Замещенные, азотсодержащие гетероциклы в качестве ингибиторов протеинкиназы р38
Номер патента: 2855
Опубликовано: 31.10.2002
Авторы: Уилсон Кейт П., Салитуро Франческо Джеральд, Су Майкл, Галулло Винсент П., Мёрко Марк А., Даффи Джон Патрик, Кочран Джон Е., Харрингтон Эдмунд Мартин, Бемис Гай В.






















Формула / Реферат
1. Соединение формулы

или его фармацевтически приемлемая соль,
где каждый из Q1 и Q2 независимо выбран из систем 5-6-членных ароматических карбоциклических или гетероциклических колец или систем 8-10-членных бициклических колец, состоящих из ароматических карбоциклических колец, ароматических гетероциклических колец или сочетания ароматического карбоциклического кольца и ароматического гетероциклического кольца;
где Q1 замещен 1-4 заместителями, которые независимо выбраны из галогена; C1-С3-алкила, возможно замещенного NR'2, OR', CO2R' или CONR'2; O-(C1-С3)-алкила, возможно замещенного NR'2, OR', CO2R' или CONR'2; NR'2; ОСF3; СF3; NO2; CO2R'; CONR'; SR'; S(O2)NR'2; SСF3; CN; N(R')C(O)R4; N(R')С(O)OR4; N(R')С(О)С(O)R4; N(R')S(O2)R4; N(R')R4; N(R4)2; OR4; OC(O)R4; ОР(O)3Н2 или N=C-NR'2, и
Q2 возможно замещен 1-4 заместителями, каждый из которых независимо выбран из галогена; C1-С3-линейного или разветвленного алкила, возможно замещенного NR'2, OR', CO2R', S(O2)NR'2, N=C-NR'2, R3 или CONR'2; O-(C1-С3)-алкила, возможно замещенного NR'2, OR', CO2R', S(O2)NR'2, N=C-NR'2, R3 или CONR'2; NR'2; ОСF3; СF3; NO2; CO2R'; CONR'; R3; OR3; NR3; SR3; C(O)R3; C(O)N(R')R3; C(O)OR3; SR'; S(O2)NR'2; SСF3; N=C-NR'2 или CN;
где R' выбран из водорода, (C1-С3)-алкила; (С2-С3)-алкенила или алкинила; фенила или фенила, замещенного 1-3 заместителями, независимо выбираемыми из галогена, метокси, циано, нитро, амино, гидрокси, метила или этила;
R3 выбран из системы 5-6-членных ароматических карбоциклических или гетероциклических колец; и
R4 означает (C1-C4)-алкил, возможно замещенный NR'2, OR', CO2R', С(О)NR'2 или SO2NR22; или систему 5-6-членных карбоциклических или гетероциклических колец, возможно замещенную NR'2, OR', CO2R', С(О)NR'2 или S(O2)NR22,
Х выбран из -S-, -O-, -S(O2)-, -S(O)-, S(O2)-N(R2)-, -N-(R2)-S(O2)-, -N(R2)-C(O)O-, -O-C(O)-N(R2)-, -C(O)-, -C(O)O-, -O-C(O)-, -C(O)-N(R2)-, - N(R2)-C(O)-, -N(R2)-, -C(R2)2- или -С(OR2)2-;
каждый R независимо выбран из водорода, -R2, -N(R2)2, -OR2, SR2, -C(O)-NR22, -S(O2)-NR22 или -С(O)-OR2, где два смежных R возможно связаны друг с другом и вместе с каждым Y, с которым они соответственно связаны, образуют 4-8 членное карбоциклическое или гетероциклическое кольцо, или когда Y означает N, R, присоединенный к нему, означает неподеленную электронную пару;
R2 выбран из водорода, (C1-С3)-алкила или (C1-С3)-алкенила; каждый возможно замещен -N(R')2, -OR', -SR', -C(O)-NR'2, -S(O2)-NR'2, -C(O)-OR' или R3,
Y выбран из N или С;
А, если присутствует, выбран из N или CR';
n равно 0 или 1, причем, когда n равно 0, связь между атомом азота в кольце и атомом углерода, связанным с группой X-Q2, означает простую связь; и
R1 выбран из водорода, (C1-С3)-алкила, ОН или O-(С1-С3)-алкила.
2. Соединение формулы

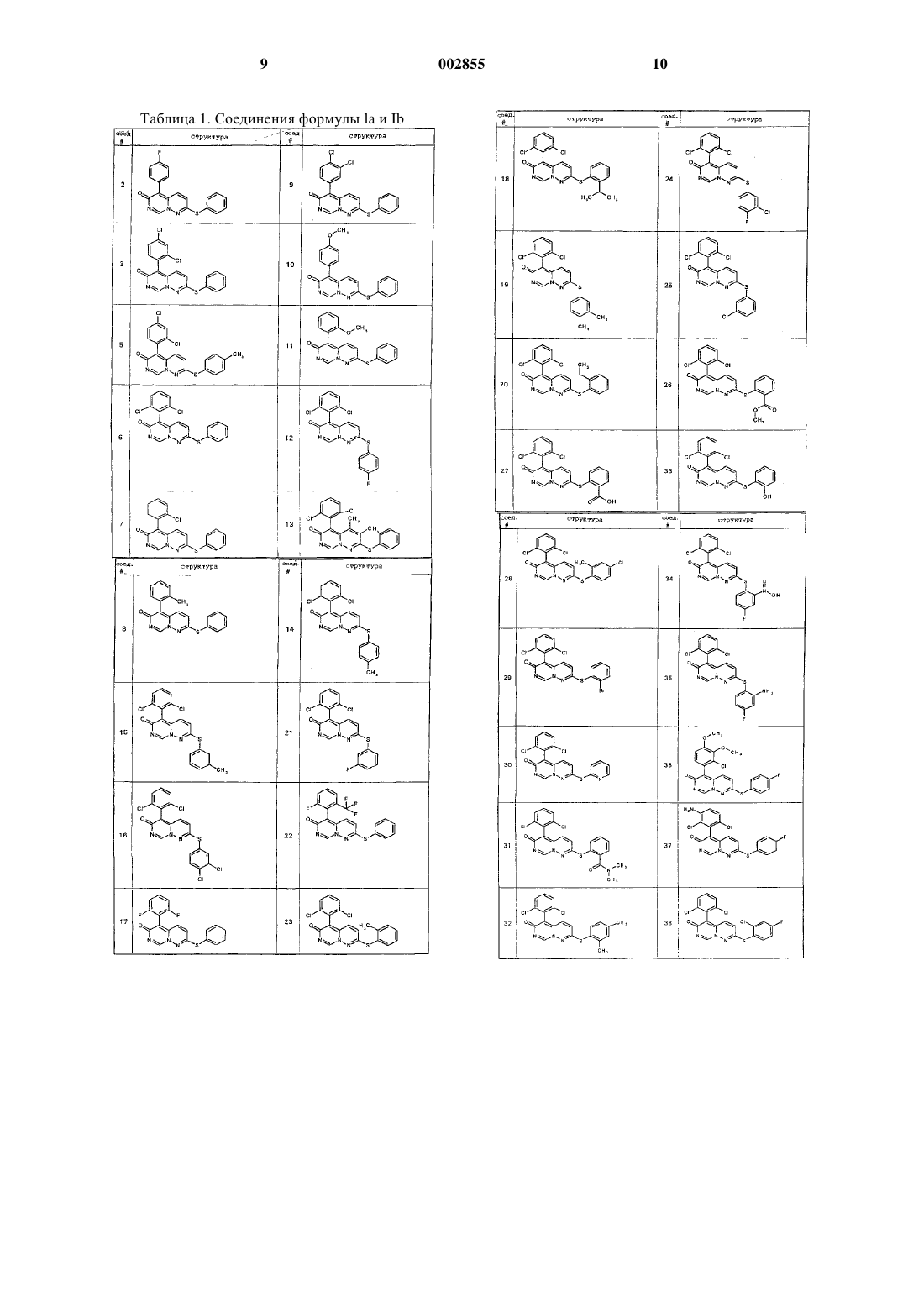
или его фармацевтически приемлемая соль, где Q1, Q2, R1, R и Y имеют значения, указанные в п.1.
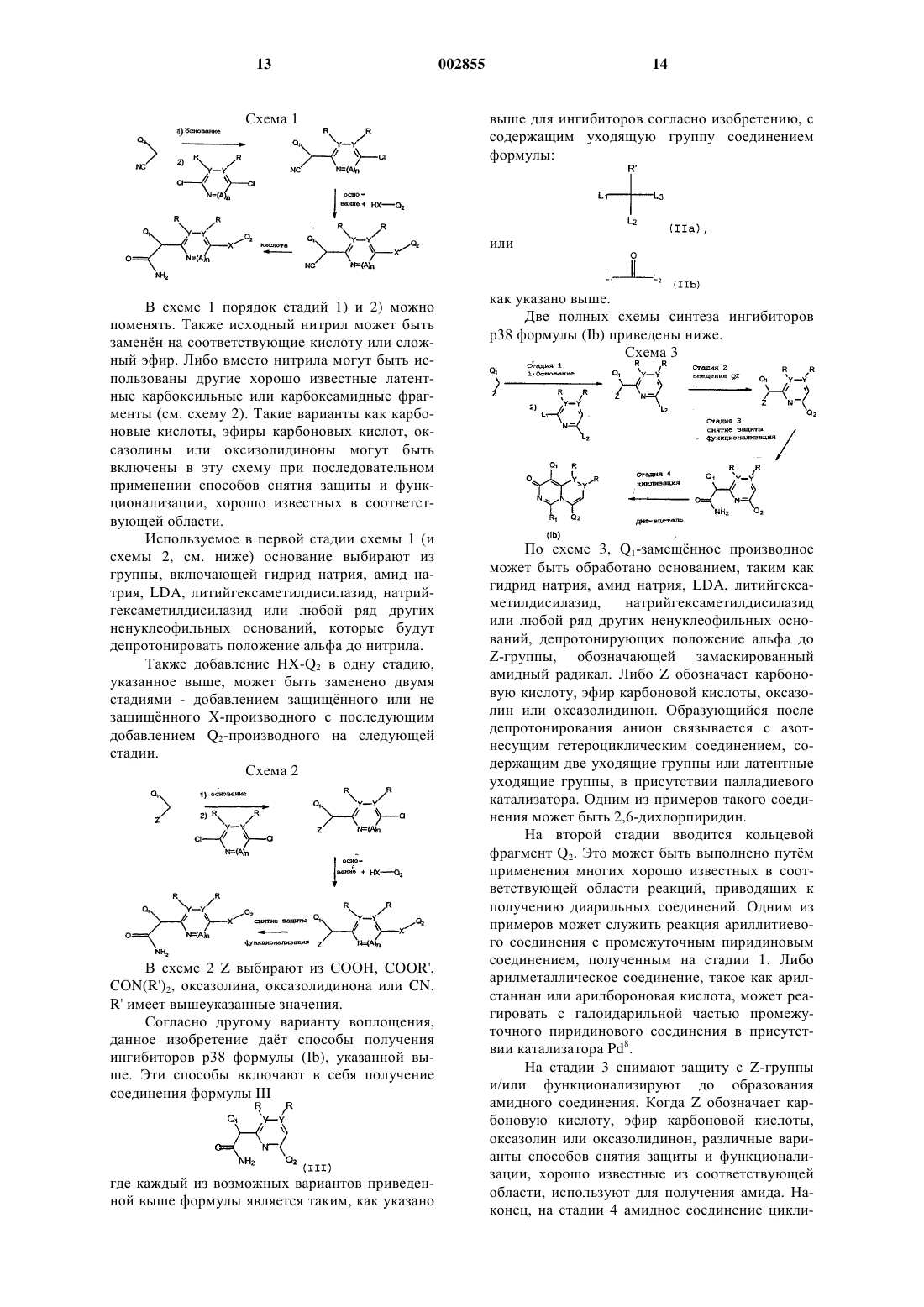
3. Соединение формулы

или его фармацевтически приемлемая соль, где A, Q1, Q2, R, R', X, Y и n имеют значения, указанные для соединений формул Iа и Ib; и R5 выбран из водорода, -CR'2OH, -C(O)R4, -C(O)OR4, -СR'2OРО3Н2 и -РО3Н2.
4. Соединение формулы

или его фармацевтически приемлемая соль, где A, Q1, Q2, R, R', R5, X, Y и n имеют значения, указанные в п.3.
5. Соединение формулы

или его фармацевтически приемлемая соль,
где Q3 означает систему 5-6-членных ароматических карбоциклических или гетероциклических колец или систему 8-10-членных бициклических колец, включающую в себя ароматические карбоциклические кольца, ароматические гетероциклические кольца или сочетание ароматического карбоциклического кольца и ароматического гетероциклического кольца, где Q3 замещен 1-4 заместителями, каждый из которых независимо выбран из галогена, (C1-С3)-алкила, возможно замещенного N(R')2, OR', CO2R' или CONR'2; O-(C1-С3)-алкила, возможно замещенного N(R')2, OR', CO2R' или CONR'2; NR'2; ОСF3; СF3; NO2; CO2R'; CONR'; SR'; S(O2)NR'2; SСF3; CN; N(R')C(O)R4; N(R')C(O)OR4; N(R')C(O)C(O)R4; N(R')S(O2)R4; N(R')R4; NR42; OR4; ОС(O)R4; ОР(О)3H2 или N=C-NR'2, и A, Q2, R, R', X, Y и n имеют значения, указанные в п.1.
6. Соединение формулы

или его фармацевтически приемлемая соль, где Q1, Q2, R, Y, X, А и n имеют значения, указанные в п.1.
7. Соединение формулы

или его фармацевтически приемлемая соль, где Q2, Q3, R, Y и n имеют значения, указанные в п.5.
8. Соединение формулы

или его фармацевтически приемлемая соль, где Q1 Q2 R, Y, X, А и n имеют значения, указанные в п.6.
9. Соединение по любому из пп.1-8, где Q1 выбран из фенила или пиридила, содержащего от 1 до 3 заместителей, независимо выбираемых из хлора, фтора, брома, -СН3, -ОСН3, -ОН, -СF3, -ОСF3, -O(СН2)2СН3, NH2, 3,4-метилендиокси, -N(CH3)2, -NН-S(O)2-фенила, -NH-C(O)O-СН2-4-пиридина, -NH-С(O)СН2-морфолина, -NH-C(O)CH2-N(СН3)2, -NH-C(О)СН2-пиперазина, -NH-C(O)СН2-пирролидина, -NH-C(O)С(O)-морфолина, -NH-C(O)С(O)-пиперазина, -NH-C(O)С(O)-пирролидина, -О-С(О)CH2-N(СН3)2 или -О-(СН2)-N(CH3)2, и где, по меньшей мере, один из указанных заместителей находится в ортоположении.
10. Соединение по п.9, уфх Q1 содержит, по меньшей мере, два заместителя, оба из которых находятся в ортоположении.
11. Соединение по п.9, где Q1 выбран из




12. Соединение по п.11, где Q1 выбран из 2-фтор-6-трифторметилфенила, 2,6-дифторфенила, 2,6-дихлорфенила, 2-хлор-4-гидроксифенила, 2-хлор-4-аминофенила, 2,6-дихлор-4-аминофенила, 2,6-дихлор-3-аминофенила, 2,6-диметил-4-гидроксифенила, 2-метокси-3,5-дихлор-4-пиридил, 2-хлор-4,5-метилендиоксифенила, или 2-хлор-4-(N-2-морфолин-ацетамид)фенила.
13. Соединение по любому из пп.1-8, где Q2 выбран из фенила или пиридила и где Q2, возможно, содержит до 3 заместителей, каждый из которых независимо выбран из хлора, фтора, брома, метила, этила, изопропила, -ОСН3, -ОН, -NH2, -СF3, -ОСF3, -SСН3, -ОСН3, -С(O)ОН, -С(O)ОСН3, -CH2NH2, -N(CH3)2, -СН2-пирролидина и -СН2OН.
14. Соединение по п.13, где, Q2 выбран из



незамещенного 2-пиридила или незамещенного фенила.
15. Соединение по п.14, где Q2 выбран из фенила, 2-изопропилфенила, 3,4-диметилфенила, 2-этилфенила, 3-фторфенила, 2-метилфенила, 3-хлор-4-фторфенила, 3-хлорфенила, 2-карбометоксифенила, 2-карбоксифенила, 2-метил-4-хлорфенила, 2-бромфенила, 2-пиридила, 2-метиленгидроксифенила, 4-фторфенила, 2-метил-4-фторфенила, 2-хлор-4-фторфенила, 2,4-дифторфенила, 2-гидрокси-4-фторфенила или 2-метиленгидрокси-4-фторфенила.
16. Соединение по любому из пп.1-8, где Х выбран из -S-, -О-, -S(O2)-, -S(O)-, -NR-, -C(R2)- или -С(O)-.
17. Соединение по п.15, где Х означает S.
18. Соединение по любому из пп.1-8, где n означает 1 и А означает N.
19. Соединение по любому из пп.1-8, где каждый Y означает С.
20. Соединение по п.19, где каждый R, связанный с Y, независимо выбран из водорода или метила.
21. Соединение по п.1, где указанное соединение выбрано из любого из соединений 2-3, или 5-53, приведенных в табл. 1.
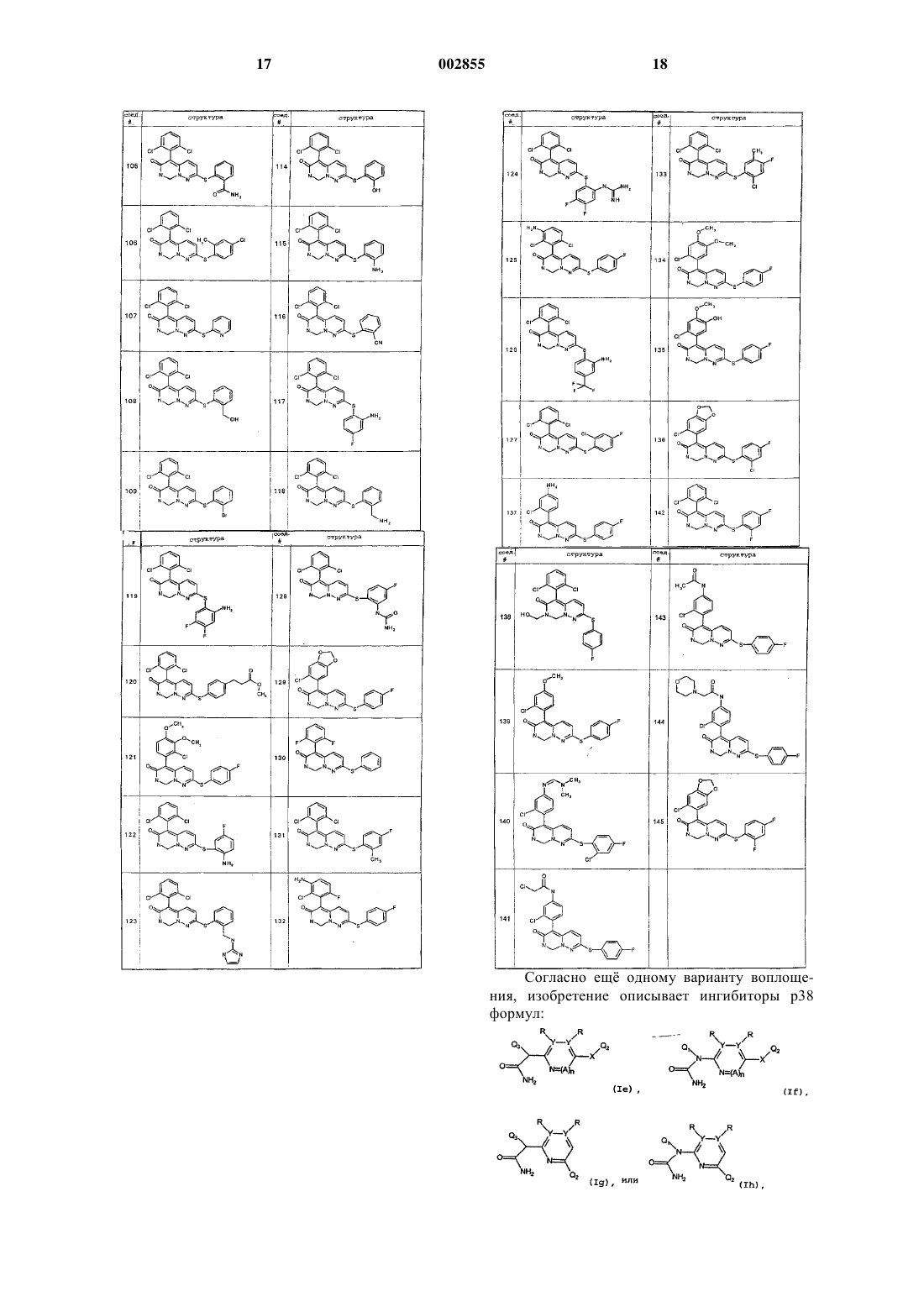
22. Соединение по п.3, где указанное соединение выбрано из любого из соединений 101-145, приведенных в табл. 2.
23. Соединение по любому из пп.5-7, где Q3 замещен 2-4 заместителями, где, по меньшей мере, один из указанных заместителей присутствует в ортоположении относительно точки присоединения Q3 к остатку ингибитора.
24. Соединение по п.23, где оба ортоположения заняты одним из указанных, независимо выбираемых заместителей.
25. Соединение по п.24, где Q3 означает моноциклическое карбоциклическое кольцо; и каждый из указанных ортозаместителей на Q3 независимо выбран из галогена или метила.
26. Соединение по п.24, где Q3 содержит от 1 до 2 заместителей в дополнение к указанным ортозаместителям, причем указанные дополнительные заместители независимо выбраны из NR'2, OR', CO2R' CN, N(R')C(O)R4; N(R')С(O)OR4; N(R')C(O)C(O)R4; N(R')S(O2)R4; N(R')R4; N(R4)2; OR4; OC(O)R4; ОР(O)3Н2 или N=C-N(R')2.
27. Соединение по п.5, где указанное соединение является соединением формулы Iе и выбрано из соединений 201 или 203-209, приведенных в табл. 3.
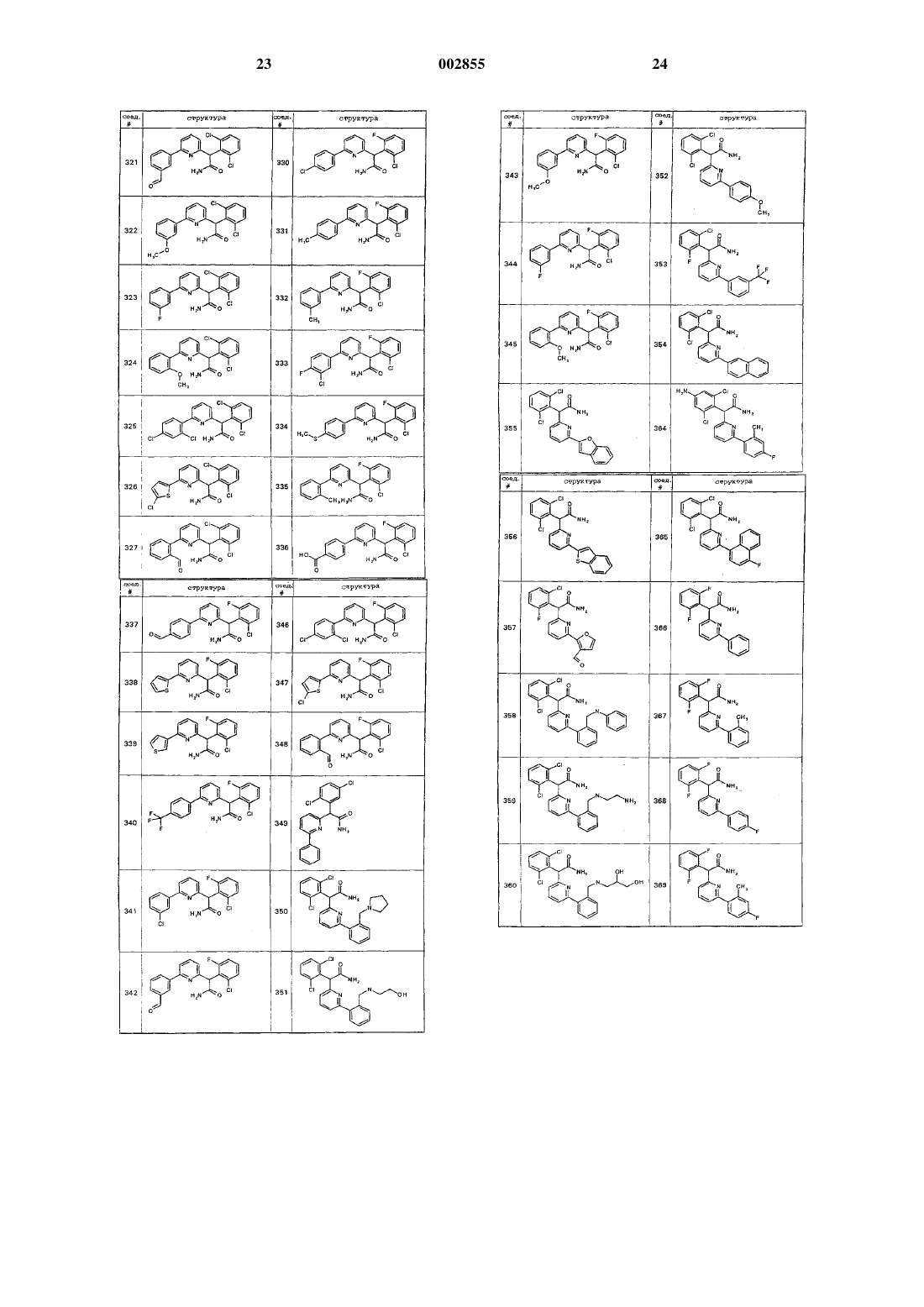
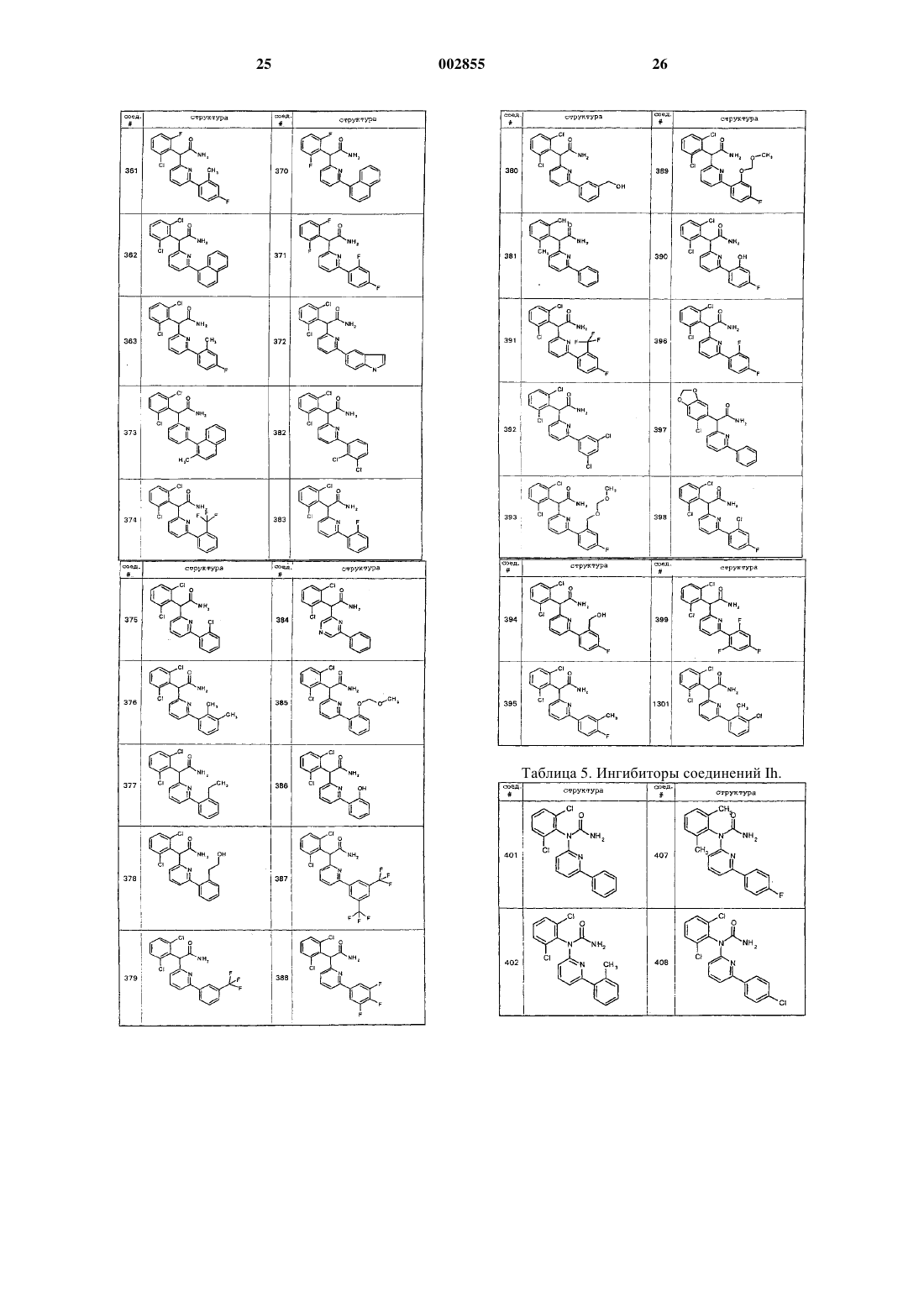
28. Соединение по п.7, где указанное соединение является соединением формулы Ig и выбрано из соединений 202/301, 302-399 или 1301, приведенных в табл. 4.
29. Соединение по п.8, где указанное соединение является соединением формулы Ih и выбрано из соединений 401-412, приведенных в табл. 5.
30. Фармацевтическая композиция, содержащая эффективное для ингибирования р38 количество соединения по любому из пп.1-8 и фармацевтически приемлемый носитель.
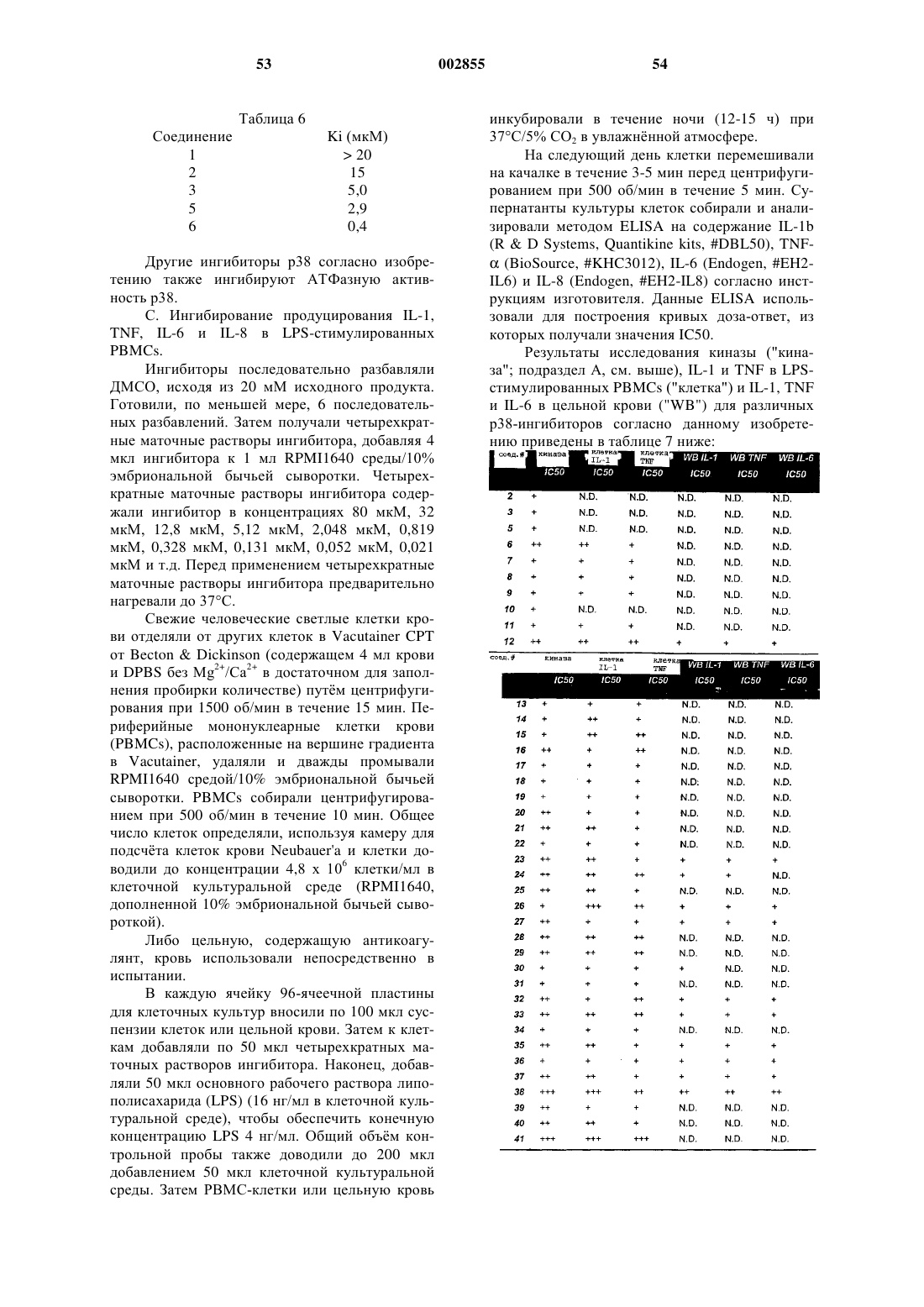
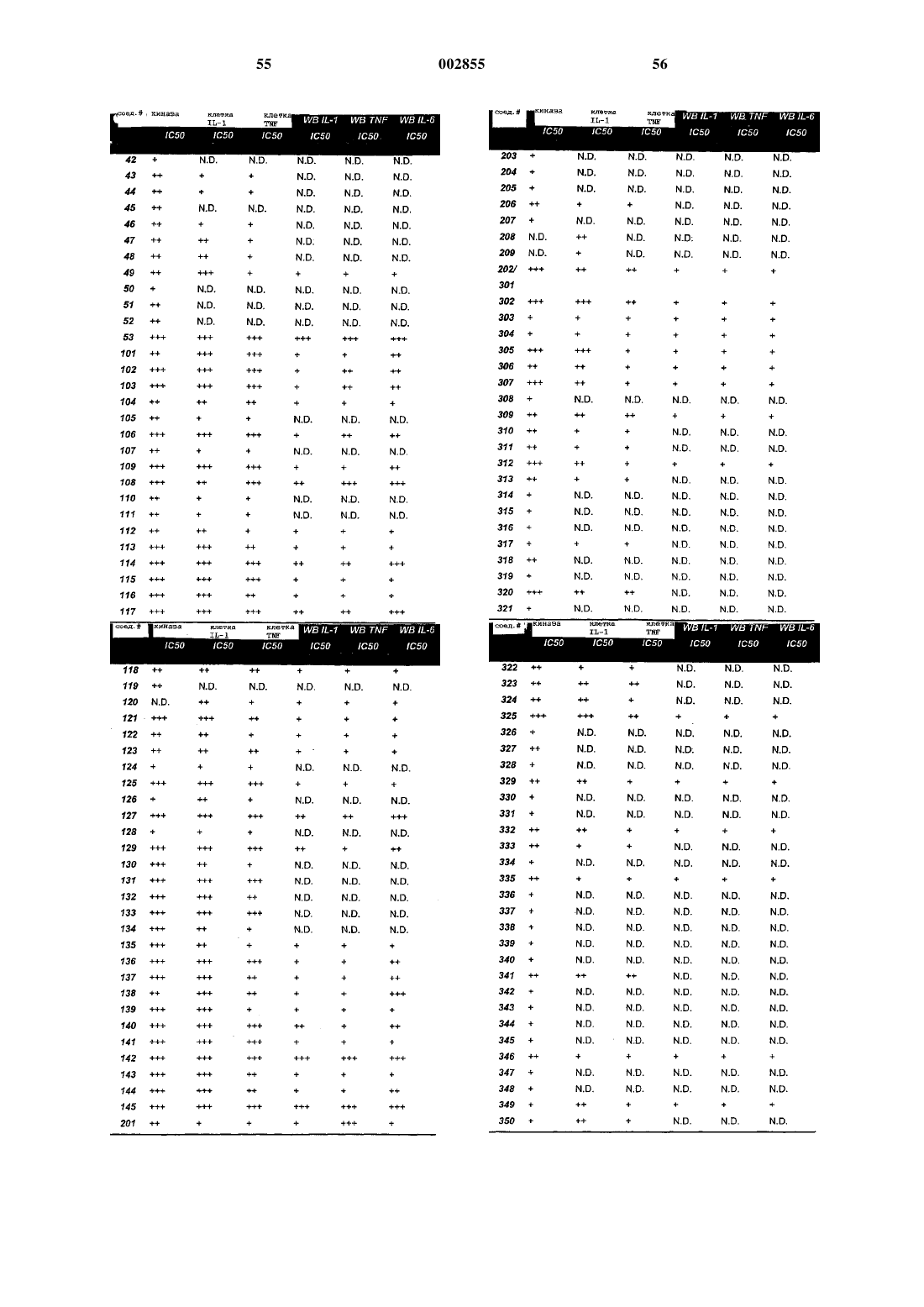
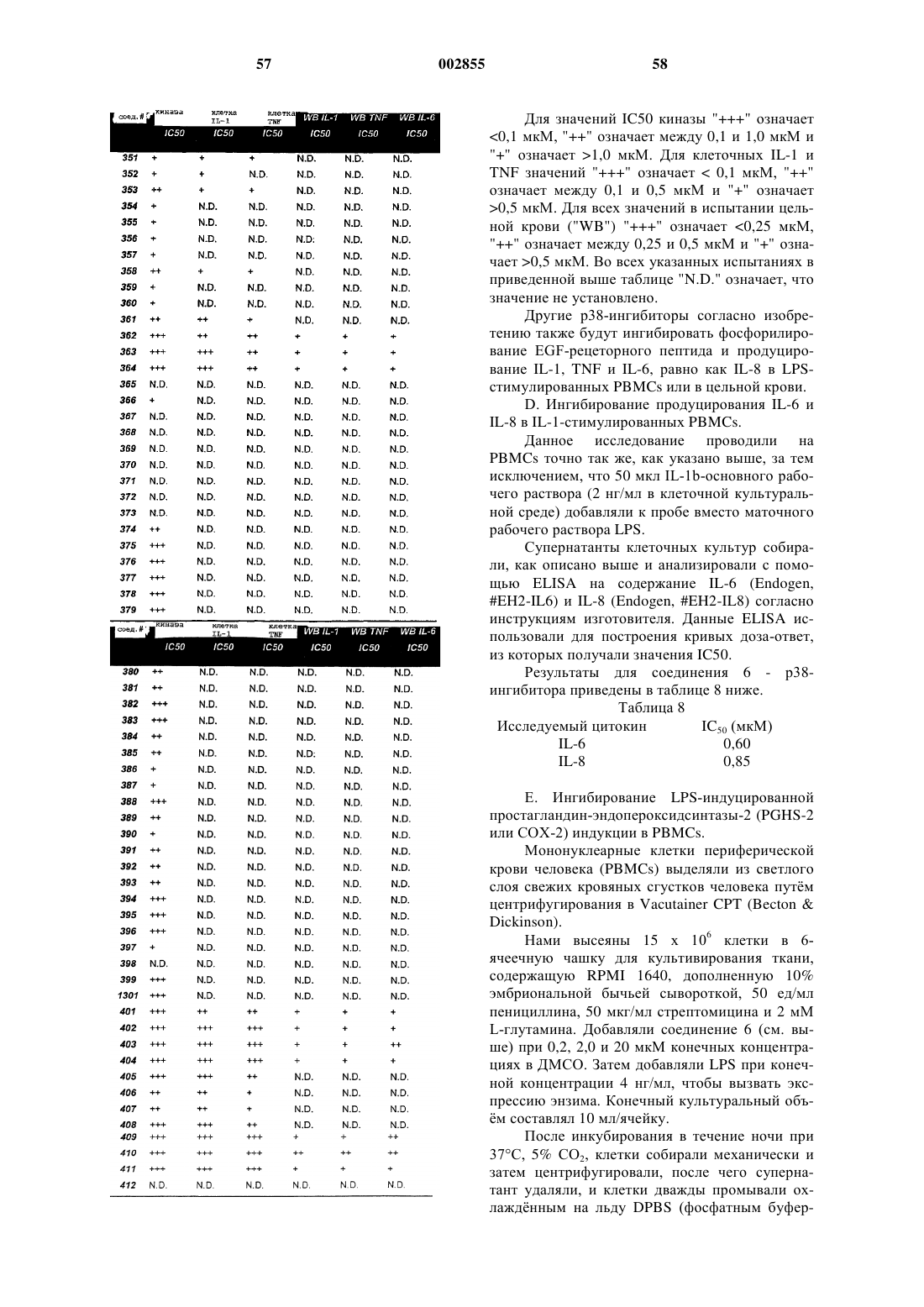
31. Способ лечения или предупреждения воспалительных заболеваний, аутоиммунных заболеваний, деструктивных заболеваний кости, пролиферативных нарушений, инфекционных заболеваний, нейродегенеративных заболеваний, аллергии, реперфузии/ишемии при мозговом ударе, сердечных приступов, ангиогенных нарушений, гипоксии органов, васкулярной гиперплазии, сердечной гипертрофии, тромбин-индуцируемой агрегации тромбоцитов или состояний, связанных с простагландин-эндопероксидазасинтазой-2 у пациентов, причем указанный способ включает в себя введение указанному пациенту композиции по п.30.
32. Способ по п.31, где указанный способ применяют для лечения или профилактики воспалительного заболевания, выбираемого из острого панкреатита, хронического панкреатита, астмы, аллергии или респираторного дистресс-синдрома у взрослых.
33. Способ по п.31, где указанный способ применяют для лечения или профилактики аутоимунного заболевания, выбираемого из гломерулонефрита, ревматоидного артрита, системной красной волчанки, склеродермии, хронического тиреоидита, болезни Грейвса, аутоиммунного гастрита, диабета, аутоиммунной гемолитической анемии, аутоиммунной нейтропении, тромбоцитопении, атопического дерматита, хронического активного гепатита, миастении gravis, рассеянного склероза, воспалительного заболевания кишечника, неспецифического язвенного колита, болезни Крона, псориаза или реакции "трансплантат против хозяина".
34. Способ по п.31, где указанный способ применяют для лечения или профилактики деструктивных заболеваний кости, выбираемых из остеоартрита, остеопороза или заболевания кости, связанного с множественной миеломой.
35. Способ по п.31, где указанный способ применяют для лечения или профилактики пролиферативных заболеваний, выбираемых из острого миелогенного лейкоза, хронического миелогенного лейкоза, метастатической меланомы, саркомы Капоши или множественной миеломы.
36. Способ по п.31, где указанный способ применяют для лечения или профилактики инфекционных заболеваний, выбираемых из сепсиса, септического шока или шигеллеза.
37. Способ по п.31, где указанный способ применяют для лечения или профилактики вирусных заболеваний, выбираемых из острого инфекционного гепатита, ВИЧ-инфекции или CMV ретинита.
38. Способ по п.31, где указанный способ применяют для лечения или профилактики нейродегенеративных заболеваний, выбираемых из болезни Альцгеймера, болезни Паркинсона, церебральной ишемии или нейродегенеративного заболевания, вызванного травматическим повреждением.
39. Способ по п.31, где указанный способ применяют для лечения или профилактики реперфузии/ишемии при мозговом ударе или миокардиальной ишемии, почечной ишемии, сердечных приступов, гипоксии органов или тромбининдуцируемой агрегации тромбоцитов.
40. Способ по п.31, где указанный способ применяют для лечения или профилактики состояний, связанных с простагландин-эндопероксидсинтазой-2, выбираемых из отека, лихорадочного состояния, аналгезии или боли.
41. Способ по п.40, где указанная боль выбрана из нервно-мышечной боли, головной боли, боли при раке, зубной боли или боли при артрите.
42. Способ по п.31, где указанный способ применяют для лечения или профилактики ангиогенных нарушений, выбираемых из твердых опухолей, глазной неоваскуляризации или детских гемангиом.
Текст
1 Данное изобретение касается ингибиторов р 38, протеинкиназы млекопитающих, вовлечнной в патологический процесс пролиферации клеток, клеточную гибель и ответную реакцию на внеклеточный раздражитель. Изобретение также связано со способами получения этих ингибиторов. К тому же, изобретение касается фармацевтических композиций, включающих в себя ингибиторы согласно изобретению, и способов применения этих композиций в лечении и профилактике различных болезней. Протеинкиназы вовлекаются в различные ответные реакции клеток на внеклеточные сигналы. Недавно было открыто семейство азотактивированных протеинкиназ (МАРК). Членами этого ряда являются Ser/Thr-киназы, активирующие субстраты путм фосфорилирования[В. Stein et al., Ann. Rep. Med. Chem., 31, pp. 28998 (1996)]. MAPKs самоактивируются под воздействием различных сигналов, включающих в себя факторы роста, цитокины, УФ-облучение и вызывающие стресс агенты. Одним из особенно интересных МАРК является р 38. р 38, известная также как цитокинподавляющий белок (CSBP), связывающий противовоспалительные лекарственные средства, иRK, была выделена из мышиных пре-В-клеток,трансфицированных липополисахаридным(LPS) рецептором СD14 и индуцированных LPS. Позднее р 38 была выделена и секвенирована, и было установлено, что у людей и мышей она кодируется кДНК. Активация р 38 наблюдалась в клетках, стимулированных стрессами, такими как обработка липополисахаридами (LPS), УФ,анизомицином или осмотическим шоком, и цитокинами, такими как IL-1 и TNF. Ингибирование р 38-киназы ведт к блокированию продуцирования как IL-1, так и TNF.IL-1 и TNF стимулируют продуцирование других провоспалительных цитокинов, таких какIL-6 и IL-8, и вовлечены в острые и хронические воспалительные заболевания и в постклимактерический остеопороз [R.B. Kimble et al., Endocrinol., 136, pp. 3054-61, 1995]. На основании этих данных полагают, что р 38, наряду с другими MAPKs, играет роль в опосредовании клеточной реакции на воспалительные раздражители, такой как аккумуляция лейкоцитов, активация макрофагов моноцитов,резорбция ткани, лихорадка, ответ в острой фазе и нейтрофилия. Вдобавок MAPKs, такие как р 38, вовлечены в нарушения, связанные с раком,тромбин-индуцируемой агрегацией тромбоцитов, иммунодефицитными расстройствами, аутоиммунными заболеваниями, клеточной гибелью, аллергиями, остеопорозом и нейродегенеративными расстройствами. Ингибиторы р 38 также вовлечены в процесс регуляции боли через ингибирование индукции простагландинэндопероксидсинтазы-2. Другие болезни, связанные с повышенной продукцией IL-1, IL-6, IL-8 или TNF, описаны в WO 96/21654. 2 Уже предпринимались попытки разработать лекарственные средства, специфически ингибирующие MAPKs. К примеру, в РСТпубликации WO 95/31451 описаны соединения пиразола, ингибирующие MAPKs, и, в частности, р 38. Однако эффективность этих ингибиторов in vivo вс ещ исследуется. Поэтому по-прежнему существует большая потребность в разработке других сильных,р 38 специфических ингибиторов, полезных в лечении различных состояний, связанных с активацией р 38. Краткое описание изобретения В настоящем изобретении эта проблема решается с помощью соединений, обнаруживающих способность к сильному и специфическому ингибированию р 38. Эти соединения имеют общую формулу где каждый из Q1 и Q2 независимо выбирают из систем 5-6-членных ароматических карбоциклических или гетероциклических колец или систем 8-10-членных бициклических колец, состоящих из ароматических карбоциклических колец, ароматических гетероциклических колец или сочетания ароматического карбоциклического кольца и ароматического гетероциклического кольца. Циклы, составляющие Q1, замещены 1-4 заместителями, каждый из которых независимо выбирают из группы, включающей в себя галоген, C1-С 3-алкил, возможно, замещнный NR'2,OR', CO2R' или CONR'2; O-(С 1-С 3)-алкил, возможно, замещнный NR'2, OR', CO2R' илиN=C-N(R')2. Циклы, составляющие Q2, могут содержать до 4 заместителей, каждый из которых независимо выбирают из группы, включающей в себя галоген, C1-С 3-линейный или разветвлнный алкил, возможно, замещнный NR'2, OR', CO2R',S(O2)N(R')2; N=C-N(R')2, R3 или CONR'2; O-(С 1 С 3)алкил; O-(С 1-С 3)алкил, возможно замещенный NR'2, OR', CO2R', S(O2)N(R')2; N=C-N(R')2,R3 или CONR'2; NR'2; ОСF3; СF3; NO2; CO2R';R' выбирают из группы, включающей в себя водород, (C1-С 3)алкил; (С 2-С 3)алкенил или алкинил; фенил или фенил, замещнный 1-3 заместителями, которые независимо выбирают из галогена, метокси, циано, нитро, амино, гидрокси, метила или этила.R3 выбирают из систем 5-6-членных ароматических карбоциклических или гетероциклических колец.SO2N(R2)2; или систему 5-6-членных карбоциклических или гетероциклических колец, возможно, замещенных N(R')2, OR', CO2R',CON(R')2 или SO2N(R2)2.-С(OR2)2-. Каждый R независимо выбирают из водорода, -R2, -N(R2)2, -OR2, SR2, -C(O)-N(R2)2-,-S(O2)-N(R2)2- или -С(О)-OR2, где два смежныхR возможно связаны друг с другом, и вместе с каждым Y, с которым они соответственно связаны, образуют 4-8-членное карбоциклическое или гетероциклическое кольцо;R2 выбирают из группы, включающей водород, (C1-С 3)алкил или (C1-С 3)алкенил; каждый член группы возможно замещн -N(R')2, -OR',SR', -C(O)-N(R')2, -S(O2)-N(R')2, -C(O)-OR' илиR1 выбирают из группы, включающей в себя водород, (C1-С 3)-алкил, ОН или О-(C1-С 3)алкил. В другом варианте воплощения изобретение дат фармацевтические композиции, содержащие ингибиторы р 38 согласно изобретению. Эти композиции могут быть использованы в способах лечения или профилактики различных заболеваний, таких как раковые, воспалительные заболевания, аутоиммунные расстройства,деструктивные заболевания кости, пролиферативные заболевания, инфекционные болезни,вирусные болезни и нейродегенеративные расстройства. Эти композиции также полезны в способах профилактики клеточной гибели и гиперплазии и, следовательно, могут быть использованы для лечения или профилактики реперфузии/ ишемии при мозговом ударе, сердечных приступов, гипоксии органов. Композиции могут также быть полезны для профилактики тромбин-индуцируемой агрегации тромбоцитов. Каждый из вышеуказанных способов также является составной частью данного изобретения. Данное изобретение касается ингибиторов р 38, имеющих общую формулу: где каждый из Q1 и Q2 независимо выбирают из систем 5-6-членных ароматических карбоциклических или гетероциклических колец или систем 8-10-членных бициклических колец, содержащих ароматические карбоциклические кольца, ароматические гетероциклические кольца или сочетание ароматического карбоциклического кольца и ароматического гетероциклического кольца. Циклы, составляющие Q1, замещены 1-4 заместителями, каждый из которых независимо выбирают из группы, включающей в себя галоген, C1-С 3-алкил, возможно, замещнный NR'2,OR', CO2R' или CONR'2; O-(С 1-С 3)-алкил, возможно, замещнный NR'2, OR', CO2R' илиN=C-N(R')2. Циклы, составляющие Q2, могут содержать до 4 заместителей, каждый из которых независимо выбирают из группы, включающей в себя галоген, C1-С 3- линейный или разветвлнный алкил, возможно, замещнный NR'2, OR', CO2R',S(O2)N(R')2; N=C-N(R')2, R3 или CONR'2, O-(C1 С 3)алкил; О-(C1-С 3)алкил, возможно, замещнный NR'2, OR', CO2R', S(O2)N(R')2; N=C-N(R')2,R3 или CONR'2; NR'2; ОСF3; СF3; NO2; CO2R';R' выбирают из группы, включающей в себя водород, (C1-С 3)алкил; (С 2-С 3)алкенил или алкинил; фенил или фенил, замещнный 1-3 заместителями, которые независимо выбирают из галогена, метокси, циано, нитро, амино, гидрокси, метила или этила.R3 выбирают из систем 5-6-членных ароматических карбоциклических или гетероциклических колец.SO2N(R2)2; или систему 5-6-членных карбоциклических или гетероциклических колец, возможно, замещенную N(R')2, OR', CO2R',CON(R')2 или SO2N(R2)2.-С(OR2)2-. Каждый R независимо выбирают из водорода, -R2, -N(R2)2, -OR2, SR2, -С(О)-N(R2)2-,-S(O2)-N(R2)2- или -С(О)-OR2, где два смежныхR, возможно, связаны друг с другом, и вместе с каждым Y, с которым они соответственно связаны, образуют 4-8-членное карбоциклическое или гетероциклическое кольцо. Специалисту в соответствующей области очевидно, что когда 2 R-компонента образуют циклическую структуру вместе сYкомпонентами, с которыми они соответственно связаны, концевой водород каждого неконденсированного R-компонента будет теряться. К примеру, если циклическая структура образуется путм связывания этих двух R-компонентов вместе, при этом один является -NH-СН 3, другой является -СН 2-СН 3, будет теряться по одному концевому водороду на каждом Rкомпоненте (выделены жирным шрифтом). Поэтому образующаяся часть циклической структуры будет иметь формулу -NH-CH2-CH2-CH2-.R1 выбирают из группы, включающей в себя водород, (C1-С 3)-алкил, ОН или О-(C1-С 3)алкил. Для специалиста в соответствующей области очевидно, что если R1 обозначает ОН,получаемый ингибитор может подвергнуться таутомеризации, превращаясь в соединение формулы которые также являются ингибиторами р 38 согласно изобретению. Согласно другому предпочтительному варианту воплощения, Q1 выбирают из фенила или пиридила, содержащего от 1 до 3 заместителей,где, по меньшей мере, один из указанных заместителей находится в орто- положении и где указанные заместители независимо выбирают из группы, включающей в себя хлор, фтор, бром,-СН 3, -ОСН 3, -ОН, -СF3, -ОСF3, -O(CH2)2CH3,NH2, 3,4-метилендиокси, -N(СН 3)2, -NH-S(О)2 фенил, -NH-C(О)O-СН 2-4-пиридин, -NН-С(O) СН 2-морфолин, -NH-C(О)CH2-N(СН 3)2, -NНС(O)СН 2-пиперазин, -NH-C(О)СН 2-пирролидин,-NH-C(О)С(О)-морфолин,-NH-С(О)С(О)-пи 002855CH2-N(СН 3)2 или, -О-(CH2)2-N(CH3)2. Ещ более предпочтительны фенил или пиридил, содержащие, по меньшей мере, 2 из вышеуказанных заместителей, из которых оба находятся в орто-положении. Некоторыми конкретными примерами предпочтительных Q1 являются: Наиболее предпочтительно, чтобы Q1 был выбран из группы, включающей в себя 2-фтор 6-трифторметилфенил, 2,6-дифторфенил, 2,6 дихлорфенил, 2-хлор-4-гидроксифенил, 2-хлор 4-аминофенил, 2,6-дихлор-4-аминофенил, 2,6 дихлор-3-аминофенил, 2,6-диметил-4-гидроксифенил, 2-метокси-3,5-дихлор-4-пиридил, 2 хлор-4,5-метилендиоксифенил или 2-хлор-4-(N2-морфолиноацетамидо)фенил. Согласно предпочтительному варианту воплощения, Q2 означает фенил или пиридил, содержащий от 0 до 3 заместителей, где каждый заместитель независимо выбирают из группы,включающей в себя хлор, фтор, бром, метил,этил, изопропил, -ОСН 3, -ОН, -NH2, -СF3, -OCF3,-SСН 3, -ОСН 3, -С(О)ОН, -С(О)ОСН 3, -CH2NH2,-N(СН 3)2, -СН 2-пирролидин и -CH2OH. Некоторыми конкретными примерами предпочтительных Q2 являются: незамещнный 2-пиридил или незамещнный фенил. Наиболее предпочтительными являются соединения, где Q2 выбирают из группы, включающей в себя фенил, 2-изопропилфенил, 3,4 диметилфенил, 2-этилфенил, 3-фторфенил, 2 метилфенил, 3-хлор-4-фторфенил, 3-хлорфенил,2-карбометоксифенил, 2-карбоксифенил, 2-метил-4-хлорфенил, 2-бромфенил, 2-пиридил, 2 метилен-гидроксифенил, 4-фторфенил, 2-метил 4-фторфенил, 2-хлор-4-фторфенил, 2,4-дифторфенил, 2-гидрокси-4-фторфенил или 2-метиленгидрокси-4-фторфенил. Согласно ещ одному предпочтительному варианту, Х означает -S-, -О-, -S(O)2-, -S(O)-,-NR-, -C(R2)- или -С(О)-. Наиболее предпочтительно, когда Х означает S. Согласно другому предпочтительному варианту воплощения, n означает 1, а А означаетN. Согласно другому предпочтительному варианту воплощения, каждый Y означает С. Согласно ещ более предпочтительному варианту воплощения, каждый Y означает С, aR, связанный с этими Y-компонентами, выбирают из водорода или метила. Некоторые конкретные ингибиторы согласно изобретению приведены ниже в таблице. где R' имеет вышеуказанные значения, или с содержащим уходящую группу реагентом формулы IIb Согласно другому варианту воплощения,данное изобретение описывает способы получения ингибиторов р 38 формулы (Iа), изображнной выше. Эти способы включают в себя реакцию соединения формулы II: где каждая из переменных в вышеуказанной формуле является такой, как указано выше для ингибиторов согласно изобретению, с содержащим уходящую группу реагентом формулы IIа где каждый из L1, L2 и L3 независимо означает уходящую группу. Применяемый в данной реакции реагент с уходящей группой используют в избытке, либо чистый, либо с совместным растворителем, таким как толуол. Реакцию проводят при температуре между 25 и 150 С. Содержащие уходящую группу реагенты формулы IIа, используемые в получении ингибиторов р 38 по данному изобретению, включают в себя диметилацеталь диметилформамида,диметилацеталь диметилацетамида, триметилортоформиат, диэтилацеталь диметилформамида и другие родственные соединения. Предпочтительно содержащим уходящую группу реагентом формулы IIа, используемым для получения ингибиторов по данному изобретению, является диметилацеталь диметилформамида. Содержащие уходящую группу реагенты формулы IIb, используемые в получении ингибиторов р 38 согласно изобретению, включают в себя фосген, карбонилдиимидазол, диэтилкарбонат и трифосген. В более предпочтительных способах получения соединений согласно изобретению используются соединения формулы II, где каждая из переменных определяется из соображений более предпочтительного и наиболее предпочтительного выбора, как сформулировано выше для соединений согласно изобретению. Поскольку источником R1 является содержащий уходящую группу реагент (C-R' или С=O), его особенности, конечно, сказываются на структуре этого реагента. Поэтому для соединений, в которых R1 означает ОН, используемым реагентом должен быть IIb. Подобным образом, когда R1 означает Н или (C1-С 3)-алкил,используемым реагентом должен быть IIа. Для получения ингибиторов, где R1 означает О-(C1 С 3)алкил, сначала получают соединение, где R1 означает ОН, с последующим алкилированием этой гидроксильной группы по стандартным методикам, таким как обработка гидридом Na в ДМФ, йодистом метиле и йодистом этиле. Непосредственные предшественники ингибиторов согласно изобретению формулы Iа(т.е., соединения формулы II) сами могут быть синтезированы по одной из указанных ниже схем синтеза: 14 выше для ингибиторов согласно изобретению, с содержащим уходящую группу соединением формулы: В схеме 1 порядок стадий 1) и 2) можно поменять. Также исходный нитрил может быть заменн на соответствующие кислоту или сложный эфир. Либо вместо нитрила могут быть использованы другие хорошо известные латентные карбоксильные или карбоксамидные фрагменты (см. схему 2). Такие варианты как карбоновые кислоты, эфиры карбоновых кислот, оксазолины или оксизолидиноны могут быть включены в эту схему при последовательном применении способов снятия защиты и функционализации, хорошо известных в соответствующей области. Используемое в первой стадии схемы 1 (и схемы 2, см. ниже) основание выбирают из группы, включающей гидрид натрия, амид натрия, LDA, литийгексаметилдисилазид, натрийгексаметилдисилазид или любой ряд других ненуклеофильных оснований, которые будут депротонировать положение альфа до нитрила. Также добавление HX-Q2 в одну стадию,указанное выше, может быть заменено двумя стадиями - добавлением защищнного или не защищнного Х-производного с последующим добавлением Q2-производного на следующей стадии. Схема 2R' имеет вышеуказанные значения. Согласно другому варианту воплощения,данное изобретение дат способы получения ингибиторов р 38 формулы (Ib), указанной выше. Эти способы включают в себя получение соединения формулы III где каждый из возможных вариантов приведенной выше формулы является таким, как указано как указано выше. Две полных схемы синтеза ингибиторов р 38 формулы (Ib) приведены ниже. Схема 3 По схеме 3, Q1-замещнное производное может быть обработано основанием, таким как гидрид натрия, амид натрия, LDA, литийгексаметилдисилазид,натрийгексаметилдисилазид или любой ряд других ненуклеофильных оснований, депротонирующих положение альфа доZ-группы, обозначающей замаскированный амидный радикал. Либо Z обозначает карбоновую кислоту, эфир карбоновой кислоты, оксазолин или оксазолидинон. Образующийся после депротонирования анион связывается с азотнесущим гетероциклическим соединением, содержащим две уходящие группы или латентные уходящие группы, в присутствии палладиевого катализатора. Одним из примеров такого соединения может быть 2,6-дихлорпиридин. На второй стадии вводится кольцевой фрагмент Q2. Это может быть выполнено путм применения многих хорошо известных в соответствующей области реакций, приводящих к получению диарильных соединений. Одним из примеров может служить реакция ариллитиевого соединения с промежуточным пиридиновым соединением, полученным на стадии 1. Либо арилметаллическое соединение, такое как арилстаннан или арилбороновая кислота, может реагировать с галоидарильной частью промежуточного пиридинового соединения в присутствии катализатора Pd8. На стадии 3 снимают защиту с Z-группы и/или функционализируют до образования амидного соединения. Когда Z обозначает карбоновую кислоту, эфир карбоновой кислоты,оксазолин или оксазолидинон, различные варианты способов снятия защиты и функционализации, хорошо известные из соответствующей области, используют для получения амида. Наконец, на стадии 4 амидное соединение цикли 15 зуется до конечного продукта с применением реагентов, таких как ДМФ-ацеталь или подобные реагенты, либо в чистом виде, либо в органическом растворителе. Схема 4 16 этих соединений приведены на схемах 5 и 6,ниже. Схема 5 Схема 4 является аналогичной, за тем исключением, что сначала, перед реакцией с Q1 исходным соединением, получают диарильное промежуточное соединение. Согласно другому варианту воплощения,изобретение описывает ингибиторы р 38, аналогичные ингибиторам формул Iа и Ib, но в которых цикл, несущий Q1 заместитель, является восстановленным. Эти ингибиторы имеют формулу: В этих схемах соединения формул Iа и Ib восстанавливают по реакции с избытком диизобутилалюминий гидрида или с эквивалентным количеством реагента до получения соединений с восстановленным циклом формул Iс или Id,соответственно. Присоединение R5-компонента, не являющегося водородом, к азоту цикла достигается реакцией соединений формул Iс или Id, указанных выше, с соответствующим реагентом (реагентами). Примеры таких модификаций приведены ниже в экспериментальном разделе. Некоторые конкретные ингибиторы согласно изобретению формулы Iс приведены ниже в таблице. где A, Q1, Q2, R, R', X, Y и n имеют значения,указанные для соединений формул Iа и Ib. Эти определения содержат все варианты воплощения каждой из этих переменных (т.е., основной,предпочтительный, более желательный и наиболее предпочтительный). R5 выбирают из водорода,-CR'2OH,-C(O)R4,-С(О)OR4,-СR'2OРО 3 Н 2, -РО 3 Н 2 и солей -РО 3 Н 2. Когда R5 не является водородом, предполагается, что образующиеся соединения служат пролекарственными формами, которые должны расщепляться in vivo с образованием соединения, где R5 является водородом. Согласно другим предпочтительным вариантам воплощения, в соединениях формулы Iс,А предпочтительно означает азот, n предпочтительно означает 1, а Х предпочтительно является серой. В соединениях формул Iс или Id, Q1 иQ2 предпочтительно являются такими же радикалами, как указано выше для этих переменных в соединениях формул Iа и Ib. Соединения формул Iс или Id могут быть получены непосредственно из соединений формул Iа и Ib, которые содержат водород, C1-С 3 алкил или C1-С 3-алкенил или алкинил в R1 положении (т.е., когда R1=R'). Схемы синтеза Согласно ещ одному варианту воплощения, изобретение описывает ингибиторы р 38 формул: 19 где A, Q1, Q2, R, X, Y и n имеют значения, указанные выше для соединений формул Iа и Ib. Эти определения содержат все варианты воплощения каждой из этих переменных (т.е., основной, предпочтительный, более желательный и наиболее предпочтительный). Более предпочтительно, чтобы Q2 в соединениях формулы Iе означал незамещнный фенил.Q3 означает систему 5-6-членных ароматических карбоциклических или гетероциклических колец, или систему 8-10-членных бициклических колец, содержащую ароматические карбоциклические кольца, ароматические гетероциклические кольца или сочетание ароматического карбоциклического кольца и ароматического гетероциклического кольца. Циклы,содержащие Q3, замещены 1-4 заместителями,каждый из которых независимо выбирают из группы, включающей в себя галоген, C1-С 3 алкил, возможно, замещнный NR'2, OR', CO2R' или CONR'2; O-(С 1-С 3)-алкил, возможно, замещнный NR'2, OR', CO2R' или CONR'2; NR'2; ОСF3; СF3; NO2; CO2R'; CONR'; SR'; S(O2)N(R')2;OR4; OC(O)R4; ОР(O)3 Н 2 или N=C-N (R')2. Согласно одному из предпочтительных вариантов воплощения, Q3 является замещнным 2-4 заместителями, где, по меньшей мере, один из указанных заместителей присутствует в ортоположении относительно точки присоединенияQ3 к остатку ингибитора. Когда Q3 означает бициклическое кольцо, 2 заместителя в ортоположении, присутствующие на кольце, являются расположенными близко (т.е., непосредственно присоединнными) к остатку молекулы ингибитора. Другие два необязательных заместителя могут присутствовать на одном из циклов. Более предпочтительно, когда оба таких орто-положения заняты одним из указанных заместителей. Согласно другому предпочтительному варианту воплощения, Q3 означает моноциклическое карбоциклическое кольцо, где каждый орто-заместитель независимо выбирают из галогена или метила. Согласно иному предпочтительному варианту воплощения, Q3 содержит 1 или 2 дополнительных заместителя, которые независимо выбирают из NR'2, OR', CO2R' , CN,N(R')C(O)R4; N(R')С(О)OR4; N(R')C(O)C(O)R4;Iе, приведенных ниже в таблице 3, или из любых Q3-фрагментов, присутствующих в соединениях Ig, приведенных в табл. 4. Соединения формулы Iе будут восприняты специалистами как непосредственные предшественники некоторых формул Iа и Iс ингибиторов р 38 согласно изобретению (т.е., тех, гдеQ1=Q3). Специалисты воспримут также соеди 002855 20 нения формулы Ig как предшественники некоторых формул Ib и Id ингибиторов р 38 согласно изобретению (т.е. тех, где Q1=Q3). Следовательно, синтез ингибиторов формулы Iе описывается вышеуказанными схемами 1 и 2, где Q1 замещн на Q3. Подобным образом, синтез ингибиторов формулы Ig описывается вышеуказанными схемами 3 и 4, где Q1 замещн на Q3. Синтезы ингибиторов формул If и Ih описаны ниже в схемах 7 и 8. Схема 7 Схема 8 описывает синтез соединений типа Ih. К примеру, обработка исходного дибромпроизводного, такого как 2,6-дибромпиридин,амином в присутствии основания, такого как гидрид натрия, дат 2-амино-6-бром-производное. Обработка этого промежуточного соединения аналогом фенилбороновой кислоты (Q2 бороновой кислотой), таким как фенилбороновая кислота в присутствии палладиевого катализатора, дат дизамещнное производное, которое может быть затем ацилировано до конечного продукта. Порядок первых двух стадий можно поменять. Не вдаваясь в теоретические подробности,заявители считают, что ди-орто-замещение в цикле Q3 ингибиторов формулы Iе и Ig и наличие азота, непосредственно связанного с циклом"сплющивание" соединения, что позволяет ему эффективно ингибировать р 38. Предпочтительным ингибитором формулыIе согласно изобретению является ингибитор, в котором А означает углерод, n равен 1, Х означает серу, каждый из Y означает углерод, каждый из R означает водород, Q3 означает 2,6 дихлорфенил и Q2 означает фенил, указанное 21 соединение упоминается как соединение 201. Предпочтительным ингибитором формулы Ig согласно изобретению является ингибитор, в котором Q3 означает 2,6-дихлорфенил, Q2 означает фенил, каждый из Y означает углерод и каждый из R означает водород. Это соединение упоминается здесь как соединение 202. Другими предпочтительными соединениями формулы Ig согласно изобретению являются соединения,перечисленные ниже в таблице 4. Предпочтительными соединениями Ih согласно изобретению являются соединения, приведенные ниже в таблице 5. Другими предпочтительными соединениями Ih являются соединения, в которых Q1 означает фенил, независимо замещнный в положениях 2 и 6 хлором или фтором; каждый из Y означает углерод; каждый из R означает водород и Q2 означает 2 метилфенил, 4-фторфенил, 2,4-дифторфенил, 2 метиленгидрокси-4-фторфенил или 2-метил-4 фторфенил. Некоторые конкретные ингибиторы формул Ie, Ig и Ih приведены ниже в таблицах. Таблица 3. Ингибиторы формулы Ie. Активность ингибиторов р 38 согласно изобретению может быть исследована in vitro, invivo или на линии клеток. Испытания in vitro включают в себя исследования по определению ингибирования либо киназной активности, либо АТФазной активности активированного р 38. Альтернативные испытания in vitro количественно оценивают способность ингибитора связываться с р 38, которая может быть измерена либо путм введения в ингибитор радиоактивной метки перед образованием связи, выделения комплекса ингибитор/р 38 и определения количества связанной радиоактивной метки, либо путм проведения конкурентного эксперимента,где новые ингибиторы инкубируют с р 38, связанным с известными радиоактивными лигандами. Исследованиями на культуре клеток ингибирующего действия соединений согласно изобретению можно определить количества TNF,IL-1, IL-6 или IL-8, продуцированных в цельной крови, или е клеточных фракциях в клетках,обработанных ингибитором, в сравнении с клетками, обработанными отрицательными контролями. Доля этих цитокинов может быть определена с применением промышленно выпускаемых ELISAs. Испытания in vivo, пригодные для определения ингибиторной активности р 38-ингибиторов согласно изобретению, состоят в подавлении отка задних лап крыс при индуцированномMycobacterium butyricum адъювантном артрите. Они описаны в J.C. Boehm et al., J. Med. Chem.,39, pp. 3929-37 (1996), упоминаемом здесь в качестве ссылки. Ингибиторы р 38 согласно изобретению могут также быть испытаны на моделях животных с артритом, резорбцией костей, 002855Therapeutics, 279, pp. 1453-61 (1996), упоминаемом здесь в качестве ссылки. Ингибиторы р 38 или их фармацевтические соли могут быть сформулированы в фармацевтические композиции для введения животным или человеку. Эти фармацевтические составы,включающие в себя количество р 38-ингибитора,эффективное для лечения или профилактики р 38-опосредованного состояния, и фармацевтически приемлемый носитель, являются другим вариантом воплощения настоящего изобретения. Термин "р 38-опосредованное состояние",здесь используемый, означает любую болезнь или вредно воздействующее обстоятельство,при которых, как известно, играет роль р 38. Он включает в себя состояния, которые известны как вызываемые избыточным продуцированиемIL-1, TNF, IL-6 и IL-8. Такие состояния включают в себя все без ограничения воспалительные болезни, аутоиммунные нарушения, деструктивные процессы кости, пролиферативные нарушения, инфекционные болезни, нейродегенеративные расстройства, реперфузию/ишемию при мозговом ударе, сердечные приступы, ангиогенные нарушения, гипоксию органов, васкулярную (сосудистую) гиперплазию, сердечную гипертрофию, тромбин-индуцируемую агрегацию тромбоцитов и состояния, связанные с простагландин-эндопероксидаза-синтазой-2. Воспалительные заболевания, которые могут быть излечены или предупреждены, включают в себя, но не в порядке ограничения, острый панкреатит, хронический панкреатит, астму, аллергию и респираторный дистресссиндром у взрослых. Аутоиммунные нарушения, которые могут быть излечены или предупреждены, включают в себя, но не в порядке ограничения, гломерулонефрит, ревматоидный артрит, системную красную волчанку, склеродермию, хронический тиреоидит, болезнь Грейвса (диффузный токсический зоб), аутоиммунный гастрит, диабет, аутоиммунную гемолитическую анемию, аутоиммунную нейтропению, тромбоцитопению, атопический дерматит, хронический активный гепатит, myasthenia gravis, рассеянный склероз,воспалительное заболевание кишечника, неспецифический язвенный колит, болезнь Крона(гранулематозную болезнь), псориаз или болезнь "трансплантат против хозяина". Деструктивные процессы кости, которые могут быть излечены или предупреждены,включают в себя, но не в порядке ограничения,остеопороз, остеоартрит и нарушения кости,связанные с множественной миеломой. Пролиферативные нарушения, которые могут быть излечены или предупреждены,включают в себя, но не в порядке ограничения, 29 острый миелогенный лейкоз, хронический миелогенный лейкоз, метастатическую меланому,саркому Капоши и множественную миелому. Ангиогенные нарушения, которые могут быть излечены или предупреждены, включают в себя солидные опухоли, глазную неоваскуляризацию, детские гемангиомы. Инфекционные болезни, которые могут быть излечены или предупреждены, включают в себя, но не в порядке ограничения, сепсис, септический шок и шигеллз. Вирусные болезни, которые могут быть излечены или предупреждены, включают в себя,но не в порядке ограничения, острые инфекционные гепатиты (включая гепатит А, гепатит В и гепатит С), ВИЧ-инфекцию и CMV-ретинит. Нейродегенеративные расстройства, которые могут быть излечены или предупреждены с помощью соединений согласно изобретению,включают в себя, но не в порядке ограничения,болезнь Альцгеймера, болезнь Паркинсона, церебральную ишемию или нейродегенеративные расстройства, вызванные травматическим повреждением."р 38-опосредованные состояния", включают в себя также, реперфузию/ишемию при мозговом ударе, сердечные приступы, миокардиальную ишемию, гипоксию органов, васкулярную гиперплазию, сердечную гипертрофию и тромбин-индуцируемую агрегацию тромбоцитов. Вдобавок ингибиторы р 38 согласно изобретению способны также к ингибированию экспрессии индуцируемых провоспалительных белков, таких как простагландин-эндопероксидсинтаза-2 (PGHS-2), упоминаемая также как циклооксигеназа-2 (СОХ-2). Поэтому другими"р 38-опосредованными состояниями" являются отк, анальгезия, лихорадочное состояние и боль, такая как нервно-мышечная боль, головная боль, боль при раке, зубная боль и боль при артрите. Болезни, которые могут быть излечены или предупреждены с помощью ингибиторов р 38 согласно изобретению, могут также быть обычно сгруппированы по цитокину (IL-1, TNF,IL-6, IL-8), который, как предполагается, отвечает за заболевание. Так, IL-1-опосредованная болезнь или состояние включает в себя ревматоидный артрит,остеоартрит, мозговой удар, эндотоксемию (наличие в крови эндотоксинов) и/или синдром токсического шока, вызванную эндотоксином воспалительную реакцию, воспалительное заболевание кишечника, туберкулез, атеросклероз,дегенерацию мышц, кахексию, псориатический артрит, синдром Рейтера, подагру, травматический артрит, артрит при коревой краснухе, острый синовит, диабет, болезнь -клеток поджелудочной железы и болезнь Aльцгеймера.TNF-опосредованная болезнь или состояние включает в себя ревматоидный артрит, рев 002855"трансплантат против хозяина", отторжения аллотрансплантата, обусловленные инфекцией жар и миалгию, вторичную от инфекции кахексию, СПИД, ARC (СПИД-связанный комплекс) или злокачественность, келоидное образование,образование рубцовой ткани, болезнь Крона,неспецифический язвенный колит или лихорадка. TNF-опосредованные болезни также включают в себя вирусные инфекции, такие как ВИЧ,CMV (цитомегаловирус), грипп и герпес; и ветеринарные вирусные инфекции, такие как лентивирусные инфекции, включающие в себя, но не в порядке ограничения, вирус конской инфекционной анемии, вирус козьего артрита,visna-вирус и maedi-вирус или ретровирусные инфекции, включающие в себя кошачий вирус иммунодефицита, бычий вирус иммунодефицита или собачий вирус иммунодефицита.IL-8-опосредованная болезнь или состояние включает в себя болезни, характеризуемые массовой инфильтрацией нейтрофильных гранулоцитов, такие как псориаз, воспалительное заболевание кишечника, астму, сердечное или почечное реперфузионное нарушение, респираторный дистресс-синдром у взрослых, тромбоз и гломерулонефрит. Вдобавок соединения согласно изобретению могут быть использованы местно для лечения или профилактики состояний, вызванных или обострнных IL-1 или TNF (фактор некроза опухоли). Такие состояния включают в себя воспаление суставов, экзему, псориаз, воспалительные состояния кожи, такие как солнечную эритему, воспалительные состояния глаз, такие как конъюнктивит, лихорадку, боль и другие состояния, связанные с воспалением. Кроме соединений согласно изобретению в композициях для лечения или профилактики вышеуказанных болезней могут также быть использованы фармацевтически приемлемые соли соединений согласно изобретению. Фармацевтически приемлемые соли соединений согласно изобретению включают в себя соли, полученные из фармацевтически приемлемых неорганических или органических кислот и оснований. Примеры подходящих кислотных солей включают в себя ацетат, адипинат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат,камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, глицерофосфат,гликолят, полусульфат, гептаноат, гексаноат, 31 гидрохлорид, гидробромид, гидроиодид, 2 гидроксиэтансульфонат, лактат, малеат, малонат, метансульфонат, 2-нафталинсульфонат,никотинат, нитрат, оксалат, пальмоат, пектинат,персульфат, 3-фенилпропионат, фосфат, пикрат,пивалат, пропионат, салицилат, сукцинат, сульфат, тартрат, тиоцианат, тозилат и ундеканоат. Другие кислоты, такие как щавелевая кислота,хотя сами по себе не являются фармацевтически приемлемыми, могут быть использованы в получении солей, пригодных в качестве промежуточных соединений в синтезе соединений согласно изобретению и их фармацевтически приемлемых кислотных аддитивных солей. Соли,полученные из подходящих оснований, включают в себя соли щелочного металла (к примеру, натрия и калия), щелочно-земельного металла (к примеру, магния), аммония и N-(Cl-4 алкил)4+. Изобретение также предусматривает кватернизацию любой из основных азотсодержащих групп указанных здесь соединений. Путм такой кватернизации могут быть получены водо- или маслорастворимые, или диспергируемые продукты. Фармацевтически приемлемые носители,которые могут быть использованы в этих фармацевтических композициях, включают в себя,но не в порядке ограничения, иониты (ионообменники), окись алюминия, стеарат алюминия,лецитин, сывороточный белок, такой как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протамин сульфат, вторичный кислый фосфорнокислый натрий, кислый (орто)фосфат калия,хлористый натрий, соли цинка, коллоидная двуокись кремния, трисиликат магния, поливинилпирролидон, вещества на основании целлюлозы,полиэтиленгликоль, карбоксиметилцеллюлоза,полиакрилаты, воски, полиэтиленполиоксипропилен-блоксополимеры, полиэтиленгликоль и ланолин. Композиции согласно изобретению могут быть введены перорально, парентерально, в виде распыляемого раствора для ингаляции, местно, ректально, через нос, трансбуккально, вагинально или через имплантированный резервуар. Термин парентерально здесь включает в себя подкожный, внутривенный, внутримышечный,интраартикулярный, внутрисуставной, интрастернальный, подоболочечный, внутрипечночный, интралезиональный или внутричерепной инъекционный или инфузионный способы введения. Предпочтительно композиции вводят перорально, внутрибрюшинно или внутривенно. Стерильные инъецируемые формы композиций согласно изобретению могут быть водной или маслянистой суспензией. Эти суспензии могут быть получены известными в соответствующей области способами с применением под 002855 32 ходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный инъецируемый препарат может также быть стерильным инъецируемым раствором или суспензией в нетоксичном парентерально приемлемом разбавителе или растворителе, к примеру, в виде раствора в 1,3-бутандиоле. В число приемлемых наполнителей и растворителей входят вода, раствор Рингера и изотонический раствор хлористого натрия. Кроме того, в качестве растворителя или суспензионной среды обычно применяются стерильные, нелетучие масла. Для этой цели может быть использовано любое лгкое нелетучее масло, в том числе моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и е глицеридные производные, применимы, особенно в полиоксиэтилированном виде, при получении растворов для инъекций, поскольку являются природными фармацевтически приемлемыми маслами, такими как оливковое масло или касторовое масло. Эти масляные растворы или суспензии могут также содержать длинноцепочечный спиртовый разбавитель или диспергатор, такой как карбоксиметилцеллюлоза или похожие диспергирующие средства, обычно используемые при составлении фармацевтически приемлемых лекарственных форм, включая эмульсии и суспензии. Другие обычно используемые сурфактанты,такие как твины, сланы и прочие эмульгирующие средства или усилители биологической доступности, обычно используемые при получении фармацевтически приемлемых тврдых,жидких или иных лекарственных форм, также могут быть использованы в целях получения композиции. Фармацевтические композиции согласно изобретению могут вводиться перорально в любой перорально приемлемой лекарственной форме, включая в себя, но не в порядке ограничения, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения обычно используемые носители включают в себя лактозу и кукурузный крахмал. Как правило, также добавляют смазывающие вещества, такие как стеарат магния. Для перорального введения в форме капсул пригодные разбавители включают в себя лактозу и сухой кукурузный крахмал. Когда водные суспензии требуются для перорального введения, активный ингредиент смешивают с эмульгирующими и суспендирующими веществами. При желании могут также быть добавлены некоторые подсластители, ароматизаторы и красители. Или же фармацевтические композиции согласно изобретению могут быть введены в форме суппозиториев для ректального введения. Они могут быть получены смешиванием агента с нераздражающим эксципиентом, который является твердым при комнатной температуре, но жидким при ректальной температуре и поэтому 33 плавится в прямой кишке, высвобождая лекарственное средство. Такие вещества включают в себя масло какао, пчелиный воск и полиэтиленгликоли. Фармацевтические композиции согласно изобретению могут также вводиться местно,особенно когда объект лечения включает в себя участки или органы, легко доступные для местного прикладывания, включая болезни глаз, кожи или нижний кишечный тракт. Легко получить подходящие композиции для местного применения для каждого из этих участков или органов. Местное применение в случае нижнего кишечного тракта может осуществляться путм разработки ректального суппозитория (смотри выше) или пригодного для клизмы состава. Могут также использоваться местные трансдермальные пластыри. Для местного применения фармацевтические композиции могут быть сформулированы в виде соответствующей мази, содержащей активный компонент, суспендированный или растворнный в одном или более носителях. Носители для местного применения соединений согласно изобретению включают в себя, но не в порядке ограничения, минеральное масло, вазелиновое масло, белый вазелин, пропиленгликоль, производное полиоксиэтилена и полиоксипропилена, эмульгирующий воск и воду. Либо фармацевтические композиции могут быть сформулированы в подходящий лосьон или крем, содержащий активные компоненты, суспендированные или растворнные в одном или более фармацевтически приемлемых носителях. Подходящие носители включают в себя, но не в порядке ограничения, минеральное масло, сорбитанмоностеарат, полисорбат 60, цетиловые сложные эфиры воска, цетеариловый спирт, 2 октилдодеканол, бензиловый спирт и воду. Для офтальмологического применения фармацевтические композиции могут быть сформулированы в виде микронизированных суспензий в изотоническом, рН-отрегулированном стерильном растворе соли или, предпочтительно, в виде растворов в изотоническом,рН-отрегулированном стерильном растворе соли, содержащих или не содержащих консервант,такой как бензилалкониумхлорид. Либо для офтальмологического применения фармацевтические композиции могут быть сформулированы в виде мази, такой как петролатум. Фармацевтические композиции согласно изобретению могут также быть введены с помощью назального аэрозоля или ингаляции. Такие составы получают способами, хорошо известными в области технологий приготовления лекарственных средств, и выпускают в форме растворов в физиологическом растворе, используя бензиловый спирт или другие подходящие консерванты, промоторы абсорбции для повышения биологической доступности, фторугле 002855 34 роды и/или другие общепринятые солюбилизирующие или диспергирующие средства. Количество ингибитора р 38, которое может быть смешано с веществами-носителями для получения разовой дозы лекарственной формы будет варьироваться в зависимости от обрабатываемого организма-хозяина, конкретного способа введения. Предпочтительно композиции должны быть сформулированы так, чтобы пациенту,принимающему эти составы, могла быть введена доза ингибитора в пределах 0,01-100 мг/кг веса тела/день. Следует также понимать, что специфическая дозировка и лечебная схема для каждого конкретного пациента будет зависеть от различных факторов, включающих в себя активность используемого конкретного соединения, возраст, вес тела, общее состояние здоровья, пол,рацион, время введения, скорость выведения из организма, сочетание лекарственных средств и тяжести конкретного заболевания, требующего лечения. Количество ингибитора также будет зависеть от конкретного соединения в композиции. Согласно другому варианту воплощения,изобретение дат способ лечения или профилактики р 38-опосредованного состояния, включающий в себя стадию введения пациенту одного из вышеуказанных фармацевтических составов. Термин "пациент", как он здесь использован, подразумевает животное, преимущественно человека. Предпочтительно этот способ используют,чтобы вылечить или предупредить состояние,отобранное из группы, включающей в себя воспалительные болезни, аутоиммунные нарушения, деструктивные процессы кости, пролиферативные нарушения, инфекционные болезни,дегенеративные расстройства, аллергию, реперфузию/ишемию при мозговом ударе, сердечные приступы, ангиогенные нарушения, гипоксию органов, васкулярную гиперплазию, сердечную гипертрофию и тромбин-индуцируемую агрегацию тромбоцитов. Согласно другому варианту воплощения,ингибиторы согласно изобретению используют,чтобы вылечить или предупредить IL-1, IL-6,IL-8 или TNF-опосредованные болезнь или состояние. Такие состояния описаны выше. В зависимости от конкретного р 38 опосредованного состояния, которое лечат или предупреждают, дополнительные лекарственные средства, которые обычно вводят для лечения или профилактики этого состояния, могут быть введены вместе с ингибиторами согласно изобретению. К примеру, при лечении пролиферативных нарушений химиотерапевтические средства или другие антипролиферативные средства можно сочетать с р 38-ингибиторами согласно изобретению. 35 Эти дополнительные средства можно вводить отдельно от содержащего р 38-ингибитор состава, в рамках схемы прима многих лекарственных средств. Или же эти средства могут входить в разовую дозу лекарственной формы,смешанные вместе с р 38-ингибитором в один состав. Последующие примеры приведены с целью более полного понимания описанного здесь изобретения. Следует понимать, что эти примеры даются только в иллюстративных целях и не предусматриваются как ограничивающие каким-либо образом данное изобретение. Пример 1. Синтез соединения 1, ингибитора р 38. В последующих 4 примерах приведены примеры синтеза некоторых соединений формулы Iа. К суспензии амида натрия, 90% (1,17 г, 30 ммоль) в сухом тетрагидрофуране (20 мл), добавляли раствор бензилцианида (2,92 г, 25,0 ммоль) в сухом тетрагидрофуране (10 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 30 мин. К реакционной смеси добавляли раствор 3,6 дихлорпиридазина (3,70 г, 25,0 ммоль) в сухом тетрагидрофуране (10 мл). После перемешивания в течение 30 мин реакционную смесь разбавляли насыщенным водным раствором бикарбоната натрия. Затем реакционную смесь экстрагировали этилацетатом. Слои разделяли, и органический слой промывали водой, раствором соли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток хроматографически очищали на силикагеле (элюент: 30% этилацетат в нгексане), получая 3,71 г (16,20 ммоль,54%) продукта в виде белого тврдого вещества. К суспензии гидрида натрия, 95% (0,14 г,6,0 ммоль) в сухом тетрагидрофуране (10 мл) добавляли тиофенол (0,66 г, 6,0 мл) при комнатной температуре. Затем реакционную смесь перемешивали в течение 10 мин. К реакционной смеси добавляли раствор продукта из стадии А.,см. выше (1,31 г, 5,72 ммоль) в абсолютном этаноле (20 мл). Затем реакционную смесь доводили до температуры кипения с обратным холодильником и перемешивали в течение одного часа. Охлажднную реакционную смесь концентрировали в вакууме. Остаток разбавляли 1 н. раствором гидроокиси натрия (10 мл), затем экстрагировали метиленхлоридом. Органическую фазу промывали водой, раствором соли, 002855 36 сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток хроматографически очищали на силикагеле (элюент: 20% этилацетат в нгексане), получая 0,66 г (2,19 мысль,40%) продукта в виде белого тврдого вещества. Смесь продукта стадии В. (0,17 г, 0,69 ммоль) и концентрированной серной кислоты (5 мл) нагревали до 100 С в течение одного часа. Раствор охлаждали, и рН доводили до 8,0 с помощью насыщенного раствора бикарбоната натрия. Реакционную смесь экстрагировали метиленхлоридом. Органический слой промывали водой, раствором соли, сушили над сульфатом магния и концентрировали в вакууме, получая 0,22 г (0,69 ммоль,100%) соединения пре-1 в виде оранжевого масла. 1 Н-ЯМР (500 МГц, CD3OD) d 7,7 (д), 7,5 Раствор пре-1 из стадии С (0,22 г, 0,69 ммоль) и диметилацеталя N,N-диметилформамида (0,18 г, 1,5 ммоль) в толуоле (5 мл) нагревали при 100 С в течение одного часа. После охлаждения полученный твердый продукт отфильтровывали и растворяли в тплом этилацетате. Продукт осаждали, добавляя по каплям диэтиловый эфир. Затем продукт отфильтровывали и промывали диэтиловым эфиром, получая 0,038 г соединения 1 (приведенного в таблице 1) в виде жлтого тврдого вещества. 1 Вышеуказанный промежуточный продукт получали способом, аналогичным описанному в примере 1 В. Это дат 0,49 г (1,5 ммоль,56%) продукта.(д). Пример 4. Получение соединения 5, ингибитора р 38. Вышеуказанный промежуточный продукт получали способом, аналогичным описанному в примере 1 С. Это дат 0,10 г (0,29 ммоль,45%) соединения пре-2. 1 Первый промежуточный продукт в синтезе соединения 6 получали способом, аналогичным описанному в примере 1 А, используя 2,6 дихлорфенилацетонитрил, что дат 2,49 г (8,38,28%) продукта. Конечную стадию синтеза соединения 6 Первый промежуточный продукт в синтезе соединения 5 получали способом, аналогичным описанному в примере 1 А, используя 2, 4 дихлорфенилацетонитрил, что дат 3,67 г (12,36 ммоль, 49%) продукта. Второй промежуточный продукт получали способом, аналогичным описанному в примере 1 В. Это дат 3,82 г (9,92 ммоль, 92%) продукта. Конечную стадию синтеза соединения 5H-ЯМР (500 МГц, CDCl) d 8,64 (с), 7,517,42 (м), 7,32-7,21 (м), 6,85 (д), 6,51 (д), 2,42 (с). Другие соединения формулы Iа согласно изобретению могут быть синтезированы аналогичным способом с применением соответствующих исходных материалов. Пример 5. Получение соединения формулы Ib, ингибитора А р 38. Пример синтеза ингибитора р 38 формулыIb согласно изобретению представлен ниже. К суспензии амида натрия, 90% (1,1 экв.) в сухом тетрагидрофуране, добавляли раствор 2,6-дихлорбензилцианида (1,0 экв.) в сухом тетрагидрофуране при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 30 мин. К реакционной смеси добавляли раствор 2,6-дихлорпиридина (1 экв.) в сухом тетрагидрофуране. Реакцию контролировали ТСХ, и после е завершения реакционную смесь разбавляли насыщенным водным раствором бикарбоната натрия. Затем реакционную смесь экстрагировали этилацетатом. Слои разделяли и органический слой промывали водой, раствором соли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток хроматографически очищали на силикагеле, получая чистый продукт. фильтровали и растворяли в тплом этилацетате. Продукт осаждали, добавляя по каплям диэтиловый эфир. Затем продукт отфильтровывали и промывали диэтиловым эфиром, получая ингибитор р 38 формулы Ib. Конечный продукт дополнительно очищали с помощью гельхроматографии на силикагеле. Другие соединения формулы Ib согласно изобретению синтезировали аналогичным способом, используя соответствующие исходные материалы. Пример 6. Синтез соединения 103, ингибитора р 38 К раствору 4-фтор-бромбензола (1 экв.) в сухом тетрагидрофуране при -78 С добавляли трет-бутиллитий (2 экв., раствор в гексане). Реакционную смесь перемешивали в течение 30 мин. К реакционной смеси добавляли раствор продукта стадии А (1 экв.) в сухом ТГФ. Затем проводили контроль реакционной смеси и медленно приводили реакционную смесь к комнатной температуре. Реакционную смесь разбавляли водой для остановки реакции и затем экстрагировали метиленхлоридом. Органическую фазу промывали водой, раствором соли, сушили над сульфатом магния и концентрировали в вакууме. Остаток хроматографически очищали на силикагеле, получая продукт. В этом примере представлен типичный синтез соединения формулы Iс. А. Соединение 12, ингибитор р 38, получали в основном, как описано в примере 4, за тем исключением, что на стадии В применяли 4 фтортиофенил. В. Соединение 12 растворяли в сухом ТГФ(5 мл) при комнатной температуре. К этому раствору добавляли гидрид диизобутилалюминия(1 М раствор в толуоле, 5 мл, 5 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Затем реакционную смесь разбавляли этилацетатом, и реакцию останавливали добавлением сегнетовой соли. Слои разделяли, и органический слой отделяли,промывали водой, раствором соли, сушили над сульфатом магния и фильтровали, получая неочищенное соединение 103. Сырой продукт подвергали хроматографии на силикагеле,элюируя 2% метанолом в метиленхлориде. Таким образом получали чистое соединение 103 Смесь продукта стадии В и концентрированной серной кислоты нагревали до 100 С в течение одного часа. Раствор охлаждали и рН доводили до 8,0 с помощью насыщенного раствора бикарбоната натрия. Реакционную смесь экстрагировали метиленхлоридом. Органический слой промывали водой, раствором соли,сушили над сульфатом магния и концентрировали в вакууме, получая продукт. Конечный продукт флэш-хроматографически очищали на силикагеле. Раствор продукта стадии С (1 экв.) и диметилацеталя N,N-диметилформамида (2 экв.) в толуоле нагревали при 100 С в течение одного часа. После охлаждения реакционную смесь(5,9 г, 31,8 ммоль), растворяли в ДМФ (20 мл) при комнатной температуре. Затем добавляли гидрид натрия (763 мг, 31,8 ммоль), что приводило к окрашиванию раствора в ярко-жлтый цвет. Через 15 мин добавляли раствор 2,5 дибромпиридина (5,0 г, 21,1 ммоль) в ДМФ (10 мл) с последующим добавлением палладийтетракис(трифенилфосфин)а (3 ммоль). Затем раствор кипятили с обратным холодильником в 41 течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом. После этого органический слой отделяли,промывали водой и затем раствором соли, сушили над сульфатом магния, фильтровали и упаривали в вакууме, получая неочищенное масло. Элюирование с флэш-хроматографической колонки с помощью 10% этилацетата в н-гексане дат продукт (5,8 г, 84%) в виде не совсем белого тврдого вещества. Полученный на стадии А бромид (194,8 мг,0,57 ммоль) растворяли в ксилоле (15 мл). К этому раствору добавляли тиофенилстаннан(200 мкл, 587 ммоль) и палладий-тетракис (трифенилфосфин) (25 мг). Раствор кипятили с обратным холодильником в течение ночи, охлаждали, фильтровали и упаривали в вакууме. Сырой продукт хроматографировали на силикагеле, элюируя метиленхлоридом, получая чистый продукт (152 мг, 72%) в виде жлтого масла. Полученный на стадии В нитрил (1,2 г,3,37 ммоль) растворяли в ледяной уксусной кислоте (30 мл). К этому раствору добавляли воду(120 мкл, 6,67 ммоль) и впоследствии четырххлористый титан (760 мкл, 6,91 ммоль), что приводило к экзотермической реакции. Затем раствор кипятили с обратным холодильником в течение двух часов, охлаждали и выливали в 1 н. НСl. Водный слой экстрагировали метиленхлоридом. Органический слой промывали обратной струй 1 н. NaOH, сушили над сульфатом магния и фильтровали через слой силикагеля. Силикагель сначала промывали метиленхлоридом для удаления не прореагировавших исходных соединений, а затем этилацетатом, получая соединение 201. Этилацетат упаривали, получая чистое соединение 201 (1,0 г, 77%). Пример 8. Синтез соединения 110, ингибитора р 38. Исходный нитрил (3,76 г, 11,1 ммоль) сначала растворяли в ледяной уксусной кислоте (20 мл). К этому раствору добавляли четыреххлористый титан (22,2 ммоль) и воду (22,2 ммоль) и кипятили с обратным холодильником в течение 1 ч. Затем реакционную смесь охлаждали и разбавляли водой/этилацетатом. Затем отделяли органический слой, промывали раствором соли 42 и сушили над сульфатом магния. После чего органический слой отфильтровывали и упаривали в вакууме. Полученный сырой продукт хроматографировали на силикагеле, элюируя 5% метанолом в метиленхлориде, получая чистый продукт в виде желтой пены (2,77 г, 70%). Полученный на стадии А амид (1,54 г, 4,3 ммоль) растворяли в толуоле (20 мл). Затем добавляли диметилацеталь N,N-диметилформамида (1,53 г, 12,9 ммоль), нагревали полученный раствор в течение 10 мин и потом оставляли охлаждаться до комнатной температуры. Затем реакционную смесь упаривали в вакууме, и остаток хроматографировали на силикагеле,элюируя 2-5% метанолом в метиленхлориде. Выделенный продукт затем растворяли в горячем этилацетате. Раствор оставляли охлаждаться, при этом происходила кристаллизация чистого продукта в виде жлтого тврдого вещества(600 мг, 40%). Дополнительное количество вещества (800 мг) выделяли из маточного раствора. Бромид из стадии В (369 мг, 1 ммоль) растворяли в ТГФ (10 мл). Затем добавляли диизобутилалюмогидрид (1,0 М раствор, 4 ммоль),перемешивали реакционную смесь при комнатной температуре в течение 10 мин и потом останавливали реакцию добавлением метанола (1 мл). После чего добавляли насыщенный раствор сегнетовой соли и смесь экстрагировали этилацетатом. Органический слой отделяли, сушили над сульфатом магния, упаривали и остаток хроматографировали на силикагеле, элюируя 13% метанолом в метиленхлориде, получая яркооранжевый тврдый продукт (85 мг, 23% выход). Полученный на стадии С бромид (35,2 мг,0,1 ммоль) растворяли в ксилоле (12 мл). К этому раствору добавляли тиофенол (0,19 ммоль) и впоследствии метилат трибутилолова (0,19 ммоль). Полученный раствор кипятили с обратным холодильником в течение 10 мин с последующим добавлением палладий-тетракис (трифенилфосфина) (0,020 ммоль). Реакционную смесь нагревали и контролировали расходова 43 ние (исчезновение) исходного бромидного вещества. Затем реакционную смесь охлаждали до комнатной температуры и пропускали через слой силикагеля. Силикагель сначала промывали метиленхлоридом для удаления избытка олово-реагента и затем 5% метанолом в этилацетате для извлечения ингибитора р 38. Фильтрат концентрировали и затем повторно хроматографировали на силикагеле, используя в качестве элюента 5% метанол в этилацетате и получая при этом чистое соединение 110 (20 мг, 52%). Пример 9. Синтез соединения 202, ингибитора р 38. Исходный нитрил (2,32 г, 12 ммоль) растворяли в ДМФ (10 мл) при комнатной температуре. Затем добавляли гидрид натрия (12 ммоль), что приводило к получению раствора,окрашенного в ярко-жлтый цвет. Через 15 мин добавляли раствор 2,6-дибромпиридина (2,36 г,10 ммоль) в ДМФ (5 мл) с последующим добавлением палладий-тетракис(трифенилфосфин)а(1,0 ммоль). Затем раствор кипятили с обратным холодильником в течение 3 ч. Потом реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом. Органический слой отделяли, промывали водой и раствором соли, сушили над сульфатом магния, фильтровали и упаривали в вакууме, получая неочищенное масло. Элюирование с флэш-хроматографической колонки с помощью 10% этилацетата в н-гексане дат продукт (1,45 г, 42%) в виде белого тврдого вещества. Полученное на стадии А бромосоединение(1,77 г, 5,2 ммоль) растворяли в толуоле (20 мл),и образовавшийся раствор дегазировали. В атмосфере азота добавляли раствор фенилборной кислоты (950 мг, 7,8 ммоль) в этаноле (4 мл) и раствор карбоната натрия (1,73 г, 14 ммоль) в воде (4 мл). Реакционную смесь кипятили с обратным холодильником в течение одного часа и затем охлаждали до комнатной температуры. Реакционную смесь разбавляли этилацетатом и промывали водой и раствором соли. Затем органический слой сушили над сульфатом магния,фильтровали и концентрировали в вакууме. Остаток очищали на силикагеле, элюируя 30% этилацетатом в гексане, получая продукт в виде белого твердого вещества (1,56 г, 88%). Нитрил, полученный на стадии В (700 мг,2,07 ммоль), растворяли в концентрированной серной кислоте (10 мл) и нагревали до 80 С в течение 1 ч. Затем реакционную смесь охлаждали до комнатной температуры и рН доводили до 8,0, используя 6 н гидроокись натрия. После чего смесь экстрагировали этилацетатом. Органический слой отделяли, сушили над сульфатом магния и упаривали в вакууме, получая соединение 202 в виде жлтой пены (618 мг, 84%). Пример 10. Синтез соединения 410. В высушенной над пламенем 100 мл круглодонной колбе к 50 мл безводного тетрагидрофурана добавляли 2,28 г (93,8 ммоль) магниевой стружки. Добавляли один кристалл йода, дающий светло-коричневый цвет. К раствору добавляли 1,5 мл из 10,0 мл (79,1 ммоль) образца 2-бром-5-фтортолуола. Раствор кипятили с обратным холодильником. Коричневый цвет блекнул, а кипение продолжалось после удаления внешнего источника нагрева, свидетельствуя об образовании реактива Гриньяра. Как только кипение затихало, добавляли другую 1,0-1,5 мл порцию бромида, вызывающую энергичное кипение. Процедуру повторяли до тех пор, пока был добавлен весь бромид. Коричневатозелный раствор нагревали снаружи до температуры кипения с обратным холодильником в течение одного часа для уверенности в завершении реакции. Раствор охлаждали в ледяной бане и шприцом добавляли к раствору 9,3 мл (81,9 ммоль) триметилбората в 100 мл тетрагидрофурана при -78 С. После добавления реактива Гриньяра колбу удаляли из охлаждающей бани и раствор перемешивали при комнатной температуре в течение ночи. Серовато-белую суспензию выливали в 300 мл H2O и летучие продукты упаривали в вакууме. Добавляли НСl (400 мл 2 н. раствора) и молочно-белую смесь перемешивали в течение одного часа при комнатной температуре. В результате осаждается белый тврдый продукт. Смесь экстрагировали диэтиловым эфиром, и органический экстракт сушили(94%) бороновой кислоты в виде белого тврдого вещества. В 100 мл круглодонной колбе 7,92 г (33,4 ммоль) 2,6-дибромпиридина растворяли в 50 мл безводного толуола, получая прозрачный, бесцветный раствор. Добавляли 4-фтор-2-метилбензолбороновую кислоту (5,09 г, 33,1 ммоль),полученную на стадии А, что приводило к образованию белой суспензии. Добавляли карбонат таллия (17,45 г, 37,2 ммоль) и впоследствии каталитическое количество (150 мг) Рd(РРh3)4. Смесь нагревали до температуры кипения с обратным холодильником в течение ночи, охлаждали и фильтровали через слой силикагеля. Кремнезм промывали CH2Cl2, и фильтрат упаривали, получая белое тврдое вещество. Тврдый продукт растворяли в минимальном количестве 50% СН 2 Сl2/гексана и хроматографировали на короткой колонке с силикагелем, используя 30% СН 2 Сl2/гексан, и получали 6,55 г(74%) 2-бром-6-(4-фтор-2-метилфенил)пиридина в виде белого тврдого вещества. В 50 мл круглодонной колбе 550 мг (2,07 ммоль) 2-бром-6-(4-фтор-2-метилфенил)пиридина, полученного на стадии В, растворяли в 30 мл безводного тетрагидрофурана, получая прозрачный, бесцветный раствор. Добавляли 2,6 дифторанилин (2,14 мл, 2,14 ммоль) и впоследствии - 112 мг (2,79 ммоль) 60% суспензии NaH в минеральном масле. Наблюдается выделение газа наряду с умеренной экзотермической реакцией. Раствор кипятили в течение ночи и затем охлаждали. Реакционную смесь выливали в 10%NH4Cl и экстрагировали CH2Cl2. Органический экстракт сушили (МgSO4) и упаривали в вакууме, получая коричневое масло, являющееся смесью продукта и исходного вещества. Вещество хроматографировали на короткой колонке с силикагелем, используя 50% СН 2 Сl2/гексан, и получали 262 мг (40%) 2-(2,6-дифторфенил)-6-(4 фтор-2-метилфенил)пиридина в виде бесцветного масла. В 100 мл круглодонной колбе 262 мг (834 ммоль) 2-(2,6-дифторфенил)-6-(4-фтор-2-метилфенил)пиридина, полученного на стадии С, растворяли в 30 мл безводного СНСl3, получая прозрачный, бесцветный раствор. Добавляли хлорсульфонилизоцианат (1,0 мл, 11,5 ммоль) и 46 светло-жлтый раствор перемешивали при комнатной температуре в течение ночи. Добавляли воду ( 30 мл), вызывающую умеренную экзотермическую реакцию и энергичное выделение газа. После перемешивания в течение ночи органический слой отделяли, сушили (MgSO4) и упаривали в вакууме, получая коричневое масло, являющееся смесью продукта и исходного вещества. Вещество хроматографировали на короткой колонке с силикагелем, используя 10%EtOAc/CH2Cl2. Полученный обратно исходный материал аналогичным образом вновь обрабатывали в условиях реакции и очищали, получая в общей сложности 205 мг (69%) карбамида в виде белого тврдого вещества. Пример 11. Синтез соединения 138. Соединение 103 (106 мг, 0,25 ммоль) растворяли в ТГФ (0,5 мл), и к этому раствору добавляли триэтиламин (35 мкл, 0,25 ммоль) и,впоследствии, избыток формальдегида (37% водный раствор, 45 мг). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Затем реакционную смесь упаривали на роторном испарителе при пониженном давлении, и остаток растворяли в метиленхлориде и переносили во флэш-колонку с силикагелем. Колонку элюировали 2% метанолом в метиленхлориде, получая чистый продукт (78 мг, 70% выход). Пример 12. Синтез соединения 103. Соединение 138 (1 эквивалент) растворяли в метиленхлориде, и к раствору добавляли триэтиламин (1 эквивалент) и, впоследствии, хлористый дибензилфосфонил (1 эквивалент). Раствор перемешивали при комнатной температуре и контролировали ТСХ расход исходного соединения. Затем слой метиленхлорида разбавляли этилацетатом и промывали 1 н. НСl, насыщенным бикарбонатом натрия и насыщеннымNaCl. Затем органический слой сушили, упаривали на роторном испарителе, и сырой продукт чистили на силикагеле. Потом чистый продукт растворяли в метаноле и снимали защиту с дибензилового эфира с применением 10% палладия на углероде в атмосфере водорода. Когда контроль реакции свидетельствовал о е завершении, катализатор отфильтровывали через целит, и фильтрат упаривали на роторном испарителе, получая фосфатный продукт. 48 силикагеле, элюируя этилацетатом в гексане в градиенте 10-25%, что дат целевой продукт Соединение 103 (210 мг, 1,05 ммоль) растворяли в ТГФ (2 мл) и охлаждали до -50 С в атмосфере азота. К этому раствору добавляли литийгексаметилдисилазан (1,1 ммоль) и впоследствии - хлористый хлорацетил (1,13 ммоль). Реакционную смесь убирали из охлаждающей бани, давая нагреться до комнатной температуры; по прошествии этого времени реакционную смесь разбавляли этилацетатом, и реакцию останавливали добавлением воды. Органический слой промывали раствором соли, сушили и упаривали досуха на роторном испарителе. Сырой продукт флэш-хроматографировали на силикагеле, используя 25% этилацетат в гексане в качестве элюента, получая 172 мг (70%) чистого целевого продукта, который в таком виде использовали в следующих реакциях. Соединение хлорацетила растворяли в метиленхлориде и обрабатывали избытком диметиламина. Реакцию контролировали ТСХ, и по завершении реакции все летучие продукты удаляли, получая целевой продукт. Пример 13. Синтез соединений 34 и 117. Нитрил из примера 5, стадии А, (300 мг,1,0 ммоль) растворяли в этаноле (10 мл), и к этому раствору добавляли тиокарбамид (80,3 мг,1,05 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 4 ч: к этому времени ТСХ показывает, что все исходное соединение израсходовалось. Реакционную смесь охлаждали, и все летучие вещества удаляли при пониженном давлении, а остаток растворяли в ацетоне (10 мл). Затем к этому раствору добавляли 2,5 дифторнитробензол (110 мкл, 1,01 ммоль) и,впоследствии, карбонат калия (200 мг, 1,45 ммоль) и воду (400 мкл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Затем реакционную смесь разбавляли метиленхлоридом (25 мл) и фильтровали через слой ваты. Все летучие продукты удаляли при пониженном давлении, и остаток подвергали флэш-хроматографии на Нитрилсодержащий продукт из стадии А(142 мг, 0,33 ммоль) смешивали с концентрированной серной кислотой (2 мл), кипятили с обратным холодильником в течение 1 ч и затем оставляли охлаждаться до комнатной температуры. После этого смесь разбавляли этилацетатом и осторожно нейтрализовали насыщенным раствором бикарбоната натрия (водным). Слои разделяли, и органический слой промывали водой, раствором соли и сушили над сульфатом магния. Смесь фильтровали и упаривали досуха. Остаток использовали в следующей стадии без дополнительной очистки (127 мг, 85% выход).Aмид, полученный на стадии В (127 мг,0,28 ммоль), растворяли в ТГФ (3 мл), и к этому раствору добавляли диметилацеталь диметилформамида (110 мкл, 0,83 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 5 мин, затем охлаждали до комнатной температуры. Все летучие продукты удаляли в вакууме, и остаток подвергали флэшхроматографии на силикагеле, элюируя 2,5% метанолом в метиленхлориде, что дат чистое целевое соединение 34 (118 мг, 92%). Раствор гексагидрата дихлорида никеля(0,84 мл/0,84 мл) добавляли к раствору соединения 34 (100,8 мг, 0,22 ммоль) в бензоле (3,4 мл),и этот раствор охлаждали до 0 С. Затем к этому раствору добавляли боргидрид натрия (49 мг,1,3 ммоль). Реакционную смесь перемешивали,давая при этом нагреться до комнатной температуры. Реакционную смесь упаривали в вакууме, и остаток подвергали флэш-хроматографии,элюируя 2% метанолом в метиленхлориде, что дат чистый целевой продукт, соединение 117 49 Продукт вышеуказанной реакции синтезировали, используя способ примера 1, стадии В,применяя хлорпиридазин (359 мг, 1,21 ммоль) и 2,4-дифтортиофенол (176 мг, 1,21 ммоль). Продукт получали после флэш-хроматографии на силикагеле (451 мг, 92%). Вышеуказанную реакцию проводили, как описано в примере 1, стадии С, используя 451 мг исходного соединения и 5 мл концентрированной серной кислоты, что дат указанный продукт (425 мг, 90%). Вышеуказанную реакцию проводили, как описано в примере 1, стадии D, используя исходный амид (410 мг, 0,96 ммоль) и диметилацеталь диметилформамида (3 ммоль). Реакционную смесь нагревали до 50 С в течение 30 мин и обрабатывали, как описано ранее. Получали соединение 53 (313 мг, 75%). Соединение 34 (213, 0,49 ммоль) растворяли в ТГФ (10 мл), охлаждали до 0 С, и к этому раствору добавляли боран в ТГФ (1 М, 0,6 ммоль). Реакционную смесь перемешивали в течение 30 мин, реакцию обрывали добавлением воды, и реакционную смесь разбавляли этилацетатом. Органический слой промывали водой и раствором соли, сушили и упаривали на роторном испарителе. Остаток очищали на силикагеле, элюируя метанолом в метиленхлориде при градиенте от 1 до 5%, что дат соединение 142(125 мг, 57%). Пример 15. Клонирование р 38-киназы в клетках насекомых. Идентифицировали два сплайсинговых варианта человеческой р 38-киназы, CSBP1 иCSBP2. Используют специфические олигонуклеотидные затравки для амплификации кодирующего участка кДНК CSBP2, применяя в качестве матрицы библиотеку Hela-клеток(Stratagene). Продукт полимеразной цепной реакции клонировали вектор pET-15b (Novagen). Бакуловирусный вектор переноса, pVL-(His)6 002855p38, создавали путм субклонирования XbalBamHI-фрагмента pET-15b-(His)6-р 38 в комплементарные сайты плазмиды pVL1392(Pharmingen). Плазмида pVL-(His)6-p38 направляет синтез рекомбинантного белка, содержащего пептид из 23 остатков (MGSSHHHHHHSSGLVPRGSHMLE, где LVPRGS обозначает сайт расщепления тромбина), гибридизованный со скелетом N-конца р 38, как подтверждено методами секвенирования ДНК и секвенирования Nконца экспрессируемого белка. Монослойную культуру клеток насекомого Spodoptera frugiperda (Sf9) (ATCC) поддерживали в средеTNM-FH (Gibco BPL), обогащенный 10% эмбриональной бычьей сывороткой, в Т-колбе при 27 С. Клетки Sf9 в логарифмической фазе котрансфицировали линейной вирусной ДНК вируса ядерного полиэдроза Autographa califonica (Pharmingen) и вектором переноса pVL(His) 6-p38, используя липофектин (Invitrogen). Индивидуальные рекомбинантные бакуловирусные клоны отбирали с помощью анализа бляшек, применяя 1% низкоплавкую агарозу. Пример 16. Экспрессия и очистка рекомбинантной р 38-киназы. Клетки (Invitrogen) Trichoplusia ni (Tn-368)Excel-405, не содержащей белка (JRH Bioscience) во встряхиваемой колбе при 27 С. Клетки при плотности 1,5 х 106 клеток/мл заражали вышеуказанным рекомбинантным бакуловирусом при множественности заражения 5. Уровень экспрессии рекомбинантного р 38 контролировали путм иммуноблоттинга, используя анти-р 38-антитело кролика (Santa Cruz Biotechnology). Клеточную массу собирали через 72 ч после заражения, когда уровень экспрессии р 38 достигал максимума. Замороженную клеточную пасту из клеток,экспрессирующих (Нis)6-меченый р 38, размораживали в 5 объмах буфера А (50 мM NaH2PO4pH 8,0, 200 мM NaCl, 2 мM -маркаптоэтанола,10% глицерина и 0,2 мМ PMSF). После механического разрушения клеток в микрофлюидизаторе лизат центрифугировали при 30 000 об/мин в течение 30 мин. Супернатант инкубировали партиями в течение 3-5 ч при 4 С с Таlоnсмолой, имеющей сродство к металлам, при соотношении 1 мл смолы к 2-4 мг ожидаемого р 38. Смолу осаждали центрифугированием при 500 об/мин в течение 5 мин и осторожно промывали порциями буфера А. Смолу суспендировали и выливали в колонку (приблизительно 2,6 х 5,0 см) и затем промывали буфером А + 5 мМ имидазола.(His)6-p38 элюировали буфером А + 100 мМ имидазола и затем диализовали в течение ночи при 4 С против 2 л буфера В, (50 мМ 51 добавлением 1,5 единиц тромбина (Calbiochem) на 1 мг р 38 и инкубированием при 20 С в течение 2-3 ч. Тромбин разрушали добавлением 0,2 мМ PMSF, и затем весь образец загружали в 2 мл колонку с бензамидин-агарозой (AmericanInternational Chemical). При непрерывном потоке фракции непосредственно загружали в 2,6 х 5,0 см колонку сQ-сефарозой (Pharmacia), предварительно уравновешенную буфером В + 0,2 мМ PMSF. р 38 элюировали 0,6 М NaCl в буфере В 20 объемами колонки в линейном градиенте. Элюированный пик белка собирали и диализовали в течение ночи при 4 С против буфера С (50 мМ HEPES рН 7,5, 5% глицерина, 50 мМ NaCl, 2 мМ DTT,0,2 мМ PMSF). Диализованный белок концентрировали вCentriprep (Ami con) до 3-4 мл и вносили в 2,6 х 100 см колонку с сефакрилом S-100HR (Pharmacia). Белок элюировали при скорости потока 35 мл/час. Фракцию основного пика собирали, доводили до 20 мМ DTT, концентрировали до 1080 мг/мл и замораживали в аликвотах при -70 С или использовали непосредственно. Пример 17. Активация р 38. Р 38 активировали, объединяя 0,5 мг/мл р 38 с 0,005 мг/мл с DD-двойным мутантом МКК 6 в буфере В + 10 мМ MgCl2, 2 мМ АТФ, 0,2 мM,Na2VО 4, в течение 30 мин при 20 С. Затем активированную смесь загружали на 1,0 х 10 см колонку MonoQ (Pharmacia) и элюировали 1,0 МNaCl в буфере В 20 объмами колонки в линейном градиенте. Активированный р 38 элюировали затем АДФ и АТФ. Фракцию активированного р 38 пика собирали и диализировали против буфера В + 0,2 мМ Na2VO4 для удаления NaCl. Диализованный белок доводили до 1,1 М фосфата калия добавлением 4,0 М основного раствора и загружали на 1,0 х 10 см колонку с HIC(Rainin Hydropore). Предварительно уравновешенную буфером D (10% глицерина, 20 мМ глицерофосфата, 2,0 мМ DTT) + 1,1 М К 2 НРO4. Белок элюировали буфером D + 50 мМ К 2 НРO4 20 объмами колонки при линейном градиенте. Дважды фосфорилированный р 38 элюировали как фракцию основного пика и собирали для диализа против буфера В + 0,2 мМ Na2VO4. Активированный р 38 хранили при -70 С. Пример 18. Испытания на р 38 ингибирование. А. Ингибирование фосфорилированияEGF-рецепторного пептида. Это испытание проводили в присутствии 10 мМ MgCl2, 25 мМ -глицерофосфата, 10% глицерина и 100 мМ HEPES-буфора при рН 7,6. При типичном определении IC50 получали основной раствор, содержащий все вышеуказанные компоненты и активированный р 38 (5 нМ). В пробирки отбирали аликвоты основного раствора. В каждую пробирку вносили фиксированный объм ДМСО или ингибитора в ДМСО(конечная концентрация ДМСО в реакционной смеси составляла 5%), перемешивали и инкубировали в течение 15 мин при комнатной температуре. К каждой пробирке добавляли EGFрецепторный пептид, KRELVEPLTPSGEAPNQALLR, фосфорил-акцептор в реакции (1) р-38 катализированной киназы, до конечной концентрации 200 мкМ. Реакцию киназы инициировали АТФ (100 мкМ) и пробирки инкубировали при 30 С. Через 30 мин реакцию останавливали добавлением равного объма 10% трифторуксусной кислоты (ТФУ). Фосфорилированный пептид количественно определяли с помощью ВЭЖХ. Отделение фосфорилированного пептида от нефосфорилированного пептида осуществляли на колонке с обращнной фазой (Deltapak, 5 мкм С 18 100D,part. no. 011795) при двойном градиенте воды и ацетонитрила, каждый из которых содержал 0,1% ТФУ. IC50 (концентрацию ингибитора,дающую 50% ингибирование) определяли по графику зависимости % остаточной активности от концентрации ингибитора. В. Ингибирование АТФазной активности. Данное испытание проводили в присутствии 10 мМ MgCl2, 25 мМ -глицерофосфата,10% глицерина и 100 мМ HEPES-буфера при рН 7,6. При типичном определении Ki величинуKm для АТФ в АТФазной активности реакции активированного р 38 определяли в отсутствие ингибитора и в присутствии двух концентраций ингибитора. Получали основной раствор, содержащий все вышеуказанные компоненты и активированный р 38 (60 нМ). В пробирки отбирали аликвоты основного раствора. В каждую пробирку вносили фиксированный объем ДМСО или ингибитора в ДМСО (конечная концентрация ДМСО в реакционной смеси составляла 2,5%), перемешивали и инкубировали в течение 15 мин при комнатной температуре. Реакцию инициировали, добавляя различные концентрации АТФ, и затем инкубировали при 30 С. Через 30 мин реакцию останавливали добавлением 50 мкл EDTA (0,1 М, конечная концентрация), рН 8,0. Продукт активности р 38 АТФазы, АЦФ, определяли количественно ВЭЖХ-анализом. Отделение АДФ от АТФ осуществляли на колонке с обращнной фазой (Supelcosil, LC-18,3 мкм, part. no. 5-8985), используя двойной градиент растворителя следующего состава: растворитель А - 0,1 М фосфатный буфер, содержащий 8 мМ бисульфата тетрабутиламмонияKi определяли из данных о скорости реакции как функцию ингибитора и концентраций АТФ. Результаты для некоторых ингибиторов согласно изобретению приведены ниже в таблице 6. Другие ингибиторы р 38 согласно изобретению также ингибируют АТФазную активность р 38. С. Ингибирование продуцирования IL-1,TNF, IL-6 и IL-8 в LPS-стимулированныхPBMCs. Ингибиторы последовательно разбавляли ДМСО, исходя из 20 мМ исходного продукта. Готовили, по меньшей мере, 6 последовательных разбавлений. Затем получали четырехкратные маточные растворы ингибитора, добавляя 4 мкл ингибитора к 1 мл RPMI1640 среды/10% эмбриональной бычьей сыворотки. Четырехкратные маточные растворы ингибитора содержали ингибитор в концентрациях 80 мкМ, 32 мкМ, 12,8 мкМ, 5,12 мкМ, 2,048 мкМ, 0,819 мкМ, 0,328 мкМ, 0,131 мкМ, 0,052 мкМ, 0,021 мкМ и т.д. Перед применением четырехкратные маточные растворы ингибитора предварительно нагревали до 37 С. Свежие человеческие светлые клетки крови отделяли от других клеток в Vacutainer CPT от BectonDickinson (содержащем 4 мл крови и DPBS без Мg2+/Са 2+ в достаточном для заполнения пробирки количестве) путм центрифугирования при 1500 об/мин в течение 15 мин. Периферийные мононуклеарные клетки крови(PBMCs), расположенные на вершине градиента в Vacutainer, удаляли и дважды промывалиRPMI1640 средой/10% эмбриональной бычьей сыворотки. PBMCs собирали центрифугированием при 500 об/мин в течение 10 мин. Общее число клеток определяли, используя камеру для подсчта клеток крови Neubauer'a и клетки доводили до концентрации 4,8 х 106 клетки/мл в клеточной культуральной среде (RPMI1640,дополненной 10% эмбриональной бычьей сывороткой). Либо цельную, содержащую антикоагулянт, кровь использовали непосредственно в испытании. В каждую ячейку 96-ячеечной пластины для клеточных культур вносили по 100 мкл суспензии клеток или цельной крови. Затем к клеткам добавляли по 50 мкл четырехкратных маточных растворов ингибитора. Наконец, добавляли 50 мкл основного рабочего раствора липополисахарида (LPS) (16 нг/мл в клеточной культуральной среде), чтобы обеспечить конечную концентрацию LPS 4 нг/мл. Общий объм контрольной пробы также доводили до 200 мкл добавлением 50 мкл клеточной культуральной среды. Затем РВМС-клетки или цельную кровь 54 инкубировали в течение ночи (12-15 ч) при 37 С/5% СО 2 в увлажннной атмосфере. На следующий день клетки перемешивали на качалке в течение 3-5 мин перед центрифугированием при 500 об/мин в течение 5 мин. Супернатанты культуры клеток собирали и анализировали методом ELISA на содержание IL-1b(RD Systems, Quantikine kits, DBL50), TNF (BioSource, KHC3012), IL-6 (Endogen, EH2IL6) и IL-8 (Endogen, EH2-IL8) согласно инструкциям изготовителя. Данные ELISA использовали для построения кривых доза-ответ, из которых получали значения IC50. Результаты исследования киназы ("киназа"; подраздел А, см. выше), IL-1 и TNF в LPSстимулированных PBMCs ("клетка") и IL-1, TNF и IL-6 в цельной крови ("WB") для различных р 38-ингибиторов согласно данному изобретению приведены в таблице 7 ниже:TNF значений означает 0,1 мкМ, означает между 0,1 и 0,5 мкМ и "+" означает 0,5 мкМ. Для всех значений в испытании цельной крови ("WB") означает 0,25 мкМ, означает между 0,25 и 0,5 мкМ и "+" означает 0,5 мкМ. Во всех указанных испытаниях в приведенной выше таблице "N.D." означает, что значение не установлено. Другие р 38-ингибиторы согласно изобретению также будут ингибировать фосфорилирование EGF-рецеторного пептида и продуцирование IL-1, TNF и IL-6, равно как IL-8 в LPSстимулированных PBMCs или в цельной крови.IL-8 в IL-1-стимулированных PBMCs. Данное исследование проводили наPBMCs точно так же, как указано выше, за тем исключением, что 50 мкл IL-1b-основного рабочего раствора (2 нг/мл в клеточной культуральной среде) добавляли к пробе вместо маточного рабочего раствора LPS. Супернатанты клеточных культур собирали, как описано выше и анализировали с помощью ELISA на содержание IL-6 (Endogen,EH2-IL6) и IL-8 (Endogen, EH2-IL8) согласно инструкциям изготовителя. Данные ELISA использовали для построения кривых доза-ответ,из которых получали значения IC50. Результаты для соединения 6 - р 38 ингибитора приведены в таблице 8 ниже. Таблица 8 Исследуемый цитокинIL-8 0,85 Е. Ингибирование LPS-индуцированной простагландин-эндопероксидсинтазы-2 (PGHS-2 или СОХ-2) индукции в PBMCs. Мононуклеарные клетки периферической крови человека (PBMCs) выделяли из светлого слоя свежих кровяных сгустков человека путм центрифугирования в Vacutainer CPT (BectonDickinson). Нами высеяны 15 х 106 клетки в 6 ячеечную чашку для культивирования ткани,содержащую RPMI 1640, дополненную 10% эмбриональной бычьей сывороткой, 50 ед/мл пенициллина, 50 мкг/мл стрептомицина и 2 мМL-глутамина. Добавляли соединение 6 (см. выше) при 0,2, 2,0 и 20 мкМ конечных концентрациях в ДМСО. Затем добавляли LPS при конечной концентрации 4 нг/мл, чтобы вызвать экспрессию энзима. Конечный культуральный объм составлял 10 мл/ячейку. После инкубирования в течение ночи при 37 С, 5% СО 2, клетки собирали механически и затем центрифугировали, после чего супернатант удаляли, и клетки дважды промывали охлажднным на льду DPBS (фосфатным буфер 59 ным раствором соли Dulbecco'a, BioWhittaker),клетки лизировали на льду в течение 10 минут в 50 мкл охлажднного лизисного буфера (20 мМTris-HCl, pH 7,2, 150 мМ NaCl, 1% Тритон-Х 100, 1% дезоксихолевая кислота, 0,1% SDS, 1 мМ EDTA, 2% апротинин (Sigma), 10 мкг/мл пепстатин, 10 мкг/мл лейпептин, 2 мМ PMSF, 1 мМ бензамидин, 1 мМ дитиотрейтол), содержащем 1 мкл бензоназы (ДНКаза от Merck). Концентрацию белка в каждом образце определяли,используя метод ВСА (Pierce) и бычий сывороточный альбумин в качестве стандарта. Затем концентрацию каждого образца доводили до 1 мг/мл с помощью охлажднного лизисного буфера. К 100 мкл лизата добавляли равный объм загружающего буфера 2xSDSPAGE, и образец кипятили в течение 5 мин. Белки (30 мкг/ряд) фракционировали по размерам в 4-20% градиенте при СДС электрофорезе в полиакриламидном геле (SDS PAGE) (Nowex) и впоследствии переносили на нитроцеллюлозную мембрану электрофоретическим способом за 2 ч при 100 мА в буфере Towbin (25 мМ Трис,192 мМ глицин), содержащем 20% метанола. Мембрану предварительно обрабатывали в течение 1 ч при комнатной температуре блокирующим буфером (5% обезжиренное сухое молоко в DPBS, дополненном 0,1% Твин-20) и промывали 3 раза в DPBS/0,1% Твин-20. Мембрану инкубировали в течение ночи при 4 С разбавленным 1:250 моноклональным антителом анти-СОХ-2 (Transduction Laboratories) в блокирующем буфере. После 3 промывок вDPBS/0,1% Твин-20 мембрану инкубировали с разбавленной 1:1000 пероксидазой хрена, конъюгированной с овечьей антисывороткой против мышиных иммуноглобулинoв (Ig) (Amersham) в блокирующем буфере в течение 1 ч при комнатной температуре. Затем мембрану снова промывали 3 раза в DPBS/0,1% Твин-20 и применяли систему детектирования ECL (SuperSignal CLHRP Substrate System, Pierce) для определения уровней экспрессии СОХ-2. Результаты вышеуказанного испытания показывают, что соединение 6 ингибирует LPSиндуцированную экспрессию PGHS-2 в PBMCs. Хотя выше представлен ряд вариантов практического воплощения изобретения, очевидно, что основные варианты исполнения могут быть видоизменены, что обеспечит другие варианты воплощения с применением способа согласно изобретению. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы 60 или его фармацевтически приемлемая соль,где каждый из Q1 и Q2 независимо выбран из систем 5-6-членных ароматических карбоциклических или гетероциклических колец или систем 8-10-членных бициклических колец, состоящих из ароматических карбоциклических колец, ароматических гетероциклических колец или сочетания ароматического карбоциклического кольца и ароматического гетероциклического кольца; где Q1 замещен 1-4 заместителями, которые независимо выбраны из галогена; C1-С 3 алкила, возможно замещенного NR'2, OR', CO2R' или CONR'2; O-(C1-С 3)-алкила, возможно замещенного NR'2, OR', CO2R' или CONR'2; NR'2; ОСF3; СF3; NO2; CO2R'; CONR'; SR'; S(O2)NR'2;Q2 возможно замещен 1-4 заместителями,каждый из которых независимо выбран из галогена; C1-С 3-линейного или разветвленного алкила, возможно замещенного NR'2, OR', CO2R',S(O2)NR'2, N=C-NR'2, R3 или CONR'2; O-(C1-С 3)алкила, возможно замещенного NR'2, OR',CO2R', S(O2)NR'2, N=C-NR'2, R3 или CONR'2;(С 2-С 3)-алкенила или алкинила; фенила или фенила, замещенного 1-3 заместителями, независимо выбираемыми из галогена, метокси, циано,нитро, амино, гидрокси, метила или этила;R3 выбран из системы 5-6-членных ароматических карбоциклических или гетероциклических колец; иSO2NR22; или систему 5-6-членных карбоциклических или гетероциклических колец, возможно замещенную NR'2, OR', CO2R', С(О)NR'2 илиS(O2)NR22,Х выбран из -S-, -O-, -S(O2)-, -S(O)-, S(O2)N(R2)-, -N(R2)-S(O2)-, -N(R2)-C(O)O-, -O-C(O)N(R2)-, -C(O)-, -C(O)O-, -O-C(O)-, -C(O)-N(R2)-,- N(R2)-C(O)-, -N(R2)-, -C(R2)2- или -С(OR2)2-; каждый R независимо выбран из водорода,-R2, -N(R2)2, -OR2, SR2, -C(O)-NR22, -S(O2)-NR22 или -С(O)-OR2, где два смежных R возможно связаны друг с другом и вместе с каждым Y, с которым они соответственно связаны, образуют 4-8 членное карбоциклическое или гетероциклическое кольцо, или когда Y означает N, R,присоединенный к нему, означает неподеленную электронную пару;
МПК / Метки
МПК: C07D 487/04, C07D 471/04, A61P 25/28, A61K 31/505, C07F 9/6561
Метки: качестве, гетероциклы, ингибиторов, протеинкиназы, замещенные, азотсодержащие
Код ссылки
<a href="https://eas.patents.su/30-2855-zameshhennye-azotsoderzhashhie-geterocikly-v-kachestve-ingibitorov-proteinkinazy-r38.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные, азотсодержащие гетероциклы в качестве ингибиторов протеинкиназы р38</a>
Замещенные пиридины в качестве избирательных ингибиторов циклооксигеназы-2

Номер патента: 1444
Опубликовано: 23.04.2001
Авторы: Фресен Ричард, Фортин Риджен, Дюб Дэниел, Готье Жак Ив, Вонг Заойин
МПК: A61P 29/00, A61K 31/44, C07D 213/34...
Метки: пиридины, замещенные, циклооксигеназы-2, качестве, ингибиторов, избирательных
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль, где R1 выбран из группы, включающей: (а) СН3, (b) NН2, (c) NНС(O)СF3, (d) NНСН3; Аr обозначает моно-, ди- или тризамещенный фенил или пиридинил (или его N-оксид), где заместители выбраны из группы, включающей: (a) водород, (b) галоген, (c) C1-6-алкокси, (d) C1-6-алкилтио, (е) CN, (f) C1-6-алкил, (g) C1-6-фторалкил, (h) N3, (i) -CO2R3, (j) гидрокси, (k) -C(R4)(R5)-OH, (l)...
N-гидрокси-2-(алкил-, арил- или гетероарилсульфанил, -сульфинил или -сульфонил)-3-замещенные алкилов, арилов или гетероариламидов в качестве ингибиторов матриксных металлопротеиназ

Номер патента: 1742
Опубликовано: 27.08.2001
Авторы: Бэйкер Жанни Леа, Венкатесан Аранапакам Мудумбай, Дэвис Жами Мари, Гросу Джордж Теодор
МПК: A61P 5/48, C07D 211/66, A61K 31/164...
Метки: матриксных, сульфинил, арил, гетероарилсульфанил, арилов, гетероариламидов, алкилов, n-гидрокси-2-(алкил, сульфонил)-3-замещенные, качестве, металлопротеиназ, ингибиторов
Формула / Реферат:
1. Соединения формулы I в которой R1 представляет собой алкил из 1-18 углеродных атомов, необязательно замещенный одной или двумя группами, независимо выбранными из R5; алкенил, содержащий 3-18 углеродных атомов с 1-3 двойными связями, необязательно замещенный одной или двумя группами, независимо выбранными из R5; алкинил, содержащий 3-18 углеродных атомов с 1-3 тройными связями, необязательно замещенный одной или двумя группами,...
Альфа-замещенные арилсульфонамидогидроксамовые кислоты в качестве ингибиторов tnf-альфа и матричных металлопротеиназ

Номер патента: 2019
Опубликовано: 24.12.2001
Автор: Паркер Дейвид Томас
МПК: C07C 311/29, C07D 213/42, A61K 31/44...
Метки: матричных, кислоты, tnf-альфа, качестве, арилсульфонамидогидроксамовые, альфа-замещенные, ингибиторов, металлопротеиназ
Формула / Реферат:
1. Соединение формулы I где Аr обозначает пиридил, R1 обозначает С1-С7алкил, циклоалкил, С1-С7алкокси-С1-С7алкил, фенил, или нафтил, или галоген-С1-С7алкил; R2 обозначает водород или С1-С7алкил; R3 и R4 независимо друг от друга обозначают водород, С1-С7алкил, С1-С7алкокси, галоген, гидрокси, ацилокси, С1-С7алкокси-С1-С7алкокси, трифторметил или циано, или R3 и R4 вместе с соседними атомами углерода обозначают С2-С7алкилендиокси, и n...
Замещенные изохинолины в качестве нервно-мышечных блокаторов ультракраткосрочного действия

Номер патента: 2224
Опубликовано: 28.02.2002
Авторы: Бигхэм Эрик Кливленд, Своринджен Рой Арчибальд, Сэймэно Винсент, Пэйтел Сэнджей Шешикент, Сэйвэриз Джон Джозеф, Борос Эрик Юджин, Бозуэлл Грейди Ивэн
МПК: A61P 41/00, A61K 31/47, C07D 217/20...
Метки: качестве, нервно-мышечных, изохинолины, блокаторов, ультракраткосрочного, замещенные, действия
Формула / Реферат:
1. Соединение формулы (I) где Х представляет собой галоген, h равно числу от 1 до 2, Y представляет собой водород или метокси, Z1 и Z2 представляют собой метил, W1 и W2 представляют собой углерод, и А представляет собой фармацевтически приемлемый анион. 2. Соединение формулы (I), которое включает в себя дихлорид...
Замещенные 2-фенилпиридины в качестве гербицидов

Номер патента: 83
Опубликовано: 25.06.1998
Авторы: Вестфален Карл-Отто, Хайстрахер Элизабет, Вальтер Хельмут, Мисслитц Ульф, Кениг Хартманн, Шэфер Петер, Клинтц Ральф, Хампрехт Герхард
МПК: A01N 43/40, C07D 213/61
Метки: качестве, 2-фенилпиридины, гербицидов, замещенные
Формула / Реферат:
1. Замещенные 2-фенилпиридины общей формулы 1 в которой переменные имеют следующее значение: n обозначает 0 или 1; R1, R3 и R4 независимо друг от друга обозначают водород, галоген, С1-C4алкил, C1-C4галогеналкил, гидрокси, C1-C4алкокси, С1-C4галогеналкокси, нитро, амино, С1-C4алкиламино, ди-(С1-C4 алкил)амино, меркапто, C1-C4алкилтио, С1-C4галогеналкилтио, циано, карбоксил, аминокарбонил, (С1-C4 алкиламино)карбонил или...