Замещённые пиримидины, композиции на их основе и применение замещенных пиримидинов в качестве ингибиторов протеинкиназ
Номер патента: 12869
Опубликовано: 30.12.2009
Авторы: Кэй Дэвид, Миллер Эндрю, Шаррьер Жан-Дамиен, Мадзей Франческа
















Формула / Реферат
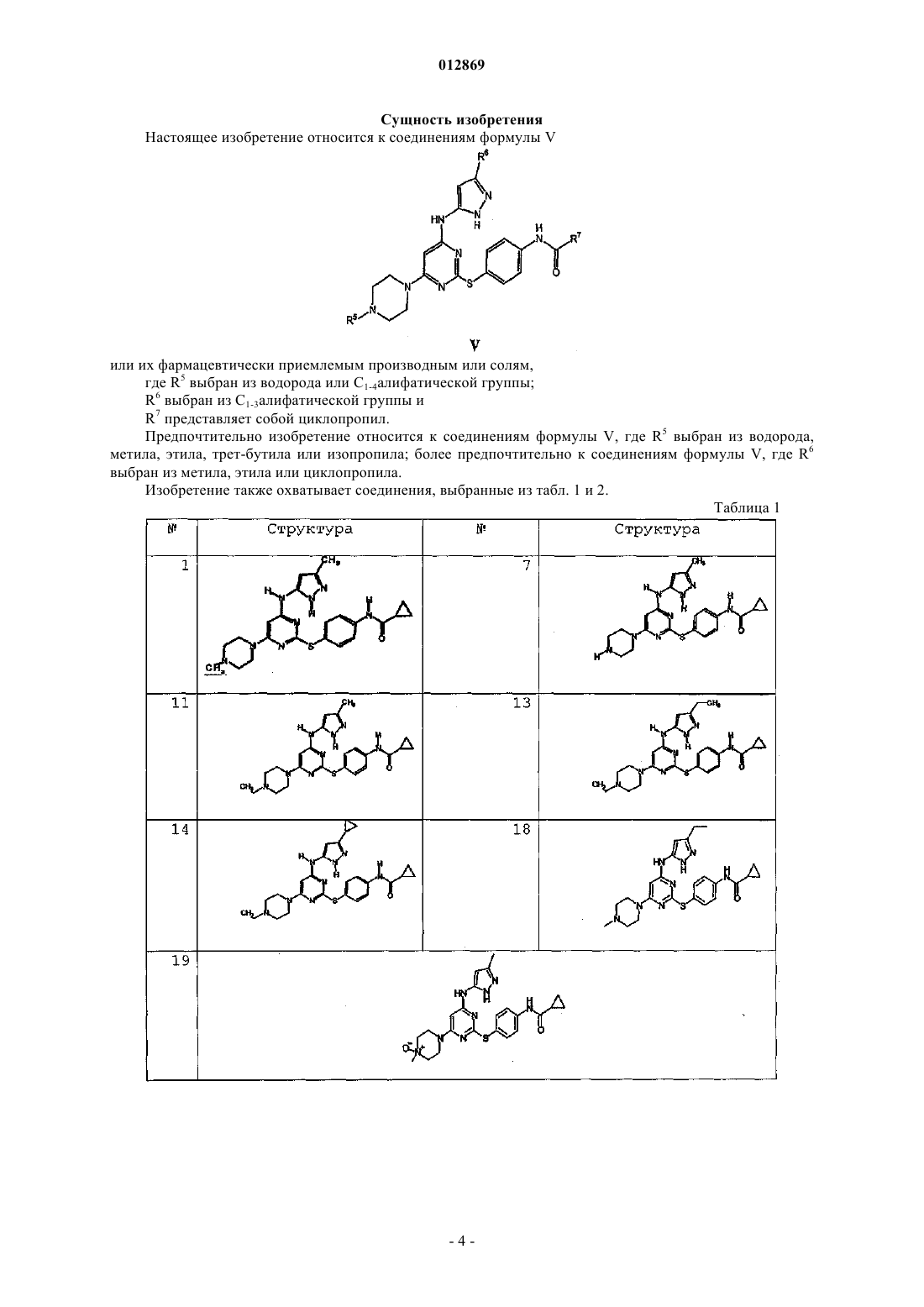
1. Соединение формулы V

или его фармацевтически приемлемые производное или соль,
где R5 выбран из водорода или C1-4алифатической группы;
R6 выбран из C1-3алифатической группы и
R7 представляет собой циклопропил.
2. Соединение по п.1, где R5 выбран из водорода, метила, этила, трет-бутила или изопропила.
3. Соединение по п.2, где R6 выбран из метила, этила или циклопропила.
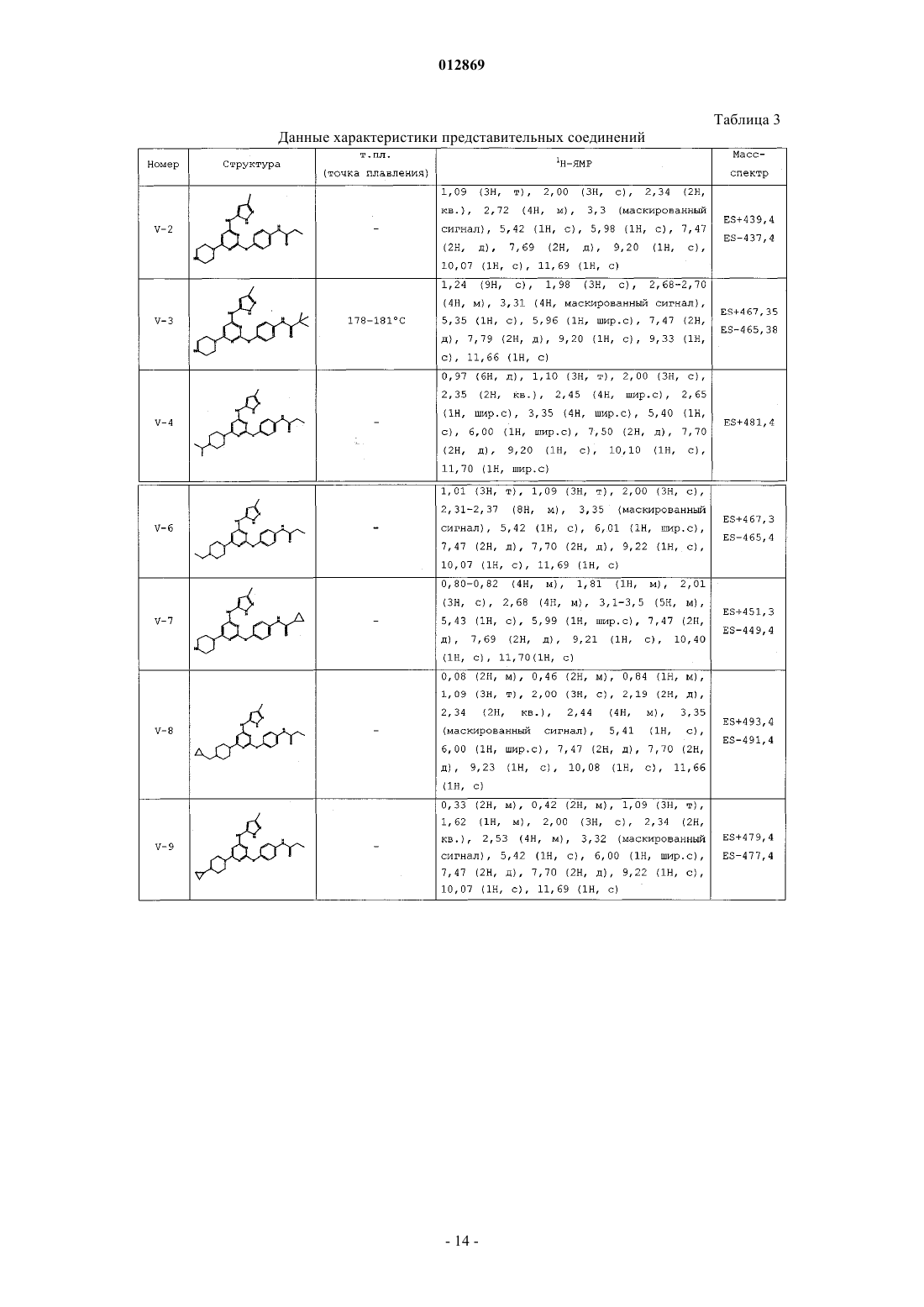
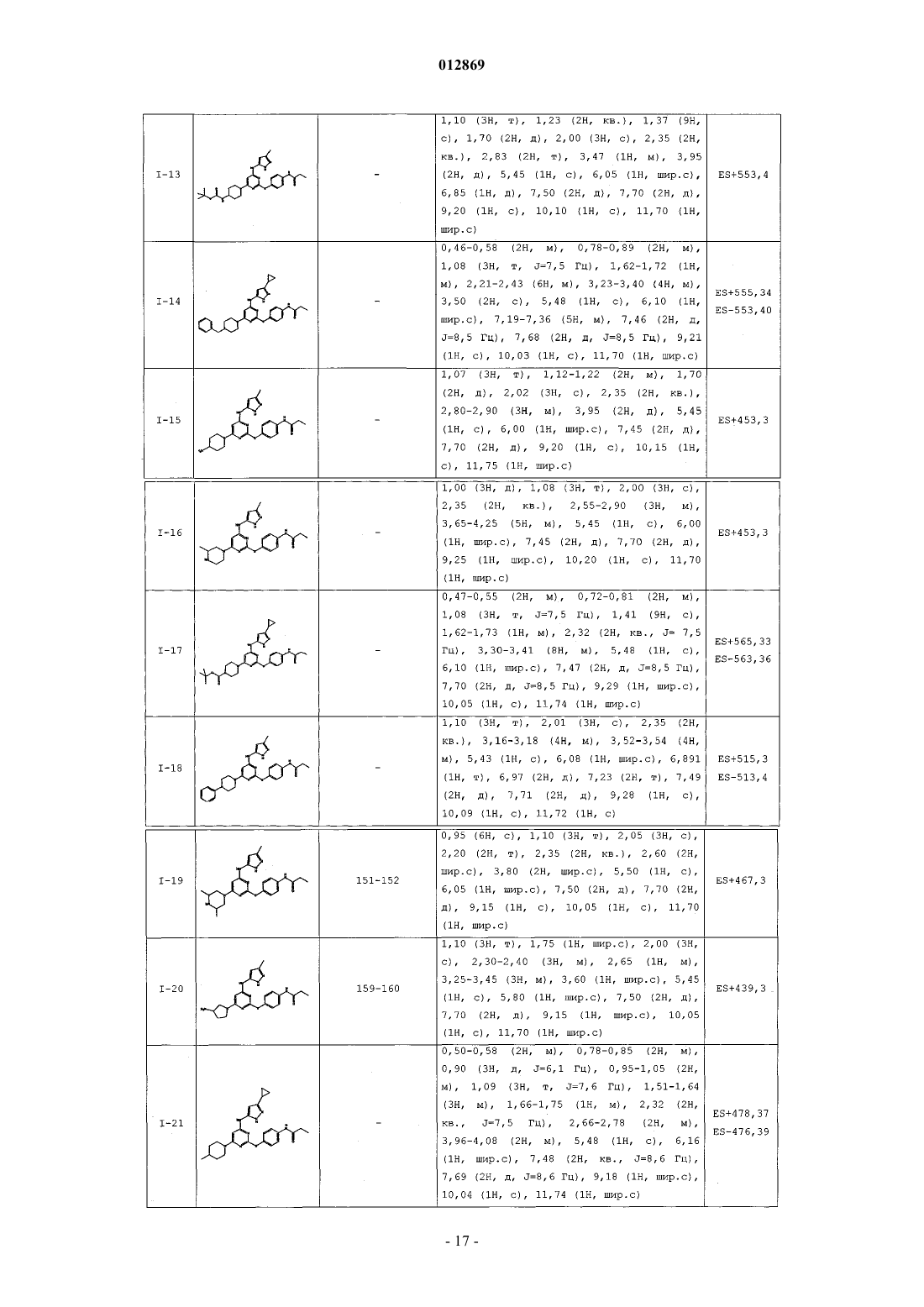
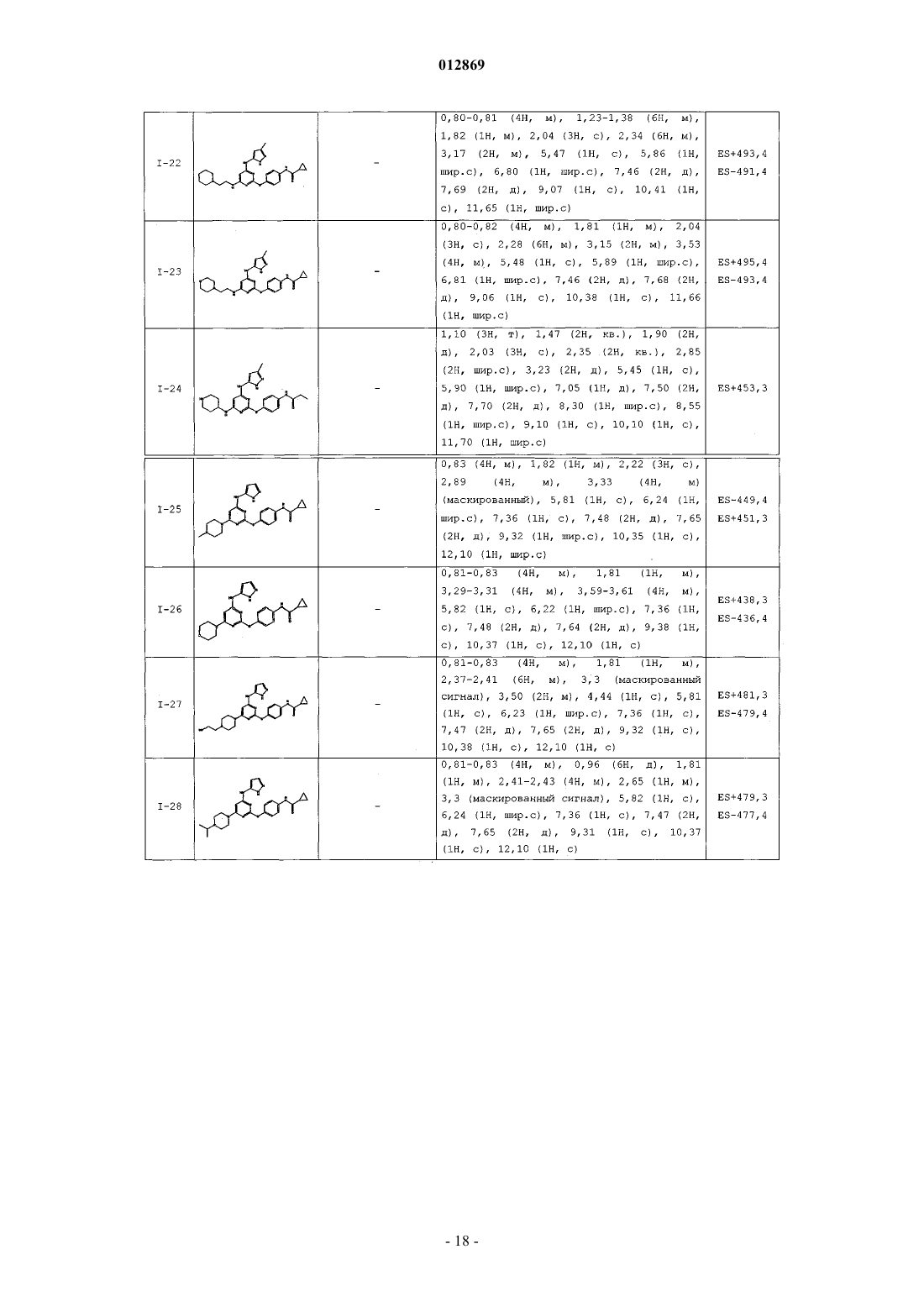
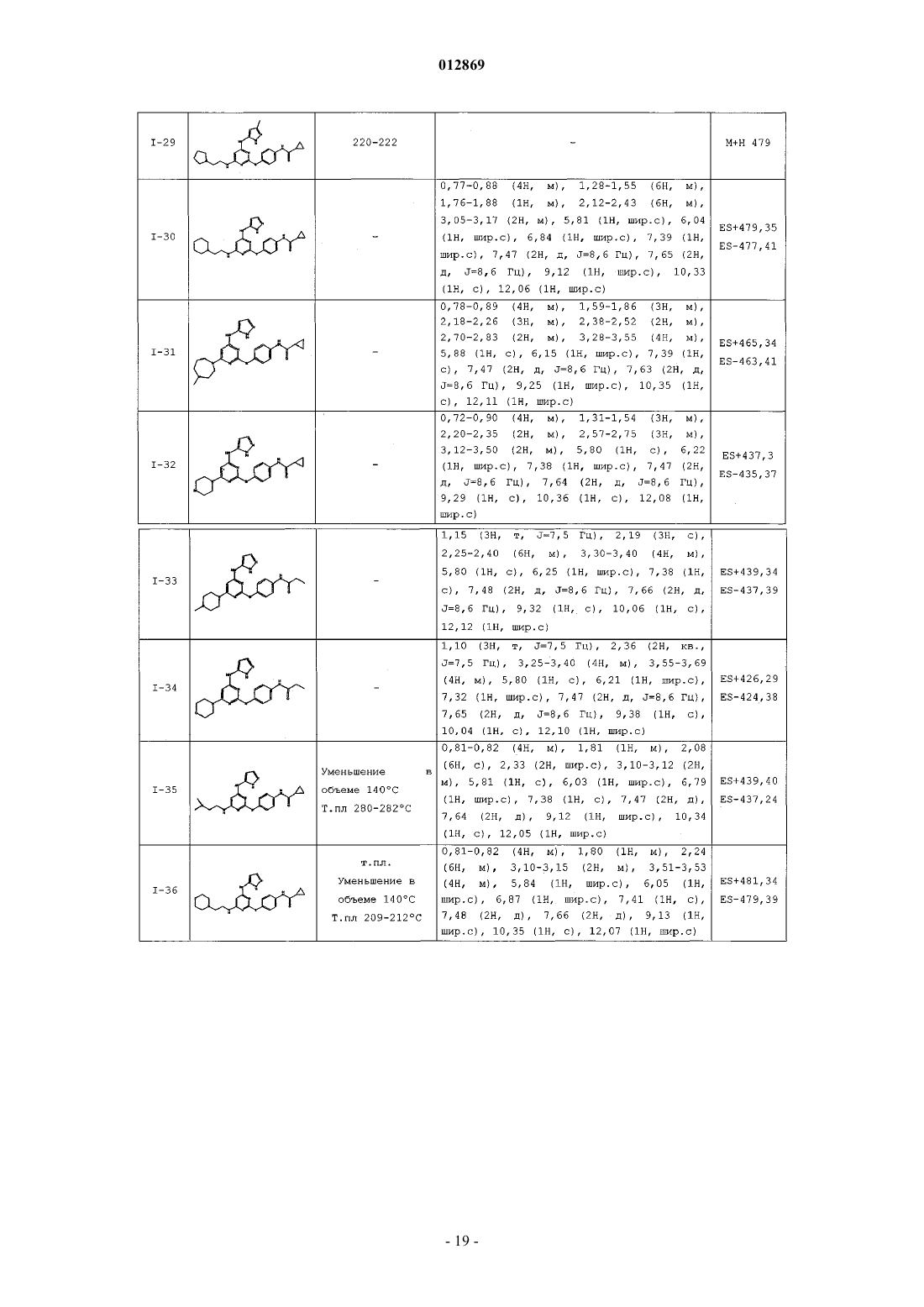
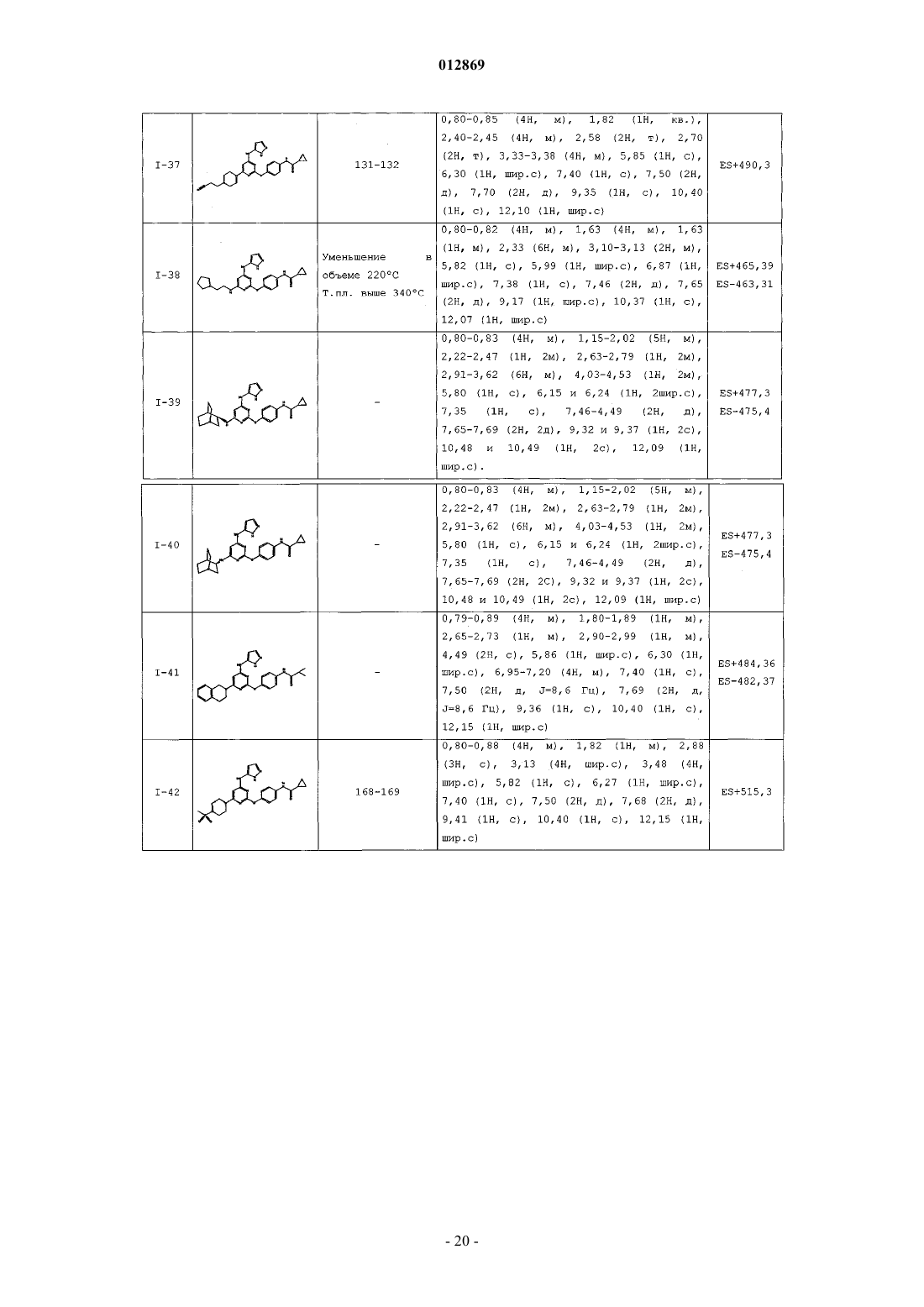
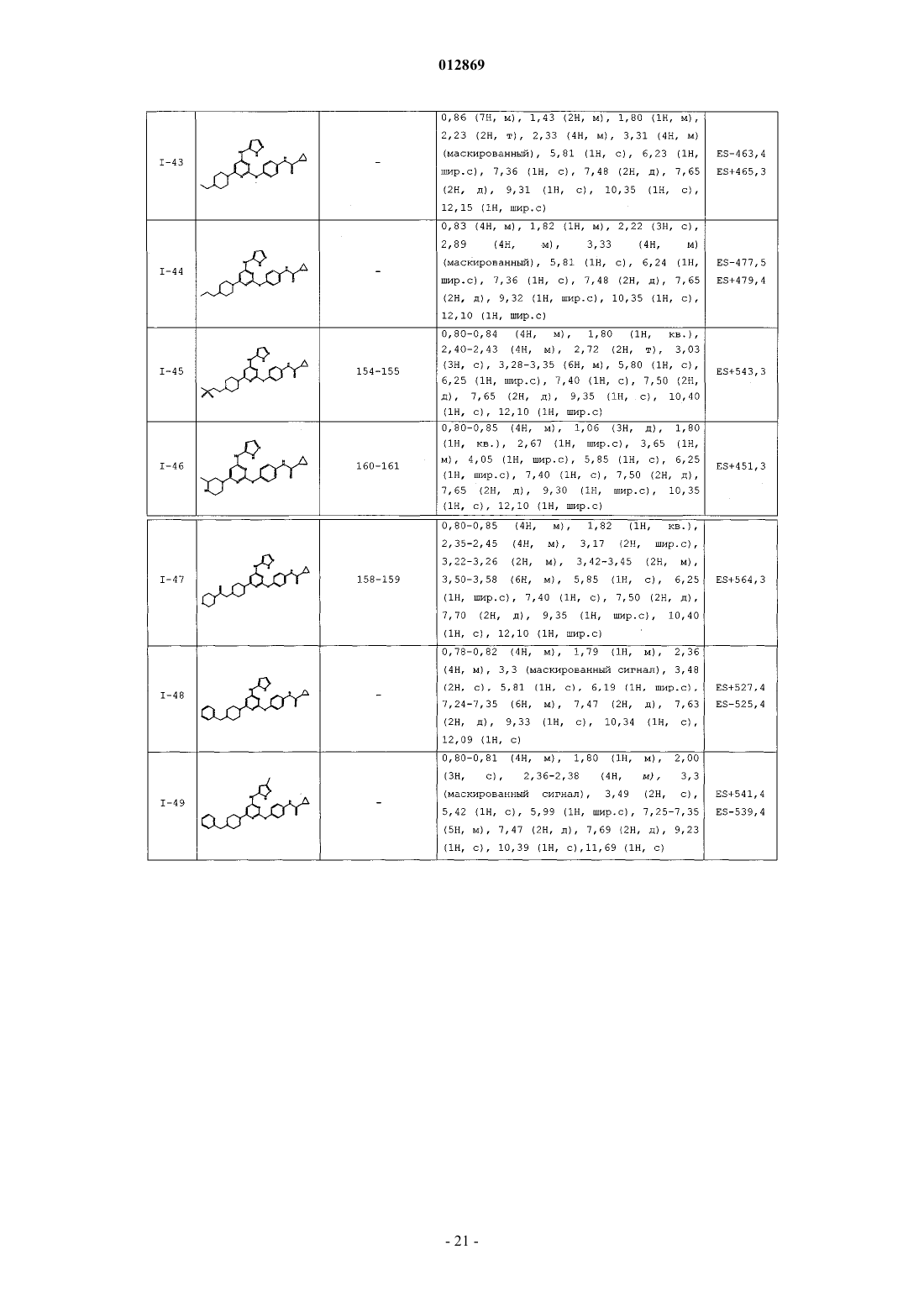
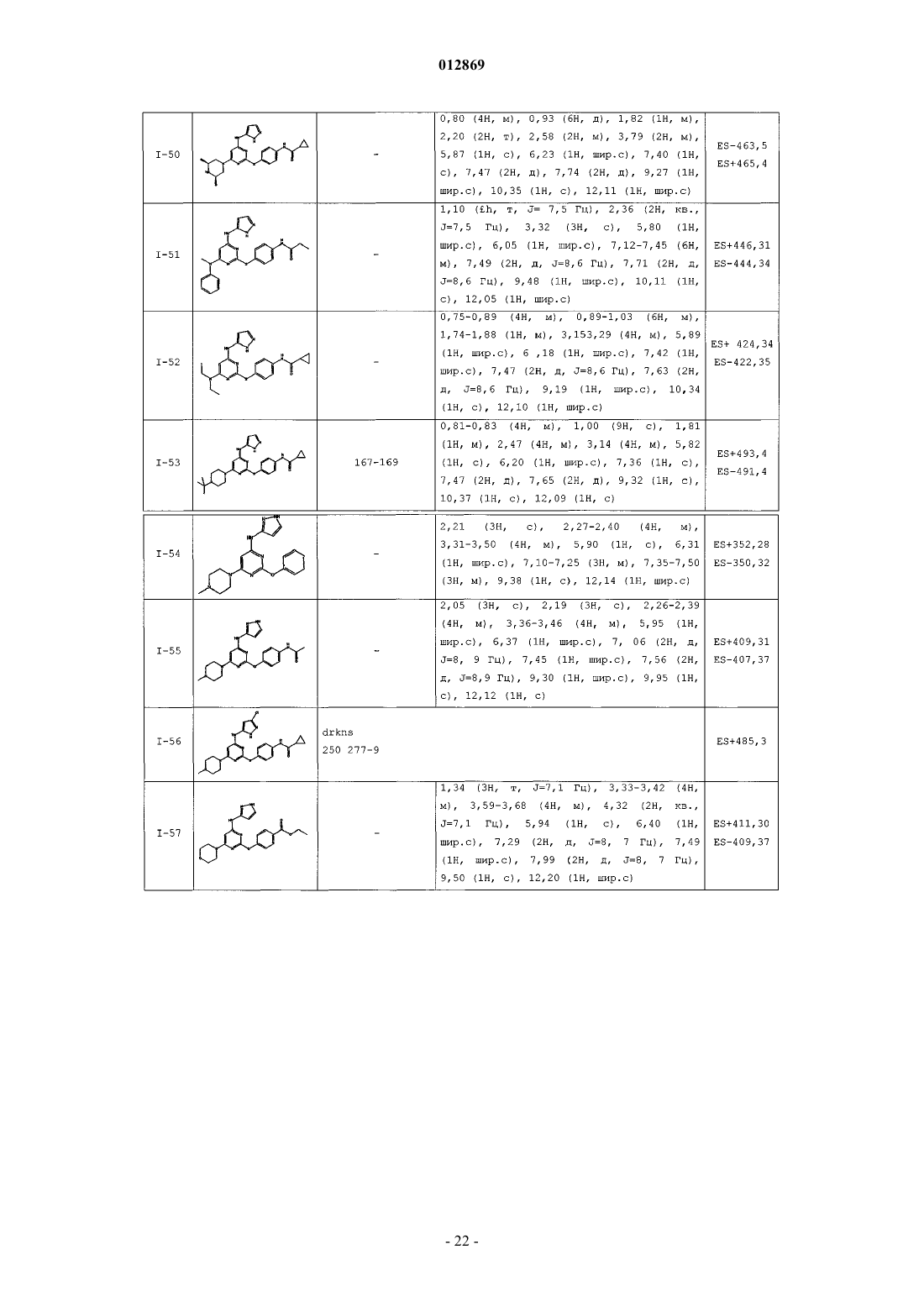
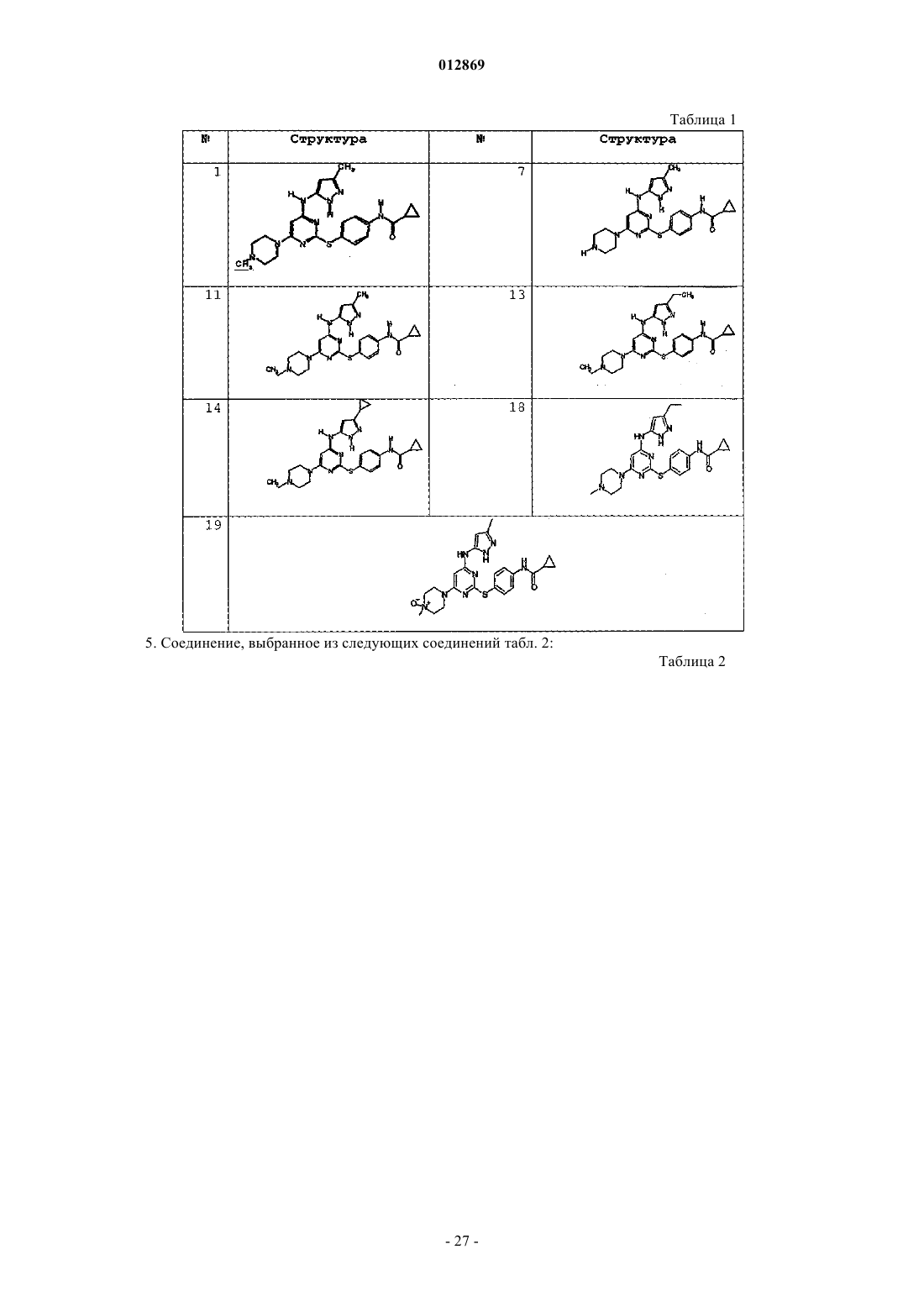
4. Соединение, выбранное из следующих соединений табл. 1:
Таблица 1

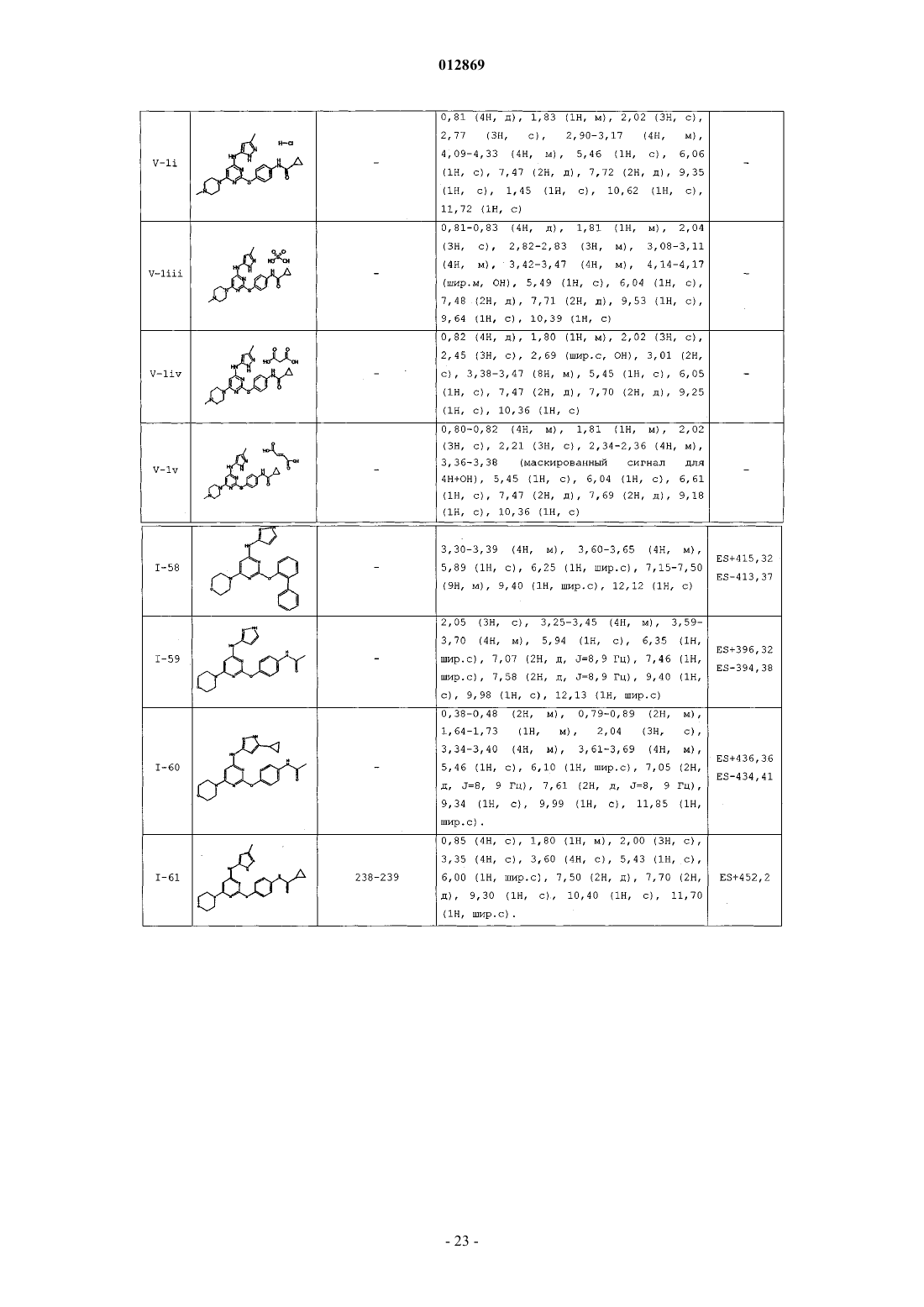
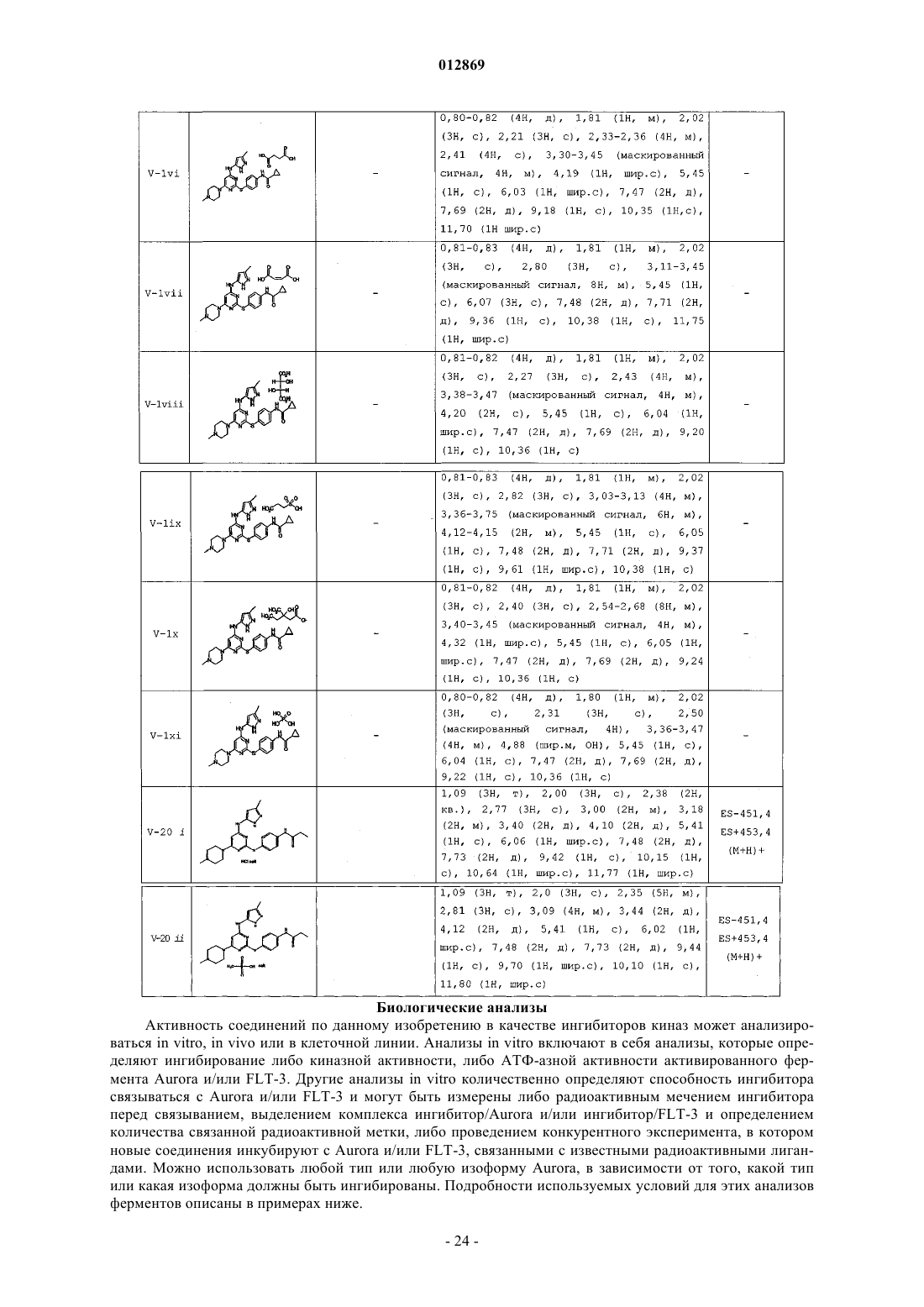
5. Соединение, выбранное из следующих соединений табл. 2:
Таблица 2

6. Композиция, содержащая соединение по п.1 и фармацевтически приемлемый носитель, адъювант или разбавитель.
7. Композиция по п.6, дополнительно содержащая антипролиферативный агент или химиотерапевтический агент.
8. Способ ингибирования Aurora-1 в биологической пробе или у пациента, включающий контактирование указанной пробы с:
(а) соединением по п.1 или
(b) композицией по п.6 или
введение пациенту:
(а) соединения по п.1 или
(b) композиции по п.6.
9. Способ ингибирования Aurora-2 в биологической пробе или у пациента, предусматривающий контактирование указанной пробы с:
(а) соединением по п.1 или
(b) композицией по п.6 или
введение пациенту
(а) соединения по п.1 или
(b) композиции по п.6.
10. Способ ингибирования Aurora-3 в биологической пробе или у пациента, предусматривающий контактирование указанной пробы с:
(а) соединением по п.1 или
(b) композицией по п.6 или
введение пациенту:
(а) соединения по п.1 или
(b) композиции по п.6.
11. Способ ингибирования FLT-3 в биологической пробе или у пациента, предусматривающий контактирование указанной пробы с:
(а) соединением по п.1 или
(b) композицией по п.6 или
введение пациенту:
(а) соединения по п.1 или
(b) композиции по п.6.
12. Способ ингибирования Aurora-1, Aurora-2, Aurora-3 и FLT-3 у пациента, включающий введение указанному пациенту:
(a) соединения по п.1 или
(b) композиции по п.6.
13. Способ лечения рака у пациента, включающий стадию введения указанному пациенту композиции по п.6.
14. Способ по п.13, включающий стадию введения указанному пациенту дополнительного химиотерапевтического или антипролиферативного агента.
15. Способ по п.13, где указанный рак выбран из меланомы, лимфомы, нейробластомы, лейкоза или рака, выбранного из рака ободочной кишки, молочной железы, легкого, почки, яичника, поджелудочной железы, почечного рака, рака ЦНС, рака шейки матки, рака предстательной железы или рака желудочно-кишечного тракта, острого миелогенного лейкоза (AML), острого лимфоцитарного лейкоза (ALL), мастоцитоза или желудочно-кишечной стромальной опухоли (GIST).
16. Способ лечения или ослабления рака у пациента, включающий стадию нарушения митоза раковых клеток ингибированием протеинкиназы Aurora соединением по п.1.
17. Способ по п.16, предусматривающий стадию введения указанному пациенту композиции по п.6.
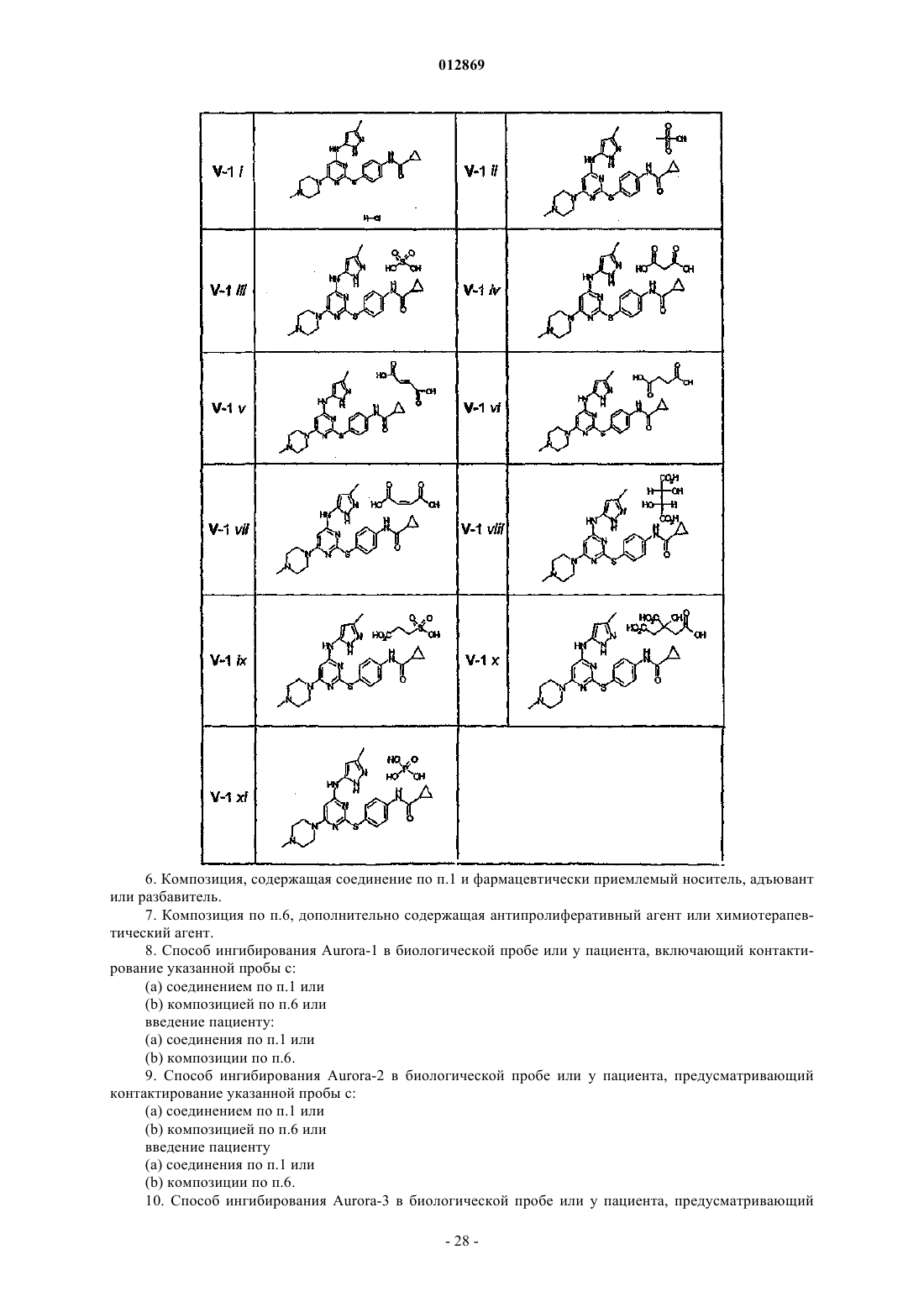
18. Соединение формулы V-1

или его фармацевтически приемлемая соль, выбранная из группы, состоящей из ацетата, адипата, альгината, аспартата, бензоата, бензолсульфоната, бисульфата, бутирата, цитрата, камфората, камфорсульфоната, циклопентанпропионата, диглюконата, додецилсульфата, этансульфоната, формиата, фумарата, глюкопентаноата, глицерофосфата, гликолата, гемисульфата, гептаноата, гексаноата, гидрохлорида, гидробромида, гидройодида, 2-гидроксиэтансульфоната, лактата, малеата, малоната, метансульфоната, 2-нафталинсульфоната, никотината, нитрата, оксалата, пальмоата, пектината, персульфата, 3-фенилпропионата, фосфата, пикрата, пивалата, пропионата, салицилата, сукцината, сульфата, тартрата, тиоцианата, тозилата, ундеканоата, натрия, калия, магния, аммония и N+(С1-4алкил)4.
19. Соединение по п.18, представляющее собой соль соединения формулы V-1 в виде сульфата.
20. Соединение по п.18, представляющее собой соль соединения формулы V-1 в виде лактата.
21. Применение соединения по п.18 в сочетании с Gleevec для производства лекарственного средства для лечения рака.
22. Применение соединения по п.18 в сочетании с Gleevec для производства лекарственного средства для лечения лейкемии.
23. Соединение формулы V

или его фармацевтически приемлемая соль,
где R5 представляет собой Н, метил, этил, н-пропил, i-пропил, циклопропил, изобутил или трет-бутил;
R6 представляет собой метил, этил, циклопропил;
R7 представляет собой метил, этил, циклопропил, трет-бутил,
при условии, что соединение не является

или его солью.
24. Соединение формулы V

или его фармацевтически приемлемая соль,
где R5 представляет собой метил;
R6 представляет собой метил;
R7 представляет собой циклопропил.
25. Соединение по пп.23, 24 в форме соли, где соль выбрана из группы, состоящей или из ее фармацевтически приемлемой соли или производного или таутомера или стереоизомера фармацевтически приемлемой соли, выбранной из группы, состоящей из ацетата, адипата, альгината, аспартата, бензоата, бензолсульфоната, бисульфата, бутирата, цитрата, камфората, камфорсульфоната, циклопентанпропионата, диглюконата, додецилсульфата, этансульфоната, формиата, фумарата, глюкопентаноата, глицерофосфата, гликолата, гемисульфата, гептаноата, гексаноата, гидрохлорида, гидробромида, гидройодида, 2-гидроксиэтансульфоната, лактата, малеата, малоната, метансульфоната, 2-нафталинсульфоната, никотината, нитрата, оксалата, пальмоата, пектината, персульфата, 3-фенилпропионата, фосфата, пикрата, пивалата, пропионата, салицилата, сукцината, сульфата, тартрата, тиоцианата, тозилата, ундеканоата, натрия, калия, магния, аммония и N+(С1-4алкил)4.
26. Соединение по п.25, представляющее собой соль соединения в виде сульфата.
27. Соединение по п.25, представляющее собой соль соединения в виде лактата.
28. Композиция, содержащая соединение по пп.25-27 и фармацевтически приемлемый носитель, адъювант или разбавитель.
29. Композиция по пп.25-27, дополнительно содержащая антипролиферативный агент или химиотерапевтический агент.
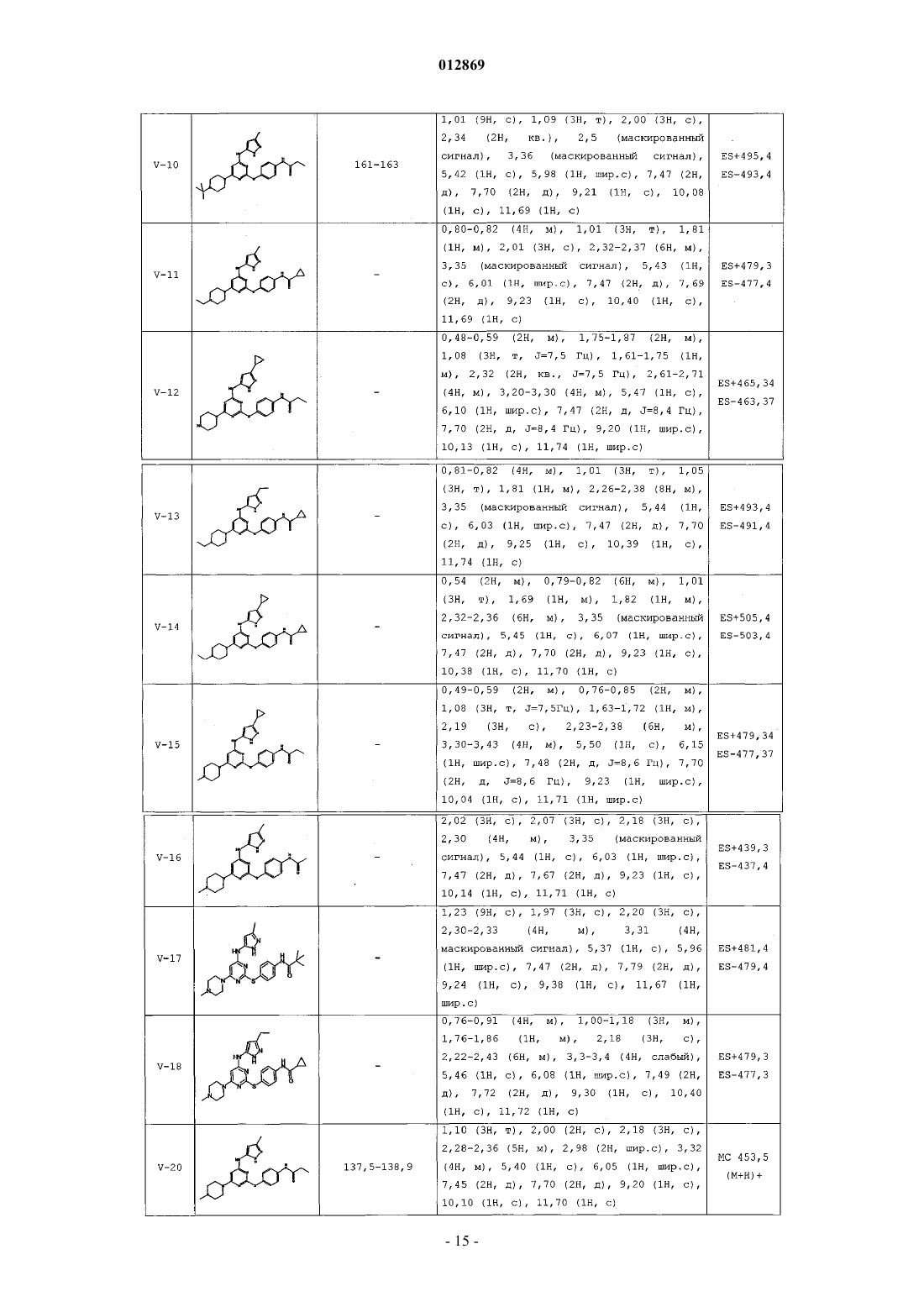
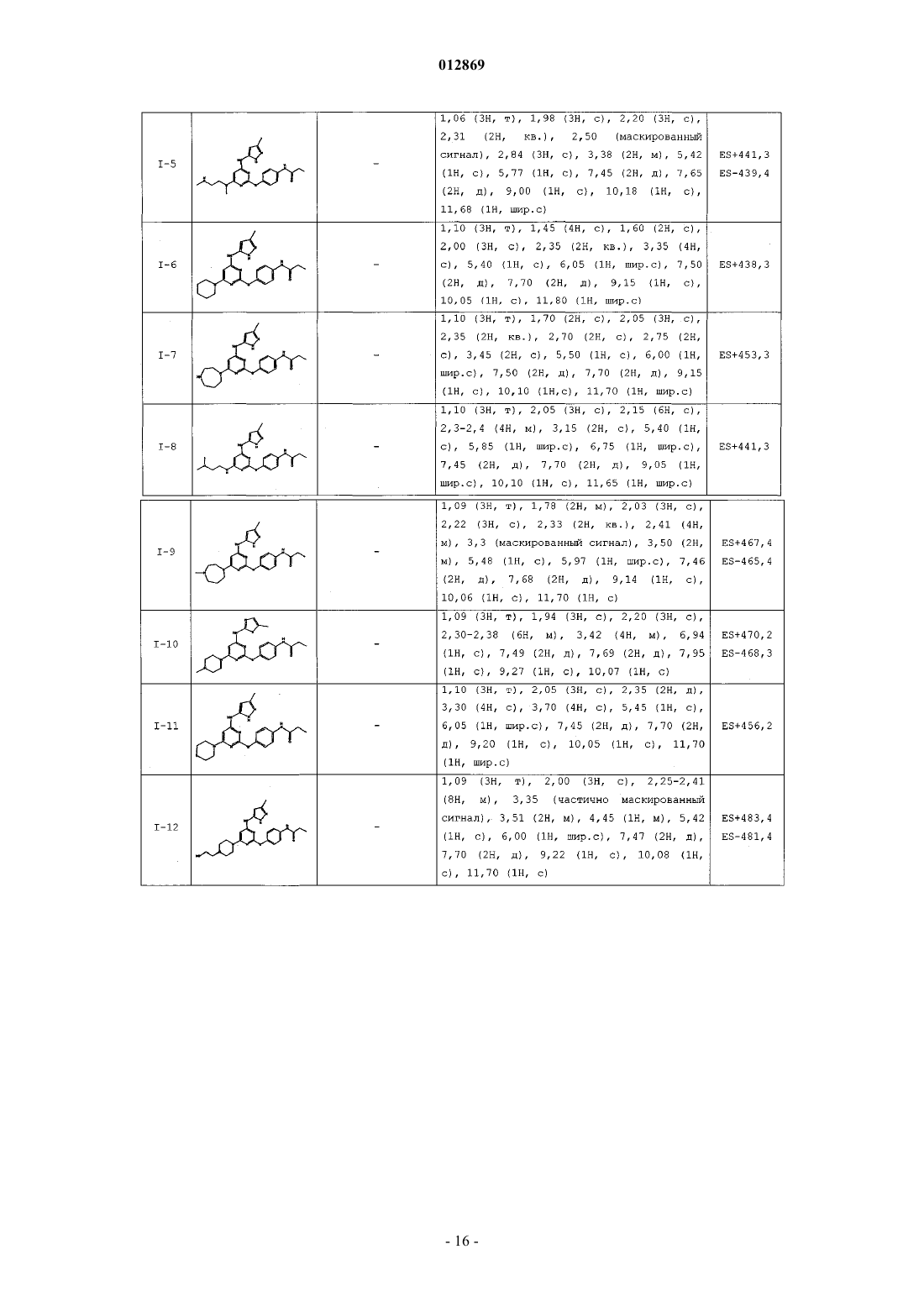
Текст
012869 Перекрестная ссылка на родственные заявки По заявке на данный патент испрашивается приоритет в соответствии с предварительной заявкой на патент США 60/390658, поданной 20 июня 2002 г., и предварительной заявкой на патент США 60/411609, поданной 18 сентября 2002 г., содержания которых включены здесь в качестве ссылки. Область техники, к которой относится изобретение Данное изобретение относится к легкому способу получения замещенных пиримидинов. Этот способ применим для получения ингибиторов протеинкиназ, в частности FLT-3 и киназ семейства Aurora,серин/треонинпротеинкиназ. Данное изобретение относится также к ингибиторам протеинкиназ FLT-3,Aurora-1, Aurora-2 и Aurora-3 и их композициям. Уровень техники Поиск новых терапевтических средств был в значительной степени облегчен в последние годы лучшим пониманием структуры ферментов и других биомолекул, ассоциированных с представляющими интерес заболеваниями. Одним важным классом ферментов, который был предметом интенсивного исследования, являются протеинкиназы. Протеинкиназы опосредуют внутриклеточную трансдукцию сигналов. Они осуществляют это путем переноса фосфорила от нуклеозидтрифосфата к белковому акцептору, который участвует в пути передачи сигнала. Имеется ряд киназ и путей, посредством которых внеклеточные и другие стимулы индуцируют различные клеточные реакции внутри клетки. Примеры таких стимулов включают в себя сигналы окружающей среды и химического стресса (например, осмотический шок, тепловой шок, ультрафиолетовую радиацию, бактериальный эндотоксин и Н 2 О 2), цитокины (например, интерлейкин-1 (IL-1) и фактор некроза опухолей альфа (TNF- и факторы роста (например, колониестимулирующий фактор гранулоцитов и макрофагов (GM-CSF) и фактор роста фибробластов (FGF. Внеклеточный стимул может действовать на одну или несколько клеточных реакций, связанных с ростом, миграцией, дифференцировкой клеток, секрецией гормонов, активацией факторов транскрипции, мышечным сокращением,метаболизмом глюкозы, регуляцией синтеза белка и регуляцией клеточного цикла. Многие заболевания ассоциированы с нарушенными клеточными реакциями, запускаемыми опосредуемыми протеинкиназами событиями. Эти заболевания включают в себя аутоиммунные заболевания, воспалительные заболевания, костные заболевания, метаболические заболевания, неврологические и нейродегенеративные заболевания, рак, сердечно-сосудистые заболевания, аллергии и астму, болезнь Альцгеймера и связанные с гормонами заболевания. Таким образом, в медицинской химии прилагались существенные усилия для нахождения ингибиторов протеинкиназ, которые являются эффективными в качестве терапевтических агентов. Семейство Aurora серин/треонинкиназ является важным для пролиферации клеток [Bischoff, J.R.Cell Biology 11, 49-54 (2001)]. Таким образом, ингибиторы семейства киназ Aurora имеют потенциал блокирования роста всех типов опухолей. Три известных члена семейства млекопитающих, Aurora-A (1), В (2) и С (3), являются высокогомологичными белками, ответственными за хромосомное расщепление, функцию митотического веретена и цитокинез. Экспрессия Aurora является низкой или недетектируемой в покоящихся клетках, с повышением экспрессии и активности до максимума во время фазы G2 и митотической фазы в клеточном цикле. В клетках млекопитающих предполагаемые субстраты Aurora включают в себя гистон Н 3, белок, участвующий в конденсации хромосом, и CENP-A, регуляторную легкую цепь миозина II, протеинфосфатазу 1, ТРХ 2, которые все необходимы для деления клеток. С его открытия в 1997 г. семейство киназ Aurora тесно связывали с онкогенезом. Наиболее привлекающим внимание доказательством этой связи является то, что сверхэкспрессия Aurora-A вызывает перерождение фибробластов грызунов (Bischoff, J.R., et al., A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 17, 3052-3065 (1998. Клетки с повышенными уровнями этой киназы содержат множественные центросомы и мультиполярные веретена и быстро становятся анеуплоидными. Онкогенная активность киназ Aurora, по-видимому, связана с генерированием такой генетической нестабильности. Действительно, наблюдали корреляцию между амплификацией локуса aurora-A и хромосомной нестабильностью в опухолях молочной железы и желудка (Miyoshi, Y.,Iwao, K., Egawa, С., and Noguchi, S. Association of centrosomal kinase STK15/BTAK mRNA expression withchromosomal instability in human breast cancers. Int. J. Cancer 92, 370-373 (2001); Sakakura, C. et al. Timoramplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation. British Journal of Cancer 84, 824-831 (2001. Сообщалось, что киназы Aurora сверхэкспрессируются в большом диапазоне опухолей человека. Повышенная экспрессия Aurora-A была обнаружена в более чем 50% колоректальных опухолей (Bischoff, J.R., et al., A homologue of Drosophila auroraof centrosorae kinase BTAK/Aurora-A in human ovarian cancer. Clinical Cancer Research 9, 1420-1426 (2003 и опухолей желудка (Sakakura, С. et al. Timor-amplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation. British Journal of Cancer 84, 824-831 (2001 и в 94% инвазивных аденокарцином млечных протоков молочной железы (Tanaka, Т., et al., Centrosomal(1999. Высокие уровни Aurora-A сообщались также в почечных, цервикальных, нейробластомных, меланомных, лимфомных, панкреатических линиях опухолевых клеток и линиях клеток опухоли предстательной железы (Bischoff, J.R., et al., A homologue of Drosophila aurora kinase is oncogenic and amplified inCancer Res. 9(3):991-7 (2003. Амплификацию/сверхэкспрессию Aurora-A наблюдают в раках мочевого пузыря человека, и амплификация Aurora-A связана с анеуплоидией и агрессивным клиническим поведением (Sen, S. et al., Amplification/overexpression of mitotic kinase gene in human bladder cancer. J. Natl.Cancer. Inst. 94(17):132909 (2002. Кроме того, амплификация локуса aurora-A (20q13) коррелирует с плохим прогнозом для пациентов с лимфоузел-отрицательным раком молочной железы (Isola, J.J., et al.cancer. American Journal of Pathology 147, 905-911 (1995. Aurora-B экспрессируется на высоком уровне во множественных линиях опухолевых клеток человека, в том числе лейкозных клетках (Katayama et al.Human AIM-1: cDNA cloning and reduced expression during endomitosis in megakaryocyte-lineage cells. Gene 244:1-7. Уровни этого фермента увеличиваются как функция стадии Duke в первичных колоректальных раках (Katayama, Н. et al. Mitotic kinase expression and colorectal cancer progression. Journal of the NationalCancer Institute 91, 1160-1162 (1999. Aurora-C, которая обычно обнаруживается только в зародышевых клетках, сверхэкспрессируется также в высоком проценте первичных колоректальных раков и в различных линиях опухолевых клеток, в том числе клетках цервикальной аденокарциномы и карциномы молочной железы (Kimura, М., Matsuda, Y., Yoshioka, Т., and Okano, Y. Cell cycle-dependent expression andcentrosomal localization of a third human Aurora/Ipl1-related protein kinase, AIK3, Journal of Biological Chemistry 274, 7334-7340 (1999); Takahashi, Т., et al. Centrosomal kinases, HsAIRk1 and HsAIRK3, are overexpressed in primary colorectal cancers. Jpn. J. Cancer Res. 91, 1007-1014 (2000. На основе известного действия киназ Aurora ингибирование их активности должно нарушать митоз,приводя к остановке клеточного цикла. Таким образом, in vivo ингибитор Aurora замедляет рост и индуцирует регресс опухоли. Повышенные уровни всех членов семейства Aurora наблюдаются в широком разнообразии линий опухолевых клеток. Киназы Aurora сверхэкспрессируются во многих опухолях человека и, как сообщается, ассоциированы с хромосомной нестабильностью в опухолях молочной железы (Miyoshi et al. 2001, 92,370-373).Aurora-2 экспрессируется на высоком уровне во множественных линиях опухолевых клеток человека, и уровни увеличиваются как функция стадии Duke в первичных колоректальных раках [Katayama, Н.et al. Mitotic kinase expression and colorectal cancer progression. Journal of the National Cancer Institute 91,1160-1162 (1999)]. Aurora-2 играет роль в регуляции точного расщепления хромосом во время митоза. Ошибочная регуляция клеточного цикла может привести к клеточной пролиферации и другим нарушениям. Было обнаружено, что в ткани рака ободочной кишки человека сверхэкспрессируется белок(1998); Kimura et al., J. Biol. Chem., 272, 13766-13771 (1997)]. Aurora-2 сверхэкспрессируется в большинстве трансформированных клеток. Bischoff et al. обнаружил высокие уровни Aurora-2 в 96% клеточных линий, полученных из опухолей легкого, ободочной кишки, почки, меланомы и молочной железы(Bischoff et al., EMBO J., 1998, 17, 3052-3065). Два интенсивных исследования показывают повышенный уровень Aurora-2 в 54 и 68% (Bischoff et al., EMBO J., 1998, 17, 3052-3065; Takahashi et al. 2000, Jpn. J.Cancer Res. 91, 1007-1014) колоректальных опухолей и в 94% инвазивных аденокарцином млечных протоков молочной железы (Tanaka et al., 1999, 59, 2041-2044). Экспрессия Aurora-1 является повышенной в клеточных линиях, полученных из опухолей ободочной кишки, молочной железы, легкого, меланомы, печени, яичника, поджелудочной железы, ЦНС, желудочно-кишечного тракта и лейкозов (Tatsuka et al. 1998, 58, 4811-4816). Высокие уровни Aurora-3 были обнаружены в нескольких линиях опухолевых клеток, хотя она ограничена яичком в нормальных тканях (Kimura et al. 1999, 274, 7334-7340). Сверхэкспрессия Aurora-3 в высоком проценте (приблизительно 50%) колоректальных раков также была документирована (Takahashiet al. 2000, Jpn. J. Cancer Res. 91, 1007-1014). В противоположность этому, семейство Aurora экспрессируется при низком уровне в большинстве нормальных тканей, за исключением тканей с высокой долей-2 012869 делящихся клеток, таких как вилочковая железа и яичко (Bischoff et al., EMBO J., 1998, 17, 3052-3065). В отношении дополнительного обзора роли киназ Aurora, которую они играют в пролиферативных нарушениях, см. Bischoff, J.R.Plowman, G.D. (The Aurora/Ipl1p kinase family: regulators of chromosome(On the role of aurora-A in centrosome function) Oncogene 21, 6175-6183 (2002). Рецепторная тирозинкиназа типа III, FLT-3, играет важную роль в поддержании, росте и развитии гемопоэтических и негемопоэтических клеток. [Scheijen, В., Griffin J.D., Oncogene, 2002, 21, 3314-3333 иReilly, J.T., British Journal of Haematology, 2002, 116, 744-757]. FLT-3 регулирует поддержание пулов стволовых клеток/клеток-предшественников, а также развитие зрелых лимфоидных и миелоидных клеток [Lyman, S., Jacobsen, S., Blood, 1998, 91, 1101-1134]. FLT-3 содержит внутренний киназный домен,который активируется после опосредованной лигандом димеризации этих рецепторов. После активации киназный домен индуцирует аутофосфорилирование рецептора, а также фосфорилирование различных цитоплазматических белков, которые способствуют размножению сигнала активации, приводящего к росту, дифференцировке и выживанию. Некоторые из стоящих далее по ходу процесса регуляторов передачи сигнала FLT-3-рецептора включают в себя PLC, PI3-киназу, Grb-2, SHIP и Src-родственные киназы [Scheijen, В., Griffin J.D., Oncogene, 2002, 21, 3314-3333]. Киназа FLT-3 играет роль во многих гемопоэтических и негемопоэтических злокачественностях. Считают, что мутации, которые индуцируют лиганднезависимую активацию FLT-3, участвуют в остром миелогенном лейкозе (AML), остром лимфоцитарном лейкозе (ALL), мастоцитозе и желудочно-кишечной стромальной опухоли (GIST). Эти мутации включают в себя изменения единственной аминокислоты в киназном домене или внутренних тандемных удвоениях, точковые мутации или делеции в рамке считывания юкстамембранного района этих рецепторов. Кроме активации мутаций, лигандзависимая (аутокринная или паракринная) стимуляция сверхэкспрессируемой киназы FLT-3 дикого типа вносит вклад в злокачественный фенотип [Scheijen, В., Griffininhibitor therapy for acute myeloid leukaemia) Cancer Cell. 1, 413-415 (2002). Три- или тетразамещенные производные пиримидинов, применимые в качестве ингибиторов киназ,известны в данной области. Обычно эти производные пиримидинов являются 2,4,6- или 2,4,5,6 замещенными, как показано ниже 2,4,5,6-замещенный пиримидин Известные способы получения таких производных пиримидинов обладают родом недостатков синтеза, такими как отсутствие возможности региоселективного введения заместителей в положении 2, 4 или 6 с высокими выходами. См. М. Botta, Nucleosides Nucleotides, 13, 8, 1994, 1769-78; М. Ban, Bioorg.-3 012869 Сущность изобретения Настоящее изобретение относится к соединениям формулы V или их фармацевтически приемлемым производным или солям,где R5 выбран из водорода или C1-4 алифатической группы;R7 представляет собой циклопропил. Предпочтительно изобретение относится к соединениям формулы V, где R5 выбран из водорода,метила, этила, трет-бутила или изопропила; более предпочтительно к соединениям формулы V, где R6 выбран из метила, этила или циклопропила. Изобретение также охватывает соединения, выбранные из табл. 1 и 2. Таблица 1-4 012869 Таблица 2 Репрезентативные соли соединений формулы V Изобретение также относится к композиции, содержащей соединение формулы V и фармацевтически приемлемый носитель, адъювант или разбавитель. Кроме того, настоящее изобретение включает варианты, когда композиция дополнительно содержит антипролиферативный агент или химиотерапевтический агент. В другом варианте изобретение относится к способу ингибирования Aurora-1 в биологической пробе или у пациента, включающему контактирование указанной пробы с (а) соединением формулы V или(b) композицией, содержащей соединение формулы V, или введение пациенту (а) соединения формулы V или (b) композиции, содержащей его. Следующее воплощение изобретения также раскрывает способ ингибирования Aurora-2 в биологической пробе или у пациента, предусматривающий контактирование указанной пробы с (а) соединением формулы V или (b) композицией, содержащей соединение формулы V, или введение пациенту (а) соединения формулы V или (b) композиции, содержащей его. В другом варианте изобретение раскрывает способ ингибирования Aurora-3 в биологической пробе или у пациента, предусматривающий контактирование указанной пробы с (а) соединением формулы V или (b) композицией, содержащей соединение формулы V, или введение пациенту (а) соединения формулы V или (b) композиции, содержащей его. Изобретение также относится к способу ингибирования FLT-3 в биологической пробе или у паци-5 012869 ента, предусматривающий контактирование указанной пробы с (а) соединением формулы V или (b) композицией, содержащей соединение формулы V, или введение пациенту (а) соединения формулы V или(b) композиции, содержащей его. В одном из вариантов изобретение раскрывает способ ингибирования Aurora-1, Aurora-2, Aurora-3 иFLT-3 у пациента, включающий введение указанному пациенту (а) соединения формулы V или (b) композицией, содержащей его. Изобретение также относится к способу лечения рака у пациента, включающему стадию введения указанному пациенту композиции, содержащей соединение формулы V. Кроме того, способ лечения рака может включать стадию введения пациенту дополнительного химиотерапевтического или антипролиферативного агента, при этом рак выбран из меланомы, лимфомы,нейробластомы, лейкоза или рака, выбранного из рака ободочной кишки, молочной железы, легкого,почки, яичника, поджелудочной железы, почечного рака, рака ЦНС, рака шейки матки, рака предстательной железы или рака желудочно-кишечного тракта, острого миелогенного лейкоза (AML), острого лимфоцитарного лейкоза (ALL), мастоцитоза или желудочно-кишечной стромальной опухоли (GIST). Изобретение также охватывает способ лечения или ослабления рака у пациента, включающий стадию нарушения митоза раковых клеток ингибированием протеинкиназы Aurora соединением формулыV. Данный способ предусматривает введение указанному пациенту композиции, содержащей соединение формулы V. В частном воплощении изобретение относится к соединению формулы V-1 или его фармацевтически приемлемой соли, выбранной из группы, состоящей из ацетата, адипата, альгината, аспартата, бензоата, бензолсульфоната, бисульфата, бутирата, цитрата, камфората, камфорсульфоната, циклопентанпропионата, диглюконата, додецилсульфата, этансульфоната, формиата, фумарата,глюкопентаноат, глицерофосфат, гликолат, гемисульфат, гептаноата, гексаноата, гидрохлорида, гидробромида, гидройодида, 2-гидроксиэтансульфоната, лактата, малеата, малоната, метансульфоната, 2 нафталинсульфоната, никотината, нитрата, оксалата, пальмоата, пектината, персульфата, 3 фенилпропионата, фосфата, пикрата, пивалата, пропионата, салицилата, сукцината, сульфата, тартрата,тиоцианата, тозилата, ундеканоата, натрия, калия, магния, аммония и N+(С 1-4 алкил)4. В предпочтительном варианте соединение формулы V-1 представляет собой сульфат или лактат. Изобретение также относится к применению соединения формулы V-1 в сочетании с Gleevec для производства лекарственного средства для лечения рака, в частности для лечения лейкемии. В частном варианте изобретение относится к соединениям формулы V или их фармацевтически приемлемым солям,где R5 представляет собой Н, метил, этил, н-пропил, i-пропил, циклопропил, изобутил, или третбутил;R7 представляет собой метил, этил, циклопропил, трет-бутил; при условии, что соединение не является или его солью. В предпочтительном варианте изобретение включает соединение формулы V или его фармацевтически приемлемую соль,где R5 представляет собой метил;R7 представляет собой циклопропил. При этом указанные соединения могут быть представлены в форме соли, где соль выбрана из группы, состоящей или из ее фармацевтически приемлемой соли или производного либо таутомера или стереоизомера фармацевтически приемлемой соли, выбранной из группы, состоящей из ацетата, адипата,альгината, аспартата, бензоата, бензолсульфоната, бисульфата, бутирата, цитрата, камфората, камфорсульфоната, циклопентанпропионата, диглюконата, додецилсульфата, этансульфоната, формиата, фумарата, глюкопентаноат, глицерофосфат, гликолат, гемисульфат, гептаноата, гексаноата, гидрохлорида,гидробромида, гидройодида, 2-гидроксиэтансульфоната, лактата, малеата, малоната, метансульфоната, 2 нафталинсульфоната, никотината, нитрата, оксалата, пальмоата, пектината, персульфата, 3 фенилпропионата, фосфата, пикрата, пивалата, пропионата, салицилата, сукцината, сульфата, тартрата,тиоцианата, тозилата, ундеканоата, натрия, калия, магния, аммония и N+(С 1-4 алкил)4. Изобретение также относится к композициям, содержащим указанные соединения в сочетании с фармацевтически приемлемым носителем, адъювантом или разбавителем и дополнительно содержащим антипролиферативный агент или химиотерапевтический агент. В соответствии с другим вариантом осуществления данное изобретение относится к композиции,содержащей соединение по данному изобретению или его фармацевтически приемлемое производное и фармацевтически приемлемый носитель, адъювант или разбавитель. Количество соединения в композициях по данному изобретению такое, которое является эффективным для детектируемого ингибирования протеинкиназы, в частности киназы Aurora и/или киназы FLT-3, в биологической пробе или у пациента. Предпочтительно композицию по данному изобретению готовят для введения пациенту, нуждающемуся в такой композиции. Наиболее предпочтительно композицию по данному изобретению готовят для перорального введения пациенту. Термин пациент в данном контексте относится к животному, предпочтительно млекопитающему и наиболее предпочтительно человеку. Термин фармацевтически приемлемый носитель, адъювант или разбавитель относится к нетоксичному носителю, адъюванту или разбавителю, который не разрушает фармакологическую активность соединения, вместе с которым его изготавливают. Фармацевтически приемлемые носители, адъюванты или разбавители, которые могут быть использованы в композициях по данному изобретению, включают в себя, но не ограничиваются ими, ионообменники, оксид алюминия, стеарат алюминия, лецитин, белки сыворотки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты,глицин, сорбиновая кислота, сорбат калия, частичные глицеридные смеси насыщенных растительных жирных кислот, воду, соли или электролиты, такие как сульфат протамина, динатрийгидрофосфат, гидрофосфат калия, хлорид натрия, соли цинка, колоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрий-карбоксиметилцеллюлозу,полиакрилаты, воски, сополимеры полиэтилена и полиоксипропилена, полиэтиленгликоль и ланолин. Термин детектируемо ингибируют в данном контексте означает измеримое изменение активности протеинкиназы между пробой, содержащей указанную композицию и протеинкиназу, и эквивалентной пробой, содержащей протеинкиназу в отсутствие указанной композиции. Термин фармацевтически приемлемые производное или соль обозначает любые нетоксичные-7 012869 соль, эфир, соль эфира или другое производное соединения по данному изобретению, которое при введении реципиенту способно обеспечивать, прямо или косвенно, соединение по данному изобретению или его ингибиторно активные метаболит или остаток. В данном контексте термин его ингибиторно активный метаболит или остаток означает, что его метаболит или остаток также является ингибитором киназы Aurora и/или киназы FLT-3. В соответствии с другим вариантом осуществления данное изобретение относится к способам получения фармацевтически приемлемой соли соединения в формуле I, I' или V, включающим стадию преобразования соединения формулы I, I' или V, полученного в соответствии со способами по данному изобретению, в желаемую фармацевтически приемлемую соль. Такие превращения хорошо известны в данной области. См., в общем виде, "Advanced Organic Chemistry", Jerry March, 4th edition, John Wiley andSons, N.Y. (1992). Фармацевтически приемлемые соли соединений по данному изобретению включают в себя соли,полученные из фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры подходящих кислотных солей включают в себя ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкопентаноат, глицерофосфат, гликолат, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат,лактат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, оксалат, пальмоат,пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, салицилат, сукцинат,сульфат, тартрат, тиоцианат, тозилат и ундеканоат. Другие кислоты, такие как щавелевая кислота, хотя они сами не являются фармацевтически приемлемыми, могут быть использованы при получении солей,которые могут использоваться в качестве промежуточных продуктов при получении соединений по данному изобретению и их фармацевтически приемлемых кислотно-аддитивных солей. Соли, полученные из подходящих оснований, включают в себя соли щелочных металлов (например,натрия и калия), соли щелочно-земельных металлов (например, магния), соли аммония и N+(С 1-4 алкил)4 соли. Данное изобретение включает также кватернизацию любой из содержащих основной азот групп описанных здесь соединений. Посредством такой кватернизации могут быть получены водорастворимые или диспергируемые в воде продукты. В табл. 2 представлены характерные соли соединений формулы V по данному изобретению. Композиции по данному изобретению могут вводиться перорально, парентерально, посредством ингаляционного спрея, местно, ректально, назально, буккально, вагинально или через имплантированный резервуар. Термин парентеральный в данном контексте включает в себя подкожный, внутривенный, внутримышечный, внутрисуставный, интрасиновиальный, внутригрудинный, внутриоболочечный,внутрипеченочный, внутрь повреждения и внутричерепной способы инъекции или инфузии. Предпочтительно эти композиции вводят перорально, внутрибрюшинно или внутривенно. Стерильные инъекционные формы композиций по данному изобретению могут быть в виде водной или масляной суспензий. Эти суспензии могут быть приготовлены в соответствии со способами, известными в данной области,использующими подходящие диспергирующие или увлажняющие агенты и суспендирующие агенты. Стерильный инъекционный препарат может также быть стерильным инъекционным раствором или суспензией в нетоксичном приемлемом для парентерального введения разбавителе или растворителе, например раствором в 1,3-бутандиоле. Среди приемлемых разбавителей и растворителей, которые могут быть использованы, можно указать воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды удобно использовать стерильные нелетучие масла. Для этой цели могут использоваться легкие нелетучие масла, в том числе синтетические моно- или диглицериды. При приготовлении инъекционных растворов могут быть использованы жирные кислоты,такие как олеиновая кислота и ее глицеридные производные, а также природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных вариантах. Эти масляные растворы или суспензии могут также содержать разбавитель, состоящий из спирта с длинной цепью, или диспергирующий агент, такой как карбоксиметилцеллюлоза или сходные диспергирующие агенты, которые обычно используют при приготовлении фармацевтически приемлемых дозированных форм, в том числе эмульсий и суспензий. Для целей приготовления могут быть также использованы другие обычно используемые поверхностно-активные вещества, такие как Твины, Спаны и другие эмульгирующие агенты и усиливающие биодоступность агенты, которые обычно используются при приготовлении фармацевтически приемлемых твердых, жидких или других дозированных лекарственных форм. Фармацевтически приемлемые композиции по данному изобретению могут вводиться перорально в любой приемлемой для перорального введения дозированной форме, в том числе, но не только, в виде капсул, таблеток, водных суспензий или растворов. В случае таблеток для перорального применения обычно используемые носители включают в себя лактозу и кукурузный крахмал. Обычно добавляют также смазывающие агенты, такие как стеарат магния. Для перорального введения в виде капсул подходящие разбавители включают лактозу и высушенный кукурузный крахмал. Когда водные суспензии не-8 012869 обходимы для перорального введения, активный ингредиент объединяют с эмульгирующими и суспендирующими агентами. Если желательно, могут быть также добавлены некоторые подслащивающие, ароматизирующие или красящие агенты. Альтернативно, фармацевтически приемлемые композиции по данному изобретению могут вводиться в виде суппозиториев для ректального введения. Они могут быть приготовлены смешиванием агента с подходящим нераздражающим эксципиентом, который является твердым при комнатной температуре, но становится жидким при ректальной температуре и, следовательно, будет расплавляться в прямой кишке с высвобождением лекарственного средства. Такие материалы включают в себя какао-масло,пчелиный воск и полиэтиленгликоли. Фармацевтически приемлемые композиции по данному изобретению могут также вводиться местно, в частности, когда мишень обработки включает в себя участки или органы, легко доступные для локального введения, в том числе глаза, кожу или нижнюю часть кишечного тракта. Подходящие композиции для локального введения могут быть легко приготовлены для каждого из этих участков и органов. Локальное применение для нижней части кишечного тракта может выполняться с использованием композиции ректального суппозитория (см. выше) или с использованием композиции подходящей клизмы. Могут быть также использованы трансдермальные пластыри. Для локальных применений фармацевтически приемлемые композиции могут быть приготовлены в виде подходящей мази, содержащей активное соединение, суспендированное или растворенное в одном или нескольких носителях. Носители для локального введения соединений по данному изобретению включают в себя, но не ограничиваются ими, минеральное масло, жидкий вазелин (вазелиновое масло),белый вазелин, пропиленгликоль, полиоксиэтилен, соединение полиоксипропилена, эмульгирующий воск и воду. Альтернативно, фармацевтически приемлемые композиции могут быть приготовлены в виде подходящих лосьона или крема, содержащих активные компоненты, суспендированные или растворенные в одном или нескольких фармацевтически приемлемых носителях. Подходящие носители включают в себя, но не ограничиваются ими, минеральное масло, моностеарат сорбитана, полисорбат 60, цетиловые эфиры, воск, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду. Для офтальмологического применения фармацевтически приемлемые композиции могут быть приготовлены в виде тонкоизмельченных суспензий в изотоническом стерильном солевом растворе с корректированным pH или предпочтительно в виде растворов в изотоническом стерильном солевом растворе с корректированным pH, либо с консервантом, таким как бензилалконийхлорид, либо без консерванта. Альтернативно, для офтальмологических применений фармацевтически приемлемые композиции могут быть приготовлены в виде мази, такой как вазелин. Фармацевтически приемлемые композиции данного изобретения могут также вводиться с использованием назального аэрозоля или ингаляции. Такие композиции готовят в соответствии со способами,хорошо известными в области приготовления фармацевтических композиций, и они могут быть приготовлены в виде растворов в солевом растворе, с использованием бензилового спирта или других подходящих консервантов, усилителей абсорбции для увеличения биодоступности, фторуглеродов и/или других общепринятых солюбилизирующих или диспергирующих агентов. Наиболее предпочтительно фармацевтически приемлемые композиции данного изобретения готовят для перорального введения. Количество соединений данного изобретения, которое может быть объединено с веществаминосителями для получения композиции в форме отдельных доз, будет варьироваться в зависимости от проходящего лечение хозяина, конкретного способа введения. Предпочтительно композиции должны быть приготовлены таким образом, чтобы доза 0,01-100 мг/кг массы тела/день ингибитора могла быть введена пациенту, получающему эти композиции. Должно быть также понятно, что конкретная доза и схема введения для любого конкретного пациента будет зависеть от различных факторов, в том числе от активности конкретного используемого соединения, возраста, массы тела, общего состояния здоровья, пола, диеты, времени введения, скорости экскреции, комбинации лекарственных средств и суждений лечащего врача и тяжести конкретного подлежащего лечению заболевания. Количество соединения данного изобретения в композиции будет также зависеть от конкретного соединения в данной композиции. В зависимости от конкретного состояния или заболевания, которое подлежит лечению или предупреждению, дополнительные терапевтические агенты, которые обычно вводят для лечения или предупреждения этого состояния, могут также присутствовать в композициях по данному изобретению. В данном контексте дополнительные терапевтические агенты, которые обычно вводят для лечения или предупреждения конкретного заболевания или состояния, известны как "подходящие для подлежащего лечению заболевания или состояния". Например, химиотерапевтические агенты или другие антипролиферативные агенты могут быть объединены с соединениями по данному изобретению для лечения пролиферативных заболеваний и рака. Примеры известных химиотерапевтических агентов включают в себя, но не ограничиваются ими,Gleevec, адриамицин, дексаметазон, винкристин, циклофосфамид, фторурацил, топотекан, таксол, интерферон и производные платины. Другие примеры агентов, с которыми могут быть также объединены-9 012869 ингибиторы по данному изобретению, включают в себя, без ограничения: средства для лечения болезни Альцгеймера, такие как Арицепт и Эксцелон; средства для лечения болезни Паркинсона, такие как LДОПА/карбидопа, энтакапон, ропинрол, прамипексол, бромокритин, перголид, тригексефендил и амантадин; агенты для лечения рассеянного склероза (MS), такие как бета-интерферон (например, Авонекс и Ребиф), Копаксон и митоксантрон; средства для лечения астмы, такие как албутерол и Сингулаир; агенты для лечения шизофрении, такие как ципрекса, риспердал, сероквел и галоперидол; противовоспалительные агенты, такие как кортикостероиды, TNF-блокаторы, IL-1 RA, азатиоприн, циклофосфамид и сульфазалазин; иммуномодуляторные и иммуносупрессивные агенты, такие как циклоспорин, такролимус, рапамицин, микофенолат мофетил, интерфероны, кортикостероиды, циклофосфамид, азатиоприн и сульфазалазин; нейротрофические факторы, такие как ингибиторы ацетилхолинэстеразы, ингибиторы МАО, интерфероны, антиконвульсанты, блокаторы ионных каналов, рилузол и агнеты против болезни Паркинсона; агенты для лечения сердечно-сосудистых заболеваний, такие как бета-блокаторы, ингибиторы АСЕ, диуретики, нитраты, блокаторы кальциевых каналов и статины; агенты для лечения заболевания печени, такие как кортикостероиды, холестирамин, интерфероны и антивирусные агенты; агенты для лечения нарушений, связанных с кровью, такие как кортикостероиды, противолейкозные агенты и факторы роста и агенты для лечения иммунодефицитных нарушений, такие как гамма-глобулин. Дополнительные примеры химиотерапевтических агентов или других антипролиферативных агентов, которые могут быть объединены с соединениями по данному изобретению для лечения пролиферативных заболеваний и рака, включают в себя, но не ограничиваются ими, например, другие терапии или антираковые агенты, которые могут быть использованы в комбинации с антираковыми агентами данного изобретения, и включают в себя хирургию, лучевую терапию (в качестве небольшого числа примеров,гамма-радиацию, нейтронную лучевую терапию, электронную лучевую терапию, протонную терапию,брахитерапию и системные радиоактивные изотопы, среди прочих), эндокринную терапию, модификаторы биологических реакций (интерфероны, интерлейкины и фактор некроза опухолей (TNF), среди прочих), гипертермию и криотерапию, агенты для ослабления любых неблагоприятных побочных эффектов (например, противорвотные средства) и другие одобренные химиотерапевтические лекарственные средства, в том числе, но не только, алкилирующие лекарственные средства (меклоретамин, хлорамбуцил, циклофосфамид, мелфалан, ифосфамид), антиметаболиты (метотрексат), антагонисты пуринов и антагонисты пиримидинов (6-меркаптопурин, 5-фторурацил, цитарабил, гемцитабин), яды митотического веретена (винбластин, винкристин, винорелбин, паклитаксел), подофиллотоксины (этопозид, иринотекан, топотекан), антибиотики (доксорубицин, блеомицин, митомицин), нитрозомочевины (кармустин,ломустин), неорганические ионы (цисплатин, карбоплатин), ферменты (аспарагиназа) и гормоны (тамоксифен, лейпролид, флутамид и мегестрол), Gleevec, адриамицин, дексаметазон и циклофосфамид. В отношении более исчерпывающего обсуждения модернизированных раковых терапий см.http://www.nci.nih.gov/, перечень одобренных FDA лекарственных средств для онкологии вhttp://www.fda.gov/cder/cancer/druglistframe.htm, и The Merck Manual, Seventeenth Ed. 1999, все содержания которых включены здесь в качестве ссылки. Количество дополнительного терапевтического агента, присутствующего в композициях данного изобретения, будет не большим, чем количество, которое вводилось бы обычно в композиции, содержащей этот терапевтический агент в виде единственного активного агента. Предпочтительно количество дополнительного терапевтического агента в описанных здесь композициях будет находиться в диапазоне приблизительно 50-100% количества, обычно присутствующего в композиции, содержащей этот агент в виде единственного терапевтически активного агента. В соответствии с другим вариантом осуществления данное изобретение относится к способу ингибирования активности киназ Aurora-1, Aurora-2, Aurora-3 и/или FLT-3 в биологической пробе, предусматривающий стадию контактирования указанной биологической пробы с соединением формулы V или композицией, содержащей указанное соединение. Термин биологическая проба в данном контексте включает в себя, без ограничения, культуры клеток или их экстракты; материал биопсий, полученный из млекопитающего, или его экстракты; и кровь, слюну, мочу, фекалии, сперму, слезы и другие жидкости тела или их экстракты. Ингибирование активности киназ Aurora-1, Aurora-2, Aurora-3 и/или FLT-3 в биологической пробе может быть использовано в различных целях, которые известны специалисту с квалификацией в данной области. Примеры таких целей включают в себя, но не ограничиваются ими, переливание крови, трансплантацию органов, хранение биологических образцов и биологические анализы. В соответствии с другим вариантом осуществления данное изобретение относится к способу ингибирования активности киназы Aurora-1 у пациента, включающему стадию введения указанному пациенту соединения формулы V или композиции, содержащей указанное соединение. В соответствии с другим вариантом осуществления данное изобретение относится к способу ингибирования активности киназы Aurora-2 у пациента, включающему стадию введения указанному пациенту соединения формулы V или композиции, содержащей указанное соединение. В соответствии с другим вариантом осуществления данное изобретение относится к способу инги- 10012869 бирования активности киназы Aurora-3 у пациента, включающему стадию введения указанному пациенту соединения формулы V или композиции, содержащей указанное соединение. В соответствии с другим вариантом осуществления данное изобретение относится к способу ингибирования активности киназы FLT-3 у пациента, включающему стадию введения указанному пациенту соединения формулы V или композиции, содержащей указанное соединение. В соответствии с другим вариантом осуществления данное изобретение относится к способу ингибирования активности киназ Aurora-1, Aurora-2, Aurora-3 и FLT-3 у пациента, включающему стадию введения указанному пациенту соединения формулы V или композиции, содержащей указанное соединение. В соответствии с другим вариантом осуществления данное изобретение относится к способу лечения или ослабления тяжести опосредованного Aurora заболевания или состояния у пациента, включающему стадию введения указанному пациенту соединения формулы V или композиции, содержащей указанное соединение. Термин "опосредованное Aurora заболевание" в данном контексте означает любое заболевание или другое пагубное состояние или заболевание, о котором известно, что в нем играет роль протеинкиназа семейства Aurora. Такие заболевания и состояния включают в себя, без ограничения, меланому, лейкоз или рак, выбранный из рака ободочной кишки, молочной железы, желудка, яичника, шейки матки, меланомы, почечного рака, рака предстательной железы, лимфомы, нейробластомы, рака поджелудочной железы, лейкоза и рака мочевого пузыря. В соответствии с другим вариантом осуществления данное изобретение относится к способу лечения рака у пациента, включающему стадию введения указанному пациенту соединения формулы V или его композиции. В соответствии с другим вариантом осуществления данное изобретение относится к способу лечения меланомы, лимфомы, нейробластомы, лейкоза или рака, выбранного их рака ободочной кишки, молочной железы, легкого, почки, яичника, поджелудочной железы, ренального рака, рака ЦНС, шейки матки, предстательной железы или рака желудочно-кишечного тракта у пациента, включающему стадию введения указанному пациенту соединения формулы V или его композиции. В соответствии с другим вариантом осуществления данное изобретение относится к способу лечения острого миелогенного лейкоза (AML), острого лимфоцитарного лейкоза (ALL), мастоцитоза или желудочно-кишечной стромальной опухоли (GIST) у пациента, включающему стадию введения указанному пациенту соединения формулы V или его композиции. Другой аспект данного изобретения относится к нарушению митоза раковых клеток у пациента,включающему стадию введения указанному пациенту соединения формулы V или его композиции. В соответствии с другим вариантом осуществления данное изобретение относится к способу лечения или ослабления тяжести рака у пациента, включающему стадию нарушения митоза раковых клеток ингибированием Aurora-1, Aurora-2 и/или Aurora-3 соединением формулы V или его композиции. В другом варианте осуществления способы по данному изобретению, по которым используются композиции, не содержащие дополнительного терапевтического агента, предусматривают дополнительную стадию отдельного введения указанному пациенту дополнительного терапевтического агента. При раздельном введении этих дополнительных терапевтических агентов они могут вводиться пациенту перед введением, последовательно с введением или после введения композиции данного изобретения. Для более полного понимания описанного здесь изобретения приводятся следующие примеры. Следует учесть, что эти примеры приведены только для иллюстративных целей и не должны рассматриваться как ограничивающие каким-либо образом данное изобретение. Примеры Общая схема- 11012869 Пример 1. 4,6-Дихлорпиримидин-2-метилсульфон (А) получен способами, по существу, одинаковыми со способами, приведенными в Koppel et al., JOC, 26, 1961, 792, следующим образом. К перемешиваемому раствору 4,6-дихлор-2-(метилтио)пиримидина (50 г, 0,26 моль) в дихлорметане (1 л) при 0 С добавляли метахлорпероксибензойную кислоту (143,6 г, 0,64 моль) в течение 20 мин. Раствору давали нагреться до комнатной температуры и перемешивали в течение 4 ч. Смесь разбавляли дихлорметаном (1,5 л) и затем обрабатывали последовательно 50% раствором Na2S2O3/NaHCO3 (2200 мл), насыщенным растворомNaHCO3 (4300 мл) и солевым раствором (200 мл), затем сушили (MgSO4). Растворитель удаляли в вакууме с получением твердого вещества не совсем белого цвета, которое вновь растворяли в EtOAC (1 л) и обрабатывали последовательно насыщенным водным раствором NaHCO3 (3300 мл) и солевым раствором (100 мл), затем сушили (MgSO4). Растворитель удаляли в вакууме с получением указанного в заголовке соединения (А) в виде белого твердого вещества (55,6 г, выход 96%). 1[4-(4,6-Дихлорпиримидин-2-илсульфанил)фенил]амид циклопропанкарбоновой кислоты (С). Суспензию соединения А (10 г, 44,04 ммоль) и (4-меркаптофенил)амид циклопропанкарбоновой кислоты (В, 8,51 г, 44,04 ммоль) в трет-бутаноле (300 мл) дегазировали откачиванием воздуха с последующей продувкой азотом. Смесь перемешивали при 90 С в атмосфере азота в течение 1 ч, затем растворитель удаляли в вакууме. Остаток растворяли в этилацетате (600 мл) и промывали водным раствором карбоната калия и хлорида натрия. Органический экстракт сушили над сульфатом магния, концентрировали до малого объема и давали кристаллизоваться. Продукт С собирали в виде бесцветных кристаллов (11,15 г, 74%). 1 Н-ЯМР ДМСО-d6,0,82-0,89 (4H, м), 1,80-1,88 (1 Н, м), 7,55 (2H, д), 7,70-7,76 (3 Н, м), 10,49 (1 Н, с); М+Н, 340. Пример 3. 4-[4-Хлор-6-(5-метил-2 Н-пиразол-3-иламино)пиримидин-2-илсульфанил]фениламид циклопропанкарбоновой кислоты (D). Смесь соединения С (1,0 г, 2,94 ммоль) и 3-амино-5-метилпиразола (314 мг, 3,23 ммоль) в диметилформамиде (6 мл) обрабатывали диизопропилэтиламином (0,614 мл, 3,53 ммоль) и йодидом натрия (530 мг, 3,53 ммоль). Смесь перемешивали в атмосфере азота при 85 С в течение 4 ч, охлаждали до комнатной температуры и разбавляли этилацетатом. Раствор промывали водой (4), сушили над сульфатом магния и концентрировали до 5 мл с получением после кристаллизации и сбора бесцветных кристаллов указанного в заголовке соединения D (920 мг, 78%). 1H-ЯМР ДМСО-d6,0,80-0,87 (4 Н, м), 1,77-1,85 (1H, м), 1,92 (1 Н, с), 5,24 (1H, шир.с), 6,47 (1 Н,шир.с), 7,55 (2 Н, д), 7,70-7,80 (2 Н, м), 10,24 (1 Н, с), 10,47 (1 Н, с), 11,92 (1 Н, с). Пример 4. 4-[4-(4-Метилпиперазин-1-ил)-6-(5-метил-2 Н-пиразол-3-иламино)пиримидин-2 илсульфанил]фениламид циклопропанкарбоновой кислоты (V-1). Соединение D (2,373 г, 5,92 ммоль) обрабатывали N-метилпиперазином (10 мл) и смесь перемешивали при 110 С в течение 2 ч. Избыток N-метилпиперазина удаляли в вакууме, затем остаток растворяли в этилацетате, промывали водным раствором бикарбоната натрия, сушили над сульфатом магния и концентрировали. Остаток кристаллизовали из метанола с получением бесцветных кристаллов желаемого продукта V-1 (1,82 г, 66%). 1- 12012869 примерах 1, 2 и 3, в н-BuOH (5 мл) обрабатывали дигидробромидом N-пропилпиперазина (887 мг,3,06 ммоль), затем диизопропилэтиламином (1,066 мл, 6,12 ммоль). Полученную смесь перемешивали при 110 С в течение 20 ч. Растворитель удаляли при пониженном давлении и остаток очищали с использованием препаративной ВЭЖХ с получением указанного в заголовке соединения. 1 Н-ЯМР (ДМСО),1,10 (3 Н, т), 2,05 (3 Н, с), 2,35 (2 Н, д), 3,30 (4 Н, с), 3,70 (4 Н, с), 5,45 (1 Н, с), 6,05N-[4-(4,6-Дихлорпиримидин-2-илокси)фенил]ацетамид. Раствор 4-ацетамидофенола (666 мг, 4,40 ммоль) в безводном ТГФ (40 мл), перемешивающийся при температуре окружающей среды, обрабатывали 60% дисперсией гидрида натрия в минеральном масле(176 мг, 4,40 ммоль). Затем реакционной смеси давали перемешиваться в течение 30 мин при температуре окружающей среды и добавляли 4,6-дихлор-2-метансульфонилпиримидин (1,0 г, 4,40 ммоль). Затем реакционную смесь перемешивали в течение еще 3 ч и разбавляли реакционную смесь насыщенным водным раствором NH4Cl и EtOAC. Органический слой отделяли, промывали насыщенным водным раствором NaCl и сушили над сульфатом натрия и затем концентрировали в вакууме. Остаток очищали колоночной хроматографией (силикагель, МеОН:CH2Cl2, 5:95) с получением указанного в заголовке соединения 1,25 г (95%) в виде твердого вещества. 1H-ЯМР (400 МГц, ДМСО-d6):2,06 (3 Н, с), 7,18 (2 Н, д, J=8,5 Гц), 7,62 (2 Н, д, J=8,5 Гц), 10,05 (1 Н,с); LS-MC: ES+=298,16, ES-=296,18. Пример 7. 4-[4-(4-Метил-4-оксипиперазин-1-ил)-6-(5-метил-2 Н-пиразол-3-иламино)пиримидин-2 илсульфанил]фениламид циклопропанкарбоновой кислоты (V-19). Соединение V-1 (1 г, 2,1 ммоль) суспендировали в дихлорметане (20 мл), охлаждали до 0 С и обрабатывали раствором дихлорметана в mCPBA в 10 аликвотах при 10-минутных интервалах (каждая аликвота состояла из 100 мг, 0,44 ммоль) в 1 мл ДХМ. Каждый раз при добавлении аликвоты этот раствор становился коричневым и постепенно возвращался к желтому цвету по мере расходования mCPBA. После расходования всего исходного материала растворитель удаляли в вакууме, и полученное оранжевое масло очищали препаративной ВЭЖХ с получением указанного в заголовке соединения в виде твердого вещества не совсем белого цвета (69 мг, 7%). 1(1 Н, с), 11,82 (1 Н, шир.с). Пример 8. Метансульфонат 4-[4-(4-метилпиперазин-1-ил)-6-(5-метил-2 Н-пиразол-3-иламино)пиримидин-2 илсульфанил]фениламида циклопропанкарбоновой кислоты (V-1ii). Соединение V-1 (515 мг, 1,11 ммоль) суспендировали в этаноле (80 мл) и нагревали с обратным холодильником при кипячении. К прозрачному раствору добавляли метансульфоновую кислоту (106 мг,1,11 ммоль) и реакционную смесь нагревали с обратным холодильником при кипячении в течение еще 10 мин. Смеси давали остыть до комнатной температуры и растворитель выпаривали, пока не начинал образовываться осадок. Затем смесь охлаждали до 0 С и полученный осадок собирали фильтрацией и затем сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (290 мг, 47%). 1(2 Н, д), 9,61 (1 Н, с), 10,41 (1 Н, шир.с), 10,80 (1 Н, с). Пример 9. Следующие соединения, представленные в табл. 3 ниже, получали в соответствии со способами по данному изобретению и способами, по существу, одинаковыми с приведенными в примерах 1-8 выше. Данные, характеризующие эти соединения, суммированы в табл. 3 ниже и включают в себя 1 Н-ЯМР,точку плавления (т. пл.) и данные масс-спектрометрии (MS). Если нет других указаний, все представленные данные 1 Н-ЯМР, приведенные в табл. 3, были получены при 400 МГц в дейтерированном диметилсульфоксиде (ДМСО-d6).- 13012869 Таблица 3 Данные характеристики представительных соединений Биологические анализы Активность соединений по данному изобретению в качестве ингибиторов киназ может анализироваться in vitro, in vivo или в клеточной линии. Анализы in vitro включают в себя анализы, которые определяют ингибирование либо киназной активности, либо АТФ-азной активности активированного фермента Aurora и/или FLT-3. Другие анализы in vitro количественно определяют способность ингибитора связываться с Aurora и/или FLT-3 и могут быть измерены либо радиоактивным мечением ингибитора перед связыванием, выделением комплекса ингибитор/Aurora и/или ингибитор/FLT-3 и определением количества связанной радиоактивной метки, либо проведением конкурентного эксперимента, в котором новые соединения инкубируют с Aurora и/или FLT-3, связанными с известными радиоактивными лигандами. Можно использовать любой тип или любую изоформу Aurora, в зависимости от того, какой тип или какая изоформа должны быть ингибированы. Подробности используемых условий для этих анализов ферментов описаны в примерах ниже.- 24012869 Пример 10. Определение Ki для ингибирования Aurora. Соединения подвергали скринингу следующим образом на их способность ингибировать Aurora с использованием стандартного ферментсвязанного анализа (Fox et al. (1998) Proyein Sci. 7, 2249). К исходного буферному раствору для анализа, содержащему 0,1 М HEPES 7,5, 10 мМ MgCl2, 1 мМ ДТТ,25 мМ NaCl, 2,5 мМ фосфоенолпируват, 3000 мМ НАДН, 30 мг/мл пируваткиназы, 10 мг/мл лактатдегидрогеназы, 40 мМ АТФ и 800 мкМ пептид (LRRASLG, American Peptide, Sunnyvale, СА) добавляли раствор в ДМСО соединения данного изобретения до конечной концентрации 30 мкМ. Полученную смесь инкубировали при 30 С в течение 10 мин. Реакцию инициировали добавлением 10 мкл исходного раствора Aurora с получением конечной концентрации 70 нМ в этом анализе. Скорости реакции получали мониторингом оптической плотности при 340 нм в течение 5-минутного времени считывания результатов при 30 С с использованием планшет-ридера BioRad Ultramark (Hercules, СА). Величины Ki определяли из данных скорости как функцию концентрации ингибитора. Было обнаружено, что соединения формулы V данного изобретения являются ингибиторамиAurora-1, Aurora-2 и Aurora-3. Пример 11. Определение Ki для ингибирования FLT-3. Соединения подвергали скринингу на их способность ингибировать активность FLT-3 с использованием радиометрического анализа связывания на фильтре. Этот анализ выполняет мониторинг включения 33 Р в субстрат поли(Glu,Tyr)4:1 (pE4Y). Реакции проводили в растворе, содержащем 100 мМ HEPES(pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 1 мМ ДТТ, 0,01% БСА и 2,5% ДМСО. Конечные концентрации субстрата в этом анализе были 90 мкМ АТФ и 0,5 мг/мл pE4Y (оба из Sigma Chemicals, St Louis, МО). Конечная концентрация соединения данного изобретения обычно была между 0,01 и 5 мкМ. Обычно проводили титрование с 12 точками приготовлением серийных разведений из исходного ДМСО-раствора тест-соединения. Реакции проводили при комнатной температуре. Готовили два раствора для анализа. Раствор 1 содержит 100 мМ HEPES (pH 7,5), 10 мМ MgCl2,25 мМ NaCl, 1 мг/мл pE4Y и 180 мкМ АТФ (содержащий 0,3 мкКи [-33 Р]АТФ для каждой реакции). Раствор 2 содержит 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 2 мМ ДТТ, 0,02% БСА и 3 нМ FLT3. Анализ проводили на 96-луночном планшете смешиванием 50 мкл каждого из раствора 1 и 2,5 мл соединения данного изобретения. Реакцию инициировали раствором 2. После инкубирования в течение 20 мин при комнатной температуре реакцию останавливали 50 мкл 20% ТХУ, содержащей 0,4 мМ АТФ. Затем весь объем реакции переносили на фильтрующий планшет и промывали 5% ТХУ с использованием харвестера 9600 из ТОМТЕС (Hamden, СТ). Количество включения 33 Р в pE4Y анализировали сцинтилляционным счетчиком для счета в микропланшетах Packard Top Count Microplate Scintillation Counter(Meriden, СТ). Данные строили с использованием программы Prism с получением IC50 или Ki. Было обнаружено, что соединения формулы V данного изобретения являются ингибиторами FLT-3. Пример 12. Определение IC50 для ингибирования Aurora в анализе с клетками Colo205. Соединения анализировали также на ингибирование пролиферации клеток. В этом анализе полную среду готовили добавлением 10% фетальной бычьей сыворотки, L-глутамина и раствора пенициллина/стрептомицина к среде RPMI 1640 (Sigma). Клетки рака ободочной кишки (клеточной линии Colo205) добавляли в 96-луночный планшет при плотности посева 1,25104 клеток/лунка/150 мкл. Раствор тест-соединения готовили в полной среде серийным разведением, тест-соединение (50 мкл) добавляли в каждую лунку. Каждый планшет содержал ряд лунок, в которые добавляли только полную среду (200 мкл), для образования контрольной группы для измерения максимальной пролиферации. Контрольную группу с носителем также добавляли на каждом планшете. Планшеты инкубировали при 37 С в течение 2 дней. Исходный раствор 3 Н-тимидина (1 мКи/мл, Amersham Pharmacia UK) разбавляли до 20 мкКи/мл в средеRPMI 1640, затем 25 мкл этого раствора добавляли в каждую лунку. Планшеты инкубировали дополнительно при 37 С в течение 3 ч и анализировали на поглощение 3 Н-тимидина с использованием жидкостного сцинтилляционного счетчика. Было обнаружено, что соединения формулы V являются ингибиторами пролиферации раковых клеток Colo205. Пример 13. Измерение пролиферации клеток в панели опухолевых и нормальных типов клеток: анализ включения 3 Н-тимидина. Анализ включения 3 Н-тимидина был выбран в качестве хорошо охарактеризованного способа определения пролиферации клеток. Клетки из нормальных тканей и большое разнообразие различных типов опухолей были выбраны для анализа. Многие опухолевые клетки были выбраны потому, что они экспрессируют высокие уровни белков Aurora (например, MCF-7, РС 3, А 375, А 549) (См. раздел 5.3.5 иBischoff et al. EMBO J. 1998 17, 3052-3065) и/или способны образовывать опухоли в голых мышах или- 25012869 крысах (например, НСТ 116, MCF-7 и MDA-MB-231). Логарифмически растущие клетки инкубировали с соединением в течение 96 ч. Для измерения пролиферации клеток за 3 ч перед окончанием этого эксперимента в каждую лунку добавляли 0,5 мкКи 3 Нтимидина. Затем клетки собирали, промывали и включенную радиоактивность считали на бета-счетчике для микропланшетов Wallac. Для определения ингибирования пролиферации, имп/мин, строили в зависимости от концентрации соединения и определяли графически IC50. В табл. 4 ниже представлены клеточные линии, использованные в описанном выше анализе пролиферации клеток. Для каждой клеточной линии определяли ингибирование пролиферации клеток и включение 3 Н-тимидина (временная точка 96 ч). Таблица 4 Клеточные линии Хотя описан ряд вариантов осуществления данного изобретения, очевидно, что эти основные примеры могут быть изменены для получения других вариантов, которые используют соединения и способы данного изобретения. Таким образом, должно быть понятно, что объем данного изобретения должен определяться прилагаемой формулой изобретения, а не конкретными вариантами осуществления, которые были представлены в качестве примеров. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы V или его фармацевтически приемлемые производное или соль,где R5 выбран из водорода или C1-4 алифатической группы;R7 представляет собой циклопропил. 2. Соединение по п.1, где R5 выбран из водорода, метила, этила, трет-бутила или изопропила. 3. Соединение по п.2, где R6 выбран из метила, этила или циклопропила. 4. Соединение, выбранное из следующих соединений табл. 1: 5. Соединение, выбранное из следующих соединений табл. 2: Таблица 2 6. Композиция, содержащая соединение по п.1 и фармацевтически приемлемый носитель, адъювант или разбавитель. 7. Композиция по п.6, дополнительно содержащая антипролиферативный агент или химиотерапевтический агент. 8. Способ ингибирования Aurora-1 в биологической пробе или у пациента, включающий контактирование указанной пробы с:(b) композиции по п.6. 9. Способ ингибирования Aurora-2 в биологической пробе или у пациента, предусматривающий контактирование указанной пробы с:(b) композиции по п.6. 10. Способ ингибирования Aurora-3 в биологической пробе или у пациента, предусматривающий- 28012869 контактирование указанной пробы с:(b) композиции по п.6. 11. Способ ингибирования FLT-3 в биологической пробе или у пациента, предусматривающий контактирование указанной пробы с:(b) композиции по п.6. 12. Способ ингибирования Aurora-1, Aurora-2, Aurora-3 и FLT-3 у пациента, включающий введение указанному пациенту:(b) композиции по п.6. 13. Способ лечения рака у пациента, включающий стадию введения указанному пациенту композиции по п.6. 14. Способ по п.13, включающий стадию введения указанному пациенту дополнительного химиотерапевтического или антипролиферативного агента. 15. Способ по п.13, где указанный рак выбран из меланомы, лимфомы, нейробластомы, лейкоза или рака, выбранного из рака ободочной кишки, молочной железы, легкого, почки, яичника, поджелудочной железы, почечного рака, рака ЦНС, рака шейки матки, рака предстательной железы или рака желудочнокишечного тракта, острого миелогенного лейкоза (AML), острого лимфоцитарного лейкоза (ALL), мастоцитоза или желудочно-кишечной стромальной опухоли (GIST). 16. Способ лечения или ослабления рака у пациента, включающий стадию нарушения митоза раковых клеток ингибированием протеинкиназы Aurora соединением по п.1. 17. Способ по п.16, предусматривающий стадию введения указанному пациенту композиции по п.6. 18. Соединение формулы V-1 или его фармацевтически приемлемая соль, выбранная из группы, состоящей из ацетата, адипата, альгината, аспартата, бензоата, бензолсульфоната, бисульфата, бутирата, цитрата, камфората, камфорсульфоната, циклопентанпропионата, диглюконата, додецилсульфата, этансульфоната, формиата, фумарата,глюкопентаноата, глицерофосфата, гликолата, гемисульфата, гептаноата, гексаноата, гидрохлорида, гидробромида, гидройодида, 2-гидроксиэтансульфоната, лактата, малеата, малоната, метансульфоната, 2 нафталинсульфоната, никотината, нитрата, оксалата, пальмоата, пектината, персульфата, 3 фенилпропионата, фосфата, пикрата, пивалата, пропионата, салицилата, сукцината, сульфата, тартрата,тиоцианата, тозилата, ундеканоата, натрия, калия, магния, аммония и N+(С 1-4 алкил)4. 19. Соединение по п.18, представляющее собой соль соединения формулы V-1 в виде сульфата. 20. Соединение по п.18, представляющее собой соль соединения формулы V-1 в виде лактата. 21. Применение соединения по п.18 в сочетании с Gleevec для производства лекарственного средства для лечения рака. 22. Применение соединения по п.18 в сочетании с Gleevec для производства лекарственного средства для лечения лейкемии. 23. Соединение формулы V или его фармацевтически приемлемая соль,где R5 представляет собой Н, метил, этил, н-пропил, i-пропил, циклопропил, изобутил или третбутил;R7 представляет собой метил, этил, циклопропил, трет-бутил,при условии, что соединение не является или его фармацевтически приемлемая соль,где R5 представляет собой метил;R7 представляет собой циклопропил. 25. Соединение по пп.23, 24 в форме соли, где соль выбрана из группы, состоящей или из ее фармацевтически приемлемой соли или производного или таутомера или стереоизомера фармацевтически приемлемой соли, выбранной из группы, состоящей из ацетата, адипата, альгината, аспартата, бензоата, бензолсульфоната, бисульфата, бутирата, цитрата, камфората, камфорсульфоната, циклопентанпропионата,диглюконата, додецилсульфата, этансульфоната, формиата, фумарата, глюкопентаноата, глицерофосфата, гликолата, гемисульфата, гептаноата, гексаноата, гидрохлорида, гидробромида, гидройодида, 2 гидроксиэтансульфоната, лактата, малеата, малоната, метансульфоната, 2-нафталинсульфоната, никотината, нитрата, оксалата, пальмоата, пектината, персульфата, 3-фенилпропионата, фосфата, пикрата, пивалата, пропионата, салицилата, сукцината, сульфата, тартрата, тиоцианата, тозилата, ундеканоата, натрия,калия, магния, аммония и N+(С 1-4 алкил)4. 26. Соединение по п.25, представляющее собой соль соединения в виде сульфата. 27. Соединение по п.25, представляющее собой соль соединения в виде лактата. 28. Композиция, содержащая соединение по пп.25-27 и фармацевтически приемлемый носитель,адъювант или разбавитель. 29. Композиция по пп.25-27, дополнительно содержащая антипролиферативный агент или химиотерапевтический агент. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2
МПК / Метки
МПК: A61K 31/506, C07D 403/14, C07D 401/14, C07D 403/12
Метки: качестве, пиримидинов, замещённые, применение, протеинкиназ, ингибиторов, композиции, замещенных, пиримидины, основе
Код ссылки
<a href="https://eas.patents.su/30-12869-zameshhyonnye-pirimidiny-kompozicii-na-ih-osnove-i-primenenie-zameshhennyh-pirimidinov-v-kachestve-ingibitorov-proteinkinaz.html" rel="bookmark" title="База патентов Евразийского Союза">Замещённые пиримидины, композиции на их основе и применение замещенных пиримидинов в качестве ингибиторов протеинкиназ</a>
Композиции на основе пирроло [2,3d] пиримидинов и их применение

Номер патента: 3604
Опубликовано: 26.06.2003
Авторы: Маккиббен Брайан, Уиттер Дэвид Дж., Кастелано Арлиндо Л.
МПК: A61P 9/00, A61K 31/505, C07D 487/04...
Метки: пиримидинов, 2,3d, основе, композиции, пирроло, применение
Формула / Реферат:
1. Соединение, имеющее формулу I где каждый из R1 и R2 независимо представляет собой атом водорода, замещенный прямой (C1-C30)алкил, замещенный разветвленный (C3-C30)алкил, замещенный (C4-C10)циклоалкил, замещенный циклопропил или замещенный или незамещенный арил; где только один из R1 и R2 может быть водородом; где, когда алкил представляет собой (C1)алкил или (C2)алкил, тогда любой заместитель фенила, если он имеется, замещен; или R1 и R2...
Применение бактерицидных пиримидинов для профилактики передачи вич половым путём и фармацевтические композиции на их основе

Номер патента: 11298
Опубликовано: 27.02.2009
Авторы: Стоффелс Пол, Де Бетюн Мари-Пьер Т.М.М.Дж., Ван Рое Енс Марсель
МПК: A61K 31/505, A61K 31/506, A61K 31/535...
Метки: пиримидинов, композиции, бактерицидных, вич, основе, фармацевтические, половым, профилактики, передачи, путём, применение
Формула / Реферат:
1. Применение 4-[[4-[(2,4,6-триметилфенил)амино]-2-пиримидинил]амино]бензонитрила или его фармацевтически приемлемой аддитивной соли для получения лекарственного микробицидного средства местного применения для профилактики (предотвращения) предачи или заражения ВИЧ, где указанные передача или заражение происходит через половую связь или связанный с ней интимный контакт между партнерами. 2. Применение по п.1, где передача или заражение происходит...
Замещённые бензазолы и их применение в качестве ингибиторов киназы raf

Номер патента: 7987
Опубликовано: 27.02.2007
Авторы: Ренхауэ Пол А., Рамуртхи Савитхри, Эмайри Пэйман, Фэнтл Уэнди, Пун Дэниел Дж., Левайн Берри Хаскелл, Сунг Леонард, Субраманиан Шарадха
МПК: C07D 401/12, C07D 401/14, A61K 31/41...
Метки: киназы, ингибиторов, бензазолы, качестве, применение, замещённые
Формула / Реферат:
1. Соединение формулы (I) где X1 и Х2 независимо выбраны из =N-, -NR4-, -О- или -S- при условии, что если X1 представляет собой -NR4-, -О- или -S-, тогда Х2 представляет собой =N-, или если Х2 представляет -NR4-, -О- или -S-, тогда X1 представляет собой =N-, и как X1, так и Х2 не являются =N-; Y представляет О или S; Y представляет О или S; А1 представляет замещенный или незамещенный С1-С12алкил, С3-С8циклоалкил, С3-С8гетероциклоалкил,...
Замещённые бензазолы и их применение в качестве ингибиторов raf-киназы

Номер патента: 11890
Опубликовано: 30.06.2009
Авторы: Ренхауэ Пол А., Рамуртхи Савинтхри, Пун Дэниел Дж., Левайн Бэрри Хэскелл, Фантл Уэнди, Субраманиан Шарадха, Амири Пэйман, Сунг Леонард
МПК: C07D 401/14, C07D 401/12, A61K 31/41...
Метки: бензазолы, ингибиторов, применение, raf-киназы, качестве, замещённые
Формула / Реферат:
1. Соединение формулы (I) где пунктирная линия представляет собой простую или двойную связь; X1 и Х2 независимо выбраны из =N-, -NR4-, -О- или -S- при условии, что если X1 представляет собой -NR4-, -О- или -S-, то Х2 представляет собой =N-, или если Х2 представляет собой -NR4-, -O- или -S-, то X1 представляет собой =N-, и оба X1 и Х2 не представляют собой =N-; Y представляет собой O; A1 представляет собой замещенный или незамещенный С3-С14арил;...
Замещённые хинолины и их применение в качестве микобактериальных ингибиторов

Номер патента: 11359
Опубликовано: 27.02.2009
Авторы: Гийемон Жером Эмиль Жорж, Паскье Элизабет Тереза Жанна
МПК: C07D 413/06, A61K 31/5355, A61P 31/10...
Метки: ингибиторов, применение, замещённые, хинолины, качестве, микобактериальных
Формула / Реферат:
1. Соединение формулы его фармацевтически приемлемые соли присоединения кислоты или основания, его четвертичные амины, его стерохимически изомерные формы, его таутомерные формы и его N-оксидные формы, в котором R1 представляет собой водород, галоген, Ar, Het, C1-6алкил, C1-6алкилокси; р представляет собой целое число, равное 1 или 2; R2 представляет собой C1-6алкилокси; R3 представляет собой Ar или Het; R4 представляет собой C1-6алкил; R5...
Предыдущий патент: Автобетононасос со складной мачтой
Следующий патент: Способ комплексной немедикаментозной коррекции функционального состояния и работоспособности человека на основе интегративной телесной технологии
Случайный патент: Растворение фильтрационной корки