Триазолиламинопиримидиновые соединения
Номер патента: 15574
Опубликовано: 31.10.2011
Авторы: Ван Янь, Сойер Джейсон Скотт, Крич Джойс З., Ху Хонг, Маниар Сачин Говиндиал, Брукс Харольд Бернс, Генри Джеймс Роберт, Макмиллен Вилльям Томас, Цзян Дэлу, Ли Хун-Юй, Слейтер Мелисса Кейт




















Формула / Реферат
1. Соединение формулы

где R1представляет собой водород, метил, циклопропил, циклопропиламино(С1-С2алкил), фтор, этокси, гидрокси, 1-(гидрокси)этил, 2-(гидрокси)(С2-С3алкокси), 2-(гидрокси)этоксиметил, 1-(хлор)этил, 1-((2-фтор)этиламино)этил, 2-(метиламино)этокси, (2-гидроксиэтил)амино, (2-гидроксиэтил)амино(С1-С2алкил), амино, амино(С1-С4алкил), амино(С2-С3алкокси), аминокарбонилметил, (1-метил)-(1-аминокарбонил)этил, (С1-С3алкил)амино(С1-С2алкил), метоксиэтиламино, N-(С1-С3алкил)-N-метиламино(С1-С2алкил), пирролидин-1-илметил, 3-(диметиламино)пирролидин-1-илметил, 3-(пирид-3-ил)пирролидин-1-илметил, 3-(амино)пирролидин-1-илметил, 3-(метиламино)пирролидин-1-илметил, (4,4-диметилоксалидин-3-ил)метил, [N-(2-гидрокси)этил-N-метил]аминометил, (азетидин-1-ил)метил, пиперидин-1-илметил, 4-(метокси)пиперидин-1-илметил, 4-(гидрокси)пиперидин-1-илметил, 4-(гидроксиметил)пиперидин-1-илметил, пиперазин-1-ил-(С1-С2алкил), 4-(метил)пиперазин-1-илметил, 3,5-(диметил)пиперазин-1-илметил или морфолин-4-илметил;
R2 представляет собой водород;
R3 представляет собой водород, метил, фтор или хлор, или R3 представляет собой амино и совместно с R2 образует пирролильное кольцо, конденсированное с указанным пиридином;
R4 представляет собой водород, метил, фтор или хлор;
R5 представляет собой водород или гидроксиметил и
R6 представляет собой водород или метил;
или фармацевтически приемлемая соль указанного соединения.
2. Соединение по п.1, где R1 представляет собой циклопропиламино(С1-С2алкил), 1-(гидрокси)этил, 2-(гидрокси)этоксиметил, 1-(хлор)этил, 1-((2-фтор)этиламино)этил, (2-гидроксиэтил)амино, (2-гидроксиэтил)амино(С1-С2алкил), амино(С1-С4алкил), аминокарбонилметил, (1-метил)-(1-аминокарбонил)этил, (С1-С3алкил)амино(С1-С2алкил), N-(С1-С3алкил)-N-метиламино(С1-С2алкил), пирролидин-1-илметил, 3-(диметиламино)пирролидин-1-илметил, 3-(пирид-3-ил)пирролидин-1-илметил, 3-(амино)пирролидин-1-илметил, 3-(метиламино)пирролидин-1-илметил, (4,4-диметилоксалидин-3-ил)метил, [N-(2-гидрокси)этил-N-метил]аминометил, (азетидин-1-ил)метил, пиперидин-1-илметил, 4-(метокси)пиперидин-1-илметил, 4-(гидрокси)пиперидин-1-илметил, 4-(гидроксиметил)пиперидин-1-илметил, пиперазин-1-ил-(С1-С2алкил), 4-(метил)пиперазин-1-илметил, 3,5-(диметил)пиперазин-1-илметил или морфолин-4-илметил;
или фармацевтически приемлемая соль указанного соединения.
3. Соединение по пп.1-2, где R1 представляет собой амино(С1-С4алкил), (С1-С3алкил)амино(C1-C2алкил), N-(С1-С3алкил)-N-метиламино(С1-С2алкил) или морфолин-4-илметил;
или фармацевтически приемлемая соль указанного соединения.
4. Соединение по пп.1-3, где
R1 представляет собой амино(С1-С4алкил) или (С1-С3алкил)амино(С1-С2алкил);
R3 представляет собой водород или фтор;
R4 представляет собой водород или фтор;
R5 представляет собой водород и
R6 представляет собой водород;
или фармацевтически приемлемая соль указанного соединения.
5. Соединение по пп.1-4, где
R1 представляет собой 1-(метиламино)этил;
R3 представляет собой фтор;
R4 представляет собой водород;
R5 представляет собой водород и
R6 представляет собой водород;
или фармацевтически приемлемая соль указанного соединения.
6. Соединение по пп.1-4, где R1 представляет собой 1-аминоэтил, R3 представляет собой фтор, R4представляет собой фтор, R5представляет собой водород и R6представляет собой водород, или фармацевтически приемлемая соль указанного соединения.
7. Соединение по пп.1-4, где R1 представляет собой 1-(метиламино)этил, R3 представляет собой фтор, R4представляет собой фтор, R5представляет собой водород и R6представляет собой водород, или фармацевтически приемлемая соль указанного соединения.
8. Фармацевтическая композиция, содержащая соединение по пп.1-7 или фармацевтически приемлемую соль указанного соединения в сочетании с фармацевтически приемлемым носителем, разбавителем или наполнителем.
9. Применение соединения по любому из пп.1-7 или фармацевтически приемлемой соли указанного соединения в качестве лекарственного средства для лечения рака.
10. Применение соединения по любому из пп.1-7 или фармацевтически приемлемой соли указанного соединения для лечения рака.
Текст
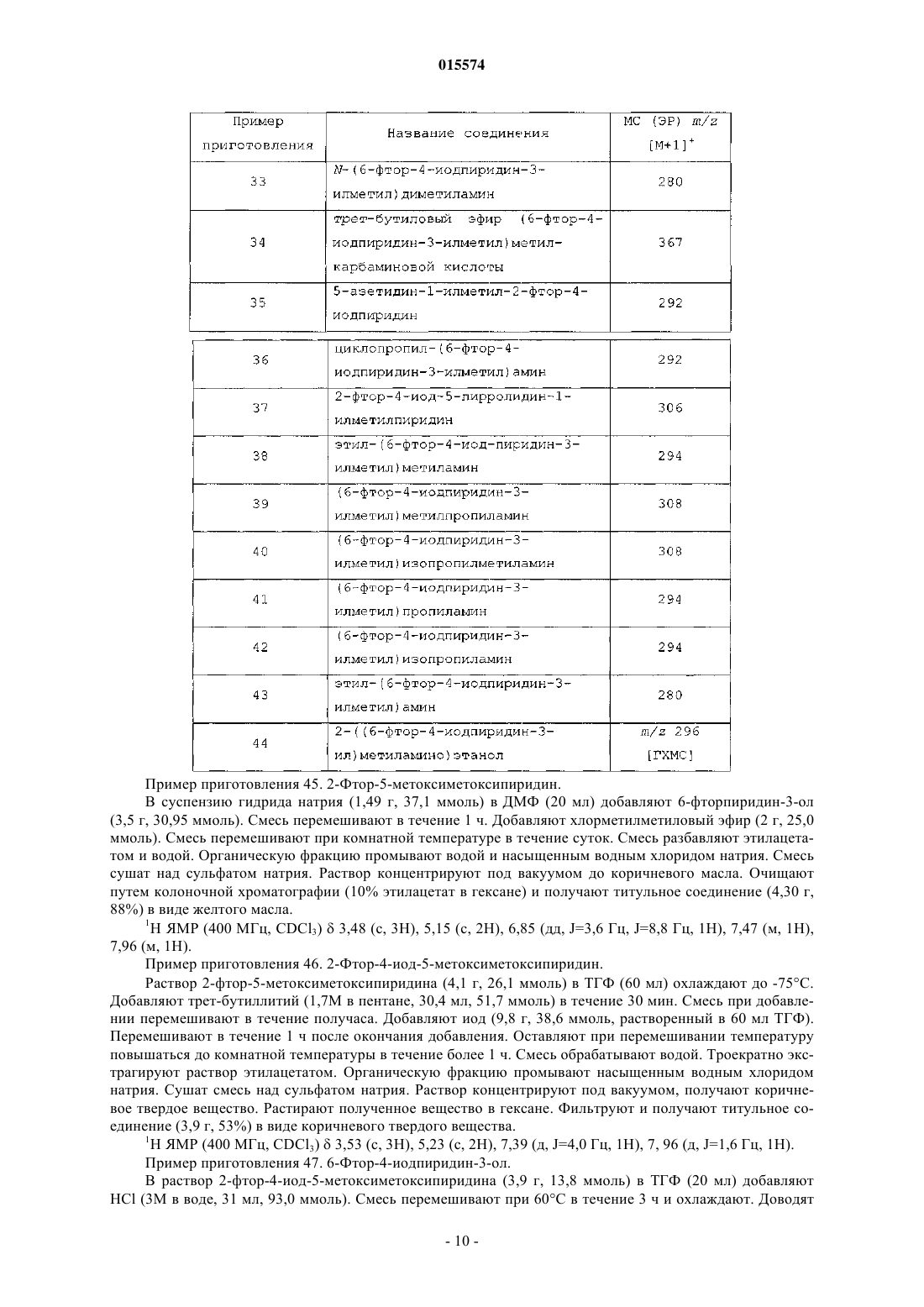
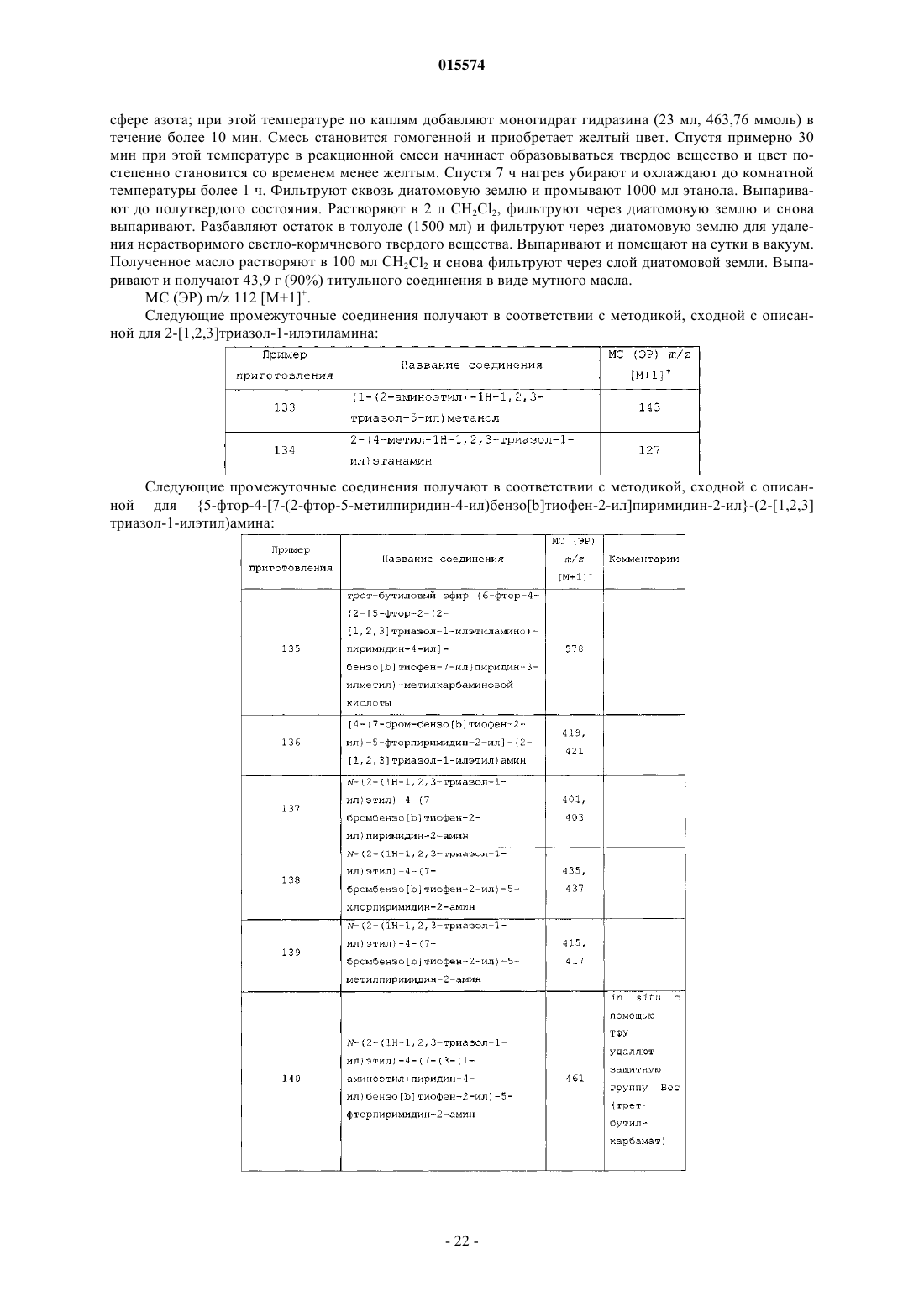
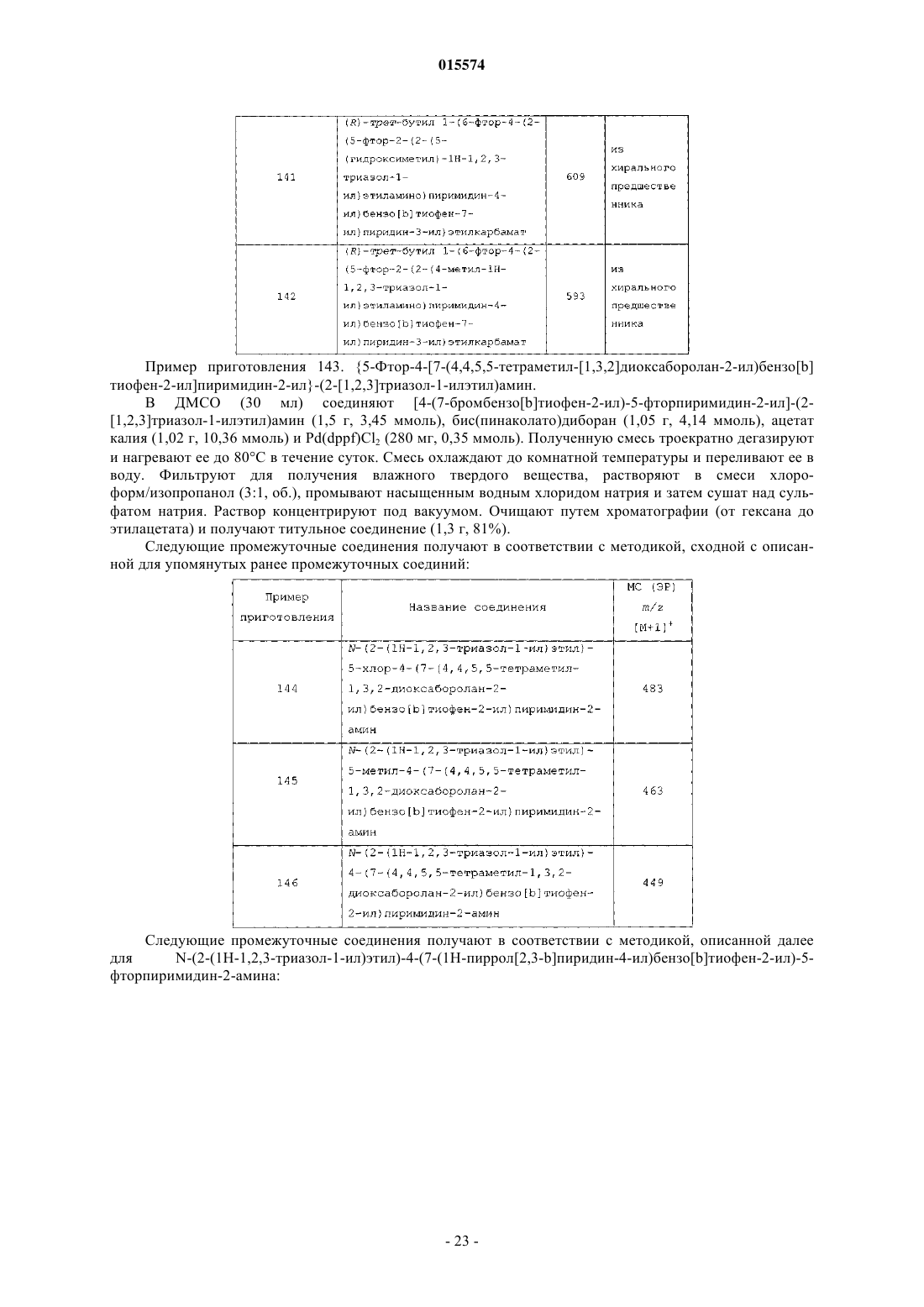
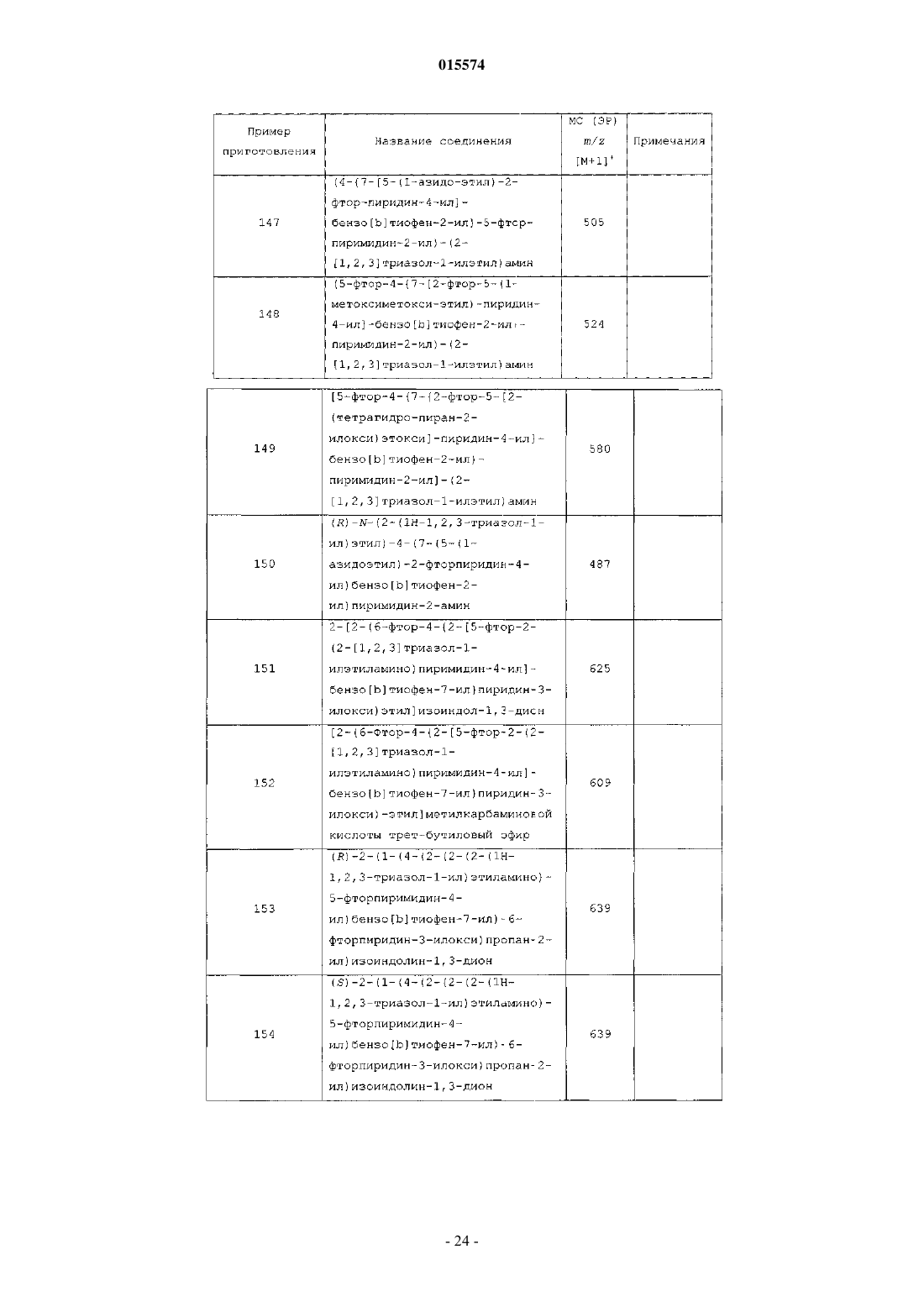
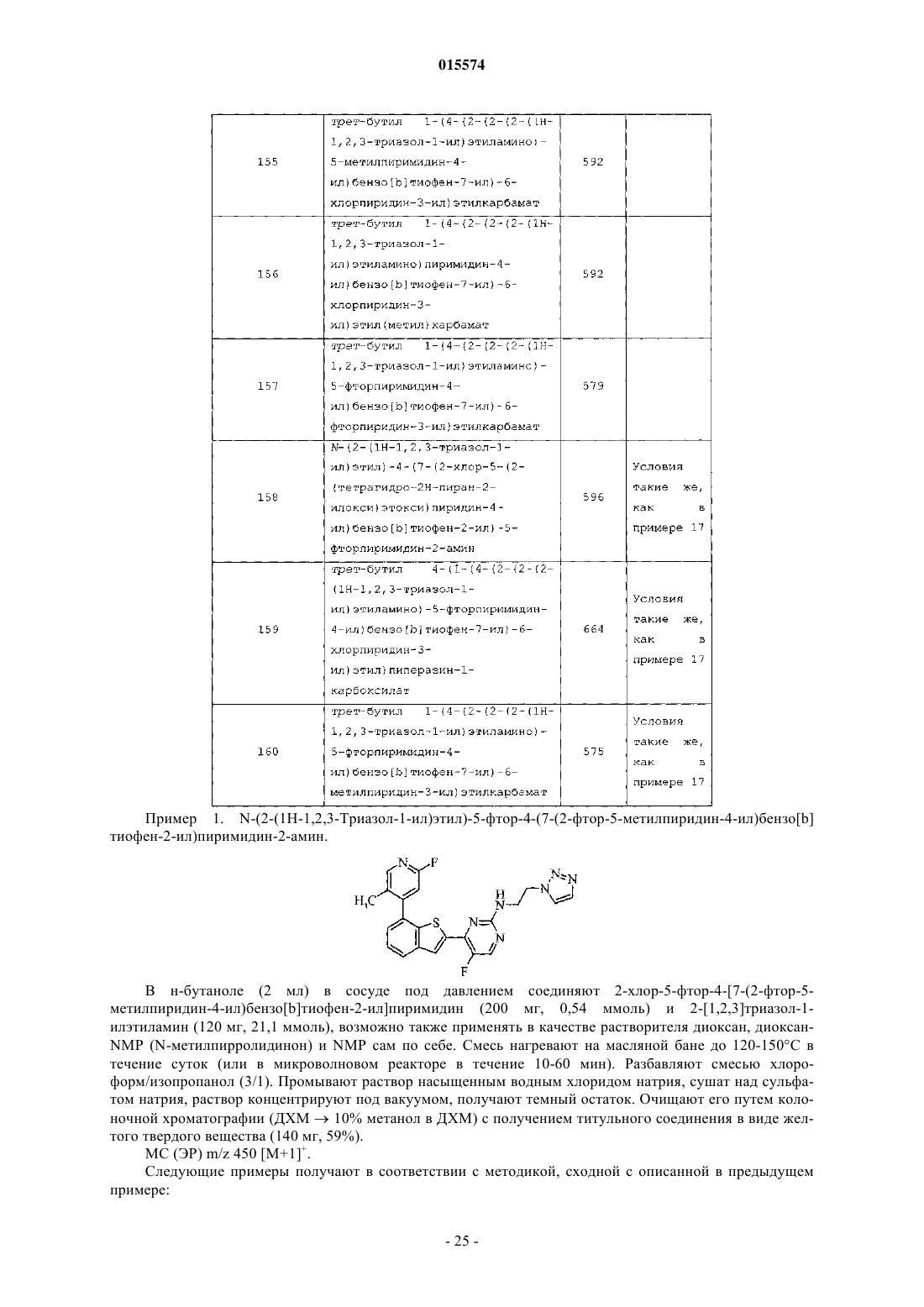
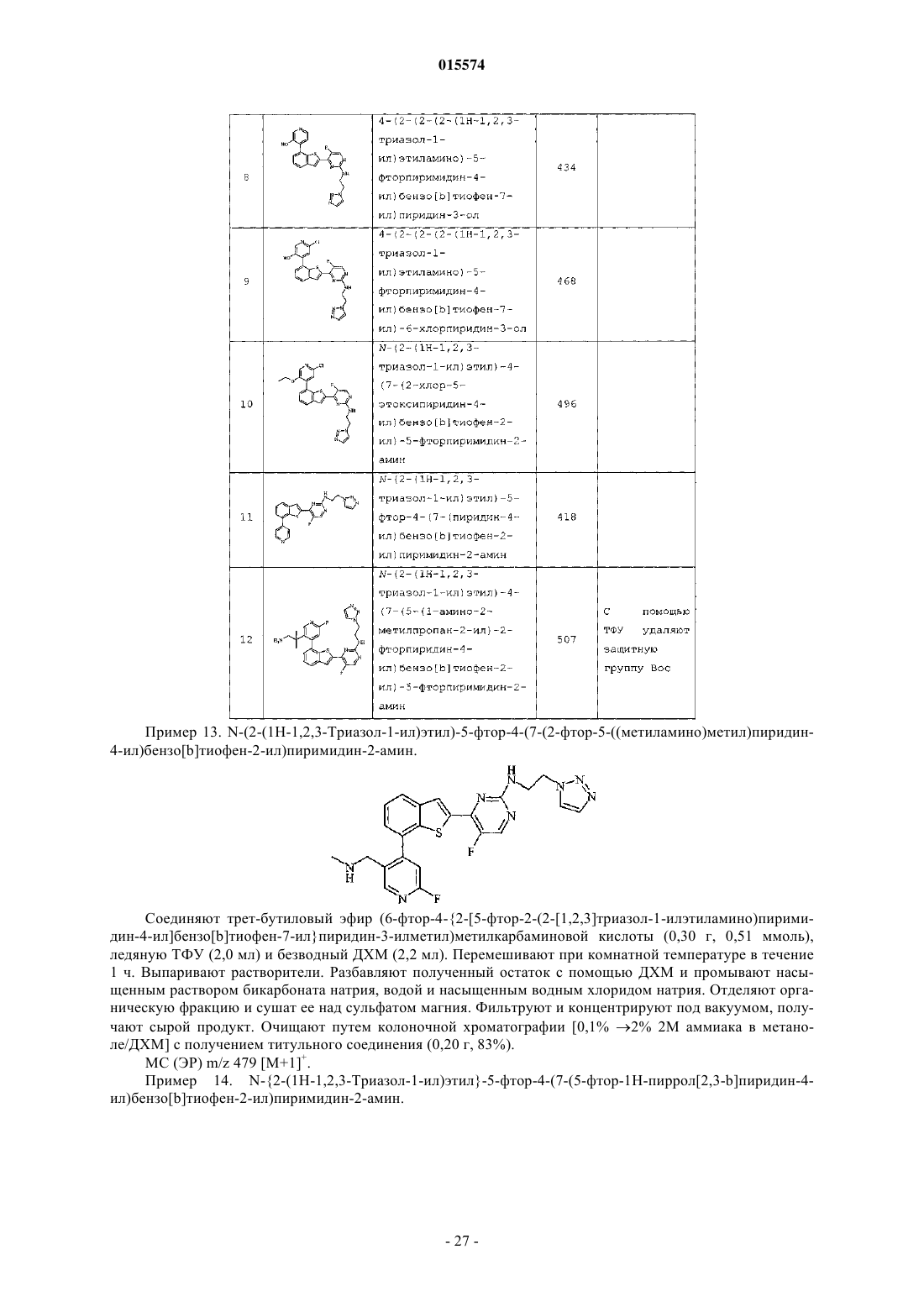
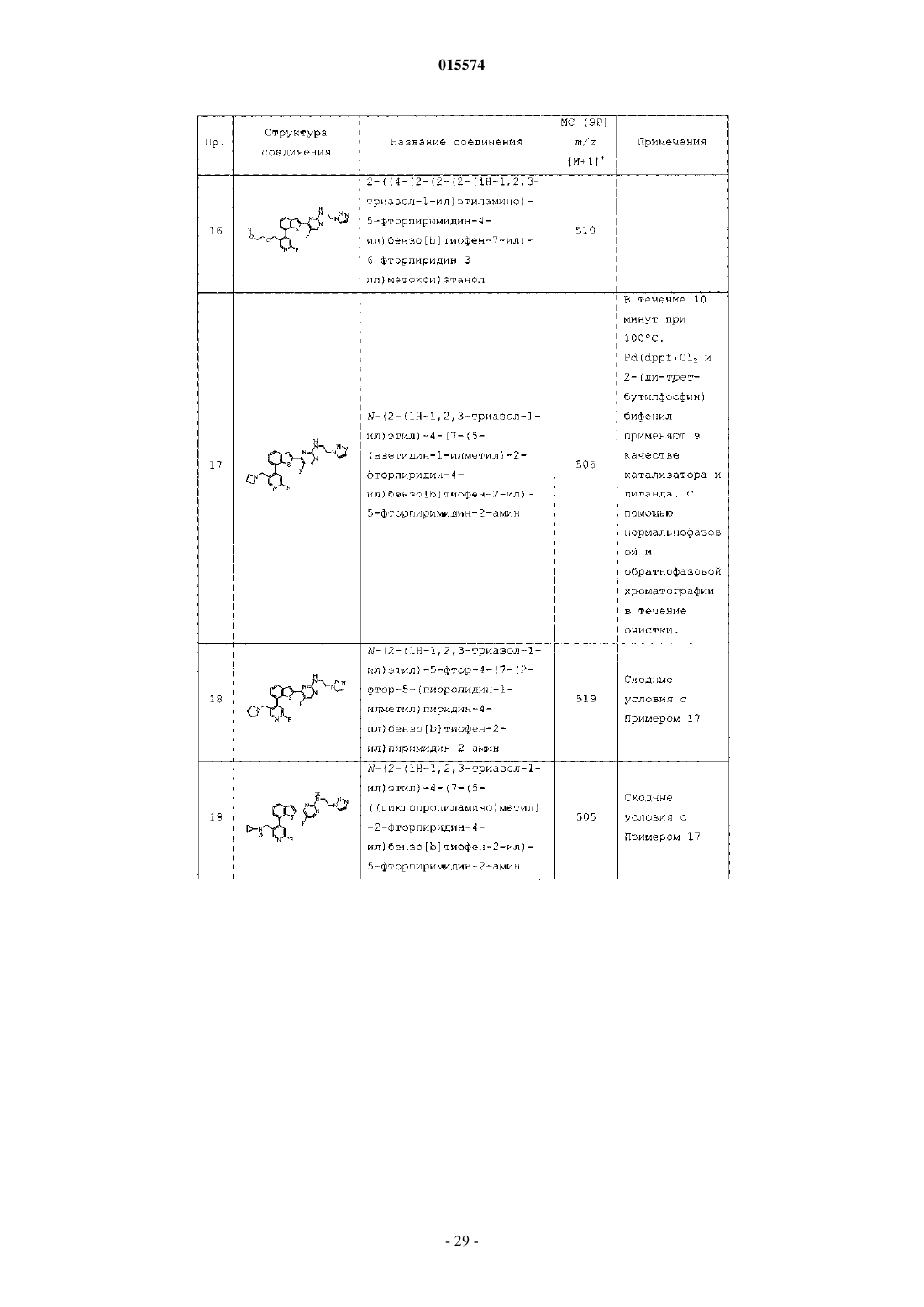
ОПИСАНИЕ ИЗОБРЕТЕНИЯ К ЕВРАЗИЙСКОМУ ПАТЕНТУ Дата публикации и выдачи патента В настоящем изобретении предложены триазолиламинопиримидиновые соединения формулы пригодные для лечения раковых заболеваний.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US)Plk1 принадлежит к небольшому семейству протеинкиназ, характеризующихся наличием фосфосерин/треонинсвязывающего домена, известного как polo-бокс домен (polo box domain). Plk1 играет определяющую роль в регуляции клеточного цикла. Полагают, что помимо других функций, Plk1 регулируют инициирование, развитие и выход из митоза, представляющего собой стадию, на которой происходит деление раковых клеток. Соответственно, блокирование Plk1 в раковых клетках предотвращает деление,или митоз, указанных клеток. Были выявлены высокоактивные противораковые агенты, препятствующие протеканию митоза, такие как алкалоиды барвинка (Navelbine), таксоиды (Taxotere) и ингибиторы топоизомеразы II (Adriamycin). Velcade представляет собой противоопухолевый агент, ингибирующий протеосому 26S. Однако указанные лекарственные препараты оказывают значительное побочное действие на нормальные, неделящиеся клетки. Ингибиторы Plk1 специфично воздействуют на делящиеся клетки и способны не вызывать нежелательного токсического действия. Ингибиторы Plk1 известны в данной области техники; см., например, WO 06/066172. Кроме того, в публикации WO 06/021548 предложены некоторые аналоги дигидроптеридинона (например, BI-2536) в качестве ингибиторов Plk1. В настоящее время BI-2536 проходит вторую фазу клинических испытаний; однако указанный препарат обладает высокой скоростью выведения (CL 1000 мл/мин), при этом существуют ограничения в отношении дозы указанного препарата, обусловленные миелосуппрессией у мужчин. В настоящее время по-прежнему существует потребность в новых соединениях, ингибирующихPlk1, обладающих более высокой активностью или улучшенными фармакокинетическими свойствами. В настоящем изобретении предложены новые триазолиламинопиримидины, которые, как полагают,подходят для клинического применения для лечения рака за счет обеспечения ингибирования Plk1. Полагают, что некоторые из указанных соединений обладают повышенной эффективностью по сравнению с соединениями, предложенными в WO 06/066172. Кроме того, полагают, что некоторые из соединений согласно настоящему изобретению обладают улучшенными фармакокинетическими свойствами, например, скоростью выведения, по сравнению с BI-2536. Далее данные по пероральной биодоступности соединений согласно настоящему изобретению, проходивших испытания, позволяют предположить, что некоторые из указанных соединений подходят для перорального введения. В настоящем изобретении предложены соединения формулы IR3 представляет собой водород, метил, фтор или хлор, или R3 представляет собой амино и совместно с R2 образует пирролильное кольцо, конденсированное с указанным пиридином;R5 представляет собой водород или гидроксиметил иR6 представляет собой водород или метил; или фармацевтически приемлемая соль указанного соединения. В настоящем изобретении предложен способ лечения ракового заболевания, выбранного из группы,включающей немелкоклеточный рак легкого, рак ротоглотки, рак пищевода, рак желудка, меланому,плоскоклеточный рак кожи, рак молочной железы, рак яичников, рак эндометрия, рак толстого кишечника, нейроглиому, глиобластому, рак щитовидной железы, рак шейки матки, рак поджелудочной железы,рак предстательной железы, гепатобластому и неходжкинскую лимфому, у млекопитающего, включаю-1 015574 щий введение млекопитающему, которое нуждается в таком лечении, эффективного количества соединения формулы I или фармацевтически приемлемой соли указанного соединения. В настоящем изобретении также предложена фармацевтическая композиция, содержащая соединение формулы I или фармацевтически приемлемую соль указанного соединения в сочетании с фармацевтически приемлемым наполнителем, носителем или разбавителем. В настоящем изобретении предложено соединение формулы I или фармацевтически приемлемая соль указанного соединения, подходящее для применения в качестве лекарственного средства. Кроме того, в настоящем изобретении предложено применение соединения формулы I или фармацевтически приемлемой соли указанного соединения для получения лекарственного препарата для лечения раковых заболеваний. В частности, указанные раковые заболевания выбраны из группы, включающей немелкоклеточный рак легкого, рак ротоглотки, рак пищевода, рак желудка, меланому, плоскоклеточный рак кожи, рак молочной железы, рак яичников, рак эндометрия, рак толстого кишечника, нейроглиому, глиобластому, рак щитовидной железы, рак шейки матки, рак поджелудочной железы, рак предстательной железы, гепатобластому и неходжкинскую лимфому. Далее в настоящем изобретении предложена фармацевтическая композиция для лечения ракового заболевания, выбранного из группы, включающей немелкоклеточный рак легкого, рак ротоглотки, рак пищевода, рак желудка, меланому, плоскоклеточный рак кожи, рак молочной железы, рак яичников, рак эндометрия, рак толстого кишечника, нейроглиому,глиобластому, рак щитовидной железы, рак шейки матки, рак поджелудочной железы, рак предстательной железы, гепатобластому и неходжкинскую лимфому, содержащая соединение формулы I или фармацевтически приемлемую соль указанного соединения в качестве активного компонента. В настоящем изобретении также предложены соединения формулыR3 представляет собой водород или галоген, или R3 представляет собой амино и совместно с R2 образует пирролильное кольцо, конденсированное с указанным пиридином;R4 представляет собой водород или галоген; или фармацевтически приемлемая соль указанного соединения. Общепринятые химические термины, используемые при описании вышеприведенных формул,употребляют в их обычном значении. Например, термин "(С 1-С 3 алкил)" означает метил, этил, пропил и изопропил. Термин "(С 1-С 2 алкил)" находится в рамках определения, данного для термина "(С 1-С 3 алкил)",и означает метил и этил. Термин "(С 2-С 3 алкокси)" означает этокси, пропокси и изопропокси. Термин "галоген" означает фтор, хлор, бром и иод. Когда заместитель присоединен через алкильную группу, такую как этил, н-пропил, изопропил, нбутил, изобутил, втор-бутил или трет-бутил, например в группах "амино(С 1-С 4 алкил)" или " (С 1-С 2 алкил) амино(С 1-С 2 алкил)", указанный заместитель (например, амино) может быть присоединен по любому из атомов углерода в составе указанного алкила или алкокси. Например, так Для специалиста в данной области техники представляется очевидным, что большинство или все соединения согласно настоящему изобретению способны образовывать соли. Соединения согласно настоящему изобретению представляют собой амины и соответствующим образом взаимодействуют с различными органическими и неорганическими кислотами с образованием фармацевтически приемлемых солей присоединения кислот. Такие фармацевтически приемлемые соли присоединения кислот и общие-2 015574 способы их получения хорошо известны в данной области техники; см., например, P. Stahl, et al., HANDBOOK OF PHARMACEUTICAL SALTS: PROPERTIES, SELECTION AND USE, (VCHA/Wiley-VCH,2002); S.M. Berge, et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciences, Vol. 66, No. 1, January 1977. Предпочтительные соединения формулы I представляют собой соединения, гдеi) R3 представляет собой водород или фтор;l) R4 представляет собой водород и фтор;q) R1 представляет собой амино (С 1-С 4 алкил) или (С 1-С 3 алкил)амино(С 1-С 2 алкил);R3 представляет собой водород или фтор;R4 представляет собой водород или фтор;R5 представляет собой водород иu) R1 представляет собой 1-(метиламино)этил, R3 представляет собой фтор, R4 представляет собой фтор, R5 представляет собой водород, R6 представляет собой водород. Приведенные ниже схемы в совокупности с препаративными примерами и примерами иллюстрируют синтез соединений согласно настоящему изобретению. Схема I. Соединение 5 на схеме 1 получают с помощью проводимой в присутствии палладия(0) реакции сочетания либо исходных соединений 1 и 2, либо исходных соединений 3 и 4. Подходящий палладиевый катализатор представляет собой такой катализатор, как тетракис(трифенилфосфин)палладий(0) или комплекс [1,1'-бис(дифенилфосфин)ферроцен]дихлорпалладия(II) [Pd(dppf)Cl2] с ДХМ (1:1). Pd(dppf)Cl2 применяют в присутствии основания, такого как карбонат натрия или карбонат калия. Указанные реакции проводят в среде растворителя, такого как тетрагидрофуран (ТГФ), диоксан и вода, в общем случае при температуре примерно от 100 до 150C с применением масляной бани или микроволнового реактора. Соединение 5 литируют диизопропиламидом лития в тетрагидрофуране (ТГФ) и триизопропилборате в situ с образованием бензо[b]тиофенбороната, с последующим проведением реакции сочетания в присутствии палладия(0) с 2,4-дихлорпиримидином (соединением 6) с образованием соединения 7. Указанный боронат в общем случае получают при низкой температуре, такой как -78C. Непосредственно после проведения указанной реакции сочетания создают приведенные выше условия для получения соединения 5. Далее с помощью реакции нуклеофильного замещения, в ходе которой соединение 7 взаимодействует с соединением 8, получают соединения согласно настоящему изобретению. Указанные реакции проводят в среде растворителя, такого как н-бутанол, диоксан и N-метилпирролидин-2-он (NMP). Реакции проводят при температуре примерно от 120 до 150C с применением масляной бани или микроволнового реактора. Используют примерно 2 экв. 1H-1,2,3-триазол-1-этанамина (соединения 8). В качестве нейтрализаторов кислоты применяют аминные основания, такие как триэтиламин и диизопропилэтиламин. Схема II. Альтернативно, соединения согласно настоящему изобретению могут быть получены с помощью реакции Сузуки, проводимой между исходными соединениями 2 и 9 или 4 и 10 в условиях, указанных выше. Поскольку для получения соединений согласно настоящему изобретению применяют две реакции сочетания, исходные соединения 9 и 10 на схеме II относятся к обратному порядку проведения реакции сочетания по сравнению со схемой I. Для опытного специалиста в данной области техники представляется очевидным, что не все заместители в соединениях согласно настоящему изобретению стабильны в условиях проведения некоторых реакций, применяемых для получения указанных соединений. Такие фрагменты могут быть введены в соединение на подходящем этапе синтеза или защищены с последующим удалением защитных групп в случае, когда это необходимо или желательно. Для опытного специалиста в данной области техники также очевидно, что защитные группы можно вводить или удалять на любом подходящем этапе синтеза соединений согласно настоящему изобретению. Способы введения и удаления защитных групп защи-4 015574 щающих азот или кислород, хорошо известны в данной области техники; см., например, Greene and Wuts,Protective Groups in Organic Synthesis, 3rd Ed., John Wiley and Sons, New York, Chapter 7 (1999). Некоторые примеры соединений согласно настоящему изобретению получают из других соединений согласно настоящему изобретению. Кроме того, для опытного специалиста в данной области техники очевидно, что во многих случаях порядок введения фрагментов не имеет существенного значения. Конкретный порядок проведения стадий, необходимых для получения соединений согласно настоящему изобретению,может зависеть от конкретных синтезируемых соединений, исходных соединений и относительной подвижности замещенных фрагментов. Некоторые соединения согласно настоящему изобретению содержат центры асимметрии. В таких случаях настоящее изобретение относится к энантиомерам и рацематам соединений. Наименования соединений в примерах были получены с помощью ChemDraw версии 10.0. Пример приготовления 1. 2-Бензо[b]тиофен-7-ил-4,4,5,5-тетраметил[1,3,2]диоксаборолан. В колбе смешивали 7-бромбензо[b]тиофен (426 мг, 2 ммоль), бис-(пинаколят)диборон (756 мг, 3 ммоль), Pd(dppf)Cl2 (81 мг, 0,1 ммоль), ацетат калия (294 мг, 3 ммоль) в диметилсульфоксиде (ДМСО)(10 мл). Через указанную смесь в течение 5 мин барботировали азот. Колбу плотно закрывали, помещали на масляную баню и нагревали при 100C в течение 4 ч. Полученную смесь разбавляли хлороформом/изопропиловым спиртом (ИПС) (3/1). Полученный раствор промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом магния. Раствор концентрировали под вакуумом до получения темного остатка. Проводили очистку с помощью колоночной хроматографии (гексан 20% этилацетата в гексане) с получением титульного соединения в виде бесцветного твердого вещества (342 мг, 66%). МС (ЭР) т/z 261 [M+1]+. Пример приготовления 2. Бензо[b]тиофен-7-бороновая кислота. Смешивали 7-бромбензо[b]тиофен (300 г, 1,41 ммоль) и триизопропилборат (403,6 г, 2,15 ммоль) в безводном ТГФ (4 л) в снабженной механической мешалкой колбе Мортона вместимостью 12 л и охлаждали в атмосфере азота на бане из сухого льда/ацетона до -70C. Добавляли по каплям н-бутиллитий(1,6 М в гексане, 714 г, 1,68 ммоль) с такой скоростью, чтобы температура реакционной смеси оставалась ниже -67,5C. По завершению добавления реакционную смесь перемешивали при указанной температуре в течение 1 ч. Убирали охлаждающую баню и медленно добавляли 4 л воды, при этом температура смеси повышалась до примерно -5C. Затем добавляли концентрированную HCl (75 мл) до достижения значения pH указанного раствора, равного примерно 2 (pH 2). Полученную суспензию перемеривали в течение 1 ч. Добавляли 5 н. водный раствор NaOH в количестве, достаточном для установления pH указанной смеси, равного примерно 12, и переносили в капельную воронку (bottom-drop funnel) вместимостью 22 л. Нижний водный слой отделяли и сохраняли, а верхний органический слой разбавляли 4 л метил-третбутилового эфира и экстрагировали 1 л 5 н. водного раствора NaOH. Водный слой отделяли, объединяли с полученным ранее водным экстрактом и помещали обратно в делительную воронку. Указанный водный слой промывали дополнительным количеством метил-трет-бутилового эфира (4 л). Вновь отделяли водный слой и переносили в трехгорлую круглодонную колбу вместимостью 12 л, снабженную механической мешалкой. Раствор охлаждали до +5C на водяной бане со льдом. Медленно добавляли концентрированную HCl до достижения значения pH раствора, равного примерно 2. Смесь перемешивали в течение 30 мин, а затем отфильтровывали образовавшееся твердое вещество. Указанное твердое вещество промывали на фильтре 2 л воды и сушили на воздухе в течение 30 мин. Затем помещали указанное твердое вещество в вакуум-сушильный шкаф и сушили под вакуумом при 50C в течение ночи. Высушенное твердое вещество суспендировали в 2 л н-гептана в течение 30 мин для устранения желтой окраски. Вновь отфильтровывали твердое вещество, сушили на воздухе в течение 30 мин, а затем в вакуумсушильном шкафу при 40C в течение ночи с образованием титульного соединения (188,8 г, 75%) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CD3OD)7,86 (д, J=8 Гц, 1H), 7,49-7,57 (м, 2H), 7,30-7,39 (м, 2H). Пример приготовления 3. 5-Бромметил-2-фтор-4-иодпиридин. В колбе соединяют 2-фтор-4-иодпиколин (10,0 г, 42,2 ммоль), N-бромсукцинимид (9,76 г, 54,8 ммоль), 2,2'-азо-бис-изобутиронитрил (3,46 г, 21,1 ммоль) и осушенный CCl4 (100 мл). Смесь нагревают до 70C в атмосфере азота в течение 16 ч. Охлаждают до комнатной температуры. Разбавляют дихлорметаном (ДХМ) и промывают водой и насыщенным водным хлоридом натрия. Фракции разделяют и сушат органическую фракцию над сульфатом магния. Концентрируют под вакуумом и получают сырой продукт. Очищают путем колоночной хроматографии (1%15% этилацетата в гексане), с получением титульного соединения (8,27 г, 62%).MC (ЭР) m/z 315 [M+1]+. Пример приготовления 4. (6-Фтор-4-иодпиридин-3-ил)ацетонитрил. В атмосфере азота в 20 мл ацетонитрила соединяют триметизилцианид (297 мг, 3,0 ммоль) и фторид тетрабутиламмония (785 мг, 3,0 ммоль). В данный раствор добавляют 5-бромметил-2-фтор-4-иодпиридин-5 015574 хлороформ/изопропанол (3/1). Раствор промывают водой и затем насыщенным водным хлоридом натрия. Сушат над сульфатом натрия. Раствор концентрируют под вакуумом. Очищают путем колоночной хроматографии (5% метанола в ДХМ) с получением титульного соединения в виде желтого воскоподобного твердого вещества (500 мг, 96%).(ДМФ) добавляют гидрид натрия (1,51 г, 62,9 ммоль) при 0C. Перемешивают смесь от 0C до комнатной температуры в течение 30 мин. Добавляют метилиодид (8,9 г, 63 ммоль) и продолжают перемешивать смесь при комнатной температуре в течение следующих 30 мин. Смесь разбавляют водой и экстрагируют ДХМ. Органическую фазу сушат над сульфатом натрия и концентрируют под вакуумом, получают маслянистый остаток. Остаток очищают путем колоночной флэш-хроматографии (FCC) (20% этилацетата в гексане) с получением титульного соединения в виде желтого твердого вещества (5 г, 82%).(10 г, 30%, 158 ммоль), 18-краун-эфир (36 мг, 0,14 ммоль), карбонат калия (2 М, 10 мл, 20 ммоль) и 10 мл этанола, получают гомогенный раствор. Смесь перемешивают в течение 3 ч. Разбавляют ее водой. Продукт экстрагируют ДХМ. Сырой продукт очищают путем жидкостной хроматографии (10% метанола в ДХМ) с получением титульного соединения в виде белого твердого вещества (250 мг, 64%).MC (ЭР) m/z 281 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 2-(6-фтор-4-иодпиридин-3-ил)ацетамида: Пример приготовления 8. 1-(6-Фторпиридин-3-ил)этанол. Бромид метилмагния (3 М расвор в эфире, 12 мл, 36 ммоль) при 0C в атмосфере азота добавляют к раствору 6-фторпиридин-3-карбальдегида (3 г, 24 ммоль) в ТГФ (20 мл). Смесь продолжают перемешивать в течение суток при комнатной температуре. Реакцию гасят 1 н. HCl, затем подщелачивают разбавленным гидроксидом аммония до pH 9. Экстрагируют продукт смесью хлороформ/изопропанол (3/1). Сушат над сульфатом натрия. Раствор концентрируют под вакуумом. Очищают путем колоночной хроматографии (10% метанол в ДХМ) с получением титульного соединения в виде бесцветного масла (2,3 г,68%). МС (ЭР) m/z 142 [M+1]+. Пример приготовления 9. 5-(1-Азидоэтил)-2-фторпиридин. В колбу на 1 л, помещенную в ледяную баню, добавляют трифенилфосфин (27,9 г, 106,3 ммоль),4,5-дихлор-3,6-диоксоциклогекса-1,4-диен-1,2-дикарбонитрил (24,12 г, 106,3 ммоль). Медленно при перемешивании добавляют ДХМ (150 мл). К темному раствору медленно добавляют азид тетра-Nбутиламмония (30,23 г, 106,3 ммоль), после чего добавляют 1-(6-фторпиридин-3-ил)этанол (10 г, 70,85 ммоль), растворенный в ДХМ (10 мл). Колбу убирают из ледяной бани и перемешивают при комнатной температуре в течение 1 ч. Растворитель удаляют на ротационном испарителе и очищают путем нормально-фазовой колоночной хроматографии 5%20% этилацетата в гексане с получением титульного соединения в виде бесцветного масла (7,75 г). ГХМС m/z 166 [M]+. Пример приготовления 10. 1-(6-Фторпиридин-3-ил)этиламин. Гидрируют 5-(1-азидоэтил)-2-фторпиридин (4,09 г, 24,59 ммоль) под давлением 60 пси в этаноле(200 мл) в присутствии PtO2 (6 мас.%). Спустя 4 ч смесь фильтруют, удаляют растворитель на ротационном испарителе и сушат полученное масло под вакуумом с получением титульного соединения (3,15 г). ГХМС m/z 140 [M]+. Пример приготовления 11. 1-(6-Хлорпиридин-3-ил)этанол. В раствор 1-(6-хлорпиридин-3-ил)этанона (10 г, 64,27 ммоль) в метаноле (100 мл) медленно добавляют тетрагидроборат натрия (1,01 г, 26,35 ммоль). Перемешивают при комнатной температуре в течение 15 мин. Реакционную смесь переливают в химический стакан, содержащий насыщенный NaHCO3 (40 мл) и затем экстрагируют водой (100 мл) и ДХМ (400 мл). Органическую фракцию промывают насыщенным водным хлоридом натрия, сушат над сульфатом натрия и концентрируют. Очищают путем нормально-фазовой хроматографии на колонках (20% этилацетат в гексане 70% этилацетат в гексане) с получением титульного соединения (7 г, 69%).MC (ЭР) m/z 158 [M+1]+. Пример приготовления 12. 2-Фтор-5-(1-метоксиметоксиэтил)пиридин. В раствор 1-(6-фторпиридин-3-ил)этанола (3,0 г, 21,3 ммоль) в ДХМ при 0C добавляют диизопропилэтиламин и хлорметоксиметан. Смесь продолжают перемешивать в течение 30 мин при 0C, затем в течение суток при комнатной температуре. Смесь разбавляют смесью хлороформ/изопропанол (3/1). Промывают раствор насыщенным водным хлоридом натрия. Сушат раствор над сульфатом натрия. Раствор концентрируют под вакуумом, получают темный остаток. Очищают путем колоночной хроматографии (гексан 20% этилацетата в гексане) и получают титульное соединение в виде бесцветного твердого вещества (3 г, 66%). МС (ЭР) m/z 186 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 2-фтор-5-(1-метоксиметоксиэтил)пиридина: Пример приготовления 14. 5-Циклопропил-2-фторпиридин. В смеси толуол/вода (20:1, 21 мл) соединяют 2-фтор-5-иодпиридин (1,12 г, 5 ммоль), циклопропилборную кислоту (645 мг, 7,5 ммоль), ацетат палладия (56 мг, 0,25 ммоль) и фосфат калия (3,2 г, 15 ммоль). Смесь нагревают до 100C в течение 4 ч. Смесь разбавляют хлороформом-изопропанолом (3:1,100 мл). Органическую фазу промывают насыщенным водным хлоридом натрия и водой. Сушат смесь над сульфатом натрия. Раствор концентрируют под вакуумом с получением коричневого масла. Очищают путем колоночной хроматографии (20% этилацетат в гексане) с получением титульного соединения в виде бледно-желтого масла (430 мг, 63%). Пример приготовления 15. 5-Циклопропил-2-фтор-3-иодпиридин. Раствор 5-циклопропил-2-фторпиридина (1,3 г, 9,5 ммоль) охлаждают в ТГФ (20 мл) в атмосфере азота до -75C на бане из сухого льда и ацетона. Добавляют диизопропиламид лития (2M в ТГФ, 6 мл, 12 ммоль) в течение 30 мин. Смесь перемешивают в течение следующих 3 ч, затем добавляют иод (2,9 г,11,4 ммоль, растворенный в 50 мл ТГФ). Оставляют перемешиваться в течение более 2 ч, затем в смесь добавляют воду (100 мл). Затем оставляют нагреваться до комнатной температуры в течение 1 ч при перемешивании. Обрабатывают смесь насыщенным раствором тиосульфата натрия (50 мл). Раствор экстрагируют эфиром. Раствор концентрируют под вакуумом. Очищают путем колоночной хроматографии(гексан 20% этилацетата в гексане) с получением титульного соединения в виде желтого масла (1,7 г,68%). 1H ЯМР (400 МГц-CDCl3)7,99 (д, J=3 Гц, 1H), 7,39 (тд, J=3,5 Гц, 1H), 6,79 (дд, J=3,8 Гц, 1H), 0,961,02 (м, 2H), 0, 63-0,69 (м, 2H). Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 5-циклопропил-2-фтор-3-иодпиридина: Пример приготовления 18. 5-Циклопропил-2-фтор-4-иодпиридин. Раствор 5-циклопропил-2-фтор-3-иодпиридина (1,7 г, 6,5 ммоль) в ТГФ (20 мл) в атмосфере азота охлаждают до -75C на бане из сухого льда и ацетона. Добавляют диизопропиламид лития (2 М в ТГФ,3,9 мл, 7,8 ммоль) в течение 30 мин. Смесь перемешивают в течение следующих 3 ч, затем добавляют воду (100 мл). Затем при перемешивании оставляют температуру повышаться до комнатной в течение 1 ч. Раствор экстрагируют эфиром. Раствор концентрируют под вакуумом с получением коричневого масла. Очищают путем колоночной хроматографии (гексан 15% этилацетат в гексане) с получением титульного соединения в виде желтого масла (1,1 г, 65%). 1H ЯМР (400 МГц-CDCl3)8,03 (дд, J=3,8 Гц, 1H), 7,99 (с, 1H), 0,91-1,00 (м, 2H), 0,71-0,78 (м, 2H). Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 5-циклопропил-2-фтор-4-иодпиридина: Пример приготовления 21. 1-(6-Фтор-4-иодпиридин-3-ил)этанол. В раствор 2-фтор-4-иод-5-(1-метоксиметоксиэтил)пиридина (1 г, 3,2 ммоль) в метаноле (10 мл) добавляют 1 н. HCl (5 мл). Смесь перемешивают в течение суток. Реакционную смесь разбавляют карбонатом натрия (2H). Продукт экстрагируют хлороформом. Органическую фазу сушат над сульфатом натрия. Раствор концентрируют под вакуумом. Сырой продукт очищают путем колоночной хроматографии (10% метанол в ДХМ) с получением титульного соединения в виде белого твердого вещества (0,75 г, 87%). МС (ЭР) m/z 268 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 1-(6-фтор-4-иодпиридин-3-ил)этанола: Пример приготовления 23. 1-(6-Фтор-4-иодпиридин-3-ил)этиловый эфир метансульфоновой кислоты. В раствор 1-( 6-фтор-4-иодпиридин-3-ил)этанола (1,5 г, 5,6 ммоль) при 0C добавляют метансульфонилхлорид (1,93 г, 16,8 ммоль) и диизопропилэтиламин (2,2 г, 16,8 ммоль) в ДХМ (50 мл). Продолжают перемешивать смесь от 0C до комнатной температуры в течение 4 ч. Реакционную смесь разбавляют разбавленным натрия карбонатом. Экстрагируют продукт хлороформом. Органическую фазу сушат над сульфатом натрия. Раствор концентрируют под вакуумом. Очищают сырой продукт путем колоночной хроматографии (20% этилацетат в гексане) с получением титульного соединения в виде белого твердого вещества (1,24 г, 64%).MC (ЭР) m/z 346 [M+1]+. Пример приготовления 24. 5-(1-Азидоэтил)-2-фтор-4-иодпиридин. В раствор 1-(6-фтор-4-иодпиридин-3-ил)этилового эфира метансульфоновой кислоты (1,2 г, 3,5 ммоль) в ДМФ (20 мл) при 0C добавляют азид натрия (0,45 г, 7 ммоль) и бромид тетра-N-бутиламмония(0,12 г, 0,4 ммоль). Смесь перемешивают при комнатной температуре в течение 42 ч. Разбавляют реакционную смесь хлороформом. Органическую фазу промывают водой и насыщенным водным хлоридом натрия. Органическую фазу сушат над сульфатом натрия. Раствор концентрируют под вакуумом. Очищают путем колоночной хроматографии (20% этилацетата в гексане) с получением титульного соединения в виде белого твердого вещества (0,77 г, 76%). 1H ЯМР (400 МГц-CDCl3)1,56 (д, J=6,8 Гц, 1H), 4,90 (м, 1H), 7,45 (д, J=3,2 Гц, 1H), 8,17 (с, 3H). Пример приготовления 25. трет-Бутил 4-(1-(6-хлор-4-иодпиридин-3-ил)этил)пиперазин-1 карбоксилат. К охлажденному льдом раствору 6-хлор-4-иодпиридин-3-ил)этанола (300 мг, 1,06 ммоль) и триэтиламина (737 мкл, 5,29 ммоль) в ацетонитриле (10 мл) по каплям добавляют раствор метансульфонового ангидрида (553 мг, 3,17 ммоль) в CH3CN (3 мл). Перемешивают в течение 40 мин при той же температуре и добавляют раствор N-трет-бутоксикарбонилпиперазина (1,97 г, 10,6 ммоль) в CH3CN (5 мл). Смесь нагревают до 60C в течение суток. Реакцию гасят насыщенным NaHCO3 (10 мл) и экстрагируют продукты этилацетатом (50 мл 3). Промывают органические фракции насыщенным водным хлоридом натрия, сушат над Na2SO4, фильтруют и удаляют растворитель с помощью ротационного испарителя. Очищают путем нормально-фазовой хроматографии на колонках (10% этилацетата в гексане 40% этилацетата в гексане) с получением титульного соединения в виде белого твердого вещества 226 мг, 47%). МС (ЭР) т/z 452 [M+1]+. Пример приготовления 26. 1-(6-Хлорпиридин-3-ил)этаноноксим. В сосуд под давлением помещают 1-(6-хлорпиридин-3-ил)этанон (3,4 г, 22,8 ммоль), гидроксиламин (50%) в воде (5,77 г, 87,4 ммоль) и 0,75 мл уксусной кислоты в 15 мл диоксана. Сосуд закрывают и нагревают смесь на масляной бане в течение 3 ч до 150C. Охлаждают смесь до комнатной температуры. Разбавляют смесью хлороформ/изопропанол (3/1), промывают водой и насыщенным водным хлоридом натрия. Фракции разделяют и сушат органическую фракцию над сульфатом натрия. Концентрируют под-8 015574 вакуумом с получением титульного соединения (3,52 г, 94%). МС (ЭР) m/z 171 [M+l]+. Пример приготовления 27. 1-(6-Хлорпиридин-3-ил)этанамин. Раствор боргидрида натрия (2,96 г, 82,65 ммоль) и тетрахлорида титана (1M в толуоле, 41,33 мл,41,33 ммоль) в 50 мл сухого 1,2-диметоксиэтана охлаждают до 0C в атмосфере N2. В раствор по каплям добавляют 1-(6-хлорпиридин-3-ил)этанон оксим (3,52 г, 20,66 ммоль). Перемешивают смесь в течение суток при комнатной температуре. Реакцию гасят 200 мл воды. Смесь подщелачивают гидроксидом аммония. Сырой продукт последовательно экстрагируют толуолом и этилацетатом. Фракции разделяют и сушат органическую фракцию над сульфатом натрия. Концентрируют под вакуумом и получают сырой продукт (1,24 г, 38%). 1H ЯМР (400 МГц, CDCl3)3,53 (с, 3H), 5,23 (с, 2H), 7,39 (д, J=4,0 Гц, 1H), 7,96 (д, J=1,6 Гц, 1H). Пример приготовления 28. 1-(6-Метилпиридин-3-ил)этанамин. Смесь 1-(6-метилпиридин-3-ил)этанона (10 г, 74 ммоль) в тетра(изопропоксиде) титана (42,1 г, 148 ммоль) и аммиака (370 ммоль, 2M в метаноле) перемешивают в атмосфере N2 в течение 6 ч при комнатной температуре. К этой смеси добавляют тетрагидроборат натрия (4,2 г, 111 ммоль) и перемешивают в течение суток. Реакцию гасят гидроксидом аммония и фильтруют реакционную смесь. Из фильтрата удаляют растворитель и экстрагируют остаток с помощью ДХМ, промывают насыщенным водным хлоридом натрия и сушат над Na2SO4, фильтруют и удаляют растворитель с получением титульного соединения в виде темно-желтого масла (8,16 г). МС (ЭР) m/z L37 [M+1]+. Пример приготовления 29. трет-Бутил 1-(6-метилпиридин-3-ил)этилкарбамат. В раствор 1-(6-метилпиридин-3-ил)этиламина (8,16 г, 59,9 ммоль) в ацетонитриле (50 мл) добавляют диизопропилэтиламин (11,6 г, 89,9 ммоль) и ди-трет-бутилдикарбонат (15,7 г, 71,9 ммоль), полученную смесь перемешивают в течение суток. Смесь промывают насыщенным NaHCO3 (200 мл) и экстрагируют с помощью ДХМ, промывают насыщенным водным хлоридом натрия и сушат над Na2SO4. Очищают путем колоночной хроматографии (5%50% этилацетат в гексане) с получением титульного соединения в виде бледно-желтого масла (7,3 г).MC (ЭР) m/z 237 [M+1]+. Пример приготовления 30. трет-Бутил 1-(4-иод-6-метилпиридин-3-ил)этилкарбамат. К раствору трет-бутиллития (51,1 мл, 87 ммоль) в ТГФ (70 мл) при -78C в атмосфере N2 с помощью иглы с двумя наконечниками добавляют раствор трет-бутил 1-(6-метилпиридин-3-ил)этилкарбамата(6,85 г, 29 ммоль) в ТГФ (20 мл). Спустя 30 мин добавляют иод (11,0 г, 43,5 ммоль) в ТГФ (25 мл) в течение 30 мин при -78C. Перемешивают в течение 1 ч и затем нагревают до комнатной температуры. Гасят реакцию водой, экстрагируют продукт этилацетатом, промывают насыщенным водным хлоридом натрия и сушат над Na2SO4. Очищают путем колоночной хроматографии 5%50% этилацетата в гексане с получением титульного соединения (620 мг).MC (ЭР) m/z 363 [M+1]+. Пример приготовления 31. (6-Фтор-4-иодпиридин-3-илметокси)этанол. К 2,5 мл этиленгликоля при 0C добавляют гидрид натрия (54 мг, 2,2 5 ммоль) и перемешивают в течение 30 мин при комнатной температуре. Добавляют 5-бромметил-2-фтор-4-иодпиридин (0,69 г, 2,14 ммоль) и нагревают до 120C в течение 30 мин. Охлаждают до комнатной температуры, разбавляют 70 мл воды и экстрагируют эфиром (350 мл). Органические фракции соединяют, промывают насыщенным водным хлоридом натрия, сушат над MgSO4 и удаляют растворитель. Очищают путем хроматографии в диэтиловом эфире с получением титульного соединения (240 мг, 38%). МС (ЭР) m/z 296 [M+1]+. Пример приготовления 32. 4-(6-Фтор-4-иодпиридин-3-илметил)морфолин. В колбе в атмосфере азота соединяют 5-бромметил-2-фтор-4-иодпиридин (6,13 г, 19,4 ммоль), морфолин (3,38 г, 38,8 ммоль) и осушенный CH3CN (100 мл). Добавляют N,N-диизопропилэтиламин (6,76 мл, 38,80 ммоль, 2 М раствор в ТГФ). Нагревают до 81C в течение 2 ч и охлаждают до комнатной температуры. Разбавляют с помощью ДХМ и промывают водой и насыщенным водным хлоридом натрия. Отделяют органическую фракцию от водной фракции и сушат над сульфатом магния. После фильтрации концентрируют под вакуумом с получением титульного соединения 6,23 г (100%). МС (ЭР) m/z 323 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 4-(6-фтор-4-иодпиридин-3-илметил)морфолина: Пример приготовления 45. 2-Фтор-5-метоксиметоксипиридин. В суспензию гидрида натрия (1,49 г, 37,1 ммоль) в ДМФ (20 мл) добавляют 6-фторпиридин-3-ол(3,5 г, 30,95 ммоль). Смесь перемешивают в течение 1 ч. Добавляют хлорметилметиловый эфир (2 г, 25,0 ммоль). Смесь перемешивают при комнатной температуре в течение суток. Смесь разбавляют этилацетатом и водой. Органическую фракцию промывают водой и насыщенным водным хлоридом натрия. Смесь сушат над сульфатом натрия. Раствор концентрируют под вакуумом до коричневого масла. Очищают путем колоночной хроматографии (10% этилацетат в гексане) и получают титульное соединение (4,30 г,88%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3)3,48 (с, 3H), 5,15 (с, 2H), 6,85 (дд, J=3,6 Гц, J=8,8 Гц, 1H), 7,47 (м, 1H),7,96 (м, 1H). Пример приготовления 46. 2-Фтор-4-иод-5-метоксиметоксипиридин. Раствор 2-фтор-5-метоксиметоксипиридина (4,1 г, 26,1 ммоль) в ТГФ (60 мл) охлаждают до -75C. Добавляют трет-бутиллитий (1,7 М в пентане, 30,4 мл, 51,7 ммоль) в течение 30 мин. Смесь при добавлении перемешивают в течение получаса. Добавляют иод (9,8 г, 38,6 ммоль, растворенный в 60 мл ТГФ). Перемешивают в течение 1 ч после окончания добавления. Оставляют при перемешивании температуру повышаться до комнатной температуры в течение более 1 ч. Смесь обрабатывают водой. Троекратно экстрагируют раствор этилацетатом. Органическую фракцию промывают насыщенным водным хлоридом натрия. Сушат смесь над сульфатом натрия. Раствор концентрируют под вакуумом, получают коричневое твердое вещество. Растирают полученное вещество в гексане. Фильтруют и получают титульное соединение (3,9 г, 53%) в виде коричневого твердого вещества. 1HCl (3 М в воде, 31 мл, 93,0 ммоль). Смесь перемешивают при 60C в течение 3 ч и охлаждают. ДоводятpH до 7 путем медленного добавления насыщенного водного раствора бикарбоната натрия. Раствор троекратно экстрагируют этилацетатом. Органическую фракцию промывают насыщенным водным хлоридом натрия. Сушат смесь над сульфатом натрия. Раствор концентрируют под вакуумом с получением титульного соединения (3,2 г, 97%) в виде желтого твердого вещества.MC (ЭР) m/z 240 [M+1]+. Пример приготовления 48. 2-[2-(6-Фтор-4-иодпиридин-3-илокси)этил]изоиндол-1,3-дион. Суспензию 6-фтор-4-иодпиридин-3-ола (1,5 г, 6,28 ммоль) охлаждают и добавляют K2CO3 (4,38 г,31,4 ммоль) в ДМФ (10 мл) при 0C в ледяной бане в атмосфере азота. Добавляют 2-(2 бромэтил)изоиндол-1,3-дион (3,19 г, 12,55 ммоль). Смесь перемешивают при комнатной температуре в течение суток. Добавляют воду. Экстрагируют этилацетатом. Раствор промывают насыщенным водным хлоридом натрия. Сушат над сульфатом натрия. Раствор концентрируют под вакуумом. Очищают путем колоночной хроматографии (от гексана до 30% этилацетата в гексане) с получением титульного соединения в виде желтого масла (1,51 г, 58%). МС (ЭР) m/z 413 [M+1]+. Пример приготовления 49. 2-(6-Фтор-4-иодпиридин-3-илокси)этиламин. В раствор 2-[2-(6-фтор-4-иодпиридин-3-илокси)этил]изоиндол-1,3-диона (440 мг, 1,06 ммоль) в этаноле (10 мл) добавляют гидразин (70 мг, 2,12 ммоль). Раствор перемешивают при комнатной температуре в течение суток. Фильтруют для удаления твердого вещества и концентрируют фильтрат, получают светло-желтое твердое вещество. Очищают путем колоночной хроматографии (10% 2 М аммиака в метаноле и метиленхлориде) с получением титульного соединения в виде светло-желтого масла (250 мг,83,8%). 1H ЯМР (400 МГц, CD3CN)2,12 (с, 2H), 3,03 (т, J=5,2 Гц, 2H), 4,10 (т, J=5,2 Гц, 2H), 7,47 (д, J=4,0 Гц, 1H), 7,69 (д, J=2,0 Гц, 1H). Пример приготовления 50. 1-(6-Фтор-4-иодпиридин-3-илокси)пропан-2-он. Суспензию 6-фтор-4-иодпиридин-3-ола (1,6 г, 6,69 ммоль) и K2CO3 (2,8 г, 2 0,1 ммоль) в ДМФ (10 мл) охлаждают до 0C на ледяной бане в атмосфере азота. Добавляют хлорацетон (0,78 г, 8,03 ммоль) в течение 30 мин. Перемешивают смесь при комнатной температуре в течение 1,5 ч. Добавляют воду и экстрагируют этилацетатом. Раствор промывают насыщенным водным хлоридом натрия. Сушат над сульфатом натрия. Раствор концентрируют под вакуумом. Очищают путем колоночной хроматографии(гексан 10% этилацетат в гексане) с получением титульного соединения (1,5 г, 76%).MC (ЭР) m/z 296 [M+1]+. Пример приготовления 51. 1-(6-Фтор-4-иодпиридин-3-илокси)пропан-2-ол. В раствор 1-(6-фтор-4-иодпиридин-3-илокси)пропан-2-она (240 мг, 0,81 ммоль) в метаноле (4 мл) медленно добавляют NaBH4 (35 мг, 0,94 ммоль). Смесь перевешивают при комнатной температуре в течение 3 ч. Добавляют 1 н. HCl и воду. Экстрагируют метиленхлоридом. Промывают органическую фракцию насыщенным водным хлоридом натрия. Сушат над сульфатом натрия. Раствор концентрируют под вакуумом, получают желтое твердое вещество. Очищают путем колоночной хроматографии (гексан 10% этилацетата в гексане) с получением титульного соединения в виде светло-желтого твердого вещества (240 мг, 99%). МС (ЭР) m/z 298 [M+1]+. Пример приготовления 52. 2-[2-(6-Фтор-4-иодпиридин-3-илокси)-1-метилэтил]изоиндол-1,3-дион. Смесь 6-фтор-4-иодпиридин-3-ола (150 мг, 0,63 ммоль), 2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил) пропилового эфира толуол-4-сульфоновой кислоты (226 мг, 0,63 ммоль) и карбоната цезия (206 мг, 0,63 ммоль) в ДМФ (2 мл) нагревают до 100C в течение 5 ч. Смесь охлаждают и добавляют воду. Экстрагируют этилацетатом. Органическую фракцию промывают насыщенным водным хлоридом натрия. Сушат над сульфатом натрия. Раствор концентрируют под вакуумом, получают светло-желтое твердое вещество. Очищают путем колоночной хроматографии (гексан 10% этилацетата в гексане) с получением титульного соединения в виде белого твердого вещества (115 мг, 43%). МС (ЭР) m/z 427 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 2-[2-(6-фтор-4-иодпиридин-3-илокси)-1-метилэтил]изоиндол-1,3-диона: Пример приготовления 55. 2-Фтор-4-иод-5-[2-(тетрагидропиран-2-илокси)этокси]пиридин. В суспензию гидрида натрия (60% дисперсия в минеральном масле, 0,1 г, 2,51 ммоль) в ДМФ (6 мл) добавляют 6-фтор-4-иодпиридин-3-ол (0,5 г, 2,09 ммоль). Смесь перемешивают в течение 1 ч. Добавляют 2-(2-бромэтокси)тетрагидропиран (0,51 г, 2,34 ммоль). Раствор перемешивают при комнатной температуре в течение суток. Смесь разбавляют этилацетатом и водой. Органическую фракцию промывают насыщенным водным хлоридом натрия и водой. Сушат смесь над сульфатом натрия. Раствор концентрируют под вакуумом. Очицают путем колоночной хроматографии (10% этилацетат в гексане) с получением титульного соединения (0,58 г, 75,5%) в виде светло-желтого масла. МС (ЭР) m/z 368 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 2-фтор-4-иод-5-[2-(тетрагидропиран-2-илокси)этокси]пиридина:-15C. Добавляют трет-бутоксид калия (8,3 г, 74 ммоль) и перемешивают при -15C в течение 30 мин. К смеси по каплям добавляют хлорметилметиловый эфир (5,81 мл, 77 ммоль) в течение 40 мин. После завершения добавления смесь перемешивают при -15C в течение дополнительного 1 ч. Удаляют ледяную баню и оставляют смесь медленно нагреваться до 15C. Смесь переливают в насыщенный водный хлорид натрия и интенсивно перемешивают в течение 10 мин. Экстрагируют полученный раствор тремя порциями этилацетата. Полученные органические фрации соединяют и промывают насыщенным водным хлоридом натрия, сушат над сульфатом натрия, фильтруют и концентрируют под вакуумом. 1H ЯМР (400 МГц, CDCl3)8,42 (д, J=3 Гц, 1H), 8,28 (д, J=5 Гц, 1H), 7,37-7,42 (м, 1H), 7,21-7,27 (м,1H), 5,20 (с, 2H), 3,49 (с, 3H). Пример приготовления 58. 2-Хлор-5-метоксиметоксипиридин. Гидрид натрия (3,7 г, 93 ммоль) суспендируют в ДМФ (50 мл) и по каплям добавляют раствор 2 хлор-5-гидроксипиридина (10 г, 77 ммоль) в ДМФ (20 мл) в течение более 45 мин. Полученный раствор перемешивают при комнатной температуре в течение 1,5 ч. По каплям добавляют хлорметилметиловый эфир (6,6 мл, 86 ммоль) в течение более 45 мин. Полученную смесь перемешивают при комнатной температуре в течение 12 ч. Разбавляют смесь этилацетатом, водой и насыщенным водным хлоридом натрия. Органический раствор отделяют и промывают тремя порциями воды, одной порцией насыщенного водного хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют под вакуумом. Сырой продукт очищают путем колоночной хроматографии на 330 г силикагеля, элюируя с градиентом от гексана до 30% этилацетата в гексане в течение более 20 мин, и затем проводят выдержку в 30% этилацетата в гексане в течение 30 мин с получением титульного соединения (10,8 г, 81%) в виде прозрачного масла. МС (ЭР) m/z 174,0 [M+1]+. Пример приготовления 59. 2-Хлор-4-иод-5-метоксиметоксипиридин. В раствор 2-хлор-5-метоксиметоксипиридина (10,8 г, 62 ммоль) в ТГФ (300 мл) по каплям при-70C в течение 10 мин добавляют трет-бутиллитий (1,7 М в пентане, 72 мл, 123 ммоль). Полученный раствор перемешивают при -70C в течение 30 мин. Добавляют по каплям в течение 30 мин раствор иода(23 г, 92 ммоль) в ТГФ (150 мл). Перемешивают полученный раствор при -70C в течение 1 ч. Удаляют ледяную баню и оставляют реакционную смесь нагреваться до комнатной температуры. Смесь разбавляют этилацетатом и водой и разделяют на фазы. Водную фазу экстрагируют двумя порциями этилацетата. Органические экстракты соединяют и промывают двумя порциями водного тиосульфата натрия, одной порцией воды и одной порцией насыщенного водного хлорида натрия. Сушат над сульфатом натрия,- 12015574 фильтруют и выпаривают. Полученное твердое вещество растирают в гексане. Вещество собирают с помощью фильтрации в вакууме и промывают полученное вещество гексаном. Сушат вещество в вакууме с получением титульного соединения 10,8 г (58%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6)8,08 (с, 1H), 7,98 (с, 1H), 5,43 (с, 2H), 3,40 (с, 3H). Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 2-хлор-4-иод-5-метоксиметокси-пиридина:(0,33 г, 0,86 ммоль) в 3 мл ДМФ добавляют гидрид натрия (620 мг, 26 ммоль). Смесь перемешивают при комнатной температуре в течение 1 ч и добавляют метилиодид (0,37 г, 2,59 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение суток. Добавляют воду и экстрагируют этилацетатом. Органическую фракцию промывают насыщенным водным хлоридом натрия. Смесь сушат над сульфатом натрия. Раствор концентрируют под вакуумом. Очищают остаток путем колоночной хроматографии (20% этилацетата в гексане) с получением титульного соединения в виде светло-желтого твердого вещества (0,13 г, 38%). 1HCl (61 мл). Полученную смесь нагревают до 60C в течение 3 ч. Смесь охлаждают до комнатной температуры и доводят pH до 7 с помощью медленного добавления насыщенного водного раствора бикарбоната натрия. Смесь экстрагируют тремя порциями этилацетата. Органические экстракты соединяют и сушат над сульфатом натрия, фильтруют и концентрируют под вакуумом с получением титульного соединения 6,8 г (98%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6)11,04 (с, 1H), 7,81-7,87 (м, 2H). Пример приготовления 63. 2-Хлор-5-этокси-4-иодпиридин. Раствор 6-хлор-4-иодпиридин-3-ола (4,9 г, 19 ммоль) и карбоната калия (8,0 г, 58 ммоль) в ДМФ (50 мл) обрабатывают этилиодидом (4,7 мл, 58 ммоль). Смесь нагревают до 60C в течение 3 ч. Смесь охлаждают до комнатной температуры и фильтруют сквозь фильтровальную бумагу. Смесь разбавляют этилацетатом и промывают 10% водным раствором лимонной кислоты. Соединяют водные растворы и экстрагируют двумя дополнительными порциями этилацетата. Соединяют органические экстракты и промывают тремя порциями воды, одной порцией насыщенного водного хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют под вакуумом с получением титульного соединения (5,1 г, 93%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6)8,00 (с, 1H), 7,93 (с, 1H), 4,18 (кв., J=7 Гц, 2H), 1,35 (т, J=7 Гц, 3H). Пример приготовления 64. 2, 2-Диметил-N-пиридин-3-илпропионамид. Круглодонную колбу на 250-мл оснащают ледяной баней, магнитной мешалкой и атмосферой N2. Добавляют 3-аминопиридин (15 г, 159 ммоль), ТГФ (60 мл), диэтиловый эфир (60 мл), триэтиламин (17,7 г, 24,4 мл, 175 ммоль). Охлаждают смесь до 0C и медленно через иглу добавляют триметилацетилхлорид (21,0 г, 14,9 мл, 175 ммоль). Перемешивают в течение суток при нагревании до комнатной температуры. Добавляют воду (100 мл), переносят в делительную воронку, экстрагируют и отбирают нижнюю водную фракцию. Сушат органическую фракцию над Na2SO4, фильтруют и концентрируют в ротационном испарителе до бесцветного масла, которое затвердевает при охлаждении. Сушат в глубоком вакууме в течение 2,5 ч с получением титульного соединения в виде светло-коричневого твердого вещества 21,56 г (76%). МС (ЭР) m/z 179 [M+1]+. Пример приготовления 65. N-(4-Иодпиридин-3-ил)-2,2-диметилпропионамид. Круглодонную колбу на 250 мл оснащают магнитной мешалкой, термопарой, баней из сухого льда в ацетоне, атмосферой N2 и воронкой для добавления. Заполняют ее 2,2-диметил-N-пиридин-3 илпропионамидом (3,0 г, 16,8 ммоль), диэтиловым эфиром (67 мл) и тетраметилендиамином (4,68 г, 6,08 мл, 40,3 ммоль). Реакционную смесь охлаждают до -78C. Медленно добавляют через стеклянную иглу н-бутиллитий (2,5 М раствор в гексане, 16,2 мл, 40,3 ммоль) в течение более 10 мин. Реакционную смесь нагревают до -13C в течение более 2 ч. Реакционную смесь охлаждают до -78C. Готовят раствор иода(I2 8,5 г, 33,6 ммоль в ТГФ (20 мл. Добавляют раствор иода в реакционную смесь чрез воронку и пере- 13015574 мешивают 2,5 ч при -68C. Реакцию гасят с помощью добавления насыщенного раствора NH4Cl (40 мл) и переносят в делительную воронку. Добавляют этилацетат (100 мл). Экстрагируют и убирают нижнюю водную фазу. Органическую фракцию промывают насыщенным раствором тиосульфата натрия (100 мл) и экстрагируют. Органическую фазу промывают насыщенным водным хлоридом натрия и экстрагируют. Органическую фазу сушат над Na2SO4 и фильтруют. Концентрируют продукт с помощью ротационного испарителя. Проводят хроматографию на силикале (80 г), элюируя с градиентом от 100% ДХМ до 70% этилацетата/30% ДХМ, получают 1,19 г (23%) титульного соединения. МС (ЭР) m/z 306 [M+1]+. Пример приготовления 66. трет-Бутиловый эфир [2-(6-фтор-4-иодпиридин-3-илокси)этил]карбаминовой кислоты. В раствор 2-(6-фтор-4-иодпиридин-3-илокси)этиламина (0,25 г, 0,89 ммоль) и ди-третбутилдикарбоната (0,29 г, 1,33 ммоль) в 5 мл ДХМ добавляют диизопропилэтиламин (0,23 г, 1,77 ммоль). Смесь перемешивают в течение суток при комнатной температуре. Смесь разбавляют ДХМ, промывают органическую фракцию насыщенным водным хлоридом натрия, сушат над сульфатом натрия, концентрируют под вакуумом, получают неочищенное масло. Очищают путем колоночной хроматографии (30% этилацетат в гексане) с получением титульного соединения (0,33 г, 97%). 1H ЯМР (400 МГц, CDCl3)1,41 (с, 9H), 3,56 (т, J=5,2 Гц, 2H), 4,10 (т, J=5,2 Гц, 2H), 5,07 (с, 1H),7,34 (д, J=3,6 Гц, 1H), 7,69 (д, J=1,6 Гц, 1H). Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для трет-бутилового эфира [2-(6-фтор-4-иодпиридин-3-илокси)этил]карбаминовой кислоты: Пример приготовления 70. трет-Бутил-1-(6-хлор-4-иодпиридин-3-ил)этилкарбамат. трет-Бутил-1-(6-хлорпиридин-3-ил)этилкарбамат (3,77 г, 14,7 ммоль) растворяют в ТГФ (60 мл). Добавляют трет-бутиллитий (1,7 М раствор в гептане, 25,9 мл, 44,0 ммоль) при -78C в атмосфере N2. Раствор перемешивают в течение 0,5 ч при -78C. Добавляют по каплям в течение 30 мин в атмосфере N2 при -78C раствор иода (5,59 г, 22,0 ммоль) в ТГФ (44 мл). Перемешивают полученный раствор при-78C в течение 1 ч, затем от -78C до комнатной температуры в течение 1 ч. Реакцию гасят водой. Экстрагируют смесь этилацетатом. Органическую фракцию промывают водой и насыщенным водным хлоридом натрия. Сушат над MgSO4. После фильтрации концентрируют и очищают сырой продукт путем жидкостной хроматографии (от 0,1 до 1% раствора 2 М NH3 в метаноле /CH2Cl2) с получением титульного соединения (1,34 г, 24%) со степенью чистоты по данным ВЭЖХ 98%. МС (ЭР) т/z 383 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для трет-бутил 1-(6-хлор-4-иодпиридин-3-ил)этилкарбамата: Пример приготовления 73. 6-Фтор-4-иодпиридин-3-амин. Растворяют трет-бутил-6-фтор-4-иодпиридин-3-илкарбамат (1,47 г, 4,35 ммоль) в ДХМ (20 мл). Добавляют трифторуксусную кислоту (ТФУ) (20 мл). Перемешивают при комнатной температуре в атмосфере N2 в течение 2 ч. Удаляют растворитель при пониженном давлении. Сушат под вакуумом, получа- 14015574 ют соль 6-фтор-4-иодпиридин-3-амина с ТФУ (1,02 г, 99%) со 100% чистотой с помощью ВЭЖХ. МС (ЭР) т/z 239 [M+1]+. Пример приготовления 74. 6-Фтор-4-иод-N-(2-(тетрагидро-2H-пиран-2-илокси)этил)пиридин-3 амин. В закрытом реакторе смешивают соль с ТФУ 6-фтор-4-иодпиридин-3-амина (0,36 г, 1,50 ммоль), 2[(2-бромэтил)окси]тетрагидро-2H-пиран (1,38 г, 6,60 ммоль), гидроксид калия (0,19 г, 3,30 ммоль), фторид калия (0,19 г, 3,30 ммоль) и иодид тетрабутиламмония (0,11 г, 0,3 ммоль) в 1,4-диоксане (1,5 мл). Реакционную смесь нагревают до 100C в течение суток. Реакционную смесь охлаждают до комнатной температуры. Гасят реакцию водой. Экстрагируют этилацетатом, промывают органическую фракцию водой и насыщенным водным хлоридом натрия. Сушат над MgSO4. После фильтрации концентрируют и очищают сырой продукт путем жидкостной хроматографии (от 0,1 до 1% раствора 2 М NH3 в метаноле/CH2Cl2) с получением титульного соединения (0,36 г, 66%) со степенью чистоты по данным ВЭЖХ,100% ВЭЖХ. МС (ЭР) m/z 367 [M+1]+. Пример приготовления 75. 2-(6-Фтор-4-иодпиридин-3-иламино)этанол. 6-Фтор-4-иод-N-(2-(тетрагидро-2H-пиран-2-илокси)этил)пиридин-3-амин (0,20 г, 0,55 ммоль) растворяют в этаноле (5,5 мл). Добавляют паратолуолсульфонат пиридиния (0,014 г, 0,055 ммоль). Реакционную смесь нагревают в течение суток до 50C. Удаляют растворитель при пониженном давлении. Очищают путем жидкостной хроматографии (от 0,1 до 1% раствора 2 М NH3 в метаноле /CH2Cl2) с получением титульного соединения (44 мг, 28%) со степенью чистоты по данным ВЭЖХ 100. МС (ЭР) m/z 283 [M+1]+. Пример приготовления 76. 6-Фтор-4-иод-N-(2-метоксиэтил)пиридин-3-амин. В закрытом реакторе смешивают соль с ТФУ 6-фтор-4-иодпиридин-3-амина (0,36 г, 1,50 ммоль), 2 хлорэтил метиловый эфир (0,31 г, 3,30 ммоль), гидроксид калия (0,19 г, 3,30 ммоль), фторид калия (0,19 г, 3,30 ммоль) и иодид тетрабутиламмония (0,11 г, 0,30 ммоль) в 1,4-диоксане (1,5 мл). Реакционную смесь в течение суток нагревают до 100C. Охлаждают реакционную смесь до комнатной температуры. Гасят реакцию водой. Экстрагируют этилацетатом, органическую фракцию промывают водой и насыщенным водным хлоридом натрия. Сушат над MgSO4. После фильтрации удаляют растворитель при пониженном давлении. Очищают сырой продукт путем жидкостной хроматографии (от 0,1 до 1% раствора 2 М NH3 в метаноле /CH2Cl2) с получением титульного соединения (0,09 г, 20%) со степенью чистоты по данным ВЭЖХ 80%. МС (ЭР) m/z 279 [M+1]+. Пример приготовления 77. трет-Бутиловый эфир [2-(6-фтор-4-иодпиридин-3-илокси)этил]метилкарбаминовой кислоты. В раствор трет-бутилового эфира [2-(6-фтор-4-иодпиридин-3-илокси)этил]карбаминовой кислоты(0,33 г, 0,86 ммоль) в 3 мл ДМФ добавляют гидрид натрия (620 мг, 26 ммоль). Смесь перемешивают при комнатной температуре в течение 1 ч и добавляют метилиодид (0,37 г, 2,59 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение суток. Добавляют воду и экстрагируют этилацетатом. Органическую фракцию промывают насыщенным водным хлоридом натрия. Смесь сушат над сульфатом натрия. Раствор концентрируют под вакуумом. Остаток очищают путем колоночной хроматографии (20% этилацетат в гексане) с получением титульного соединения в виде светло-желтого твердого вещества (0,13 г, 38%). 1H ЯМР (400 МГц, CD3CN)1,45 (с, 9H), 3,08 (с, 3H), 3,59 (т, J=5,6 Гц, 2H), 4,20 (т, J=5,6 Гц, 2H),5,07 (с, 1H), 7,38 (д, J=3,2 Гц, 1H), 7,61 (д, J=1,2 Гц, 1H). Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для трет-бутилового эфира [2-(6-фтор-4-иодпиридин-3-илокси)этил]метилкарбаминовой кислоты:- 15015574 Пример приготовления 80. (S)-трет-Бутил 1-(6-фтор-4-иодпиридин-3-ил)этил(метил)карбамат. С помощью хиральной ВЭЖХ (колонки 4,6150 мм Chiralpak AD-H, 5:95 3A/C7, 0,6 мл/мин 270 нм) разделяют трет-бутил-1-(6-фтор-4-иодпиридин-3-ил)этил(метил)карбамат (3,11 г, 8,48 ммоль), в первой фракции получают (R)-трет-бутил 1-(6-фтор-4-иодпиридин-3-ил)этил(метил)карбамат (1,09 г, 35%,99% опт.чист.) и во второй фракции - (S)-бутил-1-(6-фтор-4-иодпиридин-3-ил)этил(метил)карбамат(1,13 г, 36%, 99% опт.чист.). МС (ЭР) m/z 383 [M+1]+. Определяют абсолютную конфигурацию обоих энантиомеров с помощью спектроскопического анализа методом колебательного кругового дихроизма (Vibration Circular Dichroic, VCD). Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для (S)-трет-бутил-1-(6-фтор-4-иодпиридин-3-ил)этил(метил)карбамата: Пример приготовления 83. 4-Бензо[b]тиофен-7-ил-2-хлорпиридин. В колбе соединяют 7-бромбензо[b]тиофен (1,7 г, 12 ммоль), 2-хлор-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)пиридин (1,6 г, 7 ммоль), Pd(dppf)Cl2 (285 мг, 0,3 ммоль), 2-(ди-третбутилфосфин)бифенил (63 мг, 0,2 ммоль), карбонат натрия (2 М, 8 мл, 16 ммоль) и ТГФ (20 мл). Смесь нагревают до 100C в течение 3 ч. Разбавляют смесью хлороформ/изопропанол (3/1). Раствор промывают насыщенным водным хлоридом натрия. Сушат над сульфатом натрия. Концентрируют раствор под вакуумом, получают темный остаток. Очищают путем колоночной хроматографии (ДХМ 20% ТГФ в ДХМ) с получением титульного соединения (1,14 г, 66%) в виде желтого твердого вещества. МС (ЭР) m/z 246 [M+1]+. Пример приготовления 84. 4-Бензо[b]тиофен-7-ил-2-фтор-5-метилпиридин. В колбе соединяют 2-фтор-4-иод-5-метилпиридин (355 мг, 1,5 ммоль), 2-бензо[b]тиофен-7-ил 4,4,5,5-тетраметил[1,3,2]диоксаборолан (282 мг, 1,8 ммоль), Pd(dppf)Cl2 (61 мг, 0,07 ммоль), 2-(ди-третбутилфосфино)бифенил (13 мг, 0,04 ммоль), карбонат натрия (2 М, 1,5 мл, 3 ммоль) и ТГФ (10 мл). Смесь нагревают до 100C в течение 3 ч в масляной бане. Разбавляют смесью хлороформ/изопропанол (3/1). Раствор промывают насыщенным водным хлоридом натрия. Сушат над сульфатом натрия. Концентрируют под вакуумом, получают темный остаток. Очищают путем колоночной хроматографии (20% этилацетата в гексане) с получением титульного соединения (300 мг, 82%) в виде желтого масла. МС (ЭР) m/z 244 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 4-бензо[b]тиофен-7-ил-2-фтор-5-метилпиридина: Пример приготовления 95. 1-(4-(Бензо[b]тиофен-7-ил)пиридин-3-ил)этанол. Раствор 4-бензо[b]тиофен-7-илпиридин-3-карбальдегида (1,5 г, 6,27 ммоль в ТГФ (50 мл) охлаждают в круглодонной колбе на 250 мл до 0C и постепенно добавляют в раствор бромидметилмагния (2,38 г, 6,90 ммоль, 2,30 мл). Смесь перемешивают от 0C до комнатной температуры в течение 2 ч. Гидролизуют смесь, смешав с 1 н. HCl (200 мл). Доводят pH до 11 гидроксидом аммония. Экстрагируют смесью хлороформ/изопропанол (3/1, 200 мл). Промывают органическую фазу водой и насыщенным водным хлоридом натрия. Сушат над сульфатом натрия и концентрируют под вакуумом. Очищают путем жидкостной хроматографии (гексан 10% этилацетата в гексане, затем 20% ТГФ в ДХМ) с получением титульного соединения в виде желтого масла (1,00 г, 62%).MC (ЭР) m/z 256 [M+1]+. Пример приготовления 96. 3-(1-Азидоэтил)-4-(бензо[b]тиофен-7-ил)пиридин. Применяя методику, сходную с описанной выше для 5-(1-азидоэтил)-2-фторпиридина, с получением титульного промежуточного седиения из 1-(4-(бензо[b]тиофен-7-ил)пиридин-3-ил)этанола в виде коричневого масла (0,66 г, 93%). МС (ЭР) m/z 280 [M+1]+. Пример приготовления 97. 2-(4-(Бензо[b]тиофен-7-ил)-6-фторпиридин-3-ил)-2-метилпропан-1-амин. Тетрагидроборат натрия (296 мг, 7,83 ммоль) и тетрахлорид циркония (684 мг, 2,9 ммоль) растворяют в ТГФ (1 мл) до молочного раствора. Добавляют раствор 2-(4-бензо[b]тиофен-7-ил-6-фторпиридин 3-ил)-2-метилпропионитрила (580 мг, 1,96 ммоль) в ТГФ (20 мл) в атмосфере N2 при комнатной температуре. Смесь перемешивают при комнатной температуре в течение суток. Реакцию гасят путем переливания смеси в воду со льдом. Смесь экстрагируют смесью хлороформ/изопропанол (3/1). Подщелачивают водную фазу путем добавления разбавленного гидроксида аммония и фильтруют. Экстрагируют фильтрат хлороформом, сушат объединенную органическую фазу и концентрируют, получают бледно-желтое твердое вещество (500 мг, 84%). МС (ЭР) m/z 301 [M+1]+. Пример приготовления 98. 1-(4-(Бензо[b]тиофен-7-ил)пиридин-3-ил)этанамин.- 17015574 В раствор 3-(1-азидоэтил)-4-бензо[b]тиофен-7-илпиридина (0,66 г, 2,35 ммоль) в этаноле (10 мл),муравьиной кислоты (1,08 г, 23,54 ммоль) и гидразина (754,4 мг, 23,54 ммоль) в круглодонной колбе на 50 мл в ледяной бане добавляют никель Ренея (1,38 г, 23,5 ммоль). Смесь перемешивают при комнатной температуре в течение 1 ч и отфильтровывают никель Ренея. Разбавляют фильтрат гидроксидом аммония. Продукт реакции экстрагируют хлороформом. Органическую фазу промывают водой, сушат над сульфатом натрия и концентрируют с получением титульного соединения в виде коричневого масла (0,6 г, 100%). МС (ЭР) m/z 255 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 1-(4-(бензо[b]тиофен-7-ил)пиридин-3-ил)этанамина: Пример приготовления 101. трет-Бутил 1-(4-(бензо[b]тиофен-7-ил)пиридин-3-ил)этилкарбамат. В раствор 1-(4-бензо[b]тиофен-7-илпиридин-3-ил)этиламина (0,6 г, 2,36 ммоль) добавляют триэтиламин (477,4 мг, 4,72 ммоль) и ди-трет-бутилдикарбонат (1,03 г, 4,72 ммоль) в 1,4-диоксане (10 мл) и воде (5 мл). Смесь перемешивают при комнатной температуре в течение 1 ч. Смесь разбавляют хлороформом (100 мл), промывают насыщенным водным хлоридом натрия, сушат над сульфатом натрия и концентрируют. Очищают остаток путем жидкостной хроматографии (от 20% этилацетата в гексане до 10% метанола в ДХМ) с получением титульного соединения в виде желтого твердого вещества (0,84 г,67%). МС (ЭР) m/z 355 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для трет-бутил 1-(4-(бензо[b]тиофен-7-ил)пиридин-3-ил)этилкарбамата: Пример приготовления 105. трет-Бутил 1-(6-фтор-4-иодпиридин-3-ил)этил(2-фторэтил)карбамат. В раствор трет-бутилового эфира [1-(6-фтор-4-иодпиридин-3-ил)этил]карбаминовой кислоты (300 мг, 819 мкмоль) при 0C добавляют гидрид натрия (58,9 мг, 2,46 ммоль). Смесь перемешивают при температуре от 0C до комнатной в течение 1 ч. Добавляют 1-фтор-2-иодэтан (570 мг, 3,28 ммоль). Смесь перемешивают при комнатной температуре в течение 4 ч и затем при 50C в течение суток. Разбавляют смесью хлороформ/изопропанол (3/1, 100 мл) и промывают водой/насыщенным водным хлоридом натрия. Органическую фазу сушат над сульфатом натрия и концентрируют под вакуумом, получают сырой продукт. Сырой продукт очищают путем жидкостной хроматографии (20% этилацетат в гексане), получают 140 мг титульного соединения (41%). МС (ЭР) m/z 413 [M+1]+. Пример приготовления 106. 4-Бензо [b]тиофен-7-ил-3-метоксиметоксипиридин. Раствор A. Раствор 3-метоксиметоксипиридина (2,5 г, 18 ммоль) в диэтиловом эфире (90 мл) обрабатывают при -70C трет-бутиллитием (1,7 М в пентане, 10 мл, 18 ммоль) по каплям в течение более 10 мин. Смесь перемешивают при -70C в течение 40 мин и по каплям в течение более 5 мин добавляют раствор триизопропилбората (5 мл, 22 ммоль) в ТГФ (10 мл). Смесь перемешивают при -70C в течение 1 ч и затем удаляют ледяную баню и оставляют смесь медленно нагреваться до комнатной температуры. Раствор B. Раствор 7-бромбензо[b]тиофена (3,8 г, 18 ммоль), 2-(ди-трет-бутилфосфин)бифенила(268 мг, 0,90 ммоль), Pd(dppf)Cl2 (732 мг, 0,90 ммоль) в 1,4-диоксане (30 мл) обрабатывают 2 М водным карбонатом натрия (72 мл, 36 ммоль). Раствор нагревают до 80C, после чего раствор остывает до комнатной температуры. Раствор B по каплям обрабатывают раствором A в течение более 10 мин. Полученный раствор нагревают до 85C в течение 5 ч. Охлаждают смесь до комнатной температуры и разбавляют этилацетатом и водой. Промывают органическую фазу водой и насыщенным водным хлоридом натрия, сушат над сульфатом натрия, фильтруют и концентрируют под вакуумом. Остаток очищают путем колоночной хроматографии со 120 г силикагеля, элюируя с градиентом от ДХМ до этилацетата, с получением титульного соединения (3,8 г) содержащего некоторое количество исходного 3-метоксиметоксипиридина. 1H ЯМР (400 МГц, CDCl3)8,68 (с, 1H), 8,42 (д, J=4 Гц, 1H), 7,88 (д, J=8 Гц, 1H), 7,33-7,50 (м, 5H),5,12 (с, 2H), 3,36 (с, 3H). Пример приготовления 107. 2-Хлор-4-[7-(2-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин. В круглодонной колбе на 500 мл охлаждают раствор 4-бензо[b]тиофен-7-ил-2-хлорпиридина (13 г,53,1 ммоль) и триизопропилбората (20 г, 106 ммоль) в ТТФ (150 мл) при -70C в атмосфере азота. В охлажденный раствор добавляют диизопропиламид лития (2M в ТГФ, 53 мл, 106 ммоль) постепенно в течение 30 мин. Смесь перемешивают непрерывно в течение дополнительно 1 ч в охлаждающей бане. Смесь постепенно переносят в рефлюкс с раствором 2,4-дихлорпиримидина (12 г, 106 ммоль),Pd(dppf)Cl2 (2,2 г, 53 ммоль) и карбоната натрия (35 мл, 3 М, 106 ммоль) в ТГФ (150 мл) в течение более 30 мин. Перемешивают в рефлюксе в течение дополнительно 1 ч. Смесь охлаждают до комнатной температуры и разбавляют 500 мл хлороформа/изопропанола (3/1) и 200 мл воды. Полученное твердое вещество собирают путем фильтрации из смеси хлороформ/изопропанол/вода. Вещество промывают ДХМ и сушат под вакуумом. Фракции разделяют смесью хлороформ/изопропанол/вода. Органическую фазу промывают водой и насыщенным водным хлоридом натрия, сушат над сульфатом натрия и концентрируют под вакуумом, получают коричневый остаток. Остаток очищают путем жидкостной хроматографии(10% метанол в ДХМ) и получают дополнительный продукт. Соединяют две порции титульного соединения (13 г, 68%). МС (ЭР) т/z 358 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 2-хлор-4-[7-(2-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидина, используя соответствующие исходные материалы:- 20015574 Пример приготовления 126. 4-[2-(2-Хлор-5-фторпиримидин-4-ил)бензо[b]тиофен-7-ил]пиридин-3-ол. Раствор 2-хлор-5-фтор-4-[7-(3-метоксиметоксипиридин-4-ил)бензо[b]тиофен-2-ил]пиримидина (4 г,10 ммоль) в ТГФ (10 мл) обрабатывают 5 н. HCl (3 мл). Смесь перемешивают при комнатной температуре в течение 6 ч. Реакционную смесь концентрируют под вакуумом и разбавляют насыщенным водным бикарбонатом натрия и ДХМ. Фазы разделяют и фильтруют каждую фазу. Твердую фракцию органической фазы промывают ДХМ и получают названное соединение (300 мг) в виде светло-коричневого твердого вещества. Твердую фракцию водной фазы промывают водой и сушат с получением титульного соединение (300 мг) в виде светло-коричневого твердого вещества. Полученные вещества объединяют и получают титульное соединение (600 мг; 17%) в виде светло-коричневого твердого вещества. МС (ЭР) m/z 358 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 4-[2-(2-хлор-5-фторпиримидин-4-ил)бензо[b]тиофен-7-ил]пиридин-3-ола, используя соответствующие исходные материалы: Пример приготовления 128. 2-(2-(5-(Гидроксиметил)-1H-1,2,3-триазол-1-ил)этил)изоиндолин-1,3 дион. Смесь 2-(2-азидоэтил)изоиндолин-1,3-диона (12 г, 55,5 ммоль) и 2-пропин-1-ола (3,88 мл, 66,6 ммоль) в толуоле (50 мл) нагревают до 90C в закрытом реакторе в течение 3 дней. Охлаждают до комнатной температуры и собирают твердое вещество. Очищают путем колоночной хроматографии (от ДХМ до 2% метанола в ДХМ) и получают титульное соединение (первая фракция) в виде белого твердого вещества (4,7 г, 31%).MS (EI) m/z 273 [M+l]+. Пример приготовления 129. 2-(2-(4-(Иодметил)-1H-1,2,3-триазол-1-ил)этил)изоиндолин-1,3-дион. Смесь трифенилфосфина (0,29 г, 1,10 ммоль) и иода (0,28 г, 1,10 ммоль) в ДХМ (4 мл) перемешивают в течение 10 мин. Добавляют 1H-имидазол (0,12 г, 1,84 ммоль) и перемешивают в течение 10 мин. Добавляют 2-(2-(4-(гидроксиметил)-1H-1,2,3-триазол-1-ил)этил)изоиндолин-1,3-дион (0,2 г, 0,73 ммоль) и перемешивают в течение суток при комнатной температуре. Разбавляют ДХМ и промывают водой и насыщенным водным хлоридом натрия. Сушат над сульфатом натрия. Раствор концентрируют под вакуумом. Очищают путем хроматографии на колонках (от ДХМ до 20% этилацетата в ДХМ) и получают титульное соединение в виде желтого твердого вещества (0,22 г, 78%).MS (EI) m/z 383 [M+l]+. Пример приготовления 130. 2-(2-(4-Метил-1H-1,2,3-триазол-1-ил)этил)изоиндолин-1,3-дион. Смесь 2-(2-(4-(подметил)-1H-1,2,3-триазол-1-ил)этил)изоиндолин-1,3-диона (1 г, 2,62 ммоль) и 0,2 г 10% палладия на углероде в этаноле перемешивают (10 мл) в атмосфере водорода в течение суток. Фильтруют для удаления твердого вещества и концентрируют остаток. Его очищают путем колоночной хроматографии (от ДХМ до 20% этилацетата в ДХМ) и получают титульное соединение в виде желтого твердого вещества (0,5 г, 74%).MS (EI) m/z 257 [M+l]+. Пример приготовления 131. 2-(2-[1,2,3]Триазол-1-илэтил)изоиндол-1,3-дион. В круглодонную колбу на 5 л, оснащенную механической мешалкой, устройством для подачи азота и датчиком температуры, добавляют 1H-1,2,3-триазол (250 г, 3,51 моль), N-(2-бромэтил)фталимид (942 г,3,52 моль) и 1500 мл ДМФ; смесь охлаждают до 15C. Смесь перемешивают до тех пор, пока все твердые вещества практически не растворятся, и затем охлаждают в ледяной бане. Порциями на протяжении более 10 мин добавляют карбонат цезия (1145 г, 3,51 моль). Реакционная смесь экзотермично нагревается до 21C. Смесь оставляют перемешиваться и доходить до комнатной температуры в течение суток. Реакционную смесь переливают в колбу на 12 л, содержащую 8 л воды со льдом. Суспензию перемешивают в течение 30 мин и затем фильтруют, твердое вещество промывают 3 л воды. Сушат на воздухе в течение 2 ч. Проводят перекристаллизацию смеси региоизомеров с помощью 7 л абсолютного этанола. Твердое вещество отделяют путем фильтрации и сушат на воздухе. Снова проводят перекристаллизацию с помощью 16 л абсолютного этанола. Снова отделяют твердое вещество путем фильтрации и промывают ледяным этанолом (1000 мл). Вещество сушат в вакууме при 40C и получают титульное соединение в виде белого твердого вещества, 292,7 г (34%). МС (ЭИ) m/z 243 [M+l]+. Пример приготовления 132. 2-[1,2,3]Триазол-1-илэтиламин. В круглодонной колбе на 5 л, содержащей 2 л абсолютного этанола, растворяют 2-(2-[1,2,3]триазол 1-илэтил)изоиндол-1,3-дион (106 г, 437,59 ммоль). Смесь нагревают при перемешивании до 70C в атмо- 21015574 сфере азота; при этой температуре по каплям добавляют моногидрат гидразина (23 мл, 463,76 ммоль) в течение более 10 мин. Смесь становится гомогенной и приобретает желтый цвет. Спустя примерно 30 мин при этой температуре в реакционной смеси начинает образовываться твердое вещество и цвет постепенно становится со временем менее желтым. Спустя 7 ч нагрев убирают и охлаждают до комнатной температуры более 1 ч. Фильтруют сквозь диатомовую землю и промывают 1000 мл этанола. Выпаривают до полутвердого состояния. Растворяют в 2 л CH2Cl2, фильтруют через диатомовую землю и снова выпаривают. Разбавляют остаток в толуоле (1500 мл) и фильтруют через диатомовую землю для удаления нерастворимого светло-кормчневого твердого вещества. Выпаривают и помещают на сутки в вакуум. Полученное масло растворяют в 100 мл CH2Cl2 и снова фильтруют через слой диатомовой земли. Выпаривают и получают 43,9 г (90%) титульного соединения в виде мутного масла. МС (ЭР) m/z 112 [M+1]+. Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 2-[1,2,3]триазол-1-илэтиламина: Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для 5-фтор-4-[7-(2-фтор-5-метилпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-ил-(2-[1,2,3] триазол-1-илэтил)амина: Пример приготовления 143. 5-Фтор-4-[7-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)бензо[b] тиофен-2-ил]пиримидин-2-ил-(2-[1,2,3]триазол-1-илэтил)амин. В ДМСО (30 мл) соединяют [4-(7-бромбензо[b]тиофен-2-ил)-5-фторпиримидин-2-ил]-(2[1,2,3]триазол-1-илэтил)амин (1,5 г, 3,45 ммоль), бис(пинаколато)диборан (1,05 г, 4,14 ммоль), ацетат калия (1,02 г, 10,36 ммоль) и Pd(dppf)Cl2 (280 мг, 0,35 ммоль). Полученную смесь троекратно дегазируют и нагревают ее до 80C в течение суток. Смесь охлаждают до комнатной температуры и переливают ее в воду. Фильтруют для получения влажного твердого вещества, растворяют в смеси хлороформ/изопропанол (3:1, об.), промывают насыщенным водным хлоридом натрия и затем сушат над сульфатом натрия. Раствор концентрируют под вакуумом. Очищают путем хроматографии (от гексана до этилацетата) и получают титульное соединение (1,3 г, 81%). Следующие промежуточные соединения получают в соответствии с методикой, сходной с описанной для упомянутых ранее промежуточных соединий: Следующие промежуточные соединения получают в соответствии с методикой, описанной далее для В н-бутаноле (2 мл) в сосуде под давлением соединяют 2-хлор-5-фтор-4-[7-(2-фтор-5 метилпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин (200 мг, 0,54 ммоль) и 2-[1,2,3]триазол-1 илэтиламин (120 мг, 21,1 ммоль), возможно также применять в качестве растворителя диоксан, диоксанNMP (N-метилпирролидинон) и NMP сам по себе. Смесь нагревают на масляной бане до 120-150C в течение суток (или в микроволновом реакторе в течение 10-60 мин). Разбавляют смесью хлороформ/изопропанол (3/1). Промывают раствор насыщенным водным хлоридом натрия, сушат над сульфатом натрия, раствор концентрируют под вакуумом, получают темный остаток. Очищают его путем колоночной хроматографии (ДХМ 10% метанол в ДХМ) с получением титульного соединения в виде желтого твердого вещества (140 мг, 59%).MC (ЭР) m/z 450 [M+1]+. Следующие примеры получают в соответствии с методикой, сходной с описанной в предыдущем примере: Соединяют трет-бутиловый эфир (6-фтор-4-2-[5-фтор-2-(2-[1,2,3]триазол-1-илэтиламино)пиримидин-4-ил]бензо[b]тиофен-7-илпиридин-3-илметил)метилкарбаминовой кислоты (0,30 г, 0,51 ммоль),ледяную ТФУ (2,0 мл) и безводный ДХМ (2,2 мл). Перемешивают при комнатной температуре в течение 1 ч. Выпаривают растворители. Разбавляют полученный остаток с помощью ДХМ и промывают насыщенным раствором бикарбоната натрия, водой и насыщенным водным хлоридом натрия. Отделяют органическую фракцию и сушат ее над сульфатом магния. Фильтруют и концентрируют под вакуумом, получают сырой продукт. Очищают путем колоночной хроматографии [0,1% 2% 2 М аммиака в метаноле/ДХМ] с получением титульного соединения (0,20 г, 83%). В 2 мл смешанного растворителя из ДМФ (в других вариантах реализации - диоксан, ТГФ, ДМСО,и CH3CN) и воды (4/1, по объему) соединяют 5-фтор-4-[7-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2 ил)бензо[b]тиофен-2-ил]пиримидин-2-ил-(2-[1,2,3]триазол-1-илэтил)амин (0,105 г, 0,23 ммоль), 3-фтор 4-бром-1H-пиррол[2,3-b]пиридин (51 мг, 0,30 ммоль), восьмиводный гидроксид бария (0,21 г, 0,68 ммоль, в других воплощениях - карбонат натрия, карбонат калия, бикарбонат натрия), Pd(dppf)Cl2 (20 мг,0,025 ммоль). Реакционную смесь нагревают до 80C в течение 2,5 ч (или нагревают в микроволновой печи в течение 10-60 мин). Охлаждают до комнатной температуры. Разбавляют ее 50 мл смеси хлороформ/изопропанол (3:1, по объему), промывают ее водой и насыщенным водным хлоридом натрия и сушат над сульфатом магния. Удаляют органический растворитель, получают сырой продукт. Очищают путем колоночной хроматографии (гексанэтилацетат) с получением титульного соединения (0,05 г,47%). МС (ЭР) m/z 475 [M+1]+. Пример 15. N-(2-(1H-1,2,3-Триазол-1-ил)этил)-4-(7-(1H-пиррол[2,3-b]пиридин-4-ил)бензо[b]тиофен 2-ил)-5-фторпиримидин-2-амин. В 4 мл смешанного растворителя из ДМФ и воды (4/1, по объему) соединяют N-(2-(1H-1,2,3 триазол-1-ил)этил)-5-фтор-4-(7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензо[b]тиофен-2-ил)пиримидин-2-амин (0,28 г, 0,6 ммоль), 4-бром-1H-пиррол[2,3-b]пиридин (100 мг, 0,51 ммоль), восьмиводный гидроксид бария (0,48 г, 1,52 ммоль), Pd(dppf)Cl2 (40 мг, 0,05 ммоль). Реакционную смесь нагревают до 80C в течение 45 мин. Охлаждают ее до комнатной температуры. Разбавляют смесью хлороформ/изопропанол (3:1) 50 мл. Промывают водой и насыщенным водным хлоридом натрия и сушат над сульфатом магния. Удаляют органический растворитель и получают сырой продукт. Очищают путем колоночной хроматографии (гексанэтилацетат) с получением титульного соединения (0,17 г, 75%). МС (ЭР) m/z 457 [M+1]+. Следующие примеры получают в соответствии с методикой, сходной с описанной в предыдущем примере:
МПК / Метки
МПК: C07D 409/14, A61P 35/00, A61K 31/506
Метки: соединения, триазолиламинопиримидиновые
Код ссылки
<a href="https://eas.patents.su/30-15574-triazolilaminopirimidinovye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Триазолиламинопиримидиновые соединения</a>
Соединения-ингибиторы дипептидилпептидазы-iv, способы их получения, а также фармацевтические композиции, содержащие указанные соединения в качестве активного ингредиента

Номер патента: 12591
Опубликовано: 30.10.2009
Авторы: Хан Хее Оон, Кох Дзонг Сунг, Ким Хие Дзин, Коо Ки Донг, Ким Киоунг-Хее, Ким Мин-Дзунг, Хонг Санг Йонг, Ким Дзи Янг, Йео Донг-Дзун, Йим Хиеон Дзоо, Ким Сунгсуб, Ли Чанг-Сеок, Квон Ох Хван, Бу Сеонг Чеол, Ким Геун Тае, Ким Сунг Хо, Йеом Зи-Хо, Хур Гвонг-Чеунг, Лим Донгчул
МПК: A61P 3/10, A61K 31/452, A61K 31/444...
Метки: соединения, дипептидилпептидазы-iv, указанные, активного, содержащие, качестве, способы, соединения-ингибиторы, фармацевтические, ингредиента, композиции, получения, также
Формула / Реферат:
1. Соединение формулы (1) или его фармацевтически приемлемая соль где (А) А выбран из группы, состоящей из заместителей следующих формул со (2) по (7): где R1 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; и X представляет собой углерод или азот; где R2 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; где R3 представляет собой водород или...
Соединения 1,2,4,5-тетрагидро-3н-бензазепина, способ их получения и фармацевтические композиции, содержащие эти соединения

Номер патента: 14756
Опубликовано: 28.02.2011
Авторы: Пеглион Жан-Луи, Вийенов Николь, Дессинже Эмея, Гумен Бертран, Вилен Жан-Поль, Толлон Катрин, Шименти Стефано, Кеньяр Паскаль
МПК: C07D 223/16, A61P 9/00, A61K 31/55...
Метки: 1,2,4,5-тетрагидро-3н-бензазепина, эти, соединения, способ, фармацевтические, содержащие, получения, композиции
Формула / Реферат:
1. Соединение формулы (I)где R1представляет собой атом водорода или группу, выбранную из С3-С7 циклоалкила, бензила и линейного или разветвленного C1-C6алкила, алкильной группы, которая может быть насыщенной или ненасыщенной и может быть необязательно замещенной гидроксигруппой или С3-С7 циклоалкильной группой или одним или несколькими атомами галогена,R2, R3, R4 и R5могут быть одинаковыми или различаться, каждый выбирается из атома водорода или...
Фармкомпозиция, содержащая соединения с анти-ха активностью и соединения антагониста агрегации тромбоцитов, их применение, набор, содержащий эти соединения, способ лечения или профилактики заболеваний, сопутствующих тромбообразованию

Номер патента: 2475
Опубликовано: 27.06.2002
Авторы: Перроне Марк Х., Лидли Роберт Дж., Данвидди Кристофер Т., Юзан Андре, Кюродо Алан Х.
МПК: A61K 31/715, A61P 7/02
Метки: агрегации, активностью, лечения, применение, эти, фармкомпозиция, содержащая, тромбоцитов, способ, профилактики, тромбообразованию, заболеваний, соединения, антагониста, сопутствующих, анти-ха, содержащий, набор
Формула / Реферат:
1. Фармацевтическая композиция, содержащая фармацевтически пригодный носитель и фармацевтически эффективные количества соединения, обладающего анти-Ха активностью, и соединения антагониста агрегации тромбоцитов. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что соединение, обладающее анти-Ха активностью, является низкомолекулярно весовым гепарином. 3. Фармацевтическая композиция по п.2, отличающаяся тем, что низкомолекулярно весовой...
Диароматические соединения пропинила или диенила, промежуточные соединения, фармацевтические и косметические композиции на основе указанных соединений и применение косметических композиций.

Номер патента: 1060
Опубликовано: 30.10.2000
Автор: Бернардон Жан-Мишель
МПК: C07C 63/66, A61P 17/00, A61K 31/192...
Метки: композиции, косметических, композиций, косметические, соединений, применение, пропинила, промежуточные, указанных, основе, диенила, фармацевтические, соединения, диароматические
Формула / Реферат:
1. Диароматические соединения пропинила или диенила общей формулы (I) где R1 является (I) радикалом -СН3, (II) радикалом -СН2-O-R6, (III) радикалом -О-R6, (IV) радикалом -CO-R7, где R6 и R7 имеют значения, приведенные ниже, Аr является радикалом, отвечающим следующей формуле (а): где R5 имеет значения, приведенные ниже, Х является радикалом формулы или или где R8 и R9 имеют значения, приведенные ниже, R2 и R3,...
Четырехциклические конденсированные соединения с гетероатомами, замещенные арилом, промежуточные соединения, способы получения, композиции и методы лечения

Номер патента: 1649
Опубликовано: 25.06.2001
Автор: Гриз Тимоти А.
МПК: C07D 471/00, A61K 31/35, A61P 5/30...
Метки: промежуточные, конденсированные, арилом, замещенные, методы, соединения, гетероатомами, лечения, способы, композиции, получения, четырехциклические
Формула / Реферат:
1. Соединение формулы I или II где Y обозначает -О-, -S-, -СН2-, -СН2СН2-, -СН=СН- или -NR4-; В обозначает -СН2- или -СО-; R1 и R2 обозначают каждый независимо -Н, -ОН, -O(C1-C4алкил), -ОСОС6Н5, -ОСО(С1-С6алкил), -ОSО2(С4-С6алкил), OSO2СF3, Сl или F; n равно 1 или 2; W обозначает -СН2- или >С=O; R3 обозначает 1-пиперидинил, 2-оксо-1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 2-оксо-1-пирролидинил,...
Предыдущий патент: Применение тенектеплазы для лечения острого ишемического инсульта
Следующий патент: Закваска, способ ее получения и применение закваски
Случайный патент: Соединения галогеналкилгетероарилбензамида