Производные пиразола, фармацевтические композиции и их применение
Номер патента: 12431
Опубликовано: 30.10.2009
Авторы: Го Цзянь, Сюн Юйшэн, Лян Жуй, Броканьер Линда, Парми Эмма Р.







Формула / Реферат
1. Соединение, представленное формулой I

или его фармацевтически приемлемая соль, где
каждый R1 означает Н или выбран из группы, состоящей из:
(a) галогена или ОН,
(b) C1-6алкила или OC1-6алкила, необязательно замещенного 1-5 галогеногруппами вплоть до образования пергалогеналкильной группы;
каждый R2 выбран из R1, определенного выше, или две R2 группы, взятые вместе, могут образовывать конденсированную 5-6-членную циклическую структуру, содержащую 1-2 атома кислорода и 1-2 атома углерода, каждый из которых необязательно замещен 1-2 атомами F; и
R3 означает Н или С1-3алкил.
2. Соединение по п.1, где один R1 означает Н и другой означает Н или выбран из группы, состоящей из:
(а) галогена или ОН и
(b) C1-6алкила или OC1-6алкила, необязательно замещенного 1-5 галогеногруппами вплоть до образования пергалогеналкильной группы.
3. Соединение по п.2, где один R1 означает Н и другой означает Н или выбран из группы, состоящей из:
(а) галогена или OH и
(b) С1-4алкила или OC1-4алкила, каждый из которых необязательно замещен 1-3 галогеногруппами.
4. Соединение по п.1, где
каждый R2 означает Н или выбран из группы, состоящей из:
(а) галогена, выбранного из Cl и F,
(b) C1-6алкила или OC1-6алкила, необязательно замещенного 1-3 галогеногруппами,
или две R2 группы, взятые вместе, образуют конденсированную 5-6-членную циклическую структуру, содержащую 1-2 атома кислорода и 1-2 атома углерода, каждый из которых необязательно замещен 1-2 атомами F.
5. Соединение по п.1, где R3 означает Н или метил.
6. Соединение по п.1, где
один R1 означает Н и другой означает Н или выбран из группы, состоящей из:
(a) галогена или ОН,
(b) C1-6алкила или OC1-6алкила, необязательно замещенного 1-5 галогеногруппами вплоть до образования пергалогеналкильной группы;
каждый R2 означает Н или выбран из группы, состоящей из:
(а) галогена, выбранного из Cl и F,
(b) С1-6алкила или ОС1-6алкила, необязательно замещенного 1-3 галогеногруппами,
или две R2 группы, взятые вместе, образуют конденсированную 5-6-членную циклическую структуру, содержащую 1-2 атома кислорода и 1-2 атома углерода, каждый из которых необязательно замещен 1-2 атомами F; и
R3 означает Н или метил.
7. Соединение по п.1 или его фармацевтически приемлемая соль либо сольват, где
один R1 представляет собой Н и другой выбран из Cl, F, CF3 или OC1-3алкила;
R2 представляет собой галоген, CF3, OC1-3алкил или OCF3;
R3 означает Н или метил.
8. Соединение по п.1, выбранное из следующих таблиц:

Таблица 1


Таблица 2


Таблица 3



Таблица 4


Таблица 5


Таблица 6



или его фармацевтически приемлемая соль либо сольват.
9. Соединение по п.1, выбранное из следующей таблицы:

или его фармацевтически приемлемая соль либо сольват.
10. Соединение по п.1, представленное структурой

или его фармацевтически приемлемая соль либо сольват.
11. Соединение по п.1, представленное структурой

или его фармацевтически приемлемая соль либо сольват.
12. Соединение по п.1, представленное структурой

или его фармацевтически приемлемая соль либо сольват.
13. Соединение по п.1, представленное структурой

или его фармацевтически приемлемая соль либо сольват.
14. Соединение по п.1, представленное структурой

или его фармацевтически приемлемая соль либо сольват.
15. Фармацевтическая композиция, содержащая соединение по п.1 в комбинации с фармацевтически приемлемым носителем.
16. Применение соединения по п.1 для производства лекарственного средства для лечения сахарного диабета 2 типа у пациента-млекопитающего.
17. Применение соединения по п.1 для производства лекарственного средства для лечения гипергликемии, диабета или инсулиновой резистентности у пациента-млекопитающего.
18. Применение соединения по п.1 для производства лекарственного средства для лечения инсулиннезависимого сахарного диабета у пациента-млекопитающего.
19. Применение соединения по п.1 для производства лекарственного средства для лечения ожирения у пациента-млекопитающего.
20. Применение соединения по п.1 для производства лекарственного средства для лечения липидного расстройства, выбранного из группы, состоящей из дислипидемии, гиперлипидемии, гипертриглицеридемии, гиперхолестеринемии, низкого уровня HDL и высокого уровня LDL, у пациента-млекопитающего.
21. Применение соединения по п.1 для производства лекарственного средства для лечения атеросклероза у пациента-млекопитающего.
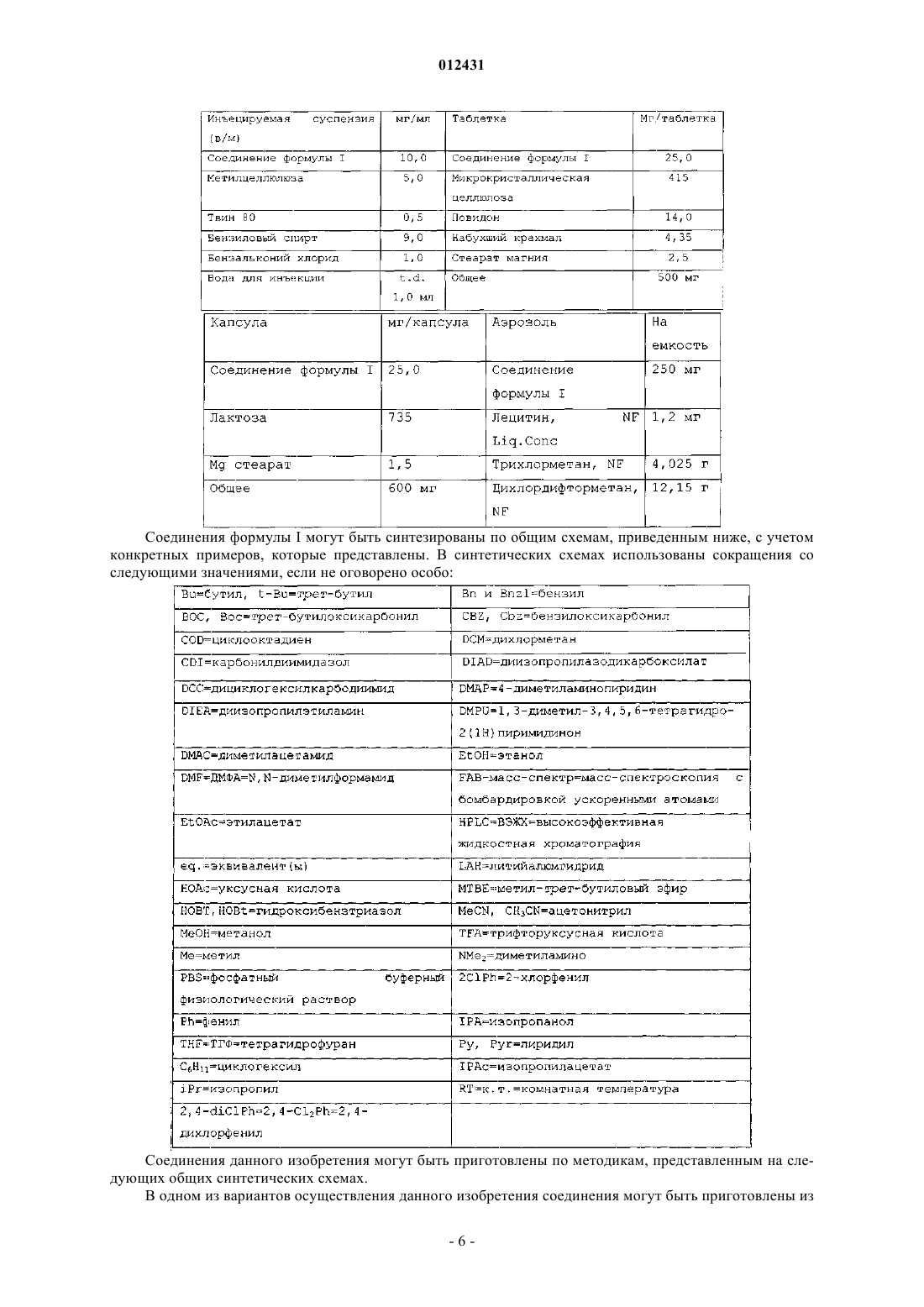
Текст
012431 Уровень техники Данное изобретение относится к производным пиразола, композициям, содержащим эти соединения, и различным способам лечения, связанным с сахарным диабетом 2 типа и родственными состояниями. Диабет относится к патологическому процессу, причинами которого являются многие факторы, и отличается повышенными уровнями глюкозы в плазме (гипергликемия) в состоянии натощак или после введения глюкозы в течение перорального теста на толерантность к глюкозе. Явно выраженный сахарный диабет (например, содержание глюкозы в крови 126 мг/дл натощак) связан с повышенной и преждевременной сердечно-сосудистой заболеваемостью и смертностью и относится прямо или косвенно к различным метаболическим состояниям, включающим изменения метаболизма липидов, липопротеинов и аполипопротеинов. У пациентов с инсулиннезависимым сахарным диабетом (сахарный диабет 2 типа), составляющих приблизительно 95% пациентов с сахарным диабетом, часто выявляют повышенные количества сывороточных липидов, таких как холестерин и триглицериды, и они имеют плохие показатели липидов крови с высокими уровнями холестерина липопротеидов низкой плотности (LDL-C) и холестерина липопротеидов высокой плотности (HDL-C). Указанные пациенты, страдающие от сахарного диабета 2 типа, находятся, таким образом, в группе повышенного риска по возникновению макрососудистых и микрососудистых осложнений, включающих коронарную болезнь сердца, удар, заболевание периферических сосудов,гипертензию (например, кровяное давление 130/80 мм Hg в состоянии покоя), нефропатию, невропатию и ретинопатию. Для пациентов с сахарным диабетом 2 типа характерны повышенные уровни инсулина в плазме по сравнению с пациентами-недиабетиками; у этих пациентов развивается резистентность к инсулиновой стимуляции метаболизма глюкозы и липидов в основных тканях, чувствительных к инсулину (мышцы,печень и жировые ткани). Таким образом, диабет 2 типа, по меньшей мере, в начале естественного развития болезни характеризуется, в основном, инсулиновой резистентностью, а не снижением образования инсулина, что приводит к недостаточному поглощению, окислению и сохранению глюкозы в мышцах,неадекватному подавлению липолиза в жировой ткани и избыточному образованию и выделению глюкозы печенью. Главный эффект сниженной чувствительности к инсулину выражается в высоких уровнях инсулина, циркулирующего в крови без соответствующего снижения глюкозы в плазме (гипергликемия). Гиперинсулинемия представляет собой фактор риска для возникновения гипертензии и может также вносить вклад в развитие сосудистых заболеваний. Глюкагон служит в качестве главного регуляторного гормона, ослабляющего эффект инсулина по ингибированию печеночного глюконеогенеза и обычно выделяется альфа-клетками в панкреатических островках в ответ на падение уровней глюкозы в крови. Данный гормон связывается со специфическими рецепторами в клетках печени, что вызывает гликогенолиз и повышение глюконеогенеза через события,опосредованные цАМР (сАМР, циклический аденозинмонофосфат). В результате этого генерируется глюкоза (например, образование печеночной глюкозы) для облегчения поддержания эугликемии путем предотвращения значительного падения уровня глюкозы в крови. В дополнение к увеличению количества циркулирующего инсулина диабетики 2 типа характеризуются повышенными количествами глюкагона в плазме и повышенными скоростями образования глюкозы в печени. Антагонисты глюкагона применяют для повышения инсулиновой чувствительности в печени, при этом снижается скорость глюконеогенеза и гликогенолиза и уменьшается скорость выделения глюкозы в печени, что приводит к снижению уровней глюкозы в плазме. В заявке WO 02/08188 от 31.01.2002 раскрыты N-замещенные индолы для лечения диабета 2 типа. Структура указанных соединений значительно отличается от соединений настоящего изобретения. В заявке WO 2004/050039 от 17.06.2004 раскрыты спироциклические производные мочевины, которые также используют для лечения диабета 2 типа. Структура указанных соединений значительно отличается от соединений настоящего изобретения. В заявке WO 2004/069158 от 19.08.2004, принадлежащей фирме Merck, описаны соединения, в состав общей формулы которых входят соединения настоящего изобретения. Однако в этом документе не раскрыты какие-либо конкретные соединения, входящие в объем настоящего изобретения. Таким образом, настоящее изобретение является селективным по отношению к раскрытому в заявкеWO 2004/069158. В заявке WO 2004/092146 от 28.10.2004 раскрыты фенилалкановые кислоты, являющиеся ингибиторами фермента РТР-1 В. Настоящая заявка имеет более ранний приоритет, чем дата публикации указанного документа. Хотя в этом документе раскрыт ряд пиразольных производных, они содержат заместители, отличающиеся по структуре от соединений настоящего изобретения. В заявке WO 2004/100875 от 25.11.2004 раскрыты замещенные бензимидазолы, которые используют для лечения диабета 2 типа. Структура указанных соединений значительно отличается от соединений настоящего изобретения.-1 012431 Сущность изобретения Данное изобретение относится к соединению, представленному формулой I или его фармацевтически приемлемой соли, где каждый R1 означает Н или выбран из группы, состоящей из:(a) галогена или ОН,(b) С 1-6 алкила или OC1-6 алкила, необязательно замещенного 1-5 галогеногруппами вплоть до образования пергалогеналкильной группы; каждый R2 выбран из R1, определенного выше, или 2 R2 группы, взятые вместе, могут образовывать конденсированную 5-6-членную циклическую структуру, содержащую 1-2 атома кислорода и 1-2 атома углерода, каждый из которых необязательно замещен 1-2 атомами F; иR3 означает Н или C1-3 алкил. Подробное описание изобретения Изобретение описано здесь в деталях с использованием терминов, определенных ниже, если не оговорено особо."Алкил", а также другие группы, имеющие префикс "алк", такие как алкокси, означает углеродные цепи, которые могут быть линейными, разветвленными или циклическими, или их комбинациями, содержащие указанное число атомов углерода. Если число не указано, то 1-10 атомов углерода предназначены для линейных или разветвленных алкильных групп. Примеры алкильных групп включают метил,этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил, октил, нонил и т.п. Циклоалкил представляет собой подкласс алкила; если не указано число атомов, то 3-10 атомов углерода предназначены для образования 1-3 карбоциклических колец, которые являются конденсированными. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, декагидронафтил и т.п."Галоген" (Halo) включает фтор, хлор, бром и йод. Когда R1 отличается от Н, то он может быть присоединен к нафтильной группе в любой доступной для присоединения точке. В самом широком аспекте данное изобретение относится к соединению, представленному формулой I или его фармацевтически приемлемой соли, где каждый R1 означает Н или выбран из группы, состоящей из:(a) галогена или ОН,(b) C1-6 алкила или OC1-6 алкила, необязательно замещенного 1-5 галогеногруппами вплоть до образования пергалогеналкильной группы; каждый R2 выбран из R1, определенного выше, или 2 R2 группы, взятые вместе, могут образовывать конденсированную 5-6-членную циклическую структуру, содержащую 1-2 атома кислорода и 1-2 атома углерода, каждый из которых необязательно замещен 1-2 атомами F; иR3 означает Н или C1-3 алкил. Другой аспект изобретения, который представляет интерес, относится к описанному выше соединению формулы I, в которой один R1 означает Н и другой означает Н или выбран из группы, состоящей из:(а) галогена или ОН, (b) C1-6 алкила или OC1-6 алкила, необязательно замещенного 1-5 галогеногруппами вплоть до образования пергалогеналкильной группы. Более конкретно, другой аспект изобретения, который представляет интерес, относится к описанному выше соединению формулы I, в которой один R1 означает Н и другой означает Н или выбран из группы, состоящей из: (а) галогена или ОН; и (b) C1-4 алкила или ОС 1-4 алкила, каждый из которых необязательно замещен 1-3 галогеногруппами. Другой аспект изобретения, который представляет интерес, относится к описанному выше соедине-2 012431 нию формулы I, в которой каждый R2 означает Н или выбран из группы, состоящей из: (а) галогена, выбранного из Cl и F, (b) C1-6 алкила или OC1-6 алкила, необязательно замещенного 1-3 галогеногруппами,или две R2 группы, взятые вместе, образуют конденсированную 5-6-членную циклическую структуру,содержащую 1-2 атома кислорода и 1-2 атома углерода, каждый из которых необязательно замещен 1-2 атомами F. Другой аспект изобретения, который представляет интерес, относится к описанному выше соединению формулы I, в которой каждый R3 означает Н или метил. Более конкретно, другой аспект изобретения, который представляет интерес, относится к описанному выше соединению формулы I, в которой один R1 означает Н и другой означает Н или выбран из группы, состоящей из:(a) галогена или ОН,(b) C1-6 алкила или OC1-6 алкила, необязательно замещенного 1-5 галогеногруппами вплоть до образования пергалогеналкильной группы; каждый R2 означает Н или выбран из группы, состоящей из: (а) галогена, выбранного из Cl и F, (b)C1-6 алкила или OC1-6 алкила, необязательно замещенного 1-3 галогеногруппами, или две R2 группы, взятые вместе, образуют конденсированную 5-6-членную циклическую структуру, содержащую 1-2 атома кислорода и 1-2 атома углерода, каждый из которых необязательно замещен 1-2 атомами F; иR3 означает Н или метил. Еще более конкретно другой аспект изобретения, который представляет интерес, относится к соединениям формулы I или их фармацевтически приемлемым солям, где один R1 представляет собой Н и другой выбран из Cl, F, CF3 или OC1-3 алкила; R2 представляет собой галоген, CF3, OC1-3 алкил или OCF3 иR3 означает Н или метил. Наиболее предпочтительные конкретные соединения по изобретению представлены в примерах,приведенных далее в описании, в частности табл. 1-6. Другой аспект изобретения, который представляет интерес, относится к фармацевтической композиции, содержащей соединение описанной выше формулы I в комбинации с фармацевтически приемлемым носителем. Другой аспект изобретения, который представляет интерес, относится к применению соединения описанной выше формулы I для производства лекарственного средства для лечения сахарного диабета 2 типа у пациента-млекопитающего. Другой аспект изобретения, который представляет интерес, относится к применению соединения описанной выше формулы I для производства лекарственного средства для лечения гипергликемии, диабета или инсулиновой резистентности у пациента-млекопитающего. Другой аспект изобретения, который представляет интерес, относится к применению соединения описанной выше формулы I для производства лекарственного средства для лечения инсулиннезависимого сахарного диабета у пациента-млекопитающего. Другой аспект изобретения, который представляет интерес, относится к применению соединения описанной выше формулы I для производства лекарственного средства для лечения ожирения у пациента-млекопитающего. Другой аспект изобретения, который представляет интерес, относится к применению соединения описанной выше формулы I для производства лекарственного средства для лечения липидного расстройства, выбранного из группы, состоящей из дислипидемии, гиперлипидемии, гипертриглицеридемии, гиперхолестеринемии, низкого уровня HDL и высокого уровня LDL, у пациента-млекопитающего. Еще один аспект изобретения, который представляет интерес, относится к применению соединения описанной выше формулы I для производства лекарственного средства для лечения атеросклероза у пациента-млекопитающего. Оптические изомеры - Диастереомеры - Геометрические изомеры - Таутомеры. Многие соединения формулы I содержат один или несколько асимметрических центров и, таким образом, существуют в виде рацематов и рацемических смесей, отдельных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. Данное изобретение включает все такие изомерные формы соединений в чистом виде и также в виде смесей. Некоторые из соединений, описанных в данной работе, содержат олефиновые двойные связи и, если не оговорено особо, предназначены для включения как Е, так и Z геометрических изомеров. Некоторые из соединений, описанных в данной работе, имеют различные точки присоединения водорода, относящиеся к таутомерам. Такой пример может представлять собой кетон или его энольную форму, известных как кето-энольные таутомеры. Индивидуальные таутомеры, а также их смеси охвачены соединениями формулы I. Соли и сольваты. Соли и сольваты соединений формулы I включены в данное изобретение. Термин "фармацевтически приемлемые соли" относится к солям, приготовленным из фармацевтически приемлемых в основном нетоксичных оснований и кислот, включающих неорганические или органические основания и неорганические или органические кислоты, а также соли, которые могут быть превращены в фармацевтически-3 012431 приемлемые соли. Соли, происходящие от неорганических оснований, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия, цинка и т.п. Особенно предпочтительны соли аммония,кальция, магния, калия и натрия. Соли, происходящие от фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включающих замещенные амины природного происхождения, циклические амины, и основные ионообменные смолы, такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин,2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, Nэтилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин,морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины, теобромин, триэтиламин,триметиламин, трипропиламин, трометамин (tromethamine) и т.п. В случае, когда соединение данного изобретения является основным, соли могут быть приготовлены из фармацевтически приемлемых нетоксичных кислот, включающих неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую,лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромисто-водородную, хлористо-водородную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую,слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, птолуолсульфоновую кислоту и т.п. Особенно предпочтительны лимонная, бромисто-водородная, хлористо-водородная, малеиновая,фосфорная, серная и винная кислоты. Сольваты, использованные в данной работе, относятся к соединению формулы I или его солям в сочетании с растворителем, таким как вода. Типичные примеры включают гидраты, полугидраты, тригидраты и т.п. При ссылках на соединения формулы I предполагается включение фармацевтически приемлемых солей и сольватов. Данное изобретение относится к способу противодействия или ингибирования образования или активности глюкагона посредством уменьшения скорости глюкогенеза и гликогенолиза и концентрации глюкозы в плазме. Соединения формулы I могут быть применены в производстве лекарственного средства для профилактического или терапевтического лечения болезненных состояний у млекопитающих, связанных с повышенными уровнями глюкозы, которое получают смешиванием соединения формулы I с материаламиносителями для воспроизведения лекарственного средства. Интервалы доз. Профилактическая или терапевтическая доза соединения формулы I будет, конечно, меняться в зависимости от природы и тяжести состояния, подлежащего лечению, выбранного конкретного соединения и способа его введения. Она будет также изменяться в соответствии с возрастом, массой и реакцией индивидуального пациента. В основном, суточный дозовый диапазон находится внутри интервала от примерно 0,001 до примерно 100, предпочтительно от примерно 0,01 до примерно 50 и более предпочтительно от примерно 0,1 до примерно 10 мг на кг массы тела, в однократных или разделенных дозах, повторяемых через определенные интервалы времени. В некоторых случаях может возникнуть потребность в применении дозировок свыше данных интервалов. Термины "эффективное количество, "противодиабетическое эффективное количество" и другие термины, появляющиеся в процессе применения, направляющего количество соединения, предназначенного для использования, относятся к предусмотренным дозовым интервалам, причем принимается в расчет любое необходимое изменение сверх указанных интервалов, устанавливаемое специалистом-медиком. Типичные дозировки соединений формулы I, а также их фармацевтически приемлемых солей и сольватов для взрослых особей находятся в интервале от примерно 0,1 мг до примерно 1,0 г в сутки,предпочтительно от примерно 1 до примерно 500 мг, в однократных или разделенных дозах, повторяемых через определенные интервалы времени. Типичные дозировки соединений, применяемых в комбинации с соединениями формулы I, являются известными, или их определение находится в компетенции специалиста в данной области с учетом описания, представленного в данной работе. При использовании внутривенного или перорального введения типичный интервал доз находится от примерно 0,001 до примерно 100 мг (предпочтительно от 0,01 до примерно 10 мг) соединения формулы I на кг массы тела в сутки и более предпочтительно от примерно 0,1 до примерно 10 мг соединения формулы I на кг массы тела в сутки. При применении в комбинации с другими средствами вышеуказанные дозировки для глюкагонового антагониста обеспечиваются вместе с обычной дозой для другого лекарственного средства. Например,когда включен DPP-IV ингибитор, такой как ингибиторы, раскрытые в патенте США 6699871 В 1,DPP-IV ингибитор может быть применен в количестве, изменяющемся от примерно 1,0 до примерно 1000 мг, предпочтительно от примерно 2,5 до примерно 250 мг и в особенности от примерно 50 до примерно 100 мг, введенных в однократных суточных дозах или в разделенных дозах, повторяемых через определенные интервалы времени, как целесообразно. Аналогично, при применении глюкагонового ан-4 012431 тагониста в комбинации с СВ 1 антагонистом/обратным агонистом, СВ 1 антагонист/обратный агонист может быть применен в количестве, изменяющемся от только примерно 0,1 вплоть до примерно 1000 мг,более конкретно в количестве, изменяющемся от примерно 1,0 до примерно 100 мг, и еще более конкретно в количестве от примерно 1,0 до примерно 10 мг, введенных в однократных суточных дозах или в разделенных дозах, повторяемых через определенные интервалы времени, как целесообразно. Примеры доз СВ 1 антагониста/обратного агониста включают 1, 2, 3, 4, 5, 6, 7, 8, 9 и 10 мг. Фармацевтические композиции. Как отмечено выше, фармацевтическая композиция содержит соединение формулы I или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель. Термин "композиция" относится к продукту, содержащему активный и инертный ингредиент(ы) (фармацевтически приемлемые наполнители), которые составляют наполнитель, а также к любому продукту, который получается, прямо или косвенно, путем комбинации, комплексообразования или агрегации любых двух или нескольких ингредиентов, или путем диссоциации одного или нескольких ингредиентов, или в результате других типов реакций или взаимодействий между ингредиентами. Предпочтительно композицию составляют из соединения формулы I в количестве, которое является эффективным для лечения, предупреждения или замедления начала сахарного диабета 2 типа, в комбинации с фармацевтически приемлемым носителем. Любой подходящий способ введения может быть использован для обеспечения млекопитающего,особенно человека, эффективной дозировкой соединения данного изобретения. Например, может быть использован пероральный, ректальный, локальный, парентеральный, глазной, легочный, назальный и т.п. способ. Примеры лекарственных форм включают таблетки, пастилки, дисперсии, суспензии, растворы,капсулы, кремы, мази, аэрозоли и т.п., при этом предпочтительны таблетки. Для приготовления пероральных композиций могут быть использованы любые обычные фармацевтические среды, такие как, например, вода, гликоли, масла, спирты, корригенты, консерванты, красители и т.п., в случае пероральных жидкостей, например суспензий, эликсиров и растворов; или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулообразующие средства,замасливатели, связующие вещества, диспергаторы и т.п., в случае пероральных твердых препаратов,например порошков, капсул и таблеток. Предпочтительными являются твердые пероральные препараты. Из-за легкости их введения таблетки и капсулы представляют собой наиболее выгодные пероральные стандартные лекарственные формы. Если требуется, то таблетки могут быть с покрытием, осуществляемым по стандартным водным или неводным методикам. В дополнение к обычным лекарственным формам, представленным выше, соединения формулы I могут быть введены способами контролируемого высвобождения действующего вещества и/или высвобождающими приспособлениями, такими как описаны в патентах США 3845770; 3916899; 3536809; 3598123; 3630200 и 4008719. Фармацевтические композиции данного изобретения, подходящие для перорального введения, могут быть представлены как отдельные дозы, такие как капсулы, саше или таблетки, причем каждая содержит предварительно фиксированное количество активного ингредиента, как порошок или гранулы или как раствор или суспензия в водной жидкости, неводной жидкости, как эмульсия масло-в-воде или эмульсия вода-в-масляной жидкости. Такие композиции могут быть приготовлены любым приемлемым фармацевтическим способом. Все такие способы включают стадию смешивания активного ингредиента(ов) с компонентами носителя. В основном, композиции приготавливают равномерным и тщательным смешиванием активного ингредиента(ов) с жидким или тонко измельченным твердым компонентом носителя и затем, если необходимо, превращением смеси в требуемую форму продукта. Например, таблетки могут быть приготовлены прессованием или формованием. Прессованные таблетки могут быть приготовлены сжатием свободнотекущего порошка или гранул, содержащих активный ингредиент(ы), необязательно смешанный с одним или несколькими наполнителями, например связующими веществами,замасливателями, разбавителями, поверхностно-активными веществами и дисперсантами. Формованная таблетка может быть сделана формованием смеси порошкового соединения, увлажненного инертной жидкостью. Желательно, если каждая таблетка может содержать, например, от примерно 0,1 мг до примерно 1,0 г активного ингредиента, и каждая капсула или саше содержит от примерно 0,1 до примерно 500 мг активного ингредиента. Далее следуют примеры фармацевтических лекарственных форм, содержащих соединения формулы I. Соединения формулы I могут быть синтезированы по общим схемам, приведенным ниже, с учетом конкретных примеров, которые представлены. В синтетических схемах использованы сокращения со следующими значениями, если не оговорено особо: Соединения данного изобретения могут быть приготовлены по методикам, представленным на следующих общих синтетических схемах. В одном из вариантов осуществления данного изобретения соединения могут быть приготовлены из где R2 и R3 принимают значения, определенные выше, и R означает алкильную группу. Соединения II, в свою очередь, могут быть приготовлены конденсацией -кетоэфира 1 и бензилгидразина 2. Соединения, такие как 1, являются коммерчески доступными, известными в литературе или могут быть удобно приготовлены многообразными способами, хорошо знакомыми специалистам в данной области. Один путь проиллюстрирован на схеме 1 и описан в публикации Clay et al., Synthesis, 1993,290. Хлорангидрид кислоты 3, который может быть коммерчески доступным или легко приготовленным из соответствующей карбоновой кислоты обработкой тионилхлоридом при повышенных температурах или оксалилхлоридом в растворителе, таком как метиленхлорид, в присутствии каталитического количества диметилформамида (ДМФА) при комнатной температуре, обрабатывают этилмалонатом калия и хлоридом магния в присутствии основания, такого как триэтиламин, в апротонном растворителе, таком как этилацетат, в течение 1-16 ч, что приводит к кетоэфиру 1. Схема 1 Бензилгидразин 2 может быть приготовлен из соответствующего карбонильного аналога конденсацией с трет-бутилкарбазатом в присутствии уксусной кислоты в неполярном растворителе, таком как толуол, при повышенной температуре в течение от 16 до 24 ч, схема 2. Промежуточный продукт 4 затем восстанавливают гидридным восстанавливающим агентом, таким как цианоборогидрид натрия, и 1 экв. п-толуолсульфоновой кислоты, которые добавляют по каплям. В альтернативном случае уксусная кислота может быть использована как сорастворитель вместо толуолсульфоновой кислоты. Реакцию проводят в полярном апротонном растворителе, таком как тетрагидрофуран (ТГФ) в течение 16-48 ч при температуре окружающей среды. После водной обработки борановый комплекс можно разложить медленным добавлением водного раствора гидроксида натрия или другого сильного основания с получением карбамата 5 (см. Calabretta et al., Synthesis, 1991, 536). Снятие защитной ВОС группы осуществляют обработкой кислотой, такой как трихлоруксусная кислота, в метиленхлориде при температуре окружающей среды в течение 0,25-2 ч. Реакция может быть проведена с или без добавления триизопропилсилана. Гидразин 2 может быть применен либо в виде его трифторацетатной соли прямо после удаления защиты, либо в виде свободного основания, которое может быть приготовлено, и продукта, выделенного в виде гидрохлорида добавлением водной хлористо-водородной кислоты и выпариванием растворителя. В случае (R3 не означает Н), когда промежуточный продукт 5 содержит хиральный центр, энантиомеры в данной точке могут быть разделены хроматографией с применением гомохиральной стационарной фазы. В альтернативном случае гидразон 4 может быть непосредственно восстановлен водородом и хиральным катализатором, таким как родий DuPHOS комплекс, как описано в публикации Burk et al., Tetrahedron, 1994, 50,4399. Растворитель, примененный для реакции, обычно был спиртом, таким как 2-пропанол, и использовался водород при повышенном давлении. Данная реакция должна дать продукт обогащенной энантиоселективности, который может быть далее очищен хиральной хроматографией, описанной выше. Схема 2-7 012431 Конденсацию -кетоэфира 1 и бензилгидразина 2, описанную на схеме 3, проводят нагреванием двух компонентов в растворителе, таком как уксусная кислота или ацетонитрил в течение 1-8 ч, получая пиразолон 6. Переход на данной стадии к эфиру -аланина 7 может быть достигнут омылением эфира 6,применяя основание, такое как водный гидроксид лития или натрия в полярном растворителе, таком как тетрагидрофуран, диоксан, метанол, этанол или смесь подобных растворителей. Конденсацию аланинового эфира 8 затем осуществляют применением 1-этил-3-(3-диметиламинопропил)карбодиимида(EDC) и 1-гидроксибензотриазола (HOBt) или бензотриазол-1-илокситриспирролидинофосфоний гексафторфосфата (РуВОР) и основания, обычно диизопропилэтиламина, в растворителе, таком как N,Nдиметилформамид (ДМФА) или метиленхлорид, в течение периода от 3 до 48 ч при температуре окружающей среды, получая соединение 7. Пиразолон 7 затем обрабатывают ангидридом трифторметансульфоновой кислоты в полярном апротонном растворителе, таком как ТГФ, в присутствии основания,такого как триэтиламин, при температуре от -78 С до комнатной температуры, получая промежуточный продукт II. Данный продукт очищают от нежелательных побочных продуктов перекристаллизацией, растиранием, препаративной тонкослойной хроматографией, флэш-хроматографией на силикагеле, как описано в публикации W.С. Still et al., J. Org. Chem., 43, 2923 (1978), или ВЭЖХ. Очистку промежуточных продуктов выполняют таким же образом. Если промежуточный продукт II является рацемическим (т.е.R3 не означает водород), то данное соединение может быть разделено хиральной ВЭЖХ с применением либо нормальной фазы, либо надкритических жидких условий. Схема 3 Конечные продукты I могут быть приготовлены взаимодействием промежуточного продукта II с соответствующей нафтилбороновой кислотой 9. Данные соединения являются коммерчески доступными или могут быть приготовлены из коммерческих продуктов. Один такой способ проиллюстрирован на схеме 4, для которой трициклический промежуточный продукт 10 приготовлен по публикации Schlosseret al., Eur. J. Org. Chem., 2001, 3991. Затем он может быть подвергнут ароматизации обработкой иодидом натрия в апротонном растворителе, таком как ацетонитрил, с последующим добавлением триметилсилилхлорида. Реакционную смесь перемешивают при температуре окружающей среды в течение периода от 1 до 5 ч для получения бромида 11. Данное соединение затем может быть превращено в бороновую кислоту обработкой бис(пинаколато)дибором, ацетатом калия и палладиевым катализатором, таким как хлорид палладия II, и лигандом, таким как дифенилфосфиноферроцен (dppf). Реакционную смесь нагревают в полярном апротонном растворителе, таком как ДМСО, в течение 1-5 ч с последующим расщеплением боронатного эфира обработкой разбавленной кислотой, такой как хлористо-водородная кислота, в растворителе, таком как ацетон, в течение длительного времени. Альтернативный путь к бороновой кислоте включает обработку нафтилгалогенида 11 сильным основанием, таким как бутиллитий, в полярном апротонном растворителе, таком как ТГФ, при низких температурах с последующим добавлением триалкилбората, такого как триметилборат. Реакционную смесь перемешивают затем 1-5 ч с нагреванием до температуры окружающей среды, затем гасят разбавленной кислотой, такой как разбавленная хлористоводородная кислота, перед выделением промежуточного продукта 9. Арилтрифлат II может быть введен во взаимодействие с бороновой кислотой 9 с применением палладиевого катализатора, такого как палладий 2-(ди-трет-бутилфосфино)бифенил или трифенилфосфин. Обычно растворитель представляет собой либо диметоксиэтан (DME), этанол, либо толуол; триэтиламин, карбонат цезия или натрия или фторид натрия также добавляют к реакции, которая может также включать воду и осуществляется при повышенных температурах и может быть проведена в микроволновом реакторе (см. Wang et al., Tet. Lett., 2000, 41, 4713 для родственных реакций перекрестного сочетания). Удаление эфира, когда R означает Me или Et, выполняют омылением с использованием основания,такого как водный гидроксид лития или натрия, в полярном растворителе, таком как тетрагидрофуран,метанол, этанол или смесь подобных растворителей. Когда R означает остаток трет-бутилового эфира,его наилучшим образом удаляют обработкой трифторуксусной кислотой в метиленхлориде в течение 0,5-3 ч при температуре окружающей среды. Продукт очищают от нежелательных побочных продуктов перекристаллизацией,растиранием,препаративной тонкослойной хроматографией,флэшхроматографией на силикагеле, как описано в публикации W.С. Still et al., J. Org. Chem., 43, 2923 (1978),или ВЭЖХ. Очистку промежуточных продуктов выполняют таким же образом. В некоторых случаях продукт реакций, описанных на схеме 4, будет далее модифицирован. Эти превращения могут включать,но без ограничения только ими, реакции замещения, восстановления, окисления, алкилирования, ацилирования и гидролиза, которые обычно известны специалистам в данной области. Альтернативный путь к соединениям (I) включает приготовление промежуточного продукта III где R1 и R3 принимают значения, определенные выше, и R означает алкильную группу. Соединения формулы III, в свою очередь, могут быть приготовлены конденсацией -кетоэфира 12 и гидразина. Соединения, такие как 12, могут быть удобно приготовлены разнообразными способами, хорошо знакомыми специалистам в данной области. Один из способов проиллюстрирован на схеме 5. Хлорангидрид кислоты 13, который может быть коммерчески доступным или легко приготовленным из соответствующей карбоновой кислоты обработкой тионилхлоридом при повышенных температурах или оксалилхлоридом в растворителе, таком как метиленхлорид, в присутствии каталитического количества диметилформамида (ДМФА) при комнатной температуре, обрабатывают этилмалонатом калия и хлоридом магния в присутствии основания, такого как триэтиламин, в апротонном растворителе, таком как этилацетат, в течение 1-16 ч, что приводит к кетоэфиру 12. Конденсацию -кетоэфира 12 осуществляют нагреванием двух компонентов в растворителе, таком как уксусная кислота или ацетонитрил, в течение 1-8 ч, получая пиразолон 13-1. Пиразолон 13-1 затем обрабатывают ангидридом трифторуксусной кислоты в полярном апротонном растворителе, таком как ТГФ, в присутствии основания, такого как триэтиламин, при температуре от -78 С до комнатной температуры, получая трифлат 14. Данное соединение затем алкилируют бензиловым спиртом 15, который приготавливают из карбонильного производного 16, омылением эфира, применяя основание, такое как водный гидроксид лития или натрия в полярном растворителе, таком как тетрагидрофуран, диоксан, метанол, этанол или смесь подобных растворителей. Конденсацию -аланинового производного 8 затем осуществляют применением 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC) и 1-гидроксибензотриазола (HOBt) или бензотриазол-1-илокситриспирролидинофосфоний гексафторфосфата (PyBOP) и основания, обычно диизопропилэтиламина, в растворителе, таком как N,N-диметилформамид (ДМФА) или метиленхлорид, в течение периода от 3 до 48 ч при температуре окружающей среды. Восстановление фрагмента кетона до спирта 15 осуществляют применением гидридного восстанавливающего агента, такого как борогидрид натрия, в полярном апротонном растворителе, таком как метанол. Схема 6 Спирт 15 конденсируется с трифлатом 14 с образованием промежуточного продукта III обработкой конденсирующим реагентом, таким как диизопропилазодикарбоксилат (DIAD), и триалкилфосфином,таким как трифенилфосфин, в неполярном апротонном растворителе, таком как метиленхлорид в течение 0,5-6 ч при температуре окружающей среды. В некоторых случаях образуются смеси региоизомеров и они могут быть разделены, так как соединение очищают от нежелательных побочных продуктов перекристаллизацией, растиранием, препаративной тонкослойной хроматографией, флэш-хроматографией на силикагеле, как описано в публикации W.С. Still et al., J. Org. Chem., 43, 2923 (1978), или ВЭЖХ. Очистку промежуточных продуктов выполняют таким же образом. Конечные продукты I затем могут быть приготовлены взаимодействием промежуточного продукта III с соответствующей арилбороновой кислотой 17. В некоторых случаях данные соединения являются коммерчески доступными, в других они могут быть приготовлены из коммерческих продуктов любыми специалистами в данной области, см. выше. Конденсацию осуществляют применением палладиевого катализатора, такого как палладий 2-(ди-третбутилфосфино)бифенил или трифенилфосфин. Обычно растворитель представляет собой либо диметоксиэтан, этанол, либо толуол, и триэтиламин, карбонат цезия или натрия или фторид натрия также добавляют к реакции, которая может также включать воду и осуществляться при повышенных температурах и может быть проведена в микроволновом реакторе. Удаление эфира, когда R=Me или Et, выполняют омылением с использованием основания, такого как водный гидроксид лития или натрия, в полярном растворителе, таком как тетрагидрофуран, диоксан, метанол, этанол или смесь подобных растворителей. Когда R означает остаток трет-бутилового эфира, его наилучшим образом удаляют обработкой трифторуксусной кислотой в метиленхлориде в течение 0,5-3 ч при температуре окружающей среды. Продукт очищают от нежелательных побочных продуктов перекристаллизацией, растиранием, препаративной тонкослойной хроматографией, флэш-хроматографией на силикагеле, как описано в публикации W.С.Still et al., J. Org. Chem., 43, 2923 (1978), или ВЭЖХ. Очистку промежуточных продуктов выполняют та- 10012431 ким же образом. Если продукт является рацемическим (т.е. R3 не означает водород), то затем данное соединение может быть разделено хиральной ВЭЖХ (HPLC) с применением либо нормальной фазы, либо надкритических жидких условий. В некоторых случаях продукт реакций, описанных на схеме 6, будет далее модифицирован. Эти превращения могут включать, но без ограничения только ими, реакции замещения, восстановления, окисления, алкилирования, ацилирования и гидролиза, которые обычно известны специалистам в данной области. В альтернативном случае модификация пиразолона 6 может быть проведена в другом порядке. Пиразолон 6 затем обрабатывают ангидридом трифторметансульфоновой кислоты (Tf2O) в полярном апротонном растворителе, таком как ТГФ, в присутствии основания, такого как триэтиламин, при температуре от -78 С до комнатной температуры, получая промежуточный продукт 18. Катализируемая палладием конденсация с соответствующей нафтилбороновой кислотой 9 может быть проведена на этой стадии применением способа, аналогичного способу, описанному выше. Конечная обработка может быть осуществлена омылением эфира 19 с использованием основания, такого как водный гидроксид лития или натрия, в полярном растворителе, таком как тетрагидрофуран, диоксан, метанол, этанол или смесь подобных растворителей. Конденсацию -аланинового производного 8 затем осуществляют применением 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC) и 1-гидроксибензотриазола (HOBt) или бензотриазол-1-илокситриспирролидинофосфоний гексафторфосфата (РуВОР) и основания, обычно диизопропилэтиламина, в растворителе, таком как N,N-диметилформамид (ДМФА) или метиленхлорид, в течение периода от 3 до 48 ч при температуре окружающей среды, получая сложный эфир конечного продукта I. Удаление эфира, когда R=Me или Et, осуществляют омылением с использованием основания, такого как водный гидроксид лития или натрия, в полярном растворителе, таком как тетрагидрофуран, диоксан,метанол, этанол или смесь подобных растворителей. Когда R означает остаток трет-бутилового эфира,его наилучшим образом удаляют обработкой трифторуксусной кислотой в метиленхлориде в течение 0,5-3 ч при температуре окружающей среды. Продукт очищают от нежелательных побочных продуктов перекристаллизацией,растиранием,препаративной тонкослойной хроматографией,флэшхроматографией на силикагеле, как описано в публикации W.С. Still et al., J. Org. Chem., 43, 2923 (1978),или ВЭЖХ. Очистку промежуточных продуктов выполняют таким же образом. Если соединение I является рацемическим (т.е. R3 не означает водород), то затем данное соединение может быть разделено хиральной ВЭЖХ с применением либо нормальной фазы, либо надкритических жидких условий. Схема 7 В некоторых случаях продукт I или предшествующий сложный эфир от реакций, описанных выше на схемах, будет далее модифицирован. Эти превращения могут включать, но без ограничения только ими, реакции замещения, восстановления, окисления, алкилирования, ацилирования и гидролиза, которые обычно известны специалистам в данной области. Одна такая модификация, показанная здесь, когда одна R2 группа представляет собой защищенный фенол, как в соединении 20 (R означает не водород),включает высвобождение спирта и последующую этерификацию, схема 8. Гидроксильная группа может быть защищена в виде силильного простого эфира, и в этом случае для реакции применяют фторидный источник, в основном фтористо-водородную кислоту или фторид тетрабутиламмония. Удаление метоксиэфирной защиты обычно осуществляют обычно обработкой соединения трибромидом бора в растворителе, таком как метиленхлорид, в течение периода 1-16 ч при температуре окружающей среды. Наконец, если спирт защищен как аллильный эфир, то защитную группу удаляют обработкой диметилбарбитуровой кислотой и палладиевым катализатором, обычно это трис(дибензилиденацетон)дипалладий(0), с лигандом, таким как 1,4-бис(дифенилфосфино)бутан, в апротонном растворителе, таком как метиленхлорид, в течение периода от 15 мин до 2 ч. См. издание "Protective Groups in Organic Synthesis", Greene,опубликованное Wiley and Sons. Свободная гидроксильная группа затем может быть модифицирована путем приготовления простых эфиров использованием спирта и конденсирующего агента, такого как диизопропилазодикарбоксилат, и трифенилфосфина в неполярном растворителе, таком как метиленхлорид, при температурах от 0 до 40 С в течение периода от 1 до 16 ч, схема 8. Промежуточный продукт 21 затем может быть превращен в целевые продукты, как описано ранее, см. выше. Альтернативный подход к соединениям (I) включает алкилирование пиразола IV где R1 и R2 принимают значения, определенные выше. Соединения IV известны в литературе или могут быть удобно приготовлены разнообразными способами, хорошо знакомыми специалистам в данной области, как описанные в публикации Katritsky et al.,Advances in Heterocyclic Chemistry, Vol. 6, p. 347-429. Один из способов проиллюстрирован на схеме 9. Эфир 22, который может быть коммерчески доступным или легко приготовленным из соответствующей карбоновой кислоты этерификацией с применением, например, метанола или этанола, содержащего кислоту, такую как серная кислота, конденсируется с анионом метилкетона 23, образуя дикетон 24. Реакцию проводят с применением основания, такого как гидрид натрия, в полярном апротонном растворителе, таком как тетрагидрофуран (ТГФ) при температуре от 0 до 25 С в течение периода от 16 до 24 ч, см.March, Advances Organic Chemistry, Vol. 3rd Ed., p. 439 и ссылки там. Соединения, такие как 23, являются коммерчески доступными или могут быть приготовлены разнообразными способами, хорошо знакомыми специалистам в данной области. Дикетон 24 затем конденсируется с гидразином в полярном растворителе, таком как метанол, который может содержать кислоту, такую как уксусная или хлористо-водородная,в течение периода от 16 до 24 ч при температуре от 0 до 25 С. Схема 9 Альтернативный путь к промежуточному продукту IV включает конденсацию алкинилкетона 25 с гидразином, как показано на схеме 2 и описано в публикации Cabarrocas et al., Tetrahedron Asymmetry,Vol. 11, p. 2483-2493, 2000 и приведенных там ссылках. Данную процедуру обычно проводят в полярном растворителе, таком как ДМФА, при температурах 0-25 С в течение 16-24 ч. Приготовление промежуточных продуктов 25 включает взаимодействие алкина 26 с амидом Вайнреба (Weinreb) карбоновой кислоты с соответствующей функциональностью при использовании затрудненного основания, такого как литийдиизопропиламид или бутиллитий, в полярном апротонном растворителе, таком как ТГФ, при-78 С. Данная реакция описана в деталях в публикации Tetrahedron Lett., Vol. 22, p. 3815, 1981. Алкины 26 являются либо коммерчески доступными, либо получены из соответствующего галогенида и алкинилмагнийиодида, см. Negishi et al., J. Org. Chem., Vol. 62, p. 8957-8960, 1997 и Org. Lett., Vol. 3, p. 31113113, 2001. Промежуточный продукт IV может быть затем превращен в соединения I, как показано на схеме 11. Алкилирование пиразола IV 4-карбоалкоксибензилбромидом может быть проведено после депротонирования пиразола основанием, таким как гидрид натрия или карбонат цезия, в полярном растворителе,обычно диметилформамиде (ДМФА), при температуре от 0 до 25 С в течение периода от 3 до 24 ч. В альтернативном случае алкилирование может быть осуществлено применением спирта 15, как описано на схеме 6, см. выше. В некоторых случаях будут образованы смеси изомеров. Их обычно можно разделить перекристаллизацией, растиранием, препаративной тонкослойной хроматографией, флэшхроматографией на силикагеле, как описано в публикации W.С. Still et al., J. Org. Chem., 43, 2923 (1978),или ВЭЖХ. Соединения, очищенные ВЭЖХ, могут быть выделены как соответствующие соли. Превращение в конечные соединения затем проводят, как описано ранее для эфира 19. В некоторых случаях продукт реакций, описанных на схеме 3, будет далее модифицирован. Эти превращения могут включать,но без ограничения только ими, реакции замещения, восстановления, окисления, алкилирования, ацилирования и гидролиза, которые обычно известны специалистам в данной области. Схема 11 Альтернативный способ, показывающий энантиоселективный путь к соединению I, раскрыт на схемах 12 и 13. Схема 12 Соединение 4a, полученное как показано на схеме 2, см. выше, восстанавливают родиевым катализатором, обычно Rh(COD)2BF4, в присутствии лиганда, такого как показан ниже, в изопропаноле, метаноле или этилацетате, получая 5 а.Ph2-F-C-P-tBu2 представляет собой катализатор Жозифоса (Josiphos), который раскрыт в патенте США 6777567 В 2 (Solvias) и коммерчески доступен от Strem. Xyl-P-Phos раскрыт в патенте США 5886182 (Synetix) и коммерчески доступен от Strem. Me-f-Ketalphos также коммерчески доступен от Как показано выше на схеме 13, коммерчески доступные соединения 27 и 28 конденсируются. Сначала соединение 28 смешивают с ТГФ-раствором трет-бутоксида калия при пониженной температуре,такой как примерно от -20 до примерно -5 С, для образования енолята (не показан). Эфир 27 добавляют с подогревом до примерно 20 С, получая дикетон 1 а. Дикетон 1a смешивают с соединением 2a в подходящем растворителе. Примеры включают EtOH,ТГФ, НОАс, ДМФА, IPA, ДМСО, DMAc, DMPU, MeCN, толуол и IPAc. Безводный LiCl добавляют для получения по региоселективному способу требуемого промежуточного продукта 19 в виде этилового эфира. Превращение данного этилового эфира в пиразолзамещенную кислоту 29 выполняют при гидролитических условиях, например, в смеси ТГФ и MeOH с NaOH при комнатной температуре. Продукт 29 в виде кислоты затем может быть выделен такими способами, как кристаллизация. Установлением рН до нейтрального значения могут быть осаждены и удалены непрореагировавший продукт и побочные продукты. Подходящие растворители для кристаллизации и смеси растворителей включают комбинации MTBE/гептан и MeOH/вода. Соединение 29 затем подвергают взаимодействию с -аланинэтиловым эфиром, HCl солью, через образование хлорангидрида кислоты (не показан), который может быть приготовлен использованием оксалил или тионилхлорида с последующим удалением HCl отгонкой. В альтернативном случае, как показано на схемах, амидирование может быть осуществлено использованием CDI как активирующего агента в подходящем растворителе, например ТГФ, при к.т. с последующим добавлением -аланина в форме этилового эфира, HCl соли, при 50 С. Для гидролиза этилового эфира добавляют основание, например NaOH, в растворителе, таком как MeOH, при к.т. Подкисление с помощью HCl дает продукт, который может быть проэкстрагирован с помощью iPAc и выделен последующей кристаллизацией из смеси ацетонитрил/H2O. Общий эксперимент: препаративную ВЭЖХ выполняли на YMC-Pack Pro C18 колонке (15020 мм внутренний диаметр (i.d. с элюированием при 20 мл/мин 0-100% ацетонитрилом в воде (0,5% TFA). Следующие примеры приведены, чтобы изобретение могло быть представлено более полно. Их не следует воспринимать как ограничение данного изобретения в любом отношении. Получение промежуточных продуктов описано ниже, они использованы в синтезах примеров 1-149. Промежуточный продукт А.CH3CN (40 мл) с последующим добавлением TMSCl (1,35 мл, 10,7 ммоль). Реакционную смесь перемешивали в течение 2,5 ч, гасили 5% Na2SO3 и экстрагировали эфиром. Эфирный раствор промывали 5%Na2SO3, насыщенным солевым раствором и сушили над Na2SO4. Неочищенный продукт хроматографировали (SiO2, гексаны), получая 2-бром-6-(трифторметокси)нафталин в виде белых кристаллов. ЯМР (500 МГц, CDCl3) : 7,36 (дд, J=2,6, 9,0 Гц, 1 Н); 7,60 (дд, J=2,0, 8,8 Гц, 1 Н); 7,63 (ушир.с, 1 Н);- 14012431 7,69 (д, J=8,8 Гц, 1 Н); 7,77 (д, J=9,0 Гц, 1.4); 8,01 (д, J=2,0 Гц, 1 Н). Стадия В. [6-(Трифторметокси)-2-нафтил]бороновая кислота. 2-Бром-6-(трифторметокси)нафталин (428 мг, 1,47 ммоль), бис(пинаколато)дибор (410 мг,1,62 ммоль) и KOAc (433 мг, 4,41 ммоль) суспендировали в ДМСО (12 мл). Смесь дезоксигенировали в вакууме периодическими поступлениями N2 с последующим добавлением катализатора PdCl2(dppf)(30 мг, 2,5 мол.%). Реакционную смесь нагревали в атмосфере N2 до 80 С в течение 2 ч. Реакционную смесь разбавляли гексаном (100 мл), промывали водой, насыщенным солевым раствором и сушили надNa2SO4. После выпаривания растворителя полученный остаток обрабатывали ацетоном (20 мл) и 2 Н HCl(5 мл) в течение 24 ч. Сырую бороновую кислоту очищали ВЭЖХ с обращенной фазой, получая [6(трифторметокси)-2-нафтил]бороновую кислоту в виде белого порошка. ЯМР (500 МГц, CDCl3) : 7,44 (дд, J=2,3, 9,0 Гц, 1 Н); 7,74 (ушир.с, 1 Н); 7,98 (д, J=8,2 Гц, 1 Н); 8,11 Стадия А. 2-Бром-7-(трифторметокси)нафталин. Данное соединение получали, используя условия для 2-бром-6-(трифторметокси)нафталина, описанные выше. ЯМР (500 МГц, CDCl3) : 7,35 (дд, J=2,4, 8,9 Гц, 1 Н); 7,58 (ушир.с, 1 Н); 7,59 (дд, J=2,0, 8,8 ц, 1 Н); 7,73 (д, J=8,8 Гц, 1 Н); 7,84 (д, J=9,0 Гц, 1 Н); 8,00 (д, J=2,0 Гц, 1 Н). Стадия В. [7-(Трифторметокси)-2-нафтил]бороновая кислота. Данное соединение получали, используя условия для [6-(трифторметокси)-2-нафтил]бороновой кислоты, описанные выше. ЯМР (500 МГц, CDCl3) : 7,74 (дд, J=2,2, 8,9 Гц, 1 Н); 7,93 (ушир.с, 1 Н); 7,97 (д, J=8,9 Гц, 1 Н); 8,02 Стадия А. 2-Бром-5-(трифторметокси)нафталин. Данное соединение получали, используя условия для 2-бром-6-(трифторметокси)нафталина, описанные выше. ЯМР (500 МГц, CDCl3) : 7,39 (пд, J=1,6, 7,8 Гц, 1 Н); 7,48 (т, J=8 Гц, 1H); 7,66 (дд, J=1,9, 9,0 Гц,1 Н); 7,69 (д, J=8,3 Гц, 1 Н); 8,01 (д, J=9,0 Гц, 1 Н); 8,05 (д, J=1,9 Гц, 1 Н). Стадия В. [5-(Трифторметокси)-2-нафтил]бороновая кислота. Данное соединение получали, используя условия для [6-(трифторметокси)-2-нафтил]бороновой кислоты, описанные выше. ЯМР (500 МГц, CDCl3) : 7,51 (дд, J=1,4, 7,6 Гц, 1 Н); 7,55 (т, J=8 Гц, 1 Н); 8,02 (д, J=8,0 Гц, 1 Н); 8,29 Стадия А. 2-Бром-8-(трифторметокси)нафталин. Данное соединение получали, используя условия для 2-бром-6-(трифторметокси)нафталина, описанные выше. ЯМР (500 МГц, CDCl3) : 7,41 (дд, J=1,9, 7,7 Гц, 1 Н); 7,47 (т, J=8 Гц, 1 Н); 7,64 (дд, J=2,0, 8,8 Гц,1 Н); 7,75 (д, J=8,7 Гц, 2 Н); 8,29 (д, J=1,9 Гц, 1 Н). Стадия В. [8-(Трифторметокси)-2-нафтил]бороновая кислота. Данное соединение получали, используя условия для [6-(трифторметокси)-2-нафтил]бороновой кислоты, описанные выше. ЯМР (500 МГц, CDCl3) : 7,48 (д, J=7,6 Гц, 1 Н); 7,59 (т, J=7,9 Гц, 1 Н); 7,88 (д, J=8,2 Гц, 1 Н); 8,05 (д,J=8,2 Гц, 1 Н); 8,40 (дд, J=1,2, 8,2 Гц, 1 Н); 8,18 (с, 1 Н). Промежуточный продукт Е.(2,2,4,4-Тетрафтор-4 Н-1,3-бензодиоксин-6-ил)бороновая кислота. Данное соединение получали, используя условия для [6-(трифторметокси)-2-нафтил]бороновой ки- 15012431 слоты, описанные выше. ЯМР (500 МГц, CDCl3) : 7,32 (д, J=8,3 Гц, 1 Н); 8,43 (д, J=8,3 Гц, 1 Н); 8,44 (с, 1 Н). Промежуточный продукт F.(2,2,3,3-Тетрафтор-2,3-дигидро-1,4-бензодиоксин-6-ил)бороновая кислота. Данное соединение получали, используя условия для [6-(трифторметокси)-2-нафтил]бороновой кислоты, описанные выше. ЯМР (500 МГц, CDCl3) : 7,30 (д, J=8,2 Гц, 1 Н); 7,96 (д, J=1,4 Гц, 1 Н); 8,01 (дд, J=1,4, 8,2 Гц, 1 Н). Промежуточный продукт G.[3-Фтор-4-(трифторметокси)фенил]бороновая кислота. Раствор 4-бром-2-фтор-1-(трифторметокси)бензола (1,0 мг, 3,9 ммоль) в ТГФ (5 мл) медленно добавляли к н-BuLi (3,0 мл, 1,6 М в гексане) в ТГФ (5 мл) при -78 С. Через 20 мин добавляли триметилборат (1,4 мл, 12 ммоль), смесь перемешивали при -78 С в течение 2 ч. Охлаждающую баню удаляли, реакционной смеси позволяли нагреваться до комнатной температуры (1-2 ч). Реакционную смесь затем гасили 2 Н HCl (10 мл) и перемешивали в продолжение ночи. Растворитель удаляли при пониженном давлении и остаток растворяли в смеси CH3CN-H2O-диоксан. ВЭЖХ с обращенной фазой, после лиофилизации, давала [3-фтор-4-(трифторметокси)фенил]бороновую кислоту в виде мелкого порошка. ЯМР (500 МГц, CDCl3) : 7,47 (м, 1 Н); 7,99 (м, 2 Н). Промежуточный продукт Н.[5-Хлор-2-(трифторметокси)фенил]бороновая кислота. Раствор H-BuLi (17 мл, 1,6 М в гексане) добавляли впрыскивающим насосом в течение одного часа к ТГФ (50 мл) раствору 1-хлор-4-(трифторметокси)бензола (5,0 г, 25,5 ммоль) и диизопропиламина(0,42 мл, 3 ммоль) при -78 С. Через 20 мин добавляли триметилборат (8 мл, 70 ммоль), смесь перемешивали при -78 С в течение 2 ч. Охлаждающую баню удаляли, реакционной смеси позволяли нагреваться до комнатной температуры (1-2 ч). Реакционную смесь затем гасили 2 Н HCl (40 мл) и перемешивали в продолжение ночи. Растворитель удаляли при пониженном давлении и остаток растворяли в смеси CH3CN-H2O-диоксан. ВЭЖХ с обращенной фазой, после лиофилизации, давала [5-хлор-2-(трифторметокси)фенил]бороновую кислоту в виде белого порошка. ЯМР (500 МГц, CDCl3) : 7,32 (д, J=8,8 Гц, 1 Н); 7,60 (дд, J=2,7, 8,8 Гц, 1 Н); 8,20 (д, J=2,7 Гц, 1 Н). Промежуточный продукт I.[3-Хлор-4-(трифторметокси)фенил]бороновая кислота. Раствор NaNO2 (2,4 г, 33 ммоль) в воде (6 мл) медленно добавляли к суспензии [3-хлор-4(трифторметокси)фенил]амина (2,95 г, 13,9 ммоль) в 20 мл 15% HCl при 0 С. Твердый продукт удаляли фильтрованием и раствор NaBF4 (2,4 г, 22 ммоль) в воде (15 мл) смешивали с фильтратом. Твердое вещество собирали фильтрованием, сушили при 40 С, получая 2,62 г диазониевой соли. ЖХ-МС (LC-MS): одинарный пик с правильным MC (223,6). Полученное выше твердое вещество затем смешивали с бис(пинаколато)дибором (2,14 г,8,4 ммоль), PdCl2 (dppf) (180 мг, 2,5%) в колбе, дезоксигенированной в вакууме периодическими поступлениями N2, с последующим добавлением MeOH (N2 продувка). Смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель выпаривали и остаток хроматографировали (SiO2, градиент 010% этилацетат в гексане), получая боратный эфир в виде масла. ЯМР (500 МГц, CDCl3) : 1,34 (с, 12 Н); 7,31 (кд, J=1,5, 8,2 Гц, 1 Н); 7,71 (дд, J=1,5, 8,2 Гц, 1 Н); 7,90(д, J=1,5 Гц, 1 Н). Боратный эфир гидролизовали в ацетоне-HCl, как описано для [6-(трифторметокси)-2 нафтил]бороновой кислоты, получая [3-хлор-4-(трифторметокси)фенил]бороновую кислоту в виде мелкого порошка. ЯМР (500 МГц, CDCl3) : 7,48 (кд, J=1,6, 8,1 Гц, 1 Н); 8,13 (дд, J=1,6, 8,1 Гц, 1 Н); 8,25 (д, J=1,6 Гц,1 Н). Стадия А. 6-(Трифторметил)-1,4-дигидро-1,4-эпоксинафталин. К 25 мл тетрагидрофурана при -78 С добавляли н-бутиллитий (13,9 мл, 22,2 ммоль), затем диизопропиламин (3,1 мл, 22,2 ммоль). Полученную смесь перемешивали при -78 С в течение 10 мин, затем медленно добавляли фуран (24 мл, 330 ммоль). К реакционной смеси добавляли 4-бромбензотрифторид(5 г, 22,2 ммоль) в виде раствора в 10 мл тетрагидрофурана, удаляли холодную баню, и смеси позволяли нагреваться до комнатной температуры на протяжении 2,5 ч. Добавляли воду, смесь выливали в гексаны,и органический слой промывали последовательно двумя порциями 1 Н HCl и одной порцией насыщенного солевого раствора. Органический слой сушили над сульфатом магния, концентрировали в вакууме и маслянистый остаток очищали флэш-колоночной хроматографией (SiO2, 5% этилацетат/гексаны), получая указанное в заголовке соединение. 1H ЯМР (500 МГц, CDCl3) : 7,51 (с, 1 Н); 7,35 (м, 2 Н); 7,10 (м, 2 Н); 5,81 (ушир.с, 2 Н). ВЭЖХ/МС: m/z=213,00 (М+1). Стадия В. 2-Бром-6-(трифторметил)-1,4-дигидро-1,4-эпоксинафталин и 2-бром-7-(трифторметил)1,4-дигидро-1,4-эпоксинафталин. 6-(Трифторметил)-1,4-дигидро-1,4-эпоксинафталин (380 мг, 1,79 ммоль) и карбонат натрия (200 мг,1,8 9 ммоль) смешивали в 11 мл четыреххлористого углерода и нагревали до 70 С. К смеси добавляли по каплям бром (288 мг, 1,80 ммоль) в виде раствора в 3 мл четыреххлористого углерода и полученную смесь нагревали при 80 С в течение 10 мин. Бледно-желтый раствор охлаждали, фильтровали через рыхлый слой сульфата натрия и концентрировали в вакууме. Полученный маслянистый остаток суспендировали в 4 мл тетрагидрофурана и добавляли к суспензии трет-бутоксида калия (638 мг, 5,4 ммоль) в 5 мл тетрагидрофурана при 50 С. После нагревания при 50 С в течение 24 ч смесь охлаждали, выливали в гексаны и промывали последовательно двумя порциями воды и одной порцией насыщенного солевого раствора. Органический слой сушили над сульфатом магния, концентрировали в вакууме и очищали препаративной ТСХ (TLC) (SiO2, 5% этилацетат/гексаны), получая указанные в заголовке соединения. 2-Бром-6-(трифторметил)-1,4-дигидро-1,4-эпоксинафталин (1H ЯМР (500 МГц, CDCl3) : 7,51 (м,2 Н); 7,40 (д, J=7,3 Гц); 7,02 (д, J=2 Гц, 1 Н); 5,84 (ушир.с, 1 Н); 5,55 (с, 1 Н и 2-бром-7-(трифторметил)1,4-дигидро-1,4-эпоксинафталин (полученные в виде 2:1 смеси с промежуточным продуктом реакции(1R,2R,3S,4S)-2,3-бибром-6-(трифторметил)-1,2,3,4-тетрагидро-1,4-эпоксинафталином). Данную смесь разделяли на следующей стадии. 1 Н ЯМР (500 МГц, CDCl3) : 7,65 (м, 2,5 Н); 7,61 (д, J=8,0 Гц, 0,5 Н); 7,52 (д, J=7,8 Гц, 0,5 Н); 7,40 (м,2 Н); 7,00 (д, J=2,1 Гц, 1 Н); 5,83 (ушир.с, 1 Н); 5,61 (с, 0,5 Н); 5,55 (с, 1H); 4,27 (м, 0,5 Н). Стадия С. 2-Бром-6-(трифторметил)нафталин. 2-Бром-6-(трифторметил)-1,4-дигидро-1,4-эпоксинафталин (624 мг, 2,14 ммоль) и иодид натрия(980 мг, 6,54 ммоль) растворяли в 13 мл сухого ацетонитрила и добавляли триметилсилилхлорид(0,823 мл, 6,54 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 3,5 ч, выливали в гексаны и органический слой промывали последовательно двумя порциями воды и одной порцией насыщенного солевого раствора. Органический слой сушили над сульфатом магния, концентрировали в вакууме и остаток очищали флэш-колоночной хроматографией (SiO2, 5% этилацетат/гексаны), получая 2-бром-6-(трифторметил)нафталин в виде белого твердого вещества. 1 Н ЯМР (500 МГц, CDCl3) : 8,15 (с, 1 Н); 8,11 (с, 1 Н); 7,89 (д, J=8,7 Гц, 1 Н); 7,83 (д, J=8,7 Гц, 1 Н); 7,70 (дд, J=1,6, 8,7 Гц, 1 Н); 7,69 (дд, J=1,8, 8,7 Гц, 1 Н). Стадия D. [6-(Трифторметил)-2-нафтил]бороновая кислота. 2-Бром-6-(трифторметил)нафталин (50 мг, 0,182 ммоль), бис(пинаколато)дибор (92 мг, 0,362 ммоль) и ацетат калия (53 мг, 0,540 ммоль) суспендировали в 2,5 мл метилсульфоксида. Смесь дезоксигенировали четырьмя порциями азота в вакууме и добавляли дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладий (II) дихлорметановый аддукт (3,7 мг, 0,0045 ммоль) и полученную смесь нагревали при 80 С в атмосфере азота в течение 1 ч. Смесь охлаждали, разбавляли этилацетатом и промывали последовательно двумя порциями воды и одной порцией насыщенного солевого раствора. Органический слой сушили над сульфатом магния, концентрировали в вакууме и остаток суспендировали в смеси 10 мл ацетона и 2 мл водной 2 Н хлористо-водородной кислоты. Полученную смесь нагревали при 60 С в течение 16 ч и сырую бороновую кислоту очищали ВЭЖХ с обращенной фазой, получая [6-(трифторметил)-2 нафтил]бороновую кислоту в виде белого порошка. 1 Н ЯМР (500 МГц, DMSO) : 8,47 (с, 1 Н); 8,38 (с, 1 Н); 8,33 (ушир.с, 2 Н); 8,14 (д, J=8,7 Гц, 1 Н); 8,07 Стадия А. 2-Бром-7-(трифторметил)нафталин. Данное соединение получали, следуя способу 2,6-изомера, описанному выше для промежуточного продукта J. 1 Н ЯМР (500 МГц, CDCl3) : 8,11 (д, J=1,6 Гц, 1 Н); 8,07 (с, 1 Н); 7,94 (д, J=8,7 Гц, 1 Н); 7,79 (д, J=9,0 Гц, 1 Н); 7,70 (дд, J=1,8, 8,9 Гц, 1H); 7,68 (дд, J=1,9, 8,9 Гц, 1 Н). Стадия В. [7-(Трифторметил)-2-нафтил]бороновая кислота. Данное соединение получали, следуя способу 2,6-изомера, описанному выше для промежуточного продукта J. 1 Н ЯМР (500 МГц, CD3OD) : 8,29 (м, 2 Н); 8,05 (д, J=8,7 Гц, 1 Н); 8,00-7,88 (м, 2 Н); 7,69 (д, J=8,5 Гц,1 Н). Промежуточный продукт L. Стадия А. 2-Бром-6-хлорнафталин. 6-Бром-2-нафтойную кислоту (4,00 г, 15,9 ммоль) обрабатывали 40 мл тионилхлорида при 80 С в течение 1 ч. Смесь концентрировали в вакууме и образовавшийся неочищенный хлорангидрид кислоты(3 г, 11,1 ммоль) смешивали с 2,2'-азобисизобутиронитрилом (731 мг, 4,45 ммоль) в 25 мл четыреххлористого углерода и 15 мл хлорбензола. Данную смесь медленно добавляли с помощью капельной воронки к смеси натриевой соли 2-меркаптопиридин-1-оксида (1,99 г, 13,7 ммоль) и 4-(диметиламино)пиридина(150 мг, 1,23 ммоль) при 100 С. После окончания добавления смесь перемешивали в течение дополнительных 4 ч, охлаждали и твердый осадок побочного продукта удаляли фильтрованием. Фильтрат концентрировали в вакууме и остаток очищали флэш-колоночной хроматографией (SiO2, гексаны), получая указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (500 МГц, CDCl3) : 8,02 (ушир.с, 1 Н); 7,83 (д, J=1,6 Гц, 1 Н); 7,72 (д, J=8,7 Гц, 1 Н); 7,66 (д,J=8,7 Гц, 1 Н); 7,60 (дд, J=2,0, 8,9 Гц, 1 Н); 7,47 (дд, J=2,1, 8,7 Гц, 1 Н). Стадия В. 2-(6-Хлор-2-нафтил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан. 2-Бром-6-хлорнафталин (205 мг, 0,849 ммоль), бис(пинаколато)дибор (432 мг, 1,70 ммоль) и ацетат калия (250 мг, 2,55 ммоль) растворяли в 12 мл метилсульфоксида. Смесь дезоксигенировали четырьмя порциями азота в вакууме и добавляли дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладий(II) дихлорметановый аддукт (70 мг, 0,085 ммоль). Полученную смесь нагревали при 80 С в атмосфере азота в течение 3 ч, затем позволяли ей находиться при температуре окружающей среды в течение 16 ч. Смесь разбавляли этилацетатом, промывали последовательно двумя порциями воды и одной порцией насыщенного солевого раствора. Органический слой сушили над сульфатом магния, концентрировали в вакууме и остаток очищали флэш-колоночной хроматографией, получая указанное в заголовке соединение. 1H ЯМР (500 МГц, DMSO) : 8,34 (с, 1 Н); 3,10 (м, 2 Н); 7,89 (д, J=8,3 Гц, 1 Н); 7,76 (д, J=8,3 Гц, 1 Н); 7,54 (дд, J=1,8, 8,7 Гц, 1 Н); 1,33 (ушир.с, 12 Н). Стадия С. (6-Хлор-2-нафтил)бороновая кислота. 2-(6-Хлор-2-нафтил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (340 мг, 1,18 ммоль) суспендировали в смеси 20 мл ацетона и 5 мл водной 2 Н хлористо-водородной кислоты и нагревали при 50 С в течение 16 ч. Продукт очищали ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение в виде белого порошка. 1 Стадия А. трет-Бутил 2-1-[4-(этоксикарбонил)фенил]этилиденгидразинкарбоксилат. Раствор трет-бутилкарбазата (13,90 г, 105 ммоль) и этил 4-ацетилбензоата (20,00 г, 0,104 ммоль) в толуоле (120 мл) перемешивали при 80 С на протяжении ночи (15 ч). трет-Бутил 2-1-[4(этоксикарбонил)фенил]этилиденгидразинкарбоксилат выделялся в виде кристаллического твердого вещества, его собирали фильтрованием смеси. ВЭЖХ/МС: m/z=307,3 (M+1)+, Rt=3,47 мин. 1 Н ЯМР (500 МГц, CDCl3) : 8,05 (2 Н, д, J=8,5 Гц), 7,88 (2 Н, д, J=8,5 Гц), 7,79 (1 Н, ушир.с), 4,41(2 Н, кв, J=7,0 Гц), 2,24 (3 Н, с), 1,58 (9 Н, с), 1,43 (3 Н, т, J=7,0 Гц). Стадия В. трет-Бутил-2-1-[4-(этоксикарбонил)фенил]этилгидразинкарбоксилат. В наполненной N2 круглодонной колбе, снабженной сывороточными колпачками и магнитной мешалкой, растворяли NaBH3CN (6,0 г, 0,095 моль) и трет-бутил-2-1-[4-(этоксикарбонил)фенил]этилиденгидразинкарбоксилат (25,6 г, 0,084 моль) в ТГФ (200 мл). Раствор моногидрата п-толуолсульфоновой кислоты (17,3 г, 0,091 моль) в ЕРА (50 мл) медленно добавляли впрыскивающим насосом. На завершение добавления требовалось примерно 10 ч. Смесь разбавляли с помощью EtOAc (200 мл) и суспензию экстрагировали насыщенным солевым раствором (150 мл). Органическую фазу отделяли, сушили (Na2SO4) и концентрировали на роторном испарителе, получая белое твердое вещество. Данное твердое вещество брали в CH2Cl2 (100 мл) и к раствору добавляли 1 Н NaOH (100 мл). Суспензию интенсивно перемешивали при к.т. в течение 1 ч и затем разбавляли CH2Cl2 (100 мл). Органическую фазу отделяли и экстрагировали 1 Н HCl (2150 мл), насыщенным солевым раствором (2150 мл), сушили(Na2SO4) и концентрировали приблизительно до 50 мл. Продукт осаждался в виде белого твердого вещества, который собирали фильтрованием и промыванием гексаном, получая трет-бутил-2-1-[4(этоксикарбонил)фенил]этилгидразинкарбоксилат. ВЭЖХ/МС: m/z=331, 3 (M+Na)+, Rt=3,24 мин. 1 Н ЯМР (500 МГц, CDCl3) : 8,03 (2 Н, д, J=8,0 Гц), 7,44 (2 Н, д, J=8,0 Гц), 5,99 (1 Н, ушир.с), 4,40(2 Н, кв, J=7,0 Гц), 4,29 (1 Н, м), 1,45 (9 Н, с), 1,41 (3 Н, т, J=7,0 Гц), 1,35 (3 Н, д, J=6,5 Гц). Стадия С. 1-[4-(Этоксикарбонил)фенил]этилгидразиний хлорид. трет-Бутил-2-1-[4-(этоксикарбонил)фенил]этилгидразинкарбоксилат (29 г, 94 ммоль) обрабатывали 100 мл TFA-DCM-триизопропилсилана (20:20:1) при комнатной температуре в течение одного часа. Смесь концентрировали при пониженном давлении, и остаток растворяли в воде (100 мл), промывали 2 раза с помощью DCM. DCM-слой снова экстрагировали 3 раза водой. К объединенному водному раствору добавляли HCl (5 Н, 20 мл) и смесь концентрировали до 50 мл. Добавляли CH3CN (50 мл) и после лиофилизации получали 22,7 г 1-[4-(этоксикарбонил)фенил]этилгидразиний хлорида. ЯМР (500 МГц, ацетон-d6) : 1,34 (т, J=7,1 Гц, 3 Н); 1,67 (д, J=6,8 Гц, 3 Н); 4,33 (кв, J=7,1 Гц, 2 Н),4,97 (кв, J=6,8 Гц, 1 Н); 7,76 (д, J=8,5 Гц, 2 Н), 7,97 (д, J=8,5 Гц, 2 Н). Стадия D. (1S)-1-[4-(Этоксикарбонил)фенил]этилгидразиний трифторацетат и (1R)-1-[4(этоксикарбонил)фенил]этилгидразиний трифторацетат. трет-Бутил-2-1-[4-(этоксикарбонил)фенил]этилгидразинкарбоксилат анализировали хиральной ВЭЖХ, применяя два типа условий. 1) Daicel колонка Chiralcel 0J, 40 С, 0,75 мл/мин, 10% EtOH/90% н-гептан: t1 6,66 мин; t2 12,25 мин. Энантиомеры разделяли на препаративной шкале, используя данную колонку (30% EtOH/70% н-гептан). 2) Daicel колонка ChiralPak AD, 40C, 0,75 мл/мин, 10% EtOH/90% н-гептан: t1 12,17 мин;t2 15,49 мин. Энантиомеры разделяли на препаративной шкале, используя данную колонку (20%EtOH/80% н-гептан). Быстро двигающийся энантиомер был одинаковым в каждом случае и, как затем было установлено, является (S)-энантиомером []D20=-120 (c1.1 MeOH), см. ниже. Также выделяли более медленный (R)-энантиомер []D20=+122 (c1.1 MeOH). Любой энантиомер может быть лишен защиты с помощью 45:45:10 TFA:DCM:TIPS (40C, 1,5 ч). Избыток реагента и растворитель выпаривали и остаток растворяли в воде. Водный раствор промывали 2 раза с помощью DCM. DCM слои снова экстрагировали дополнительным количеством воды. Объединенный водный раствор упаривали при пониженном давлении (темп. 45 С) с последующим обезвоживанием перегонкой азеотропной смеси с толуолом, получая для (S)-изомера - (1S)-1-[4(этоксикарбонил)фенил]этилгидразиний трифторацетат в виде вязкого масла. ЯМР (500 МГц, CD3OD) : 1,38 (т, J=7,1 Гц, 3 Н); 1,49 (ушир.д, J=7,0 Гц, 3 Н); 4,26 (ушир.кв, J=7,0 Гц, 1 Н); 4,37 (кв, J=7,1 Гц, 2 Н); 7,54 (д, J=8,2 Гц, 2 Н); 8,07 (д, J=8,2 Гц, 2 Н).(1R)-1-[4-(Этоксикарбонил)фенил]этилгидразиний трифторацетат может быть приготовлен таким же образом. Определение абсолютной конфигурации энантиомерных гидразинов. Абсолютную конфигурацию энантиомеров трет-бутил-2-1-[4-(этоксикарбонил)фенил]этилгидразинкарбоксилата устанавливали превращением до этил 4-[1-(2-бензоилгидразино)этил]бензоата с последующим сравнением направления оптического вращения с опубликованными данными [Burk et al., Tetrahedron, 1994, 50, 4399 - (S)-1-п-карбоэтоксифенил-1-(2-бензоилгидразино)этан (95% ее; []D20=-200,0TFA/CH2Cl2 (1:1, 10 мл) в течение 1 ч при к.т. Реакционную смесь концентрировали на роторном испарителе и остаточную TFA удаляли соиспарением от толуола. Образующийся этил 4-[(1 гидразино)этил]бензоат затем растворяли в CH2Cl2 (15 мл) и охлаждали до -78 С. Медленно добавляли раствор бензоилхлорида (365 мкл, 3,15 ммоль) и 2,6-ди-трет-бутил-4-метилпиридина (745 мг, 3,63 ммоль) в CH2Cl2 (5 мл) при -78 С. После выдерживания при -78 С в течение 3 ч реакционную смесь быстро помещали в колонку с SiO2 и элюировали смесью 30% EtOAc/гексан. Фракции, содержащие продукт, концентрировали и затем очищали с помощью ВЭЖХ с использованием колонки Kromasil С 8 (от 10 до 70% CH3CN/H2O/0,1% TFA, 12 мин) и снова на колонке с силикагелем (30% EtOAc/гексан), получая(S)-(-)-этил 4-[1-(2-бензоилгидразино)этил]бензоат готовили аналогично из более подвижного изомера трет-бутил 2-1-[4-(этоксикарбонил)фенил]этилгидразинкарбоксилата. ВЭЖХ/МС: m/z=313,4 (M+l)+, Rt=3,09 мин. Колонка Daicel Chiralcel OJ, 40 С, 0,5 мл/мин, 10% изопропанол/90% н-гептан: t=34,99 мин; Стадия А. трет-Бутил (2 Е)-2-[4-(метоксикарбонил)бензилиден]гидразинкарбоксилат. Указанное в заголовке соединение получали, следуя химии, описанной для промежуточного продукта М, вышеприведенная стадия А. ЯМР (500 МГц, CDCl3) : 1,55 (с, 9 Н); 3,92 (с, 3 Н); 7,74 (д, J=3,5 Гц, 2 Н), 7,88 (ушир.с, 1 Н); 7,96(ушир.с, 1 Н); 8,04 (д, J=8,5 Гц, 2 Н). Стадия В. трет-Бутил 2-[4-(метоксикарбонил)бензил]гидразинкарбоксилат. Указанное в заголовке соединение получали, следуя химии, описанной для промежуточного продукта M, вышеприведенная стадия В. ЯМР (500 МГц, CDCl3) : 1,46 (с, 9 Н); 3,91 (с, 3 Н); 4,06 (с, 2H); 6,03 (ушир.с, 1 Н); 7,42 (кв, J=8,3 Гц,2 Н); 8,00 (д, J=8,3 Гц, 2 Н). Стадия C. [4-(Метоксикарбонил)бензил]гидразиний хлорид. Указанное в заголовке соединение получали, следуя химии, описанной для промежуточного продукта М, вышеприведенная стадия С. ЯМР (500 МГц, CD3OD) : 3,91 (с, 3 Н); 4,19 (с, 2 Н); 7,54 (д, J=8,3 Гц, 2 Н); 8,05 (д, J=8,3 Гц, 2 Н).- 20012431 4 ч. Растворитель удаляли при пониженном давлении и остаток обрабатывали этилацетатом, промывали 2 порциями нас. NaHCO3, насыщенным солевым раствором и сушили над Na2SO4. Флэш-колоночная хроматография (SiO2, градиент 0-5% этилацетат в DCM) приводила к этил 4-1-[3-(3,5-дихлорфенил)-5 оксо-4,5-дигидро-1 Н-пиразол-1-ил]этилбензоату в виде белого твердого вещества. ТСХ (5% этилацетатDCM) Rf 0,43. ЯМР (500 МГц, CDCl3) : 1,38 (т, J=7,1 Гц, 3 Н); 1,78 (д, J=7,0 Гц, 3 Н); 3,55 (д, J=22,6 Гц, 1 Н); 3,60N-(4-1-[3-(3,5-дихлорфенил)-5-оксо-4,5-дигидро-1 Н-пиразол-1 ил]этилбензоил)аланинат. Этил 4-1-[3-(3,5-дихлорфенил)-5-оксо-4,5-дигидро-1 Н-пиразол-1-ил]этилбензоат (2,23 г, 5,50 ммоль) растворяли в смеси MeOH-диоксан (1:1, 50 мл). Добавляли раствор NaOH (0,7 г/15 мл). Смесь нагревали до 60 С в течение 1 ч. Смесь подкисляли 2 Н HCl (10 мл), растворитель удаляли и остаток сушили в вакууме, получая бледно-желтое твердое вещество (смесь кислоты продукта и NaCl). Твердое вещество суспендировали в ДМФА (15 мл), затем с DIEA (4,8 мл) и гидрохлоридом трет-бутилового эфира аланина (3 г). Затем добавляли раствор РуВОР (3,43 г) в DMF (5 мл). После перемешивания при комнатной температуре в течение 3 ч добавляли еще РуВОР (1 г) и реакционную смесь перемешивали в продолжение ночи. После добавления воды (5 мл) смесь нагревали при 60 С в течение 30 мин. Добавляли этилацетат (150 мл) и органический слой промывали 2 порциями 0,5 Н HCl, 2 порциями 5% K2CO3, 2 порциями насыщенного солевого раствора. Выпаривание растворителя давало маслянистый остаток, который после флэш-колоночной хроматографии (SiO2, 0-30% этилацетат в DCM) давал трет-бутил N-(41-[3-(3,5-дихлорфенил)-5-оксо-4,5-дигидро-1 Н-пиразол-1-ил]этилбензоил)аланинат в виде белого твердого вещества. ЯМР (500 МГц, DMSO-d6) : 1,37 (с, 9 Н); 1,78 (д, J=7,1 Гц, 3 Н); 2,45 (т, J=7,0 Гц, 2 Н); 3,42 (кв, J=7,0 Гц, 2 Н); 5,56 (кв, J=7,1 Гц, 1 Н); 5,99 (с, 1 Н); 7,30 (д, J=8,3 Гц, 2 Н); 7,47 (т, J=1,0 Гц, 1 Н), 7,73 (д, J=8,3 Гц,2 Н); 7,76 (д, J=1,9 Гц, 2H); 8,43 (т, J=5,6 Гц, 1 Н); 11,34 (с, 1 Н).N-(4-1-[3-(3,5-дихлорфенил)-5-оксо-4,5-дигидро-1 Н-пиразол-1-ил]этилбензоил)-аланинат (2,05 г, 4,05 ммоль), ТЕА (1,7 мл, 12 ммоль) растворяли в ТГФ (35 мл) при -78 С. К раствору добавляли ангидрид трифторуксусной кислоты (1,1 мл, 6,2 ммоль). Охлаждающую баню удаляли и реакционную смесь перемешивали в течение 1 ч. Реакцию гасили добавлением этилацетата, воды. Органический слой промывали 2 порциями 0,5 Н HCl, 2 порциями насыщенного солевого раствора и сушили надNa2SO4. Выпаривание растворителя и флэш-колоночная хроматография (SiO2, градиент 0-10% этилацетат в DCM) давали трет-бутил N-4-[1-(3-(3,5-дихлорфенил)-5-[(трифторметил)сульфонил]окси-1 Нпиразол-1-ил)этил]бензоилаланинат в виде бесцветного сухого тонкого слоя. ЯМР (500 МГц, CDCl3) : 1,45 (с, 9 Н); 1,97 (д, J=7,1 Гц, 3 Н); 2,53 (т, J=5,9 Гц, 2 Н); 3,67 (кв, J=5,9 Гц, 2 Н); 5,54 (кв, J=7,1 Гц, 1 Н); 6,43 (с, 1 Н); 6,86 (т, J=6,2 Гц, 1 Н); 7,33 (т, J=2,0 Гц, 1 Н); 7,36 (д, J=8,4 Гц,2 Н); 7,67 (д, J=2,0 Гц, 2 Н); 7,74 (д, J=8,4 Гц, 2 Н).N-4-[1-(3-(3,5-дихлорфенил)-5-[(трифторметил)сульфонил]окси-1 Н-пиразол-1 ил)этил]бензоилаланинат может быть разделен хиральной ВЭЖХ (ChiralPak AD колонка, условия анализа - 6% изопропанол/гептан, (S)-изомер Rt=16,1 и (R)-изомер 18,1 мин, или применение SFC хроматографии 15% MeOH:СО 2, 1,5 мл/мин - (R)-изомер Rt=5,5 и (S)-изомер 6,1 мин, препаративные условия применения SFC хроматографии 15% MeOH:CO2, 50 мл/мин). Абсолютную стереохимию двух образцов устанавливали приготовлением подлинного образца трет-бутил N-4-[(S)-1-(3-(3,5-дихлорфенил)-5[(трифторметил)сульфонил]окси-1 Н-пиразол-1-ил)этил]бензоилаланината (ChiralPak AD колонка,6% изопропанол/гептан, Rt=15,8 мин, соинъекция с выше представленным (S)-изомером Rt=16,1 мин) из(1S)-1-[4-(этоксикарбонил)фенил]этилгидразиний трифторацетата (см. выше). Данный (S)-изомер применяли на стадии D. В альтернативном случае трет-бутилN-4-[(S)-1-(3-(3,5-дихлорфенил)-5-[(трифторметил)сульфонил]окси-1 Н-пиразол-1-ил)этил]бензоилаланинат может быть приготовлен без хроматографического разделения энантиомеров из этил 4-(1S)-1-[3-(3,5-дихлорфенил)-5-оксо-4,5-дигидро-1 Нпиразол-1-ил]этилбензоата (способ С, пример 4, стадия А), использованием стадий В и С, данных выше. Стадия- 21012431 порциями N2 в вакууме. Добавляли катализатор Pd(PPh3)4 (2 мг, 10% моль) и смесь дезоксигенировали снова, прежде чем нагревали в микроволновом реакторе при 100 С в течение 10 мин. Смесь гасили 1,5 мл CH3CN:Н 2 О (3:1, с 5% TFA) и продукт выделяли препаративной ВЭЖХ с обращенной фазой. Собранный продукт обрабатывали 1 мл TFA-DCM (1:2) в течение 30 мин и остаток лиофилизировали, получая N-[4-1S)-1-3-(3,5-дихлорфенил)-5-[6-(трифторметокси)-2-нафтил]-1 Н-пиразол-1-илэтил)бензоил]-аланин в виде мелкого порошка. ЯМР (500 МГц, DMSO-d6) : 1,91 (д, J=7,0 Гц, 3 Н); 2,46 (т, J=7,0 Гц, 2 Н); 3,41 (кв, J=7,0 Гц, 2 Н); 5,78 (кв, J=7,0 Гц, 1 Н); 7,21 (д, J=8,4 Гц, 2 Н); 7,22 (с, 1 Н); 7,57 (т, J=1,9 Гц, 1 Н); 7,58-7,60 (м, 2 Н); 7,72 (д,J=8,4 Гц, 2 Н); 7,94 (д, J=1,9 Гц, 2 Н); 8,04 (ушир.с, 1 Н); 8,06 (ушир.с, 1 Н); 8,09 (д, J=9,1 Гц, 1 Н); 8,13 (д,J=8,6 Гц, 1 Н); 8,44 (т, J=5,5 Гц, 1 Н).N-[4-1R)-1-3-(3,5-Дихлорфенил)-5-[6-(трифторметокси)-2-нафтил]-1 Н-пиразол-1 илэтил)бензоил]аланин. трет-Бутил N-4-[(1R)-1-(3-(3,5-дихлорфенил)-5-[(трифторметил)сульфонил]окси-1 Н-пиразол-1 ил)этил]бензоилаланинат превращали, следуя методике примера 1, стадия D, в N-[4-1R)-1-3-(3,5 дихлорфенил)-5-[6-(трифторметокси)-2-нафтил]-1 Н-пиразол-1-илэтил)бензоил]аланин в виде мелкого порошка. ЯМР (500 МГц, DMSO-d6) : 1,91 (д, J=7,0 Гц, 3 Н); 2,46 (т, J=7,0 Гц, 2 Н); 3,4 (м, 2 Н); 5,77 (кв, J=7,0 Гц, 1 Н); 7,20 (д, J=8,2 Гц, 2 Н); 7,21 (с, 1 Н); 7,56 (т, J=2,0 Гц, 1H); 7,57-7,60 (м, 2 Н); 7,71 (д, J=8,4 Гц, 2 Н); 7,93 (д, J=2 Гц, 2 Н); 8,04 (ушир.с, 1 Н); 8,06 (ушир.с, 1 Н); 8,09 (д, J=9,1 Гц, 1 Н); 8,13 (д, J=8,6 Гц, 1 Н); 8,44 Стадия А. Этил 3-(6-метокси-2-нафтил)-3-оксопропаноат. Суспензию MgCl2 (3,5 г, 35 ммоль), этилмалоната калия (4,6 г, 30 ммоль) и триэтиламина (15 мл,105 ммоль) в сухом этилацетате (100 мл) перемешивали при 40 С на протяжении ночи. Затем к вышеуказанной смеси добавляли суспензию хлорангидрида 6-метоксинафтил-2-кислоты (4,9 г, 22,2 ммоль) в этилацетате (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2,5 ч. Реакционную смесь гасили 60 мл 2 Н HCl, перемешивали в течение 5 мин и затем промывали 2 порциями 0,5 Н HCl, 2 порциями 5% K2CO3, 2 порциями насыщенного солевого раствора. Выпаривание растворителя и вакуумная сушка давали этил 3-(6-метокси-2-нафтил)-3-оксопропаноат в виде масла. ЯМР (500 МГц, CDCl3) : 1,26 (т, J=7,1 Гц, 3 Н); 3,95 (с, 3 Н); 4,09 (с, 2 Н); 4,23 (кв, J=7,1 Гц, 2 Н); 7,15(д, J=2,5 Гц, 1 Н); 7,21 (дд, J=2,5 Гц, 9,0 Гц, 1 Н); 7,77 (д, J=8,7 Гц, 1 Н); 7,85 (д, J=9,0 Гц, 1 Н); 7,98 (дд,J=1,8 Гц, 8,7 Гц, 1 Н); 8,38 (д, J=1,8 Гц, 1 Н). В ЯМР-спектре отмечали примерно 10% енольной формы. Стадия В. 5-(6-Метокси-2-нафтил)-2,4-дигидро-3 Н-пиразол-3-он. Этил 3-(6-метокси-2-нафтил)-3-оксопропаноат (5,0 г, 18,3 ммоль) и безводный гидразин (0,63 мл,20 ммоль) кипятили с обратным холодильником в НОАс (100 мл) в течение 3 ч. Растворитель удаляли при пониженном давлении, и остаток промывали DCM и собирали фильтрованием, получая 5-(6 метокси-2-нафтил)-2,4-дигидро-3 Н-пиразол-3-он в виде не совсем белого твердого вещества. Данное соединение существует в енольной форме в ДМСО. ЯМР (500 МГц, DMSO-d6) : 3,87 (с, 3 Н); 5,95 (с, 1 Н); 7,17 (дд, J=2,7 Гц, 9,0 Гц, 1 Н); 7,31 (д, J=2,7 Гц, 1H); 7,74-7,84 (м, 3 Н); 8,11 (ушир.с, 1 Н); 9,66 (ушир.с, 1 Н); 12 (ушир., 1 Н).- 22012431 3-(6-Метокси-2-нафтил)-5-пиразолин-5-он (1,58 г, 6,58 ммоль) и пиридин (1,62 мл, 20 ммоль) растворяли в ТГФ (20 мл) при -78 С. С помощью шприца добавляли ангидрид трифторуксусной кислоты(1,68 мл, 10 ммоль). Охлаждающую баню удаляли и реакционную смесь перемешивали в течение 2 ч. Смесь охлаждали до -78 С и разбавляли этилацетатом (50 мл) и 2 Н HCl (10 мл). Этилацетатный слой промывали 2 порциями разбавленной HCl, 2 порциями насыщенного солевого раствора. Выпаривание растворителя приводило к фиолетовому остатку, который очищали колоночной хроматографией (SiO2, 02,5% этилацетат в DCM), получая 3-(6-метокси-2-нафтил)-1 Н-пиразол-5-илтрифторметансульфонат в виде белого твердого вещества. ЯМР (500 МГц, DMSO-d6) : 3,89 (с, 3 Н); 6,93 (д, J=2,2 Гц, 1 Н); 7,23 (дд, J=2,7 Гц, 8,8 Гц, 1 Н); 7,37MC C15H11F3N2O4S: Вычислено: 372,04; Найдено (М+1): 373,06. Стадия D. трет-Бутил N-(4-ацетилбензоил)аланинат. Раствор NaOH (1,7 г/12 мл) добавляли к метил 4-ацетилбензоату (5,04 г, 28,3 ммоль) в смеси MeOHдиоксан (2:1, 60 мл). После перемешивания при комнатной температуре в течение 12 ч смесь подкисляли 5 Н HCl и экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4. Выпаривание растворителя давало 4-ацетилбензойную кислоту в виде белого твердого вещества. 4-Ацетилбензойную кислоту (2,45 г, 14,9 ммоль), гидрохлорид трет-бутилового эфира -аланина(4,0 г, 22 ммоль), DIEA (3,9 мл, 22 ммоль) и DMAP (100 мг) растворяли в DCM (100 мл). Порциями добавляли EDC гидрохлорид (3,5 г, 18 ммоль). Через один час добавляли дополнительное количество EDC(0,7 г) для завершения реакции. По прошествии суммарных 3 ч реакционную смесь распределяли между этилацетатом и 0,5 Н HCl. Органический слой промывали 3 порциями 0,5 Н HCl, 2 порциями 5% K2CO3,2 порциями насыщенного солевого раствора. Выпаривание растворителя и хроматография на SiO2 (1020% этилацетат в DCM) давали трет-бутил N-(4-ацетилбензоил)аланинат в виде белого твердого вещества. ЯМР (500 МГц, CD3OD) : 1,44 (с, 9 Н); 2,57 (т, J=7,0 Гц, 2H); 2,63 (с, 3 Н); 3,61 (т, J=7,0 Гц, 2 Н); 7,89(д, J=8,5 Гц, 2 Н); 8,05 (д, J=8,5 Гц, 2 Н). Стадия E. трет-Бутил N-[4-(1-гидроксиэтил)бензоил]аланинат. Борогидрид натрия (0,28 г, 7,4 ммоль) в твердом виде добавляли к раствору трет-бутил N-(4 ацетилбензоил)аланината (2,11 г, 7,24 ммоль) в МЕОН (50 мл). После перемешивания при комнатной температуре в течение 30 мин реакцию гасили добавлением этилацетата (150 мл) и 2 Н HCl (50 мл). Органический слой промывали 2 порциями 1 Н HCl, 2 порциями насыщенного солевого раствора и сушили над Na2SO4. Выпаривание растворителя и вакуумная сушка давали трет-бутил N-[4-(1 гидроксиэтил)бензоил]аланинат в виде бесцветного масла. ЯМР (CDCl3) : 1,46 (с, 9 Н); 1,49 (д, J=6,6 Гц, 3 Н); 2,55 (т, J=6,0 Гц, 2 Н); 3,67 (кв, J=6,0 Гц, 2 Н); 4,94N-[4-(1-гидроксиэтил)бензоил]аланинат (1,2 г, 4,02 ммоль) и трифенилфосфин (1,44 г, 5,48 ммоль) суспендировали в DCM (25 мл). К суспензии медленно добавляли диизопропилазодикарбоксилат(0,87 мл, 4,38 ммоль). Смесь перемешивали в течение 2 ч и затем концентрировали до 10 мл. Данный остаток хроматографировали (SiO2, градиент 25-30% этилацетат), получая 0,727 г трет-бутил N-4-[1-(3(6-метокси-2-нафтил)-5-[(трифторметил)сульфонил]окси-1 Н-пиразол-1-ил)этил]бензоилаланинат,ЯМР (500 МГц, CDCl3) : 1,45 (с, 9 Н); 2,01 (д, J=7,1 Гц, 3 Н); 2,53 (т, J=5,9 Гц, 2 Н); 3,67 (кв, J=5,9 Гц, 2 Н); 3,94 (с, 3 Н); 5,56 (кв, J=7,1 Гц, 1 Н); 6,54 (с, 1 Н); 6,78 (ушир., 1 Н); 7,16 (ушир., 1 Н); 7,17 (дд,J=2,6 Гц, 9 Гц, 1 Н); 7,41 (д, J=8,4 Гц, 2 Н); 7,74 (д, J=8,4 Гц, 2 Н); 7,78(д, J=8,4 Гц, 1 Н); 7,79 (д, J=8,5 Гц,1 Н); 7,93 (дд, J=1,8 Гц, 8,5 Гц, 1 Н); 8,14 (д, J=1,6 Гц, 1 Н);PdCl2 (dppf) (12 мг, 0,014 ммоль) суспендировали в толуоле (0,6 мл) в стеклянном сосуде. Добавляли раствор Cs2CO3 (5 М, 25 мкл). Смесь дезоксигенировали порциями N2 в вакууме и нагревали в микроволновом реакторе до 140 С в течение 10 мин. Реакционную смесь фильтровали через стекловолокнистый слой и растворитель удаляли при пониженном давлении. Остаток растворяли в CH3CN-H2O и очищали препаративной ВЭЖХ с обращенной фазой. С полученного таким образом продукта удаляли защиту обработкой с помощью TFA-DCM (1:2, 1 мл) в течение 30 мин. Выпаривание растворителя и лиофилизация из CH3CN-H2O приводила к N-1-(4-(2-гидроксикарбонилэтиламинокарбонил)фенил)этил-3-(2,5 дихлорфенил)-5-(6-метоксинафт-2-ил)пиразолу в виде мелкого порошка. ЯМР (500 МГц, DMSO-d6) : 1,90 (д, J=6,9 Гц, 3 Н); 2,47 (т, J=7,1 Гц, 2 Н); 3,41 (кв, J=7,1 Гц, 2 Н); 3,89 (с, 3 Н); 5,79 (кв, J=6,9 Гц, 1 Н); 7,02 (с, 1 Н); 7,22 (д, J=8,4 Гц, 2 Н); 7,23 (д, J=9,0 Гц, 1 Н); 7,39 (д, J=2,6 Гц, 1 Н); 7,44 (дд, J=1,7 Гц, 8,3 Гц, 1 Н); 7,47 (дд, J=2,6 Гц, 8,6 Гц, 1 Н); 7,61 (д, J=8,6 Гц, 1 Н); 7,74 (д, J=8,4 Гц, 2 Н); 7,83 (д, J=9,0 Гц, 1 Н); 7,88 (д, J=1,7 Гц, 1 Н); 7,90 (д, J=8,6 Гц, 1 Н); 7,92 (д, J=2,6 Гц, 1 Н).MC C32H27Cl2N3O4: Вычислено: 587,14; Найдено (M+1): 588,21. Рацемический N-(4-1-[3-(2,5-дихлорфенил)-5-(6-метокси-2-нафтил)-1 Н-пиразол-1-ил]этилбензоил)аланин, полученный в примере 3, разделяли на энантиомеры хроматографией, применяя колонкуChiralPak AS (10250 мм), элюирование с помощью 40% MeOH:CO2 (0,1% TFA) при 10 мл/мин, 40 С,что давало соединение примера 82 (табл. 3) и соединение примера 106 (табл. 4). Стереохимическое представление основывали гипотетически на сравнении биологических данных с другими аналогами. Это также применимо к примерам 69, 83, 89 и 107. Общий синтез пиразолов, способ С. Пример 4. Стадия А. Этил 4-(1S)-1-[3-(3,5-дихлорфенил)-5-оксо-4,5-дигидро-1 Н-пиразол-1-ил]этилбензоат. Этил 3-(3,5-дихлорфенил)-3-оксопропаноат (4,2 г, 16,1 ммоль) и (1S)-1-[4-(этоксикарбонил)фенил]этилгидразиний трифторацетат (5,2 г, 16,1 ммоль) нагревали в сухом ацетонитриле (100 мл) до 85 С в течение 1 ч. Растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией (SiO2, 20% этилацетат в гексанах), получая этил 4-(1S)-1-[3-(3,5-дихлорфенил)-5-оксо 4,5-дигидро-1 Н-пиразол-1-ил]этилбензоат в виде белого твердого вещества. ЯМР (500 МГц, CDCl3) : 1,38 (т, J=7,1 Гц, 3 Н); 1,78 (д, J=7,0 Гц, 3 Н); 3,55 (д, J=22,6 Гц, 1 Н); 3,60MC C20H18Cl2N2O3: Вычислено: 404,07; Найдено (М+1): 405,20. Стадия В. Этил 4-[(1S)-1-(3-(3,5-дихлорфенил)-5-[(трифторметил)сульфонил]окси-1 Н-пиразол-1 ил)этил]бензоат. Этил 4-(1S)-1-[3-(3,5-дихлорфенил)-5-оксо-4,5-дигидро-1 Н-пиразол-1-ил]этилбензоат (4,93 г, 12,2 ммоль) и триэтиламин (8,5 мл, 61 ммоль) растворяли в ТГФ (100 мл) при -78 С. Добавляли ангидрид трифторуксусной кислоты (4,1 мл, 24,5 ммоль). Охлаждающую баню удаляли и реакционную смесь перемешивали в течение 1 ч. Добавляли этилацетат (100 мл) и органическую фазу промывали водой, 2 порциями 1 Н HCl, 2 порциями насыщенного солевого раствора. Флэш-колоночная хроматография (SiO2, 05% этилацетат в гексанах), давала этил 4-[(1S)-1-(3-(3,5-дихлорфенил)-5-[(трифторметил)сульфонил]окси-1 Н-пиразол-1-ил)этил]бензоат в виде бесцветного масла. ЯМР (500 МГц, CDCl3) : 1,38 (т, J=7,1 Гц, 3 Н); 1,98 (д, J=7,0 Гц, 3 Н); 4,36 (кв, J=7,1 Гц, 2 Н); 5,55(1,2 мл, 8,0 ммоль) растворяли в диметоксиэтане (40 мл) в толстостенном сосуде. Реакционную смесь продували N2 в течение 15 мин. Добавляли катализатор Pd(PPh3)4 (350 мг, 8%) и тестируемый сосуд нагревали в микроволновом реакторе до 100 С в течение 15 мин. Растворитель удаляли при пониженном давлении и остаток распределяли между этилацетатом и 1 Н HCl. Органический слой промывали 2 порциями 0,5 Н NH4Cl, 2 порциями насыщенного солевого раствора, сушили над Na2SO4 и фильтровали через рыхлый слой целита. Сырой продукт очищали колоночной хроматографией (SiO2, 10-15% этилацетат- 24012431 в гексанах), получая этил 4-(1S)-1-[3-(3,5-дихлорфенил)-5-(6-метокси-2-нафтил)-1 Н-пиразол-1 ил]этилбензоат в виде сухого тонкого слоя. ЯМР (500 МГц, CDCl3) : 1,38 (т, J=7,1 Гц, 3 Н); 1,96 (д, J=7,0 Гц, 3 Н); 3,95 (с, 3 Н); 4,37 (кв, J=7,1 Гц, 2 Н); 5,59 (кв, J=7,0 Гц, 1 Н); 6,65 (с, 1 Н); 7,17 (д, J=2,6 Гц, 1 Н); 7,20 (дд, J=2,6, 8,9 Гц, 1 Н); 7,28 (д,J=8,4 Гц, 2 Н); 7,29 (дд, J=1,8, 8,5 Гц, 1 Н); 7,30 (т, J=1,9 Гц, 1 Н); 7,63 (д, J=1,8 Гц, 1 Н); 7,67 (д, J=8,9 Гц,1 Н); 7,76 (д, J=8,5 Гц, 1 Н); 7,80 (д, J=1,9 Гц, 2 Н); 7,99 (д, J=8,4 Гц, 2 Н).MC C31H26Cl2N2O3: Вычислено: 544,13; Найдено: 545,15. Стадия D. 4-(1S)-1-[3-(3,5-Дихлорфенил)-5-(6-метокси-2-нафтил)-1 Н-пиразол-1-ил]этилбензойная кислота. Этил 4-(1S)-1-[3-(3,5-дихлорфенил)-5-(6-метокси-2-нафтил)-1 Н-пиразол-1-ил]этилбензоат (3,53 г,6,48 ммоль) растворяли в смеси MeOH-диоксан (1:1, 100 мл) и к данному раствору добавляли растворNaOH (1,2 г, избыток)/вода (10 мл). Реакционную смесь перемешивали при комнатной температуре на протяжении ночи. После концентрирования реакционной смеси до 50 мл ее подкисляли 2 Н HCl и экстрагировали этилацетатом. Органический слой промывали 2 порциями насыщенного солевого раствора и сушили над Na2SO4. Выпаривание растворителя и вакуумная сушка давали 4-(1S)-1-[3-(3,5 дихлорфенил)-5-(6-метокси-2-нафтил)-1 Н-пиразол-1-ил]этилбензойную кислоту в виде белого порошка. ЯМР (500 МГц, CDCl3) : 1,97 (д, J=7,0 Гц, 3 Н); 3,95 (с, 3 Н); 5,61 (кв, J=7,0 Гц, 1 Н); 6,66 (с, 1 Н); 7,17 (д, J=2,5 Гц, Н); 7,21 (дд, J=2,5, 9,0 Гц, 1 Н); 7,29 (дд, J=1,6, 8,4 Гц, 1 Н); 7,31 (т, J=1,9 Гц, 1 Н); 7,32 (д,J=8,4 Гц, 2 Н); 7,63 (д, J=1,6 Гц, 1 Н); 7,67 (д, J=9,0 Гц, 1 Н); 7,77 (д, J=8,4 Гц, 1 Н); 7,80 (д, J=1,9 Гц, 2 Н); 8,05 (д, J=8,4 Гц, 2 Н). Стадия Е. N-(4-(1S)-1-[3-(3,5-Дихлорфенил)-5-(6-метокси-2-нафтил)-1 Н-пиразол-1-ил]этилбензоил)аланин. 4-(1S)-1-[3-(3,5-Дихлорфенил)-5-(6-метокси-2-нафтил)-1 Н-пиразол-1-ил]этилбензойную кислоту(3,5 г, 6,76 ммоль), гидрохлорид трет-бутилового эфира -аланина (3,7 г, 20 ммоль), DIEA (3,53 мл,20 ммоль) и DMAP (40 мг, 5%) растворяли в DCM (50 мл) с последующим добавлением твердого EDCHCl (1,6 г, 8,1 ммоль). Через один час добавляли дополнительное количество EDC HCl (1,8 г). Реакцию завершали примерно через 3 ч с контролем по ЖХ-МС. К реакционной смеси добавляли этилацетат и смесь промывали 3 порциями 1 Н HCl и 2 порциями насыщенного солевого раствора. Сырой продукт затем очищали колоночной хроматографией (SiO2, 0-6% этилацетат в DCM), получая трет-бутил N-(4(1S)-1-[3-(3,5-дихлорфенил)-5-(6-метокси-2-нафтил)-1 Н-пиразол-1-ил]этилбензоил)аланинат в виде сухого пенистого вещества. ЯМР (500 МГц, DMSO-d6) : 1,36 (с, 9 Н); 1,90 (д, J=6,9 Гц, 3 Н); 2,44 (т, J=6,8 Гц, 2 Н); 3,41 (кв, J=6,8 Гц, 2 Н); 3,89 (с, 3 Н); 5,76 (кв, J=6,9 Гц, 1 Н); 7,15 (с, 1 Н); 7,21 (д, J=8,4 Гц, 2 Н); 7,22 (дд, J=2,6, 9,0 Гц, 1 Н); 7,38 (д, J=2,6 Гц, 1 Н); 7,43 (дд, J=1,9, 8,5 Гц, 1 Н); 7,55 (т, J=1,9 Гц, 1 Н); 7,71 (д, J=8,4 Гц, 2 Н); 7,81 (д,J=9,0 Гц, Н); 7,85 (д, J=1,9 Гц, 1 Н); 7,90 (д, J=8,5 Гц, 1 Н); 7,92 (д, J=1,9 Гц, 2 Н); 8,44 (т, J=5,2 Гц, 1 Н). С данного трет-бутилового эфира удаляли защиту в TFA-DCM (1:2, 200 мл) в течение 30 мин. Выпаривание растворителя и вакуумная сушка приводили к маслянистому остатку, который лиофилизировали из CH3CN:H2O (1:1, 200 мл), получая N-(4-(1S)-1-[3-(3,5-дихлорфенил)-5-(6-метокси-2-нафтил)-1 Нпиразол-1-ил]этилбензоил)аланин в виде белого порошка. ЯМР (500 МГц, DMSO-d6) : 1,90 (д, J=7,0 Гц, 3 Н); 2,47 (т, J=7 Гц, 2 Н); 3,41 (кв, J=7 Гц, 2H); 3,89 (с,3 Н); 5,76 (кв, J=7,0 Гц, 1 Н); 7,16 (с, 1 Н); 7,20 (д, J=8,4 Гц, 2 Н); 7,23 (дд, J=2,6, 9,0 Гц, 1 Н); 7,39 (д, 2,6 Гц,1 Н); 7,43 (дд, J=1,7, 8,4 Гц, 1 Н); 7,56 (т, J=1,9 Гц, 1 Н); 7,72 (д, J=8,4 Гц, 2 Н); 7,83 (д, J=9,0 Гц, 1 Н); 7,86 (д,J=l,7 Гц, 1 Н); 7,91 (д, J=8,4 Гц, 1 Н); 7,93 (д, J=1,9 Гц, 2 Н); 8,44 (т, J=5,6 Гц, 1 Н). Стадия А. Этил 3-[2-фтор-5-(трифторметил)фенил]-3-оксопропаноат. Этилмалонат калия (10,2 г, 60 ммоль), MgCl2 (6,3 г, 66 ммоль) и триэтиламин (28 мл, 200 ммоль) суспендировали в сухом этилацетате (200 мл) и нагревали при 40 С в течение 15 ч. К смеси медленно по каплям (примерно в течение одного часа) добавляли раствор 2-фтор-5-трифторметилбензоилхлорида(10 г, 44,1 ммоль) в этилацетате (40 мл). По прошествии 1 ч смесь обрабатывали 2 Н HCl (200 мл). Органический слой промывали 2 порциями 0,5 Н HCl, 2 порциями 5% K2CO3, 2 порциями насыщенного солевого раствора и сушили над Na2SO4. Выпаривание растворителя и вакуумная сушка давали этил 3-[2 фтор-5-(трифторметил)фенил]-3-оксопропаноат в виде бледно-желтого масла.- 25012431 ЯМР (500 МГц, CDCl3) : 1,25 (т, J=7,4 Гц, 3 Н); 1,32; 4,00 (д, JF-H=3 Гц, 2 Н); 4,21 (кв, J=7,4 Гц, 2 Н); 5,8; 7,22; 7,29 (т, J=10,3 Гц, 1 Н); 7,66; 7,82 (ушир., 1 Н); 8,14; 8,24 (д, J=6,4 Гц, 1 Н). Енольная форма существует примерно на 35%. Стадия B. Этил 4-1S)-1-3-[2-фтор-5-(трифторметил)фенил]-5-оксо-4,5-дигидро-1 Н-пиразол-1 илэтил)бензоат. Этил 3-[2-фтор-5-(трифторметил)фенил]-3-оксопропаноат (3 г, 9,7 ммоль) и (1S)-1-[4(этоксикарбонил)фенил]этилгидразиний трифторацетат (2,64 г, 8,2 ммоль) нагревали в сухом ацетонитриле (150 мл) до 85 С в течение 8 ч. Растворитель выпаривали и остаток очищали флэш-колоночной хроматографией (SiO2, 25% этилацетат в гексанах), получая этил 4-1S)-1-3-[2-фтор-5(трифторметил)фенил]-5-оксо-4,5-дигидро-1 Н-пиразол-1-илэтил)бензоат в виде белого твердого вещества. ЯМР (500 МГц, CDCl3) : 1,38 (т, J=7,1, 3H); 1,82 (д, 7,2 Гц, 3 Н); 3,81 (дд, J=3 Гц, 23,9 Гц, 1 Н); 3,87-78 С. Добавляли ангидрид трифторуксусной кислоты (1,1 мл, 6,6 ммоль). Охлаждающую баню удаляли и реакционную смесь перемешивали в течение 1 ч. Добавляли этилацетат (100 мл) и органическую фазу промывали водой, 2 порциями 1 Н HCl, 2 порциями насыщенного солевого раствора. Флэш-колоночная хроматография (SiO2, 0-5% этилацетат в гексанах) давала этил 4-[(1S)-1-(3-[2-фтор-5(трифторметил)фенил]-5-[(трифторметил)сульфонил]окси-1 Н-пиразол-1-ил)этил]бензоат в виде бесцветного масла. ЯМР (500 МГц, CDCl3) : 1,38 (т, J=7,1 Гц, 3 Н); 2,01 (д, J=7,0 Гц, 3 Н); 4,36 (кв, J=7,1 Гц, 2 Н); 5,59MC C22H17F7N2O5S: Вычислено: 554,07; Найдено (М+1): 555,16. Стадия D. Этил 4-(1S)-1-[3-[2-фтор-5-(трифторметил)фенил]-5-(6-метокси-2-нафтил)-1 Н-пиразол 1-ил]этилбензоат. Этил 4-[(1S)-1-(3-[2-фтор-5-(трифторметил)фенил]-5-[(трифторметил)сульфонил]окси-1 Н-пиразол-1-ил)этил]бензоат (1,05 г, 1,90 ммоль), 6-метокси-2-нафтилбороновую кислоту (0,43 г, 2,1 ммоль) и триэтиламин (0,53 мл, 3,8 ммоль) растворяли в DME (20 мл). После дезоксигенирования (циклические поступления N2 в вакууме) добавляли Pd(PPh3)4 (85 мг, 4% моль) Реакционную смесь дезоксигенировали снова и нагревали в микроволновом реакторе до 100 С в течение 15 мин. Реакционную смесь фильтровали через рыхлый слой целита, концентрировали и очищали флэш-колоночной хроматографией (SiO2,градиент 5-20% этилацетат в гексане), получая этил 4-(1S)-1-[3-[2-фтор-5-(трифторметил)фенил]-5-(6 метокси-2-нафтил)-1 Н-пиразол-1-ил]этилбензоат в виде бесцветного геля. ЯМР (500 МГц, CDCl3) : 1,38 (т, J=7,1 Гц, 3 Н); 1,99 (д, J=7,1 Гц, 3 Н); 3,96 (с, 3 Н); 4,37 (кв, J=7,1 Гц, 2 Н); 5,64 (кв, J=7,1 Гц, 1 Н); 6,87 (д, JF-H=4,2 Гц, 1 Н); 7,17 (д, J=2,5 Гц, 1 Н); 7,20 (дд, J=2,5 Гц, 8,9 Гц,1 Н); 7,25 (т, J=9,5 Гц, 1 Н); 7,30 (д, J=8,4 Гц, 2 Н); 7,32 (дд, J=1,5 Гц, 8,4 Гц, 1 Н); 7,56 (м, 1 Н); 7,66 (д, J=1,5 Гц, 1 Н); 7,68 (д, J=8,9 Гц, 1 Н); 7,77 (д, J=8,4 Гц, 1 Н); 8,00 (д, J=8,4 Гц, 2 Н); 8,48 (дд, J=2,7 Гц, 6,8 Гц, 1 Н).NaOH (2,5 г, избыток) в воде (20 мл). Реакционную смесь, которая медленно становилась прозрачной при перемешивании, оставляли на ночь. Реакционную смесь сначала концентрировали до 30 мл, подкисляли 2 Н HCl и экстрагировали этилацетатом. Органический слой промывали 2 порциями насыщенного солевого раствора и сушили над Na2SO4. Выпаривание растворителя и вакуумная сушка давали 4-(1S)-1-[3[2-фтор-5-(трифторметил)фенил]-5-(6-метокси-2-нафтил)-1 Н-пиразол-1-ил]этилбензойную кислоту в виде сухого пенистого вещества. ЯМР (500 МГц, CDCl3) : 2,00 (д, J=7,0 Гц, 3 Н); 3,95 (с, 3 Н); 5,65 (кв, J=7,0 Гц, 1 Н); 6,88 (д, JF-H=4,2 Гц, 1 Н); 7,17 (д, J=2,5 Гц, 1 Н); 7,20 (дд, J=2,5 Гц, 8,9 Гц, 1 Н); 7,26 (т, J=8,6 Гц, 1 Н); 7,31 (дд, J =1,8 Гц, 8,5 Гц, 1 Н); 7,32 (д, J=8,4 Гц, 2 Н); 7,56 (м, 1 Н); 7,66 (д, J=1,8 Гц, 1 Н); 7,68 (д, J=8,9 Гц, 1 Н); 7,77 (д, J=8,5 Гц,1 Н); 8,06 (д, J=8,4 Гц, 2 Н); 8,48 (дд, J=2,9 Гц, 6,9 Гц, 1 Н).(2,73 г, 15 ммоль) и DIEA (3,5 мл, 20 ммоль) растворяли в ДМФА (35 мл) с последующим добавлением РуВОР (2,93 г, 5,62 ммоль) в ДМФА (10 мл). Реакционную смесь перемешивали в течение 10 мин и разбавляли этилацетатом (200 мл), промывали 2 порциями 1 Н HCl, 2 порциями 5% K2CO3 и 2 порциями насыщенного солевого раствора. Растворитель выпаривали и полученный остаток очищали флэшхроматографией (SiO2, 0-6% этилацетат в DCM градиент), получая трет-бутил N-(4-(1S)-1-[3-[2-Фтор-5(трифторметил)фенил]-5-(6-метокси-2-нафтил)-1 Н-пиразол-1-ил]этилбензоил)аланинат в виде сухого пенистого вещества. ЯМР (500 МГц, DMSO-d6) : 1,35 (с, 9 Н); 1,92 (д, J=6,9 Гц, 3 Н); 2,44 (т, J=7,0 Гц, 2 Н); 3,41 (кв, J=7,0 Гц, 2 Н); 3,89 (с, 3 Н); 5,80 (кв, J=6,9 Гц, 1 Н); 6,94 (д, JF-H=3,8 Гц, 1 Н); 7,21 (д, J=8,3 Гц, 2 Н); 7,22 (дд, J=2,6 Гц, 9,0 Гц, 1 Н); 7,39 (д, J=2,6 Гц, 1 Н); 7,44 (дд, J=2,6 Гц, 8,5 Гц, 1 Н); 7,58 (т, J=9,7 Гц, 1 Н); 7,72 (д, J=8,3 Гц, 2 Н); 7,79 (м, 1 Н); 7,83 (д, J=9,0 Гц, 1 Н); 7,89 (д, J=2,6 Гц, 1 Н); 7,90 (д, J=8,5 Гц, 1 Н); 8,33 (дд, J=2,8 Гц, 6,6 Гц, 1 Н); 8,44 (т, J=5,5 Гц, NH).MC C37H35F4N3O4: Вычислено: 661,26; Найдено (М+1): 662,29. С данного трет-бутилового эфира удаляли защиту в TFA-DCM (1:2, 300 мл) при комнатной температуре в течение 30 мин. После выпаривания растворителя и вакуумной сушки остаток лиофилизировали из CH3CN:Н 2 О (1:1, 300 мл), получая N-(4-(1S)-1-[3-[2-фтор-5-(трифторметил)фенил]-5-(6-метокси-2 нафтил)-1 Н-пиразол-1-ил]этилбензоил)аланин в виде мелкого порошка.MC C33H27F4N3O4: Вычислено: 605,19; Найдено (М+1): 606,32. По методикам примеров 1-5 получены соединения, перечисленные в табл. 1-6. Таблица 1 Масс-спектрометрические данные отсутствуют. Н ЯМР данные для примера 20 - ЯМР (500 МГц, DMSO-d6) : 2,46 (т, J=7,1 Гц, 2 Н); 3,40 (кв, J=7,0 Гц, 2 Н); 3,88 (с, 3 Н); 5,62 (с, 2 Н); 7,03 (д, JFH=3,7 Гц, 1 Н); 7,11 (дд, J=8,1 Гц, 2 Н); 7,21 (дд, J=2,5, 9,1 Гц,1 Н); 7,37 (д, J=2,5 Гц, 1 Н); 7,54 (дд, J=l,9, 8,5 Гц, 1 Н); 7,60 (т, J=9,6 Гц, 1 Н); 7,72 (д, J=8,1 Гц, 2 Н); 7,79 (м,1 Н); 7,83 (д, J=9,1 Гц, 1 Н); 7,89 (д, J=8,5 Гц, 1 Н); 7,98 (ушир.с, 1 Н); 8,30 (дд, J=2,7, 6,7 Гц, 1 Н); 8,45 (т,NH, J=5,6 Гц, 1 Н). 1H ЯМР данные для примера 21 - ЯМР (500 МГц, DMSO-d6) : 1,41 (д, J=6,8 Гц, 3 Н); 2,46 (т, J=7,0 Гц, 2 Н); 3,40 (кв, J=6,8 Гц, 2 Н); 3,88 (с, 3 Н); 4,17 (кв, J=6,8 Гц, 2 Н); 5,53 (с, 2 Н); 7,00 (с, 1 Н); 7,05 (дд,J=2,0, 8,3 Гц, 1 Н); 7,11 (д, J=8,2 Гц, 2 Н); 7,17 (д, J=2,0 Гц, 1 Н); 7,21 (дд, J=2,5, 9,0 Гц, 1 Н); 7,36 (д, J=2,5 Гц, 1 Н); 7,50 (ушир.д, J=8,5 Гц 1 Н); 7,71 (д, J=3,2 Гц, 2 Н); 7,82 (д, J=9,0 Гц, 1 Н); 7,89 (д, J=8,5 Гц, 1 Н); 7,92 (ушир.с, 1 Н); 7,95 (д, J=8,3 Гц, 1 Н); 8,45 (т, NH, J=5,6 Гц, 1 Н). Таблица 2 1 Масс-спектрометрические данные отсутствуют. Н ЯМР данные для примера 25 - ЯМР (500 МГц, DMSO-d6) : 2,45 (т, J=7,l Гц, 2 Н); 3,40 (кв, J=6H ЯМР данные для примера 26 - ЯМР (500 МГц, DMSO-d6) : 2,45 (т, J=7,1 Гц, 2 Н); 3,41 (кв, J=7 Гц, 2 Н); 5,57 (с, 2 Н); 7,14 (д, J=8,2 Гц, 2 Н); 7,39 (с, 1 Н); 7,53 (т, J=1,9 Гц, 1 Н); 7,61 (ушир.д, J=8,0 Гц, 1H); 7,66 (т, J=7,9 Гц, 1 Н); 7,76 (д, J=8,3 Гц, 3 Н); 7,94 (д, J=1,9 Гц, 2 Н); 8,02 (ушир.с, 1 Н); 8,04 (д, J=8,1 Гц,1 Н); 8,18 (д, J=8,3 Гц, 1 Н); 8,47 (т, NH, J=5,6 Гц). Таблица 3
МПК / Метки
МПК: A61K 31/415, A61K 31/4155, C07D 231/12, C07D 405/04
Метки: композиции, производные, применение, пиразола, фармацевтические
Код ссылки
<a href="https://eas.patents.su/30-12431-proizvodnye-pirazola-farmacevticheskie-kompozicii-i-ih-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пиразола, фармацевтические композиции и их применение</a>
Производные бензоксазина в качестве модуляторов 5-нт-6, способ их получения, фармацевтические композиции, содержащие эти производные, и их применение

Номер патента: 9982
Опубликовано: 28.04.2008
Авторы: Кларк Робин Дуглас, Бергер Якоб, Чжао Шухай
МПК: A61K 31/536, A61P 25/18, C07D 265/36...
Метки: производные, содержащие, модуляторов, эти, бензоксазина, качестве, композиции, применение, способ, фармацевтические, получения, 5-нт-6
Формула / Реферат:
1. Соединение формулы его фармацевтически приемлемая соль или пролекарство, где m означает целое число от 0 до 3; n означает 2 или 3; р означает 2; Y означает -S(O2)-; Z1 означает N; R1 и R2 означают водород; R3 означает фенил или нафтил, которые необязательно замещены галогеном, алкилом, алкокси, гидрокси, циано, пиперазинилом, метилсульфониламино, метилсульфонилом, аминокарбонилом, или означает хинолинил, изохинолинил, тиофенил,...
Производные диазепинкарбоксамида, способ их получения, их применение в качестве лекарственных средств, фармацевтические композиции и их применение

Номер патента: 5288
Опубликовано: 30.12.2004
Авторы: Можер Жак, Бхатнагар Неерджа
МПК: A61K 31/551, C07K 5/02, C07D 487/04...
Метки: качестве, композиции, фармацевтические, производные, получения, лекарственных, способ, средств, диазепинкарбоксамида, применение
Формула / Реферат:
1. Соединения формулы (I) в которой R1 означает радикал -C(O)-R5, -SO2-R5 или -C(O)-NR6R5, в которых R6 означает атом водорода или линейный или разветвленный алкил, содержащий не более 4 атомов углерода, и R5 означает линейный или разветвленный алкил, содержащий не более 6 атомов углерода, циклоалкил, содержащий не более 6 атомов углерода, фенил, нафтил или насыщенный или ненасыщенный 5- или 6-членный гетероциклический моноциклический радикал,...
Производные бензотиадиазина, способ их получения, фармацевтические композиции и применение

Номер патента: 4053
Опубликовано: 25.12.2003
Авторы: Лестаж Пьер, Бовери Стефан, Пиротт Бернар, Де Тюллио Паскаль, Кампан Изабелла
МПК: A61K 31/549, C07D 285/24, A61P 25/18...
Метки: производные, фармацевтические, получения, применение, бензотиадиазина, композиции, способ
Формула / Реферат:
1. Соединение формулы (I) в котором X обозначает атом фтора, брома или йода, либо метильную группу, каждый из R1 и R2, которые могут быть одинаковыми или различными, представляют собой атом водорода либо линейную или разветвленную группу (C1-C6)алкил, его изомеры, когда они существуют, и его дополнительные соли с фармацевтически приемлемой кислотой. 2. Соединение формулы (I) по п.1, которое представляет собой...
Производные нафтиридина, способ их получения, их применение и содержащие их фармацевтические композиции

Номер патента: 4369
Опубликовано: 29.04.2004
Авторы: Пейман Ануширван, Рюксер Жан-Мари, Шойнеманн Карл-Хайнц, Гурве Жан-Франсуа, Гейдек Томас Р.
МПК: C07D 473/34, A61K 31/52
Метки: содержащие, получения, нафтиридина, производные, фармацевтические, композиции, способ, применение
Формула / Реферат:
1. Соединение формулы I где G представляет собой остаток формулы II -(CR1 R2)n-A-(CR1 R2)m-(CR1 R3)i-(CR1 R2)q-R4 II, A представляет собой простую связь; B представляет собой (C1-C6)алкил или гидрокси, где все группы B являются независимыми друг от друга и могут быть одинаковыми или различными; X представляет собой водород; Y представляет собой водород; Z представляет собой N; R1 и R2 представляют водород, (C1-C4)алкил, R6S(O)2NHR7 или...
Твердые фармацевтические композиции, содержащие 4 -амино-3-замещенные производные бутановой кислоты, способ их получения и применение увлажнителя для их стабилизации

Номер патента: 6553
Опубликовано: 24.02.2006
Автор: Аоматсу Акира
МПК: A61K 9/16, A61K 31/196, A61K 47/18...
Метки: увлажнителя, применение, амино-3-замещенные, фармацевтические, содержащие, твердые, способ, стабилизации, производные, получения, бутановой, композиции, кислоты
Формула / Реферат:
1. Твердая фармацевтическая композиция, содержащая 4-амино-3-замещенное производное бутановой кислоты и стабилизатор, отличающаяся тем, что в качестве 4-амино-3-замещенного производного бутановой кислоты она содержит соединение, выбранное из группы, включающей габапентин, прегабалин и их смеси, а в качестве стабилизатора - увлажнитель, выбранный из этиленгликоля, пропиленгликоля, бутиленгликоля, сорбита, глицерина и глицеринового эфира...
Предыдущий патент: Производные пропионамида, полезные в качестве модуляторов андрогенных рецепторов
Случайный патент: Способ противоточной регенерации ионитов