Соединения тетрагидроциклопента[b]индола в качестве модуляторов рецептора андрогенов
Номер патента: 15627
Опубликовано: 31.10.2011
Авторы: Джадхав Прабхакар Кондаджи, Гавардинас Константинос, Грин Джонатан Эдвард, Мэттьюз Дональд Пол


























Формула / Реферат
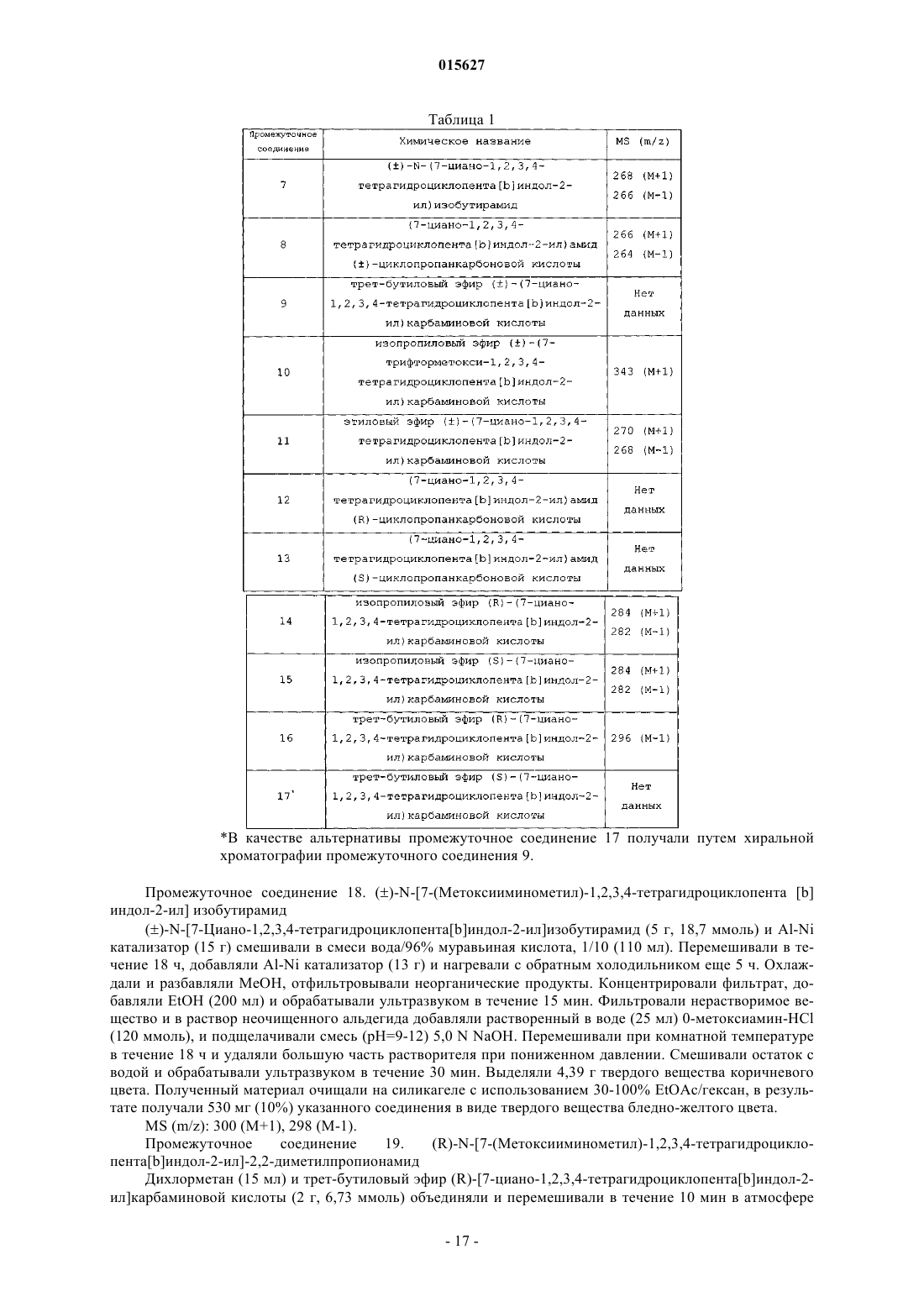
1. Соединение формулы (I)

где углеродный центр С* может быть в R, S конфигурации;
R1 представляет циано, -CH=NOCH3, -OCHF2 или -OCF3;
R2 представляет -COR2a или -SO2R2b;
R2a представляет (С1-С4)алкил, (C1-C4)алкокси, циклопропил или -NRaRb;
R2b представляет (С1-С4)алкил, циклопропил или -NRaRb;
Ra и Rb, каждый независимо, представляют собой в каждом случае Н или (C1-C4)алкил и
R3 представляет гетероарильную группу, выбранную из группы, состоящей из пиридинила, пиримидинила, пиразинила, пиридазинила, тиазолила, изотиазолила и тиадиазолила, каждый из которых необязательно замещен 1 или 2 заместителями, независимо выбранными из группы, состоящей из метила, этила, брома, хлора, фтора, -CHF2, -CF3, гидрокси, аминогруппы и -NHCH2CO2H;
или смесь его (R)- и (S)-энантиомеров, или его фармацевтически приемлемая соль.
2. Соединение или соль по п.1,
где углеродный центр С* находится в S-конфигурации, когда R2 представляет -COR2a, и в R-конфигурации, когда R2представляет -SO2R2b;
R1 представляет циано или -CH=NOCH3;
R2a представляет этил, изопропил, метокси, этокси, пропокси, изопропокси, изобутокси, трет-бутокси, циклопропил или -N(СН3)2; и R2b представляет метил, этил, пропил, циклопропил, -N(CH3)2или -N(C2H5)2; и
R3 представляет 6-фторпиридин-2-ил, пиридин-2-ил, 3-гидроксипиридин-2-ил, 6-дифторметилпиридин-2-ил, 2-аминопиридин-3-ил, 2-карбоксиметиламинопиридин-3-ил, пиримидин-4-ил, пиримидин-2-ил, 2-хлорпиримидин-4-ил, тиазол-4-ил, 2-метилтиазол-4-ил, 2-хлортиазол-4-ил, тиазол-2-ил, тиазол-5-ил, 4-аминотиазол-5-ил, пиразин-2-ил, 5-метилпиразин-2-ил, 3-хлорпиразин-2-ил, 6-метилпиразин-2-ил, 3-аминопиразин-2-ил, 3-метилпиразин-2-ил, пиридазин-3-ил, 5-бромизотиазол-3-ил, изотиазол-3-ил, 4,5-дихлоризотиазол-З-ил или [1,2,5]тиадиазол-3-ил.
3. Соединение по п.1, выбранное из группы, состоящей из изопропилового эфира (S)-(7-циано-4-пиридин-2-илметил-1,2,3,4-тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты; изопропилового эфира (S)-(7-циано-4-тиазол-5-илметил-1,2,3,4-тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты; изопропилового эфира (S)-[4-(2-аминопиридин-3-илметил)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты; (R)-N'-[4-(4-аминотиазол-5-илметил)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]-N,N-диметилсульфамида; и изопропилового эфира (S)-[4-(4-аминотиазол-5-илметил)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты, или его фармацевтически приемлемая соль.
4. Соединение или его фармацевтически приемлемая соль по п.3, представляющее собой изопропиловый эфир (S)-(7-циано-4-пиридин-2-илметил-1,2,3,4-тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты.
5. Применение соединения или его фармацевтически приемлемой соли по любому одному из пп.1-4 для получения лекарственного средства для лечения гипогонадизма, снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы, саркопении, возрастного угасания функций, задержки полового созревания у мальчиков, анемии, мужской или женской сексуальной дисфункции, эректильной дисфункции, снижения либидо, депрессии или летаргии.
6. Фармацевтическая композиция, включающая соединение или его фармацевтически приемлемую соль по любому одному из пп.1-4 в комбинации с одним или более фармацевтически приемлемыми носителями, разбавителями или эксципиентами.
Текст
Дата публикации и выдачи патента Номер заявки СОЕДИНЕНИЯ ТЕТРАГИДРОЦИКЛОПЕНТА[b]ИНДОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА АНДРОГЕНОВ Настоящее изобретение обеспечивает соединение формулы Гавардинас Константинос, Грин Джонатан Эдвард, Джадхав Прабхакар Кондаджи, Мэттьюз Дональд Пол (US) Медведев В.Н. (RU) Формула (I) или его фармацевтически приемлемую соль; фармацевтические композиции, включающие соединение формулы (I) в комбинации с приемлемыми носителем, разбавителем или эксципиентом; и способ лечения физиологических заболеваний, в частности снижения костной массы,остеопороза, остеопении или снижения мышечной массы или силы, включающий введение соединения формулы (I) или его фармацевтически приемлемой соли.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) 015627 Область техники Настоящее изобретение относится к соединениям тетрагидроциклопент[b]индола или фармацевтически приемлемым солям таких соединений, которые можно применять в качестве терапевтических агентов, а также к фармацевтическим композициям, включающим соединения или соли, к способам применения соединений или солей при лечении заболеваний у пациентов, а также к промежуточным соединениям и способам, которые можно применять для синтеза указанных соединений. Уровень техники Эндогенные стероидные андрогены оказывают глубокое влияние на множество физиологических функций. Эффекты стероидных андрогенов (например, тестостерона и 5-дигидротестостерона (DHT опосредует рецептор андрогенов (AR), и по природе эти эффекты можно охарактеризовать как анаболические или андрогенные. После связывания с андрогеном AR претерпевает изменение конформации, затем перемещается в ядро клетки, где связывается с определенными последовательностями ДНК, называющимися элементами ответа на андрогены (ARE), что приводит к запуску или подавлению транскрипции генов мишеней. Анаболические (то есть, связанные с ростом) виды действия андрогенов на ткани включают увеличение мышечной массы и силы и костной массы, а андрогенные (то есть маскулинизирующие) эффекты включают развитие вторичных мужских половых признаков, таких как внутренние репродуктивные ткани (то есть простата и семенные пузырьки), внешние половые органы (пенис и мошонка), либидо и характер оволосенения. Снижение уровня андрогенов, которое может произойти в связи с особенностями возрастной динамики, связано с серьезными последствиями как для самцов, так и для самок. Например, в процессе старения мужчин и падения уровня тестостерона, слабеют кости, повышается риск диабета и сердечнососудистых заболеваний, уменьшается соотношение мышечной и жировой массы. Для самок низкий уровень тестостерона в плазме циркулирующей крови связан с уменьшением либидо, необъяснимой усталостью, общим недомоганием и падением минеральной плотности костей у женщин в постменопаузе. Клинически основное применение терапии андрогенами состояло в лечении гипогонадизма у мужчин. Существенно, что было показано, что заместительная терапия у мужчин с гипогонадизмом также уменьшает резорбцию костей и увеличивает костную массу. Другие показания, для которых андрогены используют в клинике, включают лечение задержки полового созревания у мальчиков, анемии, первичного остеопороза и миодегенеративных заболеваний. Кроме того, заместительную терапию андрогенами недавно начали использоваться для стареющих мужчин и для регуляции мужской фертильности. Для самок терапию андрогенами в клинике используют для облегчения сексуальной дисфункции или снижения либидо. Тем не менее, терапия андрогенами имеет ограничения. Например, нежелательные побочные эффекты терапии андрогенными стероидами включают стимуляцию роста простаты и семенных пузырьков. Кроме того, с использованием андрогенов связаны стимуляция опухолей простаты и увеличение уровня специфического антигена простаты (PSA) (маркер повышенного риска рака простаты). Кроме того, оказалось, что препараты модифицированных и немодифицированных стероидных андрогенов подвержены быстрому разрушению в печени, что обуславливает плохую пероральную биодоступность и короткую продолжительность активности после парентерального введения, изменения уровней в плазме, гепатотоксичность или перекрестную реактивность с другими рецепторами стероидных гормонов (например, с рецептором глюкокортикоидов (GR), с рецептором минералкортикоидов (MR) и рецептором прогестерона (PR. Кроме того, у самок использование стероидных андрогенов может привести к оволосенению или вирилизации. Таким образом, в этой области медицины остается потребность в альтернативе терапии андрогенными стероидами, которая бы обладала полезными фармакологическими свойствами стероидных андрогенов, но с уменьшенной вероятностью или частотой типичных ограничений, связанных с терапией андрогенными стероидами. Недавние усилия по идентификации подходящей замены для стероидных андрогенов сфокусированы на идентификации тканеспецифических модуляторов рецептора андрогенов(ТМАР), которые демонстрируют различающийся профиль активности в андрогенных тканях. В частности, такие агенты предпочтительно показывают агонистичную андрогенам активность в анаболических тканях, таких как мышцы или кости, и являются только частичными агонистами или даже антагонистами в других андрогенных тканях, таких как простата или семенные пузырьки. Таким образом, задача настоящего изобретения - обеспечить нестероидные лиганды AR, которые обладают агонистической андрогенам активностью. Более конкретно, одна из задач обеспечить нестероидные агонисты андрогенов, которые связываются с AR с большей аффинностью, чем с другими рецепторами стероидных гормонов. Более конкретно, задачей является обеспечение тканеспецифических модуляторов рецептора андрогенов (ТМАР), которые демонстрируют агонистичную андрогенам активность в мышцах или костях, но лишь частично агонистичную, частично антагонистичную или антагонистичную активность в андрогенных тканях, таких как простата или семенные пузырьки. Следующие ссылки обеспечивают примеры текущего состояния области техники, имеющей отношение к настоящему изобретению:Brown, Endocrinology (2004); 145(12): 5417-5419 приводит обзор селективных нестероидных модуляторов рецептора андрогенов.Cadilla et al., Curr. Top. Med. Chem (2006); 6(3): 245-270 приводит обзор модуляторов рецептора андрогенов.Segal et al., Expert Opin. Investig. Drugs (2006); 15(4); 377-387 приводит обзор модуляторов рецептора андрогенов. В находящейся одновременно на рассмотрении международной патентной заявкеPCT/US2006/024122 раскрыты тетрагидрокарбазольные соединения в качестве модуляторов рецептора андрогенов. Краткое описание изобретения Настоящее изобретение обусловлено обнаружением того, что определенные соединения тетрагидроциклопента[b]индола, определенные формулой (I) ниже, имеют определенные профили активности,указывающие на то, что их можно применять в лечении заболеваний, чувствительных к терапии андрогенными стероидами. Соответственно, настоящее изобретение обеспечивает соединение формулы (I)Ra и Rb каждый независимо представляют собой в каждом случае Н или (C1-C4)алкил; иR3 представляет гетероарильную группу, выбранную из группы, состоящей из пиридинила, пиримидинила, пиразинила, пиридазинила, тиазолила, изотиазолила и тиадиазолила, каждый из которых необязательно замещен 1 или 2 заместителями, независимо выбранными из группы, состоящей из метила,этила, брома, хлора, фтора, -CHF2, -CF3, гидрокси, аминогруппы и -NHCH2CO2H; или его фармацевтически приемлемую соль. В другом варианте осуществления настоящее изобретение обеспечивает способ лечения гипогонадизма, снижения костной массы или плотности, остеопороза, остеопении, уменьшения мышечной массы или силы, саркопении, связанного с возрастом угасания функций, задержки половой зрелости у мальчиков, анемии, мужской или женской сексуальной дисфункции, эректильной дисфункции, уменьшении либидо, депрессии или летаргии, включающий введение пациенту при необходимости эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В более конкретном аспекте, настоящее изобретение обеспечивает способ лечения снижения костной массы или плотности,остеопороза, остеопении или снижения мышечной массы или силы. Далее настоящее изобретение обеспечивает использование соединения формулы (I) или его фармацевтически приемлемой соли в качестве терапевтического агента для лечения гипогонадизма, снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы, саркопении, возрастного угасания функций, задержки полового созревания у мальчиков, анемии, мужской или женской сексуальной дисфункции, эректильной дисфункции, снижения либидо, депрессии или летаргии. Более конкретно, настоящее изобретение обеспечивает применение соединения формулы (I) или его фармацевтически приемлемой соли в качестве терапевтического агента для лечения снижения костной массы или плотности, остеопороза, остеопении или снижения мышечной массы или силы. Кроме того,настоящее изобретение обеспечивает соединение формулы (I) или его фармацевтически приемлемую соль для применения в терапии. В другом варианте осуществления настоящее изобретение обеспечивает применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения гипогонадизма, снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы, саркопении, возрастного угасания функций, задержки полового созревания у мальчиков, анемии, мужской или женской сексуальной дисфункции, эректильной дисфункции, уменьшения либидо, депрессии или летаргии. Более конкретно, настоящее изобретение обеспечивает применение соединения формулы (I) для получения лекарственного средства для лечения снижения костной массы или плотности, остеопороза, остеопении или снижения мышечной массы или силы. Кроме того, настоящее изобретение обеспечивает фармацевтическую композицию, включающую соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с одним или более фармацевтически приемлемыми носителями, разбавителями или эксципиентами. В частности, настоящее изобретение обеспечивает фармацевтическую композицию для лечения снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы, включающую соединение-2 015627 формулы (I) или его фармацевтически приемлемую соль в комбинации с одним или более фармацевтически приемлемыми носителями, разбавителями или эксципиентами. Настоящее изобретение также включает новые промежуточные соединения и способы, которые можно применять для синтеза соединений формулы (I). Подробное описание изобретения Настоящее изобретение относится к новым соединениям тетрагидроциклопентаиндола, которые приведены выше в формуле (I). Тесты in vitro и in vivo подтверждают, что примеры соединений формулы(I) обладают профилями активности, которые позволяют предположить, что их можно применять в лечении заболеваний, чувствительных к терапии андрогенными стероидами. В частности, приведенные в качестве примеров соединения формулы (I) являются эффективными лигандами AR, действующими как агонисты рецептора андрогенов. Кроме того, приведенные в качестве примеров соединения формулы (I) связывают AR селективно по сравнению с каждым из рецепторов MR, GR и PR. Предполагается, что соединение формулы (I) или его фармацевтически приемлемую соль можно будет применять в лечении заболеваний, которые обычно лечат путем терапии андрогенами. Таким образом, способы лечения заболеваний, чувствительных к терапии андрогенами, составляют важный вариант осуществления настоящего изобретения. Такие заболевания включают гипогонадизм, снижение костной массы или плотности, остеопороз, остеопению, снижение мышечной массы или силы, саркопению, возрастное угасание функций, задержку полового созревания у мальчиков, анемию, мужскую или женскую сексуальную дисфункцию, эректильную дисфункцию, снижение либидо, депрессию и летаргию. Более конкретные заболевания, для лечения которых, как полагают, можно применять соединения формулы (I) включают снижение костной массы или плотности, остеопороз, остеопению, снижение мышечной массы или силы. Настоящее изобретение также относится к сольватам соединений формулы (I) или фармацевтически приемлемым солям соединений формулы (I). Соответственно, в настоящей заявке значение термина"Формула (I)" или любое конкретное соединение формулы (I) включает любые фармацевтически приемлемые соли и любые сольваты такого соединения или его фармацевтически приемлемую соль. Примеры фармацевтически приемлемых солей и способов их получения хорошо известны специалисту в данной области. См., например, Stahl et al., "Handbook of Pharmaceutical Salts: Properties, Selection and Use,"Entities," Organic Process Research and Development, 4: 427-435 (2000). Соединения согласно настоящему изобретению имеют один или более хиральных центров и, следовательно, могут существовать в ряде стереоизомерических конфигураций. Как следствие существования хиральных центров, соединения согласно настоящему изобретению могут представлять собой рацематы,смеси энантиомеров и индивидуальные энантиомеры, а также диастереомеры и смеси диастереомеров. Кроме изложенных далее все такие рацематы, энантиомеры и диастереомеры входят в объем настоящего изобретения. Энантиомеры соединений согласно настоящему изобретению могут быть получены, например, специалистом в данной области путем использования стандартных методов, например, описанных в J. Jacques, et al., "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, Inc., 1981, а также методов, приведенных на схемах и в примерах заявки. Термины "R" и "S" в настоящей заявке используются так, как это обычно принято в органической химии для обозначения специфических конфигураций хирального центра. Термины " " или "RS" относятся к конфигурации хирального центра, включающей рацемат. Частичный список предпочтительных вариантов и обсуждение стереохимии содержится в "Nomenclature of Organic Compounds: Principles andPractice", (J.H. Fletcher, et al., eds., 1974). Специфические стереоизомеры и энантиомеры соединений формулы I могут быть получены специалистом в данной области при использовании хорошо известных методов и процессов, таких как раскрытые в Eliel and Wilen, "Stereochemistry of Organic Compounds", John WileySons, Inc., 1994, Chapter 7; Separation of Stereoisomers, Resolution, Racemization;и Collet and Wilen, "Enantiomers, Racemates, andResolutions", John WileySons, Inc., 1981. Например, конкретные стереоизомеры и энантиомеры можно получить путем стереоспецифического синтеза с использованием энантиомерно и геометрически чистых или энантиомерно и геометрически обогащенных исходных материалов. Кроме того, специфические стереоизомеры и энантиомеры можно получить и выделить такими методами, как хроматография на хиральных неподвижных фазах, ферментативное разделение или фракционная перекристаллизация солей присоединения, образованных используемыми для этих целей реагентами. В настоящей заявке термин " (C1-C4) алкил" относится к линейной или разветвленной, одновалентной, насыщенной алифатической цепи из от 1 до 4 атомов углерода. В настоящей заявке термин " (C1-C4)алкокси" относится к атому кислорода, несущему линейную или разветвленную, одновалентную, насыщенную алифатическую цепь от 1 до 4 атомов углерода. В настоящей заявке термины "галоген", "галогенид", или "Hal" относятся к атому хлора, брома, иода или фтора, если в заявке не указано иное.-3 015627 Специалисту в данной области понятно, что некоторые гетероариальные компоненты соединений формулы (I) могут существовать в виде позиционных изомеров и таутомерных форм. Настоящее изобретение охватывает все позиционные изомеры, индивидуальные таутомерные формы, а также любые их комбинации в терминах гетероариальных компонентов соединений формулы I. относится к связи, которая выдается вперед относительно плоскости страОбозначение ницы. относится к связи, которая выдается назад относительно плоскости страОбозначение ницы назад. Обозначение относится к связи, которая существует как смесь связей, которые выдаются как вперед, так и назад относительно плоскости страницы. Для специалиста в данной области очевидно, что физиологические заболевания могут существовать в форме "хронического" состояния или "острого" эпизода. Термин "хронический" в настоящей заявке означает состояние с медленным прогрессированием и большой продолжительностью. Соответственно,хроническое состояние лечат, когда оно диагностировано и лечение продолжают на протяжении всего хода болезни. И наоборот, термин "острый" означает внезапный случай или приступ, короткое течение,за которым следует период ремиссии. Таким образом, лечение заболеваний подразумевает как острые события, так и хронические состояния. В случае острого события, соединение вводят при начале симптомов и прекращают, когда симптомы исчезают. Как описано выше, хроническое состояние лечат на протяжении всего течения заболевания. В настоящей заявке термин "пациент" относится к человеку или млекопитающему, отличному от человека, такому как собака, кошка, корова, обезьяна, лошадь, свинья или овца. Тем не менее, подразумевается, что предпочтительным пациентом, которому можно вводить соединение формулы (I) или его фармацевтически приемлемую соль, является человек. Термин "лечение" (или "лечить") в настоящей заявке включает прекращение, предотвращение, сокращение, снижение, остановку или обращение прогрессирования или тяжести симптома или заболевания. Соответственно, способы согласно настоящему изобретению охватывают как терапевтическое, так и профилактическое использование. Соединения согласно настоящему изобретению могут быть изготовлены в виде части фармацевтической композиции. Соответственно, фармацевтическая композиция, включающая соединение формулы(I) или его фармацевтически приемлемую соль в комбинации с фармацевтически приемлемым носителем, разбавителем или эксципиентом является важным вариантом осуществления настоящего изобретения. Примеры фармацевтических композиций и способов их получения хорошо известны в данной области техники. См., например, REMINGTON: THE SCIENCE AND PRACTICE OF PHARMACY (A. Gennaro, et al., eds., 19th ed., Mack Publishing (1995. Примеры композиций, включающих соединения формулы (I) включают, например: соединение формулы (I) в суспензии с 1% натриевой карбоксиметилцеллюлозой, 0,25% полисорбатом 80, и 0,05% противовспенивающего агента Antifoam 1510 (Dow Corning); и соединение формулы (I) в суспензии с 0,5% метилцеллюлозой, 0,5% лаурилсульфатом натрия, и 0,1%Antifoam 1510 в 0,01N HCl (конечный рН около 2,5-3). Предпочтительная композиция согласно настоящему изобретению включает соединение формулы (I) или его фармацевтически приемлемую соль в форме капсулы или таблетки. Соединение формулы (I) или композицию, включающую соединение формулы (I), можно вводить любым путем, который обеспечивает биодоступность соединения, включая пероральный и парентеральный пути. Специалисту в данной области понятно, что размер частицы может повлиять на растворение фармацевтического агента in vivo, которое, в свою очередь, может повлиять на всасывание агента. "Размер частицы" в настоящей заявке относится к диаметру частицы фармацевтического агента, который определяют стандартными методами, такими как рассеяние лазерного света, лазерная дифракция, рассеяниеMie, осаждения области разделения потока, спектроскопия корреляции фотонов, и тому подобное. В случае, если фармацевтические агенты имеют плохую растворимость, небольшие или уменьшенные размеры частиц могут способствовать растворению и, таким образом, увеличить всасывание агента. Amidon etal., Pharm.Research, 12; 413-420 (1995). Способы уменьшения или контроля размера частицы являются стандартными и включают перемалывание, влажное растирание, микронизацию и тому подобное. Другой способ контроля размера частицы включает получение фармацевтического агента в форме наносуспензии. Предпочтительный вариант осуществления настоящего изобретения включает соединение формулы (I) или фармацевтическую композицию, включающую соединение формулы (I), где указанное соединение имеет средний размер частицы менее чем примерно 20 мкм или размер частицы d90 (то есть,максимальный размер 90% частиц) менее чем примерно 50 мкм. Более предпочтительный вариант осуществления включает соединение формулы I, которое имеет средний размер частицы менее чем примерно 10 мкм или d90 размера частицы менее чем примерно 30 мкм. В настоящей заявке термин "эффективное количество" относится к количеству или дозе соединения формулы (I), которое при однократном или многократном введении дозы пациенту обеспечивает желаемый для пациента эффект при диагностировании или лечении. Врач-диагност, как и опытный специалист-4 015627 в данной области, легко сможет определить эффективное количество, приняв во внимание ряд факторов,таких как вид млекопитающего; его размер, возраст и общее состояние; конкретное заболевание, которое лечат; степень или остроту заболевания; ответ индивидуального пациента; конкретное вводимое соединение; способ введения; характеристики биодоступности вводимого препарата; выбранный режим дозировки; и использование любых сопутствующих лекарственных средств. При применении в комбинации со способами и применениями согласно настоящему изобретению соединения и композиции согласно настоящему изобретению можно вводить сами по себе или в комбинации со стандартными терапевтическими агентами, используемыми для лечения конкретного заболевания или состояния. Если соединения или композиции согласно настоящему изобретению используют как часть комбинации, соединения или композиции, включающей формулу (I), введение можно осуществлять раздельно или в виде части лекарственной формы, включающей терапевтический агент, с которым его предстоит комбинировать. Комбинированная терапия костной недостаточности, остеопороза или остеопении Стандартные терапевтические агенты для лечения остеопороза можно выгодно комбинировать с соединениями формулы (I) или композициями, включающими соединение формулы (I) с обеспечением дополнительных преимуществ. Стандартные агенты для лечения остеопороза включают средства заместительной гормональной терапии, такие как конъюгированный эстроген лошади (Premarin), синтетический конъюгированный эстроген (Cenestin), этерифицированный эстроген (Estratab или Menest),эстропиат (Ogen или Ortho-est); а также препараты эстрадиола для трансдермального введения, такие как Alora, Climara, Estraderm и Vivelle. Комбинированные препараты эстроген-прогестин также подходят для лечения остеопороза, включая Prempro (конъюгированный эстроген лошади и ацетат медроксипрогестерона), Premphase (конъюгированный эстрогенанд норгестимат лошади), OrthoPrefest (эстрадиол и норгестимат), Femhrt (этинил эстрадиол и норетиндрон ацетат) и Combipatch(трансдермальные эстрадиол и норетиндрон ацетат). Другие стандартные препараты против остеопороза,которые можно комбинировать с соединениями или композициями согласно настоящему изобретению,включают бифосфонаты, такие как алендронат (Fosamax), ризедронат (Actonel) и памидронат (Aredia); селективные модуляторы эстрогенового рецептора (SERM), такие как ралоксифен (Evista); кальцитонин (Calcimar или Miacalcin); паратиреоидный гормон (Forteo); кальций; витамин D; диуретики (для уменьшения выделения Са 2+); фториды; и андрогены (например, тестостерон или 5 дигидротестостерон). Таким образом, лекарственная форма для комбинированной терапии при лечении остеопороза включает: Ингредиент (А 1): соединение формулы (I); Ингредиент (А 2): один или более стандартных совместно действующих терапевтических агентов для лечения остеопороза, выбранный из группы, включающей Premarin, Cenestin, Estratab, Menest, Ogen, Ortho-est, Alora, Climara, Estraderm, Vivelle, Prempro, Premphase, Ortho-Prefest, Femhrt, Combipatch, Fosamax, Actonel, Aredia; Evista; Calcimar, Miacalcin, Forteo,кальций, витамин D, диуретики, фториды, тестостерон и 5-дигидротестостерон; и возможно Ингредиент (A3): фармацевтически приемлемый носитель, разбавитель или эксципиент. Частные аспекты изобретения Следующий список определяет несколько групп конкретных заместителей и конкретных переменных для соединений формулы (I). Следует понимать, что соединения формулы (I), имеющие такие конкретные заместители или переменные, а также способы и применение таких соединений, представляют собой частные аспекты настоящего изобретения. Таким образом, частным аспектом настоящего изобретения является реализация соединения формулы (I), где R2 и R3 имеют приведенное выше значение, и:(b) R1 представляет циано или -CH=NOCH3; или(d) R1 представляет -CH=NOCH3. Дополнительным частным аспектом настоящего изобретения является соединение формулы (I), гдеR1 и R3 имеют приведенные выше значения, и:(g) R2 представляет -SO2R2b, где R2b представляет циклопропил или -N(СН 3)2; или(h) R2 представляет -SO2R2b, где R2b представляет -N(CH3)2. Дополнительным частным аспектом настоящего изобретения является соединение формулы (I), гдеR1 и R3 имеют приведенное выше значение, и:(b) R2 представляет -SO2R2b и углеродный центр "с" находится в R-конфигурации. Дополнительным частным аспектом настоящего изобретения является соединение формулы (I), гдеR1 и R2 имеют приведенные выше значения, и:(a) R3 представляет гетероарильную группу, выбранную из группы, состоящей из пиридинила, пиримидинила, пиразинила, пиридазинила, тиазолила, изотиазолила и тиадиазолила, каждый из которых необязательно замещен 1 или 2 заместителями, независимо выбранными из группы, состоящей из метила, брома, хлора, фтора, -CHF2, гидрокси, аминогруппы и -NHCH2CO2H; или(e) R3 представляет пиридин-2-ил, 2-аминопиридин-3-ил, тиазол-5-ил или 4-аминотиазол-5-ил. Еще одним частным аспектом настоящего изобретения является соединение формулы (I), где углеродный центр "с" находится в S-конфигурации, когда R2 представляет -COR2a и в R-конфигурации, когда R2 представляет -SO2R2b;R1 представляет циано или -CH=NOCH3;R3 представляет гетероарильную группу, выбранную из группы, состоящей из пиридинила, пиримидинила, пиразинила, пиридазинила, тиазолила, изотиазолила и тиадиазолила, каждый из которых необязательно замещен 1 или 2 заместителями, независимо выбранными из группы, состоящей из метила,брома, хлора, фтора, -CHF2, гидрокси, аминогруппы и -NHCH2CO2H. В еще более частном аспекте настоящее изобретение представляет собой соединение формулы (I),где углеродный центр "с" находится в S-конфигурации, когда R2 представляет -COR2a и в Rконфигурации, когда R2 представляет -SO2R2b;R1 представляет циано или -CH=NOCH3;R3 представляет 6-фторпиридин-2-ил,пиридин-2-ил,3-гидроксипиридин-2-ил,6 дифторметилпиридин-2-ил, 2-аминопиридин-3-ил, 2-карбоксиметиламинопиридин-3-ил, пиримидин-4 ил, пиримидин-2-ил, 2-хлорпиримидин-4-ил, тиазол-4-ил, 2-метилтиазол-4-ил, 2-хлортиазол-4-ил, тиазол-2-ил, тиазол-5-ил, 4-аминотиазол-5-ил, пиразин-2-ил, 5-метилпиразин-2-ил, 3-хлорпиразин-2-ил, 6 метилпиразин-2-ил, 3-аминопиразин-2-ил, 3-метилпиразин-2-ил, пиридазин-3-ил, 5-бромизотиазол-3-ил,изотиазол-3-ил, 4,5-дихлоризотиазол-3-ил или [1,2,5]тиадиазол-3-ил. Еще более частным аспектом настоящего изобретения является соединение формулы (I), где углеродный центр "с" находится в S-конфигурации, когда R2 представляет -COR2a и в Rконфигурации, когда R2 представляет -SO2R2b;R2 представляет -COR2a или -SO2R2b, где R2a представляет изопропил, этокси, изопропокси или циклопропил; и R2b представляет циклопропил или -N(CH3)2; иR3 представляет 6-фторпиридин-2-ил, пиридин-2-ил, 2-аминопиридин-3-ил, тиазол-5-ил или 4 аминотиазол-5-ил. В еще более частном аспекте настоящее изобретение представляет собой соединение формулы (I),гдеR3 представляет пиридин-2-ил, 2-аминопиридин-3-ил, тиазол-5-ил или 4-аминотиазол-5-ил. Дополнительные частные аспекты настоящего изобретения представлены приведенными ниже соединениями формулы I(а) и формулы I(b). Подразумевается, что соединения формулы I(а) и формулыI(b), а также способы и применение таких соединений представляют собой другие аспекты настоящего изобретения. Таким образом, частный аспект настоящего изобретения предоставлен формулой I(а)R3 представляет гетероарильную группу, выбранную из группы, состоящей из пиридинила, пиримидинила, пиразинила, пиридазинила, тиазолила, изотиазолила и тиадиазолила, каждый из которых необязательно замещен 1 или 2 заместителями, независимо выбранными из группы, состоящей из метила,брома, хлора, фтора, -CHF2, гидрокси, аминогруппы и -NHCH2CO2H. Еще более частный вариант осуществления представляет собой соединение формулы I (а), гдеR1 представляет циано или -CH=NOCH3;R3 представляет 6-фторпиридин-2-ил,пиридин-2-ил,3-гидроксипиридин-2-ил,6 дифторметилпиридин-2-ил, 2-аминопиридин-3-ил, 2-карбоксиметиламинопиридин-3-ил, пиримидин-4 ил, пиримидин-2-ил, 2-хлорпиримидин-4-ил, тиазол-4-ил, 2-метилтиазол-4-ил, 2-хлортиазол-4-ил, тиазол-2-ил, тиазол-5-ил, 4-аминотиазол-5-ил, пиразин-2-ил, 5-метилпиразин-2-ил, 3-хлорпиразин-2-ил, 6 метилпиразин-2-ил, 3-аминопиразин-2-ил, 3-метилпиразин-2-ил, пиридазин-3-ил, 5-бромизотиазол-3-ил,изотиазол-3-ил, 4,5-дихлоризотиазол-3-ил или [1,2,5]тиадиазол-3-ил. Другой частный аспект осуществления представлен формулой I(а), гдеR3 представляет 6-фторпиридин-2-ил, пиридин-2-ил, 2-аминопиридин-3-ил, тиазол-5-ил или 4 аминотиазол-5-ил. Как показано, другой частный аспект осуществления настоящего изобретения предоставляет формула I(b)R1 представляет циано или -CH=NOCH3;R3 представляет гетероарильную группу, выбранную из группы, состоящей из пиридинила, пиримидинила, пиразинила, пиридазинила, тиазолила, изотиазолила и тиадиазолила, каждый из которых необязательно замещен 1 или 2 заместителями, независимо выбранными из группы, состоящей из метила,брома, хлора, фтора, -CHF2, гидрокси, аминогруппы и -NHCH2CO2H. Еще более частным осуществлением является соединение формулы I (b), гдеR1 представляет циано или -CH=NOCH3;-7 015627 Более частный аспект представлен формулой I(b), гдеR2b представляет циклопропил или -N(СН 3)2; иR3 представляет тиазол-4-ил, тиазол-2-ил, тиазол-5-ил или 4-аминотиазол-5-ил. Кроме того, следует понимать, что наиболее конкретный аспект настоящего изобретения представлен соединениями формулы (I), описанной выше, и, наиболее конкретно, соединениями: изопропиловый эфир (S)-(7-циано-4-пиридин-2-илметил-1,2,3,4-тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты; изопропиловый эфир (S)-(7-циано-4-тиазол-5-илметил-1,2,3,4-тетрагидроциклопента[b]индол-2 ил)карбаминовой кислоты; изопропиловый эфир (S)-[4-(2-аминопиридин-3-илметил)-7-циано-1,2,3,4 тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты; (R)-N'-[4-(4-аминотиазол-5-илметил)-7 циано-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]-N,N-диметилсульфамид или изопропиловый эфир(S)-[4-(4-аминотиазол-5-илметил)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты. Соединения согласно настоящему изобретению можно получить химическим путем, например при помощи путей синтеза, приведенных на схемах, а также промежуточных соединений и примеров, приведенных ниже. Тем не менее, нижеследующее обсуждение никоим образом не предполагает ограничения объема настоящего изобретения. Например, специфические стадии синтеза для каждого из описанных путей можно сочетать в различных путях или применять совместно стадии из различных схем для получения дополнительных соединений формулы (I). Заместители, если не указано иное, имеют значения, определенные ранее. Реагенты и исходные материалы легко доступны для специалиста в данной области. Другие необходимые реагенты и исходные материалы могут быть получены при помощи процедур, которые выбирали из стандартных методов органической химии и химии гетероциклов, методов, которые аналогичны синтезу известных структурно сходных соединений и процедур, описанных в примерах ниже, включая любые новые процедуры. Схема I Схема I описывает способы получения R3-CH2-X или R3-CH2-OMs, используя последовательное алкилирование соединений тетрагидроциклопента[b]индола. На схеме I, стадия А спирт формулы (2) получали путем восстановления эфира формулы (1). Эфир получали, если это необходимо, из карбоновой кислоты и хлорангидрида, используя хорошо известные в этой области способы, такие как реакция с оксалилхлоридом. Многочисленные способы восстановления карбоновых эфиров в спирты хорошо известны специалистам в данной области и могут быть найдены в тексте R.C. Larock в "Comprehensive Organic Transformations", VCH Publishers, 1989, p. 549-551. Предпочтительным способом является восстановление борогидридом лития в апротонном растворителе, таком как тетрагидрофуран, при температуре от комнатной до температуры кипячения с обратным холодильником в течение примерно от 1 до 48 ч. На схеме I, стадия В спирт формулы (2) переводили в эфир метансульфоновой кислоты формулы(3). Спирт объединяли с органическим основанием, таким как триэтиламин или диизопропилэтиламин и обрабатывали метансульфонилхлоридом в инертном растворителе, таком как дихлорметан. Реакцию проводили при температуре от 0 С до комнатной температуры в течение от 15 мин до 4 ч. Продукт выделяли с помощью технологии экстрагирования, хорошо известной специалистам в этой области. На схеме I, стадия С соединение формулы (4), где R3 является гетероарилом, галогенировали, в результате получали алкилгалогенид формулы (5). Соединение формулы (4) обрабатывали источником свободных радикалов, таким как бензоилпероксид или 1,1'-азобисизобутиронитрил или 1,1'азобис(циклогексанкарбонитрил) в четыреххлористом углероде или этилацетате с N-хлорсукцинимидом или N-бромсукцинимидом с облучением УФ-светом или без него. Предпочтительным способом является обработка 1,1'-азобис(циклогексанкарбонитрилом) или 1,1'-азобисизобутиронитрилом и Nбромсукцинимидом при температуре от комнатной до температуры кипячения с обратным холодильником в четыреххлористом углероде, примерно от 4 до 48 ч. Продукт затем можно очистить, используя-8 015627 стандартные методы, такие как фильтрация нерастворимых компонентов, с последующей хроматографией на силикагеле. Кроме того, на схеме I, стадия С, гетероарилметил формулы (4) можно хлорировать и получить алкилхлорид, в котором X представляет Cl, используя трихлоризоцианмочевую кислоту. Реакцию проводили в инертном растворителе, таком как хлороформ, и кипятили с обратным холодильником от 4 до 72 ч. Продукт выделяли путем фильтрации через слой двуокиси кремния, затем путем хроматографии. На схеме I, стадия D формилпиридин формулы (6) переводили в дифторметилпиридин формулы (7),используя трифторид бис-(2-метоксиэтил)аминосеры. Реакцию проводили в инертном растворителе, таком как дихлорметан от 4 до 24 ч и затем гасили насыщенным раствором NaHCO3. Продукт затем выделяли путем общих методов экстрагирования. На схеме I, стадия Е дифторметилпиридин формулы (7) переводили в алкилбромид формулы (5) (X представляет Br) , как ранее было описано на схеме I, стадия С. Схема II На схеме II, стадия А проводят реакцию Михаэля циклопентенона (8) с фталимидом с получением-2-(3-оксоциклопентил)изоиндол-1,3-диона (9). Реакцию проводили в метаноле/2N Na2CO3 в отношении объемов 10/1 предпочтительно при температуре окружающей среды, используя условия, подобные тем, что описаны О. Nowitzki, et. al. в Tetrahedron 1996, 52, 11799-11810. Продукт выделяли путем добавления воды и получали (9) в виде твердого вещества белого цвета. На схеме II, стадия В, -2-(3-оксоциклопентил)изоиндол-1,3-дион (9) реагировал с фенилгидразином формулы (10) в типичном синтезе индолов Фишера с получением тетрагидроциклопента[b]индола формулы (11). Специалисту понятно, что существует ряд кислотных сред для проведения синтеза индолов Фишера, включая как протонные вещества, так и кислоты Льюиса. Предпочтительные условия включают смесь кристаллической уксусной кислоты с 4N HCl в диоксане при температуре от 50 С до температуры кипячения растворителя с обратным холодильником, примерно от 4 до 24 ч. Продукт выделяли путем добавления воды, затем отфильтровывали полученное твердое вещество. Твердое вещество обрабатывали ультразвуком в метаноле, в результате получали материал достаточной чистоты. Альтернативно, реакцию проводили, используя кислоту Льюиса, такую как хлорид цинка, в количестве примерно от 2 до 4 эквивалентов. Другие предпочтительные условия для стадии В включают этанол при температуре кипячения с обратным холодильником примерно от 4 до 24 ч. Продукт выделяли и очищали фильтрацией реакционной смеси, затем хроматографией фильтрата на силикагеле. На схеме II, стадия С фталимидную группу формулы (11) отщепляли гидразином или гидратом гидразина, в результате получали аминотетрагидроциклопента[b]индол формулы (12), используя условия,которые описаны М. Alajarin, et al (Eur. J. Org. Chem. 2002, 4222-4227). Предпочтительные условия включают смесь тетрагидрофуран/этанол в отношении по объему примерно 5,5/1 при температуре от 0 до 50 С, предпочтительно при примерно комнатной температуре от 4 до 72 ч. Полученный фтальгидразид удаляли путем фильтрации и продукт выделяли путем концентрации фильтрата, и могли впоследствии очищать его путем хроматографии, используя известные в данной области методы. На схеме II, стадия D амин формулы (12) ацилировали подходящим хлорангидридом, хлороформатом, диалкилдикарбонатом или карбамоилхлоридом, в результате получали амид, карбамат или мочевину формулы (13), где R2 представляет C(O)R2a, при этом использовали хорошо известные специалисту в данной области условия. Амин объединяли с избытком органического амина основания, такого как триэтиламин или диизопропилэтиламин в инертном растворителе, таком как тетрагидрофуран, дихлорэтан или дихлорметан, N-метилпирролидинон, или N,N-диметилформамид или их смеси. Предпочтительные условия включают диизопропилэтиламин в дихлорметане в присутствии, например, изопропилхлороформата при температуре от 0 до 40 С, от 1 до 72 ч. Продукт выделяли путем добавления воды и диэтилового эфира, затем смесь перемешивали и собирали полученное твердое вещество. Если продукт обладает достаточной растворимостью в соответствующем органическом растворителе, его можно выделить методами экстракции и затем растворить в пригодном органическом растворителе, таком как гептан, и выделить путем фильтрации.-9 015627 Для специалиста в данной области понятно, что такие амины, как амины формулы (12), часто более пригодны для хранения и обращения в качестве промежуточного соединения формулы (13), если они должным образом защищены, например, группой трет-бутоксикарбонил (ВОС), где R2a представляет Отрет-бутил, или путем образования соли присоединения кислоты. Группу ВОС затем удаляют и амин ацилируют, в результате чего получают желаемый амид или карбамат соединений согласно настоящему изобретению. На схеме II, стадия Е тетрагидроциклопента[b]индол формулы (13) алкилировали R3CH2-X, где X представляет Cl или Br, или R3CH2OSO2Me (см. Схему I), в результате получали тетрагидроциклопента[b]индол формулы (14). Предпочтительные условия включают Cs2CO3 в инертном растворителе, таком как ДМФ, ДМСО или N-метилпирролидинон, при температуре от 20 до 100 С, но предпочтительно, от 45 до 60 С, от 2 до 24 ч. Продукт выделяли при помощи методов экстракции, известных специалисту в данной области, и очищали путем хроматографии на силикагеле. Альтернативно, алкилирование можно проводить, используя сильное основание, такое как гидрид натрия, гидрид калия,бис(триметилсилил)амид калия или бис(триметилсилил)амид натрия, в инертном растворителе, таком как диметилформамид, N-метилпирролидинон или тетрагидрофуран. Предпочтительные условия включают гидрид натрия в диметилформамиде при температуре от 0 до 80 С от 4 до 48 ч. Алкилированный продукт формулы (14) выделяли при помощи методов экстракции и хроматографии, известных специалисту в данной области. Схема III На схеме III, стадия А тетрагидроциклопента[b]индол формулы (15) или формулы (17) алкилировали R3CH2X, где X представляет Cl, Br или R3 CH2OSO2Me, как показано на схеме II, стадия Е, в результате получали тетрагидроциклопентаниндол формулы (16) или (18). На схеме III, стадия В фталимидную группу формулы (16) отщепляли гидратом гидразина или гидразином, в результате получали аминотетрагидроциклопента[b]индол формулы (19), как описано на схеме II, стадия С. Альтернативно, на схеме III, стадия С аминотетрагидроциклопента[b]индол формулы (19) можно получить удалением защитной группы трет-бутоксикарбонила (ВОС), защищенного амином формулы(18) . Общие условия для удаления группы ВОС хорошо известны специалистам в данной области и могут быть найдены в тексте Т. W. Green and P. G. M. Wuts в "Protective Groups in Organic Synthesis", JohnWileySons, Inc., 1991, 328-330. Предпочтительные условия включают 4 М хлороводород в диоксане при температуре примерно от 0 до 50 С в течение примерно от 10 мин до 24 ч. На схеме III, стадия D, амин формулы (19) переводили в сульфамид или сульфонамид формулы (20) в реакции с соответствующим сульфамоилом или сульфонилхлоридом соответственно, в подходящем растворителе, таком как хлороформ или дихлорметан при 50-60 С, с использованием оснований, таких как триэтиламин, основание Хенига или 1,4-диазабицикло[2,2,2]октана (DABCO). Предпочтительные условия включают хлороформ с DABCO в качестве основания. Альтернативно, (14) можно получить, как показано на схеме III, стадия Е, при реакции соответствующего хлорангидрида, хлороформата или карбамоилхлорида с амином формулы (19), как описано на схеме II, стадия D. На схеме IV, стадия А, тетрагидроциклопента[b]индол формулы (21) алкилировали, как описано на схеме II, стадия Е, в результате получали имеющий заместители тетрагидроциклопента[b]индол формулы (22). На схеме IV, стадия В, нитрил формулы (21) или (22) восстанавливали в альдегид формулы (23) или(24). Нитрил обрабатывали никель-алюминиевым катализатором в смеси растворителей вода/муравьиная кислота, в отношении от 1/10 до 1/2. Используемая муравьиная кислота может быть 98, 96 или 88%. Реакцию проводили при температуре от комнатной до температуры кипячения растворителя с обратным холодильником, примерно от 2 до 48 ч. Продукт выделяли путем добавления протонного растворителя,такого как метанол, последующей фильтрации и концентрирования фильтрата. Остаток далее очищали общими методами экстракции, например, с использованием раствора бикарбоната натрия и этилацетата,для получения альдегида, или путем ультразвуковой обработки с этанолом, и использовали без дальнейшей очистки. Кроме того, на схеме IV, стадия В, если R2 представляет трет-бутоксикарбонил (ВОС), нитрил формулы (21) или (22) затем восстанавливали, используя не кислотные условия, такие как гидрид металла в качестве восстанавливающего агента, например, гидрид диизобутилалюминия. Реакцию проводили в инертном растворителе, таком как дихлорметан, с добавлением гидрида диизобутилалюминия, а затем этилацетата, и с перемешиванием от 30 мин до 2 ч при комнатной температуре. Реакционную смесь перемешивали с 20% водным тартратом натрия в течение 1 ч и затем выделяли, используя методы экстракции. На схеме IV, стадия D альдегид формулы (23) или (25) переводили в метоксим формулы (25) или(26), соответственно. Альдегид обрабатывали солью гидрохлорида метоксиамина в этаноле или метаноле при температуре от 0 до 100 С, примерно от 2 до 24 ч, предпочтительно при комнатной температуре в течение одного часа в присутствии основания, такого как карбонат калия, карбонат натрия или гидроксид натрия. Продукт выделяли путем концентрирования, и продукт растирали в воде. Альтернативно,продукт реакции разбавляли этилацетатом, выделяли, используя методы экстракции, и впоследствии возможно (не обязательно) очищали с использованием стандартных методов, таких как хроматография. На схеме IV, стадия Е, метоксим тетрагидроциклопентаниндола формулы (25) алкилировали, в результате получали N-замещенный метоксим тетрагидроциклопентаниндола формулы (26), как описано на схеме II, стадия Е. Определение биологической активности В настоящей заявке, "Kd" относится к равновесной константе диссоциации комплекса, лигандрецептор; "Ki" относится к равновесной константе диссоциации комплекса лекарство-рецептор и является показателем концентрации лекарства, которое в равновесии связывается с половиной сайтов связывания; "IC50" относится к концентрации вещества, которая обеспечивает 50% максимального ингибирующего ответа, возможного для этого вещества, или, альтернативно, к концентрации вещества, которая обеспечивает 50% замещение связанного с рецептором лиганда; "ЕС 50" относится к концентрации вещества, которая обеспечивает 50% от максимального ответа, возможного для этого вещества; и "ED50" относится к дозе введенного терапевтического агента, которая обеспечивает 50% максимального ответа для этого агента. Тест на связывание рецептора стероидных гормонов в ядре В тесте на конкурентное связывание рецептора и лиганда для определения значений Ki использовали лизаты клеток из клеток почки зародыша человека НЕК 293 с повышенной экспрессией GR (рецептор глюкокортикоидов) человека, AR (андрогеновый рецептор) человека, MR (рецептор минералкортикоидов) человека или PR (рецептор прогестерона) человека. Вкратце, тест на конкурентное связывание стероидного рецептора проводили в буфере, содержащем 20 мМ буфер Hepes (рН=7,6), 0,2 мМ ЭДТА, 75 мМ NaCl, 1,5 мМ MgCl2, 20% глицерин, 20 мМ молибдат натрия, 0,2 мМ DTT (дитиотреитол) , 20 мкг/мл апротинина и 20 мкг/мл лейпептина. Обычно тест- 11015627 на связывание стероидного рецептора включает радиоактивно меченые лиганды, такие как 0,3 нМ [3 Н]дексаметазон для связывания с GR, 0,36 нМ [3 Н]-метилтриенолон для связывания с AR, 0,25 нМ [3 Н]альдостерон для связывания с MR и 0,29 нМ [3 Н]-метилтриенолон для связывания с PR, а также 20 мкг лизата 293-GR, 22 мкг лизата 293-AR, 20 мкг лизата 293-MR или 40 мкг лизата 293-PR на лунку. Тесты обычно проводят в 96-луночном формате. Конкурирующие тестируемые соединения добавляли в различной концентрации, колеблющейся примерно от 0,01 нМ до 10 мкМ. Неспецифическое связывание определяли в присутствии 500 нМ дексаметазона для связывания GR, 500 нМ альдостерона для связывания MR, или 500 нМ метилтриенолона для связывания AR и PR. Реакционные смеси для определения связывания (140 мкл) инкубировали в течение ночи при 4 С, затем в каждую реакционную смесь добавляли 70 мкл холодного буфера уголь-декстран (содержание на 50 мл буфера для анализа, 0,75 г активированного угля и 0,25 г декстрана). Планшеты встряхивали в течение 8 мин в орбитальном шейкере при 4 С. Планшеты затем центрифугировали при 3000 об/мин при 4 С в течение 10 мин. Аликвоту 120 мкл связавшейся реакционной смеси затем переносили на другой 96-луночный планшет и к каждой лунке добавляли 175 мкл сцинцилляционной жидкости Wallac Optiphase Hisafe 3. Планшеты закрывали и энергично встряхивали в орбитальном шейкере. После 2 ч инкубации планшеты считывали в счетчикеWallac Microbeta. Данные использовали для расчета оценочного IC50 и процента ингибирования при 10 мкМ. Kd для связывания [3 Н]-дексаметазона с GR, [3 Н]-метилтриенолона с AR, [3 Н]-альдостерона с MR или [3 Н]метилтриенолона с PR определяли путем насыщающего связывания. Значение IC50 для соединений преобразовали в Ki, используя уравнение Cheng-Prusoff. В тесте на связывание с AR, проведенному по существу в соответствии с описанным выше протоколом, примеры соединений согласно настоящему изобретению демонстрировали Ki500 нМ. Предпочтительно, соединения согласно настоящему изобретению демонстрировали Ki в тесте на связывание сAR100 нМ, и более предпочтительно 50 нМ. Кроме того, примеры соединений согласно настоящему изобретению селективно связываются с AR (более низкая Ki) по сравнению с каждым из рецепторов MR человека, GR человека и PR человека. Для демонстрации способности соединений согласно настоящему изобретению модулировать активность рецепторов стероидных гормонов (то есть агонизм, частичный агонизм, частичный антагонизм или антагонизм) проводили биологические тесты, которые позволили выявить функциональную модуляцию экспрессии целевого гена в клетках, временно трансфицированных белком внутриядерного рецептора и конструктом с геном-репортером под контролем гормон-чувствительного элемента. Растворители,реагенты и лиганды, используемые в функциональном тесте, легко доступны в коммерческих источниках или могут быть получены специалистом в данной области техники. Функциональный тест на модуляцию ядерного рецептора стероидных гормонов Клетки почки зародыша человека НЕК 293 трансфицировали рецептором стероидных гормонов и плазмидами репортерного гена, используя трансфицирующий реагент Fugene (Roche Diganostics) . Вкратце, репортерная плазмида содержала две копии ARE пробасина (андроген-чувствительный элемент 5'GGTTCTTGGAGTACT3' (SEQ ID NO:1 и промотор ТК (тимидинкиназы) репортера люциферазы кДНК ближе к 5'-концу (upstream), трансфицировали в клетки НЕК 293 вместе с плазмидой, конститутивно экспрессирующей рецептор андрогена человека (AR), используя вирусный промотор CMV (цитомегаловируса). Репортерная плазмида содержала две копии GRE (глюкокортикоид-чувствительный элемент 5'TGTACAGGATGTTCT'3 (SEQ ID NO:2 и ТК промотор репортера люциферазы кДНК ближе к 5'концу, трансфицировали вместе с плазмидой, конститутивно экспрессирующей также глюкокортикоидный рецептор человека (GR), минералкортикоидный рецептор человека (MR) или рецептор прогестерона человека (PR), используя вирусный CMV промотор. Клетки трансфицировали в колбе Т 150 см 2 со средойDMEM, дополненной очищенной на активированном угле 5% фетальной бычьей сывороткой (FBS). После инкубации в течение ночи, трансфицированные клетки обрабатывали трипсином, помещали в 96 луночные планшеты, промытые в среде DMEM, содержащей 5% FBS, очищенную на активированном угле, инкубировали 4 ч и затем подвергали действию различных концентраций тестируемых соединений,колеблющихся от примерно 0,01 нМ до 10 мкМ. В тесте в режиме антагониста для каждого соответствующего рецептора в среду добавляли низкую концентрацию агониста (0,25 нМ дексаметазон для GR,0,3 нМ метилтриенолон для AR, 0,05 нМ промегестон для PR и 0,05 нМ альдостерон для MR) . После 24 ч инкубации с тестируемыми соединениями клетки лизировали и стандартными методами определяли активность люциферазы. Для определения величины ЕС 50 данные аппроксимировали в 4-параметрической логистической кривой. Определяли процент эффективности (соединения с максимальными ответами при насыщении) или процент максимального стимулирования (соединения с максимальными ответами, которые не насыщены) относительно максимального стимулирования, полученного при действии следующих агонистов: 100 нМ метилтриенолон для теста с AR, 30 нМ промегестон для теста с PR, 30 нМ альдостерон для теста с MR и 100 нМ дексаметазон для теста с GR. Значения IC50 можно определять аналогично, используя данные теста в режиме антагониста. В режиме антагониста процент ингибирования определяли при- 12015627 сравнении активности тестируемого соединения в присутствии низкой концентрации агониста (0,25 нМ дексаметазон для GR, 0,3 нМ метилтриенолон для AR, 0,05 нМ промегестон для PR и 0,05 нМ альдостерон для MR) с ответом, производимым той же низкой концентрацией агониста в отсутствии тестируемого соединения. Тест с репортером С 2 С 12 AR/ARE В качестве индикатора агонистической активности в мышечной ткани проводили тест с репортером С 2 С 12 AR/ARE. Вкратце, миобласты мыши клетки С 2 С 12 котрансфицировали с использованием реагента Fugene. Репортерную плазмиду, содержащую GRE/ARE (глюкокортикоид-чувствительный элемент/андроген-чувствительный элемент 5'TGTACAGGATGTTCT'3 (SEQ ID NO:3) и промотор ТК кДНК репортера люциферазы кДНК ближе к 5'концу, трансфицировали с плазмидой, конститутивно экспрессирующей андрогенный рецептор (AR) человека, используя вирусный промотор CMV. Клетки трансфицировали в колбе Т 150 см 2 со средой DMEM с 4% 10% фетальной бычьей сыворотки (FBS). После 5 часов инкубации трансфицированные клетки обрабатывали трипсином, помещали в 96-луночные планшеты, промытые в среде DMEM, содержащей 10% FBS, очищенную на активированном угле, инкубировали 2 часа и затем подвергали действию различных концентраций тестируемых соединений, колеблющихся от примерно 0,01 нМ до 10 мкМ. После 48 часов инкубации с соединениями, клетки лизировали и стандартными методами определяли активность люциферазы. Для определения величины ЕС 50 данные аппроксимировали 4-параметрической модельной кривой. Определяли эффективность (в %) относительно максимального стимулирования, полученного при действии 10 нМ метилтриенолона. Специалист в данной области легко сможет разработать функциональный тест на модуляцию ядерного рецептора стероидных гормонов, подобный описанным выше. В тесте с репортером С 2 С 12AR/ARE, проведенном по существу согласно описанному выше протоколу, примеры соединений согласно настоящему изобретению демонстрировали ЕС 501000 нМ. Предпочтительно, соединения согласно настоящему изобретению демонстрировали ЕС 50 в тесте с репортером С 2 С 12 AR/ARE 100 нМ и более предпочтительно, 50 нМ.In vivo модель эффективности и селективности Самцов крыс Wistar (12 недель возраста) кастрировали (гонадоэктомия или "GDX") в соответствии с утвержденной процедурой (Charles River Labs) и оставляли группу на восемь недель. Также готовили мышей соответствующего возраста с ложной операцией. (Мыши с ложной операцией - это животные,которых подвергли тем же хирургическим процедурам, что и кастрированных животных, за исключением того, что их яички не удаляли.) Животных содержали в комнате с контролем температуры (24C) с обращенным 12-часовым циклом свет/темнота (темные 10:00/22:00) и доступными без ограничения водой и пищей. Для того чтобы продемонстрировать эффективность in vivo, соединения согласно настоящему изобретению ежедневно вводили принудительно перорально или путем подкожной инъекции кастрированным крысам в возрасте двадцати недель (вес тела около 400-450 г) . Животных случайно отбирали на основании веса тела до деления на группы, так что разница в весе между всеми обрабатываемыми группами составляла 5%. Тестируемые соединения животным вводили с использованием обычных носителей. Например, для перорального введения можно использовать 1% натриевую карбоксиметилцеллюлозу(CMC)+0,25% Tween 80 в стерильной H2O, для подкожных инъекций можно использовать 6% этиловый спирт (EtOH)+94% циклодекстран (CDX). Крыс с ложной операцией, получавших только носитель, использовали в качестве положительного контроля, в то время как кастрированных крыс, получавших только носитель, использовали в качестве отрицательного контроля. Тестируемым животным на протяжении двухнедельного периода времени перорально или подкожно вводили, например, 0,3, 1, 3, 10 или 30 мг/кг/день соединения согласно настоящему изобретению. После двухнедельной обработки в качестве индикатора активности определяли сырой вес мышцы поднимающей задний проход (LA) в тестовой группе по сравнению с сырым весом LA в группе кастрированных крыс (отрицательный контроль). Полученный в тестовой и в контрольной группах сырой вес мышцы нормировали относительно общего веса тела. В качестве индикатора тканеспецифической активности использовали сырой вес семенных пузырьков (SV) экспериментальных животных, аналогично, в сравнении с сырым весом семенных пузырьков в группе положительного контроля, с ложной операцией. Кроме того, сырой вес семенных пузырьков, полученный для тестовой и для контрольной группы, нормировали относительно общего веса тела. Дополнительно к сырому весу LA во время вскрытия выделяли левую большую берцовую кость крыс и после обнажения эпифиза тщательно удаляли окружающие кость мягкие ткани. Этот образец затем помещали в раствор, содержащий 0,2% коллагеназы в буфере Tris (pH 7,5). Полученную ферментативную вытяжку внешнего слоя надкостницы немедленно подвергали тестам на определение активности щелочной фосфатазы, индикатора анаболической активности остеобластов/кости. Вкратце, 30 мкл образца помещали в эппендорф, содержащий 200 мкл буфера для субстрата пара-нитрофенил фосфата(PNPP) (Pierce Cat 37621). Для построения стандартной кривой использовали очищенную щелочную фосфатазу (Sigma Cat. P4252), и образцы считывали на планшетном ридере при Abs405 для определения- 13015627 активности щелочной фосфатазы надкостницы (PALP). Полученные в тестовой и в контрольной группах результаты можно нормировать относительно общего веса тела. Значение эффективности в процентах (% эфф.) можно определять следующим образом:% эффективности = ( (сырой вес LA или SV или активность PALP у тестируемого животного/общий вес тела тестируемого животного) / (сырой вес LA или SV или активность PALP у контрольного животного/общий вес тела контрольного животного) ) 100. Проведя тесты, по существу, совпадающие с описанными выше, соединение из примера 74 продемонстрировало следующую активность для упомянутых ранее крыс на модели эффективности и селективности in vivo:In vivo модели заболеваний, связанных с костной недостаточностью Для демонстрации того, что соединения согласно настоящему изобретению обладают способностью лечить заболевания, связанные с потерей костной ткани, такие как остеопороз или остеопения, могут быть применены другие модели животных, хорошо известные в данной области. Примеры таких моделей приведены в Y. L. Ma et al., Japanese Journal of Bone and Mineral Metabolism 23 (Suppl.): 62-68(2005); Y.L. Ma et al., Endocrinology 144: 2008-2015 (2003); и К. Hanada et al., Biol. Pharm. Bull. 26(11): 1563-1569 (2003). Конкретное упоминание сделано для модели остеопении, вызванной недостаточностью эстрогена у самок, в результате овариэктомии, и модели остеопении, вызванной недостаточностью андрогенов у самцов в результате орхидэктомии. Модель остеопении, вызванной недостаточностью эстрогена у самок, в результате овариэктомии Девственных самок крысы Sprague Dawley в возрасте 6 месяцев (Harlan Industries, Indianapolis IN),весящих около 220 г, содержали с неограниченным доступом к пище (TD 89222 с 0,5% кальция и 0,4% фосфора, Teklad, Madison, WI) и воде. Животных подвергали двухсторонней овариэктомии (Ovx) (за исключением контрольных с ложной операцией) и затем случайным образом распределили в группы обработки по 7-8 крыс на группу. Каждый тест обычно содержал по крайней мере 2 набора контролей, включая ложную овариэктомию (Sham) и контроль с овариэктомией (Ovx), получавший носитель. Крыс Ovx оставляли для развития потери костной ткани на 1 месяц (развитие остеопении), а затем начинали лечение тестируемым соединением. Тестируемые соединения перорально принудительно вводили животнымOvx в течение 8 недель. В качестве положительного контроля подмножеству животных Ovx можно давать рекомбинантный РТН человека (1-38) (примерно 10 мкг/кг/д, подкожно). После завершения протокола тестирования, для анализа объемной минеральной плотности кости (МПК, мг/см 3) поясничного позвонка L-5 и бедренной кости использовали количественную компьютерную томографию (QCT, Norland/Stratec, Fort Atkinson, WI). Биомеханический анализ трех точек изгиба в средней части бедренной кости и нагрузку до слома в проксимальной бедренной кости выполняли, используя машину для механического испытания материалов (модель: 661,18c-01, MTS Corp, Minneapolis, MN) , и анализ проводили с использованием программного обеспечения TestWorks 4 (MTS Corp.) Модель остеопении, вызванной недостаточностью андрогена у самцов в результате орхидэктомии Самцов крысы Sprague Dawley в возрасте 6 месяцев (Harlan Industries, Indianapolis IN), весящих около 485 г, содержали с неограниченным доступом к пище (TD 89222 с 0,5% кальция и 0,4% фосфора,Teklad, Madison, WI) и воде. Животных подвергали двухсторонней орхидэктомии (Orx) (за исключением контрольных с ложной операцией) и затем случайным образом распределяли в группы обработки по 7-8 крыс на группу. Каждый тест обычно содержал по крайней мере 2 набора контролей, включая ложную орхидэктомию (Sham) и оконтроль с орхидэктомией (Orx), получавшие носитель. Крыс Orx оставляли для развития потери костной ткани на 2 месяца (развитая остеопения), а затем начинали лечение тестируемым соединением. Тестируемые соединения перорально принудительно вводили животным Orx в течение 8 недель. В качестве положительного контроля подмножеству животных Orx можно давать рекомбинантный РТН человека (1-38) (примерно 10 мкг/кг/д, подкожно). После завершения протокола тестирования, МПК позвонка и бедренной кости, а также биомеханический анализ бедренной кости можно- 14015627 выполнить так, как описано выше для модели овариэктомированных самок крысы (См. в общих чертахMa et al., JBMR 17:2256-2264 (2002), и Turner et al., Bone [Review] 14:595-608 (1993. Специалисту в данной области понятно, что вышеописанные протоколы моделей на животных можно адаптировать для использования в сочетании с соединениями и способами согласно настоящему изобретению. Приведенные ниже способы получения и примеры далее иллюстрируют изобретение и представляют типичные пути синтеза соединений Формулы (I), включая любые новые соединения, которые в общем виде описаны выше. Реагенты и исходные материалы являются легко доступными или легко могут быть синтезированы специалистом в данной области техники. Следует понимать, что способы получения и примеры изложены в целях иллюстрации, но не ограничения, и специалистом в данной области техники могут быть сделаны различные модификации. В настоящей заявке, "ТСХ" относится к тонкослойной хроматографии; "ВЭЖХ" относится к высокоэффективной жидкостной хроматографии; "GC/MS" относится к газовой хроматографии-массспектроскопии; "LC-ES/MS" относится к жидкостной хроматографии - масс-спектроскопии с электронным распылением; "Rf" относится к фактору удержания; "Rt" или "TR" относится ко времени удерживания; "5" относится к одной части на миллион относительно поля тетраметилсилана; "ТФУ" относится к трифторуксусной кислоте; "ТГФ" относится к тетрагидрофурану; "ДМФ" относится к N,Nдиметилформамиду; "ДМСО" относится к диметилсульфоксиду; "МТВЕ" относится к третбутилметиловому эфиру; "PPh3" относится к трифенилфосфину; "DEAD" относится к диэтилазодикарбоксилату; "Pd-C" относится к палладию на углероде; NaBH(OAc)3 относится к триацетоксиборогидриду натрия; "Bn" относится к бензилу; "BnNH2" относится к бензиламину;"NBS" относится к N-бромсукцинимиду; AIBN относится к 2,2'-азобисизобутиронитрилу; "ее" относится к чистоте энантиомера. Оптическое вращение определяли стандартными методами, такими как методы с использованием поляриметра. Конфигурации R или S соединений согласно настоящему изобретению можно определить стандартными методами, такими как рентгеновский анализ и корреляция с временем удержания при хиральной ВЭЖХ. Названия соединений согласно настоящему изобретению получены с использованием ChemDrawversion 7,0,1. Промежуточное соединение 1. -2-Оксоциклопентил)изоиндол-1,3-дион Циклопентанон (100 г, 1,2 моль) и фталимид (170 г, 1,2 ммоль) смешивали в МеОН (900 мл) и перемешивали в течение 18 ч при температуре окружающей среды. Перемешивали интенсивно механической мешалкой и добавляли 2 М водный Na2CO3 (80 мл). После приблизительно 2 ч образовывался плотный осадок белого цвета. Перемешивали при комнатной температуре в течение 48 ч. Собирали твердое вещество белого цвета путем вакуумной фильтрации и промывали метанолом. Твердое вещество суспендировали в воде (300 мл) и перемешивали в течение 3 ч. Собирали твердое вещество и сушили в вакуумной печи при 40 С в течение ночи, в результате получали 195 г (71%) указанного соединения в виде твердого вещества белого цвета. 1 Н ЯМР (ДМСО-d6)7,85-7,77 (м, 4 Н), 4,90 (м, 1 Н), 2,67 (ддд, 1 Н, J = 18,5, 6,2, 1,3 Гц), 2,54 (дд, 1 Н, J=18,5, 9,2 Гц), 2,45 (м, 1 Н), 2,32-2,21 (м, 3 Н); MS (m/z): 230 (М+1, слабый). Промежуточное соединение 2. -2-(1,3-Диоксо-1,3-дигидроизоиндол-2-ил)-1,2,3,4-тетрагидроциклопента[b]индол-7-карбонитрил(8,53 г, 50,3 ммоль) смешивали в НОАс (200 мл) и 4N HCl диоксане (50 мл). При механическом перемешивании реакционную смесь нагревали до 90C в течение 18 ч, затем добавляли дополнительный 4N HCl в диоксане (20 мл). Реакционную смесь нагревали до 100 С в течение 18 ч. Реакционную смесь разбавляли водой (600 мл) и собирали путем вакуумной фильтрации твердое вещество черного цвета. Твердое вещество обрабатывали ультразвуком в МеОН (200 мл) , затем собирали и сушили в вакуумной печи, в результате получали 10,94 г (66%) твердого вещества серо-коричневого цвета. MS (m/z): 328 (M+1), 326-2-(3 оксоциклопентил)изоиндол-1,3-дион смешивали в EtOH (20 мл) и нагревали с обратным холодильником в течение 14 ч. Концентрировали реакционную смесь под вакуумом и разбавляли остаток Et2O (150 мл). Смесь помещали в ультразвуковую ванну на 10 мин, затем отфильтровывали твердое вещество. Фильтрат концентрировали, в результате получали сырой продукт. Очищали материал на силикагеле (120 г) с использованием 10-60% EtOAc/гексан, в результате получали 520 мг (22%) указанного соединения в виде твердого вещества желтого цвета. 1(3100 мл) и этанола (550 мл). К этой смеси добавляли сырой 2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)1,2,3,4-тетрагидроциклопента[b]индол-7-карбонитрил (170 г, 0,52 моль) и перемешивали в течение 15 мин. Добавляли гидрат гидразина (90 мл, 1,9 моль) и перемешивали смесь при комнатной температуре в течение 16 ч. Проверяли методом LC/MS для подтверждения того, что не осталось исходного вещества. Фильтровали сырой продукт реакции под вакуумом и промывали твердое вещество ТГФ (2200 мл). Собирали исходные жидкости и удаляли растворитель под вакуумом. Очищали путем фильтрации на силикагеле (высота 1,5", очень широкий слой SiO2) с использованием (2 M NH3/MeOH)/CH2Cl2 (3-10%). Собирали содержащие продукт фракции и удаляли растворитель. Добавляли ацетонитрил (180 мл) и кипятили смесь с обратным холодильником в течение 15 мин, затем охлаждали до комнатной температуры. Твердое вещество коричневого цвета собирали путем фильтрации и сушили его под вакуумом в течение ночи при 40 С, в результате получали рацемат указанного соединения, 55 г (60%). GC-MS: 198 (M+), 196 (M-). Замечание: В этот момент либо разделяли энантиомеры рацемических аминов, либо продолжали синтез рацемического вещества и проводили хиральное разделение конечных соединений путем количественной ВЭЖХ. Промежуточное соединение 4 а. (S)-2-Амино-1,2,3,4-тетрагидроциклопента[b]индол-7-карбонитрил В колбу на 2 л, снабженную механической мешалкой и холодильником, добавляли этанол (945 мл) и -2-амино-1,2,3,4-тетрагидроциклопента[b]индол-7-карбонитрил (40 г, 0,203 ммоль). Смесь перемешивали и нагревали при 60-65C до полного растворения. Добавляли D-пироглютаминовую кислоту(28,4 г, 0,192 ммоль) и воду (55 мл). Нагревали смесь с обратным холодильником в течение 20 мин. Охлаждали до 40-45 С в течение 90 мин. Перемешивали при 40-45 С в течение одного часа и затем охлаждали до 24 С в течение 2 ч. Перемешивали при этой температуре в течение двух дополнительных часов. Выделяли кристаллическое твердое вещество путем фильтрации и промывали смесью EtOH/вода (95:5)(350 мл). Сушили твердое вещество под вакуумом при 50 С в течение суток, в результате получали 23 г соли пироглютаминовой кислоты. Выделение свободного основания: в воду (150 мл) добавляли соль пироглютаминовой кислоты и перемешивали раствор до полного растворения. Фильтровали сквозь слой диатомита. Собирали водный раствор и доводили рН до 9 путем добавления концентрированного водного аммиака. Собирали путем фильтрации твердое вещество грязно-белого цвета и сушили под вакуумом при 50 С в течение суток, в результате получали 13,5 г (35%) (S)-2-амино-1,2,3,4-тетрагидроциклопента[b]индол-7-карбонитрила,энантиомерный избыток 94% (колонка Chiracel OJ (4,6250 мм; 10 мкм); растворитель, 20% EtOH/(0,2% диметилэтиламин/гексан), проверяли относительно рацемической смеси). Специфический угол вращения: [a]D25 -68,3 (МеОН). Промежуточное соединение 4b. (R)-2-Амино-1,2,3,4-тетрагидроциклопента[b]индол-7-карбонитрил Указанное соединение получали, по существу, так, как описано для промежуточного соединения 4 а,используя L-пироглютаминовую кислоту. Продукт (энантиомерный избыток 97%) имел характерный угол вращения: [a]D25 +63 (EtOH). Промежуточное соединение 5. -7-Трифторметокси-1,2,3,4-тетрагидроциклопента[b]индол-2 иламин Получали, по существу, так, как описано для промежуточного соединения 4, используя -2-(7 трифторметокси-1,2,3,4-тетрагидроциклопента[b]индол-2-ил)изоиндол-1,3-дионена. MS (m/z): 257 (M+1),255 (М-1). Промежуточное соединение 6. Изопропиловый эфир-(7-циано-1,2,3,4 тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты В атмосфере азота смешивали -2-амино-1,2,3,4-тетрагидроциклопента[b]индол-7-карбонитрил(7,0 г, 35,5 ммоль) и диизопропилэтиламин (7,4 мл, 42,6 ммоль) в CH2Cl2 (70 мл). Добавляли изопропил хлороформат (1,0 М в толуоле, 42,6 мл, 42,6 ммоль) и перемешивали в течение ночи при комнатной температуре. Разбавляли водой (100 мл) и этиловым эфиром (50 мл), перемешивали в течение 10 мин и собирали твердое вещество. После сушки получали 7,42 г (61%) указанного соединения в виде твердого вещества желто-коричневого цвета. MS (m/z): 284 (M+1), 282 (М-1). Промежуточные соединения в табл. 1 получали, по существу, так, как описано для промежуточного соединения 6, используя соответствующий хлорангидрид, хлороформат или диалкилдикарбонат, с триэтиламином и диизопропилэтиламином в качестве основания (в качестве взаимозаменяемых вариантов). В качестве альтернативы промежуточное соединение 17 получали путем хиральной хроматографии промежуточного соединения 9. Промежуточное соединение 18. -N-[7-(Метоксииминометил)-1,2,3,4-тетрагидроциклопента [b] индол-2-ил] изобутирамид-N-[7-Циано-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]изобутирамид (5 г, 18,7 ммоль) и Al-Ni катализатор (15 г) смешивали в смеси вода/96% муравьиная кислота, 1/10 (110 мл). Перемешивали в течение 18 ч, добавляли Al-Ni катализатор (13 г) и нагревали с обратным холодильником еще 5 ч. Охлаждали и разбавляли МеОН, отфильтровывали неорганические продукты. Концентрировали фильтрат, добавляли EtOH (200 мл) и обрабатывали ультразвуком в течение 15 мин. Фильтровали нерастворимое вещество и в раствор неочищенного альдегида добавляли растворенный в воде (25 мл) 0-метоксиамин-HCl(120 ммоль), и подщелачивали смесь (рН=9-12) 5,0 N NaOH. Перемешивали при комнатной температуре в течение 18 ч и удаляли большую часть растворителя при пониженном давлении. Смешивали остаток с водой и обрабатывали ультразвуком в течение 30 мин. Выделяли 4,39 г твердого вещества коричневого цвета. Полученный материал очищали на силикагеле с использованием 30-100% EtOAc/гексан, в результате получали 530 мг (10%) указанного соединения в виде твердого вещества бледно-желтого цвета.- 17015627 азота при комнатной температуре. По каплям на протяжении 15 мин добавляли гидрид диизобутилалюминия (1 М раствор в хлористом метилене, 14,1 мл; 14,1 ммоль). В реакционную смесь добавляли этилацетат (30 мл) и перемешивали при комнатной температуре в течение 1 ч. Добавляли 20% водный раствор тартрата натрия (30 мл) и перемешивали в течение 1 ч при комнатной температуре. Органический слой отделяли и экстрагировали водный слой этилацетатом (215 мл). Органические слои объединяли и сушили (Na2SO4), фильтровали и концентрировали, в результате получали 2,2 г трет-бутилового эфира[(R)-7-формил-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты. Очищали путем хроматографии на силикагеле с использованием хлористого метилена/ацетона (95/5), в результате получали 1,3 г (64%). MS (m/z): 301 (M+l). В круглодонную колбу на 100 мл добавляли трет-бутиловый эфир [(R)-7-формил-1,2,3,4 тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты (1,00 г; 3,33 ммоль), этанол (10 мл), карбонат калия (552 мг, 4 ммоль) и О-метилгидроксиламин гидрохлорид (334 мг; 4,0 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель удаляли под вакуумом и добавляли воду(M+1). Промежуточное соединение 20. 2-Бромметил-6-фторпиридин В раствор 6-фтор-2-метилпиридина (20 мл, 0,19 моль) в EtOAc (400 мл) при комнатной температуре добавляли NBS (35,6 г, 0,20 моль). Когда температура достигала 45 С, добавляли AIBN (400 мг, 2,4 ммоль). Смесь нагревали до 65 С в течение 6 ч, затем охлаждали до комнатной температуры и добавляли гексан (1 л) . Удаляли белый осадок путем фильтрации и промывали твердое вещество смесью гексан/EtOAc (1:1). Фильтрат промывали небольшим количеством водного Na2S2O3, NaHCO3 и солевого раствора. Органические продукты сушили (Na2SO4), фильтровали, затем удаляли большую часть растворителя под вакуумом при комнатной температуре. Переносили оставшийся раствор на дистилляционную установку. Удаляли оставшийся растворитель путем дистилляции при атмосферном давлении, а затем непрореагировавшее исходное вещество удаляли при 80 мм (т.к. приблиз. 70 С; 11,2 г) и затем указанный продукт при 1 мм (т.к. приблиз. 75 С; 12,1 г, 32%). ЯМР (300 МГц; CDCl3): 7,82 (1H; дд); 7,35 (1H; дд); 6,90 (1 Н; дд); 4,50 (2 Н; с). Промежуточное соединение 21. 2-(3-Бромметилпиридин-2-ил)изоиндол-1,3-дион Указанное соединение получали с использованием метода, описанного Goswami, S. ; et al. J. Am.Chem. Soc. 1989, 111, 3425-3426, проиллюстрированного Graczyk, P. WO2004013139, 2004, в качестве исходных материалов брали 2-амино-3-пиколин и фталевый ангидрид, затем бромировали NBS (Nбромсукцинимид), получали выход 33% продукта в виде твердого вещества белого цвета. MS (m/z): 317,319 (M+1). Промежуточное соединение 22. 3-Бромметилизотиазол Из коммерчески доступного 5-амино-3-метилизотиазол гидрохлорида в соответствии с процедурой из Buttimore, D.; et al. J. Chem. Soc. 1963, 2032-2039 получали 3-метилизотиазол. Смесь 3-метилизотиазола (3,61 г, 36,6 ммоль), N-бромсукцинимида (6,8 г, 38,2 ммоль) и 1,1-азобис(циклогексанкарбонитрила) (0,18 г, 0,73 ммоль) нагревали с обратным холодильником в CCl4 (100 мл) в течение 18 ч. Охлаждали и путем фильтрации удаляли из продукта сукцинимид. Фильтрат концентрировали и очищали путем хроматографии на силикагеле с EtOAc/гексан (1/5), в результате получали 2,78 г(42,9%) продукта. 1 Н ЯМР (400 МГц, CDCl3)8,63 (д, J=5,1 Гц, 1H), 7,34 (д, J=5,1 Гц, 1 Н), 4,59 (с, 2 Н) . Промежуточное соединение 23. 5-Бром-3-бромметилизотиазол 5-Бром-3-метилизотиазол получали в соответствии со способом из Adams, A.; Slack, R. J. Chem. Soc. 1959, 3061-3072. Указанное соединение получали преимущественно так, как описано для промежуточного соединения 22 с использованием NBS и 5-бром-3-метилизотиазола. Промежуточное соединение 24. 2-Бромметил-6-дифторметилпиридин В 6-метил-2-пиридинкарбоксальдегид (2,6 г, 21,5 ммоль) в CH2Cl2 (25 мл) медленно добавляли бис(2-метоксиэтил)аминосульфуртрифторид (9,2 мл, 50 ммоль). После 18 ч осторожно переливали в стакан,содержащий насыщенный NaHCO3 (300 мл). Встряхивали вместе с водой/CH2Cl2 и разделяли. Сушили органический слой (Na2SO4) и концентрировали, в результате получали 2,11 г масла коричневого цвета. Очищали на силикагеле (50% EtOAc/гексан), в результате получали 1,16 г (38%) 2-метил-6 дифторметилпиридина в виде масла желто-коричневого цвета. Растворяли (1,0 г, 7 ммоль) в CCl4 (30 мл) и добавляли NBS (1,2 г, 6,8 ммоль) и AIBN (100 мг) . Кипятили с обратным холодильником в течение 4 ч, фильтровали и концентрировали. Полученный остаток очищали на силикагеле с использованием 5-10% EtOAc/гексан более 30 мин, в результате получали 670 мг (43%). GC/MS 221+223. Промежуточное соединение 25. 2-Хлорметил-3-хлорпиразин Указанное соединение получали с использованием процедуры хлорирования, преимущественно так,как описано в Jeromin, G. Е.; et al., DE3519364, 1986, и Russell, M. G. N.; et al. J. Med. Chem. 2005, 48,1367-1383. Растворяли 2-метил-3-хлорпиразин (24,3 г, 189 ммоль) в CHCl3 (100 мл). Добавляли бензамид- 18015627 добавляли твердую трихлоризоциануровую кислоту (17,6 г, 75,6 ммоль) и оставляли при нагревании с обратным холодильником в течение 96 ч. Охлаждали и фильтровали через 200 г силикагеля, элюировали хлористым метиленом. Очищали путем хроматографии на силикагеле с использованием градиента от 35% хлороформ/гексан до 60% хлороформ/гексан на протяжении одного часа. Получали указанное соединение в виде бесцветного масла, 5,39 г чистого указанного соединения и 9,4 г, содержащих 70% желаемого продукта. 1H-ЯМР (CDCl3)8,50 (д, 1 Н, J = 2,2 Гц), 8,37 (д, 1H, J = 2,6 Гц), 4,80 (с, 2 Н), 2,50 (с,3 Н); GC/MS М = 162+164. Промежуточные соединения 26-29 получали по существу в соответствии с процедурой, описанной для промежуточного соединения 25, с использованием 2,3-диметилпиразина, 2-хлор-4-метилпиридина, 2 метилтиазола и 3-метилпиридазина. Промежуточные соединения 30 и 31 получали по существу в соответствии с процедурами, описанными Newkome, G. R.; et al. Synthesis 1984, 676-679, с использованием 2-метилпиразин и 2,5 диметилпиразин и N-хлорсукцинимид (NCS). Промежуточное соединение 32. [1,2,5]Тиадиазол-3-карбоновая кислота Указанное соединение получали в соответствии с процедурой из Weinstock, L. М.; et al. J. Org.Chem. 1967, 32, 2823-2828. Промежуточное соединение 33. [1,2,5]Тиадиазол-3-илметанол Оксалил хлорид (11,7 г, 8,04 мл, 92,2 ммоль) и [1,2,5]тиадиазол-3-карбоновую кислоту (6,00 г, 46,1 ммоль) смешивали в CH2Cl2 (150 мл). К этой гетерогенной массе добавляли 10 капель ДМФ и перемешивали при комнатной температуре. Реакционная смесь пузырилась и постепенно становилась прозрачной в течение одного часа. Через один час реакционную смесь концентрировали под вакуумом, в результате получали хлорангидрид в виде масла коричневого цвета. Хлорангидрид растворяли в EtOH (50 мл) и перемешивали при комнатной температуре в течение одного часа, затем концентрировали под вакуумом, в результате получали этиловый эфир в виде масла коричневого цвета. Этиловый эфир растворяли в ТГФ (100 мл) и добавляли LiBH4 (2,0 М раствор в ТГФ, 46,1 мл, 92,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь переливали в водный NH4Cl (400 мл) и экстрагировали EtOAc (3150 мл). Органические продукты сушили (MgSO4) и концентрировали под вакуумом, в результате получали 4,41 г сырого продукта в виде желтого масла. Очищали на силикагеле (40 г) с использованием 30% EtOAc/гексан, в результате получали 3,71 г (69%) указанного соединения в виде масла желтого цвета. 1 Н-ЯМР (CDCl3)8,59 (с, 1 Н), 5,00(с, 2 Н). Промежуточное соединение 34. [1,2,5]Тиадиазол-3-ил-метиловый эфир метансульфоновой кислоты Указанное соединение получали в соответствии с процедурой из Yamamoto, H.; et al. Bioorg. Med.Chem. 2001, 9, 465-475. Промежуточное соединение 35. 3-Гидроксиметил-4,5-дихлоризотиазол В раствор метилового эфира 4,5-дихлоризотиазол-3-карбоновой кислоты (2,1 г, 10 ммоль) в ТГФ(60 мл) добавляли LiBH4 (2,0 М в ТГФ, 10 мл, 20 ммоль). Перемешивали при комнатной температуре в течение одного часа и затем охлаждали до 0 С. Реакцию осторожно гасили водой (10 мл), затем насыщенным водным NH4Cl (50 мл). Экстрагировали EtOAc (100 мл), затем сушили (MgSO4), фильтровали и концентрировали органические продукты, в результате получали 540 мг сырого продукта в виде сиропа оранжевого цвета. Сироп очищали на силикагеле (40 г) с использованием 5-30% EtOAc/гексан, в результате получали 310 мг (17%) указанного соединения в виде твердого вещества белого цвета. 1 Н-ЯМР(ДМСО-d6)5,55 (т, 1 Н, J = 5,9 Гц), 4,52 (д, 2 Н, J = 6,2 Гц). Промежуточное соединение 36. 2-Хлор-4-гидроксиметилтиазол Этиловый эфир 2-хлортиазол-4-карбоновой кислоты получали, по существу, так, как описано в Erlenmeyer, H.; et al. Helv. Chim. Acta 1944, 27, 1432-1436. Указанное: соединение получали по существу в соответствии с процедурой, описанной для промежуточного соединения 35 с использованием этилового эфира 2-хлортиазол-4-карбоновой кислоты. MS (m/z): 150 (M+1); 1 Н-ЯМР (CDCl3)7,16 (т, 1H, J = l,0 Гц), 4,75 (д, 2 Н, J = 0,9 Гц), 2,48 (с, 1 Н) .- 19015627 Промежуточное соединение 37. Этиловый эфир 2-амино-5-метилтиазол-4-карбоновой кислоты Указанное соединение получали в соответствии с процедурой из Henichart, J.-P.; et al. Heterocycles 1991, 32, 693-701. Промежуточное соединение 38. Этиловый эфир 5-метилтиазол-4-карбоновой кислоты В трехгорлой круглодонной колбе смешивали этиловый эфир 2-амино-5-метилтиазол-4-карбоновой кислоты (62,9 г, 338 ммоль) и ТГФ (630 мл). Нагревали с обратным холодильником, и по каплям обрабатывали реакционную смесь изоамилнитритом (52,6 г, 60,1 мл, 449 ммоль). После прекращения добавления реакционную смесь перемешивали с обратным холодильником в течение 1 ч, затем реакционную смесь концентрировали путем ротационного выпаривания (hi-vac), в результате получали 70 г сырого продукта в виде жидкого масла оранжевого цвета. Очищали на силикагеле (400 г, 20-45% EtOAc/гексан),в результате получали 39,47 г (68%) указанного соединения в виде твердого вещества желтого цвета. LCES/MS m/z 172 (M+l), TR=1,5 мин. Промежуточное соединение 39. Этиловый эфир 5-бромметилтиазол-4-карбоновой кислоты Смесь этилового эфира 5-метилтиазол-4-карбоновой кислоты (4,87 г, 28,5 ммоль) и Nбромсукцинимида (5,06 г, 28,5 ммоль) в CCl4 (100 мл) нагревали с обратным холодильником путем облучения 275 ваттной вольфрамовой лампой накаливания. Через 3 ч охлаждали до комнатной температуры и отфильтровывали твердое вещество желто-коричневого цвета. Концентрировали фильтрат путем ротационного выпаривания, в результате получали 6, 42 г сырого продукта в виде масла оранжевого цвета. Очищали на силикагеле [115 г, 0-15% (2 М NH3/MeOH)/(1:1 CH2Cl2/гексан)], в результате получали 3,61 г(51%) указанного соединения в виде желтого твердого вещества. LC-ES/MS m/z 250, 252 (М+1), TR=2,0 мин (86%). Исходное вещество: m/z 172 (М+1), TR=1,7 мин (14%). Пример 1. -N-[7-Циано-4-(6-фторпиридин-2-илметил)-1,2,3,4-тетрагидроциклопента[b]индол-2 ил]изобутирамид-N-(7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2 ил)изобутирамид (6,28 г, 18,8 ммоль), 6-фтор-2-бромметилпиридин (3,93 г, 20,7 ммоль) и Cs2CO3 (12,25 г,37,6 ммоль). Реакционную смесь нагревали при 50 С в течение 18 ч. Охлаждали, разбавляли EtOAc и промывали водой (3x200 мл). Органическую фракцию сушили (MgSO4) и концентрировали, в результате получали 7,1 г сырого вещества. Очищали путем хроматографии на силикагеле (5-20% EtOAc/CH2Cl2). Получали 4,0 г (56%) твердого вещества желто-коричневого цвета.(S)-N-[7-циано-4-(6-фторпиридин-2-илметил)-1,2,3,4 тетрагидроциклопента[b]индол-2-ил]изобутирамид Энантиомеры из примера 1 разделяли путем хиральной хроматографии с использованием Chiralpak(100%), 1 Н-ЯМР (ДМСО-d6) идентично результатам для изомера 1. Пример 2. Изопропиловый эфир В суспензию пиримидин-4-илметанола (250 мг, 2,27 ммоль) в CH2Cl2 (12 мл), добавляли N,Nдиизопропилэтиламин (475 мг, 640 мкл, 3,68 ммоль) и охлаждали до 0 С в атмосфере азота. Добавляли метансульфонилхлорид (275 мг, 186 мкл, 2,40 ммоль) и нагревали до комнатной температуры. После перемешивания при комнатной температуре в течение 1 ч, реакционную смесь загружали на картридж для- 20015627 твердофазовой экстракции Varian ChemElut CE1005 (номер партии Varian 12198006), который предварительно обрабатывали водой (2 мл). Картридж элюировали CH2Cl2 (30 мл), собирали и концентрировали органический элюент. Добавляли к элюенту ДМФ и концентрировали его под вакуумом, в результате получали раствор мезилата в ДМФ. В этот раствор добавляли изопропиловый эфир -(7-циано-1,2,3,4 тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты (495 мг, 1,75 ммоль), Cs2CO3 (1,14 г, 3,5 ммоль) и ДМФ (10 мл). Смесь нагревали до 50 С в течение 18 ч в закрытом флаконе. Разбавляли реакционную смесь EtOAc (100 мл) и промывали органические продукты водой (360 мл). Сушили органический слой над MgSO4, фильтровали и концентрировали, в результате получали 631 мг сырого продукта в виде масла красного цвета. Очищали масло на 40 г силикагеля [40-90% EtOAc/(1:1 CH2Cl2/гексан)], в результате получали 487 мг (74%) указанного соединения в виде твердого вещества белого цвета. LCMS 92% при 4,34 мин (m/z): 376 (M+1), 374 (М-1), 420 (М+НСО 2-). Пример 3. Изопропиловый эфир В охлажденный раствор (0 С) 5-(гидроксиметил)тиазола (1,22 г, 10,6 ммоль) в CH2Cl2 (50 мл) в атмосфере азота добавляли N,N-диизопропилэтиламин (2,39 г, 3,2 мл, 18,5 ммоль) и метансульфонилхлорид (1,27 г, 864 мкл, 11,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Добавляли воду (30 мл), разделяли слои, и сушили органические продукты над MgSO4. Фильтровали и добавляли к органическому слою ДМФ (10 мл). Оставшийся раствор мезилата в ДМФ концентрировали под вакуумом. К этому раствору добавляли дополнительные ДМФ (40 мл), изопропиловый эфир (S)-7-(циано-1,2,3,4-тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты (2,50 г, 8,82 ммоль) и Cs2CO3 (5,57 г, 17,6 ммоль). Перемешивали при комнатной температуре в течение 72 ч. Реакционную смесь разбавляли EtOAc (300 мл), промывали органические продукты водой (3130 мл), затем солевым раствором (100 мл). Сушили (MgSO4), фильтровали и концентрировали органические продукты,в результате получали 3,8 г сырого продукта в виде масла желтого цвета. Очищали на силикагеле [120 г,30-60% EtOAc/(1:1 CH2Cl2/гексан)] , в результате получали 2,22 г (66%) указанного соединения в виде твердого вещества белого цвета. LCMS 100% при 4,46 мин (m/z) 381 (М+Н), 425 (М+НСО 2-) . Используя описанную выше в примерах 2 или 3 процедуру, в мезилаты переводили следующие спирты: 2-гидроксиметилпиримидин,3-гидроксиметил-4,5-дихлоризотиазол,2-хлор-4 гидроксиметилтиазол, 2-гидроксиметилтиазол и 5-гидроксиметилтиазол. Используя соответствующий замещенный 1,2,3,4-тетрагидроциклопента[b]индол, получали примеры 4-53 и промежуточные соединения 40-41, показанные в табл. 2, по существу в соответствии с процедурами, описанными в примерах 1 и 2, с использованием соответствующего гетероарилметилгалогенида или гетероарилметилмезилата, которые были описаны выше или являются коммерчески доступным. Из соответствующего хирального 1,2,3,4-тетрагидроциклопента[b]индола, полученного, как описано выше,или выделенного из рацемата по существу так, как описано в примерах 1 а и 1b, получали хиральное вещество. 2-Бромметил-3-гидроксипиридин гидробромид является коммерчески доступным (Lancaster Synthesis). Данный пример получали путем хирального разделения рацемического продукта, в соответствии с процедурой, в общих чертах описанной в примерах 1a, 1b. Реакцию проводили при комнатной температуре. Альтернативная процедура для промежуточного соединения 40. Изопропиловый эфир (S)-7-циано 4-[2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)пиридин-3-илметил]-1,2,3,4-тетрагидроциклопента[b]индол 2-илкарбаминовой кислоты Изопропиловый эфир (S)-(7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты (20 г, 70,59 ммоль; энантиомерный избыток 98%, 2 ой изомер на Chiracel OJ, 0,2% DMEA в гексан/EtOH [80:20]) растворяли в диметилсульфоксиде (160 мл) и добавляли 2-(3-бромметилпиридин-2 ил) изоиндол-1, 3-дион (29,8 г, 84,71 ммоль). Смесь перемешивали до получения прозрачного раствора. Одной порцией добавляли карбонат цезия (46,4 г, 141,18 ммоль) и диметиламинопиридин (875,5 мг, 7,06 ммоль). Полученную смесь перемешивали при 22/24 С в течение 2 ч. Смесь помещали в воду (1,4 л). Полученную суспензию перемешивали в течение 30 мин и фильтровали. Полученный осадок промывали водой (100 мл). Выделенное влажное твердое вещество растворяли в дихлорметане (750 мл) и отделяли органический слой. Органический слой промывали солевым раствором и выпаривали органический растворитель. Полученное вещество очищали путем хроматографии на силикагеле, элюировали смесью гек- 27015627 сан/ацетон/CH2Cl2 (3/1/1), в результате получали 24 г (58%). MS (m/z): 520 (M+1). Пример 54. Изопропиловый эфир(S)-7-циано-4-[2-(1,3-диоксо-1,3-дигидроизоиндол-2 ил)пиридин-3-илметил]-1,2,3,4-тетрагидроциклопента[b]индол-2-илкарбаминовой кислоты (2,12 ммоль,1100 мг сырого вещества) объединяли со смесью ТГФ (22 мл) и этанола (4 мл) и перемешивали в течение 15 мин. Добавляли гидрат гидразина (0,5 мл, 10 ммоль) и перемешивали смесь при комнатной температуре в течение 18 ч. Неочищенный продукт реакции фильтровали под вакуумом и промывали твердое вещество ТГФ (50 мл). Фильтрат собирали и удаляли растворитель под вакуумом. Полученный остаток очищали путем хроматографии на силикагеле (50-100% этилацетат/CH2Cl2), в результате получали 110 мг (13%) указанного соединения. MS (m/z): 390 (M+1). Альтернативная процедура Изопропиловый эфир(S)-7-циано-4-[2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)пиридин-3 илметил]-1,2,3,4-тетрагидроциклопента[b]индол-2-илкарбаминовой кислоты (20 г; 34,64 ммоль) растворяли в тетрагидрофуране (170 мл) и этаноле (30 мл). Используя шприц, на протяжении 30 мин добавляли моногидрат гидразина (3,37 мл, 69,29 ммоль). Смесь перемешивали при 22/24 С в течение 3 ч и затем фильтровали. Осадок промывали дополнительным тетрагидрофураном (50 мл). Объединенный исходный раствор выпаривали и очищали полученный остаток путем хроматографии на силикагеле, элюировали смесью дихлорметан/(2 М NH3 в метаноле) (98:2). Содержащие чистый продукт фракции объединяли и выпаривали растворитель. Твердое вещество сушили до постоянного веса и затем добавляли этанол (50 мл). Смесь нагревали с обратным холодильником до полного растворения и затем оставляли охлаждаться до комнатной температуры в течение суток. Твердое вещество фильтровали и сушили под вакуумом до постоянного веса, в результате получали 11,45 г (84%) указанного соединения. MS (m/z): 390 (M+1). Хиральная ВЭЖХ: энантиомерный избыток 98% (Изомер 1, Chiralpak AD, EtOH/0,2% диметилэтиламин). Промежуточное соединение 42. -2-(1,3-Диоксо-1,3-дигидроизоиндол-2-ил)-4-(6-фторпиридин-2 илметил)-1,2,3,4-тетрагидроциклопента[b]индол-7-карбонитрил(6,88 г, 21,0 ммоль) и 2-бромметил-6-фторпиридин (3,99 г, 21,0 ммоль) растворяли в ДМФ (80 мл). Добавляли карбонат цезия (7,51 г, 23,1 ммоль, 1,10 экв.) и перемешивали реакционную смесь при комнатной температуре в атмосфере азота в течение 43 ч. Реакционную смесь разбавляли этилацетатом, промывали водой (3 х), сушили над безводным сульфатом натрия, фильтровали и концентрировали, в результате получали полутвердое вещество (8,10 г). Сырой продукт очищали на 120 г силикагеля на колонках,элюировали смесью от 0 до 100% этилацетат/гексан, получали 6,7 г твердого вещества желтокоричневого/коричневого цвета. Продукт суспендировали в эфире (100 мл) при комнатной температуре в течение ночи. Твердое вещество фильтровали, промывали эфиром и сушили при глубоком вакууме, в результате получали указанное соединение в виде твердого вещества желто-коричневого цвета (5,70 г,62%). LCMS 437,1 (М+1). Промежуточное соединение 43. Смесь 2-(1,З-диоксо-1,3-дигидроизоиндол-2-ил)-1,2,3,4-тетрагидроциклопента[b]индол-7 карбонитрила (5 г, 15,3 ммоль) нагревали в ДМФ (25 мл) до 40 С. Добавляли карбонат цезия (10,4 г, 32,4- 28015627 ммоль) и гидробромид 2-бромметилпиридина (4,05 г, 16 ммоль). Смесь перемешивали при 40 С в течение 24 ч. Смесь добавляли в воду (250 мл) и перемешивали в течение 1 ч. Твердое вещество фильтровали, собранное вещество сушили под вакуумом. Вещество добавляли в этанол (25 мл) и нагревали с обратным холодильником в течение 30 мин. Смесь охлаждали до 22 С и фильтровали. Твердое вещество сушили под вакуумом до постоянного веса, в результате получали 4,8 г (75%) указанного соединения. В смеси этанол (15 мл)/тетрагидрофуран (85 мл) растворяли -2-(1,3-диоксо-1,3-дигидроизоиндол 2-ил)-4-(6-фторпиридин-2-илметил)-1,2,3,4-тетрагидроциклопента[b]индол-7-карбонитрил (5,31 г, 12,2 ммоль). Добавляли моногидрат гидразина (4,43 мл, 91,2 ммоль, 7,50 экв.) и перемешивали при комнатной температуре в атмосфере азота в течение ночи. Разбавляли этилацетатом (150 мл), отфильтровывали твердое вещество белого цвета, дважды промывали органический слой 10% карбонатом калия, сушили над безводным сульфатом натрия, фильтровали и концентрировали, получая указанное соединение в виде масла оранжевого цвета (3,42 г, 91%). LCMS 307,0 (М+1), 305,0 (М-1). Промежуточное соединение 45. В ТГФ (1,3 л) и этанол (230 мл) добавляли 2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-4-пиридин-2 илметил-1,2,3,4-тетрагидроциклопента[b]индол-7-карбонитрил (77 г, 184 ммоль). Смесь перемешивали в течение 10 мин и затем добавляли моногидрат гидразина (20 мл, 400 ммоль). Смесь перемешивали при 22 С в течение 16 ч. Смесь фильтровали и выпаривали исходный раствор. Остаток растворяли в дихлорметане (300 мл) . Добавляли 4 М раствор хлороводорода в диоксане (50 мл) и перемешивали смесь в течение 2 ч. Фильтровали и выделенное твердое вещество сушили под вакуумом до постоянного веса, в результате получали 54 г (90%) указанного соединения. MS (m/z): 289 (М+1). Следующие трет-бутиловые эфиры хиральной карбаминовой кислоты, промежуточные соединения 46-51 в табл. 3 получали с использованием промежуточного соединения 16 путем алкилирования его соответствующим гетероарилметилгалогенидом или гетероарилметилмезилатом, по существу так, как описано в процедурах примеров 1 и 2.
МПК / Метки
МПК: A61P 11/00, A61P 9/10, A61P 9/04, C07D 209/58, A61P 19/02, A61K 31/404
Метки: модуляторов, рецептора, андрогенов, тетрагидроциклопента[b]индола, соединения, качестве
Код ссылки
<a href="https://eas.patents.su/30-15627-soedineniya-tetragidrociklopentabindola-v-kachestve-modulyatorov-receptora-androgenov.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения тетрагидроциклопента[b]индола в качестве модуляторов рецептора андрогенов</a>
Замещенные пиридиламидные соединения в качестве модуляторов гистаминового н3-рецептора

Номер патента: 15555
Опубликовано: 31.08.2011
Авторы: Миллз Джон Е., Ли Киев С., Виллани Фрэнк Дж., Мани Неелакандха С., Летавич Майкл А., Пандит Ченнагири Р., Кейт Джон М., Чжун Хуа
МПК: C07D 213/81, A61P 25/00, A61K 31/44...
Метки: пиридиламидные, качестве, н3-рецептора, соединения, замещенные, гистаминового, модуляторов
Формула / Реферат:
1. Соединение формулы (I)где R1представляет собой -C1-4алкил или насыщенный моноциклический циклоалкил, содержащий от 3 до 12 атомов углерода;m равно 1 или 2;X представляет собой N или CH;Y представляет собой N или CRa;Ra представляет собой -H, -Z-Ar, -CH2NRbRc, -CN или -CONRbRc;где каждый из Rb и Rc независимо представляет собой -H или -C1-4алкил;R2 представляет собой -H или -Z-Ar;при условии что один из X и Y представляет собой N, а один из Ra...
Циклотиокарбаматные производные в качестве модуляторов рецептора прогестерона

Номер патента: 4511
Опубликовано: 29.04.2004
Авторы: Терефенко Юджин А., Эдвардс Джеймс П., Фенсам Эндрю, Жи Лин, Джоунс Тодд К., Коллинз Марк А., Вроубел Джей Э., Тегли Кристофер М., Жанг Пувен
МПК: C07D 265/18, A61P 15/00, A61K 31/536...
Метки: циклотиокарбаматные, модуляторов, рецептора, качестве, производные, прогестерона
Формула / Реферат:
1. Соединение формулы где R1 и R2 представляют собой независимые заместители, выбранные из группы, состоящей из H, C1-C6алкила, замещенного C1-C6алкила, C2-C6алкенила, замещенного C2-C6алкенила, C2-C6алкинила, замещенного C2-C6алкинила, C3-C8циклоалкила, замещенного C3-C8циклоалкила, арила, замещенного арила, гетероциклила, замещенного гетероциклила, CORA и NRBCORA, или R1 и R2 сконденсированы с образованием кольца, выбранного из группы,...
Циклокарбаматные производные в качестве модуляторов рецептора прогестерона

Номер патента: 4512
Опубликовано: 29.04.2004
Авторы: Фенсам Эндрю, Джоунс Тодд К., Жанг Пувен, Жи Лин, Вроубел Джей Э., Терефенко Юджин А., Эдвардс Джеймс П., Флетчер Хорас III, Тегли Кристофер М.
МПК: A61P 15/00, A61K 31/536, C07D 265/18...
Метки: прогестерона, качестве, производные, рецептора, циклокарбаматные, модуляторов
Формула / Реферат:
1. Соединение формулы где R1 и R2 представляют собой независимые заместители, выбранные из группы, включающей H, C1-C6алкил, замещенный C1-C6алкил, C2-C6алкенил, замещенный C2-C6 алкенил, C2-C6алкинил, замещенный C2-C6алкинил, C3-C8циклоалкил, замещенный C3-C8циклоалкил, арил, замещенный арил, гетероциклил, замещенный гетероциклил CORA или NRBCORA; или R1 и R2 сконденсированы с образованием a) 3-8-членного насыщенного спироциклического...
Циклопропиламины в качестве модуляторов рецептора гистамина н3

Номер патента: 14370
Опубликовано: 29.10.2010
Авторы: Летавич Майкл А., Кэррутерс Николас И., Эллисон Бретт Д., Грайс Черил А.
МПК: C07D 211/38, A61P 25/00, A61K 31/496...
Метки: гистамина, модуляторов, рецептора, циклопропиламины, качестве
Формула / Реферат:
1. Соединение, выбранное из группы, состоящей из (4-циклопропилпиперазин-1-ил)-(4-морфолин-4-илметилфенил)метанона, (4-циклопропилпиперазин-1-ил)-[4-(4-фторпиперидин-1-илметил)фенил]метанона, (4-циклопропилпиперазин-1-ил)-(4-тиоморфолин-4-илметилфенил)метанона и (4-циклопропилпиперазин-1-ил)-[4-(2-гидроксиметилморфолин-4-илметил)фенил]метанона и их энантиомеров, гидратов, сольватов и фармацевтически приемлемых солей.2. Соединение, представляющее...
Трициклические производные пиразола в качестве модуляторов каннабиноидного рецептора

Номер патента: 11307
Опубликовано: 27.02.2009
Авторы: Лохрай Видья Бхушан, Лохрай Брадж Бхушан, Сривастава Бриджеш
МПК: A61K 31/4162, A61K 31/416, C07D 231/54...
Метки: трициклические, каннабиноидного, производные, модуляторов, качестве, пиразола, рецептора
Формула / Реферат:
1. Соединения общей формулы (I) их фармацевтически приемлемые соли, где Ar представляет собой фенильную группу, замещенную 1-2 атомами галогена; А представляет собой замещенную фенильную группу, Х выбирают из атома кислорода или атома серы, m и n представляют собой целые числа, так что m+n=2; или А представляет собой замещенную дигидротиазолильную группу; Х выбирают из -CH2- или атома серы, m и n представляют собой целые числа, так что 1_m+n_2;...
Предыдущий патент: Разделяющие композиции и способы их применения
Следующий патент: Производные тиазола и их применение
Случайный патент: Турбина для преобразования гидродинамической энергии для применения в воздушной и гидравлической окружающих средах и в среде под давлением