Ингибиторы протеазы вируса гепатита с
Номер патента: 20235
Опубликовано: 30.09.2014
Авторы: Линь Чу-Чун, Лю Чэнь-Фу, Чэнь Жун-Цзиунн, Лю Йо-Чин, Ли Куан-Юань, Чэнь Чих-Мин, Кинг Чи-Син Ричард, Ло Пинь
























Формула / Реферат
1. Соединение формулы (I)

где
R1 является С3-10циклоалкилом;
R2 является C1-6алкилом или С3-10циклоалкилом;
U является -NHSO2-;
W является -(СН2)m-, -О(СН2)n- или -SO(CH2)n-,
m равно 2 и
n равно 1;
X представляет собой -О-;
Y представляет собой

где
каждый из V и Т является -N-;
R представляет собой арил или гетероарил и
каждый из А1 и А2 независимо являются арилом или гетероарилом, каждый из которых необязательно замещен галогеном, С1-6алкилом или С1-6алкоксилом; или необязательно конденсирован с другим арилом или гетероарилом, каждый из которых необязательно замещен галогеном, С1-6алкилом или C1-6алкоксилом;
Z является -ОС(О);
где ²арил² относится к углеводородному фрагменту, содержащему одно или несколько ароматических колец;
²гетероарил² относится к фрагменту, содержащему одно или несколько ароматических колец, которые содержат по меньшей мере один гетероатом, выбранный из N, О и S.
2. Соединение по п.1, где Y является

где Ri является Н, арилом или гетероарилом, каждый арил и гетероарил необязательно замещен галогеном, C1-10алкилом или C1-10алкоксилом; каждый из Rii, Riii, Riv, Rv, Rvi, Rvii и Rviii являются независимо Н, галогеном, С1-6алкилом или C1-6алкоксилом; и Т является таким, как определено в п.1;
где ²арил² относится к углеводородному фрагменту, содержащему одно или несколько ароматических колец;
²гетероарил² относится к фрагменту, содержащему одно или несколько ароматических колец, которые содержат по меньшей мере один гетероатом, выбранный из N, О и S.
3. Соединение по п.2, где R1 является циклопропилом.
4. Соединение по п.3, где R2 является C1-5алкилом или С3-8циклоалкилом.
5. Соединение по п.2, где Y представляет собой

где каждый из Ri; Rii, Riii, Riv, Rv, Rvi, Rvii и Rviii являются такими, как определено в п.2.
6. Соединение по п.1, где R1 является циклопропилом и
R2 представляет собой C1-5алкил или С3-8циклоалкил.
7. Соединение по п.1, где соединение обладает такой стереохимией, как показано ниже

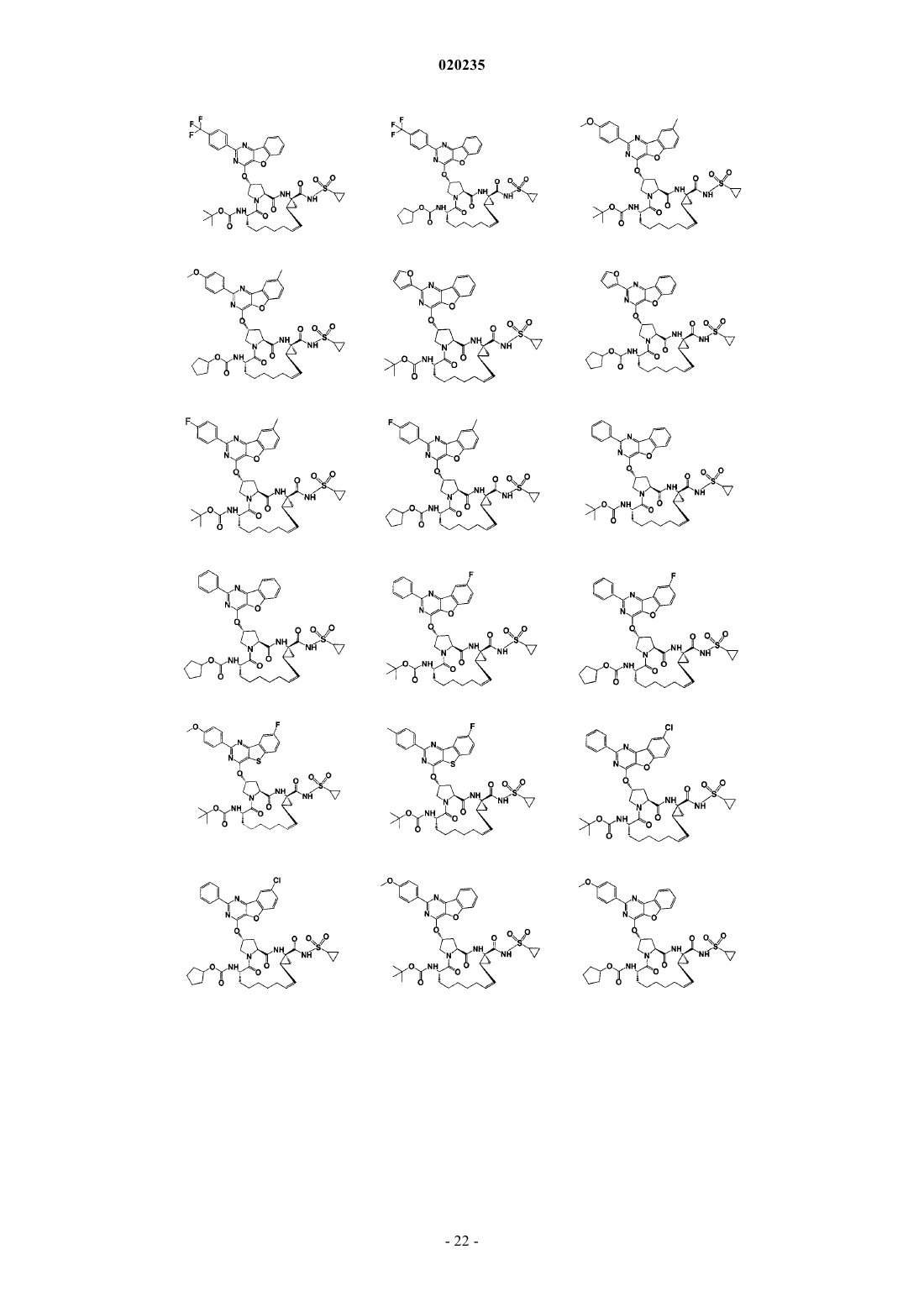
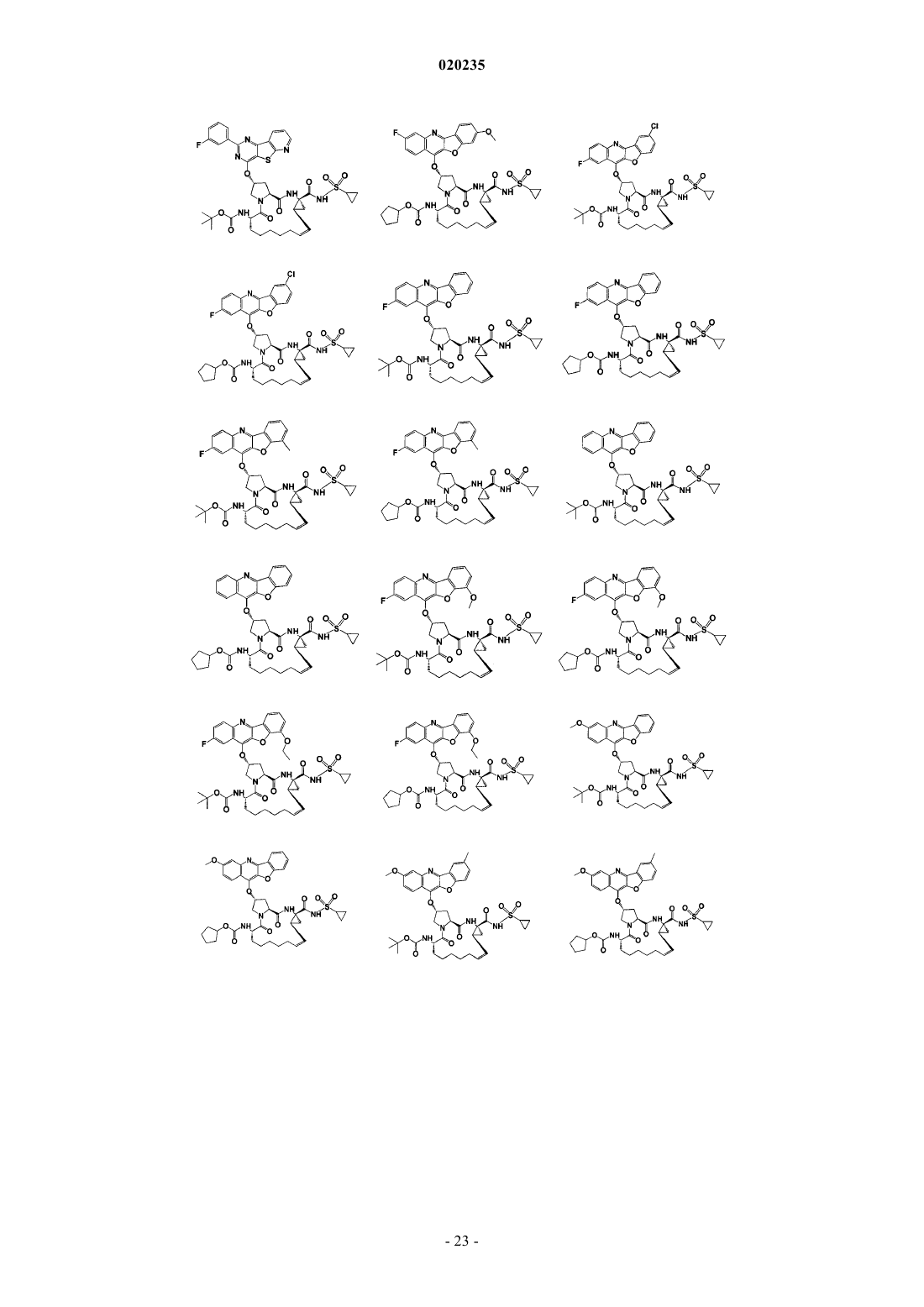
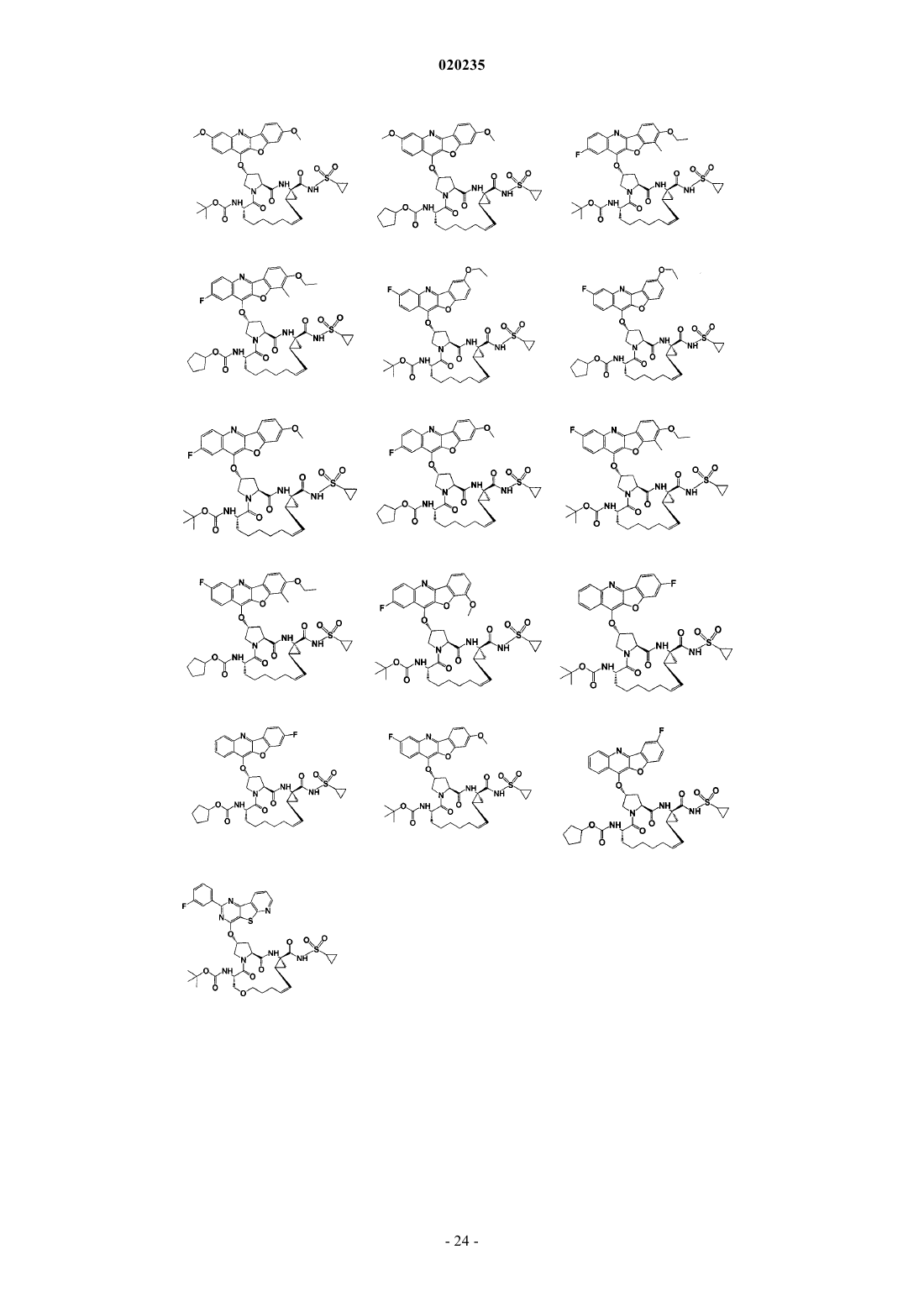
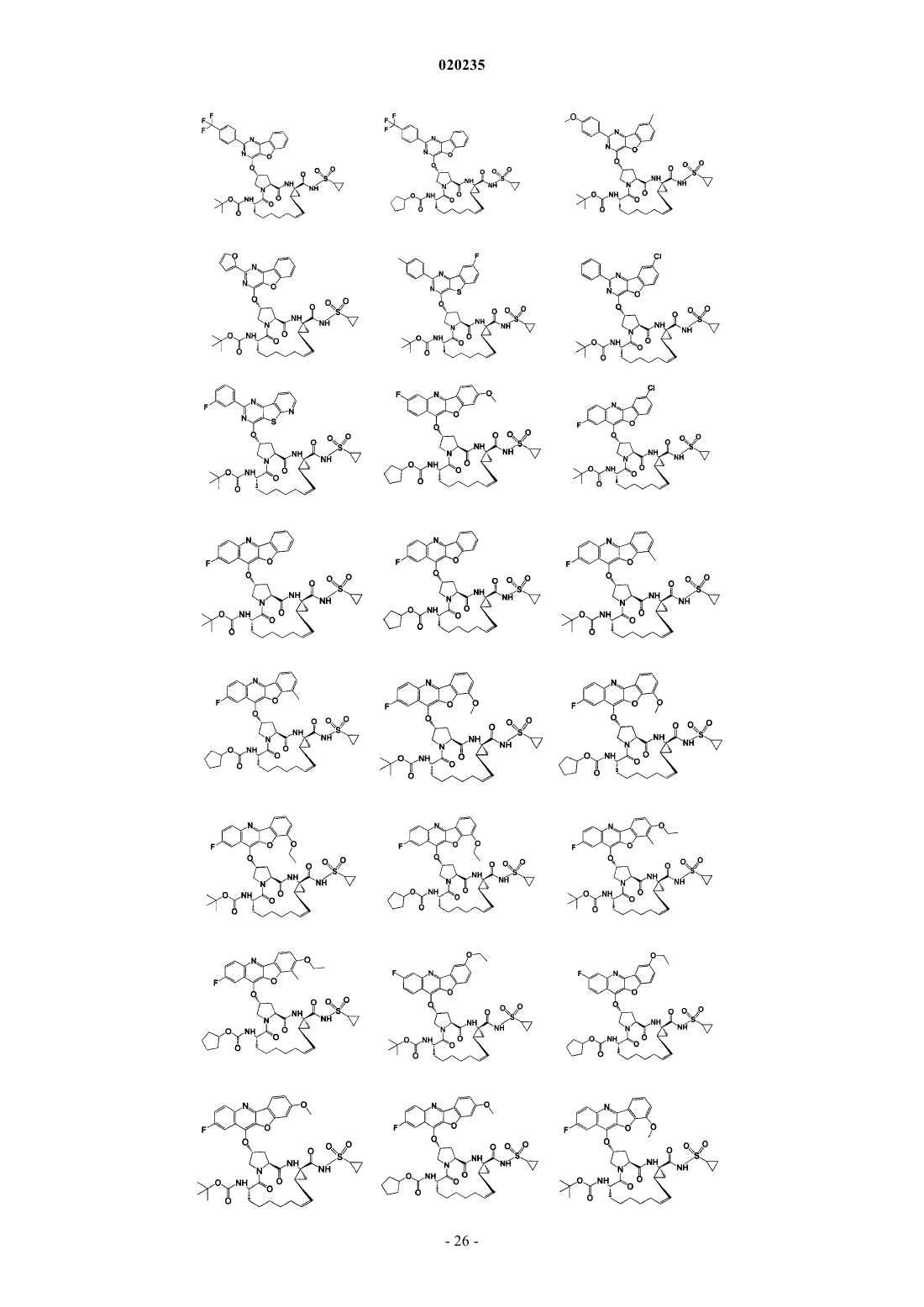
8. Соединение по п.1, где соединение представляет собой одно из соединений




9. Фармацевтическая композиция для лечения инфекции вируса гепатита С, содержащая противовирусное средство, имеющее формулу, описанную в п.1, и фармацевтически приемлемый носитель.
10. Фармацевтическая композиция по п.9, дополнительно содержащая иммуномодулирующее средство, другое противовирусное средство или ингибитор NS5B полимеразы, NS5A, NS4B или р7.
11. Способ лечения инфекции вирусного гепатита С, включающий введение нуждающемуся в этом пациенту эффективного количества соединения по п.1.
12. Способ лечения инфекции вирусного гепатита С, включающий введение нуждающемуся в этом пациенту эффективного количества соединения по п.8.
13. Соединение по п.2, где Y является

где каждый из Ri, Rii, Riii, Riv, Rv являются такими, как определено в п.2.
14. Соединение по п.13, где Ri является арилом или гетероарилом, необязательно замещенным галогеном, C1-10алкилом или C1-10алкоксилом; каждый из Rii, Riii, Riv, Rv являются независимо Н, галогеном, C1-6алкилом или C1-6алкоксилом и Т является таким, как определено в п.1;
где ²арил² относится к углеводородному фрагменту, содержащему одно или несколько ароматических колец;
²гетероарил² относится к фрагменту, содержащему одно или несколько ароматических колец, которые содержат по меньшей мере один гетероатом, выбранный из N, О и S.
15. Соединение по п.14, где Y является

где Ri является арилом или гетероарилом, необязательно замещенным C1-10алкоксилом, и каждый из Rii, Riii, Riv, Rv являются Н и Т является таким, как определено в п.1;
где ²арил² относится к углеводородному фрагменту, содержащему одно или несколько ароматических колец;
²гетероарил² относится к фрагменту, содержащему одно или несколько ароматических колец, которые содержат по меньшей мере один гетероатом, выбранный из N, О и S.
16. Соединение по п.1, где соединение представляет собой одно из соединений



Текст
ИНГИБИТОРЫ ПРОТЕАЗЫ ВИРУСА ГЕПАТИТА С Настоящее изобретение относится к макроциклическим соединениям формулы (I), в которой R1 является С 3-10 циклоалкилом; R2 является C1-6 алкилом или С 3-10 циклоалкилом; U является -NHSO2-; где каждый из V и Т является -N-; R представляет собой арил или гетероарил и каждый из A1 и А 2 независимо являются арилом или гетероарилом, каждый из которых необязательно замещен галогеном, С 1-6 алкилом или C1-6 алкоксилом; или необязательно конденсирован с другим арилом или гетероарилом, каждый из которых необязательно замещен галогеном, С 1-6 алкилом или C1-6 алкоксилом; и Z является -ОС(О). Изобретение также относится к фармацевтической композиции,содержащей данное соединение, и к способу лечения инфекции вирусного гепатита С. Перекрестная ссылка Данная заявка заявляет приоритет предварительной патентной заявки США, серийный 61/053857, поданной 16 мая 2008, содержание которой введено в данное описание посредством ссылки. Предшествующий уровень техники Вирус гепатита С (hepatitis С virus, HCV), вирус с одноцепочечной РНК положительной полярности, представляет собой основной возбудитель заболевания большинства случаев не-А, не-В гепатитов. Инфицированность HCV представляет собой проблему здоровья человека, которая привлекает всеобщее внимание. См., например, WO 05/007681; WO 89/04669; ЕР 381216; Alberti et al., J. Hepatology, 31 (Suppl. 1), 17-24 (1999); Alter J. Hepatology, 31 (Suppl. 1), 88-91 (1999) и Lavanchy J. Viral Hepatitis, 6, 35-47(1999). Гепатит, вызываемый инфекцией HCV, трудно поддается лечению, поскольку вирус способен быстро мутировать и устойчив к естественному иммунному ответу. Единственными доступными в настоящее время противо-HCV терапиями являются лечение -интерфероном, комбинацией интерферон/рибавирин и пегилированным -интерфероном. Однако доля стойкой ремиссии для интерферона или комбинации -интерферон/рибавирин составляла, как было обнаружено, менее 50%, и пациенты сильно страдали от побочных эффектов этих терапевтических средств. См., например, Walker,DDT, 4, 518-529 (1999); Weiland, FEMS Microbial. Rev., 14, 279-288 (1994) и WO 02/18369. Таким образом, сохраняется необходимость разработки более эффективных и хорошо переносимых лекарственных средств.HCV протеаза, необходимая для вирусной репликациии, содержит примерно 3000 аминокислот. Она включает нуклеокапсидный белок (С), оболочечные белки (Е 1 и Е 2) и несколько неструктурных белков (NS2, NS3, NS4a, NS5a и NS5b).NS3 белок обладает активностью сериновой протеазы и, как полагают, является существенным для вирусной репликации и инфицирующей способности. О существенной роли NS3 протеазы свидетельствует тот факт, что мутации NS3 протеазы вируса желтой лихорадки понижают инфицирующую способность вируса. См., например, Chamber et al., Proc. Natl. Acad. Sci. USA 87, 8898-8902 (1990). Также было показано, что мутации в активном центре HCV NS3 протеазы полностью подавляют HCV инфекцию у экспериментальной модели шимпанзе. См., например, Rice et al., J. Virol. 74 (4) 2046-51 (2000). Кроме того, HCV NS3 протеаза, как было обнаружено, способствует протеолизу в местах соединенийNS3/NS4a, NS4a/NS4b, NS4b/NS5a, NS5a/NS5b и, таким образом, отвечает за образование четырех вирусных белков в ходе вирусной репликации. См., например, US 2003/0207861. Следовательно, фермент HCV NS3 протеаза представляет собой привлекательную мишень для лечения HCV инфекции. Потенциальные ингибиторы NS3 HCV протеазы можно обнаружить в 02/18369,WO 00/09558, WO 00/09543, WO 99/64442, WO 99/07733, WO 99/07734, WO 99/50230, WO 98/46630, WO 98/17679, WO 97/43310, US 5990276, Dunsdon et al., Biorg. Med. Chem. Lett. 10, 1571-1579 (2000); LlinasBrunet et al., Biorg. Med. Chem. Lett. 10, 2267-2270 (2000) и S. LaPlante et al., Biorg. Med. Chem. Lett. 10,2271-2274 (2000). Сущность изобретения Данное изобретение основано на неожиданном открытии того, что определенные макроциклические соединения являются эффективными в ингибировании активности HCV NS3 и уровней HCV RNA. В одном аспекте изобретение относится к соединениям формулы (I) где каждый из V и Т является -N-;R представляет собой арил или гетероарил; и каждый из А 1 и А 2 независимо являются арилом или гетероарилом, каждый из которых необязательно замещен галогеном, С 1-6 алкилом или С 1-6 алкоксилом; или необязательно конденсирован с другим арилом или гетероарилом, каждый из которых необязательно замещен галогеном, C1-6 алкилом или C1-6 алкоксилом; иZ является -ОС(О); Термин алкил относится к насыщенному линейному или разветвленному углеводородному фрагменту, такому как -СН 3 или -СН(СН 3)2. Термин алкокси относится к -О-(C1-6 алкильному) радикалу. Термин алкенил относится к линейному или разветвленному углеводородному фрагменту, который содержит по меньшей мере одну двойную связь, такую как -СН=СН-СН 3. Термин циклоалкил относится к насыщенному циклическому углеводородному фрагменту, такому как циклогексил. Термин арил относится к углеводородному фрагменту, содержащему одно или несколько ароматических колец. Примеры арильных фрагментов включают фенил (Ph), фенилен, нафтил, нафтилен, пиренил, антрил и фенантрил. Термин гетероарил относится к фрагментам, содержащим одно или несколько ароматических колец, которые содержат по меньшей мере один гетероатом (например, N, О или S). Примеры гетероарильных фрагментов включают фурил, фурилен, флуоренил, пирролил, тиенил, оксазолил, имидазолил, тиазолил, пиридил, пиримидинил, хиназолинил, хинолил, изохинолил и индолил. Упомянутые здесь алкил,алкенил, алкинил, циклоалкил, циклоалкенил, гетероциклоалкил, гетероциклоалкенил, арил и гетероарил включают как замещенные, так и незамещенные фрагменты, если не указано иное. Возможные заместители в циклоалкиле, ариле и гетероариле включают, но не ограничиваются, C1 С 10 алкил, С 2-С 10 алкенил, С 2-С 10 алкинил, С 3-С 20 циклоалкил, С 3-С 20 циклоалкенил, C1-C20 гетероциклоалкил, C1-C20 гетероциклоалкенил, C1-С 10 алкокси, арил, арилокси, гетероарил, гетероарилокси,амино, C1-С 10 алкиламино, С 1-С 20 диалкиламино, ариламино, диариламино, C1-C10 алкилсульфонамино,арилсульфонамино, C1-C10 алкилимино, арилимино, С 1-С 10 алкилсульфонимино, арилсульфонимино, гидроксил, галоген, тио, C1-C10 алкилтио, арилтио, C1-C1 алкилсульфонил, арилсульфонил, ациламино, аминоацил, аминотиоацил, амидино, гуанидин, уреидо, циано, нитро, нитрозо, азидо, ацил, тиоацил, ацилокси, карбоксил и сложный эфир карбоновой кислоты. С другой стороны, возможные заместители в алкиле включают все из вышеупомянутых заместителей, за исключением С 1-С 10 алкила. Циклоалкил, арил и гетероарил могут также быть конденсированными друг с другом. Ниже представлены 60 иллюстративных соединений по изобретению. В другом аспекте изобретение относится к способу лечения инфекции вируса гепатита С. Способ включает введение нуждающемуся в этом пациенту эффективного количества одного или нескольких соединений формулы (I), представленных выше. В еще другом аспекте изобретение относится к фармацевтической композиции для применения в лечении HCV инфекции. Композиция содержит эффективное количество по меньшей мере одного из соединений формулы (I) и фармацевтически приемлемый носитель. Она может также включать ингибитор мишени действия, отличающейся от HCV NS3 протеазы в жизненном цикле HCV, например NS5B полимеразы, NS5A, NS4B или р 7. Примеры таких ингибиторов включают, но не ограничиваются ими, N[3-(1-циклобутилметил-4-гидрокси-2-оксо-1,2-дигидрохинолин-3-ил)-1,1-диоксо-1,4-дигидро-116 бензо[1,2,4]тиадиазин-7-ил]метансульфонамид(WO 04041818),транс-1,2-ди-4-[(фенилацетилпирролидин-2-(S)-карбонил)амино]фенилэтилен (WO 0401413) и 1-аминоадамантан (аментадин, Griffin,2004, J. Gen. Virol. 85: с. 451). Фармацевтическая композиция может дополнительно содержать иммуномодулирующее средство или второе противовирусное средство. Иммуномодулирующее средство относится к активному агенту,которое содействует иммунному ответу. Примеры иммуномодулирующих средств включают, но не ограничиваются ими, Nov-205 (Novelos Therapeutics Inc., WO 02076490) и IMO-2125 (Idera PharmaceuticalsInc., WO 05001055). Противовирусное средство относится к активному агенту, который убивает вирус или подавляет его репликацию. Примеры противовирусных средств включают, но не ограничиваются ими, рибавирин,-интерферон, пегилированный интерферон и ингибиторы HCV протеазы, такие как 2-(2-2 циклогексил-2-[(пиразин-2-карбонил)амино]ацетиламино-3,3-диметилбутирил)октагидроциклопента[с]пиррол-1-карбоновой кислоты (1-циклопропиламинооксалил-бутил)амид (Telaprevir, Vertex Pharmaceuticals Inc., WO 02018369), 3-[2-(3-трет-бутил-уреидо)-3,3-диметил-бутирил]-6,6-диметил-3-азабицикло[3.1.0]гексан-2-карбоновой кислоты (2-карбамоил-1-циклобутилметил-2-оксоэтил)амид (Boceprevir, Schering-Plough Research Institute, WO 03062265) и 4-фтор-1,3-дигидроизоиндол-2-карбоновойInc.,US2005/0267018). Также в область данного изобретения входит применение такой композиции для производства лекарственного средства для только что упомянутого лечения. Подробности одного или нескольких вариантов осуществления изобретения изложены в описании ниже. Другие характеристики, объекты и преимущества изобретения будут очевидны из описания и формулы изобретения. Подробное описание Соединения по данному изобретению можно синтезировать из коммерчески доступных исходных веществ известными в данной области способами. Например, соединения по изобретению можно получить способом, показанным на схеме 1 ниже. Схема 1 Как показано на схеме 1, мультициклическое соединение (i) сначала соединяли с N-(третбутоксикарбонил)-L-пролином (ii), с последующим метилированием до образования промежуточного продукта (iii). У промежуточного продукта (iii) снимали защиту, удаляя N-бутоксикарбонильную группу до образования N-свободного соединения (iv), которое соединяли с карбоновой кислотой (v) до получения промежуточного продукта (vi) . Промежуточный продукт (vi) гидролизовали, получая кислоту (vii),которую соединяли с аминным соединением (viii) до получения пирролидинового соединения (ix), содержащего две концевые алкенильные группы. Промежуточный продукт (ix) подвергали реакции олефинового обмена в присутствии катализатора Груббса для получения требуемого макроциклического соединения (х). Схемы 2 и 3 ниже иллюстрируют два альтернативных синтетических пути получения соединений по этому изобретению. Описанные выше способы могут также дополнительно включать стадии либо до, либо после стадий, подробно описанных на схемах 1-3, для того, чтобы ввести или удалить подходящие защитные группы для того, чтобы в конце концов сделать возможным синтез требуемых соединений. В дополнение к этому для получения требуемых соединений можно осуществить различные синтетические стадии в альтернативной последовательности. Превращения синтетической химии и методологии защитных групп(защита и снятие защиты), полезные для синтеза применяемых соединений формулы (I), известны в данной области и включают, например, те, которые описаны в R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2nd Ed.,John Wiley and Sons (1991); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, JohnSons (1995), а также в их последующие издания. Примеры 1-60 ниже обеспечивают подробные описания того, каким образом были фактически получены иллюстративные соединения 1-60. Упомянутые здесь соединения содержат неароматическую двойную связь и асимметрические центры. Таким образом, они могут существовать в качестве рацематов и рацемических смесей, отдельных энантиомеров, отдельных диастереомеров, смесей диастереомеров, таутомеров и цис- и транс-форм. Рассматриваются все такие изомерные формы. Например, соединения формул (I), представленных выше,могут обладать следующими стереохимическими конфигурациями (III) и (IV) соответственно: Соединения, описанные выше, включают соединения сами по себе, а также их соли, пролекарства и сольваты, если таковые применяются. Соль, например, можно образовать между анионом и положительно заряженной группой (например, аминогруппой) в соединении формулы (I). Подходящие анионы включают хлорид, бромид, йодид, сульфат, нитрат, фосфат, цитрат, метансульфонат, трифторацетат, ацетат, малат, тозилат, тартрат, фумарат, глутамат, глюкуронат, лактат, глутарат и малеат. Таким же образом соли можно образовать между катионом и отрицательно заряженной группой (например, карбоксилатом) в соединении формулы (I). Подходящие катионы включают ион натрия, ион калия, ион магния,ион кальция и аммониевый катион, такой как ион тетраметиламмония. Соединения формулы (I) также включают те соли, которые содержат атомы четвертичного азота. Примеры пролекарств включают сложные эфиры и другие фармацевтически приемлемые производные, которые при введении пациенту, способны давать активное соединение формулы (I). Сольваты относятся к комплексам, формируемым между активным соединением формулы (I) и фармацевтически приемлемым растворителем. Примеры фармацевтически приемлемых растворителей включают воду, этанол, изопропанол, этилацетат, уксусную кислоту и этаноламин. Также в область данного изобретения входит способ лечения HCV инфекции путем введения эффективного количества одного или нескольких соединений формулы (I) пациенту. Термин лечение относится к введению соединений пациенту с HCV инфекцией, с ее симптомами или предрасположенному к ней, с целью достижения терапевтического эффекта, например лечения, облегчения, изменения, воздействия, улучшения или предотвращения HCV инфекции, ее симптомов или предрасположенности к ней. Термин эффективное количество относится к количеству активного соединения по изобретению,которое требуется для достижения терапевтического эффекта у пациента, подвергаемого лечению. Эффективные дозировки будут варьироваться, как могут обнаружить специалисты в данной области, в зависимости от типов заболеваний, подвергаемых лечению, способа введения, применения носителя и возможности совместного применения с другим терапевтическими лечением. Для применения на практике способа по настоящему изобретению, композицию, содержащую один или несколько соединений по этому изобретению, можно ввести парентерально, орально, назально, ректально, местно или защечно. Термин парентерально, как использовано здесь, относится к подкожной,внутрикожной, внутривенной, внутримышечной, внутрисуставной, внутриартериальной, внутрисиновиальной, надчревной, внутриоболочечной, вводимой внутрь пораженных тканей или внутричерепной инъекции, а также к любым подходящим методам вливания. Стерильной инъецируемой композицией может быть любой раствор или суспензия в нетоксичном парентерально приемлемом разбавителе или растворителе, таком как раствор в 1,3-бутандиоле. Среди приемлемых средств доставки и растворителей, которые можно использовать, находятся маннит, вода,раствор Рингера и изотонический раствор хлорида натрия. В дополнение к этому отдельные масла обычно используют в качестве растворителя или суспендирующей среды (например, синтетические моно- или диглицериды). Жирная кислота, такая как олеиновая кислота, и ее глицеридные производные полезны для получения инъецируемых лекарственных средств в качестве природных фармацевтически приемлемых масел, таких как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных формах. Эти масляные растворы или суспензии могут также содержать растворитель или диспергатор на основе длинноцепочечного спирта, карбоксиметилцеллюлозу или аналогичные диспергирующие средства. Для цели составления рецептуры можно также использовать другие обычно применяемые поверхностно-активные вещества, такие как твины или спаны или другие аналогичные эмульгирующие средства или агенты, повышающие биодоступность, которые обычно используют при производстве фармацевтически приемлемых твердых, жидких или других лекарственных форм. Композиция для орального применения может представлять собой любую орально приемлемую лекарственную форму, включая капсулы, таблетки, эмульсии и водные суспензии, дисперсии и растворы. В случае таблеток общеизвестные носители включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие средства, такие как стеарат магния. Для орального применения в виде капсул пригодные разбавители включают лактозу и высушенный кукурузный крахмал. Когда орально применяют водные суспензии или эмульсии, активный ингредиент можно суспендировать или растворить в масляной фазе, соединенной с эмульгатором или суспендирующим средством. По желанию можно добавить определенные подсластители, ароматизаторы и красители. Согласно методике, хорошо известной в области составления фармацевтических рецептур, можно изготовить назальный аэрозоль или композицию для ингаляции. Например, такую композицию можно изготовить в виде раствора в солевом растворе, используя бензиловый спирт или другие подходящие консерванты, вещества, способствующие адсорбции, для того, чтобы повысить биодоступность, фторуглероды и/или другие известные в данной области солюбилизирующие или диспергирующие средства. Композицию, содержащую одно или несколько активных соединений по изобретению, можно также применять в виде суппозиториев для ректального применения. Носитель в фармацевтической композиции должен быть приемлемым в том смысле, что он является совместимым с активным ингредиентом композиции (и предпочтительно способен к стабилизации активного ингредиента) и не является опасным для лечимого пациента. В качестве фармацевтических наполнителей можно использовать один или несколько солюбилизаторов для доставки активного соединения по изобретению. Примеры других носителей включают коллоидный диоксид кремния, стеарат магния, целлюлозу, лаурилсульфат натрия и синтетический краситель DC Yellow10. Описанные выше соединения по изобретению предварительно проверяли в отношении их эффективности для лечения HCV инфекции по анализу in vitro (примеры 61 и 62 ниже) и затем подтверждали экспериментами на животных и клиническими испытаниями. Для специалистов в данной области будут также очевидны другие способы. Конкретные примеры ниже следует истолковывать только как иллюстративные и не ограничивающие остальную часть изобретения в любом, каком бы то ни было случае. Без дополнительных уточнений считается, что специалист в данной области может на основе представленного здесь описания использовать настоящее изобретение в наиболее полной степени. Все цитируемые здесь публикации введены в данное описание посредством ссылки как на единое целое. Пример 1. Синтез сложного циклопентилового эфира 4-циклопропансульфониламинокарбонил 2,15-диоксо-18-[2-(4-трифторметилфенил)бензо[4,5]фуро[3,2-d]пиримидин-4-илокси]-3,16-диазатрицикло[14.3.0.04,6]нонадец-7-ен-14-илкарбаминовой кислоты (Соединение 1) Сначала получали соединение I-3 из коммерчески доступного сложного этилового эфира 1-третбутоксикарбониламино-2-винил-циклопропанкарбоновой кислоты способом, представленным ниже: К раствору сложного этилового эфира 1-трет-бутоксикарбониламино-2-винилциклопропанкарбоновой кислоты (0,34 г, 1,3 ммоль) в ТГФ (5 мл) и метаноле (5 мл) добавляли суспензию LiOH (0,13 г, 5,3 ммоль) в воде (1,4 мл). После перемешивания в течение ночи при комнатной температуре реакцию останавливали 10% HCl (2 мл) и удаляли растворитель под вакуумом. Получившийся в результате порошок твердого вещества промывали водой (10 мл) до получения соединения I-1 (0,27 г,90%). МС m/z 249,9 (М 23); 1 Н ЯМР (CDCl3)10,35 (ушир.с, 1 Н), 5,84-5,71 (м, 1 Н), 5,29 (д, J=17,4 Гц,1 Н), 5,12 (д, J=10,2 Гц, 1 Н), 2,23-2,14 (м, 1 Н), 1,87-1,65 (м, 1 Н), 1,58-1,41 (м, 1 Н), 1,43 (с, 9 Н). Раствор соединения I-1 (0,52 г, 2,3 ммоль), 2-(1 Н-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат метанаминия (HATU, 1,7 4 г, 4,6 ммоль) и 4-диметиламинопиридин (1,39 г, 11,6 ммоль) в CH2Cl2 (40 мл) перемешивали при комнатной температуре в течение 1 ч с последующим медленным добавлением циклопропансульфонамида (0,57 г, 4,7 ммоль), диизопропилэтиламина (1,81 мл, 14,0 ммоль) и 1,8-диазабицикло[5,4,0]ундец-7-ена (1,80 г, 11,7 ммоль) в течение 15 мин. После перемешивания реакционной смеси при комнатной температуре в течение ночи удаляли растворитель под вакуумом. Остаток очищали колоночной хроматографией на силикагеле до получения соединения I-2 (0,51 г, 66%). МС m/z 353,1 (М 23); 1H ЯМР (CDCl3)9,75 (ушир.с, 1 Н), 5,64-5,51 (м, 1 Н), 5,30 (д, J=17,4 Гц, 1 Н), 5,16 (д, J=10,2 Гц,1 Н), 2,95-2,89 (м, 1 Н), 2,19-2,10 (м, 1 Н), 1,93-1,88 (м, 1 Н), 1,47 (с, 9 Н), 1,46-1,38 (м, 1 Н), 1,32-1,23 (м, 2 Н),1,15-1,00 (м, 2 Н). К раствору соединения I-2 (0,50 г, 1,5 ммоль) в МеОН (8 мл) добавляли SOCl2 (0,26 г, 2,2 ммоль) при комнатной температуре. После кипячения реакционной смеси с обратным холодильником в течение 1 ч МеОН и SOCl2 удаляли под вакуумом. Остаток растирали в порошок в пентане и фильтровали до получения промежуточного продукта I-3 в виде не совсем белого твердого вещества (0,32 г, 91%). МС m/z Раствор амида 3-аминобензофуран-2-карбоновой кислоты (1,00 г, 5,7 ммоль) и пиридина (1 мл,12,26 ммоль) в ТГФ (25 мл) перемешивали при 0 С в течение 10 мин. К получившемуся в результате раствору медленно добавляли 4-трифторметилбензоил хлорид (1,48 г, 7,1 ммоль). Затем температуру повышали до комнатной и смесь перемешивали в течение 12 ч. Потом растворитель удаляли при пониженном давлении, получившееся в результате твердое вещество собирали, промывали водой и сушили на воздухе до получения I-4 (1,92 г, 96,0%). МС: m/z 349,0 (М 1). Суспензию I-4 (1,92 г, 5,5 ммоль) и 2N NaOH (13 мл) в EtOH (25 мл) нагревали при 85 С в течение 12 ч. После охлаждения смесь подкисляли и затем удаляли EtOH. Получившееся в результате твердое вещество собирали, фильтровали, промывали водой и сушили до получения I-5 (1,71 г, 95,0%). МС m/z 331 (М 1). Раствор I-5 (1,71 г, 5,2 ммоль) и избыток оксихлорида фосфора (РОС 13) кипятили с обратным холодильником в течение 2 ч. После охлаждения и тщательного концентрирования смесь подвергали экстракции метиленхлоридом и 10% гидроксидом натрия. Органический слой сушили над MgSO4, концентрировали и кристаллизовали из CH2Cl2 и н-гексана до получения соединения I-6 (1,49 г, 82%). МС m/z 348,8, 350,9 (М 1); 1 Н ЯМР (CDCl3)8,70 (д, 2 Н), 8,34 (д, 1 Н), 7,82-7,75 (м, 4 Н), 7,57 (ддд, 1 Н). К суспензии бок-транс-4-гидрокси-L-пролина (0,53 г, 2,3 ммоль) в ДМСО (25 мл) добавляли t-BuOK(0,82 г, 5,1 ммоль) при 0 С. После того, как смеси давали нагреться до комнатной температуры и перемешивания в течение 1 ч, соединение I-6 (0,81 г, 2,3 ммоль) медленно добавляли при 10 С. Перемешивание продолжали в течение ночи. Добавляли йодометан (1,02 г, 6,9 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение дополнительных 30 мин. Реакционную смесь нейтрализовали до рН 67 10% водным раствором HCl и подвергали экстракции метиленхлоридом. Органический слой сушили над MgSO4, выпаривали под вакуумом и очищали колоночной хроматографией на силикагеле до получения соединения I-7 (1,12 г, 86%). МС m/z 557,8 (М 1); 1 Н ЯМР (CDCl3)8,63 (д, 2 Н), 8,28 (д, 1 Н), 7,80-7,74 (м, 2 Н), 7,70 (д, 2 Н), 7,51 (ддд, 1 Н). К раствору соединения I-7 (1,13 г, 2,0 ммоль) в МеОН (20 мл) добавляли SOCl2 (1,21 г, 9,8 ммоль) при комнатной температуре. Реакционную смесь кипятили с обратным холодильником в течение 1 ч и удаляли МеОН и SOCl2. Остаток растирали в порошок в пентане. Суспензию фильтровали до получения- 11020235 соединения I-8 в виде не совсем белого твердого вещества (0,87 г, 95%). МС m/z 458,1 (М 1). К раствору HATU (1,12 г, 3,0 ммоль), 1-гидроксибензотриазола (НОВТ, 0,41 г, 3,0 ммоль), 1-8 (0,86 г, 1,9 ммоль) и 2-трет-бутоксикарбониламино-нон-8-еноевой кислоты (1,21 г, 1,9 ммоль) в CH2Cl2 (40 мл) при комнатной температуре добавляли N-метилморфолин (NMM, 1,02 г, 9,9 ммоль). После перемешивания в течение ночи смесь концентрировали под вакуумом. Остаток очищали колоночной хроматографией на силикагеле до получения соединения I-9 (1,03 г, 73%). МС m/z 711,3 (М 1). К раствору соединения I-9 (1,01 г, 1,4 ммоль) в ТГФ (20 мл) добавляли 0,5 М LiOH (5,7 мл, 2,9 ммоль) при комнатной температуре. После перемешивания в течение ночи реакционную смесь нейтрализовали 10% HCl до рН 7 и концентрировали под вакуумом. Получившийся в результате остаток фильтровали и промывали водой до получения соединения I-10 (0,91 г, 92%). МС: m/z 697,3 (М 1).NMM (0,12 г, 1,2 ммоль) добавляли к раствору соединения I-3 (0,28 г, 0,4 ммоль), HATU (0,31 г, 0,8 ммоль), НОВТ (0,08 г, 0,6 ммоль) и соединения I-10 (0,09 г, 0,4 ммоль) в CH2Cl2 (10 мл) при комнатной температуре. После перемешивания в течение ночи реакционную смесь концентрировали под вакуумом. Остаток очищали колоночной хроматографией на силикагеле до получения соединения I-11 (0,10 г,85%). МС m/z 921,3 (М 1); 1 Н ЯМР (CDCl3)10,24 (с, 1 Н), 8,61 (д, 2 Н), 8,26 (д, 1 Н), 7,77 (д, 2 Н), 7,737,64 (м, 2 Н), 7,54-7,47 (м, 1 Н), 7,11 (с, 1 Н), 6,19 (д, 1 Н), 5,88-5,70 (м, 2 Н), 5,38-5,25 (м, 2 Н), 5,16 (д, 1 Н),5,00-4,90 (м, 2 Н), 4,60 (дд, 1 Н), 4,88-4,34 (м, 2 Н), 4,18-4,10 (м, 1 Н), 2,98-2,89 (м, 1 Н), 2,68 (дд, 2 Н), 2,181,96 (м, 6 Н), 1,50-1,32 (м, 7 Н), 1,28 (с, 9 Н), 1,09-1,25 (м, 2 Н). К раствору соединения I-11 (0,10 г, 0,11 ммоль) в CH2Cl2 (10 мл) добавляли катализатор HoveydaGrubbs 2nd (35 мг, 0,056 ммоль) при комнатной температуре под N2. Затем реакционную смесь перемешивали при 40 С в течение 24 ч для проведения реакции циклического метатезиса. Реакцию останавливали,и реакционную смесь очищали колоночной хроматографией до получения соединения 1 (30 мг, 31%). МС: m/z 881,3 (М 1); 1 Н ЯМР (CDCl3)10,39 (с, 1 Н), 8,59 (д, 2 Н), 8,21 (д, 1 Н), 7,77 (д, 2 Н), 7,69-7,57 (м,2 Н), 7,46 (дд, 1 Н), 7,20 (с, 1 Н), 6,12 (с, 1 Н), 5,69 (кв, 1 Н), 5,12 (д, 1 Н), 4,97 (дд, 1 Н), 4,81-4,68 (м, 2 Н),4,28-4,07 (м, 2 Н), 2,96-2,49 (м, 3 Н), 2,30 (кв, 1 Н), 1,96-1,12 (м, 14 Н), 1,08 (с, 9 Н), 0,96-0,82 (м, 2 Н). Пример 2. Синтез сложного циклопентилового эфира 4-циклопропансульфониламинокарбонил 2,15-диоксо-18-[2-(4-трифторметилфенил)бензо[4,5]фуро[3,2-d]пиримидин-4-илокси]-3,16-диазатрицикло[14.3.0.04,6]нонадец-7-ен-14-илкарбаминовой кислоты (Соединение 2) Соединение 2 получали способом, описанным ниже: К раствору соединения I-11 (0,11 г, 0,14 ммоль) в 5 мл CH2Cl2 добавляли 4N HCl в диоксане (2 мл) при комнатной температуре в течение 4 ч HCl, диоксан и CH2Cl2 удаляли выпариванием до получения неочищенного соединения I-12, которое использовали для следующей стадии без дополнительной очистки. Неочищенное вещество I-12 растворяли в 2 мл ацетонитрила и затем добавляли насыщенный водный раствор NaHCO3 (1 мл). После перемешивания в течение 10 мин, к реакционной смеси при комнатной температуре добавляли циклопентилхлороформат (0,02 г, 0,15 ммоль). Реакционную смесь перемешивали в течение еще 2 ч. После остановки реакции насыщенным водным раствором NaHCO3, смесь подвергали экстракции CH2Cl2 до получения неочищенного соединения I-13 (0,11 г, 83%). МС: m/z 921,3(М 1); 1 Н ЯМР (CDCl3)10,39 (с, 1 Н), 8,59 (д, 2 Н), 8,21 (д, 1 Н), 7,77 (д, 2 Н), 7,69-7,57 (м, 2 Н), 7,46 (дд,1 Н), 7,20 (с, 1 Н), 6,12 (с, 1 Н), 5,69 (кв, 1 Н), 5,12 (д, 1 Н), 4,97 (дд, 1 Н), 4,81-4,68 (м, 2 Н), 4,28-4,07 (м, 2 Н),2,96-2,49 (м, 3 Н), 2,30 (кв, 1 Н), 1,96-1,12 (м, 15 Н), 1,08 (с, 9 Н), 0,96-0,82 (м, 2H). Соединение 1-13 обрабатывали катализатором Hoveyda-Grubbs 2nd (35 мг, 0,056 ммоль) для проведения реакции циклического метатезиса, как описано в примере, для получения соединения 2. МС: m/z 893,3 (М 1) ; 1 Н ЯМР (CDCl3)10,36 (с, 1 Н), 8,61 (д, 2 Н), 8,23 (д, 1 Н), 7,77 (д, 2 Н), 7,69-7,43 (м, 3 Н),- 12020235 7,09 (с, 1 Н), 6,16 (с, 1 Н), 5,71 (кв, 1 Н), 5,17 (д, 1 Н), 4,98 (дд, 1 Н), 4,77 (дд, 1 Н), 4,48 (ушир.с, 1 Н), 4,63 (д,1 Н), 4,30-4,07 (м, 2 Н), 2,97-2,46 (м, 3 Н), 2,29 (кв, 1 Н), 1,96-1,06 (м, 22 Н), 0,96-0,82 (м, 2 Н). Примеры 3-52. Синтез соединений 3-52 Каждое из соединений 3-52 получали способом, аналогичным тем, которые описаны в примерах 1 и 2. Соединение 3: МС: m/z 857,3 (М 1); 1 Н ЯМР (CDCl3)10,42 (с, 1 Н), 8,41 (д, 2 Н), 7,97 (с, 1 Н), 7,517,35 (м, 2 Н), 7,20 (с, 1 Н), 7,02 (д, 2 Н), 6,10 (с, 1 Н), 5,68 (кв, 1 Н), 5,16 (д, 1 Н), 4,96 (дд, 1 Н), 4,75 (дд, 1 Н),4,65 (д, 1 Н), 4,34-4,07 (м, 2 Н), 3,90 (с, 3 Н), 2,97-2,50 (м, 3 Н), 2,51 (с, 3 Н), 2,30 (кв, 1 Н), 2,05-0,81 (м, 25 Н). Соединение 4: МС: m/z 869,3 (М 1); Соединение 5: МС: m/z 803,3 (М 1); 1 Н ЯМР (CDCl3)10,31 (с, 1 Н), 8,29 (д, 1 Н), 7,67-7,54 (м, 3 Н),7,44 (дд, 1 Н), 7,27 (д, 1 Н), 7,03 (с, 1 Н), 6,58 (дд, 1 Н), 6,09 (с, 1 Н), 5,69 (кв, 1 Н), 5,05 (д, 1 Н), 4,97 (дд, 1 Н),4,74 (дд, 1 Н), 4,63 (д, 1 Н), 4,26-4,04 (м, 2 Н), 2,95-2,20 (м, 4 Н), 1,95-1,15 (м, 14 Н), 1,08 (с, 9 Н), 0,98-0,81 (м,2 Н). Соединение 6: МС: m/z 815,3 (М 1); 1 Н ЯМР (CDCl3)10,38 (с, 1 Н), 8,29 (д, 1 Н), 7,67-7,38 (м, 4 Н),7,28-7,21 (м, 2 Н), 6,58 (с, 1 Н), 6,11 (с, 1 Н), 5,30 (кв, 1 Н), 5,26 (д, 1 Н), 5,02-4,86 (м, 1 Н), 4,80-4,68 (м, 1 Н),4,57 (д, 1 Н), 4,53-4,46 (м, 1 Н), 4,31-4,18 (м, 1 Н), 4,08 (дд, 1 Н), 2,96-2,18 (м, 4 Н), 2,04-0,82 (м, 24 Н). Соединение 7: МС: m/z 845,3 (М 1); Соединение 8: МС: m/z 857,3 (М 1); 1 Н ЯМР (CDCl3)10,33 (с, 1 Н), 8,46 (дд, 2 Н), 7,99 (с, 1 Н), 7,46 Смесь сложного этилового эфира аминотиоксоуксусной кислоты (6,00 г, 45,0 ммоль) и 2 хлорциклопентанона (5,60 г, 47,0 ммоль) в толуоле нагревали при кипячении с обратным холодильником в течение 4 ч. Полученный таким образом коричневый раствор охлаждали до комнатной температуры,разбавляли EtOAc (50 мл), промывали насыщенным водным раствором NaHCO3 (50 мл) и солевым раствором (50 мл), сушили над безводным MgSO4, фильтровали и концентрировали под вакуумом. Твердое вещество бежевого цвета очищали флеш-хроматографией на силикагелевой колонке (10% EtOAc в гексане) до получения сложного этилового эфира 5,6-дигидро-4 Н-циклопентатиазол-2-карбоновой кислоты Раствор сложного этилового эфира 5,6-дигидро-4 Н-циклопентатиазол-2-карбоновой кислоты I-14(7,5 мл, 1,5 экв.) при комнатной температуре в течение 5 ч. Смесь сушили под вакуумом до получения 5,6-дигидро-4 Н-циклопентатиазол-2-карбоновой кислоты I-15, которую использовали непосредственно на следующей стадии без дополнительной очистки. ESI-MS (М+Н+) =170,2. Раствор 4-метокси-2-амино-ацетофенона (1,67 г, 10,0 ммоль) и 5,6-дигидро-4 Н-циклопентатиазол-2 карбоновой кислоты I-15 (1,69 г, 10,0 ммоль) в пиридине (80 мл) охлаждали до -30 С, используя охлаждающую ванну. Оксихлорид фосфора (2,8 мл, 30,0 ммоль) затем добавляли по каплям в течение 15 мин. После того, как реакционную, смесь перемешивали при -30 С в течение 0,5 ч, ванну убирали, и реакционной смеси давали возможность нагреться до комнатной температуры. После перемешивания реакционной смеси в течение 2 ч ее выливали в ледяную воду. рН смеси доводили до 11 водным 2N растворомNaOH, и смесь подвергали экстракции CH2Cl2. Органический слой сушили над безводным MgSO4, фильтровали и концентрировали под вакуумом. Неочищенный продукт очищали флеш-хроматографией на колонке с силикагелем (30% EtOAc в гексане) до получения амида соединения I-16 (1,10 г, 35%) в виде твердого вещества бледно-бежевого цвета: ESI-MS (М+Н+) =317,3.t-BuOK (1,00 г, 8,8 ммоль) добавляли к суспензии амида соединения I-16 (0,71 г, 2,2 ммоль) в безводном t-BuOH (10 мл). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 2 ч, охлаждали до комнатной температуры и подкисляли добавлением HCl (4N в диоксане, 3 мл). Смесь концентрировали под вакуумом и полученный остаток выливали в раствор 10% KHSO4. После фильтрования твердое вещество промывали эфиром и водой и сушили под вакуумом до получения хинолина соединения I-17 (0,41 г, 61%) в виде твердого вещества бежевого цвета. 1 Н ЯМР (CDCl3-CD3OD)2,76-2,90 (м, 2 Н), 3,18 (т, J=6,6 Гц, 2 Н), 3,28 (т, J=6, 6 Гц, 2 Н), 3,55 (с,2 Н), 3,59 (с, 1 Н), 7,26-7,38 (м, 1 Н) , 7,46-7,72 (м, 2 Н), 8,41 (д, J=8,7 Гц, 1 Н) . ESI-MS (M+H+) =299,4. Раствор хинолина соединения I-17 (0,66 г, 2,2 ммоль), соединения пролина I-18 (0,89 г, 2,2 ммоль) и трифенилфосфина (1,2 г, 4,5 ммоль) в ДМФ (30 мл) охлаждали до 0 С. Диизопропилазодикарбоксилат(DIAD, 0,9 мл, 4,5 ммоль) добавляли по каплям в течение 15 мин. Реакционной смеси затем давали медленно нагреться до комнатной температуры и непрерывно перемешивали в течение ночи. После удаления растворителя под вакуумом смесь разбавляли CH2Cl2 (100 мл), промывали водой (100 мл) и солевым раствором (50 мл), сушили над безводным MgSO4, фильтровали и концентрировали под вакуумом. Остаток очищали флеш-хроматографией на колонке с силикагелем (50% EtOAc в гексане) до получения сложного эфира соединения I-19 (1,23 г, 82%). ESI-MS (М+Н+) =679,3. Водный раствор 2N NaOH (10 мл) добавляли к раствору сложного эфира соединения I-19 (1,91 г, с примесью трифенилфосфатоксида) в ТГФ (40 мл). Добавляли дополнительные 10 мл МеОН до получения гомогенного раствора, и получившийся в результате раствор перемешивали при комнатной температуре в течение 4 часов. Смесь подкисляли 1N HCl до рН 3 и затем дважды экстрагировали CH2Cl2. Органический слой сушили над безводным MgSO4, фильтровали и концентрировали под вакуумом. Остаток очищали флеш-хроматографией на колонке с силикагелем (10% МеОН в CH2Cl2) до получения кислоты соединения I-20 (0,94 г, 1,4 ммоль, 64% за две стадии) в виде твердого вещества желтого цвета. ESI-MS(М+Н+) =665,3. Раствор кислоты соединения I-20 (0,73 г, 1,1 ммоль), HATU (0,91 г, 2,2 ммоль) и DMAP (0,1 г, 1,1 ммоль) в CH2Cl2 (30 мл) перемешивали при комнатной температуре в течение 0,5 ч, с последующим добавлением этил-1-амино-2-винилциклопропанкарбоксилата (0,29 г, 1,1 ммоль) и DIPEA (1,2 мл, 6,7 ммоль) в CH2Cl2 (20 мл). По завершении добавления реакционную смесь перемешивали при комнатной температуре в течение еще 6 ч, разбавляли CH2Cl2 (100 мл), промывали водой (100 мл) и солевым раствором (50 мл), сушили над безводным MgSO4, фильтровали и концентрировали под вакуумом. Остаток очищали флеш-хроматографией на колонке с силикагелем (50% EtOAc в гексане) до получения сложного эфира соединения I-21 (0,72 г, 82%) в виде твердого вещества желтого цвета. ESI-MS (М+Н+) =802,3. Раствор сложного эфира соединения I-21 (0,68 г, 0,85 ммоль) в толуоле (70 мл) дегазировали азотом. При комнатной температуре добавляли катализатор Hoveyda-Grubbs 2-го поколения (0,05 г, 10% моль) . Реакционную смесь нагревали при 50 С в течение ночи. Растворитель концентрировали, и остаток очищали флеш-хроматографией на колонке с силикагелем (1% МеОН в эфире) до получения продукта I-22 (0,33 г, 50%). ESI-MS (М+Н+)=774,3. Водный раствор 2N NaOH (6 мл) добавляли к раствору сложного эфира соединения I-22 (0,33 г, 0,43 ммоль) в ТГФ (30 мл). Дополнительные 6 мл МеОН добавляли до получения гомогенного раствора и получившийся в результате раствор перемешивали при комнатной температуре в течение 2 ч. Смесь подкисляли 1N HCl до рН 3 и затем дважды экстрагировали CH2Cl2. Органический слой сушили надMgSO4, фильтровали и концентрировали под вакуумом. Неочищенное соединение I-23 использовали на следующей стадии без дополнительной очистки. ESI-MS (M+H+) =746,3. Раствор кислоты соединения I-23 (0,23 г, 0,3 ммоль), HATU (0,23 г, 0,6 ммоль) и DMAP (0,03 г, 0,3 ммоль) в ТГФ (20 мл) перемешивали при комнатной температуре в течение 0,5 ч с последующим добавлением амида циклопропансульфоновой кислоты (0,11 г, 0,9 ммоль), DIPEA (0,3 мл, 2 ммоль) и DBU (0,3 мл, 2 ммоль). После окончания добавления реакционную смесь нагревали при 50 С в течение 6 ч, раз- 16020235MgSO4, фильтровали и концентрировали под вакуумом. Остаток очищали флеш-хроматографией на колонке с силикагелем (50% EtOAc в гексане) до получения соединения 53 (0,12 г, 47%) в виде твердого вещества желтого цвета. МС: m/z 849,3 (М 1); 1 Н ЯМР (CDCl3)10,24 (с, 1 Н), 8,00 (д, 1 Н), 7,45 (с, 1 Н), 7,34 (с, 1 Н), 7,13 (с, 1 Н), 7,00 (дд, 1 Н), 5,62 Раствор 6-(3-фторфенил)-7 Н-9-тиа-1,5,7-триазафлуорен-8-она (1,83 г, 6,16 ммоль) в POCl3 (5,66 мл,61,6 ммоль) нагревали при кипячении с обратным холодильником в течение 3 ч. После удаления POCl3 в вакууме, остаток выливали в воду и гасили насыщенным NaHCO3 до рН 7, перемешивали в течение 15 мин и затем фильтровали до получения неочищенного соединения I-24 (1,82 г, 93%). ESI-MS(1,94 г, 17,28 ммоль) при комнатной температуре. После перемешивания реакционной смеси в течение 1,5 ч, соединение I-24 (1,82 г, 5,76 ммоль) растворяли в ДМСО (18,2 мл) и добавляли по каплям к реакционной смеси на ледяной бане в течение ночи. Получившуюся в результате смесь выливали в холодную воду и водный раствор подкисляли 1N HCl до рН 2 и фильтровали до получения неочищенного соединения I-25 (2,77 г, 94%). ESI-MS (М+Н+)=511,1.NMM (3,67 мл, 33,4 ммоль) добавляли к раствору соединения I-25 (3,41 г, 6,68 ммоль), HATU (5,08 г, 13,36 ммоль), HOBt (1,35 г, 10,02 ммоль) и (1-амино-2-винилциклопропанкарбонил)амида циклопропансульфоновой кислоты (1,69 г, 7,35 ммоль) в CH2Cl2 (33,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Получившуюся в результате смесь гасили насыщенным NH4Cl,экстрагировали CH2Cl2, промывали насыщенным NaHCO3 и солевым раствором, сушили над MgSO4. После концентрирования остаток очищали колоночной хроматографией на силикагеле (50% EtOAc в гексане) до получения соединения I-26 (3,20 г, 66%). ESI-MS (М+Н+)=723,2. К раствору I-26 (0,51 г, 0,69 ммоль) в CH2Cl2 (3,45 мл) добавляли CF3COOH (0,53 мл, 6,9 ммоль) при комнатной температуре. После перемешивания реакционной смеси в течение ночи раствор концентрировали до получения неочищенного соединения I-27, которое использовали для следующей стадии без дополнительной очистки. ESI-MS (M+H+) =623,2. 0,9 ммоль) и (s)-2-(трет-бутоксикарбониламино)-3-(пент-4-енилокси)пропионовой кислоты (0,25 г, 0,9 ммоль) в CH2Cl2 (3,45 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Получившуюся в результате смесь гасили насыщенным NH4Cl, экстрагировали CH2Cl2, промывали насыщенным NaHCO3 и солевым раствором, сушили над MgSO4. После концентрирования остаток очищали колоночной хроматографией на силикагеле (50% EtOAc в гексане) до получения соединения I-28(0,52 г, 85%). ESI-MS (М+Н+) =877,7. Раствор I-28 (0,52 г, 0,59 ммоль) в толуоле (59 мл) дегазировали азотом. Добавляли катализатор второго поколения Hoveyda-Grubbs 2nd (0,04 г, 10% моль) при комнатной температуре. Реакционную смесь нагревали при 50 С в течение ночи. Концентрировали растворитель и остаток очищали ТСХ (1% МеОН в эфире) до получения соединения 54 (0,07 г, 14%). ESI-MS (М+Н+)=850,2; 1 Н ЯМР (CDCl3)10,16 (с, 1 Н), 8,86-8,58 (м, 2 Н), 8,38 (д, 1 Н), 8,27 (д, 1 Н), 7,57-7,46 (м, 2 Н), 7,20(2 Н), 2,02-1,76 (м, 2 Н), 1,55-1,38 (м, 4 Н), 1,34 (с, 9 Н), 1,31-0,82 (м, 4 Н). Пример 55-60. Синтез соединений 55-60 Каждое из соединений 55-60 было получено способом, аналогичным тому, который описан в примере 54. Соединение 55: МС: m/z 804,3 (М 1); 1 Н ЯМР (CDCl3)10,19 (с, 1 Н), 8,81 (д, 1 Н), 8,37 (д, 1 Н),8,27 (д, 1 Н), 7,60-7,16 (м, 5 Н), 6,25 (с, 1 Н), 5,65 (кв, 1 Н), 5,45 (д, 1 Н), 5,21 (дд, 1 Н), 4,93 (ушир.с, 1 Н),4,74-4,61 (м, 1 Н), 4,43-4,34 (м, 1 Н), 4,13 (д, 1 Н), 3,75-3,34 (м, 4 Н), 3,08-2,88 (м, 2 Н), 2,63-2,45 (м, 2 Н),2,27-0,81 (м, 19 Н). Соединение 56: МС: m/z 804,3 (М 1); 1 Н ЯМР (CDCl3)10,06 (с, 1 Н), 8,21 (дд, 1 Н), 7,91 (д, 1 Н),7,81 (дд, 1 Н), 7,39 (дд, 1 Н), 7,30-7,22 (м, 1 Н), 7,16 (д, 1 Н), 6,24 (с, 1 Н), 5,63 (кв, 1 Н), 5,39 (д, 1 Н), 5,18 (дд,1 Н), 4,89 (дд, 1 Н), 4,68-4,58 (м, 1 Н), 4,43 (д, 1 Н), 4,13-4,06 (м, 1 Н), 4,07 (с, 3 Н), 3,72 (дд, 1 Н), 3,52 (дд,1 Н), 3,45-3,38 (м, 1 Н), 3,09-3,87 (м, 2 Н), 2,68 (дд, 1 Н), 2,53-1,91 (м, 4 Н), 1,57-1,41 (м, 6 Н), 1,37 (с, 9 Н),1,28-0,82 (м, 3 Н). Соединение 57: МС: m/z 848,2 (М 1). Соединение 58: МС: m/z 804,3 (М 1); 1 Н ЯМР (CDCl3)10,20 (с, 1 Н), 8,33 (д, 1 Н), 8,16 (дд, 1 Н),7,77 (дд, 1 Н), 7,68-7,36 (м, 6 Н), 6,22 (с, 1 Н), 5,62 (кв, 1 Н), 5,42 (д, 1 Н), 5,16 (дд, 1 Н), 4,99 (ушир.с, 1 Н),4,92 (дд, 1 Н), 4,68-4,60 (м, 1 Н), 4,37 (д, 1 Н), 4,16-4,08 (м, 1 Н), 3,73-3,35 (м, 4 Н), 3,08-2,87 (м, 2 Н), 2,692,40 (м, 2 Н), 2,32-0,81 (м, 17 Н). Соединение 59: МС: m/z 804,3 (М 1); 1 Н ЯМР (CDCl3)10,18 (с, 1 Н), 8,34 (д, 1 Н), 8,17 (дд, 1 Н),7,82 (дд, 1 Н), 7,73-7,40 (м, 4 Н), 7,35 (с, 1 Н), 6,23 (с, 1 Н), 5,63 (кв, 1 Н), 5,48 (д, 1 Н), 5,17 (дд, 1 Н), 4,99(ушир.с, 1 Н), 4,91 (дд, 1 Н), 4,74-4,62 (м, 1 Н), 4,40 (д, 1 Н), 4,17-4,08 (м, 1 Н), 3,75-3,35 (м, 4 Н), 3,08-2,87 (м,2 Н), 2,73-2,40 (м, 2 Н), 2,30-0,81 (м, 18 Н). Соединение 60: МС: m/z 838,4 (М 1). Пример 61. Ингибирование NS3/4A протеазы Экспрессия белка и очистка Плазмиду, содержащую ген, кодирующий N-концевой His6-меченный-NS4 А(21-32)-GSGS-NS3(3-181),трансформировали в Е. coli, штамм BL21(DE3)pLysS (Novagen) для сверхэкспрессии белка. Моноспецифическую колонию трансформированной BL21(DE3)pLysS культивировали в 200 мл Lauria-Bertani (LB) среде с канамицином и хлорамфениколом при 37 С в течение ночи. Бактериальную культуру переносили в 6 л LB среды (Difco), содержащей антибиотики, и инкубировали при шейкировании при 22 С. После того, как поглощение при 600 нм достигало величины 0,6, культуру стимулировали 1 мМ изопропил-1 тиоD-галактопиранозидом (IPTG) при 22 С в течение 5 ч. Впоследствии клетки, выросшие в культуре,собирали центрифугированием (6000g в течение 15 мин при 4 С). Сгустки клеток ресуспендировали в 150 мл буфера А (50 мМ HEPES, рН 7,4, 0,3 М NaCl, 0,1% (мас./об.) CHAPS, 10 мМ имидазола, 10%(об./об.) глицерина). После того, как смесь разрушали путем четырехкратного пропускания через микрофлуодайзер Microfluidizer, работающий при 30 фунтах на кв. дюйм, клеточный дербис удаляли центрифугированием (58250g в течение 30 мин при 4 С). Клеточный лизат, содержащий белки, мишени действия His6, загружали со скоростью 3 мл/мин в 25-мл колонку с Ni-NTA (Qiagen) в присутствии 10 мМ имидазола, используя систему gradiFrac (Pharmacia). Колонку промывали 10-кратным по отношению к объему колонки количеством лизирующего буфера. Связанный NS4A(21-32)-GSGS-NS3 (3-181) элюировали 8-кратным по отношению к объему колонки количеством буфера А с добавленными в него 300 мМ имидазола. Объединенные фракции дополнительно очищали на Q-Sepharose колонке, уравновешенной буфером В (50 мМ HEPES, рН 7,4, 0,1% (мас./об.) CHAPS, 10%(об./об.) глицерина, 5 мМ дитиотреитола (DTT) и 1 М NaCl). Элюант, содержащий NS4A(21-32)-GSGS-NS3 (3-181) собирали и дополнительно очищали эксклюзионной хроматографией по размерам при скорости протока 0,5 мл/мин, используя колонку с sephacryl-75 (16100 см, Pharmacia) предварительно уравновешенную буфером С (50 мМ HEPES, рН 7,4, 0,1% (мас./об.) CHAPS, 5 мМ DTT, 10% (об./об.) глицерина). Очищенный белок замораживали и хранили при -80 С до использования. Микроколоночный ВЭЖХ анализ Готовили раствор, содержащий 50 мМ Трис, рН 7,4, 100 мМ NaCl, 20% глицерина, 0,012% CHAPS,10 мМ DTT, 5 мкМ субстрата Ac-Asp-Glu-Asp(EDANS)-Glu-Glu-Abu-f-[COOAla]-Ser-Lys(DABCYL)-NH2(RET S1, ANASPEC) и 10 мкМ тестируемого соединения. 80 мкл раствора добавляли в каждую лунку 96 луночного планшета. Реакцию инициировали добавлением 20 мкл 10 нМ NS3/4A протеазы в буфере, содержащем 50 мМ Трис буфер, рН 7,4, 100 мМ NaCl, 20% глицерина, и 0,012% CHAPS. Конечная концентрация NS3/4A протеазы составила 2 нМ, что было меньше, чем Км по субстрату RET S1. Анализируемый раствор инкубировали в течение 30 мин при 30 С. Реакцию затем останавливали добавлением 100 мкл 1% TFA. Аликвоту 200 мкл переносили в каждую лунку Agilent 96-луночного планшета. Продукты реакции анализировали с использованием ВЭЖХ с обращенной фазой, описанной ниже. ВЭЖХ система включала следующее: Agilent 1100, дегазатор G1379A, двухканальный насос G1312A,автоматический пробозаборник G1367A, камеру для термостатирования колонок G1316A, детектор на основе фотодиодной матрицы G1315B, колонку: Agilent, ZORBAX Eclipse XDB-C18, 4,6 мм, 5 мкм, P/N 993967-902, термостат для колонок: при комнатной температуре, объем пробы: 100 мкл, растворитель А= вода, чистая для ВЭЖХ, + 0,09% TFA, растворитель В = ацетонитрил, чистый для ВЭЖХ + 0,09% TFA. Общее время ВЭЖХ пробега составляло 7,6 мин с линейным градиентом от 25 до 50% растворителя В в 4 мин, 50% растворителя В в течение 30 с и градиентом от 50 до 25% растворителя В в течение дополнительных 30 с. Колонку повторно уравновешивали 25% растворителем В в течение 2,6 мин перед тем как вводить следующий образец. Величину IC50 (концентрацию, при которой наблюдается 50% ингибирование NS3/4A активности) вычисляли для каждого исследуемого соединения на основании результатов ВЭЖХ. Соединения I-60 тестировали описанным выше ингибиторным анализом. Результаты показали, что 54 соединения обладали IC50 величинами ниже, чем 20 нМ, и 4 соединения обладали IC50 величинами в диапазоне 20-100 нМ. Пример 62. Протокол клеточного анализа HCV репликона Клетки, содержащие HCV репликон, выдерживали в DMEM, содержащем 10% бычьего сывороточного альбумина (FBS), 1,0 мг/мл G418 и соответствующие добавки (среда А). В первый день монослой клеток репликона обрабатывали смесью трипсин/ЭДТА, удаляли и разбавляли средой А до конечной концентрации 48000 клеток/мл. Раствор (1 мл) добавляли к каждой лунке 24-луночного культурального планшета и выращивали в течение ночи в инкубаторе культуральных тканей при 37 С с 5% CO2. Во второй день исследуемое соединение (в 100% ДМСО) серийно разбавляли DMEM, содержащим 10% FBS и соответствующие добавки (среда В). Конечную концентрацию ДМСО поддерживали при 0,2% по всем сериям разбавлений. Удаляли среду из монослоя клеток репликона и добавляли среду В, содержащую различные концентрации соединений. Среду В, не содержащую какого-либо соединения, добавляли к другим лункам, в качестве контролей, не содержащих соединений. Клетки инкубировали с соединением или с 0,2% ДМСО в среде В в течение 72 ч в инкубаторе культуральных тканей с 5% CO2 при 37 С. Затем среду удаляли и монослой клеток репликона однократно промывали PBS. Во избежание разрушения РНК к клеткам немедленно добавляли реагенты для экстракции РНК из наборов RNeasy или реагенты TRIZOL. Общую РНК экстрагировали согласно инструкции,прилагаемой производителем, с модификацией для того, чтобы улучшить эффективность экстракции и устойчивость. Наконец, общую клеточную РНК, включая РНК HCV репликона, элюировали и хранили при -80 С до следующей обработки. Количественный анализ ПЦР при комнатной температуре в режиме реального времени TaqMan проводили с двумя сериями специфических праймеров: одни были на HCV, а другие на АСТВ (бетаактин). Общую РНК добавляли в ПЦР реакции для количественного определения как HCV РНК, так и АСТВ РНК в одной и той же ПЦР лунке. Ошибки эксперимента отмечали и отклоняли на основании уровня АСТВ РНК в каждой лунке. Уровень HCV РНК в каждой лунке вычисляли согласно стандартной кривой, полученной на том же самом ПЦР планшете. Процент ингибирования уровня HCV РНК при обработке соединением вычисляли, используя ДМСО контроль или контроль без соединения, как 0% ингибирование. ЕС 50 (концентрация, при которой достигается 50% ингибирование уровня HCV РНК) вычисляли по кривой титрования для каждого заданного соединения. Соединения 1-60 тестировали по анализу активности HCV репликонов в клеточной среде. Результаты показали, что 52 соединения обладали величинами ЕС 50, ниже, чем 20 нМ, и 3 соединения обладали величинами ЕС 50 в диапазоне 20-100 нМ. Другие варианты осуществления Все признаки, раскрытые в данном описании, могут сочетаться в любой комбинации. Каждый признак, раскрытый в данном описании, можно заменить альтернативным признаком, служащим такой же,эквивалентной или аналогичной цели. Таким образом, если специально не утверждается иное, каждый раскрытый признак представляет собой только пример типичных серий эквивалентов или аналогичных признаков. Из приведенного выше описания специалист в данной области может легко установить существенные признаки настоящего изобретения и без отступления от его сущности и объема может осуществить различные изменения и модификации изобретения для его адаптации к различным применениям и условиям. Таким образом, другие варианты осуществления также входят в объем следующей формулы изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) где каждый из V и Т является -N-;R представляет собой арил или гетероарил и каждый из А 1 и А 2 независимо являются арилом или гетероарилом, каждый из которых необязательно замещен галогеном, С 1-6 алкилом или С 1-6 алкоксилом; или необязательно конденсирован с другим арилом или гетероарилом, каждый из которых необязательно замещен галогеном, С 1-6 алкилом или C1-6 алкоксилом;Z является -ОС(О); где арил относится к углеводородному фрагменту, содержащему одно или несколько ароматических колец; гетероарил относится к фрагменту, содержащему одно или несколько ароматических колец, которые содержат по меньшей мере один гетероатом, выбранный из N, О и S. 2. Соединение по п.1, где Y является где Ri является Н, арилом или гетероарилом, каждый арил и гетероарил необязательно замещен галогеном, C1-10 алкилом или C1-10 алкоксилом; каждый из Rii, Riii, Riv, Rv, Rvi, Rvii и Rviii являются независимо Н, галогеном, С 1-6 алкилом или C1-6 алкоксилом; и Т является таким, как определено в п.1; где арил относится к углеводородному фрагменту, содержащему одно или несколько ароматических колец; гетероарил относится к фрагменту, содержащему одно или несколько ароматических колец, которые содержат по меньшей мере один гетероатом, выбранный из N, О и S. где каждый из Ri; Rii, Riii, Riv, Rv, Rvi, Rvii и Rviii являются такими, как определено в п.2. 6. Соединение по п.1, где R1 является циклопропилом иR2 представляет собой C1-5 алкил или С 3-8 циклоалкил. 7. Соединение по п.1, где соединение обладает такой стереохимией, как показано ниже 8. Соединение по п.1, где соединение представляет собой одно из соединений 9. Фармацевтическая композиция для лечения инфекции вируса гепатита С, содержащая противовирусное средство, имеющее формулу, описанную в п.1, и фармацевтически приемлемый носитель. 10. Фармацевтическая композиция по п.9, дополнительно содержащая иммуномодулирующее средство, другое противовирусное средство или ингибитор NS5B полимеразы, NS5A, NS4B или р 7. 11. Способ лечения инфекции вирусного гепатита С, включающий введение нуждающемуся в этом пациенту эффективного количества соединения по п.1. 12. Способ лечения инфекции вирусного гепатита С, включающий введение нуждающемуся в этом пациенту эффективного количества соединения по п.8. 13. Соединение по п.2, где Y является где каждый из Ri, Rii, Riii, Riv, Rv являются такими, как определено в п.2. 14. Соединение по п.13, где Ri является арилом или гетероарилом, необязательно замещенным галогеном, C1-10 алкилом или C1-10 алкоксилом; каждый из Rii, Riii, Riv, Rv являются независимо Н, галогеном,C1-6 алкилом или C1-6 алкоксилом и Т является таким, как определено в п.1; где арил относится к углеводородному фрагменту, содержащему одно или несколько ароматических колец; гетероарил относится к фрагменту, содержащему одно или несколько ароматических колец, которые содержат по меньшей мере один гетероатом, выбранный из N, О и S. 15. Соединение по п.14, где Y является где Ri является арилом или гетероарилом, необязательно замещенным C1-10 алкоксилом, и каждый изRii, Riii, Riv, Rv являются Н и Т является таким, как определено в п.1; где арил относится к углеводородному фрагменту, содержащему одно или несколько ароматических колец; гетероарил относится к фрагменту, содержащему одно или несколько ароматических колец, которые содержат по меньшей мере один гетероатом, выбранный из N, О и S. 16. Соединение по п.1, где соединение представляет собой одно из соединений
МПК / Метки
МПК: C07D 207/08, C07D 207/16, A61P 31/12
Метки: гепатита, протеазы, ингибиторы, вируса
Код ссылки
<a href="https://eas.patents.su/28-20235-ingibitory-proteazy-virusa-gepatita-s.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы протеазы вируса гепатита с</a>
Ингибиторы ns3 протеазы вируса гепатита с

Номер патента: 13331
Опубликовано: 30.04.2010
Авторы: Вакка Джозеф П., Радд Майкл Т., Макинтайр Чарльз Дж., Ливертон Найджел Дж., Макколи Джон А., Олсен Дэвид Б., Холловэй М.Катарин, Людмерер Стивен У.
МПК: A61P 31/14, C07K 5/08, A61K 38/06...
Метки: протеазы, гепатита, ингибиторы, вируса
Формула / Реферат:
1. Соединение формулы Iили его фармацевтически приемлемая соль,где р и q оба равны 1;R1 является CONR10SO2R6;R2 является C1-С6алкилом или С2-С6алкенилом, где указанный алкил или алкенил необязательно замещен 1-3 атомами галогена;R3 является C1-С8алкилом или C3-С8циклоалкилом;R5 является Н;R6 является C3-С6циклоалкилом;Y является С(=O);Z является О;М является С1-С12алкиленом или С2-С12алкениленом икаждый R10 независимо является Н или...
Ингибиторы серин-протеаз, в частности ns3 протеазы вируса гепатита c (hvc)

Номер патента: 1915
Опубликовано: 22.10.2001
Авторы: Бхисетти Говинда Рао, Фармер Люк Дж., Дейнинджер Дэвид Д, Харбесон Скотт Л., Мурко Марк А., Танг Роджер Д.
МПК: A61P 1/16, C07K 5/10, A61K 38/55...
Метки: гепатита, ингибиторы, частности, вируса, серин-протеаз, протеазы, hvc
Формула / Реферат:
1. Соединение структурной формулы (II) где W является m равно 0 или 1; каждый R2 представляет собой независимо водород, алкил, алкенил, арил, аралкил, аралкенил, циклоалкил, циклоалкилалкил, циклоалкенил, циклоалкенилалкил, гетероциклил, гетероциклилалкил, гетероциклилалкенил, гетероарил или гетероаралкил, или две R2 группы, которые связаны с одним и тем же атомом азота, образуют вместе с этим атомом азота 5-7-членную моноциклическую...
Ингибиторы вируса гепатита с

Номер патента: 18793
Опубликовано: 30.10.2013
Авторы: Кэдоу Джон Ф., Белема Маконен
МПК: A61K 31/439, A61P 31/14, A61K 31/4178...
Метки: ингибиторы, вируса, гепатита
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, где L выбран из связи,R1 и R2 представляют собойили R1 представляет собойи R2 выбран изгде обозначает точку присоединения к исходной молекуле;R3 и R4 независимо выбраны из водорода и гало;каждый R5 независимо выбран из водорода;каждый R6 независимо выбран из алкила;R6a представляет собой алкил, где алкил необязательно может образовывать конденсированное 3-членное кольцо со смежным...
Ингибиторы вируса гепатита с

Номер патента: 19976
Опубликовано: 30.07.2014
Авторы: Кэдоу Джон Ф., Ст.Лорэн Дэнис Р., Снайдер Лоуренс Б., Капур Джейн, Гуд Эндрю С., Лавуа Рико, Бендер Джон А., Ромин Джефри Ли, Лопез Омар Д., Белема Маконен, Хаманн Лоуренс Г.
МПК: A61K 31/4178, A61K 31/4184, A61P 31/12...
Метки: вируса, ингибиторы, гепатита
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль,где каждая m независимо равна 0 или 1;каждая n независимо равна 0 или 1;L представляет собой связь или выбрано изгде каждая группа изображена с левым концом, присоединенным к бензимидазолу, и правым концом, присоединенным к R1;R1 выбран изкаждый R2 независимо выбран из алкила и галогена;каждый R3 представляет собой -C(O)R7;R4 представляет собой алкил или может образовывать...
Ингибиторы вируса гепатита с (hcv)

Номер патента: 15415
Опубликовано: 31.08.2011
Авторы: Линк Джон О., Граупе Михаель, Венкатарамани Чандрэсикар
МПК: A61K 31/00, A61K 47/00, A61K 31/74...
Метки: вируса, hcv, гепатита, ингибиторы
Формула / Реферат:
1. Соединение формулы (I)где Е представляет собой -COCONHR6, где R6представляет собой водород, алкил, циклоалкил, аралкил или гетероаралкил, где ароматическое кольцо необязательно замещено одним или двумя галоидами;R1 представляет собой алкил, циклоалкилалкил, где алифатические или алициклические группы в R1необязательно замещены одним или двумя радикалами Rb, которые независимо выбирают из гидрокси, алкокси, арилокси, гетероарилокси, алкилтио,...
Предыдущий патент: Система распределения текучей среды
Следующий патент: Производные хиноксалиндиона