Новые производные амида гетероциклической карбоновой кислоты
Номер патента: 11635
Опубликовано: 28.04.2009
Авторы: Саги Каталин, Фаркаш Шандор, Хорват Чилла, Надь Йожеф, Борза Иштван, Дьертьян Иштван, Галгоци Корнел, Колок Шандор





Формула / Реферат
1. Производные амида гетероциклической карбоновой кислоты формулы (I)

в которой значение
X соответствует водороду или атому галогена, гидрокси, циано, С1-С4алкилсульфонамидо, факультативно замещенной атомом галогена либо атомами галогена, C1-C4алканоиламидо, факультативно замещенной атомом галогена либо атомами галогена, арилсульфонамидогруппе,
Y соответствует -СН=группе либо атому -N=,
Z соответствует атому водорода либо атому галогена, С1-С4алкильной, С1-С4алкокси, циано, трифторметильной, трифторметоксигруппе,
а также их соли.
2. Соединение по п.1, выбранное из нижеследующей группы:
(4-бензилиденпиперидин-1-ил)-(6-гидрокси-1Н-бензоимидазол-2-ил)метанон;
(6-гидрокси-1Н-бензоимидазол-2-ил)-[4-(4-метилбензилиден)пиперидин-1-ил]метанон;
[4-(4-фторбензилиден)пиперидин-1-ил]-(6-гидрокси-1Н-бензоимидазол-2-ил)метанон;
[4-(4-хлорбензилиден)пиперидин-1-ил]-(6-гидрокси-1H-бензоимидазол-2-ил)метанон;
(4-бензилиденпиперидин-1-ил)-(6-гидрокси-1Н-индол-2-ил)метанон
и их соли.
3. Фармацевтические композиции, содержащие в своем составе эффективное количество производных амида гетероциклической карбоновой кислоты формулы (I), где значения X, Y, Z определены в п.1, либо их солей в качестве активных веществ и вспомогательные вещества, широко применяемые в фармацевтической практике, такие как носители, наполнители, разжижители, стабилизаторы, смачивающие либо эмульгирующие агенты, вещества, влияющие на рН и осмотическое давление, отдушивающие или ароматизирующие вещества, а также добавки, активирующие или доставляющие композицию.
4. Способ получения производных амида гетероциклической карбоновой кислоты формулы (I), в которой значения X, Y, Z определены в п.1, характеризующийся реакцией вторичного амина формулы (II)

где Z имеет то же значение, которое определено в случае формулы (I),
с химически активным производным карбоновой кислоты формулы (III)

в которой значения X и Y соответствуют тем значениям, которые были определены ранее в случае формулы (I),
в подходящем растворителе, с последующей необязательной трансформацией полученных таким образом производных амида гетероциклической карбоновой кислоты формулы (I),
где значения X, Y, и Z соответствуют тем значениям, которые приведены в п.1,
в другие соединения, соответствующие формуле (I), путем введения новых заместителей, и/или модификации, или удаления имеющихся заместителей, и/или путем формирования соли, и/или высвобождения соединения, соответствующего формуле (I), из состава солей с применением известных методов.
5. Способ по п.4, характеризующийся реакцией активного производного карбоновой кислоты формулы (III), в которой значения X и Y соответствуют тем значениям, которые приведены в п.1, с вторичным амином формулы (II), где значения Z соответствуют значениям, которые приведены в п.1, предпочтительно в присутствии основания.
6. Способ по п.4, характеризующийся реакцией активного производного карбоновой кислоты формулы (III), где значения X и Y соответствуют значениям, которые приведены в п.1, с вторичным амином формулы (II), где значения Z соответствуют тем значениям, которые приведены в п.1, в присутствии триэтиламина и о-бензотриазол-1-ил-N,N,N',N'-тетраметилурониумгексафторфосфата (HBTU) в диметилформамиде.
7. Способ производства фармацевтических композиций, обладающих эффектом NR2В-селективного антагониста рецептора NMDA, характеризующийся смешиванием производного амида гетероциклической карбоновой кислоты формулы (I), в которой значения X, Y, Z соответствуют тем значениям, которые приведены в п.1, либо его фармацевтически приемлемых солей в качестве активных веществ и вспомогательных веществ, широко применяемых в фармацевтической практике, таких как носители, наполнители, разжижители, стабилизаторы, смачивающие либо эмульгирующие агенты, вещества, влияющие на рН и осмотическое давление, отдушивающие или ароматизирующие вещества, а также добавки, активирующие или доставляющие композицию.
8. Способ лечения и облегчения симптомов следующих заболеваний млекопитающих, включая человека: травматического повреждения головного или спинного мозга, повреждения нервных клеток, связанных с наличием вируса иммунодефицита человека (HIV), бокового амиотрофического склероза (болезнь Шарко), толерантности и/или зависимости при лечении боли с применением синтетических наркотических препаратов (опиоидов), синдромов отмены, например, алкоголя, опиоидов или кокаина, ишемических расстройствах ЦНС, хронических нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, боли и хронических болевых состояний, таких как невропатическая боль или боль, связанная с раковым заболеванием, эпилепсии, тревожности, депрессии, мигрени, психоза, мышечного спазма, слабоумия различного происхождения, гипогликемии, дегенеративных расстройств сетчатки, глаукомы, астмы, звона в ушах, потери слуха, индуцируемой антибиотиком-аминогликозидом, характеризующийся введением нуждающемуся в лечении млекопитающему эффективного количества/количеств производного амида гетероциклической карбоновой кислоты формулы (I), в которой значения X, Y, Z соответствуют тем значениям, которые приведены в п.1, либо его фармацевтически приемлемых солей, как самих по себе, так и в комбинации с носителями, наполнителями и тому подобным, традиционно применяемыми в фармацевтической практике.
9. Применение производного амида гетероциклической карбоновой кислоты формулы (I), где значения X, Y, Z соответствуют тем значениям, которые приведены в п.1, и/или его оптических изомеров, рацемических соединений, и/или фармацевтически приемлемых солей для производства фармацевтических препаратов для лечения и облегчения симптомов следующих заболеваний млекопитающих, включая человека: травматического повреждения головного или спинного мозга, повреждения нервных клеток, связанных с наличием вируса иммунодефицита человека (HIV), бокового амиотрофического склероза (болезнь Шарко), толерантности и/или зависимости при лечении боли с применением синтетических наркотических препаратов (опиоидов), синдромов отмены, например, алкоголя, опиоидов или кокаина, ишемических расстройств ЦНС, хронических нейродегенеративных расстройств, таких как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, боли и хронических болевых состояний, таких как невропатическая боль или боль, связанная с раковым заболеванием, эпилепсии, тревожности, депрессии, мигрени, психоза, мышечного спазма, слабоумия различного происхождения, гипогликемии, дегенеративных расстройств сетчатки, глаукомы, астмы, звона в ушах, потери слуха, вызванной применением антибиотика-аминогликозида.
Текст
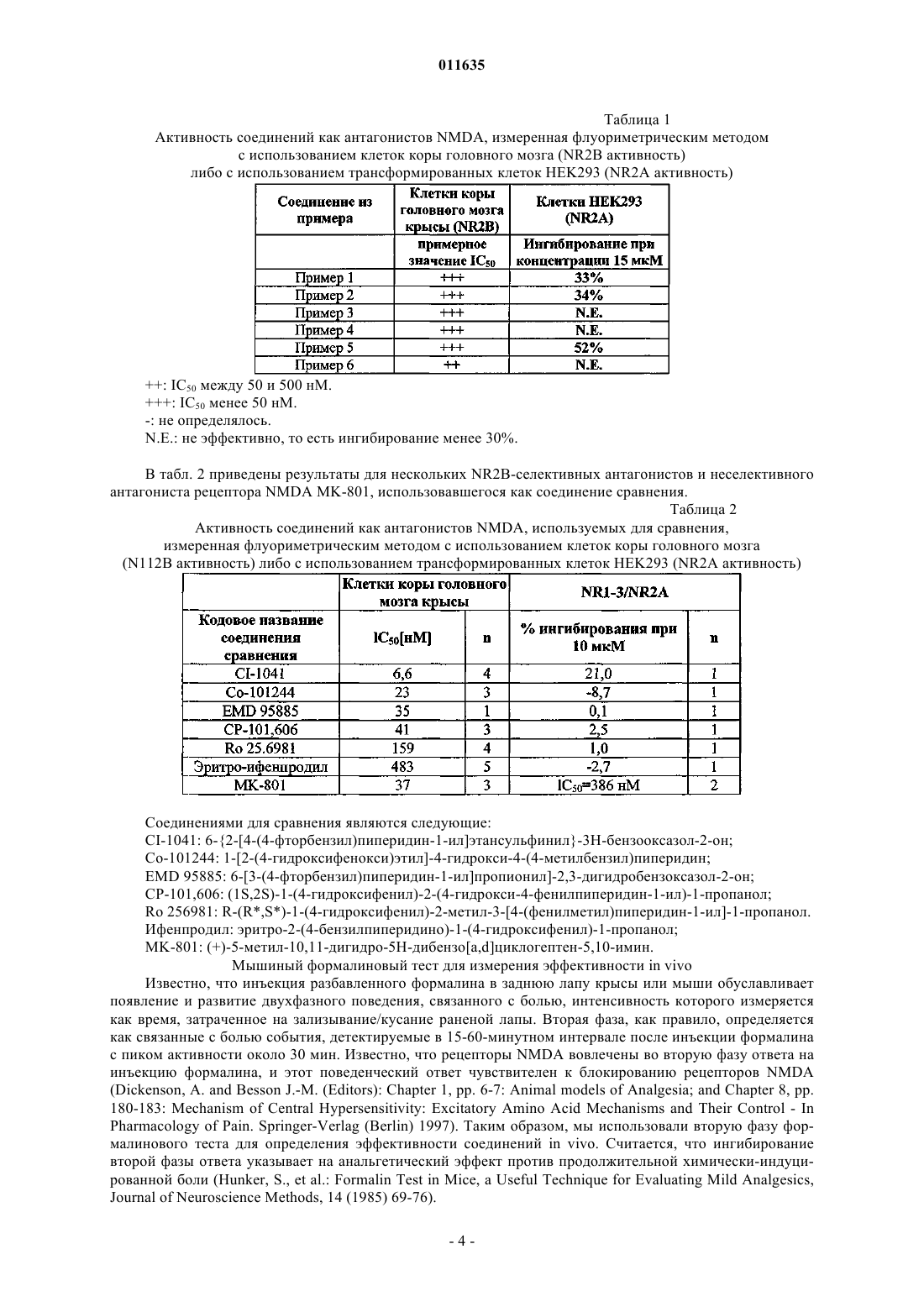
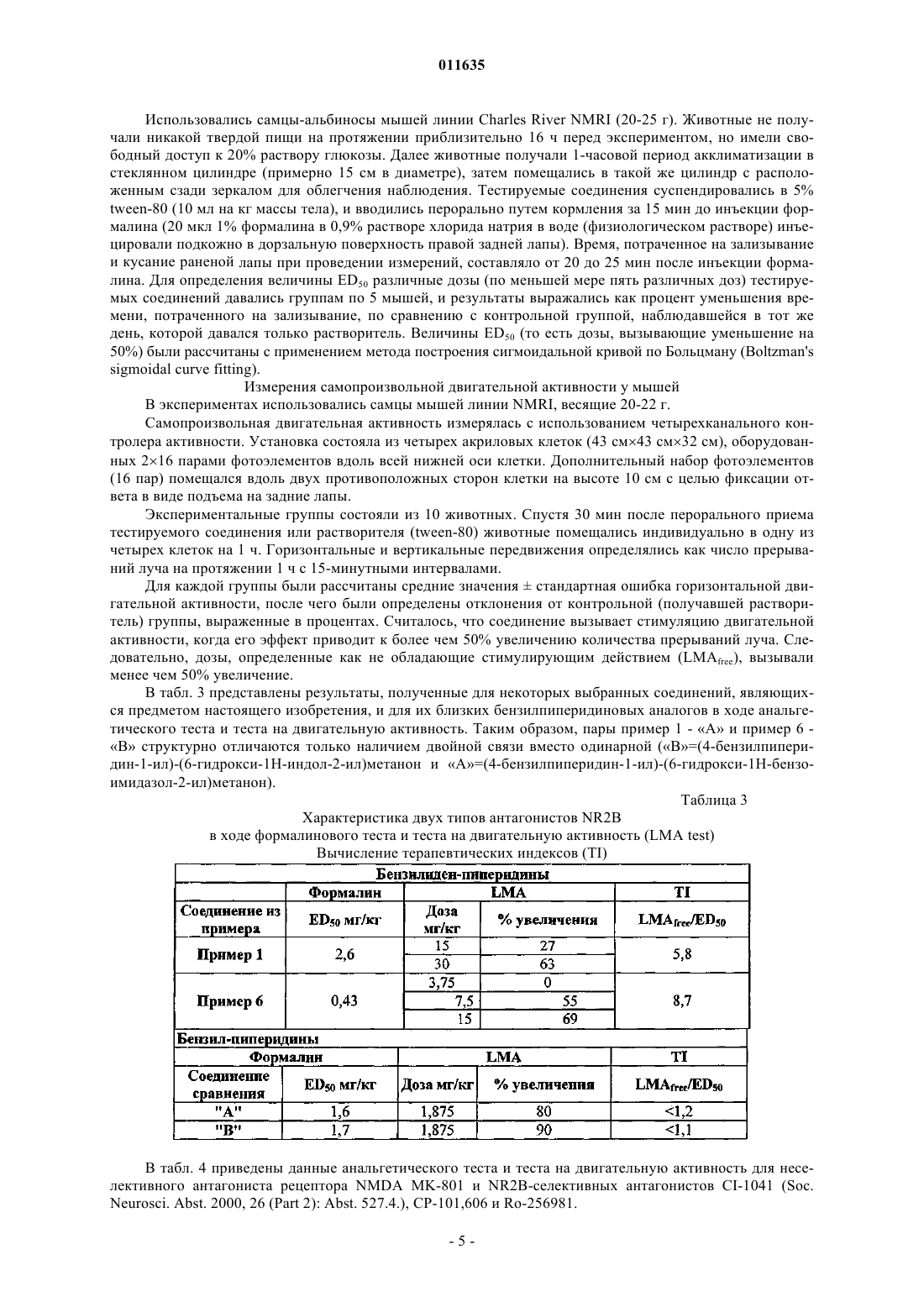
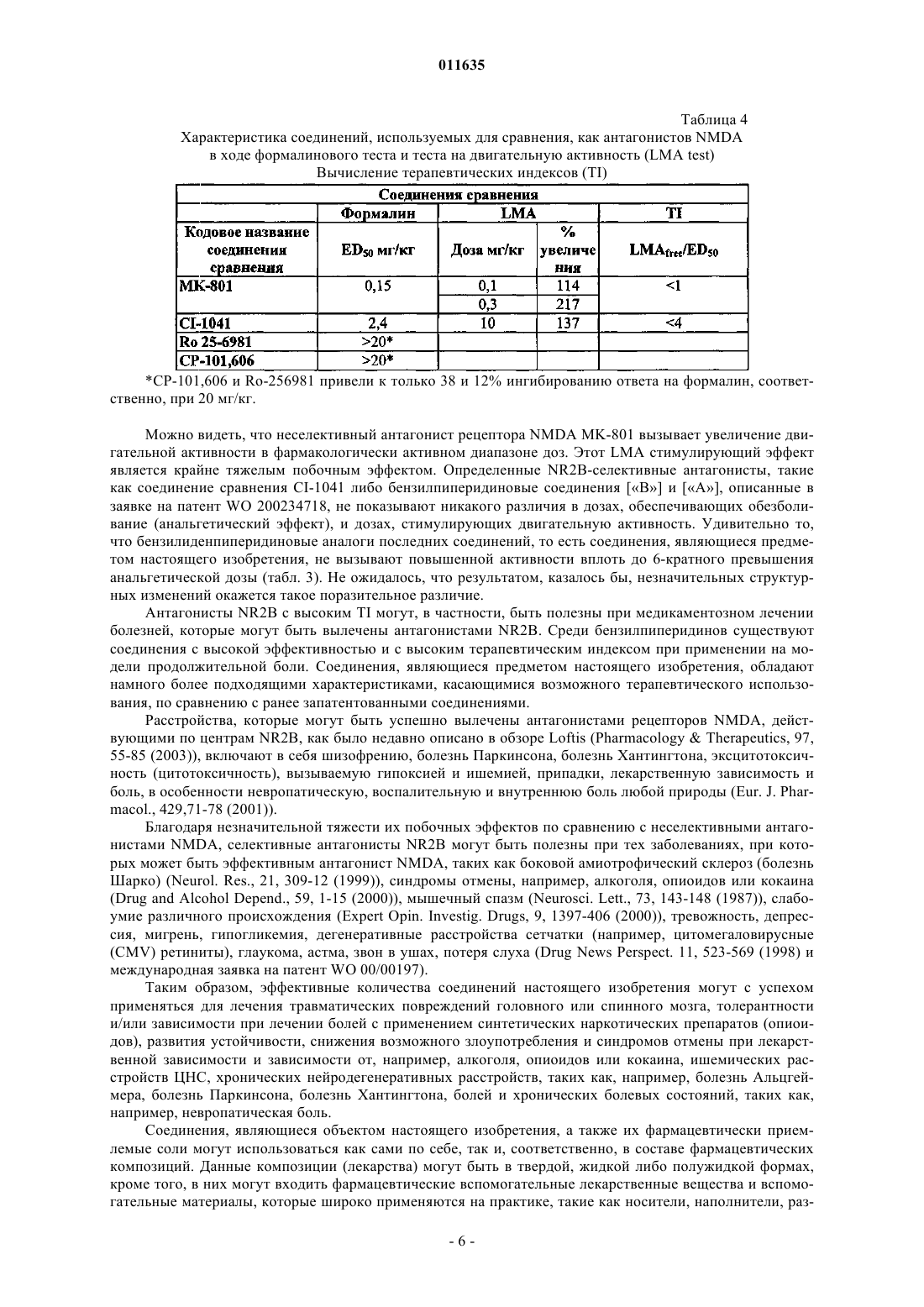
011635 Область техники, к которой относится изобретение Настоящее изобретение касается новых производных амида гетероциклической карбоновой кислоты, которые являются антагонистами NMDA рецептора либо представляют собой промежуточные продукты для их получения. Уровень техники Рецепторы N-метил-D-аспарагиновой кислоты (NMDA) представляют собой катионные каналы,управляемые лигандами, широко представленные в центральной нервной системе. Рецепторы NMDA вовлечены в рост, развитие и адаптационные изменения нейронов. Сверхактивация рецепторов NMDA глутаминовой кислотой, их естественным лигандом, может приводить к кальциевой перегрузке клеток. Это запускает каскад внутриклеточных процессов, которые изменяют функционирование клеток и, в конечном счете, могут привести к гибели нейронов. Антагонисты рецепторов NMDA могут быть использованы для лечения множества расстройств, которые сопровождаются избыточным высвобождением глутаминовой кислоты либо сверхактивацией рецепторов NMDA по любой другой причине (Curr. Opin. Investig. Drugs. 2003, 4: 826-32). Рецепторы NMDA представляют собой гетеромерные комплексы, состоящие из по меньшей мере одной субъединицы NR1 и одной или более из четырех субъединиц NR2 (NR2A-D). Как пространственное распределение в центральной нервной системе (ЦНС), так и фармакологическая чувствительность рецепторов NMDA, имеющих в своем составе различные NR2 субъединицы, являются различными. Особенно интересной является NR2B субъединица в связи с ее ограниченным распределением (наивысшие плотности в переднем мозгу и студенистом веществе спинного мозга) (Neuropharmacology, 38, 611-623(1999. Соединения, селективные для данного подтипа, являются доступными, и их эффективность была показана на животных моделях инсульта (Stroke, 28, 2244-2251 (1997 при травматических повреждениях мозга (Brain Res., 792, 291-298 (1998, болезни Паркинсона (Exp. Neurol., 163, 239-243 (2000, невропатической и воспалительной боли (Neuropharmacology, 38, 611-623 (1999. Кроме того, селективные антагонисты NR2B подтипа рецепторов NMDA могут обеспечивать терапевтическое преимущество по сравнению с неселективными антагонистами рецепторов NMDA. Неселективные антагонисты рецепторов NMDA, действующие по типу блокатора канала, фенциклидин и кетамин, индуцируют психодислептические эффекты, галлюцинации, дисфорию, кататонию и амнезию у человека. Эти серьезные неблагоприятные эффекты затрудняют их клиническое использование в качестве потенциально возможного препарата. Соединения, принадлежащие к данному классу, также обуславливают аномальное поведение у животных, например возбуждают двигательную активность, индуцируют амнезию и ухудшают координацию движений. Наличие данных серьезных эффектов у животных рассматривается как предпосылка для проявления неблагоприятных клинических побочных эффектов. Ожидается, что селективные антагонисты NR2B подтипа лишены большей части этих побочных эффектов. По итогам экспериментов по изучению поведения животных, сообщалось о том, что некоторыеBehav. Pharmacol., 14 (2003) 477-487) увеличивают двигательную активность, в то время как такой эффект не наблюдался в случае СР-101,606, другого NR2 В-селективного антагониста, и Ro 256981 - по результатам других исследователей (Neuropharmacology, 38, 611-623 (1999. Отсутствие эффекта стимулирования двигательной активности в случае СР-101,606 вплоть до уровня 56 мг/кг подкожно и 100 мг/кг внутрибрюшинно было также подтверждено другими данными (Soc. Neurosc. Abstr. 21, 439.9. 1995). Таким образом, насколько нам известно, СР-101,606 является единственным NR2B-селективным антагонистом, который, согласно сообщениям, не обладает эффектом стимулирования двигательной активности. Поскольку СР-101,606 имеет невысокую эффективность при пероральном приеме и, согласно опубликованным данным, был исследован только посредством внутривенного способа введения человеку, кроме того, он метаболизируется полиморфом CYP2D6 (Drug Metabolism and Disposition 31: 76-87), сохраняется огромная необходимость в новых антагонистах NR2B, обладающих незначительной тяжестью побочных эффектов (имеют высокий терапевтический индекс), хорошей эффективностью в случае перорального приема (биодоступностью) и имеющих хорошую перспективу для усовершенствования в терапевтических целях, в особенности с целью перорального применения. Близкие структурные аналоги производных 4-бензилиденпиперидина, соответствующих формуле(I), из литературы не известны. Насыщенные аналоги соединений, являющихся предметом настоящего изобретения, описаны в патентеWO 200234718 как селективные антагонисты NR2B подтипа NMDA. Сущность изобретения Было обнаружено, что новые производные амида гетероциклической карбоновой кислоты, соответствующие формуле (I), являющиеся предметом настоящего изобретения, представляют собой функционально активные антагонисты NMDA, селективные для рецепторов, содержащих субъединицу NR2B. Мы также обнаружили, что новые производные амида гетероциклической карбоновой кислоты обладают анальгетическими свойствами in vivo, сходными с теми, которыми обладают их насыщенные бензилпиперидиновые аналоги. Удивительно следующее: в то время как последние молекулы вызывают повышение двигательной активности при уровне, соответствующем их эффективной анальгетической дозе, соединения, являющиеся предметом настоящего изобретения, не обладают эффектом повышения двига-1 011635 тельной активности до достижения уровня, по меньшей мере, 6-кратно превышающего их анальгетическую дозу. Это свойство может обеспечить терапевтическое преимущество перед NR2B селективными антагонистами NMDA, имеющими более низкий терапевтический индекс. Осуществление изобретения Таким образом, настоящее изобретение касается, в первую очередь, новых производных амида гетероциклической карбоновой кислоты формулы (I)X соответствует водороду или атому галогена, гидрокси, циано, С 1-С 4 алкилсульфонамидо, не обязательно замещенной атомом галогена либо атомами галогена, С 1-С 4 алканоиламидо, не обязательно замещенной атомом галогена либо атомами галогена, арилсульфонамидогруппе,Y соответствует -СН= группе либо атому -N=,Z соответствует одному либо более атому водорода либо атому галогена, C1-С 4 алкильной, С 1-С 4 алкокси, циано, трифторметильной, трифторметоксигруппе,а также их солей. Кроме того, объектами настоящего изобретения являются фармацевтические композиции, содержащие в своем составе в качестве активных компонентов новые производные амида гетероциклической карбоновой кислоты формулы (I), или их оптические изомеры, рацемические соединения или соли. Следующими объектами настоящего изобретения являются способы производства новых производных амида гетероциклической карбоновой кислоты формулы (I) и фармацевтического изготовления медикаментов, содержащих в своем составе данные соединения, равно как и способы лечения с применением данных соединений, что означает введение в организм млекопитающего, включая человека, нуждающегося в лечении, эффективного количества/количеств новых производных амида гетероциклической карбоновой кислоты формулы (I), являющихся объектом настоящего изобретения, как самих по себе, так и в составе медикамента. Новые производные амида гетероциклической карбоновой кислоты формулы (I), являющиеся объектом настоящего изобретения, являются высоко эффективными и селективными антагонистами рецептора NMDA, и, более того, большая часть этих соединений являются селективными антагонистами NR2B подтипа рецепторов NMDA. Согласно настоящему изобретению производные амида карбоновой кислоты формулы (I) могут быть синтезированы посредством реакции вторичного амина формулы (П) где Z имеет то же значение, которое определено в случае формулы (I), с химически активным производным карбоновой кислоты формулы (III) где значения X и Y являются теми же, которые определены ранее для формулы (I),после чего полученные производные амида гетероциклической карбоновой кислоты формулы (I),где значения X, Y, Z соответствуют тем значениям, которые были определены в случае формулы (I), в данном случае трансформируются в другие соединения формулы (I) посредством введения новых замещающих групп, и/или модификации, или удаления групп, присутствующих изначально, и/или посредством образования соли, и/или выделения соединения из состава солей с применением известных методов. Реакция карбоновой кислоты формулы (III) и 4-бензилиденпиперидина формулы (II), то есть формирование амидной связи, осуществляется предпочтительно путем образования активного производного из карбоновой кислоты формулы (III), которое и реагирует с вторичным амином формулы (II), предпочтительно в присутствии основания. Превращение карбоновой кислоты в химически активное производное предпочтительно происходит in situ в процессе формирования амидной связи в растворителе (например, диметилформамиде, ацетонитриле, хлорированных углеводородах или углеводородах). Химически активные производные могут представлять собой хлориды кислот (полученные, например, из карбоновой кислоты и тионилхлорида),смешанные ангидриды (полученные, например, из карбоновой кислоты и изобутилхлороформата в присутствии основания, например триэтиламина), химически активные сложные эфиры (полученные, на-2 011635 пример, из карбоновой кислоты и гидроксибензтриазола и дициклогексилкарбодиимида либо о-бензотриазол-1-ил-N,N,N'N'-тетраметилурониумгексафторфосфата (HBTU) в присутствии основания, например триэтиламина). Химически активные производные образуются в диапазоне температур от комнатной температуры до 0 С. Необходимое время реакции составляет 6-20 ч. Реакционная смесь очищается с применением колоночной хроматографии с использованием Kieselgel 60 (Merck) в качестве адсорбента и соответствующего элюента. Подходящие фракции концентрируются, после чего, с целью получения чистого продукта, производится рекристаллизация из подходящего растворителя. Структуру продуктов определяют методом инфракрасной спектроскопии (IR), ядерного магнитного резонанса (ЯМР) и массспектрометрии. Способы синтеза 4-бензилиденпиперидинов формулы (II) и карбоновых кислот формулы (III) описаны в разделе Примеры. Протоколы экспериментов Экспрессия рекомбинантных рецепторов NMDA Чтобы доказать селективность наших соединений в отношении NR2B, что означает исследование их действия на рецепторы NMDAB, содержащие субъединицу NR2A, мы тестировали наиболее эффективные из них на клеточных линиях, устойчиво экспрессирующих рекомбинантные рецепторы NMDA с субъединичным составом NR1/NR2A. кДНК субъединиц NR1 и NR2A человека, субклонированные в индуцибельные экспрессионные вектора млекопитающих, были интродуцированы в HEK293 клетки, не содержащие рецепторов NMDA, с использованием опосредованного положительно заряженными липидами метода трансфекции (Biotechniques, 22, 982-987 (1997); Neurochemistry International, 43, 19-29(2003. Устойчивость к неомицину и гигромицину использовалась для отбора клонов, содержащих оба вектора, и моноклональные клеточные линии были получены на основе тех клонов, которые обеспечивали наибольший ответ на взаимодействие с NMDA. Соединения тестировались на их ингибиторную активность в отношении вызванного NMDA увеличения уровня внутриклеточного кальция, который регистрировался при флуоресцентных измерениях содержания кальция. Исследования проводились через 4872 ч после добавления индуцирующего агента. Кетамин (500 мкМ) также присутствовал в ходе индукции с целью предотвращения цитотоксичности. Определение активности антагониста NMDA in vitro посредством измерения внутриклеточной концентрации кальция с использованием сканирующего флуориметра для прочтения планшетов в культуре корковых клеток крысы Измерения внутриклеточной концентрации кальция были произведены с использованием культур первичных клеток новой коры головного мозга, полученных из 17-дневных эмбрионов крыс линииCharles River (подробное описание получения культуры клеток новой коры головного мозга см. вJohnson, M.I.; Bunge, R.P. (1992), Культуры клеток первичных периферических и центральных нейронов и нейроглии. В: Protocols for Neural Cell Culture, eds: Fedoroff, S.; Richardson A., The Humana Press Inc.,51-75). После выделения клетки были рассеяны на стандартные 96-луночные микропланшеты, после этого культуры инкубировались в атмосфере 95% воздуха, 5% СО 2 при 37 С до проведения измерений концентрации кальция. Культуры клеток использовались для измерения внутриклеточной концентрации кальция in vitro по истечении 3-7 дней. Считается, что на данной стадии клетки in vitro экспрессируют преимущественноNR2B-содержащие рецепторы NMDA (Mol. Pharmacol. 45, 846-853 (1994. Перед измерениями в клетки вводился флуоресцентный Са 2+-чувствительный краситель Fluo-4/AM (2 мкМ). Для остановки введения красителя в клетки клетки дважды промывались раствором, использовавшимся в ходе измерений (140 мМNaCl, 5 мМ KCl, 2 мМ CaCl2, 5 мМ HEPES, 5 мМ HEPES-Na, 20 мМ глюкозы, 10 мкМ глицина, рН 7,4). После промывки к культурам клеток добавлялись тестируемые соединения, растворенные в указанном выше растворе (90 мкл/лунку). Измерения внутриклеточной концентрации кальция производились с использованием сканирующего флуориметра для прочтения планшетов: увеличение флуресценции Fluo-4,которое соответствовало увеличению внутриклеточной концентрации кальция, индуцировалось посредством введения 40 мкМ NMDA. Ингибиторная активность тестируемых соединений определялась посредством измерения снижения роста концентрации кальция в присутствии различных концентраций тестируемых соединений. Кривые зависимости доза-эффект и значения IC50 определяли, используя данные, полученные по меньшей мере в трех независимых экспериментах. Ингибиторная активность соединения в одной концентрационной точке выражалась как процент ингибирования (ослабления) ответа на NMDA. По экспериментальным точкам были построены сигмоидальные кривые концентрация-ингибирование, и значенияIC50 были определены как концентрация, которая обеспечивает полумаксимальное ингибирование при использовании соединения. В табл. 1 приведены активности как антагонистов NR2B большинства эффективных соединений,являющихся предметом настоящего изобретения, которые были определены в данном эксперименте.-3 011635 Таблица 1 Активность соединений как антагонистов NMDA, измеренная флуориметрическим методом с использованием клеток коры головного мозга (NR2B активность) либо с использованием трансформированных клеток HEK293 (NR2A активность)N.E.: не эффективно, то есть ингибирование менее 30%. В табл. 2 приведены результаты для нескольких NR2 В-селективных антагонистов и неселективного антагониста рецептора NMDA MK-801, использовавшегося как соединение сравнения. Таблица 2 Активность соединений как антагонистов NMDA, используемых для сравнения,измеренная флуориметрическим методом с использованием клеток коры головного мозга(N112B активность) либо с использованием трансформированных клеток HEK293 (NR2A активность) Соединениями для сравнения являются следующие:MK-801: (+)-5-метил-10,11-дигидро-5 Н-дибензо[а,d]циклогептен-5,10-имин. Мышиный формалиновый тест для измерения эффективности in vivo Известно, что инъекция разбавленного формалина в заднюю лапу крысы или мыши обуславливает появление и развитие двухфазного поведения, связанного с болью, интенсивность которого измеряется как время, затраченное на зализывание/кусание раненой лапы. Вторая фаза, как правило, определяется как связанные с болью события, детектируемые в 15-60-минутном интервале после инъекции формалина с пиком активности около 30 мин. Известно, что рецепторы NMDA вовлечены во вторую фазу ответа на инъекцию формалина, и этот поведенческий ответ чувствителен к блокированию рецепторов NMDAPharmacology of Pain. Springer-Verlag (Berlin) 1997). Таким образом, мы использовали вторую фазу формалинового теста для определения эффективности соединений in vivo. Считается, что ингибирование второй фазы ответа указывает на анальгетический эффект против продолжительной химически-индуцированной боли (Hunker, S., et al.: Formalin Test in Mice, a Useful Technique for Evaluating Mild Analgesics,Journal of Neuroscience Methods, 14 (1985) 69-76).-4 011635 Использовались самцы-альбиносы мышей линии Charles River NMRI (20-25 г). Животные не получали никакой твердой пищи на протяжении приблизительно 16 ч перед экспериментом, но имели свободный доступ к 20% раствору глюкозы. Далее животные получали 1-часовой период акклиматизации в стеклянном цилиндре (примерно 15 см в диаметре), затем помещались в такой же цилиндр с расположенным сзади зеркалом для облегчения наблюдения. Тестируемые соединения суспендировались в 5%tween-80 (10 мл на кг массы тела), и вводились перорально путем кормления за 15 мин до инъекции формалина (20 мкл 1% формалина в 0,9% растворе хлорида натрия в воде (физиологическом растворе) инъецировали подкожно в дорзальную поверхность правой задней лапы). Время, потраченное на зализывание и кусание раненой лапы при проведении измерений, составляло от 20 до 25 мин после инъекции формалина. Для определения величины ED50 различные дозы (по меньшей мере пять различных доз) тестируемых соединений давались группам по 5 мышей, и результаты выражались как процент уменьшения времени, потраченного на зализывание, по сравнению с контрольной группой, наблюдавшейся в тот же день, которой давался только растворитель. Величины ED50 (то есть дозы, вызывающие уменьшение на 50%) были рассчитаны с применением метода построения сигмоидальной кривой по Больцману (Boltzman'ssigmoidal curve fitting). Измерения самопроизвольной двигательной активности у мышей В экспериментах использовались самцы мышей линии NMRI, весящие 20-22 г. Самопроизвольная двигательная активность измерялась с использованием четырехканального контролера активности. Установка состояла из четырех акриловых клеток (43 см 43 см 32 см), оборудованных 216 парами фотоэлементов вдоль всей нижней оси клетки. Дополнительный набор фотоэлементов(16 пар) помещался вдоль двух противоположных сторон клетки на высоте 10 см с целью фиксации ответа в виде подъема на задние лапы. Экспериментальные группы состояли из 10 животных. Спустя 30 мин после перорального приема тестируемого соединения или растворителя (tween-80) животные помещались индивидуально в одну из четырех клеток на 1 ч. Горизонтальные и вертикальные передвижения определялись как число прерываний луча на протяжении 1 ч с 15-минутными интервалами. Для каждой группы были рассчитаны средние значениястандартная ошибка горизонтальной двигательной активности, после чего были определены отклонения от контрольной (получавшей растворитель) группы, выраженные в процентах. Считалось, что соединение вызывает стимуляцию двигательной активности, когда его эффект приводит к более чем 50% увеличению количества прерываний луча. Следовательно, дозы, определенные как не обладающие стимулирующим действием (LMAfree), вызывали менее чем 50% увеличение. В табл. 3 представлены результаты, полученные для некоторых выбранных соединений, являющихся предметом настоящего изобретения, и для их близких бензилпиперидиновых аналогов в ходе анальгетического теста и теста на двигательную активность. Таким образом, пары пример 1 - А и пример 6 В структурно отличаются только наличием двойной связи вместо одинарной (В=(4-бензилпиперидин-1-ил)-(6-гидрокси-1 Н-индол-2-ил)метанон и А=(4-бензилпиперидин-1-ил)-(6-гидрокси-1 Н-бензоимидазол-2-ил)метанон). Таблица 3 Характеристика двух типов антагонистов NR2B в ходе формалинового теста и теста на двигательную активность (LMA test) Вычисление терапевтических индексов (TI) В табл. 4 приведены данные анальгетического теста и теста на двигательную активность для неселективного антагониста рецептора NMDA MK-801 и NR2B-селективных антагонистов CI-1041 (Soc.-5 011635 Таблица 4 Характеристика соединений, используемых для сравнения, как антагонистов NMDA в ходе формалинового теста и теста на двигательную активность (LMA test) Вычисление терапевтических индексов (TI) СР-101,606 и Ro-256981 привели к только 38 и 12% ингибированию ответа на формалин, соответственно, при 20 мг/кг. Можно видеть, что неселективный антагонист рецептора NMDA MK-801 вызывает увеличение двигательной активности в фармакологически активном диапазоне доз. Этот LMA стимулирующий эффект является крайне тяжелым побочным эффектом. Определенные NR2B-селективные антагонисты, такие как соединение сравнения CI-1041 либо бензилпиперидиновые соединения [В] и [А], описанные в заявке на патент WO 200234718, не показывают никакого различия в дозах, обеспечивающих обезболивание (анальгетический эффект), и дозах, стимулирующих двигательную активность. Удивительно то,что бензилиденпиперидиновые аналоги последних соединений, то есть соединения, являющиеся предметом настоящего изобретения, не вызывают повышенной активности вплоть до 6-кратного превышения анальгетической дозы (табл. 3). Не ожидалось, что результатом, казалось бы, незначительных структурных изменений окажется такое поразительное различие. Антагонисты NR2B с высоким TI могут, в частности, быть полезны при медикаментозном лечении болезней, которые могут быть вылечены антагонистами NR2B. Среди бензилпиперидинов существуют соединения с высокой эффективностью и с высоким терапевтическим индексом при применении на модели продолжительной боли. Соединения, являющиеся предметом настоящего изобретения, обладают намного более подходящими характеристиками, касающимися возможного терапевтического использования, по сравнению с ранее запатентованными соединениями. Расстройства, которые могут быть успешно вылечены антагонистами рецепторов NMDA, действующими по центрам NR2B, как было недавно описано в обзоре Loftis (PharmacologyTherapeutics, 97,55-85 (2003, включают в себя шизофрению, болезнь Паркинсона, болезнь Хантингтона, эксцитотоксичность (цитотоксичность), вызываемую гипоксией и ишемией, припадки, лекарственную зависимость и боль, в особенности невропатическую, воспалительную и внутреннюю боль любой природы (Eur. J. Pharmacol., 429,71-78 (2001. Благодаря незначительной тяжести их побочных эффектов по сравнению с неселективными антагонистами NMDA, селективные антагонисты NR2B могут быть полезны при тех заболеваниях, при которых может быть эффективным антагонист NMDA, таких как боковой амиотрофический склероз (болезнь Шарко) (Neurol. Res., 21, 309-12 (1999, синдромы отмены, например, алкоголя, опиоидов или кокаина(Drug and Alcohol Depend., 59, 1-15 (2000, мышечный спазм (Neurosci. Lett., 73, 143-148 (1987, слабоумие различного происхождения (Expert Opin. Investig. Drugs, 9, 1397-406 (2000, тревожность, депрессия, мигрень, гипогликемия, дегенеративные расстройства сетчатки (например, цитомегаловирусные(CMV) ретиниты), глаукома, астма, звон в ушах, потеря слуха (Drug News Perspect. 11, 523-569 (1998) и международная заявка на патент WO 00/00197). Таким образом, эффективные количества соединений настоящего изобретения могут с успехом применяться для лечения травматических повреждений головного или спинного мозга, толерантности и/или зависимости при лечении болей с применением синтетических наркотических препаратов (опиоидов), развития устойчивости, снижения возможного злоупотребления и синдромов отмены при лекарственной зависимости и зависимости от, например, алкоголя, опиоидов или кокаина, ишемических расстройств ЦНС, хронических нейродегенеративных расстройств, таких как, например, болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, болей и хронических болевых состояний, таких как,например, невропатическая боль. Соединения, являющиеся объектом настоящего изобретения, а также их фармацевтически приемлемые соли могут использоваться как сами по себе, так и, соответственно, в составе фармацевтических композиций. Данные композиции (лекарства) могут быть в твердой, жидкой либо полужидкой формах,кроме того, в них могут входить фармацевтические вспомогательные лекарственные вещества и вспомогательные материалы, которые широко применяются на практике, такие как носители, наполнители, раз-6 011635 жижители, стабилизаторы, увлажняющие или эмульгирующие агенты, вещества, влияющие на рН и осмотическое давление, отдушивающие или ароматизирующие вещества, а также добавки, активирующие и доставляющие композицию. Дозировка, необходимая для обеспечения терапевтического эффекта, может варьировать в широких пределах и будет оптимизироваться под индивидуальные требования в каждом конкретном случае в зависимости от стадии заболевания, состояния и массы тела нуждающегося в лечении пациента, так же,как и от чувствительности пациента к активному веществу, способа введения и частоты приема препарата в течение дня. Точная доза используемого активного вещества может быть безошибочно определена лечащим врачом, опытным в данной области и имеющим полную информацию о нуждающемся в лечении пациенте. Фармацевтические композиции, содержащие активное вещество в соответствии с настоящим изобретением, содержат, как правило, от 0,01 до 100 мг активного вещества в единичной дозированной форме. Само собой, возможно, что количество активного вещества в некоторых композициях выходит за верхние либо нижние пределы указанного выше диапазона. Твердые формы фармацевтических композиций могут представлять собой, например, таблетки,драже, капсулы, пилюли или ампулы, содержащие лиофилизированные порошки, пригодные для приготовления инъекций. Жидкие композиции представляют собой пригодные для инъекций и вливаний композиции, жидкие медикаменты, капсулы с жидким содержимым (packing fluids) и капли. Полужидкие композиции могут представлять собой мази, бальзамы, крема, взбалтываемые микстуры и суппозитории. С целью удобства применения является пригодным, если фармацевтические композиции включают в себя дозированные формы, содержащие в своем составе такое количество активного вещества, которое пригодно для однократного приема, либо нескольких повторных приемов, или приема их половины, трети либо четвертой части. Такими дозированными формами являются, например, таблетки, которые могут быть покрыты бороздками, позволяющими разламывать их пополам или на четыре части с целью приема строго необходимого количества активного вещества. Таблетки могут быть покрыты кислоторастворимым слоем для того, чтобы гарантировать высвобождение содержащегося в таблетке активного вещества после прохождения таблетки через желудок. Такие таблетки являются покрытыми энтеросолюбильной оболочкой. Подобный эффект может быть также достигнут при инкапсулировании активного вещества. Фармацевтические композиции, пригодные для перорального применения, могут содержать, например, лактозу или крахмал в качестве наполнителей,натриевую соль карбоксиметилцеллюлозы, метилцеллюлозу, поливинилпирролидон либо крахмальную пасту в качестве связующих веществ или гранулирующих агентов. Картофельный крахмал или микрокристаллическая целлюлоза добавляются как дезинтегрирующие агенты, но также могут использоваться ультраамилопектин либо формальдегид казеин. Тальк, коллоидная кремниевая кислота, стеарин, стеараты кальция или магния могут применяться в качестве антиадгезивных и смазывающих веществ. Таблетки могут быть изготовлены, например, посредством сырого гранулирования с последующим спрессовыванием. Смешанные активные вещества и наполнители, так же, как и, в данном случае, часть дезинтегрирующих веществ, подвергаются гранулированию с применением водного, спиртового либо водно-спиртового раствора связующих веществ с использованием соответствующего оборудования, после чего гранулят подвергается высушиванию. К высушенному грануляту добавляются другие дезинтегрирующие вещества, смазывающие вещества и антиадгезивные агенты, после чего смесь спрессовывается в таблетку. В данном случае таблетки изготавливаются с бороздкой посередине, что облегчает прием препарата. Таблетки могут быть изготовлены непосредственно из смеси активного вещества и подходящих вспомогательных веществ путем спрессовывания. В данном случае таблетки могут покрываться дополнительными веществами, широко применяющимися в фармакологической практике, такими как, например, стабилизаторы, ароматизаторы, красители, такие как сахар, производные целлюлозы (метил- или этилцеллюлоза, натриевая соль карбоксиметилцеллюлозы и т.д.), поливинилпирролидон, фосфат кальция, карбонат кальция, пищевые красители, пищевые добавки, ароматизирующие вещества, пигменты на основе окиси железа и т.д. В случае капсул смесь активного вещества и вспомогательных веществ заключается внутрь капсул. Жидкие композиции, предназначенные для перорального применения, такие как, например, суспензии, сиропы, эликсиры, могут изготавливаться с применением воды, гликолей, масел, спиртов, красителей и ароматизирующих веществ. Композиции, предназначенные для ректального применения, изготавливаются в виде суппозиториев или растворов для клизм. Суппозитории, помимо активного вещества, могут содержать в своем составе носитель, так называемые основы для суппозиториев (adeps pro suppository). Носителями могут являться растительные масла, такие как гидрированные растительные масла, триглицериды С 12-С 18 жирных кислот (предпочтительно носители, имеющие торговое название Witepsol). Активное вещество гомогенно смешивается с измельченной основой для суппозиториев, после чего формуются суппозитории. Композиция, предназначенная для парентерального применения, изготавливается в виде раствора для инъекций. Для приготовления раствора для инъекций активные вещества растворяются в дистилли-7 011635 рованной воде и/или в различных органических растворителях, таких как гликолиевые эфиры, в данном случае в присутствии солюбилизаторов, например полиоксиэтиленсорбитанмонолаурата, -моноолеата либо моностеарата (Tween 20, Tween 60, Tween 80). Раствор для инъекций может также содержать различные вспомогательные вещества, такие как консервирующие агенты, например этилендиаминтетраацетат, так же, как и регулирующие рН агенты и буфера и, в данном случае, местное анестезирующее вещество, например лидокаин. Раствор для инъекций, содержащий активное вещество, являющееся предметом настоящего изобретения, перед внесением в ампулы подвергается фильтрованию и стерилизуется после наполнения ампулы. Если активное вещество является гигроскопическим, в таком случае оно может быть стабилизировано посредством лиофилизации. Нижеследующие примеры иллюстрируют настоящее изобретение, никоим образом его не ограничивая. Пример 1. (4-Бензилиденпиперидин-1-ил)-(6-гидрокси-1 Н-бензоимидазол-2-ил)метанон. 1 а) 1-Бензил-4-бензилиденпиперидин. В атмосфере аргона к перемешиваемому раствору 133,2 г (704 ммоль) N-бензил-4-пиперидона(Aldrich) и 161 г (705 ммоль) диэтилового сложного эфира бензилфосфоновой кислоты (Aldrich) в 1350 мл диметилформамида при 0 С добавляется 40,5 г (60%, 37,5 ммоль) гидрида натрия. Реакционная смесь перемешивается в течение 2 ч при 20 С, затем в нее по каплям добавляется 100 мл этилового спирта,смесь выливается в 1500 мл воды и экстрагируется диэтиловым эфиром. Органический слой высушивается над сульфатом натрия и концентрируется. Неочищенный продукт используется на следующей стадии. Точка плавления: масло. 1b) 4-Бензилиденпиперидингидрохлорид. К перемешиваемому раствору ранее полученного неочищенного 1-бензил-4-бензилиденпиперидина(704 ммоль) в 2 л дихлорэтана при 0 С по каплям добавляется 80 мл (741 ммоль) 1-хлорэтилхлороформата. Реакционная смесь перемешивается при 0 С в течение 1 ч и подвергается нагреванию с обратным холодильником в течение 1 ч, после чего концентрируется, остаток растворяется в 1 л метанола и снова нагревается с обратным холодильником в течение 1 ч. Реакционная смесь концентрируется, и осадок выкристаллизовывается в присутствии ацетона с выходом, соответствующим 103,25 г (70,1%) титульного соединения. Точка плавления: 186 С (ацетон). 1 с) N-Бутил-N'-(4-метокси-2-нитрофенил)оксаламид. К суспензии 44,0 г (164 ммоль) этилового сложного эфира N-(4-метокси-2-нитрофенил)оксаламовой кислоты (J. Med. Chem., 18, 926 (1975 и 330 мл толуола при 20 С добавляется 16,8 мл (170 ммоль) н-бутиламина. Реакционная смесь перемешивается при комнатной температуре в течение 10 ч, после чего концентрируется, и осадок выкристаллизовывается в присутствии диэтилового эфира, преципитировавший продукт отфильтровывается, промывается диэтиловым эфиром и высушивается с выходом, соответствующим 45,3 г (93,3%) титульного соединения. Точка плавления: 127-128 С (диэтиловый эфир). 1d) N-(2-Амино-4-метоксифенил)-N'-бутилоксаламид. Смесь 27,0 г (91 ммоль) N-бутил-N'-(4-метокси-2-нитрофенил)оксаламида, 1200 мл метанола и 7,3 г 5% Pd/C катализатора гидрогенизируется в течение 3 ч. К реакционной смеси добавляется 600 мл ацетона. Катализатор отфильтровывается, промывается ацетоном, фильтрат концентрируется, и остаток выкристаллизовывается в присутствии диэтилового эфира с выходом, соответствующим 21,8 г (90,1%) титульного соединения. Точка плавления: 180-181 С (диэтиловый эфир). 1 е) Бутиламид 6-метокси-1 Н-бензоимидазол-2-карбоновой кислоты. В атмосфере азота при 240 С в течение 10 мин перемешивается 41,0 г (154 ммоль) N-(2-амино-4 метоксифенил)-N'-бутилоксаламида. Смесь охлаждается до комнатной температуры, затем к ней добавляется 300 мл ацетона, после чего полученная смесь перемешивается в течение 1 ч. Преципитировавший продукт отфильтровывается. Фильтрат концентрируется, и остаток смешивается со 150 мл н-гексана. Преципитировавший продукт отфильтровывается, промывается гексаном и высушивается с выходом,соответствующим 26,5 г (69,5%) титульного соединения. Точка плавления: 125-126 С (н-гексан). 1f) 6-Гидрокси-1 Н-бензоимидазол-2-карбоновая кислота. Смесь 26,0 г (105 ммоль) бутиламида 6-метокси-1H-бензоимидазол-2-карбоновой кислоты и 780 мл 48% водного раствора бромисто-водородной кислоты перемешивается при температуре 110 С в течение 8 ч, после чего подвергается нагреванию с обратным холодильником в течение 12 ч. Смесь охлаждается до комнатной температуры, преципитировавший продукт отфильтровывается, промывается водой до нейтрального рН и высушивается с выходом, соответствующим 14,3 г (76,2%) титульного соединения. Точка плавления: 206-207 С (вода). 1g) (4-Бензилиденпиперидин-1-ил)-(6-гидрокси-1 Н-бензоимидазол-2-ил)метанон (70004203). Смесь 2,0 г (11,2 ммоль) 6-гидрокси-1 Н-бензоимидазол-2-карбоновой кислоты, 3,2 мл (23,0 ммоль) триэтиламина, 2,44 г (11,1 ммоль) 4-бензилиденпиперидингидрохлорида, 4,4 г (11,6 ммоль) HBTU и 65 мл диметилформамида перемешивается в течение 16 ч при комнатной температуре. Реакционная смесь концентрируется, и остаток очищается с применением колоночной хроматографии с использованием Kieselgel 60 в качестве адсорбента (Merck) и смеси толуол:метанол=4:1 в качестве элюента, после чего продукт выкристаллизовывается из изопропанола с выходом, соответствующим 1,1 г (29,4%) титульного соеди-8 011635 нения. Точка плавления: 148 С. Пример 2. (6-Гидрокси-1 Н-бензоимидазол-2-ил)-[4-(4-метилбензилиден)пиперидин-1-ил]метанон.a) 4-(4-Метилбензилиден)пиперидингидрохлорид. Титульное соединение получают с использованием диэтилового сложного эфира (4-метилбензил) фосфоновой кислоты (Lancaster) и N-бензил-4-пиперидона в соответствии со способом, описанным в примере 1 а-b. Точка плавления: 220 С.b) (6-Гидрокси-1 Н-бензоимидазол-2-ил)-[4-(4-метилбензилиден)пиперидин-1-ил]метанон. Титульное соединение получают с использованием 6-гидрокси-1 Н-бензоимидазол-2-карбоновой кислоты (пример 1c-f) и 4-(4-метилбензилиден)пиперидингидрохлорида в соответствии со способом,описанным в примере 1g. Точка плавления: 82 С (изопропанол). Пример 3. [4-(4-Фторбензилиден)пиперидин-1-ил]-(6-гидрокси-1 Н-бензоимидазол-2-ил)метанон.a) 4-(4-Фторметилбензилиден)пиперидингидрохлорид. Титульное соединение получают с использованием диэтилового сложного эфира (4-фторбензил) фосфоновой кислоты (Tetrahedron; 55, 9, 2671 (1999 и N-бензил-4-пиперидона в соответствии со способом, описанным в примере 1a-b. Точка плавления: 183 С.b) [4-(4-Фторбензилиден)пиперидин-1-ил]-(6-гидрокси-1 Н-бензоимидазол-2-ил)метанон. Титульное соединение получают с использованием 6-гидрокси-1H-бензоимидазол-2-карбоновой кислоты (пример 1 с-f) и 4-(4-фторбензилиден)пиперидингидрохлорида в соответствии со способом, описанным в примере 1g. Точка плавления: 105-106 С (изопропанол). Пример 4. [4-(4-Хлорбензилиден)пиперидин-1-ил]-(6-гидрокси-1 Н-бензоимидазол-2-ил)метанон.a) 4-(4-Хлорбензилиден)пиперидингидрохлорид. Титульное соединение получают с использованием диэтилового сложного эфира (4-хлорбензил) фосфоновой кислоты (Lancaster) и N-бензил-4-пиперидона в соответствии со способом, описанным в примере 1 а-b. Точка плавления: 205 С.b) (6-Гидрокси-1 Н-бензоимидазол-2-ил)-[4-(4-хлорбензилиден)пиперидин-1-ил]метанон. Титульное соединение получают с использованием 6-гидрокси-1 Н-бензоимидазол-2-карбоновой кислоты (пример 1 с-f) и 4-(4-хлорбензилиден)пиперидингидрохлорида в соответствии со способом, описанным в примере 1g. Точка плавления: 88-89 С (изопропанол). Пример 5. (6-Гидрокси-1 Н-бензоимидазол-2-ил)-[4-(4-метоксибензилиден)пиперидин-1-ил]метанон.a) 4-(4-Метоксибензилиден)пиперидингидрохлорид. Титульное соединение получают с использованием диэтилового сложного эфира (4-метоксибензил) фосфоновой кислоты (Lancaster) и N-бензил-4-пиперидона в соответствии со способом, описанным в примере 1 а-b. Точка плавления: 176 С.b) (6-Гидрокси-1 Н-бензоимидазол-2-ил)-[4-(4-метоксибензилиден)пиперидин-1-ил]метанон. Титульное соединение получают с использованием 6-гидрокси-1 Н-бензоимидазол-2-карбоновой кислоты (пример 1c-f) и 4-(4-метоксибензилиден)пиперидингидрохлорида в соответствии со способом,описанным в примере 1g. Точка плавления: 208 С (изопропанол). Пример 6. (4-Бензилиденпиперидин-1-ил)-(6-гидрокси-1H-индол-2-ил)метанон. Титульное соединение получают с использованием 6-гидрокси-1H-индол-2-карбоновой кислоты (J.Chem. Soc., 1948, 1605) и 4-бензилиденпиперидингидрохлорида в соответствии со способом, описанным в примере 1g. Точка плавления: 178 С (толуол). Пример 7. Приготовление фармацевтических композиций. а) Таблетки. 0,01-50% активного вещества, соответствующего формуле (I), 15-50% лактозы, 15-50% картофельного крахмала, 5-15% поливинилпирролидона, 1-5% талька, 0,01-3% стеарата магния, 1-3% коллоидной двуокиси кремния и 2-7% ультраамилопектина смешиваются, после чего гранулируются методом сырого гранулирования и спрессовываются в таблетки.b) Драже, таблетки с пленочным покрытием. Таблетки, изготовленные описанным выше способом, покрываются слоем, состоящим из энтеролибо гастросолюбильной пленки либо из сахара и талька. Драже натираются смесью пчелиного воска с карнаубским воском.c) Капсулы. 0,01-50% активного вещества, соответствующего формуле (I), 1-5% лаурилсульфата натрия, 15-50% крахмала, 15-50% лактозы, 1-3% коллоидной двуокиси кремния и 0,01-3% стеарата магния тщательно перемешиваются, смесь пропускают через сетчатый фильтр и вносят в твердые желатиновые капсулы.d) Суспензии. Ингредиенты: 0,01-15% активного вещества, соответствующего формуле (I), 0,1-2% гидроксида натрия, 0,1-3% лимонной кислоты, 0,05-0,2% нипагина (метил 4-гидроксибензоата натрия), 0,005-0,02% нипазола, 0,01-0,5% карбопола (полиакрилата), 0,1-5% 96% этилового спирта, 0,1-1% ароматизирующего агента, 20-70% сорбитола (70% водный раствор) и 30-50% дистиллированной воды. К раствору нипагина и лимонной кислоты в 20 мл дистиллированной воды добавляют карбопол маленькими порциями при энергичном перемешивании и раствор оставляют стоять в течение 10-12 ч. За-9 011635 тем при перемешивании добавляются гидроксид натрия в 1 мл дистиллированной воды, водный раствор сорбитола и, в последнюю очередь, малиновый ароматизатор на основе этилового спирта. К этому носителю маленькими порциями добавляют активный компонент и суспендируют смесь при помощи погружаемого в смесь гомогенизатора. В завершение суспензию доводят до желаемого конечного объема дистиллированной водой и суспензию-сироп пропускают через оборудование для помола коллоидов.e) Суппозитории. Для изготовления каждого суппозитория тщательно перемешивается 0,01-15% активного вещества формулы (I) и 1-20% лактозы, после чего 50-95% основы для суппозиториев (adeps pro suppository) (например, Witepsol 4) расплавляют, охлаждают до 35 С и смесь активного компонента и лактозы вмешивают в нее, используя гомогенизатор. Полученную смесь формуют в охлаждаемых формах.f) Композиции для ампул в виде лиофилизированного порошка. 5% раствор маннита или лактозы готовят на бидистиллированной воде, применяемой для инъекций,и раствор фильтруют так, как это делается для получения стерильного раствора. 0,01-5% раствор активного компонента, имеющего формулу I, также готовят на бидистиллированной воде, применяемой для инъекций, и этот раствор фильтруют так, как это должно делаться для получения стерильного раствора. Эти два раствора смешивают в асептических условиях, вносят порциями по 1 мл в ампулы, содержание ампул лиофилизируют и ампулы запаивают под азотом. Содержимое ампул растворяют в стерильной воде или 0,9% (физиологическом) стерильном водном растворе хлористого натрия перед введением. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Производные амида гетероциклической карбоновой кислоты формулы (I)X соответствует водороду или атому галогена, гидрокси, циано, С 1-С 4 алкилсульфонамидо, факультативно замещенной атомом галогена либо атомами галогена, C1-C4 алканоиламидо, факультативно замещенной атомом галогена либо атомами галогена, арилсульфонамидогруппе,Y соответствует -СН=группе либо атому -N=,Z соответствует атому водорода либо атому галогена, С 1-С 4 алкильной, С 1-С 4 алкокси, циано, трифторметильной, трифторметоксигруппе,а также их соли. 2. Соединение по п.1, выбранное из нижеследующей группы:(4-бензилиденпиперидин-1-ил)-(6-гидрокси-1 Н-индол-2-ил)метанон и их соли. 3. Фармацевтические композиции, содержащие в своем составе эффективное количество производных амида гетероциклической карбоновой кислоты формулы (I), где значения X, Y, Z определены в п.1,либо их солей в качестве активных веществ и вспомогательные вещества, широко применяемые в фармацевтической практике, такие как носители, наполнители, разжижители, стабилизаторы, смачивающие либо эмульгирующие агенты, вещества, влияющие на рН и осмотическое давление, отдушивающие или ароматизирующие вещества, а также добавки, активирующие или доставляющие композицию. 4. Способ получения производных амида гетероциклической карбоновой кислоты формулы (I), в которой значения X, Y, Z определены в п.1, характеризующийся реакцией вторичного амина формулы (II) где Z имеет то же значение, которое определено в случае формулы (I),с химически активным производным карбоновой кислоты формулы (III) в которой значения X и Y соответствуют тем значениям, которые были определены ранее в случае формулы (I),- 10011635 в подходящем растворителе, с последующей необязательной трансформацией полученных таким образом производных амида гетероциклической карбоновой кислоты формулы (I),где значения X, Y, и Z соответствуют тем значениям, которые приведены в п.1,в другие соединения, соответствующие формуле (I), путем введения новых заместителей, и/или модификации, или удаления имеющихся заместителей, и/или путем формирования соли, и/или высвобождения соединения, соответствующего формуле (I), из состава солей с применением известных методов. 5. Способ по п.4, характеризующийся реакцией активного производного карбоновой кислоты формулы (III), в которой значения X и Y соответствуют тем значениям, которые приведены в п.1, с вторичным амином формулы (II), где значения Z соответствуют значениям, которые приведены в п.1, предпочтительно в присутствии основания. 6. Способ по п.4, характеризующийся реакцией активного производного карбоновой кислоты формулы (III), где значения X и Y соответствуют значениям, которые приведены в п.1, с вторичным амином формулы (II), где значения Z соответствуют тем значениям, которые приведены в п.1, в присутствии триэтиламина и о-бензотриазол-1-ил-N,N,N',N'-тетраметилурониумгексафторфосфата (HBTU) в диметилформамиде. 7. Способ производства фармацевтических композиций, обладающих эффектом NR2 В-селективного антагониста рецептора NMDA, характеризующийся смешиванием производного амида гетероциклической карбоновой кислоты формулы (I), в которой значения X, Y, Z соответствуют тем значениям, которые приведены в п.1, либо его фармацевтически приемлемых солей в качестве активных веществ и вспомогательных веществ, широко применяемых в фармацевтической практике, таких как носители, наполнители, разжижители, стабилизаторы, смачивающие либо эмульгирующие агенты, вещества, влияющие на рН и осмотическое давление, отдушивающие или ароматизирующие вещества, а также добавки,активирующие или доставляющие композицию. 8. Способ лечения и облегчения симптомов следующих заболеваний млекопитающих, включая человека: травматического повреждения головного или спинного мозга, повреждения нервных клеток, связанных с наличием вируса иммунодефицита человека (HIV), бокового амиотрофического склероза (болезнь Шарко), толерантности и/или зависимости при лечении боли с применением синтетических наркотических препаратов (опиоидов), синдромов отмены, например, алкоголя, опиоидов или кокаина, ишемических расстройствах ЦНС, хронических нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, боли и хронических болевых состояний, таких как невропатическая боль или боль, связанная с раковым заболеванием, эпилепсии, тревожности, депрессии,мигрени, психоза, мышечного спазма, слабоумия различного происхождения, гипогликемии, дегенеративных расстройств сетчатки, глаукомы, астмы, звона в ушах, потери слуха, индуцируемой антибиотиком-аминогликозидом, характеризующийся введением нуждающемуся в лечении млекопитающему эффективного количества/количеств производного амида гетероциклической карбоновой кислоты формулы(I), в которой значения X, Y, Z соответствуют тем значениям, которые приведены в п.1, либо его фармацевтически приемлемых солей, как самих по себе, так и в комбинации с носителями, наполнителями и тому подобным, традиционно применяемыми в фармацевтической практике. 9. Применение производного амида гетероциклической карбоновой кислоты формулы (I), где значения X, Y, Z соответствуют тем значениям, которые приведены в п.1, и/или его оптических изомеров,рацемических соединений, и/или фармацевтически приемлемых солей для производства фармацевтических препаратов для лечения и облегчения симптомов следующих заболеваний млекопитающих, включая человека: травматического повреждения головного или спинного мозга, повреждения нервных клеток, связанных с наличием вируса иммунодефицита человека (HIV), бокового амиотрофического склероза (болезнь Шарко), толерантности и/или зависимости при лечении боли с применением синтетических наркотических препаратов (опиоидов), синдромов отмены, например, алкоголя, опиоидов или кокаина,ишемических расстройств ЦНС, хронических нейродегенеративных расстройств, таких как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, боли и хронических болевых состояний, таких как невропатическая боль или боль, связанная с раковым заболеванием, эпилепсии, тревожности, депрессии, мигрени, психоза, мышечного спазма, слабоумия различного происхождения, гипогликемии, дегенеративных расстройств сетчатки, глаукомы, астмы, звона в ушах, потери слуха, вызванной применением антибиотика-аминогликозида.
МПК / Метки
МПК: A61K 31/445, C07D 401/06, A61P 25/28
Метки: карбоновой, гетероциклической, амида, кислоты, производные, новые
Код ссылки
<a href="https://eas.patents.su/12-11635-novye-proizvodnye-amida-geterociklicheskojj-karbonovojj-kisloty.html" rel="bookmark" title="База патентов Евразийского Союза">Новые производные амида гетероциклической карбоновой кислоты</a>
Новые пролекарства (n-2-пиридил-n-2-гидроксикарбонилэтил)амида 1 – метил-2-(4-амидинофениламинометил)бензимидазол-5-илкарбоновой кислоты, их получение и их применение в качестве лекарственных средств

Номер патента: 8249
Опубликовано: 27.04.2007
Авторы: Буш Ульрих, Кольбацки Флориан, Хауэль Норберт
МПК: A61K 31/245, A61P 7/02, C07D 401/12...
Метки: применение, пролекарства, получение, n-2-пиридил-n-2-гидроксикарбонилэтил)амида, метил-2-(4-амидинофениламинометил)бензимидазол-5-илкарбоновой, новые, кислоты, лекарственных, качестве, средств
Формула / Реферат:
1. Соединения общей формулы в которой а) R' обозначает атом водорода, a R обозначает метоксикарбонильную группу или б) R' обозначает атом водорода либо С1-С6алкильную группу, a R обозначает гидроксигруппу, при этом алкильные группы, содержащие более 2 атомов углерода, включают также их изомеры с разветвленной цепью, такие, например, как изопропильная, трет-бутильная и изобутильная группа, их таутомеры и их соли. 2. Соединения общей формулы I по...
Производные амида гетероарил-гексановой кислоты, их получение и их применение в качестве селективных ингибиторов связывания mip-1-альфа с его рецептором ccr1

Номер патента: 2146
Опубликовано: 24.12.2001
Авторы: Браун Мэттью Фрэнк, Посс Кристофер Стенли, Кэт Джон Чарлз
МПК: C07D 215/54, A61P 37/08, A61K 31/47...
Метки: качестве, амида, кислоты, селективных, производные, ингибиторов, mip-1-альфа, гетероарил-гексановой, применение, связывания, рецептором, получение
Формула / Реферат:
1. Соединение формулы где R1 представляет собой (С2-С9)гетероарил, возможно замещенный одним или более чем одним заместителем, независимо выбранным из группы, в которую входят водород, галогено, (C1-С6)алкил, возможно замещенный одним или более чем одним атомом фтора, гидрокси, (C1-С6)алкокси, фенил, (С2-С9)гетероарил; R2 представляет собой фенил-(CH2)m-, нафтил-(CH2)m-, (С3-С10)циклоалкил-(CH2)m-, (C1-С6)алкил или (С2-С9)гетероарил-(CH2)m-,...
Сложноэфирные производные декагидроизохинолин-3-карбоновой кислоты в качестве анальгетиков

Номер патента: 9526
Опубликовано: 28.02.2008
Автор: Орнштейн Пол Лесли
МПК: A61K 31/5377, A61P 25/00, A61K 31/4725...
Метки: анальгетиков, сложноэфирные, качестве, кислоты, декагидроизохинолин-3-карбоновой, производные
Формула / Реферат:
1. Соединение формулы где R означает С1-С20алкил, или его фармацевтически приемлемые соли. 2. Соединение по п.1, где R означает C1-С10алкил. 3. Соединение по п.2, где R означает 2-этилбутил, изобутил, 3-метилбутил, децил или этил. 4. Соединение по п.3, где R означает 2-этилбутил. 5. Соединение по п.3, где R означает изобутил. 6. Соединение по п.3, где R означает 3-метилбутил. 7. Соединение по п.3, где R означает децил. 8. Соединение по п.3, где...
Новые производные дигидроксигексановой кислоты

Номер патента: 3137
Опубликовано: 27.02.2003
Авторы: Посс Кристофер Стенли, Кэт Джон Чарлз, Браун Мэттью Фрэнк
МПК: C07C 231/00, A61K 31/498, A61P 29/00...
Метки: производные, дигидроксигексановой, кислоты, новые
Формула / Реферат:
1. Соединение формулы где указанное соединение представляет собой [4(R)-карбамоил-1(S)-(3-хлорбензил)-2(S),7-дигидрокси-7-метилоктил]амид хиноксалин-2-карбоновой кислоты; (IS)-бензил-4(R)-карбамоил-2(S),7-дигидрокси-7-метилоктил)амид 7,8-дифторхинолин-3-карбоновой кислоты; (1(S)-бензил-4(R)-карбамоил-2(S),7-дигидрокси-7-метилоктил)амид 6,7,8-трифторхинолин-3-карбоновой кислоты;...
Производные октагидро-6, 10-диоксо-6н-пиридазино [1,2-а] [1,2] диазепин-1-карбоновой кислоты, способ их получения и их применение для получения терапевтически активных соединений

Номер патента: 3280
Опубликовано: 24.04.2003
Авторы: Руссель Патрик, Ларкин Джон Патрик, Колладан Колетт, Крок Вероник
МПК: C07D 487/04
Метки: получения, 1,2, октагидро-6, применение, 1,2-а, терапевтически, диазепин-1-карбоновой, активных, соединений, кислоты, производные, 10-диоксо-6н-пиридазино, способ
Формула / Реферат:
1. Соединения общей формулы (I) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS, в формуле которых R представляет собой атом водорода, радикал алкил, содержащий от 1 до 4 атомов углерода, a R1 и R2 означают H или защитную группу аминофункции. 2. Соединения по п.1, в которых аминогруппа защищена в форме фталимида, формулы (IA1) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS. 3. Соединения формулы (I) согласно любому из...
Предыдущий патент: 3-[4-гетероциклил-1,2,3-триазол-1-ил]-n-арилбензамиды в качестве ингибиторов продуцирования цитокинов, предназначенные для лечения хронических воспалительных заболеваний
Следующий патент: Производные амида кинуреновой кислоты как антагонисты nr2b подтипа рецептора nmda
Случайный патент: Способ получения реформата с ультранизким содержанием бензола с использованием каталитической дистилляции