Гидроизоиндолиновые антагонисты рецептора тахикинина
Номер патента: 10598
Опубликовано: 30.10.2008
Авторы: Цзян Цзиньлун, Бунда Джайме Линн, Девита Роберт Дж., Миллз Сандер Г.
























Формула / Реферат
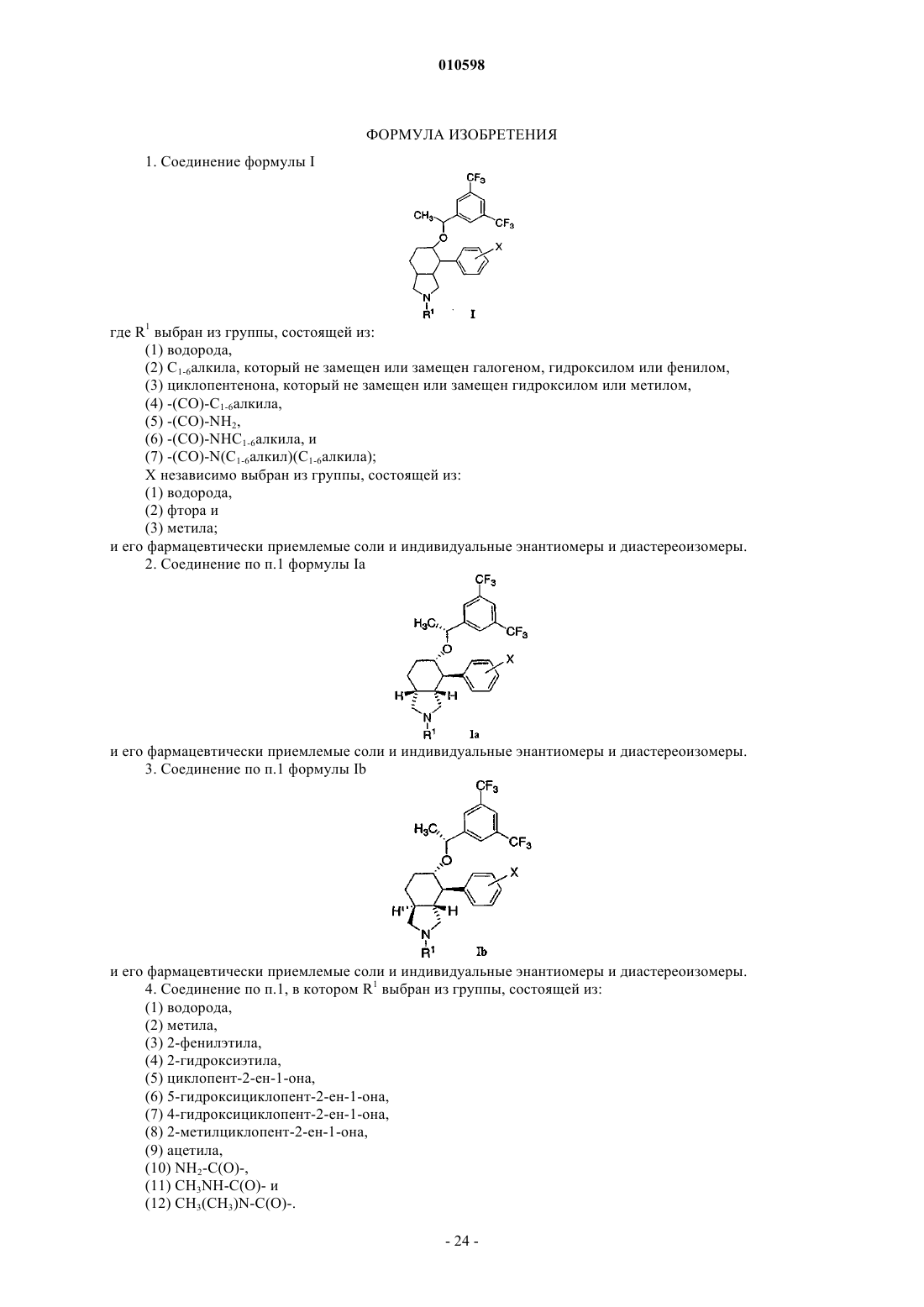
1. Соединение формулы I

где R1 выбран из группы, состоящей из:
(1) водорода,
(2) C1-6алкила, который не замещен или замещен галогеном, гидроксилом или фенилом,
(3) циклопентенона, который не замещен или замещен гидроксилом или метилом,
(4) -(СО)-C1-6алкила,
(5) -(CO)-NH2,
(6) -(СО)-NHC1-6алкила, и
(7) -(СО)-N(С1-6алкил)(C1-6алкила);
X независимо выбран из группы, состоящей из:
(1) водорода,
(2) фтора и
(3) метила;
и его фармацевтически приемлемые соли и индивидуальные энантиомеры и диастереоизомеры.
2. Соединение по п.1 формулы Ia

и его фармацевтически приемлемые соли и индивидуальные энантиомеры и диастереоизомеры.
3. Соединение по п.1 формулы Ib

и его фармацевтически приемлемые соли и индивидуальные энантиомеры и диастереоизомеры.
4. Соединение по п.1, в котором R1 выбран из группы, состоящей из:
(1) водорода,
(2) метила,
(3) 2-фенилэтила,
(4) 2-гидроксиэтила,
(5) циклопент-2-ен-1-она,
(6) 5-гидроксициклопент-2-ен-1-она,
(7) 4-гидроксициклопент-2-ен-1-она,
(8) 2-метилциклопент-2-ен-1-она,
(9) ацетила,
(10) NH2-C(O)-,
(11) CH3NH-C(O)- и
(12) CH3(CH3)N-C(O)-.
5. Соединение по п.1, где X представляет собой водород.
6. Соединение по п.1, где X представляет собой фтор.
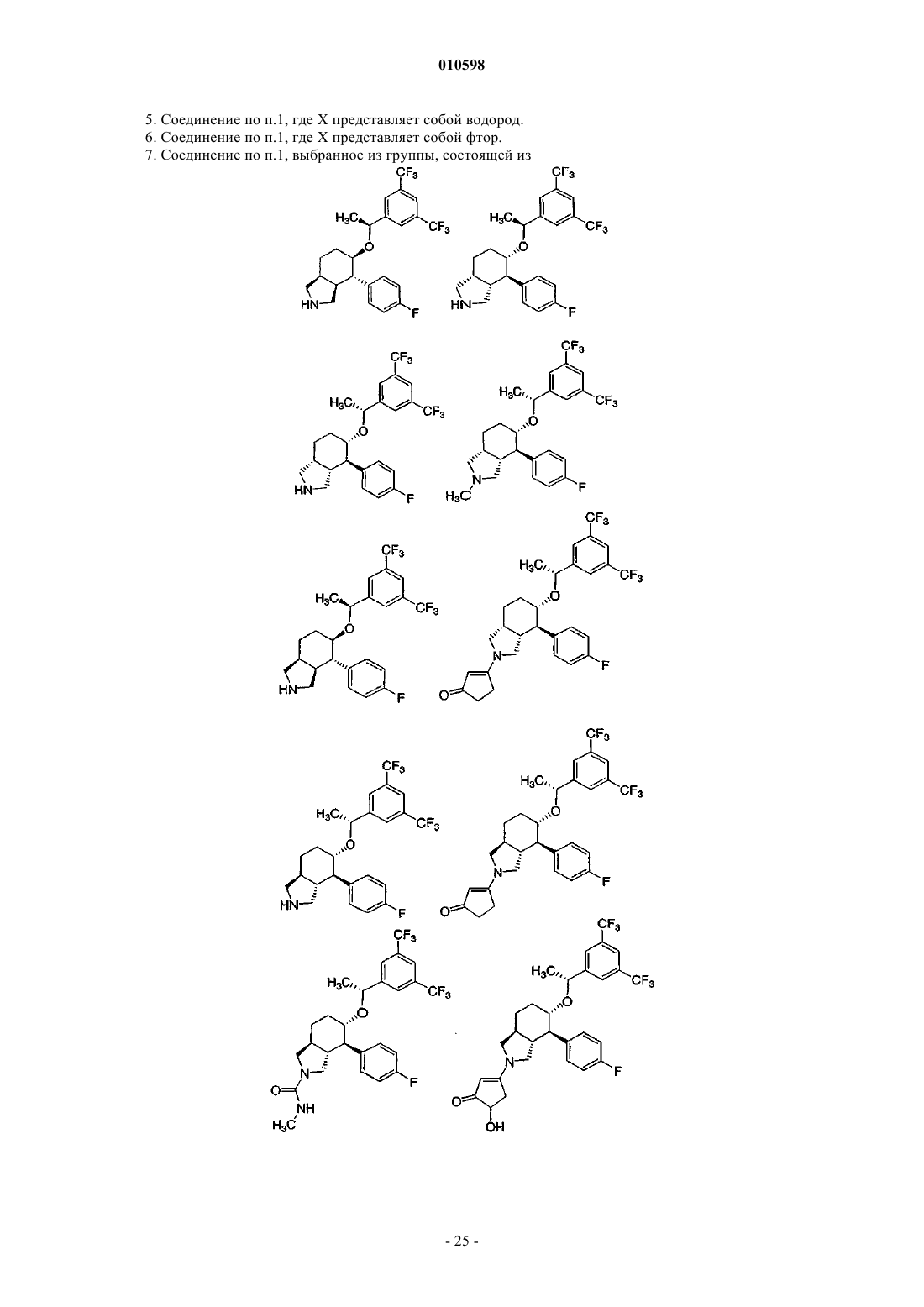
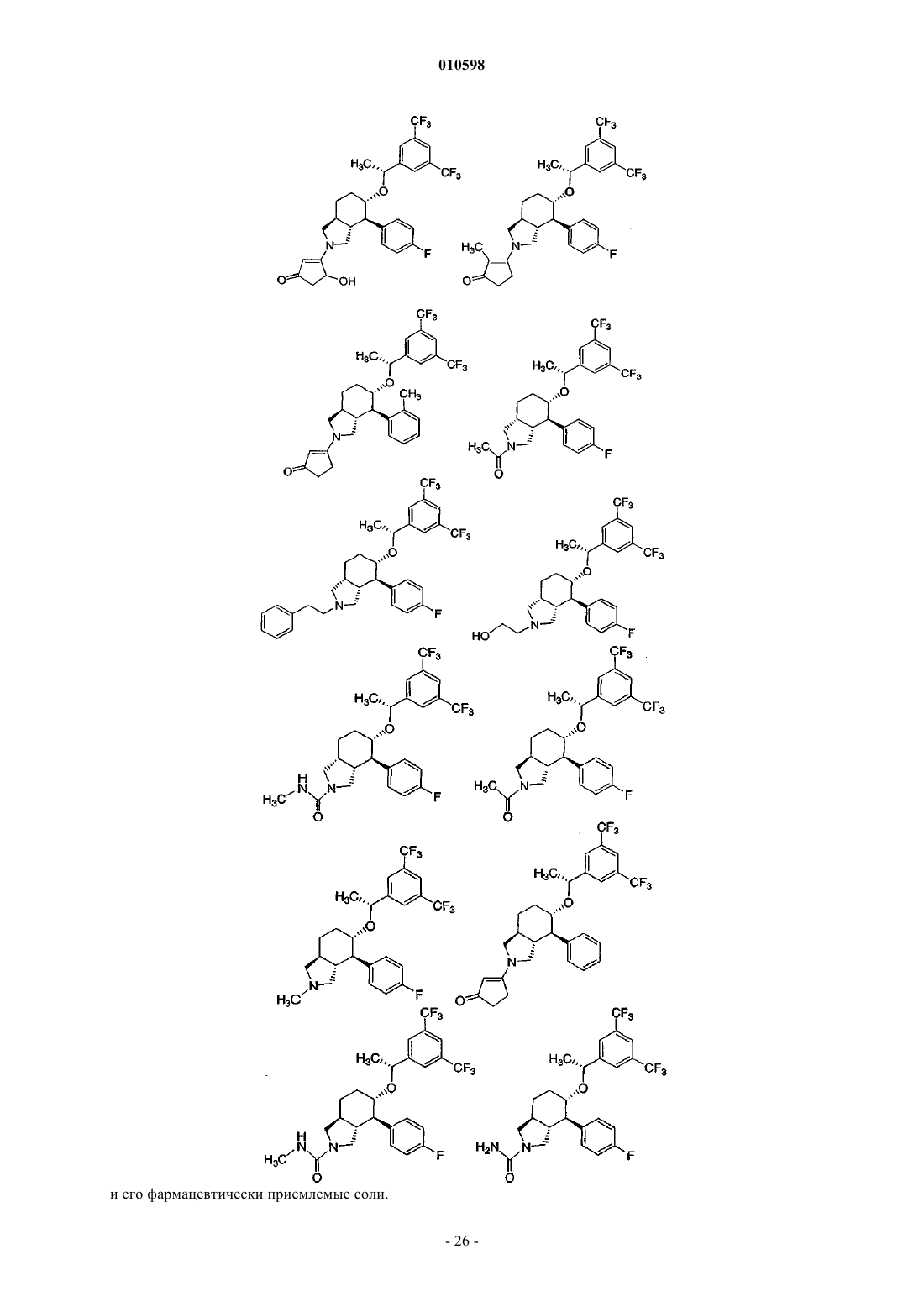
7. Соединение по п.1, выбранное из группы, состоящей из




и его фармацевтически приемлемые соли.
8. Фармацевтическая композиция, включающая инертный носитель и соединение по п.1 или его фармацевтически приемлемую соль.
9. Применение соединения по п.1 в качестве активного компонента для приготовления лекарственного средства для лечения недержания мочи.
10. Способ получения лекарственного средства для лечения физиологического нарушения, связанного с избытком тахикининов у млекопитающего, предусматривающий объединение соединения в соответствии с п.1 настоящего изобретения или его фармацевтически приемлемой соли с фармацевтическим носителем или разбавителем.
11. Соединение формулы

или его фармацевтически приемлемая соль.
12. Фармацевтическая композиция, включающая инертный носитель и соединение по п.11 или его фармацевтически приемлемую соль.
13. Применение соединения по п.11 в качестве активного компонента для приготовления лекарственного средства для лечения недержания мочи.
Текст
010598 Уровень техники Вещество Р представляет собой встречающийся в природе ундекапептид, принадлежащий к семейству пептидов тахикининов, последние называются так в силу быстрого сократительного действия на экстраваскулярные гладкомышечные ткани. Тахикинины отличаются консервативной карбоксиконцевой последовательностью. В дополнение к веществу Р известные тахикинины млекопитающих включают нейрокинин А и нейрокинин В. Настоящая номенклатура обозначает рецепторы к веществу Р, нейрокинину А и нейрокинину В как нейрокинин-1 (NK-1), нейрокинин-2 (NK-2) и нейрокинин-3 (NK-3), соответственно. Антагонисты тахикинина и, в частности, вещества Р полезны в лечении клинических состояний, которые характеризуются наличием избытка активности тахикинина, в частности, вещества Р, включая расстройства центральной нервной системы, ноцицепцию и боль, желудочно-кишечные расстройства,нарушения функции мочевого пузыря и респираторные заболевания. Краткое изложение сущности изобретения Настоящее изобретение направлено на определенные гидроизоиндолиновые соединения, которые являются полезными в качестве антагонистов рецептора нейрокинина-1 (NK-1) и на ингибиторы тахикинина и, в частности, вещество Р. Изобретение также относится к фармацевтическим препаратам, содержащим данные соединения в качестве активных ингредиентов и к применению соединений и их препаратов для лечения определенных нарушений, включая рвоту, недержание мочи, депрессию и тревогу. Подробное описание изобретения Настоящее изобретение относится к соединениям формулы I(1) водорода,(2) C1-6 алкила, который не замещен или замещен галогеном, гидроксилом или фенилом,(3) циклопентенона, который не замещен или замещен гидроксилом или метилом,(4) -(СО)-С 1-6 алкила,(5) -(CO)-NH2,(6) -(СО)-NHC1-6 алкила иX независимо выбран из группы, состоящей из:(3) метила; их фармацевтически приемлемым солям и индивидуальным энантиомерам и диастереоизомерам. Один из вариантов осуществления настоящего изобретения включает соединения формулы Ia где R1 и X определены в настоящем изобретении; и их фармацевтически приемлемые соли и их индивидуальные энантиомеры и диастереоизомеры. Еще один вариант осуществления настоящего изобретения включает соединения формулы Ib где R1 и X определены в настоящем изобретении; и их фармацевтически приемлемые соли и их индивидуальные энантиомеры и диастереоизомеры. Другой вариант осуществления настоящего изобретения включает соединения, в которых R1 выбран из группы, состоящей из:(1) водорода,(2) C1-3 алкила, который не замещен или замещен гидроксилом или фенилом,(3) циклопент-2-ен-1-она, который не замещен или замещен гидроксилом или метилом,(4) -(СО)-C1-3 алкила,(5) -(CO)-NH2,(6) -(СО)-NHC1-3 алкила и(7) -(СО)-N(C1-3 алкил)(C1-3 алкила). Согласно данному варианту осуществления настоящего изобретения предложены соединения, в которых R1 выбран из группы, состоящей из:(12) диметилацетамидо. Дополнительно в данном варианте осуществления настоящее изобретение относится к соединениям, в которых R1 представляет собой водород. Также дополнительно в данном варианте осуществления настоящее изобретение относится к соединениям, в которых R1 представляет собой метил, 2-фенилэтил или 2-гидроксиэтил. Также дополнительно в данном варианте осуществления настоящего изобретения предложены соединения, в которых R1 представляет собой который не замещен или замещен гидроксилом или метилом. Кроме того, дополнительно в данном варианте осуществления настоящее изобретение направлено на соединения, в которых R1 представляет собой ацетил, ацетамидо, метилацетамидо или диметилацетамидо. Еще один вариант осуществления настоящего изобретения включает соединения, в которых X представляет собой водород. Другой вариант осуществления настоящего изобретения включает соединения, в которых X представляет собой фтор. Еще один вариант осуществления настоящего изобретения включает соединения, в которых X представляет собой метил. Конкретные варианты осуществления настоящего изобретения включают соединение, которое выбрано из группы, состоящей из соединений, являющихся предметом примеров в настоящем изобретении и их фармацевтически приемлемых солей и их индивидуальных энантиомеров и диастереоизомеров. Соединения в соответствии с настоящим изобретением могут содержать один или более центров асимметрии и могут, таким образом, возникать в виде рацематов и рацемических смесей, отдельных энантиомеров, диастереоизомерных смесей и индивидуальных диастереоизомеров. Могут присутствовать дополнительные центры асимметрии в зависимости от природы различных заместителей в молекуле. Каждый такой центр асимметрии независимо производит два оптических изомера, и подразумевается,что все возможные оптические изомеры и диастереоизомеры в смесях и в виде чистых или частично очищенных соединений включены в пределы данного изобретения. Подразумевается, что настоящее-2 010598 изобретение охватывает все такие изомерные формы этих соединений. На формуле I показана структура класса соединений без предпочтительной стереохимии. Независимые способы синтеза этих диастереоизомеров или их хроматографического разделения, как известно из уровня техники, могут быть достигнуты путем подходящего изменения методологии, раскрытой в настоящем изобретении. Их абсолютная стереохимия может быть определена путем рентгенокристаллографии кристаллических продуктов или кристаллических интермедиатов, полученных в случае необходимости путем обработки реактивом, содержащим центр асимметрии с известной абсолютной конфигурацией. Если требуется, рацемированные смеси соединений могут быть разделены таким образом, чтобы были выделены индивидуальные энантиомеры. Разделение может быть выполнено способами, известными в уровне техники, такими, как связывание рацемической смеси соединений с энантиомерно чистым соединением для образования диастереоизомерной смеси с последующим разделением индивидуальных диастереоизомеров стандартными методами, такими как дробная кристаллизация или хроматография. Реакция связывания часто представляет собой формирование солей с применением энантиомерно чистой кислоты или основания. Затем диастереоизомерные производные могут быть преобразованы в чистые энантиомеры путем отщепления добавленного хирального остатка. Рацемическая смесь соединений может также быть разделена непосредственно с помощью хроматографических способов, использующих хиральные стационарные фазы,такие способы известны из уровня техники. Альтернативно, любой энантиомер соединения может быть получен с помощью стереоселективного синтеза с применением оптически чистых исходных материалов или реагентов известной конфигурации способами, известными из уровня техники. Существует несколько приемлемых способов присваивания названий обсуждаемым в настоящем изобретении соединениям. Например, приведенное выше соединение можно называть "(3aR,4R,5S,7aR)трет-бутил-5-гидрокси 4-фенилоктагидро-2 Н-изоиндол-2-карбоксилат" или "трет-бутил-(3aR,4R,5S,7aR)-5-гидрокси-4-фенилоктагидро-2 Н-изоиндол-2-карбоксилат". В целом, центральная структура может называться как октагидроизоиндольные, гексагидроизоиндолиновые, пергидроизоиндолиновые, гидроизоиндолиновые или гидроизоиндольные соединения. Как известно специалистам в данной области, предполагается, что галоген в контексте настоящего изобретения включает фтор, хлор, бром и йод. Аналогично C1-6, как в C1-6 алкиле, по определению идентифицирует группу, как содержащую 1, 2, 3, 4, 5 или 6 углеродов в линейной или разветвленной структуре таким образом, что С 1-8 алкил, в частности, включает метил, этил, н-пропил, изо-пропил, н-бутил,изо-бутил, трет-бутил, пентил и гексил. Группа, определенная как независимо замещенная заместителями, может быть независимо замещена множеством таких заместителей. Термин "фармацевтически приемлемые соли" обозначает соли, приготовленные из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли, полученные из неорганических оснований,включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, марганцовистые соли, марганцоватистые, калия, натрия, цинка и т.п. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли в твердой форме могут существовать в более чем одной кристаллической структуре, и могут также быть в форме гидратов. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая встречающиеся в природе замещенные амины, циклические амины и основные ионообменные смолы, такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы,прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин, и т.п. Когда соединение в соответствии с настоящим изобретением является основным, соли могут быть приготовлены из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромисто-водородную, хлористоводородную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, муциновую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, птолуолсульфоновую кислоту, и т.п. Особенно предпочтительными являются лимонная, бромистоводородная, хлористо-водородная, малеиновая, фосфорная, серная, фумаровая и винная кислоты. Следует понимать, что, как предполагается, в используемом в настоящем описании смысле, обращения к со-3 010598 единениям в соответствии с настоящим изобретением также включают фармацевтически приемлемые соли. Применение соединений, раскрытое в примерах и в данном разделе, поясняет изобретение. Конкретные соединения в настоящем изобретении включают соединение, выбранное из группы, состоящей из соединений, раскрытых в следующих примерах и их фармацевтически приемлемых солей и их индивидульных диастереоизомеров. Соединения в соответствии с настоящим изобретением полезны в профилактике и лечении широкого разнообразия клинических состояний, характеризующихся наличием избытка активности тахикинина,в частности, вещества Р. Таким образом, например, избыток активности тахикинина, в частности, вещества Р, влечет за собой разнообразные расстройства центральной нервной системы. Такие расстройства включают нарушения настроения, такие как депрессия или, в частности, депрессивные, например, единичные эпизодические или рекуррентные глубокие депрессивные расстройства и дистимические расстройства, или биполярные расстройства, например, биполярное расстройство I, биполярное расстройство II и циклотимическое расстройство; тревожные расстройства, такие как паническое расстройство с агорафобией или без нее, агорафобия без истории панического расстройства, специфичные фобии, например, специфичные фобии к животным, социальные фобии, обсессивно-компульсивное расстройство,стрессовые расстройства, включая посттравматическое стрессовое расстройство и острое стрессовое расстройство и генерализированные тревожные расстройства; шизофрения и другие психотические расстройства, например, шизофрениформные расстройства, шизоаффективные расстройства, бредовые расстройства, краткие психотические расстройства, индуцированные психотические расстройства и психотические расстройства с бредом или галлюцинациями; делирий, деменция и амнестический и другие когнитивные или нейродегенеративные расстройства, такие как болезнь Альцгеймера, сенильная деменция, деменция типа Альцгеймера, сосудистая деменция и другие деменции, например, по причине ВИЧ заболевания, травмы головы, болезни Паркинсона, болезни Хантингтона, болезни Пика, болезни Крейтцфельдта-Якоба, или по причине множественной этиологии; болезнь Паркинсона и другие экстрапирамидальные расстройства движения, такие как индуцированные лекарственным препаратом расстройства движения, например, индуцированный нейролептическим средством паркинсонизм, злокачественный нейролептический синдром, индуцированная нейролептическим средством острая дистония,индуцированная нейролептическим средством острая акатизия, индуцированная нейролептическим средством поздняя дискинезия и индуцированный лекарственным препаратом постуральный тремор; связанные с веществом расстройства, являющиеся результатом употребления алкоголя, амфетаминов (или подобных амфетамину веществ), кофеина, конопли, кокаина, галлюциногенов, летучих препаратов и аэрозольных сжатых газов, никотина, опиоидов, производных фенилглицидина, седативных средств, снотворных средств и анксиолитиков, где связанные с веществом расстройства включают зависимость и злоупотребление, интоксикацию, абстиненцию, бред при интоксикации, бред при абстиненции, персистирующую деменцию, психотические расстройства, расстройства настроения, расстройства беспокойства, половую дисфункцию и расстройства сна; эпилепсию; синдром Дауна; демиелинизирующие заболевания, такие как MS и ALS и другие нейропатологические расстройства, такие как периферическая невропатия, например диабетическая и индуцированная химиотерапией невропатия, и постгерпетическая невралгия, невралгия тройничного нерва, сегментальная или межреберная невралгия и другие невралгии; и расстройства сосудов головного мозга вследствие острого или хронического цереброваскулярного повреждения, такого как инфаркт головного мозга, субарахноидальное кровотечение или отек головного мозга. Активность тахикинина и, в частности, вещества Р, также участвует в ноцицепции и боли. В связи этим, соединения в соответствии с настоящим изобретением полезны в профилактике или лечении заболеваний и состояний, в которых преобладает боль, включая повреждение мягкой ткани и периферическое повреждение, такое как острая травма, остеоартрит, ревматоидный артрит, скелетно-мышечная боль, в частности, после травмы, спинальная боль, синдромы миофасциальной боли, головная боль, боль вследствие эпизиотомии и ожоги; глубокая и висцеральная боль, такая как сердечная боль, мышечная боль, глазная боль, ротолицевая боль, например, одонталгия, боль в животе, гинекологическая боль, например, дисменорея и родовые схватки; боль, связанная с повреждением нерва и корешка, такая как боль, связанная с нарушениями периферического нерва, например, защемление нерва и разрывы плечевого сплетения, ампутация, периферические невропатии, тригеминальная невралгия, атипичная лицевая боль, повреждение нервного корешка и арахноидит; боль, связанная с раком, часто называемая раковой болью; боль, возникающая в центральной нервной системе, такая, как боли вследствие повреждение спинного мозга или ствола головного мозга; боль в области поясницы; ишиалгия; анкилозирующий спондилит, подагра и боль шрамов. Антагонисты тахикинина и, в частности, вещества Р, могут также быть полезны в лечении респираторных заболеваний, в частности, связанных с избыточной секрецией слизи, таких, как хроническая непроходимость дыхательных путей, бронхопневмония, хронический бронхит, муковисцедоз и астма, респираторный дистресс-синдром взрослых, и бронхоспазм; воспалительные заболевания, такие как воспалительное заболевание кишки, псориаз, фиброзит, остеоартрит, ревматоидный артрит, зуд и солнечный ожог; аллергии, такие как экземы и ринит; расстройства, связанные с гиперчувствительностью, такие,-4 010598 как от сумаха ядоносного; глазные заболевания, такие как конъюнктивит, весенний конъюнктивит и т.п.; глазные состояния, связанные с пролиферацией клеток, такие, как пролиферативная витреоретинопатия; кожные заболевания, такие как контактный дерматит, аллергический дерматит, крапивница и другой экзематоидный дерматит. Антагонисты тахикинина и, в частности, вещества Р, могут также быть полезны в лечении опухолей, включая опухоли молочной железы, нейроганглиобластому и мелкоклеточные карциномы, такие как мелкоклеточный рак легких. Антагонисты тахикинина и, в частности, вещества Р, могут также быть полезны в лечении желудочно-кишечных (ЖК) нарушений, включая воспалительные нарушения и заболевания ЖК тракта, такие как гастрит, гастродуоденальные язвы, желудочные карциномы, желудочные лимфомы, нарушения, связанные с нейронным контролем внутренних органов, неспецифический язвенный колит, болезнь Крона,синдром раздраженной кишки и рвота, включая острую, отсроченную или преждевременную рвоту, такую как рвота, индуцированная химиотерапией, радиацией, токсинами, вирусными или бактериальными инфекциями, беременностью, вестибулярные расстройства, например морская болезнь, вертиго, головокружение и болезнь Меньера, хирургия, мигрень, изменения внутричерепного давления, гастроэзофагеальная рефлюксная болезнь, несварение с повышенной кислотностью, чрезмерное употребление пищи или жидкостей, повышенная кислотность желудка, кислая отрыжка или регургитация, изжога, например эпизодическая, ночная или индуцированная приемом пищи изжога, и диспепсия. Антагонисты тахикинина и, в частности, вещества Р, могут также быть полезны в лечении других различных условий, включая связанные со стрессом соматические нарушения; симпатическую рефлекторную дистрофию, такую как синдром плечо-рука; неблагоприятные иммунологические реакции, такие как отторжение трансплантированных тканей и расстройства, связанные с иммунной активацией или супрессией, такие, как системная красная волчанка; излияние плазмы, вследствие цитокиновой химиотерапии, расстройства функции мочевого пузыря, такие как цистит, гиперрефлексия детрузора мочевого пузыря, частое мочеиспускание и недержание мочи, включая профилактику или лечение сверхактивного мочевого пузыря с симптомами неотложного недержания мочи, неотложности и частоты позыва; фиброзные и коллагеновые заболевания, такие как склеродермия и эозинофильный фасциолез; расстройства кровотока, вызванные заболеваниями с расширением и сужением сосудов, такие, как стенокардия, сосудистая головная боль, мигрень и болезнь Рейнода и боль или ноцицепцию, относящуюся к или связанную с любым из предшествующих состояний, особенно передачу боли при мигрени. Соединения в соответствии с настоящим изобретением имеют также значение в лечении сочетания приведенных выше условий, в особенности в лечении сочетания послеоперационной боли и послеоперационной тошноты и рвоты. Соединения в соответствии с настоящим изобретением, в частности, полезны при профилактике или лечении рвоты, включая острую, отсроченную или преждевременную рвоту, такую как рвота, индуцированная химиотерапией, радиацией, токсинами, беременностью, вестибулярными нарушениями,движением, хирургией, мигренью и изменениями внутричерепного давления. Например, соединения в соответствии с настоящим изобретением могут быть полезны необязательно в сочетании с другими противорвотными агентами для профилактики острой и отсроченной тошноты и рвоты, связанной с начальными и повторными курсами умеренной или сильной тошнотворной химиотерапии рака, включая высокую дозу цисплатина. Особенно соединения в соответствии с настоящим изобретением полезны в лечении рвоты, индуцированной противоопухолевыми (цитотоксическими) агентами, включая обычно используемые в химиотерапии рака, и рвоты, индуцированной другими фармакологическими агентами,например, ролипрамом. Примеры таких химиотерапевтических агентов включают алкилирующие агенты, например, этилениминовые соединения, алкилсульфонаты и другие соединения с алкилирующим действием, такие как нитрозомочевины, цисплатин и дакарбазин; антиметаболиты, например, антагонисты фолиевой кислоты, пурина или пиримидина; ингибиторы митоза, например, алкалоиды барвинка и производные подофиллотоксина; и цитотоксические антибиотики. Конкретные примеры химиотерапевтических агентов описаны, например, у д. J. Stewart в Nausea and Vomiting: Recent Research and ClinicalAdvances, Eds. J. Kucharczyk et al., CRC Press Inc., Boca Raton, Florida, USA (1991), стр.177-203, в особенности, стр.188. Обычно используемые химиотерапевтические агенты включают цисплатин, дакарбазин(CCNU), доксорубицин (адриамицин), даунорубицин, прокарбазин, митомицин, цитарабин, этопозид,метотрексат, 5-фторурацил, винбластин, винкристин, блеомицин и хлорамбуцил [R. J. Gralla и др. в Cancer Treatment Reports (1984) 68(1), 163-172]. Дополнительный аспект настоящего изобретения содержит применение соединения в соответствии с настоящим изобретением для достижения хронобиологического (смещение фазы циркадного ритма) эффекта и облегчения нарушений циркадного ритма у млекопитающих. Настоящее изобретение дополнительно направлено на применение соединения в соответствии с настоящим изобретением для блокирования эффектов смещения фазы света у млекопитающего. Настоящее изобретение дополнительно направлено на применение соединения в соответствии с настоящим изобретением или его фармацевтически приемлемой соли для усиления или улучшения качества сна, а также предотвращения и лечения расстройств сна и нарушений сна у млекопитающих. В частности, настоящее изобретение обеспечивает способ усиления или улучшения качества сна путем увели-5 010598 чения эффективности сна и усиления сохранения сна. Кроме того, настоящее изобретение обеспечивает способ предотвращения и лечения расстройств сна и нарушений сна у млекопитающего, который содержит введение соединения в соответствии с настоящим изобретением или его фармацевтически приемлемой соли. Настоящее изобретение полезно для лечения расстройств сна, включая расстройства инициирования и сохранения сна (бессонницы) ("DIMS"), которые могут возникнуть по психофизиологическим причинам, как следствие психиатрических нарушений (в частности связанных с тревогой), вследствие употребления и злоупотребления наркотическими средствами и алкоголем (в частности в ходе стадий абстиненции), детские развившиеся DIMS, ночной миоклонус, фибромиалгия, мышечная боль, апноэ во сне и усталые ноги и неспецифические нарушения быстрого сна, отмечаемые при старении. Особенно предпочтительными вариантами осуществления настоящего изобретения являются лечение рвоты, недержания мочи, депрессии или тревоги путем введения соединений в соответствии с настоящим изобретением индивидууму (человеку или животному), которому требуется такое лечение. В соответствии с настоящим изобретением предложен способ получения лекарственного средства для противодействия эффекту вещества Р на клеточный рецептор или для блокады рецепторов нейрокинина-1 у млекопитающего, содержащего сочетание соединения в соответствии с настоящим изобретением с фармацевтическим носителем или разбавителем. Кроме того, настоящее изобретение относится к способу получения лекарственного средства для лечения физиологического нарушения, связанного с избытком тахикининов у млекопитающего, содержащего сочетание соединения в соответствии с настоящим изобретением с фармацевтическим носителем или разбавителем. Настоящее изобретение также относится к способу лечения или профилактики физиологических нарушений, связанных с избытком тахикининов, в особенности вещества Р, данный способ предусматривает введение пациенту, которому это требуется, соединения в соответствии с настоящим изобретением,уменьшающего количество тахикинина, или композиции, содержащей соединение в соответствии с настоящим изобретением. В используемом в настоящем описании смысле термин "лечение" или "лечить" обозначает введение соединений в соответствии с настоящим изобретением для уменьшения, улучшения или устранения симптомов или основной причины отмеченных болезненных состояний у индивидуума(человека или животного), который страдает от этого состояния или проявляет его клинические признаки. Термин "профилактика" или "предотвращать" обозначает введение соединений настоящего изобретения для уменьшения, улучшения или устранения риска или вероятности возникновения отмеченных болезненных состояний у индивидуума (человека или животного), восприимчивого или предрасположенного к данному состоянию. Соединения в соответствии с данным изобретением полезны для противодействия тахикининам, в частности веществу Р, при лечении желудочно-кишечных расстройств, расстройств центральной нервной системы, воспалительных заболеваний, боли или мигрени и астмы у млекопитающего, которому требуется такое лечение. Данная активность может быть продемонстрирована следующими анализами. Экспрессия рецептора в COS. Для быстрой экспрессии клонированного рецептора нейрокинина-1 (NK1R) человека в COS, кДНКNK1R человека клонировали в вектор экспрессии pCDM9, который был получен из pCDM8 (INVITROGEN) путем инсерции гена резистентности к ампициллину (нуклеотиды 1973-2964 из BLUESCRIPTSK+) в сайт SacII. Трансфекция 20 мкг плазмидной ДНК в 10 миллионов клеток COS была достигнута путем электропорации в 800 мкл буфера трансфекции (135 мМ NaCl, 1,2 мМ CaCl2, 1,2 мМ MgCl2,2,4 мМ K2HPO4, 0,6 мМ KH2PO4, 10 мМ глюкозы, 10 мМ HEPES рН 7,4) при 260 В и 950 мкФ с применением IBI GENEZAPPER (IBI, New Haven, CT). Клетки инкубировали в 10% фетальной телячьей сыворотки, 2 мМ глутамина, 100 ед/мл пенициллин-стептомицина, и 90% среды DMEM (GIBCO, Grand Island,NY) при 5% CO2 при 37 С в течение трех дней перед анализом. Устойчивая экспрессия в СНО. Для обеспечения устойчивой линии клеток, экспрессирующих клонированный NK1R человека,кДНК субклонировали в вектор pRcCMV (INVITROGEN). Трансфекция 20 мкг плазмидной ДНК в клетки CHO была достигнута электропорацией в 800 мкл буфера трансфекции с добавкой 0,625 мг/мл ДНК семенной жидкости сельди при 300 В и 950 мкФ с применением IBI GENEZAPPER (IBI). Трансфицируемые клетки инкубировали в средах СНО [10% фетальной телячьей сыворотки, 100 ед/мл пенициллинстептомицина, 2 мМ глутамина, 1/500 гипоксантин-тимидина (АТСС), 90% среды IMDM (JRH BIOSCIENCES, Lenexa, KS), 0,7 мг/мл G418 (GIBCO)] при 5% CO2 при 37 С, пока колонии не становились видимыми. Каждая колония была отделена и размножена. Был выбран клон клеток с самым высоким количеством NK1R человека для последующих приложений, таких как отбор лекарственного средства. Протокол анализа с использованием COS или СНО. Анализ на связывание NK1R человека, экспрессированного в клетках COS или СНО, основан на использовании в качестве радиоактивно меченного лиганда 125I-вещества Р (125I-SP от DU PONT, Boston,MA), которое конкурирует с немеченым веществом Р или любым другим лигандом за связывание сNK1R человека. Однослойные клеточные культуры COS или СНО были диссоциированы неферментативным раствором (SPECIALTY MEDIA, Lavallette, NJ) и ресуспендированы в подходящем объеме связывающего буфера (50 мМ Tris pH 7,5, 5 мМ MnCl2, 150 мМ NaCl, 0,04 мг/мл бацитрацина, 0,004 мг/мл-6 010598 лейпептина, 0,2 мг/мл BSA, 0,01 мМ фосфорамидона) так, что 200 мкл суспензии клетки дало бы начало приблизительно 10000 имп/мин специфичного связывания 125I-SP (приблизительно 50000-200000 клеток). В анализе на связывание 200 мкл клеток было добавлено в пробирку, содержащую 20 мкл 1,5-2,5 нМ 125I-SP и 20 мкл немеченого вещества Р или любого другого тестируемого соединения. Пробирки были инкубированы при 4 С или при комнатной температуре в течение 1 ч со слабым встряхиванием. Связанное радиоактивное вещество было отделено от несвязанного радиоактивного вещества с помощью фильтра GF/C (BRANDEL, Gaithersburg, MD), который был предварительно смочен 0,1% полиэтиленимином. Фильтр был три раза промыт 3 мл буфера для промывки (50 мМ Tris pH 7,5, 5 мМMnCl2, 150 мМ NaCl), и его радиоактивность была определена с помощью гамма-счетчика. Также активация фосфолипазы С за счет NK1R в клетках CHO, экспрессирующих NK1R человека, может быть измерена путем определения накопления инозитолмонофосфата, который является продуктом деградацииIP3. Клетки CHO высевают в 12-луночную плашку при 250000 клеток на лунку. После инкубирования в средах CHO в течение 4 дней, клетки загружают 0,025 мкКи/мл 3 Н-миоинозитола путем инкубирования в течение ночи. Внеклеточное радиоактивное вещество удаляют путем промывки фосфатно-буферным солевым раствором. В лунку добавляют LiCl при конечной концентрации 0,1 мМ с тестируемым соединением или без него и продолжают инкубирование при 37 С в течение 15 мин. В лунку добавляют вещество Р при конечной концентрации 0,3 нМ для активации NK1R человека. По истечении 30 мин инкубирования при 37 С, среды удаляют и добавляют 0,1 н. HCl. Каждую лунку обрабатывают ультразвуком при 4 С и экстрагируют CHCl3/метанолом (1:1). Водную фазу вносят в 1 мл ионообменную колонкуDowex AG 1X8. Колонку промывают 0,1 н. муравьиной кислотой, затем 0,025 М формиатом аммония 0,1 н. муравьиной кислотой. Инозитолмонофосфат элюируют 0,2 М формиатом аммония-0,1 н. муравьиной кислотой и подвергают количественному анализу с помощью бета-счетчика частиц. С помощью данных анализов, в частности, может быть продемонстрирована собственная активность антагониста тахикининового рецептора соединений в соответствии с настоящим изобретением. Соединения в соответствии со следующими примерами обладают активностью в указанных выше анализах в диапазоне от 0,05 нМ до 10 мкМ. Активность настоящих соединений может также быть продемонстрирована с помощью анализа, раскрытого Lei, et al., British J. Pharmacol., 105, 261-262 (1992). В соответствии с дополнительным или альтернативным аспектом настоящее изобретение обеспечивает соединение в соответствии с настоящим изобретением для применения в качестве композиции, которая может быть введена индивидууму, которому требуется снижение количества тахикинина или вещества Р в организме. Предполагается, что термин "композиция" в используемом в настоящей описании смысле охватывает продукт, содержащий определенные ингредиенты в предварительно определенных количествах или соотношениях, а также любой продукт, который получается, прямо или косвенно, из сочетания указанных ингредиентов в указанных количествах. Предполагается, что данный термин в отношении фармацевтических композиций охватывает продукт, содержащий один или более активных ингредиентов, и необязательный носитель, содержащий инертные ингредиенты, а также любой продукт, который получается, прямо или косвенно, из сочетания, комплексообразования или агрегации любых двух или более из ингредиентов, или из диссоциации одного или более из ингредиентов, или из других типов реакций или взаимодействий одного или более ингредиентов. В целом, фармацевтические композиции готовят путем равномерного и непосредственного слияния активного ингредиента с жидким носителем или мелко раздробленным твердым носителем или с обоими и, в случае необходимости, с последующим формированием продукта в требуемый препарат. В фармацевтической композиции целевое активное соединение включено в количестве, достаточном для создания требуемого действия на ход или состояние заболеваний. Соответственно, фармацевтические композиции в соответствии с настоящим изобретением охватывают любую композицию, полученную путем смешивания соединения в соответствии с настоящим изобретением и фармацевтически приемлемого носителя. Под "фармацевтически приемлемым" подразумевается носитель, разбавитель или инертный наполнитель, который должен быть совместим с другими ингредиентами препарата и не быть вредным для его реципиента. Фармацевтические композиции, предназначенные для перорального применения, могут быть приготовлены в соответствии с любым способом, известным из уровня техники для производства фармацевтических композиций, и такие композиции могут содержать один или более агентов, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей и консервантов с целью обеспечения фармацевтически привлекательных и съедобных препаратов. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми формообразующими, подходящими для производства таблеток. Данные формообразующие, например, могут представлять собой инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие агенты, например, кукурузный крахмал или альгиновую кислоту; связующие агенты, например, крахмал, желатин или гуммиарабик и смазывающие агенты, например, стеарат магния, стеариновую кислоту или тальк. Таблетки могут быть непокрытыми, или они могут быть покрыты с помощью известных методик для замедления дезинтеграции и всасывания в желудочно-кишечном тракте и, таким образом, обеспечения замедленного действия в течение более длительного периода вре-7 010598 мени. Композиции для перорального применения также могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например,карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водной или масляной средой, например, арахидным маслом, вазелиновым маслом или оливковым маслом. Водные суспензии содержат активные вещества в смеси с инертными наполнителями, подходящими для производства водных суспензий. Масляные суспензии могут быть приготовлены путем суспендирования активного ингредиента в подходящем масле. Также могут использоваться эмульсии типа "масло в воде". Диспергируемые порошки и гранулы, подходящие для приготовления водной суспензии путем добавления воды обеспечивают активный ингредиент в смеси с диспергирующим или смачивающим агентом, суспендирующим агентом и одним или более консервантами. Фармацевтические композиции настоящих соединений могут быть в форме стерильной водной или масляной суспензии для инъекции. Соединения в соответствии с настоящим изобретением можно также вводить в форме свеч для ректального введения. Для местного применения можно использовать кремы,мази, гели, растворы или суспензии и т.д., содержащие соединения в соответствии с настоящим изобретением. Соединения в соответствии с настоящим изобретением также могут быть приготовлены для введения посредством ингаляции. Соединения в соответствии с настоящим изобретением можно также вводить с помощью трансдермального пластыря способами, известными из уровня техники. Композиции, содержащие соединения в соответствии с настоящим изобретением, могут быть представлены в виде единичной дозированной формы и могут быть приготовлены любым из способов, известных из уровня техники фармакологии. Термин "единичная дозированная форма" используется для обозначения единичной дозы, в которой все активные и неактивные ингредиенты объединены в подходящей системе, такой, что пациент или человек, вводящий лекарственное средство пациенту, может открыть одну емкость или упаковку с полной содержащейся там дозой, без необходимости смешивания каких-либо компонентов из двух или более емкостей или пакетов. Типичными примерами единичной дозированной формы являются таблетки или капсулы для перорального приема, флаконы однократной дозы для инъекции или свечи для ректального введения. Предполагается, что данный список единичных дозированных форм не является ограничивающим в каком-либо случае, но представляет типичные примеры единичных дозированных форм в уровне техники фармакологии. Композиции, содержащие соединения в соответствии с настоящим изобретением, могут также быть представлены в виде набора, посредством которого два или более компонентов, которые могут представлять собой активные или неактивные ингредиенты, носители, разбавители и т.п., предоставлены с инструкциями для приготовления фактической формы дозировки пациентом или человеком, вводящим лекарственное средство пациенту. Такие наборы могут быть предоставлены с содержащимися в них всеми необходимыми материалами и ингредиентами, или они могут содержать инструкции для применения или приготовления материалов или компонентов, которые должны быть независимо получены пациентом или человеком, вводящим лекарственное средство пациенту. Под "фармацевтически приемлемым" следует понимать носитель, разбавитель или формообразующее, которые должны быть совместимыми с другими ингредиентами препарата и не быть вредными для их реципиента. Термины "введение" или "вводящий" соединение следует понимать в значении обеспечения соединения в соответствии с изобретением индивидууму, которому требуется лечение, в форме, которая может быть введена в организм данного индивидуума в терапевтически пригодной форме и терапевтически эффективном количестве, включая, без ограничений: пероральные лекарственные формы, такие как таблетки, капсулы, сиропы, суспензии и т.п.; лекарственные формы для инъекций, такие как внутривенные,внутримышечные или интраперитонеальные и т.п.; трансдермальные лекарственные формы, включая кремы, гели, порошки или пластыри; щечные лекарственные формы; порошки для ингаляции, спреи,суспензии и т.п.; и ректальные свечи. Термин "терапевтически эффективное количество" обозначает достаточное количество соединений в соответствии с настоящим изобретением в подходящей композиции и в подходящей лекарственной форме для лечения или предотвращения отмеченных болезненных состояний. Соединения в соответствии с настоящим изобретением можно вводить в сочетании с другим веществом, которое обладает дополняющим эффектом к ингибиторам тахикинина и вещества Р в соответствии с настоящим изобретением. Соответственно, в профилактике или лечении рвоты, соединение в соответствии с настоящим изобретением может быть использовано вместе с другими противорвотными агентами, в особенности с антагонистами рецептора 5 НТ 3, такими как ондансетрон, гранисетрон, трописетрон, паленосетрон и затисетрон, кортикостероидом, таким как дексаметазон, или агонистами рецептораGABAB, такими как баклофен. Аналогично, для профилактики или лечения мигрени соединение в соответствии с настоящим изобретением может быть использовано вместе с другими средствами против мигрени, такими как агонисты эрготаминов или 5 НТ 1, особенно суматриптан, наратриптан, золматриптан или ризатриптан. Учитывается, что для лечения депрессии или тревоги, соединение в соответствии с настоящим изо-8 010598 бретением может использоваться вместе с другими средствами против депрессии или тревоги, как ингибиторы обратного захвата норэпинефрина, селективные ингибиторы обратного захвата серотонина(SSRIs), ингибиторы моноаминоксидазы (MAOIs), обратимо действующие ингибиторы моноаминоксидазы (RIMAs), ингибиторы обратного захвата серотонина и норадреналина (SNRIs), антагонисты адренорецептора, атипичные антидепрессанты, бензодиазепины, агонисты или антагонисты 5-HT1A, в особенности, частичные агонисты 5-HT1A, антагонисты релизинг-фактора кортикотропина (CRF) и их фармацевтически приемлемые соли. Для лечения или профилактики расстройств пищевого поведения,включая ожирение, нейрогенную булимию и компульсивные расстройства пищевого поведения, соединение в соответствии с настоящим изобретением может быть использовано вместе с другими анорексигенными средствами. Учитывается, что для лечения или профилактики боли или ноцицепции или воспалительных заболеваний, соединение в соответствии с настоящим изобретением может быть использовано вместе с противовоспалительным или болеутоляющим средством, таким как агонист опиатов, ингибитор липоксигеназы, такой как ингибитор 5-липоксигеназы, ингибитор циклооксигеназы, такой как ингибитор циклооксигеназы-2, ингибитор интерлейкина, такой как ингибитор интерлейкина-1, антагонистNMDA, ингибитор оксида азота или ингибитор синтеза оксида азота, нестероидное противовоспалительное средство или цитокин-супрессирующее противовоспалительное средство. Учитывается, что при использовании любого сочетания, описанного в данном изобретении, как соединение в соответствии с настоящим изобретением, так и другое активное вещество (вещества) вводят пациенту в течение целесообразного промежутка времени. Соединения могут быть в одном и том же фармацевтически приемлемом носителе и поэтому могут быть введены одновременно. Они могут быть в раздельных фармацевтических носителях, например, обычных пероральных лекарственных формах, которые принимают одновременно. Термин "сочетание" также обозначает случай, в котором соединения предоставлены в раздельных лекарственных формах и их вводят последовательно. В связи с этим, в качестве примера, одна активная составляющая может быть введена в виде таблетки и затем, в пределах целесообразного промежутка времени, вторая активная составляющая может быть введена в виде пероральной лекарственной формы, такой как таблетки или быстрорастворимая пероральная лекарственная форма. Под "быстрорастворимым пероральным препаратом" подразумевается пероральная форма доставки, которая при попадании на язык пациента растворяется в пределах приблизительно 10 с. Под "целесообразным промежутком времени" подразумевается интервал времени, который не превышает приблизительно 1 ч. Таким образом, например, если первая активная составляющая представлена в виде таблетки, то вторая активная составляющая должна быть введена в течение 1 ч, либо в том же типе лекарственной формы, либо в другой лекарственной форме, которая обеспечивает эффективную доставку медикамента. Соединения в соответствии с данным изобретением могут быть введены пациентам (животным и людям), которым требуется такое лечение, в дозировках, которые обеспечивают оптимальную фармацевтическую эффективность. Учитывается, что доза, необходимая для введения в любом конкретном приложении, изменяется от пациента пациенту и определяется не только конкретно выбранным соединением или композицией, но также и путем введения, природой состояния, от которого лечат, возрастом и состоянием пациента, совместным лечением или специальными диетами, которых затем придерживается пациент и другими факторами, известными специалистам в данной области, при этом, в конечном итоге,подходящую дозировку принимают по усмотрению обслуживающего врача. В лечении состояний, связанных с избытком тахикининов, подходящий уровень дозировки соединений в соответствии с настоящим изобретением, или их фармацевтически приемлемых солей, составляет приблизительно 0,001-50 мг/кг в сутки, в частности от приблизительно 0,01 до приблизительно 25 мг/кг, например от приблизительно 0,05 до приблизительно 10 мг/кг в сутки. В целом, диапазон дозировки составляет приблизительно 0,5-1000 мг каждому пациенту в сутки, которая может быть введена в единичной или многократных дозах. Предпочтительно диапазон дозировки составляет приблизительно 0,5-500 мг каждому пациенту в сутки; более предпочтительно приблизительно 0,5-200 мг каждому пациенту в сутки; и еще более предпочтительно приблизительно 5-50 мг каждому пациенту в сутки. Конкретные дозировки соединений в соответствии с настоящим изобретением или их фармацевтически приемлемых солей для введения включают 1, 5, 10, 30, 100 и 500 мг. Фармацевтические композиции в соответствии с настоящим изобретением могут быть обеспечены в препарате, содержащем приблизительно 0,5-1000 мг активного ингредиента; более предпочтительно содержащем приблизительно 0,5-500 мг активного ингредиента; или 0,5-250 мг активного ингредиента; или 1-100 мг активного ингредиента. Конкретные фармацевтические композиции для лечения или профилактики избытка тахикининов содержат приблизительно 1, 5, 10, 30, 100 и 500 мг активного ингредиента. Несколько способов приготовления соединения в соответствии с данным изобретением проиллюстрированы в следующих примерах. Исходные материалы и необходимые интермедиаты в некоторых случаях коммерчески доступны, или могут быть приготовлены в соответствии с процедурами, известными из литературы или как проиллюстрировано в настоящем изобретении. Все спектры 1 Н ЯМР были получены на измерительной аппаратуре при напряженности поля 400 или 500 МГц.(3aR,4R,5S,7aR)-5-1(S)-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-1 Низоиндол и (3aS,4S,5R,7aS)-5-1(S)-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро 1 Н-изоиндол. Этап А. 2-(4-Фторфенил)-N-метокси-N-метилацетамид. В раствор 16,7 г (108,4 ммоль) (4-фторфенил)уксусной кислоты в сухом метиленхлориде в атмосфере азота добавляли 13,8 г (141,5 ммоль) N,O-диметилгидроксиламина, 20 мл триэтиламина, 14,2 г(119,3 ммоль) 4-диметиламинопиридина (DMAP) и 27 г (140,6 ммоль) EDC. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем переносили в делительную воронку. Смесь промывали последовательно 2 н. водн. HCl, солевым раствором, насыщенным водн. NaHCO3 и солевым раствором. Органический слой сушили над осушителем, фильтровали и выпаривали растворитель в вакууме для получения 21 г необработанного соединения в соответствии с заголовком, которое использовали без дополнительной очистки. 1 Н-ЯМР (CDCl3) : 7,26 (2 Н, м), 7,02 (2 Н, м), 3,77 (2 Н, с), 3,65 (3 Н, с), 3,21 (3 Н, с). Этап В. 1-(4-Фторфенил)бут-3-ен-2-он. В раствор 220 мл (1,0 м, 220 ммоль) винилмагнийбромида в 100 мл ТГФ, добавляли по каплям в атмосфере азота при 0 С раствор 21 г (106,6 ммоль) 2-(4-фторфенил)-N-метокси-N-метилацетамида (этап А) в 150 мл сухого эфира. Реакционную смесь перемешивали при 0 С в течение 0,5 ч, затем медленно наливали в смесь льда/2 н. водн. HCl. Получающуюся смесь разбавляли эфиром и солевым раствором,переносили в делительную воронку и отделяли органический слой. Органический слой промывали солевым раствором, сушили над осушителем, фильтровали и выпаривали растворитель в вакууме для получения 14,2 г необработанного соединения в соответствии с заголовком, которое использовали без дополнительной очистки. 1 Н-ЯМР (CDCl3) : 7,19 (2 Н, м), 7,02 (2 Н, т, J = 9,5 Гц), 6,42 (1 Н, дд, J = 14,2 Гц, J2 = 11 Гц), 6,34 (1 Н, д, J = 14,2 Гц), 5,86 (1 Н, д, J = 11 Гц), 3,87 (2 Н, с). Этап С. 1E и 1Z трет-бутил[1-(4-фторбензилиден)проп-2-ен-1-ил]оксидиметилсилан. В раствор 104 мл (104,0 ммоль, 1,2 экв.) 1,0 М раствора трет-бутоксида калия в ТГФ и 100 мл сухого ТГФ в атмосфере азота при -78 С добавляли раствор 14,2 г (86,6 ммоль, 1 экв.) 1-(4-фторфенил)бут-3 ен-2-она (этап В) и 13,0 г (86,6 ммоль) трет-бутилхлордиметилсилана в 100 мл сухого ТГФ. Реакционную смесь перемешивали при -78 С в течение 6 ч и при комнатной температуре в течение 6 ч, затем гасили путем добавления 50 мл воды. Получающуюся смесь нагревали до комнатной температуры, разбавляли 150 мл гексана, переносили в делительную воронку и отделяли органический слой. Органический слой промывали 50 мл солевого раствора, сушили над безводным сульфатом магния, фильтровали и выпаривали растворитель в вакууме для получения 20,5 г необработанных соединений в соответствии с заголовком, которые использовали без дополнительной очистки. 1 Н-ЯМР (CDCl3) : 7,52 (2 Н, м), 6,98 (2 Н, м), 6,33 (1 Н, дд, J1 = 13,2 Гц, J2=8,5 Гц), 5,97 (1 Н, с),5,52 (1 Н, д, J = 13,2 Гц), 5,17 (1 Н, д, J = 8,5 Гц). Этап D. (3aS,4R,7aR)-2-бензил-5-[трет-бутил(диметил)силил]окси-4-(4-фторфенил)-3 а,4,7,7 атетрагидро-1 Н-изоиндол-1,3(2 Н)-дион и (3aR,4S,7aS)-2-бензил-5-[трет-бутил(диметил)силил]окси-4(4-фторфенил)-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)-дион. Раствор 15 г (54,0 ммоль, 1 экв.) 1E и 1Z трет-бутил[1-(4-фторбензилиден)проп-2-ен-1 ил]оксидиметилсилана (этап С) и 12,1 г (64,6 ммоль) N-бензилмалеимида в 150 мл сухого толуола в атмосфере азота нагревали с обратным холодильником в течение 16 ч, затем охлаждали до комнатной температуры. Выпаривали растворитель в вакууме для получения 31 г необработанных соединений в соответствии с заголовком, которые содержали непрореагировавший N-бензилмалеимид, и использовали без дополнительной очистки. 1H-ЯМР (CDCl3) : 7,37-7,26 (3 Н, м), 7,22 (2 Н, м), 7,00 (2 Н, м), 6,78 (2 Н, т, J = 8,5 Гц), 5,07 (1 Н, т,J =2,3 Гц), 4,22 (1 Н, д, J = 16 Гц), 4,15 (1 Н, д, J= 16 Гц), 3,66 (1 Н, д, J = 6,5 Гц), 3,52 (1 Н, т, J = 7,0 Гц),3,14 (1 Н, м), 2,87 (1 Н, м), 2,68 (1 Н, м), 0,92 (1 Н, м), 0,78 (9 Н, с), 0,11 (3 Н, с), -0,1 (3 Н, с). Этап Е. (3aS,4S,7aS)-2-бензил-5-[трет-бутил(диметил)силил]окси-4-(4-фторфенил)-2,3,3 а,4,7,7 агексагидро-1 Н-изоиндол и (3aR,4R,7aR)-2-бензил-5-[трет-бутил(диметил)силил]окси-4-(4-фторфенил)2,3,3 а,4,7,7 а-гексагидро-1 Н-изоиндол. В колбу с круглым дном добавляли 7,3 г (192,0 ммоль, избыток) алюмогидрида лития в сухом эфире в атмосфере азота при 0 С. К получающейся смеси добавляли по каплям раствор 31 г необработанного интермедиата этапа D в 100 мл сухого метиленхлорида в атмосфере азота. Получающуюся смесь пере- 10010598 мешивали при комнатной температуре в течение 1 ч, затем аккуратно гасили при 0 С путем добавления 12 мл воды по каплям, затем 10 мл 5,0 н. водн. NaOH. Получающуюся суспензию перемешивали при комнатной температуре в течение 0,5 ч и отфильтровывали твердые вещества. Растворитель из фильтрата выпаривали в вакууме для получения необработанных соединений в соответствии с заголовком, которые использовали без дополнительной очистки. Этап F. (3aS,4S,7aS)-2-бензил-4-(4-фторфенил)октагидро-5 Н-изоиндол-5-он и (3aR,4R,7aR)-2 бензил-4-(4-фторфенил)октагидро-5 Н-изоиндол-5-он. В раствор интермедиата этапа Е в 60 мл сухого ацетонитрила в атмосфере азота при комнатной температуре добавляли 100 мл (250 ммоль) 2,5 М раствора HF в ацетонитриле. Получающуюся смесь перемешивали при комнатной температуре в течение 16 ч, затем гасили при 0 С путем добавления по каплям 120 мл 5,0 н. водн. NaOH. Ацетонитрил выпаривали в вакууме, и получающуюся водную смесь разбавляли эфиром и водой. Получающуюся смесь переносили в делительную воронку и отделяли органический слой. Водный слой экстрагировали дополнительной долей эфира. Объединенные органические слои промывали 50 мл солевого раствора, сушили над осушителем, фильтровали и выпаривали растворитель в вакууме. Остаток очищали с помощью колоночной флеш-хроматографии на силикагеле с элюциейH-ЯМР (CDCl3) : 7,27-7,23 (3 Н, м), 7,12-7,03 (2 Н, м), 3,75 (1 Н, д, J = 12,9 Гц), 3,61 (2 Н, д, J = 4,8 Гц), 3,61 (1 Н, кв, J = 14,5 Гц), 2,93 (1 Н, т, J = 8,5 Гц), 2,68-2,52 (3 Н, м), 2,43-2,33 (2 Н, м), 2,25 (1 Н, м),2,05 (2 Н, м). Этап G. (3aS,4S,5R,7aS)-2-бензил-4-(4-фторфенил)октагидро-1 Н-изоиндол-5-ол и (3aR,4R,5S,7aR)2-бензил-4-(4-фторфенил)октагидро-1 Н-изоиндол-5-ол. В раствор интермедиата (9,0 г) этапа F в атмосфере азота в сухом эфире при -78 С добавляли 1,0 М раствор алюмогидрида лития (38,3 мл) в эфире. Получающуюся смесь перемешивали при -78 С в течение 0,5 ч, затем аккуратно гасили путем добавления по каплям воды, затем 5,0 н. водн. NaOH. Получающуюся суспензию перемешивали при комнатной температуре в течение 0,5 ч и отфильтровывали твердые вещества. Растворитель из фильтрата выпаривали в вакууме для получения необработанных соединений в соответствии с заголовком в качестве основных соединений, которое использовали без дополнительной очистки. 1MS: (MH)+ 261,9. Этап H. (3aS,4S,5R,7aS)-4-(4-фторфенил)октагидро-1 Н-изоиндол-5-ол и (3aR,4R,5S,7aR)-4-(4 фторфенил)октагидро-1 Н-изоиндол-5-ол. Интермедиат этапа G подвергали гидрогенизации при 50 фунт/кв.дюйм водорода с более 10% по весу 10% Pd-C в этаноле в течение 16 ч при комнатной температуре. Катализатор фильтровали и растворитель из фильтрата выпаривали в вакууме для получения необработанных соединений в соответствии с заголовком, которые использовали без дополнительной очистки. ЭтапI. трет-Бутил-(3aS,4S,5R,7aS)-4-(4-фторфенил)-5-гидроксиоктагидро-2 Н-изоиндол-2 карбоксилат и трет-бутил-(3aR,4R,5S,7aR)-4-(4-фторфенил)-5-гидроксиоктагидро-2 Н-изоиндол-2 карбоксилат. В раствор 7,5 г (31,9 ммоль) интермедиата этапа Н в сухом метиленхлориде в атмосфере азота при комнатной температуре добавляли 9,0 г (41,5 ммоль) бикарбоната дитрет-бутила. Получающуюся смесь перемешивали при комнатной температуре в течение 16 ч, затем выпаривали растворитель в вакууме. Получающуюся смесь растворяли в метаноле и добавляли 5,0 н. водн. NaOH. Получающуюся смесь перемешивали в течение 2 ч, и удаляли метанол в вакууме. Водный остаток разбавляли EtOAc, переносили в делительную воронку и отделяли органический слой. Водный слой экстрагировали дополнительной долей EtOAc. Объединенные органические слои промывали 50 мл солевого раствора, сушили над сульфатом магния, фильтровали и выпаривали растворитель в вакууме. Остаток очищали с помощью колоночной флеш-хроматографии на силикагеле с элюцией EtOAc/гексанами (1/4) для получения 2,8 г рацемических соединений в соответствии с заголовком. 1H-ЯМР (CDCl3) : 7,22 (2 Н, м), 7,07 (2 Н, м), 3,73 (1 Н, м), 3,48-3,33 (2 Н, м), 3,21-3,10 (2 Н, м),2,51 (1 Н, м), 2,18 (1 Н, т, J = 10,7 Гц), 2,25 (1 Н, м), 1,98 (1 Н, м), 1,97-1,85 (1 Н, м), 1,63 (1 Н, м), 1,511,40 (1 Н, м), 1,49, 1,43 (9 Н, два синглета). Также выделили незначительное количество смеси цис-спирта (менее полярный): трет-бутил(3aR,4R,5R,7aR)-4-(4-фторфенил)-5-гидроксиоктагидро-2 Н-изоиндол-2-карбоксилат и трет-бутил(3aS,4S,5S,7aS)-4-(4-фторфенил)-5-гидроксиоктагидро-2 Н-изоиндол-2-карбоксилат. 1 Н-ЯМР (CDCl3) : 7,25 (2 Н, м), 7,05 (2 Н, м), 3,95 (1 Н, м), 3,50-3,20 (3 Н, м), 3,08, 2,95 (1 Н, два дублета, J = 14,3 Гц), 2,77 (1 Н, м), 2,65-2,55 (2 Н, м), 2,15 (1 Н, м), 1,82 (2 Н, м), 1,58 (1 Н, м), 1,45, 1,40(9 Н, два синглета). Этап J. трет-Бутил-(3aS,4S,5R,7aS)-5-[3,5-бис-(трифторметил)бензоил]окси-4-(4-фторфенил)- 11010598 октагидро-2 Н-изоиндол-2-карбоксилат и трет-бутил-(3aR,4R,5S,7aR)-5-[3,5-бис-(трифторметил)бензоил]окси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-карбоксилат. В раствор 0,09 г (0,26 ммоль) интермедиата этапа I в сухом метиленхлориде в атмосфере азота при комнатной температуре добавляли 0,089 г (0,32 ммоль) 3,5-бис-(трифторметил)бензоилхлорид, 0,07 млTEA и каталитическое количество DMAP. Получающуюся смесь перемешивали при комнатной температуре в течение 2 ч, затем переносили в делительную воронку, промывали насыщенным водн. NaHCO3,водн. KHSO4 и солевым раствором. Объединенные органические слои сушили над сульфатом магния,фильтровали и выпаривали растворитель в вакууме для получения 0,15 г необработанного соединения в соответствии с заголовком, которое использовали без дополнительной очистки. 1 Н-ЯМР (CDCl3) : 8,63 (1 Н, с), 8,19 (2 Н, с), 7,25 (2 Н, м), 7,00 (2 Н, м), 5,22 (1 Н, м), 3,59-3,43 (2 Н,м), 3,30-3,20 (2 Н, м), 2,83 (1 Н, т, J = 12,7 Гц), 2,62 (1 Н, м), 2,43 (1 Н, м), 2,20 (1 Н, м), 2,02 (1 Н, м), 1,901,70 (2 Н, м), 1,55, 1,47 (9 Н, два синглета). ЭтапK. трет-Бутил-(3aS,4S,7aS)-5-(1-[3,5-бис-(трифторметил)фенил]винилокси)-4-(4 фторфенил)октагидро-2 Н-изоиндол-2-карбоксилат и трет-бутил-(3aR,4R,5S,7aR)-5-(1-[3,5-бис(трифторметил)фенил]винилокси)-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-карбоксилат. В раствор 0,15 г (0,26 ммоль) интермедиата этапа J в сухом ТГФ в атмосфере азота при 0 С добавляли 2 мл 0,5 М раствора реактива Tebbe в толуоле. Получающуюся смесь перемешивали при 0 С в течение 0,5 ч, затем аккуратно гасили путем добавления по каплям 0,5 мл воды, затем 0,5 мл 5,0 н. водн.NaOH. Получающуюся суспензию разбавляли этилацетатом, перемешивали при комнатной температуре в течение 0,5 ч и отфильтровывали твердые вещества. Получающийся фильтрат перемешивали с 0,5 мл 5,0 н. водн. NaOH в течение 16 ч и твердые вещества отфильтровывали через прокладку вспомогательного фильтрующего материала. Растворитель выпаривали в вакууме для получения необработанных соединений в соответствии с заголовком, которые использовали без дополнительной очистки. 1 Н-ЯМР (CDCl3) : 7,73 (1 Н, с), 7,55 (2 Н, с), 7,30-7,18 (2 Н, м), 7,03 (2 Н, м), 4,67 (1 Н, с), 4,37 (1 Н,с), 4,25 (1 Н, м), 3,55-3,30 (3 Н, м), 3,27-3,15 (2 Н, м), 2,81 (1 Н, т, J = 12,7 Гц), 2,60 (1 Н, м), 2,40-30 (2 Н,м), 1,98 (1 Н, м), 1,83 (1 Н, м), 1,55, 1,47 (9 Н, два синглета). Этап L. трет-Бутил-(3aS,4S,5R,7aS)-5-1R-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-карбоксилат и трет-бутил-(3aR,4R,5S,7aR)-5-1S-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-карбоксилат и трет-бутил-(3aS,4S,5R,7aS)-51S-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-карбоксилат,трет-бутил-(3aR,4R,5S,-7aR)-5-1R-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро 2 Н-изоиндол-2-карбоксилат. Интермедиат этапа G подвергали гидрогенизации при 50 фунт/кв.дюйм водорода с более 10% по весу 10% Pd-C в этаноле в течение 16 ч при комнатной температуре. Катализатор фильтровали, и растворитель из фильтрата выпаривали в вакууме для получения необработанных соединений в соответствии с заголовком, которые очищали с помощью препаративной ТСХ с элюцией EtOAc/гексанами (1/3) для получения двух диастереоизомеров. Менее полярный (основной) изомер. 1 Н-ЯМР (CDCl3) : 7,73 (1 Н, с), 7,58 (2 Н, с), 7,25 (2 Н, м), 7,10 (2 Н, м), 4,05 (1 Н, м), 3,23-3,30 (3 Н,м), 3,20-3,07 (2 Н, м), 2,55 (1 Н, т, J = 10,3 Гц), 2,45 (1 Н, м), 2,33 (1 Н, м), 2,20-1,55 (3 Н, м), 1,50, 1,43 (9 Н, два синглета), 0,95 (3 Н, д, J = 6,9 Гц), 1,0-0,82 (1 Н, м). Не основной изомер. 1(3aR,4R,5S,7aR)-5-1(S)-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-1 Н-изоиндол и (3aS,4S,5R,7aS)-5-1(R)-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-1 Н-изоиндол. Менее полярный главный диастереоизомер промежуточного этапа L растворяли в сухом метиленхлориде и обрабатывали анизолом и ТФК при комнатной температуре в течение 2 ч. Растворитель выпаривали в вакууме, и остаток поглощали с помощью EtOAc. Раствор промывали водн. NaOH, затем солевым раствором, сушили над осушителем и фильтровали. Растворитель выпаривали в вакууме для получения необработанных соединений в соответствии с заголовком. 1 Н-ЯМР (CDCl3) : 7,77 (1 Н, с), 7,60 (2 Н, с), 7,28 (2 Н, м), 7,12 (2 Н, т, J = 8,2 Гц), 4,07 (1 Н, м), 3,35(3aR,4R,5S,7aR)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-1 Низоиндол. Этап A. (3aS,4S,5R,7aS)-2-Бензил-4-(4-фторфенил)октагидро-1 Н-изоиндол-5-ол и (3aR,4R,5S,7aR)2-бензил-4-(4-фторфенил)октагидро-1 Н-изоиндол-5-ол. Начиная с 3,5 г рацемической смеси (3aS,4S,5R,7aS)-2-бензил-4-(4-фторфенил)октагидро-1 Низоиндол-5-ола и (3aR,4R,5S,7aR)-2-бензил-4-(4-фторфенил)октагидро-1 Н-изоиндол-5-ола (интермедиат примера 1, этапа G), они были разделены с помощью хиральной ВЭЖХ с применением колонкиCHIRACEL AD с элюцией гексанами/EtOH (9/1) для получения первого элюируемого изомераB. трет-Бутил-(3aR,4R,5S,7aR)-4-(4-фторфенил)-5-гидроксиоктагидро-2 Н-изоиндол-2 карбоксилат. В раствор 5,36 г (15,8 ммоль) второго элюируемого изомера (3aR,4R,5S,7aR)-2-бензил-4-(4 фторфенил)октагидро-1 Н-изоиндол-5-ола (интермедиата этапа А) в 80 мл EtOH добавляли 4,31 г(19,7 ммоль) бикарбоната ди-трет-бутила и 0,5 г 10% Pd-C. Получающуюся смесь подвергали гидрогенизации при 50 фунт/кв.дюйм водорода в течение 16 ч при комнатной температуре. Катализатор фильтровали и добавляли 5 мл 5,0 н. водн. NaOH. Растворитель выпаривали в вакууме. Водный остаток разбавляли EtOAc, переносили в делительную воронку, промывали солевым раствором, сушили над осушителем, фильтровали и выпаривали растворитель в вакууме. Остаток очищали с помощью колоночной флеш-хроматографии на силикагеле с элюцией с EtOAc/гексанами (1/4) для получения соединения в соответствии с заголовком. 1H-ЯМР (CDCl3) : 7,22 (2 Н, м), 7,07 (2 Н, м), 3,73 (1 Н, м), 3,48-3,33 (2 Н, м), 3,21-3,10 (2 Н, м),2,51 (1 Н, м), 2,18 (1 Н, т, J = 10,7 Гц), 2,25 (1 Н, м), 1,98 (1 Н, м), 1,97-1,85 (1 Н, м), 1,63 (1 Н, м), 1,511,40 (1 Н, м), 1,49, 1,43 (9 Н, два синглета). Этап С. трет-Бутил-(3aR,4R,5S,7aR)-5-[3,5-бис-(трифторметил)бензоил]окси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-карбоксилат. В раствор 4,75 г (14,0 ммоль) интермедиата этапа В в сухом метиленхлориде в атмосфере азота при комнатной температуре добавляли 4,71 г (17,0 ммоль) 3,5-бис-(трифторметил)бензоилхлорида, 2,4 мл(17,3 ммоль) TEA и каталитическое количество DMAP. Получающуюся смесь перемешивали при комнатной температуре в течение 2 ч, затем переносили в делительную воронку, промывали насыщенным водн. NaHCO3 и солевым раствором. Объединенные органические слои сушили над сульфатом магния, фильтровали и выпаривали растворитель в вакууме для получения необработанного соединения в соответствии с заголовком, которое использовали без дополнительной очистки. 1 Н-ЯМР (CDCl3) : 8,63 (1 Н, с), 8,19 (2 Н, с), 7,25 (2 Н, м), 7,00 (2 Н, м), 5,22 (1 Н, м), 3,59-3,33 (2 Н,м), 3,30-3,20 (2 Н, м), 2,83 (1 Н, т, J= 12,7 Гц), 2,62 (1 Н, м), 2,43 (1 Н, м), 2,20 (1 Н, м), 2,02 (1 Н, м), 1,901,70 (2 Н, м), 1,55, 1,47 (9 Н, два синглета). ЭтапD. трет-Бутил-(3aR,4R,5S,7aR)-5-(1-[3,5-бис-(трифторметил)фенил]винилокси)-4-(4 фторфенил)октагидро-2 Н-изоиндол-2-карбоксилат. В раствор 9,5 г (14,0 ммоль) необработанного интермедиата этапа С в сухом ТГФ в атмосфере азота при 0 С добавляли 66 мл (33 ммоль) 0,5 М раствора реактива Tebbe в толуоле. Получающуюся смесь перемешивали при 0 С в течение 2 ч, затем аккуратно гасили путем добавления по каплям 7,5 мл воды,затем 7,5 мл 5,0 н. водн. NaOH. Получающуюся суспензию перемешивали при комнатной температуре в течение 0,5 ч и отфильтровывали твердые вещества. Получающийся фильтрат перемешивали с 5 мл 5,0 н. водн. NaOH в течение 16 ч и твердые вещества отфильтровывали через вспомогательный фильтрующий материал. Растворитель выпаривали в вакууме и остаток очищали с помощью колоночной хроматографии с элюцией EtOAc/гексанами (1/3) для получения соединения в соответствии с заголовком. 1- 13010598 Раствор 10,2 г необработанного интермедиата этапа D в этаноле подвергали гидрогенизации при 50 фунт/кв.дюйм водорода с более 1 г 10% Pd-C в этаноле в течение 3 ч при комнатной температуре. Катализатор фильтровали, и растворитель из фильтрата выпаривали в вакууме для получения необработанных соединений в соответствии с заголовком, которые очищали с помощью колоночной хроматографии с элюцией EtOAc/гексанами (2/3) для получения 8,9 г (15,5 моль) двух диастереоизомеров с основным (S) диастереоизомером. Данную смесь поглощали 150 мл сухого ТГФ в атмосфере азота и обрабатывали 80 мл (80 ммоль) 1,0 м раствора трет-бутоксида калия в ТГФ. Получающуюся смесь нагревали при 40 С в течение 1 ч, охлаждали до комнатной температуры и гасили путем добавления воды. Смесь разбавляли EtOAc, переносили в делительную воронку, промывали солевым раствором, сушили над осушителем, фильтровали и выпаривали растворитель в вакууме. Остаток очищали с помощью колоночной флеш-хроматографии на силикагеле с элюцией с EtOAc/гексанами (1/3) для получения менее полярного трет-бутил-(3aR,4R,5S,7aR)-5-(1S)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-карбоксилата и более полярного трет-бутил-(3aR,4R,5S,7aR)-5-(1R)-1-[3,5-бис(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-карбоксилата. 1 Н-ЯМР (CDCl3) : менее полярного изомера: : 7,73 (1 Н, с), 7,58 (2 Н, с), 7,25 (2 Н, м), 7,10 (2 Н,м), 4,05 (1 Н, м), 3,23-3,30 (3 Н, м), 3,20-3,07 (2 Н, м), 2,55 (1 Н, т, J = 10,3 Гц), 2,45 (1 Н, м), 2,33 (1 Н, м),2,20-1,55 (3 Н, м), 1,50, 1,43 (9 Н, два синглета), 0,95 (3 Н, д, J = 6,9 Гц), 1,0-0,82 (1 Н, м). 1 Н-ЯМР (CDCl3) более полярного изомера: 7,71 (1 Н, с), 7,20 (2 Н, с), 6,97 (2 Н, м), 6,85 (2 Н, м),4,47 (1 Н, м), 3,43-3,03 (4 Н, м), 2,47 (2 Н, м), 2,15 (2 Н, м), 1,92 (1 Н, т, J = 10,5 Гц), 1,80-1,57 (3 Н, м),1,50, 1,43 (9 Н, два синглета), 1,30 (3 Н, д, J = 6,9 Гц). Этап F. Хлористо-водородная соль (3aR,4R,5S,7aR)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-1 Н-изоиндола. Более полярный диастереоизомер промежуточного этапа Е (1,5 г, 2,6 ммоль) растворяли в 20 мл 4 н. HCl в диоксане и перемешивали при комнатной температуре в течение 2 ч. Растворитель выпаривали в вакууме, и остаток поглощали EtOAc. Раствор промывали водн. NaOH,затем солевым раствором, сушили над осушителем и фильтровали. Растворитель выпаривали в вакууме для получения необработанного соединения в соответствии с заголовком. Обработка с HCl в диоксане дала соль HCl. 1 Н-ЯМР (CDCl3) : 7,75 (1 Н, с), 7,37 (2 Н, с), 7,13 (2 Н, м), 6,87 (2 Н, т, J = 8,5 Гц), 4,63 (1 Н, кв, 6,5 Гц), 3,45 (1 Н, тд, J1= 4 Гц, J2 = 11,9 Гц), 3,17 (1 Н, м), 3,10 (1 Н, дд, J1 = 6,5 Гц, J2 = 9,5 Гц), 2,90 (1 Н, J = 12,7 Гц), 2,57 (2 Н, м), 2,47 (1 Н, т, J = 9,5 Гц), 2,25 (1 Н, м), 1,98 (2 Н, м), 1,68 (1 Н, м), 1,10 (3 Н, д, 6,5 Гц).(3aR,4R,5S,7aR)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)-2 метилоктагидро-1H-изоиндол. В раствор 30 мг (0,063 ммоль) (3aR,4R,5S,7aR)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4(4-фторфенил)октагидро-1 Н-изоиндола (пример 2) в 2 мл метанола добавляли 20 мг (избыток) водн. формальдегида и 40 мг ацетата натрия. Получающуюся смесь перемешивали при комнатной температуре в течение 10 мин, затем добавляли 20 мг NaBH4. Получающуюся смесь перемешивали при комнатной температуре в течение 1 ч, затем добавляли воду. Метанол выпаривали в вакууме и остаток экстрагировали эфиром (225 мл). Объединенные экстракты сушили над осушителем, фильтровали и выпаривали растворитель в вакууме. Остаток очищали с помощью препаративной ТСХ с элюцией EtOAc/MeOH (9/1) для получения соединения в соответствии с заголовком. 1(3aS,4S,5R,7aS)-5-(1S)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)-октагидро-1 Низоиндол. Этап А. трет-Бутил-(3aS,4S,5R,7aS)-4-(4-фторфенил)-5-гидроксиоктагидро-2 Н-изоиндол-2 карбоксилат. Соединение в соответствии с заголовком готовили из (3aS,4S,5R,7aS)-2-бензил-4-(4 фторфенил)октагидро-1 Н-изоиндол-5-ола (первый элюируемый изомер примера 2, этапа А) в соответствии с процедурой примера 2, этапа В. 1 Н-ЯМР (CDCl3) : 7,22 (2 Н, м), 7,07 (2 Н, м), 3,73 (1 Н, м), 3,48-3,33 (2 Н, м), 3,21-3,10 (2 Н, м),2,51 (1 Н, м), 2,18 (1 Н, т, J = 10,7 Гц), 2,25 (1 Н, м), 1,98 (1 Н, м), 1,97-1,85 (1 Н, м), 1,63 (1 Н, м), 1,511,40 (1 Н, м), 1,49, 1,43 (9 Н, два синглета). Этап В. трет-Бутил-(3aS,4S,5R,7aS)-5-[3,5-бис-(трифторметил)бензоил]окси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-карбоксилат. Соединение в соответствии с заголовком готовили из интермедиата этапа А в соответствии с процедурой примера 2, этапа С. 1(2 Н, м), 3,30-3,20 (2 Н, м), 2,83 (1 Н, т, J = 12,7 Гц), 2,62 (1 Н, м), 2,43 (1 Н, м), 2,20 (1 Н, м), 2,02 (1 Н, м),1,90-1,70 (2 Н, м), 1,55, 1,47 (9 Н, два синглета). Этап С. трет-Бутил-(3aS,4S,5R,7aS)-5-(1-[3,5-бис-(трифторметил)фенил]винилокси)-4-(4 фторфенил)октагидро-2 Н-изоиндол-2-карбоксилат. Соединение в соответствии с заголовком готовили из интермедиата этапа В в соответствии с процедурой примера 2, этапа D. 1H-ЯМР (CDCl3) : 7,73 (1 Н, с), 7,55 (2 Н, с), 7,30-7,18 (2 Н, м), 7,03 (2 Н, м), 4,67 (1 Н, с), 4,37 (1 Н,с), 4,25 (1 Н, м), 3,55-3,30 (3 Н, м), 3,27-3,15 (2 Н, м), 2,81 (1 Н, т, J = 12,7 Гц), 2,60 (1 Н, м), 2,40-2,30 (2 Н,м), 1,98 (1 Н, м), 1,83 (1 Н, м), 1,55, 1,47 (9 Н, два синглета). Этап D. трет-Бутил-(3aS,4S,5R,7aS)-5-(1S)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-карбоксилат и трет-бутил-(3aS,4S,5R,7aS)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-карбоксилат. Соединения в соответствии с заголовком готовили из интермедиата этапа С в соответствии с процедурой примера 2, этапа Е. 1 Н-ЯМР (CDCl3) : менее полярного изомера, : 7,73 (1 Н, с), 7,58 (2 Н, с), 7,25 (2 Н, м), 7,10 (2 Н,м), 4,05 (1 Н, м), 3,23-3,30 (3 Н, м), 3,20-3,07 (2 Н, м), 2,55 (1 Н, т, J = 10,3 Гц), 2,45 (1 Н, м), 2,33 (1 Н, м),2,20-1,55 (3 Н, м), 1,50, 1,43 (9 Н, два синглета), 0,95 (3 Н, д, J = 6,9 Гц), 1,0-0,82 (1 Н, м). 1 Н-ЯМР (CDCl3) более полярного изомера: 7,71 (1 Н, с), 7,20 (2 Н, с), 6,97 (2 Н, м), 6,85 (2 Н, м),4,47 (1 Н, м), 3,43-3,03 (4 Н, м), 2,47 (2 Н, м), 2,15 (2 Н, м), 1,92 (1 Н, т, J = 10,5 Гц), 1,80-1,57 (3 Н, м),1,50, 1,43 (9 Н, два синглета), 1,30 (3 Н, д, J = 6,9 Гц). Этап Е.(3aS,4S,5R,7aS)-5-(1S)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-1 Н-изоиндол. Соединение в соответствии с заголовком готовили из трет-бутил-(3aS,4S,5R,7aS)-5-(1S)-1-[3,5-бис(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-карбоксилата (этап D) в соответствии с процедурой примера 2, этапа F. Более полярный изомер. 1 Н-ЯМР (CDCl3) : 7,68 (1 Н, с), 7,20 (2 Н, с), 7,05 (2 Н, м), 6,87 (2 Н, т, J = 8,2 Гц), 4,27 (1 Н, м), 3,28 3-[(3aR,4R,5S,7aR)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро 2 Н-изоиндол-2-ил]циклопент-2-ен-1-он. В раствор 12,3 мг (0,26 ммоль) (3aR,4R,5S,7aR)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4(4-фторфенил)октагидро-1 Н-изоиндола (пример 2) в 2 мл сухого толуола добавляли 2,7 мг(0,028 ммоль) циклопентан-1,3-диона и каталитическое количество (0,5 мг) PTSA. Получающуюся смесь нагревали с обратным холодильником в течение 16 ч. Растворитель удаляли в вакууме и остаток очищали с помощью препаративной ТСХ с элюцией EtOAc/MeOH (95/5) для получения соединения в соответствии с заголовком. 1 Н-ЯМР (CDCl3) : 7,73 (1 Н, с), 7,20 (2 Н, с), 7,03-6,90 (2 Н, м), 4,98, 4,80 (1 Н, с), 4,50 (1 Н, м), 3,623,18 (2 Н, м), 3,30-3,18 (3 Н, м), 3,15, 2,97 (1 Н, д, J = 11,2 Гц), 2,68 (2 Н, м), 2,55-2,40 (4 Н, м), 2,17(3aR,4R,5S,7aS)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-1 Низоиндол. Этап А. Диэтил-4-[трет-бутил(диметил)силил]окси-3-(4-фторфенил)циклогекс-4-ен-1,2 дикарбоксилат. В раствор 37 г (чистый на 80%, 133,1 ммоль, 1 экв.) 1E и 1Z-трет-бутил-[1-(4 фторбензилиден)проп-2-ен-1-ил]оксидиметилсилана (пример 1, этап С) и 17 мл (18 г, 104,6 ммоль) диэтил (2 Е)-бут-2-ендиоата в 200 мл ксилолов в атмосфере азота нагревали при 160 С в течение 5 ч, затем охлаждали до комнатной температуры. Растворитель выпаривали в вакууме для получения маслянистой жидкости, которую использовали без дополнительной очистки. Этап В. Рацемический диэтил-(1S,2S,3R)-3-(4-фторфенил)-4-оксоциклогексан-1,2-дикарбоксилат и диэтил-(1R,2R,3S)-3-(4-фторфенил)-4-оксоциклогексан-1,2-дикарбоксилат. В раствор приведенного выше интермедиата в 30 мл ацетонитрила в атмосфере азота при комнатной температуре в пластиковой реакционной колбе добавляли 200 мл (500 ммоль) 2,5 М раствора HF в ацетонитриле. Получающуюся смесь перемешивали при комнатной температуре в течение 24 ч. Реакционную смесь добавляли в смесь 125 мл 5,0 н. водн. NaOH и 100 г льда, затем перемешивали при комнатной температуре в течение 5 мин. Получающуюся смесь разбавляли 300 мл эфира. Получающуюся смесь переносили в делительную воронку и отделяли органический слой. Водный слой насыщали NaCl, затем экстрагировали дополнительной долей эфира. Объединенный органический слой промывали солевым раствором, сушили над осушителем, фильтровали, и выпаривали растворитель в вакууме для получения 40,8 г соединений в соответствии с заголовком. 1 Н-ЯМР (CDCl3) : 7,10 (2 Н, м), 7,05 (2 Н, м), 4,23-4,15 (2 Н, м), 3,90-3,80 (3 Н, м), 3,32 (1 Н, тд,J1 = 13,0 Гц, J2 = 4,0 Гц), 3,21 (1 Н, т, J = 12,9 Гц), 2,68 (2 Н, м), 2,55 (1 Н, м), 2,07 (1 Н, м), 1,30 (3 Н, т, J = 7,2 Гц), 0,85 (3 Н, т, J = 7,2 Гц). Этап С. Рацемический диэтил-(1S,2S,3R,4S)-3-(4-фторфенил)-4-гидроксициклогексан-1,2 дикарбоксилат и диэтил-(1R,2R,3S,4R)-3-(4-фторфенил)-4-гидроксициклогексан-1,2-дикарбоксилат. В раствор 40,2 г (119,3 ммоль) интермедиата этапа В в 150 мл этанола в атмосфере азота при -78 С добавляли 4,1 г (108,5 ммоль) порошка NaBH4. Получающуюся смесь перемешивали при -78 С в течение 0,5 ч, затем при комнатной температуре в течение 2 ч. Реакционную смесь аккуратно гасили путем добавления 30 мл воды и аккуратно подкисляли 2 н. водн. HCl. Растворитель выпаривали в вакууме. Остаток растворяли в эфире, переносили в делительную воронку, промывали с насыщ. водн. NaHCO3 и солевым раствором, сушили над осушителем, фильтровали и выпаривали растворитель в вакууме для получения необработанных соединений в соответствии с заголовком, которые очищали на следующем этапе.(этап С), их разделяли с помощью препаративной хиральной ВЭЖХ с применением колонки CHIRACELAD с элюицией гептанами/i-PrOH (9/1) для получения 9,09 г требуемого первого элюируемого изомера диэтил-(1S,2S,3R,4S)-3-(4-фторфенил)-4-гидроксициклогексан-1,2-дикарбоксилата. Этап Е. (1S)-1-[3,5-бис-(трифторметил)фенил]этил-2,2,2-трихлорэтанимидоат. Раствор 25,82 г (100 ммоль) (1S)-1-[3,5-бис-(трифторметил)фенил]этанола в 200 мл сухого диэтилового эфира в атмосфере азота охлаждали во льду/водяной бане. В реакционную колбу добавляли 3 мл(20 ммоль, 0,2 экв.) чистого DBU, затем смесь перемешивали при 0 С в течение 10 мин. Медленно добавляли по каплям 15 мл (150 ммоль, 1,5 экв.) трихлорацетонитрила в течение более 15 мин. Реакцию перемешивали при 0 С в течение 2 ч, за это время она стала насыщенного желтого цвета. Летучие вещества удаляли в вакууме с применением холодной ванны (35 С) для получения светло-коричневой маловязкой жидкости, которую очищали с помощью колоночной хроматографии на силикагеле (прокладка 3"X 10") с элюицией в двух сериях гексанами/EtOAc (9/1), затем гексанами/EtOAc (4/1). Фракции продукта объединяли, и растворитель удаляли в вакууме для получения 37,5 г соединения в соответствии с заголовком в виде светло-желтой маслянистой жидкости. 1(1 Н, уш. с) ч./млн. Этап F. Диэтил-(1S,2S,3R,4S)-4-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-3-(4-фторфенил)циклогексан-1,2-дикарбоксилат. В раствор 9,09 г (26,9 ммоль) первого элюируемого изомера диэтил-(1S,2S,3R,4S)-3-(4-фторфенил)4-гидроксициклогексан-1,2-дикарбоксилата (этап D) и 21,5 г (53,5 ммоль) (1S)-1-[3,5-бис(трифторметил)фенил]этил-2,2,2-трихлорэтанимидоата (этап Е) в 250 мл циклогексана/1,2-хлорэтана(3/1) в атмосфере азота при -5 С добавляли 0,51 мл (3,58 ммоль) 54% HBF4 в эфире. Реакционную смесь перемешивали при -5-0 С в течение 2 ч, затем разбавляли эфиром. Смесь промывали насыщ. водн. NaHCO3. Органический слой сушили над осушителем, фильтровали и выпаривали растворитель в вакууме. Остаток очищали с помощью колоночной флеш-хроматографии на силикагеле с элюциейEtOAc/гексанами (1/4) для получения 9,2 г соединения в соответствии с заголовком в виде маслянистой жидкости. 1 Н-ЯМР (CDCl3) : 7,70 (1 Н, с), 7,20 (2 Н, с), 7,00 (2 Н, м), 6,85 (2 Н, т, J = 8,5 Гц), 4,43 (1 Н, кв, J = 6,0 Гц), 4,20-4,10 (2 Н, м), 3,80-3,73 (2 Н, м), 3,36 (1 Н, м), 2,90-2,76 (2 Н, м), 2,40 (1 Н, м), 2,28 (1 Н, м),1,63-1,55 (2 Н, м), 1,33 (3 Н, д, J = 6,0 Гц), 1,25 (3 Н, т, J = 7,2 Гц), 0,82 (3 Н, т, J = 7,2 Гц). Непрореагировавший исходный спирт может быть восстановлен путем промывки колонки с помощью EtOAc и многократно использован в описанной выше реакции. Этап G. [(1S,2R,3R,4S)-4-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-3-(4-фторфенил)циклогексан-1,2-диил]диметанол. В раствор 9,2 г(15,9 ммоль) диэтил-(1S,2S,3R,4S)-4-(1R)-1-[3,5-бис(трифторметил)фенил]этокси-3-(4-фторфенил)циклогексан-1,2-дикарбоксилата (этап F) в 100 мл ТГФ в атмосфере азота при комнатной температуре добавляли 2 г (112,4 ммоль, избыток) порошка LiBH4. Получающуюся смесь нагревали при 68 С в течение 2 ч, затем охлаждали до комнатной температуры. Реакционную смесь аккуратно гасили путем добавления 30 мл воды, затем экстрагировали с помощьюEtOAc. Объединенные органические экстракты сушили над осушителем, фильтровали и выпаривали растворитель в вакууме для получения 7,5 г необработанного соединения в соответствии с заголовком в виде маслянистой жидкости, которое использовали без дополнительной очистки. 1(0,1 экв.) DMAP. Реакционную смесь перемешивали при -5 С в течение 30 мин, затем гасили при данной температуре путем добавления 20 мл, насыщ. водн. NaHCO3. Смесь нагревали до комнатной температуры. Отделяли органический слой и водный слой экстрагировали дополнительными 50 мл метиленхлорида. Объединенный органический слой промывали 20 мл 2 н. водн. HCl, 30 мл насыщ. водн. NaHCO3, солевым раствором, сушили над осушителем MgSO4, фильтровали и выпаривали растворитель в вакууме- 17010598 для получения соединения в соответствии с заголовком в виде маслянистой жидкости, которое использовали без дополнительной очистки. Этап(3aR,4R,5S,7aS)-2-Бензил-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-1 Н-изоиндол. В пробирку, рассчитанную на высокое давление, поместили раствор необработанного[(1S,2R,3R,4S)-4-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-3-(4-фторфенил)циклогексан-1,2-диил]ди(метилен)диметансульфоната (этап Н) в 20 мл этанола и 1,2 мл (3 экв.) бензиламина. Пробирку, рассчитанную на высокое давление, герметично закрывали и нагревали при 150 С в масляной бане в течение 3 ч. Пробирку охлаждали до комнатной температуры и открывали. Получающуюся смесь переносили в колбу с круглым дном, и удаляли растворитель в вакууме. Остаток разбавляли 100 мл EtOAc, промывали 20 мл 5 н. водн. NaOH, сушили над осушителем MgSO4, фильтровали и выпаривали растворитель в вакууме. Остаток очищали с помощью колоночной флеш-хроматографии на силикагеле с элюциейMS: (MH)+ 566,0. Этап J. (3aR,4R,5S,7aS)-5-(1H)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-1 Н-изоиндол. В раствор (3aR,4R,5S,7aS)-2-бензил-5-(1 Н)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-1 Н-изоиндола (этап Н) в 50 мл EtOH добавляли 0,2 г (20% по весу) 10%Pd(ОН)2-C. Реакционную смесь подвергали гидрогенизации при 50 фунт/кв.дюйм в течение 16 ч при комнатной температуре. Катализатор фильтровали, и растворитель из фильтрата выпаривали в вакууме для получения соединения в соответствии с заголовком. 1 3-[(3aR,4R,5S,7aS)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-2 Низоиндол-2-ил]циклопент-2-ен-1-он. В раствор 0,73 г (1,5 ммоль) (3aR,4R,5S,7aS)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4 фторфенил)октагидро-1 Н-изоиндола (пример 6) в 25 мл сухого толуола добавляли 0,166 г (1,7 ммоль) циклопентан-1,3-диона и 0,03 г (0,15 ммоль) PTSA. Получающуюся смесь нагревали с обратным холодильником в течение 2 ч. Растворитель удаляли в вакууме, и остаток очищали с помощью препаративной ТСХ с элюцией CHCl3/2 н. NH3 в МеОН (9/1) для получения 0,49 г соединения в соответствии с заголовком. Соединение могло быть дополнительно очищено с помощью хиральной ВЭЖХ на колонкиCHIRACEL AD с элюицией гексанами/EtOH (9/1). Соединение могло быть кристаллизовано (Т. пл. = 216,5-217,5 С) из гексанов/EtOAc или гексанов/EtOH. 1 Н-ЯМР (CDCl3) : 7,71 (1 Н, с), 7,23 (2 Н, с), 7,00 (2 Н, м), 6,93 (2 Н, т, J = 8,2 Гц), 4,89, 4,48 (1 Н, с),4,47 (1 Н, м), 3,71, 3,48 (1 Н, м), 3,35 (1 Н, м), 3,28-3,17 (1 Н, м), 2,95 (1 Н, м), 2,95, 2,81 (1 Н, м), 2,68 (2 Н,м), 2,45 (2 Н, м), 2,37 (2 Н, м), 2,15 (1 Н, м), 1,93 (2 Н, м), 1,60 (1 Н, м), 1,38 (1 Н, м), 1,36 (3 Н, т, J = 6,0 Гц).(3aR,4R,5S,7aS)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)-Nметилоктагидро-2 Н-изоиндол-2-карбоксамид. В раствор 20 мг (0,042 ммоль) (3aR,4R,5S,7aS)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4(4-фторфенил)октагидро-1 Н-изоиндола (пример 6) в 2 мл сухого метиленхлорида при комнатной температуре добавляли несколько капель метилизоцианата. Получающуюся смесь перемешивали при комнатной температуре 2 ч. В реакционную смесь добавляли несколько капель 2 н. водн. NaOH. Органический слой отделяли, сушили над осушителем, фильтровали и удаляли растворитель в вакууме. Остаток очищали помощью элюции EtOAc перед ТСХ и выделяли полосу основного продукта. Остаток поглощали EtOAc и отфильтровывали твердые вещества. Растворитель фильтрата удаляли в вакууме для получения соединения в соответствии с заголовком. 1MoOPH в 2 мл сухого ТГФ в атмосфере азота при -78 С добавляли х 0,076 мл (0,15 ммоль) 2,0 М раствора KHMDS. Получающуюся смесь перемешивали при -78 С в течение 2 ч, затем гасили путем добавления насыщ. водн. NH4Cl. Смесь экстрагировали с помощью EtOAc. Объединенные органические экстракты промывали солевым раствором, сушили над осушителем и фильтровали. Остаток очищали с помощью препаративной ТСХ с элюцией CHCl3/2 н. NH3 в МеОН (9/1) для получения соединения в соответствии с заголовком в виде смеси диастереоизомеров. 1 Н-ЯМР (CDCl3) : 7,71 (1 Н, с), 7,23 (2 Н, с), 7,00 (2 Н, м), 6,93 (2 Н, т, J = 8,2 Гц), 4,85, 4,71 (1 Н, с),4,47 (1 Н, м), 4,20 (1 Н, м), 3,80-3,65 (1 Н, м), 3,37 (1 Н, м), 3,25-3,08 (1 Н, м), 3,10-2,80 (3 Н, м), 2,58 (1 Н,м), 2,50-2,30 (1 Н, м), 2,15 (1 Н, м), 1,95 (2 Н, м), 1,35 (3 Н, д, J = 6,0 Гц).MS: (МН)+ 572,5. Диастереоизомеры разделяли с помощью ВЭЖХ на колонке CHIRACEL AS элюцией гексанами/EtOH (85/15) для получения индивидуальных диастереоизомеров. Пример 10. 3-[(3aR,4R,5S,7aS)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-2 Низоиндол-2-ил]-4-гидроксициклопент-2-ен-1-он. Этап А. 4-[(3aR,4R,5S,7aS)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-ил]циклопент-4-ен-1,3-дион. Соединение в соответствии с заголовком готовили из (3aR,4R,5S,7aS)-5-(1R)-1-[3,5-бис(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-1 Н-изоиндола (пример 6) и циклопентан-1,2,4 триона в соответствии с процедурой примера 7.MS: (MH)+ 570,0. Этап В. 3-[(3aR,4R,5S,7aS)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(4-фторфенил)октагидро-2 Н-изоиндол-2-ил]-4-гидроксициклопент-2-ен-1-он. Соединение в соответствии с заголовком готовили в виде смеси диастереоизомеров из интермедиата этапа А в соответствии с процедурой примера 6, этапа С (NaBH4 в метаноле). 1 Н-ЯМР (CDCl3) ротамеры; : 7,71 (1 Н, с), 7,23 (2 Н, с), 7,00 (2 Н, м), 6,93 (2 Н, т, J = 8,2 Гц), 4,88- 19010598 4,65 (2 Н, м), 4,46 (1 Н, м), 4,07 (0,5 Н, м), 3,83 (0,5 Н, м), 3,55 (1 Н, м), 3,35 (1 Н, м), 3,28 (1 Н, м), 2,97 (2 Н, м), 2,92 (1 Н, м), 2,58 (1 Н, м), 2,43 (1 Н, м), 2,25 (1 Н, м), 2,13 (1 Н, м), 2,05-1,85 (2 Н, м), 1,70-1,55 (2 Н, м), 1,30 (3 Н, 2 д, J = 6,0 Гц).MS: (MH)+ 572,5 диастереоизомеры разделяли с помощью ВЭЖХ на колонке CHIRACEL OD с элюцией гексанами/EtOH (85/15) для получения индивидуальных диастереоизомеров. Пример 11. 3-[(3aR,4R,5S,7aS)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(2-метилфенил)октагидро 2 Н-изоиндол-2-ил]циклопент-2-ен-1-он. Этап А. 2-(2-Метилфенил)-N-метокси-N-метилацетамид. Соединение в соответствии с заголовком готовили из 2-(метилфенил)уксусной кислоты в соответствии с процедурой примера 1, этапа А. 1 Н-ЯМР (CDCl3) : 7,23 (4 Н, м), 3,83 (2 Н, с), 3,65 (3 Н, с), 3,28 (3 Н, с), 2,36 (3 Н, с). Этап В. 1-(2-Метилфенил)бут-3-ен-2-он. Соединение в соответствии с заголовком готовили из интермедиата этапа А в соответствии с процедурой примера 1, этапа В. 1H-ЯМР (CDCl3) : 7,24-7,11 (4 Н, м), 6,42 (1 Н, дд, J, = 14,2 Гц, J2 = 11 Гц), 6,34 (1 Н, д, J = 14,2 Гц),5,81 (1 Н, д, J = 11 Гц), 3,90 (2 Н, с), 2,26 (3 Н, с). Этап С. 1E и 1Z трет-бутил-[1-(2-Метилбензилиден)проп-2-ен-1-ил]оксидиметилсилан. Соединение в соответствии с заголовком готовили из интермедиата этапа В в соответствии с процедурой примера 1, этапа С. 1 Н-ЯМР (CDCl3) : 7,22-7,07 (4 Н, м), 6,40 (1 Н, дд, J1 = 13,2 Гц, J2 = 8,5 Гц), 5,85 (1 Н, с), 5,54 (1 Н,д, J = 13,2 Гц), 5,19 (1 Н, д, J = 8,5 Гц), 2,28 (3 Н, с). ЭтапD. Диэтил-4-[трет-бутил(диметил)силил]окси-3-(2-метилфенил)циклогекс-4-ен-1,2 дикарбоксилат. Соединение в соответствии с заголовком готовили из интермедиата этапа С в соответствии с процедурой примера 6, этапа А и использовали без дополнительной очистки. Этап Е. Рацемический диэтил-(1S,2S,3R)-3-(2-метилфенил)-4-оксоциклогексан-1,2-дикарбоксилат и диэтил-(1R,2R,3S)-3-(2-метилфенил)-4-оксоциклогексан-1,2-дикарбоксилат. Соединения в соответствии с заголовком готовили из интермедиата этапа D в соответствии с процедурой примера 6, этапа В. 1F. Рацемический диэтил-(1S,2S,3R,4S)-3-(2-метилфенил)-4-гидроксициклогексан-1,2 дикарбоксилат и диэтил-(1R,2R,3S,4R)-3-(2-метилфенил)-4-гидроксициклогексан-1,2-дикарбоксилат. Соединения в соответствии с заголовком из интермедиата этапа Е в соответствии с процедурой примера 6, этапа С. 1 Н-ЯМР (CDCl3) : 7,20-7,10 (4 Н, м), 4,15-4,07 (2 Н, м), 3,88-3,66 (3 Н, м), 3,09 (1 Н, т), 2,83 (2 Н, м),2,30 (3 Н, с), 2,24-2,15 (2 Н, м), 1,68 (1 Н, м), 1,57 (1 Н, м), 1,25 (3 Н, т, J = 7,2 Гц), 0,83 (3 Н, т, J = 7,2 Гц),Этап G. Диэтил-(1S,2S,3R,4S)-3-(2-метилфенил)-4-гидроксициклогексан-1,2-дикарбоксилат. Рацемическую смесь диэтил-(1S,2S,3R,4S)-3-(2-метилфенил)-4-гидроксициклогексан-1,2 дикарбоксилата и диэтил-(1R,2R,3S,4R)-3-(2-метилфенил)-4-гидроксициклогексан-1,2-дикарбоксилата(этап F) разделяли с помощью препаративной хиральной ВЭЖХ с применением колонки CHIRACEL AD с элюцией гептанами/i-PrOH (9/1) для получения требуемого первого элюируемого изомера диэтил(1S,2S,3R,4S)-3-(2-метилфенил)-4-гидроксициклогексан-1,2-дикарбоксилата в соответствии с процедурой примера 6, этапа D. Этап Н. Диэтил-(1S,2S,3R,4S)-4-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-3-(2-метилфенил)циклогексан-1,2-дикарбоксилат. Соединение в соответствии с заголовком готовили из первого элюируемого изомера диэтил(1S,2S,3R,4S)-3-(2-метилфенил)-4-гидроксициклогексан-1,2-дикарбоксилата (этап G) и (1S)-1-[3,5-бис(3 Н, с), 1,69-1,56 (2 Н, м), 1,30 (3 Н, д, J = 6,0 Гц), 1,23 (3 Н, т, J = 7,2 Гц), 0,77 (3 Н, т, J = 7,2 Гц). Непрореагировавший исходный спирт мог быть восстановлен путем промывки колонки EtOAc и многократно использован в приведенной выше реакции. Этап I. [(1S,2R,3R,4S)-4-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-3-(2-метилфенил)циклогексан-1,2-диил]диметанол. Соединение в соответствии с заголовком готовили из интермедиата этапа Н в соответствии с процедурой примера 6, этапа G. 1H-ЯМР (CDCl3) : 7,64 (1 Н, с), 7,16 (2 Н, с), 7,04-6,91 (4 Н, м), 4,24 (1 Н, кв, J = 6,0 Гц), 3,74 (1 Н,м), 3,60 (1 Н, м), 3,48 (1 Н, м), 3,35-3,20 (2 Н, м), 2,90-2,70 (2 Н, м), 2,26 (1 Н, м), 2,21 (3 Н, с), 1,85 (1 Н,м), 1,62 (1 Н, м), 1,56-1,42 (3 Н, м), 1,28 (3 Н, т, J = 6,0 Гц). Этап J. [(1S,2R,3R,4S)-4-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-3-(2-метилфенил)циклогексан-1,2-диил]ди(метилен)диметансульфонат. Соединение в соответствии с заголовком готовили из интермедиата этапа I в соответствии с процедурой примера 6, этапа Н и использовали без дополнительной очистки. Этап K. (3aR,4R,5S,7aS)-2-Бензил-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(2-метилфенил)октагидро-1 Н-изоиндол. Соединение в соответствии с заголовком готовили с промежуточного этапа J и бензиламина в соответствии с процедурой примера 6, этапа I. 1 Н-ЯМР (CDCl3) : 7,64 (1 Н, с), 7,32-7,25 (5 Н, м), 7,18 (2 Н, с), 7,04-6,97 (4 Н, м), 4,25 (1 Н, кв, J = 6,0 Гц), 3,75 (1 Н, д, J = 13,4 Гц), 3,65 (1 Н, д, J = 13,4 Гц), 3,40 (1 Н, м), 2,90 (2 Н, м), 2,54-2,30 (4 Н, м),2,22 (3 Н, с), 2,02-1,85 (3 Н, м), 1,61 (1 Н, м), 1,33 (3 Н, т, J = 6,0 Гц), 1,30 (1 Н, м). ЭтапH-ЯМР (CDCl3) : 7,64 (1 Н, с), 7,18 (2 Н, с), 7,04-6,97 (4 Н, м ), 4,25 (1 Н, кв, J = 6,5 Гц), 3,40 (1 Н,м), 3,25 (1 Н, м), 2,90-2,75 (2 Н, м), 2,63 (1 Н, т), 2,44 (1 Н, т), 2,35 (1 Н, м), 2,22 (3 Н, с), 2,05 (1 Н, м),1,86-1,72 (2 Н, м), 1,61 (1 Н, м), 1,33-1,20 (5 Н, м). Этап М. 3-[(3aR,4R,5S,7aS)-5-(1R)-1-[3,5-бис-(трифторметил)фенил]этокси-4-(2-метилфенил)октагидро-2 Н-изоиндол-2-ил]циклопент-2-ен-1-он. Соединение в соответствии с заголовком готовили из (3aR,4R,5S,7aS)-5-(1R)-1-[3,5-бис(трифторметил)фенил]этокси-4-(2-метилфенил)октагидро-1 Н-изоиндола (этап L) и циклопентан-1,3 диона в соответствии с процедурой примера 7. 1 Н-ЯМР (CDCl3)ротамеров: 7,64 (1 Н, с), 7,18 (2 Н, с), 7,04-6,97 (4 Н, м), 4,74, 4,53 (1 Н, с), 4,42 (1 Н, м), 3,68, 3,43 (1 Н, м), 3,50 (1 Н, м), 3,06 (1 Н, м), 2,93 (2 Н, м), 2,86, 2,76 (1 Н, м), 2,57 (1 Н, м), 2,40 (2 Н, м), 2,28 (3 Н, с), 2,20 (1 Н, м), 2,17 (1 Н, м), 2,10-1,94 (3 Н, м), 1,58 (1 Н, м), 1,40 (1 Н, м), 1,28 (3 Н, д,J = 6,0 Гц).MS: (MH)+ 552,43. С использованием процедур, в значительной мере сравнимых с описанными выше, приготовили соединения в соответствии со следующими примерами. С использованием процедур, в значительной мере сравнимых с описанными выше, приготовили соединения в соответствии со следующими примерами. Несмотря на то, что изобретение было описано и проиллюстрировано в отношении определенных конкретных вариантов его выполнения, специалисту в данной области будет понятно, что могут быть проделаны различные варианты, изменения, модификации, замены, исключения или добавления процедур и протоколов без отступления от сущности и объема изобретения.(1) водорода,(2) C1-6 алкила, который не замещен или замещен галогеном, гидроксилом или фенилом,(3) циклопентенона, который не замещен или замещен гидроксилом или метилом,(4) -(СО)-C1-6 алкила,(5) -(CO)-NH2,(6) -(СО)-NHC1-6 алкила, иX независимо выбран из группы, состоящей из:(3) метила; и его фармацевтически приемлемые соли и индивидуальные энантиомеры и диастереоизомеры. 2. Соединение по п.1 формулы Ia и его фармацевтически приемлемые соли и индивидуальные энантиомеры и диастереоизомеры. 3. Соединение по п.1 формулы Ib и его фармацевтически приемлемые соли и индивидуальные энантиомеры и диастереоизомеры. 4. Соединение по п.1, в котором R1 выбран из группы, состоящей из: и его фармацевтически приемлемые соли.- 26010598 8. Фармацевтическая композиция, включающая инертный носитель и соединение по п.1 или его фармацевтически приемлемую соль. 9. Применение соединения по п.1 в качестве активного компонента для приготовления лекарственного средства для лечения недержания мочи. 10. Способ получения лекарственного средства для лечения физиологического нарушения, связанного с избытком тахикининов у млекопитающего, предусматривающий объединение соединения в соответствии с п.1 настоящего изобретения или его фармацевтически приемлемой соли с фармацевтическим носителем или разбавителем. 11. Соединение формулы или его фармацевтически приемлемая соль. 12. Фармацевтическая композиция, включающая инертный носитель и соединение по п.11 или его фармацевтически приемлемую соль. 13. Применение соединения по п.11 в качестве активного компонента для приготовления лекарственного средства для лечения недержания мочи.
МПК / Метки
МПК: A61K 31/4035, C07D 209/44, A61P 25/00
Метки: рецептора, тахикинина, гидроизоиндолиновые, антагонисты
Код ссылки
<a href="https://eas.patents.su/28-10598-gidroizoindolinovye-antagonisty-receptora-tahikinina.html" rel="bookmark" title="База патентов Евразийского Союза">Гидроизоиндолиновые антагонисты рецептора тахикинина</a>
Соединения четвертичного аммония как антагонисты тахикинина

Номер патента: 2424
Опубликовано: 25.04.2002
Авторы: Монаган Сандра Марина, Алкер Дэвид, Бернс Кристофер Джон
МПК: C07D 453/02, A61K 31/439, A61P 1/06...
Метки: антагонисты, аммония, четвертичного, соединения, тахикинина
Формула / Реферат:
1. Соединение формулы где R представляет собой фенил, С3-С7 циклоалкил или гетероарил, каждый из которых является необязательно сопряженным с бензольным циклом или с С3-С7 циклоалкилом и необязательно замещенным, включая фрагмент, сопряженный с бензольным циклом или с С3-С7 циклоалкилом, 1-3 заместителями, каждый из которых независимо выбран из C1-C4 алкила, фтор(C1-C4) алкила, C1-C4 алкоксигруппы, фтор (C1-C4)алкоксигруппы, феноксигруппы,...
Производные 1-(1,2-дизамещенный пиперидинил)-4-замещенного пиперидинина как антагонисты рецепторов тахикинина.

Номер патента: 1559
Опубликовано: 23.04.2001
Авторы: Сюрлеро Доминик Луи Нестор Гилейн, Соммен Франсуа Мария, Жансенс Франс Эдуард, Ван Росбрук Ив Эмиль Мария
МПК: A61P 11/14, A61K 31/4453, C07D 211/44...
Метки: антагонисты, пиперидинил)-4-замещенного, 1-(1,2-дизамещенный, рецепторов, пиперидинина, производные, тахикинина
Формула / Реферат:
1. Соединение формулы его N-оксидная форма, фармацевтически приемлемая соль присоединения и стереохимически изомерная форма, где m=n=1; р равно 0, 1 или 2; =Q является =О или =NR3; х является ковалентной связью или бивалентным радикалом формулы -O-, -S-, -NR3-; R1 является Ar1, Аr1С1-6алкилом или ди (Ar1)С1-6алкилом, где каждая C1-6алкильная группа необязательно замещена гидроксилом, С1-4алкилокси-заместителем, оксо-заместителем или...
N-ацил-2-замещенный-4-(бензимидазолил- или имидазопиридинил -замещенные остатки)-пиперидины как антагонисты тахикинина.

Номер патента: 1247
Опубликовано: 25.12.2000
Авторы: Ленартс Йозеф Элизабет, Соммен Франсуа Мария, Сюрлеро Доминик Луи Нестор Гилейн, Жансенс Франс Эдуард
МПК: C07D 401/14, A61P 1/06, A61K 31/445...
Метки: n-ацил-2-замещенный-4-(бензимидазолил, замещенные, имидазопиридинил, остатки)-пиперидины, антагонисты, тахикинина
Формула / Реферат:
1. Соединение формулы его N-оксидная форма, фармацевтически приемлемая соль присоединения и стереохимически изомерная форма, где n равно 0, 1 или 2; m равно 1 или 2 при условии, что если m равно 2, то n равно 1; Х является ковалентной связью или бивалентным радикалом формулы -О-, -S-, -NR3-; =Q является =O или =NR3; R1 является Аr1, Аr1 C1-6 алкилом или ди(Аr1) C1-6 алкилом, где C1-6 алкильная группа необязательно замещена гидроксилом, C1-4...
Производные триазола в качестве антагонистов рецептора тахикинина

Номер патента: 7720
Опубликовано: 29.12.2006
Авторы: Савин Кеннет Аллен, Юнгхайм Луи Николас, Робертсон Майкл Алан, Ремик Дэвид Майкл, Мюль Брайан Стефен, Эмбре Эрик Джеймс, Амегадзие Альберт Кудзови, Гардинье Кевин Мэттью, Хонг Дзиан Эрик
МПК: C07D 401/04, C07D 249/06, A61K 31/4192...
Метки: антагонистов, триазола, качестве, рецептора, тахикинина, производные
Формула / Реферат:
1. Соединение формулы (I) где D означает (C1-C3)алкандиил; R1 означает фенил, который необязательно замещен от одного до трех заместителями, независимо выбираемыми из группы, состоящей из атома галогена, (C1-C4)алкила, (C1-C4)алкоксила, цианогруппы, дифторметила, трифторметила и трифторметоксигруппы; R4 означает радикал, выбираемый из группы, состоящей из где -А1-А2-А3-А4- вместе с атомами, с которыми они связаны, образуют ароматический...
Фармацевтическая композиция антагониста рецептора тахикинина в форме наночастиц

Номер патента: 7332
Опубликовано: 25.08.2006
Авторы: Бош Уилльям Г., Томпсон Карен К., Шелукар Сухас Д., Ливерсидж Элэйн
МПК: A61K 9/14, A61K 9/16, A61K 31/5377...
Метки: тахикинина, наночастиц, композиция, фармацевтическая, рецептора, антагониста, форме
Формула / Реферат:
1. Композиция в форме наночастиц, содержащая соединение 2-(R)-(1-(R)-(3,5-бис(трифтор-метил)фенил)этокси)-3-(S)-(4-фтор)фенил-4-(3-(5-оксо-1Н,4Н-1,2,4-триазоло)метилморфолин или его фармацевтически приемлемую соль, где указанное соединение имеет адсорбированный на его поверхности по меньшей мере один поверхностный стабилизатор в количестве, достаточном для поддержания эффективного размера частиц менее чем около 1000 нм. 2. Композиция по п.1, где...
Предыдущий патент: Разделительное покрытие для модельной оснастки
Следующий патент: Способ получения диоксида хлора
Случайный патент: Лифт с канатоведущим шкивом, не имеющий противовеса