Производные 2-карбамид-4-фенилтиазола, способ их получения и их применение в терапии
Номер патента: 11076
Опубликовано: 30.12.2008
Авторы: Казелла Пьер, Фресс Пьер, Жегам Самир, Флутар Даниель




Формула / Реферат
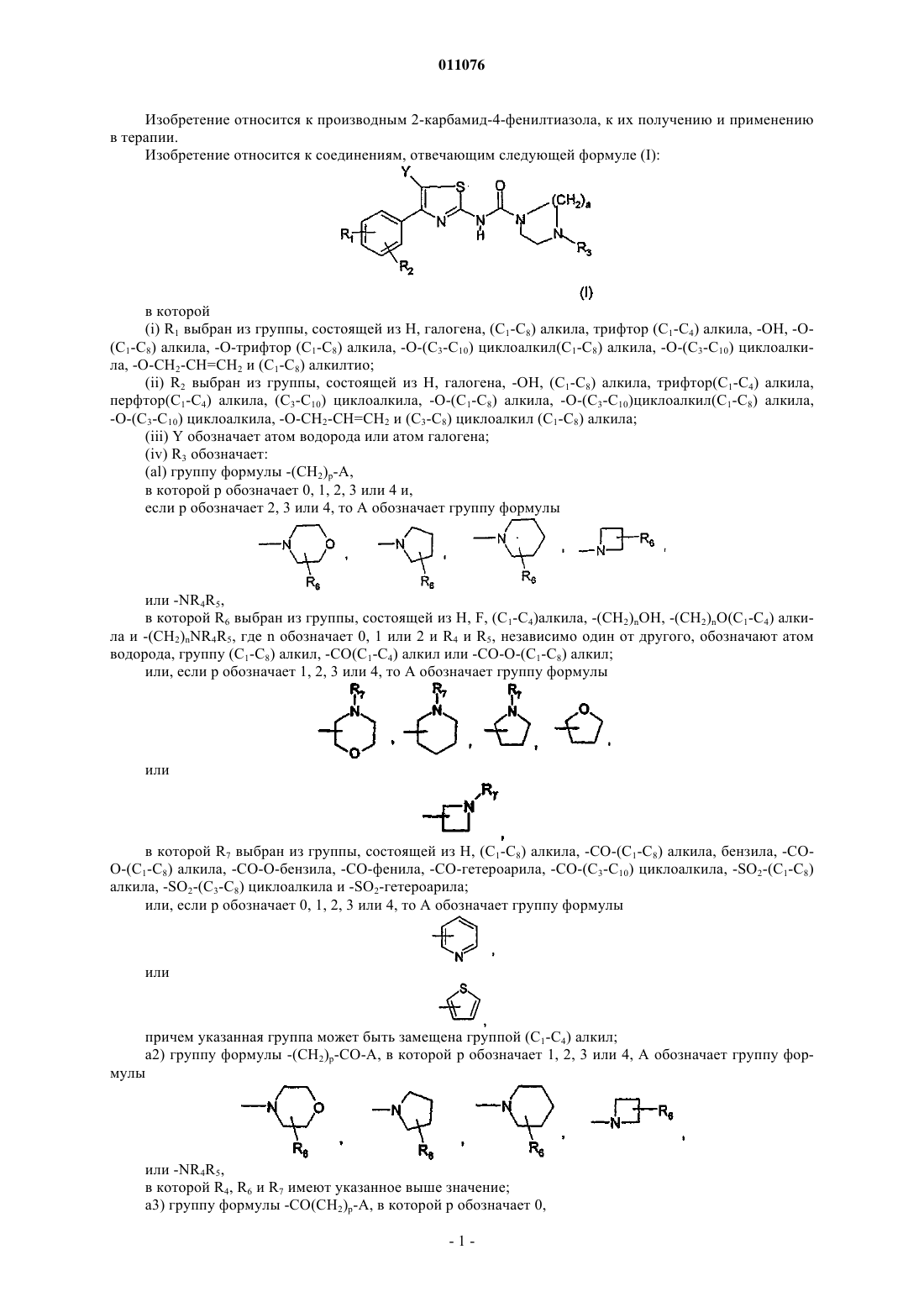
1. Соединение общей формулы (I)

в которой
(i) R1 выбран из -О-(C1-C8)алкила;
(ii) R2 выбран из группы, состоящей (C1-C8)алкила, перфтор(C1-C4)алкила, (С3-C6)циклоалкила и -О-(C1-C8)алкила;
(iii) Y обозначает атом водорода или атом галогена;
(iv) R3 обозначает:
(a1) группу формулы -(CH2)р-А, в которой р обозначает 0, 1, 2, 3 или 4, и,
если р обозначает 2, 3 или 4, то А обозначает группу формулы

или -NR4R5,
в которой R6 выбран из группы, состоящей из Н, F, (C1-С4)алкила, -(CH2)nOH, -(СН2)nO(С1-С4)алкила и -(СН2)nNR4R5, где n обозначает 0, 1 или 2 и R4 и R5 независимо один от другого обозначают атом водорода, группу (C1-C8)алкил, -СО(C1-C4)алкил или -CO-O-(C1-C8)алкил;
или, если р обозначает 1, 2, 3 или 4, то А обозначает группу формулы

или

в которой R7 выбран из группы, состоящей из Н, (C1-С8)алкила, -СО-(C1-C8)алкила, бензила, -CO-O-(C1-C8)алкила, -CO-О-бензила, -СО-фенила, -СО-гетероарила, где гетероарил выбран из группы, состоящей из пиразина, пиримидина, пиридазина, пиридина, триазина, триазола, тиазола, оксазола, пиразола, имидазола, оксопиридина, -СО-(C3-C10)циклоалкила и -SO2-(C1-С8)алкила;
или, если р обозначает 0, 1, 2, 3 или 4, то А обозначает группу формулы

или

причем указанная группа может быть замещена группой (C1-С4)алкил;
а2) группу формулы -(СН2)p-СО-А, в которой р обозначает 1, 2, 3 или 4, А обозначает группу формулы

или -NR4R5,
в которой R4, R5 и R6 имеют указанное выше значение;
а3) группу формулы -СО(CH2)р-А, в которой р обозначает 0, 1, 2, 3 или 4 и,
если р обозначает 1, 2, 3 или 4, то А обозначает группу формулы

или NR4R5,
в которой R4, R5 и R6 имеют указанное выше значение;
или, если р обозначает 0, 1, 2, 3 или 4, то А обозначает группу формулы

или

в которой R7 имеет указанное выше значение;
или, если р обозначает 0, 1, 2, 3 или 4, то А обозначает группу формулы

или

причем указанная группа может быть замещена группой (C1-С4)алкил;
а4) группу -В, в которой В обозначает группу формулы

или

в которой R7 имеет указанное выше значение;
(v) а обозначает 2 или 3;
в виде основания или аддитивной соли кислоты, а также в виде гидратов или сольватов.
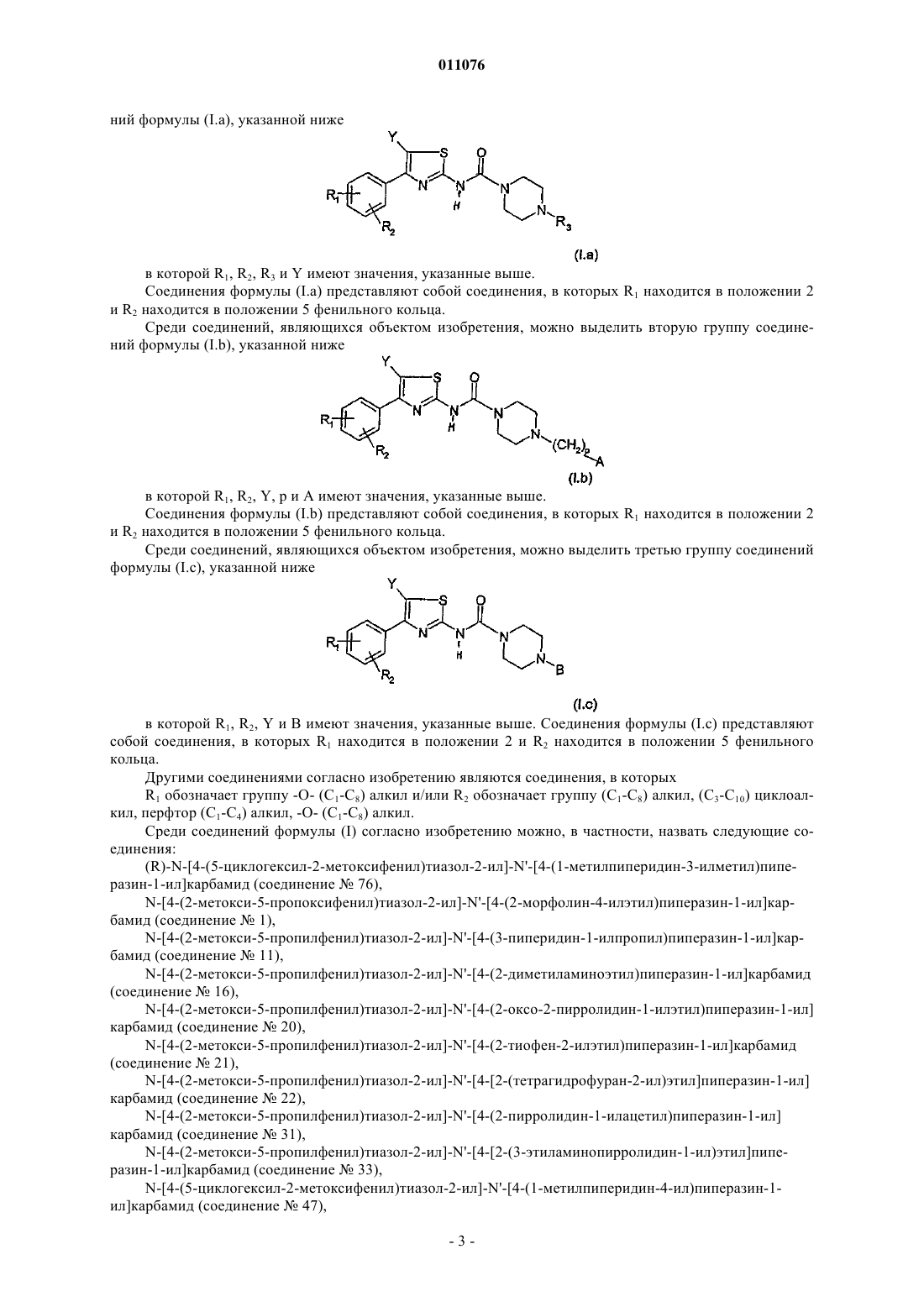
2. Соединение по п.1 формулы (I.a)

в которой R1, R2, R3 и Y имеют указанное выше значение,
в виде основания или аддитивной соли кислоты, а также в виде гидратов или сольватов.
3. Соединение по п.1 формулы (I.b)

в которой R1, R2, Y, р и А имеют указанное выше значение,
в виде основания или аддитивной соли кислоты, а также в виде гидратов или сольватов.
4. Соединение по п.1 формулы (I.c)

в которой R1, R2, Y и В имеют указанное выше значение,
в виде основания или аддитивной соли кислоты, а также в виде гидратов или сольватов.
5. Соединения по одному из пп.1 или 2, выбранные из
(R)-N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-метилпиперидин-3-илметил)пиперазин-1-ил]карбамида,
N-[4-(2-метокси-5-пропоксифенил)тиазол-2-ил]-N'-[4-(2-морфолин-4-илэтил)пиперазин-1-ил]карбамида,
N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-(3-пиперидин-1-илпропил)пиперазин-1-ил]карбамида,
N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-(2-диметиламиноэтил)пиперазин-1-ил]карбамида,
N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-(2-оксо-2-пирролидин-1-илэтил)пиперазин-1-ил]карбамида,
N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-(2-тиофен-2-илэтил)пиперазин-1-ил]карбамида,
N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-[2-(тетрагидрофуран-2-ил)этил]пиперазин-1-ил]карбамида,
N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-(2-пирролидин-1-илацетил)пиперазин-1-ил] карбамида,
N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-[2-(3-этиламинопирролидин-1-ил)этил]пиперазин-1-ил]карбамида,
N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-метилпиперидин-4-ил)пиперазин-1-ил]карбамида,
N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-бензилпиперидин-3-ил)пиперазин-1-ил]карбамида,
N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-(2-пиридин-4-илэтил)пиперазин-1-ил]карбамида,
N-[4-(2-метокси-5-пентафторэтилфенил)тиазол-2-ил]-N'-[4-(2-пирролидин-1-илэтил)пиперазин-1-ил]карбамида,
N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-изопропилпиперидин-3-ил)пиперазин-1-ил]карбамида,
N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(2-пирролидин-1-илэтил)пиперазин-1-ил]карбамида,
N-[4-(5-циклогексил-2-метоксифенил)-5-фтортиазол-2-ил]-N'-[4-(1-метилпиперидин-4-ил)пиперазин-1-ил]карбамида,
N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(2-оксо-2-пирролидин-1-илэтил)пиперазин-1-ил]карбамида,
N-[4-(5-циклогексил-2-этоксифенил)тиазол-2-ил]-N'-[4-(2-оксо-2-пирролидин-1-илэтил)пиперазин-1-ил]карбамида,
N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(2-оксо-2-морфолино-4-илэтил)пиперазин-1-ил]карбамида,
N-[4-(5-циклогексил-2-этоксифенил)тиазол-2-ил]-N'-[4-(2-оксо-2-морфолино-4-илэтил)пиперазин-1-ил]карбамида,
(S)N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-сульфометилпиперидин-3-илметил) пиперазин-1-ил]карбамида,
N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-оксо-1-(1-оксопиридин-2-ил)мет-1-илпиперидин-3-илметил)пиперазин-1-ил]карбамида.
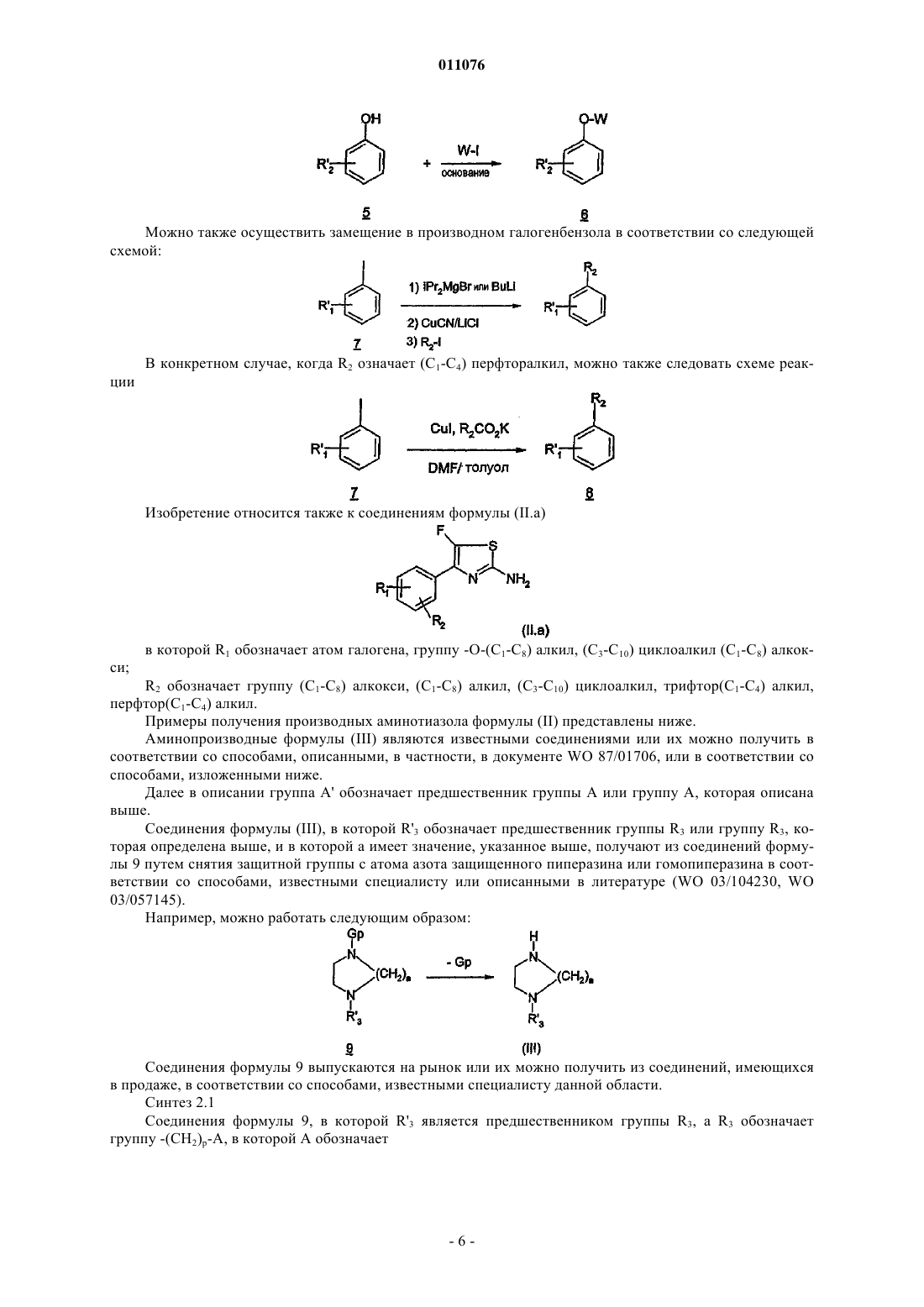
6. Способ получения соединения формулы (I) по одному из пп.1-5, включающий стадию, заключающуюся во взаимодействии соединения формулы (II) с аминопроизводным формулы (III)

в которой R1, R2, R3, Y и а имеют указанное в п.1 значение и R'3 представляет собой предшественник группы R3 или группу R3, которая имеет указанное в п.1 значение, в присутствии агента сочетания и в растворителе, таком как дихлорметан, диметилформамшф, толуол, в присутствии основания, такого как триэтиламин, K2CO3, при температуре в интервале от 0 до 100шС.
7. Соединение следующей формулы (II.а):

в которой
R1 обозначает атом галогена, группу -О-(C1-C8)алкил, -O-(C3-C10)циклоалкил(C1-C8)алкил;
R2 обозначает группу -О-(C1-C8)алкил, (C1-C8)алкил, (С3-C10)циклоалкил, трифтор(C1-C4)алкил, перфтор(C1-C4)алкил.
8. Лекарственное средство, являющееся антагонистом хемокинов, отличающееся тем, что оно содержит соединение формулы (I) по одному из пп.1-5, или аддитивную соль этого соединения с фармацевтически приемлемой кислотой, или же гидрат или сольват соединения формулы (I).
9. Фармацевтическая композиция в качестве антагониста хемокинов, отличающаяся тем, что она содержит соединение формулы (I) по одному из пп.1-5, или фармацевтически приемлемую соль, или гидрат, или сольват этого соединения, а также по меньшей мере один фармацевтически приемлемый эксципиент.
10. Применение соединения формулы (I) по одному из пп.1-5 для получения лекарственного средства, предназначенного для лечения или профилактики острых или хронических иммуно-воспалительных заболеваний и синдромов, таких как атеросклероз, аллергических заболеваний, а также заболеваний, в которых задействованы ангиогенные процессы.
11. Применение соединения формулы (I) по п.10 для получения лекарственного средства, предназначенного для лечения или профилактики раков, в которых задействованы ангиогенные процессы.
12. Применение соединения формулы (I) по одному из пп.1-5 для получения лекарственного средства, предназначенного для лечения или профилактики вирусных или бактериальных заболеваний, сердечных патологий, ожирения.
13. Применение соединения формулы (I) по одному из пп.1-5 для получения лекарственного средства,предназначенного для лечения или профилактики заболеваний, связанных с модулированием активности рецептора CCR2b.
Текст
011076 Изобретение относится к производным 2-карбамид-4-фенилтиазола, к их получению и применению в терапии. Изобретение относится к соединениям, отвечающим следующей формуле (I):(iii) Y обозначает атом водорода или атом галогена; или -NR4R5,в которой R6 выбран из группы, состоящей из Н, F, (С 1-С 4)алкила, -(CH2)nOH, -(CH2)nO(C1-C4) алкила и -(CH2)nNR4R5, где n обозначает 0, 1 или 2 и R4 и R5, независимо один от другого, обозначают атом водорода, группу (C1-C8) алкил, -CO(C1-C4) алкил или -CO-O-(C1-C8) алкил; или, если р обозначает 1, 2, 3 или 4, то А обозначает группу формулы причем указанная группа может быть замещена группой (C1-C4) алкил; а 2) группу формулы -(CH2)р-СО-А, в которой р обозначает 1, 2, 3 или 4, А обозначает группу формулы или -NR4R5,в которой R4, R6 и R7 имеют указанное выше значение; а 3) группу формулы -CO(CH2)p-А, в которой р обозначает 0,-1 011076 1, 2, 3 или 4 и,если р обозначает 1, 2, 3 или 4, то А обозначает группу формулы или -NR4R5,в которой R4, R5 и R6 имеют указанное выше значение; или, если р обозначает 0, 1, 2, 3 или 4, то А обозначает группу формулы в которой R7 имеет указанное выше значение; или, если р обозначает 0, 1, 2, 3 или 4, то А обозначает группу формулы или причем указанная группа может быть замещена группой (C1-С 4) алкил; а 4) группу -В, в которой В обозначает группу формулы или в которой R7 имеет указанное выше значение;(v) а обозначает 2 или 3. Предпочтительным галогеном является фтор. Соединения формулы (I) могут содержать один или несколько асимметричных атомов углерода. В этой связи соединения могут существовать в форме энантиомеров или диастереоизомеров. Эти энантиомеры, диастереоизомеры, а также их смеси, в том числе рацемические смеси, составляют часть изобретения. Соединения формулы (I) могут находиться в виде оснований или кислотно-аддитивных солей. Такие аддитивные соли составляют часть изобретения. Эти соли получают преимущественно с фармацевтически приемлемыми кислотами, однако соли с другими кислотами, пригодными, например, для очистки или выделения соединений формулы (I), также составляют часть изобретения. Соединения формулы (I) могут существовать также в форме гидратов или сольватов, а именно в форме ассоциаций или комбинаций с одной или несколькими молекулами воды или с растворителем. Такие гидраты и сольваты также составляют часть изобретения. В объеме настоящего изобретения подразумевают под-Ct-z, где t и z могут принимать значения от 1 до 10, углеродсодержащую цепь, которая может содержать t-z атомов углерода, например цепь C1-3, которая может содержать 1-3 атомов углерода;-Hal - атом галогена, такой как фтор, хлор, бром или иод; алкильной группой: алифатическую, насыщенную, линейную или разветвленную группу, возможно, замещенную атомом галогена. В качестве примеров можно назвать группы метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, 2-фторэтил и т.д.; циклоалкильной группой: циклическую алкильную группу. В качестве примеров можно назвать группы циклопропил, циклобутил, циклопентил, циклогексил и т.д.; алкоксильной группой: радикал -О-алкил, в котором группа алкил имеет указанные выше значения; гетероарильной группой: арильную группу, содержащую один или несколько гетероатомов, возможно, замещенную другим гетероатомом. В качестве примеров можно назвать пиразиновую, пиримидиновую, пиридазиновую, пиридиновую, триазиновую, триазольную, тиазольную, оксазольную, пиразольную, имидазольную, оксопиридиновую и т.д. группы; перфторалкильной группой: радикал алкил, определенный выше, в котором все атомы углерода замещены атомами фтора. Среди соединений, являющихся объектом изобретения, можно выделить первую группу соедине-2 011076 ний формулы (I.a), указанной ниже в которой R1, R2, R3 и Y имеют значения, указанные выше. Соединения формулы (I.a) представляют собой соединения, в которых R1 находится в положении 2 и R2 находится в положении 5 фенильного кольца. Среди соединений, являющихся объектом изобретения, можно выделить вторую группу соединений формулы (I.b), указанной ниже в которой R1, R2, Y, р и А имеют значения, указанные выше. Соединения формулы (I.b) представляют собой соединения, в которых R1 находится в положении 2 и R2 находится в положении 5 фенильного кольца. Среди соединений, являющихся объектом изобретения, можно выделить третью группу соединений формулы (I.c), указанной ниже в которой R1, R2, Y и В имеют значения, указанные выше. Соединения формулы (I.c) представляют собой соединения, в которых R1 находится в положении 2 и R2 находится в положении 5 фенильного кольца. Другими соединениями согласно изобретению являются соединения, в которыхR1 обозначает группу -О- (C1-C8) алкил и/или R2 обозначает группу (C1-C8) алкил, (C3-C10) циклоалкил, перфтор (С 1-С 4) алкил, -О- (C1-C8) алкил. Среди соединений формулы (I) согласно изобретению можно, в частности, назвать следующие соединения:(соединение 28),N-[4-(2-метокси-5-пентафторэтилфенил)тиазол-2-ил]-N'-[4-(2-пирролидин-1-илэтил)пиперазин-1 ил]карбамид (соединение 29),N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-изопропилпиперидин-3-ил)пиперазин-1 ил]карбамид (соединение 70),N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(2-пирролидин-1-илэтил)пиперазин-1-ил] карбамид (соединение 30),N-[4-(5-циклогексил-2-метоксифенил)-5-фтортиазол-2-ил]-N'-[4-(1-метилпиперидин-4-ил)пиперазин-1-ил]карбамид (соединение 59),N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(2-оксо-2-пирролидин-1-илэтил)пиперазин-1-ил]карбамид (соединение 108),N-[4-(5-циклогексил-2-этоксифенил)тиазол-2-ил]-N'-[4-(2-оксо-2-пирролидин-1-илэтил)пиперазин 1-ил]карбамид (соединение 109),N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(2-оксо-2-морфолино-4-илэтил)пиперазин 1-ил]карбамид (соединение 116),N-[4-(5-циклогексил-2-этоксифенил)тиазол-2-ил]-N'-[4-(2-оксо-2-морфолино-4-илэтил)пиперазин-1 ил]карбамид (соединение 110),(S)N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-сульфометилпиперидин-3-илметил) пиперазин-1-ил]карбамид (соединение 112),N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-оксо-1-(1-оксопиридин-2-ил)мет-1-илпиперидин-3-илметил)пиперазин-1-ил]карбамид (соединение 113). Некоторые промежуточные соединения, пригодные для получения соединений формулы (I), могут одновременно являться конечным продуктом формулы (I), как это показано в примерах, описанных ниже. И, аналогично, некоторые соединения формулы (I) согласно изобретению могут являться промежуточными соединениями, пригодными для получения соединений формулы (I) согласно изобретению. В последующем тексте описания под защитной группой Gp понимают группу, которая может, с одной стороны, защитить реакционноспособную группу, такую как гидроксильная или аминогруппа, в процессе синтеза и, с другой стороны, высвободить сохранившуюся реакционноспособную группу в конце синтеза. Примеры защитных групп, а также способы защиты и способы снятия защитных групп изложены в работе "Protective Groups in Organic Synthesis", Green et al., 2nd Edition (John WileySons,Inc., New York). Под удаляемой группой X в дальнейшем тексте понимают группу, которая может быть легко отщеплена от молекулы путем гетеролитического разрыва связи одновременно с уходом электронной пары. Эту группу можно, таким образом, легко заменить на другую группу, например, в процессе реакции замещения. Такими удаляемыми группами являются, например, атомы галогена или активированная гидроксильная группа, такая как мезильная, тозильная, трифлатная, ацетильная и т.д. Примеры удаляемых групп, а также ссылки, в которых раскрыто их получение, изложены в работе "Advances in Organic Chemistry", J.March, 3rd Edition, Wiley Interscience, p.310-316. В дальнейшем тексте под предшественником радикала R1, R2 или R3 понимают радикал R1', R2' илиR3', который может превратиться в R1, R2 и R3 в результате одной или нескольких химических реакций. Под группой Z понимают в данном описании удаляемую группу или группу, представляющую собой функциональное производное кислоты, такое как хлорангидрид кислоты, смешанный или симметричный ангидрид, или же соответствующим образом активированную кислоту, например, с помощью гексафторфосфата бензотриазол-1-илокситрис(диметиламино)фосфония (ВОР), гексафторфосфата Oбензотриазол-1-ил-N,N,N',N'-тетраметилурония (HBTU) или тетрафторбората O-бензотриазол-1-илN,N,N',N'-тетраметилурония (TBTU). Если один или несколько заместителей R1', R2' и/или R3' обозначают группу, содержащую аминоили гидроксильную функциональную группу, то последние могут быть защищены на промежуточном этапе: аминогруппа может быть защищена, например, алканоильной, бензильной, третбутоксикарбонильной (Boc), бензилоксикарбонильной или 9-флуоренилметоксикарбонильной (Fmoc) группой; гидроксильная функциональная группа может быть защищена в форме простого или сложного эфира. Соединения согласно изобретению могут быть получены в соответствии со способами, описанными в настоящей заявке. Но прежде, ниже описаны способы получения производных аминотиазола формулы (II) и способы получения аминопроизводных формулы (III), которые использованы для получения соединений формулы (I) согласно изобретению. Производные аминотиазола формулы (II) могут быть получены известными способами, такими как-4 011076 способы, описанные в документах ЕР 518731, ЕР 611766 и WO 99/15525. Согласно общему способу, когда Y=H, тиомочевину подвергают взаимодействию с галогенсодержащим кетоном формулы 1 согласно следующей реакционной схеме: Заместители R1' и R2' имеют значения, указанные выше, т.е. R1' и R2' обозначают соответственно R1 и R2, значения которых даны в отношении соединения общей формулы (I), или представляют собой предшественники радикалов R1 и R2; Hal обозначает атом галогена, предпочтительно брома, хлора или иода. Как показано на представленной выше схеме, соединения типа (II), где Y=H, a R1' и R2' имеют значения, указанные выше, могут быть превращены в соединения типа (II), где Y=F, a R1' и R2' имеют значения, указанные выше, путем взаимодействия с фторирующим агентом, таким как, например,Selectfluor, в растворителе, таком как DMF или DCM, при температуре 0-50 С. Галогенсодержащие кетоны формулы 1 могут быть получены известными для специалиста способами. Например, бромкетоны могут быть получены при действии бромом, бромидом меди (II) или фенилтриметиламмонийтрибромидом (РТТ) на производное ацетофенона формулы в которой R1' и R2' имеют значения, указанные выше, в органическом растворителе, таком как этилацетат, в хлорированном растворителе или в их смеси, или же в спирте. Если производное ацетофенона формулы 2 не выпускается в продажу, то его можно получить различными способами реакцией Фриделя-Крафтса с использованием бензола, замещенного радикалами R1' и R2', который подвергают взаимодействию с ацетилхлоридом или с уксусным ангидридом, в присутствии кислоты Льюиса, такой как, например, AlCl3 или TiCl4; действием ацетилхлорида в присутствии палладия на бензол, замещенный радикалами R1' и R2' после депротонирования бензола, например действием бутиллития с последующим добавлением хлорида цинка или иодида марганца. Эта процедура используется для получения производного ацетофенона формулы 2, в которой R1' = R2' = (С 1-С 4)перфторалкил; перегруппировкой Фриса, исходя из производного ацетоксибензола формулы и под действием кислоты Льюиса получают производное гидроксиацетофенона формулы Гидроксильная функциональная группа соответствует группе R'1, ее можно превратить в дальнейшем в группу -O-W, такую как -О-(С 1-С 8)алкил, трифторметокси, трифторэтокси, аллилокси, (С 3C10)циклоалкилметокси, (C3-C10)циклоалкилокси. Превращение R'1 в R1 может быть осуществлено либо в аминотиазоле формулы (II), либо в соединении формулы (I). Производные бензола, замещенные радикалами R1' и R2', доступны в продаже или могут быть получены известными специалисту способами. Например, для получения соединения, в котором R1 означает группу -O-W, которая описана выше,работают следующим образом: Можно также осуществить замещение в производном галогенбензола в соответствии со следующей схемой: В конкретном случае, когда R2 означает (C1-C4) перфторалкил, можно также следовать схеме реак Изобретение относится также к соединениям формулы (II.а)R2 обозначает группу (C1-C8) алкокси, (C1-C8) алкил, (C3-C10) циклоалкил, трифтор(C1-C4) алкил,перфтор(С 1-С 4) алкил. Примеры получения производных аминотиазола формулы (II) представлены ниже. Аминопроизводные формулы (III) являются известными соединениями или их можно получить в соответствии со способами, описанными, в частности, в документе WO 87/01706, или в соответствии со способами, изложенными ниже. Далее в описании группа А' обозначает предшественник группы А или группу А, которая описана выше. Соединения формулы (III), в которой R'3 обозначает предшественник группы R3 или группу R3, которая определена выше, и в которой а имеет значение, указанное выше, получают из соединений формулы 9 путем снятия защитной группы с атома азота защищенного пиперазина или гомопиперазина в соответствии со способами, известными специалисту или описанными в литературе (WO 03/104230, WO 03/057145). Например, можно работать следующим образом: Соединения формулы 9 выпускаются на рынок или их можно получить из соединений, имеющихся в продаже, в соответствии со способами, известными специалисту данной области. Синтез 2.1 Соединения формулы 9, в которой R'3 является предшественником группы R3, a R3 обозначает группу -(CH2)р-А, в которой А обозначает р обозначает 2, 3 или 4 и R4, R5, R6 имеют значения, указанные выше, могут быть получены реакцией соединения 10 с А'Н в присутствии основания, такого как K2CO3, триэтиламин или карбонат цезия, в растворителе, таком как THF, ацетонитрил, толуол или DMF, при температуре от 0 до 150 С, чтобы получить соединение 11. Соединение 11 может быть затем превращено в соединение формулы 9 путем восстановления амидной функции, например, с помощью LiAlH4, гидрида диизобутилалюминия (Dibal),ВН 3, в THF, простом эфире или толуоле при температуре 0-70 С с получением соединения формулы 9, в которой R3 обозначает группу -(СН 2)p-А Соединение формулы 10 можно получить взаимодействием монозащищенного пиперазина или гомопиперазина с помощью реактива формулы 12, указанной ниже в которой Z обозначает удаляемую группу или группу, полученную после активации карбоксильной функциональной группы кислоты, и X обозначает удаляемую группу в растворителе, таком как THF,ацетонитрил, DMF или дихлорметан, в присутствии основания, такого как K2CO3, триэтиламин, и, если Z обозначает группу ОН в присутствии реагента, активирующего кислотную функцию, такого как ВОР,TBTU или CDI, согласно следующей схеме Синтез 2.2 Соединения формулы 9, в которой R'3 представляет собой предшественник группы R3, a R3 обозначает группу -(CH2)р-А, в которой А обозначает и р обозначает 1, 2, 3 или 4, R7 имеет значения, указанные выше, могут быть получены путем взаимодействия соединения 13 с монозащищенным пиперазином или гомопиперазином, в растворителе, таком как THF, ацетонитрил, DMF или дихлорметан, в присутствии основания, такого как K2CO3, триэтиламин, и, если Z обозначает группу ОН, в присутствии реагента, активирующего кислотную функцию,такого как ВОР, TBTU или CDI, согласно следующей схеме: Полученное соединение 11 можно затем превратить в соединение формулы 9 согласно схеме, описанной выше. Соединение 13, если оно отсутствует в продаже, может быть получено путем перевода продаваемой карбоновой кислоты в ее соответствующий гомолог согласно классическим способам, таким как реакции типа Arndt-Eistert (Tetrahedron Lett., 1979, 29, 2667; "Advances in Organic Chemistry", J.March, 3rd Edition,Wiley Interscience, p.1405-1407), согласно следующей схеме: Синтез 2.3 Соединения формулы 9, в которой R'3 представляет собой предшественник группы R3, a R3 обозначает группу -(CH2)р СО-А, описанную выше, могут быть получены из соединений формулы 14, в которойZ имеет указанное выше значение, путем взаимодействия с соединением А'Н в присутствии основания,такого как K2CO3, триэтиламин или карбоната цезия, и, если Z обозначает группу -ОН, в присутствии реагента, активирующего кислотную функцию, такого как ВОР, TBTU или CDI, в растворителе, таком как, например, THF, ацетонитрил или DMF, при температуре от 0 до 150 С согласно следующей схеме: Соединения формулы 14, если они отсутствуют в продаже, можно получить из монозащищенного пиперазина или гомопиперазина и реактива 15, в котором Z имеет указанное выше значение, с использованием реакции ацилирования или присоединения пептидного типа в присутствии основания, такого какK2CO3, триэтиламин или карбонат цезия, или агента сочетания, такого как ВОР, TBTU или CDI, в растворителе, таком как, например, THF, ацетонитрил или DMF, при температуре от 0 до 150 С согласно следующей схеме: Затем соединение 16 подвергают восстановлению амидной функциональной группы, например, с помощью LiAlH4 в THF или этиловом эфире при температуре от 0 до 50 С. Полученное промежуточное соединение освобождают от защитной группы и затем окисляют до карбоновой кислоты 17, например, с помощью CrO3 или других реагентов в соответствии с "Advances in Organic Chemistry", J.March, 3rd Edition, Wiley Interscience, p.1537-1539, согласно следующей схеме: Затем соединение 17, при желании, превращают в соединение формулы 14 или используют его в форме кислоты (Z=OH). В качестве варианта соединения формулы 17 могут быть получены взаимодействием монозащищенного пиперазина или гомопиперазина с реактивом 18 формулы в которой X имеет значения, указанные выше, и R обозначает группу (С 1-С 4)алкил, алкилированием азота в присутствии основания, такого как K2CO3, триэтиламин или карбонат цезия, в растворителе, таком как THF, ацетонитрил, толуол или DMF, при температуре 25-150 С согласно следующей схеме: Полученное соединение 19 превращают затем в кислоту формулы 17 путем омыления или кислотного гидролиза, или любым другим способом, известным специалисту. Синтез 2.4 Соединения формулы 9, в которой R'3 представляет собой предшественник группы R3, a R3 обозначает группу -СО(СН 2)р-А, в которой А обозначает р обозначает 1, 2, 3, 4 и R4, R5, R6 имеют значения, указанные выше, могут быть получены с использованием способа, описанного в синтезе 2.1, описанном выше. Синтез 2.5 Соединения формулы 9, в которой R'3 представляет собой предшественник группы R3, а R3 обозначает группу -СО(CH2)р-А, в которой А обозначает р обозначает 0, 1, 2, 3, 4 и R7 имеет значения, указанные выше, могут быть получены с использованием способа, описанного в Синтезе 2,1, описанном выше. Синтез 2.6 Соединения формулы 9, в которой R'3 представляет собой предшественник группы R3, a R3 обозначает группу -В, могут быть получены взаимодействием монозащищенного пиперазина или гомопиперазина с кетоном В', являющимся предшественником В, реакцией восстановительного аминирования в присутствии восстановителя, такого как NaHB(OAc)3, NaBH3CN, в растворителе, таком как 1,2 дихлорэтан, дихлорметан, THF, при температурах в интервале 0-70 С (Synth. Commun., 1998, 261 (10),-9 011076 1897-1905, J.Org.Chem., 1992, 57(11), 3218-3225, J.Org.Chem, 1996, 61, 3849-38(52, Tetrahedron Lett., 1990,31, 5595-5598), согласно следующей схеме: Используемые кетоны В' находятся в продаже или могут быть синтезированы согласно способу,описанному в J.Org.Chem., 1989, 54, 1249-1256. Соединения формулы (I) согласно изобретению могут быть получены согласно общей схеме 1,представленной ниже. Схема 1 Как показано на схеме 1, соединения согласно изобретению получают в результате реакции присоединения производного аминотиазола формулы (II), в которой R1, R2, Y имеют указанное выше значение,с аминопроизводным формулы (III), в которой R'3 представляет собой предшественник группы R3 или группу R3, которая имеет указанное выше значение, и а имеет значение, указанное выше. В соответствии со схемой 1 производное аминотиазола формулы (II) вводят в контакт с агентом сочетания в течение 2-16 ч, затем с аминопроизводным формулы (III) в течение 0,5-4 ч. Агент сочетания может быть выбран из тех, которые известны специалисту, например фосген, ди(N-сукцинимидил)карбонат, 1,1'-карбонилдиимидазол, в соответствии со способами присоединения, изложенными в документе "Encyclopedia of Reagents for Organic Synthesis", L.A.Paquette, volume 1, p.1006;volume 4, p.2304; volume 6, p.4107. Реакцию можно проводить в различных растворителях, например в дихлорметане, диметилформамиде, толуоле, в присутствии основания, такого как триэтиламин, K2CO3, при температуре 0-100 С. Соединения согласно изобретению формулы (I), в которой R3 представляет собой группу-СО(СН 2)р-А (или -СО(CH2)р-А'), в которой А и р имеют значения, указанные выше, и А' представляет собой предшественник группы А, могут быть получены согласно следующей схеме 2. Схема 2 В соответствии со схемой 2, производное аминотиазола формулы (VIII), в которой R1, R2, Y, а и р имеют указанное выше значение и X обозначает удаляемую группу, подвергают взаимодействию с соединением А'Н, предшественником группы А, или с соединением АН, описанным выше, с получением соединения формулы (XII) (соединение формулы (I), в которой R3 представляет собой группу -СО(CH2)рА). Реакцию осуществляют в растворителе, таком как тетрагидрофуран, диметилформамид, в присутствии основания, такого как триэтиламин или K2CO3, при температуре в интервале от комнатной до 150 С в течение 1-24 ч. Соединения согласно изобретению формулы (I), в которой R3 представляет собой группу -(CH2)р-А,могут быть получены известным способом из соединения формулы (XII), описанного выше, непосредственным восстановлением карбонильной функциональной группы с помощью восстановителя, такого какRed-Al, LiAlH4, в соответствии со следующей схемой 3. В качестве варианта, соединения формулы (I) могут быть получены из соединения формулы (XII), в которой группа А защищается перед реакцией восстановления, в частности, в случае, если группа А содержит функциональные группы, несовместимые с типом используемого восстановителя. Затем, после восстановления, соединение формулы (I) получают посредством снятия защиты с группы А и возможной функционализации группы А. Соединения формулы (VIII) могут быть получены в соответствии со следующей схемой 4. Схема 4 В соответствии со схемой 4, производное аминотиазола формулы (II), описанное выше, подвергают реакции присоединения с аминопроизводным формулы (IV), в которой Gp представляет собой защитную группу, например бензильную группу или Boc, и а имеет указанные выше значения, с получением соединения формулы (V). Реакцию осуществляют в тех же условиях, которые описаны выше в отношении схемы 1. В соединении формулы (V) затем удаляют защитную группу в соответствии со способами, известными специалисту, и получают соединение формулы (VI), которое подвергают взаимодействию с соединением формулы (VII), в которой Z обозначает удаляемую группу или группу, получаемую после активации карбоксильной функциональной группы кислоты, и X является удаляемой группой, с получением соединения формулы (VIII), в которой R1, R2, Y, X и а имеют значение, указанное выше. В качестве варианта, соединения формулы (VIII) могут быть получены в соответствии со следующей схемой 5: Схема 5- 11011076 В соответствии со схемой 5, соединение формулы (VII), в которой X имеет указанное выше значение и Z обозначает удаляемую группу или группу, получаемую после активации карбоксильной функциональной группы кислоты, может сочетаться с соединением формулы (IV) путем ацилирования или присоединением пептидного типа в присутствии основания, такого как K2CO3, триэтиламин или карбонат цезия, или агента сочетания, такого как ВОР, TBTU или CDI, в растворителе, таком как, например,THF, ацетонитрил или DMF, при температуре от 0 до 150 С. Таким образом получают соединение формулы (IX). После снятия защиты с соединения формулы (IX) получают соединение формулы (X), которое подвергают затем сочетанию с аминотиазольным соединением формулы (II) в условиях, идентичных условиям, описанным в отношении схемы 1. В описанных общих схемах синтеза исходные соединения и реагенты, в отношении которых не дано описание их получения, являются продуктами, имеющимися в продаже, или описанными в литературе, или же они могут быть получены в соответствии со способами, описанными в литературе или известными специалисту. Следующие ниже примеры описывают получение соединений согласно изобретению. Эти примеры не носят ограничивающий характер и предназначены для иллюстрации настоящего изобретения. Номера соединений, иллюстрированных примерами, соответствуют номерам, указанным в таблице II, которая описывает химическую структуру и физические свойства некоторых соединений согласно изобретению. В методиках приготовления и в примерах использованы следующие сокращения: СуНех = циклогексильная группа; ТА = комнатная температура;TBTU = 2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийтетрафторборат; Получение производных аминотиазола формулы (II) Соединения формулы (II), в которой Y обозначает Н (получения 1.1-1.25), R1 и R2 находятся соответственно в положении 2 и 5 фенильного кольца. Получение 1.1. 4-(2-метокси-5-пропоксифенил)-1,3-тиазол-2-амин.A) 1-(2-гидрокси-5-пропоксифенил)этанон В сосуд объемом 500 мл помещают 10 г 2,5-дигидроксиацетофенона, суспензированные в 100 мл ацетона, добавляют 9,14 г безводного K2CO3 с последующим введением 12,4 г пропилиодида. Реакционную среду кипятят с обратным холодильником в течение 30 ч. После снижения температуры до комнатной среду фильтруют на Clite, затем концентрируют. Полученное масло коричневого цвета поглощают с помощью AcOEt, фильтруют, промывают водой, 2 М раствором HCl, затем насыщенным растворомNaCl. Органическую фазу испаряют с получением пасты черного цвета. Пасту обрабатывают хлороформом и фильтруют. Среду концентрируют до получения 11,4 г твердого вещества черного цвета. Полученный продукт поглощают абсолютным этанолом. Раствор помещают в морозильник на 10 мин, образуется осадок твердого вещества, который отделяют фильтрацией. Фильтрат концентрируют, поглощают этанолом, охлаждают в морозильнике, затем снова фильтруют. Эту операцию повторяют 4 раза и получают 8,35 г целевого соединения в виде порошка.B) 1-(2-метокси-5-пропоксифенил)этанон К раствору 35 г полученного выше твердого продукта в 350 мл DMF добавляют 49,8 г K2CO3, затем 22,4 мл метилиодида. Реакционную среду нагревают в течение 12 ч при 60 С. После снижения температуры до комнатной среду фильтруют на Celite, разбавляют эфиром и промывают 2 М раствором HCl. Водную фазу дважды экстрагируют эфиром. Объединенные органические фазы промывают разбавленным раствором гидроксида натрия, затем 2 раза водой и затем насыщенным раствором NaCl. Органическую фазу сушат над MgSO4, затем испаряют с получением 35,55 г масла коричневого цвета. Масло перегоняют при пониженном давлении и при 115 С с получением 32,8 г целевого соединения в виде масла.C) 2-бром-1-(2-метокси-5-пропоксифенил)этанон К раствору 16,4 г масла, полученного на предыдущей стадии, в 100 мл метанола прикапывают 4,8 мл брома. Среду перемешивают 30 мин при комнатной температуре, затем выпаривают. Полученное масло поглощают дихлорметаном, промывают 3 раза водой, затем сушат над MgSO4, выпаривают и по- 12011076 лучают 24,5 г масла коричневого цвета.D) 4-(2-метокси-5-пропоксифенил)-1,3-тиазол-2-амин К раствору 42 г бромкетона, полученного на предыдущей стадии, в 200 мл этанола, добавляют 24,5 г тиомочевины. Реакционную среду кипятят с обратным холодильником в течение 1 ч 30 мин. Реакционную среду помещают в холодильник на 12 ч, затем фильтруют. Отфильтрованное твердое вещество промывают сначала небольшим количеством холодного этанола, затем эфиром. Получают 25 г бромгидрата. Твердое вещество суспензируют в смеси вода/дихлорметан и подщелачивают добавлением гидроксида натрия. Водную фазу 2 раза экстрагируют дихлорметаном. Объединенные органические фазы сушат над MgSO4, затем испаряют. Полученное масло хроматографируют на силикагеле и получают 12 г целевого продукта в виде порошка. Т.пл.= 76 С. Получение 1.2. 4-(5-бутил-2-метоксифенил)-1,3-тиазол-2-амин. А) 4-бутилфенилацетат Раствор 10 г 4-н-бутилфенола, 10 мл Ас 2 О и 8 мл пиридина перемешивают при кипении с обратным холодильником в 10 мл дихлорметана. После 2 ч перемешивания реакционную среду охлаждают до комнатной температуры, разбавляют дихлорметаном, промывают водой, затем 1 М раствором HCl, насыщенным раствором CuSO4, водой и сушат над MgSO4. После выпаривания получают 10,8 г целевого соединения в виде масла.B) 1-(5-бутил-2-гидроксифенил)этанон К 5 г масла, полученного на предыдущей стадии, находящегося в емкости объемом 100 мл, прибавляют за несколько приемов 3,22 г AlCl3. Реакционную среду нагревают в течение 1 ч при температуре 130 С. После охлаждения до комнатной температуры в неочищенную реакционную среду вливают подкисленную с помощью 35% HCl ледяную воду. Среду помещают в ультразвуковую камеру. ДобавляютAcOEt и получают по истечении 15 мин солюбилизированную среду. Водную фазу экстрагируют 3 раза с помощью AcOEt, органические фазы промывают водой, затем насыщенным раствором NaCl. После сушки над MgSO4 и выпаривания получают 4,5 г масла желтого цвета.C) 1-(5-бутил-2-метоксифенил)этанон К раствору 1 г масла, полученного на предыдущей стадии, в 10 мл DMF прибавляют 1,44 г K2CO3,затем 0,648 мл метилиодида. Среду нагревают при 60 С в течение ночи. После охлаждения до комнатной температуры среду фильтруют на Celite, разбавляют эфиром и промывают 2 М раствором HCl. Водную фазу экстрагируют 2 раза эфиром. Объединенные органические фазы промывают разбавленным раствором гидроксида натрия, затем дважды промывают водой, а затем насыщенным раствором NaCl. Органическую фазу сушат над MgSO4, затем выпаривают и получают 1,27 г масла коричневого цвета. Масло очищают на хроматографической колонке и получают 0,66 г целевого продукта.D) 4-(5-бутил-2-метоксифенил)-1,3-тиазол-2-амин К раствору 0,66 г продукта, полученного на предыдущей стадии, в 10 мл метанола добавляют 0,19 мл брома. Среду перемешивают в течение 10 мин, затем выпаривают и остаток поглощают дихлорметаном. Органическую фазу промывают 3 раза водой, затем сушат над MgSO4. После выпаривания получают 0,79 г целевого продукта. Полученное соединение растворяют в 5 мл этанола в присутствии 0,46 г тиомочевины и кипятят среду с обратным холодильником в течение 2 ч 30 мин. При охлаждении до комнатной температуры выпадает осадок твердого вещества. Полученный твердый продукт промывают небольшим количеством холодного этанола, затем эфиром. Получают 0,6 г бромгидрата. Твердое вещество суспензируют в смеси вода/дихлорметан и подщелачивают путем добавления гидроксида натрия. Водную фазу 2 раза экстрагируют дихлорметаном. Объединенные органические фазы сушат над MgSO4, затем выпаривают с получением 0,34 г масла желтого цвета, которое медленно кристаллизуется. Маточные растворы упаривают, затем перемешивают в смеси вода/дихлорметан и подщелачивают путем добавления гидроксида натрия. Водную фазу 2 раза экстрагируют дихлорметаном. Объединенные органические фазы сушат над MgSO4, затем выпаривают. Полученное масло хроматографируют на силикагеле и получают 0,18 г целевого продукта. Т.пл.= 48 С. Получение 1.22. 4-(5-пентафторэтил-2-метоксифенил)-1,3-тиазол-2-амин.A) 1-метокси-4-пентафторэтилбензол В трехгорлую колбу, объемом 500 мл, снабженную насадкой Дина-Старка и холодильником, вводят в инертной атмосфере 8,3 г пентафторпропионата калия и 9,8 г CuI. Добавляют 90 мл DMF и 110 мл толуола. Среду нагревают до 140 С в атмосфере азота и отгоняют 80 мл толуола. Затем реакционную среду охлаждают до комнатной температуры и обескислораживают путем барботирования азота. Затем добавляют 6 г иоданизола и нагревают при 155 С в течение 20 ч. После возвращения к комнатной температуре среду разбавляют в 200 мл смеси вода/этиловый эфир. Среду затем фильтруют на Celite. Органическую фазу промывают 3 раза водой, сушат над MgSO4 и выпаривают с получением 4,3 г масла каштанового цвета.B) 1-(2-метокси-5-пентафторэтилфенил)этанон К раствору 3,5 г 1-метокси-4-пентафторэтилбензола в 50 мл безводного THF прибавляют при -70 С 7,4 мл 2,5 М раствора BuLi в гексане. Среду перемешивают 30 мин при -70 С, затем 45 мин при 0 С. За- 13011076 тем прибавляют 15,5 мл 1 М раствора хлорида цинка в эфире. После 10 мин перемешивания при 0 С добавляют 1,33 мл ацетилхлорида. Из среды вытесняют кислород с помощью азота и вводят 332 мг бензил(хлор)-бис(трифенилфосфин)палладия в 5 мл безводного THF. Среду перемешивают 2 ч 30 мин при 0 С, затем 72 ч при комнатной температуре. Среду выливают в 2,5 М раствор HCl, затем экстрагируют эфиром. Органическую фазу промывают с помощью 5% раствора NaHCO3 в воде, затем водой и насыщенным раствором NaCl. После сушки над MgSO4 и выпаривания неочищенную среду очищают способом флеш-хроматографии на колонке из диоксида кремния и получают 2,25 г твердого вещества белого цвета. Т.пл.=47 С. С) 4-(2-метокси-5-пентафторэтилфенил)тиазол-2-иламин К раствору 2,25 г продукта, полученного на предыдущей стадии, в 10 мл метанола прибавляют 0,5 мл брома, растворенного в 8 мл метанола. Среду перемешивают 10 мин, затем выпаривают и остаток поглощают дихлорметаном. Органическую фазу промывают 3 раза водой, затем сушат над MgSO4. После выпаривания получают 2,63 г бромсодержащего продукта. Полученное соединение растворяют в 15 мл метанола в присутствии 1,25 г тиомочевины и среду кипятят с обратным холодильником в течение 2 ч. При охлаждении до комнатной температуры выпадает в осадок твердое вещество. Полученное твердое вещество промывают этиловым эфиром. Твердое вещество суспензируют в смеси вода/дихлорметан и подщелачивают путем добавления гидроксида натрия. Водную фазу экстрагируют 2 раза дихлорметаном. Объединенные органические фазы сушат над MgSO4 и выпаривают с получением 1,63 г твердого вещества желтого цвета. Т.пл.=125 С. Получение 1.3. 4-(5-циклогексил-2-метоксифенил)-1,3-тиазол-2-амин. А) К раствору 5 г 4-циклогексилфенола в 60 мл DMF прибавляют 7,84 г K2CO3, затем 3,53 мл метилиодида. Среду нагревают при 60 С в течение ночи. После возвращения к комнатной температуре среду фильтруют на Celite, затем разбавляют эфиром и гидролизуют водой. Водную фазу подкисляют, затем экстрагируют 3 раза с помощью 50 мл эфира. Объединенные органические фазы промывают разбавленным раствором гидроксида натрия, затем 2 раза водой и потом насыщенным раствором NaCl. Органическую фазу сушат над MgSO4 и выпаривают с получением 4,31 г целевого соединения в виде твердого вещества. Т.пл.=67 С.B) 1-(5-циклогексил-2-метоксифенил)этанон Суспензию 5,6 г AlCl3 в 40 мл дихлорметана охлаждают до -10 С. Прибавляют 3 мл AcCl и 4 г соединения, полученного на предыдущей стадии. Среду перемешивают 1 ч при -10 С, затем выливают в колбу, содержащую смесь льда с 35% HCl. После декантации объединенные органические фазы сушат над MgSO4 и выпаривают с получением 4,54 г целевого соединения.C) 4-(5-циклогексил-2-метоксифенил)-1,3-тиазол-2-амин К раствору 4,5 г соединения, полученного на предыдущей стадии, в 25 мл метанола прикапывают 1,16 мл брома. Среду перемешивают 30 мин при комнатной температуре, в процессе чего она становится очень вязкой. Добавляют 5 мл метанола, а затем 3,23 г тиомочевины. Среду кипятят с обратным холодильником в течение 2 ч. После охлаждения ее до комнатной температуры выпадает осадок твердого вещества. Твердое вещество отфильтровывают, затем промывают небольшим количеством холодного метанола. Твердое вещество суспензируют в смеси вода/дихлорметан и подщелачивают путем добавления гидроксида натрия. Водную фазу экстрагируют 2 раза дихлорметаном. Объединенные органические фазы сушат над MgSO4 и выпаривают с получением 3,33 г целевого соединения в виде твердого продукта. Т.пл.=113 С. Получение 1.4. 4-(2-метокси-5-пропилфенил)-1,3-тиазол-2-амин.A) 1-(2-метокси-5-пропилфенил)этанон Суспензию 10,6 г AlCl3 в 150 мл дихлорметана охлаждают до -10 С. Прибавляют 5,7 мл AcCl и 6 г 4-пропиланизола. Среду перемешивают 30 мин при -10 С, затем выливают в колбу, содержащую смесь льда с 35% HCl. После декантации водную фазу экстрагируют 3 раза дихлорметаном, объединенные органические фазы промывают водой, затем насыщенным раствором NaCl, сушат над MgSO4 и выпаривают с получением 7,86 г масла коричневого цвета (количественный выход).B) 2-бром-1-(2-метокси-5-пропилфенил)этанон К раствору 7,86 г соединения, полученного на предыдущей стадии, в 80 мл метанола прибавляют по каплям 2,46 мл брома, растворенного в 40 мл метанола. Среду перемешивают 30 мин при комнатной температуре, затем выпаривают. Полученное масло поглощают дихлорметаном, промывают 3 раза водой, затем сушат над MgSO4 и выпаривают с получением 11,25 г (количественный выход) масла желтого цвета. С) 4-(2-метокси-5-пропилфенил)-1,3-тиазол-2-амин К раствору 8 г соединения, полученного на предыдущей стадии, в 60 мл этанола прибавляют 4,94 г тиомочевины. Среду кипятят с обратным холодильником в течение 1 ч 30 мин. Затем среду помещают в холодильник на 12 ч и после этого фильтруют. Полученное после фильтрации твердое вещество промывают небольшим количеством холодного этанола, а затем эфиром. Процедуру повторяют еще один раз. Твердое вещество суспензируют в смеси вода/дихлорметан и подщелачивают путем добавления гидроксида натрия. Водную фазу экстрагируют 2 раза дихлорметаном. Объединенные органические фазы су- 14011076 шат над MgSO4 и выпаривают с получением 4,89 г масла коричневого цвета, которое медленно кристаллизуется (67%). Маточные растворы выпаривают, затем перемешивают в смеси вода/дихлорметан и подщелачивают путем добавления гидроксида натрия. Водную фазу экстрагируют 2 раза дихлорметаном. Объединенные органические фазы сушат над MgSO4 и выпаривают. Полученное масло хроматографируют на силикагеле и получают 580 мг целевого продукта. Выход (общий)=75%. Т.пл.=84 С. Работая в соответствии с изложенными выше методиками, получают соединения формулы (II),представленные в следующей ниже табл. I. Таблица 1 Получение производных аминотиазола формулы (II). Соединения формулы (II), в которой Y обозначает атом фтора (получения 1.26 и 1.27), R1 и R2 находятся соответственно в положении 2 и 5 фенильного кольца. Получение 1.26. 4-(5-циклогексил-2-метоксифенил)-5-фтортиазол-2-иламин. К раствору 2,5 г соединения, полученного в примере получения 1.3, описанном выше, в 30 мл DMF прибавляют при 0 С 3,4 г Selectfluor и перемешивают среду в течение 2 ч при комнатной температуре. Среду гидролизуют 2 М раствором аммиака в этаноле, концентрируют, затем разбавляют водой. Сырой продукт фильтруют, твердое вещество обрабатывают дихлорметаном, промывают водой, затем 1 М раствором гидроксида натрия, а затем насыщенным раствором NaCl. После сушки органической фазы надMgSO4 и выпаривания неочищенный продукт очищают флеш-хроматографией. Получают 600 мг целевого соединения в виде порошка белого цвета. Т.пл.=159 С. Получение 1.27. 4-(5-пропил-2-метоксифенил)-5-фтортиазол-2-иламин. Соединение получают в соответствии с описанием получения 1.26 из соединения, полученного в примере получения 1.4. Т.пл.=107 С. Элементный анализ: С% 59,06 (теоретический 58.63), Н% 5,85 (теоретический 5,68), N% 10,22 (теоретический 10,52). Пример 1 (соединение n9) N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-(3-морфолин-4-илпропил)пиперазин-1-ил]-карбамид.- 16011076 Соединение общей формулы (I), в которой R1 = 2-ОМе; R2=5-н.пропил R3=3-(морфолин-4-илпропил); а=2 1.1. Получение N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-(3-морфолин-4-илпропил)пиперазин-1-ил]карбамида. К раствору 0,1 г 4-(2-метокси-5-пропилфенил)-1,3-тиазол-2-амина, полученного в примере получения 1.4, описанном выше, в 2 мл DMF прибавляют 0,18 г DSC и перемешивают среду в течение 12 ч при комнатной температуре. Добавляют 0,05 г 1-(морфолин-4-илпропил)пиперазина и перемешивают среду в течение 3 ч при комнатной температуре. Реакционную среду гидролизуют насыщенным раствором NaHCO3, затем экстрагируют с помощью DCM. Органическую фазу промывают водой, затем насыщенным раствором NaCl, затем концентрируют. После сушки над MgSO4 раствор концентрируют и очищают флеш-хроматографией на силикагеле. Твердое вещество поглощают с помощью DCM, обрабатывают 2 М раствором HCl в эфире, затем суспензию выпаривают и получают 0,077 г целевого соединения в виде его хлоргидрата. Т.пл.=229 С. Пример 2 (соединение n35) N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-[2-(3-этиламинопирролидин-1-ил)этил]-[1,4]диазепан-1-ил]карбамид. Соединение общей формулы (I), в которой R1=2-OCH3; R2=5-(CH2)2CH3; R3=4-[2-(3-этиламинопирролидин-1-ил)этил]; а=3 2.1. Получение N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-(2-хлорацетил)-[1,4]диазепан-1 ил]карбамида. К раствору 3,87 г 4-(2-метокси-5-пропилфенил)-1,3-тиазол-2-амина, полученного в примере получения 1.4, описанном выше, в 60 мл DCM прибавляют 4 г DSC и перемешивают среду в течение 12 ч при комнатной температуре. Добавляют 5,7 г 2-хлор-1-[1,4]диазепан-1-ил-этанона и 3,26 мл триэтиламина. Перемешивают среду в течение 3 ч при комнатной температуре. Среду гидролизуют насыщенным раствором NaHCO3, затем экстрагируют с помощью DCM. Органическую фазу промывают водой, затем насыщенным раствором NaCl, затем концентрируют. После сушки над MgSO4 раствор концентрируют с получением 5,9 г целевого соединения. МН+=452 при t=8,53 мин. 2.2. Получение N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-[2-(3-ацетиламинопирролидин 1-ил)ацетил]-[1,4]-диазепан-1-ил]карбамида (соединение n32). Соединение общей формулы (I), в которой R1=2-OCH3; R2=5-(CH2)2CH3; R3=4-[2-(3 ацетиламинопирролидин-1-ил)ацетил]; а=3. К раствору 2 г [4-(2-метокси-5-пропилфенил)тиазол-2-ил]амида 4-(2-хлорацетил)-[1,4]диазепан-1 карбоновой кислоты, полученного на стадии 2.1, в 10 мл ацетонитрила прибавляют 0,62 г 3 ацетамидопирролидина, затем 0,612 г K2CO3. Среду перемешивают при комнатной температуре в течение 48 ч. После фильтрации среду промывают 1 М раствором NaOH, затем водой и насыщенным раствором NaCl. После сушки над MgSO4 раствор концентрируют, затем очищают флеш-хроматографией и получают 0,95 г целевого продукта. Элементный анализ: С%=56,94; Н%=6,89; N%=14,52 (2H2O). МН+=543 при t=5,85 мин. 2.3. Получение N-[4-(2-метокси-5-пропилфенил)тиазол-2-ил]-N'-[4-[2-(3-этиламинопирролидин-1 ил)этил]-[1,4]-диазепан-1-ил]карбамида. К раствору 0,79 г [4-(2-метокси-5-пропилфенил)тиазол-2-ил]амида [4-[2-(3-ацетиламинопирролидин-1-ил)ацетил]-[1,4]диазепан-1-карбоновой кислоты, полученной на стадии 2.2, в 3 мл DCM прибавляют при 0 С 2,2 мл 65% раствора Red-Al в толуоле. После перемешивания в течение 3 ч при комнатной температуре среду концентрируют, затем поглощают с помощью DCM и промывают 1 М раствором гидроксида натрия, водой, затем насыщенным раствором NaCl. После сушки над MgSO4 органическую фазу концентрируют, затем очищают флеш-хроматографией и получают 0,27 г целевого продукта. МН+=515 при t=8,77 мин. Пример 3 (соединение n76) (R)N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-метилпиперидин-3-илметил)пиперазин-1-ил]карбамид. Соединение общей формулы (I), в которой R1=2-OCH3; R2=5-CyHex; R3=(R)4-(1-метилпиперидин-3 илметил]; а=2 3.1. Получение трет-бутилового эфира (R)-3-метансульфонилоксиметилпиперидин-1-карбоновой кислоты. К раствору 5 г трет-бутилового эфира (R)-3-гидроксиметилпиперидин-1-карбоновой кислоты в 80 мл DCM, охлажденному до 0 С, прибавляют 2,16 мл метансульфонилхлорида, затем 3,86 мл триэтиламина. Реакционную среду перемешивают в течение 1 ч 30 мин при 0 С, затем добавляют 0,7 мл триэтиламина и 0,54 мл метансульфонилхлорида. После выдерживания среды в течение 30 мин при 0 С реакционную среду гидролизуют, органическую фазу промывают два раза водой, затем насыщенным раствором NaCl и потом сушат над MgSO4. Среду выпаривают с получением 6,8 г масла бледно-желтого цвета. 3.2. Получение трет-бутилового эфира (R)-3-(4-бензилпиперазин-1-илметил)пиперидин-1 карбоновой кислоты. Неочищенный продукт, полученный на стадии 3.1, растворяют в 75 мл толуола. Прибавляют 12,16 г бензилпиперазина и затем реакционную среду помещают в герметичную емкость и нагревают в течение 5 ч при 150 С. После охлаждения до комнатной температуры среду разбавляют в смеси эфир/пентан(1/1), дважды промывают насыщенным раствором NaHCO3, дважды промывают водой, затем насыщенным раствором NaCl. После сушки над MgSO4 и выпаривания сырой продукт очищают флешхроматографией на силикателе с получением 5,73 г твердого целевого продукта. МН+=374 при t=5,26 мин. 3.3. Получение (R)-1-бензил-4-(1-метилпиперидин-3-илметил)пиперазина. К раствору 1 г LiAlH4 в 45 мл THF, охлажденному до 0 С, прибавляют 5 г соединения, полученного на стадии 3.2, растворенного в 45 мл THF. Среду перемешивают 2 ч при комнатной температуре, затем охлаждают до 0 С. Прибавляют 0,96 мл воды, затем 3 мл 5 М раствора NaOH, полученную среду фильтруют и твердое вещество промывают эфиром. Фильтрат выпаривают, поглощают остаток эфиром, промывают два раза насыщенным раствором NaHCO3, затем насыщенным раствором NaCl. После сушки над MgSO4 и выпаривания получают 3,5 г целевого продукта. МН+=288 при t=5,68 мин. 3.4. Получение (R)-1-(1-метилпиперидин-3-илметил)пиперазина. Раствор 3,46 г соединения, полученного на стадии 3.3, в 100 мл метанола гидрируют в присутствии 1,9 г катализатора Pd/C с 10% влажностью при давлении водорода 800 кПа и температуре 40 С в течение 3 ч. Среду фильтруют, затем выпаривают с получением 2,26 г бесцветного масла. 3.5. Получение (R)N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-метилпиперидин-3 илметил)пиперазин-1-ил]карбамида. Синтез этого соединения осуществляют в соответствии с процедурой, описанной в примере 1, исходя из 4-(5-циклогексил-2-метоксифенил)-1,3-тиазол-2-амина, описанного в примере получения 1.3, и из соединения, полученного на стадии 3.4. Т.пл.=108 С; []D25=-27 (с=1,05; МеОН). Пример 4 (соединение n70) N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-изопропилпиперидин-3-ил)пиперазин-1-ил]карбамид. Соединение общей формулы (I), в которой R1=2-OCH3; R2=5-CyHex; R3=4-(1-изопропилпиперидин 3-ил); а=2DCM, прибавляют 20 мл 10 гидроксида натрия. Реакционную среду перемешивают, органическую фазу декантируют, затем промывают насыщенным раствором NaCl. После сушки над MgSO4 органическую фазу концентрируют. Полученную смолу переносят в 180 мл DCE, прибавляют 10,1 г Вос-пиперазина,затем 15,9 г NaBH(OAc)3 и среду перемешивают 12 ч при комнатной температуре. Среду концентрируют,затем поглощают с помощью AcOEt. Органическую фазу промывают два раза насыщенным растворомNaHCO3, затем насыщенным раствором NaCl. После сушки над MgSO4 органическую фазу концентрируют и получают 18,63 г целевого продукта. Т.пл.-103 С. 4.2. Получение 1-(1-бензилпиперидин-3-ил)пиперазина. К раствору 9,2 г соединения, полученного на стадии 4.1, в 85 мл DCM прибавляют 30 г THF. Среду перемешивают 5 ч при комнатной температуре, затем концентрируют. Полученный сырой продукт по- 18011076 глощают DCM, затем промывают 4 раза 2 М раствором гидроксида натрия. Органическую фазу промывают насыщенным раствором NaCl. После сушки над MgSO4 органическую фазу концентрируют и получают 6,32 г целевого продукта. ЯМР 1 Н:(ppm)=7,28 (с уш. 5 Н), 3,43 (с уш. 2 Н), 2,88 (д, 1 Н), 2,70 (д, 1 Н), 2,64 (м, 4 Н), 2,43-2,22 (м,5 Н), 1,85-1,58 (м, 4 Н), 1,39 (ддд, 1 Н), 1,15 (ддд, 1 Н). 4.3. Получение N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-бензилпиперидин-3 ил)пиперазин-1-ил]карбамида (соединение n68). Соединение общей формулы (I), в которой R1=2-OCH3; R2=5-CyHex; R3=4-(1-бензилпиперидин-3 ил); а=2. Проводят процедуру, идентичную той, которая описана в примере 1, исходя из 4-(5-циклогексил-2 метоксифенил)-1,3-тиазол-2-амина, описанного в получении 1.3, и из соединения, полученного на стадии 4.2. Т.пл.=90 С. 4.4. Получение N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-пиперидин-3-ил-пиперазин-1-ил]карбамида (соединение n69). Соединение общей формулы (I), в которой R1=2-OCH3; R2=5-CyHex; R3=4-(пиперидин-3-ил); а=2. К раствору 1,69 г соединения, полученного на стадии 4.3, в 10 мл DCE прибавляют при 0 С 1,26 г хлорэтилхлорформиата. Температуру среды доводят до комнатной, затем нагревают с обратным холодильником в течение 45 мин. Среду выпаривают, затем поглощают 10 мл МеОН и нагревают с обратным холодильником в течение 1 ч. Сырой продукт фильтруют, твердое вещество промывают эфиром, затем сушат и получают 1,27 г целевого соединения в форме трихлоргидрата. Т.пл.=240 С. МН+=484 приt=6,81 мин. 4.5. Получение N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-изопропилпиперидин-3 ил)пиперазин-1-ил]карбамида (соединение n70). Соединение общей формулы (I), в которой R1=2-ОСН 3; R2=5-CyHex; R3=4-(1-изопропилпиперидин 3-ил); а=2. К раствору 0,2 г соединения, полученного на стадии 4.4, в 1,2 мл DCE прибавляют 0,05 мл ацетона,затем 0,15 г NaBH(OAc)3 и среду перемешивают 3 ч при комнатной температуре. Добавляют 0,1 мл Et3N,затем 0,1 мг NaBH(OAc)3. Среду перемешивают 12 ч при комнатной температуре. Среду концентрируют,затем поглощают AcOEt. Органическую фазу промывают два раза насыщенным раствором NaHCO3, затем насыщенным раствором NaCl. После сушки над MgSO4 органическую фазу концентрируют, затем очищают флеш-хроматографией и получают 0,11 г целевого продукта. Т.пл.=130 С. МН+=526 при t=7,06 мин. Пример 5 (соединение n74) N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-метилпиперидин-2-карбонил)пиперазин-1-ил]карбамид. Соединение общей формулы (I), в которой R1=2-OCH3; R2=5-CyHex; R3=4-(1-метилпиперидин-2 карбонил); а=2 5.1. Получение трет-бутилового эфира 4-(1-бензилоксикарбонилпиперидин-2-карбонил)пиперазин 1-карбоновой кислоты. К раствору 8 г 1-Boc-пиперазина в 80 мл ацетонитрила прибавляют при 0 С 26,6 г ВОР, 13,6 г 1(карбобензилокси)-2-пиперидинкарбоновой кислоты, затем 11,9 мл триэтиламина. Среду перемешивают при комнатной температуре в течение 12 ч, затем концентрируют. Среду поглощают AcOEt, промывают три раза насыщенным раствором Na2CO3, затем насыщенным раствором NaCl. После сушки над MgSO4 и выпаривания получают 35,4 5 г неочищенного продукта реакции. Продукт поглощают с помощью DCM,затем промывают два раза 5 М раствором гидроксида натрия, затем насыщенным раствором NaCl и сушат над MgSO4. После выпаривания твердый продукт растирают в ТВМЕ, фильтруют, промывают с помощью ТВМЕ, затем сушат и получают 17,93 г целевого соединения. Т.пл.=102 С. 5.2. Получение трет-бутилового эфира 4-(пиперидин-2-карбонил)пиперазин-1-карбоновой кислоты. К раствору 1,79 г соединения, полученного на стадии 5.1, в 14 мл EtOH прибавляют в инертной атмосфере 3,9 мл циклогексадиена, затем 1,3 г 10% катализатора Pd/C с 50% влажностью. Среду перемешивают при комнатной температуре в течение 24 ч, затем фильтруют. Фильтрат выпаривают с получением 1,02 г целевого соединения. 5.3. Получение трет-бутилового эфира 4-(1-метилпиперидин-2-карбонил)пиперазин-1-карбоновой кислоты. К раствору 0,99 г соединения, полученного на стадии 5.2, в 11 мл DCE прибавляют 0,54 мл 37% водного раствора формальдегида, затем 1,41 г NaBH(OAc)3 и среду перемешивают 12 ч при комнатной температуре. Среду разбавляют DCM, фильтруют через хлопковый фильтр. Органическую фазу промы- 19011076 вают два раза насыщенным раствором NaHCO3, затем насыщенным раствором NaCl. После сушки надMgSO4 и испарения растворителей получают 0,84 г целевого продукта. 5.4. Получение (1-метилпиперидин-2-ил)пиперазин-1-илметанона. К раствору 0,84 г соединения, полученного на стадии 5.3, в 2 мл DCM прибавляют 2 мл TFA. Среду перемешивают 6 ч при комнатной температуре. Среду выпаривают, поглощают остаток несколько раз с помощью DCM и испаряют для удаления TFA. Среду разбавляют DCM, затем обрабатывают 10% раствором NH4OH. Органическую фазу сушат над MgSO4, затем выпаривают с получением 0,15 г целевого продукта. 5.5. Получение N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-метилпиперидин-2 карбонил)пиперазин-1-ил]карбамида. Процедуру осуществляют идентично той, которая описана в примере 1, исходя из 4-(5-циклогексил 2-метоксифенил)-1,3-тиазол-2-амина, описанного в получении 1.3, и из соединения, полученного на стадии 5.4. Т.пл.=135 С. Пример 6 (соединение n88) N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-метилпиперидин-2-илметил)пиперазин-1-ил]карбамид. Соединение общей формулы (I), в которой R1=2-ОСН 3; R2=5-CyHex; R3=4-(1-метилпиперидин-2 илметил); а=2 6.1. Получение трет-бутилового эфира 4-(1-бензилоксикарбонилпиперидин-2-илметил)пиперазин-1 карбоновой кислоты. К раствору 14 г соединения, полученного на стадии 5.1, в 50 мл THF прибавляют при 0 С 162 мл мольного раствора борана в THF в течение 30 мин. Среду перемешивают при комнатной температуре в течение 24 часов, затем гидролизуют добавлением воды при 0 С. После разбавления в AcOEt среду промывают три раза насыщенным раствором Na2CO3, затем насыщенным раствором NaCl. После сушки надMgSO4 и выпаривания получают 12,6 г целевого соединения. 6.2. Получение трет-бутилового эфира 4-пиперидин-2-илметилпиперазин-1-карбоновой кислоты. К раствору 6,43 г соединения, полученного на стадии 6.1, в 50 мл EtOH прибавляют в инертной атмосфере 14,5 мл циклогексадиена, затем 3,3 г 10% катализатора Pd/C с 50% влажностью. Среду перемешивают при комнатной температуре в течение 48 ч, затем фильтруют. Фильтрат выпаривают с получением 3,63 г целевого соединения. 6.3. Получение трет-бутилового эфира 4-(1-метилпиперидин-2-илметил)пиперазин-1-карбоновой кислоты. К раствору 3,34 г соединения, полученного на стадии 6.2, в 40 мл DCE прибавляют 1,91 мл 37% водного раствора формальдегида и несколько шариков молекулярного сита 4, затем 5 г NaBH(OAc)3 и среду перемешивают 48 ч при комнатной температуре. Среду разбавляют в DCM, фильтруют через хлопковый фильтр. Органическую фазу промывают два раза насыщенным раствором NaHCO3, затем насыщенным раствором NaCl. После сушки над MgSO4 и испарения растворителей получают 3,28 г целевого продукта. 6.4. Получение 1-(1-метилпиперидин-2-илметил)пиперазина. К раствору 3,28 г соединения, полученного на стадии 6.3, в 1 мл диоксана прибавляют 1 мл 4 М раствор HCl в диоксане. Среду перемешивают 48 ч при комнатной температуре. Среду фильтруют, твердый продукт промывают эфиром, затем сушат и получают 2,9 г целевого соединения. 6.5. Получение N-[4-(5-циклогексил-2-метоксифенил)тиазол-2-ил]-N'-[4-(1-метилпиперидин-2 илметил)пиперазин-1-ил]карбамида. Процедуру осуществляют идентично той, которая описана в примере 1, исходя из 4-(5-циклогексил 2-метоксифенил)-1,3-тиазол-2-амина, описанного в получении 1.3, и из соединения, полученного на стадии 6.4. Т.пл.=103 С. Таблица II Соединения согласно изобретению были объектом фармакологических исследований, которые позволили выявить их модулирующее действие на активность хемокиновых рецепторов.- 29011076 Хемокины представляют собой низкомолекулярные белки, которые относятся в семейству провоспалительных цитокинов и участвуют в хемотаксисе лейкоцитов и эндотелиальных клеток. Хемокины регулируют многочисленные биологические процессы и ассоциированы с воспалительными нарушениями, возникающими в стрессовых состояниях, при ранениях или инфекциях; модулирование действия хемокинов позволяет обеспечить профилактику или лечение таких патологий, как астма, артрит, аллергии, аутоиммунные заболевания, атеросклероз или ангиогенез (C.D.Paavola et al., J.Biol.Chem., 1998, 273,(50), 33157-33165). Из хемокинов выделяют hMCP-1 (английское наименование Human Monocyte Chemotactic Protein),который относится к группе СС хемокинов и который является природным агонистом рецептора CCR2b. Была определена ингибирующая активность соединений согласно изобретению в отношении клеток, экспрессирующих рецептор CCR2b человека. Естественная концентрация природного агонистаhMCP-1, которая ингибирует на 50% (CI50) активность рецептора CCR2b, составляет 0,57 нМ. Соединения согласно изобретению имеют концентрацию CI50, обычно ниже 0,1 мкМ. Например, концентрацияCI50 соединения 14 составляет 0,0033 мкМ; концентрация CI50 соединения 28 составляет 0,028 мкМ; концентрация CI50 соединения 55 составляет 0,014 мкМ. Была также измерена величина ингибирования хемотаксиса в отношении моноцитарных клеток ТНР-1 человека (выпускаемых на рынок фирмой DSMZ, Германия) с использованием методики, адаптированной к той, которая описана A.Albini et al., Cancer Res., 1987, 47, 3239-3245. В этих условиях концентрация CI50 hMCP-1 составляла 6 нМ. Соединения согласно изобретению имели концентрацию CI50,главным образом, ниже 1 мкМ. Ингибирование хемотаксиса с помощью соединений согласно изобретению является подтверждением наличия у них антагонистической активности в отношении рецепторов хемокинов, в частности рецептора CCR2b. Следовательно, соединения согласно изобретению являются антагонистами в отношении активности хемокинов, в частности hMCP-1. Измерялась также ингибирующая активность соединений согласно изобретению в отношении клеток РВМС (Peripheral Blood Mononuclear Cells), инфицированных вирусом VIH-1 Bal, в соответствии с методикой, адаптированной к той, которая описана V.Dolle et col., J. Med. Chem., 2000, 43, 3949, 3962. В соответствии с этой методикой клетки РВМС были инфицированы VIH-1 Bal, затем исследуемые соединения были введены в культуральную среду на 5 суток. В соответствии с процедурой этого эксперимента определяли в супернатанте содержание обратной транскриптазы, которая коррелируется с уровнем вирусной репликации в клетках. В этих условиях продукт AZT, являющийся контрольным продуктом, ингибирующим вирусную репликацию, имел концентрацию CI50 ниже 1 мкМ. Соединения согласно изобретению также имели концентрацию CI50 ниже 1 мкМ. Например, соединение 30 имело CI50, равную 0,6 мкМ. Следовательно, соединения согласно изобретению могут быть использованы для приготовления лекарственных средств, в частности лекарственных средств, являющихся антагонистами действия хемокинов. Таким образом, в соответствии с одним из аспектов настоящее изобретение относится к лекарственным средствам, которые содержат соединение формулы (I) или аддитивную соль этого соединения с фармацевтически приемлемой кислотой, или гидрат, или сольват. Указанные лекарственные средства могут найти применение в терапии, в частности для профилактики и лечения различных патологий, таких как острые и хронические иммуно-воспалительные заболевания и синдромы, такие как атеросклероз,рестенозы, хронические легочные заболевания, в частности, COPD (chronic obstructive pulmonary disease); синдром респираторной недостаточности; бронхиальная гиперактивность; колиты; силикозы; фиброзные патологии, фиброзы легких, кистозные фиброзы; вирусные или бактериальные инфекции, СПИД, менингит, малярия, проказа, туберкулез, герпес, цитомегаловирусные инфекции; септические шоки, сепсис,эндотоксические шоки; реакции отторжения трансплантата; костные патологии, такие как остеопороз,остеоартриты; конъюнктивиты; атипичные или контактные дерматиты; экземы; гломерулонефриты; панкреатиты; язвенные колиты; ауто-иммунные заболевания, такие как ревматоидный полиартрит, рассеянный склероз, амиотрофический боковой склероз, болезнь Крона, красная волчанка, склеродермия, псориаз; болезнь Паркинсона; болезнь Альцгеймера; диабет; кахексия; ожирение; лечение боли, в частности невропатической и воспалительной; аллергические заболевания, такие как аллергические респираторные заболевания, астма, риниты,легочная гиперчувствительность, гиперчувствительность замедленного типа; заболевания и нарушения, в которых задействованы ангиогенные процессы, такие как раки (внутриопухолевый ангиогенез), ретинальные болезни (возрастная дегенерация желтого пятна: DMLA); сердечные патологии: гемодинамический шок; ишемические заболевания сердца; приступы повторной постишемической инфузии; инфаркт миокарда, коронарный тромбоз, сердечная недостаточность, стенокардия. Согласно другому аспекту настоящее изобретение относится к фармацевтическим композициям,- 30
МПК / Метки
МПК: A61P 3/04, C07D 417/12, A61P 37/00, A61P 31/00, A61P 9/00, A61K 31/427, A61K 31/551, C07D 277/40, A61K 31/496, A61P 29/00, C07D 277/44
Метки: 2-карбамид-4-фенилтиазола, терапии, способ, получения, применение, производные
Код ссылки
<a href="https://eas.patents.su/30-11076-proizvodnye-2-karbamid-4-feniltiazola-sposob-ih-polucheniya-i-ih-primenenie-v-terapii.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 2-карбамид-4-фенилтиазола, способ их получения и их применение в терапии</a>
Производные диоксан-2-алкилкарбамата, способ их получения и применение в терапии

Номер патента: 8218
Опубликовано: 27.04.2007
Авторы: Ли Адриен Так, Хорнер Кристиан, Медеско Флоренс, Даргазанли Джихад, Баз Мишель, Абуабделла Ахмед
МПК: A61K 31/335, A61P 25/08, C07D 319/06...
Метки: терапии, производные, способ, получения, применение, диоксан-2-алкилкарбамата
Формула / Реферат:
1. Соединение, соответствующее формуле (I) в которой R1 представляет собой фенильную или нафталинильную группу, возможно замещенную одним(ой) или более атомами галогена или группами гидрокси, циано, нитро, (С1-С3)алкил, (С1-С3)алкокси, трифторметил, трифторметокси, бензилокси, (С3-С6)циклоалкил-O- или (С3-С6)циклоалкил(С1-С3)алкокси; R2 представляет собой либо группу общей формулы CHR3CONHR4, в которой R3 представляет собой атом водорода или...
Производные пиперидинил- и пиперазинил- алкилкарбаматов, способы их получения и применение их в терапии

Номер патента: 9468
Опубликовано: 28.12.2007
Авторы: Пюэш Фредерик, Бюрнье Филипп, Абуабделла Ахмед, Женесс Жан, Хорнар Кристиан
МПК: A61K 31/44, A61K 31/436, A61K 31/435...
Метки: применение, терапии, получения, способы, производные, пиперидинил, пиперазинил, алкилкарбаматов
Формула / Реферат:
1. Производные пиперидинил- и пеперазинилалкилкарбаматов, соответствующие формуле (I) где А представляет собой атом азота или группу CR2, в которой R2 представляет собой атом водорода или фтора либо гидроксильную, циано, трифторметильную, C1-6алкильную или C1-6алкоксигруппу; n представляет собой целое число, равное 2 или 3, и m представляет собой целое число, равное 2, когда А представляет собой атом азота; n представляет собой целое число,...
Производные 4-арилморфолин-3-она, их получение и их применение в терапии

Номер патента: 11035
Опубликовано: 30.12.2008
Авторы: Эмон -Альт Ксавье, Проиетто Винченцо
МПК: A61P 25/00, A61P 1/00, A61K 31/537...
Метки: применение, производные, терапии, 4-арилморфолин-3-она, получение
Формула / Реферат:
1. Соединение формулы (I) в которой Ar обозначает фенил, дважды замещенный атомом галогена; R1 обозначает фенил, не замещенный или замещенный один или два раза атомом галогена; R2 обозначает пиридил; фенил, не замещенный или замещенный один или два раза одним или двумя заместителями, независимо выбранными из атома галогена, (C1-C4)алкила, (C1-C4)алкокси, трифторметильной группы, трифторметоксигруппы; кроме того, R2 может обозначать...
Производные n-[фенил(пиперидин-2-ил)метил]бензамида и их применение в терапии

Номер патента: 7225
Опубликовано: 25.08.2006
Авторы: Севрен Мирей, Роже Пьер, Медеско Флоранс, Мага Паскаль, Даргазанли Жихад, Вероник Коринн, Марабу Бенуа, Эстенн-Буту Женевьев
МПК: A61K 31/445, A61P 25/00, C07D 211/26...
Метки: производные, применение, терапии, n-[фенил(пиперидин-2-ил)метил]бензамида
Формула / Реферат:
1. Соединение в форме чистого оптического изомера (1R, 2R) или (1S, 2S) либо в форме трео-диастереоизомера, соответствующее общей формуле (I) где А представляет собой или группу общей формулы N-R1, где R1 представляет собой или атом водорода, или линейную или разветвленную (С1-С7)алкильную группу, возможно замещенную одним или более чем одним атомом фтора, или (С4-С7)циклоалкильную группу, или (С3-С7)циклоалкил(С1-С3)алкильную группу, или...
Производные 5-(пиридин-3-ил)-1-азабицикло[3.2.1]октана, их получение и применение в терапии

Номер патента: 7793
Опубликовано: 27.02.2007
Авторы: Галли Фредерик, Леклерк Одиль, Неделек Ален, Лочид Алистер
МПК: C07D 209/00, A61K 31/439, A61P 25/00...
Метки: производные, получение, применение, 5-(пиридин-3-ил)-1-азабицикло[3.2.1]октана, терапии
Формула / Реферат:
1. Соединение в форме чистого энантиомера или в форме смеси энантиомеров, соответствующее общей формуле (I) где R представляет атом галогена или фенильную группу, замещенную одной или двумя группами, выбранными из атома галогена или группы (С1-C6)алкил, (C1-C6)алкокси, нитро, трифторметил, трифторметокси, гидрокси, ацетил или метилендиокси, или группу пиперидинил, или морфолин-4-ил, или пирролидин-1-ил, или азетидин-1-ил, или азепин-1-ил, или...
Предыдущий патент: Сухая молочная смесь
Следующий патент: Устройство для розлива пенящихся напитков в открытую емкость
Случайный патент: Биологически разлагаемый состав для выращивания растений и способ его применения