Аминодигидротиазиновые производные в качестве ингибиторов bace для лечения болезни альцгеймера
Номер патента: 16955
Опубликовано: 30.08.2012
Авторы: Уотсон Брайан Морган, Шихан Скотт Мартин, Аудиа Джеймс Эдмунд, Мерготт Дастин Джеймс


























Формула / Реферат
1. Соединение формулы I

где n представляет собой 0, 1 или 2;
R1 представляет собой пиримидинил, пиразинил, необязательно замещенный хлором или фтором, или пиридинил, необязательно замещенный одним или двумя заместителями, каждый из которых независимо выбран из хлора, фтора и C1-C3-алкокси;
R2 в каждом случае независимо выбран из хлора или фтора;
R3 представляет собой водород или C1-C4-алкил, необязательно замещенный гидроксилом;
R4 представляет собой водород или C1-C3-алкил,
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где
R1 представляет собой пиримидинил, пиридинил, необязательно замещенный одним или двумя заместителями, в каждом случае независимо выбранными из хлора, фтора или метокси, или пиразинил, необязательно замещенный фтором;
R2 представляет собой хлор или фтор;
R3 представляет собой водород, метил, метил, замещенный гидроксилом, или изопропил, замещенный гидроксилом;
R4 представляет собой водород;
n представляет собой 0, 1 или 2,
или его фармацевтически приемлемая соль.
3. Соединение по любому из пп.1, 2, где
R1 представляет собой пиримидинил, пиридинил, необязательно замещенный фтором, или пиразинил, необязательно замещенный фтором;
R2 представляет собой фтор;
R3 представляет собой водород или метил;
R4 представляет собой водород;
n представляет собой 1 или 2,
или его фармацевтически приемлемая соль.
4. Соединение по любому из пп.1-3, где
R1 представляет собой пиримидинил;
R2 представляет собой фтор;
R3 представляет собой водород;
R4 представляет собой водород;
n представляет собой 2,
или его фармацевтически приемлемая соль.
5. Соединение по любому из пп.1-4, где конфигурация хирального центра, расположенного рядом с атомом азота аминотиазина, представляет собой (S), или его фармацевтически приемлемая соль.
6. Соединение 4-(2,4-дифтор-5-(пиримидин-5-ил)фенил)-4-метил-5,6-дигидро-4Н-1,3-тиазин-2-амин

или его фармацевтически приемлемая соль.
7. Соединение по п.6, где указанное соединение представляет собой (S)-4-(2,4-дифтор-5-(пиримидин-5-ил)фенил)-4-метил-5,6-дигидро-4H-1,3-тиазин-2-амин

или его фармацевтически приемлемую соль.
8. Фармацевтическая композиция, содержащая соединение по любому из пп.1-7 или его фармацевтически приемлемую соль в комбинации с фармацевтически приемлемым носителем, разбавителем или эксципиентом.
9. Способ лечения болезни Альцгеймера путем введения эффективного количества соединения по любому из пп.1-7 или его фармацевтически приемлемой соли.
10. Применение соединения по любому из пп.1-7 в терапии.
11. Применение соединения по любому из пп.1-7 для лечения болезни Альцгеймера.
12. Применение соединения по пп.1-7 или его фармацевтически приемлемой соли для производства лекарственного средства для лечения болезни Альцгеймера.
Текст
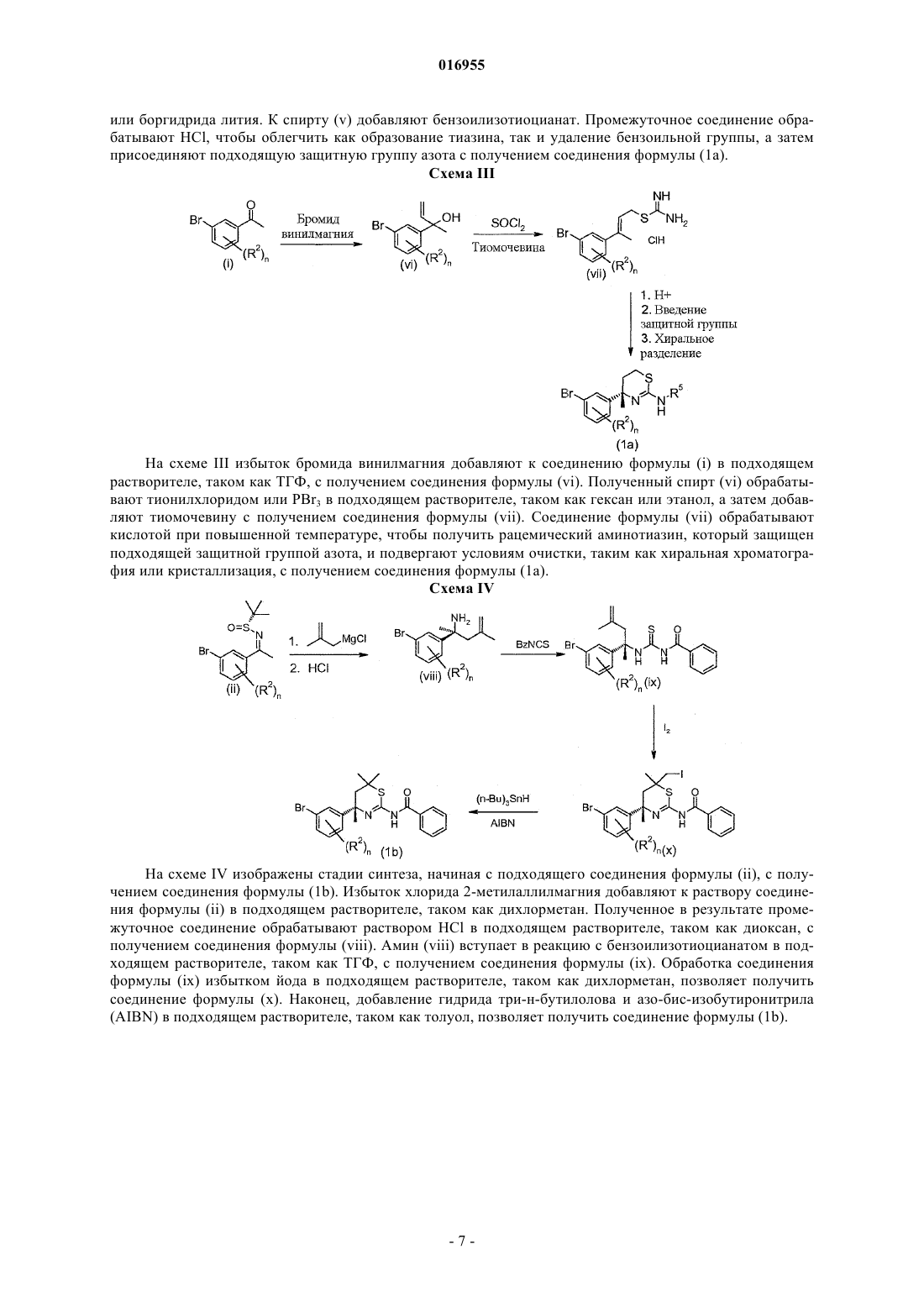
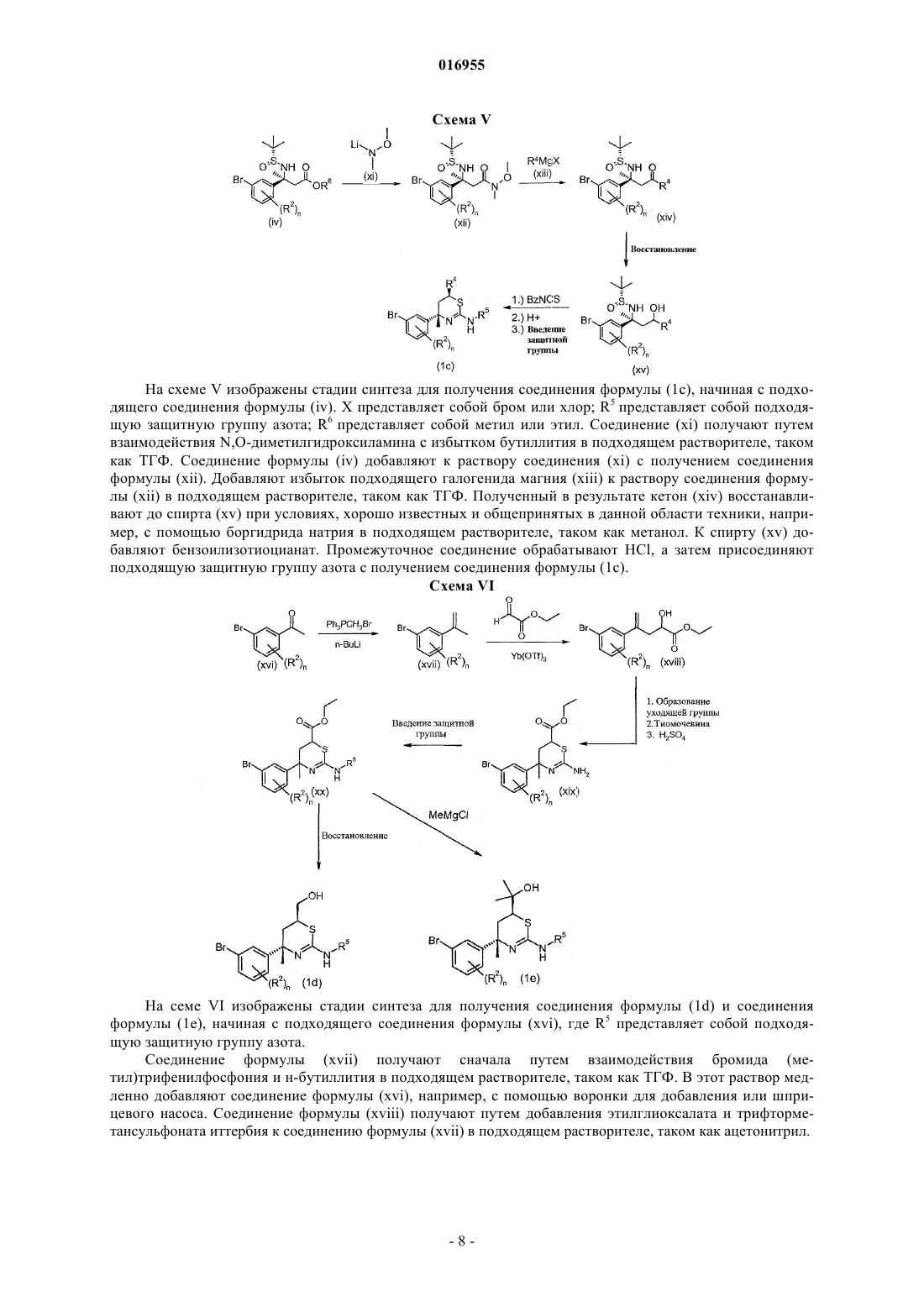
BACE ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ АЛЬЦГЕЙМЕРА Изобретение предлагает ингибиторы -секретазы или -сайта фермента, расщепляющего белокпредшественник амилоида (BACE) формулы I Аудиа Джеймс Эдмунд, Мерготт Дастин Джеймс, Шихан Скотт Мартин, Уотсон Брайан Морган (US) Медведев В.Н. (RU) способы их применения, а также промежуточные соединения и способы их получения.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) 016955 Настоящее изобретение относится к области лечения болезни Альцгеймера и других заболеваний и расстройств, в которые вовлечен амилоид-(А)-пептид, нейротоксический и сильно агрегирующий пептидный фрагмент белка-предшественника амилоида (APP). В частности, настоящее изобретение предлагает эффективные ингибиторы -секретазы или -сайта фермента, расщепляющего белокпредшественник амилоида (BACE). Было показано, что полное или частичное ингибирование BACE оказывает значительное влияние на связанные с образованием бляшек и зависимые от образования бляшек патологии в моделях на мышах, позволяя предположить, что даже небольшое уменьшение уровней А может привести к длительному значительному уменьшению массы бляшек и синаптических нарушений,таким образом обеспечивая достижение значительного терапевтического эффекта. Известные на сегодняшний день из уровня техники ингибиторы BACE представляют собой пептидомиметические аналоги переходного состояния, обычно включающие фрагмент гидроксиэтила. Хотя многие из данных соединений представляют собой эффективные ингибиторы BACE, их высокий молекулярный вес и низкая способность проникать через мембрану делают их неперспективными в качестве лекарственного средства. Поэтому от больших пептидомиметических молекул переходят к малым молекулам, таким как разнообразные гидроксиэтиламиновые каркасы, а также включающие гетероциклы каркасы. См., например, Durham и Shepherd, Current Opinion in Drug DiscoveryDevelopment, 9(6), 776791 (2006). Некоторые аминотиазиновые соединения были описаны как ингибиторы BACE вWO 2007/049532. Необходимы ингибиторы BACE, которые действенны и более эффективны, чтобы обеспечить лечение опосредованных А-пептидом расстройств, таких как болезнь Альцгеймера. Настоящее изобретение обеспечивает новые действенные и эффективные ингибиторы BACE. Настоящее изобретение предлагает соединения формулы IR1 представляет собой пиримидинил, пиразинил, необязательно замещенный хлором или фтором,или пиридинил, необязательно замещенный одним или двумя заместителями, каждый из которых независимо выбран из хлора, фтора и C1-C3-алкокси;R2 в каждом случае независимо выбран из хлора и фтора;R3 представляет собой водород или C1-C4-алкил, необязательно замещенный гидроксилом; иR4 представляет собой водород или C1-C3-алкил; или их фармацевтически приемлемую соль. Настоящее изобретение также предлагает способ лечения болезни Альцгеймера у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I. Настоящее изобретение дополнительно предлагает способ предотвращения прогрессирования легкого когнитивного нарушения в болезнь Альцгеймера у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I. Настоящее изобретение дополнительно предлагает способ предотвращения прогрессирования болезни Альцгеймера у пациента, имеющего риск ее развития, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I. Настоящее изобретение также предлагает способ ингибирования BACE у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I. Настоящее изобретение также предлагает способ ингибирования опосредованного -секретазой расщепления белка-предшественника амилоида, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I. Настоящее изобретение дополнительно предлагает способ ингибирования продукции А-пептида,включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I. Настоящее изобретение также предлагает фармацевтическую композицию, содержащую соединение формулы I в комбинации с фармацевтически приемлемым носителем, разбавителем или эксципиентом. Более того, настоящее изобретение предлагает соединение формулы I для применения в терапии, в частности для лечения болезни Альцгеймера или для предотвращения прогрессирования легкого когнитивного нарушения в болезнь Альцгеймера. Настоящее изобретение также предлагает применение соединения формулы I для производства лекарственного средства для лечения болезни Альцгеймера. Настоящее изобретение дополнительно предлагает применение соединения формулы I для производства-1 016955 лекарственного средства для предотвращения прогрессирования легкого когнитивного нарушения в болезнь Альцгеймера. Настоящее изобретение также предлагает применение соединения формулы I для производства лекарственного средства для ингибирования BACE. Настоящее изобретение дополнительно предлагает применение соединения формулы I для производства лекарственного средства для ингибирования продукции А-пептида. Дополнительно, настоящее изобретение предлагает фармацевтическую композицию, пригодную для лечения болезни Альцгеймера. Более того, настоящее изобретение предлагает фармацевтическую композицию, пригодную для предотвращения прогрессирования легкого когнитивного нарушения в болезнь Альцгеймера. Настоящее изобретение также предлагает фармацевтическую композицию, пригодную для ингибирования BACE. Более того, настоящее изобретение предлагает фармацевтическую композицию, пригодную для ингибирования опосредованного -секретазой расщепления белка-предшественника амилоида. Настоящее изобретение также предлагает фармацевтическую композицию, пригодную для лечения состояния, возникшего в результате избыточных уровней А-пептида, содержащую соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или более фармацевтически приемлемыми эксципиентами, носителями или разбавителями. Основные химические термины, используемые в формулах соединений, упомянутых выше, имеют обычные значения. Например, термин "C1-C3-алкил" относится к метилу, этилу, пропилу и изопропилу. Термин "C1-C4-алкил" относится к фрагментам метила, этила, пропила, изопропила, бутила, изобутила,втор-бутила и трет-бутила."C1-C4-алкил, необязательно замещенный гидроксилом", представляет собой C1-C4-алкильную группу, в которой один из атомов водорода заменен на молекулу гидроксила. Термин "C1-C3-алкокси" представляет собой C1-C3-алкильную группу, связанную с атомом кислорода, и относится к метокси, этокси, пропокси и изопропокси. Термин "защитная группа азота" означает группу, которая стабильна в предполагаемых условиях реакции, а также которую можно избирательно удалить реагентами и условиями реакции, при которых регенерируется амин. Такие группы хорошо известны специалисту и описаны в литературе. См., например, Greene и Wuts, Protective Groups in Organic Synthesis, третье издание, глава 7, John Wiley and SonsInc. (1999). Термин "ингибирование продукции А-пептида" в данном описании означает снижение уровней А-пептида in vivo у пациента до нормальных, если они избыточны, или ниже нормальных уровней, при необходимости. Термин "эффективное количество соединения формулы I" в данном описании означает дозу или дозы соединения формулы I, необходимые для достаточного ингибирования BACE, чтобы снизить уровни А-пептида in vivo у пациента до нормальных или ниже нормальных уровней. Термин "лечение" включает замедление или купирование прогрессирования указанного заболевания у пациента. Легкое когнитивное нарушение определено как потенциальная предшествующая фаза деменции,связанной с болезнью Альцгеймера, на основании клинического проявления и прогрессирования у пациента легкого когнитивного нарушения в деменцию Альцгеймера с течением времени. (Morris, et al., Arch.Neurol., 58, 397-405 (2001); Petersen, et al., Arch. Neurol., 56, 303-308 (1999. Термин "предотвращение прогрессирования легкого когнитивного нарушения в болезнь Альцгеймера" включает замедление, купирование или обращение прогрессирования легкого когнитивного нарушения в болезнь Альцгеймера у пациента. Для специалиста должно быть очевидно, что соединения формулы I могут существовать в таутомерных формах, изображенных на фигуре (1). Если в настоящем описа.нии указан один из отдельных таутомеров соединений формулы I, следует понимать, что упоминание включает обе таутомерные формы и все смеси указанных форм.-2 016955 Для специалиста должно быть очевидно, что соединения формулы I состоят из ядра, которое включает по меньшей мере один хиральный центр: Хотя настоящее изобретение охватывает все отдельные энантиомеры, а также смеси энантиомеров указанных соединений, включая рацематы, соединения с абсолютной конфигурацией атома, помеченного 1, показанного на фигуре (2), представляют собой предпочтительные соединения формулы I. Дополнительно, при подходящем замещении соединения с абсолютной конфигурацией атома, помеченного 2, показанного на фигуре (3), представляют собой предпочтительные соединения формулы I. Дополнительно, для специалиста должно быть очевидно, что можно создать дополнительные хиральные центры в соединениях согласно настоящему изобретению путем выбора некоторых переменных. Настоящее изобретение охватывает все отдельные энантиомеры или диастереомеры, а также смеси энантиомеров и диастереомеров указанных соединений, включая рацематы. Для специалиста также должно быть очевидно, что обозначения Кана-Ингольда-Прелога (R) или (S) для всех хиральных центров будут изменяться в зависимости от картины замещения конкретного соединения. Отдельные энантиомеры или диастереомеры можно получить с помощью хиральных реагентов или с помощью стереоселективных или стереоспецифичных методик синтеза. В качестве альтернативы,отдельные энантиомеры или диастереомеры можно выделить из смеси с помощью стандартных методик хиральной хроматографии или кристаллизации в любой удобный момент при синтезе соединений согласно настоящему изобретению. Отдельные энантиомеры и диастереомеры соединений согласно настоящему изобретению представляют собой предпочтительный вариант осуществления настоящего изобретения. Соединения согласно настоящему изобретению представляют собой амины, и они соответствующим образом реагируют с любой из множества неорганических и органических кислот с получением фармацевтически приемлемых солей присоединения кислоты. Фармацевтически приемлемые соли и обычная методика их получения хорошо известны в данной области техники. См., например, Р. Stahl, etal. Handbook of Pharmaceutical Salts: Properties, Selection and Use, (VCHA/Wiley-VCH, 2002); S.M. Berge,et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciences, vol. 66, No. 1, January 1977. Предпочтительные фармацевтически приемлемые соли представляют собой такие, которые образованы с хлористоводородной кислотой. Хотя все соединения формулы I представляют собой полезные ингибиторы BACE, некоторые классы соединений являются предпочтительными. Ниже описаны такие предпочтительные классы:c) R1 представляет собой пиридинил, необязательно замещенный одним или двумя заместителями,в каждом случае независимо выбранными из хлора, фтора или метокси;d) R1 представляет собой пиридинил, необязательно замещенный фтором или метокси;q) соединение формулы I имеет абсолютную конфигурацию (S) в хиральном центре, расположенном рядом с атомом азота в аминотиазиновом кольце;r) соединение формулы I представляет собой свободное основание;s) соединение формулы I представляет собой фармацевтически приемлемую соль;u) соединение формулы I представляет собой дихлористо-водородную соль. Предпочтительный вариант осуществления настоящего изобретения относится к соединениям формулы I, где R1 представляет собой пиримидинил, пиридинил, необязательно замещенный одним или двумя заместителя, в каждом случае независимо выбранными из хлора, фтора или метокси, или пиразинил,необязательно замещенный фтором; R2 представляет собой хлор или фтор; R3 представляет собой водород, метил, метил, замещенный гидроксилом, или изопропил, замещенный гидроксилом; R4 представляет собой водород или метил и n представляет собой 0, 1 или 2; или к фармацевтически приемлемой соли указанных соединений. В указанном варианте осуществления предпочтительно, чтобы абсолютной конфигурацией хирального центра, расположенного рядом с атомом азота аминотиазинового кольца, являлась (S) или фармацевтически приемлемая соль указанного соединения. Другой предпочтительный вариант осуществления настоящего изобретения относится к соединениям формулы I, где R1 представляет собой пиримидинил, пиридинил, необязательно замещенный одним или двумя заместителями, в каждом случае независимо выбранными из хлора, фтора или метокси, или пиразинил, необязательно замещенный фтором; R2 представляет собой хлор или фтор; R3 представляет собой водород, метил, метил, замещенный гидроксилом, или изопропил, замещенный гидроксилом; R4 представляет собой водород и n представляет собой 0, 1 или 2; или к фармацевтически приемлемой соли указанных соединений. В указанном варианте осуществления предпочтительно, чтобы абсолютной конфигурацией хирального центра, расположенного рядом с атомом азота аминотиазинового кольца, являлась (S) или фармацевтически приемлемая соль указанного соединения. Более предпочтительный вариант осуществления настоящего изобретения относится к соединениям формулы I, где R1 представляет собой пиримидинил, пиридинил, необязательно замещенный одним или двумя заместителями, в каждом случае независимо выбранными из хлора или фтора, или пиразинил, необязательно замещенный фтором; R2 представляет собой хлор или фтор; R3 представляет собой водород,метил; R4 представляет собой водород и n представляет собой 0, 1 или 2; или к фармацевтически приемлемой соли указанных соединений. В указанном варианте осуществления предпочтительно, чтобы абсолютной конфигурацией хирального центра, расположенного рядом с атомом азота аминотиазинового кольца, являлась (S) или фармацевтически приемлемая соль указанного соединения. Дополнительный вариант осуществления настоящего изобретения относится к соединениям формулы I, где R1 представляет собой пиримидинил, пиридинил, необязательно замещенный фтором или метокси, или пиразинил, необязательно замещенный фтором; R2 представляет собой фтор; R3 представляет собой водород или метил; R4 представляет собой водород; и n представляет собой 1 или 2; или к фармацевтически приемлемой соли указанных соединений. В указанном варианте осуществления предпочтительно, чтобы абсолютной конфигурацией хирального центра, расположенного рядом с атомом азота аминотиазинового кольца, являлась (S) или фармацевтически приемлемая соль указанного соединения. Наиболее предпочтительный вариант осуществления настоящего изобретения относится к соединениям формулы I, где R1 представляет собой пиримидинил, пиридинил, необязательно замещенный фтором, или пиразинил, необязательно замещенный фтором; R2 представляет собой фтор; R3 представляет собой водород или метил; R4 представляет собой водород и n представляет собой 1 или 2; или к фармацевтически приемлемой соли указанных соединений. В указанном варианте осуществления предпочтительно, чтобы абсолютной конфигурацией хирального центра, расположенного рядом с атомом азота аминотиазинового кольца, являлась (S); или фармацевтически приемлемая соль указанного соединения. Особенно предпочтительный вариант осуществления настоящего изобретения относится к соединениям формулы I, где R1 представляет собой пиримидинил; R2 представляет собой фтор; R3 представляет собой водород; R4 представляет собой водород и n представляет собой 2; или к фармацевтически приемлемой соли указанных соединений. В указанном варианте осуществления предпочтительно, чтобы абсолютной конфигурацией хирального центра, расположенного рядом с атомом азота аминотиазинового кольца, являлась (S) или фармацевтически приемлемая соль указанного соединения. Дополнительный особенно предпочтительный вариант осуществления настоящего изобретения, относящийся к соединениям формулы I, представляет собой или его фармацевтически приемлемую соль.-4 016955 Другой особенно предпочтительный вариант осуществления настоящего изобретения, относящийся к соединениям формулы I, представляет собой или его фармацевтически приемлемую соль. Соединения формулы I представляют собой ингибиторы BACE. Таким образом, настоящее изобретение также предлагает способ ингибирования BACE у пациента, который включает введение пациенту,нуждающемуся в указанном лечении, ингибирующего BACE количества соединения формулы I. Предпочтительно, чтобы пациентом, которого лечат введением соединений формулы I, являлся человек. В качестве ингибиторов BACE соединения согласно настоящему изобретению пригодны для подавления продукции А-пептида, а следовательно, для лечения расстройств, возникших в результате избыточных уровней А-пептида вследствие чрезмерной продукции и/или пониженного клиренса А-пептида. Дополнительный вариант осуществления настоящего изобретения представляет собой применение соединения формулы I для производства лекарственного средства для лечения заболевания или состояния, которое можно улучшить или предотвратить ингибированием BACE. Соединения формулы I,следовательно, считают полезными для лечения или предотвращения болезни Альцгеймера, легкого когнитивного нарушения, синдрома Дауна, наследственной церебральной геморрагии с амилоидозом голландского типа, церебральной амилоидной ангиопатии, других дегенеративных деменций, таких как деменции смешанного васкулярного и дегенеративного происхождения, деменция, связанная с болезнью Паркинсона, деменция, связанная с прогрессирующим надъядерным параличом, связанная с деменцией кортико-базальная дегенерация и тип болезни Альцгеймера с диффузными тельцами Леви. Соединения согласно настоящему изобретению можно получить множеством способов, известных в данной области техники, некоторые из которых показаны на схемах ниже. Для специалиста в данной области должно быть очевидно, что отдельные стадии на следующих схемах можно изменить, чтобы получить соединения формулы I. Конкретный порядок стадий, необходимых для получения соединений формулы I, зависит от конкретного синтезируемого соединения, исходного соединения и относительной неустойчивости содержащих заместители фрагментов. Продукты каждой стадии на схемах ниже можно выделить с помощью обычных способов, включая экстрагирование, выпаривание, осаждение, хроматографию, фильтрацию, растирание в порошок и кристаллизацию. Некоторые стереохимические центры остаются точно не определенными, и некоторые заместители были исключены из следующих схем с целью ясности, и они не предполагаются ограничивающими идею схем каким бы то ни было образом. Более того, отдельные изомеры, энантиомеры или диастереомеры можно разделить в любой удобный момент при синтезе соединений формулы I с помощью таких способов, как хиральная хроматография. Дополнительно, промежуточные соединения, описанные на следующих схемах, содержат множество защитных групп азота. Переменная защитная группа может быть такой же или отличной при каждом появлении в зависимости от конкретных условий реакции и конкретных преобразований, которые нужно осуществить. Условия введения и снятия защитных групп хорошо известны специалисту и описаны в литературе. См., например, Greene и Wuts, Protective Groups in Organic Synthesis, выше. На схемах ниже все заместители, если не указано иначе, представляют собой определенные ранее заместители. Должно быть очевидно, что соединения формул (1 а)-(1 е), (2) и (3) можно легко получить с помощью способов, которые хорошо известны и общеприняты в данной области техники, включая способы и методики, аналогичные описанным в настоящем изобретении. Необходимые исходные вещества либо коммерчески доступны, либо их можно получить из коммерчески доступных веществ с помощью способов, хорошо известных специалисту. На схеме I изображена реакция подходящего соединения любой из формул (1 а)-(1 е), где R5 представляет собой защитную группу азота, такую как ацетил, бензоил или трет-бутоксикарбонил, с подходящим соединением формулы (2) или формулы (3) с получением соединения формулы I после снятия защитных групп с промежуточного соединения (4). Соединение любой из формул (1 а)-(1 е) вступает в реакцию сочетания Сузуки с соединением формулы (2) с применением подходящего палладиевого реагента, такого как хлорид бис-(трифенилфосфин)палладия(II), тетракис-трифенилфосфинпалладий, PdCl2 или ацетат палладия(II), в присутствии подходящего основания, такого как карбонат цезия, карбонат натрия или карбонат калия. Такие реакции проводят в подходящем растворителе, таком как 1,2-диметоксиэтан, вода, этанол, ацетонитрил, диметилформамид или диоксан или их смеси. В качестве альтернативы, соединение любой из формул (1 а)-(1 е) вступает в реакцию сочетания Стилла с соединением формулы (3) с применением подходящего палладиевого реагента, такого как хлорид бис-(трифенилфосфин)палладия(II), PdCl2 или тетракис-трифенилфосфинпалладий, в присутствии подходящего вспомогательного вещества, такого как хлорид лития или фторид цезия. Такие реакции проводят в подходящем растворителе, таком как толуол или диметилформамид (ДМФА) или их смеси. На необязательной стадии фармацевтически приемлемую соль соединения формулы I можно получить путем взаимодействия подходящего свободного основания формулы I с подходящей фармацевтически приемлемой кислотой в подходящем растворителе при стандартных условиях. Дополнительно, получения такой соли можно достичь одновременно со снятием защитной группы азота. Получение такой соли хорошо известно и общепринято в данной области. Соединения формулы (1 а) можно получить двумя способами. На схемах II и III изображены стадии синтеза, начиная с подходящего соединения формулы (i), с получением соединения формулы (1 а), где R6 представляет собой метил или этил и R5 представляет собой подходящую защитную группу азота, такую как ацетил, бензоил или трет-бутоксикарбонил. Схема II На схеме II соединение формулы (i) вступает в реакцию с амидом 2-метилпропан-2-сульфиновой кислоты в присутствии Ti(OEt)4 в подходящем растворителе, таком как ТГФ, с получением соединения формулы (ii). Соединение формулы (iii) получают путем добавления n-BuLi к диизопропиламину в подходящем растворителе, таком как ТГФ. Добавляют подходящее ацетатное соединение, а затем избыток триизопропоксида хлортитана. Соединение формулы (ii) добавляют к раствору соединения формулы (iii) с получением соединения формулы (iv). Снятие защитных групп амина осуществляют с помощью способов, известных в данной области, а затем следует восстановление сложного эфира до спирта с помощью способов, хорошо известных в данной области техники, например, с применением алюмогидрида лития-6 016955 или боргидрида лития. К спирту (v) добавляют бензоилизотиоцианат. Промежуточное соединение обрабатывают HCl, чтобы облегчить как образование тиазина, так и удаление бензоильной группы, а затем присоединяют подходящую защитную группу азота с получением соединения формулы (1 а). Схема III На схеме III избыток бромида винилмагния добавляют к соединению формулы (i) в подходящем растворителе, таком как ТГФ, с получением соединения формулы (vi). Полученный спирт (vi) обрабатывают тионилхлоридом или PBr3 в подходящем растворителе, таком как гексан или этанол, а затем добавляют тиомочевину с получением соединения формулы (vii). Соединение формулы (vii) обрабатывают кислотой при повышенной температуре, чтобы получить рацемический аминотиазин, который защищен подходящей защитной группой азота, и подвергают условиям очистки, таким как хиральная хроматография или кристаллизация, с получением соединения формулы (1 а). Схема IV На схеме IV изображены стадии синтеза, начиная с подходящего соединения формулы (ii), с получением соединения формулы (1b). Избыток хлорида 2-метилаллилмагния добавляют к раствору соединения формулы (ii) в подходящем растворителе, таком как дихлорметан. Полученное в результате промежуточное соединение обрабатывают раствором HCl в подходящем растворителе, таком как диоксан, с получением соединения формулы (viii). Амин (viii) вступает в реакцию с бензоилизотиоцианатом в подходящем растворителе, таком как ТГФ, с получением соединения формулы (ix). Обработка соединения формулы (ix) избытком йода в подходящем растворителе, таком как дихлорметан, позволяет получить соединение формулы (х). Наконец, добавление гидрида три-н-бутилолова и азо-бис-изобутиронитрила(AIBN) в подходящем растворителе, таком как толуол, позволяет получить соединение формулы (1b). На схеме V изображены стадии синтеза для получения соединения формулы (1 с), начиная с подходящего соединения формулы (iv). X представляет собой бром или хлор; R5 представляет собой подходящую защитную группу азота; R6 представляет собой метил или этил. Соединение (xi) получают путем взаимодействия N,О-диметилгидроксиламина с избытком бутиллития в подходящем растворителе, таком как ТГФ. Соединение формулы (iv) добавляют к раствору соединения (xi) с получением соединения формулы (xii). Добавляют избыток подходящего галогенида магния (xiii) к раствору соединения формулы (xii) в подходящем растворителе, таком как ТГФ. Полученный в результате кетон (xiv) восстанавливают до спирта (xv) при условиях, хорошо известных и общепринятых в данной области техники, например, с помощью боргидрида натрия в подходящем растворителе, таком как метанол. К спирту (xv) добавляют бензоилизотиоцианат. Промежуточное соединение обрабатывают HCl, а затем присоединяют подходящую защитную группу азота с получением соединения формулы (1 с). Схема VI На семе VI изображены стадии синтеза для получения соединения формулы (1d) и соединения формулы (1 е), начиная с подходящего соединения формулы (xvi), где R5 представляет собой подходящую защитную группу азота. Соединение формулы (xvii) получают сначала путем взаимодействия бромида (метил)трифенилфосфония и н-бутиллития в подходящем растворителе, таком как ТГФ. В этот раствор медленно добавляют соединение формулы (xvi), например, с помощью воронки для добавления или шприцевого насоса. Соединение формулы (xviii) получают путем добавления этилглиоксалата и трифторметансульфоната иттербия к соединению формулы (xvii) в подходящем растворителе, таком как ацетонитрил.-8 016955 Соединение формулы (xix) получают с помощью трехстадийного процесса: сначала спирт соединения формулы (xviii) преобразуют в уходящую группу, например, путем реакции с ангидридом трифторметансульфоновой кислоты в присутствии подходящего аминного основания, такого как 2,6-лутидин или диизопропилэтиламин, в подходящем растворителе, таком как метиленхлорид. Добавляют избыток тиомочевины, а затем полученное в результате промежуточное соединение добавляют к избытку серной кислоты. Полученную в результате аминогруппу соединения формулы (xix) защищают подходящей защитной группой азота с помощью способов, хорошо известных и описанных в данной области техники, с получением соединения формулы (хх). Реакцию соединения формулы (хх) можно осуществить двумя способами с получением либо соединения формулы (1d), либо соединения формулы (1 е). Соединение формулы (1d) можно получить путем восстановления эфира соединения (хх) с помощью способов, хорошо известных или описанных в данной области техники, например, избытком боргидрида лития в подходящем растворителе, таком как ТГФ. Соединение формулы (1 е) можно получить путем взаимодействия соединения формулы (хх) с избытком хлорида метилмагния в подходящем растворителе, таком как ТГФ. Примеры синтеза и примеры Следующие примеры синтеза и примеры дополнительно иллюстрируют настоящее изобретение. Названия соединений согласно настоящему изобретению получали с помощью ChemDraw Ultra,версии 10,0. Сокращения, используемые в настоящем описании, имеют значения согласно Aldrichimica Acta,vol. 17, No 1, 1984. Другие сокращения имеют значения, описанные далее:SCX представляет собой сильный катионообменник; са. означает около или приблизительно;DCM представляет собой дихлорметан; ТГФ представляет собой тетрагидрофуран;Et2O представляет собой диэтиловый эфир;FRET представляет собой резонансный перенос энергии флуоресценции;RFU представляет собой относительную единицу флуоресценции;DMEM представляет собой модифицированную по способу Дульбекко среду Игла;F12 представляет собой среду Хама F12;FBS представляет собой эмбриональную сыворотку теленка. Результаты масс-спектрометрии, если не указано иначе, получали с помощью ЖХ/МС: колонкаXbridge C18 (2,1503,5 мкм) при температуре 5010 С со скоростью потока 1 мл/мин. Система элюирования представляет собой элюирование от 5 до 100% акрилонитрилом (ACN) по весу в 10 мМ бикарбонате аммония (рН 10) в течение 7,0 мин, затем выдерживание в 100% ACN в течение 1,0 мин в сочетании с ионизацией электрораспылением (диапазон сканирования 100-800 а.е.м.; шаг 0,2 а.е.м.; фрагментор 80 В; прибавление 1,0; порог 80). Некоторые соединения очищали с помощью высокоэффективной жидкостной хроматографии(ВЭЖХ), способ А: колонка Xterra RP18 (30300 мм) при температуре окружающей среды со скоростью потока 40 мл/мин. Система элюирования представляет собой или состоит из изократического градиента 0:100 (ацетонитрил: (0,1% HCl в Н 2 О в течение 1-5 мин, а затем линейного градиента от 0:100(ацетонитрил: (0,1% HCl в H2O до 50:50 (ацетонитрил: (0,1% HCl в H2O в течение 20 мин. Любые другие условия ВЭЖХ в других случаях указаны. Пример синтеза 1.Ti(OEt)4 (29,5 г, 129 ммоль, 2,0 экв.) одной порцией при температуре окружающей среды. Реакционную смесь нагревали до 70 С и оставляли для перемешивания в течение 18 ч. Реакционную смесь охлаждали до температуры окружающей среды и вливали в воду. Полученную в результате суспензию фильтровали через прокладку из диатомитовой земли и промывали этилацетатом.-9 016955 Фильтрат собирали и экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Полученный остаток очищали с помощью хроматографии на силикагеле, элюируя линейным градиентом от гексана до смеси гексан:этилацетат (3:1) в течение 20 мин, с получением указанного в заголовке соединения (выход 81%). МС (m/z): 338, 340 (М+1). Следующие соединения в табл. 1 получали, по существу, как описано для получения [1-(5-бром-2,4 дифторфенил)этилиден]амида (R)-2-метилпропан-2-сульфиновой кислоты. Таблица 1 н-Бутиллитий (41,9 мл, 105 ммоль, 2 экв.) (2,5 М в гексане) добавляли к охлажденному до -78 С раствору диизопропиламина (10,6 г, 105 ммоль, 2 экв.) в ТГФ (262 мл). Через 15 мин добавляли по каплям метилацетат (7,7 г, 105 ммоль, 2 экв.) и реакционную смесь оставляли для перемешивания в течение 30 мин. В реакционную смесь добавляли по каплям раствор триизопропоксида хлортитана (31,6 г, 115 ммоль, 2,2 экв.) в ТГФ (50 мл). После перемешивания в течение 60 мин при -78 С добавляли по каплям раствор [1-(3-бромфенил)этилиден]амида 2-метилпропан-2-сульфиновой кислоты (12,2 г,40,4 ммоль, 1 экв.) в ТГФ (50 мл). Реакционную смесь перемешивали в течение 3 ч при -78 С. Реакцию гасили насыщенным раствором хлорида аммония (100 мл), нагревали до температуры окружающей среды и разбавляли водой (100 мл). Полученную в результате суспензию фильтровали через прокладку из диатомитовой земли и промывали этилацетатом. Фильтрат собирали и экстрагировали этилацетатом. Органические слои объединяли и сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Полученный остаток очищали с помощью хроматографии на силикагеле, элюируя линейным градиентом от смеси гексан:этилацетат (5:1) до смеси гексан:этилацетат (10:7) в течение 20 мин, с получением указанного в заголовке соединения (выход 72%). МС (m/z): 412, 414 (М+1). Следующие соединения в табл. 2 получали, по существу, как описано для получения метилового эфира (S)-3-R)-2-метилпропан-2-сульфиниламино)-3-(5-бром-2,4-дифторфенил)масляной кислоты. К раствору метилового эфира (S)-3-R)-2-метилпропан-2-сульфиниламино)-3-фенилмасляной кислоты (15,5 г; 37,6 ммоль; 1 экв.) и метанола (100 мл) добавляли одной порцией хлористый водород (4 М в диоксане) (100 мл, 400 ммоль, 11 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель удаляли при пониженном давлении с получением указанного в заголовке соединения, которое использовали без дополнительной очистки (выход 95%). МС (m/z): 306, 308 (М+1). Следующие соединения в табл. 3 получали, по существу, как описано для получения гидрохлорида(40,2 г, 90,5 ммоль, 1 экв.) в ТГФ (180 мл) добавляли алюмогидрид лития (1 М в ТГФ) (118 мл,118 ммоль) в течение 45 мин, поддерживая внутреннюю температуру реакции ниже 15 С. Реакционную смесь оставляли для нагревания до температуры окружающей среды и перемешивали в течение 1,5 ч. Реакционную смесь охлаждали до 0 С и гасили добавлением по каплям воды (4,5 мл), 2 М гидроксида натрия (4,5 мл) и воды (13,6 мл). Полученное в результате твердое вещество удаляли фильтрованием и промывали этилацетатом. Фильтрат сушили над MgSO4 и фильтровали. Растворитель удаляли при пониженном давлении с получением указанного в заголовке соединения, которое использовали без дополнительной очистки (выход 71%, чистота 73%, как определено с помощью ЖХ/МС). МС (m/z): 280, 282. Пример синтеза 5. К охлажденному до 0 С раствору гидрохлорида (S)-метил-3-амино-3-(4-фторфенил)бутаноата (14 г,38,6 ммоль, 1 экв.) в ТГФ (200 мл) аккуратно добавляли боргидрид лития (1,67 г, 77,1 ммоль, 2 экв.). Через 5 мин реакционную смесь нагревали до 50 С и перемешивали. По завершении реакционную смесь охлаждали на ледяной бане и гасили путем добавления по каплям воды. Реакционную смесь подкисляли с помощью 1N HCl (100 мл). После перемешивания в течение 1 ч раствор делали основным с помощью 5N NaOH и экстрагировали дихлорметаном. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (выход 94%). МС (m/z): 244 и 246 (М+1). Следующие соединения в табл. 4 получали, по существу, в соответствии с получением (S)-3-амино 3-(3-бромфенил)бутан-1-ола. Таблица 4(50 мл) добавляли бис-(триметилсилил)трифторацетамид (8,7 г, 34 ммоль, 1 экв.). Через 2 ч добавляли по каплям бензоилизотиоцианат (5,5 г, 34 ммоль, 1 экв.). Реакционную смесь перемешивали в течение 18 ч,гасили водой и экстрагировали этилацетатом. Объединенные органические фазы экстрагировали 1N HCl и насыщенным водным NaCl. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (выход 95%,чистота 90%, как определено с помощью ЖХ/МС). МС (m/z): 443, 445 (М+1). Следующие соединения в табл. 5 получали, по существу, как описано для получения(41 г, 68 ммоль) в 1,4-диоксане (20 мл) добавляли водный раствор HCl (5N, 407 мл, 2,0 моль, 30 экв.). Полученную в результате суспензию нагревали до 100 С. После перемешивания в течение 20 ч реакционную смесь концентрировали при пониженном давлении. Полученную в результате смесь обрабатывали водным раствором HCl (5N, 407 мл, 2,0 моль) и перемешивали при 100 С в течение 18 ч. Суспензию охлаждали до 10 С и рН доводили до 10 с помощью 50% водного раствора NaOH. Полученный в результате водный раствор экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной флэш-хроматографии на силикагеле, элюируя ступенчатым градиентом от смеси гексан:ацетон (4:1) до смеси гексан:ацетон (3:1), с получением указанного в заголовке соединения (выход 57%, чистота 85%, как определено с помощью ЖХ/МС). МС (m/z): 321, 323 (М+1). Следующие соединения в табл. 6 получали, по существу, в соответствии с получением(S)-[4-(5-бром-2-фторфенил)-4-метил-5,6-дигидро-4 Н-[1,3]тиазин-2 ил]карбаминовой кислоты. Раствор (S)-1-бензоил-3-[1-(5-бром-2-фторфенил)-3-гидрокси-1-метилпропил]тиомочевины (0,79 г,1,8 ммоль) и водной HCl (5N, 25 мл, 71 ммоль) нагревали до 100 С. После перемешивания в течение 6 ч реакционную смесь охлаждали до температуры окружающей среды и давали отстояться в течение ночи. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного гидрохлорида (S)-4-(5-бром-2-фторфенил)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-2-амина. МС (m/z): 303, 305 (М+1). К раствору гидрохлорида (S)-4-(5-бром-2-фторфенил)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-2-амина в ТГФ (30 мл) и насыщенного водного бикарбоната натрия (15 мл) добавляли ди-трет-бутилдикарбонат(0,77 г, 3,5 ммоль). Через 4 ч реакционную смесь разбавляли водой, экстрагировали этилацетатом, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной флэш-хроматографии на силикагеле, элюируя линейным градиентом от гексана до смеси гексан:этилацетат (3:1), с получением указанного в заголовке соединения (выход 69%). МС (m/z): 403, 405 (М+1). Пример синтеза 9. К раствору (S)-4-(5-бром-2,4-дифторфенил)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-2-амина (14,3 г,39 ммоль 1 экв.) в 1,4-диоксане (190 мл) добавляли насыщенный водный раствор бикарбоната натрия(190 мл) и воду (30 мл) при температуре окружающей среды. Суспензию перемешивали в течение 5 мин,а затем добавляли ди-трет-бутилдикарбонат (17 г, 78 ммоль, 2 экв.). Через 1 ч реакционную смесь разбавляли водой, экстрагировали этилацетатом, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексан:этилацетат (4:1), с получением указанного в заголовке соединения (выход 78%). МС (m/z): 421, 423 (М+1). Следующие соединения в табл. 7 получали, по существу, как описано для получения (S)-трет-бутил 4-(5-бром-2,4-дифторфенил)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-2-илкарбамата. Таблица 7 К раствору 3-бромацетофенона (50 г, 250 ммоль; 1 экв.) в МТВЕ (375 мл) при 10 С добавляли по каплям бромид винилмагния (0,7 М в ТГФ, 250 ммоль; 360 мл, 1 экв.). Реакционную смесь нагревали до кипения с обратным холодильником в течение 16 ч. Реакционную смесь охлаждали и гасили насыщенным водным раствором хлорида аммония. Полученную смесь разбавляли водой, экстрагировали этилацетатом, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью хроматографии на силикагеле, элюируя ступенчатым градиен- 14016955 том от смеси гексаны:этилацетат (9:1) до смеси гексаны:этилацетат (4:1), с получением указанного в заголовке соединения (40 г, выход 42%). 1 Н ЯМР (400 МГц, CDCl3): 1,64 (с, 3 Н), 5,18 (д, J=14 Гц, 1 Н), 5,30 (д, J=23 Гц, 1 Н), 6,13 (дд, J=14,23 Гц, 1 Н), 7,21 (т, J=11 Гц, 1 Н), 7,36-7,40 (м, 2 Н), 7,63 (с, 1 Н). Пример синтеза 11. Гидрохлорид 2-[3-(3-бромфенил)бут-2-енил]изотиомочевины(20 мл) добавляли тионилхлорид (6,9 г, 58 ммоль, 2 экв.). Реакционную смесь оставляли для нагревания до температуры окружающей среды, в это время интенсивно выделялся газ. Реакционную смесь перемешивали при температуре окружающей среды до тех пор, пока газ не переставал выделяться, и растворитель удаляли при пониженном давлении. Полученный в результате остаток растворяли в ацетонитриле(100 мл). Добавляли тиомочевину (2,2 г, 29 ммоль, 1 экв.) и реакционную смесь нагревали до 50 С. Через 2 ч реакционную смесь охлаждали до температуры окружающей среды. Полученный в результате осадок собирали фильтрованием и промывали ацетонитрилом с получением указанного в заголовке соединенияHCl (53 мл, 640 ммоль, 12 экв.) нагревали до 100 С. Через 24 ч раствор охлаждали до температуры окружающей среды. Величину рН раствора доводили до 10 с помощью водного 2N NaOH и экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (выход 70%). МС (m/z): 285, 287 (М+1). Пример синтеза 13.(10 г, 35 ммоль, 1 экв.) и триэтиламина (4,3 г, 42 ммоль, 1,2 экв.) в дихлорметане (70 мл) добавляли по каплям ацетилхлорид (2,8 г, 35 ммоль, 1 экв.) в течение 5 мин. Реакционную смесь оставляли для нагревания до температуры окружающей среды. Через 1 ч реакционную смесь разбавляли дихлорметаном и экстрагировали водой. Органические слои разделяли, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексан:этилацетат (1:1), с получением указанного в заголовке соединения (выход 87%). МС (m/z): 327, 329 (М+1). Пример синтеза 14.(S)-N-[4-(3-Бромфенил)-4-метил-5,6-дигидро-4 Н-[1,3]тиазин-2-ил]ацетамид. 4-(3-Бромфенил)-4-метил-5,6-дигидро-4 Н-[1,3]тиазин-2-иламин (20 г, 61 ммоль) очищали с помощью хирального разделения ВЭЖХ [колонка: 832 см Chiralpak AD; элюент: 60:40:0,2 (изопропиловый спирт:гептаны:диметилэтиламин); поток: 350 мл/мин при УФ 260 нм]. Элюируемый вторым изомер выделяли, чтобы получить энантиомерно обогащенное указанное в заголовке соединение (выход 35%). МС (m/z): 327, 329 (М+1). К нагретому до 100 С раствору (S)-4-(5-бром-2,4-дифторфенил)-4-метил-5,6-дигидро-4 Н-1,3 тиазин-2-амина (12,6 г, 29,9 ммоль, 1 экв.) в смеси 1,2-диметоксиэтан:вода:этанол (15:7:5, 300 мл) добавляли пиримидин-5-бороновую кислоту (25 г, 203 ммоль, 6,8 экв.), а затем карбонат цезия (58 г,180 ммоль, 6 экв.) и хлорид бис-(трифенилфосфин)палладия(II) (4,2 г, 6,0 моль, 0,2 экв.). Через 40 мин реакционную смесь охлаждали до температуры окружающей среды, разбавляли водой и экстрагировали этилацетатом. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью хроматографии на силикагеле, элюируя ступенчатым градиентом от смеси гексаны:этилацетат (7:3) до смеси гексаны:этилацетат (1:1), с получением указанного в заголовке соединения (выход 67%). МС (m/z): 421 (М+1). Следующие соединения в табл. 8 получали, по существу, как описано для получения третбутилового эфира (S)-[4-(2,4-дифтор-5-пиримидин-5-илфенил)-4-метил-5,6-дигидро-4 Н-[1,3]тиазин-2 ил]карбаминовой кислоты. Таблица 8 трет-Бутоксикарбонильная группа отщепляется при условиях реакции.(S)-N-(4-(3-(2-фторпиридин-3-ил)фенил)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-2 ил)ацетамида (450 мг, 1,3 ммоль) в метаноле (40 мл) добавляли раствор K2CO3 (210 мг, 1,5 ммоль) в смеси метанол:вода (2:1, 15 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч. Растворитель удаляли при пониженном давлении и остаток растворяли в этилацетате. Этилацетатный слой промывали водой и насыщенным водным NaCl, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Полученный остаток очищали с помощью SCX колоночной хроматографии с получением указанного в заголовке соединения (выход 65%). МС (m/z): 302 (М+1). Пример синтеза 17.(S)-трет-бутил 4-(3-бром-4-фторфенил)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-2 илкарбамата (1,1 г, 2,7 ммоль) и метанола (10 мл) добавляли трифторуксусную кислоту (10 мл). Реакционную смесь нагревали до 60 С. Через 15 ч растворитель удаляли при пониженном давлении. К полученному в результате остатку добавляли воду и смесь делали основной с помощью насыщенного бикарбоната натрия. Основную водную фазу экстрагировали дихлорметаном. Органическую фазу отделяли,сушили над сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении с получением указанного в заголовке соединения (выход 62%). МС (m/z): 303, 305 (М+1). Пример синтеза 18.(S)-N-(4-(3-Бром-4-фторфенил)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-2-ил)ацетамид. К раствору (S)-4-(3-бром-4-фторфенил)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-2-амина (550 мг,1,8 ммоль, 1,0 экв.) в тетрагидрофуране (20 мл) добавляли пиридин (720 мг, 9,0 ммоль, 5 экв.) и ангидрид уксусной кислоты (220 мг, 2,2 ммоль, 1,2 экв.). Через 10 мин реакционную смесь вливали в воду и водную смесь экстрагировали дихлорметаном. Органическую фазу отделяли и промывали 1N HCl и насыщенным водным NaCl, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (выход 83%). МС (m/z) 345, 347 (М+1). Раствор трет-бутилового эфира (S)-[4-(3-бром-4-фторфенил)-4-метил-5,6-дигидро-4 Н-[1,3]тиазин-2 ил]карбаминовой кислоты (100 мг, 250 мкмоль, 1 экв.), тетракис-(трифенилфосфин)палладия (14 мг,12,40 мкмоль, 0,05 экв.) и 2-трибутилстаннилпиразина (96 мг, 250 мкмоль, 1 экв.) в диоксане (3 мл) облучали в лабораторной микроволновой печи до температуры 130 С и выдерживали в течение 20 мин. Растворитель удаляли при пониженном давлении и полученный остаток очищали с помощью хроматографии на силикагеле, элюируя линейным градиентом с понижением от гексана до смеси гексан:этилацетат(15 мл) добавляли хлорид бис-(трифенилфосфин)палладия(II) (51 мг, 72 мкмоль, 0,05 экв.) и хлорид лития (92 мг, 2,2 ммоль, 1,5 экв.). Реакционную смесь облучали в лабораторной микроволновой печи до температуры 130 С и выдерживали в течение 3 ч. Растворитель удаляли при пониженном давлении и полученный остаток очищали с помощью хроматографии на силикагеле, элюируя линейным градиентом с понижением от гексана до смеси гексан:этилацетат (1:1) в течение 20 мин, с получением указанного в заголовке соединения (выход 31%). МС (m/z): 363 (М+1). Следующее соединение в табл. 9 получали, по существу, в соответствии с получением (S)-N-(4-(4 фтор-3-(3-фторпиразин-2-ил)фенил)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-2-ил)ацетамида. Таблица 9 К охлажденному до 0 С раствору (R,Z)-N-(1-(3-бром-4-фторфенил)этилиден)-2-метилпропан-2 сульфинамида (10 г, 31 ммоль, 1,0 экв.) в дихлорметане (100 мл) медленно добавляли хлорид 2-метилаллилмагния (0,5 М в ТГФ, 250 мл, 125,92 ммоль, 4 экв.). Через 2 ч реакцию гасили насыщенным хлоридом аммония и экстрагировали этилацетатом. Растворитель удаляли при пониженном давлении и полученный остаток очищали с помощью хроматографии на силикагеле, элюируя линейным градиентом от дихлорметана до смеси 10% дихлорметан:этилацетат, с получением указанного в заголовке соединения (выход 35%). МС (m/z): 376, 378 (М+1). Пример синтеза 22.(R)-N-S)-2-(3-бром-4-фторфенил)-4-метилпент-4-ен-2-ил)-2-метилпропан-2 сульфинамида (1,8 г, 5,1 ммоль, 1 экв.) в 1,4-диоксане (6 мл) добавляли хлористый водород (4,0 М в 1,4-диоксане, 15 мл). Реакционную смесь перемешивали в течение 5 мин и растворитель удаляли при пониженном давлении. К остатку добавляли насыщенный водный бикарбонат натрия и смесь экстрагировали этилацетатом. Объединенную органическую фазу сушили над сульфатом натрия, фильтровали и- 18016955 концентрировали при пониженном давлении с получением указанного в заголовке соединения (выход 97%). 1 Н ЯМР (CDCl3, 400 МГц):7,66 (дд, 1 Н, J=6,40, 2,80 Гц), 7,37-7,33 (м, 1 Н), 7,02 (т, 1 Н, J=8,40 Гц),4,83 (с, 1 Н), 4,62 (с, 1 Н), 2,47 (д, 1 Н, J=13,2 Гц), 2,35 (д, 1 Н, J=13,2 Гц), 1,44 (с, 3 Н), 1,36 (с, 3 Н). Пример синтеза 23.(5 мл) добавляли бензоилизотиоцианат (0,63 мл, 4,6 ммоль, 1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли при пониженном давлении и полученный остаток очищали с помощью хроматографии на силикагеле, элюируя линейным градиентом от 20% дихлорметан:гексаны до 50% дихлорметан:гексаны, с получением указанного в заголовке соединения К охлажденному до 0 С раствору (S)-N-(2-(3-бром-4-фторфенил)-4-метилпент-4-ен-2 илкарбамотиоил)бензамида (1,3 г, 3,0 ммоль, 1,0 экв.) в дихлорметане (40 мл) добавляли йод (1,5 г,5,9 ммоль, 2,0 экв.). Реакционную смесь перемешивали при 0 С в течение 1 ч и постепенно нагревали до комнатной температуры. Реакционную смесь гасили насыщенным водным тиосульфатом натрия. Водный слой экстрагировали дихлорметаном. Объединенную органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (выход 82%). МС (m/z): 561, 563 (М+1). Пример синтеза 25.(0,006 г, 0,03 ммоль, 0,15 экв.) и гидрид три-н-бутилолова. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч и концентрировали при пониженном давлении. Растворитель удаляли при пониженном давлении и полученный остаток очищали с помощью хроматографии на силикагеле,элюируя линейным градиентом от 5% этилацетат:гексаны до 20% этилацетат:гексаны, с получением указанного в заголовке соединения (выход 25%). МС (m/z): 435, 437 (М+1). Пример синтеза 26.(0,301 г, 0,92 ммоль, 6,1 экв.) и хлорид бис-(трифенилфосфин)палладия(II) (0,022 г, 0,03 ммоль, 0,2 экв.). Реакционную смесь перемешивали при 97 С в течение 20 мин. После охлаждения до комнатной температуры реакционную смесь вливали в воду и экстрагировали этилацетатом. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Растворитель удаляли при пониженном давлении и полученный остаток очищали с помощью колоночной флэшхроматографии на силикагеле, элюируя линейным градиентом от дихлорметана до 15% этилацетат:дихлорметан, с получением указанного в заголовке соединения (выход 46%). МС (m/z): 435 (М+1). К охлажденному до -78 С раствору гидрохлорида N,O-диметилгидроксиламина (12 г, 130 ммоль,5,0 экв.) в ТГФ (200 мл) добавляли через трубочку н-бутиллитий (100 мл, 250 ммоль, 10 экв.) (2,5 М в гексанах). Реакционную смесь перемешивали в течение 15 мин и добавляли по каплям раствор метилового эфира (S)-3-R)-2-метилпропан-2-сульфиниламино)-3-фенилмасляной кислоты (9,5 г, 25 ммоль,1,0 экв.) в ТГФ (50 мл). Реакционную смесь нагревали до -60 С и поддерживали при этой температуре в течение 1 ч. Реакцию гасили насыщенным водным хлоридом аммония, разбавляли водой и экстрагировали этилацетатом. Органическую фазу экстрагировали водой, насыщенным водным NaCl, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (выход 60%). МС (m/z): 405, 407 (М+1). Следующие соединения в табл. 10 получали, по существу, как описано для получения(6,2 мл, 18,5 ммоль, 5,0 экв.) и реакционную смесь оставляли для нагревания до температуры окружающей среды. Через 1 ч реакционную смесь охлаждали до -78 С и гасили насыщенным водным хлоридом аммония. Полученную смесь разбавляли водой и экстрагировали этилацетатом. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (выход 95%). МС (m/z): 360, 362 (М+1). Следующие соединения в табл. 11 получали, по существу, как описано для получения [(S)-1-(3 бромфенил)-1-метил-3-оксобутил]амида (R)-2-метилпропан-2-сульфиновой кислоты.[(S)-1-(3-бромфенил)-1-метил-3 оксобутил]амида (1,34 г, 3,5 ммоль, 1,0 экв.) в метаноле (20 мл) добавляли терагидроборат натрия (1,4 г,35 ммоль, 10,0 экв.). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь осторожно гасили водой и экстрагировали этилацетатом. Органическую фазу сушили над сульфатом натрия и растворитель удаляли при пониженном давлении с получением смеси диастереомеров. Диастереомеры разделяли с помощью хроматографии на силикагеле (120 г), элюируя градиентом от (50:50) этилацетат:гексан до (100:0) этилацетат:гексан. Элюируемый вторым изомер выделяли и растворитель удаляли при пониженном давлении с получением указанного в заголовке соединения в виде отдельного диастереомера (выход 45%). МС (m/z): 362, 364 (М+1). Следующие соединения в табл. 12 получали, по существу, как описано для получения (R)-N-2S)-2(3-бромфенил)-4-гидроксипентан-2-ил)-2-метилпропан-2-сульфинамида. Таблица 12 Указанное в заголовке соединение выделяли и использовали в виде смеси диастереомеров. 3 Указанное в заголовке соединение выделяли и использовали в виде смеси диастереомеров 65:35. Раствор хлористого водорода (5 мл; 13 экв.; 20 ммоль) (4 М в диоксане) и [(S)-1-(3-бромфенил)-3 гидрокси-1-метилбутил]амида (R)-2-метилпропан-2-сульфиновой кислоты (570 мг, 1,6 ммоль, 1,0 экв.) в виде отдельного диастереомера перемешивали в течение 5 мин. Растворитель удаляли при пониженном давлении и остаток делали основным с помощью насыщенного водного бикарбоната натрия. Водную- 21016955 фазу экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения. МС (m/z): 358, 360 (М+1). Следующие соединения в табл. 13 получали, по существу, как описано для получения (S)-4-амино 4-(3-бромфенил)пентан-2-ола. Таблица 13 Указанное в заголовке соединение выделяли в виде смеси диастереомеров. К раствору (S)-4-амино-4-(3-бромфенил)пентан-2-ола (410 мг, 794 мкмоль) в виде отдельного диастереомера в ТГФ (10 мл) добавляли бис-(триметилсилил)трифторацетамид (204 мг, 0,79 ммоль). Через 1 ч добавляли по каплям бензоилизотиоцианат (259 мг, 1,7 ммоль). Реакционную смесь перемешивали в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. К полученному в результате остатку добавляли 5N хлористый водород (25 мл, 125 ммоль) и реакционную смесь нагревали до 100 С. Через 48 ч растворитель удаляли при пониженном давлении и остаток распределяли между ТГФ(20 мл) и насыщенным бикарбонатом натрия (10 мл). К смеси добавляли ди-трет-бутилдикарбонат(347 мг, 1,6 ммоль) и реакционную смесь перемешивали в течение 48 ч. Реакционную смесь разбавляли водой и экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Продукт очищали с помощью силикагеля, элюируя линейным градиентом от гексана до смеси гексан:этилацетат (5:2) в течение 20 мин, с получением указанного в заголовке соединения (выход 52%, чистота 70%, как определено с помощью ЖХ/МС). МС (m/z): 399, 401 (М+1). Пример синтеза 32. трет-Бутил-(4S,6R)-4-(3-бром-4-фторфенил)-4,6-диметил-5,6-дигидро-4 Н-1,3-тиазин-2-илкарбамат. Раствор (R)-N-2S)-2-(3-бром-4-фторфенил)-4-гидроксипентан-2-ил)-2-метилпропан-2-сульфинамида в виде смеси диастереомеров 1:5 (2 г, 1,0 экв.) в диоксане (5 мл) добавляли по каплям к раствору 4N хлористого водорода в диоксане (20 мл, 80 ммоль) при 0 С. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 5 мин. Растворитель удаляли при пониженном давлении и остаток делали основным с помощью насыщенного водного бикарбоната натрия. Водную фазу экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток растворяли в ТГФ (50 мл) и охлаждали до 0 С. Добавляли по каплям бензоилизотиоцианат (1,7 г, 10,5 ммоль). Реакционную смесь перемешивали в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в диоксане (5 мл) и переносили в толстостенный стеклянный реакционный сосуд. В раствор добавляли 5N хлористый водород(75 мл, 375 ммоль). Реакционный сосуд накрывали и нагревали до 100 С. Через 24 ч растворитель удаляли при пониженном давлении и остаток распределяли между ТГФ (20 мл) и насыщенным водным бикарбонатом натрия (10 мл). К смеси добавляли ди-трет-бутилдикарбонат (1,7 г, 7,9 ммоль) и реакционную смесь перемешивали в течение 2 ч. Реакционную смесь разбавляли водой и экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии с использованием силикагеля (340 г), элюируя градиентом от (0:100) смеси этилацетат:гексан до (50:50) смеси этилацетат:гексан в течение 25 мин. Элюируемый вторым диастереомер собирали и растворитель удаляли при пониженном давлении с получением 595 мг смеси. Пример синтеза 32. трет-Бутил-(4S,6R)-4-(3-бром-4-фторфенил)-4,6-диметил-5,6-дигидро-4 Н-1,3 тиазин-2-илкарбамат. МС (m/z): 417, 419 (М+1). Пример синтеза 32 а. К охлажденному до 0 С раствору (4S)-4-амино-4-(5-бром-2,4-дифторфенил)пентан-2-ола (1,3 г,4,4 ммоль) в виде смеси диастереомеров в ТГФ (50 мл) добавляли по каплям бензоилизотиоцианат (1,4 г,1,2 ммоль) и реакционную смесь перемешивали в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в диоксане (5 мл) и переносили в толстостенный стеклянный реакционный сосуд. К смеси добавляли 5N хлористый водород (75 мл, 375 ммоль) и реакционную смесь нагревали до 100 С. Через 36 ч растворитель удаляли при пониженном давлении и остаток растворяли в воде. Водную смесь экстрагировали этилацетатом. Водную фазу делали основной с помощью 5N NaOH и экстрагировали смесью 3:1 хлороформ:изопропанол (IPA). Фазу со смесью хлороформ:IPA сушили над сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении с получением указанного в заголовке соединения. Этилацетатный экстракт сушили над сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении. Полученный в результате остаток пропускали через уравновешенную MeOH колонку SCX, промывали метанолом, а затем элюировали 2N NH3 в MeOH (50 мл). Смыв 2N NH3 в MeOH собирали и растворитель удаляли при пониженном давлении. Остаток объединяли с указанным в заголовке соединением, полученным экстрагированием смесью хлороформ:IPA, с получением указанного в заголовке соединения в виде смеси (6R и 6S) (4S)-4-(5-бром-2,4-дифторфенил)-4,6-диметил-5,6-дигидро 4 Н-1,3-тиазин-2-амина. К смеси (6R и 6S) (4S)-4-(5-бром-2,4-дифторфенил)-4,6-диметил-5,6-дигидро-4 Н-1,3-тиазин-2 амина в ТГФ (20 мл) и насыщенного водного бикарбоната натрия (10 мл) добавляли ди-третбутилдикарбонат (900 мг, 4,1 ммоль). Через 2 ч реакционную смесь разбавляли водой и экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Продукт очищали с помощью силикагеля, элюируя линейным градиентом от гексана до смеси гексан:этилацетат (4:1) в течение 5 мин. Элюируемый вторым диастереомер собирали и растворитель удаляли при пониженном давлении с получением указанного в заголовке соединения (выход 43%). МС (m/z): 435, 437 (М+1). Пример синтеза 35. трет-Бутиловый эфир К нагретому до 100 С раствору трет-бутил (4S,6R)-4-(3-бромфенил)-4,6-диметил-5,6-дигидро-4 Н 1,3-тиазин-2-илкарбамата в смеси 1,2-диметоксиэтан:вода:этанол [(3:1,5:1), 12 мл] добавляли одной порцией пиримидин-5-бороновую кислоту(128 мг,5,9 ммоль,2,5 зкв.),хлорид бис-(трифенилфосфин)палладия(II) (29 мг, 41 мкмоль, 0,1 экв.) и карбонат цезия (1,24 г, 1,24 ммоль,3 экв.). Через 20 мин реакционную смесь охлаждали до температуры окружающей среды, разбавляли водой и экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной флэш-хроматографии на силикагеле, элюируя линейным градиентом от гексана до смеси гексан:этилацетат (5:2) в течение 20 мин, с получением указанного в заголовке соединения (выход 39%). МС (m/z): 399 (М+1). Следующие соединения в табл. 14 получали, по существу, как описано для получения третбутилового эфира Выделен в виде смеси указанного в заголовке соединения и N-4S,6R)-4-(3 бром-4-фторфенил)-4,6-диметил-5,6-дигидро-4 Н-1,3-тиазин-2-ил)бензамида. МС (m/z): 421 (М+1).(100 мл) и охлаждали до 0 С. Медленно добавляли к смеси N-бутиллитий (2,5 М в гексанах, 27,0 г,97,7 ммоль, 39,2 мл, 1,3 экв.) через воронку для добавления. Полученный в результате раствор перемешивали в течение 1 ч при 0 С. Медленно добавляли раствор 3-бромацетофенона (15,0 г, 75,3 ммоль,10,0 мл, 1,0 экв.) в тетрагидрофуране (50 мл) через воронку для добавления. Полученную в результате смесь нагревали до комнатной температуры и перемешивали в течение 3 ч. Реакционную смесь охлаждали до 0 С и гасили насыщенным водным раствором хлорида аммония. Слои разделяли в разделительной воронке и водную фазу экстрагировали гексанами. Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и давали отстояться в течение ночи при комнатной температуре. Органическую фазу сливали с осадка и концентрировали при пониженном давлении. Полученное в результате твердое вещество разбавляли гексанами и фильтровали. Осадок промывали гексанами. Фильтрат концентрировали и полученную в результате смесь очищали с помощью колоночной флэшхроматографии на силикагеле (гексаны) с получением указанного в заголовке соединения (выход 83%). 1 Н ЯМР (CDCl3, 400 МГц):7,59 (т, 1 Н, J=1,72 Гц), 7,40-7,36 (м, 2 Н), 7,18 (т, 1 Н, J=8 Гц), 5,36 (с,1 Н), 5,12-5,10 (м, 1 Н), 2,13-3,11 (м, 3 Н). Следующее соединение в табл. 15 получали, по существу, в соответствии с получением 1-бром-3(проп-1-ен-2-ил)бензола. Таблица 15(124 мл, 0,5 М) добавляли этилглиоксалат (38,1 г, 37 мл, 187 ммоль, 3 экв.) и гидрат трифторметансульфоната иттербия (7,72 г, 12,4 ммоль, 0,2 экв.). Полученную смесь перемешивали в течение ночи при комнатной температуре. Смесь концентрировали при пониженном давлении и разбавляли диэтиловым эфиром. Полученный в результате раствор промывали дважды водой. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле (330 г) двумя партиями, используя гради- 24016955 ент 0-100% смеси этилацетат/гексаны, с получением указанного в заголовке соединения (выход 87%). 1 Н ЯМР (CDCl3, 400 МГц):7,53 (т, 1 Н, J=1,9 Гц), 7,41-7,37 (дт, 1 Н), 7,33-7,30 (дт, 1 Н), 7,18 (т, 1 Н,J=7,6 Гц), 5,37 (с, 1 Н), 5,22 (с, 1 Н), 4,26-4,21 (м, 1 Н), 4,17-3,99 (м, 2 Н), 2,99 (дд, J=14,8, 4,8 Гц), 2,83-2,72 К раствору этил 4-(3-бромфенил)-2-гидроксипент-4-еноата (5,1 г, 17 ммоль) в ацетонитриле (68 мл) добавляли 2,6-лутидин (2,19 г, 20,4 ммоль, 1,2 экв.). Реакционную смесь охлаждали до 0 С и добавляли по каплям трифторметансульфоновый ангидрид (3,30 мл, 19,6 ммоль, 1,15 экв.) в течение приблизительно 5 мин. Смесь перемешивали при 0 С в течение 20 мин. Добавляли тиомочевину (2,59 г, 34,0 ммоль,2 экв.) и реакционную смесь нагревали до комнатной температуры. Через 45 мин смесь концентрировали при пониженном давлении. Полученное в результате вязкое оранжевое масло затем добавляли большой пипеткой в перемешиваемую серную кислоту (17,8 М, 8 мл) при комнатной температуре. Через 20 мин смесь добавляли по каплям в интенсивно перемешиваемый охлажденный до 0 С раствор K2CO3(приблизительно 50 г) в 50 мл H2O. Чтобы позволить перемешивание при образовании твердого вещества во время гашения, дополнительно добавляли воду. Желтовато-коричневое/оранжевое твердое вещество собирали путем фильтрования. Твердое вещество оставляли для высыхания на фильтровальной бумаге под потоком воздуха в течение 1 ч с получением указанного в заголовке соединения в виде смеси диастереомеров, которую использовали без дополнительной очистки. МС (m/z): 357, 359 (М+1). Следующее соединение в табл. 17 получали, по существу, как описано для получения этил 2-амино 4-(3-бромфенил)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-6-карбоксилата. Таблица 17(6,1 г, 17 ммоль) в 1,4-диоксане (35 мл), воде (18 мл) и насыщенном водном бикарбонате натрия (18 мл) добавляли ди-трет-бутилдикарбонат (7,42 г, 34,0 ммоль, 2 экв.). Полученную смесь перемешивали в течение приблизительно 60 ч. Реакционную смесь разбавляли водой и экстрагировали трижды CH2Cl2. Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением коричневого масла. Масло очищали с помощью колоночной флэш-хроматографии на силикагеле (150 г), элюируя градиентом от 0 до 100% смеси этилацетат:гексан, с получением указанного в заголовке соединения в виде смеси диастереомеров (выход 64%). МС (m/z): 457, 459 (М+1). Следующее соединение в табл. 18 получали, по существу, в соответствии с получением этил 4-(3-бромфенил)-2-(трет-бутоксикарбониламино)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-6-карбоксилата. Таблица 18 Этил 4-(3-бромфенил)-2-(трет-бутоксикарбониламино)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-6 карбоксилат (3,5 г, 7,7 ммоль) очищали с помощью хиральной ВЭЖХ в две стадии: (колонка: Chiralcel OJ 832 см; элюент: 1:3 (3 А спирт:гептан); поток: 400 мл/мин при УФ 240 нм), при этом брали пики 1 и 2 из 3; затем пики 1 и 2 дополнительно очищали с помощью хиральной хроматографии (колонка: ChiralcelOD 832 см; элюент 1:9 (изопропиловый спирт:гептан); поток: 400 мл/мин при УФ 240 нм). Выделение элюируемого вторым изомера позволяло получить указанное в заголовке соединение после концентрирования фракций при пониженном давлении (выход 14%). Пример синтеза 41.(25 мл) добавляли боргидрид лития (289 мг, 13,1 ммоль, 3 экв.). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 4 ч. Реакционную смесь гасили насыщенным NH4Cl. Слои разделяли и водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным водным NaCl и сушили над Na2SO4. Смесь фильтровали и концентрировали с получением светло-желтого масла. Масло очищали с помощью колоночной хроматографии на силикагеле(120 г), используя градиент от 0 до 100% смеси этилацетат:гексан, с получением указанного в заголовке соединения (выход 29%).(+/-) трет-Бутил (4S,6S)-4-(3-бромфенил)-6-(гидроксиметил)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-2 илкарбамат (870 мг, 2,1 ммоль) очищали путем хирального разделения ВЭЖХ: (колонка: Chiralpak AD 836 см 20 мкм; элюент: 100% 3 А этиловый спирт; поток: 400 мл/мин при УФ 250 нм). Элюируемый вторым изомер выделяли, чтобы получить энантиомерно обогащенное указанное в заголовке соединение К нагретому до 110 С раствору (+/-) трет-бутил (4S,6S)-4-(3-бромфенил)-6-(гидроксиметил)-4 метил-5,6-дигидро-4 Н-1,3-тиазин-2-илкарбамата (150 мг, 0,36 ммоль) в 1,2-диметоксиэтане (4,5 мл), этаноле (1,5 мл) и воде (2,3 мл) добавляли пиримидин-5-бороновую кислоту (112 мг, 0,90 ммоль, 2,5 экв.),карбонат цезия (353 мг, 1,08 ммоль, 3 экв.) и хлорид бис-(трифенилфосфин)палладия(II) (25 мг,0,036 ммоль, 0,1 экв.). Реакционную смесь перемешивали при 110 С. Через 20 мин реакционную смесь разбавляли EtOAc и H2O. Слои разделяли и водную фазу экстрагировали EtOAc. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью колоночной хроматографии на силикагеле (5% MeOH/DCM) с получением указанного в заголовке соединения (выход 29%). МС (m/z): 415 (М+1). Следующее соединение в табл. 19 получали, по существу, в соответствии с получением (+/-) третбутил(0,58 мл, 1,75 ммоль, 3,2 экв.). Через 15 мин добавляли дополнительное количество хлорида метилмагния(0,38 мл, 1,2 ммоль, 2 экв.). Через 30 мин реакционную смесь гасили насыщенным водным NH4Cl и разбавляли этилацетатом. Слои разделяли и водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным водным NaCl, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью колоночной хроматографии на силикагеле (80 г), элюируя градиентом от 0 до 100% смеси этилацетат:гексаны, с получением указанного в заголовке соединения (выход 26%). МС (m/z): 443, 445 (М+1). Следующие соединения в табл. 20 получали, по существу, в соответствии с получением (+/-) третбутил (4S,6S)-4-(3-бромфенил)-6-(2-гидроксипропан-2-ил)-4-метил-5,6-дигидро-4 Н-1,3-тиазин-2-илкарбамата.(22 мл) и воде (7 мл) добавляли пиримидин-5-бороновую кислоту (1,2 г, 9,6 ммоль, 4 экв.), хлорид бис(трифенилфосфин)палладия(II) (508 мг, 0,723 ммоль, 0,3 экв.) и карбонат цезия (2,36 г, 7,2 ммоль,3 экв.). Через 25 мин смесь охлаждали до комнатной температуры. Реакционную смесь разбавляли EtOAc и распределяли между EtOAc и водой. Водную фазу экстрагировали 3 раза EtOAc. Объединенную органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный остаток очищали с помощью колоночной хроматографии на силикагеле (80 г), элюируя EtOAc, с получением указанного в заголовке соединения Рацемическое соединение, полученное и очищенное с помощью хиральной хроматографии при условиях, описанных в примере синтеза 46.(+/-) трет-Бутил (4S,6S)-4-(4-фтор-3-(пиримидин-5 ил)фенил)-6-(2-гидроксипропан-2-ил)-4-метил 5,6-дигидро-4 Н-1,3-тиазин-2-илкарбамат (455 мг, 0,99 ммоль) очищали с помощью хирального разделения ВЭЖХ (колонка: Chiralpak AD-H 2,125 см 5 мкм; элюент: 20% EtOH/СО 2; поток: 70 мл/мин при УФ 225 нм). Элюируемый вторым изомер выделяли, чтобы получить энантиомерно обогащенное указанное в заголовке соединение (29%). МС (m/z): 461 (М+1). Примеры Пример 1.(160 мг, 0,531 ммоль) в ТГФ (4 мл) добавляли насыщенный раствор HCl в диоксане (2 мл) при 0 С. Реакционную смесь оставляли для перемешивания при комнатной температуре в течение 4 ч. Растворитель удаляли при пониженном давлении. Полученное в результате твердое вещество многократно промывали безводным эфиром и сушили при пониженном давлении с получением указанного в заголовке соединения (выход 87%). МС (m/z): 302 (М+1). Пример 2. Дигидрохлорид (S)-4-[3-(5-хлор-2-фторпиридин-3-ил)фенил]-4-метил-5,6-дигидро-4 Н-[1,3]тиазин 2-иламина 2785443, PG6-E01268-074(S)-N-4-[3-(5-хлор-2-фторпиридин-3-ил)фенил]-4-метил-5,6-дигидро-4 Н-[1,3]тиазин-2 илацетамида (430 мг, 1,1 ммоль) в трифторуксусной кислоте (50 мл) и метаноле (50 мл) перемешивали в течение 8 ч при 60 С. Растворитель удаляли при пониженном давлении. Остаток растворяли в воде и нейтрализовали с помощью насыщенного бикарбоната и экстрагировали этилацетатом. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Полученный остаток очищали с помощью колоночной флэш-хроматографии на силикагеле, элюируя этилацетатом. Полученный в результате амин растворяли в дихлорметане и барботировали раствор газообразным HCl в течение 30 с. Растворитель удаляли при пониженном давлении с получением указанного в заголовке соединения (52%). МС (m/z): 336 (М+1). Следующие соединения в табл. 22 получали, по существу, как описано для получения дигидрохлорида (S)-4-[3-(5-хлор-2-фторпиридин-3-ил)фенил]-4-метил-5,6-дигидро-4 Н-[1,3]тиазин-2-иламина.
МПК / Метки
МПК: A61P 25/28, A61K 31/541, C07D 279/06
Метки: болезни, качестве, аминодигидротиазиновые, ингибиторов, лечения, производные, альцгеймера
Код ссылки
<a href="https://eas.patents.su/30-16955-aminodigidrotiazinovye-proizvodnye-v-kachestve-ingibitorov-bace-dlya-lecheniya-bolezni-alcgejjmera.html" rel="bookmark" title="База патентов Евразийского Союза">Аминодигидротиазиновые производные в качестве ингибиторов bace для лечения болезни альцгеймера</a>
Производные терфенила для лечения болезни альцгеймера

Номер патента: 16908
Опубликовано: 30.08.2012
Авторы: Смэлт Кэсрин, Кэнсфильд Эндру, Хэррисон Ричард Джон, Вилсн Фрэнцис, Хо Чи Янг, Ридер Вэлери, Мэйджер Джереми, Рид Элисон, Буссар Сирилль, Зэнг Ен, Лёформаль Эдэляйн, Тэйлор Джесс, Хернадез-Перни Рэмэдиос, Зунузэ Михиро, Буркхардт Свенья
МПК: C07C 51/09, A61K 31/192, A61P 25/28...
Метки: лечения, производные, терфенила, болезни, альцгеймера
Формула / Реферат:
1. Соединение общей формулы (I)где X означает связь или группу -CR5R6, где R5 и R6 независимо друг от друга выбраны из группы, состоящей из Н; алкила, выбранного из группы СН3, С2Н5, изо-С3Н7, н-C3H7, изо-С4Н9, н-C4H9, втор-C4H9, трет-С4Н9;R1, R2, R3 и R4 независимо выбраны из группы, состоящей из Н; F; Cl; Br; I; CN; S(O)2R7; N(R7R8); C(O)R7; замещенного или незамещенного С1-С4-алкила и замещенного или незамещенного С1-С4-алкокси, и где...
Комбинация nmda-антагониста и ингибиторов ацетилхолинэстеразы для лечения болезни альцгеймера

Номер патента: 7632
Опубликовано: 29.12.2006
Авторы: Томсен Ларс Ликке, Педерсен Андерс Герсел
МПК: A61P 25/28, A61K 31/13, A61K 31/55...
Метки: лечения, ингибиторов, ацетилхолинэстеразы, комбинация, альцгеймера, nmda-антагониста, болезни
Формула / Реферат:
1. Применение композиции, содержащей: (a) эффективное количество ингибитора ацетилхолинэстеразы, выбранного из группы, состоящей из донепезила, такрина, ривастигмина и галантамина, или их смесей, или их фармацевтически приемлемых солей; и (b) эффективное количество NMDA-антагониста мемантина или его фармацевтически приемлемых солей, в качестве активного ингредиента при изготовлении лекарственного препарата для лечения легких форм нарушений...
2,5-бис-диамин-[ 1,4 ] бензохиноновые производные для лечения болезни альцгеймера, способ их получения и промежуточные продукты

Номер патента: 7909
Опубликовано: 27.02.2007
Авторы: Кавалли Андреа, Андризано Винченца, Мелькиорре Карло, Реканатини Маурицио, Бартолини Мануэла, Болоньези Мария Лаура
МПК: C07C 221/00, A61P 25/28, C07C 225/28...
Метки: 2,5-бис-диамин, способ, промежуточные, получения, болезни, лечения, альцгеймера, продукты, бензохиноновые, производные
Формула / Реферат:
1. 2,5-бис-Диамин-[1,4]бензохиноновое производное общей формулы (I) где R1 представляет собой заместитель, выбранный из группы, включающей водород, насыщенную или ненасыщенную, линейную или разветвленную алкильную группу из одного-пяти атомов углерода, и заместитель, обладающий электроноакцепторным индукционным свойством; R2 и R3, каждый независимо от другого, представляют собой водород или насыщенную или ненасыщенную, линейную или...
Соединения для лечения болезни альцгеймера

Номер патента: 7337
Опубликовано: 25.08.2006
Авторы: Ягоджинска Барбара, Фрескос Джон Н., Хом Рой, Мэйлард Мишель, Пулли Шон Р., Фанг Лоренс И., Тенбринк Рут Е., Джон Варгезе, Бек Джеймс П., Гэйлунас Андреа
МПК: A61K 31/33, C07C 233/40, A61K 31/165...
Метки: лечения, болезни, альцгеймера, соединения
Формула / Реферат:
1. Замещенный амин формулы (X) где R1 представляет собой -(CH2)n1-(R1-арил), где n1 представляет собой ноль или один и где R1-арил представляет собой фенил, 1-нафтил, 2-нафтил, инданил, инденил, дигидронафтил или тетралинил, который может быть замещен одним, двумя, тремя или четырьмя следующими заместителями на арильное кольцо: -F, Cl, -Br или -I, R2 представляет собой -Н, R3 представляет собой -Н, RN представляет собой RN-1-XN-, где XN...
Применение фанхинона для лечения болезни альцгеймера

Номер патента: 2526
Опубликовано: 27.06.2002
Авторы: Геролиматос Панайотис Николас, Ксилинас Мишель
МПК: A61K 31/435
Метки: альцгеймера, болезни, применение, фанхинона, лечения
Формула / Реферат:
1. Применение фанхинона для производства фармацевтической композиции для лечения или предотвращения болезни Альцгеймера. 2. Применение фанхинона и соединения или смеси соединений, выбранных из группы, включающей антиоксиданты, усилители ацетилхолина, следовые металлы, простетические группы и клиохинол, для производства фармацевтической композиции для лечения или предотвращения болезни Альцгеймера. 3. Применение по п.2, при котором антиоксидантом...
Предыдущий патент: Вращающееся перемешивающее устройство для обработки расплавленного металла
Следующий патент: Способ выемки выбросоопасного калийного маломощного пласта
Случайный патент: Тренажер специализированный для восстановления двигательных функций