одной порцией твердый борогидрид натрия (0,029 г,0,63 ммоль). Реакционную смесь охлаждали до 0 С, перемешивали в течение 1 ч, нагревали до комнатной температуры, разбавляли этилацетатом и промывали водой. Органическую фазу сушили (сульфат магния) и концентрировали в вакууме. Остаток растирали с эфиром и получали соединение О (0,077 г) в виде желтого твердого вещества, которое использовали без дополнительной очистки. Аналитические данные: 1H-ЯМР (CDCl3)3,86 (с, 3H), 3,87 (с, 3H), 5,53 (с, 1 Н), 6,79 (д, 1 Н), 7,29 (т,2 Н), 7,46 (д, 1 Н), 7,50 (с, 2 Н), 7,58 (т, 1 Н); МС: (М+Н)+=309. Получение соединения Р. К перемешиваемому раствору соединения О (1,55 г, 0,005 моль) в CH2Cl2 (40 мл) в атмосфере азота при температуре 0 С добавляли метилтиогликолят (0,54 г, 0,006 ммоль). Затем к реакционной смеси по каплям добавляли трифторуксусный ангидрид (1,42 мл, 0,01 моль). Реакционную смесь перемешивали при 0 С в течение 0,5 ч, нагревали до комнатной температуры, перемешивали в течение ночи, гасили насыщенным водным раствором бикарбоната натрия и экстрагировали в этилацетат (325 мл). Органический слой промывали водой, насыщенным солевым раствором, сушили (сульфат магния) и концентрировали в вакууме с получением соединения Р в виде желтого твердого вещества (1,75 г), которое использовали без дополнительной очистки. Аналитические данные: 1 Н-ЯМР (CDCl3)2,77 (кв., 2 Н), 3,33 (с, 3H), 3,93 (с, 3H), 4,00 (3H), 4,99 (с,1 Н), 6,96 (д, 1 Н), 7,23-7,42 (м, 2 Н), 7,47 (д, 1 Н), 7,49 (д, 1 Н), 7,64 (д, 1 Н), 7,69 (д, 1 Н), 7,72 (д, 1 Н); МС:(М+Н)+=397. Пример 92. Синтез соединения II-66. Исходя из соединения Р, это соединение получали в соответствии со способом, описанным выше для получения соединения 47, и в примере 35 для синтеза соединения I-37. Так, исходя из 0,050 мг соединения Р, при обработке аммиаком на первой стадии с последующим окислением m-CPBA на следующей стадии, получали 0,011 г соединения II-66. Аналитические данные: 1H-ЯМР (CDCl3)2,75 (д, 1 Н), 2,88 (д, 1 Н), 3,92 (с, 3H), 3,96 (с, 3H), 5,67 (с,1 Н), 6,80 (с, 1 Н), 6,94 (д, 1 Н), 7,37 (т, 1 Н), 7,45-7,52 (м, 2 Н), 7,58 (д, 1 Н), 7,64 (с, 1 Н), 7,79 (д, 1 Н); МС:(М+Н)+=420. Соединения формулы (VI) и (VIII) (табл. 2 А и 2 В) легко получают, используя подходящие циклические малеимиды. Например, циклические малеимиды, используемые для получения соединений VI-1, 2,6, 7, 8 (табл. 2 А) и VIII-1, 2, 6, 8 (табл. 2 В) коммерчески доступны. Другие циклические малеимиды известны из литературы (см., например, Bayer et al. Montash.Chem. 1997, 91, Kakiuchi et al. Chem. Lett. 1998,1001, оба полностью включены в настоящую заявку в качестве ссылки). Кроме того, на схеме 6 приведена общая схема синтеза для получения циклических имидов (С 3 и С 4), используемых в синтезе соединений VI-3, 5 (табл. 2 А) и VIII-3, 4 (табл. 2 В). Схема 6 На схеме 6 взаимодействие малеинового ангидрида (соединение G1) с подходящим амином (соединение H1, где R представляет собой -(СН 2)2 ОМе или (s)-CH(Me)CH2OH) приводит к образованию соответствующей малеиновой кислоты (соединение M1). Циклизация соединения M1 в присутствииAc2/NaOAc при комнатной температуре или в растворе толуол/триэтиламин при температуре кипения с обратным холодильником приводит к образованию соединений С 3 и С 4.- 20007781 Получение соединения С 3. К раствору малеинового ангидрида (соединение G1, 1 экв.) в уксусной кислоте добавляли по каплям 2-метоксиэтиламин (соединение H1, R=(CH2)2OMe, 1 экв.). После перемешивания в течение ночи при комнатной температуре реакционную смесь концентрировали с получением неочищенного соединения M1, которое вводили в смесь уксусного ангидрида и NaOAc (0,6 экв.). Полученную смесь перемешивали при температуре 90 С в течение 2 ч, охлаждали до комнатной температуры, гасили холодной водой и экстрагировали в эфир. Объединенные органические слои промывали насыщенным солевым раствором, сушили (сульфат магния) и концентрировали с получением соединения С 3, которое использовали сразу без дополнительной очистки. 1 Н-ЯМР (ДМСО-d6)7,02 (с, 2 Н), 3,54 (м, 2 Н), 3,44 (м, 2 Н), 3,20 (с, 3H). Получение соединения С 4. Раствор (S)-2-аминопропанола (соединение H1, R=(s)-CH(Me)CH2OH, 1 экв.) в абсолютном этаноле медленно добавляли к раствору малеинового ангидрида (соединение G1, 1 экв.). Полученную смесь перемешивали при комнатной температуре в течение ночи. Отделившееся твердое вещество отфильтровывали, промывали эфиром, поглощали толуолом и обрабатывали триэтиламином. Полученную смесь кипятили с обратным холодильником в течение 4 ч, используя ловушку Дина-Старка, охлаждали до комнатной температуры, концентрировали и пропускали через слой силикагеля (элюент: этилацетат) с получением соединения С 4. 1 Н-ЯМР (ацетон-d6):6,80 (с, 2 Н), 4,20 (м, 1 Н), 3,91 (м, 2 Н), 3,64 (м, 1 Н), 1,29 (д, 3 Н). Следующие примеры 92 а-92q синтезировали в соответствии со способом, показанным на схеме 7. Схема 7 На схеме 7 взаимодействие подходяще замещенного тиола (образованный из соответствующей соли тиоурония) с подходящим циклическим амидом в присутствии основания приводит к образованию соответствующего тиоэфира. Тиоэфир может быть окислен до соответствующего сульфоксида. Например,тиол В 1, образованный из соответствующей соли тиоурония А 1 (получена из соединения А, как показано на схеме 1), взаимодействует с метилмалеимидом (соединение С 2) в присутствии триэтиламина с образованием тиоэфира D2, который при последующем окислении перекисью водорода в ледяной уксусной кислоте образует соответствующий сульфоксид, соединение VIII (табл. 2 В). Альтернативно, тиоэфир можно непосредственно получить взаимодействием подходящей соли тиоурония с подходящим циклическим имидом в присутствии основания. Так, тиоэфир D1 непосредственно получали реакцией его соответствующей соли тиоурония А 1 с малеимидом (С 1) в присутствии 10 н. NaOH. Окисление D1 давало соответствующий сульфоксид VIII-1 (табл. 2 В). Пример 92 а. Синтез соединения VIII-1. Смесь соединения А 1 (4 г, 12,46 ммоль), 10 н. NaOH (4 мл) и воды (10 мл) перемешивали при температуре 70 С в течение 0,5 ч. Затем к реакционной смеси добавляли малеимид (соединение С 1, 1,2 г,12,37 ммоль) в этаноле (20 мл) и перемешивание продолжали при температуре 70 С в течение еще 2 ч. После охлаждения до комнатной температуры, отделившееся твердое вещество отфильтровывали, последовательно промывали водой, гексаном и простым эфиром. Фильтрат, содержащий желаемый продукт, экстрагировали в этилацетат. Объединенные органические слои последовательно промывали водой и насыщенным солевым раствором, сушили (MgSO4) и концентрировали с получением неочищенного- 21007781 продукта, который очищали флэш-хроматографией (гексан:этилацетат 1:1) с получением 0,510 г соединения D1; 1 Н-ЯМР (ДМСО-d6)11,33 (с, 1 Н), 7,90-7,32 (ряд м, 8 Н), 5,49 (с, 1 Н), 3,79 (дд, 1 Н), 2,64 (дд, 1 Н),2,24 (дд, 1 Н). В результате окисления соединения D1 перекисью водорода, как описано выше, получали указанное в заголовке соединение в виде смеси диастереомеров; Rt=10,16 мин; 1 Н-ЯМР (ДМСО-d6)11,82 (с, 0,11 Н), 11,44 (с, 0,89 Н), 8,03-7,39 (ряд м, 8 Н), 5,99 (с, 0,11 Н), 5,72 (с,0,89 Н), 4,58 (м, 0,11 Н), 3,22 (дд, 0,89 Н), 2,79 (дд, 3,32, 0,11 Н), 2,55 (дд, 0,89 Н), 1,67 (дд, 0,89 Н). МС: 312(М+Н), 334 (М+Na). Пример 92b. Синтез соединения VIII-2. Смесь соединения А 1 (1 экв.) в воде и 10 н. NaOH (4-5 экв.) перемешивали при температуре 70 С в течение 3-5 ч. Смесь охлаждали до 0 С, подкисляли разбавленной НСl и экстрагировали в простой эфир. Объединенные органические слои промывали насыщенным солевым раствором, сушили (сульфат магния) и концентрировали с получением соединения В 1, которое использовали без дополнительной очистки; 1 Н-ЯМР (ДМСО-d6)7,87-7,35 (ряд м, 8 Н), 5,21 (д, 1 Н), 3,55 (д, 1 Н). Смесь соединения В 1 (1 экв.), соединения С 2 (1 экв.) и триэтиламина в смеси этилацетат:метанол(4:1) перемешивали при комнатной температуре в течение 2-5 ч, концентрировали и очищали флэшхроматографией (гексан:этилацетат 2:1) с получением соединения D2; 1 Н-ЯМР (ДМСО-d6)7,90-7,35 (ряд м, 8 Н), 5,49 (с, 1 Н), 3,76 (м, 1 Н), 2,73 (с, 3H), 2,61 (дд, 1 Н), 2,24(дд, 1 Н). В результате окисления соединения D2 перекисью водорода, как описано выше, получали указанное в заголовке соединение в виде смеси диастереомеров; Rt=10,30 мин; 1 Н-ЯМР (ДМСО-d6)8,04-7,37 (ряд м, 8 Н), 5,98 (с, 0,08 Н), 5,77 (с, 0,92 Н), 4,61 (м, 0,08 Н), 3,31-2,50(ряд м и дд, 5,08 Н), 1,58 (дд, 0,92 Н). МС: 348 (М+Na). Пример 92 с. Синтез соединения VI-1. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=9,20 и 9,41 мин (диастереомеры); 1H-ЯМР (ДМСО-d6)11,62 и 11,53 (2 синглета, 1 Н), 7,57-7,32 (ряд м, 10 Н), 6,07 (с, 0,4 Н), 5,33 (с,0,6 Н), 3,74 (м, 0,6 Н), 3,55 (м, 0,4 Н), 3,14 (дд, 0,4 Н), 2,96 (дд, 0,6 Н), 2,82 (дд, 0,4 Н), 2,57 (дд, 0,6 Н). МС: 312 (М-Н). Пример 92d. Синтез соединения VI-2. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=8,05 и 8,17 мин (диастереомеры); 1 Н-ЯМР (ДМСО-d6)7,58-7,26 (ряд м, 10 Н), 6,04 (с, 1 Н), 3,31-2,49 (ряд м, 6 Н). МС: 350 (М + Na). Пример 92 е. Синтез соединения VI-3. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=8,38 и 8,46 мин (диастереомеры); 1 Н-ЯМР (ДМСО-d6)7,58-7,01 (ряд м, 10 Н), 6,03 (с, 0,23 Н), 5,38 (с, 0,77 Н), 3,84-3,01 (ряд м, 9,23 Н),2,61 (дд, 0,77 Н). МС: 394 (М+Na). Пример 92f. Синтез соединения VI-4. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt= 7,38 (перекрывающиеся диастереомеры); 1 Н-ЯМР (ДМСО-dб)7,58-7,33 (ряд м, 10 Н), 6,02 (с, 0,33H), 5,38 (с, 0,67 Н), 4,67 (м, 1H), 3,79-2,94(ряд м, 5,33 Н), 2,60 (дд, 0,67 Н). МС: 380 (М+Na). Пример 92g. Синтез соединения VI-5. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=7,98, 8,16 и 10,42 мин (диастереомеры); 1 Н-ЯМР (ДМСО-d6)7,58-7,35 (ряд м, 8 Н), 6,03 и 6,02 (два перекрывающихся с, 0,46 Н), 5,36 (с,0,54 Н), 4,70 (м, 0,54 Н), 4,14-2,50 (ряд м, 6 Н), 1,22-1,12 (перекрывающийся д, 3H). МС: 372(М+Н), 394(М+Na). Пример 92h. Синтез соединения VI-6. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=7,78 и 7,88 мин (диастереомеры); 1H-ЯМР (ДМСО-d6)7,63-7,22 (два м, 8 Н), 6,06 (с, 0,66 Н), 5,47 (с, 0,34 Н), 3,82 (м, 0,34 Н), 3,69 (м,0,66 Н), 3,14 (дд, 0,66 Н), 2,95 (м, 1 Н), 2,84 и 2,81 (два с, 3H), 2,9 (дд, 0,34 Н). МС: 386 (M+Na). Пример 92i. Синтез соединения VI-7. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=8,48 и 8,70 мин (диастереомеры); 1 Н-ЯМР (ДМСО-d6)11,52 (ушир., 1 Н), 7,69-7,60 (м, 4 Н), 7,29 (м, 2 Н), 6,32 (с, 0,1 Н), 5,60 (с, 0,9 Н),3,75 (м, 1 Н), 3,81 (дд, 1 Н), 2,66-2,32 (м, 1 Н). МС: 348 (М+Na).- 22007781 Пример 92j. Синтез соединения VI-8. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=8,70 мин (перекрывающиеся диастереомеры); 1 Н-ЯМР (ДМСО-d6)11,62 и 11,53 (два ушир., 1 Н), 7,74-7,25 (м, 8 Н), 6,18 и 6,13 (два с, 0,4 Н), 5,47 и 5,45 (два синглета, 0,6 Н), 3,78-2,49 (ряд м, 3H). МС: 342 (М+Na). Пример 92k. Синтез соединения VIII-3. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=10,79 и 10,98 мин (диастереомеры); 1(ряд м, 2,08 Н), 1,65 (дд, 0,5 Н). МС: 392 (М+Na). Пример 92l. Синтез соединения VIII-4. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=9,18 и 9,30 мин (диастереомеры); 1 Н-ЯМР (ДСМСО-d6)8,05-7,37 (ряд м, 8 Н), 5,96 (с, 0,09 Н), 5,77 (с, 0,91 Н), 4,80 и 4,48 (два м, 1 Н),3,57-2,49 (ряд м, 5,09 Н), 1,51 (дд, 0,91 Н). МС: 378 (М+Н), 403 (М+Na). Пример 92m. Синтез соединения VIII-5. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=10,03, 10,30, 10,42 и 11 мин (диастереомеры); 1 Н-ЯМР (ДМСО-d6)8,05-7,37 (ряд м, 8 Н), 5,95 и 5,94 (два перекрывающихся синглета, 0,36 Н),5,76 (с, 0,64 Н), 4,85-2,49 (ряд м, 6 Н), 1,29 и 1,08 (две группы перекрывающихся д, 3H). МС: 370 (М+Н),392 (М+Na). Пример 92n. Синтез соединения VIII-6. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=7,22 мин (перекрывающиеся диастереомеры); 1 Н-ЯМР (ДМСО-d6)11,23 и 10,89 (два синглета, 1 Н), 8,04-7,38 (ряд м, 8 Н), 6,00 (с, 0,37 Н), 5,77 (с,0,63 Н), 4,65 (м, 0,37 Н), 3,32 (м, 0,63 Н), 3,22 (дд, 0,37 Н), 2,86 (дд, 0,37 Н), 2,59 (дд, 0,63 Н), 1,76 (дд, 0,63 Н). МС: 328 (М+Н), 350 (М+Na). Пример 92 о. Синтез соединения VIII-7. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=13,90 и 14,12 мин (диастереомеры); 1H-ЯМР (ДМСО-d6)8,04-6,80 (ряд м, 12 Н), 5,98 (с, 0,11 Н), 5,76 (с, 0,89 Н), 4,65 (д, 0,22 Н), 4,37 (с,1,78 Н), 3,69 (с, 3H), 3,31 (м, 1 Н), 2,93 (м, 0,11 Н), 2,63 (дд, 0,89 Н), 1,78 (дд, 1 Н). МС: 554 (М+Na). Пример 92 р. Синтез соединения VIII-8. Указанное в заголовке соединение получали, используя подходящие исходные вещества и согласно методике, описанной выше. Аналитические данные: Rt=9,48 и 9,62 мин (диастереомеры); 1 Н-ЯМР(ДМСО-d6)8,04-7,09 (ряд м, 13 Н), 5,97 (с, 0,1 Н), 5,84 (с, 0,9 Н), 4,70 (м, 0,1 Н), 3,38-3,24 (м, 1 Н), 3,04 (м,0,1 Н), 2,72 (дд, 0,9 Н), 1,63 (дд, 0,9 Н). МС: 388 (М+Н), 410 (М+Na). Пример 92q. Синтез соединения VII-1. Указанное в заголовке соединение получали из соединения А 1 (схема 7), согласно методике, описанной выше для синтеза соединения VIII-1, за исключением использования 3-бром-1-фенилпирролидин 2-она вместо малеимида на первой стадии. Аналитические данные: Rt=9,36 и 9,72 мин (смесь диастереомеров); 1(м, 1 Н), 1,30 (м, 1 Н). МС: 374 (М+Н), 396 (М+Na). Следующие примеры 92r-92s синтезировали в соответствии со схемой 8. Схема 8 На схеме 8 взаимодействие подходящего тиола (соединение U) с 3-бромглутаримидом (Т) в присутствии основания приводило к получению соответствующего соединения W. В результате окисления соответствующего соединения W получали соединения VI-9 и VIII-9 соответственно. 3-Бромглутаральдегид получали в соответствии с процедурой, описанной в Японской патентной заявке 8308, 1961 г. и Японской патентной заявке 5277, 1960 г., обе полностью включены в данную заявку в качестве ссылки.- 23007781 Пример 92r. Синтез соединения VI-9. К охлажденному (0 С) раствору дифенилметилтиола (1 экв.) и 3-бромглутаримида (1 экв.) в безводном тетрагидрофуране по каплям добавляли DBU (1,8-диазабицикло[5.4.0]ундец-7-ен) (1,05 экв.). Охлаждающую баню удаляли и смесь перемешивали при комнатной температуре в течение 1-2 ч, разбавляли смесью гексан:этилацетат (1:1) и последовательно промывали водой и насыщенным солевым раствором. После сушки (сульфат магния) и выпаривания растворителя получали неочищенный продукт, который растирали с этилацетатом с получением промежуточного соединения W (где R=Ph2CH); 1H-ЯМР (ДМСО-d6)10,81 (с, 1 Н), 7,53-7,22 (ряд м, 10 Н), 5,52 (с, 1 Н), 3,3-1,81 (ряд м, 5 Н). В результате окисления соединения W перекисью водорода, следуя методике, описанной выше, получали указанное в заголовке соединение в виде смеси диастереомеров; Rt=9,20 и 9,44 мин; 1(с, 0,65 Н), 3,45-2,53 (ряд м, 5 Н). МС: 350 (М+Na). Пример 92s. Синтез соединения VIII-9. Согласно методике, описанной выше для синтеза VI-9, исходя из 9-флуоренилтиола, указанное в заголовке соединение также получали в виде смеси диастереомеров; Rt=7,18 и 7,47 мин; 1H-ЯМР (ДМСО-d6) 11,32 и 11,16 (два синглета, 1 Н), 7,99-7,35 (ряд м, 8 Н), 5,79 и 5,66 (два синглета,1 Н), 4,26 и 4,07 (два мультиплета, 1 Н), 2,70-2,10 (ряд м, 4 Н). МС: 326 (М+Н), 348 (М+Na). Пример 93. Демонстрация активности соединения I-9 по стимулированию бодрости. Использовали методику, описанную Edgar and Seidel, Journal of Pharmacology and ExperimentalTherapeutics, 283: 757-769, включенную в данную заявку полностью в качестве ссылки. Хирургическая подготовка животных. Взрослых самцов крыс Wistar (275-320 г, от Charles River Laboratories, Wilmington, MA) анестезировали (нембутал, 60 мг/кг, в/б) и хирургически подготавливали с использованием имплантатов для постоянной регистрации электроэнцефаллограммы (ЭЭГ) и регистрации электромиограммы (ЭМГ). ЭЭГ имплантаты представляли собой винтообразные стержни из нержавеющей стали (2 в лобной части (+ 3,9 АР от брегмы, 2,0 мл) и 3 в затылочной части (-6,4 АР, 5,5 мл). Два провода из нержавеющей стали с тефлоновым покрытием помещали под трапециевидной мышцей в задней части шеи для регистрации ЭМГ. Перед введением крысам все выводы были припаены к миниатюрному соединяющему устройству (Microtech, Boothwyn, PA) и стерилизованы при помощи газообразной окиси этилена. Имплантируемый блок присоединяли к черепу посредством комбинированного сцепления датчиков ЭЭГ, которое представляло собой цианоакрилат, накладываемый между герметично запаянным устройством, соединяющим импланты, и черепом, и зубной акрил (адгезив). После хирургического вмешательства в течение 3-5 дней вводили антибиотик (гентамицин). Для восстановления после операции отводили по меньшей мере 3 недели. Условия для проведения исследований. Крыс размещали по отдельности в клетки-микроизоляторы Nalgene, снабженные коммутатором,имеющим контактное кольцо с низким крутящим моментом (Biella Engineering, Irvine, CA) и специально сделанной поликарбонатной вертикальной трубой с фильтром наверху. Клетки представляли собой изолированные вентилируемые отделения из нержавеющей стали для ведения регистрации сна - бодрствования. Обеспечивали неограниченный доступ к пище и воде, а температура окружающей среды составляла 241 С. В течение всего периода испытания поддерживали 24-часовой цикл свет-темнота(свет/темнота 12-12) при помощи 4-ваттных ламп флуоресцентных ламп, расположенных примерно в 5 см от верха каждой клетки. Сила света составляла 30-35 люкс в центре каждой клетки. Животных не беспокоили в течение 3 дней как до, так и после лечения. Автоматический сбор данных. Стадии сна и бодрствования определяли при помощи SCORE, микрокомпьютерной системы физиологического мониторинга и мониторинга сна - бодрствования. Характерные особенности устройстваSCORE, испытание на грызунах и применимость для доклинической оценки лекарственных средств были широко описаны в литературе (Van Gelder, et al., 1991; Edgar, et al., 1991, 1997; Seidel, et al., 1995,включенные полностью в данную заявку в качестве ссылки). В настоящем исследовании система отслеживала усиленные показатели (X 10000) ЭЭГ (полоса пропускания 1-30 Гц, скорость преобразования в цифровую форму 100 Гц), и интегрированные данные ЭМГ (полоса пропускания 10-100 Гц, интегрирование среднеквадратичных значений). Состояния активации классифицировали в оперативном режиме как NREM сон, REM сон, бодрствование или тета-доминирующее бодрствование каждые 10 с, используя алгоритмы периода ЭЭГ и выделения амплитуды и категорий участников. Индивидуально полученные матрицы ЭЭГ-состояния активации и ЭМГ критерии дифференцировали REM сон от тетадоминирующего бодрствования (Welsh, et al., 1985, включенный в данную заявку полностью в качестве ссылки). Качество данных подтверждали дополнительно тщательной проверкой в оперативном режиме сигналов ЭЭГ и ЭМГ. Качество необработанных данных и показатели сна-бодрствования далее исследовали при сочетании графических и статистических определений данных, а также путем визуального наблюдения необработанных волновых ЭЭГ и дистрибуции интегрированных ЭМГ значений. Введение лекарственного средства и планирование эксперимента.Co., Kalamazoo, MI), или только метилцеллюлозный носитель вводили путем внутрибрюшинной инъекции при дозе 1 мл/кг. Размер выборки (n) составлял 13 животных на группу обработки. Спектральный анализ ЭЭГ. Каждые 10-секундные отрезки сигнала ЭЭГ преобразовывали в цифровые данные (100 Гц) в течение 24 ч и бодрствование оценивали, как описано ранее в Edgar and Seidel (1996), включенном в данную заявку полностью в качестве ссылки. Анализ данных и статистика. Основным регистрируемым показателем были минуты бодрствования в час. Группы обработки сравнивали с использованием анализа ANOVA с повторными измерениями, который применяли после обработки. При наличии существенного основного эффекта, для определения разницы между группами,обработанными активным соединением и контрольными группами, получавшими носитель, применяли критерий Даннета (Dunnett) (=0,05), если не указано иное. Результаты. Фиг. 1 показывает степень бодрствования у крыс, обработанных на время ноль либо 100 мг/кг (в/б) соединения I-9 (сплошная линия), либо метилцеллюлозным носителем (штриховая линия). Соединение I9 обеспечивало значительно больший период бодрствования, чем у обработанных носителем животных,который длился примерно до 110 мин после введения средства. Пример 94. Демонстрация активности соединения II-23 по стимулированию бодрости. Использовали методику на основе описания Edgar and Seidel, Journal of Pharmacology and Experimental Therapeutics, 283: 757-769, 1997, включенного в данную заявку полностью в качестве ссылки. Хирургическая подготовка животных. Взрослых самцов крыс Wistar (275-320 г, от Charles River Laboratories, Wilmington, MA) анестезировали (нембутал, 45 мг/кг, в/б) и хирургически подготавливали с использованием имплантатов для постоянной регистрации ЭЭГ и регистрации ЭМГ. ЭЭГ имплантаты были выполнены из коммерчески доступных компонентов (Plastics One, Roanoke, VA). ЭЭГ снимали при помощи винтообразных электродов из нержавеющей стали (2 в лобной части (+3,0 мм АР от брегмы 2,0 мм ML) и 2 в затылочной (-4,0 мм АР 2,0 мм ML. Два провода из нержавеющей стали с тефлоновым покрытием помещали под трапециевидной мышцей в задней части шеи для регистрации ЭМГ. Все выводы электродов вставляли в базу соединяющего устройства, и базу, винтовые электроды и провода закрепляли на черепе путем нанесения зубного акрилового адгезива. После хирургического вмешательства вводили антибиотик и крем на основе антибиотика наносили на края раны для профилактики инфекции. Регистрацию наблюдений осуществляли по меньшей мере через неделю после операции. Животных наблюдали (испытывали) в течение примерно 6-8 недель и затем умерщвляли. Условия для проведения исследований. После хирургической подготовки крыс размещали по отдельности в изолированные комнаты. По меньшей мере за 24 ч до начала эксперимента их помещали в контейнеры Nalgene (313131 см) с верхом из сплетенной проволоки, и доступ в комнату был закрыт вплоть до окончания регистрации наблюдений, за исключением моментов введения доз. Контейнеры помещали на стойку с двумя полками, по 4 на полку. Обеспечивали неограниченный доступ к пище и воде, а температура окружающей среды составляла 21 С, с влажностью 55%. Для защиты от звуков окружающей среды обеспечивали фоновый шум (68 дБ внутри контейнеров). Флуоресцентное верхнее освещение в комнате устанавливали на 24 часовой цикл свет-темнота (свет включали в 7 ч утра и выключали в 7 ч вечера). Уровень света внутри контейнеров составлял 38 и 25 люкс для верхней и нижней полок соответственно. Сбор данных. Сигналы ЭЭГ и ЭМГ передавались по кабелю в коммутатор (Plastics One), а затем на преусилители(модель 1700, А-М Systems, Carlsborg, WA). Сигналы ЭЭГ и ЭМГ усиливали (10 и 1K соответственно) и полосу пропускания фильтровали между 0,3 и 500 Гц для ЭЭГ и между 10 и 500 Гц для ЭМГ. Эти сигналы преобразовывали в цифровые данные при 128 выборок в секунду с использованием программы исследования сна ICELUS (М. Opp, U. Texas; Opp, Physiology and Behavior 63:67-74, 1998; Imeri, Mancia,and Opp, Neuroscience 92:745-749, 1999, включенные полностью в данную заявку в качестве ссылки), которую использовали вместе с программой Labview 5.1 и устройством для сбора данных (PCI-MIO-16E-4;National Instruments, Austin, TX). В день введения доз вели записи данных с 11 до 18 ч. Оценка сна/бодрствования. Стадии сна и бодрствования определяли с использованием программы ICELUS. Эта программа выводит ЭЭГ и ЭМГ данные блоками по 6 с вместе с ЭЭГ-FFT. Состояние пробуждения оценивали как бодрствование (WAK), быстрое движение глаз (REM), или медленное покачивание, или не-REM сон(NREM), в соответствии с визуальным анализом частоты и амплитудных характеристик ЭЭГ и ЭМГ активности (Opp and Krueger, American Journal of Physiology 266:R688-95, 1994; Van Gelder, et al., 1991; Edgar, et al., 1991, 1997; Seidel, et al., 1995, включенные в данную заявку полностью в качестве ссылки). По существу, активность бодрствования состоит из относительно низкоамплитудной ЭЭГ активности с от- 25007781 носительно низкой интенсивностью в низкочастотных диапазонах от 0,5 до 6 Гц, сопровождаемой от умеренного до высокого уровня ЭМГ активностью. В конкретном состоянии бодрствования ("тетабодрствование") интенсивность ЭЭГ может быть соответственно сфокусирована в диапазоне 6-9 Гц (тета), но всегда присутствует значительная ЭМГ активность. NREM сон характеризуется относительно высокоамплитудной активностью ЭЭГ с относительно более высокой интенсивностью в низкочастотных диапазонах от 0,5 до 6 Гц, сопровождаемой незначительной или нулевой ЭМГ активностью. REM сон характеризуется умеренной и постоянной амплитудой ЭЭГ, сфокусированной в области тета (диапазон 6-9 Гц), аналогичной тета бодрствования, но без ЭМГ активности. Введение лекарственного средства и планирование эксперимента. Соединения оценивали на группах из 4 или 8 крыс, которых испытывали в 2 этапа с промежутком по меньшей мере 2 дня. Для начальных исследований использовали перекрестный план так, чтобы крысы получали либо носитель, либо испытываемое соединение в течение каждого этапа. Животных распределяли псевдорандомизированным образом, чтобы они не получали одно и то же лекарственное средство дважды. Соединение II-23 суспендировали в стерильном 0,25% растворе метилцеллюлозы (рН=6,2; Upjohn Co., Kalamazoo, MI), доза составляла 30 мг/мл. Испытание проводили на 8 крысах, которых испытывали в 2 этапа с промежутком 5 дней (в общем, 7 крыс получали соединение II-23, а 6 - метилцеллюлозный носитель). Дозирование осуществляли в полдень, пока крысы преимущественно спали. Каждую крысу вынимали из ее контейнера, инъецировали внутрибрюшинно при дозе 3,33 мл/кг и возвращали обратно. Дозирование занимало примерно 8 мин. Анализ данных и статистика. Основным регистрируемым показателем были минуты бодрствования в час. Основной показатель для определения активности в этих экспериментах включал общее интегрированное время бодрствования для первых 3 ч после введения дозы в сравнении с группой, получавшей носитель. Так, у группы,получавшей носитель, типично в среднем время бодрствования составляло 20% в течение периода наблюдения или всего 0,2180=36 мин. Для показателей времени бодрствования у животных, получавших лекарственное средство и носитель, применяли непарный 2-tailed t-критерий (Statview 5.0, SAS Institute,Inc., Саrу, NC), и соединения с р 0,05 определяли как существенно стимулирующие бодрствование. Активность бодрствования также оценивали в течение последующих получасовых периодов сразу после введения доз, и индивидуальные t-критерии применяли к каждой временной точке для определения длительности существенного действия по стимулированию бодрствования. Результаты. Фиг. 2 показывает степень бодрствования у крыс, обработанных в полдень либо 100 мг/кг (в/б) соединения II-23 (заштрихованные треугольники), либо метилцеллюлозным носителем (незаштрихованные круги). Каждая точка представляет средний процент времени бодрствования для следующего получаса. Процедура введения доз обеспечивала временный (20 мин) период повышенной бодрости в обеих обработанных группах по сравнению с преддозовой базовой линией активности. Соединение II-23 обеспечивало значительно больший период бодрствования, чем у обработанных носителем животных (р 0,05). Ссылки. Следующие ссылки в той мере, в которой они обеспечивают раскрытие методик или других подробностей в дополнение к тем, которые раскрываются в данной заявке, специально включены в данную заявку во всей их полноте в качестве ссылок.Welsh, D.K., et al., Physiol Behav. 35:533-538, 1985. Хотя настоящее изобретение было описано достаточно подробно, специалистам должно быть понятно, что возможны различные изменения и модификации вариантов и предпочтительных вариантов воплощения настоящего изобретения, и что такие изменения и модификации могут быть осуществлены без отступления от сути настоящего изобретения. Предполагается, что прилагаемая формула изобретения охватывает все эквивалентные варианты, не выходящие за рамки данного изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (VI) где Ar1 и Ar2, каждый независимо, выбраны из фенила и тиенила; где каждый из Ar1 или Ar2 может быть независимо необязательно замещен 1-3 заместителями, независимо выбранными из Н, F, Cl, Br, I, -ОН, -OR7 или C1-C8 алкила;R2A представляет собой Н,R4A представляет собой Н или C1-C8 алкил,R7 представляет собой C1-C8 алкил,и его стереоизомерные формы, смеси стереоизомерных форм или фармацевтически приемлемые формы солей и сложных эфиров. 2. Соединение по п.1, где Ar1 и Ar2, каждый независимо, представляют собой незамещенный фенил или тиенил. 3. Соединение по п.2, где Ar1 и Ar2 каждый представляют собой фенил. 4. Соединение по п.1, где Q представляет собой C1 или С 2 алкилен. 5. Соединение по п.1, где R4A представляет собой Н, незамещенный C1-C6 алкил или C1-C6 алкил, замещенный OR, где R представляет собой Н или C1-C6 алкил. 6. Соединение по п.1, выбранное в соответствии с табл. 2 А. Таблица 2 А где X представляет собой связь или -O-,каждое из колец А и В вместе с атомами углерода, с которыми они связаны, независимо выбрано из бензо или тиенила;R7 представляет собой C1-C8 алкил; и его стереоизомерные формы, смеси стереоизомерных форм или фармацевтически приемлемые формы солей и сложных эфиров. 8. Соединение по п.7, где кольца А и В представляют собой бензо. 9. Соединение по п.7, где Q представляет собой C1- или С 2 алкилен. 10. Соединение по п.7, где А и В представляют собой бензо; X представляет собой связь; R2A представляет собой Н; Q представляет собой C1- или C2 алкилен; R4A представляет собой Н, OR7, фенил или 14. Применение соединения по любому из пп.1, 7 или 13 для получения лекарственного средства для лечения сонливости, усталости, болезни Паркинсона, церебральной ишемии, удара, апноэ по время сна, расстройств питания, расстройства дефицита внимания с гиперактивностью, нарушений познавательной способности или слабости; а также для стимуляции бодрствования, стимуляции аппетита или стимуляции прибавки в весе. 15. Применение по п.14, в котором лекарственное средство вводят для лечения депрессии, шизофрении или синдрома хронической усталости. 16. Фармацевтическая композиция, включающая соединение по пп.1, 7 или 13 в смеси с одним или несколькими фармацевтически приемлемыми эксципиентами для применения в лечении сонливости,усталости, болезни Паркинсона, церебральной ишемии, удара, апноэ во время сна, расстройств питания,расстройства дефицита внимания с гиперактивностью, нарушений познавательной способности или слабости; а также для стимуляции бодрствования, стимуляции аппетита или стимуляции прибавки в весе. 17. Способ лечения заболеваний или расстройств у субъекта, нуждающегося в таком лечении,включающий введение указанному субъекту терапевтически эффективного количества соединения по любому из пп.1, 7 или 13, где указанное соединение вводят для лечения сонливости, усталости, болезни Паркинсона, церебральной ишемии, удара, апноэ во время сна, расстройств питания, расстройства дефицита внимания с гиперактивностью, нарушений познавательной способности или слабости; а также для- 28007781 стимуляции бодрствования, стимуляции аппетита или стимуляции прибавки в весе. 18. Способ по п.17, где соединение вводят для лечения депрессии, шизофрении и синдрома хронической усталости. Бодрствование у крыс, обработанных соединением I-9
МПК / Метки
МПК: C07D 277/26, C07D 295/18, A61K 31/165, A61P 1/14, C07D 417/06, C07D 333/18, C07D 231/54, C07D 211/88, C07D 207/40, C07C 323/60, C07C 317/44
Метки: замещённые, тиоацетамиды
Код ссылки
<a href="https://eas.patents.su/30-7781-zameshhyonnye-tioacetamidy.html" rel="bookmark" title="База патентов Евразийского Союза">Замещённые тиоацетамиды</a>
Замещенные тиоацетамиды

Номер патента: 6135
Опубликовано: 27.10.2005
Авторы: Малламо Джон П., Чаттерджи Санкар, Данн Дерек, Миллер Мэттью С., Бэйкон Эдвард Р., Вот Джеффри Л.
МПК: A61P 1/14, A61K 31/165, C07C 323/60...
Метки: тиоацетамиды, замещенные
Формула / Реферат:
1. Соединение формулы (I-A) где Ar1 и Ar2, каждый независимо, выбраны из группы, включающей фенил, тиенил, изотиазолил, оксазолил, изоксазолил и тиазолил; Y выбран из C1-C4 алкилена, -C(R1)(R2)-, фенилена и оксазолилена, где R1 и R2, каждый независимо, представляют собой H или C1-C6 алкил; R3 и R4 являются одинаковыми или различными и каждый выбраны из группы, включающей H, C1-C6 алкил, где указанный алкил необязательно замещен OH или...
2-амино-6-(2-замещенные-4-фенокси)-замещенные-пиридины в качестве ингибиторов синтазы окиси азота

Номер патента: 3834
Опубликовано: 30.10.2003
Авторы: Волкманн Роберт Альфред, Новаковски Йоланта, Лауэ Джон Адамс III
МПК: C07D 213/73, A61K 31/4418, A61P 25/00...
Метки: азота, окиси, синтазы, качестве, ингибиторов, 2-амино-6-(2-замещенные-4-фенокси)-замещенные-пиридины
Формула / Реферат:
1. Соединение формулы где R1 и R2 выбраны независимо из группы, включающей водород, галоид, гидрокси, (C1-C6)алкокси, (C1-C7)алкил, (C2-C6)алкенил, (C2-C10)алкоксиалкил; G выбирают из группы, включающей водород, (C1-C6)алкил, (C1-C6)алкокси(C1-C3)алкил, аминокарбонил-(C1-C3)алкил-, (C1-C3)алкиламинокарбонил-(C1-C3)алкил-, ди-[(C1-C3)алкил]аминокарбонил-(C1-C3)алкил и N(R3)(R4)(C0-C4)алкил-, где R3 и R4 независимо выбирают из группы, включающей...
Замещённые анилиновые пиперидины в качестве селективных антагонистов мсн

Номер патента: 5934
Опубликовано: 25.08.2005
Авторы: Чен Чиен-Ан, Уэтзел Джон, Дзианг Ю, Лу Каи, Лагу Бхарат, Марзабади Мохаммад Р., Делеон Джон Э.
МПК: C07D 211/30, A61K 31/445
Метки: замещённые, мсн, анилиновые, антагонистов, селективных, качестве, пиперидины
Формула / Реферат:
1. Соединение, имеющее структуру где R1 представляет собой водород, прямой или разветвленный C1-C7 алкил, монофторалкил или полифторалкил, арил или гетероарил, где арил или гетероарил необязательно замещены одним или несколькими -F, -Cl, -Br, -I, -CN, -NO2, -CH3, -CF3, -COCH3, -CO2R2, фенилом, фенокси или прямым или разветвленным C1-C7 алкилом; R2 представляет собой прямой или разветвленный C3-C4 алкил или циклопропил; R3 представляет собой...
Азабициклические замещённые конденсированные гетероарильные соединения

Номер патента: 7429
Опубликовано: 27.10.2006
Авторы: Уишкэй Донн Г., Джейкобсен Эрик Джон, Рейтц Стивен Чарлз, Эккер Брэд А., Пиотроуски Дейвид У., Уокер Дэниел Патрик
МПК: A61P 25/28, A61P 25/18, A61K 31/439...
Метки: соединения, азабициклические, гетероарильные, замещённые, конденсированные
Формула / Реферат:
1. Соединение формулы I где азабицикло представляет собой W представляет собой при условии, что связь между группой -С(=Х)- и группой W может быть присоединена к любому доступному атому углерода в пределах группы W, как предусмотрено в R3, R6 и R15; X представляет собой О или S; R0 представляет собой Н, (С1-С4)алкил; каждый R1 представляет собой Н, (С1-С6)алкил, (С3-С6)циклоалкил; каждый R2 представляет собой (C1-C6)алкил, галогенированный...
Замещённые 6-(2-толил)триазолопиримидины

Номер патента: 6609
Опубликовано: 24.02.2006
Авторы: Штратманн Зигфрид, Гевер Маркус, Грамменос Вассилиос, Тормо И Бласко Хорди, Мюллер Бернд, Розе Инго, Гипзер Андреас, Гроте Томас, Райнхаймер Йоахим, Шэфер Петер, Штирль Райнхард, Рак Михаель, Шивекк Франк, Лоренц Гизела, Заутер Хуберт, Аммерманн Эберхард
МПК: A01N 43/90, C07D 487/04
Метки: 6-(2-толил)триазолопиримидины, замещённые
Формула / Реферат:
1. Замещенные 6-(2-толил)триазолопиримидины формулы I в которой R1 и R2 независимо означают водород, C1-C10алкил, C2-C10алкенил, C1-C10галогеналкил, C3-C10циклоалкил или R1 и R2 вместе с лежащим между ними атомом азота представляют 6-членное гетероциклическое кольцо, содержащее один атом азота, которое может быть замещено радикалом Ra; при этом Ra означает C1-C6алкил; R3 означает галоген, циано, C1-C10алкил, C1-C10алкокси или C(=O)A, где A...
Предыдущий патент: Кристаллические гидраты производных анилида никотиновой кислоты
Следующий патент: Новые производные пиррола в качестве фармацевтических средств
Случайный патент: Способ получения циталопрама