Производные и аналоги n-этилхинолонов и n-этилазахинолонов
Номер патента: 16470
Опубликовано: 30.05.2012
Авторы: Чжан Лихуа, Хоффман Джеймс Б., Баррос Дэвид, Джоунс Грэхем Элджин, Джордано Илариа, Дэббс Стивен, Кастро Пичел Джулия, Пирсон Нейл Дэвид, Хеннесси Алан Джозеф, Росси Джейсон Энтони, Дэвис Дэвид Томас, Ремуиньян Бланко Модесто Х., Фиандор Роман Хосе Мария, Пендрак Израиль, Майлз Тимоти Джеймс, Дэйнс Роберт А., Брукс Джеральд, Баллелл Ллуис



























Формула / Реферат
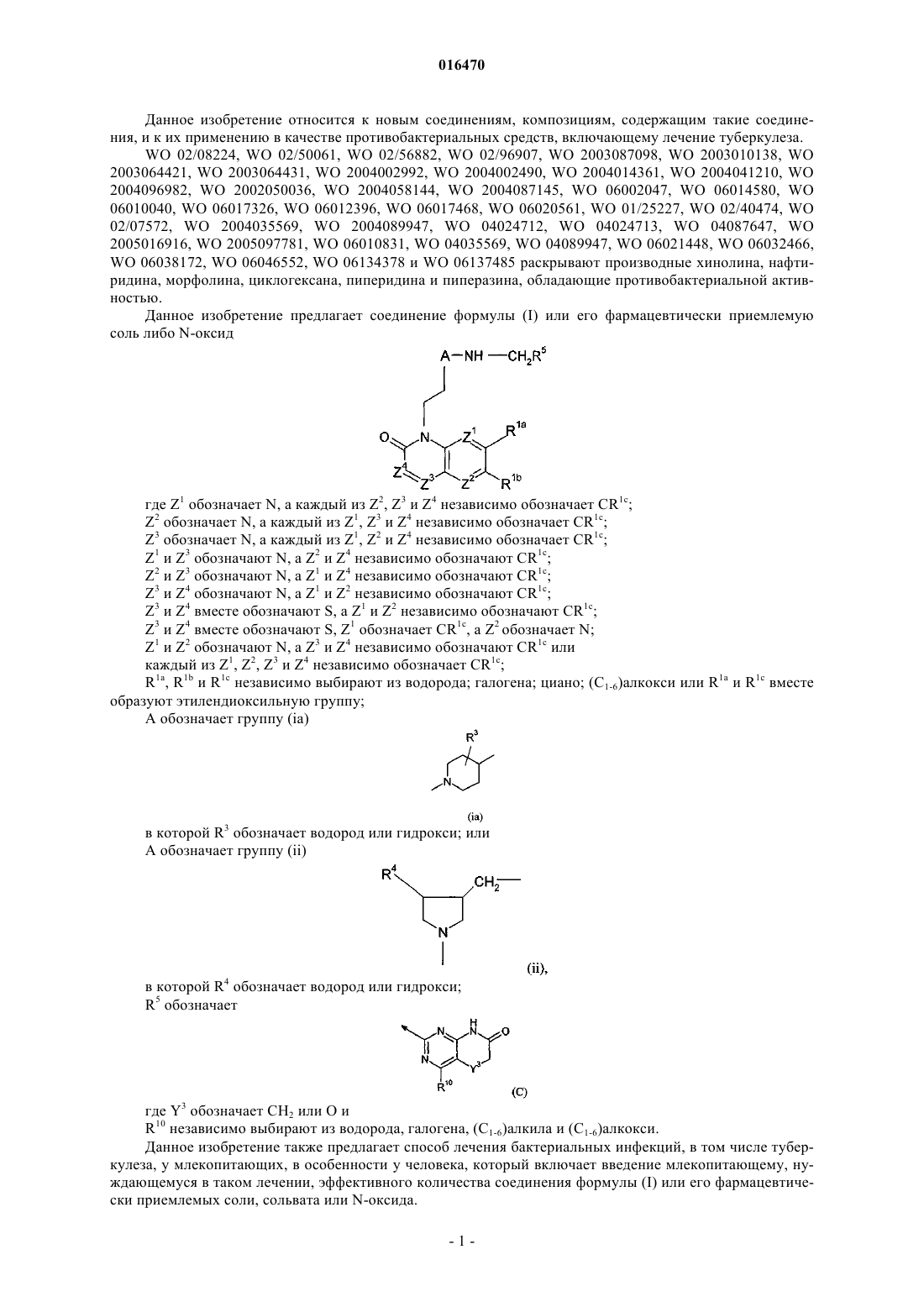
1. Соединение формулы (I) или его фармацевтически приемлемая соль либо N-оксид

где Z1 обозначает N, а каждый из Z2, Z3 и Z4 независимо обозначает CR1c;
Z2 обозначает N, а каждый из Z1, Z3 и Z4 независимо обозначает CR1c;
Z3 обозначает N, а каждый из Z1, Z2 и Z4 независимо обозначает CR1c;
Z1 и Z3 обозначают N, a Z2 и Z4 независимо обозначают CR1c;
Z2 и Z3 обозначают N, a Z1 и Z4 независимо обозначают CR1c;
Z3 и Z4 обозначают N, a Z1 и Z2 независимо обозначают CR1c;
Z3 и Z4 вместе обозначают S, a Z1 и Z2 независимо обозначают CR1c;
Z3 и Z4 вместе обозначают S, Z1 обозначает CR1c, a Z2 обозначает N;
Z1 и Z2 обозначают N, a Z3 и Z4 независимо обозначают CR1c или
каждый из Z1, Z2, Z3 и Z4 независимо обозначает CR1c;
R1a, R1b и R1c независимо выбирают из водорода; галогена; циано; (C1-6)алкокси или R1a и R1c вместе образуют этилендиоксильную группу;
A обозначает группу (ia)

в которой R3 обозначает водород или гидрокси; или
A обозначает группу (ii)

в которой R4 обозначает водород или гидрокси и
R5 обозначает

где Y3 обозначает CH2 или O, и
R10 независимо выбирают из водорода, галогена, (C1-6)алкила и (C1-6)алкокси.
2. Соединение по п.1, где R1a обозначает метокси, циано, фтор, хлор или бром, a R1b и R1c обозначают водород.
3. Соединение по п.1, где R5 представляет собой
2-замещенный 1H-пиримидо[5,4-b][1,4]оксазин-7(6H)-он;
2-замещенный 4-хлор-1H-пиримидо[5,4-b][1,4]оксазин-7(6H)-он;
2-замещенный 5,6-дигидропиридо[2,3-d]пиримидин-7(1H)-он;
2-замещенный 4-хлор-5,6-дигидропиридо[2,3-d]пиримидин-7(1H)-он;
2-замещенный 4-метил-5,6-дигидропиридо[2,3-d]пиримидин-7(1H)-он или
2-замещенный 4-метилокси-5,6-дигидропиридо[2,3-d]пиримидин-7(1H)-он

4. Соединение по любому из предшествующих пунктов, где A обозначает группу (ia), a R3 обозначает водород или гидрокси или A обозначает 3-гидроксипирролидин-4-илметил или 4-гидроксипиперидин-3-илметил.
5. Соединение по п.1, где A обозначает 4-гидроксипиперидин-3-илметил.
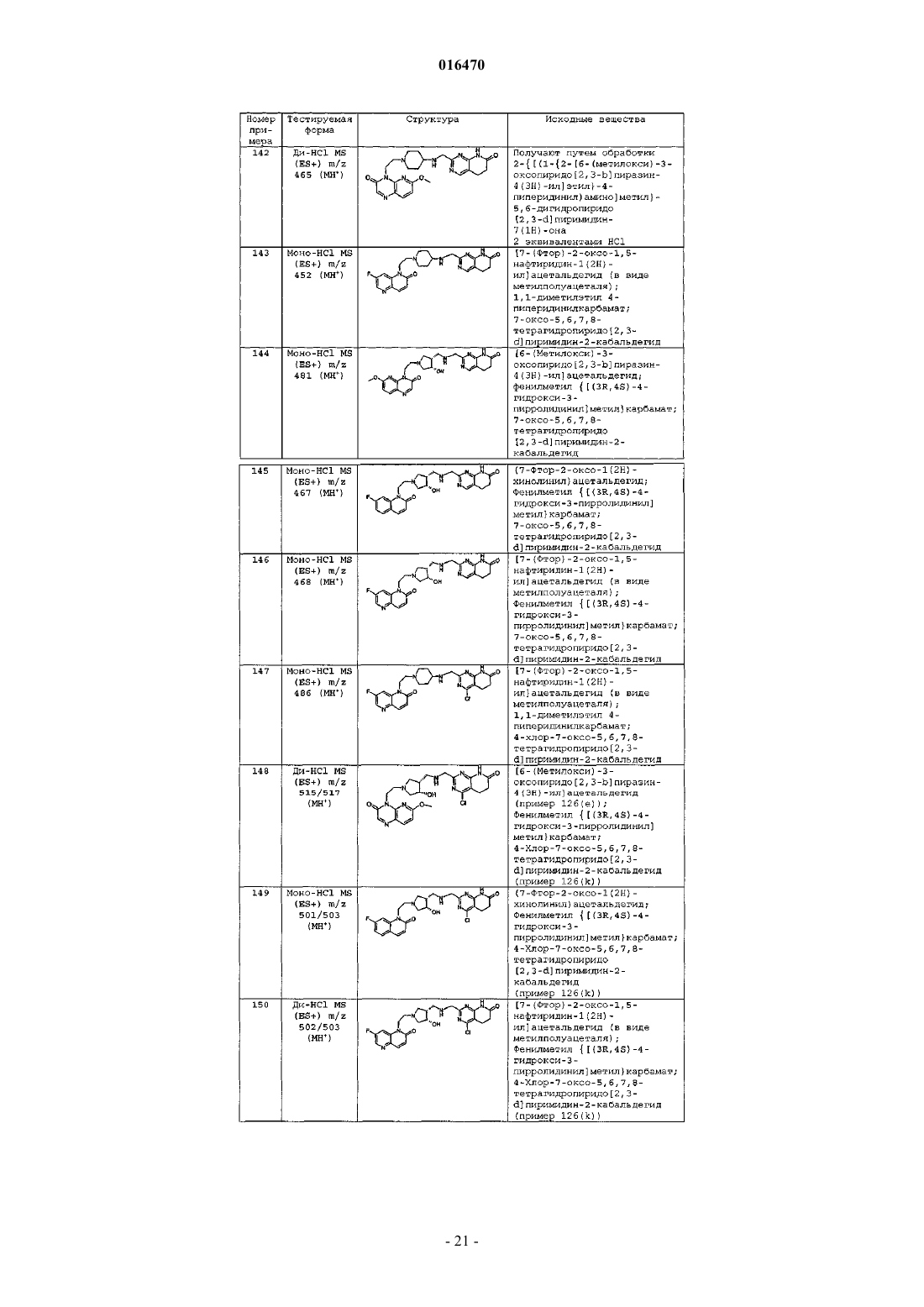
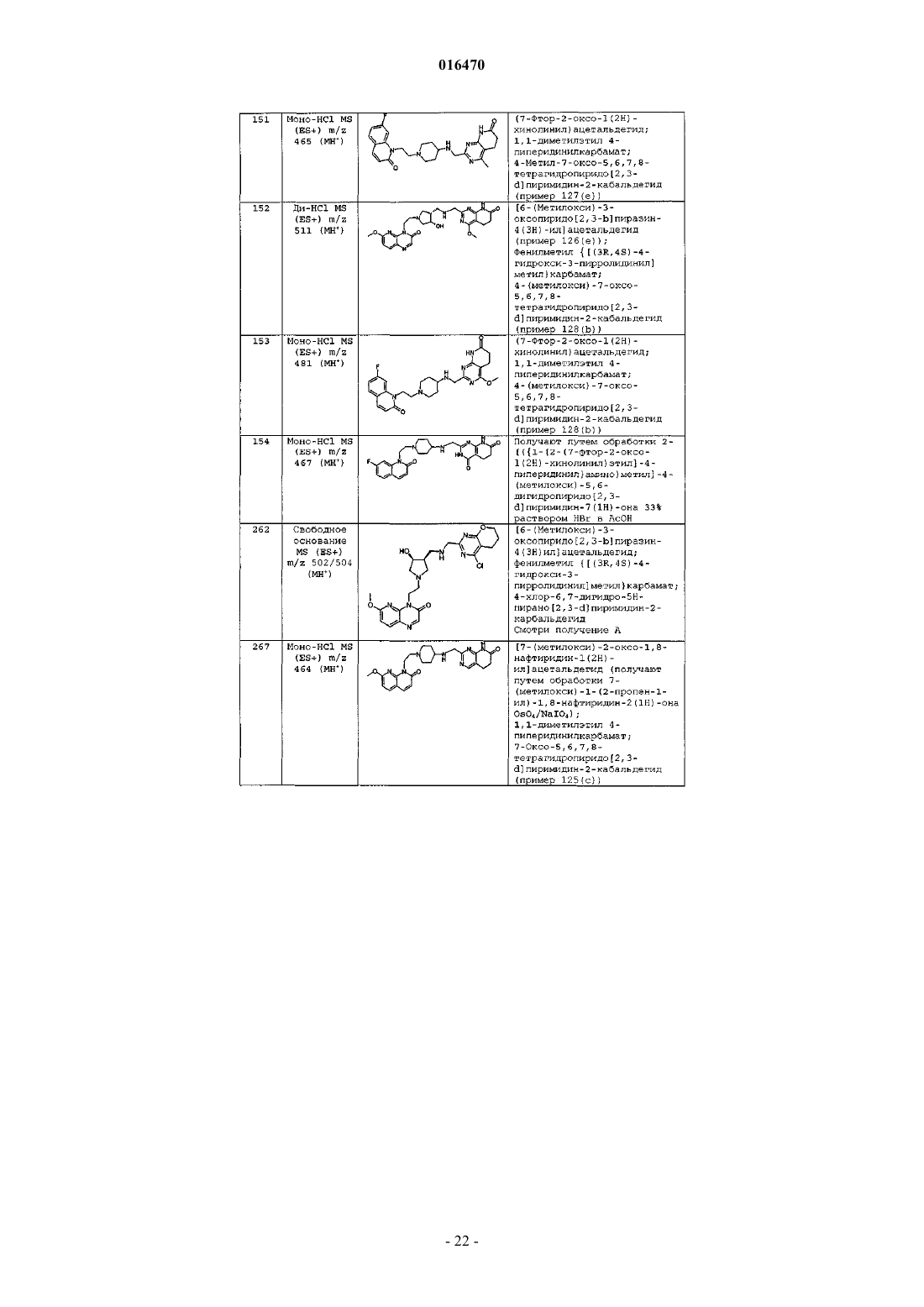
6. Соединение по п.1, выбранное из группы, включающей
2-[({1-[2-(7-фтор-2-оксо-1(2H)-хинолинил)этил]-4-пиперидинил}амино)метил]-1H-пиримидо[5,4-b][1,4]оксазин-7(6H)-он,
4-хлор-2-[({l-[2-(7-фтор-2-оксо-1(2H)-хинолинил)этил]-4-пиперидинил}амино)метил]-1H-пиримидо[5,4-b] [1,4]оксазин-7(6H)-он,
2-{[(1-{2-[6-(метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)-ил]этил}-4-пиперидинил)амино]метил}-5,6-дигидропиридо[2,3-d] пиримидин-7(1H)-он,
4-хлор-2-{[(1-{2-[6-(метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)-ил]этил}-4-пиперидинил)амино]метил}-5,6-дигидропиридо[2,3-d] пиримидин-7(1H)-он,
4-метил-2-{[(1-{2-[6-(метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)-ил]этил}-4-пиперидинил)амино]метил}-5,6-дигидропиридо[2,3-d] пиримидин-7(1H)-он,
4-(метилокси)-2-{[(1-{2-[6-(метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)-ил]этил}-4-пиперидинил)амино]метил}-5,6-дигидропиридо[2,3-d]пиримидин-7(1H)-он,
или свободное основание соединения, представленного в таблице



или фармацевтически приемлемую соль любого из перечисленных выше соединений.
7. Применение соединения по любому из предшествующих пунктов для лечения бактериальных инфекций у млекопитающих.
8. Фармацевтическая композиция, содержащая соединение по любому из предшествующих пунктов и фармацевтически приемлемый носитель.
Текст
Бициклические азотсодержащие соединения общей формулы (I) и их применение в качестве противобактериальных средств. Баллелл Ллуис, Баррос Дэвид (ES),Брукс Джеральд (GB), Кастро Пичел Джулия (ES), Дэббс Стивен (GB),Дэйнс Роберт А. (US), Дэвис Дэвид Томас (GB), Фиандор Роман Хосе Мария (ES), Джордано Илариа,Хеннесси Алан Джозеф (GB),Хоффман Джеймс Б. (US), Джоунс Грэхем Элджин, Майлз Тимоти Джеймс, Пирсон Нейл Дэвид (GB),Пендрак Израиль (US), Ремуиньян Бланко Модесто Х. (ES), Росси Джейсон Энтони, Чжан Лихуа (Лили)(71)(73) Заявитель и патентовладелец: ГЛЭКСО ГРУП ЛИМИТЕД (GB) 016470 Данное изобретение относится к новым соединениям, композициям, содержащим такие соединения, и к их применению в качестве противобактериальных средств, включающему лечение туберкулеза.WO 02/08224, WO 02/50061, WO 02/56882, WO 02/96907, WO 2003087098, WO 2003010138, WO 2003064421, WO 2003064431, WO 2004002992, WO 2004002490, WO 2004014361, WO 2004041210, WO 2004096982, WO 2002050036, WO 2004058144, WO 2004087145, WO 06002047, WO 06014580, WO 06010040, WO 06017326, WO 06012396, WO 06017468, WO 06020561, WO 01/25227, WO 02/40474, WO 02/07572, WO 2004035569, WO 2004089947, WO 04024712, WO 04024713, WO 04087647, WO 2005016916, WO 2005097781, WO 06010831, WO 04035569, WO 04089947, WO 06021448, WO 06032466,WO 06038172, WO 06046552, WO 06134378 и WO 06137485 раскрывают производные хинолина, нафтиридина, морфолина, циклогексана, пиперидина и пиперазина, обладающие противобактериальной активностью. Данное изобретение предлагает соединение формулы (I) или его фармацевтически приемлемую соль либо N-оксидZ1 и Z2 обозначают N, a Z3 и Z4 независимо обозначают CR1c или каждый из Z1, Z2, Z3 и Z4 независимо обозначает CR1c;R1a, R1b и R1c независимо выбирают из водорода; галогена; циано; (C1-6)алкокси или R1a и R1c вместе образуют этилендиоксильную группу; в которой R3 обозначает водород или гидрокси; или в которой R4 обозначает водород или гидрокси;R10 независимо выбирают из водорода, галогена, (C1-6)алкила и (C1-6)алкокси. Данное изобретение также предлагает способ лечения бактериальных инфекций, в том числе туберкулеза, у млекопитающих, в особенности у человека, который включает введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы (I) или его фармацевтически приемлемых соли, сольвата или N-оксида.-1 016470 Данное изобретение также предлагает применение соединения формулы (I) или его фармацевтически приемлемых соли, сольвата или N-оксида в производстве лекарственного средства для лечения бактериальных инфекций, в том числе туберкулеза, у млекопитающих. Данное изобретение также предлагает фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемые соль, сольват или N-оксид и фармацевтически приемлемый носитель. В отдельном аспекте каждый R1a, R1b и R1c независимо обозначает водород, метокси, циано или галоген. В некоторых воплощениях только одна группа из R1a, R1b или R1c отличается от водорода. В отдельном воплощении R1a обозначает метокси, циано или галоген, такой как фтор, хлор или бром, a R1b или R1c обозначают водород. В альтернативном воплощении R1b обозначает группу, отличную от водорода, например фтор. В других воплощениях две группы R1a, R1b или R1c отличаются от водорода. Предпочтительно R1a обозначает фтор, a R1b или R1c обозначает группу, отличную от водорода, например фтор, этил или метокси. В следующих воплощениях Z1 обозначает R1c, a R1a и R1c вместе образуют этилендиоксильную группу. Конкретные примеры R3 включают водород или гидрокси. В следующем аспекте R3 находится в 3- или 4-положении, более предпочтительно в 3-положении. В более предпочтительном аспекте A обозначает (ia), a R3 находится в 3-положении, более предпочтительно в цис-положении по отношению к группе NH. В отдельных воплощениях A обозначает группу (ia), a R3 обозначает водород или гидрокси. Более предпочтительно, если А обозначает 3-гидроксипиперидин-4-иламино, имеет место конфигурация (3R,4S). В отдельном аспекте, если A обозначает (ii), R4 обозначает H или OH. Если A обозначает 3 гидроксипирролидин-4-илметил, в отдельном аспекте конфигурация представляет собой (3S,4S). В некоторых воплощениях R5 обозначаетR10 независимо выбран из водорода, галогена, (C1-6)алкила и (C1-6)алкокси. Более предпочтительно 10 гдеобозначает точку присоединения. В данном описании термин "алкил" включает группы с линейными или разветвленными цепями,например метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, т-бутил, пентил и гексил. Термин "алкенил" следует интерпретировать аналогичным образом. Термин галоген включает фтор, хлор, бром и иод. Соединения данного изобретения содержат гетероциклильную группу и могут существовать в виде двух или более таутомерных форм в зависимости от природы гетероциклильной группы; все указанные таутомерные формы входят в объем настоящего изобретения. Некоторые соединения данного изобретения можно получить путем кристаллизации или перекристаллизации из растворителей, таких как водные и органические растворители. В таких случаях могут образовываться сольваты. В объем данного изобретения входят стехиометрические сольваты, в том чис-2 016470 ле гидраты, а также соединения, содержащие разные количества воды, которые могут образовываться в результате обработки, такой как лиофилизация. Кроме того, следует понимать, что такие фразы, как "соединение формулы (I) или его фармацевтически приемлемые соль либо N-оксид" охватывают соединение формулы (I), N-оксид соединения формулы (I), фармацевтически приемлемую соль соединения формулы (I) или их любое фармацевтически приемлемое сочетание. Так, в качестве неограничивающего примера, используемого в данном документе с целью иллюстрации, "соединение формулы (I) или его фармацевтически приемлемая соль" может включать фармацевтически приемлемую соль соединения формулы (I). Поскольку соединения формулы (I) предназначаются для применения в фармацевтических композициях, очевидно, что в отдельных воплощениях они предоставляются в практически чистом виде и имеют чистоту, которая составляет, например, по меньшей мере 60%, предпочтительно по меньшей мере 75%, более предпочтительно по меньшей мере 85% и наиболее предпочтительно по меньшей мере 98%(% приводятся в массовых соотношениях). Неочищенные препараты соединений можно использовать для получения более чистых форм, используемых в фармацевтических композициях; указанные менее чистые препараты соединений могут содержать по меньшей мере 1%, предпочтительно по меньшей мере 5% и более предпочтительно от 10 до 59% соединения формулы (I) или его фармацевтически приемлемых соли, сольвата или N-оксида. Конкретные соединения данного изобретения включают соединения, упомянутые в примерах, а также их фармацевтически приемлемые N-оксиды, соли. Фармацевтически приемлемые соли вышеупомянутых соединений формулы (I) включают кислотно-аддитивные соли или соли четвертичного аммония, например соли соединений и минеральных кислот, таких как хлористо-водородная, бромисто-водородная, серная, азотная или фосфорная, или органических кислот, таких как уксусная, фумаровая, янтарная, малеиновая, лимонная, бензойная, птолуолсульфоновая, метансульфоновая, нафталинсульфоновая или винная. Соединения формулы (I) также могут быть получены в виде N-оксидов. Данное изобретение охватывает все указанные производные. Некоторые соединения формулы (I) могут существовать в виде оптических изомеров, например диастереомеров, а также смесей изомеров в любых соотношениях, например рацемических смесей. Данное изобретение включает все указанные формы, в особенности чистые изомерные формы. Разные изомерные формы можно разделить или отделить одну от другой с помощью традиционных методов или какой-либо конкретный изомер можно получить с помощью традиционных способов синтеза или с помощью стереоспецифического или ассиметричного синтеза. Кроме того, некоторые соединения формулы(I) могут существовать в виде полиморфных форм, которые также входят с объем данного изобретения. В следующем аспекте данное изобретение предлагает способ получения соединений формулы (I), а также их фармацевтически приемлемых солей, сольватов или N-оксидов, указанный способ включает взаимодействие соединения формулы (II) с соединением формулы (III) где R20 обозначает UR5, или группу, способную превращаться в UR5, a R2' обозначает R2, или группу, способную превращаться в R2, где Z1, Z2, Z3, Z4, A, R1a, R1b, R2, U и R5 имеют значения, указанные в формуле (I), и затем необязательно, или при необходимости, превращение R20 и R2' в UR5 и R2, взаимопревращение некоторых вариабельных групп и/или получение фармацевтически приемлемых соли, сольвата или N-оксида. Взаимодействие представляет собой восстановительное алкилирование (описание способов его проведения можно найти, например, в Smith, M.B.; March, J.M. Advanced Organic Chemistry, WileyInterscience 2001), его проводят с использованием подходящего восстанавливающего реагента, такого как цианоборгидрид натрия (в смеси метанол/хлороформ/уксусная кислота), триацетоксиборгидрид или (полистирилметил)триметиламмония цианоборгидрид. Если амин находится в виде соли хлористоводородной кислоты, предпочтительно забуферивать реакционную смесь избытком ацетата натрия. Для облегчения образования промежуточного имина также можно использовать молекулярные сита 3A. Соединение формулы (II) может быть получено в виде полуацеталя. Один из R20 и R2' может представлять собой N-защитную группу, такую как т-бутоксикарбонил,бензилоксикарбонил, 9-флуоренилметилоксикарбонил или трифторацетил. Такую группу можно удалить несколькими способами, хорошо известными специалистам в данной области (примеры данных способов-3 016470 можно найти в "Protective Groups in Organic Synthesis, T.W. Greene and P.G.M. Wuts, Wiley-Interscience,1999), например путем традиционного кислого гидролиза (например, с использованием смесей трифторуксусная кислота/дихлорметан, хлористо-водородная кислота/дихлорметан/метанол) или в смеси карбонат калия/метанол, после чего свободный амин превращают в NR2UR5 с помощью традиционных способов, таких как образование амида или сульфонамида, с использованием ацильного производногоR5COW, в случае соединений, в которых U обозначает CO, или в которых U обозначает CH2, путем алкилирования алкилгалогенидом R5CH2-галогенид в присутствии основания, ацилирования/восстановления с использованием ацильного производного R5COW, или путем восстановительного алкилирования альдегидом R5CHO в традиционных условиях (см., например, Smith, M.B.; March, J.M. Advanced OrganicChemistry, Wiley-Interscience 2001). Подходящие условия включают применение цианоборгидрида (в смеси метанол/хлороформ/уксусная кислота). Если амин (III) находится в виде соли хлористоводородной кислоты, для забуферивания реакционной смеси можно использовать ацетат натрия. Альтернативным восстанавливающим средством является триацетоксиборгидрид натрия. Альтернативно, соединение формулы (III) можно заменить на соединение H-A-OH. После стадии взаимодействия с соединением (II) гидроксильную группу можно окислить до циклического кетона с помощью подходящего окисляющего средства, такого как перииодинан Десса-Мартина (1,1,1-трис(ацетилокси)-1,1-дигидро-1,2-бензиодоксол-3-(1H)-он. Кетон подвергают взаимодействию с аминомHN(R20)R2' путем традиционного восстановительного алкилирования. Подходящие реагенты, содержащие нужную группу R5, включают известные соединения, или соединения, которые можно получить с помощью известных способов, см., например, WO 02/08224, WO 02/50061, WO 02/56882, WO 02/96907, WO 2003087098, WO 2003010138, WO 2003064421, WO 2003064431, WO 2004002992, WO 2004002490, WO 2004014361, WO 2004041210, WO 2004096982, WO 2002050036, WO 2004058144, WO 2004087145, WO 2006014580, WO 2004/035569, WO 2004/089947, WO 2003082835, WO 2002026723, WO 06002047, WO 06010040, WO 06017326, WO 06012396, WO 06017468,WO 06020561, WO 06132739, WO 06134378, WO 06137485 и ЕР 0559285. Соединение формулы (II) можно получить с помощью нижеследующей схемы 1: Соединения формулы (IV) можно получить путем аллилирования соединений типа (V) в традиционных условиях (см., например, Smith, M.B.; March, J.M. Advanced Organic Chemistry, Wiley-Interscience 2001). Превращение (IV) в (II) можно осуществить путем обработки озоном или тетраоксидом осмия и периодатом натрия в традиционных условиях (см., например, Smith, M.B.; March, J.M. Advanced OrganicChemistry, Wiley-Interscience 2001). Соединение формулы (IV) также можно получить с помощью нижеследующей схемы 2: Превращение соединения формулы (VII) в четвертичную соль (VI) можно осуществить путем обработки аллилиодидом в традиционных условиях (см., например, Smith, M.B.; March, J.M. Advanced Organic Chemistry, Wiley-Interscience 2001). Затем можно получить соединение (IV) путем окисления соединения (VI) с использованием K3[Fe(CN)6] (см., например, Baxter, P.N.W.; Khoury, R.G.; Lehn, J.M.;Baum, G.; Fenske, D. Chemistry- A European Journal (2000), 6(22), 4140). Соединения формулы (V), в которых Z1 обозначает CH, можно получить с помощью нижеследующей схемы 3: Бромсодержащее производное (IX) можно гидрировать с использованием Pd/C с получением соединения (VIII). После деметилирования с помощью HBr получают соединение (V). В альтернативном аспекте данное изобретение предлагает способ получения соединений формулы(I), а также их фармацевтически приемлемых солей, сольватов и/или N-оксидов, указанный способ включает взаимодействие соединения формулы (IIA) с соединением формулы (IIIA) или (IIIB): где R2' обозначает R2, или группу, способную превращаться в R2, a Y обозначает H или уходящую группу, где Z1, Z2, Z3, Z4, A, R1a, R1b, R2, U и R5 имеют значения, указанные в формуле (I), и затем необязательно, или при необходимости, превращение R2' в R2, взаимопревращение некоторых вариабельных групп и/или получение фармацевтически приемлемых соли, сольвата или N-оксида. Взаимодействие представляет собой восстановительное алкилирование, алкилирование или ацилирование, как описано выше. Соединения формулы (IIA) можно получить путем взаимодействия описанных выше соединений формул (II) и (III), где R20 обозначает водород. Альтернативно соединения формулы (IIA), где Z1 и Z3, оба, обозначают атомы азота, можно получить с помощью нижеследующей схемы 4: Превращение соединения формулы (XII) в (XI) осуществляют в традиционных условиях (см., например, Smith, M.B.; March, J.M. Advanced Organic Chemistry, Wiley-Interscience 2001). Затем можно получить соединение (X) из соединения (XI) путем каталитического гидрирования в традиционных условиях (см., например, Smith, M.B.; March, J.M. Advanced Organic Chemistry, Wiley-Interscience 2001). Соединение (X) можно превратить в соединение (IIA) путем селективного алкилирования этилбромацетатом,термической циклизации с последующим окислением диоксидом магния или кислородом в традиционных условиях (см., например, Smith, M.B.; March, J.M. Advanced Organic Chemistry, Wiley-Interscience 2001). Соединения формулы (II), в которых Z1 и Z3 оба обозначают атомы азота, можно получить с помощью модифицированной схемы 4, в которой соединение формулы (XII) взаимодействует с диметилацеталем аминоацетальдегида. После каталитического гидрирования, селективного алкилирования этилбромацетатом и термической циклизации получают диметилацеталь формулы (II), который можно превратить в альдегид (II) путем обработки трифторуксусной кислотой. Превращение R1a' в R1a и взаимопревращения R1a, R1b, R1c, R2, A и R5 осуществляют в традиционных условиях. Например, алкоксикарбонил R1a' путем гидролиза можно превратить в карбокси R1a, который, в свою очередь, можно превратить в аминокарбонил и циано R1a с помощью традиционных способов. Галоген R1a можно вводить с помощью традиционных реакций галогенирования, например хлорсодержащую группу можно ввести в R1b путем хлорирования хлорсукцинимидом в уксусной кислоте. Подходящие традиционные защитные группы, которые можно удалить с соединений, содержащих необязательно защищенную гидроксильную группу, не вызывая нарушений в остальной части молекулы, включают ацильные и алкилсилильные группы. N-защитные группы удаляют традиционными методами. Например, метокси R1a, R1b или R1c превращают в гидрокси R1a, R1b или R1c путем обработки литием и дифенилфосфином (общий способ, описанный Ireland et al., J. Amer. Chem. Soc., 1973, 7829) или HBr. Алкилирование гидроксильной группы подходящим алкильным производным, несущим уходящую группу, такую как галогенид, дает замещенный алкокси R1a, R1b или R1c. Галогенсодержащий R1a превращают в другой R1a традиционными способами, например в гидрокси алкилтиол (через тиол) и амино его превращают посредством реакций сочетания, катализируемых металлами, например, с использованием меди, как описано в Synlett (2003), 15, 2428-2439 and Angewandte Chemie, International Edition, 2003,42(44), 5400-5449.-5 016470 Соединения формул HA-N(R20)R2', (V), (VII) и (IX) представляют собой известные соединения, или соединения, которые можно получить с помощью известных способов, например хиназолинон и хиназолины можно получить стандартными способами, как описано T.A. Williamson in Heterocyclic Compounds,6, 324 (1957) Ed. R.C. Elderfield. Пиридазины можно получить с помощью способов, аналогичных описанным в Comprehensive Heterocyclic Chemistry, volume 3, Ed A.J. Boulton and A. McKillop, a нафтиридины можно получить с помощью способов, аналогичных описанным в Comprehensive Heterocyclic Chemistry, volume 2, Ed A.J. Boulton and A. McKillop. 4-Галогеновые производные, такие как соединение (IX), являются коммерчески доступными, или их можно получить с помощью известных в данной области способов. Соединение, содержащее бром в 4 положении, можно получить из хинолин- или нафтиридин-4-она путем взаимодействия с трибромидом фосфора (PBr3) в ДМФ. 4-Хлорхиназолин получают из соответствующего хиназолин-4-она путем взаимодействия с оксихлоридом фосфора (POCl3) или пентахлоридом фосфора, PCl5. Соединения формулы HA-N(R20)R2', (V), (VII) и (IX) описаны, например, в WO 2004/035569, WO 2004/089947, WO 02/08224, WO 02/50061, WO 02/56882, WO 02/96907, WO 2003087098, WO 2003010138,WO 2003064421, WO 2003064431, WO 2004002992, WO 2004002490, WO 2004014361, WO 2004041210,WO 2004096982, WO 2002050036, WO 2004058144, WO 2004087145, WO 2003082835, WO 2002026723,WO 06002047, WO 06014580, WO 06134378 и WO 06137485. Как показано на схеме 5, гидроксиаминометилпирролидины формулы (III) (A обозначает (ii), X обозначает CR4R8, W1 обозначает связь, W2 и W3, оба, обозначают CH2, R4 и R7 обозначают H, a R8 обозначает OH) можно получить из дважды защищенного хирального промежуточного соединения (XV), разделенного методом препаративной ВЭЖХ. Бензилоксикарбонильную защитную группу удаляют путем гидрирования с получением соединения (XIV) и функциональную аминогруппу превращают в трифторацетамид (XIII). т-Бутоксикарбонильную (Boc) защитную группу удаляют с помощью HCl, получая гидрохлорид пирролидина (III). Промежуточное соединение (XV) можно получить с помощью общего способа, изображенного на схеме 6(c) бензилоксикарбонилсукцинимид, Et3N, дихлорметан, комнатная температура. В соответствии со схемой 7 аминометилпирролидин формулы (III) (A обозначает (ii), X обозначаетCR4R8, W1 обозначает связь, W2 и W3, оба, обозначают CH2, все R4, R7 и R8 обозначают H) можно получить из коммерчески доступного Boc-защищенного аминометилпирролидина и превратить его в трифторацетамид. Аминометилморфолин, промежуточное соединение формулы (III) (A обозначает (ii), X обозначаетO, каждый из W1, W2 и W3 обозначает CH2), можно получить из хирального дихлорбензильного промежуточного соединения (XX) (WO 2003082835) (схема 8) путем защиты функциональной аминогруппы защитной группой Boc (XIX), с последующим удалением дихлорбензильной группы гидрированием с получением (III), защитой N-атома морфолина бензилоксикарбонильной группой (чтобы обеспечить возможность очистки хроматографическими методами) (XVIII) и гидрированием с получением целевого морфолинового производного (III). Способ получения пиримидинилоксазинонового элемента (R5 (C), где Y3 = O, R10 = H) изображен на схеме 9 Подходящим образом защищенный этилгликолят (в данном примере THP-защищенное соединение 1) формилируют, используя этилформиат и основание, такое как NaH, в ТГФ или диэтиловом эфире. Затем промежуточное соединение формиленолят 2 сразу подвергают взаимодействию с амидином, в данном случае (2E)-3-фенил-2-пропенимидамидом 3, получая пиримидинон 4. Пиримидинон 4 превращают в трифторметансульфоновый эфир (5), который, в свою очередь, подвергают взаимодействию с аммиаком в подходящем растворителе, таком как 1,4-диоксан, с получением амина 6. Затем с соединения 6 удаляют THP-защитную группу с помощью раствора кислоты в метаноле и получают аминоспирт 7. После обработки соединения 7 основанием и эфиром галогенуксусной кислоты в спиртовом растворителе,таком как абсолютный этанол, непосредственно получают бициклическое промежуточное соединение 8. Такое превращение можно проводить с использованием основания, такого как трет-бутоксид калия, и алкилирующего реагента этилбромацетата. В качестве основания вместо описанного здесь алкоксида можно использовать амин, такой как триэтиламин (примеры подобных синтезов описаны в N.V. Sazonovand T.S. Safonova, Khimiya Geterotsiklicheskikh Soedinenii, 1971, 1285-1288). Затем получают целевой промежуточный альдегид 9 путем окислительного расщепления боковой цепи фенилэтенила. Такое рас-7 016470 щепление можно осуществить, например, путем взаимодействия соединения 8 с NaIO4 в смеси 1,4 диоксан-вода, в присутствии каталитического количества OsO4. Целевое превращение также можно осуществить другими методами, такими как озонолиз. Пиримидиндигидропиридоновый альдегид (R5 (C), где Y3 = CH2, a R10 = Cl) можно получить по способу, изображенному на схеме 10 В результате взаимодействия диметилмалоната (10) с этилакрилатом (11) получают сложный триэфир 12. Конденсация соединения 12 с (2E)-3-фенил-2-пропенимидамидом (3) в присутствии основания дает дигидроксипиримидин 13. Для проведения данного превращения можно использовать триэтиламин в EtOH, однако предпочтительные условия включают применение NaOMe в MeOH. Следует отметить,что в последнем случае образуется метиловый сложный эфир соединения 13 (R = Me), тогда как в первом случае сохраняется этиловый сложный эфир. Для проведения остальных стадий синтеза можно использовать любой сложный эфир, как метиловый, так и этиловый. После обработки соединения 13 POCl3 получают дихлорпиримидин 14. Нагревание соединения 14 в герметично закрытой пробирке в присутствииNH4OH, как правило, дает смесь компонентов 15, 16 и 17 с преобладанием соединений 15 и 16. Затем промежуточное соединение 15 можно превратить в соединение 16 путем обработки K2CO3 в MeOH. Кроме того, соединение 17 можно преобразовать в соединение 15 (R = Et) путем обработки этанольным раствором HCl. Получение альдегида 18 осуществляют путем окислительного расщепления олефиновой боковой цепи соединения 16 с использованием либо OsO4, либо NaIO4, или путем озонолиза. На схеме 11 показан подходящий способ удаления заместителя хлора, присутствующего в соединении 18, с получением дез-хлоральдегида 20 (R5 (C), где Y3 = CH2, a R10 = H). Чтобы осуществить такое удаление, вначале защищают альдегидную группу соединения 18 путем образования диметилацеталя с использованием п-толуолсульфоновой кислоты (p-TsOH) и MeOH, и получают соединение 19. Затем хлор удаляют гидрированием, используя катализатор Pd-C в атмосфере H2. Путем обработки водным раствором кислоты, например раствором ТФУ в воде, удаляют защиту с альдегидной группы, получая соединение 20. Схема 12 иллюстрирует способ получения аналогов, содержащих альтернативные заместители в 4 положении пиримидинового цикла, например R5 (C), где Y3 = CH2, a R10 = OMe или Me. Данные аналоги можно получить из ранее описанного промежуточного соединения 16 с помощью ряда хорошо известных способов. На схеме 13 изображен способ получения 4-метоксильных и 4-метильных производных,однако подобные или другие способы можно использовать для введения широкого ряда заместителей. Как показано ниже, соединение 16 можно обработать NaOMe в метаноле при нагревании с обратным холодильником с получением метоксисодержащего промежуточного соединения 21A. Соединение 21B,содержащее метильную группу, можно получить из соединения 16 путем катализируемого Pd взаимодействия с метилбороновой кислотой. Свободную альдегидную функциональную группу получают путем окислительного расщепления олефиновой боковой цепи с помощью таких методов, как озонолиз,или путем взаимодействия с OsO4 и NaIO4, с получением соединений 22A и 22B. На схеме 13 показано получение пиримидинового оксазинонового альдегидного соединения, необходимого для получения соединений, содержащих R5(C), где Y3 = O, a R10 = Cl, с использованием в качестве исходного соединения диметилдиазомалоната (23), по способу Peace, Carman, Wulfman, Synthesis,658-661, (1971). Взаимодействие соединения 23 с этилгликолятом, катализируемое родием, дает замещенный малонат 24. Соединение 25, содержащее пиримидиновую циклическую систему, получают путем взаимодействия соединения 24 с (2E)-3-фенил-2-пропенимидамидом (3) и метоксидом натрия. Промежуточное соединение 25 выделяют в виде карбоновой кислоты, поскольку метиловый эфир гидролизуется в условиях взаимодействия с метоксидом натрия. Обработка соединения 25 POCl3 с последующим добавлением MeOH дает дихлорид-метиловый эфир 26. Замену одного из атомов хлора на аммиак осуществляют путем обработки соединения 26 NH4OH, при этом также получают первичный амид, который затем превращают в этиловый эфир 27 с использованием HCl и EtOH. Получение оксазинонового цикла проводят путем обработки соединения 27 основанием, таким как K2CO3, в полярном растворителе, таком как ДМФ. Как правило, для полного превращения в бициклическую систему 28 требуется нагревание. Превращение в альдегид можно осуществить путем окислительного расщепления 2-фенилэтенильной боковой цепи, в данном конкретном примере боковой цепи соединения 28, в результате взаимодействия с OsO4 и NaIO4, с получением альдегида 29. Более подробное описание способов получения соединений формулы (I) можно найти в примерах. Противобактериальные/противотуберкулезные соединения данного изобретения, подходящие для введения любым удобным способом и применяющиеся в медицине или ветеринарии, можно получить по аналогии с другими противобактериальными/противотуберкулезными соединениями. Фармацевтические композиции данного изобретения включают композиции, адаптированные для перорального, местного или парентерального применения, которые можно использовать для лечения бактериальной инфекции, в том числе туберкулеза, у млекопитающих, включая людей. Можно получить композиции, подходящие для любого способа введения. Композиции могут находиться в виде таблеток, капсул, порошков, гранул, пастилок, свечей, кремов или жидких препаратов, таких как пероральные или стерильные парентеральные растворы или суспензии. Местные композиции настоящего изобретения могут быть получены, например, в виде мазей, кремов или лосьонов, глазных мазей и глазных или ушных капель, пропитанных перевязочных средств и аэрозолей, и могут содержать подходящие традиционные добавки, такие как консерванты, растворители,облегчающие проникновение лекарственного средства, а также мягчительные средства, применяющиеся для получения мазей и кремов. Композиции также могут содержать совместимые традиционные носители, такие как основы для кремов или мазей, и этиловый или олеиловый спирт в случае лосьонов. Такие носители могут составлять примерно от 1 до 98% композиции. Чаще они составляют примерно до 80% композиции. Таблетки и капсулы для перорального введения могут представлять собой унифицированные лекарственные формы и могут содержать традиционные носители, такие как связывающие средства, например патока, гуммиарабик, желатин, сорбит, трагакант или поливинилпирролидон; наполнители, например лактоза, сахар, кукурузный крахмал, фосфат кальция, сорбит или глицин; смазывающие средства для таблетирования, например стеарат магния, тальк, полиэтиленгликоль или оксид кремния; дезинтегрирующие средства, например картофельный крахмал; или приемлемые увлажняющие средства, такие как лаурилсульфат натрия. На таблетки можно нанести покрытие с помощью способов, хорошо извест-9 016470 ных в области нормальной фармацевтической практики. Пероральные жидкие препараты могут находиться в виде, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров,или они могут быть предоставлены в виде сухого продукта, предназначенного для разбавления водой или другой подходящей средой перед применением. Такие жидкие препараты могут содержать традиционные добавки, такие как суспендирующие средства, например сорбит, метилцеллюлоза, сироп глюкозы,желатин, гидроксиэтилцеллюлоза, карбоксиметилцеллюлоза, гель стеарата алюминия или гидрированные пищевые жиры, эмульгирующие средства, например лецитин, моноолеат сорбитана или гуммиарабик; неводные среды (которые могут включать пищевые масла), например миндальное масло, жирные сложные эфиры, например глицерина, пропиленгликоля или этилового спирта; консерванты, например метил- или пропил-п-гидроксибензоат или сорбиновая кислота, и, при желании, традиционные ароматизирующие или окрашивающие средства. Свечи могут содержать традиционные основы для свечей, например кокосовое масло или другой глицерид. Жидкие стандартные лекарственные формы для парентерального введения получают с использованием соединения и стерильной среды, причем предпочтительной средой является вода. Соединение можно либо суспендировать, либо растворить в среде, в зависимости от вида используемой среды и концентрации. При получении растворов соединение можно растворить в воде для инъекций и стерилизовать фильтрованием, после чего полученный раствор вносят в подходящий флакон или ампулу и герметично закрывают. Предпочтительно, чтобы в среде были растворены такие средства, как локальный анестетик, консервант и забуферивающие средства. Для повышения стабильности композицию можно заморозить после внесения во флакон и удалить воду под вакуумом. Затем сухой лиофилизированный порошок герметично закрывают во флаконе и поставляют его вместе с флаконом, содержащим воду для инъекций, чтобы восстановить жидкий препарат перед применением. Парентеральные суспензии получают практически таким же способом, за исключением того, что соединение не растворяют, а суспендируют в среде, и поэтому невозможно проводить стерилизацию фильтрованием. Соединение можно стерилизовать путем обработки этиленоксидом с последующим суспендированием в стерильной среде. Чтобы обеспечить однородное распределение соединения в состав композиции можно ввести поверхностно-активное вещество или увлажняющее средство. Композиции могут содержать от 0,1 мас.%, предпочтительно 10-60 мас.% активного вещества в зависимости от способа введения. Если композиции представлены в виде стандартных лекарственных форм, каждая форма предпочтительно содержит 50-1000 мг активного ингредиента. Доза, используемая для лечения взрослых людей, предпочтительно варьирует от 100 до 3000 мг в день, например 1500 мг в день, в зависимости от способа и частоты введения. Такая доза соответствует 1,5-50 мг/кг в день. Предпочтительно доза составляет от 5 до 30 мг/кг в день. Соединение формулы (I) может входить в состав композиций как единственное терапевтическое средство или в сочетании с другими противобактериальными средствами, включающими противотуберкулезные соединения. Если другое противобактериальное средство представляет собой -лактам, можно также использовать ингибитор -лактамазы. Соединения формулы (I) можно использовать для лечения бактериальных инфекций, вызываемых широким рядом организмов, включающих как грамотрицательные, так и грамположительные организмы,таких как инфекции верхних и/или нижних дыхательных путей, инфекции кожи и мягких тканей и/или инфекции мочевых путей. Соединения формулы (I) также можно использовать для лечения туберкулеза,вызываемого Mycobacterium tuberculosis. Некоторые соединения формулы (I) могут обладать активностью против нескольких организмов. Такую активность можно определить с помощью способов, описанных в данном документе. Нижеследующие примеры иллюстрируют способы получения некоторых соединений формулы (I),а также активность конкретных соединений формулы (I) против разных бактериальных организмов, в том числе Mycobacterium tuberculosis. Примеры и экспериментальная часть Общие сведения. Используемые в примерах сокращения: фунт/кв. дюйм = фунты на квадратный дюйм (1 фунт/кв. дюйм = 0,069 бар); КТ/к.т. = комнатная температура;ES = масс-спектрометрия с электрораспылением; ЖХМС = жидкостная хроматомасс-спектрометрия; ВЭЖХ = высокоэффективная жидкостная хроматография (вр. уд. относится к времени удерживания). Некоторые реагенты также имеют сокращенные названия. ДХМ - дихлорметан, ДМФ - диметилформамид, ДМСО - диметилсульфоксид, MeOH - метанол, ТФУ - трифторуксусная кислота, ТГФ - тетрагидрофуран, Pd/C - катализатор палладий-на-угле, Boc - трет-бутоксикарбонил, EtOH - этанол, dppf - 1,1'- 10016470 бис-(дифенилфосфино)ферроцен, EDC - гидрохлорид N-[3-(диметиламино)пропил]этилкарбодиимида,HOBt - 1-гидроксибензотриазол. Спектры протонного ядерного магнитного резонанса (1H ЯМР) регистрируют при 400 или 250 мГц,значения химических сдвигов приводят в миллионных доляхпо отношению к внутреннему стандарту тетраметилсилану (ТМС). В спектрах ЯМР используют следующие сокращения: с = синглет, д = дублет,т = триплет, кв = квартет, м = мультиплет, дд = двойной дублет, дт = двойной триплет, явн = явный, шир= широкий. J обозначает константу взаимодействия ЯМР, измеряемую в Герцах. CDCl3 обозначает дейтериохлороформ, ДМСО-d6 обозначает гексадейтериодиметилсульфоксид, a CD3OD обозначает тетрадейтериометанол. Масс-спектры получают, используя метод ионизации электрораспылением (ES). Все значения температуры указаны в градусах Цельсия. Celite представляет собой вспомогательное фильтровальное вещество, состоящее из обработанного кислотой диатомового кремнезема, и является торговым знаком Manville Corp., Denver, Colorado. MDAP или Mass directed autoprep = масс-контролируемая препаративная ВЭЖХ (с использованием масс-спектрометра ZQ (Waters. Получение комплекса триэтенилбороксин-пиридин описано в Kerins, Fergal; O'Shea, Donea F.; J. Org. Chem. (2002) 67(14) 4968. MPкарбонат представляет собой макропористый триэтиламмония метилполистиролкарбонат (ArgonautTechnologies). Amberlyst A21 представляет собой слабоосновную, макропористую смолу, содержащую алкиламиновые функциональные группы,зарегистрированный торговый знак RohmHaas Co. SCX представляет собой ионообменную колонку, содержащую сильную катионообменную смолу (бензолсульфоновая кислота), поставляемую Varian, USA. Chiralpak IA, Chiralpak AS-H и Chiralcel OD представляют собой хиральные колонки для ВЭЖХ на основе полисахаридов (Chiral Technologies Inc.). КолонкаChiralpak AS-H содержит амилозу трис-[(S)-альфа-метилбензилкарбамат], нанесенную на частицы оксида кремния размером 5 мкм. Колонка Chiralpak IA содержит оксид кремния для препаративных колонок(размер частиц 5 мкм, вн. д. 21 ммдлина 250 мм), на котором иммобилизована амилоза трис-(3,5 диметилфенилкарбамат). Колонки Chiralpak AD и AD-H содержат оксид кремния для препаративных колонок (размер частиц AD-H 5 мкм и размер частиц AD 10 мкм, вн. д. 21 ммдлина 250 мм; размер частиц AD 20 мкм, вн. д. 101 ммдлина 250 мм), покрытый амилозой трис-(3,5-диметилфенилкарбамат)(Chiral Technologies USA). Измеряемые значения времени удерживания зависят от конкретных условий,применяющихся в хроматографических процедурах. Если данные значения приводятся ниже в примерах,они указывают на порядок элюирования. Реакции, в которых участвуют гидриды металлов, включающие гидрид лития, гидрид лития алюминия, гидрид диизобутилалюминия, гидрид натрия, боргидрид натрия, триацетоксиборгидрид натрия,цианоборгидрид (полистиролметил)триметиламмония, проводят в атмосфере аргона или другого инертного газа. Опытным химикам следует понимать, что ссылки на способы получения, проводимые в соответствии с общим способом других способов получения, могут охватывать вариации стандартных параметров,таких как время, температура, условия обработки, незначительные изменения количеств реагентов и др. Пример 61. 6-([3S,4S)-4-Гидрокси-1-2-[7-(метилокси)-2-оксо-1(2H)-хинолинил]этил-3 пирролидинил)метил]аминометил)-2H-пиридо[3,2-b][1,4]тиазин-3(4H)-она дигидрохлорид(3S,4S)-3-гидрокси-4[([(фенилметил)окси]карбониламино)метил]-1-пирролидинекарбоксилата (синтез описан в WO 2006002047, способ получения 24(c), изомер E1 -1,1-диметилэтил цис-3-гидрокси-4[([(фенилметил)окси]карбониламино)метил]-1-пирролидинкарбоксилата) (1 г, 2,85 ммоль) в ДХМ (50 мл) добавляют трифторуксусную кислоту (50 мл) и перемешивают в течение 2 ч. Реакционную смесь концентрируют и помещают в глубокий вакуум на 3 ч. Соль ТФУ растворяют в ДХМ (50 мл), добавляют смолу MP-карбонат (4 г, 11,4 ммоль) и перемешивают в течение ночи. Реакционную смесь фильтруют и концентрируют, получая указанное в заголовке соединение в виде бледно-желтого масла (840 мг, 100%). МС (ES+) m/z 251,3 (MH+).(10 мл) и безводном MeOH (2 мл) со шпателем твердого карбоната натрия. Реакционную смесь перемешивают в атмосфере азота 1 ч, затем добавляют триацетоксиборгидрид натрия (2,03 г, 9,12 ммоль) и перемешивают в течение ночи. Реакционную смесь концентрируют и после колоночной хроматографии(c) 1-2-[(3S,4S)-3-(Аминометил)-4-гидрокси-1-пирролидинил]этил-7-(метилокси)-2(1H)-хинолинон. К раствору фенилметил [3S,4S)-4-гидрокси-1-2-[7-(метилокси)-2-оксо-1(2H)-хинолинил]этил-3 пирролидинил)метил]карбамата (1 г, 2,21 ммоль) добавляют 20% Pd(OH)2/C, дегазируют и подвергают воздействию H2 под давлением 1 атм в течение 2 ч. Реакционную смесь фильтруют через целит и концентрируют с получением указанного в заголовке соединения в виде желтого масла (622 мг, 89%).(146 мг, 0,460 ммоль) и 3-оксо-3,4-дигидро-2H-пиридо[3,2-b][1,4]тиазин-6-карбоксальдегид (описание синтеза можно найти в WO 2004058144, пример 7(d (98 мг, 0,506 ммоль) объединяют в безводном ДХМ(5 мл) и безводном MeOH (1 мл) со шпателем твердого карбоната натрия. Реакционную смесь перемешивают в атмосфере азота в течение 18 ч, затем добавляют триацетоксиборгидрид натрия (306 мг, 1,38 ммоль) и перемешивают 1 ч. Реакционную смесь концентрируют и очищают колоночной хроматографией (90:10:1 ДХМ:MeOH:NH4OH), получая свободное основание указанного в заголовке соединения в виде бледно-желтого масла (91 мг, 40%).H ЯМР (400 МГц, CD3OD)м.д. 3,43-3,55 (м, 3H), 3,58 (с, 2H), 3,68 (с, 3H), 3,73 (с, 3H), 4,00 (с,4H), 4,38 (с, 2H), 4,65 (с, 1H), 4,76 (т, J=5,94 Гц, 3H), 6,59 (д, J=9,35 Гц, 1H), 7,02 (дд, J=8,59, 2,02 Гц, 1H),7,07 (с, 1H), 7,16 (д, J=7,83 Гц, 1H), 7,70 (д, J=8,59 Гц, 1H), 7,85 (д, J=7,83 Гц, 1H), 7,92 (д, J=9,35 Гц, 1H). Дигидрохлорид получают путем добавления 92 мкл 4 н. раствора HCl в 1,4-диоксане к раствору свободного основания. Пример 121. 4-(2-4-[(2,3-Дигидро[1,4]диоксино[2,3-c]пиридин-7-илметил)амино]-1-пиперидинилэтил)-6-(метилокси)-1,2,4-бензотриазин-3(4H)-она гидрохлорид(a) 6-(Метилокси)-4-(2-пропен-1-ил)-1,2,4-бензотриазин-3(4H)-он. К раствору 6-хлор-4-(2-пропен-1-ил)-1,2,4-бензотриазин-3(4H)-она 1-оксида (способ получения описан в примере 60(c (420 мг, 1,77 ммоль) в MeOH (10 мл) добавляют раствор метоксида натрия (25% мас./об., 8,84 ммоль, 1,9 мл), после чего реакционную смесь перемешивают при к.т. в течение 2 ч. Затем реакционную смесь обрабатывают водой (100 мл) и экстрагируют ДХМ (2100 мл). Объединенные органические фазы сушат, упаривают, неочищенный остаток очищают хроматографией на силикагеле, используя градиент 0-5% MeOH/ДХМ, затем перетирают с диэтиловым эфиром (50 мл) и получают целевое соединение в виде желтой пены (205 мг, 53%). МС (ES+) m/z 218 (MH+).(b) [6-(Метилокси)-3-оксо-1,2,4-бензотриазин-4(3H)-ил]ацетальдегид (в виде метилполуацеталя). 6-(Метилокси)-4-(2-пропен-1-ил)-1,2,4-бензотриазин-3(4H)-он (75 мг, 0,346 ммоль) растворяют в 1,4-диоксане (5 мл) и воде (2 мл). Добавляют периодат натрия (185 мг, 0,865 ммоль) и затем тетраоксид осмия (0,1 мл 4% водного раствора). Смесь перемешивают при к.т. в течение 4 ч, после чего 1,4-диоксан упаривают в вакууме, оставшуюся водную фазу экстрагируют 20% MeOH/ДХМ (3200 мл). Органические экстракты объединяют, сушат над безводным сульфатом магния, фильтруют и упаривают при пониженном давлении, получая 6-[6-(метилокси)-3-оксо-1,2,4-бензотриазин-4(3H)-ил]ацетальдегид (преимущественно в виде метилполуацеталя) в виде желтого масла с примесями (89 мг, 117%). МС (ES+) m/z 220 (MH+), 252 (метилполуацетальН+).(89 мг, 0,346 ммоль) и 1,1-диметилэтил 4-пиперидинилкарбамата (69 мг, 0,346 ммоль) в хлороформе (5 мл) и MeOH (0,5 мл) перемешивают в течение 2 ч, после чего добавляют NaBH(OAc)3 (220 мг, 1,038 ммоль). Реакционную смесь перемешивают в течение 0,5 ч и добавляют насыщенный водный растворNaHCO3 (20 мл). Затем реакционную смесь экстрагируют 20% MeOH в ДХМ (3100 мл). Объединенные- 12016470 органические фазы сушат, упаривают, неочищенный остаток очищают хроматографией на силикагеле,используя градиент 0-5% MeOH/ДХМ, и получают целевое соединение (48 мг, 34%). МС (ES+) m/z 404 (MH+).(d) 4-[2-(4-Амино-1-пиперидинил)этил]-6-(метилокси)-1,2,4-бензотриазин-3(4H)-она дигидрохлорид. К раствору 1,1-диметилэтил (1-2-[6-(метилокси)-3-оксо-1,2,4-бензотриазин-4(3H)-ил]этил-4 пиперидинил)карбамата (48 мг, 0,119 ммоль) в хлороформе (2 мл) добавляют 4 М HCl в 1,4-диоксане (2 мл). Реакционную смесь перемешивают при к.т. в течение 1 ч, после чего упаривают, перетирают с этилацетатом и получают целевое соединение в виде желтого масла, которое используют без дополнительной очистки (42 мг, 94%). МС (ES+) m/z 304 (MH+).(e) Указанное в заголовке соединение. Смесь 4-[2-(4-амино-1-пиперидинил)этил]-6-(метилокси)-1,2,4-бензотриазин-3(4H)-она дигидрохлорид (42 мг, 0,112 ммоль) в хлороформе (2 мл) и MeOH (0,1 мл) обрабатывают триэтиламином (50 мкл,0,358 ммоль) и перемешивают в течение 0,25 ч, после чего добавляют 2,3-дигидро[1,4]диоксино[2,3c]пиридин-7-карбоксальдегид (синтез описан в WO 2004058144, пример 2(с), или WO 03/087098, пример 19(d (18 мг, 0,112 ммоль). Реакционную смесь перемешивают в течение 0,5 ч и добавляют NaBH(OAc)3(71 мг, 0,336 ммоль). Реакционную смесь перемешивают в течение 1 ч после чего добавляют насыщенный водный раствор NaHCO3 (10 мл). Затем реакционную смесь экстрагируют 20% MeOH в ДХМ (3200 мл). Объединенные органические фазы сушат и упаривают, после чего неочищенный остаток очищают хроматографией на силикагеле, используя градиент 0-20% MeOH/ДХМ, и получают указанное в заголовке соединение в виде свободного основания (26 мг, 51%). МС (ES+) m/z 453 (MH+). 1(1H, с), 8,29 (1H, д, J=9 Гц). Свободное основание указанного в заголовке соединения превращают в соль HCl путем растворения полученного свободного основания в смеси 1:1 ДХМ:MeOH с добавлением 1 экв. 4 М раствора HCl в 1,4-диоксане. Затем смесь упаривают досуха. Пример 123. 2-[(1-[2-(7-Фтор-2-оксо-1(2H)-хинолинил)этил]-4-пиперидиниламино)метил]-1Hпиримидо[5,4-b][1,4]оксазин-7(6H)-она гидрохлорид К раствору 4-хлор-2-[(l-[2-(7-фтор-2-оксо-1(2H)-хинолинил)этил]-4-пиперидиниламино)метил]1H-пиримидо[5,4-b][1,4]оксазин-7(6H)-она (способ получения описан в примере 124 (j (67 мг, 0,13 ммоль) в метаноле (3 мл) добавляют NaHCO3 (40 мг) и затем 10% палладий-на-угле в качестве катализатора (30 мг). Полученную смесь перемешивают при комнатной температуре при давлении водорода 1 атм (баллон) в течение 24 ч. Реакционную смесь фильтруют через нейлоновый фильтр, неочищенный остаток очищают хроматографией (силикагель), используя градиент 0-20% MeOH/ДХМ, и получают указанное в заголовке соединение в виде свободного основания (10 мг, 16%).(1H, дд, J=10,5, 2 Гц), 7,50 (1H, дд, J=10,5 Гц, 2 Гц), 7,68 (1H, д, J=10 Гц), 7,9 (1H, с). Свободное основание указанного в заголовке соединения превращают в соль HCl путем растворения в 1:1 ДХМ:MeOH с добавлением 1 экв. 4 М HCl в 1,4-диоксане. Затем смесь упаривают досуха. Пример 124. 4-Хлор-2-[(l-[2-(7-фтор-2-оксо-1(2H)-хинолинил)этил]-4-пиперидиниламино)метил]1H-пиримидо[5,4-b][1,4]оксазин-7(6H)-она гидрохлорид(a) Диметил [2-(этилокси)-2-оксоэтил]оксипропандиоат. К раствору диметил диазопропандиоата (4 г, 25 ммоль), полученного по способу Peace, Carman,Wulfman, Synthesis, 658-661, (1971), в ДХМ (10 мл) добавляют этилгликолят (1,2 мл, 12,8 ммоль) и затем димер ацетата родия(II) (2 г, 20 мол.%). Реакционную смесь перемешивают при комнатной температуре в течение 24 ч. Полученную суспензию фильтруют через слой Celite и растворитель удаляют в вакууме. Неочищенный остаток очищают колоночной хроматографией (силикагель), используя градиент 0-60%EtOAc/гексаны, и получают целевой продукт в виде масла (3 г, 97%). МС (ES+) m/z 235 (MH+).(b) (4-Гидрокси-6-оксо-2-[(Е)-2-фенилэтенил]-1,6-дигидро-5-пиримидинилокси)уксусная кислота. К раствору диметил [2-(этилокси)-2-оксоэтил]оксипропандиоата (3 г, 12,8 ммоль) в MeOH (10 мл) при комнатной температуре добавляют (2E)-3-фенил-2-пропенимидамид (1,87 г, 12,8 ммоль) (способ получения описан в примере 3(g и затем NaOMe (8,3 г, 38,4 ммоль; 25% раствор в метаноле). Полученную смесь перемешивают при комнатной температуре в течение 24 ч. Растворитель удаляют в вакууме и полученное твердое вещество используют на следующей стадии без дополнительной очистки. МС (ES+) m/z 288 (MH+).(4-гидрокси-6-оксо-2-[(E)-2-фенилэтенил]-1,6-дигидро-5-пиримидинилокси)уксусной кислоте (12,8 ммоль) добавляют POCl3 (8 мл, 76,9 ммоль) и затем N,N-диметиланилин (1,7 мл, 12,8 ммоль). Полученную смесь нагревают при 120C в запечатанной пробирке в течение 3 ч. Полученную смесь охлаждают до 0C и гасят холодным метанолом. Растворитель удаляют в вакууме, неочищенный остаток очищают хроматографией (силикагель), используя градиент 0-30% EtOAc/гексаны, и получают целевой продукт в виде твердого вещества (1,3 г, 30% с 2 стадий). МС (ES+) m/z 340 (MH+).(d) 2-(4-Амино-6-хлор-2-[(E)-2-фенилэтенил]-5-пиримидинилокси)ацетамид. К раствору метил (4,6-дихлор-2-[(E)-2-фенилэтенил]-5-пиримидинилокси)ацетата (1,1 г, 3,24 ммоль) в 1,4-диоксане (10 мл) добавляют концентрированный NH4OH (2 мл, 20 экв.). Полученную смесь нагревают при 65C в течение 4 ч. После охлаждения до комнатной температуры растворитель упаривают при пониженном давлении и остаток экстрагируют 10% раствором метанола в ДХМ (3300 мл). Органические экстракты объединяют, сушат над безводным MgSO4, фильтруют и концентрируют, получая твердое вещество, которое очищают колоночной хроматографией (силикагель) с использованием градиента 0-10% метанола в ДХМ, и получают целевой продукт в виде твердого вещества (0,6 г, 61%). МС (ES+) m/z 305 (MH+). Кроме того, получают 0,3 г 4-хлор-2-[(E)-2-фенилэтенил]-6H-пиримидо[5,4-b][1,4]оксазин-7(8H)она.(e) Этил (4-амино-6-хлор-2-[(E)-2-фенилэтенил]-5-пиримидинилокси)ацетат. Газообразный хлористый водород барботируют через раствор 2-(4-амино-6-хлор-2-[(E)-2 фенилэтенил]-5-пиримидинилокси)ацетамида (0,6 г, 1,97 ммоль) в этаноле (20 мл). Полученную смесь нагревают при 100C в течение 3 ч. Смесь упаривают при пониженном давлении и получают целевой продукт в виде твердого вещества (0,55 г, 84%), которое используют без дополнительной очистки. МС (ES+) m/z 334 (MH+).(f) 4-Хлор-2-[(E)-2-фенилэтенил]-6H-пиримидо[5,4-b][1,4]оксазин-7(8H)-он. К раствору этил (4-амино-6-хлор-2-[(E)-2-фенилэтенил]-5-пиримидинилокси) ацетата (0,55 г, 1,65 ммоль) в ДМФ (5 мл) добавляют твердое вещество K2CO3 (0,46 г, 3,3 ммоль) и полученную смесь нагревают при 75C в течение 1 ч. Растворитель упаривают при пониженном давлении и остаток экстрагируют 10% раствором метанола в ДХМ (3100 мл). Органические экстракты объединяют, сушат над безводнымMgSO4, фильтруют и упаривают при пониженном давлении, после чего получают твердое вещество, которое очищают колоночной хроматографией (силикагель), используя градиент 0-10% метанол в ДХМ, и получают целевой продукт в виде твердого вещества (0,47 г, 99%). МС (ES+) m/z 288 (MH+).(g) 4-Хлор-7-оксо-6,7-дигидро-1H-пиримидо[5,4-b][1,4]оксазин-2-карбальдегид. К раствору 4-хлор-2-[(E)-2-фенилэтенил]-6H-пиримидо[5,4-b][1,4]оксазин-7(8H)-она (0,45 г, 1,16 ммоль) в 1,4-диоксане (25 мл) и воде (10 мл) добавляют NaIO4 (1,26 г, 4,4 ммоль) вместе с каталитическим количеством OsO4 (0,36 мл, 4 мас.% в воде). Полученную смесь перемешивают при комнатной температуре в течение 5 ч. Растворитель упаривают при пониженном давлении и остаток экстрагируют 10% раствором метанола в ДХМ (350 мл). Органические экстракты объединяют, сушат над безводнымMgSO4, фильтруют и концентрируют при пониженном давлении, получая твердое вещество, которое очищают колоночной хроматографией (силикагель) с использованием градиента 0-10% метанол в ДХМ и получают целевой продукт в виде светло-желтого твердого вещества (0,28 г, 84%). МС (ES+) m/z 214 (MH+). 1(7-Фтор-2-оксо-1(2H)-хинолинил)ацетальдегид (0,50 г, 2,41 ммоль) (способ получения описан в примере 88 (a объединяют с 1,1-диметилэтил 4-пиперидинилкарбаматом (0,48 г, 2,41 ммоль) в смеси 1:1 MeOH/ДХМ (20 мл). Добавляют избыток Na2SO4 в качестве осушающего средства и раствор перемешивают при температуре окружающей среды в течение 16 ч. Добавляют NaBH(OAc)3 (1,53 г, 7,23 ммоль) и реакционную смесь перемешивают еще 2 ч. Раствор концентрируют на силикагеле в вакууме, после чего неочищенный остаток очищают колоночной хроматографией (силикагель), используя градиент- 14016470 ДХМ/ДХМ:MeOH:NH4OH (90:10:1), и получают целевой продукт в виде желтого твердого вещества(i) 1-[2-(4-Амино-1-пиперидинил)этил]-7-фтор-2(1H)-хинолинона гидрохлорид. К раствору 1,1-диметилэтил 1-[2-(7-фтор-2-оксо-1(2H)-хинолинил)этил]-4-пиперидинилкарбамата (0,67 г, 1,74 ммоль) в ДХМ (20 мл) добавляют 4 М раствор HCl в 1,4-диоксане (2,18 мл, 8,71 ммоль) и раствор перемешивают при температуре окружающей среды в течение 16 ч. Раствор концентрируют в вакууме, получая целевой продукт в виде не совсем белого твердого вещества (0,55 г, 98%). ЖХМС: m/z 290,0 (MH+).(j) Указанное в заголовке соединение. К раствору 1-[2-(4-амино-1-пиперидинил)этил]-7-фтор-2(1H)-хинолинона гидрохлорида (0,40 г, 0,12 ммоль) в ДХМ (5 мл) и метаноле (2 мл) добавляют 4-хлор-7-оксо-6,7-дигидро-1H-пиримидо[5,4b][1,4]оксазин-2-карбальдегид (0,026 г, 0,12 ммоль), затем NaHCO3 (0,1 г, 1,2 ммоль) и безводный Na2SO4 в качестве осушающего средства. Полученную смесь перемешивают при комнатной температуре в течение 24 ч, после чего добавляют NaBH(OAc)3 (80 мг, 0,36 ммоль). Реакционную смесь перемешивают в течение 1 ч. Реакционную смесь концентрируют и остаток экстрагируют 20% MeOH в ДХМ (320 мл). Объединенные органические фазы сушат (MgSO4), упаривают, неочищенный остаток очищают хроматографией (силикагель), используя градиент 0-20% MeOH/ДХМ, и получают указанное в заголовке соединение в виде свободного основания (24 мг, 40%). 1MC (ES+) m/z 487 (MH+). Свободное основание указанного в заголовке соединения превращают в соль HCl путем растворения в смеси 1:1 ДХМ:MeOH с добавлением 1 экв. 4 М HCl в 1,4-диоксане. Затем смесь упаривают досуха. Пример 125. 2-[(1-2-[6-(Метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)-ил]этил-4-пиперидинил)амино]метил-5,6-дигидропиридо[2,3-d]пиримидин-7(1H)-она гидрохлорид(a) 2-[бис-(Метилокси)метил]-4-хлор-5,8-дигидропиридо[2,3-d]пиримидин-7(6H)-он. К раствору 4-хлор-7-оксо-5,6,7,8-тетрагидропиридо[2,3-d]пиримидин-2-карбальдегида (способ получения описан в примере 126 (k (1,43 г, 6,78 ммоль) в MeOH добавляют п-TsOHH2O (0,13 г, 0,68 ммоль). Раствор нагревают с обратным холодильником в течение 2,5 ч и затем охлаждают до температуры окружающей среды. Раствор концентрируют в вакууме, получая целевой продукт, который используют без дополнительной очистки. ЖХМС: m/z 257,9 (MH+).(предположительно 6,78 ммоль), растворенному в MeOH, добавляют 10% Pd/C (0,15 г). Раствор перемешивают в атмосфере H2 (баллон) в течение ночи. Pd/C отфильтровывают и раствор концентрируют в вакууме. Неочищенный остаток очищают колоночной хроматографией (силикагель), используя градиент ДХМ/ДХМ:MeOH:NH4OH (90:10:1), и получают целевой продукт в виде белого твердого вещества (0,873 г, 58% с 2 стадий). ЖХМС: m/z 223,9 (MH+).(c) 7-Оксо-5,6,7,8-тетрагидропиридо[2,3-d]пиримидин-2-карбальдегид. К раствору 2-[бис-(метилокси)метил]-5,8-дигидропиридо[2,3-d]пиримидин-7(6H)-она (0,873 г, 3,91 ммоль) в смеси 1:1 H2O/ацетон (10 мл) добавляют п-TsOHH2O (0,074 г, 0,391 ммоль) и реакционную смесь нагревают до 80C в течение 3 дней с дополнительным количеством п-TsOHH2O (0,20 г). После исчезновения исходного вещества раствор концентрируют в вакууме, получая целевой продукт (1,023 г). ЖХМС: m/z 178,0 (MH+).(d) Указанное в заголовке соединение. К раствору гидрохлорида 4-[2-(4-амино-1-пиперидинил)этил]-6-(метилокси)пиридо[2,3-b]пиразин 3(4H)-она (способ получения описан в примере 126(m (0,600 г, 1,98 ммоль) в смеси 1:1 MeOH/ДХМ добавляют 7-оксо-5,6,7,8-тетрагидропиридо[2,3-d]пиримидин-2-карбальдегид (0,350 г, 1,98 ммоль),NaHCO3 (0,831 г, 9,90 ммоль) и избыток Na2SO4. Раствор перемешивают при температуре окружающей среды в течение ночи, после чего добавляют NaBH(OAc)3 (1/68 г, 7,92 ммоль). Полученный раствор перемешивают еще 1 ч и концентрируют на силикагеле в вакууме, после чего неочищенный остаток очи- 15016470 щают колоночной хроматографией (силикагель), используя градиент ДХМ/ДХМ:MeOH:NH4OH(90:1:0,1), и получают целевой продукт в виде свободного основания (0,396 г, 43%). ЖХМС: m/z 465,2 (MH+). 1H ЯМР (400 МГц, CDCl3)1,42-1,53 (м, 2H), 1,90 (д, J=10,86 Гц, 2H), 2,18 (т, J=10,61 Гц, 2H), 2,522,60 (м, 1H), 2,66-2,77 (м, 5H), 2,93 (т, J=7,71 Гц, 2H), 3,07 (д, J=11,62 Гц, 2H), 3,98 (с, 2H), 4,00-4,03 (м,3H), 4,52-4,62 (м, 2H), 6,70 (д, J=8,84 Гц, 1H), 7,99 (д, J=8,59 Гц, 1H), 8,12 (с, 1H), 8,34 (с, 1H). Свободное основание указанного в заголовке соединения превращают в соль HCl путем добавления 1 экв. 1 М HCl в эфире. Пример 126. 4-Хлор-2-[(1-2-[6-(метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)-ил]этил-4 пиперидинил)амино]метил-5,6-дигидропиридо[2,3-d]пиримидин-7(1H)-она гидрохлорид(a) N-[2,2-бис-(Метилокси)этил]-6-(метилокси)-3-нитро-2-пиридинамин. Смесь 2-хлор-6-(метилокси)-3-нитропиридина (10 г, 53 ммоль), диметилацеталя аминоацетальдегида (5,6 г, 6,2 мл, 53 ммоль) и карбоната калия (7,4 г, 53 ммоль) в ацетонитриле (100 мл) и ДМФ (10 мл) нагревают при 40C в течение 30 мин. Смесь фильтруют и экстрагируют ДХМ и насыщенным соляным раствором. Органический экстракт наносят на колонку с оксидом кремния и затем элюируют 0-100% этилацетатом в гексане, получая желтое твердое вещество (12,4 г, 90%). МС (+ve ионизация электрораспылением) m/z: 258 (MH+).(b) N2-[2,2-бис-(Метилокси)этил]-6-(метилокси)-2,3-пиридиндиамин. Раствор N-[2,2-бис-(метилокси)этил]-6-(метилокси)-3-нитро-2-пиридинамина (2,5 г, 10 ммоль) в метаноле гидрируют при 50 фунт/кв. дюйм в течение 0,5 ч в присутствии 10% палладия на угле (0,9 г). Смесь фильтруют, упаривают и подвергают азеотропной перегонке с хлороформом, получая темное масло (2,2 г). МС (+ve ионизация электрораспылением) m/z: 228 (MH+).(5 мл) перемешивают при комнатной температуре в течение ночи. Смесь фильтруют и упаривают. Остаток растворяют в этилацетате, после чего промывают водой и насыщенным соляным раствором. Органический экстракт концентрируют, наносят на колонку с оксидом кремния и затем элюируют 0-100% этилацетатом в гексане, получая продукт (2,5 г, 82%). МС (+ve ионизация электрораспылением) m/z: 314 (MH+).(d) 4-[2,2-бис-(Метилокси)этил]-6-(метилокси)пиридо[2,3-b]пиразин-3(4H)-он. Смесь этил N-[2-[2,2-бис-(метилокси)этил]амино-6-(метилокси)-3-пиридинил]глицината (1 г, 3,2 ммоль) и карбоната калия (1,3 г, 9,6 ммоль) в ДМФ (64 мл, 0,05 М) нагревают при 105-110C в течение 2 ч. Смесь охлаждают до комнатной температуре, после чего добавляют MnO2 (0,8 г, 11 ммоль) и реакционную смесь перемешивают в течение 18 ч. Реакционную смесь фильтруют, концентрируют, остаток растворяют в этилацетате и промывают водой. Органический экстракт концентрируют, добавляют на колонку с оксидом кремния и затем элюируют 0-100% этилацетатом в гексане, получая целевое соединение (0,78 г, 78%). МС (+ve ионизация электрораспылением) m/z: 266 (MH+).(3 мл) добавляют к 4-[2,2-бис-(метилокси)этил]-6(метилокси)пиридо[2,3-b]пиразин-3(4H)-ону (0,9 г, 3,4 ммоль) в воде (3 мл) при комнатной температуре и перемешивают в течение 2 ч. Реакционную смесь концентрируют, остаток очищают колоночной хроматографией (силикагель), используя градиент 0-10% MeOH/ДХМ/1% NH4OH, и получают продукт в виде смеси альдегида и полуацеталя (0,6 г, 80%). МС (+ve ионизация электрораспылением) m/z: 220 (MH+).(f) 3-Этил 1,1-диметил 1,1,3-пропантрикарбоксилат. К раствору диметилмалоната (2,5 г, 18,9 ммоль) в безводном ТГФ (20 мл) добавляют NaH (0,038 г,0,95 ммоль, 60% в минеральном масле). Реакционную смесь перемешивают при температуре окружающей среды в течение 15 мин. Этилакрилат (1,02 мл, 9,45 ммоль) растворяют в безводном ТГФ (1 мл) в отдельной колбе и затем добавляют по каплям в течение 30 мин к раствору диметилмалоната. Реакционную смесь перемешивают при температуре окружающей среды в течение ночи и концентрируют в вакууме. Остаток растворяют в EtOAc, промывают насыщенным раствором NH4Cl и насыщенным соляным раствором. Органическую фазу сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищен- 16016470 ный остаток очищают колоночной хроматографией (силикагель), используя градиент EtOAc/гексаны, и получают целевое соединение (1,68 г, 77%). 1(g) (2E)-3-Фенил-2-пропенимидамид. Циннамонитрил (25,0 г, 194 ммоль) растворяют в EtOH. Раствор охлаждают до 0C и газообразныйHCl барботируют через раствор в течение 30 мин. Раствор перемешивают при температуре окружающей среды в течение 1 ч и концентрируют в вакууме. Остаток растворяют в EtOH (100 мл), охлаждают до 0C и через другую воронку по каплям добавляют раствор NH3 в MeOH (7 М, 69 мл, 484 ммоль). По окончании добавления раствор оставляют нагреваться до температуры окружающей среды и перемешивают в течение ночи, после чего отфильтровывают образовавшийся NH4Cl. Раствор концентрируют в вакууме и полученный продукт используют без дополнительной очистки (28,6 г неочищенного). ЖХМС: m/z 147,4 (MH+).(h) Этил 3-4-гидрокси-6-оксо-2-[(E)-2-фенилэтенил]-1,6-дигидро-5-пиримидинилпропаноат. 3-Этил 1,1-диметил 1,1,3-пропантрикарбоксилат (1,65 г, 7,11 ммоль) и (2E)-3-фенил-2 пропенимидамид (1,04 г, 7,11 ммоль) объединяют в EtOH (36 мл). Добавляют триэтиламин (1,98 мл, 14,2 ммоль) и раствор нагревают с обратным холодильником в течение 3 ч, при этом по данным ЖХМС изменения не наблюдаются. Раствор охлаждают до комнатной температуры, обрабатывают NaOMe вMeOH (1,0 мл, 5,33 ммоль, 25-30% мас./мас. раствор) и затем нагревают с обратным холодильником в течение 3 ч. Добавляют две другие порции NaOMe в MeOH (21,0 мл) и раствор нагревают с обратным холодильником в течение ночи. По истечении данного времени образуется желтый осадок, который отфильтровывают. Маточный раствор подкисляют до pH 2 с помощью 1 н. HCl, после чего раствор концентрируют в вакууме. Полученное вещество объединяют с желтым твердым веществом и используют без дополнительной очистки. ЖХМС: m/z 315,2 (MH+).(i) Этил 3-4,6-дихлор-2-[([E])-2-фенилэтенил]-5-пиримидинилпропаноат. Неочищенный этил 3-4-гидрокси-6-оксо-2-[(E)-2-фенилэтенил]-1,6-дигидро-5-пиримидинилпропаноат (7,1 ммоль) растворяют в POCl3 (25 мл) и к раствору медленно добавляют N,N-диметиланилин(0,862 г, 0,9 мл, 7,1 ммоль). Затем реакционную смесь нагревают с обратным холодильником в течение 2 ч. После охлаждения до температуры окружающей среды полученный раствор осторожно и медленно добавляют в воду со льдом, чтобы погасить избыток POCl3. Смесь экстрагируют EtOAc (3) и концентрируют в вакууме. Неочищенный остаток очищают колоночной хроматографией (силикагель), используя градиент EtOAc/гексаны, и получают целевое соединение в виде желтого твердого вещества (0,48 г,19% с 2 стадий). ЖХМС: m/z 351,4 (MH+).(j) 4-Хлор-2-[(E)-2-фенилэтенил]-5,8-дигидропиридо[2,3-d]пиримидин-7(6H)-он. К раствору этил 3-4,6-дихлор-2-[(E)-2-фенилэтенил]-5-пиримидинилпропаноата (0,42 г, 1,19 ммоль) в 1,4-диоксане (5 мл) добавляют конц. NH4OH (3,5 мл). Реакционную смесь нагревают при 75C в герметично закрытой пробирке в течение ночи. Раствор концентрируют в вакууме, разбавляют водой и экстрагируют EtOAc/ДХМ. Органический слой промывают насыщенным соляным раствором, сушат надNa2SO4 и концентрируют в вакууме. Неочищенный остаток очищают колоночной хроматографией (силикагель) и получают целевое соединение (0,072 г, 21%). ЖХМС: m/z 286,2 (MH+). Кроме того, получают 3-4-амино-6-хлор-2-[(E)-2-фенилэтенил]-5-пиримидинилпропанамид (0,175 г). ЖХМС: m/z 303,3 (MH+). 3-4-Амино-6-хлор-2-[(E)-2-фенилэтенил]-5-пиримидинилпропанамид (0,175 г, 0,58 ммоль) растворяют в EtOH и газообразный HCl барботируют через раствор до насыщения. Раствор нагревают с обратным холодильником в течение 2 ч, охлаждают до температуры окружающей среды и концентрируют в вакууме. Остаток растворяют в воде, нейтрализуют до pH 9 с помощью раствора K2CO3 и экстрагируютEtOAc (3). Органические слои объединяют, сушат над Na2SO4, фильтруют и концентрируют в вакууме,получая этил 3-4-амино-6-хлор-2-[(E)-2-фенилэтенил]-5-пиримидинилпропаноат в виде белого твердого вещества. ЖХМС: m/z 332,2 (MH+). Затем полученный продукт растворяют в ДМФ (5 мл), обрабатывают K2CO3 (0,16 г, 1,16 ммоль) и нагревают при 75C в течение 30 мин. Раствор охлаждают, разбавляют водой и экстрагируют Et2O (3). Органический слой сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный остаток очищают колоночной хроматографией (силикагель), используя ДХМ/(ДХМ:MeOH:NH4OH) 90:10:1, и получают еще 0,11 г целевого соединения. ЖХМС: m/z 286,2 (MH+).- 17016470 ммоль) и каталитическое количество OsO4 (1 мл, 4% водный раствор), после чего раствор перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь концентрируют в вакууме, разбавляют водой и экстрагируют 10% MeOH/ДХМ (4). Органические слои объединяют, сушат надNa2SO4, фильтруют и концентрируют в вакууме. Неочищенный остаток очищают колоночной хроматографией (силикагель), используя градиент ДХМ/ДХМ:MeOH:NH4OH (90:10:1), и получают целевое соединение (0,05 г, 44%). ЖХМС: m/z 212,0 (MH+).[6-(Метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)-ил]ацетальдегид (0,250 г, 1,14 ммоль) объединяют с 1,1-диметилэтил 4-пиперидинилкарбаматом (0,229 г, 1,14 ммоль) в смеси 1:1 MeOH/ДХМ. Добавляют избыток Na2SO4 в качестве осушающего средства и раствор перемешивают при температуре окружающей среды в течение ночи. Добавляют NaBH(OAc)3 (0,724 г, 3,42 ммоль) и реакционную смесь перемешивают еще 2 ч. Раствор концентрируют на силикагеле в вакууме, неочищенный остаток очищают колоночной хроматографией (силикагель), используя градиент ДХМ/ДХМ:MeOH:NH4OH (90:10:1), и получают целевое соединение (0,271 г, 59%). ЖХМС: m/z 404,6 (MH+).(m) 4-[2-(4-Амино-1-пиперидинил)этил]-6-(метилокси)пиридо[2,3-b]пиразин-3(4H)-он. К раствору 1,1-диметилэтил (1-2-[6-(метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)-ил]этил-4 пиперидинил)карбамат (0,27 г, 0,67 ммоль) в ДХМ добавляют раствор HCl в 1,4-диоксане (1,68 мл, 6,7 ммоль, 4 М раствор). Реакционную смесь перемешивают при температуре окружающей среды в течение 3 ч. Реакционный раствор концентрируют в вакууме и получают соль гидрохлорид. Гидрохлорид помещают в смесь 1:1 MeOH/ДХМ. Затем полученный раствор обрабатывают смолойMP-карбонат (10 экв.; Argonaut Technologies Inc.) и перемешивают в течение 1 ч. Смолу отфильтровывают, раствор концентрируют в вакууме и получают свободное основание в виде не совсем белого твердого вещества (0,22 г, количественный выход). ЖХМС: m/z 304,3 (MH+).(0,064 г, 0,213 ммоль) в смеси 1:1 MeOH/ДХМ добавляют 4-хлор-7-оксо-5,6,7,8-тетрагидропиридо[2,3d]пиримидин-2-карбальдегид (0,045 г, 0,213 ммоль) и избыток Na2SO4. Раствор перемешивают при температуре окружающей среды в течение ночи и затем добавляют NaBH(OAc)3 (0,135 г, 0,639 ммоль). Полученный раствор перемешивают еще 2 ч, концентрируют на силикагеле в вакууме, неочищенный остаток очищают колоночной хроматографией (силикагель), используя градиент ДХМ/ДХМ:MeOH:NH4OH(90:10:1), и получают указанное в заголовке соединение в виде свободного основания (0,062 г, 58%). ЖХМС: m/z 499,6 (MH+). 1H ЯМР (400 МГц, CDCl3)1,48-1,59 (м, 2H), 1,96 (д, J=15,92 Гц, 2H), 2,28 (с, 2H), 2,58-2,69 (м, 1H),2,73-2,84 (м, 5H), 3,03-3,15 (м, 4H), 4,01-4,06 (м, 4H), 4,54-4,65 (м, 2H), 6,66-6,76 (м, 1H), 7,96-8,05 (м,1H), 8,09-8,15 (м, 1H). Часть свободного основания указанного в заголовке соединения (27 мг) превращают в соль HCl путем растворения свободного основания в смеси 1:1 ДХМ:MeOH с добавлением 1 экв. 1 М HCl в эфире. Затем смесь упаривают досуха (получают 25 мг). Пример 127. 4-Метил-2-[(1-2-[6-(метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)-ил]этил-4 пиперидинил)амино]метил-5,6-дигидропиридо[2,3-d]пиримидин-7(1H)-она гидрохлорид(31,0 г, 143 ммоль), после чего раствор перемешивают при комнатной температуре в течение 2 дней. Раствор становится темным, и темно-зеленое твердое вещество отфильтровывают. Раствор концентрируют в вакууме, разбавляют водой, подкисляют до pH 2 с помощью 6 н. HCl и образовавшийся желтый осадок отфильтровывают. Водный маточный раствор экстрагируют EtOAc. Собирают образовавшееся в процессе экстракции дополнительное количество желтого осадка. Объединенное желтое твердое вещество сушат в вакууме и используют без дополнительной очистки (12,1 г, 59%). ЖХМС: m/z 301,0 (MH+).(b) Метил 3-4,6-дихлор-2-[(E)-2-фенилэтенил]-5-пиримидинилпропаноат. Метил 3-4-гидрокси-6-оксо-2-[(E)-2-фенилэтенил]-1,6-дигидро-5-пиримидинилпропаноат раство- 18016470 ряют в POCl3 (75 мл), обрабатывают N,N-диметиланилином (4,85 г, 40 ммоль) и нагревают с обратным холодильником в течение 2 ч. После охлаждения до температуры окружающей среды полученный раствор осторожно и медленно добавляют в воду со льдом, чтобы погасить избыток POCl3. Смесь экстрагируют EtOAC (2), сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный остаток очищают колоночной хроматографией (силикагель), используя градиент EtOAC/гексаны, и получают целевой продукт в виде желтого твердого вещества (3,04 г, 23%). ЖХМС: m/z 337,2 (MH+).(c) 4-Хлор-2-[(E)-2-фенилэтенил]-5,8-дигидропиридо[2,3-d]пиримидин-7(6H)-он. К раствору метил 3-4,6-дихлор-2-[(E)-2-фенилэтенил]-5-пиримидинил]пропаноата (3,04 г, 9,02 ммоль) в 1,4-диоксане (100 мл) добавляют конц. NH4OH (20 мл). Реакционную смесь нагревают при 60C в герметично закрытой пробирке в течение 16 ч. Раствор концентрируют в вакууме, разбавляют водой и экстрагируют EtOAc. Органический слой промывают насыщенным соляным раствором, сушат надNa2SO4 и концентрируют. Неочищенный остаток очищают колоночной хроматографией (силикагель),используя градиент ДХМ/ДХМ:MeOH:NH4OH (90:10:1), и получают желтое твердое вещество (1,69 г),состоящее из 4-хлор-2-[(E)-2-фенилэтенил]-5,8-дигидропиридо[2,3-d]пиримидин-7(6H)-она (ЖХМС: m/z 285,9 (MH+ и метил 3-4-амино-6-хлор-2-[(E)-2-фенилэтенил]-5-пиримидинилпропаноата (ЖХМС: m/z 317,9(MH+. К объединенным продуктам, (1,69 г) растворенным в ДМФ (20 мл), добавляют K2CO3 (0,74 г, 5,3 ммоль) и раствор нагревают при 70C в течение 30 мин. Раствор концентрируют в вакууме и очищают колоночной хроматографией (силикагель), используя градиент ДХМ/ДХМ:MeOH:NH4OH (90:10:1), и получают целевой продукт в виде не совсем белого твердого вещества (0,92 г, 36% с 2 стадий). ЖХМС: m/z 285,9 (MH+).(d) 4-Метил-2-[(E)-2-фенилэтенил]-5,8-дигидропиридо[2,3-d]пиримидин-7(6H)-он. 4-Хлор-2-[(E)-2-фенилэтенил]-5,8-дигидропиридо[2,3-d]пиримидин-7(6H)-он (0,434 г, 2,05 ммоль) растворяют в ДМФ (5 мл) и вносят во флакон для СВЧ. Добавляют MeB(OH)2 (0,273 г, 4,56 ммоль),Pd(Ph3P)2Cl2 (0,107 г, 0,152 ммоль) и K2CO3 (1,05 г, 7,61 ммоль), после чего флакон закрывают. Реакционную смесь нагревают в микроволновой печи при 140C в течение 10 мин. Реакционную смесь концентрируют на силикагеле, очищают колоночной хроматографией (силикагель), используя градиент ДХМ/ДХМ:MeOH:NH4OH (90:10:1), и получают целевой продукт в виде не совсем белого твердого вещества (0,431 г, 74%). ЖХМС: m/z 265,9 (MH+).(e) 4-Метил-7-оксо-5,6,7,8-тетрагидропиридо[2,3-d]пиримидин-2-карбальдегид. 4-Метил-2-[(E)-2-фенилэтенил]-5,8-дигидропиридо[2,3-d]пиримидин-7(6H)-он (0,43 г, 1,63 ммоль) растворяют в ДХМ (20 мл) и раствор охлаждают до -78C. Газообразный озон барботируют через раствор, пока его цвет не станет темно-синим. После перемешивания в течение еще 10 мин при -78C одной порцией добавляют метилсульфид (1,0 мл). Раствор оставляют нагреваться до температуры окружающей среды в течение ночи. Раствор концентрируют на оксиде кремния, очищают колоночной хроматографией(силикагель), используя градиент ДХМ/ДХМ:MeOH:NH4OH (90:10:1), и получают целевой продукт в виде желтого твердого вещества (0,178 г, 49%). ЖХМС: m/z 191,9 (MH+).(f) Указанное в заголовке соединение. К раствору гидрохлорида 4-[2-(4-амино-1-пиперидинил)этил]-6-(метилокси)пиридо[2,3-b]пиразин 3(4H)-она (0,097 г, 0,287 ммоль) в смеси 1:1 MeOH/ДХМ (16 мл) добавляют 4-метил-7-оксо-5,6,7,8 тетрагидропиридо[2,3-d]пиримидин-2-карбальдегид (0,064 г, 0,287 ммоль), NaHCO3 (0,12 г, 1,44 ммоль) и избыток Na2SO4. Раствор перемешивают при температуре окружающей среды в течение 16 ч и добавляют NaBH(OAc)3 (0,182 г, 0,861 ммоль). Полученный раствор перемешивают еще 2 ч, концентрируют на силикагеле, неочищенный остаток очищают колоночной хроматографией (силикагель), используя градиент ДХМ/ДХМ:MeOH:NH4OH (90:10:1), и получают свободное основание целевого продукта в виде желтоватой маслянистой пленки (0,072 г, 53%). ЖХМС: m/z 479,2 (MH+). 1H ЯМР (400 МГц, CDCl3)м.д. 1,41-1,51 (м, 2H), 1,91 (д, J=11,12 Гц, 2H), 2,20 (т, J=10,61 Гц, 2H),2,45 (с, 3H), 2,49-2,59 (м, 1H), 2,68-2,79 (м, 4H), 2,93 (т, J=7,71 Гц, 2H), 3,06 (д, J=11,62 Гц, 2H), 3,92 (с,2H), 4,04 (с, 3H), 4,54-4,63 (м, 2H), 6,72 (д, J=8,59 Гц, 1H), 8,01 (д, J=8,59 Гц, 1H), 8,14 (с, 1H). Свободное основание указанного в заголовке соединения превращают в соль HCl путем растворения свободного основания в смеси 1:1 ДХМ:MeOH с добавлением 1 экв. 4 М HCl в 1,4-диоксане. Затем смесь упаривают досуха. Пример 128. 4-(Метилокси)-2-[(1-2-[6-(метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)-ил]этил-4 пиперидинил)амино]метил-5,6-дигидропиридо[2,3-d]пиридин-7(1H)-она гидрохлорид(a) 4-(Метилокси)-2-[(E)-2-фенилэтенил]-5,8-дигидропиридо[2,3-d]пиримидин-7(6H)-он. К суспензии 4-хлор-2-[(E)-2-фенилэтенил]-5,8-дигидропиридо[2,3-d]пиримидин-7(6H)-она (0,45 г,1,58 ммоль) в MeOH (10 мл) добавляют NaOMe (0,094 г, 1,74 ммоль). Реакционную смесь нагревают с обратным холодильником в течение 3 ч, после чего добавляют еще 0,10 г NaOMe и продолжают нагревание с обратным холодильником. Данную процедуру повторяют еще два раза в течение 9 ч. После полного исчезновения исходного вещества (ЖХМС) реакционную смесь концентрируют на силикагеле, очищают колоночной хроматографией (силикагель), используя градиент ДХМ/ДХМ:MeOH:NH4OH(90:10:1), и получают целевой продукт в виде не совсем белого твердого вещества (0,404 г, 91%). ЖХМС: m/z 282,2 (MH+).(b) 4-(Метилокси)-7-оксо-5,6,7,8-тетрагидропиридо[2,3-d]пиримидин-2-карбальдегид. 4-(Метилокси)-2-[(E)-2-фенилэтенил]-5,8-дигидропиридо[2,3-d]пиримидин-7(6H)-он (0,40 г, 1,44 ммоль) растворяют в ДХМ (20 мл). Раствор охлаждают до -78C, после чего через раствор барботируютO3, пока цвет раствора не станет темно-синим. Раствор перемешивают еще 10 мин при -78C и затем одной порцией добавляют метилсульфид (1,0 мл). Раствор оставляют нагреваться до температуры окружающей среды в течение 2 дней. Раствор концентрируют на оксиде кремния, очищают колоночной хроматографией (силикагель), используя градиент ДХМ/ДХМ:MeOH:NH4OH (90:10:1), и получают целевой продукт в виде желтого твердого вещества (0,216 г, 72%). ЖХМС: m/z 207,6 (MH+).(c) Указанное в заголовке соединение. К раствору гидрохлорида 4-[2-(4-амино-1-пиперидинил)этил]-6-(метилокси)пиридо[2,3-b]пиразин 3(4H)-она (0,105 г, 0,309 ммоль) в смеси 1:1 MeOH/ДХМ (16 мл) добавляют 4-(метилокси)-7-оксо-5,6,7,8 тетрагидропиридо[2,3-d]пиримидин-2-карбальдегид (0,074 г, 0,309 ммоль), NaHCO3 (0,13 г, 1,55 ммоль) и избыток Na2SO4. Раствор перемешивают при температуре окружающей среды в течение 16 ч и затем добавляют NaBH(OAc)3 (0,196 г, 0,927 ммоль). Полученный раствор перемешивают еще 2 ч и концентрируют на силикагеле в вакууме, после чего неочищенный остаток очищают колоночной хроматографией(силикагель), используя градиент ДХМ/ДХМ:MeOH:NH4OH (90:10:1), и получают свободное основание целевого продукта в виде желтоватой маслянистой пленки (0,114 г, 75%). ЖХМС: m/z 495,3 (MH+). 1H ЯМР (400 МГц, CDCl3)1,41-1,51 (м, 2H), 1,85-1,96 (м, 2H), 2,19 (т, J=10,61 Гц, 2H), 2,55 (ддд,J=14,21, 10,29, 4,04 Гц, 2H), 2,65 (т, J=7,58 Гц, 2H), 2,71-2,78 (м, 2H), 2,83 (т, J=7,71 Гц, 2H), 3,06 (д,J=11,62 Гц, 2H), 3,84-3,90 (м, 2H), 3,98-4,01 (м, 3H), 4,01-4,04 (м, 3H), 4,54-4,62 (м, 2H), 6,72 (д, J=8,59 Гц,1H), 8,01 (д, J=8,59 Гц, 1H), 8,14 (с, 1H), 8,73 (с, 1H). Свободное основание указанного в заголовке соединения превращают в соль HCl путем растворения свободного основания в смеси 1:1 ДХМ:MeOH с добавлением 1 экв. 4 М HCl в 1,4-диоксане. Затем смесь упаривают досуха. Если не указано иначе, соединения примеров 142-154 и 262, 267, 275 и 277-281 получают из указанных в таблице исходных веществ с помощью общего способа, описанного в примере 121(c)-(e) для третбутоксикарбонилзащищенных центральных элементов, или с помощью общего способа, описанного в примере 61(b)-(d) для бензилоксикарбонилзащищенных центральных элементов.(a) 3-Этил 1,1-диметил 1,1,3-пропантрикарбоксилат. К раствору диметилмалоната (2,5 г, 18,9 ммоль) в безводном ТГФ (20 мл) добавляют NaH (0,038 г,0,95 ммоль, 60% в минеральном масле). Реакционную смесь перемешивают при температуре окружающей среды в течение 15 мин. В отдельной колбе этилакрилат растворяют в безводном ТГФ (1 мл) и затем добавляют по каплям в течение 30 мин к раствору диметилмалоната. Реакционную смесь перемешивают при температуре окружающей среды в течение 16 ч и концентрируют в вакууме. Остаток растворяют вEtOAc (этилацетат) и промывают насыщенным раствором NH4Cl и насыщенным соляным раствором. Органическую фазу сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный остаток очищают колоночной хроматографией (силикагель), используя градиент EtOAc/гексаны, и получают целевое соединение в виде бесцветного масла (1,68 г, 77%). 1(b) (2E)-3-Фенил-2-пропенимидамид. Циннамонитрил (25,0 г, 194 ммоль) растворяют в EtOH (этанол) (200 мл). Раствор охлаждают до 0C, после чего через раствор в течение 30 мин барботируют газообразный HCl. Раствор перемешивают при температуре окружающей среды в течение 16 ч и концентрируют в вакууме. Остаток растворяют вEtOH (100 мл), охлаждают до 0C и через другую воронку по каплям добавляют раствор NH3/MeOH (7M, 69 мл, 484 ммоль). После окончания добавления раствор оставляют нагреваться до температуры окружающей среды и полученный NH4Cl отфильтровывают. Раствор концентрируют в вакууме и получают белое твердое вещество, которое используют без дополнительной очистки (26 г неочищенного вещества). ЖХМС: m/z 147,4 (MH+).(c) Этил 3-4-гидрокси-6-оксо-2-[(E)-2-фенилэтенил]-1,6-дигидро-5-пиримидинилпропаноат. 3-Этил 1,1-диметил 1,1,3-пропантрикарбоксилат (1,65 г, 7,11 ммоль) и (2E)-3-фенил-2 пропенимидамид (1,04 г, 7,11 ммоль) объединяют в EtOH (36 мл). Добавляют триэтиламин (1,98 мл, 14,2 ммоль) и раствор нагревают с обратным холодильником в течение 3 ч, при этом по данным ЖХМС изменения отсутствуют. Раствор охлаждают до комнатной температуры и обрабатывают NaOMe в MeOH(1,0 мл, 5,33 ммоль, 25 мас.% раствор), после чего раствор нагревают с обратным холодильником в течение еще 4 ч. Добавляют другую порцию NaOMe в MeOH (1,0 мл, 5,33 ммоль, 25 мас.% раствор) и раствор нагревают с обратным холодильником в течение 16 ч. По истечении указанного времени образовавшийся желтый осадок отфильтровывают. Маточный раствор подкисляют до pH 2 с помощью 1 н. HCl и затем раствор концентрируют в вакууме. Полученное вещество объединяют с желтым твердым веществом и используют без дополнительной очистки. ЖХМС: m/z 315,2 (MH+).(d) Этил 3-4,6-дихлор-2-[(E)-2-фенилэтенил]-5-пиримидинилпропаноат. Неочищенный этил 3-4-гидрокси-6-оксо-2-[(E)-2-фенилэтенил]-1,6-дигидро-5-пиримидинилпропаноат растворяют в POCl3 (25 мл), после чего к раствору медленно добавляют N,N-диметиланилин (0,9 мл, 7,1 ммоль). Затем реакционную смесь нагревают с обратным холодильником в течение 2 ч. После охлаждения до температуры окружающей среды полученный раствор осторожно и медленно добавляют в воду со льдом, чтобы погасить избыток POCl3. Смесь экстрагируют EtOAc (3), сушат над Na2SO4,фильтруют и концентрируют в вакууме. Неочищенный остаток очищают колоночной хроматографией(силикагель), используя градиент EtOAc/гексаны, и получают целевое соединение в виде желтого твердого вещества (0,48 г, 19% с 2 стадий). ЖХМС: m/z 351,4 (MH+).(10 мл) при -78C добавляют 1 н. раствор гидрида лития алюминия в ТГФ (2,0 мл, 2,0 ммоль). Реакционную смесь перемешивают в атмосфере азота в течение 1 ч и затем оставляют нагреваться до 0C. После выдерживания в течение 2 ч при 0C реакционную смесь гасят путем добавления 5 мл метанола и 2 мл воды. Реакционную смесь концентрируют в вакууме и остаток распределяют между EtOAc и насыщенным соляным раствором. Водный слой экстрагируют EtOAc (3), сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая продукт в виде желтого твердого вещества (275 мг, 86%), которое используют без дополнительной очистки. ЖХМС: m/z 309,0, 311,0 (MH+).(4 мл) добавляют K2CO3 (0,50 г, 3,6 ммоль). Суспензию нагревают до 60C и оставляют перемешиваться в течение 16 ч в атмосфере азота. Реакционную смесь охлаждают до к.т., концентрируют, чтобы удалить большую часть ДМФ, и остаток распределяют между EtOAc и водой. Водный слой дополнительно экстрагируют EtOAc (2), промывают насыщенным соляным раствором, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный остаток очищают колоночной хроматографией (силикагель),используя градиент EtOAc/гексаны, и получают целевое соединение в виде желтого твердого вещества(g) 4-Хлор-6,7-дигидро-5 Н-пирано[2,3-d]пиримидин-2-карбальдегид. 4-Хлор-2-[(E)-2-фенилэтенил]-6,7-дигидро-5 Н-пирано[2,3-d]пиримидин (0,21 г, 0,79 ммоль) растворяют в смеси 2:1 1,4-диоксан/вода (6 мл) и охлаждают до 0C. Добавляют NaIO4 (0,50 г, 2,4 ммоль) и каталитическое количество OsO4 (0,25 мл, 4% водный раствор), после чего раствор перемешивают при температуре окружающей среды в течение 2 ч. Реакционный раствор разбавляют водой (10 мл) и экстрагируют EtOAc (4). Органические слои объединяют, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный остаток очищают колоночной хроматографией (силикагель), используя ДХМ:MeOH (90:10), и получают целевое соединение в виде не совсем белого твердого вещества (0,055 г,36%).(3RS,5SR)-3-[(1,1-диметилэтил)(диметил)силил]окси-5-[(1,3-диоксо-1,3 дигидро-2H-изоиндол-2-ил)метил]-1-пиперидинкарбоксилату (0,92 г, 1,8 ммоль) в 10 мл ТГФ при 0C добавляют 1 н. раствор фторида тетрабутиламмония в ТГФ (4 мл, 4,0 ммоль). Реакционную смесь оставляют перемешиваться в атмосфере азота и нагреваться до комнатной температуры в течение 2 ч. Реакционную смесь распределяют между EtOAc и насыщенным водным раствором NH4Cl. Органическую фазу отделяют, промывают насыщенным водным раствором NaHCO3 и насыщенным соляным раствором, сушат над Na2SO4 и концентрируют при пониженном давлении. Неочищенный остаток очищают хроматографией на силикагеле, используя градиент 5-100% EtOAc/гексаны, и получают целевое соединение (380 мг, 54%) в виде бесцветного масла. МС (ES+) m/z 395,1 (MH+).Ag2O (1,15 г, 5,0 ммоль). Реакционную смесь оставляют перемешиваться при комнатной температуре в атмосфере азота в течение 48 ч. Реакционную смесь фильтруют через слой Celite, промывая 25 млCH2Cl2, и концентрируют при пониженном давлении. Неочищенный остаток очищают хроматографией на силикагеле, используя градиент 10-100% EtOAc/гексаны, и получают целевое соединение (210 мг,53%) в виде бесцветного масла. МС (ES+) m/z 409,3 (MH+).(c) цис-2-[(3RS,5SR)-5-(Метилокси)-3-пиперидинил]метил-1 Н-изоиндол-1,3(2H)-дион. К цис-фенилметил (3RS,5SR)-3-[(1,3-диоксо-1,3-дигидро-2H-изоиндол-2-ил)метил]-5-(метилокси)1-пиперидинкарбоксилату (210 мг, 0,51 ммоль) в EtOH (20 мл) добавляют Pd/C (50 мг, 10%). Реакционную смесь гидрируют в аппарате Парра при давлении H2 45 фунт/кв. дюйм в течение 3 ч. Водород заменяют на N2 и раствор фильтруют через слой Celite, чтобы удалить катализатор, который промывают дополнительным количеством EtOH (50 мл). Фильтрат концентрируют при пониженном давлении и получают целевое соединение (125 мг, 90%) в виде бесцветного масла, которое используют без дополнительной очистки. МС (ES+) m/z 274,9 (MH+).(d) 2-[3RS,5SR)-5-(Метилокси)-1-2-[6-(метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)-ил]этил 3-пиперидинил)метил]-1 Н-изоиндол-1,3(2H)-дион. К цис-2-[(3RS,5SR)-5-(метилокси)-3-пиперидинил]метил-1H-изоиндол-1,3(2H)-диону (125 мг,0,43 ммоль) в MeOH (1 мл) и CHCl3 (5 мл) добавляют [6-(метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)ил]ацетальдегид (способ получения описан в примере 126(е (100 мг, 0,50 ммоль), после чего реакционную смесь оставляют перемешиваться при комнатной температуре в атмосфере N2 в течение 4 ч. Добавляют Na(OAc)3BH (210 мг, 1,0 ммоль) и реакционную смесь оставляют перемешиваться при комнатной температуре в атмосфере азота в течение еще 14 ч. Реакционную смесь распределяют между насыщенным водным раствором NaHCO3 (5 мл) и CH2Cl2(20 мл), водный слой дополнительно экстрагируют CH2Cl2 (20 мл) и объединенные органические слои сушат над Na2SO4. Растворители удаляют, неочищенный остаток очищают хроматографией на силикагеле, используя градиент 0-10% MeOH/ДХМ, и получают целевое соединение (110 мг, 55%) в виде бесцветного масла. МС (ES+) m/z 478,2 (MH+).(e) цис-4-2-[(3RS,5SR)-3-(Аминометил)-5-(метилокси)-1-пиперидинил]этил-6-(метилокси)пиридо[2,3-b]пиразин-3(4H)-он. К 2-[3RS,5SR)-5-(метилокси)-1-2-[6-(метилокси)-3-оксопиридо[2,3-b]пиразин-4(3H)-ил]этил-3 пиперидинил)метил]-1 Н-изоиндол-1,3(2H)-диону (110 мг, 0,23 ммоль) в EtOH (5 мл) добавляют безводный гидразин (0,5 мл, 15,0 ммоль). Реакционную смесь нагревают до 40C и оставляют перемешиваться в атмосфере азота в течение 24 ч. Реакционную смесь фильтруют через слой Celite, после чего фильтрат распределяют между EtOAc (50 мл) и водой (10 мл). Водный слой дополнительно экстрагируют EtOAc(225 мл) и объединенные органические слои сушат над Na2SO4. Растворители удаляют и получают целевое соединение (62 мг, 77%) в виде желтовато-коричневого твердого вещества, которое используют без дополнительной очистки. МС (ES+) m/z 348,0 (MH+). Способ получения C. [7-(Метилокси)-2-оксо-1(2H)-хиноксалинил]ацетальдегид.(a) 7-(Метилокси)-2(1H)-хиноксалинон и 6-(метилокси)-2(1H)-хиноксалинон. Смесь 4-(метилокси)-1,2-бензолдиамина (10 г, 72,5 ммоль) и этилглиоксалата (50% в толуоле, 15,2 мл, 74,2 ммоль) в этаноле (400 мл) нагревают с обратным холодильником в течение 2 ч, охлаждают и держат в охлажденном состоянии в течение ночи. Твердое вещество отфильтровывают, промывают охлажденным на льду этанолом, сушат и получают смесь 7-(метилокси)-2(1H)-хиноксалинона и 6(метилокси)-2(1H)-хиноксалинона (в соотношении приблизительно 1:1, 9,99 г). После концентрирования маточного раствора и охлаждения на льду получают вторую порцию смеси изомеров (в соотношении приблизительно 2:1), которую собирают, промывают и сушат, как указано выше (1,11 г).(b) 7-(Метилокси)-2-(2-пропен-1-илокси)хиноксалин (O-аллил 1) и 6-(метилокси)-2-(2-пропен-1 илокси)хиноксалин (O-аллил 2). Смесь 7-(метилокси)-2(1H)-хиноксалинона и 6-(метилокси)-2(1H)-хиноксалинона (11,1 г, 63,1 ммоль) перемешивают с аллилиодидом (6,25 мл, 69,6 ммоль) и карбонатом калия (26,2 г, 189,8 ммоль) в диметилформамиде (200 мл) при комнатной температуре в течение 2,5 ч и затем упаривают. Остаток растворяют в дихлорметане и воде, после чего фазы разделяют. Органическую фазу экстрагируют дихлорметаном, сушат и упаривают. Хроматографируют на оксиде кремния, элюируя 0-100% эфир/петролейный эфир, и получают вначале изомер 1 О-аллила (1,18 г, 9%), затем изомер 2 O-аллила(1,99 г, 15%), и в конце смесь двух изомеров N-аллила, 7-(метилокси)-1-(2-пропен-1-ил)-2(1H)хиноксалинона и 6-(метилокси)-1-(2-пропен-1-ил)-2(1H)-хиноксалинона (9,18 г, 67%).(c) 7-(Метилокси)-1-(2-пропен-1-ил)-2(1H)-хиноксалинон. Изомер 1 O-аллила (1,18 г, 5,45 ммоль) нагревают с обратным холодильником в присутствии тетракис(трифенилфосфин)палладия(0) (120 мг) в толуоле (25 мл) в течение 3 ч. Растворитель упаривают, остаток хроматографируют на оксиде кремния, элюируя 50-100% этилацетат/гексан, и получают 7(метилокси)-1-(2-пропен-1-ил)-2(1H)-хиноксалинон (1,03 г, 87%).(d) Указанное в заголовке соединение. Смесь 7-(метилокси)-1-(2-пропен-1-ил)-2(1H)-хиноксалинона (1,03 г, 4,75 ммоль) и периодата натрия (4,68 г, 21,8 ммоль) в 2-бутаноле (20 мл) и воде (40 мл) обрабатывают тетраоксидом осмия (4% раствор в воде, 1,1 мл) и перемешивают в течение 3,75 ч. Бутанол упаривают, остаток разбавляют водой и экстрагируют дихлорметаном, затем смесью дихлорметан/ТГФ. Экстракты сушат и упаривают, после чего остаток хроматографируют на оксиде кремния, элюируя 50-100% этилацетат/гексан, и получают альдегид (0,68 г, 66%).(a) 5-(3-Гидроксипропил)-2-[(E)-2-фенилэтенил]-4(1H)-пиримидинон. К -валеролактону (2,0 г, 20 ммоль) в безводном диэтиловом эфире (20 мл) добавляют этилформиат(1,6 мл; 21 ммоль) и гидрид натрия (1,0 г 60% мас.:мас. дисперсии в масле, 25 ммоль). Реакционную смесь оставляют перемешиваться при комнатной температуре в атмосфере азота в течение 45 мин. Добавляют раствор (2E)-3-фенил-2-пропенимидамида (2,92 г, 20 ммоль) в EtOH (25 мл), после чего смесь нагревают до 70C и перемешивают при данной температуре в течение 4 ч. Реакционную смесь охлаждают до комнатной температуры, добавляют воду (50 мл) и экстрагируют EtOAc (2200 мл). Объединенные органические экстракты сушат Na2SO4 и удаляют растворители, после чего неочищенные остатки очищают колоночной хроматографией на силикагеле, используя градиент 0-10% MeOH/ДХМ. Фракции,содержащие продукт, концентрируют и получают целевое соединение (2,3 г, 45%). МС (ES+) m/z 257 (MH+).(b) 2-[(E)-2-Фенилэтенил]-6,7-дигидро-5 Н-пирано[2,3-d]пиримидин. К 5-(3-гидроксипропил)-2-[(E)-2-фенилэтенил]-4(1H)-пиримидинону (1,0 г, 4,0 ммоль) в ТГФ (20 мл) добавляют трифенилфосфин (1,6 г, 6,0 ммоль) и диэтилазодикарбоксилат (1,0 г, 6,0 ммоль). Реакционную смесь оставляют перемешиваться при комнатной температуре в атмосфере азота в течение 14 ч. Реакционную смесь распределяют между водой (20 мл) и EtOAc (100 мл), после чего водную фазу дополнительно экстрагируют EtOAc (250 мл). Объединенные органические экстракты сушат Na2SO4, растворители удаляют, неочищенный остаток очищают колоночной хроматографией на силикагеле, исполь- 26016470 зуя градиент 0-2,5% MeOH в ДХМ, и получают целевое соединение (0,81 г, 84%). МС (ES+) m/z 239 (MH+).(с) 6,7-Дигидро-5 Н-пирано[2,3-d]пиримидин-2-карбальдегид. 2-[(E)-2-Фенилэтенил]-6,7-дигидро-5 Н-пирано[2,3-d]пиримидин (0,5 г, 2,1 ммоль) растворяют в ДХМ (20 мл) и через реакционную смесь в течение 15 мин барботируют O3 при -78C. Затем в течение 10 мин барботируют азот, чтобы удалить избыток O3, и реакционную смесь гасят диметилсульфидом (2,0 мл, 32,2 ммоль). Реакционную смесь оставляют нагреваться до к.т. и перемешивают еще 14 ч. Затем все растворители удаляют и получают целевое соединение (0,28 г, 80%), которое используют без дополнительной очистки. МС (ES+) m/z 165 (MH+). Способ получения E. 2-Оксо-2H-пирано[2,3-b]пиридин-7-карбальдегид.(a) 3-(Гидроксиметил)-6-метил-2(1H)-пиридинон. 2-Гидрокси-6-метилпиридин-3-карбоновую кислоту (2 г, 13 ммоль) растворяют в сухом ТГФ (80 мл) и полученный раствор охлаждают до -70C. Затем в течение 20 мин добавляют LiAlH4 (32 мл, 32 ммоль). Реакционную смесь нагревают до комнатной температуры и затем нагревают при 70C в течение 1 ч. После охлаждения до 0C к реакционной смеси добавляют H2O (2 мл), 10% NaOH (4 мл) и в конце еще H2O (2 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 ч. Добавляют Na2SO4 (6 г), после чего смесь перемешивают еще 30 мин, фильтруют и промывают MeOH. Фильтрат концентрируют досуха и получают целевой продукт в виде твердого вещества (2 г, 100%). ЖХМС (ES+) m/z 140 (MH+).(80 мл) и MeOH (5 мл), после чего добавляют MnO2 (3,7 г, 43 ммоль). Реакционную смесь нагревают при 57C в течение 18 ч. Реакционную смесь фильтруют через слой Celite, концентрируют и получают твердое вещество (1,8 г, 90%). ЖХМС (ES+) m/z 138 (MH+).(c) 7-Метил-2H-пирано[2,3-b]пиридин-2-он. К 6-метил-2-оксо-1,2-дигидро-3-пиридинкарбальдегиду (318 мг, 2,3 ммоль) в Ac2O (3 мл) добавляют Et3N (1 мл). Реакционную смесь нагревают при 120C в течение 18 ч. Реакционную смесь концентрируют, полученное вещество растворяют в MeOH, загружают на силикагеле и упаривают. После очистки колоночной хроматографией (силикагель) с использованием градиента гексаны/этилацетат (0-100%EtOAc) получают целевой продукт в виде твердого вещества (80 мг, 30%). ЖХМС (ES+) m/z 162 (MH+).(d) Указанное в заголовке соединение. К раствору 7-метил-2H-пирано[2,3-b]пиридин-2-она (160 мг, 0,98 ммоль) в 1,4-диоксане (12 мл) в реакционном флаконе для СВЧ добавляют SeO2 (328 мг, 3,0 ммоль). Реакционный флакон закрывают и смесь нагревают в микроволновой печи при 160C в течение 90 мин. Реакционную смесь разбавляют ДХМ, фильтруют через слой Celite и концентрируют. Полученное вещество растворяют в ДХМ, загружают на слой силикагеля и упаривают. После очистки колоночной хроматографией (силикагель) с использованием градиента ДХМ/MeOH (0-10% MeOH) получают целевой продукт в виде бледно-желтого твердого вещества (120 мг, 40%). ЖХМС (ES+) m/z 176 (MH+). Способ получения F. 7-Оксо-6,7-дигидро-1H-пиримидо[5,4-b][1,4]оксазин-2-карбальдегид.(a) 2-[(E)-2-Фенилэтенил]-5-(тетрагидро-2H-пиран-2-илокси)-4(1H)-пиримидинон NaH (0,38 г, 9,5 ммоль, 60% в жидком парафине) добавляют медленно к раствору этил (тетрагидро-2H-пиран-2 илокси)ацетата (полученного путем обработки этилгидроксиацетата 3,4-дигидро-2H-пираном и TsOH)(1,0 г, 5,3 ммоль) и сухого этилформиата (3,9 г, 53 ммоль) в ТГФ (20 мл). Реакционную смесь перемешивают при комнатной температуре в течение 15 мин и затем нагревают при 65C в течение 45 мин. Реакционную смесь концентрируют досуха и получают бледно-желтое твердое вещество. Твердое вещество добавляют к раствору (2E)-3-фенил-2-пропенимидамида (0,78 г, 5,3 ммоль) в MeOH/EtOH (20 мл/20 мл),после чего смесь нагревают при 80C в течение 4 ч. Полученное вещество выливают в ДХМ (10 мл), содержащий силикагель (3 г), и упаривают. После очистки колоночной хроматографией (силикагель) с использованием градиента MeOH/ДХМ (0-10%) получают целевой продукт в виде бледно-желтого твердого вещества (1 г, 63%). ЖХМС: m/z 299 (MH+).(b) 2-[(E)-2-Фенилэтенил]-5-(тетрагидро-2H-пиран-2-илокси)-4-пиримидинила трифторметансульфонат. К суспензии 2-[(E)-2-фенилэтенил]-5-(тетрагидро-2H-пиран-2-илокси)-4(1H)-пиримидинона (2,04 г,6,84 ммоль) в ДХМ (25 мл) добавляют пиридин (1,22 мл, 15,05 ммоль). После охлаждения до -78C по каплям медленно добавляют тирфторметансульфоновый ангидрид (1,38 мл, 8,2 ммоль). Температуру реакционной смеси поддерживают при -78C в течение 10 мин, после чего охлаждающую баню заменяют на баню со льдом и реакционную смесь перемешивают еще 0,5 ч. Реакционную смесь выливают в воду и- 27016470 водную фазу экстрагируют ДХМ. Органическую фазу затем промывают водой, насыщенным водным раствором NaHCO3 и насыщенным соляным раствором. Органическую фазу сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая темно-красное масло, которое непосредственно используют на следующей стадии. ЖХМС: m/z 431,0 (MH+).(c) 2-[(E)-2-Фенилэтенил]-5-(тетрагидро-2H-пиран-2-илокси)-4-пиримидинамин. Неочищенный 2-[(E)-2-фенилэтенил]-5-(тетрагидро-2H-пиран-2-илокси)-4-пиримидинила трифторметансульфонат (6,8 ммоль) подвергают взаимодействию с 0,5 М раствором аммиака в 1,4-диоксане (136 мл) в сосуде для реакций под давлением при 60C в течение 24 ч. Реакционную смесь концентрируют в вакууме, остаток помещают в ДХМ и промывают водой, насыщенным водным раствором NaHCO3 и насыщенным соляным раствором. Органическую фазу сушат над Na2SO4, фильтруют и концентрируют. Неочищенный остаток очищают колоночной хроматографией (силикагель), используя градиентMeOH/ДХМ, и получают целевое соединение в виде желтовато-коричневого твердого вещества (1,28 г,63% с двух стадий). ЖХМС: m/z 298,0 (MH+).(d) 4-Амино-2-[(E)-2-фенилэтенил]-5-пиримидинола гидрохлорид. 2-[(E)-2-Фенилэтенил]-5-(тетрагидро-2H-пиран-2-илокси)-4-пиримидинамин (1,28 г, 4,3 ммоль) суспендируют в MeOH (25 мл) и нагревают в масляной бане при 50C до полного растворения. К полученному раствору добавляют 4 М HCl в 1,4-диоксане (0,11 мл, 0,43 ммоль) и реакционную смесь нагревают при 50C в течение 1,5 ч. В данный момент времени ЖХМС показывает незначительное развитие реакции, поэтому добавляют еще 1,1 мл 4 М HCl/1,4-диоксан и нагревание продолжают в течение 3 ч. Реакционную смесь оставляют охлаждаться до комнатной температуры, при этом выпадает белый осадок. Растворитель удаляют в вакууме и полученное желтовато-коричневое твердое вещество сушат в глубоком вакууме в течение ночи, получая 1,08 г (100%, для соли HCl). Данное вещество используют без дополнительной очистки. ЖХМС: m/z 214,0 (MH+).(e) 2-[(E)-2-Фенилэтенил]-1H-пиримидо[5,4-b][1,4]оксазин-7(6H)-он. К суспензии гидрохлорида 4-амино-2-[(E)-2-фенилэтенил]-5-пиримидинола (250 мг, 1,0 ммоль) в абсолютном этаноле (5 мл) добавляют трет-бутоксид калия (224 мг, 2,0 ммоль) при комнатной температуре. После перемешивания в течение 5 мин по каплям добавляют этилбромацетат (0,11 мл, 1,0 ммоль) и реакционную смесь перемешивают в течение 18 ч. Растворитель упаривают и остаток помещают в смесь 10% MeOH-CHCl3 с небольшим количеством воды. Слои разделяют и водную фазу экстрагируют 10%MeOH-CHCl3 (3). Объединенные органические экстракты концентрируют и полученное твердое вещество перетирают с EtOAc. Белое твердое вещество собирают фильтрацией, получая 106 мг (42%). ЖХМС: m/z 254,0 (MH+).(f) Указанное в заголовке соединение. К суспензии 2-[(E)-2-фенилэтенил]-1H-пиримидо[5,4-6][1,4]оксазин-7(6H)-она (106 мг, 0,418 ммоль) в 1,4-диоксане (12 мл) и воде (3 мл) добавляют NaIO4 (357 мг, 1,67 ммоль) и OsO4 (0,1 мл, 4 мас.%, в воде), после чего реакционную смесь перемешивают при комнатной температуре. Через 2 ч дополнительно добавляют 3 мл 1,4-диоксана и 180 мг NaIO4. Всего через 7,5 ч реакционную смесь закрывают и хранят в морозильнике в течение выходных дней в конце недели. После нагревания до комнатной температуры добавляют дополнительное количество OsO4 (0,1 мл, 4 мас.%, в воде) и реакционную смесь перемешивают еще 4 ч. Растворитель упаривают и получают белое твердое вещество, которое растворяют в ДХМ и воде. Водный слой экстрагируют 10% MeOH-ДХМ (6). Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют, получая светлое желтовато-коричневое твердое вещество (92 мг), которое дополнительно не очищают. ЖХМС: m/z 180,0 (MH+). Способ получения G. 7-Оксо-5,6,7,8-тетрагидропиридо[2,3-d]пиримидин-2-карбальдегид.(a) Диэтил -формилглутарата натриевая соль. Получают из диэтилглутарата и этилформиата в присутствии металлического натрия в диэтиловом эфире с помощью описанного в литературе способа (J. Bigs, P. Sykes, J. Chemical Society (1959), 1849-55). Получают твердое вещество (28 г, 45%). ЖХМС (ES+) m/z 238 (MH+).(0,61 г, 4,2 ммоль), полученный в примере 126(g), объединяют в EtOH (20 мл) и MeOH (10 мл) и нагревают с обратным холодильником в течение 12 ч. Растворитель удаляют, полученное твердое вещество перетирают с эфиром, фильтруют и получают целевой продукт в виде желтого твердого вещества (0,75 г,56%). Маточный раствор содержит дополнительное количество продукта, но его не выделяют. ЖХМС (ES+) m/z 299 (MH+).(30 мл) нагревают при 120C в течение 4 ч. Растворитель удаляют в вакууме. Неочищенный остаток очищают колоночной хроматографией (силикагель), используя градиент EtOAc/гексаны (30-50%), и получают целевое соединение в виде желтого твердого вещества (6,6 г, 53%). ЖХМС (ES+) m/z 317 (MH+).(d) 2-[(E)-2-Фенилэтенил]-5,8-дигидропиридо[2,3-d]пиримидин-7(1H)-он. В герметичную колбу объемом 320 мл при комнатной температуре добавляют этил 3-4-хлор-2[(E)-2-фенилэтенил]-5-пиримидинилпропаноат (6,6 г, 20,83 ммоль) и раствор аммиака в MeOH (59,5 мл,417 ммоль, 7 М раствор). Колбу герметично закрывают и реакционную смесь нагревают при 110C в течение 7 ч. Через 3 ч осаждается твердое вещество. Затем реакционную смесь перемешивают при комнатной температуре в течение ночи и нагревают в течение еще 5 ч. Реакционную смесь фильтруют через воронку Бюхнера, полученное твердое вещество промывают этанолом и сушат. Получают твердое вещество (2 г, 36,3%). ЖХМС (ES+) m/z 252 (MH+).(e) 7-Оксо-5,6,7,8-тетрагидропиридо[2,3-d]пиримидин-2-карбальдегид. В круглодонную колбу добавляют 2-[(E)-2-фенилэтенил]-5,8-дигидропиридо[2,3-d]пиримидин 7(6H)-он (600 мг, 2,39 ммоль) в ДМФ (10 мл) и метаноле (10 мл). Через раствор при комнатной температуре в течение 20 мин барботируют озон, после чего избыток озона удаляют потоком азота. Добавляют диметилсульфид (1,77 мл, 23,9 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 24 ч. Растворитель упаривают и полученное твердое вещество растворяют в 20% MeOH-ДХМ. Неочищенный продукт наносят на колонку с силикагелем и элюируют 0-20% MeOH-ДХМ. Из собранных фракций выделяют указанное в заголовке соединение (400 мг, выход 95%). ЖХМС (ES+) m/z 177 (MH+). 1H ЯМР (400 МГц, ДМСО-d6)2,6 (т, 2H), 3,0 (т, 2H), 8,7 (с, 1H), 9,8 (с, 1H), 10,27 (шир.с, 1H). Если не указано иначе, соединения примеров 310, 312-315 получают из указанных в таблице исходных веществ с помощью общего способа, описанного в примере 121(c)-(e) для трет-бутоксикарбонилзащищенных центральных элементов, или с помощью общего способа, описанного в примере 61(b)-(d) для бензилоксикарбонилзащищенных центральных элементов.
МПК / Метки
МПК: A61P 31/04, C07D 497/04, C07D 491/04, A61P 31/06, C07D 519/00, C07D 513/04, C07D 498/04
Метки: n-этилазахинолонов, n-этилхинолонов, производные, аналоги
Код ссылки
<a href="https://eas.patents.su/30-16470-proizvodnye-i-analogi-n-etilhinolonov-i-n-etilazahinolonov.html" rel="bookmark" title="База патентов Евразийского Союза">Производные и аналоги n-этилхинолонов и n-этилазахинолонов</a>
Тетраазабензо[е]азуленовые производные и их аналоги

Номер патента: 10888
Опубликовано: 30.12.2008
Авторы: Хаммонд Марлис, Эллиотт Ричард Луис, Камерон Кимберли О'киф
МПК: A61P 43/00, C07D 487/04, A61K 31/551...
Метки: производные, тетраазабензо[е]азуленовые, аналоги
Формула / Реферат:
1. Соединение формулы (I) где каждый из А, В, X и D представляет собой -C(R5)-; и каждый из Е и G представляет собой -N-, R1 выбран из группы, состоящей из (С2-С6)алкила, группировок галогензамещенный (С1-C6)алкил-, (C6-C10)арила, группировок (C6-C10)арил(С1-C6)алкил-, гетероарил-А, гетероарил-А(С1-C6)алкил- и полностью насыщенный (C3-С7)циклоалкил(С1-C6)алкил-, и когда ни один из R6 и R7 не представляет собой фенилметил-, тогда R1 выбран из...
Производные триптамина и их аналоги, и содержащие их фармацевтические композиции

Номер патента: 5918
Опубликовано: 30.06.2005
Авторы: Зисапел Нава, Лаудон Моше
МПК: A61K 31/404, C07D 209/12
Метки: аналоги, производные, содержащие, фармацевтические, триптамина, композиции
Формула / Реферат:
1. Соединение формулы (I) являющееся основным, где каждый из R1, R2 и R3, независимо, представляет собой водород, галоген, C1-4-алкил, C1-4-алкокси, NR'R", N(R')C(:O)Rш, нитрогруппу, арил, арил-C1-4-алкил или арил-C1-4-алкокси, Rш представляет собой C1-4-алкил или арил, и каждый из R' и R" являются независимо H или C1-4-алкилом, или R' = R" = ClCH2CH2, или NR'R" представляет собой насыщенное 3-8-членное гетероциклическое кольцо; m означает 0-4;...
Фармацевтические композиции, содержащие бета-лапахон или его производные или аналоги, и способы их применения

Номер патента: 7075
Опубликовано: 30.06.2006
Авторы: Редди Дашаратха Дж., Дзианг Зивей
МПК: A61K 31/335, A61K 31/35
Метки: аналоги, содержащие, композиции, фармацевтические, производные, способы, бета-лапахон, применения
Формула / Реферат:
1. Фармацевтическая композиция, включающая терапевтически эффективное количество b -лапахона или его производного или аналога и фармацевтически приемлемый водорастворимый носитель. 2. Фармацевтическая композиция по п.1, которая включает комплекс или раствор терапевтически эффективного количества b -лапахона или его производного или аналога и фармацевтически приемлемого водорастворимого носителя. 3. Фармацевтическая композиция по п.1...
Проостровковые пептиды человека, их производные и аналоги и способы их применения

Номер патента: 13821
Опубликовано: 30.08.2010
Авторы: Упхэм Лорейн В., Леветан Клареса С.
МПК: A61K 38/08, A61K 38/10, A61P 3/10...
Метки: человека, производные, применения, способы, аналоги, проостровковые, пептиды
Формула / Реферат:
1. Изолированный проостровковый пептид человека, включающий аминокислотную последовательность, выбранную из SEQ ID NO:3 и SEQ ID NO:7.2. Проостровковый пептид человека по п.1, включающий соединение, конъюгированное с проостровковым пептидом человека, в котором соединение выбрано из альбумина, трансферрина и полиэтиленгликоля.3. Проостровковый пептид человека по п.2, в котором соединение является полиэтиленгликолем.4. Проостровковый пептид...
4-((феноксиалкил)тио)феноксиуксусные кислоты и их аналоги

Номер патента: 12241
Опубликовано: 28.08.2009
Авторы: Ван Айхуа, Диэнджелис Алан Р., Чжан Жуй, Ко Джи-Хун
МПК: A61K 31/192, A61K 31/277, A61K 31/381...
Метки: 4-((феноксиалкил)тио)феноксиуксусные, аналоги, кислоты
Формула / Реферат:
1. Соединение формулы (I) где X выбран из ковалентной связи или О, Y представляет собой S или О; ---W--- представляет собой группу, выбранную из -CH=, -СН2-; Z выбран из О, CH и СН2, при условии, что когда Y представляет собой О, Z представляет собой О; R1 представляет собой H или C1-3алкил; R2 представляет собой H или галоген; R3 представляет собой H; R4 представляет собой С1-5алкил, замещенный галогеном; R5 представляет собой необязательно...
Предыдущий патент: Замороженное кондитерское изделие
Случайный патент: Непрерывный способ карбонилирования этилена