Бензимидазолтиофеновые соединения в качестве ингибиторов polo-подобных киназ (plk)
Номер патента: 14111
Опубликовано: 29.10.2010
Авторы: Чеунг Муи, Эммит Кайл Аллен, Салович Джеймс Майкл







Формула / Реферат
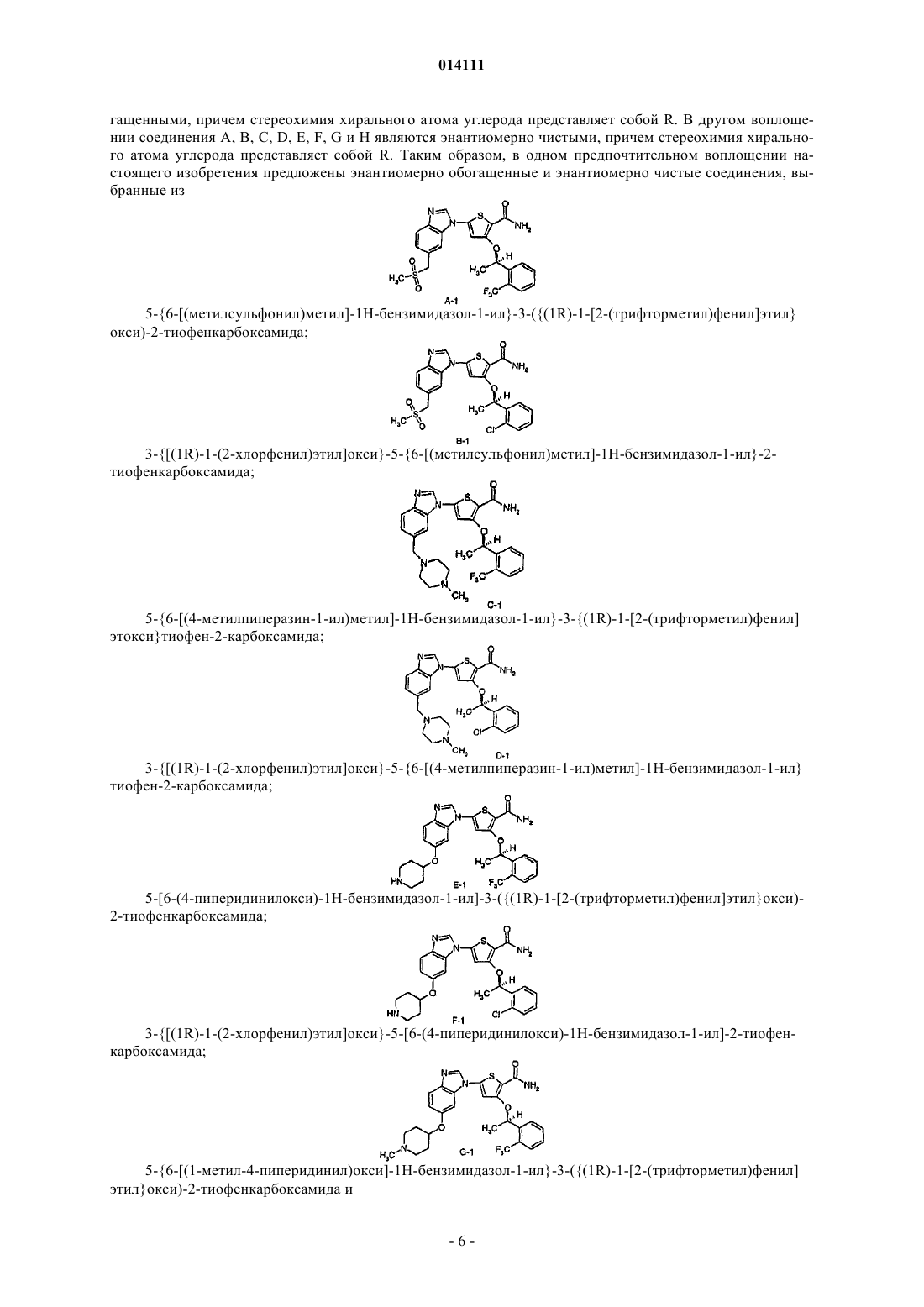
1. Соединение

2. Фармацевтически приемлемая соль соединения

3. Фармацевтическая композиция, содержащая соединение по п.1 или соль по п.2 и фармацевтически приемлемый носитель, разбавитель или эксципиент.
4. Фармацевтическая композиция по п.3, где указанная композиция энантиомерно обогащена изображенным R-изомером по отношению к соответствующему S-изомеру.
5. Фармацевтическая композиция по п.3, где указанная композиция содержит по меньшей мере 90 мас.% изображенного R-изомера по отношению к соответствующему S-изомеру.
Текст
Чеунг Муи, Эммит Кайл Аллен, Салович Джеймс Майкл (US) Представитель: В изобретении предложены бензимидазолтиофеновые соединения, фармацевтические композиции, содержащие их, способы их получения и их применение в качестве фармацевтических агентов (формулы А-Н). 014111 Предшествующий уровень техники Настоящее изобретение относится к новым бензимидазолтиофеновым соединениям, к фармацевтическим композициям, содержащим эти соединения, и к применению этих соединений в терапии.Polo-подобные киназы ("PLK") представляют собой сохранившиеся за время эволюции сериновые/треониновые киназы, которые играют важную роль в регуляции процессов в клеточном цикле. PLK играют роль во вступлении в митоз и в выходе из митоза в разных организмах от дрожжей до клеток млекопитающих. PLK включают PLK1, PLK2, PLK3 и PLK4. Сверхэкспрессия PLK1, по-видимому, в большой степени связана с неопластическими клетками(включая рак). Опубликованное исследование показало высокие уровни экспрессии РНК PLK1 в более чем 80% опухолей легкого и молочной железы при слабой экспрессии или ее отсутствии в соседней нормальной ткани. Несколько исследований показали взаимосвязь между экспрессией PLK, гистологической стадией и прогнозом при некоторых типах рака. Обнаружены существенные корреляции между процентным содержанием PLK-позитивных клеток и гистологической стадией рака яичников и эндометрия (Р 0,001). В этих исследованиях отмечено, что PLK сильно экспрессируется в инвазивных клетках карциномы эндометрия, и что это могло бы отражать уровень злокачественности и пролиферации при карциноме эндометрия. Используя анализ RT-PCR (полимеразная цепная реакция с обратной транскриптазой), сверхэкспрессию PLK обнаруживали в 97% карцином пищевода и 73% карцином желудка по сравнению с соответствующими нормальными тканями. Кроме того, пациенты с высокими уровнями сверхэкспрессии PLK при карциноме пищевода являлись группой со значительно худшим прогнозом,чем пациенты с низкими уровнями сверхэкспрессии PLK. При раковых заболеваниях головы и шеи повышенная экспрессия мРНК PLK1 наблюдалась в большинстве опухолей; анализ Каплана-Мейера показал, что пациенты с умеренными уровнями экспрессии PLK1 жили дольше, чем пациенты с высокими уровнями экспрессии PLK1. Анализ пациентов с немелкоклеточной карциномой легкого показал аналогичные результаты в отношении экспрессии PLK1. В публикации РСТ WO 2004/014899, SmithKline Beecham, раскрыты новые бензимидазолтиофеновые соединения формулы IN, О и S, или 5-6-членный гетероарил, имеющий 1, 2, 3 или 4 гетероатома, выбранных из N, О и S, причем каждый из них, возможно, замещен 1-2 заместителями, выбранными из группы, состоящей из галогено, алкила, оксо, групп -ОН, -R2-OH, -О-алкил, -R2-О-алкил, -NH2, -N(Н)алкил, -N(алкил)2, -CN и -N3;Q1 представляет собой группу формулы -(R2)a-(Y1)b-(R2)c-R3;a, b и с являются одинаковыми или разными, и каждый из них независимо равен 0 или 1, и по меньшей мере один из а или b равен 1;Q2 представляет собой группу формулы -(R2)aa-(Y2)bb-(R2)cc-R4 или две соседние группы Q2 выбраны из группы, состоящей из алкила, алкенила, -OR7, -S(O)fR7 и 7 8-NR R , и вместе с атомами углерода, с которыми они связаны, они образуют C5-6 циклоалкил, С 5-6 циклоалкенил, фенил, 5-7-членный гетероцикл, имеющий от 1 до 2 гетероатомов, выбранных из N, О и S, или 5-6-членный гетероарил, имеющий от 1 до 2 гетероатомов, выбранных из N, О и S; аа, bb и сс являются одинаковыми или разными, и каждый из них независимо равен 0 или 1; каждый из Y1 и Y2 является одинаковым или разным и независимо выбран из группы, состоящей из-О-, -S(O)f-, -N(R7)-, -С(О)-, -ОС(О)-, -СО 2-, -C(O)N(R7)-, -C(O)N(R7)S(O)2-, -OC(O)N(R7)-, -OS(O)2-,-S(O)2N(R7)-, -S(O)2N(R7)C(O)-, -N(R7)S(O)2-, -N(R7)C(O)-, -N(R7)CO2- и -N(R7)C(O)N(R7)-; каждый из R2 является одинаковым или разным и независимо выбран из группы, состоящей из алкилена, алкенилена и алкинилена; каждый из R3 и R4 является одинаковым или разным, и каждый независимо выбран из группы, состоящей из Н, галогено, алкила, алкенила, алкинила, -C(O)R7, -C(O)NR7R8, -CO2R7, -C(S)R7, -C(S)NR7R8,-1 014111 где кольцо А выбрано из группы, состоящей из С 5-10 циклоалкила, С 5-10 циклоалкенила, арила, 5-10 членного гетероцикла, имеющего 1, 2 или 3 гетероатома, выбранных из N, О и S, и 5-10-членного гетероарила, имеющего 1, 2 или 3 гетероатома, выбранных из N, О и S каждый d равен 0 или 1; е равен 0, 1, 2, 3 или 4; каждый R6 является одинаковым или разным и независимо выбран из группы, состоящей из Н, галогено, алкила, алкенила, алкинила, циклоалкила, циклоалкенила, Ph, Het, -CH(OH)-R2-OH, -C(O)R7,-CO2R7, -CO2-R2-Ph, -CO2-R2-Het, -C(O)NR7R8, -C(O)N(R7)C(O)R7, -C(O)N(R7)CO2R7, -C(O)N(R7)C(O)R5 выбран из группы, состоящей из Н, галогено, алкила, циклоалкила, OR7, -S(O)fR7, -NR7R8,-NHC(O)R7, -NHC(O)NR7R8 и NHS(O)2R7,f равен 0, 1 или 2; и каждый R7 и каждый R8 являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из Н, алкила, алкенила, алкинила, циклоалкила и циклоалкенила; где, когда R1 представляет собой -СО 2 СН 3 и n равен 0, тогда Q1 не является -ОН; или их фармацевтически приемлемая соль, сольват или физиологически функциональное производное. Также раскрыты фармацевтические композиции, содержащие эти соединения, способы их получения и способы лечения состояний, опосредованных PLK, с применением этих соединений Краткое изложение сущности изобретения Согласно первому аспекту изобретения предложено соединение, выбранное из:-2 014111 гдеуказывает хиральный атом углерода; и его фармацевтически приемлемые соли или сольваты. В другом аспекте предложено энантиомерно обогащенное соединение, выбранное из А, В, С, D, E,F, G и Н, где стереохимия хирального атома углерода представляет собой R. В третьем аспекте изобретения предложена фармацевтическая композиция, содержащая соединение, выбранное из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов. В одном воплощении фармацевтическая композиция дополнительно содержит фармацевтически приемлемый носитель, разбавитель или эксципиент. В четвертом аспекте изобретения предложен способ лечения состояния, опосредованного PLK, у млекопитающего, нуждающегося в этом. Данный способ включает введение млекопитающему терапевтически эффективного количества соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов. В пятом аспекте изобретения предложен способ лечения поддающегося лечению новообразования у млекопитающего, нуждающегося в этом. Данный способ включает введение млекопитающему терапевтически эффективного количества соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов. Поддающееся лечению новообразование может быть выбрано из группы, состоящей из рака молочной железы, рака толстой кишки, рака легкого, включая мелкоклеточный рак легкого и немелкоклеточный рак легкого, рака простаты, рака эндометрия, рака желудка, меланомы, рака яичника, рака поджелудочной железы, плоскоклеточной карциномы, карциномы головы и шеи, карциномы пищевода, гепатоцеллюлярного рака и гематологических злокачественных заболеваний,таких как острые лейкозы и агрессивные лимфомы. В шестом аспекте данного изобретения предложен способ лечения рака молочной железы у млекопитающего, нуждающегося в этом. Данный способ включает введение млекопитающему терапевтически эффективного количества соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов. В седьмом аспекте данного изобретения предложен способ лечения рака яичника у млекопитающего, нуждающегося в этом. Данный способ включает введение млекопитающему терапевтически эффективного количества соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов. В восьмом аспекте данного изобретения предложен способ лечения немелкоклеточного рака легкого у млекопитающего, нуждающегося в этом. Данный способ включает введение млекопитающему терапевтически эффективного количества соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов. В девятом аспекте данного изобретения предложен способ лечения рака простаты у млекопитающего, нуждающегося в этом. Данный способ включает введение млекопитающему терапевтически эффективного количества соединения, выбранного из А, В, С, D, E, F, G и Н, и его фармацевтически приемлемых солей или сольватов. В десятом аспекте данного изобретения предложен способ лечения гематологического злокачественного заболевания у млекопитающего, нуждающегося в этом. Данный способ включает введение млекопитающему терапевтически эффективного количества соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов. В другом аспекте данного изобретения предложен способ лечения состояния, характеризующегося неадекватной пролиферацией клеток. Данный способ включает приведение клетки в контакт с терапевтически эффективным количеством соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов. В другом аспекте настоящего изобретения предложен способ ингибирования пролиферации клетки. Данный способ включает приведение клетки в контакт с некоторым количеством соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов. В другом аспекте настоящего изобретения предложен способ ингибирования митоза в клетке. Данный способ включает введение в клетку некоторого количества соединения, выбранного из А, В, С, D, E,F, G и Н и их фармацевтически приемлемых солей или сольватов. В другом аспекте настоящего изобретения предложено соединение, выбранное из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов, для применения в терапии. В еще одном аспекте настоящего изобретения предложено соединение, выбранное из А, В, С, D, E,F, G и Н и их фармацевтически приемлемых солей или сольватов, для применения в лечении состояния,опосредованного PLK, у млекопитающего, нуждающегося в этом. В еще одном аспекте настоящего изобретения предложено соединение, выбранное из А, В, С, D, E,F, G и Н и их фармацевтически приемлемых солей или сольватов, для применения в лечении поддающегося лечению новообразования у млекопитающего. В еще одном аспекте настоящего изобретения предложено соединение, выбранное из А, В, С, D, E,F, G и Н и их фармацевтически приемлемых солей или сольватов, для применения в лечении рака молочной железы, рака яичника, немелкоклеточного рака легкого, рака простаты или гематологического злокачественного заболевания у млекопитающего.-3 014111 В другом аспекте настоящего изобретения предложено соединение, выбранное из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов, для применения в лечении состояния, характеризующегося неадекватной пролиферацией клеток. В еще одном аспекте настоящего изобретения предложено соединение, выбранное из А, В, С, D, E,F, G и Н и их фармацевтически приемлемых солей или сольватов, для применения в ингибировании пролиферации клетки. В еще одном аспекте настоящего изобретения предложено соединение, выбранное из А, В, С, D, E,F, G и Н и их фармацевтически приемлемых солей или сольватов, для применения в ингибировании митоза в клетке. В еще одном аспекте настоящего изобретения предложено применение соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов, для изготовления лекарственного средства для лечения состояния, опосредованного PLK, у млекопитающего. В еще одном аспекте настоящего изобретения предложено применение соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов, для изготовления лекарственного средства для лечения новообразования, поддающегося лечению, у млекопитающего. В еще одном аспекте настоящего изобретения предложено применение соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов, для изготовления лекарственного средства для лечения рака молочной железы, рака яичника, немелкоклеточного рака легкого,рака простаты или гематологического злокачественного заболевания у млекопитающего. В еще одном аспекте настоящего изобретения предложено применение соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов, для изготовления лекарственного средства для лечения состояния, характеризующегося неадекватной пролиферацией клеток у млекопитающего. В еще одном аспекте настоящего изобретения предложено применение соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов, для изготовления лекарственного средства для ингибирования пролиферации клетки. В еще одном аспекте настоящего изобретения предложено применение соединения, выбранного из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов, для изготовления лекарственного средства для ингибирования митоза в клетке. В еще одном аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение, выбранное из А, В, С, D, E, F, G и Н и их фармацевтически приемлемых солей или сольватов для применения в лечении новообразования, поддающегося лечению, такого как рак молочной железы, рак яичника, немелкоклеточный рак легкого, рак простаты или гематологические злокачественные заболевания у млекопитающего. Подробное описание изобретения При использовании в данном описании изобретения "соединение(я) по изобретению" или "соединение(я), выбранное(ые) из А, В, С, D, E, F, G и Н" означает соединение(я), выбранное(ые) из А, В, С, D, E,F, G и Н, или энантиомерно обогащенное(ые) соединение(я), выбранное(ые) из А, В, С, D, Е, F, G и Н,или его(их) фармацевтически приемлемую соль или сольват. При использовании в данном описании изобретения "соединение(я), выбранное(ые) из А-1, В-1, С 1, D-1, Е-1, F-1, G-1 и Н-1" означает соединение(я), выбранное(ые) из А-1, В-1, С-1, D-1, Е-1, F-1, G-1 и Н-1, или его(их) фармацевтически приемлемую соль или сольват. При использовании в данном описании изобретения термин "возможно" означает, что событие(я),описанное далее, может происходить или может не происходить, и включает как событие(я), которое(ые) происходит(ят), так и событие(я), которое(ые) не происходит(ят). Соединения по данному изобретению существуют в стереоизомерных формах (например, они содержат один или более чем один хиральный или асимметрический атом углерода). Термин "хиральный" относится к молекуле, которая не может быть наложена на свое зеркальное отображение. Термин "ахиральный" относится к молекуле, которая может быть наложена на свое зеркальное отображение. Термин "стереоизомеры" относится к соединениям, которые имеют одинаковый химический состав,но отличаются по расположению атомов или групп в пространстве. Стереоизомеры могут быть оптическими изомерами или геометрическими изомерами. Оптические изомеры включают как энантиомеры,так и диастереомеры. "Энантиомер" представляет собой один из пары оптических изомеров, содержащих хиральный атом углерода, молекулярная конфигурация которого имеет лево- и правосторонние (хиральные) формы. То есть "энантиомер" относится к каждому из пары оптических изомеров соединения, которые являются неналагающимися зеркальными отображениями друг друга. "Диастереомер" является одним из пары оптических изомеров соединения с двумя или более центрами асимметрии, молекулы которых не являются зеркальными отображениями друг друга. Номенклатура хирального центра определяется (R)-(S)-системой. Обозначено ли конкретное соединение согласно данной системе как "R-" или "S-" энантиомер, зависит от природы атомов или групп, которые связаны с хиральным углеродом. Энантиомеры отличаются по их поведению в отношении плоскополяризованного света, то есть по их оптической активности. Говорят, что энантиомер, который вращает плоскополяризованный свет по-4 014111 направлению часовой стрелки, является правовращающим, и его обозначают символом "d" или "(+)" для положительного вращения. Говорят, что энантиомер, который вращает плоскость поляризованного света в направлении против часовой стрелки, является левовращающим, и его обозначают символом "I" или"(-)" для отрицательного вращения. Не существует корреляции между конфигурацией энантиомеров и направлением, в котором они вращают плоскость поляризованного света. Также не существует необходимой корреляции между обозначением (R) и (S) и направлением вращения плоскости поляризованного света. Оптическую активность, или направление вращения плоскополяризованного света, энантиомера соединения по изобретению можно определить, используя стандартные методики. Термины "рацемат" и "рацемическая смесь" при использовании в данном описании изобретения относятся к смеси (R)- и (S)-оптических изомеров (например энантиомеров) соединения в равном, т.е. 50:50, соотношении. Термин "энантиомерно обогащенный" при использовании в данном описании изобретения относится к препаратам, содержащим смесь оптических изомеров, в которой количество одного энантиомера больше, чем количество другого. Таким образом, термин "энантиомерно обогащенный" относится к смесям оптических изомеров, где соотношение энантиомеров составляет более чем 50:50. Энантиомерно обогащенное соединение содержит более 50 мас.% одного энантиомера по отношению к другому. Например, энантиомерно обогащенный 5-6-[(метилсульфонил)метил]-1 Н-бензимидазол-1-ил-3-1R)-1[2-(трифторметил)фенил]этилокси)-2-тиофенкарбоксамид относится к композиции, содержащей более 50 мас.% (R)-энантиомера данного соединения по отношению к (S)-энантиомеру. В одном воплощении энантиомерно обогащенное соединение содержит по меньшей мере 75 мас.% одного энантиомера по отношению к другому. В другом воплощении энантиомерно обогащенное соединение содержит по меньшей мере 80 мас.% одного энантиомера по отношению к другому. В одном конкретном воплощении энантиомерно обогащенное соединение содержит по меньшей мере 85 мас.% одного энантиомера по отношению к другому. Термин "энантиомерно чистое" при использовании в данном описании изобретения относится к энантиомерно обогащенным соединениям, содержащим по меньшей мере 90 мас.% одного энантиомера по отношению к другому. В одном воплощении энантиомерно чистое соединение содержит по меньшей мере 95 мас.% одного энантиомера по отношению к другому. В одном конкретном воплощении энантиомерно чистое соединение содержит по меньшей мере 99 мас.% одного энантиомера по отношению к другому. Согласно настоящему изобретению предложены соединения, выбранные из гдеуказывает хиральный атом углерода; и их фармацевтически приемлемых солей и сольватов. В одном конкретном воплощении соединения А, В, С, D, E, F, G и Н являются энантиомерно обо-5 014111 гащенными, причем стереохимия хирального атома углерода представляет собой R. В другом воплощении соединения А, В, С, D, Е, F, G и Н являются энантиомерно чистыми, причем стереохимия хирального атома углерода представляет собой R. Таким образом, в одном предпочтительном воплощении настоящего изобретения предложены энантиомерно обогащенные и энантиомерно чистые соединения, выбранные из 3-[(1R)-1-(2-Хлорфенил)этил]окси-5-6-[(1-метил-4-пиперидинил)окси]-1 Н-бензимидазол-1-ил 2-тиофенкарбоксамида; и их фармацевтически приемлемых солей и сольватов. Специалистам в данной области очевидно, что соединения по настоящему изобретению можно использовать в форме их фармацевтически приемлемых солей или сольватов. Фармацевтически приемлемые соли соединений по настоящему изобретению (или их энантиомерно обогащенные или энантиомерно чистые формы) включают обычные соли, образованные из фармацевтически приемлемых неорганических или органических кислот или оснований, а также четвертичные аммониевые соли. Более конкретные примеры подходящих солей кислот включают соли соляной, бромисто-водородной, серной, фосфорной, азотной, перхлорной, фумаровой, уксусной, пропионовой, янтарной, гликолевой, муравьиной, молочной, малеиновой, винной, лимонной, пальмовой, малоновой, гидроксималеиновой, фенилуксусной,глутаминовой, бензойной, салициловой, фумаровой, толуолсульфоновой, метансульфоновой (мезилат),нафталин-2-сульфоновой, бензолсульфоновой, гидроксинафтойной, иодисто-водородной, яблочной, стериновой, дубильной кислот и подобные им. Другие кислоты, такие как щавелевая, хотя сами и не являются фармацевтически приемлемыми, могут быть полезны в получении солей, пригодных в качестве промежуточных соединений в получении соединений по изобретению и их фармацевтически приемлемых солей. Конкретные примеры подходящих основных солей включают натриевые, литиевые, калиевые, магниевые, алюминиевые, кальциевые, цинковые, N',N-дибензилэтилендиаминовые, хлорпрокаиновые, холиновые, диэтаноламиновые, этилендиаминовые, N-метилглюкаминовые и прокаиновые соли. Термин "сольват" при использовании в данном описании изобретения относится к комплексу переменной стехиометрии, образованному растворенным веществом (соединением по изобретению или его энантиомерно обогащенной или энантиомерно чистой формой) и растворителем. Растворители, в качестве примера, включают воду, метанол, этанол или уксусную кислоту. Способы получения фармацевтически приемлемых солей и сольватов соединений по изобретению являются традиционными в данной области техники. Смотрите, например, Burger's Medicinal ChemistryAnd Drug Discovery 5th Edition, Vol. 1: Principles And Practice. Как будет очевидно специалистам в данной области в способах получения соединений по изобретению, описанных ниже, некоторые промежуточные соединения альтернативно могут находиться в форме фармацевтически приемлемых солей или сольватов соединения. Эти термины в том виде, как они применяются к любому промежуточному соединению, используемому в способе получения соединений по изобретению, имеют те же самые значения, что и указаны выше в отношении соединений по изобретению. Способы получения фармацевтически приемлемых солей и сольватов таких промежуточных соединений известны в данной области и аналогичны способу получения фармацевтически приемлемых солей и сольватов соединений по изобретению. Соединения по настоящему изобретению типично представляют собой ингибиторы PLK. Под ингибитором PLK подразумевается соединение, которое демонстрирует pIC50 более 6 в анализе ингибирования PLK, описанном ниже в примерах, или IC50 менее 10 мкМ в анализе с метиленовым синим или в анализе ингибирования роста Cell-Titre Glo, описанных ниже в примерах; более конкретно, ингибитор PLK представляет собой соединение, которое демонстрирует pIC50 более 7 или IC50 менее 1 мкМ, с использованием способов, описанных в примерах, приведенных ниже. Согласно настоящему изобретению дополнительно предложены соединения по изобретению для применения в лечении животного, например млекопитающего, такого как человек. В частности, в настоящем изобретении предложены соединения для применения в лечении состояния, опосредованногоPLK. В настоящем изобретении также предложены соединения для применения в лечении новообразования, поддающегося лечению. Термин "новообразование, поддающееся лечению" определен ниже. В частности, в настоящем изобретении предложены соединения для применения в лечении ряда солидных опухолей, включая, но не без ограничения ими, рак молочной железы, рак яичника, немелкоклеточный рак легкого, рак простаты и гематологические злокачественные заболевания, включая, без ограничения ими, острые (миелоидные и лимфоидные) лейкозы и агрессивные лимфомы. Согласно настоящему изобретению предложены соединения для применения в лечении состояния,характеризующегося неадекватной пролиферацией клеток. Согласно настоящему изобретению также предложены соединения для применения в ингибировании пролиферации клетки. Согласно настоящему изобретению также предложены соединения для применения в ингибировании митоза в клетке. Согласно настоящему изобретению предложены способы лечения некоторых состояний или заболеваний, все из которых включают стадию введения терапевтически эффективного количества соединения по изобретению. Термин "лечение" при использовании в данном описании изобретения относится к-7 014111 облегчению определенного состояния, к устранению или ослаблению симптомов этого состояния, к замедлению или устранению развития состояния и к предупреждению или к задерживанию повторного возникновения данного состояния у субъекта, страдавшего от него ранее. Термин "терапевтически эффективное количество" при использовании в данном описании изобретения означает количество соединения по изобретению, которое является достаточным у субъекта, которому его вводят, чтобы вызвать тот биологический или медицинский ответ клеточной культуры, ткани,системы, животного (включая человека), которого добиваются, например, исследователь или практикующий врач. Например, терапевтически эффективное количество соединения по изобретению для лечения состояния, опосредованного PLK, представляет собой количество, достаточное для лечения состояния, опосредованного PLK, у субъекта. Аналогично, терапевтически эффективное количество соединения по изобретению для лечения новообразования, поддающегося лечению, представляет собой количество, достаточное для лечения новообразования, поддающегося лечению, у субъекта. В одном воплощении настоящего изобретения терапевтически эффективное количество соединения по изобретению представляет собой количество, достаточное для ингибирования митоза клетки. В одном воплощении настоящего изобретения терапевтически эффективное количество соединения по изобретению представляет собой количество, достаточное для регуляции, модуляции, связывания или ингибирования PLK. Точное терапевтически эффективное количество соединения по изобретению будет зависеть от ряда факторов, включая, без ограничения ими, возраст и массу субъекта, которого лечат, конкретное расстройство, требующее лечения, и его тяжесть, природу препарата и путь введения, и, в конечном счете,будет определять по усмотрению лечащего врача или ветеринара. Обычно соединение по изобретению будут давать для лечения в интервале от 0,1 до 200 мг/кг массы тела реципиента (животного) в сутки или на дозу или на цикл лечения и более типично в интервале от 1 до 100 мг/кг массы тела в сутки или на дозу или на цикл лечения. Приемлемые дозировки могут составлять от примерно 0,1 до примерно 2000 мг в сутки, на дозу или цикл лечения и предпочтительно от примерно 0,1 до примерно 500 мг в сутки, на дозу или цикл лечения. В качестве одного аспекта настоящего изобретения предложены способы регуляции, модуляции,связывания или ингибирования PLK для лечения состояний, опосредованных PLK, в частности PLK1."Регуляция, модуляция, связывание или ингибирование PLK" относится к регуляции, модуляции, связыванию или ингибированию активности PLK, в частности PLK1, а также к регуляции, модуляции, связыванию или ингибированию сверхэкспрессии PLK, в частности PLK1. Такие состояния включают некоторые новообразования (включая раковые заболевания и опухоли), которые ассоциированы с PLK, в частности с PLK1, и состояния, характеризующиеся неадекватной пролиферацией клеток. В настоящем изобретении предложен способ лечения состояния, опосредованного PLK, в частностиPLK1, который включает введение животному терапевтически эффективного количества соединения по изобретению. Этот способ и другие способы по настоящему изобретению являются полезными для лечения животного, такого как млекопитающее, и, в частности, людей. Состояния, опосредованные PLK, известны в данной области и включают, без ограничения ими, новообразования и состояния, характеризующиеся неадекватной пролиферацией клеток. В настоящем изобретении также предложен способ лечения новообразования, поддающегося лечению (рака или опухоли), у животного, такого как млекопитающее (например, человек), нуждающегося в этом, причем данный способ включает введение данному животному терапевтически эффективного количества соединения по изобретению. "Новообразование, поддающееся лечению" при использовании в данном описании изобретения относится к новообразованиям, которые поддаются лечению ингибиторомPLK, известны в данной области и включают как первичные, так и метастатические опухоли и раковые заболевания. Например, поддающиеся лечению новообразования, входящие в объем настоящего изобретения, включают, без ограничения ими, рак молочной железы, рак толстой кишки, рак легкого (включая мелкоклеточный рак легкого и немелкоклеточный рак легкого), рак простаты, рак эндометрия, рак желудка, меланому, рак яичника, рак поджелудочной железы, плоскоклеточную карциному, карциному головы и шеи, карциному пищевода, гепатоцеллюлярную карциному и гематологические злокачественные заболевания, включая, без ограничения ими, острые лейкозы и агрессивные лимфомы. В одном конкретном воплощении настоящего изобретения предложен способ лечения рака молочной железы у животного, такого как млекопитающее (например, человек), нуждающегося в этом, путем введения терапевтически эффективного количества соединения по настоящему изобретению В другом конкретном воплощении настоящего изобретения предложен способ лечения рака яичника у животного, такого как млекопитающее (например, человек), нуждающегося в этом, путем введения терапевтически эффективного количества соединения по настоящему изобретению. В другом конкретном воплощении согласно настоящему изобретению предложен способ лечения немелкоклеточного рака легкого у животного, такого как млекопитающее (например, человек), нуждающегося в этом, путем введения терапевтически эффективного количества соединения по настоящему изобретению. В другом конкретном воплощении согласно настоящему изобретению предложен способ лечения рака простаты у животного, такого как мле-8 014111 копитающее (например, человек), нуждающегося в этом, путем введения терапевтически эффективного количества соединения по настоящему изобретению. В другом конкретном воплощении согласно настоящему изобретению предложен способ лечения острого лейкоза, включая острый миелоидный лейкоз и острый лимфоидный лейкоз, у животного, такого как млекопитающее (например человек), нуждающегося в этом, путем введения терапевтически эффективного количества соединения по настоящему изобретению. В другом конкретном воплощении согласно настоящему изобретению предложен способ лечения агрессивной лимфомы у животного, такого как млекопитающее (например человек), нуждающегося в этом, путем введения терапевтически эффективного количества соединения по настоящему изобретению. При лечении таких поддающихся лечению новообразований соединения по данному изобретению можно использовать по одному или с получением аддитивных или синергических эффектов с одним или более другими соединениями по изобретению, или в комбинации с некоторыми существующими химиотерапиями и/или с другими терапиями против новообразований. Кроме того, соединения по данному изобретению можно использовать для восстановления эффективности одного или более других соединений по данному изобретению, некоторых существующих химиотерапий и/или других терапий против новообразований. Термин "терапии против новообразований" при использовании в данном описании изобретения включает, без ограничения ими, цитотоксическую химиотерапию, гормональную терапию,целевые киназные ингибиторы, терапевтические моноклональные антитела, хирургию и лучевую терапию. Согласно настоящему изобретению также предложен способ лечения состояния, характеризующегося неадекватной пролиферацией клеток, у животного, такого как млекопитающее (например, человек),нуждающегося в этом. Данный способ включает введение терапевтически эффективного количества соединения по настоящему изобретению. Под "неадекватной пролиферацией клеток" подразумевается пролиферация клеток в результате неадекватного роста клеток, пролиферация клеток в результате избыточного деления клеток, пролиферация клеток в результате деления клеток с повышенной скоростью, пролиферация клеток в результате неадекватного выживания клеток и/или клеточная пролиферация в нормальной клетке, происходящая с нормальной скоростью, которая, тем не менее, является нежелательной. Состояния, характеризующиеся неадекватной пролиферацией клеток, включают, без ограничения ими,новообразования, пролиферативные расстройства кровеносных сосудов, фиброзные расстройства, мезангиально-клеточные пролиферативные расстройства и воспалительные/иммунноопосредованные заболевания. Пролиферативные расстройства кровеносных сосудов включают артрит и рестеноз. Фиброзные расстройства включают цирроз печени и атеросклероз. Мезангиально-клеточные пролиферативные расстройства включают гломерулонефрит, злокачественный нефросклероз и гломерулопатии. Воспалительные/иммуноопосредованные расстройства включают псориаз, заживление застарелых ран, отторжение органотрансплантата, синдромы тромбозной микроангиопатии и нейродегенеративные заболевания. Остеоартрит и другие заболевания, зависимые от пролиферации остеокластов, связанные с избыточной резорбцией кости, являются примерами состояний, характеризующихся неадекватной пролиферацией клеток, при которых пролиферация клеток происходит в нормальных клетках с нормальной скоростью, но, тем не менее, является нежелательной. Согласно настоящему изобретению также предложен способ ингибирования пролиферации клетки,причем данный способ включает приведение данной клетки в контакт с количеством соединения по изобретению, достаточным для ингибирования пролиферации клетки. В одном конкретном воплощении данная клетка представляет собой неопластическую клетку. В одном конкретном воплощении данная клетка представляет собой неадекватно пролиферирующую клетку. Термин "неадекватно пролиферирующая клетка" при использовании в данном описании изобретения относится к клеткам, которые растут неадекватно (ненормально), к клеткам, которые делятся избыточно или с повышенной скоростью, к клеткам, которые неадекватно (ненормально) выживают, и/или к нормальным клеткам, которые пролиферируют с нормальной скоростью, но для которых пролиферация является нежелательной. Неопластические клетки (включая раковые клетки) являются примером неадекватно пролиферирующих клеток, но они не являются единственными неадекватно пролиферирующими клетками.PLK является необходимой для митоза клетки и, соответственно, считается, что соединения по изобретению являются эффективными для ингибирования митоза. "Ингибирование митоза" относится к ингибированию входа в фазу М клеточного цикла, к ингибированию нормального хода фазы М клеточного цикла, как только клетка вошла в фазу М, и к ингибированию нормального выхода из фазы М клеточного цикла. Таким образом, соединения по настоящему изобретению могут ингибировать митоз путем ингибирования вхождения клетки в митоз, путем ингибирования прохождения клетки через митоз или путем ингибирования выхода клетки из митоза. В одном аспекте настоящего изобретения предложен способ ингибирования митоза в клетке, включающий введение в клетку количества соединения по изобретению,достаточного для ингибирования митоза. В одном конкретном воплощении данная клетка представляет собой неопластическую клетку. В одном конкретном воплощении данная клетка представляет собой неадекватно пролиферирующую клетку. Согласно настоящему изобретению также предложено применение соединения по изобретению для-9 014111 изготовления лекарственного средства для лечения состояния, опосредованного PLK, у животного, такого как млекопитающее (например, человек). Согласно настоящему изобретению дополнительно предложено применение соединения для изготовления лекарственного средства для лечения поддающегося лечению новообразования у животного. В частности, согласно настоящему изобретению предложено применение соединения для изготовления лекарственного средства для лечения рака молочной железы у животного. Согласно настоящему изобретению также предложено применение соединения для изготовления лекарственного средства для лечения рака яичника у животного. Согласно настоящему изобретению предложено применение соединения для изготовления лекарственного средства для лечения немелкоклеточного рака легкого у животного. Согласно настоящему изобретению также предложено применение соединения для изготовления лекарственного средства для лечения рака простаты у животного. Согласно настоящему изобретению предложено применение соединения для изготовления лекарственного средства для лечения острого лейкоза (включая острый миелоидный и острый лимфоидный лейкоз) у животного. Согласно настоящему изобретению предложено применение соединения для изготовления лекарственного средства для лечения агрессивной лимфомы у животного. Согласно настоящему изобретению дополнительно предложено применение соединения для изготовления лекарственного средства для лечения состояния, характеризующегося неадекватной пролиферацией клеток. Согласно настоящему изобретению дополнительно предложено применение соединения для изготовления лекарственного средства для ингибирования пролиферации клетки. Согласно настоящему изобретению дополнительно предложено применение соединения для изготовления лекарственного средства для ингибирования митоза в клетке. Хотя для применения в терапии терапевтически эффективное количество соединения по изобретению, возможно, может быть введено в виде необработанного химического соединения, обычно оно представлено в виде активного ингредиента фармацевтической композиции или препарата. Соответственно,согласно данному изобретению дополнительно предложена фармацевтическая композиция, содержащая соединение по изобретению. Данная фармацевтическая композиция может дополнительно содержать один или более чем один фармацевтически приемлемый носитель, разбавитель и/или эксципиент. Данные носитель(и), разбавитель(и) и/или эксципиент(ы) должны быть приемлемыми в смысле совместимости с другими ингредиентами данного препарата и не вредными для их реципиента. Согласно другому аспекту данного изобретения также предложен способ получения фармацевтического препарата, включающий смешивание соединения по изобретению с одним или более чем одним фармацевтически приемлемым носителем, разбавителем и/или эксципиентом. Фармацевтические препараты могут быть представлены в стандартной лекарственной форме, содержащей заданное количество активного ингредиента на стандартную дозу. Такая единица может содержать терапевтически эффективную дозу соединения по изобретению или часть терапевтически эффективной дозы, так что многократные стандартные лекарственные формы можно вводить в заданное время для достижения желательной терапевтически эффективной дозы. Предпочтительными композициями со стандартными дозами являются препараты, содержащие суточную дозу или субдозу активного ингредиента, как здесь изложено выше, или ее подходящую долю. Кроме того, такие фармацевтические препараты можно получить любым способом, хорошо известным в фармацевтической области. Фармацевтические препараты могут быть адаптированы для введения любым подходящим путем,например пероральным (включая буккальный или сублингвальный), ректальным, назальным, местным(включая подкожный, внутримышечный, внутривенный или внутрикожный) путем. Такие препараты могут быть получены любым способом, известным в области фармации, например путем сочетания активного ингредиента с носителем(ями) или с эксципиентом(ами). Фармацевтические препараты, адаптированные для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы или таблетки; порошков или гранул; растворов или суспензий в водных или неводных жидкостях, пищевых пен или кремов; жидких эмульсий масло-вводе или жидких эмульсий вода-в-масле. Например, для перорального введения в форме таблетки или капсулы активный лекарственный компонент можно объединять с пероральным, нетоксичным, фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и тому подобное. Порошки получают путем измельчения данного соединения до подходящего мелкого размера и смешивания с аналогично измельченным фармацевтическим носителем, таким как пищевой углеводород, такой как, например, крахмал или манит. Также могут присутствовать корригент, консервант, диспергирующий агент и краситель Капсулы изготавливают путем получения порошковой смеси, как описано выше, и заполнения ей формованных желатиновых оболочек. Перед операцией заполнения в порошковую смесь можно добавить скользящие смазывающие вещества, такие как коллоидный диоксид кремния, тальк, стеарат магния,стеарат кальция и твердый полиэтиленгликоль. Также можно добавить разрыхлитель или солюбилизирующий агент, такой как агар-агар, карбонат кальция или карбонат натрия, для улучшения доступности лекарственного средства при проглатывании данной капсулы. Кроме того, когда это желательно или необходимо, в данную смесь также можно включать подхо- 10014111 дящие связующие вещества, смазки, разрыхлители и красители. Подходящие связующие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические камеди, такие как аравийская камедь, трагакантовая камедь или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воска и тому подобное. Смазки, используемые в этих лекарственных формах, включают натрия олеат, натрия стеарат, магния стеарат, натрия бензоат, натрия ацетат, натрия хлорид и тому подобное. Разрыхлители включают, без ограничения ими,крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и тому подобное. Таблетки готовят в виде препарата, например, путем получения порошковой смеси, гранулирования или комкования, добавления смазки и разрыхлителя и прессования в таблетки. Порошковую смесь получают путем смешивания подходящим образом измельченного соединения с разбавителем или с основой, как описано выше, и,возможно, со связующим веществом, таким как карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, с замедлителем растворения, таким как парафин, с ускорителем всасывания, таким как четвертичная соль, и/или с абсорбционным агентом, таким как бентонит, каолин или дикальция фосфат. Порошковую смесь можно гранулировать путем увлажнения со связующим веществом, таким как сироп, крахмальная паста, акациевая камедь или растворы целлюлозных или полимерных веществ, и продавливания через сетку. В качестве альтернативы гранулированию, порошковую смесь можно прогнать через таблетировочную машину, в результате чего получают не полностью сформированные куски,разламывающиеся на гранулы. Гранулы можно смазать для предотвращения прилипания к формующим таблетки пуансонам путем добавления стеариновой кислоты, стеаратной соли, талька или минерального масла. Смазанную смесь затем прессуют в таблетки. Соединения по настоящему изобретению также можно объединять с легкосыпучим инертным носителем и непосредственно прессовать в таблетки без прохождения через стадии гранулирования или комкования. Можно обеспечить прозрачное или непрозрачное защитное покрытие, состоящее из изолирующего покрытия из шеллака, покрытия из сахара или полимерного вещества и глянцевого покрытия из воска. В эти покрытия можно добавлять красители для различения разных стандартных дозировок. Пероральные жидкости, такие как растворы, сиропы и эликсиры, можно получать в стандартных лекарственных формах так, что данное количество содержит заданное количество активного ингредиента. Сиропы можно получать путем растворения соединения в подходящим образом ароматизированном водном растворе, тогда как эликсиры получают путем применения нетоксичного спиртового наполнителя. Суспензии можно приготовить в виде препарата путем диспергирования соединения в нетоксичном наполнителе. Также можно добавлять солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и полиоксиэтиленсорбитоловые эфиры, консерванты, корригирующие добавки, такие как масло перечной мяты или природные подсластители, или сахарин, или другие искусственные подсластители и тому подобное. Когда это уместно, препараты в виде стандартной лекарственной формы для перорального введения можно микроинкапсулировать. Препарат также может быть приготовлен для продления или замедления высвобождения, например, путем нанесения покрытия или заделывания частиц вещества в полимеры,воск или тому подобное. Соединения по данному изобретению также можно вводить в виде липосомных систем доставки,таких как маленькие однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Липосомы можно сформировать из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Соединения по данному изобретению также можно доставлять, используя моноклональные антитела в качестве индивидуальных носителей, с которыми связаны молекулы соединения. Соединения также можно связать с растворимыми полимерами в качестве нацеливаемых носителей лекарственного средства. Такие полимеры могут включать пептиды, поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный остатками пальмитоила. Кроме того, соединения можно связать с классом биоразлагаемых полимеров, полезных для достижения контролируемого высвобождения лекарственного средства, например с полимолочной кислотой, полиэпсилон-капролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и со сшитыми или амфипатическими блок-сополимерами гидрогелей. Фармацевтические препараты, адаптированные для трансдермального введения, могут быть представлены в виде отдельных пластырей, предназначенных для того, чтобы оставаться в тесном контакте с эпидермисом реципиента в течение длительного периода времени. Например, активный ингредиент может быть доставлен из пластыря ионтофорезом, как в общем виде описано в Pharmaceutical Research,3(6):318 (1986). Фармацевтические препараты, адаптированные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел. Для лечения глаз или других наружных тканей, например рта и кожи, препараты предпочтительно наносят в виде мази или крема для местного применения. При приготовлении в виде препарата мази, активный ингредиент можно использовать либо с парафиновой, либо со смешивающейся с водой основой- 11014111 мази. Альтернативно, активный ингредиент можно приготовить в виде крема с кремовой основой маслов-воде или вода-в-масле. Фармацевтические препараты, адаптированные для местных введений в глаз, включают глазные капли, где активный ингредиент растворен или суспендирован в подходящем носителе, особенно в водном растворителе. Фармацевтические препараты, адаптированные для местного введения в рот, включают лепешки,пастилки и полоскания для полости рта. Фармацевтические препараты, адаптированные для ректального введения, могут быть представлены в виде суппозиториев или клизм. Фармацевтические препараты, адаптированные для назального введения, где носитель представляет собой твердое вещество, включают грубый порошок, имеющий размер частиц, например, в интервале от 20 до 500 мкм, который вводят способом, при котором производится втягивание воздуха через нос, т.е. посредством быстрой ингаляции через носовой ход из контейнера с порошком, удерживаемого вплотную к носу. Подходящие препараты, данный носитель является жидкостью, для введения в виде назального спрея или в виде назальных капель включают водные или масляные растворы активного ингредиента. Фармацевтические препараты, адаптированные для введения путем ингаляции, включают присыпки или аэрозоли, которые могут быть образованы посредством различных типов дозирующих аэрозольных баллончиков, небулайзеров или инсуффляторов. Фармацевтические препараты, адаптированные для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или спреевых препаратов. Фармацевтические препараты, адаптированные для парентерального введения, включают водные и неводные стерильные инъецируемые растворы, которые могут содержать антиоксиданты, буферы, бактериостатические средства и растворенные вещества, которые делают данный препарат изотоничным крови предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Препараты могут быть представлены в однодозовых или многодозовых контейнерах, например в запаянных ампулах и сосудах, и могут храниться в сублимированном (лиофилизированном) состоянии, которое требует только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед применением. Инъекционные растворы и суспензии, приготовленные для немедленного приема, могут быть получены из стерильных порошков,гранул и таблеток. Следует понимать, что в дополнение к ингредиентам, конкретно упомянутым выше, препараты могут включать другие агенты, традиционные в данной области, имеющие отношение к типу рассматриваемого препарата, например агенты, подходящие для перорального введения, могут включать корригенты. В вышеописанных способах лечения и применениях соединение по изобретению можно использовать само по себе, в комбинации с одним или более чем одним другим соединением по изобретению, или в комбинации с другими терапевтическими агентами и/или в комбинации с другими антинеопластическими терапиями. В частности, в способах лечения состояний, опосредованных PLK, и в способах лечения новообразований, поддающихся лечению, предусматривается комбинация с другими химиотерапевтическими агентами, а также комбинация с хирургической терапией и с лучевой терапией. Термин "химиотерапевтический" при использовании в данном описании изобретения относится к любому химическому агенту, оказывающему терапевтический эффект на субъекта, которому его вводят. "Химиотерапевтические" агенты включают, без ограничения ими, антинеопластические агенты, анальгетики и противорвотные средства. Термин "антинеопластические агенты" при использовании в данном описании изобретения включает как цитостатические, так и цитотоксические агенты, такие как, но без ограничения ими, агенты для цитотоксической химиотерапии, гормональной терапии, нацеленные ингибиторы киназы и терапевтические моноклональные антитела. Комбинированные терапии согласно настоящему изобретению, таким образом, включают введение по меньшей мере одного соединения по изобретению и применение по меньшей мере одного другого способа лечения рака. В одном воплощении комбинированные терапии согласно настоящему изобретению включают введение по меньшей мере одного соединения по изобретению и по меньшей мере одного другого химиотерапевтического агента. В одном конкретном воплощении настоящее изобретение включает введение по меньшей мере одного соединения по изобретению и по меньшей мере одного антинеопластического агента. В качестве дополнительного аспекта, согласно настоящему изобретению предложены способы лечения и применения, как описано выше, которые включают введение соединения по изобретению вместе с по меньшей мере одним химиотерапевтическим агентом. В одном конкретном воплощении данный химиотерапевтический агент представляет собой антинеопластический агент. В другом воплощении настоящего изобретения предложена фармацевтическая композиция, как описано выше, дополнительно содержащая по меньшей мере один другой химиотерапевтический агент, более конкретно данный химиотерапевтический агент представляет собой антинеопластический агент. Обычно любой химиотерапевтический агент, который обладает активностью против поддающегося лечению новообразования, которое лечат, можно использовать в комбинации с соединениями по изобре- 12014111 тению, при условии, что этот конкретный агент клинически совместим с терапией, использующей соединение по данному изобретению. Типичные антинеопластические агенты, полезные в настоящем изобретении, включают, но без ограничения ими, агенты против микротрубочек, такие как дитерпеноиды и алкалоиды барвинка; координационные комплексы платины; алкилирующие агенты, такие как азотистые иприты, оксазафосфорины, алкилсульфонаты, нитрозомочевины и триазены; антибиотики, такие как антрациклины, актиномицины и блеомицины; ингибиторы топоизомеразы II, такие как эпиподофиллотоксины; антиметаболиты, такие как аналоги пурина и пиримидина, и антифолатные соединения; ингибиторы топоизомеразы I, такие как камптотецины; гормоны и аналоги гормонов; ингибиторы путей трансдукции сигналов; ингибиторы ангиогенеза на основе нерецепторной тирозинкиназы; иммунотерапевтические агенты; проапоптозные агенты и ингибиторы передачи сигнала в клеточном цикле. Агенты против микротрубочек или антимитотические агенты представляют собой фазоспецифичные агенты, активные против микротрубочек опухолевых клеток на протяжении фазы М или фазы митоза клеточного цикла. Примеры агентов против микротрубочек включают, без ограничения ими, дитерпеноиды и алкалоиды барвинка. Примеры дитерпеноидов включают, без ограничения ими, паклитаксел и его аналог доцетаксел. Примеры алкалоидов барвинка включают, без ограничения ими, винбластин, винкристин и винорелбин. Координационные комплексы платины представляют собой нефазоспецифичные антинеопластические агенты, которые взаимодействуют с ДНК. Комплексы платины поступают в опухолевые клетки, подвергаются гидратации и образуют внутри межнитевые поперечные связи с ДНК, что приводит к неблагоприятным биологическим эффектам на опухоль. Примеры координационных комплексов платины включают, без ограничения ими, оксалиплатин, цисплатин и карбоплатин. Алкилирующие агенты представляют собой нефазоспецифичные антинеопластические агенты и сильные электрофилы. Обычно алкилирующие агенты образуют, посредством алкилирования ковалентные связи с ДНК через нуклеофильные группировки молекулы ДНК, такие как фосфатные, амино и гидроксильные группы. Такое алкилирование нарушает функцию нуклеиновой кислоты, что приводит к гибели клетки. Примеры алкилирующих агентов включают, без ограничения ими, азотистые иприты, такие как циклофосфамид, мелфалан и хлорамбуцил; алкилсульфонаты, такие как бусульфан; нитрозомочевины, такие как кармустин; и триазены, такие как дакарбазин. Антибиотические химиотерапевтические агенты представляют собой нефазоспецифичные агенты,которые связываются или встраиваются в ДНК. Обычно такое действие приводит к стабильным комплексам ДНК или к разрыву нити, что нарушает обычную функцию нуклеиновых кислот, приводя к гибели клетки. Примеры антибиотических антинеопластических агентов включают, без ограничения ими,актиномицины, такие как дактиномицин, антрациклины, такие как даунорубицин и доксорубицин; и блеомицины. Ингибиторы топоизомеразы II включают, без ограничения ими, эпиподофиллотоксины. Эпиподофиллотоксины представляют собой фазоспецифические антинеопластические агенты, происходящие из растения мандрагора. Эпиподофиллотоксины обычно воздействуют на клетки в фазах S иG2 клеточного цикла путем образования трехкомпонентного комплекса с топоизомеразой II и ДНК, что вызывает разрывы нити ДНК. Разрывы нити накапливаются, и следует гибель клетки. Примеры эпиподофиллотоксинов включают, без ограничения ими, этопозид и тенипозид. Антиметаболические неопластические агенты представляют собой фазоспецифичные антинеопластические агенты, которые действуют на S-фазе (синтез ДНК) клеточного цикла, ингибируя синтез ДНК или ингибируя синтез пуриновых или пиримидиновых оснований и, таким образом, ограничивая синтез ДНК. Соответственно, S-фаза не протекает, и следует гибель клетки. Примеры антиметаболических антинеопластических агентов включают, без ограничения ими, фторурацил, метотрексат, цитарабин, меркаптопурин и тиогуанин. Камптотецины, включающие камптотецин и производные камптотецина, имеются в наличии или находятся в разработке в качестве ингибиторов топоизомеразы I. Считается, что цитотоксическая активность камптотецинов имеет отношение к их ингибиторной активности в отношении топоизомеразы I. Примеры камптотецинов включают, без ограничения ими, иринотекан, топотекан и различные оптические формы 7-(4-метилпиперазинометилен)-10,11-этилендиокси-20-камптотецина. Гормоны и гормональные аналоги являются полезными соединениями для лечения раковых заболеваний, при которых существует связь между гормоном(ами) и ростом и/или отсутствием роста раковой опухоли. Примеры гормонов и гормональных аналогов, считающихся полезными в лечении новообразований, включают,без ограничения ими, адренокортикостероиды, такие как преднизон и преднизолон, которые полезны в лечении злокачественной лимфомы и острого лейкоза у детей; аминоглютетимид и другие ингибиторы ароматазы, такие как анастрозол, летразол, воразол и экземестан, полезные в лечении адренокортикальной карциномы и гормонозависимой карциномы молочной железы, содержащей рецепторы эстрогена; прогестрины, такие как мегестрола ацетат, полезные в лечении гормонозависимого рака молочной железы и карциномы эндометрия; эстрогены, андрогены и антиандрогены, такие как флутамид, нилутамид,бикалутамид, ципротерона ацетат, и 5-редуктазы, такие как финастерид и дутастерид, полезные в лечении карциномы простаты и доброкачественной гипертрофии простаты; антиэстрогены, такие как тамоксифен, торемифен, ралоксифен, дролоксифен и йодоксифен, полезные в лечении гормонозависимой кар- 13014111 циномы молочной железы; и гонадотропин-высвобождающий гормон (GnRH) и его аналоги, такие как гозерелина ацетат и леупролид, которые стимулируют высвобождение лютинизирующего гормона (LH) и/или фолликулстимулирующего гормона (FSH) при кратковременном или периодическом применении,но приводят к подавлению LH и FSH при долговременном применении, показанном для лечения карциномы простаты и гормонозависимой карциномы молочной железы. Ингибиторы пути трансдукции сигнала представляют собой те ингибиторы, которые блокируют или ингибируют химический процесс, который вызывает внутриклеточное изменение. При использовании здесь такое изменение представляет собой пролиферацию клетки, выживание, ангиогенез или дифференциацию. Ингибиторы трансдукции сигнала, полезные в настоящем изобретении, включают ингибиторы рецепторных тирозинкиназ, нерецепторных тирозинкиназ, блокаторы SH2/SH3 доменов, ингибиторы сериновых/треониновых киназ, фосфатидил-инозитол-3 киназ, мио-инозитольной передачи сигнализов и онкогенов Ras. Некоторые протеин-тирозинкиназы катализируют фосфорилирование специфических тирозильных остатков в различных белках, вовлеченных в регуляцию клеточного роста. Такие протеин-тирозинкиназы можно грубо классифицировать как рецепторные или нерецепторные киназы. Рецепторные тирозинкиназы представляют собой трансмембранные белки, имеющие внеклеточный лигандсвязывающий домен, трансмембранный домен и тирозинкиназный домен. Рецепторные тирозинкиназы вовлечены в регуляцию роста клетки и иногда называются рецепторами факторов роста. Было показано, что неадекватная или неконтролируемая активация многих из этих киназ, т.е. аберрантная киназная активность рецептора фактора роста, например за счет сверхэкспрессии или мутации, приводит к неконтролируемому клеточному росту. Соответственно, аберрантная активность таких киназ связана со злокачественным ростом ткани. Следовательно, ингибиторы таких киназ могли бы обеспечить способы лечения рака. Рецепторы факторов роста включают, например, рецептор эпидермального фактора роста(EGFR, ErbB2 и ErbB4), рецептор тромбоцитарного фактора роста (PDGFR), рецептор фактора роста эндотелия сосудов (VEGFR), тирозинкиназы с иммуноглобулиноподобными и с гомологичными эпидермальным фактором роста доменами (TIE-2), рецептор инсулиноподобного фактора роста-I (IGF-I), рецепторы макрофагального колониестимулирующего фактора (cfms), BTK, ckit, cmet, рецепторы фактора роста фибробластов (FGF), рецепторы Trk (TrkA, TrkB и TrkC), рецепторы эфрина (Eph) и рецептор протоонкогена RET. Некоторые ингибиторы рецепторов факторов роста находятся в разработке и включают антагонисты лигандов, антитела, ингибиторы тирозинкиназы, антисмысловые олигонуклеотиды и аптамеры. Рецепторы факторов роста и агенты, ингибирующие функцию рецептора фактора роста, описаны,например, в Kath, John С, Exp. Opin. Ther. Patents (2000) 10(6):803-818; Shawver et al. DDT. Vol. 2, No. 2.Chemotherapy, Ed. Workman, Paul and Kerr, David, CRC Press 1994, London. Тирозинкиназы, которые не являются рецепторными киназами факторов роста, называются нерецепторными тирозинкиназами. Нерецепторные тирозинкиназы, полезные в настоящем изобретении, которые представляют собой мишени или потенциальные мишени антинеопластических лекарственных средств, включают cScr, Lck, Fyn, Yes, Jak, cAbl, FAK (киназа фокальной адгезии), тирозинкиназу Брутонса и Bcr-Abl. Такие нерецепторные киназы и агенты, которые ингибируют функцию нерецепторной тирозинкиназы, описаны в Sinh, S. and Corey, S.J., (1999) Journal of Hematotherapy and Stem Cell Research 8 (5): 465-80; и Bolen, J.B., Brugge, J.S., (1997) Annual Review of Immunology, 15: 371-404. Блокаторы доменов SH2/SH3 представляют собой агенты, которые нарушают связывание доменаSH2 или SH3 у ряда ферментов или адапторных белков, включая субъединицу р 85 Pl3-K, киназы Srcсемейства, адапторные молекулы (She, Crk, Nek, Grb2) и Ras-GAP. Домены SH2/SH3 в качестве мишеней для противораковых лекарственных средств обсуждаются в Smithgall, Т.Е. (1995), Journal of Pharmacological and Toxicological Methods, 34(3) 125-32. Ингибиторы сериновых/треониновых киназ, включая блокаторы МАР-киназного каскада, которые включают блокаторы Raf-киназ (Rafk), митоген- или внеклеточно регулируемой киназы (MEK) и экстраклеточные регулируемых киназ (ERK); и блокаторы членов семейства протеинкиназы С (PKC), включая блокаторы подтипов РКС (альфа, бета, гамма, эпсилон, мю, лямбда, йота, зета), семейства киназ lkB(IKKa, IKKb), семейства киназ РКВ, членов семейства киназ Akt и рецепторных киназ TGF-бета. Такие сериновые/треониновые киназы и их ингибиторы описаны в Yamamoto, Т., Taya, S., Kaibuchi, K., (1999),Journal of Biochemistry. 126 (5) 799-803; Brodt, P, Samani, A., and Navab, R. (2000), Biochemical Pharmacology, 60. 1101-1107; Massague, J., Weis-Garcia, F. (1996) Cancer Surveys. 27:41-64; Philip, P.A., and Harris,A.L. (1995), Cancer Treatment and Research. 78: 3-27, Lackey, K. et al. Bioorganic and Medicinal ChemistryLetters, (10), 2000, 223-226; и Martinez-Lacaci, L., et al., Int. J. Cancer (2000), 88(1), 44-52. Ингибиторы членов семейства фосфатидилинозитол-3-киназы, включающие блокаторы Pl3-киназы,ATM, DNA-PK и Ku, также могут быть полезными в комбинации с настоящим изобретением. Такие киназы обсуждаются в Abraham, R.T. (1996), Current Opinion in Immunology. 8 (3) 412-8; Canman, C.E., Lim,D.S. (1998), Oncogene 17 (25) 3301-3308; Jackson, S.P. (1997), International Journal of Biochemistry and CellBiology. 29 (7):935-8; и Zhong, H. et al., Cancer Res, (2000) 60(6), 1541-1545. Также полезными в комбинации с настоящим изобретением являются ингибиторы мио- 14014111 инозитольной передачи сигналов, такие как блокаторы фосфолипазы С и аналоги миоинозитола. Такие ингибиторы передачи сигналов описаны в Powis, G., and Kozikowski A., (1994) New Molecular Targets forCancer Chemotherapy ed., Paul Workman and David Kerr, CRC Press 1994, London. Другой группой ингибиторов пути трансдукции сигналов, полезных в комбинации с настоящим изобретением, являются ингибиторы онкогена Ras. Такие ингибиторы включают ингибиторы фарнезилтрансферазы, геранил-геранилтрансферазы и СААХ-протеиназ, а также антисмысловые олигонуклеотиды, рибозимы и иммунотерапию. Было показано, что такие ингибиторы блокируют активацию Ras в клетках, содержащих мутант Ras дикого типа, действуя, таким образом, как антипролиферативные агенты. Ингибирование онкогена Ras обсуждается в Scharovsky, O.G., Rozados, V.R., Gervasoni, S.I., Matar, P.(2000), Journal of Biomedical Science. 7(4) 292-8; Ashby, M.N. (1998), Current Opinion in Lipidology. 9(2)99102; и BioChim. Biophys. Acta, (1989) 1423(3):19-30. Как упомянуто выше, антитела, связывающиеся с лигандом рецепторной киназы, также могут служить в качестве ингибиторов трансдукции сигнала. Эта группа ингибиторов пути трансдукции сигнала включает применение гуманизированных антител к внеклеточному лигандсвязывающему домену рецепторных тирозинкиназ. Например, EGFR-специфичное антитело Imclone С 225 (смотрите Green, M.C. et al.,Monoclonal Antibody Therapy for Solid Tumors, Cancer Treat. Rev., (2000), 26(4), 269-286); ErbB2-антителоMice, Cancer Res. (2000) 60, 5117-5124). Ингибиторы рецепторных киназ, вовлеченных в ангиогенез, также могут найти применение в настоящем изобретении. Ингибиторы ангиогенеза, связанного с VEGFR и TIE2, обсуждаются выше в связи с ингибиторами трансдукции сигнала (оба рецептора представляют собой рецепторные тирозинкиназы). В комбинации с соединениями по настоящему изобретению можно использовать другие ингибиторы. Например, анти-VEGF антитела, которые не распознают VEGFR (рецепторная тирозинкиназа), но связываются с лигандом; низкомолекулярные ингибиторы интегрина (альфабета 3), которые будут ингибировать ангиогенез; эндостатин и ангиостатин (не-RTK) также могут быть полезными в комбинации с ингибиторами PLK. В комбинации с соединениями по данному изобретению также могут быть полезны агенты, используемые в иммунотерапевтических схемах. В комбинации по настоящему изобретению также можно использовать агенты, используемые в проапоптозных схемах (например bcl-2-антисмысловые олигонуклеотиды). Члены семейства белков Bcl-2 блокируют апоптоз. Повышающая регуляция bcl-2, следовательно, связана с хеморезистентностью. Исследования показали, что эпидермальный фактор роста (EGF) стимулирует антиапоптозные члены bcl-2-семейства (например mcl-1). Соответственно, стратегии, разработанные для понижающей регуляции экспрессии bcl-2 в опухолях продемонстрировали клиническую пользу и в настоящее время находятся на фазах II/II клинических исследований, а именно bcl-2 антисмысловой олигонуклеотид G3139 от Genta. Такие проапоптозные стратегии с использованием стратегии антисмысловых олигонуклеотидов в отношении bcl-2 обсуждаются в Water J.S. et al., J. Clin. Oncol. 18:1812-1823 (2000); и Kitada S. et al., Antisense Res. Dev. 4:71-79 (1994). Ингибиторы сигнализации в клеточном цикле ингибируют молекулы, вовлеченные в контроль клеточного цикла. Циклинзависимые киназы (CDK) и взаимодействующие с ними циклины контролируют продвижение по клеточному циклу у эукариот. Скоординированная активация и инактивация разных комплексов циклин/CDK необходима для нормального продвижения по клеточному циклу. Несколько ингибиторов сигнализации в клеточном цикле находятся в разработке. Например, примеры циклинзависимых киназ, включая CDK2, CDK4 и CDK6, и их ингибиторы описаны, например, в Rosania, et al., Exp.Opin. Then Patents, 10(2):215-230 (2000). В одном воплощении способы по настоящему изобретению включают введение животному соединения по изобретению в комбинации с ингибитором пути трансдукции сигнала, в частности с гефитинибом (IRESSA). Способы и применения с использованием этих комбинаций могут включать введение соединения по изобретению и другого химиотерапевтического/антинеопластического агента либо последовательно в любом порядке, либо одновременно в отдельных или в комбинированных фармацевтических композициях. При комбинировании в том же самом препарате очевидно, что два данных соединения должны быть стабильными и совместимыми друг с другом и с другими компонентами данного препарата и могут быть приготовлены в виде препарата для введения. При приготовлении в виде отдельного препарата их можно представить в любом удобном препарате способом, известным для таких соединений в данной области техники. Когда соединение по изобретению используют в комбинации с химиотерапевтическим агентом, доза каждого соединения может отличаться от дозы при использовании данного соединения самого по себе. Подходящие дозы легко оценят специалисты в данной области. Подходящая доза соединения(ий) по изобретению и другого(их) терапевтически активного(ых) агента(ов) и относительное время введения- 15014111 будут выбраны так, чтобы добиться желательного комбинированного терапевтического эффекта, и они находятся в пределах компетенции и свободного выбора лечащего врача. Соединения по данному изобретению можно удобно получить способами, описанными в примерах,следующих ниже. В настоящем изобретении также предложены меченные радиоактивным изотопом соединения по изобретению и биотинилированные соединения по изобретению и их варианты, присоединенные к твердой подложке. Меченные радиоактивным изотопом соединения по изобретению и биотинилированные соединения по изобретению можно получить с использованием традиционных методик. Например, меченные радиоактивным изотопом соединения по изобретению можно получить путем взаимодействия соединения по изобретению с газом тритием в присутствии подходящего катализатора с получением меченных радиоактивным изотопом соединений по изобретению. В одном воплощении данные соединения являются тритированными. Меченные радиоактивным изотопом соединения по изобретению и биотинилированные соединения по изобретению являются полезными в анализах для идентификации соединений, которые ингибируютPLK, для идентификации соединений для лечения состояния, опосредованного PLK, для лечения новообразований, поддающихся лечению, для лечения состояний, характеризующихся неадекватной пролиферацией, для ингибирования пролиферации клетки и для ингибирования митоза в клетке. Соответственно, согласно настоящему изобретению предложен способ анализа для идентификации таких соединений, включающий стадию специфического связывания меченного радиоактивным изотопом соединения по изобретению или биотинилированного соединения по изобретению с белком-мишенью или с клеточными гомогенатами. Более конкретно, подходящие способы анализа будут включать анализы конкурентного связывания. Меченные радиоактивным изотопом соединения по изобретению и биотинилированные соединения по изобретению и их варианты, присоединенные к твердой подложке, можно использовать в анализах в соответствии со способами, традиционными в данной области. Следующие примеры предназначены только для иллюстрации и не предназначены для ограничения объема данного изобретения каким-либо способом, данное изобретение определяется следующей ниже формулой изобретения. Следующие сокращения, используемые в примерах, имеют значения, приведенные ниже: г - грамм(ы); мг - миллиграмм(ы); моль - моль(и); ммоль - миллимоль(и); н. - нормальный; л - литр(ы); мл - миллилитр(ы); мкл - микролитр(ы); ч - час(ы); мин - минута(ы); С - градусы ЦельсияXANTPHOS (4,5-бис(дифенилфосфино)-9,9-диметилксантен) представляет собой имеющийся в продаже катализатор от Aldrich. Реактивы имеются в продаже или получены согласно методикам, описанным в литературе. В следующих структурах "Me" относится к группе -СН 3. Промежуточный пример 1. Метил-5-амино-3-1R)-1-[2-(трифторметил)фенил]этилокси)-2 тиофенкарбоксилат Суспензию трифенилфосфина на полимерной подложке (62,36 г, 2,21 ммоль/г, 137,8 ммоль) в DCM(1,0 л) перемешивали при комнатной температуре в течение 10 мин. Смесь охлаждали до 0 С. Добавляли метил-3-гидрокси-5-нитро-2-тиофенкарбоксилат (20,00 г, 98,44 ммоль), который может быть получен способом, аналогичным описанному в литературе (Barker, J.M.; Huddleston, P.R.; Wood, M.L.; Burkitt,S.A. Journal of Chemical Research (Miniprint) 2001, 1001-1022), затем добавляли (1S)-1-[2-(трифторметил) фенил]этанол (26,20 г, 137,8 ммоль) и ди-трет-бутилазодикарбоксилат (31,73 г, 137,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 21,25 ч, затем фильтровали через фриттованную воронку и концентрировали. Остаток обрабатывали 4 н. HCl в 1,4-диоксане (300 мл) и перемешивали при комнатной температуре в течение 3 ч. Смесь затем гасили добавлением 3 н. гидроксида натрия (300 мл) и насыщенного водного NaHCO3 (200 мл). Смесь экстрагировали DCM (3250 мл). Объединенные органические фракции сушили над MgSO4, фильтровали и концентрировали на силикагеле. Очистка колоночной хроматографией (от 0 до 25% EtOAc:гексаны) давала 36,08 г (98%) соединения,указанного в заголовке, в виде желтого масла. 1 Н ЯМР (300 МГц, CDCl3):7.82 (d, 1H, J=7,8 Гц), 7.68 (d,1H, J=7,8 Гц), 7.59 (t, 1H, J=7,4 Гц), 7,46 (s, 1H), 7.42 (t, 1H, J= 7,6 Гц), 5.77 (q, 1H, J=6,1 Гц), 3.94 (s, 3H),1.74 (d,3H,J=6,1 Гц). Стадия Б. Метил-5-амино-3-1R)-1-[2-(трифторметил)фенил]этилокси)-2-тиофенкарбоксилат В колбу, оснащенную температурным датчиком, верхней механической мешалкой, обратным холодильником и воронкой для добавления, добавляли порошок железа (26,84 г, 480,6 ммоль) и уксусную кислоту (130 мл). Суспензию железа/уксусной кислоты механически перемешивали и нагревали до внутренней температуры 50 С. В воронку для добавления добавляли раствор метил-5-нитро-3-1R)-1-[2(трифторметил)фенил]этилокси)-2-тиофенкарбоксилата (36,08 г, 96,13 ммоль) в уксусной кислоте (160 мл). Затем раствор в воронке для добавления добавляли по каплям к суспензии железа/уксусной кислоты с такой скоростью, чтобы внутренняя температура поддерживалась на уровне ниже 60 С (общее время добавления - 2,5 ч). Реакционную смесь охлаждали до комнатной температуры, разбавляли DCM (500 мл) и затем гасили добавлением 6 н. гидроксида натрия (750 мл) и насыщенного водного NaHCO3 (200 мл). Затем всю смесь фильтровали через слой целита для удаления нерастворимого вещества, промывая целит дополнительным количеством DCM (250 мл). Разделяли водную и органическую фракции. Водную фракцию экстрагировали ЕЮАс (2400 мл). Органические фракции объединяли, сушили над MgSO4,фильтровали и концентрировали с получением 30,66 г (92%) соединения, указанного в заголовке, в виде оранжевого твердого вещества. 1 Н ЯМР (300 МГц, CDCl3):7.89 (d, 1 Н, J=7,7 Гц), 7.62 (d, 1 Н, J=7,7 Гц),7.56 (t, 1 Н, J=7,7 Гц), 7.36 (t, 1 Н, J=7,7 Гц), 5.72 (s, 1H), 5.65 (q, 1H, J=6,3 Гц), 4.26 (br s, 2H), 3.80 (s, 3H),1.66 (d, 3 Н, J=6,3 Гц), MS (APCI) (масс-спектрометрия с химической ионизацией при атмосферном давлении): 368,00 [M+Na]+. Промежуточный пример 2. Метил-5-амино-3-[(1R)-1-(2-хлорфенил)этил]окси-2-тиофенкарбоксилат К смеси 3-гидрокси-4-нитробензойной кислоты (5,0 г, 27,3 ммоль) в 1,2-дихлорэтане (100 мл) добавляли триметилборат (4,9 мл, 43,7 ммоль), затем добавляли диэтилэтерат трифторида бора (5,5 мл, 43,7 ммоль). Затем медленно по каплям добавляли боран-пиридиновый комплекс (4,1 мл, 41,0 ммоль). Реакционную смесь перемешивали в течение 4 ч при комнатной температуре, затем охлаждали до 0 С и гасили МеОН (10 мл). Смесь концентрировали под вакуумом и остаток переносили в толуол (200 мл), затем экстрагировали 1 н. водным гидроксидом натрия (3100 мл). Объединенные водные слои доводили до рН 1,0 путем добавления 12 н. HCl, затем экстрагировали EtOH (3250 мл). Объединенные органические слои промывали водой, рассолом, сушили над MgSO4 и концентрировали под вакуумом с получением 4,55 г (98%) соединения, указанного в заголовке, в виде светло-желтого твердого вещества. 1 Н ЯМР (400 МГц, DMSO-d6):10.87 (s, 1H), 7.85 (d, 1H, J=8,6 Гц), 7.08 (s, 1H), 6.88 (dd, 1H, J=1,19, 8,51 Гц), 5.43 (s,1H), 3.33 (s, 2H). Стадия Б. 3-Гидрокси-4-нитробензилпивалат Смесь 5-(гидроксиметил)-2-нитрофенола (11,35 г, 67,15 ммоль) и 3-(2,2-диметилпропаноил)-1,3 тиазолидин-2-тиона (15,0 г, 73,89 ммоль), которую можно получить способом, аналогичным методике,описанной в литературе (Yamada, S. Tetrahedron Letters, 1992, 33, 2171-2174), перемешивали в толуоле(670 мл) при 100 С в течение 40 ч, затем охлаждали до комнатной температуры. Реакционную смесь концентрировали под вакуумом до объема приблизительно 200 мл и образующуюся взвесь фильтровали через фильтровальную бумагу, промывая твердое вещество холодным толуолом. Затем фильтрат концентрировали под вакуумом и очищали хроматографией на силикагеле, элюируя градиентом от 0 до 20%EtOAc/гексаны с получением 11,09 г (65%) соединения, указанного в заголовке, в виде прозрачного желтого масла. 1 Н ЯМР (400 МГц, DMSO-d6):11.05 (s, 1H), 7.87 (d, 1H, J=8,42 Гц), 7.06 (s, 1 Н), 6.90 (dd, 1 Н,J=1,46, 8,42 Гц), 5.09 (s, 2H), 1.18 (s, 9H). Стадия В. 4-Нитро-3-[(трифторметил)сульфонил]оксибензил пивалатN,N-диизопропилэтиламин (15,5 мл, 88,9 ммоль). Реакционную смесь перемешивали в течение 45 мин при 0 С, затем в течение 45 мин при комнатной температуре. Затем концентрировали реакционную смесь в вакууме и очищали хроматографией на силикагеле, элюируя градиентом от 5 до 20% EtOAc/гексаны с получением 16,87 г (99%) соединения, указанного в заголовке, в виде не совсем белого твердого вещества. 1 Н ЯМР (400 МГц, DMSO-d6):8.36 (d, 1H, J=8,42 Гц), 7.75-7.69 (m, 2H), 5.27 (s, 2H), 1.19 (s, 9H). Стадия Г. Метил 5-[(5-[(2,2-диметилпропаноил)окси]метил-2-нитрофенил)амино]-3-1R)-1-[2(трифторметил)фенил]этилокси)тиофен-2-карбоксилат(900 мг, 6,5 ммоль) перемешивали в толуоле (5,2 мл) при 100 С в течение 2 ч, затем охлаждали до комнатной температуры и фильтровали через целит, промывая EtOAc и DCM. Фильтрат концентрировали под вакуумом и очищали хроматографией на силикагеле, элюируя градиентом от 5 до 25% EtOAc/гексан с получением 1,26 г (84%) соединения, указанного в заголовке, в виде красного масла. 1 Н ЯМР (400 МГц,DMSO-d6):9.75 (s, 1H), 8.09 (d, 1H, J=8,6 Гц), 7.89 (d, 1H, J=7.87 Гц), 7.69-7.78 (m, 2H), 7.52 (t, 1H, J= 7.59 Гц), 7.34 (s, 1H), 7.01 (dd, 1H, J=1,46, 8.60 Гц), 6.62 (s, 1 Н), 5.70-5.75 (m, 1 Н), 5.07 (s, 2H), 3.74 (s, 3 Н),1.58 (d, 3 Н, J=6,22 Гц), 1.13 (s, 9H). Стадия Д. Метил-5-[(2-амино-5-[(2,2-диметилпропаноил)окси]метилфенил)амино]-3-1R)-1-[2(трифторметил)фенил]этилокси)тиофен-2-карбоксилат Смесь метил-5-[(5-[(2,2-диметилпропаноил)окси]метил-2-нитрофенил)амино]-3-1R)-1-[2-(трифторметил)фенил]этилокси)тиофен-2-карбоксилата (2,42 г, 4,17 ммоль) и платины (сульфидированная,5 мас.%, на углероде) (811 мг, 0,21 ммоль) в EtOAc (30 мл) добавляли в реакционный сосуд высокого давления. Реакционную смесь вакуумировали и продували газообразным N2, затем подавали газообразный Н 2 под давлением 50 фунт/кв.дюйм 344,738 кПа в течение 1 ч. Реакционную смесь фильтровали через целит, промывая EtOAc. Фильтрат концентрировали под вакуумом с получением 2,27 г (99%) соединения, указанного в заголовке, в виде желтовато-коричневого твердого вещества. 1 Н ЯМР (400 МГц,DMSO-d6):8.62 (s, 1H), 7.84 (d, 1H, J=7,87 Гц), 7.72 (dd, 2H, J=7,60, 13,09 Гц), 7.50 (t, 1H, J=7.60 Гц),7.01 (d, 1H, J=1,46 Гц), 6.88 (dd, 1H, J=1,74, 8,15 Гц), 6.68 (d, 1H, J=8,24 Гц), 5.83 (s, 1H), 5.59-5.65 (m,1H), 4.97 (s, 2H), 4.85 (s, 2H), 3.64 (s, 3 Н), 1.55 (d, 3 Н, J =6,23 Гц), 1.11 (s, 9 Н); MS (ESI): 551 [М+Н]+. Стадия Е. Метил-5-(6-[(2,2-диметилпропаноил)окси]метил-1 Н-бензимидазол-1-ил)-3-1R)-1-[2(трифторметил)фенил]этилокси)тиофен-2-карбоксилат К смеси метил-5-[(2-амино-5-[(2,2-диметилпропаноил)окси]метилфенил)амино]-3-1R)-1-[2(трифторметил)фенил]этилокси)тиофен-2-карбоксилата (2,27 г, 4,13 ммоль) в триэтилортоформиате (10 мл, 60,2 ммоль) и DCM (3 мл) добавляли пиридиния паратолуолсульфонат (100 мг, 0,4 ммоль). Реакционную смесь перемешивали при 40 С в течение 1 ч, затем охлаждали до комнатной температуры. Всю реакционную смесь загружали на силикагель и очищали хроматографией на силикагеле, элюируя градиентом от 0 до 50% EtOAc/гексаны с получением 2,0 г (86%) соединения, указанного в заголовке, в виде светлого желтовато-коричневого твердого вещества. 1 Н ЯМР (400 МГц, DMSO-d6):8.65 (s, 1H), 7.99 (d,1H, J=7,87 Гц), 7.75-7.80 (m, 2 Н), 7.72 (d, 1H, J =7,87 Гц), 7.63 (s, 1H), 7.53 (t, 1H, J=7,60 Гц), 7.40 (s, 1H),7.35 (d, 1H, J=8,42 Гц), 5.96 (q, 1H, J=6,10 Гц), 5.21 (s, 2H), 3.83 (s, 3 Н), 1.65 (d, 3 Н, J=6,23 Гц), 1.16 (s,9 Н); MS (ESI): 561 [М+Н]+. Стадия Ж. Метил-5-[6-(гидроксиметил)-1 Н-бензимидазол-1-ил]-3-1R)-1-[2-(трифторметил)фенил]этилокси)тиофен-2-карбоксилат К перемешиваемому раствору метил-5-(6-[(2,2-диметилпропаноил)окси]метил-1 Н-бензимидазол 1-ил)-3-1R)-1-[2-(трифторметил)фенил]этилокси)тиофен-2-карбоксилата (5,21 г, получен из другой партии с использованием методики, аналогичной примеру 1, способ 1, стадия Е, 9,30 ммоль) в МеОН (24 мл) добавляли 0,5 М гидроксид натрия в МеОН (24,0 мл, 12 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 72 ч, затем гасили уксусной кислотой (2 мл). Реакционную смесь разбавляли DCM (350 мл) и наполовину насыщенным водным раствором рассола (150 мл). Водный слой экстрагировали DCM (250 мл). Объединенные органические слои сушили над MgSO4 и концентрировали под вакуумом с получением 4,40 г (99%) соединения, указанного в заголовке, в виде светло-желтого твердого вещества. 1 Н ЯМР (400 МГц, DMSO-d6):8.58 (s, 1 Н), 7.99 (d, 1H, J=7,87 Гц), 7.69-7.81 (m, 3 Н),7.51-7.58 (m, 2H), 7.38 (s, 1H), 7.30 (d, 1H, J= 8,42 Гц), 5.96 (q, 1H, J=6,10 Гц), 5.30 (t, 1H, J=5,77 Гц), 4.62 К перемешиваемому раствору метил-5-[6-(гидроксиметил)-1 Н-бензимидазол-1-ил]-3-1R)-1-[2(трифторметил)фенил]этилокси)тиофен-2-карбоксилата (1,47 г, 3,08 ммоль) и трифенилфосфина (1,05 г,4,01 ммоль) в DCM (30 мл) добавляли N-хлорсукцинимид (0,53 г, 4,01 ммоль). Реакционную смесь затем нагревали до температуры дефлегмации и перемешивали в течение 20 мин, затем охлаждали до комнатной температуры. Реакционную смесь разбавляли DCM (400 мл) и наполовину насыщенным водным раствором рассола (150 мл). Водный слой затем экстрагировали DCM. Объединенные органические слои сушили над Na2SO4, концентрировали под вакуумом и очищали хроматографией на силикагеле, элюируя градиентом от 10 до 60% EtOAc/гексаны с получением 1,4 г (92%) соединения, указанного в заголовке, в виде белого твердого вещества. 1 Н ЯМР (400 МГц, DMSO-d6):8.65 (s, 1H), 7.99 (d, 1H, J=7,87 Гц), 7.727.81 (m, 3 Н), 7.69 (s, 1H), 7.54 (t, 1H, J=7,69 Гц), 7.43 (d, 1H, J= 8,42 Гц), 7.38 (s, 1H), 5.97 (q, 1H, J=6,10 Гц), 4.91 (s, 2H), 3.84 (s, 3 Н), 1.66 (d, 3 Н, J=6,23 Гц); MS (ESI): 495 [М+Н]+. Стадия И. Метил-5-6-[(метилтио)метил]-1 Н-бензимидазол-1-ил-3-1R)-1-[2-(трифторметил)фенил]этилокси)тиофен-2-карбоксилат К перемешиваемой смеси метил-5-[6-(хлорметил)-1 Н-бензимидазол-1-ил]-3-1R)-1-[2-(трифторметил)фенил]этилокси)тиофен-2-карбоксилата (200 мг, 0,40 ммоль) в N,N-диметилформамиде (2,5 мл) добавляли тиометилат натрия (37 мг, 0,52 ммоль). Реакционную смесь перемешивали в течение 30 мин,затем разбавляли EtOAc, промывали водой (5), рассолом, сушили над Na2SO4, концентрировали под вакуумом и очищали хроматографией на силикагеле, элюируя градиентом от 0 до 60% EtOAc/гексаны с получением 147 мг (72%) соединения, указанного в заголовке, в виде белого твердого вещества. MS Смесь метил-5-6-[(метилтио)метил]-1 Н-бензимидазол-1-ил-3-1R)-1-[2-(трифторметил)фенил] этилокси)тиофен-2-карбоксилата (144,0 мг, 0,28 ммоль) и 7 н. аммиака в МеОН (18 мл, 126,0 ммоль) добавляли в стеклянную реакционную колбу высокого давления. Колбу герметично закрывали, затем нагревали до 80 С в течение приблизительно 16 ч. Колбу охлаждали до комнатной температуры, открывали и реакционную смесь концентрировали под вакуумом, затем очищали хроматографией на силикагеле,элюируя градиентом от 0 до 3% MeOH/DCM с 1% гидроксидом аммония с получением 130 мг (93%) соединения, указанного в заголовке, в виде бело-золотистого твердого вещества. 1 Н ЯМР (400 МГц,DMSO-d6):8.49 (s, 1H), 7.93 (d, 1H, J= 7,68 Гц), 7.85 (br s, 1H), 7.80-7.75 (m, 2H), 7.69 (d, 1H, J=8,23 Гц),7.56 (t, 1H, J=7,68 Гц), 7.39 (s, 1H), 7.29 (d, 1H, J=8,42 Гц), 7.15 (br s, 1H), 7.08 (s, 1H), 5.94 (m, 1 Н), 3.79 (s,2 Н), 1.93 (s, 3 Н), 1.75 (d, 3 Н, J=6,22 Гц); MS (ESI): 492 [М+Н]+. Стадия Л. 5-6-[(Метилсульфонил)метил]-1 Н-бензимидазол-1-ил-3-1R)-1-[2-(трифторметил) фенил]этилокси)тиофен-2-карбоксамид(3,0 мл) добавляли мета-хлорпероксибензойную кислоту (70 мг, 0,31 ммоль). Реакционную смесь перемешивали в течение 30 мин, нагревали до комнатной температуры и перемешивали в течение 15 мин,затем концентрировали под вакуумом. Остаток разбавляли хлороформом и промывали водным насыщенным NaHCO3, водой, сушили над Na2SO4, концентрировали под вакуумом и очищали хроматографией на силикагеле, элюируя градиентом от 0 до 5% MeOH/DCM с 1%-ным гидроксидом аммония, с получением 78 мг (96%) соединения, указанного в заголовке, в виде белого твердого вещества. 1 Н ЯМР (400 МГц, DMSO-d6):8.57 (s, 1H), 7.95 (d, 1H, J=7,87 Гц), 7.85 (br s, 1H), 7.80-7.73 (m, 3 Н), 7.69 (s, 1 Н), 7.55 Смесь метил-5-[6-(хлорметил)-1 Н-бензимидазол-1-ил]-3-1R)-1-[2-(трифторметил)фенил]этил окси)тиофен-2-карбоксилата (4,53 г, полученного из другой партии с использованием методики, аналогичной методике, описанной в примере 1, способ 1, стадия 3, 9,17 ммоль), натриевой соли метансульфиновой кислоты (2,81 г, 27,5 ммоль) и этанола (40,0 мл) добавляли в стеклянную реакционную колбу высокого давления. Колбу герметично закрывали, затем нагревали до 85 С в течение приблизительно 16 ч. Колбу охлаждали до комнатной температуры, открывали и реакционную смесь концентрировали под вакуумом, затем очищали хроматографией на силикагеле, элюируя градиентом от 5 до 35% EtOAc/гексана с получением 4,54 г (92%) соединения, указанного в заголовке, в виде светло-желтого твердого вещества. 1 Н ЯМР (400 МГц, DMSO-d6):8.67 (s, 1H), 8.01 (d, 1H, J=7,87 Гц), 7.82-7.70 (m, 4 Н), 7.53 (t, 1H,J=7,69 Гц), 7.45 (s, 1H), 7.40 (d, 1H, J=8,42 Гц), 5.95 (m, 1 Н), 4.63 (m, 2 Н), 3.83 (s, 3H), 2.90 (s, 3H), 1.65 (d,3H, J=6,204 Гц); MS (ESI): 539 [М+Н]+. Стадия Б. 5-6-[(Метилсульфонил)метил]-1 Н-бензимидазол-1-ил-3-1R)-1-[2-(трифторметил) фенил]этилокси)-тиофен-2-карбоксамид Смесь метил-5-6-[(метилсульфонил)метил]-1 Н-бензимидазол-1-ил-3-1R)-1-[2-(трифторметил) фенил]этилокси)-2-тиофенкарбоксилата (4,53 г, 8,42 ммоль) и 7 н. аммиака в МеОН (250,0 мл, 1,75 моль) добавляли в стеклянную реакционную колбу высокого давления. Колбу герметично закрывали, затем нагревали до 85 С в течение приблизительно 36 ч. Колбу охлаждали до комнатной температуры, открывали и реакционную смесь объединяли со второй партией метил-5-6-[(метилсульфонил)метил]-1 Нбензимидазол-1-ил-3-1R)-1-[2-(трифторметил)фенил]этилокси)-2-тиофенкарбоксилата (4,11 г, 7,63 ммоль), которую также обрабатывали 7 н. аммиаком в МеОН (200,0 мл, 1,40 моль) в стеклянной реакционной колбе высокого давления при 85 С в течение приблизительно 36 ч, затем охлаждали до комнатной температуры и открывали колбу. Объединенные реакционные смеси концентрировали под вакуумом,затем очищали хроматографией на силикагеле, элюируя градиентом от 0 до 5% MeOH/DCM с 1%-ным гидроксидом аммония с получением 7,47 г (89%) соединения, указанного в заголовке, в виде не совсем белого твердого вещества. MS (ESI): 524 [М+Н]+. Пример 2. 3-[(1R)-1-(2-Хлорфенил)этил]окси-5-(6-[(метилсульфонил)-метил]-1 Н-бензимидазол-1 ил)-2-тиофенкарбоксамид К смеси 5-бром-1 Н-бензимидазола (43,78 г, 222,0 ммоль) в хлороформе (800 мл) добавляли Nметилимидазол (44,5 мл, 560,0 ммоль), затем добавляли метил-2-хлор-3-оксо-2,3-дигидро-2-тиофенкарбоксилат (44,8 г, 233,0 ммоль). Реакционную смесь перемешивали в течение 20 ч при комнатной температуре, затем добавляли N-метилимидазол (18,0 мл, 226,0 ммоль), затем трет-бутилдиметилсилилхлорид(36,8 г, 245,0 ммоль). Реакционную смесь перемешивали в течение 1 ч, затем гасили МеОН и вливали вDCM и воду. Водный слой экстрагировали DCM (3). Объединенные органические слои затем сушили над Na2SO4, концентрировали и разделяли хроматографией на силикагеле, элюируя градиентом от 50 до 75% 25%-ного EtOAc в гексане/гексане с получением 25,18 г (24%) соединения, указанного в заголовке. К перемешиваемому раствору метил-5-(6-бром-1 Н-бензимидазол-1-ил)-3-[(1,1-диметилэтил)(диметил)силил]окси-2-тиофенкарбоксилата (25,18 г, 53,9 ммоль) в THF (540,0 мл) добавляли 1,0 М тетрабутиламмония фторид в THF (60,0 мл, 60,0 ммоль). Реакционную смесь перемешивали в течение 1,5 ч,затем добавляли насыщенный водный NH4Cl (200 мл). Образующуюся взвесь перемешивали в течение 15 мин, затем разбавляли водой (750 мл) и EtOAc (1,0 л). Водный слой отделяли, и его рН доводили до 3,0 добавлением 1 М водной HCl. Затем экстрагировали водный слой EtOAc (3). Объединенные органические слои промывали водной 0,1 М HCl, рассолом, сушили над MgSO4 и концентрировали под вакуумом с Суспензию трифенилфосфина на полимерной подложке (53,0 г, 2,04 ммоль/г, 108 ммоль), (1S)-1-[2(трифторметил)фенил]этанола (15,4 г, 81,0 ммоль) и метил-5-(6-бром-1 Н-бензимидазол-1-ил)-3 гидрокситиофен-2-карбоксилата (промежуточный пример 3) (19,0 г, 53,9 ммоль) в DCM (750 мл) перемешивали при комнатной температуре в течение 10 мин. Взвесь затем обрабатывали ди-третбутилазодикарбоксилатом (24,8 г, 108 ммоль). Реакционную смесь перемешивали в течение 3 ч, затем пропускали через фильтровальную бумагу, промывая твердые вещества смолы DCM и МеОН. Фильтрат концентрировали под вакуумом и очищали хроматографией на силикагеле, элюируя градиентом от 5 до 50% EtOAc/гексана с получением 23,8 г (84%) соединения, указанного в заголовке, в виде бледножелтого твердого вещества. 1 Н ЯМР (400 МГц, DMSO-d6):8.63 (s, 1H), 7.97 (d, 1H, J=7,87 Гц), 7.80-7.71 К смеси 3-метил-5-(6-бром-1 Н-бензимидазол-1-ил)-3-(1R)-1-[2-(трифторметил)фенил]этокси тиофен-2-карбоксилата (23,8 г, 45,4 ммоль), калия винилтрифторбората (7,25 г, 54,5 ммоль) и триэтиламина (6,3 мл, 45,4 ммоль), перемешиваемой при комнатной температуре в н-пропаноле (230 мл) добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) дихлорметана (750 мг, 0,91 ммоль). Смесь затем нагревали до температуры дефлегмации и перемешивали в течение 3 ч, затем охлаждали до комнатной температуры, вливали в воду и экстрагировали EtOAc (3). Объединенные органические слои промывали рассолом, сушили над MgSO4, концентрировали под вакуумом и очищали хроматографией на силикагеле, элюируя градиентом от 10 до 40% EtOAc/гексана с получением 17,06 г(80%) соединения, указанного в заголовке, в виде желтой твердой пены. 1 Н ЯМР (400 МГц, DMSO-d6):8.59 (s, 1H), 7.98 (d, 1H, J= 7,87 Гц), 7.80-7.71 (m, 3H), 7.59 (s, 1H), 7.56-7.52 (m, 2H), 7.39 (s, 1H), 6.85 (dd,1H, J=10,98 и 17,75 Гц), 6.00 (q, 1H, J=6,10 Гц), 5.86 (d, 1 Н, J=17,56 Гц), 5.31 (d, 1H, J=10,98 Гц), 3.83 (s,3H), 1.65 (d, 3H, J=6,04 Гц); MS (ESI): 473 [M+H]+. Стадия В. Метил-5-[6-(1,2-дигидроэтил)-1 Н-бензимидазол-1-ил]-3-(1R)-1-[2-(трифторметил)фенил]этокситиофен-2-карбоксилат- 24014111 К перемешиваемому раствору метил-3-(1R)-1-[2-(трифторметил)фенил]этокси-5-(6-винил-1 Нбензимидазол-1-ил)тиофен-2-карбоксилата (17,06 г, 36,1 ммоль) в 360 мл смеси ацетон/вода (3:1) добавляли 4-метилморфолина N-оксид (5,1 г, 43,4 ммоль), затем 2,5 мас.% раствор тетроксида осмия в 2 метил-2-пропаноле (10,0 мл, 0,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч, затем гасили водным (насыщенным) сульфитом натрия. Смесь экстрагировали EtOAc(3). Объединенные органические слои промывали рассолом, сушили над MgSO4, концентрировали под вакуумом и очищали хроматографией на силикагеле, элюируя градиентом от 1 до 8% MeOH/DCM с 1%ным гидроксидом аммония с получением 16,72 г (92%) соединения, указанного в заголовке, в виде бледно-желтой твердой пены. 1 Н ЯМР (400 МГц, DMSO-d6):8.59 (d, 1H, J=1,46 Гц), 7.98 (d, 1 Н, J=7,87 Гц),7.80-7.68 (m, 3H), 7.59-7.52 (m, 2 Н), 7.36-7.31 (m, 2 Н), 5.95 (q, 1 Н, J=6,10 Гц), 5.37 (t, 1 Н, J=3,66 Гц), 4.764.64 (m, 2 Н), 3.83 (s, 3H), 3.46-3.42 (m, 2 Н), 1.65 (d, 3H, J=6,04 Гц); MS (ESI): 507 [М+Н]+. Стадия Г. Метил-5-(6-формил-1 Н-6 ензимидазол-1-ил)-3-(1R)-1-[2-(трифторметил)фенил]этокси тиофен-2-карбоксилат К раствору метил-5-[6-(1,2-дигидроэтил)-1 Н-бензимидазол-1-ил]-3-(1R)-1-[2-(трифторметил)фенил]этокситиофен-2-карбоксилата (16,72 г, 33,0 ммоль) в смеси 1:1:1 DCM/вода/МеОН (220 мл) добавляли перйодат натрия (10,58 г, 49,5 ммоль). Образующуюся суспензию перемешивали в течение 1 ч, затем разбавляли водой и EtOAc. Водный слой экстрагировали EtOAc (3). Объединенные органические слои промывали рассолом, сушили над MgSO4 и концентрировали под вакуумом с получением 14,76 г(9,9 г, 46,7 ммоль). Реакционную смесь перемешивали в течение 1,5 ч, затем добавляли 5%-ный водныйK2CO3, пока рН не достигал приблизительно 8. Смесь затем разбавляли EtOAc и водой. Водный слой экстрагировали EtOAc (3). Объединенные органические слои промывали рассолом, сушили над Na2SO4,концентрировали под вакуумом и очищали хроматографией на силикагеле, элюируя градиентом от 1 до 8% MeOH/DCM с 1%-ным гидроксидом аммония с получением 15,82 г (91%) соединения, указанного в заголовке, в виде бледно-желтой твердой пены. 1 Н ЯМР (400 МГц, DMSO-d6):8.57 (s, 1H), 7.98 (d, 1H,J= 7,87 Гц), 7.80-7.68 (m, 3H), 7.54 (t, 1H, J=7,59 Гц), 7.46 (s, 1H), 7.33-7.28 (m, 2H), 5.97 (q, 1H, J=6,16 Гц), 3.83 (s, 3H), 3.55 (s, 2 Н), 2.45-2.20 (m, 8 Н), 2.13 (s, 3H), 1.66 (d, 3H, J= 6,04 Гц); MS (ESI): 559 Смесь метил-5-6-[(4-метилпиперазин-1-ил)метил]-1 Н-бензимидазол-1-ил-3-(1R)-1-[2-(трифторметил)фенил]этокситиофен-2-карбоксилата (15,82 г, 28,35 ммоль) и 7 н. аммиака в МеОН (250 мл, 1,75 моль) добавляли в стеклянную реакционную колбу высокого давления. Колбу герметично закрывали,- 25014111 затем нагревали до 80 С в течение приблизительно 40 ч. Колбу охлаждали до комнатной температуры,открывали и реакционную смесь концентрировали под вакуумом, затем очищали хроматографией на силикагеле, элюируя градиентом от 2 до 8% MeOH/DCM с 1%-ным гидроксидом аммония с получением 14,11 г (92%) соединения, указанного в заголовке, в виде белой твердой пены. 1 Н ЯМР (400 МГц, DMSOd6):8.49 (s, 1H), 7.93 (d, 1 Н, J=7,87 Гц), 7.86 (br s, 1 Н), 7.80-7.75 (m, 2H), 7.68 (d, 1 Н, J=8,23 Гц), 7.56 (t,1 Н, J=7,68 Гц), 7.33 (s, 1 Н), 7.28(d, 1 Н, J=8,42 Гц), 7.15 (br s, 1H),7.06(s, 1H), 5.94 (q, 1H, J=6,10 Гц), 3.52 Раствор 3-бром-4-нитрофенола (20,0 г, 91,7 ммоль) в DCM (475 мл) перемешивали при 0 С. Добавляли диизопропилэтиламин (19,2 мл, 110,0 ммоль), затем добавляли по каплям раствор хлорметилметилового эфира (7,7 мл, 100,9 ммоль) в DCM (25 мл). Реакционную смесь перемешивали при 0 С в течение 1 ч, затем нагревали до комнатной температуры и гасили водой (150 мл). Смесь вливали в рассол (150 мл) и водный слой экстрагировали EtOAc (3). Объединенные органические слои промывали рассолом,сушили над MgSO4, концентрировали под вакуумом и разделяли хроматографией на силикагеле (330 г),элюируя градиентом от 0 до 25% EtOAc/гексан с получением 20,0 г (83%) соединения, указанного в заголовке, в виде прозрачного оранжевого масла. 1 Н ЯМР (400 МГц, DMSO-d6):8.07 (d, 1H, J=8,97 Гц),7.50 (d, 1 Н, J=2,20 Гц), 7.21 (dd, 1 Н, J=8,97 и 2,38 Гц), 5.34 (s, 2H), 3.39 (s, 3H). Стадия Б. Метил-5-[(5-[(метилокси)метил]окси-2-нитрофенил)амино]-3-1R)-1-[2-(трифторметил)фенил]этилокси)-2-тиофенкарбоксилат К перемешиваемому раствору метил-5-амино-3-1R)-1-[2-(трифторметил)фенил]этилокси)-2 тиофенкарбоксилата (1,32 г, 3,82 ммоль) и 2-бром-4-[(метилокси)метил]окси-2-нитробензола (1,0 г,3,82 ммоль) в диоксане (20 мл) добавляли трис(дибензилиденацетон)дипалладий(0) (70,0 мг, 0,076 ммоль) и XANTPHOS (97,0 мг, 0,17 ммоль), затем карбонат цезия (6,2 г, 19,0 ммоль). Смесь нагревали до 60 С и перемешивали в течение 12 ч, затем охлаждали до комнатной температуры, разбавляли EtOAc и фильтровали через целит, промывая твердые вещества EtOAc и DCM. Фильтрат концентрировали под вакуумом и разделяли хроматографией на силикагеле (120 г), элюируя градиентом от 5 до 35% В реакционную колбу высокого давления для гидрирования добавляли метил-5-[(5-[(метилокси) метил]окси-2-нитрофенил)амино]-3-1R)-1-[2-(трифторметил)фенил]этилокси)-2-тиофенкарбоксилат(сульфидированную) (740 мг, 0,19 ммоль) и триметилортоформиат (40 мл). Колбу продували N2 (газ),помещали под вакуум (3), затем продували Н 2 (газ), помещали под вакуум (3). Затем подавали газообразный Н 2 под давлением 50 фунтов/кв.дюйм (344,738 кПа) в течение 3 ч. Реакционную смесь затем фильтровали через целит, промывая твердые вещества EtOAc и DCM. Затем фильтрат концентрировали под вакуумом с получением 1,92 г (100%) соединения, указанного в заголовке, в виде светло-желтой твердой пены. 1 Н ЯМР (400 МГц, DMSO-d6):8.49 (s, 1H), 7.98 (d, 1H, J=8,05 Гц), 7.79-7.66 (m, 3H), 7,53(130 мл) добавляли 1 н. HCl в воде (65 мл, 65,0 ммоль). Реакционную смесь нагревали до 35 С и перемешивали в течение 72 ч, затем охлаждали до комнатной температуры. Реакционную смесь вливали в DCM(500 мл) и добавляли воду (100 мл). Смесь нейтрализовали до рН 7 добавлением водного (насыщенного)NaHCO3. Водный слой затем экстрагировали DCM (1) и EtOAc (1). Объединенные органические слои сушили над MgSO4, концентрировали под вакуумом и разделяли хроматографией на силикагеле (120 г),элюируя градиентом от 10 до 60% EtOAc/гексана с получением 6,9 г (92%) соединения, указанного в заголовке, в виде пенистого твердого вещества светло-лососевого цвета. 1 Н ЯМР (400 МГц, DMSO-d6):9.62 (s, 1H), 8.41 (s, 1H), 8.00 (d, 1H, J=7,87 Гц), 7.80-7.71 (m, 2H), 7.56-7.51 (m, 2H), 7.38 (s, 1H), 7.05 (d,1H, J=2,01 Гц), 6.81 (dd, 1H, J=8,70 и 2,11 Гц), 5,95 (m, 1H), 3.83 (s, 3H), 1.64 (d, 3H, J= 6,23 Гц); MS (ESI): 463 [M+H]+. Стадия Д. Метил-3-1R)-1-[2-(трифторметил)фенил]этилокси)-5-(6-[(трифторметил)сульфонил] окси-1 Н-бензимидазол-1-ил)-2-тиофенкарбоксилат К перемешиваемому охлажденному (0 С) раствору метил-5-(6-гидрокси-1 Н-бензимидазол-1-ил)-31R)-1-[2-(трифторметил)фенил]этилокси)-2-тиофенкарбоксилата (2,49 г, 5,38 ммоль) и N-фенилтрифторметана сульфонамида (2,06 г, 5,76 ммоль) в DCM (30 мл) добавляли диизопропилэтиламин (2,0 мл,11,5 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 12 ч. Затем концентрировали реакционную смесь под вакуумом и хроматографировали на силикагеле (120 г), элюируя градиентом от 5 до 40% EtOAc/гексана с получением 3,12 г (98%) соединения,указанного в заголовке, в виде светло-желтого твердого вещества. 1 Н ЯМР (400 МГц, DMSO-d6):8.76 К смеси метил-3-1R)-1-[2-(трифторметил)фенил]этилокси)-5-(6-[(трифторметил)сульфонил] окси-1 Н-бензимидазол-1-ил)-2-тиофенкарбоксилата (20,69 г, получен из другой партии с использованием методики, аналогичной примеру 3, способ 2, стадия Е, 34,83 ммоль), калия винилтрифторбората (5,6 г,42,10 ммоль) и триэтиламина (4,85 мл, 34,86 ммоль), перемешиваемой при комнатной температуре в нпропаноле (175 мл), добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) дихлорметана (570 мг, 0,70 ммоль). Затем нагревали смесь до температуры дефлегмации и перемешивали в течение 3 ч, затем охлаждали до комнатной температуры, вливали в воду и экстрагировали EtOAc (3). Объединенные органические слои промывали рассолом, сушили над MgSO4, концентрировали под вакуумом и очищали хроматографией на силикагеле, элюируя градиентом от 10 до 50% EtOAc/гексана с получением 12,98 г (79%) соединения, указанного в заголовке, в виде светло-желтой твердой пены. MS Соединение, указанное в заголовке, можно получить в соответствии с методикой, аналогичной методике из примера 3, способ 1, стадия Е. Стадия К. 5-6-[(4-Метилпиперазин-1-ил)метил]-1 Н-бензимидазол-1-ил-3-(1R)-1-[2-(трифторметил)фенил]этокситиофен-2-карбоксамид К перемешиваемому раствору метил-5-[6-(хлорметил)-1 Н-бензимидазол-1-ил]-3-1R)-1-[2(трифторметил)фенил]этилокси)тиофен-2-карбоксилата (150 мг, 0,30 ммоль) в диоксане (1,0 мл) добавляли N-метилпиперазин (50 мкл, 0,45 ммоль). Реакционную смесь нагревали при 60 С в течение 18 ч,охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток растворяли в EtOAc и воде. Водный слой экстрагировали EtOAc. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия, концентрировали под вакуумом и очищали хроматографией на силикагеле,элюируя градиентом от 0 до 10% MeOH/DCM с 1%-ным гидроксидом аммония с получением 134 мг(79%) соединения, указанного в заголовке, в виде белого твердого вещества. MS (ESI): 559 [М+Н]+. Стадия Б. 5-6-[(4-Метилпиперазин-1-ил)метил]-1 Н-бензимидазол-1-ил-3-(1R)-1-[2-(трифторме- 28014111 тил)фенил]этокситиофен-2-карбоксамид Раствор 3-бром-4-нитробензальдегида (7,97 г, 34,6 ммоль), который получали способом, аналогичным методике, описанной в литературе (Katritzky, A.R.; Xie, L. Tetrahedron Letters, 1996, 37, 347-350),триметилортоформиата (11,4 мл, 104 ммоль) и гидрата паратолуолсульфоновой кислоты (329 мг, 1,73 ммоль) в МеОН (69 мл) кипятили с обратным холодильником в течение 3 ч. Реакционную смесь затем гасили добавлением насыщенного водного гидроксида аммония (1 мл) и концентрировали на силикагеле. Очистка колоночной хроматографией (градиент от 10 до 25% EtOAc тексаны) давала 8,76 г (92%) соединения, указанного в заголовке, в виде оранжевого масла. 1 Н ЯМР (300 МГц, CDCl3):7.89 (m, 2H), 7.59 Раствор трис(дибензилиденацетон)дипалладия(0) (117 мг, 0,127 ммоль), 9,9-диметил-4,5-бис (дифенилфосфино)ксантена (162 мг, 0,280 ммоль), метил-5-амино-3-1R)-1-[2-(трифторметил)фенил]этил окси)-2-тиофенкарбоксилата (2,31 г, 6,69 ммоль), 4-[бис(метилокси)метил]-2-бром-1-нитробензола (1,76 г, 6,37 ммоль) и карбоната цезия (10,39 г, 31,89 ммоль) в 1,4-диоксане (25 мл) получали в круглодонной колбе в атмосфере N2. Колбу освобождали от содержимого, заполняли три раза N2 и затем перемешивали при 60 С в течение 16 ч. Реакционную смесь затем разбавляли тетрагидрофураном (100 мл) и концентрировали на силикагеле. Очистка колоночной хроматографией (градиент от 5 до 75% EtOAc/гексан) давала 2,79 г (81%) соединения, указанного в заголовке, в виде красной пены. 1 Н ЯМР (300 МГц, CDCl3):9.63 (br s, 1H), 8.21 (m, 1H), 7.94 (m, 1H), 7.62 (m, 2H), 7.48 (s, 1H), 7.40 (m, 1H), 7.02 (m, 1 Н), 6.47 (s,1 Н), 5.73 (q, 1 Н, J=6,2 Гц), 3.88 (s, 3H), 3.34 (s, 1 Н), 3.31 (s, 3H), 3.28 (s, 3H), 1.72 (d, 3H, J=6,2 Гц); MS К раствору метил-5-(5-[бис(метилокси)метил]-2-нитрофениламино)-3-1R)-1-[2-(трифторметил) фенил]этилокси)-2-тиофенкарбоксилата (2,71 г, 5,01 ммоль) в триметилортоформиате (50 мл) в колбе Фишера-Портера (Fisher-Porter) добавляли пиридиния пара-толуолсульфонат (126 мг, 0,501 ммоль) и сульфидированную платину на углероде (5 мас.% Pt, 977 мг, 0,250 ммоль Pt). Смесь гидрировали на гидрогенизационной установке Фишера-Портера при давлении водорода 50 фунтов/кв. дюйм (344,738 кПа),пока не прекращалось поглощение водорода (17 ч). Реакционную смесь фильтровали через фильтр из
МПК / Метки
МПК: C07D 409/04, A61K 31/4184, A61P 35/00
Метки: plk, бензимидазолтиофеновые, соединения, киназ, polo-подобных, ингибиторов, качестве
Код ссылки
<a href="https://eas.patents.su/30-14111-benzimidazoltiofenovye-soedineniya-v-kachestve-ingibitorov-polo-podobnyh-kinaz-plk.html" rel="bookmark" title="База патентов Евразийского Союза">Бензимидазолтиофеновые соединения в качестве ингибиторов polo-подобных киназ (plk)</a>
Птеридиноны в качестве ингибиторов киназ

Номер патента: 5287
Опубликовано: 30.12.2004
Авторы: Денни Уильям Эликзэндер, Рукасл Гордон Уильям, Тугуд Питер Лоренс, Крамер Джеймс Бернард, Мак Намара Деннис Джозеф, Добрусин Эллен Майра, Шоуолтер Хауард Дэниэл Холлис
МПК: A61K 31/505, A61P 35/00, C07D 475/00...
Метки: качестве, птеридиноны, киназ, ингибиторов
Формула / Реферат:
1. Соединение формулы и его фармацевтически приемлемые соли, эфиры, амиды и пролекарства, где R2 представляет собой (CH2)nарил и (CH2)nгетероарил, где каждая из вышеупомянутых арильных и гетероарильных групп может быть не замещена или замещена 1-5 группами-заместителями, выбранными из (а) галогена; (б) амино, алкиламино и диалкиламино; (в) C1-C10алкокси, аминоалкокси, алкиламиноалкокси и диалкиламиноалкокси; (г) гидрокси; (д) алканоила,...
Замещенные 4-аминотиазолы в качестве ингибиторов циклинзависимых киназ

Номер патента: 3527
Опубликовано: 26.06.2003
Авторы: Ли Лин, Дувади Рохит К., Ксиао Вей, Янг Йи, Чонг Уэсли К.М., Чу Шао Сонг
МПК: C07D 277/42, A61K 31/427, A61P 35/00...
Метки: 4-аминотиазолы, замещенные, качестве, киназ, ингибиторов, циклинзависимых
Формула / Реферат:
1. Соединение формулы I где R1 - замещенная или незамещенная группа, выбранная из C1-6-алкила; фенила; 5-9-членного гетероцикла, содержащего в качестве гетероатомов N, O, S; циклоалкила; (C1-6-алкил)карбонила; (C1-6-алкил)арила; и, в случае если R1 замещен, каждый заместитель независимо представляет собой галоген; галогеналкил; C1-6-алкил; C1-6-алкоксил; амино; нитро; простой тиоэфир; циано; амидо; карбоксил; сульфонамидную группу; кето-группу;...
Производные пиразолохиназолина: способ получения и применение в качестве ингибиторов киназ

Номер патента: 10904
Опубликовано: 30.12.2008
Авторы: Тракванди Габриелла, Получчи Паоло, Вианелло Паола, Д`алессио Роберто, Певарелло Паоло, Панцери Акилле, Квартьери Франческа, Вульпетти Анна, Фергюсон Рон, Ролетто Фульвия, Фанчелли Даниеле, Браска Мария Габриелла
МПК: A61K 31/519, A61P 35/00, C07D 487/04...
Метки: способ, применение, ингибиторов, производные, получения, пиразолохиназолина, качестве, киназ
Формула / Реферат:
1. Производное пиразолохиназолина, представленное формулой (Ia) или (Ib) или его фармацевтически приемлемая соль, где R обозначает водород или группу, выбранную из амино, линейного или разветвленного C1-C6алкила, C3-C10циклоалкила, арила, арилC1-C6алкила, гетероциклила или гетероциклилC1-C6алкила; X обозначает простую связь или двухвалентный радикал, выбранный из -NR'-, -CONR'-, -NH-CO-NH-, -О- или -S-, где R' обозначает водород или группу,...
Производные аминоиндазола, активные в качестве ингибиторов киназ, способ их получения и содержащая их фармацевтическая композиция

Номер патента: 7430
Опубликовано: 27.10.2006
Авторы: Амичи Раффаэлла, Вульпетти Анна, Мартина Катия, Салом Барбара, Д'анелло Маттео
МПК: C07D 231/56, A61K 31/416, A61P 35/00...
Метки: качестве, аминоиндазола, содержащая, получения, активные, производные, ингибиторов, композиция, киназ, способ, фармацевтическая
Формула / Реферат:
1. Способ лечения заболеваний, обусловленных и/или связанных с измененной (нарушенной) активностью протеинкиназ, включающий введение нуждающемуся в этом млекопитающему эффективного количества производного аминоиндазола, представленного формулой (I) где R выбирают из группы, состоящей из -NHCOR', -NHCONHR', -NHCONR'R", -NHSO2R' или -NHCOOR', где R' и R" каждый независимо выбран из прямого или разветвленного C1-С6алкила, С2-С6 алкенила или...
Аминокислотные соединения и их применение в качестве ингибиторов nep, ace и ece

Номер патента: 5004
Опубликовано: 28.10.2004
Авторы: Беннжан Каролина, Ренар Пьер, Рок Бернар П., Фурнье-Залуски Мари-Клод, Скальбер Элизабет, Ингимбер Никола, Пора Эрве
МПК: C07D 209/20, A61K 31/33, A61P 43/00...
Метки: качестве, ингибиторов, применение, соединения, аминокислотные
Формула / Реферат:
1. Соединения формулы (I) в которой n представляет собой коэффициент, в котором 0_n_3, m представляет собой коэффициент, в котором 0_m_6, R3 и R4 совместно образуют с двумя атомами углерода, которые их несут, фенильную группу, которая незамещена или замещена от 1 до 3 одинаковыми или различными группами, выбираемыми из алкила, алкенила, алкинила, алкокси, гидрокси, алкилтио, меркапто, циано, нитро, амино, алкиламино, диалкиламино,...
Предыдущий патент: Компоновки и способы сварки разнородных материалов
Следующий патент: Моноклональное антитело к миостатину и способы его применения
Случайный патент: Способ нанесения покрытия на соединение между двумя трубами