Гидрохлорид 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила
Номер патента: 13686
Опубликовано: 30.06.2010
Авторы: Петерс Йозеф, Стевенс Пол Теодор Агнес, Вандекрюйс Рогер Петрус Гереберн, Стапперс Альфред Элизабет, Копманс Алекс Херман


























Формула / Реферат
1. Твердая фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения формулы (I-а)

или его N-оксида.
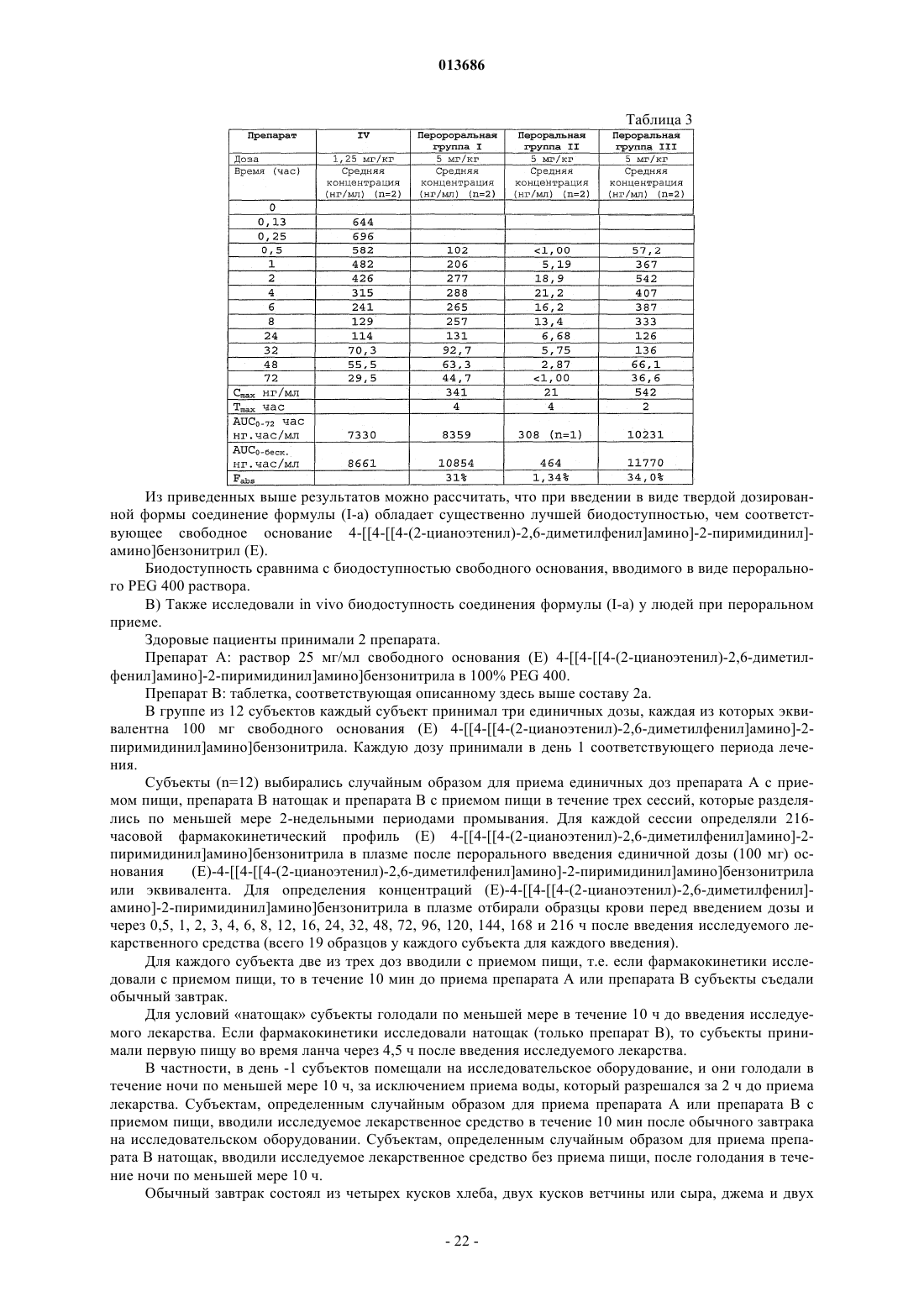
2. Фармацевтическая композиция по п.1, в которой соединение формулы (I-а) представляет собой полиморфную форму А, характеризующуюся пиками порошковой рентгеновской дифракции в положениях два тета 9,7±0,2°, 13,5±0,2° и 15,0±0,2°.
3. Фармацевтическая композиция по п.2, в которой полиморфная форма А дополнительно характеризуется пиками порошковой рентгеновской дифракции в положениях два тета 9,1±0,2°, 11,0±0,2°, 14,6±0,2°, 22,0±0,2°, 25,0±0,2°, 25,3±0,2° и 26,7±0,2°.
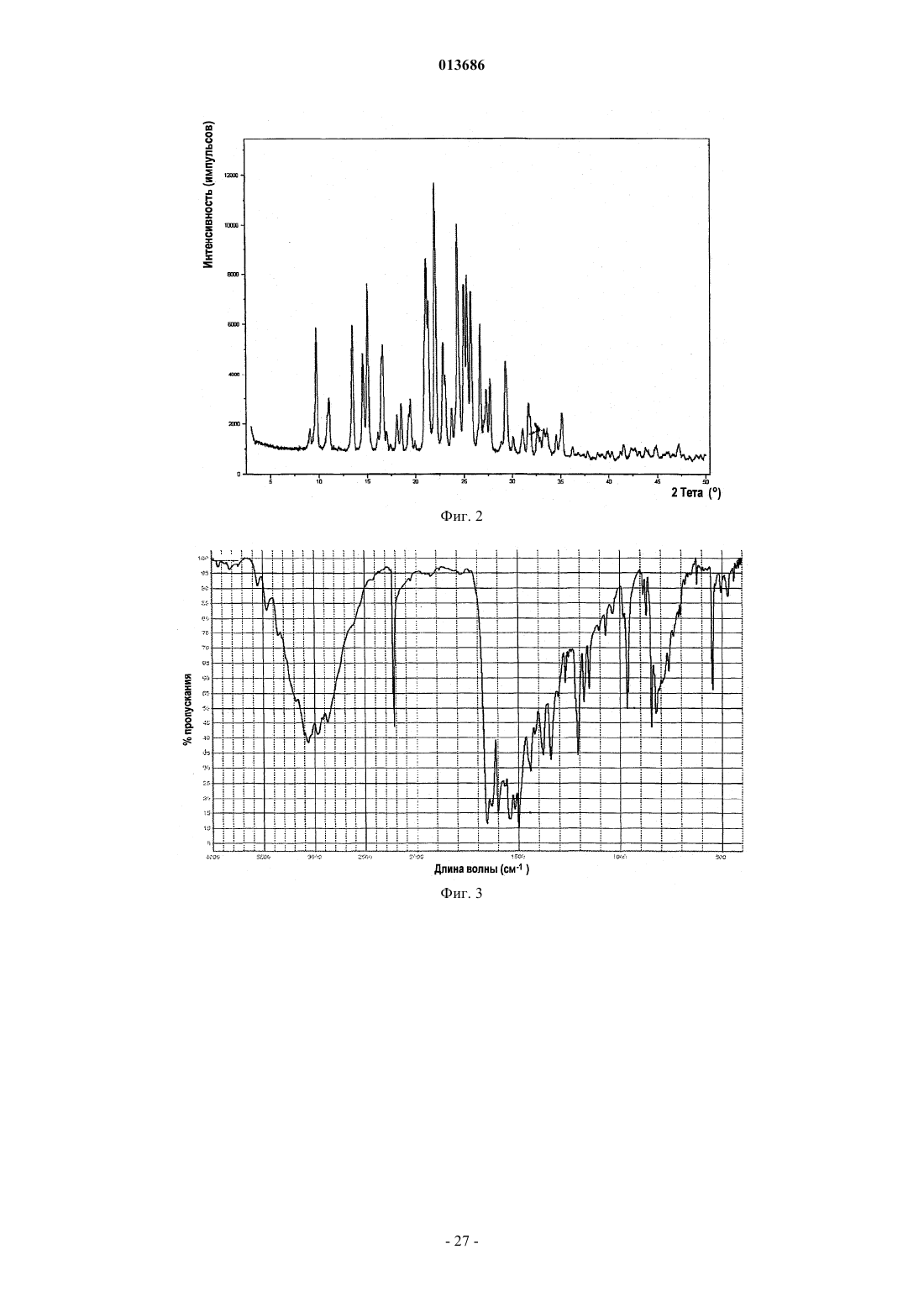
4. Фармацевтическая композиция по п.1, в которой соединение формулы (I-а) представляет собой полиморфную форму В, характеризующуюся пиками порошковой рентгеновской дифракции в положениях два тета 4,5±0,2°, 8,8±0,2° и 12,5±0,2°.
5. Фармацевтическая композиция по п.4, в которой полиморфная форма В дополнительно характеризуется пиками порошковой рентгеновской дифракции в положениях два тета 10,3±0,2°, 14,7±0,2°, 20,6±0,2°, 22,2±0,2 и 26,1±0,2°.
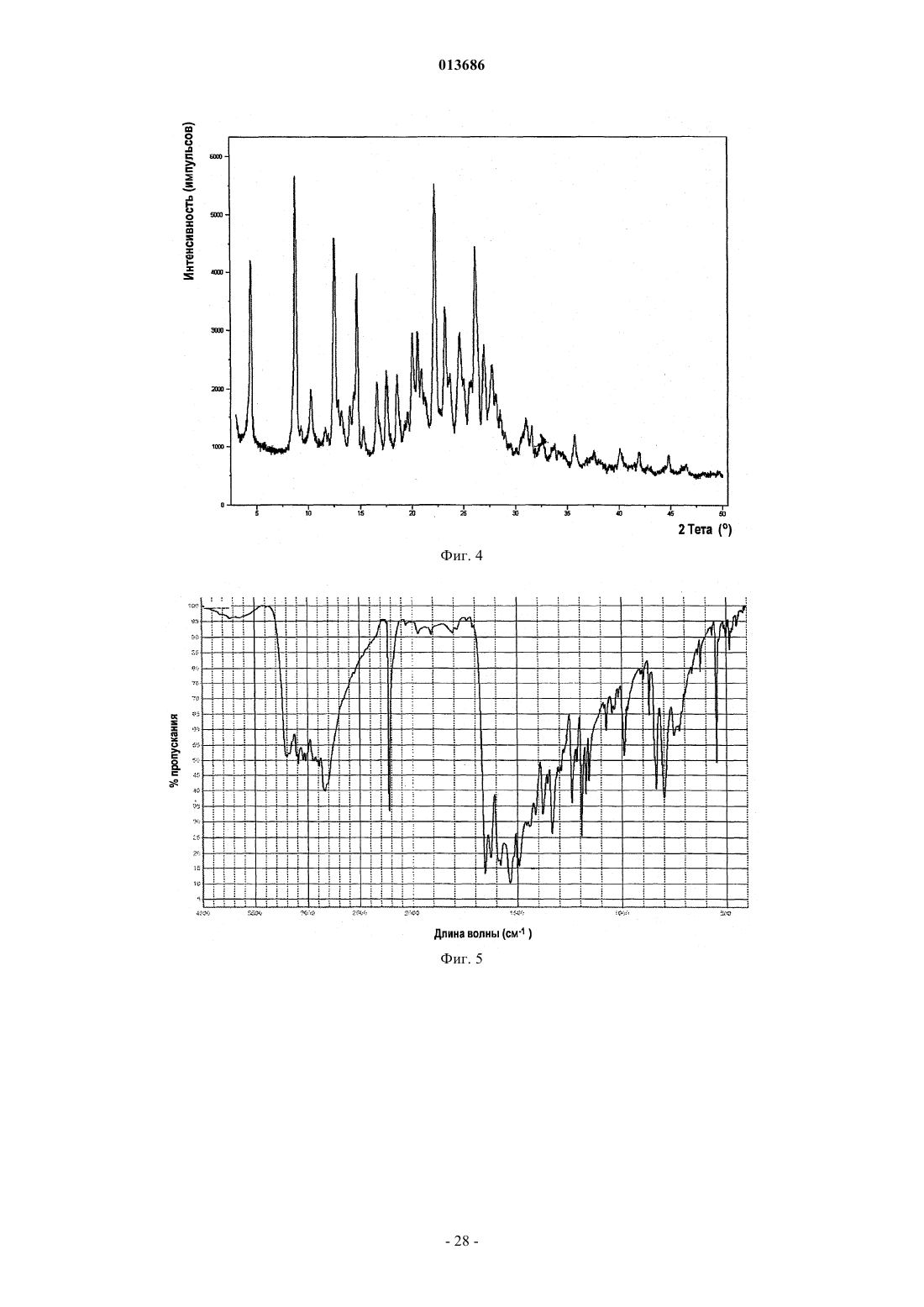
6. Фармацевтическая композиция по п.1, в которой соединение формулы (I-а) представляет собой полиморфную форму С, характеризующуюся пиками порошковой рентгеновской дифракции в положениях два тета 11,9+0,2°, 14,3±0,2° и 22,3±0,2°.
7. Фармацевтическая композиция по п.6, в которой полиморфная форма С дополнительно характеризуется пиками порошковой рентгеновской дифракции в положениях два тета 12,8±0,2°, 18,5±0,2°, 21,2±0,2°, 24,3±0,2 и 26,0±0,2°.
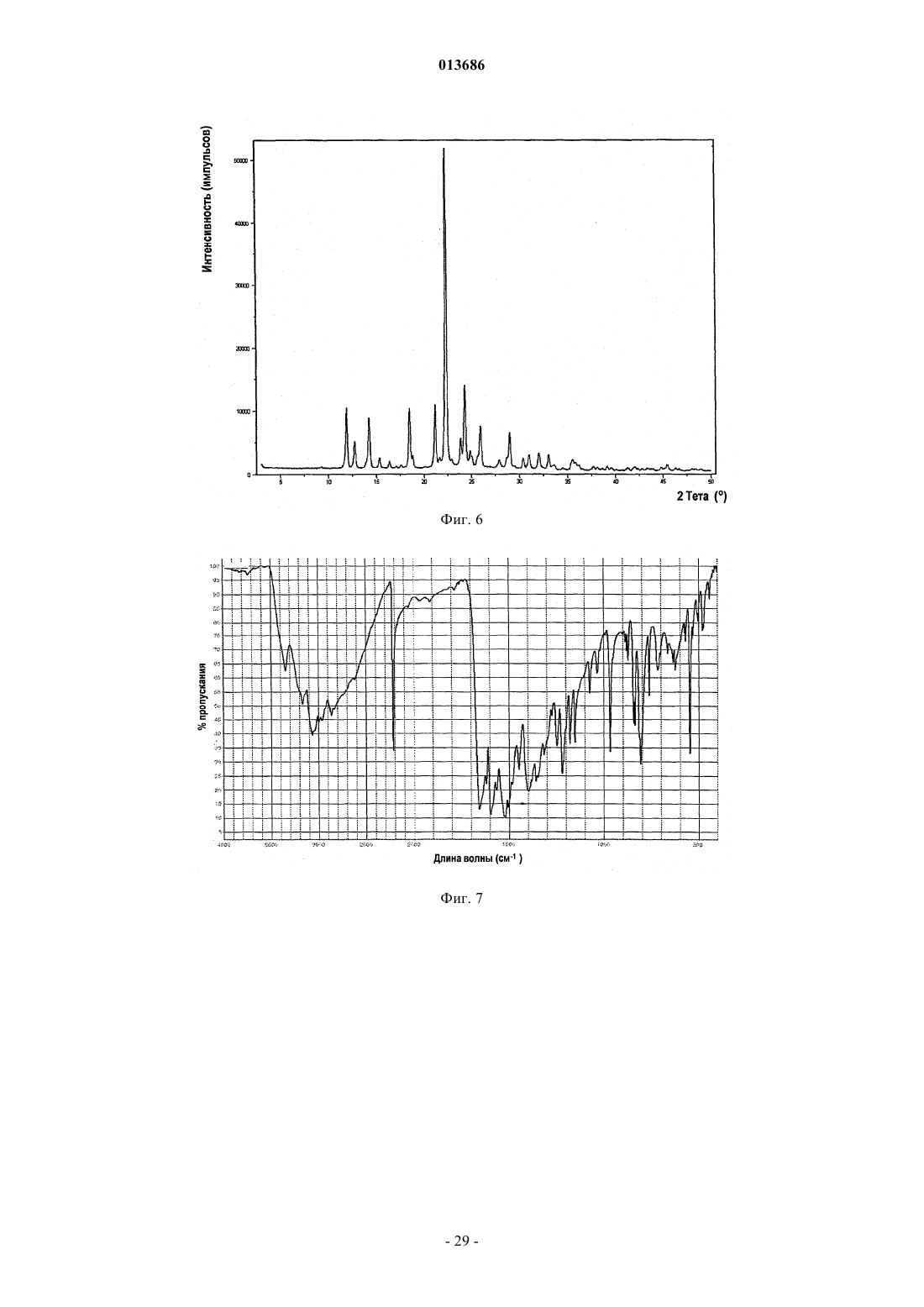
8. Фармацевтическая композиция по п.1, в которой соединение формулы (I-а) представляет собой полиморфную форму D, характеризующуюся пиками порошковой рентгеновской дифракции в положениях два тета 6,6±0,2°, 11,6±0,2° и 17,1±0,2°.
9. Фармацевтическая композиция по п.8, в которой полиморфная форма D дополнительно характеризуется пиками порошковой рентгеновской дифракции в положениях два тета 15,0±0,2°, 19,2±0,2°, 20,5±0,2°, 21,6±0,2 и 29,8±0,2°.
10. Фармацевтическая композиция по любому из пп.1-3, в которой соединение формулы (I-a) представляет собой полиморфную форму А, характеризующуюся ФТ ИК-спектром с типичными полосами поглощения примерно при 2217, 1652, 1497, 1435, 1338, 1199 и 550 см-1.
11. Фармацевтическая композиция по п.10, в которой полиморфная форма А дополнительно характеризуется ФТ ИК-спектром с типичными полосами поглощения примерно при 1631, 1596, 1537, 1504, 1249, 1214, 1179, 1152 и 1070 см-1.
12. Фармацевтическая композиция по любому из предшествующих пунктов, где композиция является пригодной для перорального введения.
13. Фармацевтическая композиция по любому из предшествующих пунктов, где композиция дополнительно включает увлажняющий агент.
14. Фармацевтическая композиция по п.13, в которой увлажняющий агент представляет собой эфир полиэтиленсорбита и жирной кислоты.
15. Фармацевтическая композиция по любому из предшествующих пунктов, где композиция находится в форме таблетки.
16. Фармацевтическая композиция по п.15, которая имеет пленочное покрытие.
17. Фармацевтическая композиция по любому из предыдущих пунктов, имеющая следующий состав:
(а) 5-50% активного ингредиента;
(b) 0,01-5% увлажняющего агента;
(c) 40-92% разбавителя;
(d) 0-10% полимера;
(e) 2-10% разрыхлителя;
(f) 0,1-5% глиданта;
(g) 0,1-1,5% лубриканта.
18. Фармацевтическая композиция по любому из предыдущих пунктов, включающая количество активного ингредиента, которое эквивалентно 25 мг соответствующего свободного основания (осн. экв).
19. Фармацевтическая композиция по любому из пп.1-3, 10 или 11, где фармацевтическая композиция находится в форме таблетки, включающей ядро таблетки, имеющее состав:

20. Фармацевтическая композиция по любому из пп.1-3, 10 или 11, где фармацевтическая композиция находится в форме таблетки, включающей ядро таблетки, имеющее состав:

21. Фармацевтическая композиция по любому из пп.1-17, включающая количество активного ингредиента, которое эквивалентно 50 мг соответствующего свободного основания (осн. экв.).
22. Фармацевтическая композиция по любому из пп.1-3, 10 или 11, где фармацевтическая композиция находится в форме таблетки, включающей ядро таблетки, имеющее состав:

23. Фармацевтическая композиция по любому из пп.1-17, включающая количество активного ингредиента, которое эквивалентно 75 мг соответствующего свободного основания (осн. экв.).
24. Фармацевтическая композиция по любому из пп.1-3, 10 или 11, где фармацевтическая композиция находится в форме таблетки, включающей ядро таблетки, имеющее состав:

25. Фармацевтическая композиция по любому из пп.1-17, включающая количество активного ингредиента, которое эквивалентно 100 мг соответствующего свободного основания (осн. экв.).
26. Фармацевтическая композиция по любому из пп.1-3, 10 или 11, где фармацевтическая композиция находится в форме таблетки, включающей ядро таблетки, имеющее состав:

27. Фармацевтическая композиция по любому из пп.1-17, включающая количество активного ингредиента, которое эквивалентно 150 мг соответствующего свободного основания (осн. экв.).
28. Фармацевтическая композиция по любому из пп.1-3, 10 или 11, где фармацевтическая композиция находится в форме таблетки, включающей ядро таблетки, имеющее состав:

29. Способ получения фармацевтической композиции по любому из предыдущих пунктов, включающий следующие стадии:
(i) сухое перемешивание активного ингредиента и части разбавителя;
(ii) получение связующего раствора посредством растворения связующего и увлажняющего агента в растворителе для связующего раствора;
(iii) распыление связующего раствора, полученного на стадии (ii), на смесь, полученную на стадии (i);
(iv) сушка влажного порошка, полученного на стадии (iii), с последующим просеиванием и необязательным перемешиванием;
(v) смешивание оставшейся части разбавителя, разрыхлителя и необязательного глиданта со смесью, полученной на стадии (iv);
(vi) необязательное добавление лубриканта к смеси, полученной на стадии (v);
(vii) прессование таблеток из смеси, полученной на стадии (vi);
(viii) необязательное нанесение пленочного покрытия на таблетку, полученную на стадии (vii).
30. Применение соединения формулы (I-а), как определено по п.1, для производства композиции по пп.1-28 для лечения или профилактики ВИЧ-инфекции.
31. Фармацевтическая композиция по любому из пп.1-28, в которой частицы соединения формулы (I-а) имеют размер менее чем 50 мкм.
32. Фармацевтическая композиция по п.31, в которой частицы соединения формулы (I-а) имеют размер менее чем 25 мкм.
33. Фармацевтическая композиция по любому из пп.1-11, в которой частицы соединения формулы (I-а) имеют размер менее чем 50 мкм.
34. Фармацевтическая композиция по п.33, в которой частицы соединения формулы (I-а) имеют размер менее чем 25 мкм.
Текст
013686 Настоящее изобретение относится к фармацевтической композиции, содержащей соль гидрохлорида 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, и ее получению. В WO 03/16306 раскрываются производные пиримидина, ингибирующие ВИЧ-репликацию, среди которых 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил и его фармацевтически приемлемые соли. В WO 04/0162581 раскрываются способы получения 4-4-4-(2 цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила. 4-4-4-(2-Цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил, в частности Е-изомер, обладает превосходной активностью по ингибированию ВИЧ-репликации относительно дикого типа ВИЧ, а также штаммов ВИЧ с лекарственной и мультилекарственной устойчивостью (т.е. штаммов, которые стали устойчивы относительно известного в данной области лекарства (лекарств. Таким образом, данное соединение потенциально может быть хорошим кандидатом для разработки лекарственного средства для лечения ВИЧ-инфекции. Однако высокая фармакологическая активность, хороший фармакологический профиль - это не единственный фактор, который определяет лекарственную способность соединения. Хороший кандидат в лекарства предпочтительно должен быть стабилен химически, а также физически; должен иметь приемлемый профиль токсичности; должен иметь приемлемую биодоступность. Биодоступность соединения влияет на дозу соединения, которую необходимо ввести пациенту для достижения терапевтически эффективной концентрации соединения. Соединения, имеющие низкую биодоступность, необходимо вводить при более высоких дозах по сравнению с соединениями, имеющими более высокую биодоступность. Возможные последствия необходимости более высоких доз могут включать повышенный риск неблагоприятных эффектов; увеличение размера дозированной формы; увеличение частоты введения. Данные факторы могут влиять на соблюдение курса антиретровирусной терапии. Строгое соблюдение курса лечения является одним из наиболее важных факторов, влияющих на эффективность лечения ВИЧ. Увеличение частоты приема дозы и увеличение размера таблетки может вести к снижению строгости соблюдения курса лечения и, следовательно, снижать эффективность терапии. Таким образом, составляя лекарственное средство для лечения ВИЧ, предпочтительно иметь активное соединение с приемлемой биодоступностью. Биодоступность соединения, предназначенного для перорального приема, зависит от растворимости соединения в воде, а также проницаемости соединения (его способности абсорбироваться через кишечную мембрану). Научной основой для классификации лекарственных веществ на базе их растворимости в воде и кишечной проницаемости является Биофармацевтическая Система Классификации или BCS. СогласноBCS, лекарственные вещества классифицируют следующим образом: класс 1: высокая растворимость - высокая проницаемость; класс 2: низкая растворимость - высокая проницаемость; класс 3: высокая растворимость - низкая проницаемость; класс 4: низкая растворимость - низкая проницаемость. Соединения с низкой растворимостью или низкой проницаемостью (класс 2-4) могут иметь низкую биодоступность при пероральном введении. Свободное основание 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил может быть классифицировано как соединение BCS-класса 2 и, таким образом, иметь низкую растворимость в воде. 4-4-4-(2-Цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил демонстрирует низкую растворимость не только в воде, но также в кислой среде. Следовательно, при пероральном введении в виде обычной твердой дозированной формы можно ожидать низкую биодоступность. Имея дело с соединением BCS-класса 2, предназначенным для перорального введения, специалист в области фармацевтической технологии сосредотачивает внимание на выяснении возможностей повышения растворимости соединений, например, получая подходящую соль. Данным путем следуют в случае 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила. Полученные соли, по-видимому, имеют только незначительно улучшенную растворимость в воде и в HCl. Полученные соли еще принадлежат BCS-классу 2. Таким образом, для полученных солей также можно ожидать низкую биодоступность. Неожиданно, в настоящее время обнаружено, что соль гидрохлорид 4-4-4-(2-цианоэтенил)-2,6 диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, в частности его Е-изомер, имеет значительно улучшенную биодоступность in vivo по сравнению с свободным основанием. Фактически настоящая соль, вводимая в виде твердой дозированной формы, имеет in vivo биодоступность, которая сравнима с биодоступностью свободного основания, вводимого перорально в виде раствора PEG 400. Благодаря повышенной биодоступности in vivo, соль гидрохлорид можно получить без необходимости применения методик комплексообразования.-1 013686 Обнаружено также, что соль гидрохлорид по настоящему изобретению является негигроскопичной и химически и физически стабильной в различных условиях влажности и температуры. Описание чертежей На фиг. 1 представлен ИК-спектр полиморфной формы А соли (Е) 4-4-4-(2-цианоэтенил)-2,6 диметилфенил]амино]-2-пиримидинил]амино]бензонитрилHCl. На фиг. 2 представлена картина рентгеновской дифракции порошка полиморфной формы А соли(Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрилHCl. На фиг. 3 представлен ИК-спектр полиморфной формы В соли (Е) 4-4-4-(2-цианоэтенил)-2,6 диметилфенил]амино]-2-пиримидинил]амино]бензонитрилHCl в сухом состоянии. На фиг. 4 представлена картина рентгеновской дифракции порошка полиморфной формы В соли(Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрилHCl в сухом состоянии. На фиг. 5 представлен ИК-спектр полиморфной формы С соли (Е) 4-4-4-(2-цианоэтенил)-2,6 диметилфенил]амино]-2-пиримидинил]амино]бензонитрилHCl. На фиг. 6 представлена картина рентгеновской дифракции порошка полиморфной формы С соли(Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрилHCl. На фиг. 7 представлен ИК-спектр псевдополиморфной формы D соли (Е) 4-4-4-(2-цианоэтенил)2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрилHCl. На фиг. 8 представлена картина рентгеновской дифракции порошка псевдополиморфной формы D соли (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрилHCl. Настоящее изобретение относится к соли гидрохлорида (HCl) 4-4-4-(2-цианоэтенил)-2,6 диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, его N-оксида или стереохимически изомерной формы. Таким образом, настоящее изобретение относится, в частности, к соединению формулы (I) его N-оксиду или стереохимически изомерной форме. Предполагается, что N-оксидные формы настоящего соединения формулы (I) включают соединения формулы (I), в которых один или несколько третичных атомов азота окислены до так называемого Nоксида. Используемое здесь выше выражение "стереохимически изомерные формы" определяет все возможные стереоизомерные формы, которые может иметь соединение формулы (I) и N-оксиды. Если не упоминается или не указано по-другому, химическое название соединения обозначает смесь всех возможных стереохимически изомерных форм, а также каждую из индивидуальных изомерных форм соединения формулы (I) и его N-оксидов, по существу, не содержащих других изомеров. Очевидно, подразумевается, что стереохимически изомерные формы соединения формулы (I) включены в область данного изобретения. Соединение формулы (I) может существовать в 2 стереохимических конфигурациях по двойной связи цианоэтенильной цепи, а именно Е-конфигурации (Entgegen=напротив) (Е-изомер) и Zконфигурации (Zusammen=вместе) (Z-изомер). Обозначения Е и Z хорошо известны специалисту в данной области. Конкретный вариант соединения формулы (I) представляет Е-изомер, т.е. соединение формулы (I-а) Другой конкретный вариант соединения формулы (I) представляет Z-изомер, т.е. соединение формулы (I-b) Когда бы здесь ни упоминался Е-изомер, подразумевается чистый Е-изомер или любая изомерная-2 013686 смесь Е- и Z-изомеров, в которой Е-изомер преобладает, т.е. изомерная смесь, содержащая более 50%,или, в частности, более 80% Е-изомера, или еще конкретнее 90% Е-изомера. Особый интерес представляет Е-изомер, по существу не содержащий Z-изомера. Выражение по существу не содержащий в данном контексте обозначает E-Z-смеси без или почти без Z-изомера, например изомерные смеси, содержащие более 90%, в частности 95%, или даже 98%, или 99% Е-изомера. Когда бы здесь ни упоминался Z-изомер, подразумевается чистый Z-изомер или любая изомерная смесь Z- и Е-изомеров, в которой Z-изомер преобладает, т.е. изомерная смесь, содержащая более 50% или, в частности, более 80% Z-изомера или, еще конкретнее, более 90% Z-изомера. Особый интерес представляет Z-изомер, по существу не содержащий Е-изомера. Выражение по существу не содержащий в данном контексте обозначает E-Z-смеси без или почти без Е-изомера, например изомерные смеси, содержащие более 90%, в частности 95%, или даже 98%, или 99% Z-изомера. Полиморфные формы настоящих солей также входят в объем настоящего изобретения. Полиморфные формы фармацевтических соединений могут представлять интерес для тех, кто занимается разработкой подходящей дозированной формы, так как если полиморфная форма не сохраняется постоянной в течение клинических исследований и исследования стабильности, то нельзя сопоставлять точную используемую или отмеренную дозу при переходе от одной серии к другой. Получив фармацевтическое соединение для применения, важно распознать полиморфную форму, доставленную в каждую дозированную форму, чтобы убедиться, что в процессе получения используется одинаковая форма и что в каждую дозу включается одинаковое количество лекарства. Таким образом, надо обязательно убедиться, что присутствует одна полиморфная форма или некоторая известная комбинация полиморфных форм. Кроме того, некоторые полиморфные формы могут демонстрировать повышенную термодинамическую стабильность и могут больше, чем другие полиморфные формы, подходить для включения в фармацевтические препараты. Как здесь принято, полиморфные формы соединения по данному изобретению представляют собой одинаковую химическую сущность, но в разной кристаллической структуре. Формы с присоединенным растворителем (сольваты), которые способны образовывать соли по настоящему изобретению, также входят в область настоящего изобретения. Примерами таких форм являются, например, гидраты, алкоголяты и подобные. Сольваты также обозначаются здесь как псевдополиморфные формы. Предпочтительна безводная соль. Конкретным вариантом настоящего изобретения является конкретная полиморфная или псевдополиморфная форма соединения формулы (I-а), т.е. (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрилHCl. Первая индивидуальная полиморфная форма соединения формулы (I-а) обозначена здесь как форма А (см. фиг. 1 и 2). Вторая индивидуальная форма соединения формулы (I-а) обозначена здесь как форма В. Форма В может присутствовать в двух состояниях, сухом состоянии (полиморфная форма) и увлажненном состоянии (псевдополиморфная форма). Приведены только характеристики формы В в сухом состоянии(см. фиг. 3 и 4). Третья индивидуальная полиморфная форма соединения формулы (I-а) обозначена здесь как форма С (см. фиг. 5 и 6). Четвертая индивидуальная псевдополиморфная форма соединения формулы (I-а) обозначена здесь как форма D (см. фиг. 7 и 8). Предпочтительной полиморфной формой соединения формулы (I-а) является форма А. Когда бы здесь далее ни использовалось выражение "соединение формулы (I), (I-а) или (I-b)", подразумевается, что оно также включает N-оксидные формы, стереохимически изомерные формы и полиморфные или псевдополиморфные формы. Особый интерес представляет стереохимически чистая форма соединения формулы (I). Предпочтительным соединением формулы (I) является соединение формулы (I-а). Соединения формулы (I), (I-а) или (I-b) можно получить посредством взаимодействия соответствующего свободного основания с соляной кислотой (HCl) в присутствии подходящего растворителя, такого как, например, подходящая кислота, например уксусная кислота. Соединения формулы (I), (I-а) или (I-b) обладают антиретровирусной активностью. Они способны ингибировать репликацию ВИЧ, в частности ВИЧ-1. ВИЧ (вирус иммунодефицита человека) представляет собой этиологический агент синдрома приобретенного иммунодефицита (СПИД) у людей. Вирус ВИЧ преимущественно инфицирует клетки Т-4 человека и разрушает их или изменяет их обычную функцию,особенно координацию иммунной системы. В результате, инфицированный пациент постоянно имеет пониженное количество клеток Т-4, которые, кроме того, ведут себя аномально. Следовательно, иммунологическая защитная система не способна бороться с инфекциями и новообразованиями, и ВИЧинфицированный субъект обычно умирает от условно-патогенных инфекций, таких как пневмония, или от раковых заболеваний. Другие состояния, ассоциированные с ВИЧ-инфицированием, включают тромбоцитопению, саркому Капоши и инфекцию центральной нервной системы, характеризующиеся прогрессирующей демиелинизацией, результатом которой является слабоумие и такие симптомы, как про-3 013686 грессирующая дизартрия, атаксия и дезориентация. Кроме того, ВИЧ-инфицирование также ассоциируется с периферической невропатией, прогрессирующей генерализованной лимфаденопатией (PGL) и СПИД-связанным комплексом (ARC). Настоящие соединения также демонстрируют активность против ВИЧ-штаммов с лекарственной устойчивостью и множественной лекарственной устойчивостью, в частности штаммов ВИЧ-1 с лекарственной устойчивостью и множественной лекарственной устойчивостью, более конкретно, настоящие соединения демонстрируют активность против ВИЧ-штаммов, в особенности ВИЧ-1 штаммов, которые обладают благоприобретенной устойчивостью к одному или нескольким известным в данной области ненуклеозидным ингибиторам обратной транскриптазы. Известные в данной области ненуклеозидные ингибиторы обратной транскриптазы представляют собой ненуклеозидные ингибиторы обратной транскриптазы, отличные от настоящих соединений, и, в частности, коммерческие ненуклеозидные ингибиторы обратной транскриптазы. Активность 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила по ингибированию ВИЧ-репликации описана в заявке WO 03/16306, которая включена здесь в виде ссылки. Благодаря своим антиретровирусным свойствам, в частности своим анти-ВИЧ свойствам, главным образом, своей ингибирующей репликацию ВИЧ-1 активности, настоящие соединения пригодны для лечения ВИЧ-инфицированных индивидуумов и для профилактики данных инфекций. Вообще, соединения настоящего изобретения могут быть полезны при лечении теплокровных млекопитающих, инфицированных вирусами, существование которых опосредовано или зависит от фермента обратной транскриптазы. Состояния, которые можно предотвратить или лечить соединениями настоящего изобретения,главным образом, состояния, ассоциированные с ВИЧ и другими патогенными ретровирусами, включают СПИД, СПИД-связанный комплекс (ARC), прогрессирующую генерализованную лимфаденопатию(PGL), а также хронические заболевания центральной нервной системы, вызванные ретровирусами, такие как, например, ВИЧ-опосредованное слабоумие и рассеянный склероз. Следовательно, соединения формулы (I), (I-а) или (I-b) можно применять в качестве лекарственного средства. Таким образом, соединения настоящего изобретения можно использовать в качестве лекарственных средств против указанных выше состояний. Указанное применение в качестве лекарственного средства или способ лечения включает введение ВИЧ-инфицированным субъектам количества, эффективного для борьбы с состояниями, ассоциированными с ВИЧ и другими патогенными ретровирусами, главным образом, с ВИЧ-1. В частности, настоящие соединения можно использовать в производстве лекарственного средства для лечения или профилактики ВИЧ-инфицирования, предпочтительно для лечения ВИЧинфицирования. С учетом применимости настоящих соединений обеспечен также способ лечения млекопитающих,включая людей, или способ профилактики теплокровных млекопитающих, включая людей, страдающих от вирусных инфекций, в особенности от ВИЧ-инфекций. Указанный способ включает введение, предпочтительно пероральное введение, млекопитающим, включая людей, эффективного количества соли по настоящему изобретению. Благодаря более высокой биодоступности настоящих соединений по сравнению с соответствующим свободным основанием, можно получить терапевтически эффективные уровни в плазме при введении фармацевтической композиции, содержащей меньшее количество соли по сравнению с необходимым количеством соответствующего свободного основания. Следовательно, можно уменьшить размер фармацевтической композиции или снизить частоту дозирования. Таким образом, настоящее изобретение также касается фармацевтической композиции, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения формулы (I), (I-а) или (I-b). Настоящее изобретение также касается фармацевтической композиции, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения формулы (I), (I-а) или (I-b), при условии, что композиция не содержит ни эмтрицитабина, ни тенофовирдиизопроксилфумарата. В частности, настоящее изобретение также касается фармацевтической композиции, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения формулы (I), (I-а) или (I-b), при условии, что композиция не включает один или несколько нуклеозидных ингибиторов обратной транскриптазы и/или один или несколько нуклеотидных ингибиторов обратной транскриптазы. Для целей введения настоящие соединения формулы (I), (I-а) или (I-b) можно приготовить в виде разнообразных фармацевтических композиций. В качестве подходящих композиций можно указать все композиции, обычно используемые для системного приема лекарств. Для получения фармацевтических композиций по данному изобретению эффективное количество соединения формулы (I), (I-а) или (I-b) в качестве активного ингредиента объединяют в гомогенную смесь с фармацевтически приемлемым носителем, причем указанный носитель может иметь широко разнообразные формы в зависимости от формы требуемого для введения препарата. Данные фармацевтические композиции желательно иметь в стан-4 013686 дартном дозированном виде, подходящем, в частности, для перорального приема. Например, при получении композиций в виде дозированной формы для перорального приема можно использовать любую из обычных фармацевтических сред, например воду, гликоли, масла, спирты и подобные, в случае жидких препаратов для перорального приема, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, разбавители, лубриканты, связующие, разрыхлители и подобные в случае порошков, пилюль, капсул и таблеток. Из-за простоты введения таблетки и капсулы представляют собой наиболее преимущественные стандартные дозированные формы для перорального приема, в которых, очевидно, используют твердые фармацевтические носители. Носитель для парентеральных композиций обычно включает стерильную воду, по меньшей мере большую часть, хотя можно включать другие ингредиенты, например, для содействия растворимости. Например, можно приготовить растворы для инъекций, в которых носитель включает физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Можно также приготовить суспензии для инъекций, в которых можно использовать подходящие жидкие носители, суспендирующие агенты и подобные. Включены также препараты в виде твердых форм, которые предназначены для превращения в жидкие формы незадолго до использования. В композициях, подходящих для чрескожного введения,носитель необязательно включает агент, увеличивающий проникание, и/или подходящий увлажняющий агент, необязательно объединенные с подходящими добавками любой природы при незначительном содержании, причем указанные добавки не оказывают существенного вредного эффекта на кожу. Указанные добавки могут способствовать введению в кожу и/или могут быть полезны для получения требуемых композиций. Данные композиции можно вводить различными способами, например посредством трансдермального пластыря, точечно, в виде мази. Соли по настоящему изобретению также можно вводить посредством ингаляции или инсуффляции,применяя методы и препараты, используемые в данной области для введения данным способом. Таким образом, вообще, соли настоящего изобретения можно вводить в легкие в виде раствора, суспензии или сухого порошка. Любая система, разработанная для доставки растворов, суспензий или сухих порошков посредством пероральной или носовой ингаляции либо инсуффляции, подходит для введения настоящих соединений. Соединения настоящего изобретения можно также принимать локально в виде капель, в частности глазных капель. Указанные глазные капли могут быть в виде раствора или суспензии. Любая система,разработанная для доставки растворов или суспензий, такая как глазные капли, подходит для введения настоящих соединений.WO 2004/069812, которая включена здесь в виде ссылки, описывает способность производных пиримидина, среди которых 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил и его фармацевтически приемлемые соли, предотвращать ВИЧ-инфицирование при половых сношениях или интимном контакте между партнерами. Таким образом, настоящее изобретение также относится к фармацевтической композиции в виде, адаптированном для применения на том месте, где может иметь место половое сношение или интимный контакт, таком как гениталии, прямая кишка, рот,руки, низ живота, верх бедер, в особенности влагалище и рот, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента эффективное количество соединения формулы (I), (I-а) или (I-b). В частности, настоящее изобретение также относится фармацевтической композиции в виде,адаптированном для применения на месте, где может иметь место половое сношение или интимный контакт, таком как гениталии, прямая кишка, рот, руки, низ живота, верх бедер, в особенности влагалище и рот, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента эффективное количество соединения формулы (I), (I-а) или (I-b), при условии, что композиция не содержит ни эмтрицитабина, ни тенофовирдиизопроксилфумарата. Более конкретно, настоящее изобретение также относится к фармацевтической композиции в виде, адаптированном для применения на месте, где может иметь место половое сношение или интимный контакт, таком как гениталии, прямая кишка, рот, руки,низ живота, верх бедер, в особенности влагалище и рот, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента эффективное количество соединения формулы (I), (I-а) или (Ib), при условии, что композиция не включает один или несколько нуклеозидных ингибиторов обратной транскриптазы и/или один или несколько нуклеотидных ингибиторов обратной транскриптазы. В качестве подходящих специально адаптированных композиций можно указать все композиции, обычно применяемые во влагалище, прямой кишке, во рту и на коже, такие как, например, гели, желе, кремы, мази,пленки, губки, пены, внутривлагалищные кольца, цервикальные колпачки, суппозитории для ректального или влагалищного применения, влагалищные, ректальные или буккальные таблетки, полоскания для рта. Для получения таких фармацевтических композиций эффективное количество активного ингредиента объединяют в гомогенную смесь с фармацевтически приемлемым носителем, причем указанный носитель может иметь широкое разнообразие форм в зависимости от формы введения. Для увеличения времени удержания такой фармацевтической композиции на месте применения может быть полезно включить в композицию биоадгезионный агент, в частности биоадгезионный полимер. Биоадгезионный агент можно определить как материал, который прилипает к живой биологической поверхности, такой как,например, слизистая мембрана или ткань кожи.-5 013686 Таким образом, настоящее изобретение также относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента эффективное количество соединения формулы (I), (I-а) или (I-b), характеризующейся тем, что данная фармацевтическая композиция является биоадгезионной относительно места применения. Предпочтительным местом применения является влагалище, прямая кишка, рот или кожа, наиболее предпочтительным является влагалище. В частности, настоящее изобретение также относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента эффективное количество соединения формулы (I), (I-а) или (I-b), характеризующейся тем, что данная фармацевтическая композиция является биоадгезионной относительно места применения при условии, что композиция не содержит ни эмтрицитабина, ни тенофовирдиизопроксилфумарата. Более конкретно, настоящее изобретение также относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента эффективное количество соединения формулы (I), (I-а) или (I-b), характеризующейся тем, что данная фармацевтическая композиция является биоадгезионной относительно места применения при условии, что композиция не включает один или несколько нуклеозидных ингибиторов обратной транскриптазы и/или один или несколько нуклеотидных ингибиторов обратной транскриптазы. Особенно полезно составлять указанные выше фармацевтические композиции в виде стандартной дозированной формы для простоты введения и единообразия дозировки. Используемая здесь стандартная дозированная форма представляет физически дискретные единицы, подходящие в качестве разовых доз,причем каждая единица содержит предварительно определенное количество активного ингредиента, рассчитанное таким образом, чтобы производить желательный терапевтический эффект, в ассоциации с необходимым фармацевтическим носителем. Примерами таких стандартных дозированных форм являются таблетки (включая таблетки с насечкой или в оболочке), капсулы, пилюли, пакетики с порошками, пластинки, суппозитории, растворы или суспензии для инъекций и подобные, и их отдельные множества. Точная дозировка и частота приема зависят от конкретного состояния, подлежащего лечению, тяжести подлежащего лечению состояния, возраста, веса, пола, степени заболевания и общего физического состояния конкретного пациента, а также от другого лечения, которое может принимать индивидуум, что хорошо известно специалистам в данной области. Кроме того, очевидно, что указанное эффективное ежедневное количество можно уменьшать или увеличивать в зависимости от реакции принимающего лечение субъекта и/или в зависимости от оценки лечащего врача, прописывающего соединения данного изобретения. Пациенты могут принимать фармацевтические композиции настоящего изобретения в любое время дня независимо от приема пищи. Предпочтительно принимать настоящие композиции на сытый желудок. Предпочтительный вариант настоящего изобретения относится к пероральной фармацевтической композиции, т.е. фармацевтической композиции, подходящей для перорального введения, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения формулы (I), (I-а) или (I-b). В частности, настоящее изобретение относится к пероральной фармацевтической композиции, т.е. фармацевтической композиции, подходящей для перорального введения, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения формулы (I), (I-а) или (I-b), при условии, что композиция не содержит ни эмтрицитабина, ни тенофовирдиизопроксилфумарата; более конкретно,фармацевтической композиции, подходящей для перорального введения, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения формулы (I), (I-а) или (I-b), при условии, что композиция не включает один или несколько нуклеозидных ингибиторов обратной транскриптазы и/или один или несколько нуклеотидных ингибиторов обратной транскриптазы. В частности, пероральная фармацевтическая композиция представляет собой твердую фармацевтическую композицию для перорального приема, более конкретно таблетку или капсулу, еще конкретнее таблетку. Согласно настоящему изобретению таблетку можно приготовить в форме таблетки для приема один раз в день. Предпочтительно, чтобы фармацевтические композиции настоящего изобретения содержали такие количества соединения формулы (I), (I-а) или (I-b), которые эквивалентны примерно 5-500 мг соответствующего свободного основания 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, его Е- или Z-изомера, более предпочтительно примерно 10-250 мг соответствующего свободного основания, еще предпочтительнее примерно 20-200 мг соответствующего свободного основания. Предпочтительно, чтобы настоящие фармацевтические композиции содержали такие количества соединения формулы (I), (I-а) или (I-b), которые эквивалентны 25, 50, 75, 100 или 150 мг соответствующего свободного основания (осн. экв.). Используемое здесь выше или далее выражение "примерно" в отношении численных значений х означает, например, х 10%. Размер частиц соединения формулы (I), (I-а) или (I-b) предпочтительно составляет меньше 50 мкм,-6 013686 более предпочтительно меньше 25 мкм, еще предпочтительнее меньше 20 мкм. Более предпочтительным является размер частиц примерно 15 мкм или меньше, или примерно 12 мкм или меньше, или примерно 10 мкм или меньше, или примерно 5 мкм или меньше. Наиболее предпочтителен размер частиц в диапазоне от примерно 0,2 до примерно 15 мкм или от примерно 0,2 до примерно 10 мкм. Фармацевтические композиции по настоящему изобретению предпочтительно включают увлажняющий агент. Что касается увлажняющего агента, в композициях данного изобретения можно использовать любой из физиологически допустимых увлажняющих агентов, подходящих для использования в фармацевтической композиции. В данной области хорошо известно, что увлажняющий агент является амфифильным соединением; он содержит полярные гидрофильные фрагменты, а также неполярные гидрофобные фрагменты. Термины "гидрофильный" или "гидрофобный" являются относительными. Относительную гидрофильность или гидрофобность увлажняющего агента можно выразить его значением гидрофильно-липофильного баланса (значением HLB). Увлажняющие агенты с низким значением HLB категоризированы как "гидрофобные" увлажняющие агенты, тогда как увлажняющие агенты с высоким значением HLB категоризированы как "гидрофильные" увлажняющие агенты. Эмпирическим правилом является: увлажняющие агенты, имеющие значения HLB больше примерно 10, обычно считаются гидрофильными увлажняющими агентами; увлажняющие агенты, имеющие значения HLB ниже примерно 10, обычно считаются гидрофобными увлажняющими агентами. Настоящие композиции предпочтительно содержат гидрофильный увлажняющий агент. Следует принимать во внимание, что значение HLB увлажняющего агента является только грубым показателем гидрофильности/гидрофобности увлажняющего агента. Значение HLB конкретного увлажняющего агента может меняться в зависимости от способа определения HLB; может меняться в зависимости от его коммерческого источника; меняется от партии к партии. Специалист в данной области может легко идентифицировать гидрофильные увлажняющие агенты, подходящие для использования в фармацевтических композициях по настоящему изобретению. Увлажняющий агент настоящего изобретения может быть анионным, катионным, цвиттерионным или неионным увлажняющим агентом, последний является предпочтительным. Увлажняющий агент настоящего изобретения также может представлять собой смесь двух или более увлажняющих агентов. Подходящие увлажняющие агенты для использования в композициях по настоящему изобретению перечислены ниже. Следует подчеркнуть, что указанный список увлажняющих агентов является только иллюстративным, репрезентативным и неисчерпывающим. Таким образом, изобретение не ограничено увлажняющими агентами, перечисленными ниже. В настоящих композициях можно также использовать смеси увлажняющих агентов. Подходящие увлажняющие агенты, которые можно использовать в настоящем изобретении, включают: а) моноэфиры полиэтиленгликоля и жирных кислот, включая эфиры лауриновой кислоты, олеиновой кислоты, стеариновой кислоты, рицинолевой кислоты и подобных с PEG 6, 7, 8, 9, 10, 12, 15, 20, 25,30, 32, 40, 45, 50, 55, 100, 200, 300, 400, 600 и подобными, например PEG-6 лаурат или стеарат, PEG-7 олеат или лаурат, PEG-8 лаурат, или олеат, или стеарат, PEG-9 олеат или стеарат, PEG-10 лаурат, или олеат, или стеарат, PEG-12 лаурат, или олеат, или стеарат, или рицинолеат, PEG-15 стеарат или олеат,PEG-20 лаурат, или олеат, или стеарат, PEG-25 стеарат, PEG-32 лаурат, или олеат, или стеарат, PEG-30 стеарат, PEG-40 лаурат, или олеат, или стеарат, PEG-45 стеарат, PEG-50 стеарат, PEG-55 стеарат, PEG100 олеат или стеарат, PEG-200 олеат, PEG-400 олеат, PEG-600 олеат (увлажняющие агенты, принадлежащие к данной группе, известны, например, как Cithrol, Algon, Kessco, Lauridac, Mapeg, Cremophor,Emulgante, Nikkol, Myrj, Crodet, Albunol, Lactomul);b) диэфиры полиэтиленгликоля и жирных кислот, включающие диэфиры лауриновой кислоты,стеариновой кислоты, пальмитиновой кислоты, олеиновой кислоты и подобных с PEG-8, 10, 12, 20, 32,400 и подобными, например PEG-8 дилаурат или дистеарат, PEG-10 дипальмитат, PEG-12 дилаурат, или дистеарат, или диолеат, PEG-20 дилаурат, или дистеарат, или диолеат, PEG-32 дилаурат, или дистеарат,или диолеат, PEG-400 диолеат или дистеарат (увлажняющие агенты, принадлежащие к данной группе,известны, например, как Mapeg, Polyalso, Kessco, Githrol);d) полиэтиленгликольглицериновые эфиры жирных кислот, такие как, например, PEG-20 глицериллаурат, или глицерилстеарат, или глицерилолеат, PEG-30 глицериллаурат или глицерилолеат, PEG-15 глицериллаурат, PEG-40 глицериллаурат и подобные (увлажняющие агенты, принадлежащие к данной группе, известны, например, как Tagat, Glycerox L, Capmul);e) продукты трансэтерификации спирт-масло, включающие сложные эфиры спиртов или полиспиртов, таких как глицерин, пропиленгликоль, этиленгликоль, полиэтиленгликоль, сорбит, пентаэритрит и подобные, с природными и/или гидрированными маслами или растворимыми в маслах витаминами, такими как касторовое масло, гидрированное касторовое масло, витамин А, витамин D, витамин Е, вита-7 013686 мин K, пищевое растительное масло, например кукурузное масло, оливковое масло, арахисовое масло,косточковое пальмовое масло, косточковое абрикосовое масло, миндальное масло и подобные, напримерPEG-20 - касторовое масло, или гидрированное касторовое масло, или глицериды кукурузного масла, или глицериды миндального масла, PEG-23 - касторовое масло, PEG-25 - гидрированное касторовое масло или триолеат, PEG-35 - касторовое масло, PEG-30 - касторовое масло или гидрированное касторовое масло, PEG-38 - касторовое масло, PEG-40 - касторовое масло, или гидрированное касторовое масло, или косточковое пальмовое масло, PEG-45 - гидрированное касторовое масло, PEG-50 - касторовое масло или гидрированное касторовое масло, PEG-56 - касторовое масло, PEG-60 касторовое масло, или гидрированное касторовое масло, или глицериды кукурузного масла, или глицериды миндального масла, PEG80 - гидрированное касторовое масло, PEG-100 - касторовое масло или гидрированное касторовое масло,PEG-200 - касторовое масло, PEG-8 - каприловые/каприновые глицериды, PEG-6 - каприловые/каприновые глицериды, лауроилмакрогол-32 глицерид, стеароилмакроголглицерид, токоферил PEG1000 сукцинат (TPGS) (увлажняющие агенты, принадлежащие к данной группе, известны, например, какf) полиглицеридные производные жирных кислот, включая эфиры полиглицерина и жирных кислот,такие как, например, полиглицерил-10 лаурат, или олеат, или стеарат, полиглицерил-10 моно- и диолеат,полиглицерилполирицинолеат и подобные (увлажняющие агенты, принадлежащие к данной группе, известны, например, как Nikkol Decaglyn, Caprol или Polymuls);g) производные стерина, включающие полиэтиленгликольные производные стерина, такие как PEG24 холестериновый эфир, PEG-30 холестанол, PEG-25 фитостерин, PEG-30 соевый стерин и подобныеj) сложные эфиры сахаров, такие как, например, дистеарат/моностеарат сахарозы, моностеарат, или монопальмитат, или монолаурат сахарозы и подобные (увлажняющие агенты, принадлежащие к данной группе, известны, например, как эфир Sucro, Crodesta, монолаурат сахарозы);m) ионные увлажняющие агенты, включая катионные, анионные и цвиттерионные ПАВ, такие как соли жирных кислот, например олеат натрия, лаурилсульфат натрия, лаурилсаркозинат натрия, диоктилсульфосукцинат натрия, миристат натрия, пальмитат натрия, sodium state, рицинолеат натрия и подобные; такие как желчные соли, например холат натрия, таурохолат натрия, гликохолат натрия и подобные; такие как фосфолипиды, например яичный/соевый лецитин, гидроксилированный лецитин, лизофосфатидилхолин, фосфатидилхолин, фосфатидилэтаноламин, фосфатидилглицерин, фосфатидилсерин и подобные; такие как сложные эфиры фосфорной кислоты, например фосфат диэтаноламмонийполиоксиэтилен-10 олеилового эфира, продукты этерификации жирных спиртов или этоксилатов жирных спиртов фосфорной кислотой или фосфорным ангидридом; такие как карбоксилаты, например сукцинилированные моноглицериды, стеарилфумарат натрия, стеароилпропиленгликоль гидросукцинат, моно/диацетилированные сложные эфиры винной кислоты и моно- и диглицеридов, сложные эфиры лимонной кислоты и моно- и диглицеридов, глицериллактоэфиры жирных кислот, лактильные эфиры жирных кислот, стеароил-2-лактилат кальция/натрия, стеароиллактилат кальция/натрия, соли альгинаты,пропиленгликольальгинат, (простой эфир) карбоксилаты и подобные; такие как сульфаты и сульфонаты,например этоксилированные алкилсульфаты, алкилбензолсульфаты, альфа-олефинсульфонаты, ацилизэтионаты, ацилтаураты, алкилглицериловый эфир сульфонаты, динатрийоктилсульфосукцинат, динатрийундециленамидо-МЕА-сульфосукцинат и подобные; такие как катионные увлажняющие агенты, напри-8 013686 мер гексадецилтриаммонийбромид, децилтриметиламмонийбромид, цетилтриметиламмонийбромид, додециламмонийхлорид, соли алкилбензилдиметиламмония, соли диизобутилфеноксиэтоксидиметилбензиламмония, соли алкилпиридиния, бетаины (лаурилбетаин), этоксилированные амины (полиоксиэтилен-15 кокосовый амин) и подобные. Если в приведенном выше списке подходящих увлажняющих агентов перечислены различные возможности, например PEG-20 олеиловый эфир, или цетиловый эфир, или стеариловый эфир, это означает,что имеется в виду PEG-20 олеиловый эфир, и PEG-20 цетиловый эфир, и PEG-20 стеариловый эфир. Таким образом, например, выражение PEG-20 - касторовое масло, или гидрированное касторовое масло,или глицериды кукурузного масла, или глицериды миндального масла следует толковать как PEG-20 касторовое масло, и PEG-20 - гидрированное касторовое масло, и PEG-20 - глицериды кукурузного масла, и PEG-20 глицериды миндального масла. Предпочтительными увлажняющими агентами в настоящих композициях являются лаурилсульфат натрия, диоктилсульфосукцинат натрия или такие увлажняющие агенты, которые принадлежат к группе эфиров полиэтиленгликольсорбитана и жирных кислот, например увлажняющие агенты, известные какTween, например Tween 20, 60, 80. Наиболее предпочтительным увлажняющим агентом является Tween 20. В композициях по данному изобретению увлажняющий агент предпочтительно присутствует при концентрации от примерно 0,01 до примерно 5 мас.% относительно общей массы композиции, предпочтительно от примерно 0,1 до примерно 3 мас.%, более предпочтительно от примерно 0,1 до примерно 1 мас.%. Количество увлажняющего агента, используемое в настоящих композициях, может зависеть от количества присутствующего в композиции соединения формулы (I), (I-а) или (I-b) или от размера частиц соединения формулы (I), (I-а) или (I-b). Большее количество или меньший размер частиц может потребовать больше увлажняющего агента. Согласно настоящему изобретению в случае твердой фармацевтической композиции для перорального приема, такой как таблетка или капсула, композиция может также дополнительно содержать органический полимер. Органический полимер можно использовать в производстве композиции в качестве связующего. Органический полимер, используемый в композициях по данному изобретению, может быть любым из физиологически приемлемых водорастворимых синтетических, полусинтетических или несинтетических органических полимеров. Таким образом, полимер может быть, например, природным полимером, таким как полисахарид или полипептид либо их производные, или синтетическим полимером, таким как полиалкиленоксид (например, PEG), полиакрилат, поливинилпирролидон и др. Конечно, можно также использовать смешанные полимеры, например блок-сополимеры и гликопептиды. Удобно, если полимер имеет молекулярную массу в диапазоне от 500 Да до 2 МДа и кажущуюся вязкость от 1 до 15000 мПас в 2% водном растворе при 20 С. Например, можно выбрать водорастворимый полимер из группы, включающей алкилцеллюлозы, например метилцеллюлозу; гидроксиалкилцеллюлозы, например гидроксиметилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу и гидроксибутилцеллюлозу; гидроксиалкилалкилцеллюлозы, например гидроксиэтилметилцеллюлозу и гидроксипропилметилцеллюлозу (например, НРМС 2910 15 мПас; НРМС 2910 5 мПас); карбоксиалкилцеллюлозы, например карбоксиметилцеллюлозу; соли щелочных металлов и карбоксиалкилцеллюлозы, например натрий-карбоксиметилцеллюлозу; карбоксиалкилалкилцеллюлозы, например карбоксиметилэтилцеллюлозу,сложные эфиры карбоксиалкилцеллюлозы; крахмалы, например крахмал 1551; пектины, например натрий-карбоксиметиламилопектин; производные хитина, например хитозан; гепарин и гепариноиды; полисахариды, например альгиновую кислоту, ее соли со щелочными металлами и аммонием; каррагенаны, галактоманнаны, трагакант, агар-агар, аравийскую камедь, гуаровую смолу и ксантановую смолу; полиакриловые кислоты и их соли; полиметакриловые кислоты и их соли, метакрилатные сополимеры; поливиниловый спирт; поливинилпирролидон, сополимеры поливинилпирролидона с винилацетатом; полиалкиленоксиды, например полиэтиленоксид и полипропиленоксид, и сополимеры этиленоксида и пропиленоксида, например полоксамеры и полоксамины. Для приготовления композиций согласно настоящему изобретению также подходят не перечисленные полимеры, которые являются фармацевтически приемлемыми и имеют подходящие физико-9 013686 химические свойства, которые определены здесь выше. Предпочтительными органическими полимерами являются крахмал, поливинилпирролидон или простые эфиры целлюлозы, например PVP K29-32, PVP K90, метилцеллюлоза, гидроксипропилцеллюлоза, гидроксиэтилметилцеллюлоза или гидроксипропилметилцеллюлоза (НРМС). Указанная НРМС содержит достаточное количество гидроксипропильных и метоксигрупп для обеспечения растворимости в воде. НРМС, имеющие степень метоксизамещения от примерно 0,8 до примерно 2,5 и молярного гидроксипропилзамещения от примерно 0,05 до примерно 3,0, обычно являются водорастворимыми. Степень метоксизамещения указывает среднее число метилэфирных групп,присутствующих на ангидроглюкозных звеньях молекулы целлюлозы. Молярное гидроксипропилзамещение указывает среднее число молей пропиленоксида, которые прореагировали с каждым ангидроглюкозным звеном молекулы целлюлозы. Предпочтительной НРМС является гипромелоза 2910 (15 мПас) или гипромелоза 2910 (5 мПас), в особенности гипромелоза 2910 (15 мПас). Гидроксипропилметилцеллюлоза - это принятое в США название для гипромелозы (см. Martindale, The Extra Pharmacopoeia, 29thedition, page 1435). В четырехзначном номере "2910" первые две цифры представляют примерное процентное содержание метоксигрупп, третья и четвертая цифры представляют примерное процентное содержание гидроксипропоксигрупп; 15 или 5 мПас являются показателями кажущейся вязкости 2% водного раствора при 20 С. В подходящих композициях данного изобретения органический полимер может присутствовать в количестве примерно до 10 мас.%, предпочтительно от примерно 0,1 до примерно 5%, более предпочтительно от примерно 0,5 до примерно 3 мас.% (относительно общей массы композиции). Согласно настоящему изобретению в случае твердой фармацевтической композиции для перорального приема, такой как таблетка или капсула, композиция может также дополнительно содержать разбавитель и/или глидант. Фармацевтически приемлемые разбавители включают карбонат кальция, двухосновный фосфат кальция, дигидрат двухосновного фосфата кальция, трехосновный фосфат кальция, сульфат кальция,микрокристаллическую целлюлозу, в том числе окремненную микрокристаллическую целлюлозу, порошкообразную целлюлозу, декстраты, декстрин, декстрозный наполнитель, фруктозу, каолин, лактитол,безводную лактозу, моногидрат лактозы, маннит, сорбит, крахмал, предварительно желатинированный крахмал, хлорид натрия, сахарозу, прессуемый сахар, сахарную глазурь, высушенную распылением смесь моногидрата лактозы и микрокристаллической целлюлозы (75:25), доступную коммерчески какMicrocelac, совместно обработанную, высушенную распылением смесь микрокристаллической целлюлозы и коллоидного диоксида кремния (98:2), доступную коммерчески как Prosolv. Предпочтительными являются моногидрат лактозы, микрокристаллическая целлюлоза или окремненная микрокристаллическая целлюлоза. Фармацевтически приемлемые глиданты включают тальк, коллоидный диоксид кремния, крахмал,стеарат магния. Предпочтителен коллоидный диоксид кремния. В случае таблеток композиция может также дополнительно включать разрыхлитель и лубрикант. Фармацевтически приемлемые разрыхлители включают крахмал, ионообменные смолы, напримерAmberlite, сшитый поливинилпирролидон, модифицированную целлюлозную смолу, например натрийкросскармелозу (например, Ac-di-Sol), натрий-крахмалгликолат, натрий-карбоксиметилцеллюлозу,додецилсульфат натрия, модифицированный кукурузный крахмал, микрокристаллическую целлюлозу,магнийалюминийсиликат, альгиновую кислоту, альгинат, порошкообразную целлюлозу. Фармацевтически приемлемые лубриканты включают стеарат магния, стеарат кальция, стеариновую кислоту, тальк, полиэтиленгликоль, лаурилсульфат натрия, лаурилсульфат магния. Кроме того, таблетки по настоящему изобретению могут включать другие необязательные наполнители, такие как, например, ароматизаторы, подсластители и красители. Твердые фармацевтические композиции согласно настоящему изобретению могут включать (считая от общей массы композиции):(d) от 0,1 до 5% глиданта. Таблетки согласно настоящему изобретению могут включать (считая от общей массы ядра таблетки):- 10013686 На таблетки по настоящему изобретению необязательно можно нанести пленочное покрытие, следуя известным в данной области методикам нанесения покрытия. Таблетки с пленочным покрытием легче заглатывать, чем непокрытые ядра таблеток, обычно их легче отличить от других таблеток (в частности, если пленочные покрытия содержат краситель или пигмент), они могут иметь пониженную липкость и, кроме того, иметь повышенную стабильность (увеличенный срок хранения), например, благодаря тому, что покрытие может защищать активный ингредиент от влияния света. Предпочтительно, чтобы пленочное покрытие представляло собой покрытие для быстрого высвобождения. Пленочные покрытия могут включать пленкообразующий полимер и необязательно пластификатор или пигмент. Примером подходящего пленкообразующего полимера является гидроксипропилметилцеллюлоза и примером подходящего пластификатора является полиэтиленгликоль, например макрогол 3000 или 6000 или триацетин. Коммерчески доступные подходящие покрытия для фармацевтических таблеток хорошо известны специалисту в данной области. Предпочтительно, чтобы пленочное покрытие было непрозрачным пленочным покрытием. Примером подходящего покрытия является Opadry, в частности порошковое покрытиеOpadry II White. Таблетки по настоящему изобретению можно получить прямым прессованием или влажным гранулированием. Таким образом, настоящее изобретение относится также к способу получения таблетки, содержащей соединение формулы (I), (I-а) или (I-b), включающему следующие стадии:(i) сухое перемешивание активного ингредиента, разрыхлителя и необязательного глиданта с разбавителем;(ii) необязательное смешивание лубриканта со смесью, полученной на стадии (i);(iii) прессование таблеток из смеси, полученной на стадии (i) или на стадии (ii), в сухом состоянии; и(iv) необязательное нанесение пленочного покрытия на таблетку, полученную на стадии (iii). Настоящее изобретение также относится к способу получения таблетки, содержащей соединение формулы (I), (I-а) или (I-b), включающему следующие стадии:(i) сухое перемешивание активного ингредиента и части разбавителя;(ii) получение связующего раствора посредством растворения связующего и увлажняющего агента в растворителе для связующего раствора;(iii) распыление связующего раствора, полученного на стадии (ii), на смесь, полученную на стадии(iv) сушка влажного порошка, полученного на стадии (iii), с последующим просеиванием и необязательным перемешиванием;(v) смешивание оставшейся части разбавителя, разрыхлителя и необязательного глиданта со смесью, полученной на стадии (iv);(vi) необязательное добавление лубриканта к смеси, полученной на стадии (v);(vii) прессование таблеток из смеси, полученной на стадии (vi);(viii) необязательное нанесение пленочного покрытия на таблетку, полученную на стадии (vii). Специалист в данной области представляет наиболее подходящее оборудование, которое следует использовать для описанных выше способов. Специалист в данной области может модифицировать описанный выше общий способ получения таблеток по настоящему изобретению, например, добавляя некоторые ингредиенты не на тех стадиях, которые указаны выше, а на других. Настоящее соединение формулы (I), (I-а) или (I-b) можно использовать само по себе или в комбинации с другими терапевтическими агентами, такими как антивирусные агенты, антибиотики, иммуномодуляторы или вакцины для лечения вирусных инфекций. Их можно также использовать сами по себе или в комбинации с другими профилактическими агентами для профилактики вирусных инфекций. Настоящие соединения можно использовать в вакцинах и способах для защиты индивидуумов от вирусных инфекций в течение продолжительного периода времени. Соединения можно использовать в таких вакцинах сами по себе или вместе с другими антивирусными агентами способом, согласующимся с обычным применением ингибиторов обратной транскриптазы в вакцинах. Таким образом, настоящие соединения можно объединять с фармацевтически приемлемыми адъювантами, обычно используемыми в вакцинах, и вводить в профилактически эффективных количествах для защиты индивидуумов в течение продолжительного периода времени от ВИЧ-инфицирования. Также можно использовать в качестве лекарственного средства комбинацию антиретровирусного соединения и соединения формулы (I), (I-а) или (I-b). Таким образом, настоящее изобретение относится также к продукту, содержащему (а) соединение формулы (I), (I-а) или (I-b) и (b) одно или несколько других антиретровирусных соединений, в качестве комбинированного препарата для одновременного, раздельного или последовательного применения при анти-ВИЧ-лечении. Различные лекарства можно объединять в едином препарате вместе с фармацевтически приемлемыми носителями. Таким образом, настоящее изобретение также относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель, (а) терапевтически эффективное количество соединения формулы (I), (I-а) или (I- 11013686b) и (b) один или несколько антиретровирусных агентов. В частности, данное изобретение относится также к продукту, содержащему (а) соединение формулы (I), (I-а) или (I-b) и (b) одно или несколько других антиретровирусных соединений, в виде комбинированного препарата для одновременного, раздельного или последовательного применения при анти-ВИЧ-лечении, при условии, что композиция не содержит ни эмтрицитабина, ни тенофовирдиизопропоксилфумарата. Более конкретно, данное изобретение относится также к продукту, содержащему (а) соединение формулы (I), (I-а) или (I-b) и (b) одно или несколько других антиретровирусных соединений, в виде комбинированного препарата для одновременного, раздельного или последовательного применения при анти-ВИЧ-лечении, при условии, что указанное одно или несколько других антиретровирусных соединений отличны от нуклеозидных ингибиторов обратной транскриптазы и/или нуклеотидных ингибиторов обратной транскриптазы. Различные лекарства можно объединять в едином препарате вместе с фармацевтически приемлемыми носителями. Таким образом, настоящее изобретение также относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель, (а) терапевтически эффективное количество соединения формулы (I), (Iа) или (I-b) и (b) один или несколько антиретровирусных агентов. В частности, данное изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель, (а) терапевтически эффективное количество соединения формулы (I), (I-а) или (I-b) и (b) один или несколько антиретровирусных агентов, при условии, что композиция не содержит ни эмтрицитабина, ни тенофовирдиизопропоксилфумарата. Данное изобретение относится также к фармацевтической композиции,содержащей фармацевтически приемлемый носитель, (а) терапевтически эффективное количество соединения формулы (I), (I-а) или (I-b) и (b) один или несколько других антиретровирусных агентов, при условии, что указанное одно или несколько антиретровирусных соединений отличны от нуклеозидных ингибиторов обратной транскриптазы и/или нуклеотидных ингибиторов обратной транскриптазы. Указанные другие антиретровирусные соединения могут представлять собой известные антиретровирусные соединения, такие как сурамин, пентамидин, тимопентин, кастаноспермин, декстран (декстрансульфат), фоскарнет-натрий (тринатрийфосфоноформиат); нуклеозидные ингибиторы обратной транскриптазы, например зидовудин (3'-азидо-3'-деокситимидин, AZT), диданозин (2',3'-дидеоксиинозин;(2',3'-дидегидро-3'-деокситимидин, d4T), абакавир, абакавирсульфат, эмтрицитабин -)FTC), рацемический FTC и подобные; ненуклеозидные ингибиторы обратной транскриптазы, такие как невирапин (11 циклопропил-5,11-дигидро-4-метил-6 Н-дипиридо-[3,2-b:2',3'-е][1,4]диазепин-6-он), эфавиренц, делавирдин, ТМС-120, ТМС-125 и подобные; соединения TIBO (тетрагидроимидазо[4,5,1-jk][1,4]бензодиазепин 2(1 Н)-он и -тион)типа, например (S)-8-хлор-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил)имидазо[4,5,1-jk][1,4]бензодиазепин-2(1 Н)-тион; соединения типа -АРА (-анилинофенилацетамида), например-[(2-нитрофенил)амино]-2,6-дихлорбензолацетамид и подобные; ингибиторы транс-активирующих белков, такие как ТАТ-ингибиторы, например RO-5-3335, или REV-ингибиторы и. подобные; ингибиторы протеазы, например индинавир, ритонавир, саквинавир, лопинавир (АВТ-378), нелфинавир, ампренавир,ТМС-114, BMS-232632, VX-175 и подобные; ингибиторы слияния, например Т-20, Т-1249 и подобные; антагонисты рецепторов CXCR4, например AMD-3100 и подобные; ингибиторы вирусной интегразы; ингибиторы обратной транскриптазы нуклеотидного типа, например тенофовир, тенофовир дифосфат,тенофовир дизопроксил фумарат и подобные; ингибиторы рибонуклеотидной редуктазы, например гидроксикарбамид и подобные; CCR5 антагонисты, например анкривирок, аплавирок гидрохлорид, викривирок. Принимая соединения настоящего изобретения с другими антивирусными агентами, целью которых являются различные события в вирусном жизненном цикле, можно усилить терапевтический эффект данных соединений. Комбинационные терапии, которые описаны выше, оказывают синергический эффект при ингибировании ВИЧ-репликации, благодаря тому, что каждый компонент комбинации действует на разные сайты ВИЧ-репликации. Используя такие комбинации, можно уменьшить дозировку принимаемого обычного антиретровирусного агента, которая требуется для достижения желательного терапевтического или профилактического эффекта, по сравнению с приемом данного агента при монотерапии. Данные комбинации могут снизить или исключить побочные эффекты обычной антиретровирусной монотерапии, не влияя на антивирусную активность агентов. Данные комбинации снижают возможности резистентности к терапиям с применением одного агента, при этом снижая до минимума любую ассоциированную токсичность. Данные комбинации могут также увеличить эффективность обычного агента без повышения ассоциированной токсичности. Соединения настоящего изобретения можно также принимать в комбинации с иммуномодулирующими агентами, например левамизолом, бропиримином, антителом против человеческого альфаинтерферона, интерфероном-альфа, интерлейкином 2, метионин энкефалином, диэтилдитиокарбаматом,фактором некроза опухоли, налтрексоном и подобными; антибиотиками, например пентамидинизетиоратом и подобными; холинергическими агентами, например такрином, ривастигмином, донепезилом, галантамином и подобными; NMDA-блокаторами каналов, например мемантином, для профилактики или борьбы с инфицированием и заболеваниями или симптомами заболеваний, связанными с ВИЧ- 12013686 инфекциями, такими как СПИД и ARC, например слабоумием. Хотя настоящее изобретение фокусируется на применении настоящих соединений с целью профилактики или лечения ВИЧ-инфекций, настоящие соединения можно также использовать в качестве ингибиторных агентов для других вирусов, которые зависят от тех же обратных транскриптаз для обязательных событий в их жизненном цикле. Экспериментальная часть А. Синтез соединения формула (I-a).a) 10,99 кг (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила и 57 л уксусной кислоты (2 л/моль) нагревают до 90 С в сосуде для получения продукции. Раствор фильтруют при 95 С и промывают 3 л уксусной кислоты (0,21 л/моль). Добавляют 2,973 л соляной кислоты (1,1 моль/моль) при 80 С. При 85 С медленно добавляют 60 л воды (2 л/моль). Смесь медленно охлаждают до 25 С, промывают два раза 5,4 л воды и сушат при 50 С. Полученный продукт размалывают. Выход: соединение формулы (I-а), форма А.b) Примерно 150 мг соединения формулы (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2 пиримидинил]амино]бензонитрилHCl и 500 мл пропанона нагревают в химическом стакане до кипячения с обратным холодильником. Полученную фракцию оставляют кристаллизоваться при комнатной температуре. Растворитель выпаривают под током воздуха до получения сухого продукта. Выход: соединение формулы (I-а), форма В.c) 73,29 кг (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила и 300 л уксусной кислоты (2 л/моль) нагревают до 104 С в сосуде для получения продукции. Раствор фильтруют при 100 С. Добавляют 19,8 л соляной кислоты (1,1 моль/моль) при 91,4 С. При 70 С медленно добавляют 150 л воды (2 л/моль). Смесь медленно охлаждают до 20 С, промывают два раза 75 л воды и сушат при 75 С. Полученный продукт размалывают. Выход: соединение формулы (I-а), форма С.d) 10,99 кг (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила и 57 л уксусной кислоты (2 л/моль) нагревают до 93 С в сосуде для получения продукции. Раствор фильтруют при 100 С и промывают 3 л уксусной кислоты (0,21 л/моль). Добавляют 2,973 л соляной кислоты (1,1 моль/моль) при 85 С. Медленно добавляют 60 л воды (2 л/моль) при температуре 85-65 С. Смесь медленно охлаждают до 19,5 С, промывают два раза 5,4 л воды и сушат при 50 С. Полученный продукт размалывают. Смешивают 200 мг продукта с 1 мл воды и суспендируют в течение 1 дня при комнатной температуре. Выход: соединение формулы (I-а), форма D. В. Идентификация соединения формулы (I-а). Результаты идентификации форм А, В, С и D методами инфракрасной спектроскопии и рентгеновской дифракции порошка (XRPD) представлены ниже. Результаты дифференционной сканирующей калориметрии (ДСК) для формы А также приведены ниже. Инфракрасная спектрометрия: KBr-дисперсия. Подлежащее анализу соединение смешивают с галогенидом щелочного металла и прессуют таблетку (Ph. Eur.). Рентгеновский дифракционный анализ порошка. Рентгеновский дифракционный анализ порошка (XRPD) проводят на дифрактометре Philips X'PertPRO MPD PW3050/60 с генератором PW3040. Прибор оснащен рентгеновской трубкой Cu LFFPW3373/00. Подлежащее анализу соединение намазывают на держатель образца с нулевым фоном. Параметры прибора Дифференциальная сканирующая калориметрия. Примерно 3 мг соединения, подлежащего анализу, переносят в стандартную алюминиевую кювету для образца ТА-установки. Кювету закрывают подходящей крышкой и регистрируют ДСК-кривую на ТА-установке Q1000 MTDSC, снабженной охлаждающим блоком RCS. Используют следующие параметры: Результаты. Форма А - ИК. Форма А характеризуется ФТ ИК-спектром с типичными полосами поглощения примерно при 2217,1652, 1497, 1435, 1338, 1199 и 550 см-1. Дополнительные полосы поглощения наблюдают при 1631, 1596, 1537, 1504, 1249, 1214, 1179, 1152 и 1070 см-1 (см. фиг. 1). Форма А - XRPD. Форма А характеризуется типичными дифракционными пиками в положениях два тета 9,70,2 о,13,50,2 и 15,00,2. Форма А дополнительно характеризуется пиками рентгеновской дифракции порошка в положениях два тета 9,10,2, 11,00,2, 14,60,2, 22,00,2, 25,00,2, 25,30,2 и 26,70,2(см. фиг. 2) (могут происходить вариации интенсивности вследствие процессов, которые влияют на интенсивности, главным образом, это история обработки образца). Форма А - ДСК. Форма А плавится с разложением. Плавление с разложением начинается примерно при 250 С и происходит примерно при 286 С. Форма В. Форма В может быть представлена двумя состояниями: сухим состоянием и увлажненным состоянием. Приведены только характеристики формы В в сухом состоянии. Форма В - ИК. Форма В характеризуется спектром ФТ ИК с типичными полосами поглощения примерно при 2227,2220, 1599, 1500, 1440, 1341, 1209, 549 и 544 см-1. Дополнительные полосы поглощения наблюдают примерно при 1656, 1538, 1518, 1270, 1179, 1152 и 1070 см-1 (см. фиг. 3). Форма В - XRPD. Форма В характеризуется типичными дифракционными пиками в положениях два тета 4,50,2,8,80,2 и 12,50,2. Форма В дополнительно характеризуется пиками рентгеновской дифракции порошка в положениях два тета 10,30,2, 14,70,2, 20,60,2, 22,20,2 и 26,10,2 (см. фиг. 4) (могут происходить вариации интенсивности вследствие процессов, которые влияют на интенсивности, главным образом, это история обработки образца). Форма С - ИК. Форма С характеризуется спектром ФТ ИК с типичными полосами поглощения примерно при 2221,1654, 1502, 1239, 1193 и 546 см-1. Дополнительные полосы поглощения наблюдают примерно при 1627, 1580, 1537, 1492, 1216, 1173,- 14013686 1157 и 1084 см-1 (см. фиг. 5). Форма С - XRPD. Форма С характеризуется типичными дифракционными пиками в положениях два тета 11,90,2,14,30,2 и 22,30,2. Форма С дополнительно характеризуется пиками рентгеновской дифракции порошка в положениях два тета 12,80,2, 18,50,2, 21,20,2, 24,30,2 и 26,00,2 (см. фиг. 6) (могут происходить вариации интенсивности вследствие процессов, которые влияют на интенсивности, главным образом, это история обработки образца). Форма D - ИК Форма D характеризуется спектром ФТ ИК с типичными полосами поглощения примерно при 2218,1657, 1506, 1448, 1357, 1220 и 547 см-1. Дополнительные полосы поглощения наблюдают примерно при 1620, 1597, 1565, 1247, 1214, 1179 1152 и 1073 см-1 (см. фиг. 7). Форма D - XRPD. Форма D характеризуется типичными дифракционными пиками в положениях два тета 6,60,2,11,60,2 и 17,10,2. Форма D дополнительно характеризуется пиками рентгеновской дифракции порошка в положениях два тета 15,00,2, 19,20,2, 20,50,2, 21,60,2 и 29,80,2 (см. фиг. 8) (могут происходить вариации интенсивности вследствие процессов, которые влияют на интенсивности, главным образом, это история обработки образца). С. Данные по растворимости. В табл. 1 представлены данные по растворимости свободного основания (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила и соединения формулы (I-а). Таблица 1 Свободное основание, а также соль HCl имеют слабую растворимость в воде, а также в 0,01N HCl. Свободное основание и соль HCl можно классифицировать как соединения BCS-класса 2. Растворимость свободного основания существенно повышается в PEG 400.D. Данные по стабильности. а) Химическая стабильность. Соединение (I-а) (форма А) хранят в различных условиях влажности и температуры. После хранения соль анализируют методом высокоэффективной жидкостной хроматографии (ВЭЖХ) на содержание примесей. Результаты собраны в табл. 2. Можно сделать вывод, что данное соединение химически стабильно. Таблица 2 Примечание: - = не исследовано,КТ = комнатная температура,ОВ = относительная влажность. Обнаружено также, что данное соединение не является гигроскопичным.b) Физическая стабильность. Исследовали стабильность кристаллической структуры соединения формулы (I-а) (форма А) после хранения в течение шести недель при различных условиях влажности и температуры. Применяли такие же условия, как описанные в табл. 2. После хранения соединение анализировали методом инфракрасной спектроскопии. Наблюдали отсутствие изменений в кристаллической структуре, это показывает, что соединение является кристаллографически стабильным. Исследовали также стабильность соединения формулы (I-а) (форма А) после хранения в течение 1 года при 5 С и 25 С/80% ОВ. Обнаружено, что данное соединение физически стабильно. Не присутствует в конечной таблетке. Указанные выше таблетки получают, растворяя гипромелозу или поливинилпирролидон и полисорбат 20 в очищенной воде (сколько требуется), с последующим распылением указанного раствора на псевдоожиженный порошок, состоящий из смеси формы А и моногидрата лактозы. Полученные гранулы- 20013686 сушат, просеивают и смешивают с микрокристаллической целлюлозой или окремненной микрокристаллической целлюлозой, натрий-кросскармелозой и необязательно коллоидным диоксидом кремния. После добавления стеарата магния порошковую смесь прессуют в виде таблеток, а затем наносят на таблетки пленочное покрытие, используя суспензию порошкового покрытия Opadry II White в очищенной воде. В указанных выше композициях микрокристаллическая целлюлоза предпочтительно представляет собой Avicel PH101, натрий-кросскармелоза предпочтительно представляет собой Ас-Di-Sol; окремненная микрокристаллическая целлюлоза предпочтительно представляет собой ProsolvHD90; поливинилпирролидон предпочтительно представляет собой PVP K29-32.F. Исследование in vivo биодоступности. А) Для исследования in vivo биодоступности соединения формулы (I-а) проводят исследование на самцах гончих собак. Сравнивают биодоступность соединения формулы (I-а) после перорального введения с биодоступностью свободного основания после внутривенного введения. Препарат, используемый для внутривенного введения, представляет собой раствор свободного основания (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила в смеси 75% PEG 400/25% стерильная вода, который вводят при дозе 1,25 мг/кг. Для перорального введения используют следующие препараты:PEG 400 раствор свободного основания (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2 пиримидинил]амино]бензонитрила (группа I); капсула (размер 0; красная крышка-красный корпус), содержащая 1,61% (мас./мас.) свободного основания (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила,0,18% (мас./мас.) лаурилсульфата натрия, 0,18% (мас./мас.) диоксида кремния, 91,97% (мас./мас.) гранулированного моногидрата лактозы (группа II); капсула (размер 0; красная крышка-красный корпус), содержащая 8,36% (мас./мас.) соединения формулы (I-а), 0,18% (мас./мас.) лаурилсульфата натрия, 0,18% (мас./мас.) диоксида кремния, 91,28%(мас./мас.) гранулированного моногидрата лактозы (группа III) (% мас./мас. от содержимого капсулы). Различные препараты вводят перорально при дозе 5 мг осн. экв./кг. Препараты готовят на основе предварительно определенной массы тела животного. Точная вводимая доза рассчитывается с использованием массы тела непосредственно перед дозированием и составляет в среднем 5 мг осн. экв./кг. Отбирают образцы крови (4 мл на EDTA) из яремной вены через 0 (= до введения дозы), 0,5, 1, 2, 4,6, 8, 24, 32, 48 и 72 ч после введения дозы. Сразу после отбора крови образцы крови помещают на тающий лед и защищают от света. Образцы крови центрифугируют примерно при 1900g в течение 10 мин и при 5 С для отделения плазмы. Образцы плазмы отделяют, переносят во вторую пробирку в течение 2 ч от момента отбора образцов крови и хранят при температуре -18 С до исследования. Образцы постоянно защищают от света и помещают на тающий лед или в температуру -18 С. Определяют концентрации в плазме 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2 пиримидинил]амино]бензонитрила (Е), применяя утвержденный способ ЖХ-МС/МС. ЖХ-МС/МС анализ проводят на установке API-3000 МС/МС (Applied Biosystems), которая соединена с ВЭЖХ-помпой(Agilent) и автосемплером (Interscience). Рассчитывают средние (n=2) концентрации в плазме для каждого препарата и времени отбора образца. Определяют пиковые концентрации в плазме (Cmax), соответствующие пиковым временам (Tmax), иAUC0-t (где t - момент времени, соответствующий последней определяемой концентрации, выше предела количественного определения). Рассчитывают область под кривой, экстраполированной до бесконечности (AUC0-беск.), как сумму AUC0-t и Ct/, гдепредставляет константу скорости элиминации, определяют, применяя логарифмически-линейный регрессионный анализ данных конечная концентрация в плазме - время. Для всех препаратов рассчитывают средние (n=2) ПК-параметры. Оценку абсолютной биодоступности (Fabs) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила (Е) получают посредством деления нормализованного по дозе среднего значения AUC0-беск. после перорального введения на нормализованное по дозе среднее значение AUC0-беск. после внутривенного введения, и так для всех пероральных препаратов. Результаты, собранные в описанном выше исследовании, суммированы в табл. 3. Из приведенных выше результатов можно рассчитать, что при введении в виде твердой дозированной формы соединение формулы (I-а) обладает существенно лучшей биодоступностью, чем соответствующее свободное основание 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил (Е). Биодоступность сравнима с биодоступностью свободного основания, вводимого в виде перорального PEG 400 раствора. В) Также исследовали in vivo биодоступность соединения формулы (I-а) у людей при пероральном приеме. Здоровые пациенты принимали 2 препарата. Препарат А: раствор 25 мг/мл свободного основания (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила в 100% PEG 400. Препарат В: таблетка, соответствующая описанному здесь выше составу 2 а. В группе из 12 субъектов каждый субъект принимал три единичных дозы, каждая из которых эквивалентна 100 мг свободного основания (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2 пиримидинил]амино]бензонитрила. Каждую дозу принимали в день 1 соответствующего периода лечения. Субъекты (n=12) выбирались случайным образом для приема единичных доз препарата А с приемом пищи, препарата В натощак и препарата В с приемом пищи в течение трех сессий, которые разделялись по меньшей мере 2-недельными периодами промывания. Для каждой сессии определяли 216 часовой фармакокинетический профиль (Е) 4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2 пиримидинил]амино]бензонитрила в плазме после перорального введения единичной дозы (100 мг) основания(Е)-4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила или эквивалента. Для определения концентраций (Е)-4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила в плазме отбирали образцы крови перед введением дозы и через 0,5, 1, 2, 3, 4, 6, 8, 12, 16, 24, 32, 48, 72, 96, 120, 144, 168 и 216 ч после введения исследуемого лекарственного средства (всего 19 образцов у каждого субъекта для каждого введения). Для каждого субъекта две из трех доз вводили с приемом пищи, т.е. если фармакокинетики исследовали с приемом пищи, то в течение 10 мин до приема препарата А или препарата В субъекты съедали обычный завтрак. Для условий натощак субъекты голодали по меньшей мере в течение 10 ч до введения исследуемого лекарства. Если фармакокинетики исследовали натощак (только препарат В), то субъекты принимали первую пищу во время ланча через 4,5 ч после введения исследуемого лекарства. В частности, в день -1 субъектов помещали на исследовательское оборудование, и они голодали в течение ночи по меньшей мере 10 ч, за исключением приема воды, который разрешался за 2 ч до приема лекарства. Субъектам, определенным случайным образом для приема препарата А или препарата В с приемом пищи, вводили исследуемое лекарственное средство в течение 10 мин после обычного завтрака на исследовательском оборудовании. Субъектам, определенным случайным образом для приема препарата В натощак, вводили исследуемое лекарственное средство без приема пищи, после голодания в течение ночи по меньшей мере 10 ч. Обычный завтрак состоял из четырех кусков хлеба, двух кусков ветчины или сыра, джема и двух- 22013686 чашек кофе без кофеина или чая с молоком и/или сахаром. Эта пища съедалась за 20 мин под наблюдением медицинской сестры или штатного сотрудника. Для всех субъектов исследуемое лекарственное средство вводили вместе примерно с 200 мл воды утром с 9 до 11 ч. Через 2 ч после введения дозы всем субъектам разрешали прием воды. Ланч подавали через 4,5 ч после введения дозы и обед подавали через 10 ч после приема дозы. После обеда субъектам разрешали возобновить их обычное питание. Субъектов освобождали от исследовательского оборудования на день 2 после отбора 24-часовой фармакокинетической пробы и возвращали на данное оборудование через 8 ч и снова в дни 3, 4, 5, 6, 7, 8 и 10 для дальнейших определений. Более подробно, для концентраций определения (Е)-4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила в плазме отбирали образец крови до введения дозы и через 0,5, 1, 2, 3, 4, 6, 8, 12, 16, 24, 32,48, 72, 96, 120, 144, 168 и 216 ч после введения исследуемого лекарственного средства (всего 19 образцов для каждого субъекта и каждого введения). Для каждого индивидуального субъекта между введениями доз существовал по меньшей мере 2-недельный временной интервал. Биоанализ(Е)-4-4-4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила в человеческой плазме проводят утвержденным ЖХ-МС/МС способом. В табл. 4 собраны результаты in vivo исследования на человеке. Таблица 4 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Твердая фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения формулы (I-а) или его N-оксида. 2. Фармацевтическая композиция по п.1, в которой соединение формулы (I-а) представляет собой полиморфную форму А, характеризующуюся пиками порошковой рентгеновской дифракции в положениях два тета 9,70,2, 13,50,2 и 15,00,2. 3. Фармацевтическая композиция по п.2, в которой полиморфная форма А дополнительно характеризуется пиками порошковой рентгеновской дифракции в положениях два тета 9,10,2, 11,00,2,14,60,2, 22,00,2, 25,00,2, 25,30,2 и 26,70,2. 4. Фармацевтическая композиция по п.1, в которой соединение формулы (I-а) представляет собой полиморфную форму В, характеризующуюся пиками порошковой рентгеновской дифракции в положениях два тета 4,50,2, 8,80,2 и 12,50,2. 5. Фармацевтическая композиция по п.4, в которой полиморфная форма В дополнительно характеризуется пиками порошковой рентгеновской дифракции в положениях два тета 10,30,2, 14,70,2,20,60,2, 22,20,2 и 26,10,2. 6. Фармацевтическая композиция по п.1, в которой соединение формулы (I-а) представляет собой полиморфную форму С, характеризующуюся пиками порошковой рентгеновской дифракции в положениях два тета 11,9+0,2, 14,30,2 и 22,30,2. 7. Фармацевтическая композиция по п.6, в которой полиморфная форма С дополнительно характеризуется пиками порошковой рентгеновской дифракции в положениях два тета 12,80,2, 18,50,2,21,20,2, 24,30,2 и 26,00,2. 8. Фармацевтическая композиция по п.1, в которой соединение формулы (I-а) представляет собой полиморфную форму D, характеризующуюся пиками порошковой рентгеновской дифракции в положениях два тета 6,60,2, 11,60,2 и 17,10,2. 9. Фармацевтическая композиция по п.8, в которой полиморфная форма D дополнительно характеризуется пиками порошковой рентгеновской дифракции в положениях два тета 15,00,2, 19,20,2,- 23013686 20,50,2, 21,60,2 и 29,80,2. 10. Фармацевтическая композиция по любому из пп.1-3, в которой соединение формулы (I-a) представляет собой полиморфную форму А, характеризующуюся ФТ ИК-спектром с типичными полосами поглощения примерно при 2217, 1652, 1497, 1435, 1338, 1199 и 550 см-1. 11. Фармацевтическая композиция по п.10, в которой полиморфная форма А дополнительно характеризуется ФТ ИК-спектром с типичными полосами поглощения примерно при 1631, 1596, 1537, 1504,1249, 1214, 1179, 1152 и 1070 см-1. 12. Фармацевтическая композиция по любому из предшествующих пунктов, где композиция является пригодной для перорального введения. 13. Фармацевтическая композиция по любому из предшествующих пунктов, где композиция дополнительно включает увлажняющий агент. 14. Фармацевтическая композиция по п.13, в которой увлажняющий агент представляет собой эфир полиэтиленсорбита и жирной кислоты. 15. Фармацевтическая композиция по любому из предшествующих пунктов, где композиция находится в форме таблетки. 16. Фармацевтическая композиция по п.15, которая имеет пленочное покрытие. 17. Фармацевтическая композиция по любому из предыдущих пунктов, имеющая следующий состав:(g) 0,1-1,5% лубриканта. 18. Фармацевтическая композиция по любому из предыдущих пунктов, включающая количество активного ингредиента, которое эквивалентно 25 мг соответствующего свободного основания (осн. экв). 19. Фармацевтическая композиция по любому из пп.1-3, 10 или 11, где фармацевтическая композиция находится в форме таблетки, включающей ядро таблетки, имеющее состав: 20. Фармацевтическая композиция по любому из пп.1-3, 10 или 11, где фармацевтическая композиция находится в форме таблетки, включающей ядро таблетки, имеющее состав: 21. Фармацевтическая композиция по любому из пп.1-17, включающая количество активного ингредиента, которое эквивалентно 50 мг соответствующего свободного основания (осн. экв.). 22. Фармацевтическая композиция по любому из пп.1-3, 10 или 11, где фармацевтическая композиция находится в форме таблетки, включающей ядро таблетки, имеющее состав: 23. Фармацевтическая композиция по любому из пп.1-17, включающая количество активного ингредиента, которое эквивалентно 75 мг соответствующего свободного основания (осн. экв.). 24. Фармацевтическая композиция по любому из пп.1-3, 10 или 11, где фармацевтическая композиция находится в форме таблетки, включающей ядро таблетки, имеющее состав: 25. Фармацевтическая композиция по любому из пп.1-17, включающая количество активного ингредиента, которое эквивалентно 100 мг соответствующего свободного основания (осн. экв.). 26. Фармацевтическая композиция по любому из пп.1-3, 10 или 11, где фармацевтическая композиция находится в форме таблетки, включающей ядро таблетки, имеющее состав: 27. Фармацевтическая композиция по любому из пп.1-17, включающая количество активного ингредиента, которое эквивалентно 150 мг соответствующего свободного основания (осн. экв.). 28. Фармацевтическая композиция по любому из пп.1-3, 10 или 11, где фармацевтическая композиция находится в форме таблетки, включающей ядро таблетки, имеющее состав: 29. Способ получения фармацевтической композиции по любому из предыдущих пунктов, включающий следующие стадии:(i) сухое перемешивание активного ингредиента и части разбавителя;(ii) получение связующего раствора посредством растворения связующего и увлажняющего агента- 25013686 в растворителе для связующего раствора;(iii) распыление связующего раствора, полученного на стадии (ii), на смесь, полученную на стадии(iv) сушка влажного порошка, полученного на стадии (iii), с последующим просеиванием и необязательным перемешиванием;(v) смешивание оставшейся части разбавителя, разрыхлителя и необязательного глиданта со смесью, полученной на стадии (iv);(vi) необязательное добавление лубриканта к смеси, полученной на стадии (v);(vii) прессование таблеток из смеси, полученной на стадии (vi);(viii) необязательное нанесение пленочного покрытия на таблетку, полученную на стадии (vii). 30. Применение соединения формулы (I-а), как определено по п.1, для производства композиции по пп.1-28 для лечения или профилактики ВИЧ-инфекции. 31. Фармацевтическая композиция по любому из пп.1-28, в которой частицы соединения формулы(I-а) имеют размер менее чем 50 мкм. 32. Фармацевтическая композиция по п.31, в которой частицы соединения формулы (I-а) имеют размер менее чем 25 мкм. 33. Фармацевтическая композиция по любому из пп.1-11, в которой частицы соединения формулы(I-а) имеют размер менее чем 50 мкм. 34. Фармацевтическая композиция по п.33, в которой частицы соединения формулы (I-а) имеют размер менее чем 25 мкм.
МПК / Метки
МПК: C07D 239/20, A61K 31/505, A61P 31/18, C07D 239/02
Метки: 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, гидрохлорид
Код ссылки
<a href="https://eas.patents.su/30-13686-gidrohlorid-4-4-4-2-cianoetenil-26-dimetilfenilamino-2-pirimidinilaminobenzonitrila.html" rel="bookmark" title="База патентов Евразийского Союза">Гидрохлорид 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила</a>
Фумарат 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила

Номер патента: 11036
Опубликовано: 30.12.2008
Авторы: Стапперс Альфред Элизабет, Копманс Алекс Херман, Вандекрюйс Рогер Петрус Гереберн, Петерс Йозеф, Стевенс Пол Теодор Агнес
МПК: A61P 31/18, A61K 31/505
Метки: 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, фумарат
Формула / Реферат:
1. Соединение формулы (I) его N-оксид или стереохимически изомерная форма. 2. Соединение по п.1, где указанное соединение представляет собой 3. Соединение по п.1 или 2 для использования в качестве лекарственного средства. 4. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и в качестве активного ингредиента - терапевтически эффективное количество соединения по п.1 или 2. 5. Фармацевтическая композиция по п.4, где...
Способы получения 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила

Номер патента: 7953
Опубликовано: 27.02.2007
Авторы: Гийемон Жером Эмиль Жорж, Виллемс Йоаннес Йозефус Мария, Медар Барт Петрус Анна Мария Йозеф, Жанссен Поль Адриан Ян, Хэрес Ян, Лендерс Рубен Герардус Жорж, Схилс Дидье Филипп Робер, Паскье Элизабет Тереза Жанна
МПК: C07C 253/20, C07C 253/30, A61K 31/505...
Метки: способы, 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, получения
Формула / Реферат:
1. Способ получения 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила формулы (I) его N-оксида, фармацевтически приемлемой кислотно-аддитивной соли, четвертичного амина или стереохимически изомерной формы, который включает осуществление взаимодействия промежуточного соединения формулы (II) его подходящей кислотно-аддитивной соли или стереохимически изомерной формы с промежуточным соединением формулы (III) его...
Гидрохлорид 5-[4-[2-(n-метил-n-(2-пиридил)амино)этокси] бензил] тиазолидин-2, 4-диона

Номер патента: 5115
Опубликовано: 28.10.2004
Автор: Крэйг Эндрю Симон
МПК: C07D 417/12, A61P 3/00, A61K 31/44...
Метки: 5-[4-[2-(n-метил-n-(2-пиридил)амино)этокси, бензил, 4-диона, тиазолидин-2, гидрохлорид
Формула / Реферат:
1. Соединение 5-[4-[2-(N-метил-N-(2-пиридил)амино)этокси]бензил]тиазолидин-2,4-дион, гидрохлорид, отличающееся тем, что оно (i) имеет инфракрасный спектр, содержащий пики при приблизительно 1745, 1516, 1257, 1056 и 803 см-1; (ii) имеет порошковую рентгенограмму (XRPD), содержащую пики при приблизительно 10,1, 13,4, 17,2, 22,2 и 29,4ш 2q ; и/или (iii) имеет спектр комбинационного рассеяния, содержащий пики при приблизительно 1314, 1242,...
Производные замещенного 2-пиримидинил-6, 7, 8, 9-тетрагидропиримидо [1,2-а]пиримидин-4-она и 7-пиримидинил-2,3-дигидроимидазо[1, 2-а]пиримидин-5( 1н )она для нейродегенеративных нарушений

Номер патента: 7165
Опубликовано: 25.08.2006
Авторы: Ларденуа Патрик, Галле Тьерри, Йеш Филипп, Неделек Ален, Саади Мурад, Локхид Алистер, Маргери Северен, Словински Франк
МПК: C07D 487/04, A61P 25/28, A61K 31/519...
Метки: 7-пиримидинил-2,3-дигидроимидазо[1, нарушений, замещенного, производные, 9-тетрагидропиримидо, 2-пиримидинил-6, она, 1,2-а]пиримидин-4-она, нейродегенеративных, 2-а]пиримидин-5
Формула / Реферат:
1. Производное пиримидона, представленное формулой (I), или его соль, или его сольват, или его гидрат где X представляет собой два атома водорода, атом серы, атом кислорода или C1-2алкильную группу и атом водорода; Y представляет собой связь, этениленовую группу, этиниленовую группу, атом кислорода, атом серы, сульфонильную группу, сульфоксидную группу, карбонильную группу, атом азота, возможно замещенный C1-6алкильной группой, фенильной или...
Фармацевтическая композиция с контролируемым высвобождением, содержащая гидрохлорид трамадола, и способ ее получения

Номер патента: 6438
Опубликовано: 29.12.2005
Авторы: Гаттнар Ондрей, Варга Иван, Земанек Марьян, Разус Любослав, Седларова Гелена
МПК: A61P 25/04, A61K 9/22, A61J 3/10...
Метки: композиция, трамадола, гидрохлорид, высвобождением, фармацевтическая, контролируемым, получения, способ, содержащая
Формула / Реферат:
1. Фармацевтическая композиция с контролируемым высвобождением, содержащая гидрохлорид трамадола, отличающаяся тем, что она содержит от 100 до 200 мг активного ингредиента в смеси с тонкоизмельченными сложными эфирами глицерина с высшими жирными кислотами в количестве от 28 до 47 мас.%, солями фосфорной кислоты и щелочно-земельных металлов в количестве от 20 до 41 мас.%, неионными винилпирролидоновыми полимерами в количестве от 1,15 до 1,75...
Предыдущий патент: Сульфаматные и сульфамидные производные для лечения эпилепсии и родственных расстройств
Следующий патент: Способ извлечения глинозема
Случайный патент: Сеть и способ передачи данных в системах трубопроводов