Новые пиперазиновые производные
Номер патента: 6079
Опубликовано: 25.08.2005
Авторы: Макглинн Молли Энн, Глэйдью Роналд Пол, Посс Кристофер Стенли, Браун Мэттью Фрэнк, Бламберг Лора Кук






Формула / Реферат
1. Соединение формулы

или его фармацевтически приемлемая соль, где
a равно 1;
b равно 1;
c равно 1;
d равно 1;
e равно 1;
j равно 1;
X представляет собой C(O);
Y представляет собой CH2;
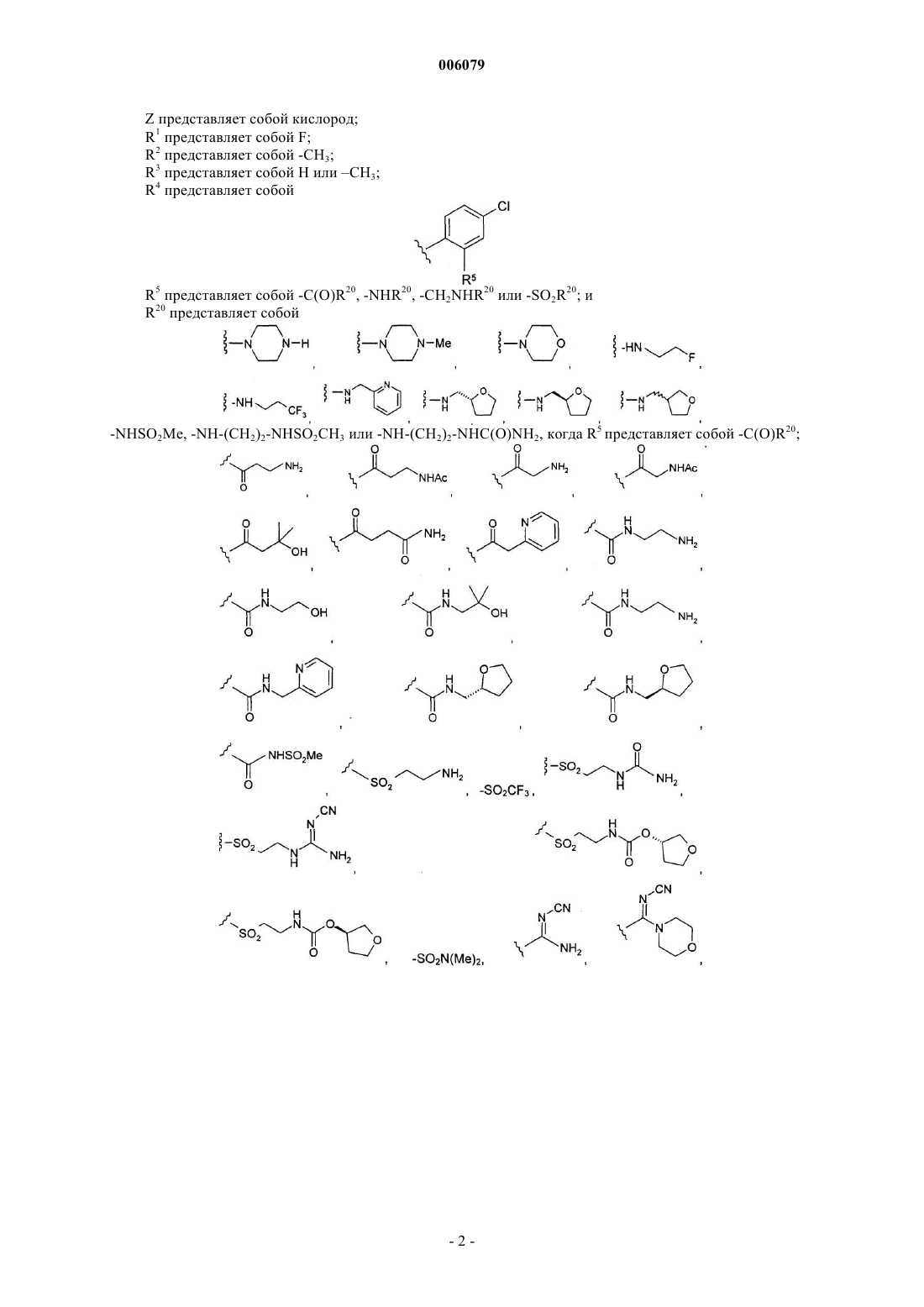
Z представляет собой кислород;
R1 представляет собой F;
R2 представляет собой -CH3;
R3 представляет собой H или -CH3;
R4 представляет собой

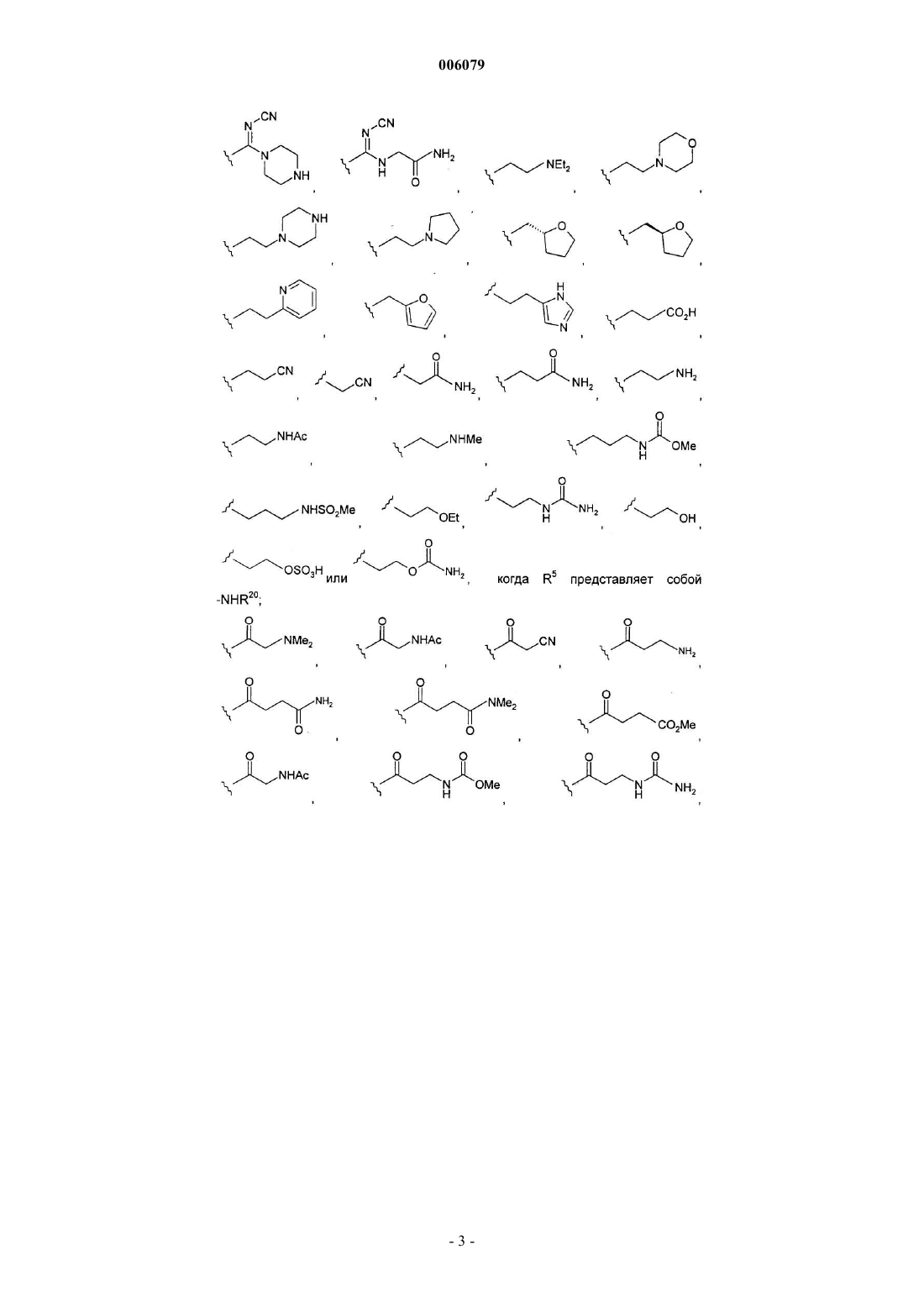
R5 представляет собой -C(O)R20, -NHR20, -CH2NHR20 или -SO2R20; и
R20 представляет собой

-NHSO2Me, -NH-(CH2)2-NHSO2CH3 или -NH-(CH2)2-NHC(O)NH2, когда R5 представляет собой -C(O)R20;



когда R5 представляет собой -CH2NHR20; -NHCH3, -NH2,

когда R5 представляет собой -SO2R20.
2. Фармацевтическая композиция для лечения или предупреждения расстройства или состояния, выбранного из аутоиммунных заболеваний, ревматоидного артрита, юношеского диабета I типа, волчанки, воспалительного заболевания кишечника, ретробульбарного неврита, псориаза, рассеянного склероза, ревматической полимиалгии, увеита и васкулита, острых и хронических воспалительных состояний, остеоартрита, респираторного дистресс-синдрома взрослых, респираторного дистресс-синдрома детей, ишемического реперфузионного повреждения, гломерулонефрита и хронической обструктивной болезни легких (ХОБЛ), аллергических состояний, астмы и атопического дерматита, воспаления, связанного с инфекцией, вирусного воспаления, гриппа, гепатита и синдрома Гийена-Барре (острый первичный идиопатический полирадикулоневрит), хронического бронхита, хронического или острого отторжения трансплантата ткани, клетки и твердого органа, ксенотрансплантации, атеросклероза, рестеноза, ВИЧ инфективности (использование корецептора) и гранулематозных заболеваний, саркоидоза, проказы и туберкулеза, и осложнений, связанных с раками, множественной миеломой; ограничения продукции цитокинов и/или ФНО в воспалительных сайтах как следствие уменьшения клеточной инфильтрации; для лечения заболеваний и/или застойной сердечной недостаточности, связанных с ФНО и ИЛ-1, и для лечения эмфиземы легких или одышки, связанной с ней, эмфиземы; ВИЧ-1, ВИЧ-2, ВИЧ-3; цитомегаловируса (CMV), аденовирусов, вирусов герпеса (Herpes zoster (вирус опоясывающего лишая) и Herpes simplex (вирус простого герпеса)), для лечения осложнений, связанных с инфекцией, когда такая инфекция индуцирует продукцию вредных воспалительных цитокинов и/или ФНО, грибкового менингита, повреждения суставной ткани, гиперплазии, образования паннуса и костной резорбции, псориатического артрита, печеночной недостаточности, бактериального менингита, синдрома Кавасаки, инфаркта миокарда, острой печеночной недостаточности, болезни Лайма, септического шока, рака, травмы и малярии, у млекопитающего, содержащая количество соединения по п.1 или его фармацевтически приемлемой соли или пролекарства, эффективное при лечении или предупреждении такого расстройства или состояния, и фармацевтически приемлемый носитель.
3. Фармацевтическая композиция для лечения или предупреждения расстройства или состояния, которое можно лечить или предупреждать посредством ингибирования связывания хемокина с рецептором CCR1 у млекопитающего, содержащая количество соединения по п.1 или его фармацевтически приемлемой соли, эффективное при лечении или предупреждении такого расстройства или состояния, и фармацевтически приемлемый носитель.
4. Способ лечения или предупреждения расстройства или состояния, выбранного из аутоиммунных заболеваний, ревматоидного артрита, юношеского диабета I типа, волчанки, воспалительного заболевания кишечника, ретробульбарного неврита, псориаза, рассеянного склероза, ревматической полимиалгии, увеита и васкулита, острых и хронических воспалительных состояний, остеоартрита, респираторного дистресс-синдрома взрослых, респираторного дистресс-синдрома детей, ишемического реперфузионного повреждения, гломерулонефрита и хронической обструктивной болезни легких (ХОБЛ), аллергических состояний, астмы и атопического дерматита, воспаления, связанного с инфекцией, вирусного воспаления, гриппа, гепатита и синдрома Гийена-Барре (острый первичный идиопатический полирадикулоневрит), хронического бронхита, хронического или острого отторжения трансплантата ткани, клетки и твердого органа, ксенотрансплантации, атеросклероза, рестеноза, ВИЧ инфективности (использование корецептора) и гранулематозных заболеваний, саркоидоза, проказы и туберкулеза, и осложнений, связанных с раками, множественной миеломой; ограничения продукции цитокинов и/или ФНО в воспалительных сайтах как следствие уменьшения клеточной инфильтрации; лечения заболеваний и/или застойной сердечной недостаточности, связанных с ФНО и ИЛ-1, и лечения эмфиземы легких или одышки, связанной с ней, эмфиземы; ВИЧ-1, ВИЧ-2, ВИЧ-3; цитомегаловируса (CMV), аденовирусов, вирусов герпеса (Herpes zoster (вирус опоясывающего лишая) и Herpes simplex (вирус простого герпеса)), лечения осложнений, связанных с инфекцией, когда такая инфекция индуцирует продукцию вредных воспалительных цитокинов и/или ФНО, грибкового менингита, повреждения суставной ткани, гиперплазии, образования паннуса и костной резорбции, псориатического артрита, печеночной недостаточности, бактериального менингита, синдрома Кавасаки, инфаркта миокарда, острой печеночной недостаточности, болезни Лайма, септического шока, рака, травмы и малярии, у млекопитающего, при котором млекопитающему, нуждающемуся в таком лечении или предупреждении, вводят количество соединения по п.1 или его фармацевтически приемлемой соли или пролекарства, которое является эффективным при лечении или предупреждении такого расстройства или состояния.
5. Способ лечения или предупреждения расстройства или состояния, которое можно лечить или предупреждать посредством действия в качестве антагониста рецептора CCR1 у млекопитающего, при котором млекопитающему, нуждающемуся в таком лечении или предупреждении, вводят количество соединения по п.1 или его фармацевтически приемлемой соли, которое является эффективным при лечении или предупреждении такого расстройства или состояния.
Текст
006079 Предпосылки изобретения Настоящее изобретение относится к новым пиперазиновым производным, к способам их применения и фармацевтическим композициям, содержащим их. Соединения по изобретению являются сильнодействующими и избирательными ингибиторами связывания хемокина с его рецептором CCR1, обнаруженным на воспалительных и иммуномодуляторных клетках (предпочтительно лейкоцитах и лимфоцитах). Рецептор CCR1 также иногда называют CC-CKR1 рецептором. Эти соединения также ингибируют индуцированный МIР-1 (и родственными хемокинами,которые, как показано, взаимодействуют с CCR1 (например RANTES и МСР-3 хемотаксис ТНР-1 клеток и человеческих лейкоцитов, и они потенциально являются полезными для лечения или предупреждения аутоиммунных заболеваний (таких как ревматоидный артрит, юношеский диабет I типа, волчанка,воспалительное заболевание кишечника, ретробульбарный неврит, псориаз, рассеянный склероз, ревматическая полимиалгия, увеит и васкулит), острых и хронических воспалительных состояний (таких как остеоартрит, респираторный дистресс синдром взрослых, респираторный дистресс синдром детей, ишемическое реперфузионное повреждение и гломерулонефрит), аллергических состояний (таких как астма и атопический дерматит), инфекции, связанной с воспалением (такой как вирусное воспаление (включая грипп и гепатит) и синдром Гийена-Барре (острый первичный идиопатический полирадикулоневрит,хронического бронхита, ксенотрансплантации, отторжения ткани при трансплантации (хронического и острого), отторжения трансплантата органа (хронического и острого), атеросклероза, рестеноза, ВИЧ инфективности (использование корецептора) и гранулематозных заболеваний (включая саркоидоз, проказу и туберкулез) и осложнений, связанных с некоторыми раками, такими как множественная миелома. Соединения этой серии могут также ограничивать продуцирование цитокинов в воспалительных сайтах,включая, но не ограничиваясь ими, ФНО (фактор некроза опухоли) и ИЛ-1 (интерлейкин-1), как следствие уменьшения клеточной инфильтрации, принося пользу при заболеваниях, связанных с ФНО и ИЛ-1,включая застойную сердечную недостаточность, эмфизему легких или одышку, связанную с ней, эмфизему; HIV-1 (ВИЧ-1), HIV-2 (ВИЧ-2), HIV-3 (ВИЧ-3); цитомегалогеновирус (CMV), аденовирусы, вирусы герпеса (Herpes zoster (вирус опоясывающего лишая) и Herpes simplex (вирус простого герпеса. Они могут также приносить пользу при осложнениях, связанных с инфекцией, где такая инфекция индуцирует продуцирование вредных воспалительных цитокинов, таких как ФНО, например, при грибковом менингите, повреждении суставной ткани, гиперплазии, образовании паннуса и костной резорбции, псориатическом артрите, печеночной недостаточности, бактериальном менингите, синдроме Кавасаки, инфаркте миокарда, острой печеночной недостаточности, болезни Лайма, септическом шоке, раке, травме и малярии. МIР-1 и RANTES представляют собой растворимые хемотактические пептиды (хемокины), которые продуцируются воспалительными клетками, в частности CD8+ лимфоцитами, полиморфноядерными нейтрофилами (ПМН) и макрофагами, J. Biol. Chem., 270 (30) 29671-29675 (1995). Эти хемокины действуют посредством индукции миграции и активации ключевых воспалительных и иммуномодуляторных клеток. Повышенные уровни хемокинов обнаружены в синовиальной жидкости пациентов с ревматоидным артритом, пациентов с хроническим и острым отторжением ткани от трансплантата и в носовых секретах пациентов с аллергическим ринитом после воздействия аллергена (Teran, et al J. Immunol., 1806-1812 (1996) и Kuna et al., J. Allergy Clin. Immunol. 321 (1994. Антитела, которые препятствуют взаимодействию хемокин/рецептор посредством нейтрализации МIР-1 или прерывания гена,обеспечили прямое доказательство роли МIР-1 и RANTES в заболевании посредством ограничения рекрутмента моноцитов и CD8+ лимфоцитов (Smith et al., J. Immunol. 153, 4704 (1994) и Cook et al., Science, 269, 1583 (1995. Вместе эти данные демонстрируют, что антагонисты CCR1 рецептора были бы эффективны для лечения некоторых заболеваний, основанных на иммунитете. Эти соединения, описанные здесь, являются сильнодействующими и избирательными антагонистами CCR1 рецептора. Краткое изложение сущности изобретения Настоящее изобретение также относится к соединению формулы или его фармацевтически приемлемой соли, где а равно 1;Y представляет собой СН 2; когда R5 представляет собой SO2R20. Настоящее изобретение также относится к фармацевтически приемлемым солям присоединения кислоты соединений формулы I. Кислоты, которые используют для получения фармацевтически приемлемых солей присоединения кислоты вышеупомянутых основных соединений по данному изобретению,представляют собой те, которые образуют нетоксичные соли присоединения кислоты, то есть соли, содержащие фармакологически приемлемые анионы, такие как соли гидрохлорид, гидробромид, гидроиодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, ацетат, лактат, цитрат, кислый цитрат, тартрат,битартрат, сукцинат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, паратолуолсульфонат и памоат [т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)]. Данное изобретение также относится к солям присоединения основания соединений формулы I. Химические основания, которые можно использовать в качестве реагентов для получения фармацевтически приемлемых солей основания тех соединений формулы I, которые являются кислыми по своей природе, представляют собой те, которые образуют нетоксичные соли основания с такими соединениями. Такие нетоксичные соли основания включают в себя, но не ограничены ими, соли, полученные от таких фармакологически приемлемых катионов, как катионы щелочных металлов (например калия и натрия) и катионы щелочно-земельных металлов (например кальция и магния), соли аммония или соли присоединения водорастворимого амина, такого как N-метилглюкамин-(меглумин), и низшего алканоламмония и другие соли основания фармацевтически приемлемых органических аминов. Соединения по данному изобретению могут содержать олефиноподобные двойные связи. Когда присутствуют такие связи, соединения по изобретению существуют в виде цис- и транс-конфигураций и в виде их смесей. Настоящее изобретение также относится к соединениям формулы I, в которых любой из атомов водорода может быть возможно заменен дейтерием. Соединения по изобретению включают в себя все конформационные изомеры (например цис- и транс-изомеры) и все оптические изомеры соединений формулы (I) (например, энантиомеры и диастереомеры), а также рацемические, диастереомерные и другие смеси таких изомеров. Настоящее изобретение также относится к фармацевтической композиции для лечения или предупреждения расстройства или состояния, выбранного из аутоиммунных заболеваний, ревматоидного артрита, юношеского диабета I типа, волчанки, воспалительного заболевания кишечника, ретробульбарного неврита, псориаза, рассеянного склероза, ревматической полимиалгии, увеита и васкулита, острых и хронических воспалительных состояний, остеоартрита, респираторного дистресс-синдрома взрослых,-4 006079 респираторного дистресс-синдрома детей, ишемического реперфузионного повреждения, гломерулонефрита и хронической обструктивной болезни легких (ХОБЛ), аллергических состояний, астмы и атопического дерматита, воспаления, связанного с инфекцией, вирусного воспаления, гриппа, гепатита и синдрома Гийена-Барре (острый первичный идиопатический полирадикулоневрит), хронического бронхита,хронического или острого отторжения трансплантата ткани, клетки и твердого органа, ксенотрансплантации, атеросклероза, рестеноза, ВИЧ инфективности (использование корецептора) и гранулематозных заболеваний, саркоидоза, проказы и туберкулеза, и осложнений, связанных с раками, множественной миеломой; ограничения продукции цитокинов и/или ФНО в воспалительных сайтах как следствие уменьшения клеточной инфильтрации; для лечения заболеваний и/или застойной сердечной недостаточности, связанных с ФНО и ИЛ-1, и для лечения эмфиземы легких или одышки, связанной с ней, эмфиземы; ВИЧ-1, ВИЧ-2, ВИЧ-3; цитомегаловируса (CMV), аденовирусов, вирусов герпеса (Herpes zoster (вирус опоясывающего лишая) и Herpes simplex (вирус простого герпеса, для лечения осложнений, связанных с инфекцией, когда такая инфекция индуцирует продукцию вредных воспалительных цитокинов и/или ФНО, грибкового менингита, повреждения суставной ткани, гиперплазии, образования паннуса и костной резорбции, псориатического артрита, печеночной недостаточности, бактериального менингита,синдрома Кавасаки, инфаркта миокарда, острой печеночной недостаточности, болезни Лайма, септического шока, рака, травмы и малярии, у млекопитающего, содержащая количество соединения формулы I или его фармацевтически приемлемой соли или пролекарства, эффективное при лечении или предупреждении такого расстройства или состояния, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к фармацевтической композиции для лечения или предупреждения расстройства или состояния, которое можно лечить или предупреждать посредством ингибирования связывания хемокина с рецептором CCR1 у млекопитающего, содержащая количество соединения формулы I или его фармацевтически приемлемой соли, эффективное при лечении или предупреждении такого расстройства или состояния, и фармацевтически приемлемый носитель. Примерами таких расстройств и состояний являются те, которые перечислены в предыдущем параграфе. Настоящее изобретение также относится к способу лечения или предупреждения расстройства или состояния, выбранного из аутоиммунных заболеваний, ревматоидного артрита, юношеского диабета I типа, волчанки, воспалительного заболевания кишечника, ретробульбарного неврита, псориаза, рассеянного склероза, ревматической полимиалгии, увеита и васкулита, острых и хронических воспалительных состояний, остеоартрита, респираторного дистресс синдрома взрослых, респираторного дистресс синдрома детей, ишемического реперфузионного повреждения, гломерулонефрита и хронической обструктивной болезни легких (ХОБЛ), аллергических состояний, астмы и атопического дерматита, воспаления,связанного с инфекцией, вирусного воспаления, гриппа, гепатита и синдрома Гийена-Барре (острый первичный идиопатический полирадикулоневрит), хронического бронхита, хронического или острого отторжения трансплантата ткани, клетки и твердого органа, ксенотрансплантации, атеросклероза, рестеноза, ВИЧ инфективности (использование корецептора) и гранулематозных заболеваний, саркоидоза, проказы и туберкулеза, и осложнений, связанных с раками, множественной миеломой; ограничения продукции цитокинов и/или ФНО в воспалительных сайтах как следствие уменьшения клеточной инфильтрации; лечения заболеваний и/или застойной сердечной недостаточности, связанных с ФНО и ИЛ-1, и лечения эмфиземы легких или одышки, связанной с ней, эмфиземы; ВИЧ-1, ВИЧ-2, ВИЧ-3; цитомегаловируса (CMV), аденовирусов, вирусов герпеса (Herpes zoster (вирус опоясывающего лишая) и Herpes simplex (вирус простого герпеса, лечения осложнений, связанных с инфекцией, когда такая инфекция индуцирует продукцию вредных воспалительных цитокинов и/или ФНО, грибкового менингита, повреждения суставной ткани, гиперплазии, образования паннуса и костной резорбции, псориатического артрита,печеночной недостаточности, бактериального менингита, синдрома Кавасаки, инфаркта миокарда, острой печеночной недостаточности, болезни Лайма, септического шока, рака, травмы и малярии, у млекопитающего, при котором млекопитающему, нуждающемуся в таком лечении или предупреждении, вводят количество соединения формулы I или его фармацевтически приемлемой соли или пролекарства,которое является эффективным при лечении или предупреждении такого расстройства или состояния. Настоящее изобретение также относится к способу лечения или предупреждения расстройства или состояния, которое можно лечить или предупреждать посредством действия в качестве антагониста рецептора CCR1 у млекопитающего, при котором млекопитающему, нуждающемуся в таком лечении или предупреждении, вводят количество соединения формулы I или его фармацевтически приемлемой соли,которое является эффективным при лечении или предупреждении такого расстройства или состояния. Подробное описание изобретения Следующие реакционные схемы иллюстрируют получение соединений по настоящему изобретению. Если не указано иначе, а, b, с, d, e, j, R1, R2, R3 и R4 в реакционных схемах и в последующем обсуждении определены, как выше.(цианогуанидино),аминокарбонил(C1-С 6)алкил(цианогуанидино),(C1-С 6)алкиламинокарбонил(С 1 С 6)алкил(цианогуанидино),C1-С 6)алкил)2 аминокарбонил(С 1-С 6)алкил(цианогуанидино),(С 2-С 9) гетероциклоалкила, амино(C1-С 6)алкиламино, (С 1-С 6)алкиламино(C1-С 6)алкиламино, С 1-С 6)алкил)2 амино(C1-С 6)алкиламино, (C2-С 9)гетероариламино, уреидо(С 1-С 6)алкиламино, (С 1-С 6)алкилуреидо(C1 С 6)алкиламино, С 1-С 6)алкил)2 уреидо(С 1-С 6)алкиламино, (C1-С 6)алкилсульфониламино(С 1-С 6)алкиламино,(C1-С 6)алкоксикарбониламино(C1-С 6)алкиламино,(С 2-С 9)гетероциклоалкилоксикарбониламино(С 1-С 6)алкиламино, (С 2-С 9)гетероарилоксикарбониламино(С 1-С 6)алкиламино, аминокарбонил(C1 С 6)алкиламино,цианогуанидино(С 1-С 6)алкиламино,(С 2-С 9)гетероарил(С 1-С 6)алкиламино,(С 2 С 9)гетероциклоалкиламино, (C1-С 6)алкилкарбониламино, галогено(C1-С 6)алкилкарбониламино, (C1 С 6)алкоксикарбониламино, уреидо, (C1-С 6)алкилуреидо, C1-С 6)алкил)2 уреидо, амино, (C1-С 6)алкиламино, (С 3-С 10)циклоалкиламино, C1-С 6)алкил)2 амино, гидрокси(С 1-С 6)алкиламино, (С 6-С 10)ариламино,(С 6-С 10)арил(С 1-С 6)алкиламино, (C1-С 6)алкилкарбониламино, (С 6-С 10)арилкарбониламино, C1-С 6) алкилкарбонил)С 1-С 6)алкил)амино,(С 3-С 10)циклоалкил(C1-С 6)алкил)амино,(C1-С 6)алкоксикарбониламино, (C1-С 6)алкоксикарбонил(C1-С 6)алкилкарбониламино, C1-С 6)алкоксикарбонил)С 1-С 6)алкил)амино,(C1-С 6)алкилсульфониламино,C1-С 6)алкилсульфонил)C1-С 6)алкил)амино,(С 6-С 10) арилсульфониламино, С 6-С 10)арилсульфонил)C1-С 6)алкил)амино, (C2-С 9)гетероциклоалкиламино, (C2 С 9)гетероариламино, галогено(C1-С 6)алкиламино, (С 6-С 10)ариламино, (С 6-С 10)арил(C1-С 6)алкиламино,аминокарбонил(C1-С 6)алкиламино, C1-С 6)алкиламинокарбонил(C1-С 6)алкиламино, (карбокси(C1-С 6) алкил)амино, C1-С 6)алкоксикарбонил(C1-С 6)алкиламино, (амино(С 1-С 6)алкил)амино, (гидрокси(C1-С 6) алкиламино, уреидо, (С 1-С 6)алкилуреидо, С 1-С 6)алкил)2 уреидо, (С 6-С 10)арилуреидо, С 6-С 10) арил)2 уреидо, (С 6-С 10)арил(С 1-С 6)алкилуреидо, гелогено(C1-С 6)алкилуреидо, (галогено(C1-С 6)алкил)C1 С 6)алкил)уреидо, C1-С 6)алкоксикарбонил(C1-С 6)алкил)уреидо и глицинамидо. Если не указано иначе, алкильная и алкенильная группы, на которые ссылаются здесь, а также алкильные группировки других групп, на которые ссылаются здесь, (например алкокси), могут быть нормальными или разветвленными, и они могут также быть циклическими (например циклопропил, циклобутил, циклопентил, циклогексил или циклогептил), или быть нормальными или разветвленными и содержать циклические группировки. Если не указано иначе, галоген включает в себя фтор, хлор, бром и йод.(С 3-С 10)Циклоалкил, когда его используют здесь, относится к циклоалкильным группам, содержащим от нуля до двух уровней ненасыщения, таким как циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, 1,3-циклогексадиен, циклогептил, циклогептенил, бицикло[3.2.1]октан, норборнанил и так далее.(С 2-С 9)Гетероциклоалкил, когда его используют здесь, относится к пирролидинилу, тетрагидрофуранилу, дигидрофуранилу, тетрагидропиранилу, пиранилу, тиопиранилу, азиридинилу, оксиранилу, ме-7 006079 тилендиоксилу, хроменилу, барбитурилу, изоксазолидинилу, 1,3-оксазолидин-3-илу, изотиазолидинилу,1,3-тиазолидин-3-илу, 1,2-пиразолидин-2-илу, 1,3-пиразолидин-1-илу, пиперидинилу, тиоморфолинилу,1,2-тетрагидротиазин-2-илу, 1,3-тетрагидротиазин-3-илу, тетрагидротиадиазинилу, морфолинилу, 1,2 тетрагидродиазин-2-илу, 1,3-тетрагидродиазин-1-илу, тетрагидроазепинилу, пиперазинилу, хроманилу и так далее.(С 2-С 9)Гетероарил, когда его используют здесь, относится к фурилу, тиенилу, тиазолилу, пиразолилу, изотиазолилу, оксазолилу, изоксазолилу, пирролилу, триазолилу, тетразолилу, имидазолилу, 1,3,5 оксадиазолилу, 1,2,4-оксадиазолилу, 1,2,3-оксадиазолилу, 1,3,5-тиадиазолилу, 1,2,3-тиадиазолилу, 1,2,4 тиадиазолилу, пиридилу, пиримидилу, пиразинилу, пиридазинилу, 1,2,4-триазинилу, 1,2,3-триазинилу,1,3,5-триазинилу, пиразоло[3,4-d]пиридинилу, циннолинилу, птеридинилу, пуринилу, 6,7-дигидро-5 Н[1]-пиридинилу, бензо[b]тиофенилу, 5,6,7,8-тетрагидрохинолин-3-илу, бензоксазолилу, бензотиазолилу,бензизотиазолилу, бензизоксазолилу, бензимидазолилу, тианафтенилу, изотианафтенилу, бензофуранилу, изобензофуранилу, изоиндолилу, индолилу, индолизинилу, индазолилу, изохинолилу, хинолилу,фталазинилу, хиноксалинилу, хиназолинилу, бензоксазинилу и так далее. Арил, когда его используют здесь, относится к фенилу или нафтилу. Термин уреидо, как его используют здесь, относится к амино-карбонил-амино группировке. Термин ацетил, как его используют здесь, относится к алкил-карбонил группировке, в которой алкил определен, как выше. Термин цианогуанидино, как его используют здесь, относится к функциональной группе, имеющей следующую формулу: В реакции 1 получения А соединение формулы XXXV, где b составляет 0, 1 или 2, превращают в соответствующее соединение формулы XXXIV взаимодействием XXXV с этилдиаминовым соединением формулы (R3)j-этилдиамин в присутствии апротонного растворителя, такого как диэтиловый эфир. Реакционную смесь нагревают до флегмообразования в течение периода времени между примерно 1 ч и при- 13006079 мерно 12 ч. В реакции 2 получения А соединение формулы XXXIV превращают в соответствующее соединение формулы XXXIII восстановлением XXXIV восстанавливающим агентом, таким как боргидрид натрия, в кипящем с обратным холодильником протонном растворителе, таком как этанол. В реакции 3 получения А соединение формулы XXXIII превращают в соответствующее соединение формулы XXX взаимодействием XXXIII с бензальдегидным соединением формулы в присутствии основания, такого как триэтиламин, и восстанавливающего агента, такого как триацетоксиборгидрид натрия, в апротонном растворителе, таком как 1,2-дихлорэтан. Реакционную смесь перемешивают при комнатной температуре в течение периода времени между примерно 1 ч и примерно 4 ч,предпочтительно примерно 2 ч. В реакции 1 получения Б соединение формулы XXII, где b составляет 0, 1 или 2, превращают в соответствующее соединение формулы XXI взаимодействием XXIII с бензальдегидным соединением формулы в присутствии основания, такого как триэтиламин, восстанавливающего агента, такого как боргидрид натрия, и апротонного растворителя, такого как 1,2-дихлорэтан. Реакционную смесь перемешивают при комнатной температуре в течение периода времени между примерно 1 ч и примерно 4 ч, предпочтительно примерно 2 ч. В реакции 2 получения Б соединение формулы XXI превращают в соответствующее соединение формулы XX сначала взаимодействием соединения формулы где j равно 0, 1 или 2, с 4-метилморфолином и изобутилхлорформиатом в присутствии полярного апротонного растворителя, такого как тетрагидрофуран, с последующим взаимодействием образованного таким образом промежуточного соединения с соединением формулы XXI. Реакционную смесь, образованную таким образом, перемешивают в течение ночи при комнатной температуре. В реакции 3 получения Б соединение формулы XX превращают в соответствующее пиперизин-2,5 дионовое соединение формулы XIX обработкой XX трифторуксусной кислотой в присутствии полярного апротонного растворителя, такого как метиленхлорид. Реакционную смесь перемешивают при комнатной температуре в течение периода времени между примерно 1 ч и примерно 4 ч, предпочтительно примерно 2 ч. В реакции 4 получения Б соединение формулы XIX превращают в соответствующее соединение формулы XVIII восстановлением XIX восстанавливающим агентом, таким как алюмогидрид лития. Эту реакцию проводят при температуре между примерно -10 и примерно 10 С, предпочтительно примерно 0 С, в течение периода времени между примерно 10 мин и примерно 90 мин, предпочтительно примерно 40 мин. В реакции 1 получения В соединение формулы XXV превращают в соответствующее соединение формулы XXIV взаимодействием XXV с амином формулы NHR18R19, где R18 и R19, каждый независимо,выбран из водорода, азотсодержащей (C2-С 9)гетероциклоалкильной или (С 2-С 9)гетероарильной группы,либо (C1-С 6)алкила, возможно замещенного гидрокси, аминокарбонилом, (C1-С 6)алкиламинокарбонилом,C1-С 6)алкил)2 карбонилом, карбокси, (C1-С 6)алкилсульфониламино, (C1-С 6)алкоксикарбониламино,аминосульфонилом, (C1-С 6)алкиламиносульфонилом, C1-С 6)алкил)2 аминосульфонилом, (С 6-С 10)алкокси, (С 2-С 9)гетероарилом, (С 2-С 9)гетероциклоалкилом, (С 1-С 6)алкилкарбониламино, C1-С 6)алкилкарбонил)C1-С 6)алкил)амино, циано, уреидо, (C1-С 6)алкилуреидо, С 1-С 6)алкил)2 уреидо, цианогуанидино, (C1-С 6)алкилцианогуанидино и C1-С 6)алкил)2 цианогуанидино, либо R18 и R19 взяты вместе с атомом азота, к которому они присоединены, с образованием (С 2-С 9)гетероарильной или (С 2 С 9)гетероциклоалкильной группы, в присутствии полярного апротонного растворителя, такого как метиленхлорид. Реакционную смесь перемешивают при комнатной температуре в течение периода времени между примерно 1 ч и примерно 24 ч, предпочтительно примерно 12 ч. В реакции 2 получения В соединение формулы XXIV превращают в соответствующее соединение формулы XXIII взаимодействием XXIV с тиофенолом в присутствии основания, такого как гидрид на- 14006079 трия, и полярного апротонного растворителя, такого как диметилформамид. Реакционную смесь нагревают до флегмобразования в течение периода времени между примерно 1 ч и примерно 10 ч, предпочтительно примерно 4 ч. В реакции 3 получения В соединение формулы XXV превращают в соответствующее соединение формулы XXXVIII взаимодействием XXV с цианатом натрия в присутствии пиридина и полярного апротонного растворителя, такого как ацетонитрил. Реакционную смесь перемешивают при комнатной температуре в течение периода времени между примерно 2 ч и примерно 18 ч, предпочтительно примерно 10 ч. Затем добавляют амин формулы H2N-C(O)-NR18R19 и реакционную смесь, образованную таким образом, перемешивают при комнатной температуре в течение периода времени между примерно 2 ч и примерно 24 ч, предпочтительно примерно 8 ч. В реакции 4 получения В соединение формулы XXXVIII превращают в соответствующее соединение формулы XXXVII в соответствии с методикой, описанной выше в реакции 2 получения В. В реакции 1 схемы 1 соединение формулы XXX превращают в соответствующее соединение формулы X взаимодействием XXX с соединением формулы A-(X)c-(Y)d-A, где А представляет собой хлоро или бромо, в присутствии основания, такого как триэтиламин, и полярного апротонного растворителя,такого как метиленхлорид. Реакционную смесь перемешивают при температуре между примерно -10 С и примерно 10 С в течение периода времени между примерно 15 мин и примерно 90 мин, предпочтительно примерно 30 мин. В реакции 2 схемы 1 соединение формулы X превращают в соответствующее соединение формулыI взаимодействием X с соединением формулы H-(Z)e-R4, где е составляет 1, a Z представляет собой кислород, в присутствии карбоната калия, иодида калия и апротонного растворителя, такого как бутанон. Реакционную смесь нагревают до флегмообразования в течение периода времени между примерно 4 ч и примерно 8 ч, предпочтительно примерно 6 ч. В реакции 1 схемы 2 соединение формулы XXX превращают в соответствующее соединение формулы I взаимодействием XXX с соединением формулы A-(X)c-(Y)d-(Z)e-R4, где А представляет собой хлоро или бромо, в присутствии основания, такого как триэтиламин, и полярного апротонного растворителя, такого как метиленхлорид. Реакционную смесь перемешивают при температуре между примерно-10 и примерно 10 С в течение периода времени между примерно 15 мин и примерно 90 мин, предпочтительно примерно 30 мин. В реакции 1 схемы 3 соединение формулы X превращают в соответствующее соединение формулыXII в соответствии с методикой, описанной выше в реакции 2 схемы 1. В реакции 2 схемы 3 соединение формулы XII превращают в соответствующее соединение формулы XI взаимодействием XII с моногидратом гидроксида лития в присутствии метанола, тетрагидрофурана и воды. Реакционную смесь перемешивают в течение ночи при комнатной температуре. В реакции 3 схемы 3 соединение формулы XI превращают в соответствующее соединение формулыII взаимодействием XI с амином в присутствии 4-диметиламинопиридина, 1-(3-диметиламинопропил)-3 этилкарбодиимина и полярного апротонного растворителя, такого как метиленхлорид. Полученную в результате реакционную смесь перемешивают в течение ночи при комнатной температуре. В реакции 1 схемы 4 соединение формулы X превращают в соответствующее соединение формулыXV в соответствии с методикой, описанной выше в реакции 2 схемы 1. В реакции 2 схемы 4 соединение формулы XV превращают в соответствующее соединение формулы XIV гидрогенизацией XV в присутствии катализатора, такого как платина на угле, и полярного протонного растворителя, такого как этанол. Реакцию проводят при давлении между примерно 206,84 кПа(30 фунт/кв.дюйм) и примерно 275,79 кПа (40 фунт/кв.дюйм), предпочтительно примерно 241,32 кПа (35 фунт/кв.дюйм), в течение периода времени между примерно 15 мин и примерно 1 ч, предпочтительно 30 мин. В реакции 3 схемы 4 для образования мочевины соединение формулы XIV превращают в соответствующее соединение формулы V сначала взаимодействием XIV с 4-нитрофенилхлорформиатом в присутствии основания, такого как пиридин, и полярного апротонного растворителя, такого как метиленхлорид, с последующим взаимодействием образованного таким образом промежуточного соединения с амином. Реакционной смеси, образованной таким образом, дают перемешиваться в течение ночи при комнатной температуре. Для образования сульфонамида соединение формулы XIV подвергают взаимодействию с сульфонилхлоридным соединением формулы R16-Cl в присутствии основания, такого как триэтиламин, и полярного апротонного растворителя, такого как метиленхлорид. Реакционную смесь перемешивают в течение ночи при температуре окружающей среды. Для образования цианогуанидина соединение формулы XIV сначала обрабатывают гидридом натрия в апротонном растворителе, таком как тетрагидрофуран, с последующим взаимодействием образованного таким образом промежуточного соединения с диметил-N-цианодитиоиминокарбонатом. Реакционную смесь, образованную таким образом,нагревают до флегмообразования в течение ночи. Промежуточное соединение N-циано-S-метилизотиомочевину затем подвергают взаимодействию с амином в присутствии полярного протонного растворителя, такого как метанол. Для образования амида соединение формулы XIV подвергают взаимодействию с кислотой, такой как 3-трет-бутоксикарбониламинопропионовая кислота, в присутствии N- 15006079 метилморфолина, О-бензотриазол-1-ил-N,N,N,N-тетраметилурония гексафторфосфата и полярного апротонного растворителя, такого как метиленхлорид. В реакции 1 схемы 5 соединение формулы X превращают в соответствующее соединение формулыXVI, где k составляет 0, 1, 2, 3 или 4, в соответствии с методикой, описанной выше в реакции 2 схемы 1. В реакции 2 схемы 5 соединение формулы XVI превращают в соответствующее соединение формулы VII взаимодействием XVI с амином формулы R16R17N, где R16 и R17, каждый независимо, представляет собой водород, азотсодержащую (С 2-С 9)гетероциклоалкильную или (С 2-С 9)гетероарильную группу,либо (C1-С 6)алкил, возможно замещенный гидрокси, аминокарбонилом, (С 1-С 6)алкиламинокарбонилом,C1-С 6)алкил)2 карбонилом, карбокси, (C1-С 6)алкилсульфониламино, (C1-С 6)алкоксикарбониламино,аминосульфонилом, (C1-С 6)алкиламиносульфонилом, C1-С 6)алкил)2 аминосульфонилом, (С 6-С 10)алкокси, (С 2-С 9)гетероарилом, (C2-С 9)гетероциклоалкилом, (C1-С 6)алкилкарбониламино, C1-С 6)алкилкарбонил)C1-С 6)алкил)амино, циано, уреидо, (С 1-С 6)алкилуреидо, C1-С 6)алкил)2 уреидо, цианогуанидино, (С 1-С 6)алкилцианогуанидино и C1-С 6)алкил)2 цианогуанидино, в присутствии раствора дихлорэтана/уксусной кислоты в соотношении 10:1. Реакционную смесь перемешивают при комнатной температуре в течение периода времени между примерно 30 мин и примерно 2 ч, предпочтительно примерно 1 ч. Затем к этой смеси добавляют восстанавливающий агент, такой как цианоборгидрид натрия, и реакционной смеси дают перемешиваться в течение ночи при комнатной температуре. Когда R16 и/или R17 представляет(ют) собой водород, соединение формулы VII можно дополнительно подвергнуть взаимодействию в соответствии с методикой, описанной выше в реакции 3 схемы 4, с получением мочевин, сульфонамидов, цианогуанидино или амидов. В реакции 1 схемы 6 соединение формулы X превращают в соответствующее соединение формулыXXXIX в соответствии с методикой, описанной выше в реакции 2 схемы 1. В реакции 2 схемы 6 соединение формулы X превращают в соответствующее соединение формулы ХХХХ в соответствии с методикой, описанной выше в реакции 2 схемы 1. В реакции 1 схемы 7 кислое соединение формулы XXXVI превращают в соответствующее соединение формулы XXXII обработкой XXXVI тионилхлоридом в чистом виде или в апротонном растворителе при комнатной температуре в течение периода времени между примерно 1 ч и примерно 24 ч, предпочтительно 1 ч. Хлорангидрид, образованный таким образом, растворяют в полярном апротонном растворителе с соединением формулы (H3CO)(H3C)NHHCl в присутствии аминного основания, такого как триэтиламин. Реакционную смесь перемешивают при комнатной температуре в течение периода времени между примерно 1 ч и примерно 48 ч, предпочтительно примерно 12 ч. В реакции 2 схемы 7 амидное соединение формулы XXXII превращают в соответствующее соединение формулы XXXI взаимодействием XXXII с (C2-С 9)гетероариллитиевым реагентом в присутствии полярного апротонного растворителя при температуре между примерно -100 С и комнатной температурой предпочтительно примерно -78 С. Полученную в результате реакционную смесь перемешивают в течение периода времени между примерно 1 ч и примерно 24 ч, предпочтительно примерно 12 ч, при температуре между примерно -78 С и примерно 50 С, предпочтительно примерно 20 С. Если не указано иначе, давление каждой из вышеуказанных реакций не является критическим. Обычно реакции следует проводить при давлении от примерно одной до примерно трех атмосфер, предпочтительно при давлении окружающей среды (примерно одна атмосфера). Соединения формулы I, которые являются основными по своей природе, способны к образованию широкого разнообразия различных солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемы для введения животным, часто желательно на практике сначала выделить соединение формулы I из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто превратить последнюю обратно в соединение в форме свободного основания обработкой щелочным реагентом, и последовательно превратить это свободное основание в фармацевтически приемлемую соль присоединения кислоты. Соли присоединения кислоты основных соединений по данному изобретению легко получают обработкой основного соединения, по существу,эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как метанол или этанол. При осторожном выпаривании растворителя получают желаемую твердую соль. Кислоты, которые используют для получения фармацевтически приемлемых солей присоединения кислоты основных соединений по данному изобретению, представляют собой те кислоты, которые образуют нетоксичные соли присоединения кислоты, то есть соли, содержащие фармакологически приемлемые анионы, такие как соли гидрохлорид, гидробромид, гидроиодид, нитрат, сульфат или бисульфат,фосфат или кислый фосфат, ацетат, лактат, цитрат или кислый цитрат, тартрат или битартрат, сукцинат,малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат и памоат (то есть 1,1'-метилен-бис-(2 гидрокси-3-нафтоат. Те соединения формулы I, которые являются также кислыми по своей природе, способны к образованию солей основания с различными фармакологически приемлемыми катионами. Примеры таких солей включают в себя соли щелочных или щелочно-земельных металлов, и, в частности, соли натрия и калия. Все эти соли получают с помощью общепринятых методик. Химические основания, которые ис- 16006079 пользуют в качестве реагентов для получения фармацевтически приемлемых солей оснований по данному изобретению, представляют собой те основания, которые образуют нетоксичные соли основания с описанными здесь кислыми соединениями формулы I. Эти нетоксичные соли основания включают в себя те соли, которые получены от таких фармакологически приемлемых катионов, как натрий, калий, кальций и магний и так далее. Эти соли можно легко получить обработкой соответствующих кислых соединений водным раствором, содержащим желаемые фармакологически приемлемые катионы, а затем выпариванием полученного в результате раствора до сухости, предпочтительно при пониженном давлении. Альтернативно их также можно получить смешиванием растворов кислых соединений и желаемого алкоксида щелочного металла вместе в низших спиртах, а затем выпариванием полученного в результате растворадо сухости тем же способом, как ранее. В любом случае предпочтительно используют стехиометрические количества реагентов, чтобы гарантировать завершение реакции и максимальные выходы продукта. Соединения формулы I и их фармацевтически приемлемые соли (которые здесь и далее также называют все вместе активные соединения) являются сильнодействующими антагонистами рецепторовCCR1. Эти активные соединения являются полезными при лечении или предупреждении аутоиммунных заболеваний (таких как ревматоидный артрит, юношеский диабет I типа, воспалительное заболевание кишечника, ретробульбарный неврит, псориаз, рассеянный склероз, ревматическая полимиалгия, увеит и васкулит), острых и хронических воспалительных состояний (таких как остеоартрит, респираторный дистресс синдром взрослых, респираторный дистресс синдром детей, ишемическое реперфузионное повреждение и гломерулонефрит), аллергических состояний (таких как астма и атопический дерматит),инфекции, связанной с воспалением (таким как вирусное воспаление (включая грипп и гепатит) и синдром Гийена-Барре (острый первичный идиопатический полирадикулоневрит, хронического бронхита,ксенотрансплантации, отторжения ткани при трансплантации, атеросклероза, рестеноза, ВИЧ инфективности (использование корецептора) и гранулематозных заболеваний (включая саркоидоз, проказу и туберкулез). Активность соединений по изобретению можно оценить в соответствии с методиками, известными специалистам в данной области техники. Примеры признанных способов определения CCR1 индуцированной миграции можно найти в Coligan, J.E., Kruisbeek, A.M., Margulies, D.H., Shevach, E.M., Strober, W.editors: Current Protocols In Immunology, 6.12.1-6.12.3. (John Wiley and Sons, NY, 1991). Один конкретный пример того, как определить активность соединения для ингибирования миграции, подробно описан ниже. Анализ хемотаксиса Способность соединений ингибировать хемотаксис к различным хемокинам можно оценить с использованием стандартных 48- или 96-луночных камер Бойдена с 5-микронным поликарбонатным фильтром. Все реагенты и клетки можно готовить в стандартной питательной среде для культуры тканейRPMI (BioWhitikker Inc.), в которую добавлен 1 мг/мл бычьего сывороточного альбумина. Кратко, MIP1 (Peprotech, Inc., P.O. Box 275, Rocky Hill NJ) или другие тестируемые агонисты помещают в нижние камеры камеры Бойдена. Затем накладывают поликарбонатный фильтр и верхнюю камеру закрывают. Таким образом определяют количество выбранного агониста, которое дает максимальное количество хемотаксиса в этой системе (например 1 нМ для MIP-1 должно быть достаточно). Затем в верхние камеры можно добавить ТНР-1 клетки (АТСС TIB-202), первичные моноциты человека или первичные лимфоциты, выделенные с помощью стандартных методик, в трехкратной повторности вместе с различными концентрациями тестируемого соединения. Разбавления соединения можно получить, используя стандартные серологические методики, и их смешивают с клетками перед добавлением в камеру. После соответствующего периода инкубации при 37 С (например 3,5 ч для ТНР-1 клеток, 90 мин для первичных моноцитов) камеру удаляют, клетки в верхней камере отсасывают, верхнюю часть фильтра осушают, и число мигрирующих клеток можно определить согласно следующему способу. Для ТНР-1 клеток камеру (96-луночный вариант, производимый Neuroprobe) можно центрифугировать, чтобы стряхнуть клетки с нижней камеры, и число клеток можно количественно подсчитать против стандартной кривой по изменению цвета красителя флуороцеина диацетата. Для первичных моноцитов человека или лимфоцитов фильтр можно окрасить красителем DifQuik (American Scientific Products), и число мигрирующих клеток можно определить микроскопически. Число клеток, мигрирующих в присутствии соединения, делят на число клеток, мигрирующих в контрольных лунках (без соединения). Частное представляет собой % ингибирования для соединения,которое затем можно нанести на график, используя стандартные графические методики, против концентрации использованного соединения. Точку 50% ингибирования затем определяют, используя анализ линейного схождения для всех протестированных концентраций. Линейное схождение для всех точек данных должно иметь коэффициент корреляции (R квадрат)90%, чтобы считать анализ достоверным. Все соединения по изобретению, которые были протестированы, имели ИК 50 менее чем 25 мкМ в анализе хемотаксиса.- 17006079 Препараты композиций по настоящему изобретению можно изготовлять общепринятым способом,используя один или более чем один фармацевтически приемлемый носитель. Таким образом, активные соединения по изобретению можно включать в препараты для перорального, трансбуккального, интраназального, парентерального (например, внутривенного, внутримышечного или подкожного) или ректального введения, либо в форме, подходящей для введения путем ингаляции или инсуффляции. Активные соединения по изобретению можно также включать в препараты для замедленной доставки. Для перорального введения фармацевтические композиции могут принимать форму, например,таблеток или капсул, полученных общепринятыми способами с фармацевтически приемлемыми эксципиентами, такими как связывающие агенты (например, прежелатинизированный кукурузный крахмал,поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители (например, лактоза, микрокристаллическая целлюлоза или фосфат кальция); смазывающие агенты (например, стеарат магния, тальк или кремнезем); разрыхлители (например, картофельный крахмал или крахмальный гликолят натрия); либо увлажняющие агенты (например, лаурил сульфат натрия). Таблетки могут быть покрыты оболочкой с помощью способов, хорошо известных в данной области техники. Жидкие препараты для перорального введения могут принимать форму, например, растворов, сиропов или суспензий, либо они могут быть представлены в виде сухого продукта для разведения водой или другим подходящим растворителем перед применением. Такие жидкие препараты можно изготовлять общепринятыми способами с фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сироп сорбита, метилцеллюлоза или гидрогенизированные пищевые жиры); эмульгирующие агенты (например лецитин или аравийская камедь); неводные растворители (например, миндальное масло, жирные эфиры или этиловый спирт); и консерванты (например, метил или пропилпарагидроксибензоаты или сорбиновая кислота). Для трансбуккального введения композиция может принимать форму таблеток или лепешек, изготовленных общепринятым способом. Активные соединения по изобретению можно включать в препараты для парентерального введения путем инъекции, включая использование общепринятых методик катетеризации или инфузии. Препараты для инъекции могут быть представлены в стандартной лекарственной форме, например в ампулах или в многодозовых контейнерах, с добавленным консервантом. Композиции могут принимать такие формы,как суспензии, растворы или эмульсии в масляных или водных растворителях, и могут содержать такие агенты, используемые в технологии приготовления лекарства, как суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно активный ингредиент может находиться в порошкообразной форме для разведения подходящим растворителем, например стерильной апирогенной водой,перед применением. Активные соединения по изобретению можно также включать в ректальные композиции, такие как суппозитории или удерживающие клизмы, например содержащие общепринятые основы для суппозиториев, такие как масло какао или другие глицериды. Для интраназального введения или введения путем ингаляции активные соединения по изобретению удобно доставлять в форме раствора или суспензии из нагнетаемого контейнера с аэрозолем, который сжимается или нагнетается пациентом, либо в форме представления аэрозоля из герметичного контейнера или распылителя, с использованием подходящего пропеллента, например дихлордифторметана,трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае герметичного аэрозоля стандартную дозу можно определить путем обеспечения клапана для доставки отмеренного количества. Герметичный контейнер или распылитель может содержать раствор или суспензию активного соединения. В капсулы и картриджи (изготовленные, например, из желатина) для использования в ингаляторе или инсуффляторе можно включать порошкообразную смесь соединения по изобретению и подходящую порошкообразную основу, такую как лактоза или крахмал. Предполагаемая доза активных соединений по изобретению для перорального, парентерального или трансбуккального введения среднему взрослому человеку для лечения состояний, перечисленных выше (например ревматоидного артрита), составляет от 0,1 до 1000 мг активного ингредиента на стандартную дозу, которую следует вводить, например, от 1 до 4 раз в сутки. Аэрозольные препараты для лечения состояний, перечисленных выше (например, ревматоидного артрита), у среднего взрослого человека предпочтительно изготовляют таким образом, чтобы каждая отмеренная доза или толчок аэрозоля содержала от 20 до 1000 мкг соединения по изобретению. Общая суточная доза при использовании аэрозоля должна находиться в диапазоне от 0,1 до 1000 мг. Введение можно осуществлять несколько раз в сутки, например 2, 3, 4 или 8 раз, давая, например, 1, 2 или 3 дозы каждый раз. Активные агенты можно включать в препараты для замедленной доставки в соответствии со способами, хорошо известными обычным специалистам в данной области техники. Примеры таких препаратов можно найти в патентах США 3538214, 4060598, 4173626, 3119742 и 3492397. Соединения по изобретению можно также применять в комбинированной терапии с другими терапевтическими агентами, но не ограничиваясь ими, такими как иммуносупрессирующие агенты Т-клеток,такие как рапамицин, циклоспорин А и FK-506, с агентами, истощающими стероиды, такими какCellcept, или с классическими противовоспалительными агентами (например, ингибиторами циклооксигеназы/липоксигеназы), такими как тенидап, аспирин, ацетаминофен, напроксен и пироксикам. Следующие примеры иллюстрируют получение соединений по настоящему изобретению. Данные ЯМР представлены в частях на миллион (S) и относятся к сигналу синхронизации дейтерия из образца растворителя (дейтериохлороформ, если не указано иначе). Коммерческие реагенты использовали без дополнительной очистки. ТГФ относится к тетрагидрофурану. ДМФ относится к N,N-диметилформамиду. Хроматография относится к колоночной хроматографии, осуществляемой с использованием 32-63 мм силикагеля и выполняемой в условиях давления азота(флэш-хроматография). Масс-спектры низкого разрешения (МСНР) записывали либо на Hewlett Packard 5989 с использованием химической ионизации (аммоний), либо Fisons (или Micro Mass) с использованием платформы химической ионизации атмосферного давления (APCI), в которой используют смесь ацетонитрил/вода в соотношении 50:50 с 0,1% муравьиной кислотой в качестве ионизирующего агента. Комнатная температура или температура окружающей среды относится к 20-25 С. Все неводные реакции проводили в атмосфере азота для удобства и максимизации выходов. Названия соединений по изобретению даны с помощью версии Autonom 2.0 PC-batch от Beilstein Informationssysteme GmbH (ISBN 389536-976-4). Пример 1.(2R,5S)-2-[4-Хлор-2-(пиперазин-1-карбонил)фенокси]-1-[4-(4-фторбензил)-2,5-диметилпиперазин-1 ил]этанон. Метиловый эфир (R)-2-(4-фторбензиламино)пропионовой кислоты. К раствору гидрохлорида метилового эфира (R)-2-аминопропионовой кислоты (25 г, 179 ммоль) и 4-фторбензальдегида (23 мл, 215 ммоль) в 1,2-дихлорэтане (200 мл) добавляли триэтиламин (25 мл, 179 ммоль). Полученную в результате смесь перемешивали в течение 2 ч при температуре окружающей среды, после чего добавляли ацетоксиборгидрид натрия (57 г, 268 ммоль) четырьмя порциями. Полученную в результате смесь перемешивали в течение ночи при температуре окружающей среды. Эту реакционную смесь нейтрализовали разбавленным водным раствором гидроксида натрия и экстрагировали дихлорметаном (2 х). Органические слои объединяли, высушивали над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматографией на силикагеле получили соединение, указанное в заголовке (34,4 г, выход 91%). Метиловый эфир(2R,5S)-2-[(2-трет-бутоксикарбониламинопропионил)-(4-фторбензил)амино] пропионовой кислоты. К раствору (R)-2-трет-бутоксикарбониламинопропионовой кислоты (37 г, 195 ммоль) в сухом тетрагидрофуране (250 мл) при 0 С добавляли 4-метилморфолин (21,5 мл, 195 ммоль), а затем изобутилхлорформиат (25,3 мл, 195 ммоль). Этой реакционной смеси давали нагреться до температуры окружающей среды и перемешивали в течение 2 ч. После этого добавляли метиловый эфир (S)-2-(4-фторбензиламино)пропионовой кислоты (34,4 г, 162 ммоль). Полученную в результате смесь перемешивали в течение ночи при температуре окружающей среды. Эту реакционную смесь фильтровали через подложку из целита, и фильтрационную лепешку промывали этилацетатом. Фильтрат концентрировали в вакууме,разбавляли этилацетатом и промывали водой и рассолом. Органические фазы высушивали над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматографией на силикагеле получили соединение, указанное в заголовке (43,2 г, выход 70%).(2R,5S)-1-(4-Фторбензил)-3,6-диметилпиперазин-2,5-дион. К раствору метилового эфира (2R,5S)-2-[(2-трет-бутоксикарбониламинопропионил)-(4-фторбензил)амино]пропионовой кислоты (43 г, 382 ммоль) в дихлорметане (120 мл) при 0 С добавляли трифторуксусную кислоту (60 мл). Этой реакционной смеси давали нагреться до температуры окружающей среды и перемешивали в течение 2 ч. Эту реакционную смесь охлаждали до 0 С и медленно гасили добавлением 3 Н водного гидроксида натрия до щелочного рН. Полученную в результате смесь экстрагировали дихлорметаном (2 х). Объединенные органические фазы высушивали над сульфатом магния, фильтровали и концентрировали в вакууме с получением соединения, указанного в заголовке (22 г, выход 78%).- 19006079 тетрагидрофуране (160 мл) при 0 С добавляли раствор алюмогидрида лития (1M в тетрагидрофуране,373 мл, 373 ммоль) по каплям в течение 40 мин. Затем эту реакционную смесь кипятили с обратным холодильником в течение 4 ч, охлаждали до температуры окружающей среды и медленно гасили водой. Полученную в результате смесь фильтровали через подложку из целита и фильтрационную лепешку промывали этилацетатом. Фильтрат затем концентрировали, разбавляли этилацетатом и промывали насыщенным водным кислым карбонатом натрия. Органический слой отделяли, высушивали над сульфатом магния, фильтровали и концентрировали в вакууме с получением соединения, указанного в заголовке (17,7 г, выход 91%).(2R,5S)-2-Хлор-1-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]этанон. К раствору (2R,5S)-1-(4-фторбензил)-2,5-диметилпиперазина (2,5 г, 11,25 ммоль) в сухом дихлорметане (11 мл) при 0 С добавляли триэтиламин (1,57 мл, 11,2 ммоль), а затем хлорацетилхлорид (0,858 мл, 11,2 ммоль). Полученную в результате реакционную смесь затем перемешивали в течение 30 мин. Затем эту реакционную смесь фильтровали через подложку из целита, промывали дихлорметаном и полученный в результате фильтрат концентрировали с получением желтого масла. Хроматографией на силикагеле получили соединение, указанное в заголовке (2,84 г, выход 86%). Метиловый эфир(2,54 г, 18,4 ммоль) и иодид калия (1,52 г, 9,2 ммоль). Полученную в результате смесь перемешивали при кипячении с обратным холодильником в течение 6 ч. Затем эту реакционную смесь охлаждали, разбавляли этилацетатом и промывали рассолом. Органические фазы высушивали над сульфатом магния, фильтровали и концентрировали в вакууме с получением оранжевого масла. Хроматографией на силикагеле получили соединение, указанное в заголовке (4,1 г, выход 100%).(2R,5S)-5-Хлор-2-2-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]-2-оксоэтоксибензойная кислота. К раствору метилового эфира (2R,5S)-5-хлор-2-2-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]-2 оксоэтоксибензойной кислоты (4,12 г, 9,18 ммоль) в тетрагидрофуране (10 мл), метаноле (10 мл) и воде(4 мл) добавляли моногидрат гидроксида лития (1,93 г, 45,9 ммоль). Полученную в результате смесь перемешивали в течение ночи при температуре окружающей среды. Затем эту реакционную смесь концентрировали, разбавляли 1 н. соляной кислотой и экстрагировали дихлорметаном (2 х). Органические слои объединяли, высушивали над сульфатом магния, фильтровали и концентрировали с получением белой пены. Растиранием в дихлорметане и диэтиловом эфире получили соединение, указанное в заголовке(0,039 г, 0,318 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (0,061 г, 0,318 ммоль) и трет-бутил-1-пиперазинкарбоксилат (0,041 г, 0,222 ммоль). Полученную в результате смесь перемешивали в течение ночи при температуре окружающей среды. Затем ее разбавляли дихлорметаном и промывали рассолом. Органические фазы высушивали над сульфатом магния, фильтровали и концентрировали с получением прозрачного масла. Хроматографией на силикагеле получили соединение, указанное в заголовке (0,110 г, выход 85%).(2R,5S)-2-[4-Хлор-2-(пиперазин-1-карбонил)фенокси]-1-[4-(4-фторбензил)-2,5-диметилпиперазин-1 ил]этанон. К раствору трет-бутилового эфира (2R,5S)-4-(5-xлop-2-2-[4-(4-фтopбензил)-2,5-диметилпиперазин-1-ил]-2-оксоэтоксибензоил)пиперазин-1-карбоновой кислоты (0,110 г, 0,182 ммоль) в дихлорэтане (10 мл) добавляли трифторуксусную кислоту (5 мл). Эту реакционную смесь перемешивали в течение 2 ч при температуре окружающей среды. Затем эту реакционную смесь разбавляли дихлорметаном и промывали 1 н. водным гидроксидом натрия. Органические фазы высушивали над сульфатом магния,фильтровали и концентрировали с получением соединения, указанного в заголовке (0,080 г, выход 87%). Соединения, указанные в заголовках примеров 2-12, получили способом, аналогичным описанному в примере 1.(2R)-3-Амино-N-(5-хлор-2-2-[4-(4-фторбензил)-2-метилпиперазин-1-ил]-2-оксоэтоксифенил) пропионамид. трет-Бутиловый эфир [2-(5-хлор-2-2-[4-(4-фторбензил)-2-метилпиперазин-1-ил]-2-оксоэтокси фенилкарбамоил)этил]карбаминовой кислоты. К раствору 2-(2-амино-4-хлорфенокси)-1-[4-(4-фторбензил)-2-метилпиперазин-1-ил]этанона (0,066 г, 0,17 ммоль) в метиленхлориде (2 мл) при температуре окружающей среды добавляли Nметилморфолин (0,025 мл, 0,23 ммоль), 3-трет-бутоксикарбониламинопропионовую кислоту (0,044 г,0,23 ммоль) и О-бензотриазол-1-ил-N,N,N',N'-тетраметилурония гексафторфосфат (0,076 г, 0,20 ммоль). Полученный раствор перемешивали при температуре окружающей среды в течение 60 ч, затем концентрировали. Радиальной хроматографией (2 мм пластинка) получили соединение, указанное в заголовке(2R)-3-Амино-N-(5-хлор-2-2-[4-(4-фторбензил)-2-метилпиперазин-1-ил]-2-оксоэтоксифенил) пропионамид. К раствору трет-бутилового эфира [2-(5-хлор-2-2-[4-(4-фторбензил)-2-метилпиперазин-1-ил]-2 оксоэтоксифенилкарбамоил)этил]карбаминовой кислоты (0,110 г, 0,2 ммоль) в метиленхлориде (3 мл) добавляли трифторуксусную кислоту (0,50 мл). Эту реакционную смесь перемешивали в течение 2 ч при температуре окружающей среды, затем разбавляли насыщенным водным кислым карбонатом натрия. Эту смесь экстрагировали метиленхлоридом и объединенные органические фазы высушивали над суль- 21006079 фатом магния, фильтровали и концентрировали в вакууме с получением соединения, указанного в заголовке (0,069 г). Соединения, указанные в заголовках примеров 14-19, получили способом, аналогичным описанному в примере 13.(2R,5S)-2-(4-Хлор-2-нитрофенокси)-1-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]этанон. К раствору метилового эфира (2R,5S)-5-хлор-2-2-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]-2 оксоэтоксибензойной кислоты (1,0 г, 3,35 ммоль) в бутаноне (35 мл) добавляли 2-нитро-4-хлорфенол(0,639 г, 3,69 ммоль), карбонат калия (0,925 г, 6,7 ммоль) и иодид калия (0,556 г, 3,35 ммоль). Эту реакционную смесь нагревали с обратным холодильником в течение ночи. Затем эту реакционную смесь охлаждали, разбавляли водой и экстрагировали этилацетатом. Объединенные органические фазы высушивали над сульфатом магния, фильтровали и концентрировали с получением оранжевого масла. Хроматографией на силикагеле получили соединение, указанное в заголовке (1,35 г, выход 93%).(2R,5S)-2-(2-Амино-4-хлорфенокси)-1-[4-(4-фторбензил)-2.5-диметилпиперазин-1-ил]этанон. К раствору (2R,5S)-2-(4-хлор-2-нитрофенокси)-1-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил] этанона (2,2 г, 5,05 ммоль) в этаноле (50 мл) в колбе Парра добавляли 5% платину на угле (2,2 г). Эту реакционную смесь обрабатывали газообразным водородом (241,32 кПа (35 фунт/кв.дюйм в течение 30 мин. Затем эту реакционную смесь фильтровали через целит и промывали этанолом. Фильтрат концентрировали с получением коричнево-рыжей пены. Хроматографией на силикагеле получили соединение,указанное в заголовке (1,42 г, выход 70%). 4-Нитрофениловый эфир (2R,5S)-(5-хлор-2-2-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]-2 оксоэтоксифенил)карбаминовой кислоты.- 22006079 К раствору (2R,5S)-2-(2-амино-4-хлорфенокси)-1-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил] этанона (0,150 г, 0,37 ммоль) в дихлорметане (7 мл) добавляли пиридин (0,066 мл, 0,82 ммоль), а затем 4 нитрофенилхлорформиат (0,075 г, 0,41 ммоль). Эту реакционную смесь перемешивали при температуре окружающей среды в течение 3,5 ч. Реакционную смесь концентрировали с последующей хроматографией на силикагеле с получением соединения, указанного в заголовке (0,153 г, выход 74%).(2R,5S)-1-(2-Аминоэтил)-3-(5-хлор-2-2-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]-2-оксоэтоксифенил)мочевина. К раствору 4-нитрофенилового эфира (2R,5S)-(5-хлор-2-2-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]-2-оксоэтоксифенил)карбаминовой кислоты (0,206 г, 0,37 ммоль) в сухом метаноле (6 мл) добавляли этилдиамин (0,05 мл, 0,814 ммоль). Эту реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Эту реакционную смесь концентрировали и подвергали хроматографии на силикагеле с получением соединения, указанного в заголовке (0,115 г, выход 63%). Соединения, указанные в заголовках примеров 21-27, получили способом, аналогичным описанному в примере 20.(2R)-2-Аминоэтансульфоновой кислоты (5-хлор-2-2-[4-(4-фторбензил)-2-метилпиперазин-1-ил]-2 оксоэтоксифенил)амид. 2-(1,3-Диоксо-1,3-дигидроизоиндол-2-ил)этансульфоновой кислоты (5-хлор-2-2-[4-(4-фторбензил)2-метилпиперазин-1-ил]-2-оксоэтоксифенил)амид. К раствору 2-(2-амино-4-хлорфенокси)-1-[4-(4-фторбензил)-2-метилпиперазин-1-ил]этанона (0,050 г, 0,13 ммоль) в метиленхлориде (1 мл) при температуре окружающей среды добавляли триэтиламин(0,027 мл, 0,19 ммоль) и 2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этансульфонилхлорид (0,045 г, 0,17 ммоль). Эту реакционную смесь перемешивали в течение ночи при температуре окружающей среды. Добавляли дополнительное количество триэтиламина (0,027 мл, 0,19 ммоль) и 2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этансульфонилхлорида (0,045 г, 0,17 ммоль). Эту реакционную смесь перемешивали в течение 1 ч, затем добавляли дополнительное количество триэтиламина (0,055 мл, 0,34 ммоль). Эту реакционную смесь перемешивали в течение 1 ч, затем добавляли дополнительное количество 2-(1,3 диоксо-1,3-дигидроизоиндол-2-ил)этансульфонилхлорида (0,090 г, 0,34 ммоль). После перемешивания в течение 1 дополнительного ч, эту реакционную смесь обрабатывали насыщенным водным кислым карбонатом натрия и экстрагировали метиленхлоридом (3 х). Объединенные органические фазы высушивали над сульфатом магния, фильтровали и концентрировали в вакууме. Очисткой с помощью радиальной хроматографии (2 мм пластинка) получили соединение, указанное в заголовке (0,030 г).(2R)-2-Аминоэтансульфоновой кислоты (5-хлор-2-2-[4-(4-фторбензил)-2-метилпиперазин-1-ил]-2 оксоэтоксифенил)амид. К раствору 2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этансульфоновой кислоты (5-хлор-2-2-[4-(4 фторбензил)-2-метилпиперазин-1-ил]-2-оксоэтоксифенил)амида (0,030 г, 0,048 ммоль) в ЕtOН (1 мл) при температуре окружающей среды добавляли гидрат гидразина (0,025 мл). Эту реакционную смесь перемешивали в течение ночи при температуре окружающей среды, затем разбавляли водой и экстрагировали метиленхлоридом (2 х). Объединенные органические фазы промывали насыщенным водным солевым раствором и высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме. Очисткой с помощью радиальной хроматографии (2 мм пластинка) получили соединение, указанное в заголовке (0,014 г). Соединения, указанные в заголовках примеров 29-34, получили способом, аналогичным описанному в примере 28.(2R)-N-(5-Хлор-2-2-[4-(4-фторбензил)-2-метилпиперазин-1-ил]-2-оксоэтоксифенил)цианогуанидин. 1-(5-Хлор-2-2-[4-(4-фторбензил)-2-метилпиперазин-1-ил]-2-оксоэтоксифенил)-N-циано-S-метилизотиомочевина. К раствору 2-(2-амино-4-хлорфенокси)-1-[4-(4-фторбензил)-2-метилпиперазин-1-ил]этанона (0,30 г,0,77 ммоль) в тетрагидрофуране (5 мл) при температуре окружающей среды добавляли гидрид натрия(0,029 г, 1,22 ммоль) и эту реакционную смесь перемешивали в течение 30 мин. К этой смеси добавлялиS,S1-диметил-N-цианодитиоиминокарбонат (0,168 г, 1,15 ммоль), и эту смесь нагревали с обратным холодильником в течение ночи. Эту реакционную смесь охлаждали и гасили насыщенным водным хлоридом аммония. Эту смесь экстрагировали ЕtOАс и объединенные органические фазы высушивали надNa2SO4, фильтровали и концентрировали в вакууме. Хроматографией на силикагеле получили соединение, указанное в заголовке (0,350 г).(2R)-N-(5-Хлор-2-2-[4-(4-фторбензил)-2-метилпиперазин-1-ил]-2-оксоэтоксифенил)цианогуанидин. К раствору 1-(5-хлор-2-2-[4-(4-фторбензил)-2-метилпиперазин-1-ил]-2-оксоэтоксифенил)-Nциано-S-метилизотиомочевины (0,045 г, 0,092 ммоль) в ЕtOН (1 мл) добавляли гидроксид аммония (0,100 мл), и полученный в результате раствор встряхивали при 60 С в течение ночи. Эту сырую реакционную смесь очищали непосредственно с помощью радиальной хроматографии (2 мм пластинка) с получением соединения, указанного в заголовке (0,027 г). Соединения, указанные в заголовках примеров 36-38, получили способом, аналогичным описанному в примере 35.(2R,5S)-5-Хлор-2-2-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]-2-оксоэтоксибензальдегид. К раствору 2-хлор-1-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]этанона (2,87 г, 9,6 ммоль) в ДМФ (20 мл) добавляли 5-хлорсалицилальдегид (1,65 г, 10,5 ммоль), карбонат калия (2,64 г, 19,2 ммоль) и иодид калия (1,59 г, 9,6 ммоль). Полученную в результате смесь нагревали до 100 С в течение 12 ч. Эту реакционную смесь охлаждали, разбавляли насыщенным водным рассолом и экстрагировали этилацетатом (3 х). Объединенные органические фазы высушивали над сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме с получением сырого продукта. Очисткой с помощью хроматографии на силикагеле (15% EtOAc/гексаны) получили соединение, указанное в заголовке (3,40 г, выход 85%).(2R,5S)-5-хлор-2-2-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]-2-оксоэтокси бензальдегида (0,100 г, 0,25 ммоль) в смеси дихлорэтана и уксусной кислоты в соотношении 10:1 (2,2 мл) добавляли (диэтиламино)этиламин (0,088 мл, 0,625 ммоль), и полученный в результате раствор перемешивали в течение 1 ч при температуре окружающей среды. К этому раствору добавляли цианоборгидрид натрия (0,0094 г, 0,15 ммоль), и эту реакционную смесь перемешивали в течение ночи при температуре окружающей среды. При завершении добавляли воду и эту смесь подщелачивали твердым бикарбонатом натрия (рН 10). Продукт экстрагировали дихлорметаном (2 х) и диэтиловым эфиром (2 х). Объединенные органические фазы высушивали над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматографией на силикагеле получили соединение, указанное в заголовке (0,039 г, х 30% выход). Соединения, указанные в заголовках примеров 40-62, получили способом, аналогичным описанному в примере 39.(2R,5S)-5-хлор-2-2-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]-2-оксоэтокси бензальдегида (1,86 г, 4,44 ммоль) в МеОН (20 мл) добавляли ацетат аммония (3,42 г, 44 ммоль) и циано- 27006079 боргидрид натрия (0,195 г, 3,1 ммоль). Полученную в результате смесь перемешивали в течение ночи при температуре окружающей среды. Эту реакцию гасили концентрированной соляной кислотой и концентрировали в вакууме. Остаток растворяли в воде и подщелачивали водным 3 н. NaOH (pH10). Продукт экстрагировали дихлорметаном (2 х) и этилацетатом (2 х). Объединенные органические фазы высушивали над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматографией на силикагеле получили соединение, указанное в заголовке (1,29 г, выход 69%).(0,021 г, 0,15 ммоль) и триэтиламин (0,036 мл, 0,36 ммоль). После того, как эту реакционную смесь перемешивали в течение 48 ч, раствор разбавляли насыщенным водным бикарбонатом натрия и экстрагировали дихлорметаном (3 х). Объединенные органические фазы высушивали над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматографией на силикагеле получили соединение, указанное в заголовке (0,063 г, выход 80%). Соединения, указанные в заголовках примеров 64-85, получили способом, аналогичным описанному в примере 63.(2R,5S)-5-хлор-2-2-[4-(4-фторбензил)-2,5-диметилпиперазин-1-ил]-2-оксоэтокси бензиламина (0,200 г, 0,477 ммоль) в метиленхлориде (10 мл) добавляли пиридин (0,077 мл, 0,954 ммоль) и 4-нитрофенилхлорформиат (0,097 г, 0,525 ммоль). Полученную в результате смесь перемешивали в течение 1 ч при температуре окружающей среды, а затем концентрировали в вакууме. Остаток (0,055 г,0,094 ммоль) растворяли в метаноле (1 мл). Добавляли N-ацетилэтилендиамин (0,019 мл, 0,188 ммоль) и эту реакционную смесь перемешивали при комнатной температуре в течение ночи. Эту реакционную смесь концентрировали в вакууме и хроматографией на силикагеле получили соединение, указанное в заголовке (0,019 г, выход 27%). Соединения, указанные в заголовках примеров 87-90, получали способом, аналогичным описанному в примере 86.
МПК / Метки
МПК: A61K 31/495, C07D 295/185, A61P 29/00
Метки: производные, новые, пиперазиновые
Код ссылки
<a href="https://eas.patents.su/30-6079-novye-piperazinovye-proizvodnye.html" rel="bookmark" title="База патентов Евразийского Союза">Новые пиперазиновые производные</a>
Пиперазиновые производные и их применение в качестве противовоспалительных агентов

Номер патента: 4038
Опубликовано: 25.12.2003
Авторы: Вэй Го Пин, Чжэн Вэй, Чаннам Амин Ф., Моррис Майкл М., Сюй Вэй, Букман Брэд О., Мей Карен Б., Хесселгессер Джоузеф Е., Хоурук Ричард, Ислам Имадул, Нг Хауард П., Лян Мейна, Бауман Джон Г., Монахан Шон Д.
МПК: A61K 31/496, A61P 29/00, C07D 241/04...
Метки: пиперазиновые, противовоспалительных, качестве, применение, производные, агентов
Формула / Реферат:
1. Соединение формулы (Ia) где R1a обозначает один или несколько заместителей, выбранных независимо друг от друга из группы, включающей гало, (C1-C8)алкил, (C3-C10)циклоалкил, (C3-C10)циклоалкиламино(C1-C8)алкил, гало(C1-C8)алкил, гидрокси(C2-C8)алкенил, гидрокси(C2-C8)алкинил, (гидрокси)-(необязательно замещенный фенил или нафтил) (C1-C8)алкил, циано(C1-C8)алкил, гало(C1-C8)алкилкарбониламино(C1-C8)алкил, (C1-C8)алкокси(C1-C8)алкил,...
Фармацевтические композиции, содержащие производные 3-аминоазетидина, новые производные и способ их получения

Номер патента: 4649
Опубликовано: 24.06.2004
Авторы: Ашар Даниель, Букерель Жан, Филош Брюно, Иттэнжер Огюстэн, Майерс Майкл, Бушар Эрве, Гризони Серж
МПК: C07D 205/04, A61K 31/397, A61P 25/00...
Метки: фармацевтические, способ, получения, новые, содержащие, композиции, производные, 3-аминоазетидина
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы в которой R1 обозначает радикал -NHCOR4 или -N(R5)-Y-R6, Y обозначает CO или SO2, R2 и R3, одинаковые или разные, обозначают либо ароматический радикал, выбранный из фенила, нафтила и инденила, которые могут быть незамещенными или замещенными одним или несколькими заместителями: галоген, алкил, алкокси, формил, гидрокси, трифторметил, трифторметокси,...
Новые производные дигидроксигексановой кислоты

Номер патента: 3137
Опубликовано: 27.02.2003
Авторы: Кэт Джон Чарлз, Браун Мэттью Фрэнк, Посс Кристофер Стенли
МПК: C07C 231/00, A61K 31/498, A61P 29/00...
Метки: новые, дигидроксигексановой, кислоты, производные
Формула / Реферат:
1. Соединение формулы где указанное соединение представляет собой [4(R)-карбамоил-1(S)-(3-хлорбензил)-2(S),7-дигидрокси-7-метилоктил]амид хиноксалин-2-карбоновой кислоты; (IS)-бензил-4(R)-карбамоил-2(S),7-дигидрокси-7-метилоктил)амид 7,8-дифторхинолин-3-карбоновой кислоты; (1(S)-бензил-4(R)-карбамоил-2(S),7-дигидрокси-7-метилоктил)амид 6,7,8-трифторхинолин-3-карбоновой кислоты;...
Новые производные камптотецина

Номер патента: 2211
Опубликовано: 28.02.2002
Авторы: Савада Сейго, Фурута Томио, Иаегаси Такаси, Огава Таканори
МПК: A61K 31/4745, A61P 35/00, C07D 471/14...
Метки: производные, новые, камптотецина
Формула / Реферат:
Производные камптотецина общей формулы (1) где R1 является атомом водорода или алкильной группой с 1-6 атомами углерода, R2 означает одинаковые или различные присутствующие в количестве от 0 до 4 алкильные группы с 1-6 атомами углерода, атом галогена, алкоксильную или гидроксильную группу, R3 является низшей алкиламино, динизшей алкиламино, ариламино, циклической амино или низшей алкоксильной группами, и их соли. ...
Лекарственные средства, содержащие производные полигидроксиалкилпиразина, новые производные полигидроксиалкилпиразина и их получение

Номер патента: 3642
Опубликовано: 28.08.2003
Авторы: Эвер Мишель, Филош Брюно, Башиард Жорж, Миньяни Серж, Карри Жан-Кристоф
МПК: A61P 3/10, C07D 241/12, A61K 31/495...
Метки: содержащие, средства, получение, полигидроксиалкилпиразина, лекарственные, производные, новые
Формула / Реферат:
1. Лекарственное средство, содержащее в качестве активного начала, по меньшей мере, одно соединение формулы в которой либо R2 обозначает цепь -CH2-(CHOH)2-CH3, a R3 обозначает атом водорода, либо R2 обозначает атом водорода, a R3 обозначает цепь -CH2-(CHOH)2-CH3, или один из его стереоизомеров или одну из его солей с фармацевтически приемлемой минеральной или органической кислотой. 2. Лекарственное средство по п.1, содержащее в качестве...
Предыдущий патент: Устройство и способ для преобразования солнечной энергии
Следующий патент: Замещенные в положении 6 индолиноны, их получение и их применение в качестве лекарственных средств
Случайный патент: Способ определения точки входа текучего компонента в трубопровод