Транс-4-[[(5s)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2h-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1н-1-бензазепин-1-ил]метил]циклогексанкарбоновая кислота
Номер патента: 20600
Опубликовано: 30.12.2014
Авторы: Фрэнк Скотт Алан, Ремик Дэвид Майкл, Чэнь Синьчао, Педерсен Стивен Уэйн







Формула / Реферат
1. Соединение, которое представляет собой транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновую кислоту, гидрат или фармацевтически приемлемую соль указанного соединения.
2. Соединение по п.1, отличающееся тем, что катион фармацевтически приемлемой соли выбран из натрия, калия, магния, кальция, цинка или трет-бутиламмония.
3. Соединение по п.1, которое представляет собой гидрат транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты.
4. Соединение по п.1 или 3, которое представляет собой гидрат транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты в кристаллической форме, характеризуемой порошковой рентгеновской дифрактограммой, полученной с применением источника CuKα(l=1,54056 Å), содержащей пики при:
a) 7,5, 9,2, 10,7 и 15,5±0,2° 2θ, или
b) 7,5, 9,2, 10,7, 13,8, 15,0, 15,5 и 19,5±0,2° 2θ, или
c) 7,5, 9,2, 10,7, 13,8, 11,3, 15,0, 15,5, 17,7, 19,5 и 25,1±0,2° 2θ.
5. Соединение по любому из пп.1 или 3, которое представляет собой гидрат транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты в кристаллической форме, характеризуемой спектром ЯМР твердого тела, содержащим пики, относительно адамантана (δ=29,5 м.д.), при:
a) 175,6, 168,0, 61,1, 21,2 и 18,3±0,2 м.д., или
b) 175,6, 168,0, 145,6, 144,8, 61,1, 45,0, 21,2 и 18,3±0,2 м.д., или
c) 175,6, 168,0, 145,6, 144,8, 139,9, 136,3, 61,1, 53,0, 49,8, 45,0, 21,2 и 18,2±0,2 м.д.
6. Соединение, которое представляет собой транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2Н-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1Н-1-бензазепин-1-ил]метил]циклогексанкарбоновую кислоту или ее фармацевтически приемлемую соль.
7. Фармацевтическая композиция для лечения дислипидемии, содержащая транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновую кислоту, гидрат или гидрат в кристаллической форме по любому из пп.1-6.
8. Фармацевтическая композиция по п.7, содержащая гидрат транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты в кристаллической форме.
9. Фармацевтическая композиция по п.7, содержащая более чем примерно 80% гидрата транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты в кристаллической форме.
10. Соединение, представляющее собой гемиэтанолсольват геми-трет-бутиламиновой соли транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты в кристаллической форме, характеризуемой порошковой рентгеновской дифрактограммой, полученной с применением источника CuKα (l=1,54056 Å), содержащей пики при:
a) 5,5, 9,0, 14,3, 22,0 и 22,5±0,2° 2θ, или
b) 5,5, 9,0, 14,3, 17,5, 18,2, 19,4, 20,6, 22,0 и 22,5±0,2° 2θ, или
c) 5,5, 9,0, 13,2, 13,6, 14,3, 15,2, 17,5, 18,2, 19,4, 19,8, 20,6, 22,0 и 22,5±0,2° 2θ.
11. Соединение, представляющее собой муравьинокислый сольват транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты в кристаллической форме, характеризуемой порошковой рентгеновской дифрактограммой, полученной с применением источника CuKα (l=1,54056 Å), содержащей пики при:
a) 15,4, 16,9, 18,2 и 18,6±0,2° 2θ, или
b) 15,4, 15,7, 16,9, 18,2, 18,6, 19,5, 22,8, 25,7 и 25,5±0,2° 2θ, или
c) 13,0, 13,9, 15,4, 15,7, 16,9, 16,4, 18,2, 18,6, 19,5, 20,8, 22,8, 25,7 и 25,5±0,2° 2θ.
12. Соединение, представляющее собой уксусно-кислый сольват транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты в кристаллической форме, характеризуемой порошковой рентгеновской дифрактограммой, полученной с применением источника CuKα(l=1,54056 Å), содержащей пики при:
a) 12,9, 15,1, 18,4, 19,4 и 20,8±0,2° 2θ, или
b) 12,9, 13,8, 15,1, 16,4, 17,8, 18,4, 19,4, 20,1 и 20,8±0,2° 2θ, или
c) 11,0, 12,9, 13,8, 15,1, 15,6, 16,4, 17,8, 18,4, 19,4, 20,1, 20,8 и 21,7±0,2° 2θ.
13. Соединение, представляющее собой изопропанольный сольват трет-бутиламиновой соли транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты в кристаллической форме, характеризуемой порошковой рентгеновской дифрактограммой, полученной с применением источника CuKα (l=1,54056 Å), содержащей пики при:
a) 5,6, 11,3, 12,6 и 17,9±0,2° 2θ или
b) 5,6, 8,0, 11,3, 12,6, 17,9, 20,4 и 24,1±0,2° 2θ.
14. Фармацевтическая композиция для лечения дислипидемии, содержащая соединение по любому из пп.1-6 и 10-13 и по меньшей мере один фармацевтически приемлемый носитель, наполнитель или разбавитель.
15. Фармацевтическая композиция по п.14, содержащая гидрат транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2Н-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты.
16. Фармацевтическая композиция по п.14 или 15, содержащая гидрат транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты в кристаллической форме.
17. Применение соединения по любому из пп.1-6 и 10-13 для получения лекарственного средства для лечения дислипидемии.
18. Применение соединения по любому из пп.1-6 и 10-13 для получения лекарственного средства для лечения атеросклероза.
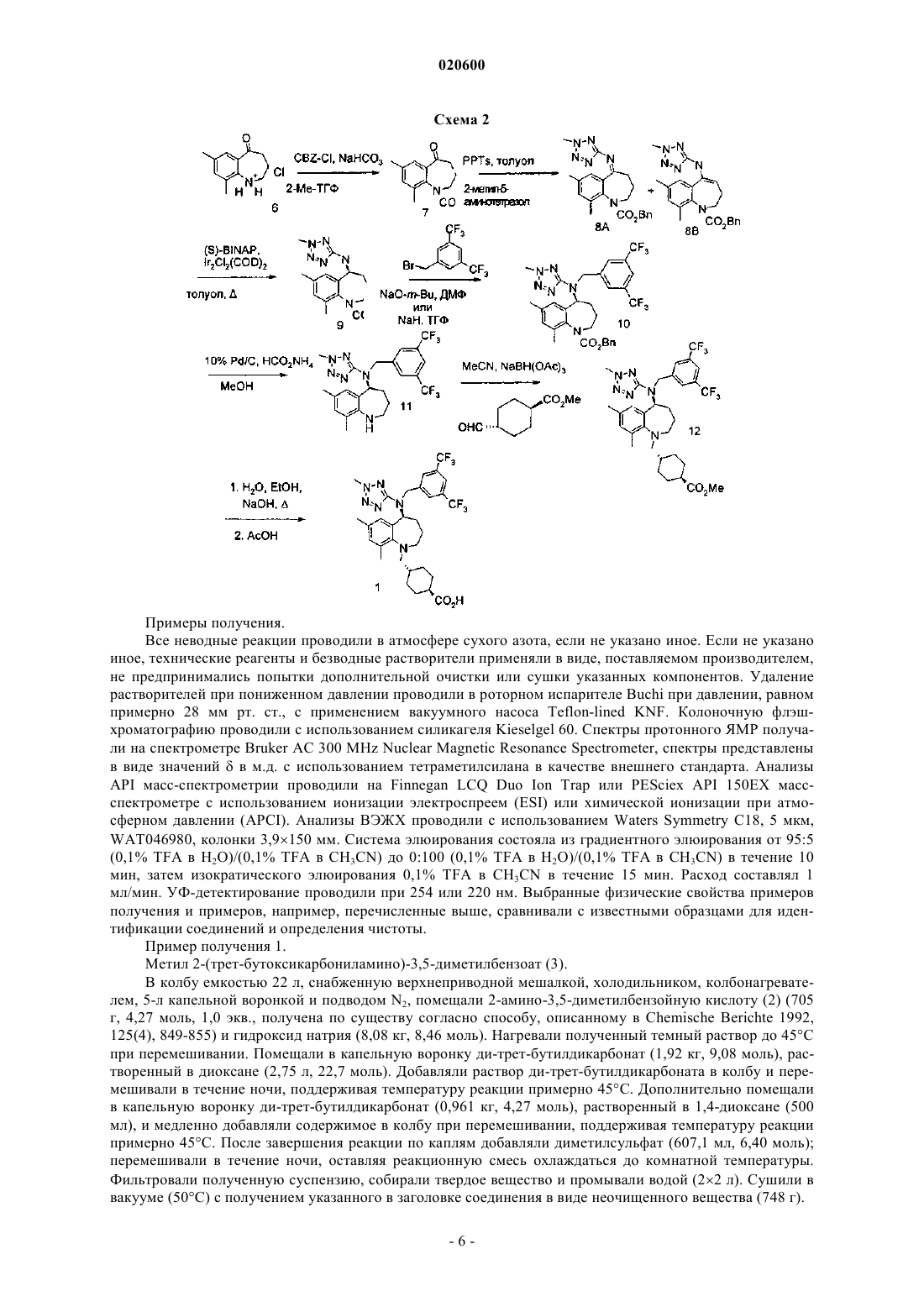
19. Способ получения транс-4-[[(5S)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2H-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1H-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты или ее фармацевтически приемлемой соли, включающий деэтерификацию соединения формулы II

где R выбран из C1-4алкила, С1-4галогеналкила, С3-6циклоалкила, С1-4алкил-С3-6циклоалкила, фенила или С1-5алкилфенила,
с обеспечением соединения формулы I или его фармацевтически приемлемой соли

20. Способ по п.19, дополнительно включающий конденсацию соединения формулы III

с последующим получением соединения формулы I или его фармацевтически приемлемой соли.
21. Соединение, имеющее структуру

где R выбран из С1-4алкила, С1-4галогеналкила, С3-6циклоалкила, С1-4алкил-С3-6циклоалкила, фенила или С1-5алкилфенила.
Текст
Чэнь Синьчао, Фрэнк Скотт Алан,Ремик Дэвид Майкл, Педерсен Стивен Уэйн (US) названные транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5 ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислотой, в виде их свободной кислоты или фармацевтически приемлемой соли, гидрата и гидрата в кристаллической форме; фармацевтические составы и способы их применения.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Настоящее изобретение относится к транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2 метил-2 Н-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоте, фармацевтически приемлемым солям и кристаллическим формам указанного соединения, а также к их получению и способам лечения с применением указанного соединения. Дислипидемия является основным фактором риска развития сердечно-сосудистых заболеваний(ССЗ). Низкое содержание в плазме крови холестерина липопротеинов высокой плотности (ЛПВП-X) вкупе с нормальным или повышенным содержанием холестерина липопротеинов низкой плотности(ЛПНП-Х) является значительным фактором риска развития атеросклероза и связанной с ним ишемической болезни сердца. Белок-переносчик эфиров холестерила (или холестерина) (СЕТР) представляет собой гликопротеин, который способствует обмену холестериловых эфиров в ЛПВП на триглицериды в богатых триглицеридами липопротеинах. Конечным результатом активности СЕТР является снижение уровня ЛПВП-холестерина и повышение уровня ЛПНП-холестерина. Указанное влияние на свойства липопротеина, как полагают, является проатерогенным, в частности, у субъектов, у которых липидный профиль свидетельствует о повышенном риске развития ИБС. В опубликованной международной заявке WO 06/002342 предложены конкретные соединения,имеющие приведенную ниже структуру, где R1-R6 описаны в указанной заявке, подходящие для лечения сердечно-сосудистых заболеваний. Несмотря на известность приведенного выше решения, по-прежнему существует большая потребность в эффективных соединениях, подходящих для лечения сердечно-сосудистых заболеваний, включая атеросклероз и/или дислипидемию. Существует необходимость получения соединений, эффективных для лечения сердечнососудистого заболевания при пероральном дозировании, которые являются стабильными в кислой среде,например, в желудке. Также необходимо, чтобы после введения соединения проявляли биодоступность и/или пероральное действие, достаточно высокие для обеспечения более эффективного лечения. Настоящее изобретение направлено на удовлетворение указанных потребностей и обеспечивает соединение, подходящее для лечения сердечно-сосудистых заболеваний, включая, но не ограничиваясь ими, дислипидемию и атеросклероз. В настоящем изобретении предложено соединение, проявляющее особенно преимущественные и неожиданные свойства. Физические и фармакологические свойства соединения согласно настоящему изобретению делают его особенно подходящим для введения в состав таблеток для перорального дозирования. Конкретные преимущественные свойства включают, среди прочего, повышенную стабильность, растворимость и/или биодоступность. В настоящем изобретении предложено соединение, которое представляет собой транс-4-(5S)-53,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9 диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновую кислоту (идентифицированную согласно ее названию в Chemical Abstracts (в настоящем описании обозначенную как ВССА), имеющую структуру формулы I, приведенную ниже, и фармацевтически приемлемые соли указанного соединения. Соединение - ВССА - может представлять собой свободную кислоту (далее свободная кислота ВССА) или ее фармацевтически приемлемую соль в виде сольвата (далее ВССАсольват) и гидрата (далее ВССАгидрат). Сольватные молекулы включают воду (в виде гидрата), метанол, этанол, муравьиную кислоту, уксусную кислоту и изопропанол. В настоящем изобретении предложено соединение, которое представляет собой ВССА, его фармацевтически приемлемую соль, гидрат или сольват. Согласно одному из вариантов реализации предложено соединение ВССА в виде свободной кислоты. Согласно другим вариантам реализации предложена ВССА из гидрата ВССА или сольвата ВССА (свободная кислота или соль) в кристаллической форме. Согласно другим вариантам реализации в настоящем изобретении предложено превращение ВССА из гидрата ВССА или сольвата ВССА в аморфную твердую форму. В настоящем изобретении предложено соединение, которое представляет собой гидрат ВССА в кристаллической форме, характеризуемой порошковой рентгеновской дифрактограммой, полученной с применением источника CuK (=1,54056 ), которая содержит пики при: а) 7,5, 9,2, 10,7 и 15,50,2 2;b) 7,5, 9,2, 10,7, 13,8, 15,0, 15,5 и 19,50,2 2 или с) 7,5, 9,2, 10,7, 13,8, 11,3, 15,0, 15,5, 17,7, 19,5 и 25,10,2 2. Согласно другому варианту реализации в настоящем изобретении предложено соединение, которое представляет собой гидрат ВССА в кристаллической форме, характеризуемой спектром ЯМР твердого тела, содержащим пики, относительно адамантана (=29,5 м.д.), при: а) 175,6, 168,0, 61,1, 21,2 и 18,30,2 м.д.;b) 175,6, 168,0, 145,6, 144,8, 61,1, 45,0, 21,2 и 18,30,2 м.д. или с) 175,6, 168,0, 145,6, 144,8, 139,9, 136,3, 61,1, 53,0, 49,8, 45,0, 21,2 и 18,20,2 м.д. Согласно другим вариантам реализации предложена ВССА в виде гидрата ВССА в кристаллической форме, характеризуемой по меньшей мере одним из следующего: а) порошковой рентгеновской дифрактограммой, полученной с применением источника CuK(=1,54056 ), которая содержит пики при 7,5, 9,2, 10,7 и 15,50,2 2, илиb) спектром ЯМР твердого тела, содержащим пики, относительно адамантана (=29,5 м.д.) при 175,6, 168,0, 61,1, 21,2 и 18,30,2 м.д. В настоящем изобретении предложено соединение, которое представляет собой гемиэтанолсольват геми-трет-бутиламиновой соли ВССА в кристаллической форме, характеризуемой порошковой рентгеновской дифрактограммой, полученной с использованием источника CuK (=1,54056 ), содержащей пики при: а) 5,5, 9,0, 14,3, 22,0 и 22,50,2 2, илиb) 5,5, 9,0, 14,3, 17,5, 18,2, 19,4, 20,6, 22,0 и 22,50,2 2, или с) 5,5, 9,0, 13,2, 13,6, 14,3, 15,2, 17,5, 18,2, 19,4, 19,8, 20,6, 22,0 и 22,50,2 2. В настоящем изобретении предложено соединение, которое представляет собой муравьинокислый сольват ВССА в кристаллической форме, характеризуемой порошковой рентгеновской дифрактограммой, полученной с применением источника CuK (=1,54056 ), содержащей пики при: а) 15,4, 16,9, 18,2 и 18,60,2 2, илиb) 15,4, 15,7, 16,9, 18,2, 18,6, 19,5, 22,8, 25,7 и 25,50,2 2, или с) 13,0, 13,9, 15,4, 15,7, 16,9, 16,4, 18,2, 18,6, 19,5, 20,8, 22,8, 25,7 и 25,50,2 2. В настоящем изобретении предложено соединение, которое представляет собой уксуснокислый сольват ВССА в кристаллической форме, характеризуемой порошковой рентгеновской дифрактограммой, полученной с применением источника CuK (=1,54056 ), которая содержит пики при: а) 12,9, 15,1, 18,4, 19,4 и 20,80,2 2, илиb) 12,9, 13,8, 15,1, 16,4, 17,8, 18,4, 19,4, 20,1 и 20,80,2 2, или с) 11,00, 12,9, 13,8, 15,1, 15,6, 16,4, 17,8, 18,4, 19,4, 20,1, 20,8 и 21,70,2 2. В настоящем изобретении предложено соединение, которое представляет собой изопропанолсольват трет-бутиламиновой соли ВССА в кристаллической форме, характеризуемой порошковой рентгеновской дифрактограммой, полученной с применением источника CuK (=1,54056 ), которая содержит пики при: а) 5,6, 11,3, 12,6 и 17,90,2 2 илиb) 5,6, 8,0, 11,3, 12,6, 17,9, 20,4 и 24,10,2 2. Согласно другому варианту реализации в настоящем изобретении предложена ВССА или ее фармацевтически приемлемая соль в виде сольвата, где сольват выбран из воды (также называемый гидратом),метанола, этанола, изопропанола, муравьиной кислоты или уксусной кислоты. Мольное отношение ВССА и сольвата может составлять примерно от 1:0,3 до 1:1, более предпочтительно примерно от 1:0,5 до 1:10,2 (ВССА или соль:сольват). Предпочтительные сольваты включают воду, изопропанол и этанол. Согласно другому варианту реализации в настоящем изобретении предложена ВССА в виде фармацевтически приемлемой соли. Предпочтительные катионы фармацевтически приемлемой соли могут быть выбраны из натрия, калия, магния, кальция, цинка или трет-бутиламина (или трет-бутиламмония). Более предпочтительными катионами являются натрий, кальций и трет-бутиламин. В настоящем изобретении предложена, по существу, чистая ВССА в виде свободной кислоты ВССА или ее фармацевтически приемлемых солей, гидрата ВССА, сольвата ВССА (или гидрата ВССА) и сольвата соли ВССА в кристаллической форме (в отдельности "названная форма ВССА"). Используемый в настоящем описании термин "по существу, чистый" относится к композиции, содержащей более чем 80% (мас./мас.) названной формы ВССА, предпочтительно более чем 95% (мас./мас.) названной формы ВССА, еще более предпочтительно более чем 98% (мас./мас.) названной формы ВССА. Согласно особенно предпочтительному аспекту в настоящем изобретении предложен, по существу, чистый гидрат ВССА в кристаллической форме. В настоящем изобретении предложена фармацевтическая композиция, содержащая ВССА или ее фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый носитель,наполнитель или разбавитель. В выбранных вариантах осуществления фармацевтическая композиция содержит, по существу, чистую ВССА в виде свободной кислоты ВССА или ее фармацевтически приемлемой соли, сольвата ВССА, сольвата соли ВССА или гидрата ВССА в кристаллической форме. Согласно особенно предпочтительному аспекту фармацевтическая композиция содержит, по существу, чистый гидрат ВССА в кристаллической форме. В настоящем изобретении также предложен способ лечения сердечно-сосудистого заболевания у пациента, при этом способ включает введение пациенту эффективного количества ВССА. В предпочтительных вариантах реализации способ включает введение ВССА в виде гидрата ВССА. В других вариантах реализации способ включает введение сольвата ВССА в кристаллической форме или гидрата ВССА в кристаллической форме. В настоящем изобретении предложено применение ВССА (в виде свободной кислоты ВССА или ее фармацевтически приемлемой соли, сольвата соли ВССА, гидрата ВССА или сольвата ВССА в кристаллической форме или гидрата ВССА в кристаллической форме) согласно настоящему изобретению для получения лекарственного средства для лечения сердечно-сосудистых заболеваний, включающих, но не ограничивающихся ими, дислипидемию и атеросклероз. В настоящем изобретении предложена ВССА (в виде свободной кислоты ВССА; сольвата соли ВССА, гидрата ВССА или сольвата ВССА в кристаллической форме согласно настоящему изобретению) в качестве лекарственного средства. В настоящем изобретении также предложена ВССА (в виде свободной кислоты ВССА, или сольвата соли ВССА, гидрата ВССА, сольвата ВССА в кристаллической форме,в кристаллической форме согласно настоящему изобретению) для применения в терапии. В настоящем изобретении предложена ВССА (в виде свободной кислоты ВССА, или гидрата ВССА, сольвата соли ВССА или сольвата ВССА в кристаллической форме согласно настоящему изобретению) для применения для лечения сердечно-сосудистых заболеваний, включающих, но не ограничивающихся ими, дислипидемию и атеросклероз. В настоящем изобретении также предложено соединение, имеющее структуру, приведенную ниже где R выбран из С 1-4 алкила, С 3-6 циклоалкила, С 1-4 алкил-С 3-6 циклоалкила, фенила и C1-5 алкилфенила,с обеспечением соединения формулы I или его фармацевтически приемлемой соли. Предпочтительные группы R включают С 1-4 алкил, С 1-4 галогеналкил, С 3-6 циклоалкил, С 1-4 алкил-С 3-6 циклоалкил, фенил и С 1-5 алкилфенил. Особенно предпочтительные группы R включают метил, этил, фенил и бензил. Термин "С 1-4 алкил" или "С 1-5 алкил" в настоящем описании включает линейную или разветвленную алкильную цепь, содержащую от 1 до 4 или от 1 до 5 атомов углерода соответственно. Термин "галогеналкил" относится к алкильной группе, содержащей один или более галоген, присоединенныйк одному или более атому углерода. Как отмечено выше, алкильная группа может быть линейной или разветвленной. Предпочтительными галогенами являются фтор, хлор и бром. Фтор является особенно предпочтительным. Согласно другом варианту реализации в настоящем изобретении предложено соединение, которое представляет собой или его фармацевтически приемлемая соль. В настоящем изобретении предложен способ получения ВССА. Способ включает деэтерификацию приведенного ниже соединения формулы II где R выбран из С 1-4 алкила, С 1-4 галогеналкила, С 3-6 циклоалкила, С 1-4 алкил-С 3-6 циклоалкила, фенила или C1-5 алкилфенила,с обеспечением соединения формулы I или его фармацевтически приемлемой соли для получения ВССА или ее фармацевтически приемлемой соли. Предпочтительные группы R включают С 1-4 алкил, фенил и С 1-5 алкилфенил. Особенно предпочтительные группы R включают метил, этил, фенил и бензил. Способ также может включать снятие защиты с заместителя, представляющего собой защищенную карбоксильную группу, при циклогексильной группе. Примеры различных функциональных защитных групп для кислот, способы получения защищенных кислот и способы снятия защиты с кислот можно найти в "Protecting Groups in Organic Synthesis", 3rd Ed. Greene, T.W., Wuts, P.G.M., Eds., John Wiley andSons, New York, 1999. Специалисты в данной области техники понимают, что в дополнение к карбоновой кислоте и защищенной карбоновой кислоте можно применять другие функциональные группы, которые могут быть легко превращены в карбоновую кислоту, вместо карбоновой кислоты или защищенной кислоты. Такие функциональные группы, примеры получения и превращения указанных групп в карбоновые кислоты можно найти в "Comprehensive Organic Transformations: A Guide to Functional Group Preparations" by Larock. R.C., Wiley VCH, 1999 и в "March's Advanced Organic Chemistry, Reactions, Mechanismsand Structure", Smith, M.B., and March, J., Wiley-Interscience, 6th Ed. 2007. Согласно другому варианту реализации в настоящем изобретении предложен способ, описанный выше и дополнительно включающий конденсацию представленного соединения формулы III где R описан выше,с получением соединения формулы I или его фармацевтически приемлемой соли. Как отмечено выше, карбоновую кислоту, сложный эфир или защищенную кислоту, обозначенную как CO2R в структуре, представленной непосредственно выше, можно заменить на функциональную группу, которую можно превратить в карбоновую кислоту или защищенную кислоту. Предпочтительно соединение, являющееся эффективным ингибитором СЕТР, должно быть стабильно химически и термически, иметь достаточную растворимость для простоты введения и приготовления в виде состава и сохранять достаточную активность. Кроме того, соединения должны проявлять достаточно высокую биодоступность для обеспечения количества соединения in vivo для эффективного лечения. Указанные свойства, проявляемые ВССА, не изучались и не могли быть предсказаны исходя из уровня техники. Гидрат ВССА можно хранить при температуре окружающей среды с минимальным или очень низ-4 020600 ким разложением. Соединение, представляющее собой гидрат ВССА в кристаллической форме, начинает переходить из формы сольвата и/или плавиться при температуре более примерно 50 С, определенной путем дифференциальной сканирующей калориметрии, что делает его приемлемым для традиционных промышленных процессов, например измельчения. Также ВССА, изопропанолсольват третбутиламиновой соли ВССА, гемиэтанолсольват геми-трет-бутиламиновой соли ВССА и гидрат ВССА в кристаллической форме являются негигроскопичными при хранении при температуре окружающей среды. Кроме того, фармацевтически приемлемые соли, такие как соли кальция или цинка, по существу,являются негигроскопичными при хранении при температуре окружающей среды. ВССА в виде свободной кислоты, сольвата или гидрата в кристаллической форме можно получать согласно представленным далее способам, в целом проиллюстрированным далее на схемах 1, 2, 3 и 4, в частности, описанных в представленных далее примерах получения и примерах. В настоящем описании используют следующие сокращения:TFA относится к трифторуксусной кислоте; ТГФ относится к тетрагидрофурану; КТ относится к комнатной температуре. Если не указано иное, наименования и нумерация соединений, приведенных в настоящем описании,получены при помощи CHEMDRAW ULTRA AUTONOM version 7.0.1 или Symyx Draw version 3.2. Схема 1 Примеры получения. Все неводные реакции проводили в атмосфере сухого азота, если не указано иное. Если не указано иное, технические реагенты и безводные растворители применяли в виде, поставляемом производителем,не предпринимались попытки дополнительной очистки или сушки указанных компонентов. Удаление растворителей при пониженном давлении проводили в роторном испарителе Buchi при давлении, равном примерно 28 мм рт. ст., с применением вакуумного насоса Teflon-lined KNF. Колоночную флэшхроматографию проводили с использованием силикагеля Kieselgel 60. Спектры протонного ЯМР получали на спектрометре Bruker AC 300 MHz Nuclear Magnetic Resonance Spectrometer, спектры представлены в виде значенийв м.д. с использованием тетраметилсилана в качестве внешнего стандарта. АнализыAPI масс-спектрометрии проводили на Finnegan LCQ Duo Ion Trap или PESciex API 150EX массспектрометре с использованием ионизации электроспреем (ESI) или химической ионизации при атмосферном давлении (APCI). Анализы ВЭЖХ проводили с использованием Waters Symmetry C18, 5 мкм,WAT046980, колонки 3,9150 мм. Система элюирования состояла из градиентного элюирования от 95:5(0,1% TFA в Н 2 О)/(0,1% TFA в CH3CN) до 0:100 (0,1% TFA в Н 2 О)/(0,1% TFA в CH3CN) в течение 10 мин, затем изократического элюирования 0,1% TFA в CH3CN в течение 15 мин. Расход составлял 1 мл/мин. УФ-детектирование проводили при 254 или 220 нм. Выбранные физические свойства примеров получения и примеров, например, перечисленные выше, сравнивали с известными образцами для идентификации соединений и определения чистоты. Пример получения 1. Метил 2-(трет-бутоксикарбониламино)-3,5-диметилбензоат (3). В колбу емкостью 22 л, снабженную верхнеприводной мешалкой, холодильником, колбонагревателем, 5-л капельной воронкой и подводом N2, помещали 2-амино-3,5-диметилбензойную кислоту (2) (705 г, 4,27 моль, 1,0 экв., получена по существу согласно способу, описанному в Chemische Berichte 1992,125(4), 849-855) и гидроксид натрия (8,08 кг, 8,46 моль). Нагревали полученный темный раствор до 45 С при перемешивании. Помещали в капельную воронку ди-трет-бутилдикарбонат (1,92 кг, 9,08 моль), растворенный в диоксане (2,75 л, 22,7 моль). Добавляли раствор ди-трет-бутилдикарбоната в колбу и перемешивали в течение ночи, поддерживая температуру реакции примерно 45 С. Дополнительно помещали в капельную воронку ди-трет-бутилдикарбонат (0,961 кг, 4,27 моль), растворенный в 1,4-диоксане (500 мл), и медленно добавляли содержимое в колбу при перемешивании, поддерживая температуру реакции примерно 45 С. После завершения реакции по каплям добавляли диметилсульфат (607,1 мл, 6,40 моль); перемешивали в течение ночи, оставляя реакционную смесь охлаждаться до комнатной температуры. Фильтровали полученную суспензию, собирали твердое вещество и промывали водой (22 л). Сушили в вакууме (50 С) с получением указанного в заголовке соединения в виде неочищенного вещества (748 г). Пример получения 2. Метил 2-(трет-бутоксикарбонил-(4-этокси-4-оксобутил)амино)-3,5-диметилбензоат (4). Помещали в 22-л колбу, снабженную верхнеприводной мешалкой, колбонагревателем, холодильником и подводом N2, ДМФ (10 л), этил-4-бромбутират (1,07 кг, 787,8 мл, 5,32 моль), карбонат цезия (2,92 кг, 22,5 моль) и метил 2-(трет-бутоксикарбониламино)-3,5-диметилбензоат (1000,0 г, 3,54 моль). Нагревали полученную смесь до примерно 55 С и перемешивали в течение примерно 48 ч. Охлаждали, отфильтровывали твердое вещество и промывали твердое вещество МТБЭ (24 л). Объединяли фильтрат и промывные воды МТБЭ в 50-л колбе и охлаждали до менее чем примерно 5 С. Добавляли воду (6 л) для гашения реакции. Разделяли слои. Промывали водный слой МТБЭ (3 л), объединяли органические слои и промывали полученный органический раствор солевым раствором (23 л). Сушили органический слой над Na2SO4, фильтровали и промывали собранные твердые вещества МТБЭ с получением 1,582 кг указанного в заголовке соединения. Пример получения 3. трет-Бутил 4-этил 7,9-диметил-5-оксо-2,3,4,5-тетрагидро-1 Н-бензо[b]азепин-1,4-дикарбоксилат (5 А) и трет-бутил 4-метил 7,9-диметил-5-оксо-2,3,4,5-тетрагидро-1 Н-бензо[b]азепин-1,4-дикарбоксилат (5 В). Помещали в 22-л колбу, снабженную верхнеприводной мешалкой, термопарой, 5-л капельной воронкой, подводом азота и охлаждающей баней, метил 2-(трет-бутоксикарбонил-(4-этокси-4 оксобутил)амино)-3,5-диметилбензоат (700 г, 1,78 моль), растворенный в ТГФ (3,5 л) и охлаждали до менее чем 5 С. Помещали в капельную воронку 1 M раствор трет-бутоксида калия в ТГФ (KO т-Bu, 3,56 моль, 1 M) и по каплям добавляли к охлажденному раствору ТГФ, поддерживая температуру реакции равной окружающей температуре (5 С). После завершения добавления оставляли реакционную смесь нагреваться до температуры окружающей среды. После завершения реакции охлаждали смесь до менее чем 10 С и медленно добавляли 2,5 M HCl с получением смеси с рН, составляющим менее чем примерно 3. Добавляли МТБЭ (4 л) и перемешивали, затем отделяли органический слой от водного слоя. Экстрагировали водный слой в МТБЭ (2 л). Объединяли органические слои и последовательно промывали солевым раствором (23 л). Сушили органический слой над MgSO4, фильтровали и промывали собранное твердое вещество МТБЭ. Объединяли отфильтрованные растворы и удаляли растворитель в вакууме с получением смеси указанных в заголовке соединений в виде оранжевой маслянистой жидкости (670 г). Пример получения 4. Гидрохлорид 7,9-диметил-3,4-дигидро-1 Н-бензо[b]азепин-5(2 Н)-она (6). Помещали в 5-л коблу, снабженную верхнеприводной мешалкой, колбонагревателем, термопарой,подводом азота и тефлоновыми трубками, смесь трет-бутил 4-этил 7,9-диметил-5-оксо-2,3,4,5 тетрагидро-1 Н-бензо[b]азепин-1,4-дикарбоксилата и трет-бутил 4-метил 7,9-диметил-5-оксо-2,3,4,5 тетрагидро-1 Н-бензо[b]азепин-1,4-дикарбоксилата (1286 г, 3,56 моль), растворенную в IPA (2 л), и нагревали полученную смесь до 50 С. В отдельной 12-л колбе, оборудованной верхнеприводной мешалкой,колбонагревателем, термопарой и подводом азота с газоочистителем, добавляли 5 н. NaOH (1 л) и H2O (3 л). Затем добавляли HCl (конц., 1,87 л, 21,8 моль) в 12-л колбу и нагревали до 50 С. Переносили содержимое 5-л колбы в 12-л колбу через тефлоновые трубки, продувая N2 для удаления выделяемых в результате реакции газов. После добавления нагревали полученную смесь до 80 С и продолжать продуватьN2. После завершения реакции оставляли реакционную смесь охлаждаться до менее чем 20 С. Добавляли МТБЭ (4 л) и доводили рН до нейтрального при помощи водного NaOH. Переносили полученную реакционную смесь в 22-л колбу и разделяли слои. Промывали водный слой МТБЭ (22 л). Объединяли органические промывные воды и промывали солевым раствором (2 л), сушили над MgSO4, фильтровали и промывали МТБЭ. Концентрировали фильтрат до темной маслянистой жидкости. Растворяли в IPA (8 объемов) и переносили в 12-л колбу, снабженную верхнеприводной мешалкой, термопарой, 1-л капельной воронкой и подводом N2. Помещали в капельную воронку HCl (конц., 765 мл) и по каплям добавляли в течение примерно 1 ч к раствору в IPA. Перемешивали полученную суспензию в течение примерно 1-2 ч, фильтровали, промывали твердые вещества холодным IPA (3500 мл) и сушили твердое вещество в течение ночи при примерно 50 С с получением 561 г указанного в заголовке соединения. Пример получения 5. Бензил 7,9-диметил-5-оксо-2,3,4,5-тетрагидро-1 Н-бензо[b]азепин-1-карбоксилат (7). Помещали в 22-л колбу, снабженную верхнеприводной мешалкой, термопарой, 3-л капельной воронкой, ловушкой, охлаждающей баней и подводом N2, гидрохлорид 7,9-диметил-3,4-дигидро-1 Нбензо[b]азепин-5(2 Н)-она (1000 г, 4,43 моль). Добавляли 2-метилтетрагидрофуран (6,44 кг, 7,5 л, 74,5 моль) и перемешивали беловатую суспензию. Охлаждали до 5-15 С. Добавляли воду (2,5 л, 138,8 моль) иNa2CO3 (1,12 кг, 3 моль) в капельную воронку, затем в течение 25 мин быстро по каплям добавляли к реакционной смеси. Помещали в 2-л капельную воронку бензилхлорформиат (91,59 кг, 8,86 моль), по каплям добавляли к реакционной смеси, поддерживая температуру реакции менее чем примерно 15 С. Переносили полученную смесь в колбу, снабженную холодильником, нагревали до 25-25 С и перемешивали в течение примерно 50 ч. Охлаждали до примерно 15 С, добавляли HCl (5 M, до достижения рН,равного примерно 5). Разделяли слои. Экстрагировали водный слой в метилтетрагидрофуране (4 л), объ-7 020600 единяли органические слои и промывали водой (4 л). Концентрировали органический слой при примерно 40 С. Добавляли IPA (4 л), затем концентрировали до объема 2 л. Переносили в 12-л колбу, снабженную верхнеприводной мешалкой, термопарой, колбонагревателем, холодильником, 2-л капельной воронкой и подводом N2. Нагревали содержимое колбы до примерно 70-80 С и добавляли гептан (5 л). Медленно охлаждали до КТ в течение ночи, вносили при необходимости затравку в смесь для начала кристаллизации указанного в заголовке соединения. Охлаждали и собирали твердое вещество, промывали твердое вещество холодным гептаном и сушили в вакууме при 50 С с получением 1316 г указанного в заголовке соединения. Пример получения 6. Смесь(Z)-бензил 7,9-диметил-5-(2-метил-2 Н-тетразол-5-илимино)-2,3,4,5-тетрагидро-1 Нбензо[b]азепин-1-карбоксилата (8 А) и (Е)-бензил 7,9-диметил-5-(2-метил-2 Н-тетразол-5-иламино)-2,3 дигидро-1 Н-бензо[b]азепин-1-карбоксилата (8B). Помещали в 12-л колбу, снабженную верхнеприводной мешалкой, колбонагревателем, термопарой,холодильником, ловушкой Дина-Старка и подводом N2, бензил 7,9-диметил-5-оксо-2,3,4,5-тетрагидро 1 Н-бензо[b]азепин-1-карбоксилат (920 г, 2,84 моль) и толуол (400 мл). Добавляли толуол (500 мл) в ловушку Дина-Старка и начинали перемешивание. Добавляли 2-метил-5-аминотетразол (563,8 г, 5,69 моль) и PPTS (364,8 г, 0,5 моль) и кипятили полученную смесь с обратным холодильником. Добавляли холодную воду (5 л) и выливали в раствор NaHCO4 (850 г), отслеживая рН для предотвращения подкисления смеси. Разделяли слои и промывали водный слой толуолом (4 л). Объединяли органические слои и промывали водой (24 л), сушили над Na2SO4, фильтровали и промывали твердое вещество толуолом. Концентрировали фильтрат в вакууме при примерно 55 С с получением 1,186 кг смеси титульных соединений. Пример получения 7.(4,04 г,10 ммоль),S-(-)-2,2'-бис(дифенилфосфино)-1,1'-бинафтил S)-BINAP, 60 мг, 96,3 мкмоль), KI (415 мг, 24 ммоль, при необходимости KI можно удалить) и дихлор-бис-1,2,5,6-эта)-1,5-циклоокстадиен)дииридий (Ir2Cl2 (COD)2 (927 мг, 40,2 мкмоль. Продували автоклав азотом. Поддерживая бескислородную среду, добавляли дегазированный толуол (50 мл, 472,8 ммоль) и герметизировали автоклав. Нагревали до 100 С при 500 psi(3447,37 кПа) Н 2 в течение 66 ч. Охлаждали реакционную смесь до КТ и спускали давление. Отфильтровывали твердые вещества и собирали органическую смесь. Промывали органическую смесь водой (225 мл), сушили над Na2SO4 и фильтровали. Собирали фильтрат и удаляли растворитель в вакууме с получением указанного в заголовке соединения (3,99 г, 88,7% э.и. определен путем ВЭЖХ). Пример получения 8.(S)-Бензил 5-3,5-бис-(трифторметил)бензил)(2-метил-2 Н-тетразол-5-ил)амино)-7,9-диметил 2,3,4,5-тетрагидро-1 Н-бензо[b]азепин-1-карбоксилат (10). Помещали в ампулу (S)-бензил 7,9-диметил-5-(2-метил-2 Н-тетразол-5-иламино)-2,3,4,5-тетрагидро 1 Н-бензо[b]азепин-1-карбоксилат (100 мг, 246 мкмоль), NaH (23 мг, 60% в минеральном масле, 575,1 мкмоль) и ТГФ (9,2 мл, 24,6 ммоль) (в качестве альтернативы, вместо NaH и ТГФ можно применять третбутоксид калия в ДМФ). Перемешивали реакционную смесь при КТ в течение 10 мин. Добавляли 3,5 бис-(трифторметил)бензилбромид (173,8 мг, 566,2 мкмоль) по каплям в течение 30 мин. Примерно через 19 ч добавляли NaH (10 мг, 60% в минеральном масле) и дополнительно перемешивали в течение 1 ч. Разделяли реакционную смесь в EtOAc (50 мл) и солевом растворе (225 мл). Собирали органические слои и сушили над Na2SO4. Фильтровали, а затем концентрировали фильтрат с получением маслянистой жидкости. Разделяли маслянистую жидкость в ACN (50 мл) и гептане (225 мл). Собирали слой в ACN; сушили с получением указанного в заголовке соединения в виде маслянистой жидкости (189 мг). Пример получения 9.(S)-N-(3,5-бис-(трифторметил)бензил)-7,9-диметил-N-(2-метил-2 Н-тетразол-5-ил)-2,3,4,5 тетрагидро-1 Н-бензо[b]азепин-5-амин (11). Помещали в 1-л колбу формиат аммония (39,0 г, 618,0 ммоль) и 10% Pd/C (3,91 г), и (S)-бензил 53,5-бис-(трифторметил)бензил)(2-метил-2 Н-тетразол-5-ил)амино)-7,9-диметил-2,3,4,5-тетрагидро-1 Нбензо[b]азепин-1-карбоксилат (39,1 г, 61,8 ммоль), растворенный в МеОН (391 мл, 966 моль). Перемешивали полученную суспензию при КТ и контролировали путем ВЭЖХ. После завершения реакции фильтровали и собирали твердое вещество. Промывали твердое вещество МеОН (100 мл). Объединяли отфильтрованные растворы и органические промывные воды, а затем сушили над Na2SO4. Фильтровали и концентрировали досуха. Растворяли в МеОН (92 мл) и добавляли затравочные кристаллы указанного в заголовке соединения. Охлаждали и оставляли на ночь с получением 14,03 г, 45,5% указанного в заголовке соединения, 99,92% э.и., определенный с помощью ВЭЖХ.(Транс)-метил 4-S)-5-3,5-бис-(трифторметил)бензил)(2-метил-2 Н-тетразол-5-ил)амино)-7,9 диметил-2,3,4,5-тетрагидро-1 Н-бензо[b]азепин-1-ил)метил)циклогексанкарбоксилат (12). Помещали в колбу, снабженную верхнеприводной мешалкой, датчиком температуры, подводом азота,(S)-N-(3,5-бис-(трифторметил)бензил)-7,9-диметил-N-(2-метил-2 Н-тетразол-5-ил)-2,3,4,5 тетрагидро-1 Н-бензо[b]азепин-5-амин (5 г, 10,03 ммоль) и триацетоксиборгидрид натрия (3,19 г, 15,05 ммоль) и ацетонитрил (40 мл). Опускали колбу в ледяную баню для охлаждения суспензии до менее чем примерно 5 С, затем добавляли (транс)-метил 4-формилциклогексанкарбоксилат (2,99 г, 17,57 ммоль,получен по существу согласно способу И.Н. Упи с соавторами (Houpis, I. N. et al.), Tetrahedron Let. 1993,34(16), 2593-2596 и JP49048639), растворенный в ТГФ (10 мл), через шприц, поддерживая реакционную смесь при температуре, равной менее чем примерно 5 С. Оставляли реакционную смесь нагреваться до КТ и перемешивали в течение ночи. Добавляли NH4Cl (25 мл, 50% насыщенный водный раствор) и отделяли водный слой от органического слоя. рН органического слоя должен составлять примерно 5,5. Нагревали органический слой до примерно 45 С и добавляли воду (16 мл). Добавляли затравочный кристалл указанного в заголовке соединения и охлаждали до примерно 35 С. Собирали полученное твердое вещество путем фильтрования и промывали ACN. Сушили с получением 5,80 г указанного в заголовке соединения. Схема 3 Альтернативный способ получения ВССА Пример получения 12. Метил 7,9-диметил-5-оксо-2,3,4,5-тетрагидро-1 Н-1-бензазепин-1-карбоксилат. Стадия 1. Вакуумировали 3000-л эмалированный реактор до менее чем -0,08 МПа, а затем наполняли N2 до нормального давления, повторяли операцию трижды. В атмосфере N2 помещали в реактор 2,4 диметиланилин (300,00 кг) и триэтиламин (873,0 кг) при перемешивании. Нагревали смесь до 70-75 С, а затем добавляли этил 4-бромбутират (604,0 кг) со скоростью 20-25 кг/ч через 500-л капельную воронку,поддерживая температуру 70-75 С. Перемешивали смеси при 70-75 С в течение 2 ч, а затем исследовали завершение реакции путем ГХ. Реакцию считали завершенной, если содержание 2,4-диметиланилина двух последовательно взятых образцов составляло менее 3%, а содержание примеси (этил 4-2,4 диметилфенил)(3-(пропионилокси)пропил)амино)бутаноат) составляло менее 10%. Через 56 ч содержание примеси составляло менее 10%, но содержание 2,4-диметиланилина составляло более 3%, поэтому добавляли дополнительное количество этил 4-бромбутирата (6,0 кг) к смеси, затем, через 17,5 ч, содержание анилина оставалось выше 3%, но реакцию гасили. Охлаждали реакционную смесь до 15-25 С и переносили в два 5000-л эмалированных реактора. В каждый реактор добавляли воду (1200,0 кг) и толуол (1044,0 кг). Перемешивали смесь в течение 1 ч перед разделением. Объединяли органические фазы двух реакторов и дважды промывали водой (1200,0 кг 2). Отбирали органическую фазу для подтверждения того, что содержание триэтиламина составляло менее 23%. Концентрировали органическую фазу при 50-60 С при пониженном давлении (-0,08 МПа) до завершения выхода фракций и достижения мас.% триэтиламина, равного менее 0,1%, и KF, равного менее 1%. Охлаждали смесь до 20-30 С и использовали продукт (этил 4-[(2,4-диметилфенил)амино]бутаноат) непосредственно на стадии 2. Газовая хроматография (ГХ): колонка: НР-5, длина 30 м 0,32 мм ВД 0,25 мкм пленка или эквивалент колонки; газ-носитель: газообразный гелий; расход 1,90 мл/мин; время цикла 22,5 мин; Программа: начальная температура: 50 С (на старте), начальное время: 2,00 мин; скорость изменения температуры: 1; скорость: 20; конечная температура: 260; конечное время: 10,00; скорость изменения температуры 2: 0,0 (на финише); температура старта 250 С; температура детектирования: 300 С. Времена удерживания: 1) (13,56 мин) этил 4-[(2,4-диметилфенил)амино]бутаноат; 2) (17,40) этил 42,4-диметилфенил)(3-(пропионилокси)пропил)амино)бутаноат; 3) (8,38 мин) 2,4-диметиланилин. Стадия 2. Помещали в 2000-л эмалированный реактор толуол (742,0 кг) и этил 4-[(2,4 диметилфенил)амино]бутаноат (171,0 кг). Перемешивали смесь и добавляли карбонат натрия (77,0 кг) по частям. Поддерживали температуру при 20-25 С и добавляли метилхлорформиат (96,2 кг) со скоростью 18 кг/ч. Перемешивали смесь при 20-25 С и исследовали через 1 ч путем ГХ. Реакцию считали завершенной, если содержание этил 4-[(2,4-диметилфенил)амино]бутаноата составляло менее 1%. Переносили смесь в 5000-л эмалированный реактор и промывали 2000-л реактор толуолом (75 кг). Добавляли растворNaOH (87,2 кг), метанол (934,0 кг) и воду (1482,0 кг) со скоростью 360-400 кг/ч, поддерживая температуру 20-25 С. Затем нагревали смесь до 55-65 С и перемешивали при 55-60 С. Через 1 ч исследовали образец путем ВЭЖХ для определения содержания этил 4-[(2,4-диметилфенил)амино]бутаноата. Охлаждали смесь до 20-30 С, выдерживали 1 ч, затем разделяли. Концентрировали водную фазу при 40-50 С при пониженном давлении (равном 0,09 МПа или менее) до отгонки 929 кг метанола. Охлаждали смесь до 15-25 С, добавляли воду (256,5 кг), перемешивали в течение 0,5 ч и экстрагировали в дихлорметане (342,0 кг 2). Понижали температуру до 0-5 С и, поддерживая, добавляли конц.HCl (269,0 кг) со скоростью 40-50 кг/ч для доведения рН до 1-2. Нагревали смесь до 15-25 С и перемешивали в течение 1 ч, поддерживая указанную температуру. Разделяли, экстрагировали водную фазу в дихлорметане (684,0 кг 2) и объединяли органические фазы. Промывали органическую фазу дважды 0,5% HCl (342,0 кг 2), а затем дважды конц. HCl (3,42 кг). Концентрировали органическую фазу при пониженном давлении (равным -0,08 МПа или менее; 40-50 С) до прекращения выделения фракций. Добавляли дихлорметан (250,0 кг) к остатку (4-[(2,4-диметилфенил)метоксикарбониламино]бутановой кислоте) (пример получения 11) и непосредственно применяли на следующей стадии. 190,0 кг (раствор 407,4 кг); выход 98,5%; чистота 97,8%. ВЭЖХ: колонка: Waters XTerra MS C18; 4,6150 мм; 3,5 мкм; детектирование 230 нм; расход 1,0 мл/мин; температура 25 С; изократическая мобильная фаза: А: ACN; В: Н 2 О+0,1% Н 3 РО 4 (об./об.). Время удерживания: (13,86 мин) 4-[(2,4-диметилфенил)метоксикарбониламино]бутановая кислота. Стадия 3. Вакуумировали 3000-л эмалированный реактор до менее чем -0,08 МПа, заполняли газообразным азотом до нормального давления, операцию повторяли 3 раза. Помещали дихлорметан (1900,0 кг), 4-[(2,4-диметилфенил)метоксикарбониламино]бутановую кислоту (190,0 кг) и ДМФ (11,4 кг) и перемешивали. Охлаждали смесь до -5-0 С и, поддерживая при указанной температуре, добавляли тионилхлорид (85,3 кг) со скоростью 18 кг/ч. Перемешивали смесь при -5-0 С. Через 1 ч исследовали реакцию путем ВЭЖХ для определения содержания 4-[(2,4-диметилфенил)метоксикарбониламино]бутановой кислоты, которые должно составлять менее 1%. При отборе образцов добавляли образцы к метанолу и оценивали путем ВЭЖХ. Концентрировали смесь при 40-45 С при нормальном давлении до прекращения выделения фракций, затем охлаждали до 15-25 С. Затем концентрировали при 40-45 С при пониженном давлении (равном -0,08 МПа или менее) до прекращения выделения фракций. Разбавляли дихлорметаном(1031,0 кг). По каплям добавляли раствор дихлорметана (1030,0 кг) и безводного гексагидрата хлорида алюминия (287,2 кг) при 30-35 С. Перемешивали смесь при 35-45 С в течение 2 ч, а затем исследовали путем ВЭЖХ до достижения содержания метил-7,9-диметил-5-оксо-2,3,4,5-тетрагидро-1 Н-1-бензазепин 1-карбоксилата, равного более чем 80% (отбирали образец смеси, добавляли его в метанол и отправляли для детектирования ВЭЖХ). Гасили смесь смесью воды (1140,0 кг) и льда (570,0 кг), поддерживали температуру 0-10 С и перемешивали в течение 1 ч. Нагревали смесь до 15-25 С, перемешивали в течение 0,5 ч, выдерживали 0,5 ч и разделяли. Промывали органическую фазу водой (814,0 кг 2). Добавляли силикагель (380,0 кг) к органической фазе и перемешивали смесь в течение 1 ч. Фильтровали смесь, промывали осадок дихлорметаном (339,0 кг) и объединяли фильтраты. Концентрировали фильтрат при 40-45 С при пониженном давлении (равном -0,08 МПа или менее) до получения 150-200 л смеси. Добавляли гептан (190,0 кг), охлаждали смесь до 15-20 С рециркулирующей водой, затем охлаждали до 0-5 С при помощи солевого раствора. Перемешивали смесь при указанной температуре для кристаллизации. Фильтровали смесь, сушили осадок при 35-40 С в сушильной камере с получением 80,9 кг твердого вещества грязно-белого цвета. ВЭЖХ 97,9%. Хранили в сухом герметичном месте в защитной атмосфере азота. Процедура ВЭЖХ: аналогична для стадии 2 выше. Время удерживания (14,71 мин) метил 7,9 диметил-5-оксо-2,3,4,5-тетрагидро-1 Н-1-бензазепин-1-карбоксилат. Пример получения 13. Метил-5,5-диметокси-7,9-диметил-2,3,4,5-тетрагидро-1 Н-бензазепин-1-карбоксилат. Стадия 4. К раствору метил 7,9-диметил-5-оксо-2,3,4,5-тетрагидро-1 Н-1-бензазепин-1-карбоксилата(1000 г; 1,00 экв.; 4,04 моль) в метаноле (2,50 л) и триэтоксиметане (9,10 моль; 995,42 мл; 965,55 г), перемешиваемому при помощи верхнеприводной мешалки при температуре окружающей среды в защитной атмосфере азота добавляли Amberlyst 15 (100 г). Нагревали реакционную смесь до 50 С и выдерживали температуру в течение 1 ч. Через 1 ч реакция завершалась. Охлаждали реакционную смесь до температуры окружающей среды, отфильтровывали гранулыKOH (1 л) при перемешивании при помощи верхнеприводной мешалки, с последующим добавлением Н 2 О (1500 мл). Перемешивали суспензию в течение 20 мин, а затем фильтровали через 32-см керамический фильтр с полипропиленовой подложкой. Промывали осадок 41 л воды и фильтровали в вакууме досуха. Сушили в вакуумируемом сушильном шкафу в течение ночи при 60 С, после чего измельчали твердое вещество и оставляли сушить при 60 С на ночь. Выход неочищенного вещества после сушки: 1235 г. Растворяли неочищенные твердые вещества в 5 объемах гептана (6 л), нагревали до 70 С и перемешивали в течение 15 мин при 70 С. Охлаждали раствор до 55 С, после чего происходило осаждение реакционной смеси. Медленно продолжали охлаждение, продукт начинал выпадать в осадок при 45 С. Продолжали медленно охлаждать до комнатной температуры, затем оставляли суспензию перемешиваться на ночь. Фильтровали суспензию продукта через подложку полипропилена. Промывали осадок 2500 мл гептана. Фильтровали в вакууме, затем помещали в вакуумируемый сушильный шкаф при 45 С досуха (4 дня). 766 г. Чистота 99,9%, определена ВЭЖХ. ВЭЖХ: Zorbax Bonus-RP 504,6 мм, 1,8 мкм; 2 мл/мин, 40 С, от 10-30% ACN в течение 12 мин до 95% через 14 мин, выдержка 2 мин, элюент для повторного установления равновесия 10 мМ NH4Ac. Время удерживания=13,77 мин. Пример получения 14. Метил-(5S)-7,9-диметил-5-[(2-метил-2 Н-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-1 Н-1-бензазепин 1-карбоксилат. Стадия 5. Обеспечивали сухость водородного реактора путем сушки N2 в течение 1-2 ч. Добавляли 2-метил-2H-тетразол-5-амин (52,0 г), метил 5,5-диметокси-7,9-диметил-2,3,4,5-тетрагидро-1 Н-1 бензазепин-1-карбоксилат (140,0 г), Ir2Cl2(COD)2 (0,080 г), TBAI (1,76 г), (1S)-(+)-10 камфорсульфокислоту (2,2 г) и (S)-дифторфосфин (0,163 г) в реактор. Закачивали в реактор 5 psi (34,47 кПа) азота. Добавляли 600 мл толуола (через который в течение 75 мин продували N2) через трубку/азот. Продували реактор дважды 20 psi (137,87 кПа) азота и не опускали давление ниже 5 psi (34,47 кПа). Закачивали 500 psi (3447,37 кПа) водорода в реактор и медленно нагревали до 115 С. Выдерживали при указанной температуре в течение ночи. Охлаждали до 50 С и отбирали образец для анализа ВЭЖХ. Для хиральной ВЭЖХ разбавляли образец толуолом, промывали бикарбонатом натрия, сушили над Na2SO4,разбавляли смесью гептан/этанол. Выливали раствор в толуоле в делительную воронку и добавляли 140 мл этилацетата для переведения всех веществ в делительную воронку и сохранения всего раствора в процессе обработки. Промывали раствор 420 мл 1 М раствора NaOH (органический слой выглядел мутным; водный слой прозрачный). Разделяли слои и промывали органический слой 420 мл Н 2 О. Перегоняли органический слой при атмосферном давлении до достижения 1,5 объемов толуола (2,5 объема оставшегося раствора). Охлаждали раствор до 60 С и медленно в течение 3 мин при 60 С добавляли 700 мл гептана (5 объемов) и нагревали полученный прозрачный раствор при 60 С в течение ночи при перемешивании верхнеприводной мешалкой (150 об/мин). Через 15 мин количество твердого вещества увеличивалось. Утром охлаждали суспензию, фильтровали, собирали твердое вещество и сушили в вакуумируемом сушильном шкафу при 60 С в течение 3 ч. 126,97 г, ВЭЖХ: чистота 99,4%, хиральная ВЭЖХ: 94,6% э.и. ВЭЖХ: Zorbax Bonus-RP 504,6 мм, 1,8 мкм; 2 мл/мин, 40 С, от 10-30 ACN в течение 12 мин, до 95% через 14 мин, выдержка 2 мин, элюент для повторного установления равновесия 10 мМ NH4Ac. Время удерживания=11,36 мин. Хиральная ВЭЖХ: Chiralpak IA 2504,6 мм, 2 мл/мин, 40 С, от 5% IPA в течение 11 мин до 50%(5S)-7,9-диметил-N-(2-метилтетразол-5-ил)-2,3,4,5-тетрагидро-1 Н-1-бензазепин-5-амин. Стадия 6. Объединяли метил-(5S)-7,9-диметил-5-[(2-метил-2 Н-тетразол-5-ил)амино]-2,3,4,5 тетрагидро-1 Н-1-бензазепин-1-карбоксилат (20,1 г), NaOH (12,10 г) и этанол (80 мл) в 250-мл тефлоновой колбе с верхнеприводной мешалкой. Продували азотом в течение 10 мин, кипятили с обратным холодильником (85 С) в течение 5,5 ч. Охлаждали до комнатной температуры на холодной водяной бане. Медленно при комнатной температуре добавляли смесь уксусная кислота/вода (17,5 мл/53 мл) и переносили в 500 мл колбу, заполнив примерно на половину (30 мин общего времени добавления). Вносили затравку 5 мг (5S)-7,9-диметил-N-(2-метилтетразол-5-ил)-2,3,4,5-тетрагидро-1 Н-1-бензазепин-5-амина. Нагревали до 50 С (в течение примерно 15 мин до указанной температуры). Добавляли 54 мл воды в течение примерно 30 мин. На следующий день охлаждали на ледяной бане, фильтровали и промывали смесью метанол:вода (1:1, 230 мл). Сушили влажный осадок в вакууме при 60 С с получением 13,99 г (5S)7,9-диметил-N-(2-метилтетразол-5-ил)-2,3,4,5-тетрагидро-1 Н-1-бензазепин-5-амина. Анализ ВЭЖХ: 99,4%, время удерживания 4,84 мин. Хиральная ВЭЖХ: хиральная чистота 98,5%. Способ ВЭЖХ: Zorbax SB-C8 754,6 мм, 3,5 мкм, 2 мл/мин, 40 С, длина волны 225 нм, от 5% ацетонитрила (ACN) в течение 2 мин до 95% ACN через 10 мин и выдержка 1 мин. Хиральная ВЭЖХ: Chiralpak AD-H 1504, мм, 5 мкм, 1 мл/мин, 30 С, изократичный режим 50% этанола в гептане. Пример получения 16.(5S)-N-[3,5-бис-(трифторметил)бензил]-7,9-диметил-N-(2-метил-2 Н-тетразол-5-ил)-2,3,4,5 тетрагидро-1 Н-1-бензазепин-5-амин. Стадия 7. Дегазировали все жидкости перед добавлением в реакционную смесь путем насыщения каждого раствора за счет добавления N2 под поверхность раствора. В 250-мл колбу с подводом азота добавляли (5S)-7,9-диметил-N-(2-метилтетразол-5-ил)-2,3,4,5-тетрагидро-1 Н-1-бензазепин-5-амин (18,36 ммоль; 5,00 г) и толуол (15,00 мл). По каплям в течение 30 мин добавляли гексаметилдисилазид калия(19,28 ммоль; 38,55 мл) в виде 0,5 М раствора в толуоле. Наблюдали выделение тепла до 21,5 С. Перемешивали реакционную смесь в течение 30 мин. Через 30 мин добавляли 1-(хлорметил)-3,5-бис(трифторметил)бензол (25,71 ммоль; 25,7 мл) в виде 1 М раствора в толуоле. Наблюдали выделение тепла до 25,5 С. Перемешивали реакционную смесь при комнатной температуре в течение 16 ч. Промывали реакционную смесь водой (220 мл). Объединяли водные слои и повторно экстрагировали в толуоле(125 мл). Объединяли слои в толуоле, концентрировали досуха и перекристаллизовывали остаток из 92 мл 60% 1-пропанола в воде. Перемешивали кристаллизовавшийся продукт в течение 2 ч при 0 С, фильт- 12020600 ровали и промывали 10 мл 50% 1-пропанола в воде. Сушили выделенный продукт в вакууме при 45 С. 7,735 г. Данные ВЭЖХ показали 98,3% чистоты, время удерживания=10,62 мин. Градиентная ВЭЖХ: колонка Zorbax Bonus RP 5 мкм, 4,6150 см, Поток=2 мл/мин; Длина волны 225 нм; температура колонки=40 С; Растворитель А=вода; Растворитель В=ACN; Время 0 мин 85% А 15% В; 12 мин 10% А 90% В; 13 мин 10% А 90% В; 13,5 мин 85% А 15% В. Пример получения 17. Метил-транс-4-[(5S)-5-[3,5-бис-(трифторметил)бензил](2-метил-2 Н-тетразол-5-ил)амино-7,9 диметил-2,3,4,5-тетрагидро-1 Н-1-бензазепин-1-ил]метилциклогексанкарбоксилат. Стадия 12. Охлаждали суспензию (5S)-N-[3,5-бис-(трифторметил)бензил]-7,9-диметил-N-(2-метил 2H-тетразол-5-ил)-2,3,4,5-тетрагидро-1 Н-1-бензазепин-5-амина (3,40 г, 6,82 ммоль), триацетоксиборгидрата натрия (3,01 г, 13,64 ммоль) и ACN (28 мл) до (-12)-(-10)С на бане лед/ацетон и добавляли раствор(транс)-4-формилциклогексанкарбоксилат (2,03 г, 11,93 ммоль) в толуоле (18,5 мл) в течение 30 мин через шприцевой насос (0,66 мл/мин). Продолжали перемешивание реакционной смеси на бане лед/ацетон в течение 2 ч. Добавляли 44 мл 10% (по массе) NH4Cl в воде к реакционной смеси и перемешивали в течение 30 мин при комнатной температуре. Останавливали перемешивание, образовывались два слоя. Разделяли слои, концентрировали органический слой с получением одного объема растворителя. Добавляли 24 мл этанола и концентрировали с получением твердого вещества (или до одного объема). Добавляли 24 мл этанола, 2 мл воды и нагревали до 60 С (в виде суспензии). Добавляли 2 мл воды по каплям. Оставляли суспензию охлаждаться до комнатной температуры и перемешивали в течение ночи при комнатной температуре. После перемешивания в течение ночи при комнатной температуре охлаждали суспензию до -10 С в течение 0,5 ч, затем фильтровали. Промывали отфильтрованные твердые вещества 3 мл смеси EtOH:вода (4:1) с температурой -10 С. Сушили твердые вещества в течение ночи в вакуумируемом сушильном шкафу при 50 С с получением 4,24 г метил-транс-4-[(5S)-5-[3,5-бис(трифторметил)бензил](2-метил-2 Н-тетразол-5-ил)амино-7,9-диметил-2,3,4,5-тетрагидро-1 Н-1 бензазепин-1-ил]метилциклогексанкарбоксилата в виде бесцветного твердого вещества. Данные ВЭЖХ показали 99,75% чистоты. Анализ ВЭЖХ: Zorbax Bonus RP 150 мм 4,6 мм, 3,5 мкм, 30C, УФ-детектирование 260 нм, поток: 2,0 мл/мин, градиент: А=0,05% TFA в Н 2 О, В=0,05% TFA в ACN; от 0 мин 95% А до 30 мин 0% А до 30,5 мин 95% А до 35 мин 95% А. Метил-транс-4-[(5S)-5-[3,5-бис-(трифторметил)бензил](2-метил-2 Нтетразол-5-ил)амино-7,9-диметил-2,3,4,5-тетрагидро-1 Н-1-бензазепин-1 ил]метилциклогексанкарбоксилат элюирует через 23,33 мин, цис-изомер элюирует через 23,05 мин. Пример получения 18. Аддукт бисульфата натрия и (транс)-метил-4-формилциклогексанкарбоксилата. Стадия 8. Помещали 12-л трехгорлую круглодонную колбу в охлаждающую ванну, снабженную механической мешалкой, термопарой с дисплеем, подводом азота и трубкой с осушителем. В колбу помещали (транс)-4-метоксикарбонилциклогексанкарбоновую кислоту (450 г). В колбу помещали дихлорметан (2,25 л) в атмосфере азота. В колбу добавляли оксалилхлорид (364 г, растворенный в CH2Cl2 (100 мл через капельную воронку при температуре окружающей среды. Перемешивали реакционную смесь в течение 10 мин при комнатной температуре. В колбу добавляли каталитическое количество ДМФ (1,82 г, растворенный в CH2Cl2 (10 мл через капельную воронку при температуре окружающей среды. Перемешивали реакционную смесь при температуре, равной менее чем 20 С, в течение 2 ч. Исследовали прохождение реакции путем ГХ: [DBI 30 м 0,25 мм, 0,5 мкм], удаляли образец перемешиваемого раствора при температуре,равной менее чем 20 С. Время удерживания(транс)-4 хлоркарбонилциклогексанкарбоксилата=7,3 мин; Rt (транс)-4-метоксикарбонилциклогексанкарбоновой кислоты=7,75 мин. Реакцию считали завершенной, если оставалось 3,0% исходного вещества (транс)-4 метоксикарбонилциклогексанкарбоновой кислоты. Стадия 9. Концентрировали реакционную смесь при пониженном давлении при температуре, равной менее чем 35 С. Собирали избыток оксалилхлорида вместе с дистиллятом; применяли щелочную ловушку для предотвращения поступления паров кислоты в вакуумную систему. Совместно выпаривали остаток и ТГФ (2900 мл). Разбавляли остаток ТГФ (4,5 л) и 2,6-лутидином (321 г). Переносили в автоклавируемый реактор для гидрогенирования. Помещали 5% Pd на активированном угле (45 г суспензии в ТГФ (500 мл в реакционную смесь. Продували реактор газообразным азотом (от 20 до 30 psi) (137,89206,84 кПа) от 2 до 3 раз. Продували реактор газообразным водородом (от 20 до 30 psi) (137,89-206,84 кПа) от 2 до 3 раз. Перемешивали реакционную смесь в атмосфере водорода (от 50 до 60 psi) (344,73413,68 кПа) в течение 15 ч при 30-35 С. Через 15 ч удаляли газообразный водород в вытяжной шкаф и продували реактор газообразным азотом (от 20 до 30 psi) (137,89-206,84 кПа) от 2 до 3 раз. Исследовали реакцию путем ГХ. Rt (транс)-метил-4-формилциклогексанкарбоксилата=6,46 мин; Rt (транс)-4 хлоркарбонилциклогексанкарбоксилата=7,3 мин. Реакцию считали завершенной, если оставалось менее чем 1,0% (транс)-4-хлоркарбонилциклогексанкарбоксилата. Фильтровали реакционную смесь через подложку целита в атмосфере азота. Концентрировали значительную часть растворителя при пониженном давлении при температуре, равной менее чем 35 С. Раз- 13020600 бавляли остаток МТБЭ (1,8 л) и переносили в делительную воронку. Промывали органический раствор водой (2,25 л) и разделяли слои. Повторно экстрагировали водную фазу МТБЭ (21,8 л) и объединяли все органические слои. Промывали 0,5 н. водным раствором хлороводородной кислоты (12,25 л). Промывали органическую фазу насыщенным водным раствором бикарбоната натрия (12,5 л). Промывали органическую фазу солевым раствором (12,5 л). Сушили органическую фазу над сульфатом магния и фильтровали смесь через стекловолоконный фильтр. Концентрировали фильтрат при пониженном давлении на бане с температурой, равной менее чем 35 С. Выделяли неочищенное вещество в виде бесцветной маслянистой жидкости и применяли без дополнительной очистки. Стадия 10. В ампулу, содержащую 0,49 г бисульфита натрия, добавляли 1 мл воды. Помещали ампулу в ультразвуковую баню для растворения бисульфита натрия. Добавляли 5 мл ТГФ в ампулу (образуется двухфазный раствор без твердых веществ). Добавляли указанный двухфазный раствор ТГФ и водного бисульфита натрия в 50-мл колбу, снабженную магнитной мешалкой, содержащую (транс)-4 формилциклогексанкарбоксилат(транс)-4 формилциклогексанкарбоксилат имеет только 80% чистоту, поэтому применяли 0,8 экв. NaHSO3). Дополнительно разбавляли раствор 5 мл ТГФ, и в течение 1 мин образовывалась очень вязкая кристаллическая масса. Кипятили кристаллическую массу с обратным холодильником. Разбавляли смесь 35 мл ТГФ. Кристаллы представляли собой хлопья. Перемешивали при комнатной температуре в течение нескольких часов, затем фильтровали, промывали 20 мл ТГФ и сушили на фильтре в течение 1 ч (1,27 г). Сушили в течение ночи в вакуумируемом сушильном шкафу при 40 С с получением 1,19 г аддукта бисульфата натрия и (транс)-метил-4-формилциклогексанкарбоксилата в виде бесцветного твердого вещества. Пример получения 19.(Транс)-4-формилциклогексанкарбоксилат. Стадия 11. Добавляли аддукт бисульфата натрия и(транс)-метил-4 формилциклогексанкарбоксилата (3,92 г, 11,93 ммоль) к смеси карбоната натрия (5,06 г, 47,72 ммоль),толуола (17 мл) и воды (33 мл) и перемешивали при комнатной температуре. Через 1 ч перемешивания при комнатной температуре разделяли слои и промывали слой в толуоле 15 мл воды. Применяли указанный раствор в толуоле, который содержит (транс)-4-формилциклогексанкарбоксилат на стадии 12. Пример 1. Транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5-ил)амино]-2,3,4,5 тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновая кислота, аморфное твердое вещество. Помещали в колбу (транс)-метил 4-S)-5-3,5-бис-(трифторметил)бензил)(2-метил-2 Н-тетразол-5 ил)амино)-7,9-диметил-2,3,4,5-тетрагидро-1 Н-бензо[b]азепин-1-ил)метил)циклогексанкарбоксилат (149 мг). Добавляли МеОН (6 мл) и 1,0 н. NaOH (3,0 мл). Нагревали полученную смесь до примерно 60 С. Исследовали реакцию путем ТСХ. Через 7 ч или после завершения реакции исходного сложного эфира охлаждали смесь до примерно 0 С и гасили 1 н. HCl. Разбавляли смесь EtOAc (60 мл). Последовательно промывали смесь водой (20 мл), солевым раствором (20 мл) и сушили над Na2SO4. Фильтровали и концентрировали фильтрат. Очищали с помощью флэш-хроматографии с применением CH2Cl2. Удаляли растворитель с получением указанного в заголовке соединения (125 мг) в виде белого твердого вещества. Пример 2. Транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5-ил)амино]-2,3,4,5 тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновая кислота, аморфное твердое вещество. Помещали в 500-мл трехгорлую колбу, снабженную механической мешалкой, капельной воронкой и термопарой, воду (265 мл) и охлаждали на ледяной бане. Растворяли ВССА (22,0 г) в 40 мл ацетона во второй колбе и добавляли смесь в трехгорлую колбу через капельную воронку. Дополнительно промывали колбу 4 мл ацетона и оставляли полученную смесь перемешиваться, поддерживая температуру примерно 0-1 С. Собирали полученное твердое вещество, дважды промывали водой, затем сушили при 40 С в течение ночи с получением 22,3 г белого порошка. Пример 3. Транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5-ил)амино]-2,3,4,5 тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновая кислотагидрат. Продували колбу, снабженную верхнеприводной мешалкой, датчиком температуры, подводом азота, азотом, затем, поддерживая атмосферу азота, в колбу добавляли (транс)-метил 4-S)-5-3,5-бис(трифторметил)бензил)(2-метил-2 Н-тетразол-5-ил)амино)-7,9-диметил-2,3,4,5-тетрагидро-1 Нбензо[b]азепин-1-ил)метил)циклогексанкарбоксилат (1,0 кг, 1,53 моль), МеОН (10 л) и 2 М NaOH (1,53 л,3,06 моль). Кипятили полученную смесь с обратным холодильником при примерно 68 С. Примерно через 4 ч оставляли реакционную смесь охлаждаться до КТ и перемешивали в течение ночи. Фильтровали смесь и собирали фильтрат. Добавляли смесь МеОН:вода (500 мл, 1:1 (об./об., а затем медленно добавляли уксусную кислоту (260 мл) для начала осаждения. Собирали полученное твердое вещество в виде смеси ВССА, гидрата ВССА и метанолсольвата ВССА. Сушили твердое вещество в сушильном шкафу при 40 С. Помещали в 2-л колбу, снабженную холодильником, термопарой, колбонагревателем и механической мешалкой, твердую ВССА (109 г), МеОН (900 мл) и воду (10 мл). Кипятили полученную суспензию с обратным холодильником в атмосфере азота. После растворения всех твердых веществ прекращали нагрев и оставляли смесь охлаждаться до температуры окружающей среды. При необходимости можно добавлять затравочный кристалл гидрата ВССА в кристаллической форме. Собирали твердое вещество. Последовательно промывали твердое вещество смесью метанол:вода (9:1 (об./об.), 100 мл) и водой (1 л). Сушили твердое вещество в вакууме при примерно 40 С с получением 100 г белого твердого вещества. Затем повторно суспендировали твердое вещество в смеси изопропанол:вода (1:1 (об./об.), 1 л). Собирали полученное твердое вещество, промывали водой (500 мл) и сушили в течение ночи в вакууме при примерно 40 С с получением 60 г указанного в заголовке соединения. Анализ по Карлу Фишеру 2,53% воды, элементный анализ: C31H36F6N6O2H2O теоретический (%) С 56,70, Н 5,83, N 12,80 экспериментальный (%): С 56,59, Н 5,28, N 12,55. ЯМР твердого тела. Спектры 13 С ЯМР с кроссполяризацией и вращением под магическим углом (CP/MAS) (твердотельный ЯМР или ТТЯМР) кристаллического гидрата ВССА в кристаллической форме получали на ЯМР спектрометре Bruker Avance II 400 MHz с рабочей частотой для углерода, равной 100,622 МГц, оборудованном 4-мм датчиком для двойного резонанса Bruker (K299552). Подавление боковых полос TOSS применяли вместе с кроссполяризацией с применением спиновой развязки SPINAL64 (95,4 Вт) и Н-ядерного импульса КП RAMP100. Параметры получения спектров представлены далее: ширина 90 r.f. импульса протонов 2,5 мкс, время взаимодействия составляло 1,5 мс, продолжительность импульса составляла 20 с, MAS частота 5 кГц, спектральная ширина 30 кГц, время снятия спектра составляло 34 мс, количество сканов 3,844. Типичные резонансы 13 С ЯМР гидрата ВССА включают: 175,6, 168,0, 145,6, 144,8, 143,5,139,9, 136,3, 132,8, 132,1, 129,2, 127,3, 126,2, 122,7, 121,0, 61,1, 53,0, 49,8, 45,0, 40,2, 38,7, 31,4, 30,4, 29,0,27,8, 27,0, 21,2, 18,30,2 м.д. Химические сдвиги соотносили со сдвигом адамантана (=29,5 м.д.), полученным в отдельном эксперименте. Пример 4. Гидрат и этанолсольват транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Нтетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты в кристаллической форме. Помещали в колбу (транс)-метил 4-S)-5-3,5-бис-(трифторметил)бензил)(2-метил-2 Н-тетразол-5 ил)амино)-7,9-диметил-2,3,4,5-тетрагидро-1 Н-бензо[b]азепин-1-ил)метил)циклогексанкарбоксилат (12,95 кг), EtOH (129,5 л) и 2 М NaOH (9,9 л, 2 экв.), перемешивали полученную смесь в течение примерно 10 мин, после чего нагревали реакционную смесь до примерно 40-45 С в течение 6 ч. Исследовали реакцию путем ВЭЖХ. Затем добавляли уксусную кислоту (3,75 кг), затем воду (15,5 мл) и затравку гидрата ВССА в кристаллической форме. После перемешивания в течение примерно 2 ч охлаждали реакционную смесь до КТ и дополнительно перемешивали в течение 2 ч. Собирали полученные твердые вещества; промывали твердые вещества смесью EtOH:вода (1:1, 226 л) и сушили в течение ночи с получением указанного в заголовке соединения (15,87 кг, влажное вещество) в виде смеси этанолсольвата и гидрата. Пример 5. Транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5-ил)амино]-2,3,4,5 тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновая кислота в кристаллической форме. Помещали в колбу транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5 ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновую кислоту(15,87 кг) и воду (158,8 л). Перемешивали полученную смесь при КТ в течение 2 ч. Затем фильтровали полученную смесь и собирали твердые вещества. Сушили твердые вещества в течение ночи. Полученные твердые вещества растворяли в МеОН, нагретом до примерно 65-70 С, в течение 2 ч с получением прозрачного раствора. Фильтровали прозрачный раствор, затем охлаждали фильтрат до примерно 0-5 С для начала кристаллизации. Собирали полученные кристаллы и сушили в течение ночи. Суспендировали кристаллическое вещество в воде (158,8 л) и перемешивали в течение примерно 2 ч при КТ. Собирали кристаллическое твердое вещество; сушили в глубоком вакууме при 40-45 С с получением твердого вещества, которое содержит воду в количестве, равном от 2,7 до 3,1%, определенном по способу Карла Фишера. Пример 6. Кристаллизация гидрата транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Нтетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1 ил]метил]циклогексанкарбоновой кислоты. Помещали в 250-мл колбу, снабженную верхнеприводной мешалкой, этанол (100 мл), (транс)-метил 4-S)-5-3,5-бис-(трифторметил)бензил)(2-метил-2 Н-тетразол-5-ил)амино)-7,9-диметил-2,3,4,5- 15020600 тетрагидро-1 Н-бензо[b]азепин-1-ил)метил)циклогексанкарбоксилат (10,0 г, 15,32 ммоль) и NaOH (2 М,15,0 мл, 30,0 ммоль). Нагревали полученную смесь до примерно 40 С и перемешивали при указанной температуре в течение примерно 4 ч. Оставляли реакционную смесь охлаждаться до КТ. Добавляли уксусную кислоту (2,7 мл, 45,4 ммоль) и нагревали до 40 С. Медленно добавляли воду (40 мл) в течение 2,5 ч с получением вязкой белой суспензии. Продолжали нагревание в течение 1,5 ч, затем оставляли суспензию охлаждаться до КТ. Собирали твердое вещество путем фильтрования и последовательно промывали твердое вещество смесью этанол:вода (20 мл, 1:1), водой (20 мл). Суспендировали твердое вещество в воде (70 мл); собирали твердое вещество путем фильтрования; затем промывали твердое вещество водой (220 мл). Повторяли указанную операцию с водной суспензией дважды. Собирали полученное твердое вещество и сушили при 60 С с получением указанного в заголовке соединения в виде белого твердого вещества (6,1 г). Пример 7. Кристаллизация гидрата транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Нтетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты. Помещали в сцинтилляционную ампулу ВССА (60 мг) и добавляли 1,5 мл МеОН. Нагревали прозрачный раствор до примерно 45 С. Добавляли 1,5 мол.экв. муравьиной кислоты в 100 мкл воды с образованием белой суспензии. Добавляли 0,5 мл МеОН к белой суспензии и осторожно нагревали до примерно 55 С. Через несколько часов охлаждали суспензию до КТ, собирали белое кристаллическое твердое вещество путем вакуумного фильтрования и оставляли твердое вещество сушиться на воздухе. Анализ указанного твердого вещества путем ЯМР раствора не показал наличия растворителя муравьиной кислоты. Кристаллическая форма может быть дополнительно охарактеризована дифференциальным термальным/термогравиметрическим анализом, который показал, что кристаллическая форма имеет процент потерь летучих компонентов 2,6 в диапазоне от 38 до 133 С, указанные компоненты представляют собой воду, что определено путем ТГА-МС, при этом испарение летучих компонентов начинается при 81 С. Дифференциальный термический/термогравиметрический анализ. Дифференциальный термический/термогравиметрический анализ проводили на приборе для ДТА и ТГА Mettler Toledo (Модели TGA/SDTA 851). Образцы нагревали в герметичных алюминиевых кюветах с отверстиями от 25 до 300-350 С со скоростью 10 С/мин с подводом азота 50 мл/мин. Температуру ТГА калибровали при помощи индиевого/алюминиевого стандарта, Тпл=156,6 и 660,3 С. Калибровку массы проводили при помощи поставляемых производителем стандартов и подтверждали на процессе десольватации дигидрата цитрата натрия.XRD (Рентгеновская дифрактометрия) спектрографический анализ. Спектр XRD получали на рентгеновском дифрактометре Bruker-AXS D4 Endeavor с применением источника CuK (=1,54056, Мощность: 40 кВ, 50 мА) и детектора Vantec. Данные собирали в диапазоне 4-40 2 с шагом 0,009 2 и временем шага 0,5 с. Способ: USP 29 941, поправку на боковое смещение проводили при помощи пиков при 8,853 или 17,759 2 внутреннего стандарта. Список основных пиков, в 2, представлен далее в табл. 1. В области кристаллографии хорошо известно, что для любой данной кристаллической формы относительные интенсивности пиков дифракции могут различаться вследствие предпочтительной ориентации, возникающей в результате таких факторов, как морфология кристалла. Если эффекты предпочтительной ориентации присутствуют, интенсивности пиков изменяются, но характеристические положения пиков полиморфа остаются неизменными. См., например, The United States Pharmacopeia 23, NationalFormulary 18, p. 1843-1844, 1995. Кроме того, в области кристаллографии также известно, что для любой данной кристаллической формы угловые положения пиков могут незначительно меняться. Например, положения пиков могут сдвигаться вследствие изменения температуры, при которой анализируют образец, замены образца или присутствия или отсутствия внутреннего стандарта. В настоящем случае погрешность положения пика, равная 0,2 2, учитывает указанные потенциальные изменения, не препятствуя однозначной идентификации кристаллических солей согласно настоящему изобретению. Хорошо известным и принятым способом исследования кристаллических форм в литературе является способ Финка. В способе Финка применяют четыре наиболее интенсивные линии для первоначального исследования, а затем следующие четыре наиболее интенсивные линии. Пример 8. Метанолсольват транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5 ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты. Суспендировали ВССА (4 г) в 4 мл метанола и кипятили с обратным холодильником. Добавляли примерно 50 мл метанола с получением тонкой суспензии. Охлаждали смесь до комнатной температуры и выдерживали в течение дня. Выделяли твердый продукт путем вакуумного фильтрования и хранили в камере с метанолом для защиты указанного метастабильного кристалла от влаги. Пример 9. Этанолсольват транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5 ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты. Суспендировали ВССА (3 г) в 10 мл этанола в течение нескольких часов. Выделяли твердый продукт путем вакуумного фильтрования и хранили в этаноле для защиты указанного метастабильного кристалла от влаги. Пример 10. Муравьинокислый сольват транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Нтетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты. Растворяли ВССА в 7 мл изопропанола. К раствору добавляли муравьиную кислоту (3 мл). Добавляли воду до точки помутнения (4 мл) при комнатной температуре. Нагревали суспензию до 70 С в течение 6 ч, затем охлаждали до комнатной температуры. Выделяли твердый продукт путем вакуумного фильтрования и сушки на воздухе. Получали спектр XRD согласно описанию примера 7. В табл. 2, представленной далее, перечислены пики, полученные в спектре XRD. Таблица 2(1,5 г) в 10 мл гептана. Нагревали суспензию до 50 С. Добавляли 1 мл уксусной кислоты, и суспензия становилась прозрачной. Добавляли 10 мл гептана и охлаждали до комнатной температуры. Выделяли твердый продукт путем вакуумного фильтрования и сушки на воздухе. Получали спектр XRD аналогично описанию примера 7. В табл. 3, представленной далее, перечислены пики, полученные в спектре XRD. Таблица 3 Пример 12. Натриевая соль транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5 ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты. Добавляли в 500-мл сосуд гидрат транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил 2 Н-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1 ил]метил]циклогексанкарбоновой кислоты (20,16 г), этанол SDA 3 А (200,94 мл). Перемешивали с применением магнитной мешалки и добавляли 1 н. гидроксид натрия (31,95 г). Получали прозрачный раствор. Сушка распылением. Применяли распылительную сушилку (Niro SD Micro Spray Dryer) со скоростью потока газа-осушителя 32,5 кг/ч, 2,8 бар, расход газа-распылителя 2,0 кг/ч, 0,3 бар, температура на входе (автоматическое управление) 115 С. После достижения температуры на входе, равной 115 С, и температуре на выходе (109 С), начинали подавать смесь 83:17 (мас./мас.) этанол:вода на распылитель. Настройка насоса 3,0, капли не попадали на стенки/выход камеры. Оставляли систему для достижения термического равновесия - выход=104 С. Меняли смесь этанол:вода (при комнатной температуре) на образец для сушки в растворе этанол/вода. После сушки распылением получали 13,51 г. Пример 13. Магниевая соль транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5 ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты. Растворяли натриевую соль транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Нтетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты (2 г) в 5 мл метанола. Растворяли ацетат магния (335 мг) в 20 мл воды. Добавляли по каплям при комнатной температуре раствор ацетата магния при интенсивном перемешивании, а затем нагревали до 65 С. Происходило осаждение. При повышенной температуре дополнительно добавляли 10 мл воды. Охлаждали суспензию до комнатной температуры и выделяли твердый продукт путем вакуумного фильтрования и промывали водой. Сушили продукт в вакуумируемом сушильном шкафу при комнатной температуре. Кальциевые и цинковые соли ВССА можно получать с применением способов, аналогичных получению магниевой соли ВССА. Калиевую соль можно получать с применением способа, аналогичного получению натриевой соли ВССА. Пример 14. Сольват транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5-ил)амино]2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты и третбутиламина в изопропиловом спирте (1:1:1). Устанавливали 1-л трехгорлую круглодонную колбу, снабженную механической мешалкой, устройством управления температурой, капельной воронкой и колбонагревателем. Суспендировали гидрат транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Нтетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1 ил]метил]циклогексанкарбоновой кислоты (50 г) в гептане (500 мл). Нагревали суспензию до 69,1 С и добавляли трет-бутиламин (5,57 г) в виде раствора в изопропиловом спирте (70 мл) и дополнительно промывали 5 мл изопропилового спирта. Раствор образуется непосредственно после добавления. В течение добавления температура снижалась до 63 С, но поднималась обратно до 69 С после завершения добавления. На кончике шпателя вносили затравку трет-бутиламин транс-4-(5S)-5-3,5-бис(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1 бензазепин-1-ил]метил]циклогексанкарбоновой кислоты. Начинала образовываться суспензия - продолжали перемешивание при 69 С. Начинали охлаждать реакционную смесь до 55 С. Примерно через 1 ч образовывалась тонкая суспензия. Продолжали охлаждение до 45 С. Примерно через 1 ч снижали температуру суспензии до 35 С. Примерно через 2 ч удаляли источник нагрева и оставляли охлаждаться до температуры окружающей среды. Примерно через 3 ч фильтровали суспензию (температура составляла 21 С) и промывали 100 мл гептана. Сушили в вакууме в течение примерно 1,5 ч. Сушили в течение выходных в вакууме при 33 С с получением 55,9 г. Получали спектр XRD согласно описанию примера 7. В табл. 4, представленной далее, перечислены пики, полученные на спектре XRD. Пример 15. Гемиэтанолсольват геми-трет-бутиламиновой соли транс-4-(5S)-5-3,5-бис(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1 бензазепин-1-ил]метил]циклогексанкарбоновой кислоты (1:1:2) в кристаллической форме. Суспендировали гидрат транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Нтетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновой кислоты (50,5 г) в гептане (404,00 мл) с механическим перемешиванием верхнеприводной мешалкой. Добавляли этанол (25,25 мл) и нагревали до 55 С. Образовывался раствор. Добавляли трет-бутиламин(2,81 г) при 55 С. Вносили затравку геми-трет-бутиламингемиэтанолсольвата транс-4-(5S)-5-3,5-бис(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1 Н-1 бензазепин-1-ил]метил]циклогексанкарбоновой кислоты (1:1:2) и происходило осаждение. Поддерживали температуру при 50 С. В течение нескольких минут образовывалась тонкая суспензия с выделением тепла до 60 С. Продолжали перемешивание и охлаждали до 50 С. Примерно через 5,5 ч выключали нагрев и оставляли охлаждаться до температуры окружающей среды. Примерно через 3 ч фильтровали,промывали колбу частью маточного раствора, а затем промывали осадок 100 мл гептана. Сушили в течение ночи в вакууме при 45 С. 50,9 г. Получали спектр XRD согласно описанию примера 7. В табл. 5,представленной далее, перечислены пики, полученные на спектре XRD. Пример 16. Гидрат транс-4-(5S)-5-3,5-бис-(трифторметил)фенил]метил](2-метил-2 Н-тетразол-5-ил)амино]2,3,4,5-тетрагидро-7,9-диметил-1 Н-1-бензазепин-1-ил]метил]циклогексанкарбоновая кислоты. Стадия 13 схемы 3: добавляли 2 М раствор NaOH (9,9 л) к метил-транс-4-[(5S)-5-[3,5-бис(трифторметил)бензил](2-метил-2 Н-тетразол-5-ил)амино-7,9-диметил-2,3,4,5-тетрагидро-1 Н-1 бензазепин-1-ил]метилциклогексанкарбоксилату (12,95 кг) при комнатной температуре и перемешивали реакционную смесь в течение 10 мин. Перемешивали реакционную смесь при 40-45 С в течение 6 ч. Исследовали реакцию путем ВЭЖХ. К смеси добавляли уксусную кислоту (3,75 кг), затем воду (1,2 объема), вносили затравку гидрата ВССА и перемешивали в течение 2 ч при 40-45 С. Добавляли воду (2,8 объема) к реакционной смеси и перемешивали в течение 2 ч при 40-45 С. Охлаждали реакционную смесь до комнатной температуры, перемешивали в течение 2 ч и фильтровали. Собирали твердые вещества,промывали смесью этанол:H2O (1:1, 22 объема) и сушили на фильтре под давлением, что приводило к получению 15,87 кг твердого вещества, применяемого на стадии 2. Добавляли метанол к твердому веществу, полученному на стадии 1, и перемешивали при 65-70 С в течение 2 ч с получением прозрачного раствора. Фильтровали через плоский фильтр под давлением во второй реактор, перемешивали фильтрат в течение 2 ч при 0-5 С, фильтровали и сушили на фильтре под давлением в течение ночи. Перемешивали перекристаллизованное твердое вещество в воде (10 объемов) при комнатной температуре в течение 2 ч, фильтровали и сушили на фильтре под давлением. Сушили в сушильном шкафу в глубоком вакууме при 40-45 С с получением содержания воды, равного от 2,7 до 3,1%, определенного путем анализа Карла Фишера, что приводило к получению 11,80 кг вещества. Анализ Карла Фишера=3,16%. Масс-спектроскопия ES-API в режиме детектирования положительных ионов=639,30; в режиме детектирования отрицательных ионов=637,20. Химическая стабильность. В табл. 6 перечислена химическая стабильность свободной кислоты ВССА, а также выбранных примеров 31, 89, 153 и 175, описанных в WO 06/002342, пронумерованных далее как соединения 13, 14,15 и 16 соответственно. Представленные далее данные показывают, что свободная кислота ВССА имеет преимущественные свойства за счет увеличенной стабильности в кислой среде, которая значительно выше по сравнению с соединениями 13, 14 и 16. Указанная улучшенная стабильность свободной кислоты ВССА не изучена и не может быть предсказана при рассмотрении WO 06/002342. Получение образцов: 1) образец соединения: 0,9 мг/мл в ACN; 2) 100 мкл образца + 700 мкл ACN + 1 мл среды (0,1 н. HCl, 50 мМ PO4, рН 8 и 0,3% Н 2 О 2); 3) помещали ампулы в нагретый (40 С) автодозатор, впрыскивали с 4-часовыми интервалами в течение 16 или 24 ч. Параметры автодозатора для хроматографии. Колонка: Alltech Alltima Phenyl, 3 мкм, 4,6150 мм или Waters XTerra MS C18, 3,5 мкм, 4,6150 мм,температура колонки: 50 С, впрыскиваемый объем: 10 мкл. Детектирование: УФ 226, 254 или 266 нм (в зависимости от соединения). Расход: 1,5 мл/мин. Мобильная фаза: 0,1% TFA в 50% воды/50% ACN (может требоваться коррекция % ACN для достижения пика времени удерживания соединения 5 мин), конечная концентрация образца 0,05 мг/мл; содержание ACN 44%. Определение IV и РО фармакокинетики ингибиторов СЕТР у самцов крыс линии Спраг-Доули для сравнения. Далее представлено определение фармакокинетических параметров, включая пероральную биодоступность, после введения единственного 1 мг/кг внутривенного болюса или 3 мг/кг пероральной дозы соединения самцам крыс линии Спраг-Доули (n=4) в перекрестном исследовании. Внутривенный носитель представлял собой 20% микроэмульсия солютола/80% деионизованной воды. Пероральный носитель представлял собой повидон USP 10%/SLS 0,5%/QS деионизованной воды. Из передней лапы после внутривенного введения отбирали образцы крови через 0,08, 0,25, 0,5, 1, 2, 5, 8, 12 и 24 ч после введения дозы. Из передней лапы после перорального введения отбирали образцы крови через 0, 0,25, 0,5, 1, 2, 5,8, 12 и 24 ч после введения дозы. Плазму получали путем центрифугирования, помещали в лед и замораживали до переноса образцов в сухом льду в Bioanalytical Systems, Inc. (BASi; West Lafayette, IN) для биоаналитического анализа. Концентрации ВССА определяли путем ЖХ/МС/МС, фармакокинетические параметры высчитывали с применением аттестованного программного обеспечения (Watson, Version 7.1). Для оценки биодоступности можно применять другие носители, включая: 1% (мас./об.) карбоксиметилцеллюлозы натрия, 0,25% (мас./об.) полисорбата 80 (Tween 80) и 0,05% Dow Corning Antifoam 1510US в очищенной воде, или 1% (мас./об.) карбоксиметилцеллюлозы натрия, 0,5% (мас./об.) лаурилсульфата натрия и 0,05% Dow Corning Antifoam 1510-US в очищенной воде, каждый из которых можно применять с обработкой ультразвуком, способствующей растворению соединений, или без нее. Данные, представленные в табл. 7, показывают, что свободная кислота ВССА обладает преимуществами, заключающимися в проявлении 4-кратной биодоступности по сравнению с соединением 15(пример 153 WO 06/002342). Указанная повышенная биодоступность не изучена и не может быть предсказана при рассмотрении WO 06/002342. 3 мг/кг РО (объем дозы 2 мл/кг), 1 мг/кг IV (объем дозы 1 мл/кг). Исследования. Представленные далее способы и результат(ы) анализа, показывающие применимость и эффективность соединения и/или способов согласно настоящему изобретению, представлены для иллюстрации и не должны рассматриваться в качестве ограничения. Анализ ингибитора СЕТР in vitro: анализ SPA. Сцинтилляционный анализ сближения (SPA) in vitro применяли для оценки способности соединений согласно настоящему изобретению ингибировать перенос радиомеченных холестериновых эфиров между ЛПВП и ЛПНП. В указанном анализе изучают ингибирование переноса [3 Н]холестериновых эфиров от ЛПВП (Amersham) в биотинилированные ЛПНП (Amersham) источником СЕТР. Источник СЕТР для указанного анализа может продуцироваться клетками AV-12, которые созданы для экспрессии человеческого СЕТР. Радиомеченный холестериновый эфир переносили в основный буфер HEPES-NaCl, через 30 мин инкубирования реакцию останавливали и биотинилированные ЛПНП связывали с покрытыми стрептавидином/сцинтиллянтом гранулами для SPA (Amersham). Радиоактивный сигнал измеряли в 96 луночном сцинтилляционном TopCounter производства Packard в полностью открытом виде. Снижение радиоактивного сигнала ЛПНП по сравнению со стандартом показывает способность соединений ингибировать активность СЕТР. В качестве альтернативы можно применять другие источники СЕТР для возникновения переноса радиомеченного холестеринового эфира в указанном анализе. Например, в качестве источника СЕТР в указанном анализе можно применять эндогенный СЕТР плазмы человека, СЕТР мышей, которые экспрессируют СЕТР человека и эндогенный СЕТР хомяков. В указанном анализе можно применять буферы, отличные от основного HEPES-NaCl буфера, например человеческую плазмы, плазму мышей или Tris-буфер, которые содержат высокое количество альбумина. Специалисты в данной области техники понимают, что для отслеживания активности СЕТР в указанном анализе можно применять другие источники радиоактивности. Дополнительно, в указанном анализе можно применять радиомеченный ЛПНП. Исследование активности СЕТР in vivo. Для определения активности соединений in vivo можно применять сирийских золотистых хомяков,которые экспрессируют эндогенный СЕТР. Исследуемые соединения вводили перорально в 30 мг/кг дозах в носителе, состоящем из 79,5% кукурузного масла, 20% олеиновой кислоты и 0,5% Lubrafil группе трансгенных мышей, которые экспрессируют СЕТР человека (самки или самцы СЕТР и Аро А 1 гетерозиготные мыши Taconic, Germantown, NY). Можно получать образцы крови/плазмы в различное время после введения дозы в диапазоне от 4 до 48 ч. Активность СЕТР можно определять путем способа, аналогичного описанному выше при анализе активности СЕТР in vitro, с модификацией, заключающейся в том, что плазму обработанных животных применяли в указанном анализе в качестве источника СЕТР.In vivo активность ВССА согласно указанному анализу представлена далее в табл. 8. Исследование липидов плазмы in vivo. Активность соединений согласно настоящему изобретению in vivo можно оценивать путем сравнения уровня увеличения ЛПВП-холестерина данным количеством соединения у СЕТР-содержащих животных по сравнению с контрольным опытом. Исследуемые соединения вводили согласно представленному выше описанию анализа активности СЕТР in vivo. Образцы крови получали в различное время после введения дозы в диапазоне от 4 до 24 ч. Кровь оставляли для свертывания, получали сыворотку свернувшейся крови путем центрифугирования. Содержание ЛПВП-холестерина в сыворотке можно определять путем известных способов с применением ЛПВП-Х-положительных реагентов (Roche/Hitachi, Indianapolis, IN) и клинического химического анализатора (Roche/Hitachi, Indianapolis, IN). Дополнительно,липиды сыворотки можно анализировать ферментными способами. Липиды в ЛПОНП, ЛПНП и ЛПВП фракциях анализировали путем ферментных способов после осаждения или эксклюзионной хроматографии. Примеры повышения уровня ЛПВП-холестерина через 8 ч после введения аморфной ВССА представлены далее в табл. 8.
МПК / Метки
МПК: A61K 31/55, C07D 403/12, A61P 9/10
Метки: кислота, транс-4-[[(5s)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2h-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1н-1-бензазепин-1-ил]метил]циклогексанкарбоновая
Код ссылки
<a href="https://eas.patents.su/30-20600-trans-4-5s-5-35-bis-triftormetilfenilmetil2-metil-2h-tetrazol-5-ilamino-2345-tetragidro-79-dimetil-1n-1-benzazepin-1-ilmetilciklogeksankarbonovaya-kislota.html" rel="bookmark" title="База патентов Евразийского Союза">Транс-4-[[(5s)-5-[[[3,5-бис-(трифторметил)фенил]метил](2-метил-2h-тетразол-5-ил)амино]-2,3,4,5-тетрагидро-7,9-диметил-1н-1-бензазепин-1-ил]метил]циклогексанкарбоновая кислота</a>
Твердые формы (s)-этил-2-амино-3-(4-(2-амино-6-((r)-1-(4-хлор-2-(3-метил-1h-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропаноата и способы их применения

Номер патента: 17275
Опубликовано: 30.11.2012
Авторы: Канамарлапуди Раманаиах К., Де Поль Сьюзан, Чжан Хаймин, Беднарз Марк С., Перлберг Анетт
МПК: A61K 31/506, A61P 25/00, A61P 1/00...
Метки: способы, твердые, применения, s)-этил-2-амино-3-(4-(2-амино-6-((r)-1-(4-хлор-2-(3-метил-1h-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропаноата, формы
Формула / Реферат:
1. Кристаллический (S)-этил-2-амино-3-(4-(2-амино-6-((R)-1-(4-хлор-2-(3-метил-1Н-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропаноат или его фармацевтически приемлемая соль.2. Соединение по п.1, которое представляет собой кристаллический (S)-этил-2-амино-3-(4-(2-амино-6-((R)-1-(4-хлор-2-(3-метил-1Н-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропаноат.3. Соединение по п.2, которое имеет температуру плавления,...
Способ получения производных 1,3,4,5 – тетрагидро -2н-3- бензазепин -2- она

Номер патента: 7743
Опубликовано: 29.12.2006
Авторы: Бриго Даньель, Лерести Жан-Мишель, Суви Жан-Клод, Лекув Жан-Пьер
МПК: C07D 223/16, A61P 9/00, A61K 31/55...
Метки: бензазепин, получения, производных, тетрагидро, 1,3,4,5, 2н-3, способ
Формула / Реферат:
1. Способ синтеза соединений формулы (I) в которой R1 и R2, которые могут быть одинаковыми или разными, каждый представляет собой линейную или разветвленную (С1-С8)алкоксигруппу или образуют вместе с атомом углерода, к которому они присоединены, 1,3-диоксановое, 1,3-диоксолановое или 1,3-диоксепановое кольцо, в котором соединение формулы (VI) в которой R1 и R2 имеют значения, указанные выше, подвергают каталитической реакции гидрогенизации в...
Гидраты 2-хлор-5-[3,6-дигидро-3-метил-2,6-диоксо-4-(трифторметил)-1-(2н)-пиримидинил]-4-фтор-n-[[метил-(1-метилэтил)амино]сульфонил]бензамида

Номер патента: 15757
Опубликовано: 30.12.2011
Авторы: Кокс Герхард, Михель Альфред, Райнхард Роберт, Зайтц Вернер, Цагар Цирилл, Веверс Ян-Хендрик, Майер Гуидо, Зиверних Бернд, Лёр Зандра, Вольф Бернд, Сакселл Хейди Эмилиа, Эрк Петер, Кайл Михаэль, Хампрехт Герхард, Шмидт Томас, Гебхардт Йоахим
МПК: C07D 239/54, A01N 43/48
Метки: гидраты, 2-хлор-5-[3,6-дигидро-3-метил-2,6-диоксо-4-(трифторметил)-1-(2н)-пиримидинил]-4-фтор-n-[[метил-(1-метилэтил)амино]сульфонил]бензамида
Формула / Реферат:
1. Гидраты 2-хлор-5-[3,6-дигидро-3-метил-2,6-диоксо-4-(трифторметил)-1-(2Н)-пиримидинил]-4-фтор-N-[[метил-(1-метилэтил)амино]сульфонил]бензамида.2. Гидраты по п.1, содержащие от 0,8 до 1,2 моль воды в пересчете на 1 моль 2-хлор-5-[3,6-дигидро-3-метил-2,6-диоксо-4-(трифторметил)-1-(2Н)-пиримидинил]-4-фтор-N-[[метил-(1-метилэтил)амино]сульфонил]бензамида.3. Гидраты по п.1 или 2 с пиком плавления в пределах от 100 до 140°С.4. Гидрат по одному из...
Кристаллическая форма 2-хлор-5-[3,6-дигидро-3-метил-2,6-диоксо-4-(трифторметил)-1-(2н)-пиримидинил]-4-фтор-n-[[метил-(1-метилэтил) амино]сульфонил]бензамида

Номер патента: 15518
Опубликовано: 31.08.2011
Авторы: Михель Альфред, Цагар Цирилл, Зиверних Бернд, Вольф Бернд, Шмидт Томас, Хампрехт Герхард, Кайл Михаэль, Эрк Петер, Лёр Зандра, Гебхардт Йоахим, Зайтц Вернер, Майер Гуидо, Кокс Герхард, Сакселл Хейди Эмилиа, Райнхард Роберт, Веверс Ян-Хендрик
МПК: C07D 239/54, A01N 43/48
Метки: амино]сульфонил]бензамида, 2-хлор-5-[3,6-дигидро-3-метил-2,6-диоксо-4-(трифторметил)-1-(2н)-пиримидинил]-4-фтор-n-[[метил-(1-метилэтил, форма, кристаллическая
Формула / Реферат:
1. Кристаллическая, по существу, не содержащая растворитель форма II 2-хлор-5-[3,6-дигидро-3-метил-2,6-диоксо-4-(трифторметил)-1-(2Н)-пиримидинил]-4-фтор-N-[[метил-(1-метилэтил)амино]сульфонил]бензамида, которая в рентгеновской порошковой дифрактограмме при 25°С и Cu-Ka-излучении показывает по меньшей мере два из следующих приведенных как 2q-значения отражений: 6,3±0,3°, 9,4±0,3°, 10,9±0,3°, 11,9±0,3°, 12,6±0,3°, 15,0±0,3°, 15,8±0,3°,...
Полиморфная форма 2-(r) – (1- (r) – (3,5-бис (трифторметил) фенил) этокси) -3- (s) – (4-фтор) фенил – 4 – (3 – (5-оксо-1h, 4h-1, 2, 4 – триазоло) метил)морфолина в качестве антагониста рецептора тахикинина

Номер патента: 2405
Опубликовано: 25.04.2002
Авторы: Макколи Джеймс, Крокер Луис
МПК: C07D 265/32, A61K 31/5375, A61P 25/28...
Метки: форма, трифторметил, метил)морфолина, 4h-1, тахикинина, 4-фтор, рецептора, антагониста, 5-оксо-1h, качестве, полиморфная, триазоло, фенил, 3,5-бис, 2-(r, этокси
Формула / Реферат:
1. Полиморфная форма соединения 2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фтор)фенил-4-(3-(5-оксо-1Н,4Н-1,2,4-триазоло)метил)морфолина, обозначенная как форма I, по существу, отличающаяся рентгеновской порошковой дифрактограммой с характерными отражениями приблизительно 12,0, 15,3, 16,6, 17,0, 17,6, 19,4, 20,0, 21,9, 23,6, 23,8 и 24,8ш (2 тэта). 2. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и...
Предыдущий патент: Фунгицидные смеси, содержащие замещенные 1-метилпиразол-4-илкарбоксанилиды и абамектин
Следующий патент: Способ дезинфекции или стерилизации изделия
Случайный патент: Защитная нить или полоса, включающая ориентированные магнитные частицы, находящиеся в краске, и способ и средства для ее получения