Аминопиримидиновые противораковые соединения
Номер патента: 21801
Опубликовано: 30.09.2015
Авторы: Чэнь Синь, Дун Ханьцин, Форман Кеннет, Ли Ань-Ху, Захлер Роберт, Гупта Рамеш К., Столз Кэтрин М., Аппари Рама Деви, Шерман Дэн, Крю Эндрю П., Чилукури Рамеш, Ферраро Катерина, Волк Брайан














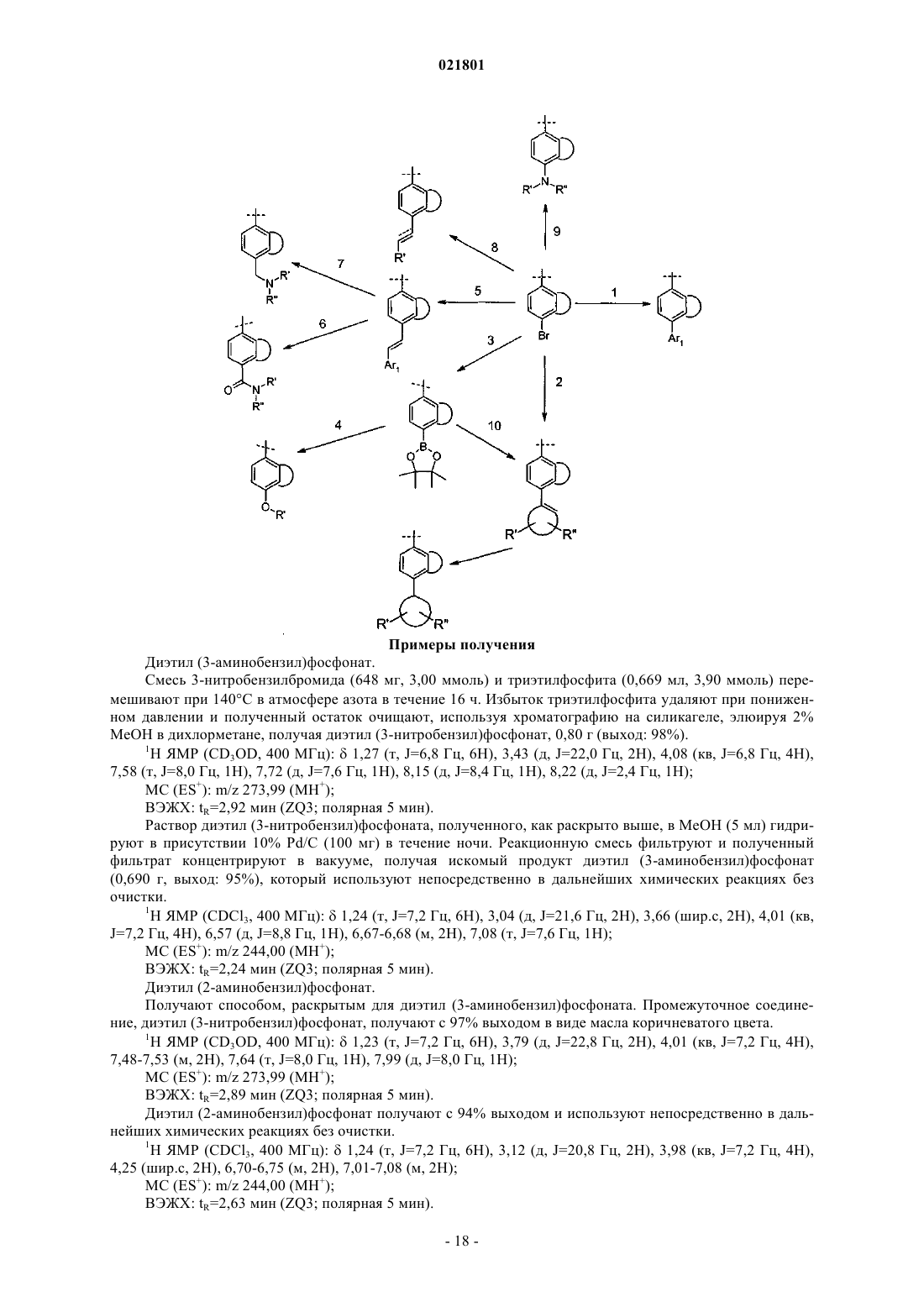











Формула / Реферат
1. Соединение формулы 1а

где Х1 представляет собой СН2, расположенный в мета-положении относительно R6;
Х2 представляет собой -(СН2)0-1-;
R представляет собой

или

А представляет собой СН или N;
R1 представляет собой галоген или -CF3;
R2 представляет собой -Р(О)R9R10;
R3 представляет собой -S(O)2R11, -S(О)2NR11R12, -C(O)NR11R12, -C(O)OR11, -NR11S(O)2R12 или -NR11R12;
R4 представляет собой -x-y-z, где х представляет собой С4-6циклоалкил, С6арил, С4-6гетероарил, содержащий по меньшей мере один гетероатом N, С4-6гетероциклоалкил, содержащий по меньшей мере один гетероатом N; у отсутствует или представляет собой С4-6гетероциклоалкил, содержащий по меньшей мере один гетероатом N; и z отсутствует или представляет собой C1-3алкил, необязательно замещенный 1-2 гидрокси- или С1-6алкоксигруппой, или z представляет собой гидрокси- или -C(O)О-С0-3алкил; или -x-y-z представляет собой С0-6алкокси, С2-6алкенил, С2-6алкинил или С0-6алкил, каждый, необязательно замещенные 1-2 гидроксигруппой;
R5 представляет собой С1-2алкил, необязательно замещенный 1-3 независимыми гидроксигруппами;
R6 представляет собой водород, галоген или С1-3алкокси;
R9 и R10 представляют собой независимо С0-4алкокси;
R11 и R12 представляют собой независимо С0-6алкил, которые, взятые вместе при любом из своих атомов, могут образовывать моноциклический кольцевой фрагмент, содержащий от 5 до 6 углеродов в кольце, содержащий 1-2 гетероатома N; и
R11 или R12 могут независимо, взятые вместе по любому из своих атомов с кольцом, к которому присоединен R3, образовывать кольцо,
или его фармацевтически приемлемая соль.
2. Соединение или его фармацевтически приемлемая соль по п.1, где
Х2 представляет собой СН2;
R представляет собой

R1 представляет собой Cl, Br или -CF3;
R3 представляет собой -N(CH3)S(О)2СН3 или -C(O)NHCH3 и
R6 представляет собой метокси.
3. Соединение или его фармацевтически приемлемая соль по п.2, где
А представляет собой СН;
R1 представляет собой -CF3 и
R3 представляет собой -C(O)NHCH3.
4. Соединение или его фармацевтически приемлемая соль по п.1 формулы

где R6 представляет собой метокси;
R9 и R10 независимо представляют собой С0-3алкокси.
5. Соединение или его фармацевтически приемлемая соль по п.1, где
Х2 представляет собой СН2;
R представляет собой

R1 представляет собой Br, Cl или -CF3;
R4 представляет собой -x-y-z, где х представляет собой С4-6циклоалкил, фенил, пиразолил, пирролидинил, пиперидинил или пиперазинил; у отсутствует или представляет собой пиперидинил или пиперазинил и z отсутствует или представляет собой C1-3алкил, необязательно замещенный 1-2 гидроксигруппами, или z представляет собой гидрокси- или -С(O)O-С0-3алкил; или -x-y-z представляет собой С0-6алкокси, С2-6алкенил, С2-6алкинил или С0-6алкил, причем каждый необязательно замещен 1-2 гидроксигруппами;
R5 представляет собой метил и
R6 представляет собой метокси.
6. Соединение или его фармацевтически приемлемая соль по п.5, где R9 и R10 независимо представляют собой С0-3алкокси.
7. Соединение или его фармацевтически приемлемая соль по п.6, где R4 представляет собой С4-6циклоалкил, который необязательно замещен гидрокси, пиперазинил, который необязательно замещен N-метилом, -C(O)О-С0-3алкил или С1-3алкил, необязательно замещенный 1-2 гидроксигруппами.
8. Соединение или его фармацевтически приемлемая соль по п.5, где
R1 представляет собой -CF3 и
R4 представляет собой -x-y-z, где х представляет собой С4-6циклоалкил, фенил, пиразолил, пирролидинил, пиперидинил или пиперазинил; у отсутствует и z отсутствует или представляет собой С1-3алкил, необязательно замещенный 1-2 гидроксигруппами, гидрокси- или -С(O)O-С0-3алкил.
9. Соединение или его фармацевтически приемлемая соль по п.1, которое представляет собой диэтил (3-метокси-4-{[4-{[2-(метилкарбамоил)фенил]амино}-5-(трифторметил)пиримидин-2-ил]амино}бензил)фосфонат.
10. Соединение или его фармацевтически приемлемая соль по п.1, которое представляет собой гидрохлорид диэтил (4-{[4-{[7-(транс-4-гидроксициклогексил)-2-метил-3-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}-5-(трифторметил)пиримидин-2-ил]амино}-3-метоксибензил)фосфоната.
11. Соединение или его фармацевтически приемлемая соль по п.1, которое представляет собой диэтил (3-метокси-4-{[4-({2-метил-7-[транс-4-(4-метилпиперазин-1-ил)циклогексил]-3-оксо-2,3-дигидро-1H-изоиндол-4-ил}амино)-5-(трифторметил)пиримидин-2-ил]амино}бензил)фосфонат.
12. Соединение или его фармацевтически приемлемая соль по п.1, которое представляет собой 2-{2-[3-(диметилфосфиноилметил)фениламино]-5-трифторметилпиримидин-4-иламино}-N-метилбензамид.
13. Соединение или его фармацевтически приемлемая соль по п.1, которое представляет собой 2-{[2-{[4-(диметилфосфорил)фенил]амино}-5-(трифторметил)пиримидин-4-ил]амино}-N-метилбензамид.
14. Фармацевтическая композиция для лечения рака, опосредованного, по меньшей мере частично, киназой фокальной адгезии (FAK), содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по п.1, составленная с одним или более фармацевтическими носителями.
Текст
Соединения формулы (1a), определенные в настоящем описании, фармацевтически приемлемые соли, способы синтеза, промежуточные соединения, составы и способы лечения ими заболеваний,включая раковые заболевания, опосредованные, по меньшей мере частично, FAK. 021801 Заявка на данное изобретение испрашивает приоритет заявок США 61/182898 (1 июня 2009 г.) и 61/298349 (26 января 2010 г.), содержание которых включено в настоящее описание во всей своей полноте посредством ссылки. Область техники, к которой относится изобретение, и уровень техники Настоящее изобретение относится в значительной степени к лечению раковых заболеваний, к прицельной терапии, к некоторым химическим соединениям, препаратам и способам лечения опухолей и раковых заболеваний указанными соединениями. Киназа фокальной адгезии (FAK) представляет собой цитоплазмическую тирозинкиназу, которая играет главную роль в трансдукции сигнала, передаваемого интегринами, семейством гетеродимерных рецепторов для клеточной адгезии. FAK и интегрины сгруппированы в перимембранных структурах,называемых адгезионными бляшками. Система сигнализации FAK через ERK, PI3K и p130cas играет важную роль в пролиферации, выживании и миграции раковых клеток. Сообщалось, что сверхэкспрессия pFAK и/или FAK наблюдается во многих раковых опухолях. Усиление пролиферации опухолевых клеток in vivo наблюдалось после индуцирования экспрессии FAK в астроцитомных клетках человека. Cary et al., J. Cell Sci., 109:1787-94 (1996).FAK сверхэкспрессируется в случае раковых заболеваний простаты, молочной железы, щитовидной железы, толстой кишки, при меланоме и при раке мозга и легких, причем уровень экспрессии FAK непосредственно коррелирует с опухолями, демонстрирующими наиболее агрессивный фенотип. Weiner et al.,Lancet, 342(8878):1024-25 (1993); Owens et al., Cancer Res., 55:2752-55 (1995); Maung et al., Oncogene,18:6824-28 (1999); Wang et al., J. Cell Sci., 113:4221-30 (2000). FAK проявляет высокую активность в эпителиальных и мезенхимальных опухолях человека, таких как меланома, лимфома и множественная миелома. Повышенное содержание FAK коррелирует с повышенной инвазивностью и повышенной способностью к метастазированию рака. Ингибирование сигнальной системы FAK in vitro индуцирует остановку клеточного роста, снижает подвижность и может вызвать гибель клеток. Было показано, что KD-FAK и DN-FAK ингибируют рост опухоли in vivo. FAK известна также как PTK2. В гепатоцитах TGF вызывает Src-зависимую активацию FAK; и существует доказательство того,что сигнальная система FAK необходима для транскрипционной повышающей регуляции мезенхимальных маркеров и маркеров инвазивности и для делокализации связанного с мембраной Е-кадхерина. Exp.Cell Res., 314, 143-52 (2008). В ряде публикаций раскрыты соединения, которые, как считают, обладают активностью в отношении ингибирования FAK или других киназ, например Cancer Res., 68(6), 1935-1944 (2008), US 6649608,US 6878697, US 7109335, US 7109337, US 7122670, US 7230098, US 7351712, US 7514446, US 7521457,US 2004/0220177, US 2005/0124637, US 2005/0203114, US 2005/0256144, US 2005/0256145,US 2006/0252748, US 2007/0015207, US 2007/0203161, US 2008/0039447, US 2008/0132504,US 2008/0176881, US 2008/0182840, US 2008/0293708, US 2009/0054395, US 2009/0149438,WO 2001/64655, WO 2001/070741, WO 02/096888, WO 2006/021454, WO 2007/021937, WO 2008/051547,WO 2008/094602, WO 2008/094737, WO 2008/129380, WO 2009/020990, WO 2009/071535,WO 2009/105498, WO 2009/143389. Существует необходимость в создании ингибиторов FAK, обладающих потенциалом в отношении достижения клинического и регуляторного одобрения для лечения заболеваний, таких как рак. Сущность изобретения В некоторых аспектах настоящее изобретение относится к соединениям формулы 1 а, как представлено далее: где X1 представляет собой СН 2, расположенный в мета-положении относительно R6; Х 2 представляет собой -(CH2)0-1-;; А представляет собой СН или N;R1 представляет собой галоген или CF3;R4 представляет собой -x-y-z, где х представляет собой С 4-6 циклоалкил, С 6 арил, С 4-6 гетероарил, содержащий по меньшей мере один гетероатом N, С 4-6 гетероциклоалкил, содержащий по меньшей мере один гетероатом N; у отсутствует или представляет собой С 4-6 гетероциклоалкил, содержащий по меньшей мере один гетероатом N; и z отсутствует или представляет собой C1-3 алкил, необязательно замещенный 1-2 гидрокси или C1-6 алкоксигруппой, или z представляет собой гидрокси- или -C(O)О-С 0-3 алкил; или -x-y-z представляет собой С 0-6 алкокси, С 2-6 алкенил, С 2-6 алкинил или С 0-6 алкил, каждый, необязательно замещенные 1-2 гидроксигруппой;R9 и R10 представляют собой независимо С 0-4 алкокси;R11 и R12 представляют собой независимо С 0-6 алкил, которые, взятые вместе при любом из своих атомов, могут образовывать моноциклический кольцевой фрагмент, содержащий от 5 до 6 углеродов в кольце, содержащий 1-2 гетероатома N; иR11 или R12 могут независимо, взятые вместе по любому из своих атомов с кольцом, к которому присоединен R3, образовывать кольцо. В некоторых аспектах соединения настоящего изобретения представляют собой ингибиторы киназ,включая FAK. В некоторых аспектах соединения настоящего изобретения представляют собой селективные ингибиторы FAK. В некоторых аспектах настоящее изобретение включает способы лечения пролиферативных заболеваний, включая раковые заболевания, опосредованные, по меньшей мере частично, FAK, одной или в комбинационных режимах с другими агентами. Настоящее изобретение включает соединения и их соли, любые их физические формы, включая сольваты и гидраты, приготовление соединений, промежуточные соединения и фармацевтические композиции и их лекарственные формы. Подробное описание изобретения Соединения. В некоторых аспектах настоящее изобретение включает соединения представленной выше формулы 1 а, где (подрод 1):R1 представляет собой галоген или -CF3;X1 представляет собой СН 2, расположенный в мета-положении относительно R6; Х 2 представляет собой -(CH2)0-1-;R9 и R10 независимо представляют собой С 0-4 алкокси;R11 и R12 независимо представляют собой С 0-6 алкил, которые, взятые вместе при любом из своих атомов, могут образовывать моноциклический кольцевой фрагмент, содержащий от 5 до 6 углеродов в кольце, содержащий 1-2 гетероатома N; иR11 или R12 могут независимо, взятые вместе по любому из своих атомов с кольцом, к которому присоединен R3, образовывать кольцо;R4 представляет собой -x-y-z, где х представляет собой С 4-6 циклоалкил, С 6 арил, С 4-6 гетероарил, содержащий по меньшей мере один гетероатом N, С 4-6 гетероциклоалкил, содержащий по меньшей мере один гетероатом N; у отсутствует или представляет собой С 4-6 гетероциклоалкил, содержащий по меньшей мере один гетероатом N; и z отсутствует или представляет собой C1-3 алкил, необязательно замещенный 1-2 гидрокси или С 1-6 алкоксигруппой, или z представляет собой гидрокси- или -С(О)О-С 0-3 алкил; или -x-y-z представляет собой С 0-6 алкокси, С 2-6 алкенил, С 2-6 алкинил или С 0-6 алкил, каждый, необязательно замещенные 1-2 гидроксигруппой,или его фармацевтически приемлемая соль. В некоторых вариантах формулы 1 а или подрода 1-4 (подрод 5): Х 2 представляет собой СН 2;-2 021801 В некоторых вариантах формулы 1 а или подрода 4-7 (подрод 8): А представляет собой СН;R3 представляет собой -C(O)NHCH3. В некоторых вариантах формулы 1 а или подрода 1 (подрод 9) соединение или соль по п.1 имеет формулу (подрод 9):R9 и R10 независимо представляют собой С 0-3 алкокси. В некоторых вариантах формулы 1 а или подрода 4 (подрод 10): Х 2 представляет собой СН 2;R4 представляет собой -x-y-z, где х представляет собой С 4-6 циклоалкил, фенил, пиразолил, пирролидинил, пиперидинил или пиперазинил; у отсутствует или представляет собой пиперидинил или пиперазинил и z отсутствует или представляет собой C1-3 алкил, необязательно замещенный 1-2 гидроксигруппами, или z представляет собой гидрокси- или -C(O)О-С 0-3 алкил; или -x-y-z представляет собой С 0-6 алкокси, С 2-6 алкенил, С 2-6 алкинил или С 0-6 алкил, причем каждый необязательно замещен 1-2 гидрокси;R6 представляет собой метокси. В некоторых вариантах формулы 1 а или подрода 10 (подрод 11) R9 и R10 независимо представляют собой С 0-3 алкокси. В некоторых вариантах формулы 1 а или подрода 10 или 11 (подрод 12) R4 представляет собой С 4-6 циклоалкил, который необязательно замещен гидрокси, пиперазинил, который необязательно замещен N-метилом, -С(О)O-С 0-3 алкил, или C1-3 алкил, необязательно замещенный 1-2 гидроксигруппами. В некоторых вариантах формулы 1 а или подродов 10 или 11 (подрод 13):R4 представляет собой -x-y-z, где х представляет собой С 4-6 циклоалкил, фенил, пиразолил, пирролидинил, пиперидинил или пиперазинил; у отсутствует и z отсутствует или представляет собой C1-3 алкил,необязательно замещенный 1-2 гидроксигруппами, гидрокси- или -C(O)О-С 0-3 алкил. В некоторых вариантах формулы 1 а или любого его подрода соединение присутствует в виде вещества в практически чистой форме. В некоторых вариантах соединение выбирают из любого одного из приведенных здесь примеров или его фармацевтически приемлемых солей. Каждое из приведенных выше определений переменных включает любой их набор, и соединения формулы 1 а включают любые комбинации таких переменных или подгруппы переменных. В некоторых аспектах настоящее изобретение включает любые приведенные здесь примеры соединений и их фармацевтически приемлемые соли. Настоящее изобретение включает соединения и их соли и их физические формы, приготовление соединений, полезные промежуточные соединения и фармацевтические композиции и их лекарственные формы. Соединения настоящего изобретения и термин "соединение" в формуле изобретения включают любые фармацевтически приемлемые соли или сольваты и любые аморфные или кристаллические формы или таутомеры, независимо от того, указаны они конкретно в контексте или нет. Настоящее изобретение включает изомеры соединений. Соединения, которые могут содержать один или более асимметрических атомов углерода, могут существовать в виде двух или более стереоизомеров. Если соединение настоящего изобретения содержит алкенильную или алкениленовую группу,возможны геометрические цис/транс (или Z/E) изомеры. Если полученное соединение содержит, например, кетогруппу или группу оксима или ароматический фрагмент, может существовать таутомерный изомеризм ("таутоизомеризм"). Отдельное соединение может существовать в виде изомеров более одно-3 021801 го типа. Настоящее изобретение включает любые стереоизомеры, даже если они конкретно не представлены, индивидуально, а также смеси, геометрические изомеры и их фармацевтически приемлемые соли. Если соединение или стереоцентр указаны или изображены без определенной стереохимии, их следует рассматривать как включающие все возможные индивидуальные изомеры, конфигурации и их смеси. Таким образом, образец соединения, содержащего смесь стереоизомеров, охватывает как упоминание стереоизомеров, так и упоминание без определения стереохимии. Также включены любые цис/трансизомеры или таутомеры раскрытых соединений. В объем настоящего изобретения включены все стереоизомеры, геометрические изомеры и таутомерные формы соединений изобретения, включая соединения, демонстрирующие более одного типа измеризма и смеси одного или более из них. Если существует таутомер соединения формулы (I), полученное соединение формулы (I) настоящего изобретения включает любые возможные таутомеры и их фармацевтически приемлемые соли и их смеси, за исключением случаев, когда конкретно указано иное. Соединения настоящего изобретения не ограничены соединениями, в которых все атомы существуют в их природном изотопном виде. Настоящее изобретение включает соединения, в которых один или более атомов водорода, углерода или других атомов заменены другими их изотопами. Такие соединения могут быть полезны в качестве исследовательских или диагностических инструментов при фармакокинетических исследованиях метаболизма и в анализах связывания. Ссылка на соединение или атом в соединении включает изотопологи, т.е. образцы, в которых атом или соединение отличается только в отношении изотопного обогащения и/или в положении изотопного обогащения. В качестве примера, в некоторых случаях может оказаться желательным заменить один или более атомов водорода дейтерием (D) или заменить атом углерода изотопом 13 С. Другие примеры изотопов, которые можно вводить в соединения настоящего изобретения, включают изотопы водорода, хлора, фтора, йода, азота, кислорода, фосфора и серы. Некоторые меченные изотопами соединения настоящего изобретения можно использовать при исследовании распределения лекарств в субстратах и/или тканях. Замещения тяжелыми изотопами, такими как дейтерий, могут предоставить некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например увеличивая время полужизни или уменьшая необходимые дозировки, и, следовательно, могут оказаться предпочтительными в некоторых обстоятельствах. Замещения эмитирующими позитроны изотопами можно использовать в исследованиях с помощью позитронной эмиссионной топографии (PET) для исследования рецепторной занятости субстрата. Кроме того, соединения могут быть аморфными или могут существовать или их можно получить в различных кристаллических формах или полиморфах, включая сольваты и гидраты. Настоящее изобретение включает любые такие представленные здесь формы, при любой степени чистоты. Указание на соединение per se означает полученное соединение любой неконкретизированной стереохимии, физической формы и независимо от того, связано ли оно с растворителем или водой. Соединения настоящего изобретения могут существовать как в сольватированной, так и в несольватированной формах. Термин "сольват" используют здесь для определения молекулярного комплекса,включающего соединение настоящего изобретения и одну или более молекул фармацевтически приемлемого растворителя, например этанола. Термин "гидрат" используют, если растворителем служит вода. Фармацевтически приемлемые сольваты в соответствии с настоящим изобретением включают гидраты и сольваты, в которых растворитель кристаллизации может быть изотопно замещенным, например D2O,d6-ацетон, d6-ДМСО. Кроме того, в объем настоящего изобретения включены комплексы, такие как клатраты, комплексы включения лекарство-хозяин, где в противоположность вышеуказанным сольватам лекарство и хозяин присутствуют в стехиометрическом или нестехиометрическом количествах. Включены также комплексы лекарственных средств, содержащих два или более органических и/или неорганических компонента, которые могут быть в стехиометрическом или нестехиометрическом количестве. Образованные комплексы могут быть ионизированными, частично ионизированными или неионизированными. Настоящее изобретение включает пролекарства соединений настоящего изобретения, которые могут, после введения пациенту, превращаться в соединения настоящего изобретения, например, в результате гидролитического расщепления. Пролекарства в соответствии с настоящим изобретением можно,например, получить в результате замены соответствующих функциональных групп, присутствующих в соединениях настоящего изобретения, определенными фрагментами, которые хорошо известны специалистам как "про-фрагменты", известными специалистам способами. Особенно благоприятными производными и пролекарствами настоящего изобретения являются такие, которые повышают биодоступность соединений, когда такие соединения вводят пациенту, улучшают доставку исходного соединения в конкретное биологическое место, повышают растворимость, позволяя осуществлять введение путем инъекций, изменяют метаболизм или изменяют скорость экскреции. Фармацевтически приемлемые соли соединений настоящего изобретения можно легко получить,смешивая вместе растворы полученного соединения и нужной кислоты или основания, в зависимости от ситуации. Соль можно высадить из раствора и собрать фильтрованием или можно выделить, выпаривая-4 021801 растворитель. Степень ионизации соли может варьироваться от полностью ионизированной соли до почти неионизированной. Соединения, которые являются основными, могут образовывать соли с широким кругом органических и неорганических кислот. Кислотами, которые можно использовать для получения фармацевтически приемлемых солей присоединения кислот таких основных соединений, являются те, которые образуют приемлемые соли присоединения кислот. Если соединение настоящего изобретения является основным, его соответствующие соли можно легко получить из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают, например, уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, муравьиную, фумаровую, глюконовую, глутамовую, бромисто-водородную, хлористо-водородную, изетионовую,молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памоевую,пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую кислоты и т.п. Другие соли представляют собой аспартат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камсилат,эдисилат, глуцептат, глюкуронат, гексафторфосфат, хибензат, гидробромид/бромид, гидройодид/йодид,малонат, метилсульфат, нафтилат, 2-напсилат, никотинат, оротат, оксалат, пальмитат, гидрофосфат, дигидрофосфат, фосфат, сахарат, стеарат, тартрат, тозилат и трифторацетат. Если полученное соединение настоящего изобретения является кислым, его соответствующие соли можно легко получить из фармацевтически приемлемых оснований, включая неорганические основания и органические основания. Соли, полученные из таких неорганических оснований, включают соли алюминия, аммония, кальция, соли одновалентной и двухвалентной меди, соли двухвалентного и трехвалентного железа, лития, магния, марганца (ic и ous), калия, натрия, цинка и т.п. Соли, полученные из фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, а также циклических аминов и замещенных аминов, таких как встречающихся в природе и синтезированных замещенных аминов. Другие фармацевтически приемлемые органические основания,с которыми можно образовывать соли, включают ионообменные смолы, такие как, например, аргинин,бетаин,кофеин,холин,N',N'-дибензилэтилендиамин,диэтиламин,2-диэтиламиноэтанол,2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, смолы полиаминов, прокаин, пурины, теобром, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Другие примеры включают бензатин, диоламин, глицин, меглумин и оламин. Синтез Настоящее изобретение включает раскрытые здесь соединения, промежуточные соединения, примеры и способы синтеза. Фосфонаты. Схема 1 Получение диалкилфосфонатов можно осуществить множеством способов. Например, один диалкилфосфонат можно превратить в другой, отличающийся диалкилфосфонат, как представлено на схеме 1. Гидролиз диалкилфосфоната в кислотных условиях, таких как концентрированная хлористоводородная кислота, приводит к получению соответствующей фосфоновой кислоты. Другие кислотные реагенты можно использовать для осуществления такой трансформации, включая, но ими не ограничиваясь, HBr и HBr/НОАс. Указанную трансформацию можно также осуществить, используя щелочные условия, например обрабатывая NaOH/MeOH или используя реагенты, такие как, но ими не ограничиваясь, TMSI, TMSBr, TMSCl/NaI и NaI в растворителях, таких как ацетон, ацетонитрил, DCM, хлороформ и диоксан. На схемах X-Y представляет собой соответствующий арил, аралкил или другой фрагмент, как раскрыто в настоящем описании, содержащий специфицированную фосфорную группу. В частности, Y соответствует Х 1 в формуле 1. Каждый R, Rw, Rv представляет собой фосфорный заместитель, как раскрыто и определено в настоящем описании, такой как R9, R10 в формуле 1. Схема 2-5 021801 Арилфосфоновые кислоты можно получить из арилгалогенидов/трифлатов, используя катализаторы - переходные металлы и используя реакцию с аддуктами фосфиновой кислоты с последующим окислением реагентами, такими как перхлорат или перекись водорода (схема 2). Гидролиз диалкилфосфоната до фосфоновой кислоты обычно происходит через промежуточный алкилгидрофосфонат (схема 1), который можно выделить, если остановить реакцию до осуществления полной конверсии. Использование условий основного гидролиза, в частности, приводит к эффективному выделению промежуточного соединения алкилгидрофосфоната. Полученные таким образом фосфоновые кислоты и алкилгидрофосфонаты можно активировать рядом способов до реакции со спиртами, получая необходимые диалкилфосфонаты или алкилалкилфосфинаты соответственно. Один из способов активирования включает галогенирование, например используя такие реагенты, как оксалилхлорид, тионилхлорид, оксихлорид фосфора и оксибромид фосфора или в чистом виде или в соответствующих растворителях, таких как DCM и DCE. При желании, к полученной смеси можно добавлять каталитические количества ДМФ. Обработка полученных таким образом фосфиновых дигалогенидов спиртами приводит к получению искомых диалкилфосфонатов. Другой способ активации включает образование "активного сложного эфира", используя реакцию с реагентами, такими как DCC и РуВОР. Специалистам должно быть известно множество других связующих реагентов для активации фосфоновых кислот аналогичным образом. Использование условий Митсунобу, напримерPPh3/DEAD или эквивалентных реагентов, представляет собой следующий способ активации системы фосфоновая кислота/алкилгидрофосфонат, которая после обработки соответствующим спиртом обеспечивает получение нужных диалкилфосфонатов или алкилалкилфосфинатов соответственно. Фосфоновые кислоты можно также превратить в диалкилфосфонаты, используя кислотный катализ, например TsOH, в присутствии алкилового спирта. Схема 3 Диалкилфосфонаты можно также получить, используя способ Михаэлиса-Арбузова, как представлено на схеме 3, в котором алкилгалогениды или сульфонаты (например, Hal=Br, I, Cl, OMs; Y=CR7R8) нагревают с триалкилфосфитами. Схема 4 Фосфонаты, содержащие Р-водород, можно подвергнуть взаимодействию с алкилгалогенидами или сульфонатами (например, Hal=Cl, Br, I, OMs; Y=CR7R8) в присутствии основания, включая, но ими не ограничиваясь, Na2CO3, K2CO3, NaOH, Cs2CO3, Et3N, DIPEA и DBU, и в подходящих растворителях, например в ацетонитриле, ДМФ, диоксане, DMA, получая Р-замещенные продукты в результате химических реакций SN2-типа (схема 4). Кроме того, фосфонаты, содержащие Р-водород, можно подвергнуть взаимодействию с арилгалогенидами или трифлатами (например, Hal=Cl, Br, I, OTf; Y=связь), используя условия с катализаторами на основе переходных металлов. Примеры таких условий включают, но ими не ограничиваются, Pd2(dba)3/XantPhos/Cs2CO3/диоксан. Как и в случае многих реакций, катализируемыхPd(0) или другими переходными металлами, выбор катализатора, лиганд катализатора, основание, растворитель и температура могут быть важными переменными, используемыми для оптимизации таких реакций. Фосфинаты. Схема 5 Алкилфосфинаты можно получить множеством способов, раскрытых выше для алкилфосфонатов. Например, используя условия Михаэлиса-Арбузова, где алкилдиалкоксифосфин подвергают взаимодействию с алкилгалогенидом или сульфонатом (например, Hal=Br, I, Cl, OMs; Y=CR7R8). Аналогично фосфонатам, катализируемые переходными металлами реакции фосфинатов, содержащих Р-водород, также приводят к получению производных фосфинатов при взаимодействии с арилгалогенидами или трифлатами (например, Hal=Br, I, Cl, OMs; Y=связь). Реакция фосфинатов, содержащих Р-водород, с алкилгалогенидами или сульфонатами (например, Hal=Cl, Br, I, OTf; Y=Y=CR7R8) в присутствии основания также происходит в результате SN2 реакций по типу реакций с фосфонатами (схема 6). Фосфиноксиды. Схема 7 Фосфиноксиды можно получить, используя несколько способов, включая способ через реакцию диалкилфосфонатов с углеродными нуклеофилами, включая, но ими не ограничиваясь, реагенты Гриньяра Как и в случае фосфонатов и фосфинатов, оксиды диалкилфосфинов, содержащие Р-водород, можно подвергнуть взаимодействию с алкилгалогенидами или сульфонатами (например, Hal=Br, I, Cl, OMs;Y=CR7R8) в основных условиях или арилгалогенидами (например, Hal=Br, I, Cl, OTf; Y=связь) с катализаторами на основе переходных металлов с образованием Р-замещенных производных (схема 8). Схема 9 Другие способы получения оксидов фосфинов включают окисление фосфинов окисляющими агентами, такими как, но ими не ограничиваясь, перекись водорода (схема 9). Схема 10 Кроме того, диалкилалкоксифосфины можно подвергнуть взаимодействию с алкилгалогенидами или сульфонатами (например, Hal=Br, I, Cl, OMs; Y=CR7R8) с образованием оксидов фосфинов в результате химических реакций типа Михаэлиса-Арбузова (схема 10). Схема 11 Фосфонаты можно алкилировать по альфа-углероду, используя депротонирование сильными основаниями, неограничивающими примерами которых являются t-BuOK, n-BuLi, NaH и LDA, затем обрабатывая углеродными электрофилами, например MeI, ДМФ и хлороформиатами (схема 11) (Rm и/или Rn может быть метилом). Регулирование стехиометрии компонентов реакции может привести к получению моно- или диалкилированных продуктов. Кроме того, последовательное использование основания, электрофила 1, основания и электрофила 2 является другим средством для эффективного регулирования диалкилирования. 5-Замещенные производные 2,4-диаминопиримидина. 2,4-Диаминопиримидины можно синтезировать, используя несколько подходов, получая целевые молекулы, представляющие фармакологический интерес. Далее приведены неограничивающие примеры различных подходов, которые специалисты могут использовать для получения целевых молекул, и примеры типов, раскрытых в данном документе. Коммерчески доступные 5-замещенные 2,4-дихлорпиримидины (А) можно подвергнуть взаимодействию непосредственно с анилинами или аминами в SNAr реакциях, получая смеси монозамещенных продуктов Bi и Bii (схема 12). В зависимости от используемых условий и природы анилина/амина и X группы, может образовываться преобладающий изомер. В таких ситуациях или в условиях минимальной региоселективности изомеры можно разделить (полностью или частично), используя хроматографию и/или кристаллизацию. Отнесение структур к каждому чистому изомеру можно осуществить, используя ЯМР эксперименты, особенно используя НМВС эксперименты (гетероядерная многополосная корреляция), которые могут обнаружить 3-связанную Н-С корреляцию между C4-NH и С 5-С в случае 4-замещенного продукта, что не наблюдается для C2-NH изомера. Альтернативно, смесь региоизомеров(Bi/Bii) можно подвергнуть взаимодействию со вторым анилином/амином, получая смесь продуктовCi/Cii, которую можно разделить, используя такие методики, как препаративная ВЭЖХ (MDP), получая чистые изомеры. Отнесение структуры можно осуществить путем сравнения спектральных данных с теми изомерами, данные для которых были получены описанными выше способами, или непосредственно,используя ЯМР эксперименты. Кроме SNAr замещений групп хлора в положениях 2- и 4-, как указано выше, специалистам известно, что другие группы также могут служить в качестве подходящих уходящих групп, которые можно заменить аминами и анилинами в соответствующих условиях. Примеры замещаемых групп включают,но ими не ограничиваются, алкилтио, алкилсульфонил, бром, трихлорметил, фтор, сульфонилокси иN-бензотриазолилокси и в каждом случае уходящие группы в 2- и 4-положении могут быть одинаковыми или различными. Действительно, в некоторых обстоятельствах может оказаться предпочтительным, чтобы замещаемые группы в 2- и 4-положении были различными, так как это предоставляет возможность извлечь преимущество из различия потенциалов уходящих групп и, таким образом, регулировать региохимию SNAr реакции. Схема 13 Другой подход к получению 2-анилино, 4-анилино или 2,4-дианилинопиримидинов включает арилирование аминопиримидина, катализируемое переходными металлами. В типичном способе 2- или 4-аминопиримидин нагревают с арил- или гетероарилбромидом или йодидом в присутствии йодида меди(I), этилендиаминового лиганда и основания - карбоната калия в диоксане. Другими, обычно используемыми реагентами являются Ph2-пентадиенон-Pd, NaOPh и XantPhos. В зависимости от природы X группы в 5-положении (что соответствует R1 в формуле 1) такие реакции можно осуществлять для 2,4-диаминопиримидинов и при этом наблюдается некоторое предпочтение для одной или другой аминогруппы. Ra может соответствовать аминогруппе NH-X2R формулы 1 или предшественнику; Ar-Rb может соответствовать замещенной фенильной группе формулы 1; Rc может соответствовать аминогруппе формулы 1 или предшественнику; Ar-Rd или HetAr-Rd могут соответствовать NH-X2R формулы 1 или-8 021801 предшественнику. Кроме рефункционализации активированной пиримидиновой системы, нужные производные можно также получить путем конструирования самого пиримидинового кольца. Схема 14 Например, реакция соответствующим образом замещенного гуанидина с соответствующим образом замещенным -формиловым эфиром, -(диметиламино)пропеноатом или -(алкокси)пропеноатом приводит к получению гидроксипиримидина, как указано на представленной выше схеме. Такие гидроксипиримидины можно хлорировать, используя реагенты, такие как POCl3, с образованием промежуточных соединений, аналогичных тем, которые раскрыты на схеме 17 (Bii). Q' замещение в реагентах может быть таким же, как финальная R1 группа формулы 1 (Q) или может отличаться, как требуется для обеспечения успешного осуществления реакций циклизации и/или галогенирования. Если Q' отличается от R1, взаимное превращение Q' в R1 можно осуществить, используя множество способов, известных специалистам,неограничивающий перечень которых раскрыт далее в описании. Схема 15 Другие способы получения соответствующим образом функционализированных производных пиримидина включают модификацию коммерчески доступных урацила или тиоурацила. Например,S-метилирование тиоурацила с использованием йодометана и основания приводит к получению промежуточного соединения, которое можно функционализировать по С 5 положению, чтобы ввести Q или предшественник Q, Q'. Пример такой С 5-функционализации представляет собой галогенирование, с использованием брома в уксусной кислоте, N-йодосукцинимида в ДМФ или N-хлорсукцинимида в уксусной кислоте, что приводит к введению в положение С 5 группы брома, йода или хлора соответственно. Такие группы, как CF3 можно ввести, используя реакцию с CF3I в присутствии FeSO4, Н 2 О 2 и ДМСО. Аналогичные реакции по положению С 5 можно также использовать для самого урацила и производных, превращенных в 4-хлор (для производных тиоурацила), или 2,4-дихлорпиримидиновых производных, используя реакцию с такими реагентами, как POCl3. Взаимное превращение функциональных групп. Различные функциональные группы, существующие в целевых молекулах и примерах (например, X в 5-положении пиримидиновой системы или на подвешенных N-арильных или N-бензильных группах),можно ввести, соответствующим образом выбирая исходные вещества, или если финальная функциональная группа недоступна непосредственно, используя указанный процесс, или если такая функциональность может ставить под угрозу последующие реакции для создания целевой молекулы, можно использовать альтернативные функциональные группы и затем превратить в конечные целевые функциональные группы различными способами и в такие моменты последовательности реакций, которые легко могут определить специалисты. Например, неограничивающий список таких трансформаций включает следующие превращения:CNCO2H (конц. H2SO4),Ar/HetAr-BrAr/HetAr-NR'R" (Pd2(dba)3, DPPF, HNR'R"),Ar/HetAr-I/BrAr/HetAr-CF3 (CF3CO2Na, CuI, NMP). Примеры взаимного превращения других функциональных групп (FGI), относящиеся к созданию анилиновых и бензиламиновых синтонов, используют в SNAr химических реакциях с 2,4-дихлорпиримидинами или пиримидинами с другими заменяемыми уходящими группами, как показано далее. Схема 16 Как показано на схеме 16, нитро- или азидобензолы можно восстановить до нужных соединений анилина в различных условиях. Типичное гидрирование в присутствии Pd/C катализатора в растворителях, таких как метанол, этанол, этилацетат, приведет к получению нужного продукта. В случае восстановления азида можно эффективно использовать условия Стаудингера и Ph3P. Многие предшественники анилина коммерчески доступны для превращения в сам анилин. Производные N-ацила, такие как амиды,можно гидролизовать в кислотных или щелочных условиях, получая анилин. В случае карбаматов, например трет-бутоксикарбонилом (Boc) защищенных анилинов, ацильную группу можно удалить, используя HCl в растворителях, таких как диоксан, или используя TFA в DCM. Fmoc защищенные анилины требуют щелочных условий, обычно пиперидина в ДМФ, чтобы удалить ацильный фрагмент. Если R представляет собой электроноакцепторную группу и/или если ароматическая система представляет собой п-дефицитный гетероцикл, фтор (или другой галоген или трифлат), идеально конъюгированный с указанным фтором, можно заменить в SNAr условиях самим аммиаком или эквивалентом аммиака или предшественником. Арил- или гетероарилгалогениды и трифлаты можно также подвергнуть взаимодействию в условиях катализа на основе переходных металлов с эквивалентами аммиака или предшественниками, чтобы обеспечить введение азотной функциональной группы. Специалистам известно большое число катализаторов, лигандов, оснований и растворителей, цитированных в обширной литературе и которые коммерчески доступны для таких превращений. Схема 17 Бензиламины как синтоны в SNAr реакциях с 2,4-дихлорпиримидинами или пиримидинами с другими заменяемыми 2- и 4-уходящими группами можно также получить, используя ряд других подходов.- 10021801 На схеме 22 представлены несколько репрезентативных примеров некоторых более часто используемых способов. Первичные амиды можно восстановить до целевых бензиламинов, используя восстанавливающие агенты, такие как комплексы LiAlH4 и борана; альдегиды претерпевают восстановительное аминирование под действием эквивалентов аммиака в присутствии восстанавливающих агентов, таких какNaCN(BH3) и Na(OAc)3BH; бензилгалогениды претерпевают типичные SN2 замещения эквивалентами или предшественниками аммиака и бензонитрилы можно восстановить, используя ряд восстанавливающих агентов, примерами которых являются никель Рэнея и LiAlH4. Пример синтеза диалкил (4-аминобензил)фосфонатов из других диалкилфосфонатов. Схема 18 Примеры диалкил (4-аминобензил)фосфонатов можно получить в соответствии со схемой 18. Например, коммерчески доступный диэтил (4-нитробензил)фосфонат I можно гидролизовать до соответствующей фосфоновой кислоты II, используя концентрированную хлористо-водородную кислоту. Указанную кислоту можно превратить в дихлорид фосфоновой кислоты III, используя оксалилхлорид и реакцию с алкиловыми спиртами, получая диалкилфосфонаты IV. Нитрогруппу можно восстановить множеством способов, включая гидрирование в присутствии палладиевого катализатора, получая диалкил (4 аминобензил)фосфонаты V. Пример синтеза диалкил (3-аминобензил)фосфонатов с использованием реакции МихаэлисаАрбузова. Схема 19 Диалкилбензилфосфонаты можно также получить, используя реакцию Михаэлиса-Арбузова, нагревая триалкилфосфиты VI с 1-бромметил-3- или 4-нитробензолом VII, получая диалкил (3- или 4-нитробензил)фосфонаты VIII непосредственно (схема 19). Нитрогруппу можно восстановить, используя различные условия, известные специалистам, включая, но ими не ограничиваясь, каталитическое гидрирование в присутствии палладия-на-угле, получая диалкил (3- или 4-аминобензил)фосфонаты IX. Пример синтеза диалкил (3- и 4-аминофенил)фосфонатов, используя реакцию Pd(0) сочетания. Схема 20 Диалкил 3- или 4-аминофенилфосфонаты можно получить, как представлено на схеме 20. Катализируемые палладием реакции п-или м-бромнитробензола XI с диэтилфосфонатом X приводят к получе- 11021801 нию диэтил 3- или 4-нитрофенилфосфонатов XII. Указанные фосфонаты можно гидролизовать до соответствующих фосфоновых кислот XIII, используя концентрированную хлористо-водородную кислоту, и затем обрабатывают тионилхлоридом, получая дихлориды фосфоновой кислоты XIV. Реакция указанных дихлоридов со спиртами приводит к получению диалкил 3- или 4-нитрофенилфосфонатных эфиров XV,которые можно восстановить до соответствующих анилинов XVI путем гидрирования. Пример синтеза алкил (3- и 4-аминобензил)алкилфосфинатов с помощью реакции МихаэлисаАрбузова. Схема 21 Примеры алкилфосфинатных промежуточных соединений можно получить в соответствии со схемой 21, используя реакцию Михаэлиса-Арбузова. Например, п- или м-нитробензилбромиды XVIII можно нагревать с чистым диэтоксиметилфосфином XVII (т.е. R=Et, Rw=Me), получая этилметил (4- или 3-нитробензил)фосфинат XIX. Полученные нитросоединения можно восстановить различными способами, включая гидрирование в присутствии палладиевого катализатора, получая соответствующие аминобензилфосфинаты XX, в данном случае этил (3- или 4-аминобензил)метилфосфинат. Пример синтеза алкилгидрофосфонатов и фосфоновых кислот с помощью гидролиза диалкилфосфонатов. Схема 22 Как представлено на схеме 22, алкилгидрофосфонаты XXI и фосфоновые кислоты XXII можно синтезировать из соответствующих диэтилфосфонатов, используя гидролиз концентрированной хлористоводородной кислотой. Остановка реакции до наступления полного гидролиза позволяет выделить как алкилгидрофосфонат, так и фосфоновую кислоту, используя такие хроматографические методики, как препаративная ВЭЖХ. Пример синтеза диалкил (3- и 4-аминобензил)фосфиноксидов с помощью реакции МихаэлисаАрбузова. Схема 23 Диалкилбензилфосфиноксиды можно получить в соответствии со схемой 23, используя реакцию диалкилфосфинхлорида XXIII, например хлордиметилфосфина с алкоксидом, например этоксидом натрия, с образованием промежуточного соединения, алкилдиалкилфосфинита XXIV, в данном примере этилдиметилфосфинита. Такой промежуточный фосфинит можно подвергнуть взаимодействию без выделения или очистки с бензилгалогенидом, например 3- или 4-нитробензилбромидом XVIII при повышенных температурах, используя реакцию Михаэлиса-Арбузова, получая нужные диалкилбензилфосфиноксиды XXV, в данном случае диметил (3- или 4-нитробензил)фосфиноксид. Нитрогруппу этого примера можно восстановить, используя различные условия, например гидрирование в присутствии катализатора, палладия-на-угле, получая диалкил (3- и 4-аминобензил)фосфиноксиды XXVI. Пример синтеза диалкил (3- и 4-аминофенил)фосфиноксидов в реакции сочетания опосредованной Диалкил(3- и 4-аминофенил)фосфиноксиды можно получить, как представлено на схеме 24. Реакция диэтилфосфоната XXVII с алкилмагнийбромидом (например, метилмагнийбромидом) при температуре от 0 С до комнатной температуры приводит к получению диалкилфосфиноксида XXVIII (например,диметилфосфиноксида), который можно подвергнуть реакции сочетания с палладиевым катализатором с 3- или 4-нитройодобензолом XXIX, получая диалкил (3- или 4-нитрофенилфосфин)оксид XXX (например, диметил (3- или 4-нитрофенилфосфин)оксид). Нитрогруппу можно восстановить множеством способов, включая каталитическое гидрирование, получая нужные анилиновые промежуточные соединения Диалкилбензилфосфонаты, такие как диэтил(4-нитробензил)фосфонат I, можно моноалкилировать по бензильному углероду, используя реакцию с сильным основанием, неограничивающим примером которого является LDA, с последующим введением подходящего алкилгалогенида, такого как йодометан. Полученный таким образом моноалкилированный продукт XXXII можно алкилировать второй раз по бензильному углероду через депротонирование гидридом натрия и реакцию с алкилгалогенидом, таким как йодометан, получая производные XXXIII. В случае, представленном на схеме, нитропроизводные продукты можно восстановить до соответствующих анилинов XXXIV и XXXV такими способами, как каталитическое гидрирование. Пример синтеза производных 2,3-дигидро-1 Н-изоиндол-1-она. Схема 26 Производные аминолактама можно получить в соответствии со схемой 26. Метил 2-метил-6 нитробензоат можно бромировать по бензильному положению NBS, получая производное XXXVI. Полученный сложный эфир можно циклизовать, используя метиламин, получая нитролактам XXXVII, который можно восстановить до производного анилина XXXVIII. Бромирование XXXVIII при низкой температуре приводит к получению 7-амино-4-бром-2-метил-2,3-дигидро-1 Н-изоиндол-1-она XXXIX. Полученное вещество можно подвергнуть взаимодействию с производными 4-хлорпиримидина и затем осуществить реакции, известные специалистам в данной области, которые превращают бромфункциональные группы в другие используемые группы, неограничивающими примерами которых являются алкокси,алкил, арил, гетероарил, гетероциклил и циклоалкил. Пример способа получения 2-замещенных N-метил-N-[3-([5-(трифторметил)пиримидин-4 ил]аминометил)пиридин-2-ил]метансульфонамидов, например N-[3-([2-(4-[(диэтилфосфорил)метил]фениламино)-5-(трифторметил)пиримидин-4-ил]аминометил)пиридин-2-ил]-N-метилметансульфонамид. Реакция 2,4-дихлор-5-(трифторметил)пиримидина XL с N-[3-(аминометил)пиридин-2-ил]-Nметилметансульфонамидом (WO 2007/072158, WO 2008/115369) в присутствии диизопропилэтиламина приводит к получению раствора N-[3-([2-хлор-5-(трифторметил)пиримидин-4-ил]аминометил)пиридин-2-ил]-N-метилметансульфонамида XLIa и его региоизомера N-[3-([4-хлор-5-(трифторметил)пиримидин-2-ил]аминометил)пиридин-2-ил]-N-метилметансульфонамида XLIb. При перемешивании в крупных масштабах при 0 С происходит осаждение чистого 4-замещенного изомера XLIa. Реакция образца чистой смеси N-[3-([2-хлор-5-(трифторметил)пиримидин-4-ил]аминометил)пиридин-2 ил]-N-метилметансульфонамида с анилинами, такими как 4-[(диэтилфосфорил)метил]анилин, в присутствии TFA и TFE при микроволновом облучении приводит к получению 2-замещенных N-метил-N-[3([5-(трифторметил)пиримидин-4-ил]аминометил)пиридин-2-ил]метансульфонамидов Реакция 2,4-дихлор-5-(трифторметил)пиримидина XL с N-[3-(аминометил)пиридин-2-ил]-Nметилметансульфонамидом (WO 2007/072158, WO 2008/115369) в присутствии диизопропилэтиламина приводит к получению смеси N-[3-([2-хлор-5-(трифторметил)пиримидин-4-ил]аминометил)пиридин-2 ил]-N-метилметансульфонамида и его региоизомера N-[3-([4-хлор-5-(трифторметил)пиримидин-2 ил]аминометил)пиридин-2-ил]-N-метилметансульфонамида XLIa/XLIb. Реакция указанной смеси с анилинами, такими как коммерчески доступный диэтил(4-аминобензил)фосфонат, в присутствии TFA и TFE при микроволновом облучении приводит к получению 2-замещенных N-метил-N-[3-([5(трифторметил)пиримидин-4-ил]аминометил)пиридин-2-ил]метансульфонамидов и региоизомеров, которые можно разделить, используя препаративную ВЭЖХ, получая, например, диэтил [4-(4-[(2[метил(метилсульфонил)амино]пиридин-3-илметил)амино]-5-(трифторметил)пиримидин-2 иламино)бензил]фосфонат (пример 1).- 14021801 Пример способа получения 2-замещенныхXL с коммерчески доступными 2-амино-N-метилбензамидом и диизопропилэтиламином при комнатной температуре в течение ночи приводит к получению смеси 2-[2-хлор-5-(трифторметил)пиримидин-4-ил]амино-N-метилбензамида и 2-[4-хлор-5-(трифторметил)пиримидин-2-ил]амино-N-метилбензамида (XLIIIa/XLIIIb). Тщательное растирание смеси с метиленхлоридом приводит к получению чистого образца целевого 4-замещенного продукта XLIIIa. Концентрирование маточных растворов и хроматографическая очистка полученного остатка на силикагеле с использованием 5% EtOAc в метиленхлориде в качестве элюента, выделение менее полярного соединения приводит к получению дополнительно чистого 4-замещенного изомера плюс смеси изомеров, которую, при желании, можно подвергнуть дальнейшей хроматографической обработке. Реакция чистого 2-[2-хлор-5-(трифторметил)пиримидин-4-ил]амино-N-метилбензамидаXLIIIa с анилинами, такими как диэтил(4-аминобензил)фосфонат, в присутствии TFA и TFE при микроволновом облучении приводит к получению нужных продуктов (например, диэтил(4-[4-[2-(метилкарбамоил)фенил]амино-5-(трифторметил)пиримидин-2-ил]аминобензил)фосфоната,пример 20). Пример способа получения 2-замещенныхXL с коммерчески доступными 1-[3-(метилсульфонил)фенил]метанамином и диизопропилэтиламином при 0 С в течение ночи приводит к получению смеси 2-хлор-N-[3-(метилсульфонил)бензил]-5-(трифторметил)пиримидин-4-амина и 4-хлор-N-[3-(метилсульфонил)бензил]-5-(трифторметил)пиримидин-2-аминаXLIVa/XLIVb. Сырую смесь кристаллизуют из метиленхлорида, получая образец чистого нежелательного 2-замещенного изомера XLIVb. Маточные растворы подвергают хроматографической обработке на силикагеле, элюируя смесью 1:1 гексан:этилацетат, получая дополнительно чистый образец 2-замещенного изомера плюс образец требуемого 2-хлор-N-[3-(метилсульфонил)бензил]-5-(трифторметил)пиримидин-4-амина XLIVa. Реакция этого последнего изомера с анилинами, такими как диэтил(4-аминобензил)фосфонат, в присутствии TFA и TFE при микроволновом облучении приводит к получению целевого продукта (например,диэтил- 15021801 Пример способа получения 2-замещенныхXL подвергают взаимодействию с 1-[3-(метилсульфинил)фенил]метанамингидрохлоридом (Bioorg. Med. Chem. Lett. 2005, 15, 4136-4142) и диизопропилэтиламином при 0 С в течение ночи, получая смесь 2-хлор-N-[3-(метилсульфинил)бензил]5-(трифторметил)пиримидин-4-амина и 4-хлор-N-[3-(метилсульфинил)бензил]-5-(трифторметил)пиримидин-2-амина XLVa/XLVb. Образец чистого, искомого 4-замещенного изомера XLVa выделяют как неполярный компонент смеси хроматографией на силикагеле, элюируя смесью 1:3 гексан:этилацетат. Реакция этого последнего изомера с анилинами, такими как диэтил(4-аминобензил)фосфонат, в присутствии TFA и TFE при микроволновом облучении приводит к получению целевого продукта (например,диэтил(4-[4-[3-(метилсульфинил)бензил]амино-5-(трифторметил)пиримидин-2 ил]аминобензил)фосфоната, пример 47). Пример способа получения 2-[5-хлор/бром-2-(ариламино)пиримидин-4-ил]амино-Nметилбензамидов. 5-Хлор- и 5-бром-2,4-диаминопроизводные пиримидины можно получить в результате реакций,аналогичных тем, что были ранее раскрыты для 5-трифторметильных вариантов, например, как представлено на схеме 12, используя 5-хлор- или 5-бром-варианты XLIIIa, а именно XLVIa и XLVIIa соответственно. Схема 27 Другие соединения настоящего изобретения можно получить через соединения-предшественники,соответствующие R формулы 1, в соответствии с представленными далее способами, где неограничивающими вариантами R могут быть: Исходными промежуточными соединениями могут быть любые бромарильные или гетероарильные предшественники R, которые могут быть присоединены к пиримидиновому ядру на любой стадии, как раскрыто в настоящем описании, через амин. Общее конденсированное кольцо представляет собой произвольное, необязательно замещенное кольцо или другой заместитель, как раскрыто выше для R. Реакция 1 на схеме 27 изображает реакцию перекрестного сочетания с использованием палладия с любой арильной или гетероарильной группой (Ar1) в условиях реакции Сузуки с соответствующим бораном или боронатом, описанными в примере 63 и в других приведенных в настоящем описании примерах или в любых подходящих вариантах. Реакции 2, 3 и 10 также изображают Pd реакции сочетания с использованием палладия, используемые для достижения дальнейшего разнообразия различных циклических заместителей. Исходное соединение брома можно обработать аналогично реакции 2 соответствующим образом функционализированным циклическим боронатом, как в соединении 232F. В соответствующих случаях полученное соединение можно восстановить таким способом, как гидрирование, соединение 232 Е. Альтернативно, функциональные группы реагентов можно возвратить, как в реакциях 3 и 10, представленных в соединении 205 Е.R1 и R" представляют собой любые заместители, как раскрыто в настоящем описании. Боронатный продукт реакции 3 можно функционализировать, как в реакции 4, в условиях, таких как в примере 205, используя обработку KHF2, с последующим алкилированием. В соответствии с реакциями 5-7 аминоалкильную и амидную функциональные группы можно ввести в группу R. Как раскрыто в соединении 109 Е, соответствующий арилбромид можно присоединить к фенэтилбороновой кислоте с последующим восстановительным аминированием или присоединением амида, как в примерах 116 и 117. Модификации согласно реакции 8 можно использовать для введения ненасыщенного алкинового спейсера для функциональной группы, как в примерах 75-77, используяPd-опосредованное присоединение алкина. Далее, бром можно заменить амином, как, например, в примерах 199 и 208. Естественно, специалистам будет понятно, что такие реакции являются общими и что указанные примеры не являются ограничивающими. Примеры получения Диэтил (3-аминобензил)фосфонат. Смесь 3-нитробензилбромида (648 мг, 3,00 ммоль) и триэтилфосфита (0,669 мл, 3,90 ммоль) перемешивают при 140 С в атмосфере азота в течение 16 ч. Избыток триэтилфосфита удаляют при пониженном давлении и полученный остаток очищают, используя хроматографию на силикагеле, элюируя 2% МеОН в дихлорметане, получая диэтил (3-нитробензил)фосфонат, 0,80 г (выход: 98%). 1 Н ЯМР (CD3OD, 400 МГц):1,27 (т, J=6,8 Гц, 6 Н), 3,43 (д, J=22,0 Гц, 2 Н), 4,08 (кв, J=6,8 Гц, 4 Н),7,58 (т, J=8,0 Гц, 1 Н), 7,72 (д, J=7,6 Гц, 1 Н), 8,15 (д, J=8,4 Гц, 1 Н), 8,22 (д, J=2,4 Гц, 1 Н); МС (ES+): m/z 273,99 (МН+); ВЭЖХ: tR=2,92 мин (ZQ3; полярная 5 мин). Раствор диэтил (3-нитробензил)фосфоната, полученного, как раскрыто выше, в МеОН (5 мл) гидрируют в присутствии 10% Pd/C (100 мг) в течение ночи. Реакционную смесь фильтруют и полученный фильтрат концентрируют в вакууме, получая искомый продукт диэтил (3-аминобензил)фосфонат(0,690 г, выход: 95%), который используют непосредственно в дальнейших химических реакциях без очистки. 1 Н ЯМР (CDCl3, 400 МГц):1,24 (т, J=7,2 Гц, 6 Н), 3,04 (д, J=21,6 Гц, 2 Н), 3,66 (шир.с, 2 Н), 4,01 (кв,J=7,2 Гц, 4 Н), 6,57 (д, J=8,8 Гц, 1 Н), 6,67-6,68 (м, 2 Н), 7,08 (т, J=7,6 Гц, 1 Н); МС (ES+): m/z 244,00 (МН+); ВЭЖХ: tR=2,24 мин (ZQ3; полярная 5 мин). Диэтил (2-аминобензил)фосфонат. Получают способом, раскрытым для диэтил (3-аминобензил)фосфоната. Промежуточное соединение, диэтил (3-нитробензил)фосфонат, получают с 97% выходом в виде масла коричневатого цвета. 1 Н ЯМР (CD3OD, 400 МГц):1,23 (т, J=7,2 Гц, 6 Н), 3,79 (д, J=22,8 Гц, 2 Н), 4,01 (кв, J=7,2 Гц, 4 Н),7,48-7,53 (м, 2 Н), 7,64 (т, J=8,0 Гц, 1 Н), 7,99 (д, J=8,0 Гц, 1 Н); МС (ES+): m/z 273,99 (МН+); ВЭЖХ: tR=2,89 мин (ZQ3; полярная 5 мин). Диэтил (2-аминобензил)фосфонат получают с 94% выходом и используют непосредственно в дальнейших химических реакциях без очистки. 1- 18021801 Диэтил [1-(4-аминофенил)этил]фосфонат. 2 М LDA в ТГФ (1,83 мл, 3,66 ммоль) добавляют по каплям в атмосфере N2 к перемешиваемому раствору диэтил (4-нитробензил)фосфоната (1,00 г, 3,66 ммоль) в ТГФ (10 мл) при -78 С. Через 1 ч по каплям добавляют йодометан (0,456 мл, 7,32 ммоль) и полученную смесь перемешивают при -78 С в течение 30 мин, затем оставляют нагреваться до комнатной температуры в течение 3-4 ч. Затем медленно добавляют воду (10 мл), затем EtOAc (20 мл). Слои разделяют и водную фазу экстрагируют EtOAc(320 мл). Органические фазы объединяют, сушат (MgSO4), концентрируют в вакууме и полученный остаток очищают, используя хроматографию на силикагеле, элюируя 10% EtOAc в гексанах, получая 180 мг чистого диэтил [1-(4-нитрофенил)этил]фосфоната (выход: 17%) и 680 мг смеси диэтилH ЯМР (CD3OD, 400 МГц):1,19 (т, J=7,2 Гц, 3 Н), 1,29 (т, J=7,2 Гц, 3 Н), 1,57 (д, J=7,2 Гц, 1,5 Н),1,61 (д, J=7,2 Гц, 1,5 Н), 3,52 (кв, J=7,6 Гц, 0,5 Н), 3,57 (кв, J=7,6 Гц, 0,5 Н), 3,93-4,00 (м, 2 Н), 4,06-4,10 (м,2 Н), 7,60 (дд, J=2,4, 8,8 Гц, 2 Н), 8,20 (дд, J=1,2, 8,8 Гц, 2 Н); МС (ES+): m/z 288,09 (МН+); ВЭЖХ: tR=1,36 мин (СЭЖХ TOF, полярная). Раствор диэтил [1-(4-нитрофенил)этил]фосфоната (180 мг) в МеОН (5 мл) гидрируют в присутствии 10% Pd/C (50 мг) в течение 4 ч. Реакционную смесь фильтруют и полученный фильтрат концентрируют в вакууме, получая 150 мг целевого диэтил [1-(4-аминофенил)этил]фосфоната (выход: 94%), который используют непосредственно в дальнейших химических реакциях без очистки. 1 Н ЯМР (CDCl3, 400 МГц):1,16 (т, J=7,2 Гц, 3 Н), 1,27 (т, J=7,2 Гц, 3 Н), 1,50 (д, J=7,6 Гц, 1,5 Н),1,53 (д, J=7,6 Гц, 1,5 Н), 3,01-3,10 (м, 1 Н), 3,76-3,82 (м, 1 Н), 3,89-3,93 (м, 1 Н), 3,97-4,03 (м, 2 Н), 6,66-6,68(м, 2 Н), 7,12-7,15 (м, 2 Н); МС (ES+): m/z 258,04 (МН+); ВЭЖХ: tR=2,20 мин (ZQ3; полярная 5 мин). Диэтил [2-(4-аминофенил)пропан-2-ил]фосфонат. Раствор диэтил [1-(4-нитрофенил)этил]фосфоната (206 мг, 0,717 ммоль) в ТГФ (5 мл) при комнатной температуре в атмосфере N2 обрабатывают порциями NaH (60% в минеральном масле, 143 мг,3,58 ммоль). Полученную смесь перемешивают в течение 5 мин, затем добавляют по каплям йодометан(0,268 мл, 4,30 ммоль) и полученную смесь перемешивают в течение еще 5 ч. Медленно добавляют воду(10 мл), затем добавляют EtOAc (20 мл). Слои разделяют и водную фазу экстрагируют EtOAc (320 мл). Органические фазы объединяют, сушат (MgSO4), концентрируют в вакууме и полученный остаток очищают, используя хроматографию на силикагеле, элюируя 5% МеОН в дихлорметане, получая 103 мг целевого диэтил [2-(4-нитрофенил)пропан-2-ил]фосфоната (выход: 48%). 1 Н ЯМР (CD3OD, 400 МГц):1,22 (т, J=7,2 Гц, 6 Н), 1,64 (с, 3 Н), 1,69 (с, 3 Н), 3,94-4,01 (м, 4 Н), 7,78(дд, J=2,4, 9,2 Гц, 2 Н), 8,21 (дд, J=0,8, 8,8 Гц, 2 Н); МС (ES+): m/z 302,11 (МН+); ВЭЖХ: tR=1,44 мин (СЭЖХ TOF, полярная). Диэтил [2-(4-нитрофенил)пропан-2-ил]фосфонат (103 мг), полученный, как раскрыто выше, гидрируют в присутствии 10% Pd/C (50 мг) в МеОН (5 мл) в течение 4 ч. Полученную смесь фильтруют и полученный фильтрат концентрируют в вакууме, получая 92,5 мг целевого диэтил [2-(4 аминофенил)пропан-2-ил]фосфоната (выход: 99%), который используют в дальнейших химических реакциях без очистки. 1 Н ЯМР (CDCl3, 400 МГц):1,20 (т, J=7,2 Гц, 6 Н), 1,54 (с, 3 Н), 1,59 (с, 3 Н), 3,65 (шир.с, 2 Н), 3,803,94 (м, 2 Н), 6,66 (д, J=8,8 Гц, 2 Н), 7,32 (дд, J=2,4, 8,8 Гц, 2 Н); МС (ES+): m/z 272,02 (МН+); ВЭЖХ: tR=2,45 мин (ZQ3; полярная 5 мин). 3-[(Диметилфосфорил)метил]анилин. Раствор хлордиметилфосфина (250 мг, 2,60 ммоль) в ТГФ (0,7 мл) добавляют по каплям к перемешиваемой суспензии этоксида натрия (194 мг, 2,85 ммоль) в ТГФ (0,8 мл) при 0 С. Полученную смесь перемешивают при комнатной температуре в течение 30 мин, затем обрабатывают 1-(бромметил)-3 нитробензолом (560 мг, 2,59 ммоль). Затем полученную смесь перемешивают в атмосфере азота при 10 С в запаянной ампуле в течение 2 ч, концентрируют в вакууме и полученный остаток очищают, используя на силикагеле, элюируя 5% МеОН в дихлорметане, получая 153 мг диметил(3 нитробензил)фосфаноксида (выход: 28%). 1 Н ЯМР (CDCl3, 400 МГц):1,53 (д, J=12,4 Гц, 6 Н), 3,27 (д, J=14,0 Гц, 2 Н), 7,55 (т, J=7,6 Гц, 1 Н),7,71 (д, J=7,6 Гц, 1 Н), 8,11 (д, J=2,0 Гц, 1 Н), 8,16 (дд, J=0,8, 7,2 Гц, 1 Н); МС (ES+): m/z 214,00 (MH+); ВЭЖХ: tR=2,16 мин (ZQ3; полярная 5 мин). Диметил(3-нитробензил)фосфаноксид (153 мг), полученный, как раскрыто выше, гидрируют в присутствии 10% Pd/C (100 мг) в МеОН (5 мл) в течение ночи. Полученную смесь фильтруют и полученный- 19021801 фильтрат концентрируют в вакууме, получая 128 мг целевого 3-[(диметилфосфорил)метил]анилина (выход: 96%), который используют в дальнейших химических реакциях без очистки. 1 Н ЯМР (CDCl3, 400 МГц):1,44 (д, J=12,8 Гц, 6 Н), 3,06 (д, J=15,6 Гц, 2 Н), 3,81 (шир.с, 2 Н), 6,556,58 (м, 3 Н), 7,08 (т, J=7,6 Гц, 1 Н); МС (ES+): m/z 184,02 (МН+); ВЭЖХ: tR=0,58 и 0,90 мин (ZQ3; полярная 5 мин). 4-[(Диметилфосфорил)метил]анилин. Получают способом, раскрытым выше для получения 3-[(диметилфосфорил)метил]анилина, используя 1-(бромметил)-4-нитробензол. Промежуточное нитросоединение,диметил(4 нитробензил)фосфаноксид, получают с 21% выходом. 1 Н ЯМР (CDCl3, 400 МГц):1,51 (д, J=12,8 Гц, 6 Н), 3,27 (д, J=14,4 Гц, 2 Н), 7,47 (дд, J=2,4, 8,8 Гц,2 Н), 8,22 (д, J=8,4 Гц, 2 Н); МС (ES+): m/z 214,00 (МН+); ВЭЖХ: tR=2,17 мин (ZQ3; полярная 5 мин). 4-[(Диметилфосфорил)метил]анилин получают с 95% выходом и используют непосредственно в дальнейших химических реакциях без очистки. 1 Н ЯМР (CDCl3, 400 МГц):1,41 (д, J=12,4 Гц, 6 Н), 3,05 (д, J=15,2 Гц, 2 Н), 3,74 (шир.с, 2 Н), 6,65 (д,J=7,2 Гц, 2 Н), 6,99 (дд, J=2,0, 8,0 Гц, 2 Н); МС (ES+): m/z 184,01 (МН+); ВЭЖХ: tR=0,56 и 0,75 мин (ZQ3; полярная 5 мин). 3-[(Диэтилфосфорил)метил]анилин. Получают способом, раскрытым выше для получения 3-[(диметилфосфорил)метил]анилина, используя хлордиэтилфосфин. Промежуточное нитросоединение, диэтил(3-нитробензил)фосфаноксид, получают с 80% выходом. 1 Н ЯМР (CD3OD, 400 МГц):1,13 (т, J=7,6 Гц, 3 Н), 1,17 (т, J=8,0 Гц, 3 Н), 1,73-1,85 (м, 4 Н), 3,40 (д,J=13,2 Гц, 2 Н), 7,60 (т, J=7,6 Гц, 1 Н), 7,12 (д, J=7,6 Гц, 1 Н), 8,16 (д, J=8,0 Гц, 1 Н), 8,23 (дд, J=1,6, 7,6 Гц,1 Н);MC (ES+): m/z 242,01 (MH+); ВЭЖХ: tR=2,45 мин (ZQ3; полярная 5 мин). Производное анилина, 3-[(диэтилфосфорил)метил]анилин, получают с выходом 97% и используют непосредственно в дальнейших химических реакциях без очистки. 1 Н ЯМР (CDCl3, 400 МГц):1,12-1,20 (м, 6 Н), 1,62-1,71 (м, 4 Н), 3,03 (д, J=14,4 Гц, 2 Н), 3,72 (шир.с,2 Н), 6,56-6,62 (м, 3 Н), 7,08 (т, J=7,6 Гц, 1 Н);MC (ES+): m/z 212,02 (МН+); ВЭЖХ: tR=1,62 мин (ZQ3; полярная 5 мин). 3-[(Диэтилфосфорил)метил]анилин. Получают способом, раскрытым выше для получения 3-[(диметилфосфорил)метил]анилина, используя хлордиэтилфосфин и 1-(бромметил)-4-нитробензол. Промежуточное нитросоединение, диэтил(4 нитробензил)фосфаноксид, получают с 70% выходом. 1 Н ЯМР (CD3OD, 400 МГц):1,13 (т, J=7,6 Гц, 3 Н), 1,17 (т, J=8,0 Гц, 3 Н), 1,74-1,84 (м, 4 Н), 3,40 (д,J=13,6 Гц, 2 Н), 7,55 (дд, J=1,6, 8,8 Гц, 2 Н), 8,21 (д, J=8,4 Гц, 2 Н);MC (ES+): m/z 241,99 (МН+); ВЭЖХ: tR=2,45 мин (ZQ3; полярная 5 мин). Производное анилина, 4-[(диэтилфосфорил)метил]анилин, получают с выходом 97% и используют непосредственно в дальнейших химических реакциях без очистки. 1 Н ЯМР (CDCl3, 400 МГц):1,12-1,20 (м, 6 Н), 1,59-1,68 (м, 4 Н), 3,02 (д, J=14,4 Гц, 2 Н), 6,64 (д,J=8,8 Гц, 2 Н), 7,02 (дд, J=1,6, 8,0 Гц, 2 Н);MC (ES+): m/z 212,02 (МН+); ВЭЖХ: tR=1,14 мин (ZQ3; полярная 5 мин). 3-[(Дипропан-2-илфосфорил)метил]анилин. Получают способом, раскрытым выше для получения 3-[(диметилфосфорил)метил]анилина, используя дипропан-2-илфосфинистый хлорид. Промежуточное нитросоединение,(3-нитробензил)(дипропан-2-ил)фосфаноксид, получают с 75% выходом. 1 Н ЯМР (CD3OD, 400 МГц):1,13-1,21 (м, 12 Н), 2,08-2,18 (м, 2 Н), 3,42 (д, J=11,6 Гц, 2 Н), 7,58 (т,J=8,0 Гц, 1 Н), 7,78 (д, J=7,2 Гц, 1 Н), 8,14 (д, J=8,0 Гц, 1 Н), 8,29 (д, J=1,6 Гц, 1 Н);MC (ES+): m/z 270,04 (МН+); ВЭЖХ: tR=2,74 мин (ZQ3; полярная 5 мин). Производное анилина, 3-[(дипропан-2-илфосфорил)метил]анилин, получают с выходом 91% и используют непосредственно в дальнейших химических реакциях без очистки. 1 Н ЯМР (CD3OD, 400 МГц):1,12-1,19 (м, 12 Н), 2,05-2,13 (м, 2 Н), 3,12 (д, J=12,8 Гц, 2 Н), 6,60-6,70- 20021801 МС (ES+): m/z 240,04 (МН+); ВЭЖХ: tR=2,07 мин (ZQ3; полярная 5 мин). 4-[(Дипропан-2-илфосфорил)метил]анилин. Получают способом, раскрытым выше для получения 3-[(диметилфосфорил)метил]анилина, используя дипропан-2-илфосфинистый хлорид и 1-(бромметил)-4-нитробензол. Промежуточное нитросоединение, (4-нитробензил)(дипропан-2-ил)фосфаноксид, получают с 85% выходом. 1 Н ЯМР (CD3OD, 400 МГц):1,14-1,21 (м, 12 Н), 2,08-2,18 (м, 2 Н), 3,41 (д, J=12,8 Гц, 2 Н), 7,61 (дд,J=2,0, 8,8 Гц, 2 Н), 8,19 (д, J=8,8 Гц, 2 Н); МС (ES+): m/z 270,04 (МН+); ВЭЖХ: tR=2,74 мин (ZQ3; полярная 5 мин). Производное анилина, 4-[(дипропан-2-илфосфорил)метил]анилин, получают с выходом 92% и используют непосредственно в дальнейших химических реакциях без очистки. 1 Н ЯМР (CD3OD, 400 МГц):1,10-1,17 (м, 12 Н), 2,02-2,10 (м, 2 Н), 3,10 (д, J=12,4 Гц, 2 Н), 6,69 (д,J=8,4 Гц, 2 Н), 7,07 (дд, J=2,0, 8,0 Гц, 2 Н); МС (ES+): m/z 240,04 (МН+); ВЭЖХ: tR=1,91 мин (ZQ3; полярная 5 мин). Диметилфосфаноксид. Диэтилфосфит (1,29 мл, 10 ммоль) медленно добавляют по каплям к раствору метилмагнийбромида в ТГФ (1,4 М, 21,4 мл, 30 ммоль) при 0 С. После перемешивания при комнатной температуре в течение 5 ч осторожно добавляют насыщенный водный раствор бикарбоната натрия (10 мл), затем добавляют МеОН (10 мл). Полученное твердое вещество белого цвета удаляют фильтрованием и полученный фильтрат концентрируют в вакууме, получая целевой диметилфосфаноксид в виде прозрачного масла (0,45 г,выход: 58%). Полученное вещество затем сушат, подвергают азеотропной перегонке с толуолом для удаления воды, перед использованием в последующих химических реакциях. 1 Н ЯМР (D2O, 400 МГц):1,68 (дд, J=4,0, 14,4 Гц, 6 Н), 7,09 (д.кв, J=4,0, 489,2 Гц, 1 Н). 4-(Диметилфосфорил)анилин. Дегазированную суспензию 1-йодо-4-нитробензола (90 мг, 0,36 ммоль), диметилфосфаноксида(29,2 мг, 0,38 ммоль), бис-(дибензилиденацетон)палладия (3,31 мг, 0,0036 ммоль), XantPhos (6,27 мг,0,0108 ммоль) и Cs2CO3 (165 мг, 0,506 ммоль) в сухом диоксане (0,4 мл) нагревают в запаянной ампуле при 90 С в течение 4 ч. Охлажденную смесь фильтруют, полученный фильтрат концентрируют в вакууме и полученный остаток подвергают хроматографической обработке на силикагеле, элюируя 5% МеОН в дихлорметане, получая 26 мг диметил(4-нитрофенил)фосфаноксида (выход: 36%). 1H ЯМР (CD3OD, 400 МГц):1,86 (д, J=13,6 Гц, 6 Н), 8,06 (дд, J=8,8, 10,8 Гц, 2 Н), 8,38 (дд, J=1,6,8,8 Гц, 2 Н); МС (ES+): m/z 199,96 (МН+); ВЭЖХ: tR=1,96 мин (ZQ3; полярная 5 мин). Диметил(4-нитрофенил)фосфаноксид, полученный, как раскрыто выше, гидрируют в присутствии 10% Pd/C (15 мг) в МеОН (5 мл) в течение 2 ч. Затем смесь фильтруют и фильтрат концентрируют в вакууме, получая 19 мг целевого 4-(диметилфосфорил)анилина (выход: 86%), который используют непосредственно в дальнейших химических реакциях без очистки. 1 Н ЯМР (CDCl3, 400 МГц):1,67 (д, J=12,8 Гц, 6 Н), 4,11 (шир.с, 2 Н), 6,73 (дд, J=2,0, 8,4 Гц, 2 Н),7,48 (дд, J=8,4, 11,2 Гц, 2 Н); МС (ES+): m/z 170,04 (МН+); ВЭЖХ: tR=1,23 мин (ZQ3; полярная 5 мин). 3-(Диметилфосфорил)анилин. Получают способом, раскрытым выше для получения 4-(диметилфосфорил)анилина, используя 1-йодо-3-нитробензол. Промежуточное нитросоединение, диметил(3-нитрофенил)фосфаноксид, получают с 27% выходом. 1 Н ЯМР (CD3OD, 400 МГц):1,87 (д, J=13,6 Гц, 6 Н), 7,83 (дт, J=2,4, 7,6 Гц, 1 Н), 8,19 (т, J=8,8 Гц,1 Н), 8,45 (д, J=8,0 Гц, 1 Н), 8,66 (дт, J=1,6, 12,0 Гц, 1 Н);MC (ES+): m/z 199,96 (MH+); ВЭЖХ: tR=2,06 мин (ZQ3; полярная 5 мин). Производное анилина, 3-(диметилфосфорил)анилин, получают с выходом 96% и используют непосредственно в дальнейших химических реакциях без очистки. 1- 21021801 кипении с обратным холодильником в течение ночи. Полученную смесь фильтруют, полученный фильтрат концентрируют в вакууме и остаток подвергают хроматографической обработке на силикагеле, элюируя смесью петролейный эфир:EtOAc 5:1, получая 60 г диэтил (4-нитрофенил)фосфоната (выход: 94%). Смесь диэтил (4-нитрофенил)фосфоната (150 мг, 0,58 ммоль), EtOH (40 мл) и 10% Pd/C (30 мг) гидрируют при давлении водорода 50 фунт/кв.дюйм (345 кПа) при 40 С в течение 6 ч. После охлаждения до комнатной температуры реакционную смесь фильтруют и полученный фильтрат концентрируют в вакууме, получая 130 мг целевого диэтил (4-аминофенил)фосфоната (выход: 98%), который используют непосредственно в дальнейших химических реакциях без очистки. 1 Н ЯМР (CDCl3, 400 МГц):1,17-1,27 (м, 6 Н), 3,91-4,07 (м, 4 Н), 6,62 (дд, J=3,6, 8,4 Гц, 2 Н), 7,51 (дд,J=8,4, 12,8 Гц, 2 Н); МС: m/z 230,05 [МН+]. Диэтил (3-аминофенил)фосфонат. Диэтил(3-нитрофенил)фосфонат получают способом,раскрытым выше для диэтил (4-нитрофенил)фосфоната, используя м-бромнитробензол (выход: 49%). 1 Н ЯМР (CDCl3, 400 МГц):1,35 (т, J=6,8 Гц, 6 Н), 4,10-4,23 (м, 4 Н), 7,68 (м, 1 Н), 8,15 (м, 1 Н), 8,39(50 мл) нагревают при кипении с обратным холодильником в течение 12 ч. Затем полученную смесь фильтруют и полученный фильтрат концентрируют в вакууме, получая сырой продукт, который очищают, используя препаративную ВЭЖХ, получая 320 мг целевого диэтил (3-аминофенил)фосфоната (выход: 69%). 1 Н ЯМР (ДМСО-d6, 400 МГц):1,20 (м, 6 Н), 3,91-4,00 (м, 4 Н), 5,39 (шир.с, 2 Н), 6,73-6,81 (м, 2 Н),6,91 (дд, J=1,6, 16,4 Гц, 1 Н), 7,11-7,16 (м, 1 Н); МС: m/z 230,10 [МН+].(водн.) перемешивают при кипении с обратным холодильником в течение ночи. Избыток HCl удаляют при пониженном давлении и полученный продукт используют в дальнейших химических реакциях без очистки (46,8 г, выход: 100%).(3-Нитрофенил)фосфоновая кислота. Получают способом, раскрытым для (4-нитрофенил)фосфоновой кислоты, используя диэтил (3-нитрофенил)фосфонат (выход: 76%). Дихлорид (4-нитрофенил)фосфоновой кислоты. Раствор (4-нитрофенил)фосфоновой кислоты (1,0 г, 5 ммоль) в SOCl2 (30 мл) перемешивают при кипении с обратным холодильником в течение 5 ч. Полученную реакционную смесь концентрируют при пониженном давлении досуха и сырой дихлорид (4-нитрофенил)фосфоновой кислоты выделяют, используя непосредственно в дальнейших химических реакциях без очистки. Дихлорид (3-нитрофенил)фосфоновой кислоты. Получают способом, раскрытым для дихлорида (4-нитрофенил)фосфоновой кислоты, используя(3-нитрофенил)фосфоновую кислоту. Дипропан-2-ил (4-нитрофенил)фосфонат. Раствор дистиллированного сухого пропан-2-ола (20 мл) и триэтиламина (1,0 г, 10 ммоль) добавляют по каплям к дихлориду (4-нитрофенил)фосфоновой кислоты (1,2 г, 5 ммоль) при 0 С. Полученную смесь перемешивают при кипении с обратным холодильником в течение ночи, смесь фильтруют и фильтрат концентрируют при пониженном давлении, получая сырой продукт, который очищают хроматографией на силикагеле, элюируя смесью петролейный эфир:EtOAc: 5:1, получая 360 мг целевого дипропан 2-ил (4-нитрофенил)фосфоната (выход: 25%). 1 Н ЯМР (CDCl3, 400 МГц):1,18 (д, J=6,0 Гц, 6 Н), 1,33 (д, J=6,4 Гц, 6 Н), 4,65-4,73 (м, 2 Н), 7,93 (дд,J=8,8, 12,8 Гц, 2 Н), 8,22 (дд, J=3,2, 8,8 Гц, 2 Н). Дипропан-2-ил (3-нитрофенил)фосфонат. Получают способом, раскрытым для дипропан-2-ил (4-нитрофенил)фосфоната, используя дихлорид(3-нитрофенил)фосфоновой кислоты (выход: 41%). 1 Н ЯМР (CDCl3, 400 МГц):1,19 (д, J=6,0 Гц, 6 Н), 1,34 (д, J=6,4 Гц, 6 Н), 4,68-4,73 (м, 2 Н), 7,60 (м,1 Н), 8,08 (дд, J=7,6, 12,8 Гц, 1 Н), 8,32 (д, J=8,4 Гц, 1 Н), 8,57 (д, J=14,0 Гц, 1 Н). Следующие два фосфонатных эфира получают в соответствии с раскрытым выше способом получения дипропан-2-ил (4-нитрофенил)фосфоната и дипропан-2-ил (3-нитрофенил)фосфоната, используя соответствующий дихлорид фосфоновой кислоты и спирт. 5,5-Диметил-2-(4-нитрофенил)-1,3,2-диоксафосфинан 2-оксид. Выход: 75%. 1 Н ЯМР (CDCl3, 400 МГц):1,07 (с, 3 Н), 1,10 (с, 3 Н), 3,84 (дд, J=11,2, 13,6 Гц, 2 Н), 4,28 (дд, J=9,6,- 22021801 10,8 Гц, 2 Н), 7,95-8,00 (м, 2 Н), 8,26-8,29 (м, 2 Н). 5,5-Диметил-2-(3-нитрофенил)-1,3,2-диоксафосфинан 2-оксид. Выход: 76%. 1 Н ЯМР (CDCl3, 400 МГц):1,07 (с, 3 Н), 1,13 (с, 3 Н), 3,86 (дд, J=11,2, 13,6 Гц, 2 Н), 4,31 (дд, J=9,6,10,8 Гц, 2 Н), 7,66 (д, J=4,0 Гц, 1 Н), 8,12 (д, J=12,8 Гц, 1 Н), 8,38 (м, 1 Н), 8,61 (д, J=14,4 Гц, 1 Н). Дипропан-2-ил (4-аминофенил)фосфонат. Смесь дипропан-2-ил (4-нитрофенил)фосфоната (287 мг, 1,0 ммоль), i-PrOH (30 мл) и 10% Pd/C(30 мг) гидрируют при давлении водорода 50 фунт/кв.дюйм (345 кПа) при 40 С в течение 6 ч. Реакционную смесь затем фильтруют и полученный фильтрат концентрируют в вакууме, получая 238 мг целевого дипропан-2-ил (4-аминофенил)фосфоната (выход: 92%), который используют непосредственно в дальнейших химических реакциях без очистки. 1 Н ЯМР (ДМСО-d6, 400 МГц):1,11 (д, J=6,0 Гц, 6 Н), 1,21 (д, J=6,4 Гц, 6 Н), 4,38-4,43 (м, 2 Н), 5,74(шир.с, 2 Н), 6,70-6,77 (м, 2 Н), 6,58 (д, J=8,0 Гц, 1 Н), 7,10-7,25 (м, 1 Н); МС: m/z 257,97 [МН+]. 4-(5,5-Диметил-2-оксидо-1,3,2-диоксафосфинан-2-ил)анилин. Получают способом, раскрытым для дипропан-2-ил (4-аминофенил)фосфоната, используя 5,5-диметил-2-(4-нитрофенил)-1,3,2-диоксафосфинан 2-оксид. Выход: 41%; 1 Н ЯМР (ДМСО-d6, 400 МГц):0,97 (с, 3 Н), 1,02 (с, 3 Н), 3,78 (м, 2 Н), 4,08 (м, 2 Н), 5,91 (шир.с, 2 Н),6,61 (дд, J=3,6, 8,4 Гц, 2 Н), 7,34 (м, 2 Н); МС: m/z 242,10 [МН+]. 3-(5,5-Диметил-2-оксидо-1,3,2-диоксафосфинан-2-ил)анилин. Получают способом, раскрытым для дипропан-2-ил (4-аминофенил)фосфоната, используя 5,5-диметил-2-(3-нитрофенил)-1,3,2-диоксафосфинан 2-оксид. Выход: 63%; 1 Н ЯМР (ДМСО-d6, 400 МГц):0,94 (с, 3 Н), 1,07 (с, 3 Н), 3,84 (дд, J=9,6, 10,8 Гц, 2 Н), 4,07 (дд,J=11,2, 14,0 Гц, 2 Н), 5,43 (шир.с, 2 Н), 6,75-6,78 (м, 2 Н), 6,88 (д, 1 Н), 7,16 (м, 1 Н); МС: m/z 242,11 [МН+]. Этил (4-амино-3-метоксибензил)метилфосфинат. Смесь (3-метокси-4-нитрофенил)метанола (0,5 г, 2,73 ммоль) и тионилхлорида (0,3 мл, 4,09 ммоль) нагревают при кипении с обратным холодильником в течение 12 ч. Затем полученную смесь концентрируют в вакууме, получая 0,55 г 4-(хлорметил)-2-метокси-1-нитробензола (выход: 100%), который используют в дальнейших химических реакциях без очистки. 1 Н ЯМР (CD3OD, 400 МГц):4,00 (с, 3 Н), 4,60 (с, 2 Н), 7,05 (дд, J=8,34, 1,77 Гц, 1 Н), 7,14 (д,J=1,52 Гц, 1 Н), 7,86 (д, J=8,34 Гц, 1 Н); МС (ES+): m/z 202,0264 [МН+] (TOF, полярная). Смесь 4-(хлорметил)-2-метокси-1-нитробензола (1,1 г, 5,46 ммоль) и диэтилметилфосфонита(0,89 г, 1,2 ммоль) нагревают при 100 С в течение 16 ч в запаянной ампуле. Реакционную смесь концентрируют в вакууме и полученный сырой остаток очищают, используя систему ISCO Combiflash, элюируя 0-5% МеОН в DCM в качестве элюента, получая 0,641 г этил (3-метокси-4-нитробензил)метилфосфината(выход: 43% выход). МС (ES+): m/z 273,99 [МН+] (ZQ3; полярная 5 мин). К раствору этил (3-метокси-4-нитробензил)метилфосфината (0,64 г, 2,34 ммоль) в этаноле (5,0 мл) добавляют палладий 10 вес.% на активированном угле (0,25 г). Реакционную смесь откачивают, продувают газообразным водородом (3) и оставляют при перемешивании в атмосфере водорода в течение 16 ч. Реакционную смесь фильтруют через слой целита и полученный фильтрат концентрируют в вакууме, получая 0,54 г этил (4-амино-3-метоксибензил)метилфосфината (выход: 95%). Полученное соединение используют в последующих реакциях без дополнительной очистки. МС (ES+): m/z 244,01 [МН+] (ZQ3; полярная 5 мин). Диэтил (4-амино-3-метоксибензил)фосфонат. Указанное соединение получают способом, раскрытым выше для получения этил (4-амино-3 метоксибензил)метилфосфината, используя триэтилфосфит на первой стадии. МС (ES+): m/z 274,01 [МН+] (ZQ3; полярная 5 мин). Метил 2-(бромметил)-6-нитробензоат (XXXVI). Раствор метил 2-метил-6-нитробензоата (TL (1996), 37, 5425, 15,6 г, 80 ммоль), NBS (21,4 г, 120- 23021801 ммоль) и перекиси бензоила (200 мг, 0,82 ммоль) в 1,2-дихлорэтане (250 мл) нагревают при кипении с обратным холодильником в течение 8 ч. Полученную реакционную смесь концентрируют досуха и сырую смесь очищают, используя хроматографию на силикагеле, элюируя смесью 2% этилацетат/гексаны,получая 10,5 г метил 2-(бромметил)-6-нитробензоата (XXXVI, выход: 65%). 1 Н ЯМР (CDCl3, 400 МГц):3,98 (с, 3 Н), 4,57 (с, 2 Н), 7,59 (дд, 1 Н, J=7,8, 8,4 Гц), 7,78 (д, 1 Н,J=7,8 Гц), 8,1 (д, 1 Н, J=8,4 Гц). 2-Метил-7-нитро-2,3-дигидро-1 Н-изоиндол-1-он (XXXVII). Раствор метиламина в этаноле (10 мл, 80 ммоль, 8 М раствора в этаноле) добавляют к раствору метил 2-(бромметил)-6-нитробензоата (8,1 г, 29,6 ммоль) в ТГФ (30 мл). После перемешивания в течение 2 ч полученную реакционную смесь концентрируют досуха и добавляют воду (30 мл) при быстром перемешивании. Полученное твердое вещество выделяют фильтрованием и сушат, получая 4,35 г 2-метил-7 нитро-2,3-дигидро-1 Н-изоиндол-1-она (XXXVII, выход: 78%). 1(8:2) гидрируют при давлении водорода 40 фунт/кв.дюйм (275,8 кПа) в присутствии 5% Pd/C (500 мг) до тех пор, пока не прекращается поглощение Н 2. Реакционную смесь фильтруют и фильтрат сушатNBS (2,3 г, 12,96) добавляют к холодному (от -8 до -10 С) раствору 7-амино-2-метил-2,3-дигидро 1 Н-изоиндол-1-она (2 г, 12,4 ммоль) в DCM (40 мл) и смесь перемешивают при температуре от -8 до-10 С в течение 1 ч. Затем к реакционной смеси добавляют водный 10% раствор тиосульфата натрия(30 мл) и перемешивание продолжают в течение еще 20 мин. Слои разделяют и водный слой экстрагируют DCM (220 мл). Объединенные органические экстракты промывают водой (340 мл) и солевым раствором (30 мл) и органический слой сушат (Na2SO4) и концентрируют в вакууме, получая 3,2 г сырого продукта. Полученное вещество тщательно растирают с этилацетатом (10 мл), получая 2,2 г чистого 7-амино-4-бром-2-метил-2,3-дигидро-1 Н-изоиндол-1-она (XXXIX, выход: 74%). 1 Н ЯМР (CDCl3, 400 МГц):3,14 (с, 3 Н), 4,20 (с, 2 Н), 5,20 (шир.с, 2 Н), 5,49 (д, 1 Н, J=8,4 Гц), 5,79(д, 1 Н, J=8,4 Гц). 2-Хлор-N-[3-(метилсульфонил)бензил]-5-(трифторметил)пиримидин-4-амин (XLIVa). Диизопропилэтиламин (31,5 мл, 180 ммоль) добавляют к суспензии 2,4-дихлор-5 трифторметилпиримидина (XL, 15,0 г, 69,4 ммоль) и 1-[3-(метилсульфонил)фенил]метанамингидрохлорида (13,3 г, 60 ммоль) в ТГФ (300 мл) при 0 С и полученную смесь перемешивают при комнатной температуре в течение ночи. Затем полученную смесь выпаривают досуха, полученный остаток переносят в метиленхлорид (100 мл) и полученный раствор промывают водой (100 мл), сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме. Полученный сырой остаток переносят в горячий метиленхлорид (35 мл), затем оставляют при перемешивании в течение ночи при комнатной температуре. Образовавшееся твердое вещество выделяют фильтрованием (9,4 г) и определяют как чистый образец более полярной компоненты смеси продуктов,(4-хлор-N-[3-(метилсульфонил)бензил]-5-(трифторметил)пиримидин-2-амин XLIVb). Маточные растворы адсорбируют на силикагеле и подвергают хроматографической обработке на силикагеле, элюируя смесью гексан:этилацетат 50:50,получая 8,5 г чистого неполярного соединенияXLIVa: 1 Н ЯМР (ДМСО-d6):8,63 (т, J=5,9 Гц, 1 Н), 8,46 (с, 1 Н), 7,92 (с, 1 Н), 7,83 (дт, J=7,1, 1,8 Гц,1 Н), 7,66 (дт, J=7,7, 1,8 Гц, 1 Н), 7,63 (т, J=7,3 Гц, 1 Н), 4,72 (д, J=6,0 Гц, 2 Н), 3,20 (с, 3 Н); 13 С ЯМР (ДМСО-d6):162,58, 158,33, 155,80 (кв, J=5 Гц), 140,89, 139,87, 132,40, 129,56, 125,86,125,67, 123,42 (кв, J=272 Гц), 105,24 (кв, J=32 Гц), 43,65, 43,51. НМВС трехсвязанная корреляция наблюдается между С 4-N-протоном на 8,63 м.д. и С 5-углеродом на 105 м.д. 2-Хлор-N-[3-(метилсульфинил)бензил]-5-(трифторметил)пиримидин-4-амин (XLVa). Получают способом, раскрытым выше для получения 2-хлор-N-[3-(метилсульфонил)бензил]-5(трифторметил)пиримидин-4-амина (XLIVa), используя 2,4-дихлор-5-трифторметилпиримидин (XL,324 мг, 1,5 ммоль), 1-[3-(метилсульфинил)фенил]метанамингидрохлорид (249 мг, 1,2 ммоль) и диизопропилэтиламин(10 мл). Чистый целевой 2-хлор-N-[3-(метилсульфинил)бензил]-5-(трифторметил)пиримидин-4-амин (XLVa, 162 мг) выделяют хроматографически как неполярную компоненту, элюируя смесью гексан:этилацетат 25:75. 1 Н ЯМР (ДМСО-d6):8,61 (т, J=5,7 Гц, 1 Н), 8,44 (с, 1 Н), 7,67 (с, 1 Н), 7,49-7,58 (м, 2 Н), 7,45 (дт,- 24021801J=6,6, 2,0 Гц, 1 Н), 4,70 (д, J=5,9 Гц, 2 Н), 2,72 (с, 3 Н); 13 С ЯМР (ДМСО-d6):162,59, 158,35, 155,74 (кв, J=5 Гц), 146,49, 139,73, 129,47, 129,24, 123,43 (кв,J=272 Гц), 122,40, 122,36, 105,16 (кв, J=32 Гц), 43,84, 43,23. НМВС трехсвязанная корреляция наблюдается между С 4-N-протоном на 8,61 м.д. и С 5-углеродом на 105 м.д. 2-[2-Хлор-5-(трифторметил)пиримидин-4-ил]амино-N-метилбензамид (XLIIIa). Раствор 2,4-дихлор-5-трифторметилпиримидина(XL,9,5 г,44,0 ммоль) и 2-амино-N-метилбензамида (6,0 г, 40 ммоль) в ТГФ (50 мл) обрабатывают DIPEA (15 мл, 8,6 ммоль) и полученную смесь перемешивают при комнатной температуре в течение ночи. Полученную смесь продуктов концентрируют в вакууме и полученный остаток тщательно растирают с метиленхлоридом, получая твердое вещество белого цвета, которое представляет собой чистый образец неполярной компоненты смеси (2-[2-хлор-5-(трифторметил)пиримидин-4-ил]амино-N-метилбензамид XLIIIa). Метиленовые маточные растворы концентрируют и полученный остаток подвергают хроматографической обработке на силикагеле, элюируя 5% EtOAc в метиленхлориде, получая следующий образец чистого неполярного изомера (0,6 г), образец, содержащий 90% неполярного плюс 10% полярного изомера (XLIIIb, 0,7 г) и 1:1 смесь обоих изомеров (3 г). Полный выход обоих изомеров: 9,3 г (70%).XLIIIa: 1H ЯМР (ДМСО-d6):12,06 (с, 1 Н), 8,86 (кв, J=4,3 Гц, 1 Н), 8,68 (с, 1 Н), 8,38 (д, J=8,2 Гц,1 Н), 7,78 (дд, J=7,8, 1.2 Гц, 1 Н), 7,59 (тд, J=7,9, 1,3 Гц, 1 Н), 7,26 (тд, J=7,6, 0,9 Гц, 1 Н), 2,80 (д, J=4,5 Гц,3 Н); 13 С ЯМР (ДМСО-d6):168,60, 162,19, 156,78 (кв, J=5 Гц), 156,42, 137,52, 131,57, 128,02, 123,14 (кв,J=272 Гц), 123,87, 122,31, 107,25 (кв, J=32 Гц), 26,24. В экспериментах НМВС трехсвязанная корреляция наблюдается между С 4-N-протоном на 12,06 м.д. и С 5-углеродом на 107 м.д. Диэтил (4-[4-хлор-5-(трифторметил)пиримидин-2-ил]амино-3-метоксибензил)фосфонат. 1,0 М раствор дихлорида цинка в эфире (4,07 мл, 4,07 ммоль) добавляют к раствору 2,4-дихлор-5-трифторметилпиримидина (0,803 г, 3,70 ммоль) в 1,2-дихлорэтане (6,7 мл) и t-BuOH(6,7 мл). Через 30 мин добавляют диэтил (4-амино-3-метоксибензил)фосфонат (1,01 г, 3,70 ммоль), затем добавляют триэтиламин (0,567 мл, 4,07 ммоль), поддерживая при этом температуру при 25 С. Реакционную смесь оставляют при перемешивании при комнатной температуре в течение ночи, перед тем, как гасят насыщенным водным раствором NaHCO3 (10 мл). Полученную смесь экстрагируют EtOAc (15 мл) и органический слой промывают солевым раствором (10 мл), сушат над безводным NaSO4, фильтруют и концентрируют при пониженном давлении, получая масло желтого цвета. Полученное сырое вещество вначале очищают, используя систему ISCO Combiflash, элюируя 05% МеОН в DCM в качестве элюента,затем используя препаративную ВЭЖХ(м, 4 Н), 6,90-6,96 (м, 1 Н), 7.03 (м, J=2,00 Гц, 1 Н), 8,02 (д, J=8,34 Гц, 1 Н), 8,62 (с, 1 Н); МС (ES+). Этил (4-аминобензил)метилфосфинат. Раствор п-нитробензилбромида (2,4 г, 11 ммоль) в диэтоксиметилфосфине (2,0 г, 15 ммоль) перемешивают при 140 С в запаянной ампуле в атмосфере азота в течение 16 ч. Реакционную смесь концентрируют в вакууме и полученный остаток очищают, используя хроматографию на силикагеле, элюируя 25% EtOAc в гексане, получая 2,4 г, 87% выход, этил метил(4-нитробензил)фосфината. 1 Н ЯМР (400 МГц, хлороформ-d):м.д. 1,29 (т, J=7,1 Гц, 3 Н), 1,44 (д, J=13,9 Гц, 3 Н), 3,25 (д,J=17,7 Гц, 2 Н), 3,96-4,18 (м, 2 Н), 7,47 (дд, J=9,0, 2,4 Гц, 2 Н), 8,20 (д, J=8,1 Гц, 2 Н); МС (ES+): m/z = 244,0745 [МН+]; ВЭЖХ: tR=1,13 мин (TOF МС: полярная 3 мин). К раствору полученного вещества (2,4 г, 9,9 ммоль) в МеОН (10 мл) добавляют палладий на активированном угле (10 вес.%) (0,52 г, 0,49 ммоль) и откачивают и продувают водородом (3). Полученную смесь перемешивают в атмосфере водорода в течение 16 ч при комнатной температуре, затем фильтруют через слой целита и концентрируют в вакууме, получая 2,0 г (выход: 95%) указанного в заголовке соединения. 1 Н ЯМР (400 МГц, хлороформ-d):м.д. 1,29 (т, J=7,1 Гц, 3 Н), 1,34 (д, J=13,6 Гц, 3 Н), 3,04 (д,J=17,4 Гц, 2 Н), 3,97-4,12 (м, 2 Н), 6,65 (д, J=7,8 Гц, 2 Н), 7,04 (дд, J=8,6, 2,5 Гц, 2 Н); МС (ES+): m/z = 214,0976 [МН+]; ВЭЖХ: tR=0,76 мин (TOF МС: полярная 3 мин). Этил (4-[4-хлор-5-(трифторметил)пиримидин-2-ил]аминобензил)метилфосфинат. Получают, как раскрыто выше для диэтил (4-[4-хлор-5-(трифторметил)пиримидин-2-ил]амино-3 метоксибензил)фосфоната, используя этил (4-аминобензил)метилфосфинат вместо диэтил (4-амино-3 метоксибензил)фосфоната. После хроматографии полученный продукт кристаллизуют из этилацетата,получая 1,1 г (выход: 61%) указанного в заголовке соединения. Н ЯМР (400 МГц, ДМСО-d6):м.д. 10,64 (с, 1 Н), 8,79 (д, J=0,51 Гц, 1 Н), 7,62 (д, J=8,34 Гц, 2 Н),7,25 (дд, J=2,27, 8,59 Гц, 2 Н), 3,86-4,01 (м, 2 Н), 3,15 (д, J=17,68 Гц, 2 Н), 1,31 (д, J=13,64 Гц, 3 Н), 1,19 (т,J=6,95 Гц, 3 Н); МС (ES+): m/z = 396,0708 [МН+]; ВЭЖХ: tR=1,41 мин (СЭЖХ TOF МС: полярная 3 мин). Диэтил (4-[4-хлор-5-(трифторметил)пиримидин-2-ил]амино-3-метоксибензил)фосфонат. 1,0 М раствор дихлорида цинка в эфире (4,07 мл, 4,07 ммоль) добавляют к раствору коммерчески доступного 2,4-дихлор-5-трифторметилпиримидина (0,803 г, 3,70 ммоль) в 1,2 дихлорэтане (6,7 мл) иt-BuOH (6,7 мл). Через 30 мин добавляют диэтил (4-амино-3-метоксибензил)фосфонат (1,01 г,3,70 ммоль), затем добавляют триэтиламин (0,567 мл, 4,07 ммоль), поддерживая при этом температуру при 25 С. Реакционную смесь оставляют при перемешивании при комнатной температуре в течение ночи перед тем, как гасят насыщенным водным раствором NaHCO3 (10 мл). Полученную смесь экстрагируют EtOAc (15 мл) и органический слой промывают солевым раствором (10 мл), сушат над безводнымNaSO4, фильтруют и концентрируют при пониженном давлении, получая масло желтого цвета. Полученное сырое вещество вначале очищают, используя систему ISCO Combiflash, элюируя 05% МеОН вDCM в качестве элюента, затем используя препаративную ВЭЖХ (MDP), получая диэтил (4-[4-хлор-5(трифторметил)пиримидин-2-ил]амино-3-метоксибензил)фосфонат, 1,09 г (выход: 70%). 1 Н ЯМР (CD3OD, 400 МГц):1,28 (т, J=7,07 Гц, 6 Н), 3,23-3,29 (м, 2 Н), 3,90-3,95 (м, 3 Н), 4,01-4,13(транс-изомер). К раствору этил 4-(2-метил-7-нитро-1-оксо-2,3-дигидро-1 Н-изоиндол-4-ил)циклогекс-3-ен-1 карбоксилата (1,03 г, 2,99 ммоль) в EtOH (2,6 мл, 45 ммоль) добавляют палладий 10 вес.% на активированном угле (0,1:0,9 палладий:уголь, 0,11 г, 0,10 ммоль). Реакционную смесь откачивают и продувают газообразным водородом (3). Реакционную смесь оставляют при перемешивании в атмосфере водорода при комнатной температуре в течение 16 ч. Реакционную смесь фильтруют через слой целита. Полученный фильтрат собирают и затем концентрируют в вакууме, получая масло. Два изомера разделяют, используя способ SFC очистки, получая 544 мг (выход: 58%) цис- и 232 мг (выход: 25%) транс-изомеров,указанных в заголовке соединений. Цис-изомер: 1 Н ЯМР (400 МГц, метанол-d):м.д. 7,06 (д, J=8,34 Гц, 1 Н), 6,61 (д, J=8,34 Гц, 1 Н),4,37 (с, 2 Н), 4,20 (кв, J=7,07 Гц, 2 Н), 3,11 (с, 3 Н), 2,72 (д, J=2,53 Гц, 1 Н), 2,46-2,56 (м, 1 Н), 2,22-2,29 (м,2 Н), 1,60-1,75 (м, 6 Н), 1,29 (т, 3 Н); МС (ES+): m/z = 317,14/319,18 (100/15) [МН+]; ВЭЖХ: tR=1,41 мин (Micromass TOF: полярная 3 мин). транс-Изомер: 1 Н ЯМР (400 МГц, метанол-d):7,15 (д, J=8,34 Гц, 1 Н), 6,62 (д, J=8,34 Гц, 1 Н), 4,38(3) и подвергают микроволновому облучению при 100 С в течение 30 мин. Сырую смесь концентрируют в вакууме до твердого вещества и очищают, используя систему органической очистки Combiflash Rf 4 [120 г, колонка RediSep нормальная фаза, GOLD Silica Flash Column, сухая загрузка, градиент элюирования: 040% EtOAc в DCM], получая 1,03 г (выход: 63%) целевого продукта. 1 Н ЯМР (400 МГц, Хлороформ-d):7,71 (д, J=8,08 Гц, 1 Н), 7,45 (д, 1 Н), 5,93-5,99 (м, 1 Н), 4,20 (кв,J=7,07 Гц, 2 Н), 2,65-2,75 (м, 1 Н), 2,49-2,56 (м, 2 Н), 2,36-2,49 (м, 2 Н), 2,16-2,24 (м, 1 Н), 1,87-2,00 (м, 1 Н),1,54 (шир.с, 3 Н), 1,31 (т, J=7,20 Гц, 3 Н); МС (ES+): m/z = 346,15/347,15 (100/15) [МН+]; ВЭЖХ: tR=1,43 мин (СЭЖХ TOF; полярная 3 мин). 2-Метил-7-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,3-дигидро-1 Н-изоиндол-1-он. К дегазированному азотом раствору диоксана (375 мл) добавляют Pd2(dba)3 (2,02 г, 2,21 ммоль) и трициклогексилфосфин (2,48 г, 8,85 ммоль) и перемешивают в течение 30 мин. К полученной смеси добавляют 4-бром-2-метил-7-нитро-2,3-дигидроизоиндол-1-он (20,0 г, 73,8 ммоль), бис-(пинаколато)дибор(24,4 г, 96 ммоль) и KOAc (11,58 г, 118,1 ммоль) и нагревают при кипении с обратным холодильником в- 26021801 течение 6 ч. Охлаждают до комнатной температуры и выпавшее в осадок твердое вещество отфильтровывают. Фильтрат выпаривают досуха и остаток тщательно растирают с диизопропиловым эфиром(100 мл) и фильтруют, получая 2-метил-7-нитро-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-2,3 дигидроизоиндол-1-он в виде твердого вещества желтого цвета (16,0 г, 68%). 1 Н ЯМР (CDCl3, 500 МГц):1,34 (с, 12 Н), 3,24 (с, 3 Н), 4,58 (с, 2 Н), 7,68 (д, J=7,5 Гц, 1 Н), 8,06 (д,J=7,5 Гц, 1 Н). 4-Бром-2-метил-7-нитро-2,3-дигидро-1 Н-изоиндол-1-он. К холодной суспензии 4-бром-2-метил-2,3-дигидроизоиндол-1-она (60 г, 265 ммоль) в концентрированной серной кислоте (60 мл) добавляют предварительно охлажденную смесь концентрированной азотной кислоты (12,5 мл, 265 ммоль) и концентрированной серной кислоты (60 мл) в течение 20 мин. Реакционную смесь перемешивают в течение 30 мин при 0 С и 2 ч при комнатной температуре. Реакционную смесь выливают на смесь воды и льда (300 мл). Образовавшийся осадок отфильтровывают, промывают водой (3100 мл) и суспендируют в изопропаноле (200 мл). Полученную взвесь нагревают на паровой бане и охлаждают. Твердое вещество отфильтровывают и сушат на воздухе, получая 53 г (74%) указанного в заголовке соединения. 1 Н ЯМР (CDCl3, 300 МГц):3,22 (с, 3 Н), 4,36 (с, 2 Н), 7,67 (д, J=8,4 Гц, 1 Н), 7,78 (д, J=8,4 Гц, 1 Н). 4-Бром-2-метил-2,3-дигидро-1 Н-изоиндол-1-он. Раствор метиламина в этаноле (10 мл, 80 ммоль, 8 М раствор в этаноле) добавляют к раствору метил 3-бром-2-(бромметил)бензоата в ТГФ (30 мл). После перемешивания в течение 2 ч полученную реакционную смесь концентрируют досуха и добавляют воду (30 мл) при быстром перемешивании. Полученный твердый продукт выделяют фильтрованием и сушат, получая искомый продукт. Метил 3-бром-2-(бромметил)бензоат. Указанное соединение получают из коммерческой 3-бром-2-метилбензойной кислоты, получая вначале метиловый эфир, затем осуществляя NBS бромирование. Этил 4-[(трифторметил)сульфонил]оксициклогекс-3-ен-1-карбоксилат. 1,50 М литийдиизопропиламид моно(тетрагидрофуран) в циклогексане (2,15 мл, 3,23 ммоль) добавляют к раствору 4-(этоксикарбонил)циклогексанона (0,500 г, 2,94 ммоль) в ТГФ (19,1 мл, 235 ммоль) при-78 С и оставляют при перемешивании в течение 1 ч, после чего добавляют трифторметансульфоновый ангидрид (0,912 г, 3,23 ммоль). Реакционную смесь оставляют при перемешивании, пока она нагревается до комнатной температуры. Реакцию гасят водой. Органический слой разбавляют EtOAc и промывают водой (2) и солевым раствором (1), сушат над сульфатом натрия, фильтруют и затем концентрируют в вакууме. Сырую смесь очищают, используя систему органической очистки Combiflash Rf 4 [12 гRediSep нормальная фаза, Silica Flash Column, сухая загрузка, градиент элюирования: 015% EtOAc в гептане], получая 0,30 г (выход: 33%) целевого продукта. Продукт переносят на следующую стадию без какой-либо дополнительной очистки. МС (ES+): m/z = 304,05/305,05 (100/65) [МН+]; ВЭЖХ: tR=1,37 мин (СЭЖХ TOF; полярная 3 мин). 7-Амино-4-(транс-4-гидроксициклогексил)-2-метил-2,3-дигидро-1 Н-изоиндол-1-он (транс-изомер) и 7-амино-4-(цис-4-гидроксициклогексил)-2-метил-2,3-дигидро-1 Н-изоиндол-1-он (цис-изомер). Раствор 7-амино-2-метил-4-(4-оксоциклогексил)-2,3-дигидро-1 Н-изоиндол-1-она(1,35 г,5,23 ммоль) растворяют в MeOH (30 мл) и охлаждают до 0 С, затем добавляют боргидрид натрия(0,198 г, 5,23 ммоль). Реакционную смесь перемешивают при 0 С в течение 1 ч. Реакцию гасят насыщенным водным раствором NH4Cl, разбавляют EtOAc, промывают солевым раствором и сушат над безводным сульфатом натрия. Полученное соединение очищают, используя ISCO Combiflash, элюируя от 0 до 4% MeOH в EtOAc, получая транс-изомер и цис-изомер. транс-Изомер: 1H ЯМР (400 МГц, ДМСО-d6):7,05 (д, J=8,34 Гц, 1 Н), 6,51 (д, J=8,34 Гц, 1 Н), 5,83(1,64 г, 5,42 ммоль) в ТГФ (60 мл) добавляют водный раствор 3,0 М HCl (1,81 мл), полученную смесь перемешивают при комнатной температуре в течение ночи. Смесь при 0 С разбавляют водой и подщелачивают до pH 10, используя 1 М NaOH. Водную смесь экстрагируют EtOAc (3) и объединенные органические фракции промывают солевым раствором, сушат над сульфатом натрия, фильтруют и концентрируют, получая указанное в заголовке соединение.(1,7 г, 5,7 ммоль) в EtOH (100 мл) добавляют палладий 10 вес.% на активированном угле (0,60 г,0,57 ммоль). Реакционную смесь откачивают и продувают водородом (3) и перемешивают в течение ночи при комнатной температуре. Реакционную смесь фильтруют через слой целита и концентрируют в вакууме, получая указанное в заголовке соединение. 1[1,1'-бис-(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (1:1) (0,57 г, 0,69 ммоль) в 1,4-диоксане (40 мл) и Н 2 О (10 мл), откачивают и снова заполняют азотом (3) и осуществляют микроволновое облучение при 100 С в течение 30 мин. Реакционную смесь разделяют между EtOAc и солевым раствором и выделяют. Водный слой экстрагируют EtOAc (3) и объединенные органические фракции сушат над сульфатом натрия, фильтруют и концентрируют. Соединение очищают, используя ISCO(4-[4-хлор-5(трифторметил)пиримидин-2-ил]амино-3-метоксибензил)фосфоната, используя коммерчески доступный диэтил(4-аминобензил)фосфонат вместо диэтил (4-амино-3-метоксибензил)фосфоната. После хроматографической обработки полуочищенный продукт кристаллизуют из этилацетата, получая указанное в заголовке соединение. 1 Н ЯМР (400 МГц, метанол-d4):1,22-1,32 (м, 6 Н), 3,17-3,28 (м, 2 Н), 3,98-4,10 (м, 4 Н), 7,30 (дд,J=8,72, 2,65 Гц, 2 Н), 7,65 (д, J=7,83 Гц, 2 Н), 8,63 (с, 1 Н). 7-Амино-2-метил-4-[цис-4-(4-метилпиперазин-1-ил)циклогексил]-2,3-дигидро-1 Н-изоиндол-1-он(транс-изомер). К раствору 7-амино-2-метил-4-(4-оксоциклогексил)-2,3-дигидро-1 Н-изоиндол-1-она (100,0 мг,0,3871 ммоль) в 1,2-дихлорэтане (2,0 мл) добавляют 1-метилпиперазин (58,16 мг, 0,5807 ммоль) и триацетоксиборгидрид натрия (164,1 мг, 0,7743 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 24 ч. Реакционную смесь гасят, используя NaHCO3 (10 мл) и экстрагируют EtOAc(20 мл). Органический слой промывают солевым раствором (10 мл), сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении, получая масло светло-желтого цвета. Полученное сырое вещество очищают, используя хроматографическую колонку с силикагелем и систему Combiflash Rf, используя DCM/7 M NH3 в MeOH (100:0-90:10) в качестве элюента, получая цис-изомер (76,5 мг, 58%, первый элюируемый продукт) и транс-изомер (30,0 мг, 23%, второй элюируемый продукт). цис-Изомер: 1H ЯМР (CDCl3, 400 МГц):1,42-1,56 (м, 4 Н), 1,82-1,96 (м, 2 Н), 2,06 (д, J=11,87 Гц,2 Н), 2,27 (шир.с, 1 Н), 2,34 (с, 3 Н), 2,38-2,78 (м, 9 Н), 3,14 (с, 3 Н), 4,31 (с, 2 Н), 5,08 (с, 2 Н), 6,58 (д,J=8,34 Гц, 1 Н), 7,18 (д, J=8,34 Гц, 1 Н); МС (ES+): m/z = 343,25 [МН+]. транс-Изомер: 1 Н ЯМР (CDCl3, 400 МГц):1,35-1,75 (м, 7 Н), 1,91 (д, J=12,13 Гц, 2 Н), 2,03-2,13 (м,2 Н), 2,34 (с, 3 Н), 2,47-2,82 (м, 7 Н), 3,14 (с, 3 Н), 4,28 (с, 2 Н), 5,09 (с, 2 Н), 6,56 (д, J=8,34 Гц, 1 Н), 7,09 (д,J=8,34 Гц, 1 Н); МС (ES+): m/z = 343,26 [МН+]. 4-Хлор-N-(2-метоксифенил)-5-(трифторметил)пиримидин-2-амин. Указанное соединение получают способом, аналогичным способу получения диэтил (4-[4-хлор-5(трифторметил)пиримидин-2-ил]амино-3-метоксибензил)фосфоната, используя коммерчески доступные- 28021801 исходные соединения (2,4-дихлор-5-(трифторметил)пиримидин и 2-метоксианилин). Сырой продукт из указанной реакции перекристаллизовывают, используя MeCN, получая твердое вещество белого цвета,610 мг, 46% выход. 1 Н ЯМР (CDCl3): 3,93 (с, 3 Н), 6,94 (дд, J=8,1, 1,5 Гц, 1 Н), 7,03 (тд, J=7,8, 1,5 Гц, 1 Н), 7,10 (тд, J=7,6,1,8 Гц, 1 Н), 8,14 (шир.с, 1 Н), 8,41 (дд, J=8,1, 1,8 Гц, 1 Н), 8,56 (с, 1 Н); МС (ES+): m/z = 304,00 [MH+]; ВЭЖХ: tR=3,89 мин (ZQ3; полярная 5 мин). Примеры Пример 1. Диэтил [4-(4-[(2-[метил(метилсульфонил)амино]пиридин-3-илметил)амино]-5(трифторметил)пиримидин-2-иламино)бензил]фосфонат.N-[3-([(2-Хлор-5-(трифторметил)пиримидин-4-ил)]аминометил)пиридин-2-ил]-Nметилметансульфонамид (XLIa). N-[3-(аминометил)пиридин-2-ил]-N-метилметансульфонамид (5 г,23,25 ммоль) добавляют по каплям к раствору 2,4-дихлор-5-трифторметилпиримидина (XL, 6,5 г,30 ммоль) в MeOH (80 мл) при 0 С. Через 30 мин через шприц добавляют DIPEA (8,25 мл, 47,5 ммоль) и перемешивание продолжают в течение 1 ч при 0 С. Выпавший в осадок твердый продукт (3 г) собирают фильтрованием, и, по данным ТСХ, он представляет собой чистую неполярную компоненту исходной смеси (XLIa). Полученный фильтрат выпаривают и полученный остаток подвергают хроматографической обработке на силикагеле, элюируя 5% EtOAc в DCM, получая другую порцию чистого неполярного изомера (0,3), смесь двух изомеров (1,7 г) и чистый полярный изомер (XLIb, 1,3 г).XLIa: 1 Н ЯМР (ДМСО-d6):8,54 (т, J=5,7 Гц, 1 Н), 8,46 (дд, J=5,2, 1,6 Гц, 1 Н), 8,45 (с, 1 Н), 7,77 (дд,J=7,8, 1,6 Гц, 1 Н), 7,44 (дд, J=7,7, 4,8 Гц, 1 Н), 4,74 (д, J=5,2 Гц, 2 Н), 3,28 (с, 3 Н), 3,12 (с, 3 Н). Неотнесенные пики:1,29 (м). НМВС эксперименты свидетельствуют о трехсвязанной Н-С корреляции между С 4-N-протоном на 8,54 м.д. и С 5-углеродом, которая наблюдается только для региоизомера, как представлено выше. Раствор 1:1 смеси N-[3-([(2-хлор-5-(трифторметил)пиримидин-4-ил)]аминометил)пиридин-2-ил]N-метилметансульфонамидаN-[3-([(4-хлор-5-(трифторметил)пиримидин-2 ил)]аминометил)пиридин-2-ил]-N-метилметансульфонамида (XLIb) (30 мг, 0,08 ммоль) и коммерчески доступного диэтил (4-аминобензил)фосфоната (22,1 мг, 0,091 ммоль) в смеси трифторуксусной кислоты(6,8 мкл, 0,1 ммоль) и трифторэтанола (0,5 мл) в атмосфере N2, перемешивают при 105 С в течение 75 мин в микроволновом реакторе Biotage. Растворители удаляют в вакууме и полученную смесь разделяют, используя препаративную ВЭЖХ (MDP), получая 14,2 мг диэтил [4-(4-[(2[метил(метилсульфонил)амино]пиридин-3-илметил)амино]-5-(трифторметил)пиримидин-2 иламино)бензил]фосфоната (пример 1) (выход: 31%) и 9,5 мг региоизомера XLIIb (выход: 21%). 1 Н ЯМР (ДМСО-d6, 400 МГц):1,14 (т, J=7,2 Гц, 6 Н), 3,08 (д, J=21,2 Гц, 2 Н), 3,16 (с, 3 Н), 3,17 (с,3 Н), 3,88-3,94 (м, 4 Н), 4,81 (с, д, J=6,0 Гц, 2 Н), 7,01 (д, J=6,8 Гц, 2 Н), 7,36-7,38 (м, 2 Н), 7,41 (дд, J=4,8,7,6 Гц, 1 Н), 7,66 (д, J=6,4 Гц, 2 Н), 8,26 (д, J=0,8 Гц, 1 Н), 8,43 (дд, J=1,6, 4,4 Гц, 1 Н), 9,60 (шир.с, 1 Н); МС (ES+): m/z = 603,43 (МН+); ВЭЖХ: tR=3,18 мин (ZQ3; полярная 5 мин).
МПК / Метки
МПК: A61K 31/497, C07D 403/12, A61P 35/00, C07D 401/12, C07D 403/14
Метки: аминопиримидиновые, противораковые, соединения
Код ссылки
<a href="https://eas.patents.su/30-21801-aminopirimidinovye-protivorakovye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Аминопиримидиновые противораковые соединения</a>
Противораковые комбинации

Номер патента: 8056
Опубликовано: 27.02.2007
Авторы: Уилсон Уилльям Роберт, Сиим Бронуин Гей
МПК: A61K 31/19, A61K 31/505, A61K 31/475...
Метки: комбинации, противораковые
Формула / Реферат:
1. Способ лечения рака, который включает введение млекопитающему, включая человека, нуждающемуся в таком лечении, эффективного количества DMXAA, или ее фармацевтически приемлемой соли, или ее сложного эфира и одновременно или последовательно введение эффективного количества соединения, выбираемого из ингибиторов топоизомеразы I и гемцитабина. 2. Способ по п.1, где в качестве соединения, выбираемого из ингибиторов топоизомеразы I, используют...
Комбинация, включающая комбретастатин и противораковые средства

Номер патента: 6316
Опубликовано: 27.10.2005
Автор: Биссери Мари-Кристин
МПК: A61P 35/00, A61K 31/4745
Метки: комбинация, включающая, комбретастатин, средства, противораковые
Формула / Реферат:
1. Фармацевтическая комбинация, содержащая эффективное количество противоракового соединения, выбранного из группы, состоящей из таксанов, алкалоидов барвинка, алкилирующих агентов, антиметаболитов, эпидофиллоптоксинов и антибиотиков в сочетании с эффективным количеством комбретастатина для лечения солидных опухолей, где указанный комбретастатин обладает следующей формулой: 2. Комбинация по п.1, где указанный комбретастатин находится в виде...
Индукторы апоптоза, содержащие их противораковые и канцеростатические средства, способ индуцирования апоптоза, полисахариды и способы их получения

Номер патента: 3902
Опубликовано: 30.10.2003
Авторы: Накаяма Синдзи, Ю Фу-Гонг, Катаяма Каору, Томинага Таканари, Кодзима Каору, Икай Кацусиге, Китано Хидео, Сакай Такеси, Наканиси Йосикуни, Симанака Казуо, Като Икуносин, Кимура Хитоми
МПК: C08B 37/00, A61K 31/715, A61P 35/00...
Метки: канцеростатические, получения, способ, средства, индукторы, полисахариды, противораковые, способы, содержащие, апоптоза, индуцирования
Формула / Реферат:
1. Индуктор апоптоза, включающий сульфированный(ые) фукозусодержащий(ие) полисахарид(ы) и/или продукт(ы) его(их) расщепления, причем указанный(ые) сульфированный(ые) фукозусодежащий(ие) полисахарид(ы) имеет(ют) следующие физико-химические свойства: (а) составляющий его(их) сахарид содержит уроновую кислоту и (б) будучи расщепленным(и) фукоиданазой, продуцируемой Flavobacterium sp. SA-0082 (FERM BP-5402), образует, по крайней мере, одно из...
2-аминопиримидиновые модуляторы рецептора н4 гистамина, способ их получения, фармацевтические композиции и способ их применения для модуляции рецептора h4 гистамина

Номер патента: 16133
Опубликовано: 28.02.2012
Авторы: Вэй Дзяньмэй, Чавез Франк, Волин Рональд Л., Лю Цзин, Мани Неелакандха С., Цай Хой, Фитцджералд Энн Е., Савалль Брэд М., Венэйбл Дженнифер Д., Риззолио Мишель К., Смит Дебора М., Эдвардс Джеймс П., Винер Даниэль К.
МПК: A01N 43/40, A61K 31/445
Метки: способ, 2-аминопиримидиновые, получения, композиции, гистамина, рецептора, модуляции, применения, модуляторы, фармацевтические
Формула / Реферат:
1. Химическое соединение, выбранное из соединений формулы (I)в которойR1 обозначает:a) C1-6алкил, в случае необходимости замещенный -OC1-4алкилом, -CF3 или -O-(моноциклическим циклоалкилом);b) бензил, -CH2-(моноциклический гетероарил) (где гетероарил выбран из пиридинила, тиенила и тетрагидрофуранила) или фенетил, каждый из которых может быть в случае необходимости замещен галогеном;c) моноциклический циклоалкил, -(CH2)0-1-тетрагидрофуранил или...
Соединения 1,2,4,5-тетрагидро-3н-бензазепина, способ их получения и фармацевтические композиции, содержащие эти соединения

Номер патента: 14756
Опубликовано: 28.02.2011
Авторы: Шименти Стефано, Гумен Бертран, Пеглион Жан-Луи, Вийенов Николь, Толлон Катрин, Дессинже Эмея, Вилен Жан-Поль, Кеньяр Паскаль
МПК: A61P 9/00, C07D 223/16, A61K 31/55...
Метки: эти, способ, соединения, получения, композиции, фармацевтические, 1,2,4,5-тетрагидро-3н-бензазепина, содержащие
Формула / Реферат:
1. Соединение формулы (I)где R1представляет собой атом водорода или группу, выбранную из С3-С7 циклоалкила, бензила и линейного или разветвленного C1-C6алкила, алкильной группы, которая может быть насыщенной или ненасыщенной и может быть необязательно замещенной гидроксигруппой или С3-С7 циклоалкильной группой или одним или несколькими атомами галогена,R2, R3, R4 и R5могут быть одинаковыми или различаться, каждый выбирается из атома водорода или...