Трициклические азотсодержащие соединения и их применение в качестве бактерицидных средств
Номер патента: 18817
Опубликовано: 30.10.2013
Авторы: Фиандор Роман Хосе Мария, Барфут Кристофер, Ремуинан-Бланко Модесто Хесус, Алемпарте-Галлардо Карлос, Баррос-Агирре Давид, Хеннесси Алан Джозеф, Пирсон Нейл Дэвид, Качо-Искердо Моника



























Формула / Реферат
1. Соединение формулы (I), его фармацевтически приемлемая соль

где один из Z1 и Z2 представляет собой CH или N, а другой представляет собой CH;
U представляет собой группу, выбранную из фенила, пиридила, пиридазинила, тиазолила и тиофенила;
n равно 0 или 1;
R5 и R6 независимо выбраны из галогена, CF3, OCF3, С1-3алкила, С1-3алкокси, нитро и циано; или R5 может представлять собой группу -CmH2m-A, где m принимает значения от 1 до 5, фрагмент -CmH2m- может представлять собой фрагмент с прямой или разветвленной цепью и А выбран из OH, OR7 и OCOR7, где R7 представляет собой С1-5алкил.
2. Соединение по п.1, где в формуле (I) U представляет собой группу, выбранную из фенила, пиридила, пиридазинила, тиазолила и тиофенила.
3. Соединение по п.1, где когда n равно 0, R5 представляет собой CF3, OCF3, Cl, Br или NO2.
4. Соединение по п.1, где n=1, один из R5 и R6 представляет собой Cl, а другой представляет собой Cl, CH3, C2H5, CN, CF3 или OCF3.
5. Соединение по п.1, где n=1 и один из R5 и R6 представляет собой F, а другой представляет собой Cl, CF3, CN или СН3.
6. Соединение по п.1, где n=1 и один из R5 и R6 представляет собой СН3, а другой представляет собой Br, СН3, CF3, CN или NO2.
7. Соединение по п.1, где R5 представляет собой -СН2-OH, n равно 1 и R6 представляет собой Cl.
8. Соединение по п.1, где U выбран из группы, включающей фенил, пиридил и пиридазинил, n равно 0 и R5 находится в пара-положении U относительно связи между U и группой CH2, к которой он присоединен.
9. Соединение по п.1, где U выбран из группы, включающей фенил и пиридил, n равно 1 и один из R5 и R6 находится в пара-положении, а другой находится в мета-положении U относительно связи между U и группой CH2, к которой он присоединен.
10. Соединение по п.1, где абсолютная стереохимия соединения формулы (I) представлена формулой (IB)

11. Соединение, которое выбрано из следующих соединений:
(1R)-1-[(4-{[(5-хлор-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(2R)-2-[(4-{[(5-хлор-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-{[4-({[6-(трифторметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(2R)-2-[(4-{[(3,4-дихлорфенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
(2R)-2-{[4-({[4-фтор-3-(трифторметил)фенил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
5-{[(1-{[(2R)-3,8-диоксо-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-2-ил]метил}-4-пиперидинил)амино]метил}-2-фторбензонитрил;
(2R)-2-[(4-{[(4-фтор-3-метилфенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-[(4-{[(5-хлор-4-метил-2-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[(4-{[(5-хлор-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-{[4-({[6-(трифторметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-[(4-{[(5-бром-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[(4-{[(5,6-диметил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
2-[(4-{[(5,6-диметил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(5,6-дихлор-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-{[4-({[4-хлор-3-(трифторметил)фенил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-{[4-({[6-(трифторметил)-3-пиридазинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-{[4-({[6-(трифторметил)-3-пиридазинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
гидрохлорид 2-{[4-({[6-(трифторметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-диона;
2-{[4-({[6-(трифторметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(4-хлорфенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(3-хлорфенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(4-метил-3-нитрофенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(5-бром-4-метил-2-тиенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(3,4-диметилфенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(5-хлор-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-({4-[({4-[(трифторметил)окси]фенил}метил)амино]-1-пиперидинил}метил)-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-{[4-({[4-(трифторметил)фенил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(4-бромфенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
гидрохлорид 2-[(4-{[(5-хлор-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-диона;
(1R)-1-[(4-{[(3,4-дихлорфенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
5-{[(1-{[(1R)-4,9-диоксо-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-1-ил]метил}-4-пиперидинил)амино]метил}-3-метил-2-пиридинкарбонитрил;
2-[(4-{[(6-фтор-5-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(4-хлор-3-метилфенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(4-бром-2-тиенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(3,4-дихлорфенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(2-хлор-1,3-тиазол-5-ил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(4-нитрофенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-{[4-({[3-хлор-4-(метилокси)фенил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(5-бром-2-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(5-бром-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(5-хлор-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(3-фтор-4-метилфенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(3,4-дифторфенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
(2R)-2-{[4-({[6-(трифторметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
гидрохлорид (2R)-2-{[4-({[6-(трифторметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-диона;
(2S)-2-[(4-{[(3,4-дихлорфенил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
(2S)-2-{[4-({[6-(трифторметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
3-хлор-5-{[(1-{[(1R)-4,9-диоксо-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-1-ил]метил}-4-пиперидинил)амино]метил}-2-пиридинкарбонитрил;
дигидрохлорид 3-хлор-5-{[(1-{[(1R)-4,9-диоксо-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-1-ил]метил}-4-пиперидинил)амино]метил}-2-пиридинкарбонитрила;
(2R)-2-[(4-{[(5-бром-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
2-[(4-{[(5-фтор-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
(2S)-2-[(4-{[(5-хлор-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
(1R)-1-[(4-{[(6-этил-5-фтор-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
гидрохлорид (1R)-1-[(4-{[(5-бром-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-диона;
(1R)-1-{[4-({[5-хлор-6-(гидроксиметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
2-{[4-({[4-метил-3-(метилокси)фенил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
гидрохлорид (1R)-1-{[4-({[6-(трифторметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-диона;
(1R)-1-{[4-({[5-метил-6-(трифторметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-{[4-({[5-бром-6-(трифторметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-{[4-({[5-хлор-6-(1-гидрокси-1-метилэтил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[(4-{[(5-фтор-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-{[4-({[3-хлор-4-(гидроксиметил)фенил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
дигидрохлорид 3-хлор-5-{[(1-{[(2R)-3,8-диоксо-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-2-ил]метил}-4-пиперидинил)амино]метил}-2-пиридинкарбонитрила;
(2R)-2-[(4-{[(5,6-дихлор-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
(2R)-2-{[4-({[5-хлор-6-(гидроксиметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион;
(2-хлор-4-{[(1-{[(1R)-4,9-диоксо-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-1-ил]метил}-4-пиперидинил)амино]метил}фенил)метилацетат;
(1R)-1-{[4-({[5-хлор-6-(1-гидроксиэтил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[(4-{[(6-хлор-5-метил-3-пиридазинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион;
(1R)-1-[(4-{[(6-хлор-5-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион и
гидрохлорид (2R)-2-{[4-({[5-метил-6-(трифторметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-диона.
12. Соединение по п.11, которое представляет собой (1R)-1-[(4-{[(5-хлор-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион.
13. Соединение по п.11, которое представляет собой (2R)-2-[(4-{[(5-хлор-6-метил-3-пиридинил)метил]амино}-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2а,5,8а-триазааценафтилен-3,8-дион.
14. Соединение по п.11, которое представляет собой (1R)-1-{[4-({[6-(трифторметил)-3-пиридинил]метил}амино)-1-пиперидинил]метил}-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион.
15. Фармацевтическая композиция для лечения туберкулеза у млекопитающих, включающая соединение по п.1 и один или несколько фармацевтически приемлемых носителей, наполнителей или разбавителей.
16. Применение соединения по п.1 для лечения туберкулеза у млекопитающих.
17. Применение соединения по п.1 для производства лекарственного средства для лечения туберкулеза у млекопитающих.
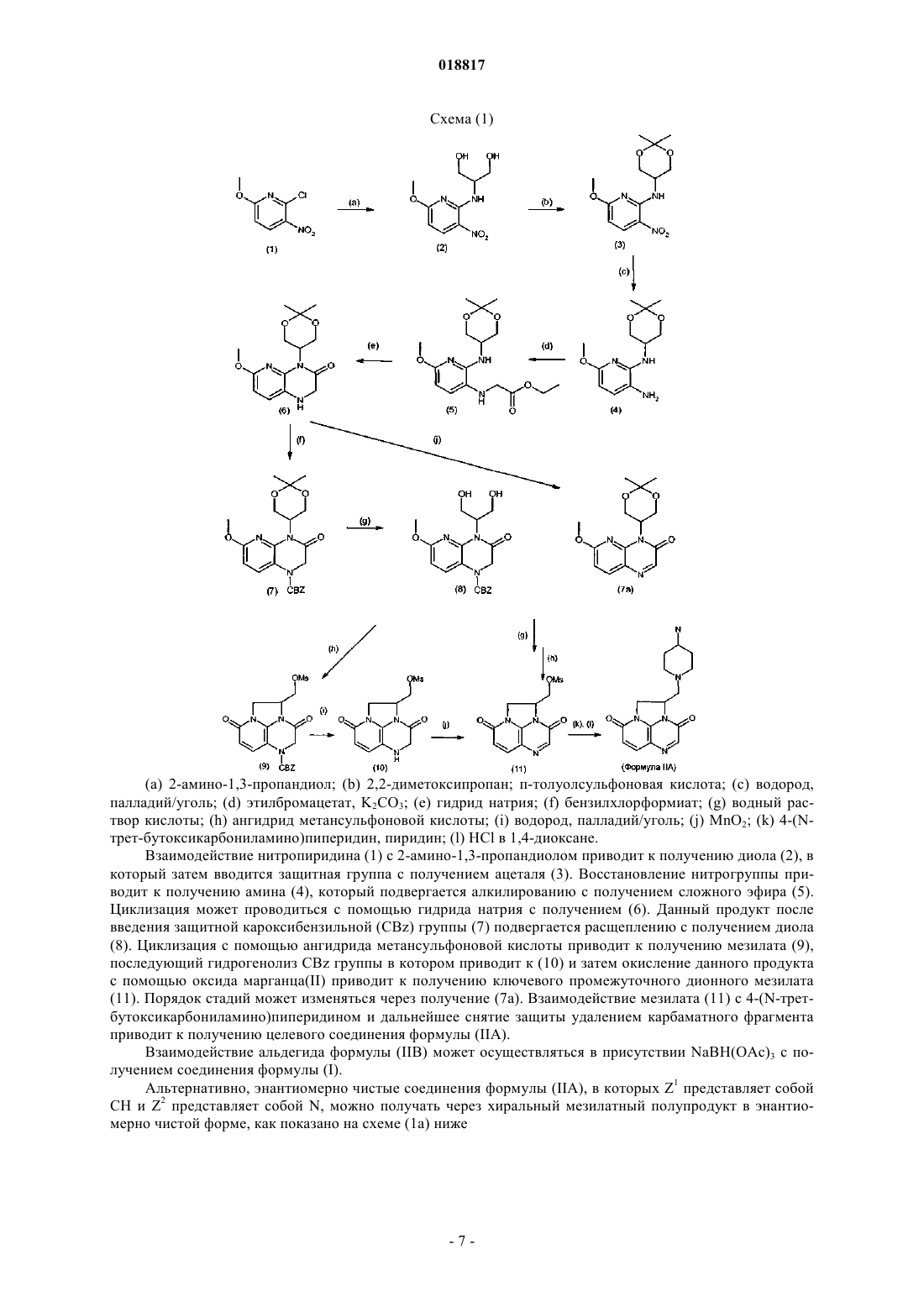
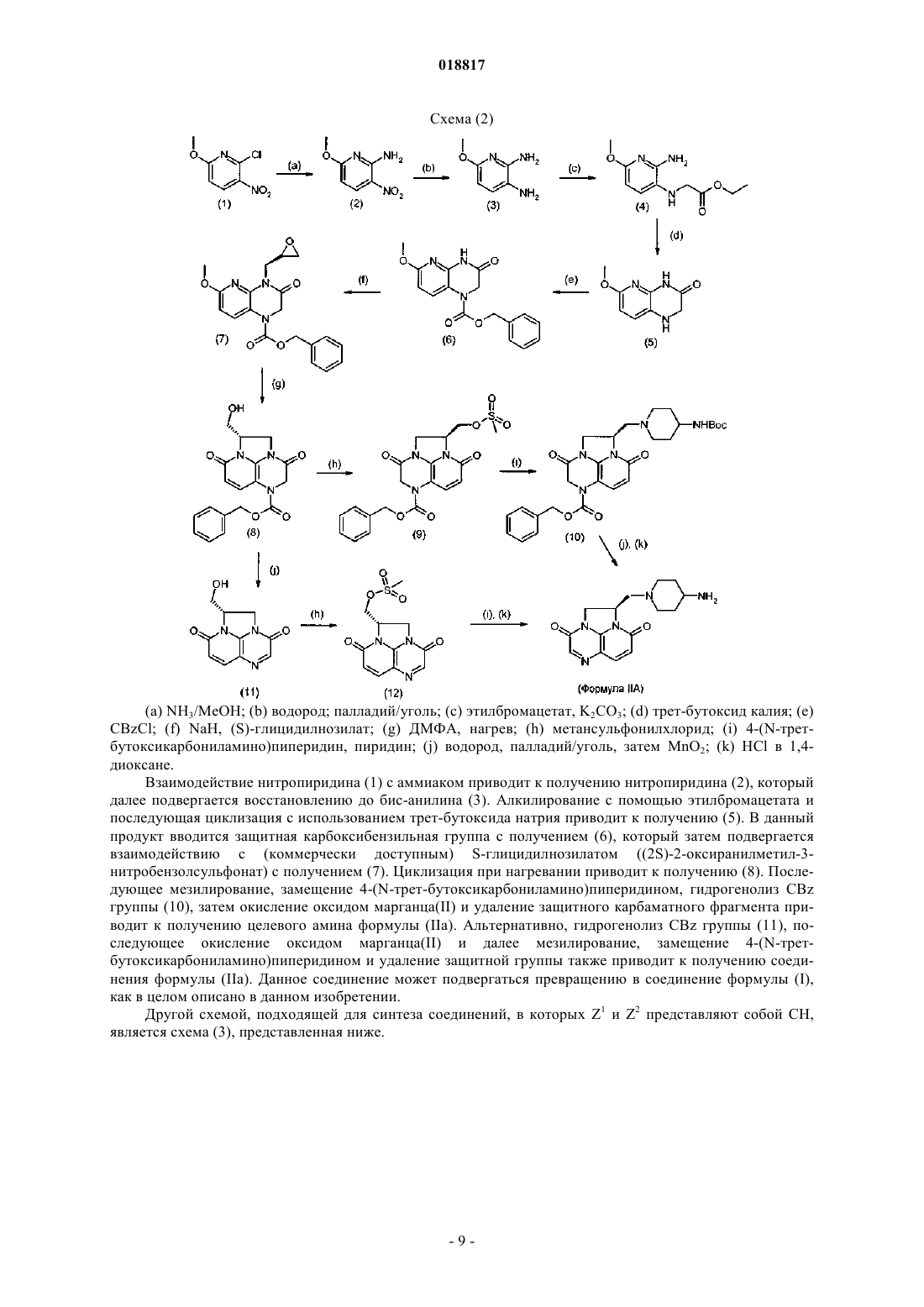
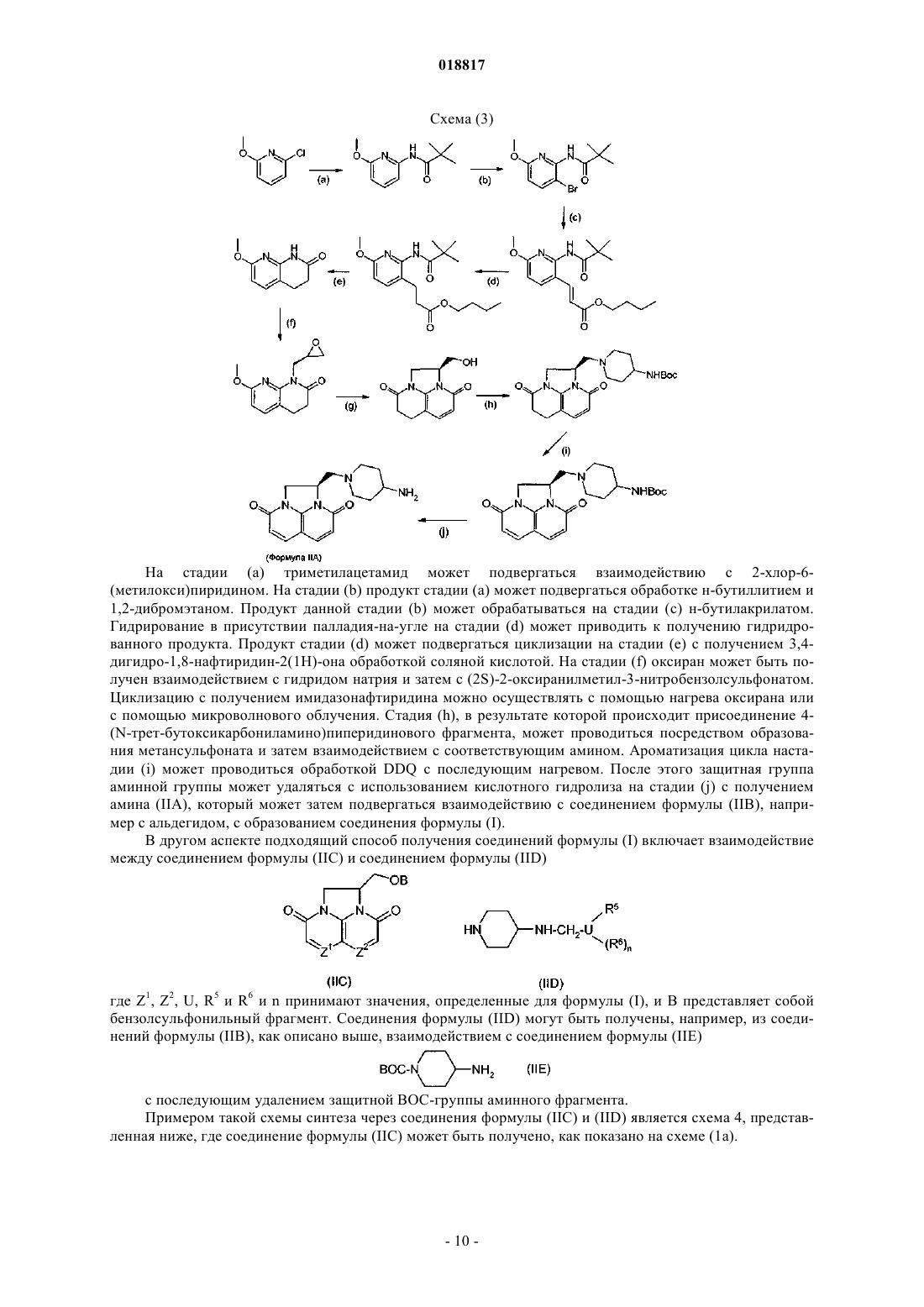
18. Способ получения соединения по п.1, включающий взаимодействие амина формулы (IIA) и соединения формулы (IIB)

где Z1, Z2, U, R5 и R6 и n принимают значения, определенные для формулы (I), и W представляет собой альдегидный фрагмент -CH=O, причем такое взаимодействие представляет собой реакцию восстановительного аминирования.
19. Способ получения соединения по п.1, включающий взаимодействие амина формулы (IIA) и соединения формулы (IIB)

где Z1, Z2, U, R5 и R6 и n принимают значения, определенные для формулы (I), или W представляет собой бромметильный фрагмент -CH2Br.
Текст
ТРИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ БАКТЕРИЦИДНЫХ СРЕДСТВ Соединения формулы (I), их фармацевтически приемлемые соли или N-оксиды (показана относительная стереохимия), фармацевтические композиции, включающие указанные соединения,их применение в терапии, в частности для лечения туберкулеза, а также способы получения указанных соединений. Алемпарте-Галлардо Карлос (ES),Барфут Кристофер (GB), БарросАгирре Давид, Качо-Искердо Моника,Фиандор Роман Хосе Мария (ES),Хеннесси Алан Джозеф (GB), Пирсон Нейл Дэвид (US), Ремуинан-Бланко Модесто Хесус (ES) Медведев В.Н. (RU)(71)(73) Заявитель и патентовладелец: ГЛЭКСО ГРУП ЛИМИТЕД (GB) Область техники, к которой относится изобретение Данное изобретение относится к соединениям, композициям, содержащим указанные соединения,их применению в терапии, включая применение в качестве бактерицидных средств, например для лечения туберкулеза, и к способам получения таких соединений. Уровень техники В РСТ патентных публикациях WO 02/08224, WO 02/50061, WO 02/56882, WO 02/96907, WO 2003087098, WO 2003010138, WO 2003064421, WO 2003064431, WO 2004002992, WO 2004002490, WO 2004014361, WO 2004041210, WO 2004096982, WO 2002050036, WO 2004058144, WO 2004087145, WO 2006002047, WO 2006014580, WO 2006010040, WO 2006017326, WO 2006012396, WO 2006017468, WO 2006020561, WO 2006081179, WO 2006081264, WO 2006081289, WO 2006081178, WO 2006081182, WO 01/25227, WO 02/40474, WO 02/07572, WO 2004024712, WO 2004024713, WO 2004035569, WO 2004087647, WO 2004089947, WO 2005016916, WO 2005097781, WO 2006010831, WO 2006021448, WO 2006032466, WO 2006038172, WO 2006046552, WO 2006099884, WO 2006105289, WO 2006081178, WO 2006081182, WO 2006134378, WO 2006137485, WO 2007016610, WO 2007081597, WO 2007071936, WO 2007115947, WO 2007118130, WO 2007122258, WO 2007138974, WO 2008006648, WO 2008003690 и WO 2008009700 описываются производные хинолина, нафтиридина, морфолина, циклогексана, пиперидина и пиперазина, а также трициклические соединения с конденсированными циклами, которые обладают бактерицидной активностью. В публикации WO 2004104000 описываются трициклические соединения с конденсированными циклами, которые могут селективно воздействовать на каннабиноидные рецепторы. Синтетические лекарственные средства для лечения туберкулеза (ТБ) доступны уже в течение полувека, но заболеваемость этой болезнью продолжает возрастать во всем мире. В 2004 г. выявлено 24500 человек с этим развитым активным заболеванием, и около 5500 человек ежедневно умирают от ТБ (World Health Organization, Global Tuberculosis Control: Surveillance, Planning,Financing. WHO Report 2006, Geneva, Switzerland, ISBN 92-4 156314-1). Одновременное инфицирование ВИЧ стимулирует повышение заболеваемости (Williams, B.G.; Dye, С. Science, 2003, 301, 1535), и причина смерти 31% пациентов со СПИД на африканском континенте может быть отнесена к ТБ (Corbett, E.L.;Watt, C.J.; Catherine, J.; Walker, N.; Maher D.; Williams, B.G.; Raviglione, M.C.; Dye, C. Arch. Intl. Med.,2003, 163, 1009, Septkowitz, A.; Raffalli, J.; Riley, Т.; Kiehn, Т.Е.; Armstrong, D. Clin. Microbiol. Rev. 1995,8, 180). С появлением полирезистентных штаммов Mycobacterium tuberculosis (MDR-TB) масштаб проблемы увеличился. Прошло уже более десяти лет с тех пор, как ВОЗ объявила ТБ "глобальной угрозой здоровью" (World Health Organization, Global Tuberculosis Control: Surveillance, Planning, Financing. WHOReport 2006, Geneva, Switzerland, ISBN 92-4 156314-1). Проблемы противотуберкулезной терапии и предупреждения заболевания туберкулезом хорошо известны. Доступная на сегодняшний день вакцина BCG была внедрена в 1921 г. и уже не в состоянии защищать большинство взрослых людей. В настоящее время пациенты, которые становятся инфицированными активным заболеванием, получают в качестве комбинированного терапевтического лечения исониазид, рифампин, пиразинамид и этамбутол в течение двух месяцев, а затем в течение дополнительных четырех месяцев продолжают получать исониазид и рифампин. Необходимо ежедневное введение препаратов, и невысокая эффективность препаратов приводит к появлению и распространению полирезистентных штаммов, лечение которых требует повышения интенсивности лечения. В недавно опубликованном обзоре подробно обсуждаются многие аспекты ТБ, такие как патогенез, эпидемиология, а также лекарственные средства и разработанные вакцины, которые используются до настоящего времени(Nature Medicine, Vol. 13(3), pages 263-312). Таким образом, существует острая потребность в менее длительных курсах лечения с применением более активных лекарственных средств, которые можно принимать с меньшей частотой, то есть существует потребность в лекарственных средствах, эффективных в отношении полирезистентных штаммов ТБ(MDR-TB). Следовательно, существует потребность в открытии и разработке новых химических соединений для лечения ТБ (разработанные в последнее время основные химические структуры представлены в обзоре Ballell, L.; Field, R.A.; Duncan, K.; Young, R.J. Antimicrob. Agents Chemother. 2005, 49, 2153). Подробное описание изобретения В соответствии с одним аспектом настоящее изобретение предоставляет соединение формулы (I),его фармацевтически приемлемую соль или N-оксид где один из Z1 и Z2 представляет собой CH или N, а другой представляет собой CH;R5 и R6 независимо выбраны из галогена, CF3, OCF3, С 1-3 алкила, С 1-3 алкокси, нитро и циано; или R5 может представлять собой группу -CmH2m-A, где m принимает значения от 1 до 5, фрагмент -CmH2m- может представлять собой фрагмент с прямой или разветвленной цепью и A выбран из OH, OR7 и OCOR7,где R7 представляет собой С 1-5 алкил. Изобретение предоставляет также фармацевтическую композицию, включающую соединение формулы (I) или его фармацевтически приемлемую соль и один или несколько фармацевтически приемлемых носителей, наполнителей или разбавителей. Изобретение предоставляет также соединение формулы (I) или его фармацевтически приемлемую соль для применения в терапии. Изобретение предоставляет также соединение формулы (I), его фармацевтически приемлемую соль для применения в лечении туберкулеза у млекопитающих. Изобретение предоставляет также соединение формулы (I) или его фармацевтически приемлемую соль для применения в лечении бактериальных инфекций млекопитающих, в частности человека. Изобретение предоставляет также применение соединений формулы (I) или его фармацевтически приемлемой соли в производстве лекарственного средства для применения в лечении туберкулеза у млекопитающих. Изобретение предоставляет также применение соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарственного средства для применения в лечении бактериальных инфекций млекопитающих. Изобретение предоставляет также фармацевтическую композицию, включающую соединение формулы (I) или его фармацевтически приемлемую соль и один или несколько фармацевтически приемлемых носителей, наполнителей или разбавителей, для применения в лечении туберкулеза у млекопитающих, в частности у человека. Изобретение предоставляет также фармацевтическую композицию, включающую соединение формулы (I) или его фармацевтически приемлемую соль и один или несколько фармацевтически приемлемых носителей, наполнителей или разбавителей, для применения в лечении бактериальных инфекций млекопитающих, в частности человека. В одном варианте осуществления изобретения, когда n=0, R5 представляет собой CF3, OCF3, Cl, Br или NO2. В еще одном варианте осуществления изобретения n=1 и один из R5 и R6 представляет собой Cl, а другой представляет собой Cl, CH3, С 2 Н 5, CN, CF3 или OCF3. В еще одном варианте осуществления изобретения n=1 и один из R5 и R6 представляет собой F, а другой представляет собой Cl, CF3, CN, CH3 или С 2 Н 5. В еще одном варианте осуществления изобретения n=1 и один из R5 и R6 представляет собой СН 3, а другой представляет собой Br, СН 3, CF3, CN или NO2. В еще одном варианте осуществления изобретения n равно 0 и R5 представляет собой CF3. В еще одном варианте осуществления изобретения n равно 1 и R5 и R6 представляют собой Cl и СН 3. В еще одном варианте осуществления изобретения n равно 1 и R5 и R6 представляют собой СН 3 иCF3. В еще одном варианте осуществления изобретения n равно 1 и R5 и R6 представляют собой Cl и CN. В еще одном варианте осуществления изобретения m равно 1 и А представляет собой -OH, так чтоR5 представляет собой -СН 2-OH, n равно 1 и R6 представляет собой Cl. В еще одном варианте осуществления изобретения, когда U представляет собой группу, выбранную из фенила, пиридила или пиридазинила и n=0, тогда R5 находится в пара-положении U относительно связи между U и группой CH2, к которой он присоединен. В еще одном варианте осуществления изобретения, когда U представляет собой группу, выбранную из фенила или пиридила, и n=1, один из R5 и R6 находится в пара-положении, а другой - в метаположении U относительно связи между U и группой CH2, к которой он присоединен. Один или несколько представленных выше структурных вариантов осуществления изобретения могут быть представлены в соединении формулы (I). В варианте осуществления изобретения абсолютная стереохимия соединения формулы (I) или (IA) представлена формулой (IB)-2 018817 Соединения формулы (I) могут существовать в форме солей, сольватов, и формула (I) включает в себя эти формы. В соответствии с одним аспектом соединения, применимые в настоящем изобретении, включают соединения, упомянутые в примерах, их фармацевтически приемлемые соли, сольваты. В соответствии с другим аспектом соединения, применимые в данном изобретении, включают следующие соединения:(1R)-1-[(4-[(6-хлор-5-метил-3-пиридинил)метил]амино-1-пиперидинил)метил]-1,2-дигидро 4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион и гидрохлорид (2R)-2-[4-([5-метил-6-(трифторметил)-3-пиридинил]метиламино)-1-пиперидинил] метил-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона. Некоторые соединения формулы (I) могут существовать в форме оптических изомеров, например в форме смесей изомеров всех соотношений, в частности рацемических смесей. Изобретение включает все такие формы, в частности чистые изомерные формы. Например, изобретение включает энантиомеры. Различные изомерные формы могут выделяться или отделяться одна от другой стандартными методами,или любой заданный изомер может быть получен стандартными методами синтеза или методами стереоспецифического или асимметрического синтеза. Термины и определения Термин "С 1-3 алкил", когда используется в данном описании, относится к алкильной группе с прямой или разветвленной цепью, содержащей от 1 до 3 атомов углерода. Примеры С 1-3 алкильных групп включают метил, этил, н-пропил, изопропил. Термин "галоген", когда используется в данном описании, относится к атому фтора, хлора, брома и йода. В соответствии с одним аспектом термин "галоген", когда используется в данном описании, относятся к атому фтора, хлора и брома. Термин "С 1-3 алкокси", когда используется в данном описании, относится к алкоксигруппе с прямой или разветвленной цепью, содержащей от 1 до 3 атомов углерода. Примеры С 1-3 алкоксигрупп включают метокси, этокси, пропокси и изопропокси. Термин "соединения согласно изобретению", когда используется в данном описании, означает соединение формулы (I), его фармацевтически приемлемую соль или N-оксид. Термин "соединение согласно изобретению" означает любое одно из соединений согласно изобретению, как определено выше. Кроме того, будет понятно, что такие фразы как "соединение формулы (I), его фармацевтически приемлемая соль или N-оксид" или "соединения согласно изобретению", как это подразумевается, включают соединение формулы (I), фармацевтически приемлемую соль или N-оксид соединения формулы (I) или их любое фармацевтически приемлемое сочетание. Таким образом, в качестве примера, используемого в данном описании для иллюстративной цели, но без ограничения, фраза "соединение формулы (I),его фармацевтически приемлемая соль или N-оксид" включает фармацевтически приемлемую соль соединения формулы (I), которая представлена как сольват, или данная фраза может включать смесь соединения формулы (I) и соли соединения формулы (I). Будет дополнительно приниматься во внимание, что все кристаллические формы, полиморфы и энантиомеры соединений согласно изобретению или их смеси рассматриваются как относящиеся к области настоящего изобретения. За исключением особо оговоренных случаев (например, когда показана абсолютная стереохимия) для соединений согласно изобретению, которые содержат стереоцентры и,следовательно, которые могут образовывать энантиомеры, соединение содержит смесь энантиомеров в соотношении 1:1, т.е. представляет собой рацемическую смесь энантиомеров. Энантиомеры могут разделяться с использованием стандартных способов, таких как хиральная ВЭЖХ. Некоторые соединения согласно настоящему изобретению могут подвергаться кристаллизации или перекристаллизации из растворителей, таких как водные и органические растворители. В таких случаях-5 018817 могут образовываться сольваты. Данное изобретение включает в своем объеме стехиометрические сольваты, включая гидраты, а также соединения, содержащие изменяемые количества воды, которые могут быть получены, например, лиофилизацией. Поскольку соединения формулы (I) предназначены для применения в фармацевтических композициях, будет понятно, что в особых вариантах осуществления изобретения они могут быть получены, по существу, в чистой форме, например с чистотой по меньшей мере 60%, более приемлемо с чистотой по меньшей мере 75%, еще более приемлемо с чистотой по меньшей мере 85%, особенно приемлемо с чистотой по меньшей мере 98% (% из расчета на массу относительно общей массы). Неочищенные препараты соединений могут использоваться для получения более чистых форм, используемых в фармацевтических композициях; эти менее чистые препараты соединений должны содержать по меньшей мере 1%,более приемлемо по меньшей мере 5%, еще более приемлемо от 10 до 59% соединения формулы (I), его фармацевтически приемлемой соли. Фармацевтически приемлемые соли соединений формулы (I) включают кислотно-аддитивные или четвертичные аммониевые соли, например их соли с минеральными кислотами, такими как соляная,бромисто-водородная, серная, азотная или фосфорная кислоты, или с органическими кислотами, такими как уксусная, фумаровая, янтарная, малеиновая, лимонная, бензойная, п-толуолсульфоновая, метансульфоновая, нафталинсульфоновая или винная кислоты. В соответствии с одним аспектом изобретения соль соединения формулы (I) представляет собой дигидрохлоридную соль. В соответствии с другим аспектом соль соединения формулы (I) представляет собой дигидрохлоридную соль. Соединения формулы (I) также могут быть получены в виде N-оксида. Изобретение относится ко всем таким солям. Получение соединений В дополнительных аспектах изобретения предоставлены способы получения соединений формулы(I) и их фармацевтически приемлемых солей или N-оксидов. В соответствии с одним аспектом подходящий способ включает взаимодействие между аминным соединением формулы (IIA) и соединением формулы (IIB) где Z1, Z2, U, R5 и R6 и n принимают значения, определенные для формулы (I), и W представляет собой альдегидный фрагмент -CH=O, причем данная реакция представляет собой реакцию восстановительного аминирования, которая обычно проводится в присутствии восстановителя, такого как боргидрид натрия. Такие реакции могут проводиться в органическом растворителе, таком как ДХМ/MeOH, при температуре окружающей среды. См., например, Smith, M.B.; March, J.M. Advanced Organic Chemistry, Wiley-Interscience. Соединения формулы (IIA) в форме соли, например в форме гидрохлоридной соли, могут применяться в данном способе для получения соединений формулы (I) в форме соли. Соединения формулы (IIA) могут быть получены в соответствии с различными схемами получения. Одна такая схема, подходящая для получения соединений, в которых Z1 представляет собой CH и 2Z представляет собой N, представлена ниже как схема (1).(a) 2-амино-1,3-пропандиол; (b) 2,2-диметоксипропан; п-толуолсульфоновая кислота; (с) водород,палладий/уголь; (d) этилбромацетат, K2CO3; (e) гидрид натрия; (f) бензилхлорформиат; (g) водный раствор кислоты; (h) ангидрид метансульфоновой кислоты; (i) водород, палладий/уголь; (j) MnO2; (k) 4-(Nтрет-бутоксикарбониламино)пиперидин, пиридин; (l) HCl в 1,4-диоксане. Взаимодействие нитропиридина (1) с 2-амино-1,3-пропандиолом приводит к получению диола (2), в который затем вводится защитная группа с получением ацеталя (3). Восстановление нитрогруппы приводит к получению амина (4), который подвергается алкилированию с получением сложного эфира (5). Циклизация может проводиться с помощью гидрида натрия с получением (6). Данный продукт после введения защитной кароксибензильной (CBz) группы (7) подвергается расщеплению с получением диола(8). Циклизация с помощью ангидрида метансульфоновой кислоты приводит к получению мезилата (9),последующий гидрогенолиз CBz группы в котором приводит к (10) и затем окисление данного продукта с помощью оксида марганца(II) приводит к получению ключевого промежуточного дионного мезилата(11). Порядок стадий может изменяться через получение (7 а). Взаимодействие мезилата (11) с 4-(N-третбутоксикарбониламино)пиперидином и дальнейшее снятие защиты удалением карбаматного фрагмента приводит к получению целевого соединения формулы (IIA). Взаимодействие альдегида формулы (IIB) может осуществляться в присутствии NaBH(OAc)3 с получением соединения формулы (I). Альтернативно, энантиомерно чистые соединения формулы (IIA), в которых Z1 представляет собойCH и Z2 представляет собой N, можно получать через хиральный мезилатный полупродукт в энантиомерно чистой форме, как показано на схеме (1 а) ниже(a) EtOH, кипячение с обратным холодильником; (b) TBS-Cl, (с) цинк, уксусная кислота; (d) этилбромацетат, K2CO3; е) NaH; (f) водород, палладий/уголь; (g) MnO2; (h) ангидрид метансульфоновой кислоты; (i) ТФУК; (j) ангидрид метансульфоновой кислоты или бензолсульфонилхлорид. Взаимодействие 2-хлор-6-(метилокси)-3-нитропиридина с хиральным амином (2) приводит к получению промежуточного продукта (3). Введение защитной группы (3) с помощью трет-бутилдиметилсилилхлорида приводит к получению (4). Восстановление нитрогруппы приводит к получению амина (5), который подвергается алкилированию с получением сложного эфира (6). Циклизация (6) может осуществляться с помощью гидрида натрия, и последующая обработка водородом над катализатором палладий/уголь приводит к получению промежуточного продукта (7). Окисление оксидом марганца(II) и обработка ангидридом метансульфоновой кислоты приводит к получению(8). Удаление защитной группы из этого промежуточного продукта может осуществляться с помощью ТФУК с получением (9), и взаимодействие полученного соединения с ангидридом метансульфоновой кислоты или бензолсульфонилхлоридом приводит к получению (10) в форме энантиомерно чистого соединения. Полученный мезилат или бензолсульфонат (10) может затем подвергаться превращению в амин формулы (IIa) и, наконец, в целевое соединение формулы (I), как в общем описано здесь. Другой схемой, подходящей для получения соединений, в которых Z1 представляет собой N и Z2 представляет собой СН, является схема (2), представленная ниже.CBzCl; (f) NaH, (S)-глицидилнозилат; (g) ДМФА, нагрев; (h) метансульфонилхлорид; (i) 4-(N-третбутоксикарбониламино)пиперидин, пиридин; (j) водород, палладий/уголь, затем MnO2; (k) HCl в 1,4 диоксане. Взаимодействие нитропиридина (1) с аммиаком приводит к получению нитропиридина (2), который далее подвергается восстановлению до бис-анилина (3). Алкилирование с помощью этилбромацетата и последующая циклизация с использованием трет-бутоксида натрия приводит к получению (5). В данный продукт вводится защитная карбоксибензильная группа с получением (6), который затем подвергается взаимодействию с (коммерчески доступным) S-глицидилнозилатом 2S)-2-оксиранилметил-3 нитробензолсульфонат) с получением (7). Циклизация при нагревании приводит к получению (8). Последующее мезилирование, замещение 4-(N-трет-бутоксикарбониламино)пиперидином, гидрогенолиз CBz группы (10), затем окисление оксидом марганца(II) и удаление защитного карбаматного фрагмента приводит к получению целевого амина формулы (IIa). Альтернативно, гидрогенолиз CBz группы (11), последующее окисление оксидом марганца(II) и далее мезилирование, замещение 4-(N-третбутоксикарбониламино)пиперидином и удаление защитной группы также приводит к получению соединения формулы (IIa). Данное соединение может подвергаться превращению в соединение формулы (I),как в целом описано в данном изобретении. Другой схемой, подходящей для синтеза соединений, в которых Z1 и Z2 представляют собой СН,является схема (3), представленная ниже. На стадии (а) триметилацетамид может подвергаться взаимодействию с 2-хлор-6(метилокси)пиридином. На стадии (b) продукт стадии (а) может подвергаться обработке н-бутиллитием и 1,2-дибромэтаном. Продукт данной стадии (b) может обрабатываться на стадии (с) н-бутилакрилатом. Гидрирование в присутствии палладия-на-угле на стадии (d) может приводить к получению гидридрованного продукта. Продукт стадии (d) может подвергаться циклизации на стадии (e) с получением 3,4 дигидро-1,8-нафтиридин-2(1 Н)-она обработкой соляной кислотой. На стадии (f) оксиран может быть получен взаимодействием с гидридом натрия и затем с (2S)-2-оксиранилметил-3-нитробензолсульфонатом. Циклизацию с получением имидазонафтиридина можно осуществлять с помощью нагрева оксирана или с помощью микроволнового облучения. Стадия (h), в результате которой происходит присоединение 4(N-трет-бутоксикарбониламино)пиперидинового фрагмента, может проводиться посредством образования метансульфоната и затем взаимодействием с соответствующим амином. Ароматизация цикла настадии (i) может проводиться обработкой DDQ с последующим нагревом. После этого защитная группа аминной группы может удаляться с использованием кислотного гидролиза на стадии (j) с получением амина (IIA), который может затем подвергаться взаимодействию с соединением формулы (IIB), например с альдегидом, с образованием соединения формулы (I). В другом аспекте подходящий способ получения соединений формулы (I) включает взаимодействие между соединением формулы (IIC) и соединением формулы (IID) где Z1, Z2, U, R5 и R6 и n принимают значения, определенные для формулы (I), и В представляет собой бензолсульфонильный фрагмент. Соединения формулы (IID) могут быть получены, например, из соединений формулы (IIB), как описано выше, взаимодействием с соединением формулы (IIE) с последующим удалением защитной ВОС-группы аминного фрагмента. Примером такой схемы синтеза через соединения формулы (IIC) и (IID) является схема 4, представленная ниже, где соединение формулы (IIC) может быть получено, как показано на схеме (1 а). Солевые формы соединений формулы (I) и (IA), например гидрохлориды, могут быть получены обработкой соответствующих свободных оснований кислотой, такой как соляная кислота, или при получении соединений в присутствии такой кислоты. Большое количество реагентов формулы (IIB), содержащих необходимую группу R5 и необязательную группу R6, являются известными в данной области соединениями (см., например, коммерческие источники, указанные в табл. 1) или могут быть получены аналогично известным соединениям (см., например, WO 02/08224, WO 02/50061, WO 02/56882, WO 02/96907, WO 2003087098, WO 2003010138, WO 2003064421, WO 2003064431, WO 2004002992, WO 2004002490, WO 2004014361, WO 2004041210, WO 2004096982, WO 2002050036, WO 2004058144, WO 2004087145, WO 06002047, WO 06014580, WO 06010040, WO 06017326, WO 06012396, WO 06137485, WO 06017468, WO 06020561 и EP 0559285). Использование указанных схем, альтернативных реагентов для получения альтернативных заместителей, таких как R2, R5, R6 и т.д., будет понятно специалисту в данной области техники. Дополнительные подробности получения соединений формулы (I) представлены в приведенных далее примерах. Препараты и способы введения Бактерицидные и/или противотуберкулезные соединения согласно данному изобретению могут быть введены в препараты для введения для применения в здравоохранении или ветеринарии любым удобным способом, аналогичным для других бактерицидных или противотуберкулезных соединений. Фармацевтические композиции согласно изобретению включают фармацевтические композиции в форме, адаптированной для перорального, местного или парентерального применения, и могут применяться для лечения бактериальных инфекций или инфекций Mycobacterium tuberculosis у млекопитающих, включая людей. Соединения могут быть введены в препарат для введения любым способом, подходящим для противобактериального и/или противотуберкулезного терапевтического лечения. Композиции могут быть в форме таблеток, капсул, порошков, гранул, пастилок, кремов или жидких препаратов, таких как стерильные растворы или суспензии для перорального или парентерального введения. Препараты для местного введения согласно настоящему изобретению могут быть представлены,например, в виде мазей, кремов или лосьонов, глазных мазей или глазных или ушных капель, импрегнированных перевязочных материалов и аэрозолей и могут содержать подходящие стандартные добавки,такие как консерванты, растворители для способствования проникновению лекарственных средств и смягчители в мазях и кремах. Препараты также могут содержать совместимые стандартные носители, такие как основы для кремов и мазей, и этанол или олеиновый спирт для лосьонов. Такие носители могут присутствовать в препарате в количестве, составляющем от примерно 1 до примерно 98% препарата. Чаще они будут составлять примерно 80% препарата. Препараты для перорального введения могут представлять собой, например, таблетки или капсулы лекарственной формы стандартной дозы и могут содержать традиционные наполнители, такие как связующие агенты, например сироп, гуммиарабик, желатин, сорбит, трагакант или поливинилпирролидон; заполнители, например лактозу, сахар, кукурузный крахмал, фосфат кальция, сорбит или глицин; смазывающие вещества для производства таблеток, например стеарат магния, тальк, полиэтиленгликоль или диоксид кремния; дезинтегрирующие агенты, например кукурузный крахмал; или приемлемые смачивающие агенты, такие как натрийлаурилсульфат. Таблетки могут быть покрыты оболочкой в соответствии с методами, хорошо известными в традиционной фармацевтической практике. Препараты для перорального введения также могут быть представлены в жидкой форме, например в форме водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров, или могут быть представлены в форме сухого продукта для смешения с водой или другим подходящим разбавителем перед применением. Такие жидкие препараты могут содержать стандартные добавки, такие как суспендирующие агенты, например сорбит, метилцеллюлозу, сироп глюкозы, желатин, гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия или способные подвергаться гидрированию пищевые жиры; эмульгаторы, например лецитин, сорбитанмоноолеат или гуммиарабик; неводные разбавители(которые могут включать пищевые масла), маслянистые сложные эфиры, такие как глицерин, пропиленгликоль, или этиловый спирт; консерванты, например метил- или пропил-п-гидроксибензоат или сорбиновую кислоту, и, если нужно, традиционные добавки, улучшающие вкус, или красители. Суппозитории будут содержать традиционные основы для суппозиториев, например масло какао- 11018817 или другой глицерид. Лекарственные формы стандартной дозы для парентерального введения получают с использованием соединения и стерильного разбавителя, причем предпочтительным разбавителем является вода. Соединение, в зависимости от разбавителя и используемой концентрации, может суспендироваться либо растворяться в разбавителе. При приготовлении растворов соединение может растворяться в воде для инъекции и стерилизоваться фильтрованием перед заполнением полученным раствором подходящего пузырька или ампулы и герметизацией. Преимущественно в разбавителе могут растворяться такие добавки, как местные анестетики, консерванты и буферные агенты. Для повышения стабильности композиция может подвергаться замораживанию после ее внесения в пузырек и удалению воды в вакууме. Сухой лиофилизованный порошок после этого герметично закрывается в пузырьке и одновременно может поставляться пузырек воды для инъекции для восстановления жидкости перед применением. Суспензии для парентерального введения получают, по существу, таким же образом, но с тем отличием, что соединение не растворяется, а суспендируется в разбавителе и стерилизация может проводиться не во время фильтрации. Соединение может стерилизоваться посредством выдерживания в этиленоксиде перед суспендированием в стерильном разбавителе. Преимущественно для способствования однородному распределению соединения в препарате в композицию включается поверхностно-активное вещество или смачивающий агент. В зависимости от способа введения композиции могут содержать 0,1 мас.%, предпочтительно от 10 до 60 мас.% соединения согласно изобретению. Когда композиция включает лекарственные формы стандартных доз, каждая стандартная доза будет предпочтительно содержать от 25 до 1000 мг активного ингредиента. Дозировка при применении для лечения взрослых людей будет предпочтительно находиться в интервале от 100 до 3000 мг в день, например 1500 мг в день, в зависимости от способа и частоты введения. Подходящая дозировка составляет от 5 до 30 мг/кг в сутки. Соединение формулы (I), его фармацевтически приемлемая соль или N-оксид может быть единственным терапевтическим средством в композициях согласно изобретению, или оно может присутствовать в препарате в сочетании с одним или несколькими дополнительными терапевтическими средствами. Таким образом, изобретение предоставляет в дополнительном аспекте комбинацию, включающую соединение формулы (I), его фармацевтически приемлемую соль или N-оксид вместе с одним или несколькими дополнительными терапевтическими средствами. Дополнительное терапевтическое средство, упомянутое выше, представляет собой, например, лекарственное средство, применимое для лечения туберкулеза у млекопитающего. Примеры таких терапевтических средств включают исониазид, этамбутол, рифампин, пиразинамид, стрептомицин, капреомицин, ципрофлоксацин и клофазимин. Когда соединение формулы (I), его фармацевтически приемлемая соль или N-оксид применяется в комбинации с одним или несколькими дополнительными терапевтическими средствами, доза соединения или лекарственного средства может отличаться от той дозы, которая применяется при введении одного соединения или лекарственного средства. Подходящие дозы будут легко подбираться специалистом данной области техники. Следует представлять, что количество соединения согласно изобретению и одного или нескольких дополнительных терапевтических средств, необходимых для применения в лечении, будет изменяться в зависимости от природы состояния, подлежащего лечению, и возраста и состояния пациента и будет, в конечном итоге, определяться лечащим врачом или ветеринаром. Комбинации могут быть удобно представлены для применения в форме фармацевтического препарата. В дополнительном аспекте настоящего изобретения предоставлена фармацевтическая комбинация,включающая соединение формулы (I), его фармацевтически приемлемую соль или N-оксид вместе с одним или несколькими дополнительными терапевтическими средствами и один или несколько фармацевтически приемлемых носителей, наполнителей или разбавителей. Отдельные компоненты таких комбинаций могут вводиться последовательно или одновременно в раздельных или объединенных фармацевтических препаратах посредством традиционного способа введения. Когда введение является последовательным, соединение согласно настоящему изобретению либо одно или несколько дополнительных терапевтических средств может вводиться первым. Когда введение является одновременным, комбинация может вводиться либо в одной фармацевтической композиции,либо в разных фармацевтических композициях. Следует представлять, что при введении в одном препарате соединение и другие лекарственные средства должны быть стабильными и совместимыми с каждым другим и прочими компонентами препарата. При введении в раздельных препаратах соединения согласно изобретению и другие лекарственные средства могут быть предоставлены в любом удобном препарате, традиционно таком, которые известны для таких соединений в данной области техники. Соединение формулы (I) может представлять собой единственное терапевтическое средство в композициях согласно изобретению или представлять одно соединение из комбинации с одним другим или несколькими бактерицидными и/или противотуберкулезными соединениями. Если другое бактерицидное средство представляет собой -лактам, тогда может также применяться ингибитор -лактамазы. Соединения формулы (I) также могут применяться в лечении бактериальных инфекций, вызываемых широким спектром организмов, включая грамотрицательные и грамположительные организмы. Не- 12018817 которые соединения формулы (I) могут быть активными в отношении более чем одного организма. Это может определяться способами, описанными в данном изобретении. Все публикации, включая, но без ограничения, патенты и заявки на патенты, упомянутые в данном описании, введены в описание в виде ссылки, как если бы каждая отдельная публикация была конкретно и отдельно указана для введения в виде ссылки во всей ее полноте. Приведенные далее примеры иллюстрируют получение некоторых соединений формулы (I) и активность некоторых соединений формулы (I) в отношении различных бактериальных организмов и организмов, которые известны как вызывающие туберкулез. Примеры и экспериментальная часть Используемые аббревиатуры. АсОН - уксусная кислота,Ас 2 О - уксусный ангидрид,Bn - бензил,BOC - N-трет-бутоксикарбонил,ВОС - ди-трет-бутилдикарбонат ангидрид,CH3CN - ацетонитрил,CBz - карбобензил,Celite - вспомогательная фильтрующая добавка, состоящая из диатомового диоксида кремния(торговая марка Manville Corp., Denver, Colorado),CF3TMS - (трифторметил)триметилсилан,DME - диметоксиэтан,ДХМ - дихлорметан,DDQ - 2,3-дихлор-5,6-дициано-1,4-бензохинон (реагент атомаризации),DIBAL-H - гидрид диизобутилалюминия,ДМФА - диметилформамид,ДМСО-d6 - дейтерированный диметилсульфоксид,ДМСО - диметилсульфоксид,ES МС - электрораспылительная масс-спектрометрия,EtOAc - этилацетат,EtOH - этанол,Et3N - триэтиламин,ч - час, часы,ВЭЖХ - высокоэффективная жидкостная хроматография,ЖХМС - жидкостная хроматографическая масс-спектроскопия,MeOH - метанол,МР- макропористый карбонат триэтиламмонийметилполистиролкарбонат (Argonaut Technologies),Ms - метансульфонил (мезил),NaBH(OAc)3 - триацетоксиборгидрид натрия,NMP - N-метилпирролидон (растворитель),ЯМР - спектроскопия ядерно-магнитного резонанса,АсО - ацетокси,Pd/C - катализатор - палладий-на-угле,Pd2(dba)3 - трис-(дибензилиденацетон)дипалладий,rt - комнатная температура,SCX - представляет собой ионообменную колонку Cartridge, содержащую прочную катионообменную смолу (бензолсульфоновая кислота), доступна от Varian, USA,TBS-Cl - трет-бутилдиметилсилилхлорид,ТВМЕ - простой метил-трет-бутиловый эфир,ТФУК - трифторуксусная кислота,ТГФ - тетрагидрофуран,УФ - ультрафиолетовое излучение. Записывают спектры протонного ядерного магнитного резонанса и химические сдвиги в нижнюю область, фиксируют в частях на миллионотносительно тетраметилсилана (ТМС), который используется в качестве внутреннего стандарта. Аббревиатуры, используемые для данных ЯМР, имеют следующие значения: c = синглет, д = дуплет, т = триплет, кв = квартет, м = мультиплет, дд = дуплет дуплетов,дт = дуплет триплетов, арр = мнимый, уш. = уширенный. Масс-спектры получают с использованием методов электрораспылительной (ES) ионизации. Все температуры приводятся в градусах Цельсия. Специалисту данной области техники понятно, что ссылки на синтез, проводимый аналогичным образом или в соответствии с общим способом, другие способы получения могут включать изменение обычных параметров, таких как продолжительность реакции, температура, условия обработки, незначительные изменения в количествах реагентов и т.д.- 13018817 Реакции, в которых задействованы гидриды металлов, включая гидрид лития, алюмогидрид лития,гидрид диизобутилалюминия, гидрид натрия, боргидрид натрия и триацетоксиборгидрид натрия, проводятся в атмосфере аргона. Экспериментальные примеры, представленные ниже, получение соединений, обозначенных как соединения примеров 1, 2, 9, 11, 12, 17, 51, 59, 60, 71 и 75, представлены как типичные примеры соединений согласно настоящему изобретению и различных схем получения, описанных в данном изобретении. Другие соединения согласно настоящему изобретению, полученные аналогичными способами, представлены в табл. 1. Пример соединения 1. Синтез гидрохлорида (1R)-1-[(4-[(5-хлор-6-метил-3-пиридинил)метил]амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-диона в соответствии со схемой получения (3)(1,488 г, 1,625 ммоль) и Xantphos (4,5-бис(дифенилфосфино)-9,9-диметилксантен) (1,880 г, 3,249 ммоль) в сухом дегазированном 1,4-диоксане (800 мл) в атмосфере аргона обрабатывают ультразвуком в течение 0,25 ч и затем 2-хлор-6-(метилокси)пиридином (19,32 мл, 162,494 ммоль). После этого смесь кипятят с обратным холодильником в течение 24 ч. Смесь упаривают, обрабатывают водой (1 л) и трижды экстрагируют ДХМ (1 л, затем 2500 мл). Объединенные органические слои сушат (MgSO4), упаривают и хроматографируют (элюирование: 50-100% ДХМ/40-60 петролейный эфир, затем 0-5% MeOH/ДХМ), получая указанное в заголовке соединение в виде твердого вещества желтого цвета (25,191 г, 121,111 ммоль,75%). Фракции, содержащие загрязняющие примеси, снова хроматографируют на колонке (элюирование,как описано выше), получая дополнительное количество продукта (4,990 г, 23,990 ммоль, 15%). Общий выход 90%.(450 мл) в трехгорлой колбе объемом 1 л, снабженной внешним термометром, в атмосфере аргона охлаждают до -78C и в течение 15 мин обрабатывают н-бутиллитием (232 мл, 581,847 ммоль), затем раствору дают возможность нагреться до 0C и перемешивают раствор при 0C в течение 7 ч. После этого смесь снова охлаждают до -78C, обрабатывают в течение 10 мин 1,2-дибромэтаном (27,3 мл, 317 ммоль), затем раствору дают возможность нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 30 мин, в процессе чего весь твердый продукт, который образовался,снова растворяется. На данной стадии происходит выделение газа, поэтому к одному из трех горл колбы присоединяют барботер. Затем осторожно в течение 10 мин добавляют воду (100 мл). После этого добавляют дополнительное количество воды (500 мл) и смесь экстрагируют Et2O (3500 мл). После этого объединенные органические растворители сушат (MgSO4), фильтруют, выпаривают, получая сырой продукт. Полученный продукт растворяют в теплом EtOAc (100 мл) и оставляют на ночь в холодильнике. Полученный твердый продукт, который выпадает в виде кристаллов, отфильтровывают, промывают охлажденным на ледяной бане Et2O (20 мл) и сушат в вакууме, получая продукт в виде твердого белого вещества (45,660 г, 159,011 ммоль, 60%). Фильтрат упаривают и остаток хроматографируют (элюирование: 025% EtOAc/40-60 петролейный эфир), получая очищенное исходное вещество (7,264 г, 34,9 ммоль) и продукт в виде твердого белого вещества (8,038 г, 27,992 ммоль, 10%). Продукт перекристаллизации и хроматографии идентифицируют с помощью ЯМР и ЖХ-МС и объединяют.(47,1 мл, 329 ммоль) и дициклогексилметиламином (64,5 мл, 302 ммоль). Реакционную смесь нагревают до 80C в течение 4 ч и затем до 120C в течение 3 ч. Затем реакционную смесь упаривают, добавляют воду (1000 мл) и смесь экстрагируют Et2O (3500 мл). Объединенные органические растворители сушат(MgSO4), фильтруют, выпаривают, получая сырой продукт. Продукт растворяют в ДХМ (300 мл), хроматографируют (элюирование: 10-30% EtOAc:40-60 петролейный эфир) и затем сушат в вакууме, получая продукт в виде твердого белого вещества (87,412 г, 95%).(43,706 г, 131 ммоль) в этаноле (450 мл) в атмосфере аргона при комнатной температуре обрабатывают палладием-на-угле (10% паста) (5,0 г, 47,0 ммоль) и затем перемешивают при комнатной температуре в атмосфере водорода с давлением 1 атм (101325 Па) в течение 90 ч. После этого реакционную смесь фильтруют через тонкий слой кизельгура, промывая продукт дополнительным количеством этанола (200 мл). Затем растворитель выпаривают, получая продукт в виде твердого вещества желтого цвета (43,549 г,99%).(e) 7-(Метилокси)-3,4-дигидро-1,8-нафтиридин-2(1H)-он. Смесь бутил-3-[2-[(2,2-диметилпропаноил)амино]-6-(метилокси)-3-пиридинил]пропаноата (86,01 г,256 ммоль) и соляной кислоты (500 мл, 3000 ммоль) (6 М, водный раствор) нагревают до 80C и выдерживают при данной температуре в течение 6 ч. Реакционный раствор охлаждают, обрабатывают водой(500 мл), переносят в коническую колбу объемом 5 л и осторожно нейтрализуют твердым K2CO3 (требуется примерно 250 г) (наблюдают энергичное вспенивание). После этого смесь экстрагируют 20%MeOH/ДХМ (3500 мл). Затем объединенные органические слои сушат (MgSO4), фильтруют, упаривают,получая сырой продукт в виде твердого вещества желтого цвета (35,84 г, 79%).(f) 7-(Метилокси)-1-[(2R)-2-оксиранилметил]-3,4-дигидро-1,8-нафтиридин-2(1H)-он. Раствор 7-(метилокси)-3,4-дигидро-1,8-нафтиридин-2(1H)-она (4,974 г, 27,9 ммоль) в ДМФА (100 мл) при 0C в атмосфере аргона обрабатывают гидридом натрия (60%, 1,340 г, 33,5 ммоль) и полученную смесь перемешивают при 0C в течение 20 мин. Реакционную смесь после этого обрабатывают (2S)-2 оксиранилметил-3-нитробензолсульфонатом (7,60 г, 29,3 ммоль), затем дают возможность смеси медленно нагреться до комнатной температуры и перемешивают смесь при комнатной температуре в течение 1 ч. Добавляют воду (5 мл). Реакционную смесь упаривают, добавляют насыщенный водный растворNaHCO3 (500 мл) и смесь экстрагируют ДХМ (3500 мл). После этого объединенные органические слои сушат (MgSO4), фильтруют, упаривают, получая сырой продукт.(g) (1S)-1-(гидроксиметил)-1,2,5,6-тетрагидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-дион. Раствор 7-(метилокси)-1-[(2R)-2-оксиранилметил]-3,4-дигидро-1,8-нафтиридин-2(1H)-она (1,167 г,4,98 ммоль) в ДМФА (20 мл) в атмосфере аргона нагревают до 120C и выдерживают при данной температуре в течение 6 ч. После этого реакционную смесь упаривают и хроматографируют (элюирование: 020% MeOH/ДХМ), получая продукт в виде твердого вещества оранжевого цвета (339 мг, 31%).[ES MC] m/z 221 (MH+, 100%). Альтернативно, реакционную смесь можно обрабатывать СВЧ при 160C в течение 40 мин.(1S)-1-(гидроксиметил)-1,2,5,6-тетрагидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9 диона (1,909 г, 8,67 ммоль) в ДХМ (100 мл) при 0C в атмосфере аргона обрабатывают Et3N (1,450 мл,10,40 ммоль) и затем метансульфонилхлоридом (0,743 мл, 9,54 ммоль), после этого дают возможность смеси нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 1 ч. Реакционную смесь обрабатывают насыщенным водным раствором NaHCO3 (100 мл) и экстрагируют ДХМ (2100 мл). После этого объединенные органические слои сушат (MgSO4), фильтруют, упаривают,получая сырой полупродукт [(2S)-4,9-диоксо-1,2,8,9-тетрагидро-4H,7 Н-имидазо[1,2,3-ij]-1,8-нафтиридин 2-ил]метилметансульфонат. Полученный промежуточный продукт растворяют в сухом CH3CN (100 мл),обрабатывают пиридином (1,402 мл, 17,34 ммоль) и 1,1-диметилэтил 4-пиперидинилкарбаматом (3,47 г,17,34 ммоль), нагревают до 70C и выдерживают при данной температуре в течение 20 ч. После этого добавляют дополнительное количество 1,1-диметилэтил-4-пиперидинилкарбамата (3,47 г, 17,34 ммоль) и пиридина (1,402 мл, 17,34 ммоль), смесь нагревают до температуры образования флегмы (нагрев останавливают при 95C) и реакционную смесь перемешивают при данной температуре в течение дополнительных 4 ч. Реакционную смесь упаривают, обрабатывают насыщенным водным раствором NaHCO3(200 мл) и полученную смесь экстрагируют ДХМ (3200 мл). После этого объединенные органические слои сушат (MgSO4), фильтруют, упаривают, получая сырой продукт в виде твердого вещества коричневого цвета.(i) 1,1-Диметилэтил-(1-[(1R)-4,9-диоксо-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-1 ил]метил-4-пиперидинил)карбамат. Раствор 1,1-диметилэтил-(1-[(2R)-4,9-диоксо-1,2,8,9-тетрагидро-4H,7 Н-имидазо[1,2,3-ij]-1,8 нафтиридин-2-ил]метил-4-пиперидинил)карбамата (5,710 г, 14,19 ммоль) в 1,4-диоксане (50 мл) при комнатной температуре обрабатывают DDQ (4,83 г, 21,28 ммоль), затем нагревают до 120C и выдержи- 15018817 вают при данной температуре в течение 1 ч. После этого реакционную смесь охлаждают до комнатной температуры. Реакционную смесь обрабатывают насыщенным водным раствором K2CO3 (5%, 1000 мл) и экстрагируют ДХМ (3500 мл). Объединенные органические слои сушат (MgSO4), фильтруют, упаривают, получая сырой продукт в виде твердого вещества коричневого цвета. Реакцию повторяют, используя дополнительную порцию карбамата (2,889 г, 7,18 ммоль) в 1,4-диоксане (50 мл) и DDQ (2,444 г, 10,77 ммоль). После завершения реакции полученную реакционную смесь обрабатывают, как описано выше, и объединенные остатки хроматографируют (элюирование: 0-100% EtOAc:40-60 петролейный эфир, затем 0-20% MeOH:EtOAc), получая продукт в виде твердого вещества коричневого цвета (1,532 г, 18%).(1R)-1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин 4,9-дион. Раствор 1,1-диметилэтил-(1-[(1R)-4,9-диоксо-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин 1-ил]метил-4-пиперидинил)карбамата (1,532 г, 3,83 ммоль) в хлороформе (20 мл) в атмосфере аргона при комнатной температуре обрабатывают 4 М раствором HCl в 1,4-диоксане (10 мл, 40,0 ммоль) и перемешивают при комнатной температуре в течение 0,25 ч. После этого добавляют MeOH (20 мл) и реакционную смесь перемешивают в течение дополнительных 0,25 ч. Затем реакционную смесь упаривают и растирают с Et2O (20 мл). Полученное твердое вещество сушат в вакууме, получая сырой продукт в виде твердого вещества коричневого цвета (1,443 г, 101%).[ES MC] m/z 301 (MH+, 100%). Дигидрохлорид 1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-диона, полученный в соответствии с данной общей методикой (пример 5(a)-(j, анализируют с помощью хиральной ВЭЖХ (Chiralpak AS-H (5 мкм и обнаруживают единственный энантиомер, который предположительно представляет собой R-энантиомер. Сырой дигидрохлорид растворяют в MeOH и воде (1 мл) и вносят в SCX колонку (20 г) (предварительно обработанную MeOH, объем которого равен двум объемам колонки). После этого колонку элюируют (под действием силы тяжести), используя (i) MeOH, (ii) 0,5 М раствор аммиака в MeOH. Подходящие фракции объединяют и упаривают при пониженном давлении, получая 1,0 г (88%) указанного в заголовке соединения в виде твердого вещества бежевого цвета.(1R)-1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-диона (119 мг, 0,396 ммоль) и 5-хлор-6-метил-3-пиридинкарбальдегида (синтез см. в WO 2006/137485 А 1 - пример 256) (61,6 мг, 0,396 ммоль) в смеси ДХМ (1,5 мл) и MeOH (1,5 мл) перемешивают при комнатной температуре в течение 2 ч. Затем добавляют NaBH(OAc)3 (252 мг, 1,189 ммоль) и полученную смесь перемешивают при комнатной температуре. Ход реакции контролируют с помощью ЖХМС и, когда наблюдают полную конверсию, реакцию гасят 10% NaHCO3 и смесь экстрагируют ДХМ. Органический слой сушат (MgSO4), фильтруют и концентрируют. Очистка флэш-хроматографией (Flashmaster II; картридж: силикагель, 5 г; элюирование: смеси ДХМ и MeOH) приводит к получению 88 мг (0,2 ммоль, 51%) ожидаемого продукта в форме свободного основания.[ES MC] m/z 440 (MH+). Полученное свободное основание растворяют в MeOH (9 мл). К раствору по каплям добавляют HCl(4 М раствор в 1,4-диоксане, 0,050 мл, 0,2 ммоль). Смесь перемешивают при комнатной температуре в течение 20 мин и растворитель выпаривают, используя MeOH и гексан в качестве вспомогательных растворителей для удаления диоксана. Гидрохлорид (1R)-1-[(4-[(5-хлор-6-метил-3-пиридинил)метил]амино-1-пиперидинил)метил]-1,2 дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-диона (92,5 мг, 0,194 ммоль, 49,0%) получают в виде твердого не совсем белого вещества. 1- 16018817 Пример 1 А. Синтез соединения примера 1 - гидрохлорида (1R)-1-[(4-[(5-хлор-6-метил-3 пиридинил)метил]амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин 4,9-диона через солевую форму соединения формулы (IIA).(i) Дигидрохлорид (1R)-1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]1,8-нафтиридин-4,9-диона. Раствор 1,1-диметилэтил-(1-[(1R)-4,9-диоксо-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин 1-ил]метил-4-пиперидинил)карбамата (см. пример l(i) выше) (4,282 г, 10,69 ммоль) в CHCl3 (20 мл) в атмосфере Ar при комнатной температуре обрабатывают HCl (4M раствор в 1,4-диоксане, 20 мл, 80 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 15 мин, после этого добавляют MeOH (20 мл). Реакционную смесь перемешивают в течение 15 мин, затем упаривают и остаток растирают с Et2O (20 мл). Полученное твердое вещество сушат в вакууме, получая дигидрохлорид(4,55 г, 12,21 ммоль, 100%) в виде порошка желтого цвета.(ii) Целевое соединение: гидрохлорид (1R)-1-[(4-[(5-хлор-6-метил-3-пиридинил)метил]амино-1 пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-диона. Раствор дигидрохлорида (1R)-1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3ij]-1,8-нафтиридин-4,9-диона (63,3 мг, 0,170 ммоль) и Et3N (0,022 мл, 0,154 ммоль) в CHCl3 (3 мл) перемешивают при комнатной температуре в течение 15 мин, после этого к смеси добавляют 5-хлор-6-метил 3-пиридинкарбальдегид (синтез см. в WO 2006/137485 A1, пример 256) (24 мг, 0,154 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 1 ч, затем добавляютNaBH(OAc)3 (98 мг, 0,463 ммоль) и полученную смесь перемешивают при комнатной температуре в течение ночи. Ход реакции контролируют с помощью ЖХМС, когда наблюдают полную конверсию, реакцию гасят 10% NaHCO3 и смесь несколько раз экстрагируют ДХМ. Органический слой сушат (MgSO4),фильтруют и концентрируют. Очистка флэш-хроматографией (картридж: силикагель, 5 г; элюирование: смеси ДХМ и MeOH), получая 9 мг (13%) ожидаемого продукта в виде свободного основания. Продукт растворяют в MeOH (2 мл). К полученному раствору по каплям добавляют HCl (1 М раствор в Et2O, 0,02 мл, 0,02 ммоль). Смесь перемешивают при комнатной температуре в течение 20 мин, растворитель выпаривают. Гидрохлорид (1R)-1-[(4-[(5-хлор-6-метил-3-пиридинил)метил]амино-1-пиперидинил)метил]1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-диона (92,5 мг, 0,194 ммоль, 49%) получают в виде порошка желтого цвета. 1[ES MC] m/z 440 (MH+). Соединение примера 2. Синтез гидрохлорида (2R)-2-[(4-[(5-хлор-6-метил-3-пиридинил)метил] амино-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона в соответствии со схемой получения (1).(a) 2-[6-(Метилокси)-3-нитро-2-пиридинил]амино-1,3-пропандиол. 6-Метокси-2-хлор-3-нитропиридин (36,94 г, 195,9 ммоль) и 2-аминопропан-1,3-диол (35,65 г, 391,3 ммоль) в течение 3 ч перемешивают в этаноле (500 мл) при кипячении с обратным холодильником в атмосфере аргона. Смеси дают возможность охладиться до комнатной температуры и оставляют на ночь. Растворитель частично удаляют при пониженном давлении (до примерно 150 мл) и полученную взвесь ярко-желтого цвета выливают в ледяную воду (1,5 л) при энергичном перемешивании. Смесь перемешивают в течение 1 ч, затем фильтруют с подсосом при охлаждении. Твердый продукт промывают ледяной водой (200 мл) и сушат на воздухе, получая указанное в заголовке соединение в виде твердого вещества ярко-желтого цвета (45,03 г, 94%). ЖХМС показывает наличие целевого продукта (93%) с примесью 7% исходного вещества. Продукт используют на следующей стадии без дополнительной очистки.(b) N-(2,2-диметил-1,3-диоксан-5-ил)-6-(метилокси)-3-нитро-2-пиридинамин. К 2-[6-(метилокси)-3-нитро-2-пиридинил]амино-1,3-пропандиолу (53,93 г, 228,7 ммоль) в 2,2 диметоксипропане (900 мл) при перемешивании в атмосфере аргона добавляют моногидрат птолуолсульфоновой кислоты (1,00 г). Смесь перемешивают при комнатной температуре в течение ночи,получая прозрачный раствор желтого цвета. Раствор разбавляют ДХМ (1 л) и полученный раствор обрабатывают насыщенным водным раствором NaHCO3 (20 мл) и твердым NaHCO3 (20 г) при энергичном перемешивании (наблюдается вспенивание). Смесь энергично перемешивают в течение 20 мин, после чего оставшуюся воду абсорбируют добавлением безводного Na2SO4. Смесь фильтруют с подсосом и твердые продукты промывают ДХМ (500 мл). Объединенные фильтрат и промывные растворы упаривают при пониженном давлении, получая твердое вещество желтого цвета, в которое добавляют петролейный эфир (40-60), и полученную смесь перемешивают в течение выходных дней. Твердый продукт выделяют фильтрацией с подсосом, промывают петролейным эфиром (40-60) и сушат на воздухе, получая указанное в заголовке соединение в виде твердого вещества ярко-желтого цвета (57,83 г, 92%).N-(2,2-диметил-1,3-диоксан-5-ил)-6-(метилокси)-3-нитро-2-пиридинамин (35,00 г, 123,6 ммоль) разделяют на 2 аликвоты, каждую из которых переносят в 1,4-диоксан (500 мл) и гидрируют над 10% Pdна-угле (паста, 1:1 мас./мас., с водой, 4,00 г) в атмосфере водорода с давлением 1 атм (101325 Па) при комнатной температуре в течение 18 ч. Смеси фильтруют через целит (Celite) с подсосом, используя атмосферу аргона и минимизируя контакт продукта с воздухом. Твердые продукты промывают 1,4 диоксаном и объединенные фильтрат и промывные растворы упаривают при пониженном давлении, получая указанное в заголовке соединение в виде масла насыщенного пурпурного цвета. Полученный продукт сразу используют на следующей стадии.(d) Этил-N-[2-[(2,2-диметил-1,3-диоксан-5-ил)амино]-6-(метилокси)-3-пиридинил]глицинат. Сырой N2-(2,2-диметил-1,3-диоксан-5-ил)-6-(метилокси)-2,3-пиридиндиамин (123,6 ммоль) растворяют в безводном ДМФА (500 мл) в атмосфере аргона, добавляют безводный K2CO3 (37,56 г), затем этилбромацетат (12,31 мл). Смесь перемешивают при комнатной температуре в течение ночи. Растворитель удаляют при пониженном давлении и полученную взвесь красновато-коричневого цвета распределяют между ДХМ (1,2 л) и водой (300 мл). Органическую фазу отделяют и промывают водой (300 мл),сушат над Na2SO4, фильтруют и упаривают при пониженном давлении, получая масло темно-красного цвета, которое переносят в минимальное количество ДХМ и очищают колоночной хроматографией (диоксид кремния; элюирование: 5-60% EtOAc в петролейном эфире (40-60. Подходящие фракции объединяют и упаривают при пониженном давлении, получая указанное в заголовке соединение в виде масла темно-оранжевого цвета (35,42 г, 84%).(e) 4-(2,2-Диметил-1,3-диоксан-5-ил)-6-(метилокси)-1,4-дигидропиридо[2,3-b]пиразин-3(2H)-он. Этил-N-[2-[(2,2-диметил-1,3-диоксан-5-ил)амино]-6-(метилокси)-3-пиридинил]глицинат (35,42 г,104,4 ммоль) растворяют в сухом ТГФ (500 мл) и полученный раствор по каплям в течение 2 ч добавляют к охлажденной суспензии (0C) гидрида натрия (4,173 г, 60% мас./мас., дисперсия в масле) в сухом ТГФ (500 мл) в атмосфере аргона. В процессе добавления цвет суспензии изменяется от оранжевого до зеленого. Смесь перемешивают при 0C в течение дополнительных 15 мин, затем дают возможность смеси нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 1 ч. Смесь охлаждают до 0C и осторожно при энергичном перемешивании добавляют насыщенный растворNH4Cl (15 мл) (наблюдается вспенивание). После прекращения вспенивания полученной смеси дают возможность нагреться до комнатной температуры, смесь перемешивают в течение 4 ч, затем разбавляютEtOAc (500 мл) и фильтруют с подсосом. Твердые продукты промывают EtOAc (300 мл) и объединенные фильтрат и промывные растворы упаривают при пониженном давлении, получая твердое вещество темно-коричневого цвета. Продукт перемешивают с петролейным эфиром (40-60) (500 мл) и EtOAc (20 мл) и фильтруют с подсосом, получая твердое вещество коричневого цвета более светлого оттенка, продукт промывают петролейным эфиром (40-60) (100 мл) и сушат на воздухе, получая указанное в заголовке соединение (25,37 г, 82%).(f) 4-(2,2-Диметил-1,3-диоксан-5-ил)-6-(метилокси)пиридо[2,3-b]пиразин-3(4H)-он. 4-(2,2-Диметил-1,3-диоксан-5-ил)-6-(метилокси)-1,4-дигидропиридо[2,3-b]пиразин-3(2H)-он (25,37 г) и активированный диоксид марганца (120 г) перемешивают в ДХМ (500 мл) при комнатной температуре в течение 2 ч. Смесь фильтруют с подсосом и твердые продукты промывают ДХМ (2100 мл). Объединенные фильтрат и промывные растворы упаривают при пониженном давлении, получая пену коричневого цвета; продукт очищают колоночной хроматографией (диоксид кремния; элюирование: 0-100%EtOAc в петролейном эфире (40-60. Подходящие фракции объединяют и упаривают при пониженном давлении, получая указанное в заголовке соединение в виде твердого вещества светло-желтоватокоричневого цвета (17,40 г, 69%).(200 мл) (в растворе появляются переходящие голубой и зеленый цвета) и затем раствор, который приобрел светло-желтую окраску, перемешивают при комнатной температуре в течение 1 ч. Смесь концентрируют до примерно 300 мл на роторном испарителе, используя охлаждающую водную баню (в осадок выпадает некоторое количество твердого вещества), затем энергично перемешивают с добавлением небольшими порциями твердого гидрокарбоната натрия (осторожно: вспенивание) до достижения значения рН полученной смеси примерно 8. Полученное твердое вещество желтого цвета собирают фильтрацией с подсосом, промывают водой (220 мл) и сушат на воздухе, получая указанное в заголовке соединение в виде аморфного твердого вещества желтого цвета (13,805 г, 91%).(h) (3,8-Диоксо-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-2-ил)метилметансульфонат. В круглодонную колбу объемом 1 л загружают 4-[2-гидрокси-1-(гидроксиметил)этил]-6(метилокси)пиридо[2,3-b]пиразин-3(4H)-он (11,330 г, 45,1 ммоль). Добавляют безводный хлороформ (280 мл), затем Et3N (31,4 мл, 225 ммоль) и ангидрид метансульфоновой кислоты (31,4 г, 180 ммоль), получая раствор темного желтовато-коричневого цвета. В процессе добавления ангидрида метансульфоновой кислоты происходит экзотермическая реакция, теплоты которой достаточно для того, чтобы вызвать вскипание растворителя. Смесь кипятят с обратным холодильником при энергичном перемешивании в атмосфере аргона в течение 4,5 ч. Смеси дают возможность охладиться до комнатной температуры, разбавляют ДХМ до примерно 600 мл и промывают водой (200 мл). Органическую фазу отделяют, водную фазу экстрагируют ДХМ (2200 мл). Объединенные органические экстракты сушат над безводнымNa2SO4, фильтруют и упаривают при пониженном давлении, получая сырой мезилат в виде масла темнокоричневого цвета. Продукт оставляют на ночь при 40-60 под смесью бензина (200 мл) и ДХМ (50 мл). Образующееся твердое вещество выделяют фильтрацией с подсосом, промывают смесью керосин:ДХМ(4:1, 250 мл) и сушат на воздухе, получая указанное в заголовке соединение в виде твердого аморфного вещества коричневого цвета (6,950 г, 52%).(6,950 г, 23,38 ммоль) растворяют в сухом CH3CN (200 мл), полученную смесь обрабатывают пиридином(7,55 мл, 94,0 ммоль) и затем 1,1-диметилэтил-4-пиперидинилкарбаматом (10,30 г, 51,4 ммоль). Смесь кипятят с обратным холодильником при перемешивании в атмосфере аргона в течение 3 ч, затем при 50C в течение выходных. После этого смесь перемешивают при 90C в течение 2 ч, затем летучие соединения удаляют при пониженном давлении и остаток распределяют между ДХМ (600 мл) и водой (100 мл). Органическую фазу отделяют и водную фазу экстрагируют ДХМ (2200 мл). Объединенные органические экстракты промывают водой (2100 мл), сушат над безводнымNa2SO4, фильтруют и упаривают при пониженном давлении, получая твердое вещество темного желтовато-коричневого цвета; продукт переносят в минимальное количество 5% раствора MeOH в ДХМ и хроматографируют (элюирование: 0-10% MeOH в ДХМ). Подходящие фракции объединяют и упаривают при пониженном давлении, получая указанное в заголовке соединение в виде твердого аморфного вещества бледного желтовато-коричневого цвета (5,444 г, 57%).(j) 2-[(4-Амино-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-дион (рацемический и энантиомерный синтез 1 и 2). Способ А (рацемический синтез). 1,1-Диметилэтил-1-[(3,8-диоксо-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-2-ил)метил]-4 пиперидинил)карбамат (1,630 г, 4,06 ммоль) суспендируют в ДХМ (30 мл) и к полученной суспензии добавляют 4 М раствор HCl в 1,4-диоксане (15 мл), получая суспензию ярко-желтого цвета (наблюдают выделение газа). Смесь ярко-желтого цвета выдерживают при комнатной температуре в течение 1 ч. ЖХМС показывает отсутствие исходного вещества. Растворители удаляют при пониженном давлении и остаток сушат при пониженном давлении в течение ночи, получая дигидрохлоридную соль указанного в заголовке соединения в виде твердого аморфного вещества желтовато-коричневого цвета (1,760 г ( теоретического выхода для дигидрохлорида вследствие присутствия растворителя. Часть сырого дигидрохлорида (0,513 г) растворяют в смеси MeOH (4 мл) и воды (1 мл) и вносят вSCX колонку (10 г) (предварительно обработанную MeOH, взятым в количестве, равном 2 объемам колонки). После этого колонку элюируют под действием силы тяжести, используя (i) MeOH (250 мл), (ii) 0,5M аммиак в MeOH (фракции 350 мл). Подходящие фракции объединяют, упаривают при пониженном давлении, получая сырое соединение, указанное в заголовке, в виде твердого аморфного вещества- 19018817 желтовато-коричневого цвета (410 мг), которое содержит не растворимые в MeOH примеси, не выявляемые с помощью ЖХМС (возможно NH4Cl). Продукт встряхивают с MeOH (30 мл) и суспензию фильтруют. Твердый продукт промываютMeOH (20 мл), фильтрат и промывные растворы объединяют и упаривают при пониженном давлении,получая указанное в заголовке соединение (360 мг, 87%).[ES MC] m/z 302 (MH+). Способ В. 1,1-Диметилэтил-1-[(3,8-диоксо-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-2-ил)метил]-4 пиперидинилкарбамат (9,735 г, 24,25 ммоль) суспендируют в ДХМ (90 мл) и к полученной суспензии добавляют 4 М раствор HCl в 1,4-диоксане (45 мл), получая суспензию ярко-желтого цвета (наблюдают выделение газа). Смесь ярко-желтого цвета перемешивают при комнатной температуре в течение 1 ч. Растворители удаляют при пониженном давлении, получая сырой дигидрохлорид в виде твердого аморфного вещества ярко-желтого цвета (10,420 г), который содержит остаточное количество растворителя. Рацемический дигидрохлорид (10,42 г) разделяют на два энантиометра с помощью препаративной ВЭЖХCH3CN:MeOH:изопропиламин). Значениеравно 3,1 и основное разделение наблюдают в продолжении всех 3 циклов. Перекрывающая фракция отсутствует, и оба энантиомера (в виде свободных оснований) выделяют с выходом 99,8% ее каждый.(k) Целевое соединение: гидрохлорид (2R)-2-[(4-[(5-хлор-6-метил-3-пиридинил)метил]амино-1 пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона. Раствор (2R)-2-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8 диона (63,4 мг, 0,210 ммоль) и 5-хлор-6-метил-3-пиридинкарбальдегида (синтез см. в WO 2006/137485 А 1, пример 256) (32,7 мг, 0,210 ммоль) в смеси ДХМ (1,5 мл) и MeOH (0,5 мл) перемешивают при комнатной температуре в течение 30 мин. Затем к смеси добавляют NaBH(OAc)3 (312 мг, 1,473 ммоль) и полученную смесь перемешивают при комнатной температуре. Ход реакции контролируют с помощью ЖХМС и, когда наблюдают полную конверсию, реакцию гасят 10% NaHCO3 и экстрагируют ДХМ. Органический слой сушат (MgSO4), фильтруют и концентрируют. Очистка флэш-хроматографией (Flashmaster II; картридж: силикагель, 2 г; элюирование: смеси ДХМ и MeOH) приводит к получению 34,6 мг (0,078 ммоль, 37%) ожидаемого продукта в виде свободного основания. Продукт растворяют в MeOH (6 мл). К раствору по каплям добавляют HCl (4 М раствор в 1,4-диоксане, 0,019 мл, 0,078 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 35 мин. После этого растворитель выпаривают, используя MeOH и диоксан для способствования удалению диоксана. Гидрохлорид(2R)-2-[(4-[(5-хлор-6-метил-3-пиридинил)метил]амино-1 пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона (60,4 мг, 0,127 ммоль) получают в виде твердого вещества желтого цвета. 1[ES MC] m/z 441 (MH+). Соединение примера 2. Синтез гидрохлорида (2R)-2-[(4-[(5-хлор-6-метил-3-пиридинил)метил] амино-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона с использованием схемы получения (4).- 20018817 порциями в течение 10 мин добавляют Et3N (1,27 л, 9,11 моль). Затем порциями в течение 10 мин добавляют раствор 2-хлор-6-метокси-3-нитропиридина (672,7 г, 3,567 моль) в EtOH (0,6 л). Смесь нагревают до 78C и выдерживают при данной температуре в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и добавляют при перемешивании к смеси лед/Н 2 О (8,4 л). Смесь экстрагируютEtOAc (9 л+6 л+3 л). Экстракты промывают насыщенным раствором соли (6 л), сушат над MgSO4 (900 г), фильтруют и упаривают, получая (2R)-2-[6-(метилокси)-3-нитро-2-пиридинил]амино-3[(фенилметил)окси]-1-пропанол (1373,1 г, 115%) в виде масла оранжевого цвета.(2R)-2-[6-(метилокси)-3-нитро-2-пиридинил]амино-3-[(фенилметил)окси]-1 пропанола (675,8 г) в ДМФА (2,37 л) небольшими порциями добавляют хлор(1,1 диметилэтил)диметилсилан (382,0 г). Затем небольшими порциями добавляют имидазол (331,2 г) и полученную смесь перемешивают при комнатной температуре в течение 1,5 ч. Реакционную смесь упаривают и полученную смолу оранжевого цвета распределяют между EtOAc (3 л) и H2O (3 л). Слои разделяют и водный слой экстрагируют EtOAc (2,5+2 л). Органические слои объединяют, сушат над MgSO4 (500 г),фильтруют и упаривают, получая масло оранжевого цвета. Сырой продукт очищают колоночной хроматографией (диоксид кремния, 10 кг; элюирование: гептан:EtOAc 9:1), получая N-1S)-2-[(1,1 диметилэтил)(диметил)силил]окси-1-[(фенилметил)окси]метилэтил)-6-(метилокси)-3-нитро-2-пиридин (774,7 г, 100%) в виде твердого вещества ярко-желтого цвета.N-1S)-2-[(1,1-диметилэтил)(диметил)силил]окси-1-[(фенилметил)окси]метил этил)-6-(метилокси)-3-нитро-2-пиридинамина (642,6 г, 1,436 моль) в MeOH (12 л) добавляют суспензию порошкообразного цинка (938 г, 14,35 моль) в MeOH (4 л). К смеси по каплям в течение 30 мин добавляют уксусную кислоту (411 мл, 7,18 моль), поддерживая температуру ниже 36C. После завершения добавления полученную смесь перемешивают в течение 15 мин. Реакционную смесь фильтруют через целит и осадок на фильтре промывают EtOAc (8 л). Смесь промывают раствором соли (8 л) и слои разделяют. Водную фазу экстрагируют EtOAc (8+6 л), затем органические фазы объединяют, промывают насыщенным раствором NaHCO3 (8 л), раствором соли (8 л), сушат над MgSO4 (1 кг) и фильтруют. Фильтрат выпаривают, получая 478 г (80%) N2-1S)-2-[(1,1-диметилэтил)(диметил)силил]окси-1[(фенилметил)окси]метилэтил)-6-(метилокси)-2,3-пиридиндиамин в виде вязкого масла темнокрасного цвета.(d) Этил N-[2-[1S)-2-[(1,1-диметилэтил)(диметил)силил]окси-1-[(фенилметил)окси]метил этил)амино]-6-(метилокси)-3-пиридинил]глицинат. К раствору N2-1S)-2-[(1,1-диметилэтил)(диметил)силил]окси-1-[(фенилметил)окси]метил этил)-6-(метилокси)-2,3-пиридиндиамина (673,1 г, 1,612 моль) в ДМФА (10 л) добавляют K2CO3 (490,1 г,3,546 моль). К смеси в течение 15 мин добавляют бромацетат этила (196,6 мл, 1,77 моль) и реакционную смесь перемешивают при комнатной температуре. Через 3 ч реакцию гасят H2O (10 л) и смесь экстрагируют EtOAc (10 л). Слои разделяют и водный слой экстрагируют EtOAc (7 л). Органические слои объединяют, промывают H2O (7 л) и раствором соли (7 л), сушат над MgSO4 (1 кг), фильтруют и упаривают. Полученное темное масло (853 г) очищают колоночной хроматографией (диоксид кремния, 9 кг; элюирование: гептан и EtOAc), получая этил-N-[2-[1S)-2-[(1,1-диметилэтил)(диметил)силил]окси-1[(фенилметил)окси]метилэтил)амино]-6-(метилокси)-3-пиридинил]глицинат (672,0 г, 83%) в виде темного масла.(e) 4-1S)-2-([(1,1-Диметилэтил)(диметил)силил]окси)-1-[(фенилметил)окси]метилэтил)-6(метилокси)-1,4-дигидропиридо[2,3-b]пиразин-3(2H)-он. К раствору NaH (50,5 г, 57-63% дисперсия в минеральном масле, 1,26 моль) в ТГФ (8,5 л) добавляют этил-N-[2-[1S)-2-[(1,1-диметилэтил)(диметил)силил]окси-1-[(фенилметил)окси]метилэтил) амино]-6-(метилокси)-3-пиридинил]глицинат (577,8 г, 1,147 моль) при -5C. После завершения добавления полученную смесь нагревают до комнатной температуры и перемешивают в течение 30 мин. Реакцию гасят добавлением насыщенного водного раствора NH4Cl (610 мл) и полученную смесь перемешивают в течение 10 мин. К смеси добавляют Na2SO4 (1 кг), полученную смесь перемешивают в течение 10 мин и затем фильтруют через целит. Осадок на фильтре промывают EtOAc (5 л). Фильтрат выпаривают и полученное масло темно-пурпурного цвета очищают колоночной хроматографией (элюирование: смеси гептана и EtOAc), получая 477 г (91%) 4-1S)-2-[(1,1-диметилэтил)(диметил)силил]окси-1[(фенилметил)окси]метилэтил)-6-(метилокси)-1,4-дигидропиридо[2,3-b]пиразин-3(2H)-он в виде вязкого масла пурпурного цвета.- 21018817 бар (500000 Па) в течение 3,5 ч. Реакционную смесь фильтруют через целит и фильтрат упаривают, получая 4-[(1S)-2-[(1,1-диметилэтил)(диметил)силил]окси-1-(гидроксиметил)этил]-6-(метилокси)-1,4 дигидропиридо[2,3-b]пиразин-3(2H)-он (191,2 г, 95%) в виде твердого вещества темно-голубого цвета.(g) 4-[(1S)-2-([(1,1-Диметилэтил)(диметил)силил]окси)-1-(гидроксиметил)этил]-6-(метилокси)пиридо[2,3-b]пиразин-3(4H)-он. Раствор 4-[(1S)-2-[(1,1-диметилэтил)(диметил)силил]окси-1-(гидроксиметил)этил]-6-(метилокси)-1,4 дигидропиридо[2,3-b]пиразин-3(2H)-он (494,1 г, 1,34 моль) и MnO2 (584,4 г, 6,72 моль) в ДХМ (18 л) перемешивают при комнатной температуре в течение 1 ч. Смесь фильтруют через целит и фильтрат упаривают, получая твердое вещество пурпурного/белого цвета. Сырой продукт очищают колоночной хроматографией(h) (2S)-2-([(1,1-Диметилэтил)(диметил)силил]оксиметил)-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-дион. К раствору 4-[(1S)-2-[(1,1-диметилэтил)(диметил)силил]окси-1-(гидроксиметил)этил]-6(метилокси)пиридо[2,3-b]пиразин-3(4H)-она (1 г, 2,74 ммоль) в CHCl3 (50 мл) добавляют Et3N (0,953 мл,6,84 ммоль) и затем раствор ангидрида метансульфоновой кислоты (0,953 г, 5,47 ммоль) в CHCl3 (50 мл). Реакционную смесь кипятят с обратным холодильником при перемешивании в течение 5 ч. Сырую реакционную смесь разбавляют водой, промывают раствором NaCl, сушат (MgSO4), фильтруют и концентрируют. Очистка флэш-хроматографией (картридж: силикагель, 40 г; элюирование: смеси ДХМ и MeOH),получая (2S)-2-([(1,1-диметилэтил)(диметил)силил]оксиметил)-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-дион (0,806 г, 88%) в виде твердого вещества желтовато-коричневого цвета. 1(2S)-2-([(1,1-диметилэтил)(диметил)силил]оксиметил)-1,2-дигидро-3H,8H-2 а,5,8 атриазааценафтилен-3,8-диона (42,6 г, 128 ммоль) и ионообменной смолы Dowex 50X2-200 (50 г, 128 ммоль) в MeOH (1000 мл) перемешивают при комнатной температуре в течение 15 ч. Реакционную смесь фильтруют, получая (2S)-2-(гидроксиметил)-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-дион(39,6 мл, 284 ммоль). Реакционную смесь осторожно кипятят с обратным холодильником при 42C в течение 2 ч. Затем реакционную смесь охлаждают до 5C и к смеси по каплям добавляют 2(диметиламино)этанол (7,30 мл, 72,5 ммоль). Неочищенную смесь перемешивают при комнатной температуре в течение 30 мин, затем добавляют ТВМЕ (600 мл). Полученный твердый продукт собирают фильтрацией. Твердый продукт суспендируют в 100 мл 1N HCl, промывают водой и ТВМЕ и сушат, получая [(2S)-3,8-диоксо-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-2-ил]метилбензолсульфонат (37,04 г, 85%) в виде твердого вещества светло-желтого цвета. 1(k) Целевое соединение: гидрохлорид (2R)-2-[(4-[(5-хлор-6-метил-3-пиридинил)метил]амино-1 пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона. К суспензии [(2S)-3,8-диоксо-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-2-ил]метилбензолсульфонат (14,5 г, 40,3 ммоль) в CH3CN (100 мл) при 0C добавляют N-[(5-хлор-6-метил-3 пиридинил)метил]-4-пиперидинамин (синтез см. в примере получения 11) (11,61 г, 48,4 ммоль) и N,Nдиизопропилэтиламин (7,03 мл, 40,3 ммоль). Реакционную смесь перемешивают при 77C в течение 6 ч. Реакционной смеси дают возможность охладиться до комнатной температуры и смесь охлаждают до 0C на ледяной бане. Выпавшее в осадок твердое вещество собирают фильтрацией в вакууме и промываютCH3CN, получая первый фильтрат. Затем выделенное твердое вещество снова промывают Н 2 О и ТВМЕ,получая второй фильтрат. Полученный твердый продукт собирают, получая первую порцию (2R)-2-[(4-[(5-хлор-6-метил-3 пиридинил)метил]амино-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8 диона (3,5 г, 20%), который представляет собой твердое вещество желтого цвета. Первый фильтрат концентрируют в вакууме и растворяют в ДХМ. К полученному раствору добав- 22018817 ляют 2N HCl до достижения значения pH в интервале 1-2 и органическую фазу экстрагируют и утилизируют. К водной фазе добавляют 10% раствор NaHCO3 до достижения рН значения 9, органическую фазу экстрагируют ДХМ, промывают раствором NaCl, сушат (MgSO4), фильтруют и концентрируют. Очистка флэш-хроматографией (картридж: силикагель, 50 г; элюирование: смеси ДХМ и MeOH) приводит к получению второй порции (2R)-2-[(4-[(5-хлор-6-метил-3-пиридинил)метил]амино-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона (2,09 г, 12%). Второй фильтрат экстрагируют ДХМ и ДХМ/MeOH 95/5, сушат (Na2SO4), фильтруют и концентрируют,получая третью порцию(2R)-2-[(4-[(5-хлор-6-метил-3-пиридинил)метил]амино-1 пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона в форме свободного основания (7,75 г, 44%). Общий выход 76%.[ES MC] m/z 441 (MH+). К раствору свободного основания (3,5 г, 7,94 ммоль) в ДХМ (80 мл) по каплям добавляют HCl (4 М раствор в 1,4-диоксане, 2,0 мл, 8,0 ммоль). Смесь перемешивают при комнатной температуре в течение 1 ч, растворитель выпаривают в вакууме и сырой продукт диспергируют в смеси гексан/ДХМ. Твердый продукт собирают фильтрацией, получая гидрохлорид (2R)-2-[(4-[(5-хлор-6-метил-3-пиридинил)метил] амино-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона (3,2 г, 84%) в виде твердого вещества желтого цвета. 1(1R)-1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-диона (см. выше стадию (j) примера соединения 1; 400 мг, 1,33 ммоль) и 6-(трифторметил)-3 пиридинкарбальдегида (233 мг, 1,33 ммоль, от Apollo) в смеси ДХМ (5 мл) и MeOH (5 мл) перемешивают при комнатной температуре в течение 2 ч. После этого добавляют NaBH(OAc)3 (1,7 г, 7,99 ммоль) и полученную смесь перемешивают при комнатной температуре. Ход реакции контролируют с помощью ЖХМС. Добавляют дополнительное количество альдегида (47 мг, 0,27 ммоль) и NaHB(OAc)3 (421 мг,1,98 ммоль) и, когда наблюдают полную конверсию, реакцию гасят 10% NaHCO3 и экстрагируют ДХМ. Органический слой сушат (MgSO4), фильтруют и концентрируют. Очистка флэш-хроматографией (картридж: силикагель, 70 г; элюирование: смеси ДХМ и MeOH) приводит к получению 463 мг (75%) ожидаемого продукта в форме свободного основания. Продукт растворяют в MeOH (50 мл). К раствору по каплям добавляют HCl (4 М раствор в 1,4-диоксане, 0,25 мл, 1 ммоль). Смесь перемешивают при комнатной температуре в течение 20 мин, растворитель выпаривают. Получают гидрохлорид (1R)-1-[4-([6(трифторметил)-3-пиридинил]метиламино)-1-пиперидинил]метил-1,2-дигидро-4H,9H-имидазо[1,2,3ij]-1,8-нафтиридин-4,9-диона (238 мг, 0,480 ммоль, 36%) в виде твердого вещества бежевого цвета. 1[ES MC] m/z 460 (MH+). Пример соединения 11. Синтез (1R)-1-[4-([6-(трифторметил)-3-пиридинил]метиламино)-1 пиперидинил]метил-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-дион в соответствии со схемой получения (2)(a) 6-(Метилокси)-3-нитро-2-пиридин. Раствор/суспензию 2-хлор-6-(метилокси)-3-нитропиридина (65,7 г, 348 ммоль) в 2 М растворе аммиака в MeOH (500 мл, 1000 ммоль) и водном растворе аммиака (500 мл, 348 ммоль) перемешивают при 65C в течение 18 ч. Реакционный раствор охлаждают, твердый продукт собирают фильтрацией, промывают водой (2100 мл). Твердый продукт сушат в вакуумной печи при 40C в течение ночи, получая продукт в виде твердого вещества ярко-желтого цвета (52,14 г, 84% чистота в соответствии с данными ЯМР, 74%).(b) 6-(Метилокси)-2,3-пиридиндиамин. 6-(Метилокси)-3-нитро-2-пиридинамин (26 г, 129 ммоль) суспендируют в EtOH (500 мл) при комнатной температуре в атмосфере аргона и затем обрабатывают палладием-на-угле (15 г, 14,10 ммоль)(10% паста). Реакционную смесь перемешивают в атмосфере водорода с давлением 1 атм (101325 Па) в течение ночи. Реакционную смесь фильтруют через тонкий слой целита (Celite) и осадок на фильтре промывают EtOH (500 мл). EtOH выпаривают, получая продукт в виде масла пурпурного цвета (20,68 г,слабо загрязненное).CH3CN (500 мл) при комнатной температуре в атмосфере аргона и затем обрабатывают K2CO3 (24,38 г,176 ммоль) и этилбромацетатом (18,13 мл, 163 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. После этого CH3CN удаляют в вакууме. Реакцию повторяют, используя дополнительное количество 6-(метилокси)-2,3-пиридиндиамина (20,68 г, чистота 87%, 129 ммоль) вCH3CN (500 мл), K2CO3 (23,23 г) и этилбромацетата (17,27 г), реакционную смесь снова перемешивают при комнатной температуре в течение ночи и после этого CH3CN удаляют в вакууме. Остатки распределяют между водой (1 л) и EtOAc (1 л) и слои разделяют. Водный слой экстрагируют еще раз EtOAc (1 л) и объединенные органические экстракты сушат над MgSO4, фильтруют и упаривают, получая масло пурпурного цвета (64 г). Масло обрабатывают ДХМ (300 мл) и нерастворимые примеси удаляют фильтрацией. Раствор в ДХМ загружают в колонку (диоксид кремния, 800 г; элюирование: 0-2% MeOH/ДХМ), получая 40,6 г указанного в заголовке продукта в виде твердого вещества коричневого цвета (ЖХМС и ЯМР подтверждают 75% содержание указанного в заголовке соединения и 15% циклизованного продукта 6-(метилокси)-1,4-дигидропиридо[2,3-b]пиразин-3(2H)-она) и 6,4 г циклизованного продукта 6(метилокси)-1,4-дигидропиридо[2,3-b]пиразин-3(2H)-она в виде твердого вещества пурпурного цвета.(d) 6-(Метилокси)-1,4-дигидропиридо[2,3-b]пиразин-3(2H)-он. Этил-N-[2-амино-6-(метилокси)-3-пиридинил]глицинат (40,6 г, 135 ммоль) растворяют в ТГФ (1 л) при комнатной температуре в атмосфере аргона и обрабатывают трет-бутоксидом калия (15,17 г, 135 ммоль). Смесь перемешивают в течение 2 ч при комнатной температуре, затем добавляют насыщенный раствор NH4Cl (500 мл) и ТГФ выпаривают. К остатку добавляют воду (500 мл), затем 20% MeOH/ДХМ(1 л); нерастворимые вещества собирают фильтрацией, промывают Et2O и сушат в вакуумной печи при 40C в течение ночи, получая целевой продукт в виде твердого вещества желтого цвета (15,3 г): ЖХМС и ЯМР подтверждают данный продукт (в соответствии с данными ЯМР присутствует 9% окисленного соединения). Две фазы переносят в делительную воронку и разделяют. Водный слой дважды экстрагируют дополнительным количеством 20% MeOH/ДХМ (2500 мл), объединенные органические экстракты сушат над MgSO4, фильтруют и упаривают, полученное твердое вещество коричневого цвета промывают избытком Et2O, получая дополнительное количество указанного в заголовке соединения в виде твердого вещества бледно-зеленого цвета (7,7 г): ЖХМС и ЯМР подтверждают данный продукт (в соответствии с данными ЯМР присутствует 20% окисленного вещества).[ES MC] m/z 180 (MH+). Альтернативная методика. Этил-N-[2-амино-6-(метилокси)-3-пиридинил]глицинат (16,2 г, 72 ммоль) растворяют в тетрагидрофуране (500 мл) и охлаждают до 0C (охлаждение на ледяной бане) в атмосфере аргона. После этого раствор обрабатывают трет-бутоксидом калия (1 М в ТГФ, 80 мл, 80 ммоль). Спустя 1,5 ч реакционную- 24018817 смесь обрабатывают АсОН (80 ммоль) и упаривают, получая твердое вещество темной окраски. Продукт растирают с водой (200 мл), фильтруют и сушат в вакууме (13 г, количественный выход), можно использовать на следующей стадии без дополнительной очистки.(600 мл)/NaHCO3 (насыщенный раствор) (200 мл) при энергичном перемешивании при комнатной температуре добавляют бензилхлорформиат (5,31 мл, 37,2 ммоль). Спустя 45 мин реакцию завершают. Слои разделяют и органический слой сушат над MgSO4, фильтруют и упаривают, получая указанный в заголовке продукт в виде твердого не совсем белого вещества (11 г, 99%).(f) Фенилметил-6-(метилокси)-4-[(2R)-2-оксиранилметил]-3-оксо-3,4-дигидропиридо[2,3-b]пиразин 1(2H)-карбоксилат. Фенилметил-6-(метилокси)-3-оксо-3,4-дигидропиридо[2,3-b]пиразин-1(2H)-карбоксилат (11 г, 35,1 ммоль) растворяют в ДМФА (300 мл) при комнатной температуре в атмосфере аргона, получая раствор желтого цвета. Полученный раствор охлаждают на ледяной бане и обрабатывают гидридом натрия (1,685 г, 42,1 ммоль). Раствору дают возможность нагреться до комнатной температуры. Спустя 20 мин к раствору добавляют (2S)-2-оксиранилметил-3-нитробензолсульфонат (9,56 г, 36,9 ммоль). Спустя 1 ч все исходные вещества полностью расходуются, поэтому реакционную смесь обрабатывают насыщенным раствором NaHCO3 (350 мл) и водный слой экстрагируют ДХМ (3400 мл). Объединенные органические слои сушат над MgSO4, фильтруют и упаривают, получая масло светло-коричневого цвета (16,93 г). Продукт используют на следующей стадии без дополнительной очистки.(g) Фенилметил-(1S)-1-(гидроксиметил)-3,8-диоксо-1,2,3,4-тетрагидро-5H,8H-2 а,5,8 а-триазааценафтилен-5-карбоксилат. Фенилметил-6-(метилокси)-4-[(2R)-2-оксиранилметил]-3-оксо-3,4-дигидропиридо[2,3-b]пиразин 1(2H)-карбоксилат (сырой продукт, 15,93 г, приблизительно 32,8 ммоль) растворяют в ДМФА (250 мл) при комнатной температуре, раствор нагревают до 130C и выдерживают при данной температуре в течение 2 ночей и затем при 120C в течение одной ночи. Реакция завершается, ДМФА выпаривают и остаток обрабатывают смесью вода/раствор соли (350/50 мл) и ДХМ (500 мл). Слои разделяют и водный слой экстрагируют еще раз ДХМ (500 мл). Объединенные органические экстракты сушат над MgSO4, фильтруют и упаривают до масла коричневого цвета, которое сушат в высоком вакууме в течение выходных. Сырой продукт очищают хроматографией (диоксид кремния; элюирование с градиентом: 0-10%MeOH/ДХМ), получая указанное в заголовке соединение в виде золотистой пены (3,6 г, 30,9%).(h) (1S)-1-(Гидроксиметил)-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-дион. Фенилметил-(1S)-1-(гидроксиметил)-3,8-диоксо-1,2,3,4-тетрагидро-5H,8H-2 а,5,8 а-триазааценафтилен-5-карбоксилат (1,6 г, 4,50 ммоль) растворяют в этаноле (100 мл) при комнатной температуре и затем обрабатывают палладием-на-угле (10% паста) (1 г, 0,940 ммоль). Смесь перемешивают при комнатной температуре в атмосфере водорода с давлением 1 атм (101325 Па) в течение 3 ч. После этого реакционную смесь фильтруют через тонкий слой целита и осадок на фильтре промывают дополнительным количеством этанола. Продукт элюируют ДМФА (400 мл) и ДМФА выпаривают, получая твердое вещество коричневого цвета (780 мг). Твердый продукт суспендируют в 30% MeOH/ДХМ (150 мл), перемешивают с диоксидом марганца (1,174 г, 13,51 ммоль) при комнатной температуре в течение 5 ч и затем фильтруют через тонкий слой целита, который промывают 20% MeOH/ДХМ (100 мл). Растворители выпаривают, получая указанное в заголовке соединение в виде твердого вещества коричневого цвета (750 мг, 76%).(1S)-1-(гидроксиметил)-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-дион (750 мг, 3,42 ммоль) суспендируют в сухом ДХМ (100 мл) при комнатной температуре в атмосфере аргона и затем обрабатывают Et3N (0,572 мл, 4,11 ммоль). После этого смесь охлаждают на бане со смесью воды со льдом. После этого добавляют метансульфонилхлорид (0,293 мл, 3,76 ммоль) и реакционной смеси дают возможность нагреться до комнатной температуры. Спустя 50 мин исходное вещество полностью расходуется, полученную смесь промывают насыщенным раствором NaHCO3 (100 мл). Водный слой экстрагируют смесью 20% MeOH/ДХМ (2100 мл); объединенные органические экстракты сушат над MgSO4,фильтруют и упаривают, получая продукт в виде пены коричневого цвета (1,05 г, 90% чистоты в соответствии с ЖХМС).[(1S)-3,8-диоксо-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-1-ил]метилметансульфо- 25018817 ната (1,05 г, 3,53 ммоль) в сухом CH3CN (50 мл) при комнатной температуре в атмосфере аргона обрабатывают пиридином (0,343 мл, 4,24 ммоль), затем 1,1-диметилэтил-4-пиперидинилкарбаматом (0,884 г,4,24 ммоль). Смесь нагревают до 70C и выдерживают при данной температуре в течение 1,5 ч, затем при 90C в течение 3 ч. ЖХМС показывает наличие 25% продукта. Поэтому добавляют 0,5 экв. пиридина и 0,5 экв. 1,1-диметилэтил-4-пиперидинилкарбамата, реакционную смесь нагревают до 90C и выдерживают при данной температуре в течение ночи, затем перемешивают при комнатной температуре в течение 2 дней. Реакцию завершают. Растворитель выпаривают и остаток распределяют между насыщенным раствором NaHCO3 и 20% MeOH/ДХМ (100 мл/100 мл). Слои разделяют и водный слой снова экстрагируют 20% MeOH/ДХМ (2100 мл). Объединенные органические экстракты сушат над MgSO4, фильтруют и упаривают, получая 1,7 г сырого продукта, который очищают хроматографией (диоксид кремния; элюирование с градиентом: 0-5% MeOH/ДХМ), получая продукт в виде твердого вещества желтого цвета(1R)-1-[(4-Амино-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8 дион. Раствор 1,1-диметилэтил-(1-[(1R)-3,8-диоксо-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-1 ил]метил-4-пиперидинил)карбамата (0,57 г, 1,420 ммоль) в хлороформе (7 мл) при комнатной температуре обрабатывают HCl (4 М раствор в 1,4-диоксане, 7 мл). Твердое вещество выпадает в осадок и полученную смесь перемешивают при комнатной температуре. Спустя 0,5 ч добавляют некоторое количествоMeOH для растворения большей части твердого продукта, затем толуол и все растворители удаляют, получая продукт в виде твердого вещества желтого цвета. Сырой дигидрохлорид растворяют в смеси MeOH (4 мл) и воды (1 мл) и вносят в SCX колонку (10 г)(предварительно обработанную MeOH, взятым в количестве, равном 2 объемам колонки). После этого колонку элюируют (i) MeOH (250 мл), (ii) 0,5 М аммиака в MeOH (350 мл). Подходящие фракции объединяют и упаривают при пониженном давлении, получая 385 мг (90%) указанного в заголовке соединения.(l) Целевое соединение примера (1R)-1-[4-([6-(трифторметил)-3-пиридинил]метиламино)-1 пиперидинил]метил-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-дион. Раствор (1R)-1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8 диона (10,7 мг, 0,036 ммоль) и 6-(трифторметил)-3-пиридинкарбальдегида (6,22 мг, 0,036 ммоль) в смеси ДХМ (0,5 мл) и MeOH (0,5 мл) перемешивают при комнатной температуре в течение ночи. После этого добавляют NaBH(OAc)3 (22,58 мг, 0,107 ммоль). Полученную смесь перемешивают при комнатной температуре, добавляют дополнительное количество NaBH(OAc)3 в виде нескольких порций до тех пор, пока исходное вещество не исчезнет в соответствии с ЖХМС. Реакцию гасят добавлением 10% NaHCO3 и экстрагируют ДХМ. Органический слой сушат (MgSO4), фильтруют и концентрируют. Сырую реакционную смесь очищают флэш-хроматографией (Flashmaster II; картридж: силикагель,500 мг; элюирование: смеси ДХМ и MeOH). Полученный твердый продукт растирают с гексаном и фильтруют с использованием вакуума, получая 4 мг (8,69 мкмоль, 24,47%) (1R)-1-[4-([6(трифторметил)-3-пиридинил]метиламино)-1-пиперидинил]метил-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона в виде твердого вещества коричневого цвета. 1[ES MC] m/z 461 (MH+). Соединение примера 12. Синтез гидрохлорида (1R)-1-[(4-[(5-бром-6-метил-3-пиридинил)метил] амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-диона в соответствии со схемой получения (3)(1R)-1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8 нафтиридин-4,9-диона (синтез см. в примере 1(j), 66,1 мг, 0,220 ммоль) и 5-бром-6-метил-3 пиридинкарбальдегида (синтез см. в примере получения 7 ниже) (44 мг, 0,172 ммоль) в смеси ДХМ (2,0 мл) и MeOH (0,1 мл) перемешивают при комнатной температуре в течение ночи. Затем к смеси добавляют NaBH(OAc)3 (140 мг, 0,661 ммоль) и смесь перемешивают при комнатной температуре. Ход реакции- 26018817 контролируют с помощью ЖХМС и, когда наблюдают полную конверсию, реакцию гасят 10% NaHCO3 и экстрагируют ДХМ. Органический слой сушат (MgSO4), фильтруют и концентрируют. Очистка флэш-хроматографией (Flashmaster II; картридж: силикагель, 2 г; элюирование: смеси гексана и EtOAc, затем смесь ДХМ и MeOH) приводит к получению 69,5 мг (0,14 ммоль, 83%) ожидаемого продукта в виде свободного основания. Продукт растворяют в MeOH (3 мл). К раствору по каплям добавляют HCl (3 М раствор в MeOH, 38 мкл, 0,114 ммоль). Смесь перемешивают при комнатной температуре в течение 20 мин, растворитель выпаривают. Гидрохлорид (1R)-1-[(4-[(5-бром-6-метил-3 пиридинил)метил]амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин 4,9-диона (47 мг, 0,09 ммоль, 80%) получают в виде твердого вещества бледно-желтого цвета. 1[ES MC] m/z 484 (MH+). Соединение примера 17. Синтез 2-[4-([6-(трифторметил)-3-пиридазинил]метиламино)-1 пиперидинил]метил-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-дион в соответствии со схемой получения (1)(синтез см. в примере 2(j (56,3 мг, 0,187 ммоль), K2CO3 (34,4 мг, 0,249 ммоль) и 3-(бромметил)-6(трифторметил)пиридазина (синтез см. в примере получения 1 ниже, 30 мг, 0,124 ммоль) в CH3CN (1 мл) и MeOH (0,1 мл) перемешивают при комнатной температуре в течение ночи. ЖХМС показывает присутствие в смеси оставшегося аминпроизводного. Добавляют избыток 3-(бромметил)-6-(трифторметил)пиридазина (25 мг). Смесь перемешивают при комнатной температуре в течение 1 ч. Твердый продукт отфильтровывают и растворитель выпаривают. Очистка флэш-хроматографией (Flashmaster II; элюирование: смеси ДХМ и MeOH) приводит к получению 2-[4-([6-(трифторметил)-3-пиридазинил]метиламино)-1-пиперидинил]метил-1,2-дигидро 3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона (17 мг, 30%). 1[ES MC] m/z 462 (MH+). Соединение примера 51. Синтез гидрохлорида (2R)-2-[(4-[(5-бром-6-метил-3-пиридинил)метил] амино-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона в соответствии со схемой получения (1). Раствор (2R)-2-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8 диона (синтез см. в примере 2(j), 187 мг, 0,622 ммоль) и 5-бром-6-метил-3-пиридинкарбальдегида (синтез см. в примере получения 7 ниже) (97 мг, 0,485 ммоль) в смеси ДХМ (10 мл) и MeOH (0,1 мл) перемешивают при комнатной температуре в течение ночи. Затем к смеси добавляют NaBH(OAc)3 (396 мг, 1,867 ммоль) и смесь перемешивают при комнатной температуре. Ход реакции контролируют с помощью ЖХМС и, когда наблюдают полную конверсию, реакцию гасят 10% NaHCO3 и экстрагируют ДХМ. Органический слой сушат (MgSO4), фильтруют и концентрируют. Очистка флэш-хроматографией (Flashmaster II; картридж: силикагель, 5 г; элюирование: смеси ДХМ и MeOH) приводит к получению 140 мг (0,288 ммоль, 60%) ожидаемого продукта в виде свободного основания. Продукт растворяют в MeOH (10 мл). К раствору по каплям добавляют HCl (3 М раствор вMeOH, 0,071 мл, 0,213 ммоль). Смесь перемешивают при комнатной температуре в течение 20 мин, рас- 27018817 творитель выпаривают. Гидрохлорид (2R)-2-[(4-[(5-бром-6-метил-3-пиридинил)метил]амино-1 пиперидинил)метил]-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона (95 мг, 0,173 ммоль, 81%) получают в виде твердого вещества бледно-желтого цвета. 1[ES MC] m/z 485 (MH+). Соединение примера 59. Синтез гидрохлорида (1R)-1-[4-([6-(трифторметил)-3-пиридинил]метил амино)-1-пиперидинил]метил-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-диона в соответствии со схемой (3).(1R)-1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8 нафтиридин-4,9-диона (синтез см. в примере l(j), 400 мг, 1,33 ммоль) и 6-(трифторметил)-3 пиридинкарбальдегида (233 мг, 1,33 ммоль) в смеси ДХМ (5 мл) и MeOH (5 мл) перемешивают при комнатной температуре в течение 2 ч. Затем к смеси добавляют NaBH(OAc)3 (1,7 г, 7,99 ммоль) и полученную смесь перемешивают при комнатной температуре. Ход реакции контролируют с помощью ЖХМС. Добавляют дополнительное количество альдегида (47 мг, 0,27 ммоль) и NaBH(OAc)3 (421 мг, 1,98 ммоль) и, когда наблюдают полную конверсию, реакцию гасят 10% NaHCO3 и экстрагируют ДХМ. Органический слой сушат (MgSO4), фильтруют и концентрируют. Очистка флэш-хроматографией (картридж: 70 г,силикагель; элюирование: смеси ДХМ и MeOH) приводит к получению 463 мг (75%) предполагаемого продукта в виде свободного основания. Продукт растворяют в MeOH (50 мл). К раствору по каплям добавляют HCl (4 М раствор в 1,4-диоксане, 0,25 мл, 1 ммоль). Смесь перемешивают при комнатной температуре в течение 20 мин, растворитель выпаривают. Гидрохлорид (1R)-1-[4-([6-(трифторметил)-3 пиридинил]метиламино)-1-пиридинил]метил-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9 диона (238 мг, 0,480 ммоль, 36%) получают в виде твердого вещества бежевого цвета. 1(1R)-1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9-диона (пример 1 стадия (j): 70,5 мг, 0,235 ммоль), 5-метил-6-(трифторметил)-3 пиридинкарбальдегида (пример получения 12, 37 мг, 0,196 ммоль) и сульфата магния (безводный) (58,9 мг, 0,489 ммоль) в ДХМ (7 мл) перемешивают в течение ночи при комнатной температуре. ЖХМС сырого продукта показывает наличие в нем имина и отсутствие исходного вещества. Время удерживания целевого соединения равно 2,59. [M+H]+ 472,2. Затем добавляют триацетоксиборгидрид натрия (124 мг,0,587 ммоль) и полученную смесь перемешивают в течение 3 ч. ЖХМС показывает наличие имина, поэтому добавляют 1 экв. триацетоксиборгидрида натрия (41,5 мг, 0,196 ммоль). После этого в органическую фазу добавляют насыщенный водный раствор бикарбоната натрия до достижения pH значения 9. Органическую фазу экстрагируют ДХМ и отделяют, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией (Flash-Master,картридж: силикагель, 2 г; элюирование: ДХМ/MeOH 5%), получая три порции продукта (9,3 мг). 1 Н ЯМР подтверждает наличие указанного в заголовке соединения, и ЖХМС также согласуется с наличием указанного в заголовке соединения. [M+H]+ 474, время удерживания = 1,81, чистота 95%. Соединение примера 71. Синтез (2-хлор-4-[(1-[(1R)-4,9-диоксо-1,2-дигидро-4H,9H-имидазо[1,2,3ij]-1,8-нафтиридин-1-ил]метил-4-пиперидинил)амино]метилфенил)метилацетата (последняя стадия). Данный пример иллюстрирует применение соединения формулы (IIB), в котором W представляет собой бромметильный фрагмент.(1R)-1-[(4-амино-1-пиперидинил)метил]-1,2-дигидро-4H,9H-имидазо[1,2,3-ij]-1,8-нафтиридин-4,9 дион (пример 1, стадия l(j), 34 мг, 0,113 ммоль) растворяют в ацетонитриле (5 мл), к полученному раствору добавляют [4-(бромметил)-2-хлорфенил]метилацетат (34,6 мг, 0,125 ммоль), карбонат калия (31,3 мг, 0,226 ммоль) и иодид натрия (0,848 мг, 5,66 мкмоль) и реакционную смесь оставляют на ночь при комнатной температуре. На следующее утро ЖХМС показывает полную конверсию исходного вещества,растворитель удаляют в вакууме. Полученный сырой продукт растворяют в ДХМ и несколько раз промывают водой. Органические слои сушат над сульфатом натрия, фильтруют и летучие растворители уда- 28018817 ляют в вакууме. Очистку проводят с помощью флэш-хроматографии (flash master II, Si II 2 г; элюирование: ДХМ-MeOH (0-20%, получая 10 мг указанного в заголовке соединения в виде твердого вещества желтого цвета. Соединение примера 75. Синтез гидрохлорида (2R)-2-[4-([5-метил-6-(трифторметил)-3 пиридинил]метиламино)-1-пиперидинил]метил-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8 диона. К раствору(2R)-2-[4-([5-метил-6-(трифторметил)-3-пиридинил]метиламино)-1-пиперидинил]метил-1,2-дигидро-3H,8H-2 а,5,8 а-триазааценафтилен-3,8-диона (соединение примера 60: 80 мг,0,169 ммоль) в ДХМ (2 мл) при 0C добавляют HCl (4 М в 1,4-диоксане) (0,042 мл, 0,169 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 40 мин. Затем растворитель выпаривают в вакууме и сырой продукт диспергируют в смеси гексан/ДХМ. Твердый осадок выделяют фильтрацией в вакууме и промывают ДХМ и гексаном, получая 42 мг твердого вещества бежевого цвета. 1H ЯМР подтверждает указанный в заголовке продукт. ЖХМС подтверждает укзанный в заголовке продукт[M+H]+ 475, время удерживания (RT) = 2,58, чистота 95%. В табл. 1 показан источник соединения формулы IIB, используемый при получении соединений примеров. В тех случаях, когда эти соединения являются неизвестными соединениями, их получение описано ниже. Другие соединения примеров, указанных выше, получают с использованием аналогичной методики с незначительными модификациями и выделяют либо в виде свободного основания, либо в виде гидрохлорида. Далее представлены примеры получения различных соединений формулы IIB, используемых в примерах получения соединений выше. Пример получения 1. 3-(Бромметил)-6-(трифторметил)пиридазин (используемый для получения соединений примеров 17 и 18).(а) 3-Йод-6-метилпиридазин. К раствору 3-хлор-6-метилпиридазина (500 мг) в HI (3 мл) добавляют NaI (782 мг). Реакционную смесь нагревают до 40C и выдерживают при данной температуре в течение 4 ч, затем до 70C и выдерживают при данной температуре в течение ночи. Выпавшее в осадок твердое вещество желтого цвета собирают фильтрацией. Маточную жидкость подщелачивают с помощью твердого NaOH, экстрагируют ДХМ, сушат над MgSO4, фильтруют, концентрируют и очищают хроматографией (силикагель; элюирование: EtOAc/гексан), получая 171,7 мг (20%) указанного в заголовке соединения в виде твердого белого вещества, которое является почти чистым согласно ВЭЖХ. Осадок желтого цвета подщелачивают, используя эту же водную фазу, экстрагируют ДХМ, сушат над MgSO4, фильтруют и концентрируют, получая 579,1 мг (68%) чистого указанного в заголовке соединения в виде твердого белого вещества. 1(b) 3-Метил-6-(трифторметил)пиридазин. Смесь CuI (263 мг) и KF (80 мг) нагревают в вакууме в течение 30 мин до появления зеленоватой окраски. Систему заполняют N2 и последовательно добавляют раствор 3-йод-6-метилпиридазин (300 мг) в ДМФА (1,25 мл), N-метилпирролидинон (1,25 мл) и триметил(трифторметил)силан (185 мкл). Наблюдают проявление темно-коричневой окраски. Смесь перемешивают при комнатной температуре в течение 5 дней, реакция проходит не полностью. Добавляют CuI (263 мг), KF (80 мг) и CF3TMS (185 мкл) и полученную смесь перемешивают в течение ночи. Добавляют дополнительное количество CuI (135 мг),KF (40 мг) и CF3TMS (90 мкл), смесь перемешивают в течение еще одного дня при комнатной температуре, после чего исходное вещество почти исчезает. Добавляют водный раствор аммиака и ТВМЕ и фазы разделяют. Водную фазу промывают ТВМЕ. Органические экстракты объединяют и последовательно промывают водным раствором аммиака, 1 М HCl, насыщенным водным раствором NaHCO3 и раствором соли, сушат над Na2SO4, фильтруют и концентрируют, получая 70 мг (35%) указанного в заголовке соединения. 1 Н-ЯМР (, м.д., CDCl3): 7,72 (1H, д), 7,53 (1H, д), 2,85 (3H, с).
МПК / Метки
МПК: A61K 31/4745, C07D 471/16, A61P 31/06
Метки: средств, азотсодержащие, соединения, трициклические, применение, бактерицидных, качестве
Код ссылки
<a href="https://eas.patents.su/30-18817-triciklicheskie-azotsoderzhashhie-soedineniya-i-ih-primenenie-v-kachestve-baktericidnyh-sredstv.html" rel="bookmark" title="База патентов Евразийского Союза">Трициклические азотсодержащие соединения и их применение в качестве бактерицидных средств</a>
Трициклические азотсодержащие соединения в качестве антибактериальных агентов

Номер патента: 15821
Опубликовано: 30.12.2011
Авторы: Пирсон Нейл Дэвид, Дэвис Дэвид Эван, Джордано Илариа, Хеннесси Алан Джозеф, Дэвис Дэвид Томас
МПК: A61K 31/4985, A61P 31/00, C07D 471/16...
Метки: соединения, качестве, азотсодержащие, агентов, антибактериальных, трициклические
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль и/или N-оксидв которой Z1 и Z2независимо выбраны из CH и N;R1a и R1b независимо выбраны из водорода; галогена; циано; (C1-6)алкила; (C1-6)алкилтио; трифторметила; трифторметокси; карбокси; гидрокси, в случае необходимости замещенного (C1-6)алкилом или (C1-6)алкоксизамещенным (C1-6)алкилом; (C1-6)алкоксизамещенного (C1-6)алкила; гидрокси(C1-6)алкила; аминогруппы, в случае...
Азабициклические соединения, способ их получения и их применение в качестве лекарственных средств, в частности, в качестве антибактериальных средств

Номер патента: 4920
Опубликовано: 28.10.2004
Авторы: Роулендс Дэвид Ален, Лампила Максим, Асзоди Жозеф, Фроментэн Клод
МПК: A61K 31/529, A61P 31/04, C07D 487/08...
Метки: применение, получения, соединения, частности, средств, качестве, лекарственных, азабициклические, антибактериальных, способ
Формула / Реферат:
1. Соединение общей формулы (I) или одна из его солей с основанием или с кислотой в которой R1 означает атом водорода, радикал COOH, CN, COOR, CONR6R7, (CH2)n'R5 или радикал где R выбирают из группы, состоящей из алкила с 1-6 атомами углерода, возможно замещенного пиридилом или карбамоилом; -CH2-алкенила, содержащего в целом 3-9 атомов углерода; арила с 6-10 атомами углерода или аралкила с 7-11 атомами углерода, причем арильное или...
Трициклические соединения и их применение в качестве модуляторов глюкокортикоидного рецептора

Номер патента: 16001
Опубликовано: 30.01.2012
Авторы: Ху Сяо, Джером Кевин Диуэйн, Рукер Пол Винсент, Олсон Лиза, Веббер Рональд Кит, Чэн Хэнмяо, Обукович Марк Джерард
МПК: A61K 31/44, A61P 1/00, A61P 11/06...
Метки: качестве, трициклические, применение, глюкокортикоидного, рецептора, соединения, модуляторов
Формула / Реферат:
1. Соединение формулы (I)или его соль.2. Соединение по п.1, представляющее собой соединение формулыили его соль.3. Соединение по п.2, представляющее собой соединение формулы4. Хлористо-водородная соль соединения по п.1.5. Фармацевтическая композиция, включающая соединение по п.1 или его соль и носитель.6. Способ модулирования глюкокортикоидного рецептора, включающий контактирование глюкокортикоидного рецептора с соединением по п.1 или с его...
Трициклические соединения, обладающие активностью в отношении интегринов, в частности, в отношении интегрина альфаvбета 3, способ их получения и промежуточные соединения, используемые в этом способе,их применение в качестве медикаментов и содержащие их фармацевтические композиции.

Номер патента: 2271
Опубликовано: 28.02.2002
Авторы: Кнолле Йохен, Венер Фолькмар, Гадек Томас Р., Карниато Дени, Штильц Ханс-Ульрих, Гурвест Жан-Франсуа, Питти Роберт М., Тетш Жан-Жорж, Бодари Сара С., Бернар Серж, Макдауэлл Роберт С.
МПК: A61P 9/10, C07C 281/12, A61K 31/19...
Метки: применение, композиции, медикаментов, используемые, отношении, активностью, способ, альфаvбета, способе,их, частности, качестве, трициклические, соединения, содержащие, промежуточные, интегринов, фармацевтические, этом, получения, интегрина, обладающие
Формула / Реферат:
1. Соединения общей формулы (I) в которой R1 обозначает группу -О-[А]-[В]-COR6, в которой R6 обозначает -ОН, C1-С6алкокси, -О-СН2-СН(ОН)-СН2OН, [A] обозначает группу C1-С6алкилен, возможно замещенный оксогруппой, [B] обозначает радикал -CH(Z)- или простую связь, Z обозначает группу -NHCO2Rc, или -NHSO2Rc, где Rc обозначает радикал фенил(C1-С4)алкил-, хинолинил или пиридинилимидазолил(C1-С4)алкил-, R2 и R3, одинаковые или разные, обозначают атом...
Азотсодержащие гетероароматические соединения в качестве ингибиторов фактора ха, фармацевтическая композиция и способ лечения

Номер патента: 3056
Опубликовано: 26.12.2002
Авторы: Каччиола Джозеф, Февиг Джон Мэттью, Пинто Дональд Джозеф Филлип, Кван Мими Лайфен, Пруитт Джеймс Рассел, Орват Майкл Джеймс, Хан Кви, Росси Карен Анита
МПК: A61P 7/02, A61K 31/41, C07D 207/34...
Метки: качестве, способ, композиция, ингибиторов, лечения, гетероароматические, ха, азотсодержащие, фармацевтическая, фактора, соединения
Формула / Реферат:
1. Соединение формулы I или его стереоизомер или фармацевтически приемлемая соль, где кольцо М содержит, кроме J, 0-3 атома N, при условии, что если М содержит 2 атома N, тогда R1b отсутствует, и если М содержит 3 атома N, тогда R1a и R1b отсутствуют; J представляет собой N или NH; D выбран из CN, C(=NR8)NR7R9, NHC(=NR8)NR7R9, NR8CH(=NR7), C(O)NR7R8 и (CR8R9)tNR7R8, при условии, что D присоединен к Е в мета- или пара-положении относительно G, Е...
Предыдущий патент: Способы перфорирования с использованием вязкоупругих поверхностно-активных текучих сред и связанные с ними составы
Следующий патент: Удаление зараженных фузариозом зерен из зерновой культуры
Случайный патент: Плоская текстильная упрочняющая лента для трубы и труба, упрочненная такой лентой