Способ синтеза ингибиторов hmg-coa редуктазы





Формула / Реферат
1. Способ получения соединения формулы IX

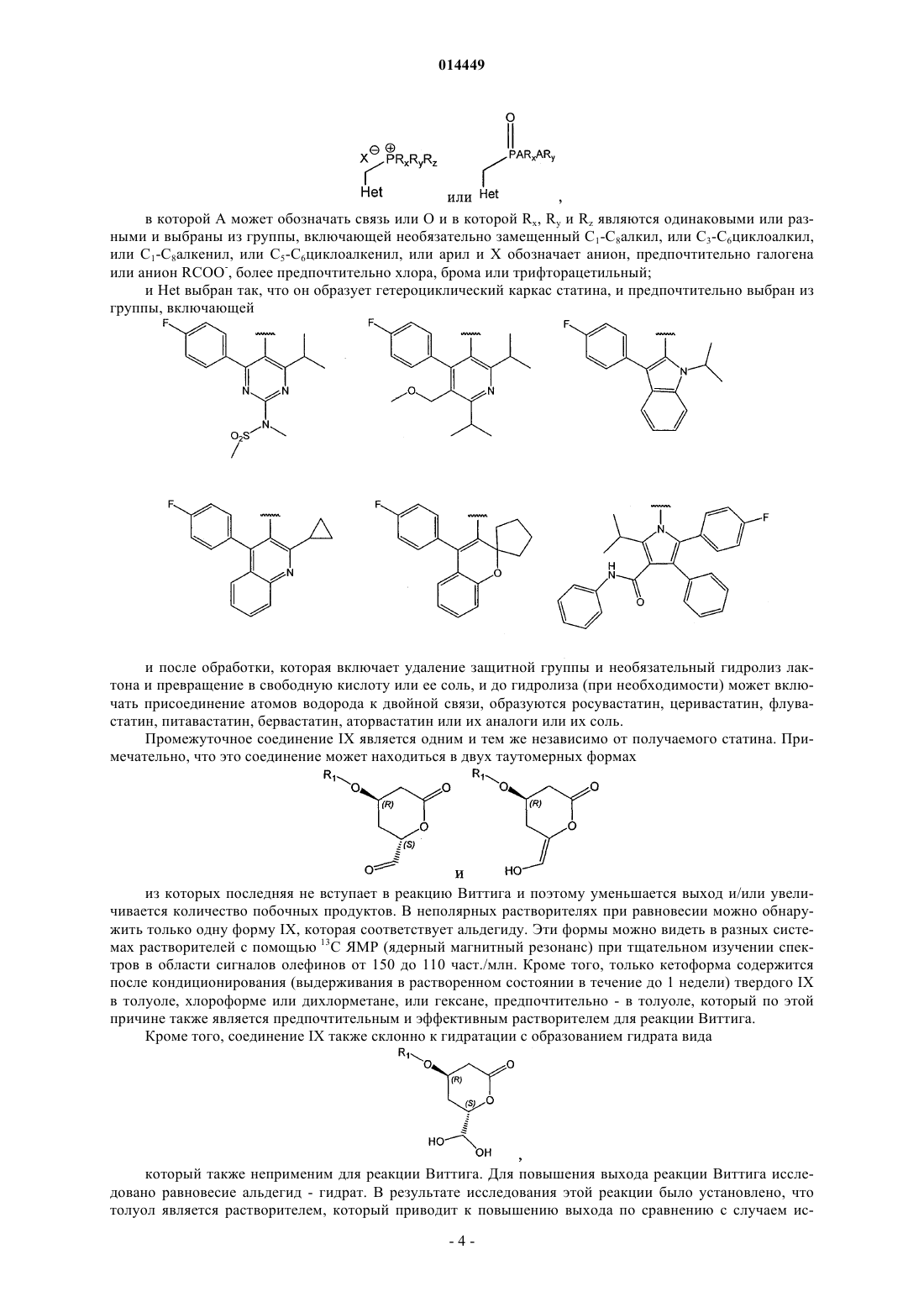
в которой R1 обозначает защитную группу,
из соединения формулы VII

в которой R4 обозначает (необязательно замещенный галогеном, С1-С8алкилом, арилом, С1-С8алкилокси или арилокси) С1-С4алкил.
2. Способ по предыдущему пункту, в котором соединение формулы VII

получают из соединения формулы VIa

в которой X" обозначает галоген, С1-С8алкилсульфонил или арилсульфонил.
3. Способ по п.1, в котором соединение формулы IX

в которой R1 является таким, как определено выше,
получают способом, включающим одну или более стадий, выбранных из:
а) превращения соединения формулы VII в соединение формулы VIII

b) превращения соединения формулы VIII путем окисления в соединение формулы IX

4. Способ по предыдущему пункту, который дополнительно включает стадии:
a) необязательно, превращения соединения формулы VIa, в которой X" обозначает С1-С8алкилсульфонил или арилсульфонил, в соединение формулы VIa, в которой X" обозначает галоген; и
b) превращения соединения формулы VIa

в которой X" обозначает галоген, С1-С8алкилсульфонил или арилсульфонил,
в соединение формулы VII

в которой R4 обозначает (необязательно замещенный галогеном, С1-С8алкилом, арилом, С1-С8алкилокси или арилокси) С1-С4алкил.
5. Способ по одному из пп.2 и 4, в котором соединение формулы VIa

в которой X" обозначает галоген и R1 обозначает защитную группу,
получают с помощью следующих стадий:
а) (необязательно) взаимодействие алкил-3(S)-гидрокси-4-хлорбутирата формулы I,
![]()
в которой R2 обозначает С1-С8алкил или С5-С7циклоалкил или, альтернативно, -COOR2также может образовать амид формулы -CONRaRb, в которой Ra и Rb могут независимо обозначать Н, С1-С8алкил или С5-С7циклоалкил, арил или совместно с N могут образовать гетероцикл,
с йодидом с образованием алкил-3(S)-гидрокси-4-йодбутирата формулы II
![]()
в которой R2 является таким, как определено выше;
b) введение защитной группы в соединение формулы II с получением защищенного производного формулы III

в которой R1 и R2являются такими, как определено выше;
с) взаимодействие соединения формулы III с винилмагнийгалогенидом в присутствии галогенида меди(I) и производного фосфита формулы

в которой все R', R" и R'" являются одинаковыми или разными и обозначают С1-С4алкил, С5-С7циклоалкил или арил,
с получением алкена формулы IV, в которой R1 и R2являются такими, как определено выше

d) гидролиз соединения формулы IV с получением соединения формулы V

e) взаимодействие соединения формулы V, в которой R1является таким, как определено выше, с источником галогена в присутствии NaHCO3 и
f) (необязательно) разделение смеси диастереоизомеров, полученной на предыдущей стадии.
6. Способ по п.4, в котором стадию b) проводят с использованием ацилирующего реагента, выбранного из группы, включающей NaOAc, LiOAc, KОАс, CsOAc, AgOAc, CuOAc, Mg(OAc)2, Ca(OAc)2, R4NOAc.
7. Способ по п.3, в котором стадию а) проводят путем дезацетилирования оловоорганическим соединением, выбранным из группы, включающей дибутилоловооксид или [t-Bu2SnOH(Cl)]2, или путем реакции с ферментом, выбранным из группы, включающей свиную панкреатическую липазу, липазу MY, липазу PS, липазу Al, липазу Candida и алкалазу, или с реагентом, выбранным из группы, включающей гуанидин и гуанидин/гуанидинийнитрат, HBF4´Et2O/MeOH и BF3´Et2O/MeCN, DBU/MeOH, гидразин/MeOH и гидразингидрат/ТГФ, цианид/МеОН, I2/МеОН, и/или где стадию b) проводят с помощью реакции окисления, выбранной из группы, включающей реакции окисления при катализе диметилсульфоксидом (окисление по Шверну: пара ДМСО-(COCl)2), методика Пфитцнера-Моффата: пара ДМСО-дициклогексилкарбодиимид (ДЦК), методика Парика-Деринга: пара ДМСО-SO3´Ру), реакции окисления при катализе N-оксоаммониевыми соединениями (пара (2,2,6,6-тетраметил-1-пиперидинилоксил (TEMPO) - окислитель), реакции окисления органическим гипервалентным соединением йода, выбранным из группы, включающей перйодинан Десса-Мартина (ПДМ) и о-йодоксибензойную кислоту (IBX или SIBX), реакции окисления содержащими хром(VI) окислителями, выбранными из группы, включающей реагент Коллинза (CrO3´Ру2), пиридинийдихромат (ПДХ) (пара ПДХ-активированные молекулярные сита 4Å), пиридинийхлорхромат (ПХХ), реакции окисления производными марганца, выбранными из группы, включающей MnO2 и BaMnO4, или реакции окисления тетра-н-пропиламмонийперрутенатом: Pr4N+ RuO4- (ТПАП).
8. Способ получения соединения формулы

или его соли, амида или лактона,
в которой Het выбран из группы, включающей

Включающий:
а) получение промежуточного продукта формулы VII

в которой R4 обозначает (необязательно замещенный галогеном, С1-С8алкилом, арилом, С1-С8алкилокси или арилокси) С1-С4алкил, и
b) получение промежуточного продукта формулы IX

в которой R1 обозначает защитную группу,
из промежуточного продукта формулы VII и
с) взаимодействие промежуточного продукта формулы IX с соединением формулы

в которой Rx, Ry и Rz являются одинаковыми или разными и выбраны из группы, включающей С1-С8алкил, или С3-С6циклоалкил, или С1-С8алкенил, или С5-С6циклоалкенил, или арил,
X- обозначает анион и
Het является таким, как определено выше, и
необязательно включающий одну или более последующих стадий, на которых соединение формулы X

в которой R1 и Het являются такими, как определено выше,
превращают в соединение формулы XI

в которой R1 и Het являются такими, как определено выше,
или его соль, амид или лактон.
9. Способ по предыдущему пункту, в котором соединение формулы VII

получают из соединения формулы VIa

в которой X" обозначает галоген, С1-С8алкилсульфонил или арилсульфонил.
10. Способ получения росувастатина, характеризующийся тем, что он включает стадии:
а) (необязательно) взаимодействие алкил-3(S)-гидрокси-4-хлорбутирата формулы I
![]()
в которой R2 обозначает С1-С8алкил или С5-С7циклоалкил или, альтернативно, -COOR2также может образовать амид формулы -CONRaRb, в которой Ra и Rb могут независимо обозначать Н, С1-С8алкил или С5-С7циклоалкил, арил или совместно с N могут образовать гетероцикл,
с йодидом с образованием алкил-3(S)-гидрокси-4-йодбутирата формулы II

в которой R2 является таким, как определено выше;
b) (необязательно) введение защитной группы в соединение формулы II с получением защищенного производного формулы III

в которой R1 и R2являются такими, как определено выше;
с) взаимодействие соединения формулы III с винилмагнийгалогенидом в присутствии галогенида меди(I) и фосфита формулы

в которой все R', R" и R'" являются одинаковыми или разными и обозначают С1-С4алкил, С5-С7циклоалкил или арил,
с получением алкена формулы IV

в которой R1 и R2являются такими, как определено выше;
d) гидролиз соединения формулы IV с получением соединения формулы V

e) взаимодействие соединения формулы V, в которой R1является таким, как определено выше, с источником галогена в присутствии NaHCO3 с получением соединения формулы VI;
f) (необязательно) разделение смеси диастереоизомеров, полученной на предыдущей стадии, с получением соединения формулы VIa

в которой X" обозначает галоген и R1 является таким, как определено выше;
g) превращение соединения формулы VIa в соединение формулы VII

в которой R1 является таким, как определено выше, и R4 выбран из группы, включающей (необязательно замещенный галогеном, или алкокси, или арилокси) С1-С4алкил;
h) превращение соединения формулы VII в соединение формулы VIII

в которой R1 является таким, как определено выше;
i) превращение соединения формулы VIII путем окисления в соединение формулы IX

в которой R1 является таким, как определено выше;
j) взаимодействие указанного соединения формулы IX с соединением формулы

в которой Rx, Ry и Rz являются одинаковыми или разными и выбраны из группы, включающей С1-С8алкил, или С3-С6циклоалкил, или С1-С8алкенил, или С5-С6циклоалкенил, или арил,
X- обозначает анион; и
k) удаление защитной группы R1, необязательно очистку и превращение полученного соединения в кальциевую соль.
11. Способ по любому из пп.9 и 11, в котором анионом X-является галогенидный или алканоатный анион.
12. Применение для синтеза статинов промежуточного продукта формулы

в которой R1 обозначает С1-С8триалкилсилил, С1-С8диалкиларилсилил, С1-С8алкилдиарилсилил, где алкилы могут быть одинаковыми или разными, и R4обозначает (необязательно замещенный галогеном, С1-С8алкилом, арилом, С1-С8алкилокси или арилокси) С1-С4алкил.
13. Соединение формулы

в которой R1 обозначает С1-С8триалкилсилил, С1-С8диалкиларилсилил, C1-С8алкилдиарилсилил, где алкилы и арилы могут быть одинаковыми или разными, и R4 обозначает (необязательно замещенный галогеном, С1-С8алкилом, арилом, С1-С8алкилокси или арилокси) С1-С4алкил.
14. Соединение формулы

в которой R1 обозначает С1-С8триалкилсилил, С1-С8диалкиларилсилил, C1-С8алкилдиарилсилил, где алкилы и арилы могут быть одинаковыми или разными.
Текст
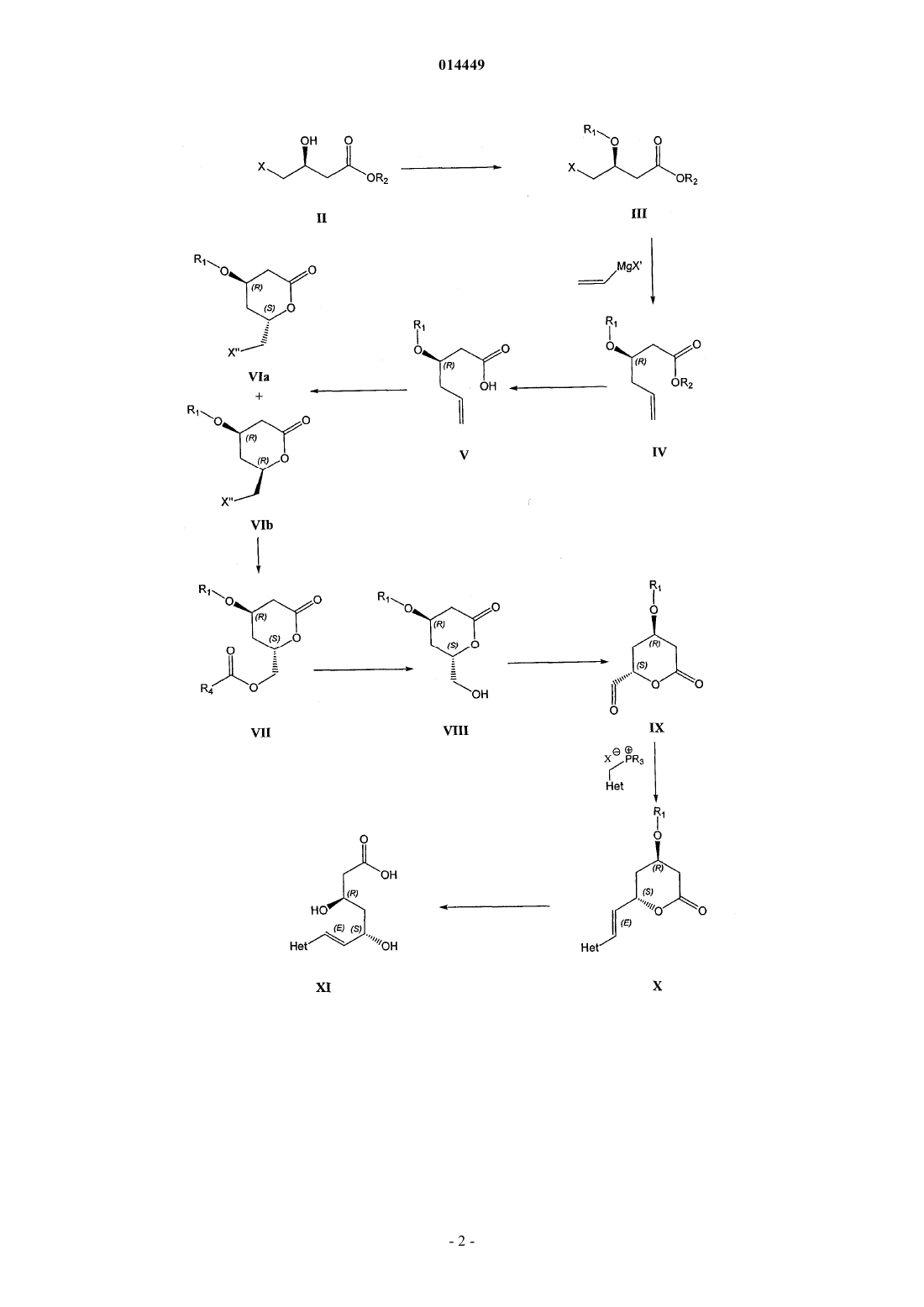
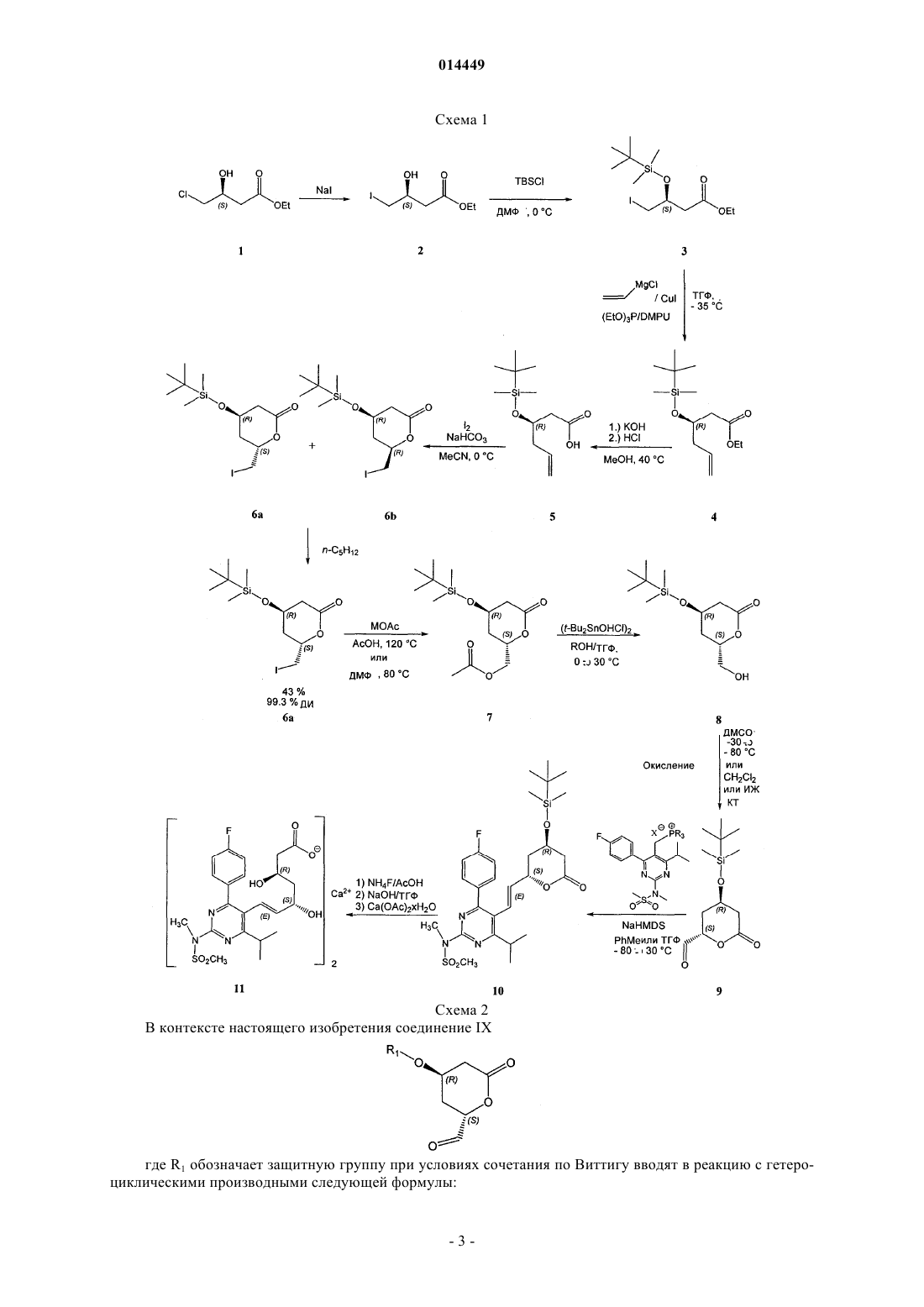
СПОСОБ СИНТЕЗА ИНГИБИТОРОВ HMG-CoA РЕДУКТАЗЫ В изобретении описан новый синтез статинов с использованием реакции Виттига между гетероциклическим ядром статина и лактонизованной боковой цепью, уже обладающей необходимой стереохимической конфигурацией. Разделение диастереоизомеров проводят в начале синтеза. 014449 Область техники, к которой относится изобретение Настоящее изобретение относится к способу получения ингибиторов HMG-СоА редуктазы, также известных под названием "статины", в особенности росувастатина. Точнее настоящее изобретение относится к общим промежуточным продуктам, которые можно использовать для получения всех статинов. Уровень техники Предполагают, что причиной биологической активности статинов, типичные представители которых могут быть выбраны из группы, включающей росувастатин, церивастатин, аторвастатин, флувастатин, питавастатин, бервастатин и их аналоги или правастатин, симвастатин, ловастатин и их аналоги,является их структура, содержащая соответственно фрагмент гептеновой или гептановой кислоты (свободной кислоты, соли или лактона), связанный с ароматическим ядром, и в особенности их стереохимическая конфигурация, в особенности конфигурация хиральных атомов, представленная на приведенной ниже формуле их типичного примера, аниона росувастатина Методики синтеза статинов, например, описаны в US 4625039 и WO 05/047276. Описание изобретения Настоящее изобретение относится к новому способу синтеза статинов, при использовании которого один хиральный центр уже содержится в легко доступном исходном соединении, а другой вводят в начале синтеза. Согласно изобретению предложен регулируемый способ, в котором фрагмент IX, обладающий необходимой стереохимической конфигурацией где R1 обозначает защитную группу; вводят в реакцию с подходящей фосфониевой солью или фосфитом каркаса статина. Важно синтезировать этот фрагмент из (S) предшественника стереоселективным образом. Это обеспечивается с помощью реакции с использованием нового промежуточного продукта VII где R4 обозначает (необязательно замещенный) алкил. Таким образом, единственную стадию выделения энантиомера проводят при разделении соединений где X" обозначает галоген или алкил- или арилсульфонил; которую проводят в начале синтеза. Кроме того, согласно изобретению синтез этих соединений-предшественников проводят так, чтобы обеспечить высокий выход необходимого (4R,6S)-дисатереоизомера. Общий путь синтеза представлен на приведенной ниже схеме 1 и конкретный пример использования силильной защитной группы и пример синтеза Са соли росувастатина представлен на схеме 2. Схема 2 В контексте настоящего изобретения соединение IX где R1 обозначает защитную группу при условиях сочетания по Виттигу вводят в реакцию с гетероциклическими производными следующей формулы: в которой А может обозначать связь или О и в которой Rx, Ry и Rz являются одинаковыми или разными и выбраны из группы, включающей необязательно замещенный С 1-С 8 алкил, или С 3-С 6 циклоалкил,или С 1-С 8 алкенил, или C5-С 6 циклоалкенил, или арил и X обозначает анион, предпочтительно галогена или анион RCOO-, более предпочтительно хлора, брома или трифторацетильный; и Het выбран так, что он образует гетероциклический каркас статина, и предпочтительно выбран из группы, включающей и после обработки, которая включает удаление защитной группы и необязательный гидролиз лактона и превращение в свободную кислоту или ее соль, и до гидролиза (при необходимости) может включать присоединение атомов водорода к двойной связи, образуются росувастатин, церивастатин, флувастатин, питавастатин, бервастатин, аторвастатин или их аналоги или их соль. Промежуточное соединение IX является одним и тем же независимо от получаемого статина. Примечательно, что это соединение может находиться в двух таутомерных формах из которых последняя не вступает в реакцию Виттига и поэтому уменьшается выход и/или увеличивается количество побочных продуктов. В неполярных растворителях при равновесии можно обнаружить только одну форму IX, которая соответствует альдегиду. Эти формы можно видеть в разных системах растворителей с помощью 13 С ЯМР (ядерный магнитный резонанс) при тщательном изучении спектров в области сигналов олефинов от 150 до 110 част./млн. Кроме того, только кетоформа содержится после кондиционирования (выдерживания в растворенном состоянии в течение до 1 недели) твердого IX в толуоле, хлороформе или дихлорметане, или гексане, предпочтительно - в толуоле, который по этой причине также является предпочтительным и эффективным растворителем для реакции Виттига. Кроме того, соединение IX также склонно к гидратации с образованием гидрата вида который также неприменим для реакции Виттига. Для повышения выхода реакции Виттига исследовано равновесие альдегид - гидрат. В результате исследования этой реакции было установлено, что толуол является растворителем, который приводит к повышению выхода по сравнению с случаем ис-4 014449 пользования ТГФ (тетрагидрофуран), при использовании которого в условиях равновесия примерно половина соединения находится в форме альдегида и половина - в форме гидрата и поэтому ТГФ, который обычно используют в качестве растворителя для реакции Виттига, непригоден, и авторы настоящего изобретения установили, что в хлорированных растворителях, таких как хлороформ, дихлорметан, в гексане и предпочтительно - толуоле равновесие смещено в сторону альдегида. Настоящее изобретение относится к простому способу получения промежуточного продукта IX изIVa, который, в свою очередь, получают только за 5 стадий из имеющегося в продаже (S)-этил-3 гидрокси-4-хлорбутирата. Точнее,(4R,6S)-4-(трет-бутилдиметилсилилокси)-6-(йодметил)тетрагидропиран-2-он (6 а) при суммарном выходе искомого стереоизомера, равном 23-31%, который намного выше, чем выход для известных методик. Промежуточное соединение IX, применяющееся при осуществлении способа, предлагаемого в настоящем изобретении, получают из соединения VIa (4R,6S)-4-(защищенная гидроксигруппа)-6(галогенметил)-тетрагидропиран-2-он или его производного (X" = алкилсульфонил или арилсульфонил) путем превращения через соединение формулы VII (сложный эфир, такой как пивалоат, хлорацетат, дихлорацетат, трихлорацетат, трифторацетат, метоксиацетат, трифенилметоксиацетат, феноксиацетат и его производные, фенилацетат и его производные, дифенилацетат, 3-фенилпропионат, пентеноат, 4 оксопентеноат, пентеноат, предпочтительно - ацетат) в соединение формулы VIII и окисления в соединение формулы IX. Это превращение в соединение формулы VII, в которой R4 обозначает необязательно замещенный алкил, необходимо, поскольку попытки прямого превращения галогенпроизводного в гидроксипроизводное приводили или к разложению, или к раскрытию лактонного кольца. Образование необходимой стереохимической конфигурации обеспечивается на последней стадии синтеза промежуточного соединения VI, которое получают катализируемой галогеном циклизацией соединения V с использованием молекулярных галогенов, таких как йод, бром или хлор, в качестве источника галогенсодержащих электрофилов. Для этой реакции также можно использовать альтернативные источники галогенов. Чаще всего используют галогениды или оксогалогениды щелочных или щелочноземельных металлов, такие как KI, KI3, Ca(OCl)2, интергалогены, такие как монохлорид йода (I-Cl), монобромид йода (I-Br), которые являются более реакционноспособными, чем элементарный йод, и реагенты, содержащие галоген(I), такие как йодонийацетат (I-ОАс), N-йодсукцинимид (NIS), Nбромсукцинимид (NBS), биспиридинйодонийтетрафторборат (Py2IBF4). Для проведения реакции циклизации также применимы гипервалентные галогенидные электрофилы, такие как диацетоксийодбензол,бис(трифторацетокси)йодбензол, гидрокси(тозилокси)йодбензол (реагент Козера). Однако для превращения V в смесь (R,S)- и (R,R)-диастереоизомеров лактона VIa и VIb предпочтительно использовать молекулярный йод. Обычно реакцию проводят таким образом, чтобы (R,S)-диастереоизомер образовывался в большем количестве, чем (R,R)-диастереоизомер, и при необходимости (R,S)-диастереоизомер выделяют из смеси и получают оптически чистое соединение VIa. X" также может быть замещенным, так чтобы X" являлся подходящим заместителем, предпочтительно - галогеном, цианогруппой, алкилсульфонилом или арилсульфонилом, наиболее предпочтительно - йодом. Соединение V можно получить из соединения формулы III по реакции с подходящим реагентом Гриньяра (винилмагнийгалогенидом) в котором X' обозначает галоген, предпочтительно хлор, предпочтительно в присутствии ортофосфита и галогенида меди(I), что приводит к соединению формулы IV, которое гидролизуют в соединение формулы V. Для всех синтезов подходящим исходным веществом является алкил-3(S)-гидрокси-4-хлорбутиратI, в котором R2 предпочтительно обозначает С 1-С 8 алкил или С 5-С 7 циклоалкил, который необязательно может быть замещенным, предпочтительно алкилом или арилом, альтернативно исходным веществом может быть производное I, где например, -COOR2 может представлять собой амид формулы -CONRaRb, в которой Ra и Rb могут независимо обозначать Н, необязательно замещенный С 1-С 8 алкил или С 5 С 7 циклоалкил, арил или вместе с атомом N образовывать гетероцикл. Термин "замещенный" в настоящем описании означает, что замещенный фрагмент содержит один или большее количество заместителей, которые предпочтительно выбраны из группы, включающей ацил, алкил, алкенил, циклоалкил, циклоалкенил, арил, гетероарил, галоген, нитрогруппу, аминогруппу, алкоксигруппу. Если не указано иное,то в настоящем описании алкил и арил предпочтительно означают алкил, содержащий до 12 атомов углерода, предпочтительно содержащий 1, 2, 3, 4, 5, 6 или 7 атомов углерода, и арил, содержащий до 3 конденсированных ароматических колец, которые могут содержать один или большее количество гетероатомов, более предпочтительно фенил, который может быть дополнительно замещенным. Однако можно использовать любое галогенпроизводное или аналогичное соединение. Предпочтительно использовать йодсодержащее соединение или превратить указанное соединение в его йодпроизводное формулы II, в которой X обозначает I и R2 является таким, как определено выше. На следующей стадии группу ОН соединения формулы II защищают с помощью любого подходящего защищающего реагента и получают соединение формулы III, в которой R1 обозначает подходящую защитную группу, предпочтительно силил, более предпочтительно С 1-С 8 триалкилсилил, С 1 С 8 диалкиларилсилил, С 1-С 8 алкилдиарилсилил, где алкилы могут быть одинаковыми или разными, предпочтительно, если арил представляет собой фенил и алкилы содержат от 1 до 4 атомов С. Описанный выше способ приводит к получению статинов, выбранных из группы, включающей росувастатин, церивастатин, флувастатин, питавастатин, бервастатин, аторвастатин и их аналоги, которые можно включить в фармацевтическую композицию. Эти статины предпочтительнее статинов, полученных альтернативными способами, в которых разделение стереоизомеров проводят на более поздних стадиях. Известно, что стереоизомеры трудно удалять, однако методики очистки, использующиеся после проводимого вначале отделения соединения формулы VIa и VIb все же дают возможность удаления части нежелательных стереоизомеров. Поэтому пациенты, которым вводят указанные композиции, получит меньше нежелательных стереоизомеров, что приведет к уменьшению количества побочных эффектов. В одном варианте осуществления настоящего изобретения получают соединение (4R,6S)-4-(третбутилдиметилсилилокси)-6-(йодметил)тетрагидропиран-2-он (6 а), соответствующее общей формуле VIa,в которой R1 обозначает трет-бутилдиметилсилил, и превращают в подходящий предшественник статина. Например, (6 а) можно превратить в (2S,4R)-4-(трет-бутилдиметилсилилокси)-6-оксотетрагидро-2Hпиран-2-карбальдегид (9), соответствующий общей формуле IX, в которой R1 обозначает третбутилдиметилсилил, который затем можно ввести в реакцию сочетания с подходящей гетероциклической системой, в предпочтительном варианте осуществления - с пиримидиновой системой, и после удаления защитной группы и раскрытия лактонового кольца и превращения в соль, в предпочтительном варианте осуществления образуется росувастатин. Одна методика получения (6 а) с использованием метил-2-цианоацетата в качестве исходного вещества описана в публикации J. Chem. Soc, Perkin Trans. 1, 1991, 133-140. Хлорсодержащий аналог (соединение формулы VIa, в которой X" обозначает С 1) можно получить по ферментативной методике. 3(R)(трет-Бутилдиметилсилилокси)-5-гексеноат (4), описанный в публикации J. Antibiot. 2002, 55, 147-154,можно превратить в (R)-3-(трет-бутилдиметилсилилокси)-5-гексеновую кислоты (5) по методике, описанной в публикации Tetrahedron Lett. (43)2002,4381-4384. На первой стадии синтеза (S)-этил-3-гидрокси-4-йодбутират (2) получают из этил-3(S)гидроксихлорбутирата (1) по реакции с NaI. Подходящие растворители выбраны из числа амидов, предпочтительно - выбраны из группы, включающей: N,N-диметилформамид - (ДМФ), N,N-диметилацетамид(ДМСО); ацетонитрил; низшие спирты; кетоны, предпочтительно - ацетон. Реакцию можно проводить при температуре от 58 до 90 С. Предпочтительно при 60 С и проводить в течение от половины дня до большего количества дней, предпочтительно в течение от 41 (при 90 С) до 120 ч (при 60 С). На второй стадии вводят защитную группу и таким образом получают (S)-этил-3-(третбутилдиметилсилилокси)-4-йодбутират(3) из этил-3(S)-гидрокси-4-йодбутирата и третбутил(хлор)диметилсилана (TBSC1). Содержание последнего может составлять менее 1,5 моль на 1 моль бутирата. Реакцию предпочтительно проводить в присутствии основания, выбранного из группы, включающей амины, имидазолы и пиридины, предпочтительно - имидазола в растворителях, таких как амиды(ДМФ, ДМА, ГМФТА, NMP, ДМПМ, ТММ), ДМСО, нитрилы (ацетонитрил), хлорированные углеводороды (дихлорметан, хлороформ), ароматические углеводороды (толуол), предпочтительно - в ДМФ. Реакцию можно проводить при температуре от 0 до 10 С и предпочтительно проводить в присутствии NaI. Предпочтительно - при 0 С. Реакцию проводят в течение от 1 ч до дня, предпочтительно в течение от 12 до 17 ч. На третьей стадии этил-3(R)-(трет-бутилдиметилсилилокси)-5-гексеноат (4) получают из винилмагнийгалогенида и (S)-этил-3-(трет-бутилдиметилсилилокси)-4-йодбутирата, используют растворители,такие как амиды (ДМФ, ДМА, ГМФТА, NMP, ДМПМ, ТММ) или ДМСО, предпочтительно N,N'диметилприпиленмочевину (ДМПМ), предпочтительно в присутствии галогенида меди, предпочтительно - CuI и ортофосфита формулы в которой все R', R" и R'" являются одинаковыми или разными и обозначают С 1-С 4 алкил, С 5 С 7 циклоалкил или арил, который необязательно может быть замещенным, предпочтительно - С 1 С 4 алкил, фенил, бензил, наиболее предпочтительно - Р(OEt)3. Реакцию можно проводить при температуре от -45 до -25 С. Предпочтительно - при -40 С. Реакцию можно проводить с винилмагнийхлоридом,бромидом или йодидом, предпочтительно с винилмагнийхлоридом. Реакцию проводят в течение до 1 дня, предпочтительно в течение от 3 до 5 ч и затем останавливают путем прибавления насыщенного водного раствора NH4Cl при температуре от -10 до 0 С. Неочищенный продукт экстрагируют несмешивающимся с водой растворителем и органический раствор промывают разбавленными кислотами, такими как H2SO4, HCl, Н 3 РО 4 и т.п. На каждой из этих трех стадий продукты обычно выделяют путем экстракции несмешивающимися с водой растворителями, такими как простые эфиры, такие как: Et2O (диэтиловый эфир), i-Pr2O (диизопропиловый эфир), t-BuMeO (трет-бутилметиловый эфир), или алканы, такие как пентан, гексан, гептан,или хлорированные углеводороды, такие как метиленхлорид, предпочтительно с помощью t-BuMeO. Продукт можно очистить перегонкой в вакууме при подходящей температуре (т.е. 70-90 С на первой стадии, 70-95 С на второй стадии, 55-80 С на третьей стадии) и давлении, равном 0,100-0,500 мбар. На четвертой стадии пока еще не выделенное соединение, (R)-3-(трет-бутилдиметилсилилокси)-5 гексеновую кислоту (5), получают гидролизом этил-3(R)-(трет-бутилдиметилсилилокси)-5-гексеноата щелочью в растворителе, таком как МеОН. Реакцию можно проводить при температуре от 0 до 80 С. Предпочтительно - при 40 С. Реакцию можно проводить в спиртах, ТГФ, амидных растворителях или в смеси этих растворителей с водой. Предпочтительно - в спиртах. Реакцию проводят в течение минут или часов, предпочтительно в течение от 0,5 до 3 ч. Гидролиз сложного эфира можно проводить с использованием NaOH, КОН, LiOH, CsOH, Ca(OH)2 или Ва(ОН)2 в качестве основания. Предпочтительно - с помощью КОН. После гидролиза подкисление до рН 2 можно проводить разбавленными кислотами, такими как HCl, H2SO4, H3PO4 и т. п. Предпочтительно - с помощью HCl. И в этом случае подходящей методикой выделения является экстракция простыми эфирами и алканами, описанная выше, предпочтительно с помощью t-BuMeO. На пятой стадии (R)-3-(трет-бутилдиметилсилилокси)-5-гексеновую кислоту (5) с помощью йода превращают в смесь (4R,6S)-4-(трет-бутилдиметилсилилокси)-6-(йодметил)-тетрагидропиран-2-она и(4R,6R)-4-(трет-бутилдиметилсилилокси)-6-(йодметил)тетрагидропиран-2-она (6 а и 6b) в виде бледножелтого твердого вещества. Это твердое вещество перекристаллизовывают, предпочтительно - несколько раз, из н-пентана и получают (4R,6S)-4-(трет-бутилдиметилсилилокси)-6-(йодметил)тетрагидропиран-2 он (ДИ 99,3 %) (6 а) в виде бесцветных иголок с выходом 43%. Эта простая стадия придает молекулам необходимую хиральность. Реакцию можно проводить при температуре от -10 до 10 С. Предпочтительно при 0 С. И в этом случае можно использовать экстракцию простыми эфирами и алканами, как и выше,предпочтительно с помощью t-BuMeO. Оптически чистое соединение, (4R,6S)-4-(третбутилдиметилсилилокси)-6-(йодметил)тетрагидропиран-2-он, можно выделить в промышленном масштабе с помощью ВЭЖХ (высокоэффективная жидкостная хроматография). Диастереоизомеры (в предпочтительном варианте осуществления начальным является соотношение 6 а:6b = 4:1) можно разделить при комнатной температуре на колонке с диоксидом кремния с нормальной фазой (PHENOMENEX 4,6150 мм, dp = 5 мкм) с использованием смесей гексана с t-BuMeO разного состава в качестве подвижной фазы. Выражение "оптически чистое соединение" означает, что средний диастереоизомерный избыток(ДИ) составляет более 96 %, предпочтительно более 99 %, более предпочтительно более 99,7 % (ДИ 99% означает, что определенное с помощью ВЭЖХ среднее соотношение составляет 99,5 %/0,5 %. Оптически чистое соединение, (4R,6S)-4-(трет-бутилдиметилсилилокси)-6-(йодметил)тетрагидропиран-2-он, также альтернативно получают из (4R,6S)-4-(трет-бутилдиметилсилилокси)-6-(хлорметил)-тетрагидропиран-2 она (VIa, Х"=Cl), который можно получить из не содержащего защитной группы (4R,6S)-6-(хлорметил)4-гидрокситетрагидропиран-2-она. (4R,6S)-6-(Хлорметил)-4-гидрокситетрагидропиран-2-он можно получить с помощью ферментативной однореакторной тандемной альдольной реакции, катализируемой дезоксирибозо-5-фосфатальдолазой (ДЕРА) с последующей стадией химического окисления, как описано в публикации Proc. Natl. Acad. Sci. USA 2004, 101, 5788-5793. Аналогичную методику можно использовать, если R1 обозначает защитную группу, не являющуюся приведенной в качестве примера третбутилдиметилсилильной. На шестой стадии реакции 2S,4R)-4-(трет-бутилдиметилсилилокси)-6-оксотетрагидро-2H-пиран 2-ил)метил ацетат(4R,6S)-4-(трет-бутилдиметилсилилокси)-6-(йодметил)тетрагидропиран-2-она. Реакцию можно проводить при температуре от 0 до 130 С. Предпочтительно при 120 С. Реакцию можно проводить в амидных растворителях, выбранных из группы, включающей(ДМСО) и уксусную кислоту (АсОН). Предпочтительно - в АсОН. Реакцию проводят в течение до одного дня, предпочтительно - в течение от 1 до 17 ч. Ацилирование (замену йода) можно проводить с помощью ацилирующего реагента, выбранного из группы, включающей NaOAc, KOAc, LiOAc, CsOAc,AgOAc, CuOAc, Са(ОАс)2, Mg(OAc)2 и R4NOAc, использующегося в качестве нуклеофильного реагента. Предпочтительно - с помощью LiOAc или AgOAc. Выделение неочищенного продукта путем экстракции можно проводить с помощью AcOEt, простых эфиров и алканов, как и выше, предпочтительно - с помощью t-BuMeO. На седьмой стадии реакции 2S,4R)-4-(трет-бутилдиметилсилилокси)-6-оксотетрагидро-2H-пиран 2-ил)метил ацетат (7) дезацетилируют с получением (4R,6S)-4-(трет-бутилдиметилсилилокси)-6(гидроксиметил)-тетрагидропиран-2-она (8). Реакцию можно проводить с использованием гидроксидов щелочных металлов, но предпочтительным является наиболее селективный реагент. Дезацетилирование также можно проводить с помощью ферментов: свиной панкреатической липазы, липазы MY, липазы PS,липазы Al, липазы Candida и алкалазы, или реагентов, выбранных из группы, включающей гуанидин и гуанидин/гуанидинийнитрат, HBF4Et2O/MeOH и BF3Et2O/MeCN, DBU/MeOH, гидразин/МеОН и гидразингидрат/ТГФ, цианид/МеОН, I2/МеОН или оловосодержащих катализаторов, таких как димеры диалкилхлорстаннилгидроксидов, такие как [t-Bu2SnOH(Cl)]2. Димеры диалкилхлорстаннилгидроксидов являются предпочтительными и [t-Bu2SnOH(Cl)]2 является наиболее предпочтительным. Реакцию с использованием [t-Bu2SnOH(Cl)]2 можно проводить при температуре от 0 до 40 С. Предпочтительно - при 25 С. Реакцию можно проводить в спиртах, таких как: МеОН, EtOH, i-PrOH, или в смесях этих спиртов с простыми эфирами, такими как ТГФ, Et2O, i-Pr2O, t-BuMeO. Для реакции дезацетилирования можно использовать 5-15 мол.% оловосодержащего катализатора. Реакцию проводят в течение до 1 дня, предпочтительно в течение 4-17 ч. Выделение продукта можно провести путем кристаллизации. Для этой цели,как и выше, можно использовать алканы или простые эфиры, предпочтительно гексан. На восьмой стадии реакции (2S,4R)-4-(трет-бутилдиметилсилилокси)-6-оксотетрагидро-2R-пиран 2-карбальдегид(4R,6S)-4-(трет-бутилдиметилсилилокси)-6-(гидроксиметил)тетрагидропиран-2-она (8) с помощью подходящей методики окисления, такой как методики окисления при катализе диметилсульфоксидом (окисление по Шверну: пара ДМСО-(COCl)2), методика ПфитцнераМоффата: пара ДМСО-дициклогексилкарбодиимид (ДЦК), методика Парика-Деринга: пара ДМСОSO3 Ру), методики окисления при катализе N-оксоаммониевыми соединениями, пара (2,2,6,6 тетраметил-1-пиперидинилоксил (TEMPO) - окислитель), методики окисления органическим гипервалентным соединением йода, таким как перйодинан Десса-Мартина (ПДМ) или о-йодоксибензойная кислота (IBX или SIBX), методики окисления содержащими хром(VI) окислителями, такими как реагент Коллинза (CrO3 Ру 2), пиридинийдихромат (ПДХ) (пара ПДХ-активированные молекулярные сита 4),пиридинийхлорхромат (ПХХ), методики окисления производными марганца, такими как MnO2 иBaMnO4, методики окисления тетра-н-пропиламмонийперрутенатом: Pr4N+RuO4- (ТПАП). Реакции можно проводить при температуре от 0 до 40 С (перйодинан Десса-Мартина и пара ПДХ-активированные молекулярные сита 4) и от -80 до -40 С (окисление по Шверну). Реакцию можно проводить в CHCl3,CH2Cl2, ионной жидкости (ИЖ), такой как 1-бутил-3-метилимидазолийтетрафторборат (BMIMBF4) или ДМСО. Реакцию проводят в течение 1-24 ч. Выделение неочищенного продукта путем экстракции можно проводить с помощью AcOEt, простых эфиров и алканов, как и выше. Предпочтительно с помощьюPhMe, MTBE или AcOEt. Аналогичные условия проведения реакции также можно использовать в способах, входящих в объем настоящего изобретения, когда R1 не обозначает использующийся в настоящем изобретении третбутилдиметилсилил и R2 не обозначает использующийся в настоящем изобретении Et. В связи с этим длительность описанных в настоящем изобретении реакций может быть изменена и обычно она зависит от условий проведения реакции, в особенности от температуры, использующихся растворителей и наличия катализатора. Полученные соединения общей формулы IX можно использовать для синтеза статинов. Последующие стадии реакции будут разными в зависимости от конечного синтезируемого соединения. Реакцию сочетания по Виттигу для соединения формулы IX проводят в присутствии сильного основания, предпочтительно - амида или силазана, наиболее предпочтительно - выбранного из группы,включающей натриевую соль гексаметилдисилазана, калиевую соль гексаметилдисилазана, литиевую соль гексаметилдисилазана, диизопропиламид лития, гидрид натрия, бутиллитий и реагенты Гриньяра,при температуре от - 80 до 40 С в органическом растворителе или смеси органических растворителей,предпочтительно - в толуоле или смеси другого органического растворителя с толуолом или тетрагидрофураном и способ может дополнительно включать обработку реакционной смеси, включающую стадии:(необязательно) концентрирование реакционной смеси; подкисление реакционной смеси в присутствии воды и экстрагирование продукта несмешивающимся с водой органическим растворителем; (необязательно) промывка раствора продукта в органическом растворителе водой, водным раствором соли щелочного металла или соли аммония и/или водным раствором неорганической кислоты; (необязательно)-8 014449 промывка раствора продукта в органическом растворителе смесью вода/полярный апротонный органический растворитель; (необязательно) сушка раствора осушающим агентом; концентрирование раствора с получением остатка, предпочтительно - выпариванием; и очистка остатка с помощью хроматографии на колонке с диоксидом кремния. В предпочтительном варианте осуществления, относящемся к росувастатину, на следующей стадии реакции (2S,4R)-4-(трет-бутилдиметилсилилокси)-6-оксотетрагидро-2H-пиран-2-карбальдегид (9) при условиях сочетания по Виттигу (в присутствии основания) вводят в реакцию с 4-(4-фторфенил)-6 изопропил-2-(N-метилметилсульфонамидо)пиримидин-5-ил)метил)трифенилфосфонийгалогенидом или любой другой 4-(4-фторфенил)-6-изопропил-2-(N-метилметилсульфонамидо)пиримидин-5 ил)метил)фосфониевой солью или альтернативно диизопропил (4-(4-фторфенил)-6-изопропил-2[метил(метилсульфонил)амино]-5-пиримидинилметилфосфонатом или любым другим (4-(4 фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]-5-пиримидинилметилфосфонатным сложным эфиром и получают N-(5-E)-2-2S,4R)-(трет-бутилдиметилсилилокси)-6-оксотетрагидро-2Hпиран-2-ил)винил)-4-(4-фторфенил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамид (10). В качестве основания можно использовать литиевую соль гексаметилдисилазана (LiHMDS), калиевую соль гексаметилдисилазана (KHMDS), натриевую соль гексаметилдисилазана (NaHMDS), диизопропиламид лития (ДАЛ), гидрид натрия, бутиллитий или реагенты Гриньяра, предпочтительно можно использовать натриевую соль гексаметилдисилазана и реакцию можно проводить в простых эфирах, выбранных из группы, включающей ТГФ, Et2O, i-Pr2O, t-BuMeO; алканах, выбранных из группы, включающей пентан,гексан, гептан, толуоле или в смесях этих растворителей. Предпочтительными растворителями являются безводный толуол и тетрагидрофуран. Реакцию можно проводить при температуре от -80 до 40 С. Предпочтительно - от 0 до 30 С. Реакцию проводят в течение 1-12 ч. Выделение неочищенного продукта путем экстракции можно проводить с помощью AcOEt, простых эфиров или алканов, как и выше. Предпочтительно - с помощью t-BuMeO. Силильную защитную группу можно удалить и лактон раскрыть и получить свободную кислоту росувастатина или ее соль, необязательно с амином, которую можно превратить в полукальциевую соль. Удаление защитной группы можно проводить при температуре от 0 до 80 С. Предпочтительно - от 25 до 60 С в подходящем растворителе, предпочтительно в растворителе, выбранном из группы, включающей спирты, ТГФ, ацетонитрил, метилтетрагидрофуран, диоксан, CH2Cl2, более предпочтительно - в спиртах и ТГФ. Можно использовать обычные реагенты, применяющиеся для удаления защитных групп,такие как фторид аммония, FeCl3, TMSCl/HF2H2O хлорэтилхлорформиат (ХЭФ), Ph3PCH2COMeBr. Раскрытие лактона предпочтительно проводить в смеси ТГФ/H2O состава от 4:1 до 2:1, а также в чистом ТГФ при температуре от 20 до 60 С с помощью подходящей щелочи, такой как NaOH, КОН, аммиак или амины. Гидролиз проводят в течение от 30 мин (при 60 С) до 2 ч (при 20 С). После этого выпаривание растворителей при пониженном давлении можно проводить при температуре от 10 до 50 С и превращение в кальциевую соль, предпочтительно путем прибавления Са(ОАс)2H2O, можно проводить при температуре от 0 до 40 С и его можно прибавлять одной порцией или по каплям в течение от 5 до 60 мин. После прибавления Са(ОАс)2H2O суспензию можно перемешивать при температуре от 0 до 40 С в течение от 30 мин до 2 ч. Для получения других статинов (2S,4R)-4-(трет-бутилдиметилсилилокси)-6-оксотетрагидро-2Hпиран-2-карбальдегид (9) при аналогичных условиях сочетания по Виттигу вводят в реакцию с подходящим реагентом и затем при необходимости проводят гидрирование. Статин, содержащий азот, такой как аторвастатин, можно получить аналогичным образом путем использования соединения формулы VIa, в которой X" обозначает цианогруппу, путем превращения в амин и циклизации с помощью подходящего производного с получением промежуточного продукта, который после обработки можно превратить в указанный статин, содержащий азот. Так (6 а) в присутствии цианида можно превратить в 2-2R,4R)-4-(трет-бутилдиметилсилилокси)-6-оксотетрагидро-2H-пиран-2 ил)ацетонитрил и последующее восстановление цианогруппы и последующая конденсация с циклизацией аминогруппы с помощью соответствующего предшественника дает статин, содержащий азот, предпочтительно производное аторвастатина. Ниже кратко описаны объекты настоящего изобретения: Первым главным объектом настоящего изобретения является способ получения соединения формулы VIa в которой X" обозначает галоген, предпочтительно - йод; и R1 обозначает защитную группу, предпочтительно - силил или бензил [более предпочтительно - выбранную из группы, включающей необязательно замещенный C1-С 8 триалкилсилил, С 1-С 8 диалкиларилсилил, С 1-С 8 алкилдиарилсилил, где алкилы могут быть одинаковыми или разными], включающий стадии: в которой R2 обозначает необязательно замещенный С 1-С 8 алкил или С 5-С 7 циклоалкил, или, альтернативно -COOR2 также может образовать амид формулы -CONRaRb, в которой Ra и Rb могут независимо обозначать Н, необязательно замещенный С 1-С 8 алкил или С 5-С 7 циклоалкил, арил или совместно с N могут образовать гетероцикл, с йодидом с образованием алкил-3(S)-гидрокси-4-йодбутирата формулы II в которой R2 является таким, как выше; введение защитной группы в соединение формулы II с получением защищенного производного формулы III в которой R1 и R2 являются такими, как выше; введение соединения формулы III в реакцию с винилмагнийгалогенидом (предпочтительно - винилмагнийхлоридом) в присутствии галогенида меди(I) и фосфита формулы в которой все R', R" и R'" являются одинаковыми или разными и обозначают С 1-С 4 алкил, С 5 С 7 циклоалкил или арил, который необязательно может быть замещенным,с получением алкена формулы IV в которой R1 и R2 являются такими, как выше гидролиз (предпочтительно - щелочью с последующим подкислением) соединения формулы IV с получением соединения формулы V введение в реакцию соединения формулы V, в которой R1 является таким, как выше, с источником галогена (предпочтительно - выбранным из группы, включающей: йод, бром, хлор, галогениды и оксогалогениды щелочных металлов и щелочно-земельных металлов, интергалогены, галогенацетаты, Nйодсукцинимид (NIS), N-бромсукцинимид (NBS), биспиридинйодонийтетрафторборат и гипервалентные галогенсодержащие электрофильные реагенты) в присутствии NaHCO3; и (необязательно) разделение смеси диастереоизомеров, полученной на предыдущей стадии. В одном объекте настоящего изобретения соединение формулы VIa выделяют с оптической чистотой, определенной с помощью ВЭЖХ, превышающей 99% ДИ. Другим объектом настоящего изобретения является применение растворителя, выбранного из группы, включающей t-BuMeO, i-Pr2O, пентан, гексан, гептан, циклопентан, циклогексан, AcOEt, метиленхлорид, хлороформ и их смесь (смеси) при осуществлении любого из способов, описанных выше, и применение вакуумной перегонки при осуществлении способа очистки полученных соединений при осуществлении любого из способов, заявленных выше. В дополнение к описанному выше способу получения соединения VI вторым главным объектом настоящего изобретения дополнительно является способ получения соединения формулы IX- 10014449 в которой R1 является таким, как определено выше, из соединения формулы VIa в которой X" обозначает галоген, алкилсульфонил или арилсульфонил и в которой эти сульфонилпроизводные можно получить из галогенсодержащих соединений по обычным методикам. Более предпочтительно, если в последнем описанном способе соединение формулы IX в которой R1 является таким, как определено выше, получают способом, включающим одну или большее количество стадий, выбранных из группы, включающейg) превращение соединения формулы VIah) превращение соединения формулы VII в соединение формулы VIIIi) превращение соединения формулы VIII путем окисления в соединение формулы IX Характерными особенностями соответствующих стадий является то, что стадию g) проводят с использованием ацилирующего реагента, выбранного из группы, включающей NaOAc, LiOAc, KOAc,CsOAc, AgOAc, CuOAc, Mg(OAc)2, Са(ОАс)2, R4NOAC; стадию h) проводят путем дезацетилирования оловоорганическим соединением, выбранным из группы, включающей дибутилоловооксид и [tBu2SnOH(Cl)]2, или по реакции с ферментом, выбранным из группы, включающей свиную панкреатическую липазу, липазу MY, липазу PS, липазу Al, липазу Candida и алкалазу, или с реагентом, выбранным из группы, включающей гуанидин и гуанидин/гуанидинийнитрат, HBF4Et2O/MeOH и BF3Et2O/MeCN,- 11014449DBU/MeOH, гидразин/MeOH и гидразингидрат/ТГФ, цианид/МеОН, I2/МеОН; стадию i) проводят с помощью методики окисления, выбранной из группы, включающей: методики окисления при катализе диметилсульфоксидом (окисление по Шверну: пара ДМСО-(COCl)2), методика Пфитцнера-Моффата: пара ДМСО-дициклогексилкарбодиимид (ДЦК), методика Парика-Деринга: пара ДМСО-SO3Py), методики окисления при катализе N-оксоаммониевыми соединениями (пара (2,2,6,6-тетраметил-1 пиперидинилоксил (TEMPO) - окислитель), методики окисления органическим гипервалентным соединением йода, выбранным из группы, включающей перйодинан Десса-Мартина (ПДМ) и ойодоксибензойную кислоту (IBX или SIBX), методики окисления содержащими хром(VI) окислителями,выбранными из группы, включающей реагент Коллинза (CrO3 Ру 2), пиридинийдихромат (ПДХ) (пара ПДХ-активированные молекулярные сита 4), пиридинийхлорхромат (ПХХ), методики окисления производными марганца, выбранными из группы, включающей MnO2 и BaMnO4, и методики окисления тетра-н-пропиламмонийперрутенатом: Pr4N+ RuO4- (ТПАП). Еще одним объектом настоящего изобретения является способ получения соединения формулы включающий получение промежуточного продукта формулы VIa в которой R1 и X" являются такими, как определено выше,способом, который является первым главным объектом настоящего изобретения, описанным выше получение промежуточного продукта формулы IX в которой R1 является таким, как определено выше, из промежуточного продукта формулы VIa способом, который является вторым главным объектом настоящего изобретения, описанным выше; и введение промежуточного продукта формулы IX в реакцию с соединением формулы в которой А может обозначать связь или О,и в которой Rx, Ry и Rz являются одинаковыми или разными и выбраны из группы, включающей необязательно замещенный С 1-С 3 алкил, или С 3-С 6 циклоалкил, или С 1-С 8 алкенил, или С 5-С 6 циклоалкенил,или арил,X- обозначает анион (предпочтительно - галогенидный или алканоатный) и Het является таким, как определено выше, и необязательно включающий одну или большее количество последующих стадий,на которых соединение формулы X в которой R1 и Het являются такими, как определено выше, или его соль, амид или лактон. Предпочтительно, если Het обозначает гетероциклический каркас росувастатина формулы Другим предпочтительным объектом настоящего изобретения является способ получения росувастатина, характеризующийся тем, что он включает стадии (необязательно) введение в реакцию алкил 3(S)-гидрокси-4-хлорбутирата формулы I в которой R2 обозначает необязательно замещенный С 1-С 8 алкил или С 5-С 7 циклоалкил, или, альтернативно -COOR2 также может образовать амид формулы -CONRaRb, в которой Ra и Rb могут независимо обозначать Н,необязательно замещенный С 1-С 8 алкил или С 5-С 7 циклоалкил, арил или совместно с N могут образовать гетероцикл, с йодидом с образованием алкил-3(S)-гидрокси-4-йодбутирата формулы II(необязательно) введение защитной группы в соединение формулы II с получением защищенного производного формулы III в которой R1 и R2 являются такими, как выше; введение соединения формулы III в реакцию с винилмагнийгалогенидом в присутствии галогенида меди(I) и фосфита формулы: в которой все R', R" и R"' являются одинаковыми или разными и обозначают С 1-С 4 алкил, С 5 С 7 циклоалкил или арил, который необязательно может быть замещенным,с получением алкена формулы IV в которой R1 и R2 являются такими, как выше; гидролиз соединения формулы IV с получением соединения формулы V введение в реакцию соединения формулы V, в которой R1 является таким, как выше, с источником галогена в присутствии NaHCO3 с получением соединения формулы VI;(необязательно) разделение смеси диастереоизомеров, полученной на предыдущей стадии, с получением соединения формулы VIa в которой X" обозначает галоген и R1 является таким, как выше; превращение соединения формулы VIa в соединение формулы VII в которой R1 является таким, как выше, и R4 выбран из группы, включающей (необязательно галоген-, или алкокси-, или арилоксизамещенный) С 1-С 4 алкил; превращение соединения формулы VII в соединение формулы VIII- 14014449 превращение соединения формулы VIII путем окисления в соединение формулы IX, в которой R1 является таким, как выше; введение указанного соединения формулы IX в реакцию с соединением формулы в которой Rx, Ry и Rz являются одинаковыми или разными и выбраны из группы, включающей необязательно замещенный С 1-С 8 алкил, или С 3-С 6 циклоалкил, или С 1-С 8 алкенил, или С 5-С 6 циклоалкенил,или арил,X- обозначает анион; (предпочтительно - галогенидный или алканоатный) и удаление защитной группы R1 необязательно очистку и превращение полученного соединения в кальциевую соль. Объектом настоящего изобретения также является применение для синтеза статинов промежуточного продукта XI характеризующегося тем, что он получен из промежуточного продукта формулы VIa в которой X" обозначает галоген, арилсульфонил или алкилсульфонил и R1 обозначает защитную группу и также применение для синтеза статинов промежуточного продукта формулы в которой R1 обозначает необязательно замещенный С 1-С 8 триалкилсилил, С 1-С 8 диалкиларилсилил,С 1-С 8 алкилдиарилсилил, где алкилы могут быть одинаковыми или разными и R4 обозначает (необязательно галоген-, алкил-, арил-, алкилокси- или арилоксизамещенный) С 1-С 4 алкил. Новые соединения формулы в которой R1 обозначает необязательно замещенный С 1-С 8 триалкилсилил, C1-С 8 диалкиларилсилил,С 1-С 8 алкилдиарилсилил, где алкилы и арилы могут быть одинаковыми или разными и R4 обозначает (не- 15014449 обязательно галоген-, алкил-, арил-, алкилокси- или арилоксизамещенный) С 1-С 4 алкил, также являются объектами настоящего изобретения, также как и их применение в качестве промежуточных продуктов для синтеза статинов, предпочтительно росувастатина, предпочтительно, если R4 обозначает СН 3,С(СН 3)3, CH2Cl, CHCl2, CCl3, CF3, CH2OCH3, CH2OCPh3, CH2OPh, CH2Ph, CHPh2, CH2CH2Ph,СН=СНСН 2 СН 3 или СН 2 СН 2(С=O)СН 3), более предпочтительно, если R1 обозначает третбутилдиметилсилил и R4 обозначает СН 3. Объектом настоящего изобретения также являются соединения формулы (и его применение для синтеза статинов, предпочтительно росувастатина) в которой R1 обозначает необязательно замещенный С 1-С 8 триалкилсилил, C1-С 8 диалкиларилсилил,С 1-С 8 алкилдиарилсилил, где алкилы и арилы могут быть одинаковыми или разными, предпочтительно - в которой R1 обозначает трет-бутилдиметилсилил. К этому также относится способ превращения соединения формулы характеризующийся растворением в хлороформе, дихлорметане, гексане или толуоле, предпочтительно - толуоле, предпочтительно в течение более 24 ч, более предпочтительно более 150 ч. Более предпочтительным объектом настоящего изобретения является способ получения соединения формулы X включающий реакцию Виттига, в котором соединение формулы IX в которой А может обозначать связь или О и в которой Rx, Ry и Rz являются одинаковыми или разными и выбраны из группы, включающей необязательно замещенный С 1-С 8 алкил, или С 3-С 6 циклоалкил,- 16014449 или С 1-С 8 алкенил, или С 5-С 6 циклоалкенил, или арил,R1 обозначает защитную группу,X обозначает анион и Het выбран так, что он образует гетероциклический каркас статина, характеризующийся тем, что реакцию проводят в растворителе, выбранном из группы, включающей хлороформ,дихлорметан и гексан, и предпочтительно в толуоле. Предпочтительно, если Het выбран из группы, включающей Другим предпочтительным объектом настоящего изобретения является способ получения росувастатина включающий реакцию Виттига, в котором соединение формулы IX в которой А может обозначать связь или О и в которой Rx, Ry и Rz являются одинаковыми или разными и выбраны из группы, включающей необязательно замещенный С 1-С 8 алкил, или С 3-С 6 циклоалкил,или С 1-С 8 алкенил, или С 5-С 6 циклоалкенил, или арил,R1 обозначает защитную группу,X обозначает анион и Het выбран так, что он образует гетероциклический каркас статина,характеризующийся тем, что реакцию проводят в толуоле. Описанная выше реакция Виттига предпочтительно характеризуется тем, что соединение формулыIX растворяют в толуоле по меньшей мере за 6 ч до проведения реакции и/или тем, что ее проводят в- 17014449 присутствии сильного основания при температуре от -80 до 40 С, предпочтительно от 0 до 40 С, более предпочтительно - от 10 до 35 С. Предпочтительно, если сильное основание выбрано из группы, включающей амиды металлов и силазаны, гидриды металлов, алкилпроизводные лития и арилпроизводные лития, более предпочтительно - из группы, включающей: литиевую соль гексаметилдисилазана, натриевую соль гексаметилдисилазана, калиевую соль гексаметилдисилазана, диизопропиламид лития, гидрид натрия, бутиллитий и реагенты Гриньяра. В одном объекте настоящего изобретения описанный выше способ дополнительно включает обработку реакционной смеси, включающую указанную ниже последовательность стадий:(необязательно) концентрирование реакционной смеси; подкисление реакционной смеси в присутствии воды и экстрагирование продукта несмешивающимся с водой органическим растворителем; (необязательно) промывка раствора продукта в органическом растворителе водой, водным раствором соли щелочного металла или соли аммония и/или водным раствором неорганической кислоты;(необязательно) промывка раствора продукта в органическом растворителе смесью вода/полярный апротонный органический растворитель;(необязательно) сушка раствора осушающим агентом; концентрирование раствора с получением остатка, предпочтительно выпариванием; и очистка остатка. Предпочтительными объектами настоящего изобретения являются статины, выбранные из группы,включающей росувастатин, церивастатин, флувастатин, питавастатин, бервастатин, аторвастатин и их аналоги, характеризующиеся тем, что они получены способом, описанным в настоящем изобретении, и фармацевтическая композиция, включающая фармацевтически приемлемый носитель и статин, выбранный из группы, включающей кальциевую соль росувастатина, натриевую соль флувастатина, кальциевую соль аторвастатина, полученный способом, в котором применяется промежуточный продукт формулы в которой X обозначает галоген и R1 обозначает защитную группу. Различные варианты осуществления настоящего изобретения представлены в приведенных ниже примерах. Пример 1. Этил-3(S)-гидрокси-4-йодбутират (2) К раствору этил-3(S)-гидроксихлорбутирата (1) (639,0 г, 3,84 моль) в сухом ацетоне (7,7 л) прибавляют безводный Nal (2300 г, 15,34 моль). Смесь энергично перемешивают при 58-60 С в течение 120 ч в атмосфере аргона. Ацетон отгоняют при пониженном давлении при 40-60 С. Остаток разбавляют водой(5,75 л) и затем прибавляют насыщенный раствор Na2S2O3 (1,53 л) и t-BuMeO (2,3 л). Смесь энергично перемешивают при температуре окружающей среды в течение 30 мин. Затем органический слой отделяют и водную фазу дополнительно экстрагируют с помощью t-BuMeO (21,15 л). Объединенные органические слои промывают водой (800 мл) и сушат (MgSO4). Выпаривание растворителя при пониженном давлении (20 мбар) при 40 С дает 949,9 г (96%) этил-3(S)-гидрокси-4-йодбутирата (2) в виде желтого масла (чистота по данным ГХ - 85,9%). Продукт, обладающий чистотой по данным ГХ (газовая хроматография), равной 98-99%, можно получить перегонкой в вакууме (73-89 С при 0,180-0,330 мбар) неочищенного продукта. 1 Н ЯМР (300 МГц, CDCl3):4,17 (q, J = 7,2 Гц, 2 Н), 3,99 (m, 1H), 3,34 (dd, J = 10,3 и 5,2 Гц, 1 Н),3,28 (dd, J = 10,3 и 5,7 Гц, 1H), 3,21 (br s, 1H), 2,67 (dd, J = 16,5 и 4,3 Гц, 1H), 2,58 (dd, J = 16,5 и 7,9 Гц,1H), 1,27 (t, J = 7,2 Гц, 3 Н). 13 С ЯМР (75 МГц, CDCl3):171,7, 67,4,61,0,40,7, 14,1, 12,0. Пример 2. Этил-3(S)-(трет-бутилдиметилсилилокси)-4-йодбутират (3) К раствору имидазола (252,3 г, 7,36 моль) в сухом ДМФ (7,6 л) в атмосфере аргона при температуре окружающей среды прибавляют этил-3(S)-гидрокси-4-йодбутират (2) (949,6 г, 3,68 моль, чистота по данным ГХ - 85,9 %) и безводный NaI (1106 г, 7,36 моль). Суспензию охлаждают до 0 С и порциями прибавляют трет-бутил(хлор)диметилсилан (838 г, 5,56 моль). Реакционную смесь перемешивают при 0 С в- 18014449 течение 1,5 ч и затем перемешивают в течение 15,5 ч при температуре от 0 С до температуры окружающей среды. Затем смесь охлаждают до 0 С и прибавляют Н 2 О (4,2 л). После 2 ч перемешивания прибавляют дополнительное количество Н 2 О (6,2 л) и насыщенный раствор Na2S2O3 (0,5 л). Продукт экстрагируют с помощью t-BuMeO. Объединенные органические слои промывают водой и сушат (MgSO4). Эфир полностью удаляют при пониженном давлении (20 мбар) при 60 С и получают желтый маслообразный остаток. Этот остаток дополнительно очищают перегонкой в вакууме (80-89 С при 0,150-0,310 мбар) и получают 1193,5 г (97%) этил-3(S)-(трет-бутилдиметилсилилокси)-4-йодбутирата (3) в виде бледножелтого масла (чистота по данным ГХ - 96,1%). 1 Н ЯМР (300 МГц, CDCl3):4,13 (qd, J = 7,2 и 2,1 Гц, 2 Н), 4,01 (m, 1H), 3,28 (dd, J = 10,2 и 4,2 Гц, 1 Н),3,24 (dd, J = 10,2 и 6,0 Гц, 1 Н), 2,66 (dd, J = 15,3 и 4,8 Гц, 1H), 2,51 (dd, J = 15,3 и 7,2 Гц, 1 Н), 1,26 (t, J = 7,2 Гц,3 Н), 0,87 (s, 9H), 0,10 (s, 3 Н), 0,05 (s, 3 Н). 13 С ЯМР (75 МГц, CDCl3):170,8, 68,3, 60,5, 42,5, 25,6, 17,9, 14,1, 12,9, -4,6, -5,0. Пример 3. Этил-3(R)-(трет-бутилдиметилсилилокси)-5-гексеноат (4) К суспензии CuI (83,64 г, 437,0 ммоль) в сухом ТГФ (875 мл) в атмосфере аргона при температуре от -44 до-31 С в течение 15 мин при энергичном перемешивании прибавляют винилмагнийхлорид (1,9 М в ТГФ, 460,0 мл, 874,0 ммоль). Полученную темную взвесь перемешивают в течение 15 мин и при -42 С одной порцией прибавляют ДМПМ (112 г, 874,0 ммоль) и затем при -40 С по каплям прибавляют (в течение 5 мин) Р(OEt)3 (160,1 мл, 874,0 ммоль). Полученную смесь перемешивают в течение 30 мин и при температуре от -40 до -36 С в течение 15 мин прибавляют раствор (S)-этил-3-(трет-бутилдиметилсилилокси)-4-йодбутирата (3) (162,7 г, 437,0 ммоль) в ТГФ (220 мл). Перемешивание продолжают в течение 1 ч при температуре от -40 до -35 С и затем смеси дают нагреваться до 11 С в течение 3,5 ч. Реакцию останавливают при -2 С (насыщенный растворt-BuMeO. Частичное выпаривание растворителя при пониженном давлении дает желтый раствор, который промывают с помощью 0,1 М H2SO4, водой и сушат (MgSO4). Эфир полностью удаляют при пониженном давлении (15 мбар) при 80 С и получают желтый маслообразный остаток. Этот остаток дополнительно очищают перегонкой в вакууме (64-72 С при 0,10-0,44 мбар) и получают 80,8 г (67,8%) этил 3(R)-(трет-бутилдиметилсилилокси)-5-гексеноата (4) в виде бесцветного масла (чистота по данным ГХ 96%). 1 Н ЯМР (300 МГц, CDCl3):5,81 (ddt, J = 17,8, 9,6 и 7,2 Гц, 1 Н), 5,11-5,03 (m, 2H), 4,21 (квинтет, J = 6,8 Гц, 1 Н), 4,12 (qt, J = 7,1 и 1,5 Гц, 2 Н), 2,43 (d, J = 7,1 Гц, 1 Н), 2,43 (d, J = 5,4 Гц, 1H), 2,31-2,25 (m, 2H),1,26 (t, J = 7,2 Гц, 3 Н), 0,87 (s, 9H), 0,07 (s, 3 Н), 0,04 (s, 3 Н). 13 С ЯМР (75 МГц, CDCl3):171,7, 134,1, 117,6, 68,9, 60,2, 42,1, 42,1, 25,7, 17,9, 14,1,-4,6,-5,0. Пример 4. (R)-3-(трет-Бутилдиметилсилилокси)-5-гексеновая кислота (5) К раствору этил-3(R)-(трет-бутилдиметилсилилокси)-5-гексеноата (4) (80,60 г, 295,8 ммоль) в МеОН (500 мл) прибавляют 40 % раствор КОН (150 мл). Смесь перемешивают в течение 2 ч при 40 С. Затем смесь охлаждают до температуры окружающей среды и МеОН удаляют при пониженном давлении(20 мбар) при 42 С. Полученное коричневое твердое вещество растворяют в Н 2 О (700 мл). Раствор промывают с помощью t-BuMeO (1340 мл + 3215 мл) и затем подкисляют с помощью 4 н. HCl (222 мл) до рН = 2. Желтое масло, которое отделяется от воды, экстрагируют с помощью t-BuMeO (6110 мл). Объединенные органические слои промывают водой и сушат (MgSO4). Эфир полностью удаляют при пониженном давлении (15 мбар) при 40 С и получают оранжево-красный маслообразный остаток (68,2 г, 94,3%). Этот остаток дважды фильтруют через тонкий слой диоксида кремния с использованием t-BuMeO в качестве растворителя и получают (R)-3-(трет-бутилдиметилсилилокси)-5-гексеновую кислоту (5) (67,1 г, 93%) в виде желтого масла (чистота по данным ГХ - 97,3%). 1 Н ЯМР (300 МГц, CDCl3):11,03 (br s, 1H), 5,80 (ddt, J = 17,8, 9,5 и 7,2 Гц, 1H), 5,13-5,11 (m, 1H),5,08-5,06 (m, 1H), 4,20 (квинтет, J = 6,5 Гц, 1 Н), 2,54 (dd, J = 15,1 и 5,1 Гц, 1H), 2,46 (dd, J = 15,1 и 7,1 Гц,1H), 2,33-2,28 (m, 2H), 0,88 (s, 9H), 0,09 (s, 3 Н), 0,07 (s, 3 Н). 13 С ЯМР (75 МГц, CDCl3):178,1, 133,8, 118,1, 68,8, 42,0, 41,8, 25,7, 17,9, -4,5, -5,0. К раствору (R)-3-(трет-бутилдиметилсилилокси)-5-гексеновой кислоты (5) (68,00 г, 278,2 ммоль) в сухом MeCN (950 мл) при температуре окружающей среды прибавляют безводный NaHCO3 (708,3 г,8,347 моль). При перемешивании суспензию охлаждают до 0 С. Затем при энергичном перемешивании к суспензии одной порцией прибавляют йод (212,9 г, 834,7 ммоль). Реакционную смесь перемешивают при 0 С в течение 4 ч и затем прибавляют t-BuMeO ИЛИ i-Pr2O (410 мл) и насыщенный раствор Na2S2O3 (820 мл). Органический слой отделяют и водную фазу дополнительно экстрагируют с помощью tBuMeO ИЛИi-Pr2O (5200 мл). Объединенные органические слои сушат (MgSO4). Растворители в основном удаляют при пониженном давлении (20 мбар) при 40 С и получают оранжевый маслообразный остаток. Этот остаток растворяют в t-BuMeO или i-Pr2O (200 мл) и раствор дополнительно промывают насыщенным раствором Na2S2O3 (2100 мл) и водой (2100 мл). Органический слой сушат (MgSO4). Растворитель полностью удаляют при пониженном давлении (20 мбар) при 50 С и получают желтый маслообразный остаток(99,96 г, 97%), который затвердевает при температуре ниже 10 С и получают 77:23 смесь (4R,6S)-4-(третбутилдиметилсилилокси)-6-(йодметил)тетрагидропиран-2-она (6 а) и (4R,6R)-4-(трет-бутилдиметилсилилокси)-6-(йодметил)тетрагидропиран-2-она (6b) в виде бледно-желтого твердого вещества. Неочищенную смесь растворяют в подвижной фазе (гексан и t-BuMeO) и при комнатной температуре последовательно инжектируют в прибор для ВЭЖХ с использованием колонки с диоксидом кремния с нормальной фазой (PHENOMENEX 4,6150 мм, dp = 5 мкм) и элюируют в изократическом режиме. Активные фракции (соответствующие всему пику) препаративного разделения собирают и анализируют с помощью того же прибора и обнаруживают, что 97% площади пика соответствует (6 а) и 0,15% соответствует (6b) и что содержатся некоторые другие примеси. В качестве альтернативы хроматографическому разделению это твердое вещество 7 раз перекристаллизовывают из н-пентана и получают 43,6 г (42,6%) (4R,6S)-4-(трет-бутилдиметилсилилокси)-6(йодметил)тетрагидропиран-2-она (6 а) (ДИ 99,3%, ВЭЖХ) в виде бесцветных иголок. Температура плавления = 64 С (пик ДСК (дифференциальная сканирующая калориметрия). 1 Н ЯМР (300 МГц, CDCl3) : 4,60 (ddt, 1H, J= 11,3, 5,0, 3,2 Гц, 6-Нах), 4,35 (br квинтет, 1H, J = 3,5 Гц,4-Heq), 3,40 (d, 2 Н, J= 5,0 Гц, CH2I), 2,59 (d, 2H, J= 3,5 Гц, 3-СН 2), 2,10 (dddd, 1H,J= 13,9,3,9,3,2, 1,7 Гц, 5Heq), 1,76 (ddd, 1H, J= 13,7, 11,3,2,1 Гц, 5-Hax), 0,89 (s, 9H, SiC (CH3)3), 0,09, (s, 6H, Si(CH3)2). 13 С ЯМР (75 МГц, CDCl3) : 169,0, 73,9, 63,1, 38,7, 36,2, 25,5, 17,7, 8,6, -5,1, -5,1. Пример 6. 2S,4R)-4-(трет-Бутилдиметилсилилокси)-6-оксотетрагидро-2H-пиран-2-ил)метил ацетат (7)(40,00 г, 108,0 ммоля) в АсОН (660 мл) прибавляют AgOAc (20,03 г, 118,8 ммоля). Затем полученную смесь нагревают при 125 С в течение 6 ч. Реакционную смесь фильтруют через диатомитовую фильтрующую среду (Celite). Полученный фильтрат выпаривают и получают остаток. К этому остатку при- 20014449 бавляют EtOAc (500 мл) и воду (600 мл). Органический слой отделяют и водный слой повторно промывают с помощью EtOAc (5150 мл). Объединенные органические слои промывают водой (4300 мл),рассолом (5300 мл) и сушат над безводным MgSO4, фильтруют и концентрируют при пониженном давлении и получают 30,28 г (92,6%) 2S,4R)-4-(трет-бутилдиметилсилилокси)-6-оксотетрагидро-2H-пиран 2-ил)метилацетата (7) в виде желтого масла (чистота по данным ВЭЖХ - 98%). 1 Н ЯМР (300 МГц, CDCl3):4,93 (m, 1H), 4,37 (m, 1H), 4,30 (dd, J = 12 Гц, J = 3 Гц, 1 Н), 4,21 (dd, J = 12 Гц, J = 5 Гц, 1H), 2,62 (d, J = 4 Гц, 2H), 2,11 (s, 3H), 1,84-1,80 (m, 2H), 0,89 (s, 9H), 0,09, 0,09 (2s, 6H). 13 2S,4R)-4-(трет-Бутилдиметилсилилокси)-6-оксотетрагидро-2H-пиран-2-ил)метилацетат (7) (8,36 г,27,64 ммоль) и [t-Bu2SnOH(Cl)]2 (1,577 г, 2,764 ммоль) растворяют в смеси МеОН/ТГФ (280 мл). Реакционную смесь перемешивают при 23-25 С в течение 27 ч. Затем растворитель удаляют при пониженном давлении и образовавшийся остаток очищают с помощью хроматографии на силикагеле (элюирование смесью t-BuMeO/гексан) и получают неочищенный продукт в виде белого твердого вещества (5,59 г,78%). Перекристаллизация из н-гексана дает (3,90 г, 54%) (4R,6S)-4-(трет-бутилдиметилсилилокси)-6(гидроксиметил)тетрагидропиран-2-она (8) в виде белых иголок. Температура плавления = 102 С (пик ДСК). 1 Н ЯМР (300 МГц, CDCl3) : 4,80 (m, 1H), 4,38 (m, 1 Н), 3,91 (dd, J = 12 Гц, J = 3 Гц,1 Н), 3,66 (dd, J = 12 Гц, J = 5 Гц,1 Н), 2,60 (d,J = 4 Гц, 2 Н), 2,31 (bs, 1H), 1,97-1,75 (m, 2H), 0,88 (s, 9H), 0,09, 0,08 (2s, 6H). 13 Смесь (4R,6S)-4-(трет-бутилдиметилсилилокси)-6-(гидроксиметил)тетрагидропиран-2-она (8) (150 мг, 0,58 ммоль) и перйодинана Десса-Мартина (380 мг, 0,86 ммоль) в CH2Cl2 (15 мл) перемешивают при температуре окружающей среды в течение 3 ч. Смесь разбавляют с помощью t-BuMeO (20 мл), промывают насыщенным раствором Na2S2O3, насыщенным раствором NaHCO3, сушат (MgSO4) и концентрируют и получают 130 мг (87%) неочищенного (2S,4R)-4-(трет-бутилдиметилсилилокси)-6 оксотетрагидро-2H-пиран-2-карбальдегида (9), который используют на следующей стадии без дополнительной очистки. 1C ЯМР (75 МГц, CDCl3) : 199,4, 168,0, 79,2, 62,9, 39,6, 31,4, 25,6, 17,9, -4,9. Гидрат (9) обладает следующими спектрами ЯМР: 1 Н ЯМР (300 МГц, ТГФ-d8) : 5,27 (d, J = 6 Гц, 1H, ОН), 5,19 (d, J= 6 Гц, 1 Н, ОН), 4,90-4,85 (m, 1 Н),4,44-4,38 (m, 2 Н), 2,58 (dd, J= 17 Гц, J= 4 Гц,1 Н), 2,44-2,36 (m, 1H), 1,92-1,87 (m, 2 Н), 0,91 (s, 9H), 0,10, (s,6H). 13 С ЯМР (75 МГц, ТГФ-d8) : 168,7, 91,7, 79,0, 65,1, 40,3, 31,0, 26,2, 18,7, -4,8, -4,8. При перемешивании к холодной (-30 С) суспензии 4-(4-фторфенил)-6-изопропил-2-(Nметилметилсульфонамидо)пиримидин-5-ил)метил)трифенилфосфонийбромида (376 мг, 0,55 ммоль) в сухом тетрагидрофуране (10 мл) прибавляют литиевую соль гексаметилдисилазана в ТГФ (0,42 мл 1,33 М раствора, 0,55 ммоля). Реакционную смесь перемешивают в течение 30 мин, охлаждают до -78 С и обрабатывают раствором(2S,4R)-4-(трет-бутилдиметилсилилокси)-6-оксотетрагидро-2H-пиран-2 карбальдегида (9) (130 мг, 0,50 ммоля) в 5 мл тетрагидрофурана. Через 60 мин раствор нагревают до температуры окружающей среды, перемешивают в течение 10 мин и обрабатывают насыщенным раствором хлорида аммония. Водную фазу экстрагируют с помощью t-BuMeO (210 мл) и объединенные органические слои сушат и концентрируют. Остаток очищают с помощью хроматографии на силикагеле(элюирование смесью t-BuMeO/гексан) и получают 190 мг (65%) N-(5-E)-2-2S,4R)-4-(третбутилдиметилсилилокси)-6-оксотетрагидро-2H-пиран-2-ил)винил)-4-(4-фторфенил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамида (10) в виде белого аморфного твердого вещества. Пример b) При перемешивании к суспензии 4-(4-фторфенил)-6-изопропил-2-(N-метилметилсульфонамидо)пиримидин-5-ил)метил)трибутилфосфоний-2,2,2-трифторацетата (260 мг, 0,4 ммоль) при комнатной температуре в сухом толуоле (4 мл) порциями в течение 10 мин прибавляют натриевую соль гексаметилдисилазана в толуоле (0,67 мл 0,6 М раствора, 0,4 ммоль). Реакционную смесь перемешивают в течение 60 мин и при комнатной температуре обрабатывают раствором (2S,4R)-4-(третбутилдиметилсилилокси)-6-оксотетрагидро-2H-пиран-2-карбальдегида (9) (105 мг, 0,40 ммоля) в 13 мл сухого толуола. После 24 ч перемешивания при комнатной температуре раствор обрабатывают насыщенным раствором хлорида аммония или водой. Водную фазу экстрагируют с помощью t-BuMeO (210 мл) и объединенные органические слои сушат и концентрируют. Остаток очищают с помощью хроматографии на силикагеле (элюирование смесью t-BuMeO/гексан) и получают 104 мг (45%) N-(5-E)-22S,4R)-4-(трет-бутилдиметилсилилокси)-6-оксотетрагидро-2H-пиран-2-ил)винил)-4-(4-фторфенил)-6 изопропилпиримидин-2-ил)-N-метилметансульфонамида (10) в виде белого аморфного твердого вещества. 1 Н ЯМР (300 МГц, CDCl3) : 7,62 (dd, J = 9 Гц, J = 5 Гц, 2 Н), 7,09 (t, J =9 Гц, 2 Н), 6,69 (dd, J = 16 Гц,J = 1 Гц, 1H), 5,49 (dd, J = 16 Гц, J = 6 Гц, 1 Н), 5,22-5,16 (m, 1H), 4,29-4,27 (m, 1H), 3,56 (s, 3 Н), 3,50 (s,3 Н), 3,32 (септет, 1 Н), 2,61-2,59 (m, 2 Н), 1,80-1,73 (m, 1 Н), 1,64-1,54 (m, 1H), 1,26 (d, J= 1 Гц, 6 Н), 0,87 (s,9 Н), 0,07, 0,06 (2s, 6H). 13 С ЯМР (75 МГц, CDCl3) : 174,9, 169,5, 163,5, 163,2 (d, JC-F = 250 Гц), 157,4, 134,7, 134,1 (d, JC-F = 3N-(5-E)-2-2S,4R)-4-(трет-бутилдиметилсилилокси)-6 оксотетрагидро-2H-пиран-2-ил)винил)-4-(4-фторфенил)-6-изопропилпиримидин-2-ил)-N-метилметансульфонамида (10) (190 мг, 0,33 ммоль) в 3 мл безводного тетрагидрофурана прибавляют раствор фторида аммония (73 мг, 1,97 ммоль)/АсОН (2 мл) в ТГФ. Реакционную смесь нагревают до 60 С, перемешивают в течение 5 ч, обрабатывают с помощью 3 мл водного раствора хлорида аммония и несколько раз экстрагируют с помощью t-BuMeO. Объединенные органические слои промывают водой, сушат и концентрируют. Остаток растворяют в 3 мл 4:1 смеси ТГФ/Н 2 О. Прозрачный раствор нагревают до 30 С и порциями прибавляют 8,0 М NaOH (0,044 мл, 0,35 ммоль). Реакционную смесь перемешивают при 30 С в течение 2 ч и получают прозрачный желтый раствор. Затем ТГФ полностью удаляют при пониженном давлении (20 мбар) при 40 С. Оставшийся водный раствор разбавляют с помощью Н 2 О до 1,5 мл и промывают с помощью AcOEt (21 мл). После отделения от органического слоя водную фазу отгоняют при пониженном давлении (20 мбар) при 40 С для полного удаления растворенного AcOEt. Оставшийся прозрачный раствор росувастатината натрия (1,3 мл) разбавляют с помощью Н 2 О до 1,5 мл и нагревают до 40 С. При энергичном перемешивании к раствору росувастатината натрия в течение 5 мин при 40 С по каплям прибавляют Са(ОАс)2H2O (44 мг, 0,25 ммоль в 0,3 мл H2O) для осаждения кальциевой соли росувастатина. После окончания прибавления суспензию перемешивают в течение еще 30 мин при 40 С. Белый осадок отфильтровывают. Затем влажное белое твердое вещество суспендируют в Н 2 О (1 мл) и энергично перемешивают в течение 1 ч при 20 С. Нерастворившийся осадок собирают фильтрованием,промывают с помощью Н 2 О (1 мл) и сушат в вакууме при 40 С и получают 143 мг (87 %) кальциевой соли росувастатина (11) в виде белого порошкообразного вещества. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы IX в которой R4 обозначает (необязательно замещенный галогеном, С 1-С 8 алкилом, арилом, С 1 С 8 алкилокси или арилокси) С 1-С 4 алкил. 2. Способ по предыдущему пункту, в котором соединение формулы VII получают из соединения формулы VIa в которой X" обозначает галоген, С 1-С 8 алкилсульфонил или арилсульфонил. 3. Способ по п.1, в котором соединение формулы IX в которой R1 является таким, как определено выше,получают способом, включающим одну или более стадий, выбранных из: а) превращения соединения формулы VII в соединение формулы VIIIb) превращения соединения формулы VIII путем окисления в соединение формулы IX 4. Способ по предыдущему пункту, который дополнительно включает стадии:a) необязательно, превращение соединения формулы VIa, в которой X" обозначает С 1 С 8 алкилсульфонил или арилсульфонил, в соединение формулы VIa, в которой X" обозначает галоген; иb) превращение соединения формулы VIa в которой R4 обозначает (необязательно замещенный галогеном, С 1-С 8 алкилом, арилом, С 1 С 8 алкилокси или арилокси) С 1-С 4 алкил. 5. Способ по одному из пп.2 и 4, в котором соединение формулы VIa в которой X" обозначает галоген и R1 обозначает защитную группу,получают с помощью следующих стадий: а) (необязательно) взаимодействие алкил-3(S)-гидрокси-4-хлорбутирата формулы I, в которой R2 обозначает С 1-С 8 алкил или С 5-С 7 циклоалкил или, альтернативно, -COOR2 также может образовать амид формулы -CONRaRb, в которой Ra и Rb могут независимо обозначать Н, С 1-С 8 алкил или С 5-С 7 циклоалкил, арил или совместно с N могут образовать гетероцикл,с йодидом с образованием алкил-3(S)-гидрокси-4-йодбутирата формулы IIb) введение защитной группы в соединение формулы II с получением защищенного производного формулы III в которой R1 и R2 являются такими, как определено выше; с) взаимодействие соединения формулы III с винилмагнийгалогенидом в присутствии галогенида меди(I) и производного фосфита формулы в которой все R', R" и R'" являются одинаковыми или разными и обозначают С 1-С 4 алкил, С 5 С 7 циклоалкил или арил,с получением алкена формулы IV, в которой R1 и R2 являются такими, как определено вышеd) гидролиз соединения формулы IV с получением соединения формулы Ve) взаимодействие соединения формулы V, в которой R1 является таким, как определено выше, с источником галогена в присутствии NaHCO3 иf) (необязательно) разделение смеси диастереоизомеров, полученной на предыдущей стадии. 6. Способ по п.4, в котором стадию b) проводят с использованием ацилирующего реагента, выбранного из группы, включающей NaOAc, LiOAc, KOAc, CsOAc, AgOAc, CuOAc, Mg(OAc)2, Ca(OAc)2,R4NOAc. 7. Способ по п.3, в котором стадию а) проводят путем дезацетилирования оловоорганическим соединением, выбранным из группы, включающей дибутилоловооксид или [t-Bu2SnOH(Cl)]2, или путем реакции с ферментом, выбранным из группы, включающей свиную панкреатическую липазу, липазу MY,липазу PS, липазу Al, липазу Candida и алкалазу, или с реагентом, выбранным из группы, включающей гуанидин и гуанидин/гуанидинийнитрат, HBF4Et2O/MeOH и BF3Et2O/MeCN, DBU/MeOH, гидразин/MeOH и гидразингидрат/ТГФ, цианид/МеОН, I2/МеОН, и/или где стадию b) проводят с помощью реакции окисления, выбранной из группы, включающей реакции окисления при катализе диметилсульфоксидом (окисление по Шверну: пара ДМСО-(COCl)2), методика Пфитцнера-Моффата: пара ДМСОдициклогексилкарбодиимид (ДЦК), методика Парика-Деринга: пара ДМСО-SO3 Ру), реакции окисления при катализе N-оксоаммониевыми соединениями (пара (2,2,6,6-тетраметил-1-пиперидинилоксил(TEMPO) - окислитель), реакции окисления органическим гипервалентным соединением йода, выбран- 25014449 ным из группы, включающей перйодинан Десса-Мартина (ПДМ) и о-йодоксибензойную кислоту (IBX или SIBX), реакции окисления содержащими хром(VI) окислителями, выбранными из группы, включающей реагент Коллинза (CrO3 Ру 2), пиридинийдихромат (ПДХ) (пара ПДХ-активированные молекулярные сита 4 ), пиридинийхлорхромат (ПХХ), реакции окисления производными марганца, выбранными из группы, включающей MnO2 и BaMnO4, или реакции окисления тетра-нпропиламмонийперрутенатом: Pr4N+ RuO4- (ТПАП). 8. Способ получения соединения формулы включающий: а) получение промежуточного продукта формулы VIIb) получение промежуточного продукта формулы IX в которой R1 обозначает защитную группу,из промежуточного продукта формулы VII и с) взаимодействие промежуточного продукта формулы IX с соединением формулы в которой Rx, Ry и Rz являются одинаковыми или разными и выбраны из группы, включающей С 1 С 8 алкил, или С 3-С 6 циклоалкил, или С 1-С 8 алкенил, или С 5-С 6 циклоалкенил, или арил,X- обозначает анион иHet является таким, как определено выше, и необязательно включающий одну или более последующих стадий, на которых соединение формулы X в которой R1 и Het являются такими, как определено выше,или его соль, амид или лактон. 9. Способ по предыдущему пункту, в котором соединение формулы VII получают из соединения формулы VIa в которой X" обозначает галоген, С 1-С 8 алкилсульфонил или арилсульфонил. 10. Способ получения росувастатина, характеризующийся тем, что он включает стадии: а) (необязательно) взаимодействие алкил-3(S)-гидрокси-4-хлорбутирата формулы I в которой R2 обозначает С 1-С 8 алкил или С 5-С 7 циклоалкил или, альтернативно, -COOR2 также может образовать амид формулы -CONRaRb, в которой Ra и Rb могут независимо обозначать Н, С 1-С 8 алкил или С 5-С 7 циклоалкил, арил или совместно с N могут образовать гетероцикл,с йодидом с образованием алкил-3(S)-гидрокси-4-йодбутирата формулы IIb) (необязательно) введение защитной группы в соединение формулы II с получением защищенного в которой R1 и R2 являются такими, как определено выше; с) взаимодействие соединения формулы III с винилмагнийгалогенидом в присутствии галогенида меди(I) и фосфита формулы в которой все R', R" и R'" являются одинаковыми или разными и обозначают С 1-С 4 алкил, С 5 С 7 циклоалкил или арил,с получением алкена формулы IVd) гидролиз соединения формулы IV с получением соединения формулы Ve) взаимодействие соединения формулы V, в которой R1 является таким, как определено выше, с источником галогена в присутствии NaHCO3 с получением соединения формулы VI;f) (необязательно) разделение смеси диастереоизомеров, полученной на предыдущей стадии, с получением соединения формулы VIag) превращение соединения формулы VIa в соединение формулы VIIh) превращение соединения формулы VII в соединение формулы VIIIi) превращение соединения формулы VIII путем окисления в соединение формулы IXj) взаимодействие указанного соединения формулы IX с соединением формулы в которой Rx, Ry и Rz являются одинаковыми или разными и выбраны из группы, включающей С 1 С 8 алкил, или С 3-С 6 циклоалкил, или С 1-С 8 алкенил, или С 5-С 6 циклоалкенил, или арил,X- обозначает анион; иk) удаление защитной группы R1, необязательно очистку и превращение полученного соединения в кальциевую соль. 11. Способ по любому из пп.9 и 11, в котором анионом X- является галогенидный или алканоатный анион. 12. Применение для синтеза статинов промежуточного продукта формулы в которой R1 обозначает С 1-С 8 триалкилсилил, С 1-С 8 диалкиларилсилил, С 1-С 8 алкилдиарилсилил, где алкилы могут быть одинаковыми или разными, и R4 обозначает (необязательно замещенный галогеном,С 1-С 8 алкилом, арилом, С 1-С 8 алкилокси или арилокси) С 1-С 4 алкил. 13. Соединение формулы в которой R1 обозначает С 1-С 8 триалкилсилил, С 1-С 8 диалкиларилсилил, C1-С 8 алкилдиарилсилил, где алкилы и арилы могут быть одинаковыми или разными, и R4 обозначает (необязательно замещенный галогеном, С 1-С 8 алкилом, арилом, С 1-С 8 алкилокси или арилокси) С 1-С 4 алкил. 14. Соединение формулы в которой R1 обозначает С 1-С 8 триалкилсилил, С 1-С 8 диалкиларилсилил, C1-С 8 алкилдиарилсилил, где алкилы и арилы могут быть одинаковыми или разными.
МПК / Метки
МПК: C07F 7/18
Метки: hmg-cоa, редуктазы, способ, ингибиторов, синтеза
Код ссылки
<a href="https://eas.patents.su/30-14449-sposob-sinteza-ingibitorov-hmg-coa-reduktazy.html" rel="bookmark" title="База патентов Евразийского Союза">Способ синтеза ингибиторов hmg-coa редуктазы</a>
Комбинации ингибиторов протеин фарнезилтрансферазы и hmg koa редуктазы и их применение для лечения рака

Номер патента: 3860
Опубликовано: 30.10.2003
Авторы: Лиопольд Джюдит, Ньютон Роджер Шофильд
МПК: A61K 31/4155, C07D 233/54, A61P 35/00...
Метки: редуктазы, рака, комбинации, фарнезилтрансферазы, применение, лечения, ингибиторов, протеин
Формула / Реферат:
1. Фармацевтическая композиция, включающая соединение формулы I в которой R21 означает водород или C1-C6алкил; RQ означает n равен 0 или 1; A означает -CORa, -CO2Ra', -CONHRa', -CSRa, -C(S)ORa', -C(S)NHRa', -SO2Ra, -CONRaRa", -C(O)SRa или -C(S)NRaRa"; Ra, Ra' и Ra" означают независимо C1-C6алкил, -(CH2)m-циклоалкил, -(CH2)m-арил или -(CH2)m-гетероарил; каждое m независимо равно от 0 до 3; R1, R2 и R4 означают независимо...
Способ синтеза ингибиторов циклооксигеназы-2

Номер патента: 2975
Опубликовано: 26.12.2002
Авторы: Дэвис Ян В., Пай Филип Дж., Ларсен Роберт Д., Россен Кай, Корли Эдвард Г.
МПК: A61K 31/44, C07D 211/00
Метки: синтеза, циклооксигеназы-2, способ, ингибиторов
Формула / Реферат:
1. Способ синтеза соединения формулы I в которой присутствует 0-2 группы R; каждый из R, R' независимо представляет C1-10-алкил, С6-10-арил, аралкил, галоген, -S(O)mH, -S(O)mC1-6-алкил, -S(О)m-арил, нитро, амино, C1-6-алкиламино, ди-С1-6-алкиламино, -S(O)mNH2, -S(О)mNH-C1-6-алкил, -S(О)mNHC(О)СF3 и циано, причем алкильная и арильная группы и алкильная и арильная части аралкила, -S(О)m-C1-6-алкила, -S(О)m-арила, C1-6-алкиламино,...
Способ синтеза ингибиторов протеазы вич

Номер патента: 3022
Опубликовано: 26.12.2002
Авторы: Танг Роджер Деннис, О'каллахан Джон, Сингх Хардев, Робертсон Марк Стюарт, Макги Стивен, Дейнинджер Дейвид Д., Роджерс Кейт, Рут Стивен Джон, Эл-Фархан Эмил
МПК: C07D 307/20, A61P 31/18
Метки: вич, ингибиторов, способ, синтеза, протеазы
Формула / Реферат:
1. Способ получения соединения формулы (I) при котором 1) п-нитрофенилсульфонильную группу подвергают взаимодействию с соединением формулы (А) в котором Р является защитной группой амина; 2) снимают защиту с соединения, полученного на стадии (1); 3) соединение, полученное на стадии (2), подвергают взаимодействию с тетрагидрофурилоксикарбонильной группой; либо соединение, полученное на стадии (2), подвергают взаимодействию с фосгеном или его...
Новый способ синтеза соединений ( 2s, 3аs, 7as )-1-[ (s)-аланил]октагидро -1н-индол-2-карбоновой кислоты и их применение для синтеза периндоприла

Номер патента: 9062
Опубликовано: 26.10.2007
Авторы: Ланглуа Паскаль, Дюбюффе Тьерри
МПК: C07K 5/06, C07D 209/42, C07K 5/02...
Метки: синтеза, 1, кислоты, периндоприла, новый, 3аs, применение, s)-аланил]октагидро, 1н-индол-2-карбоновой, способ, соединений
Формула / Реферат:
1. Способ синтеза соединений (I) в которой R1 представляет собой атом водорода или бензильную группу, a R2 - это защитная группа для группы амина, представляющая собой трет-бутоксикарбонильную группу, характеризующийся тем, что 1-(1-циклогексен-1-ил)пирролидин формулы (III) подвергают реакции с соединением серина формулы (IV) в которой R1 является таким, как определено для формулы (I), a R3 - это защитная группа для группы амина,...
Новый способ синтеза (2s, 3аs, 7аs )-пергидроиндол-2-карбоновой кислоты и её сложных эфиров, а также их применение для синтеза периндоприла

Номер патента: 7948
Опубликовано: 27.02.2007
Авторы: Дюбюффе Тьерри, Ланглуа Паскаль
МПК: C07D 209/42
Метки: сложных, периндоприла, кислоты, 3аs, способ, эфиров, применение, также, новый, пергидроиндол-2-карбоновой, синтеза
Формула / Реферат:
1. Способ синтеза соединений формулы (I) в которой R представляет собой атом водорода или защитную группу для кислотной группы, характеризующийся тем, что 1-(1-циклогексен-1-ил)пирролидин формулы (III) подвергают реакции с соединением формулы (IV) в которой R является таким, как определено для формулы (I), a R' представляет собой защитную группу для аминовой группы, которая является отличной от R, с получением соединения формулы (V) в...
Предыдущий патент: Способ промышленной очистки исходного потока титана
Следующий патент: Производные трициклических n-гетероарилкарбоксамидов, содержащих группу бензимидазола, их получение и применение в терапии
Случайный патент: Состав с пролонгированным высвобождением, используемый с действующим началом, представляющим собой гемитартрат зольпидема