Пролекарства триптолида
Номер патента: 21135
Опубликовано: 30.04.2015
Авторы: Салуя Ашок К., Виккерс Селвин М., Патил Сатиш Пракаш, Чугх Рохит, Георг Ингрид Гунда




















Формула / Реферат
1. Соединение формулы I

где каждый R1 представляет собой Н или (C1-C6)алкил;
каждый R2 представляет собой Н или (C1-C6)алкил;
n представляет собой 1, 2 или 3 и
каждый Х+ представляет собой фармацевтически приемлемый органический катион или неорганический катион.
2. Соединение по п.1, представляющее собой соединение формулы Ia

где Х+ представляет собой фармацевтически приемлемый органический катион или неорганический катион.
3. Соединение по п.1, где R1 представляет собой Н.
4. Соединение по п.1 или любому из пп.2-3, где каждый Х+ представляет собой Н.
5. Соединение по п.1 или любому из пп.2-3, где каждый Х+ представляет собой литий, натрий, калий, магний, кальций, барий, цинк или алюминий.
6. Соединение по п.1 или любому из пп.2-3, где каждый Х+ имеет формулу HY+, где Y представляет собой аммиак, триэтиламин, трометамин, триэтаноламин, этилендиамин, глюкамин, N-метилглюкамин, глицин, лизин, орнитин, аргинин, этаноламин или холин.
7. Соединение по любому из пп.1-3, где каждый Х+ представляет собой Na+.
8. Соединение, представляющее собой динатриевую соль 14-O-фосфонооксиметилтриптолида, динатриевую соль 14-O-фосфонооксиэтилтриптолида или динатриевую соль 14-O-фосфонооксипропилтриптолида.
9. Соединение, представляющее собой динатриевую соль 14-O-фосфонооксиметилтриптолида.
10. Фармацевтическая композиция, содержащая соединение формулы I по любому из пп.1-9 в комбинации с фармацевтически приемлемым носителем.
11. Способ лечения рака у млекопитающего, включающий введение указанному млекопитающему соединения формулы I по любому из пп.1-9.
12. Способ ингибирования роста раковых клеток в экспрессирующей HSP70 злокачественной опухоли у млекопитающего, включающий введение указанному млекопитающему эффективного для ингибирования количества соединения формулы I по любому из пп.1-9.
Текст
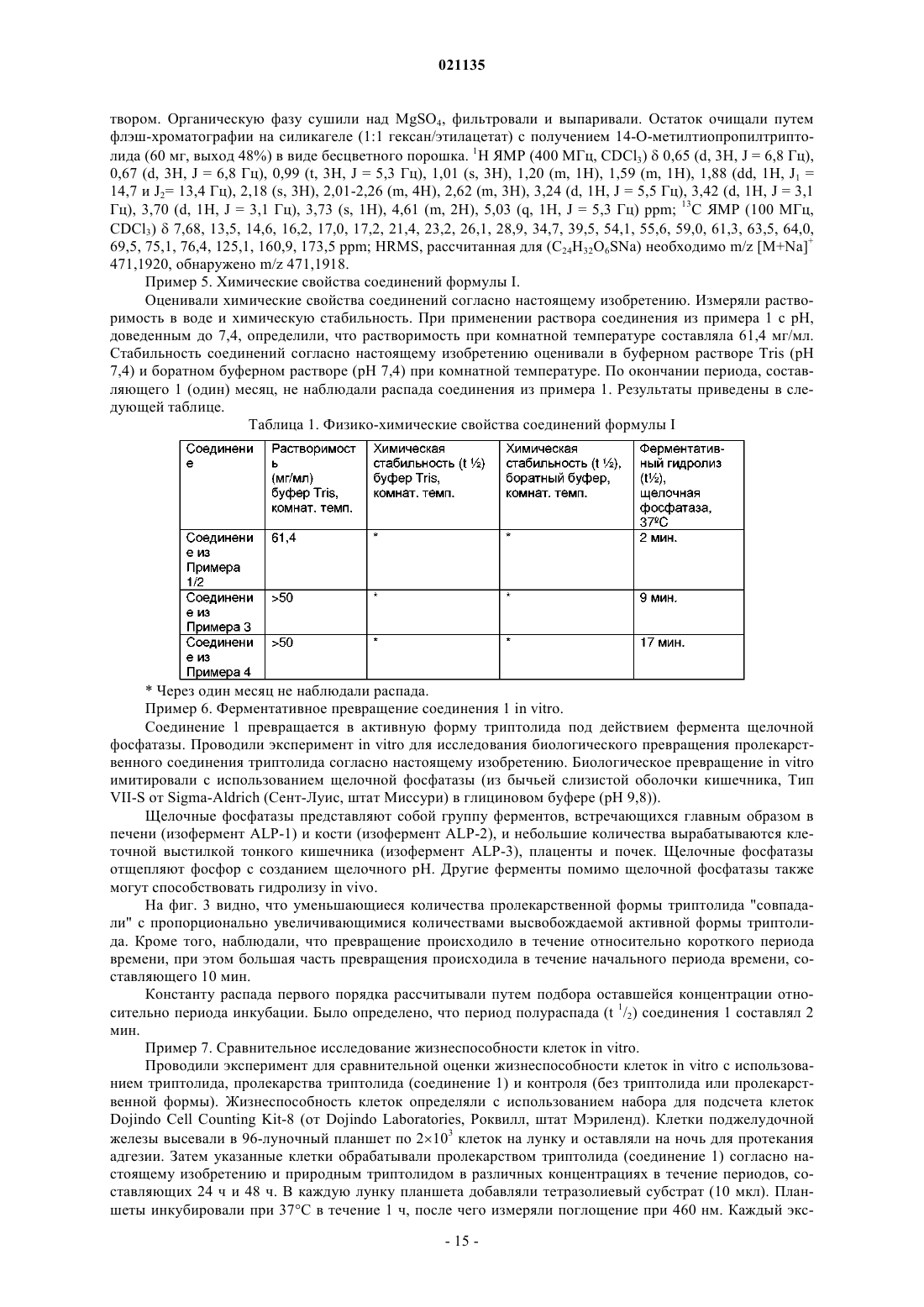
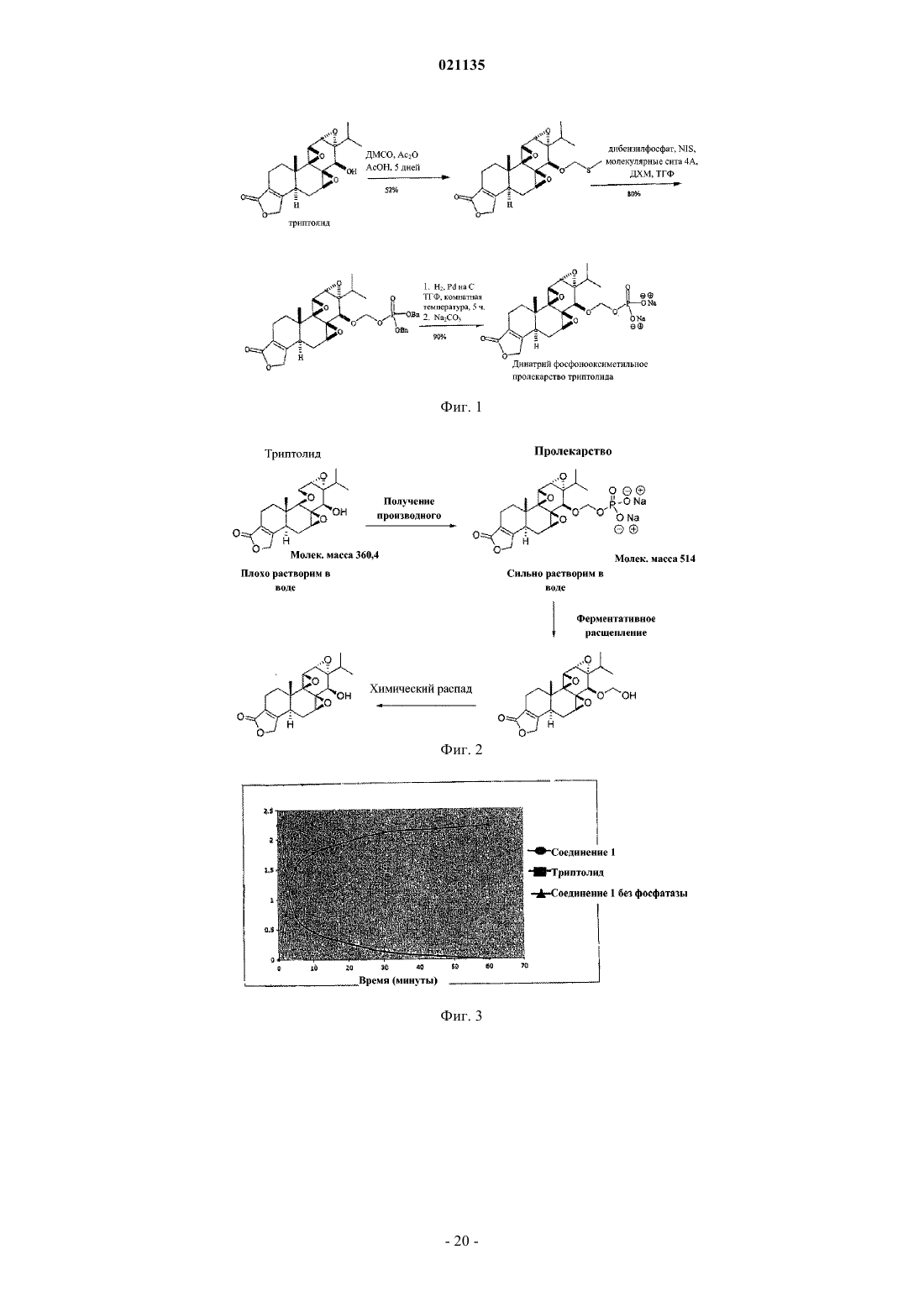
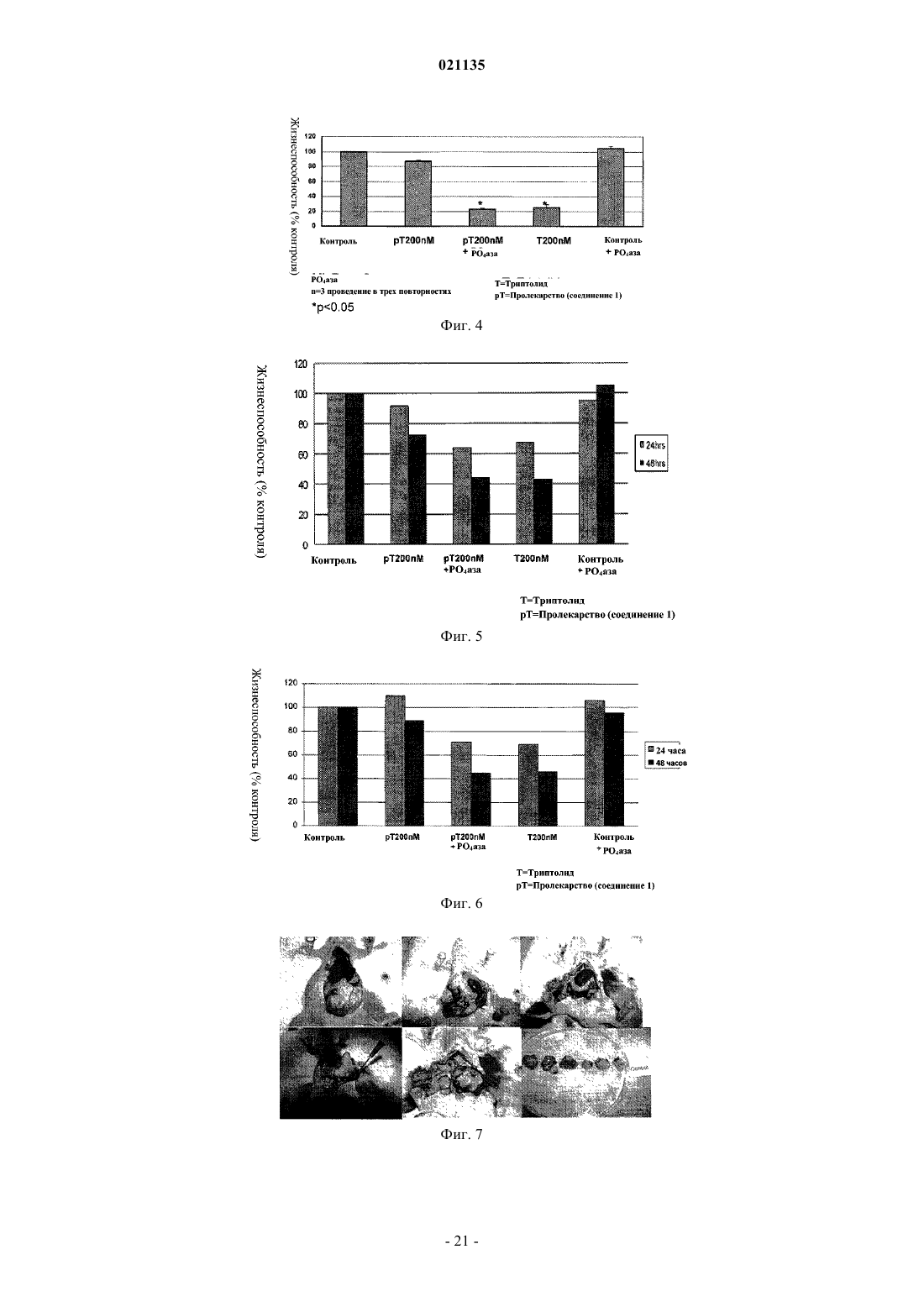
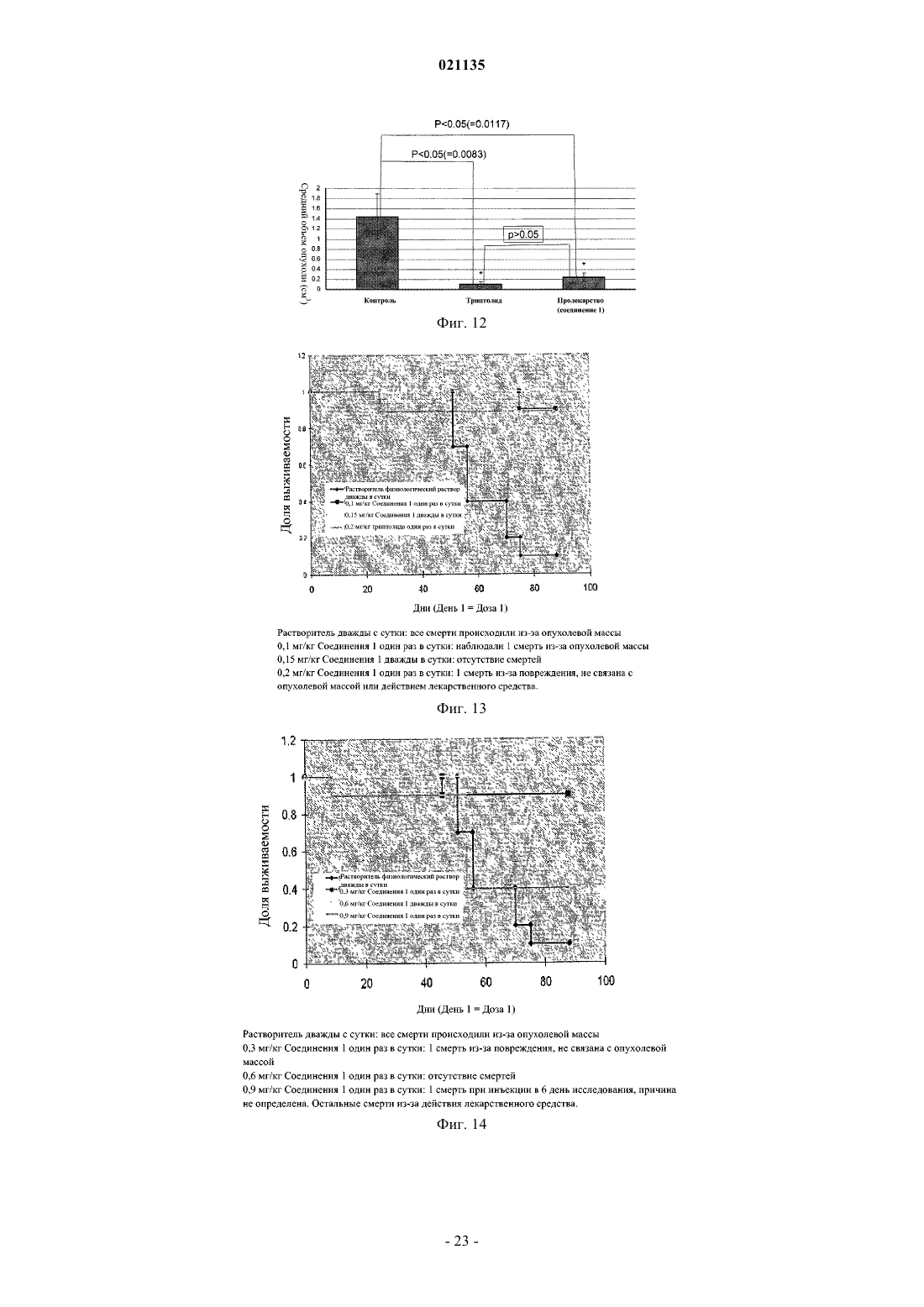
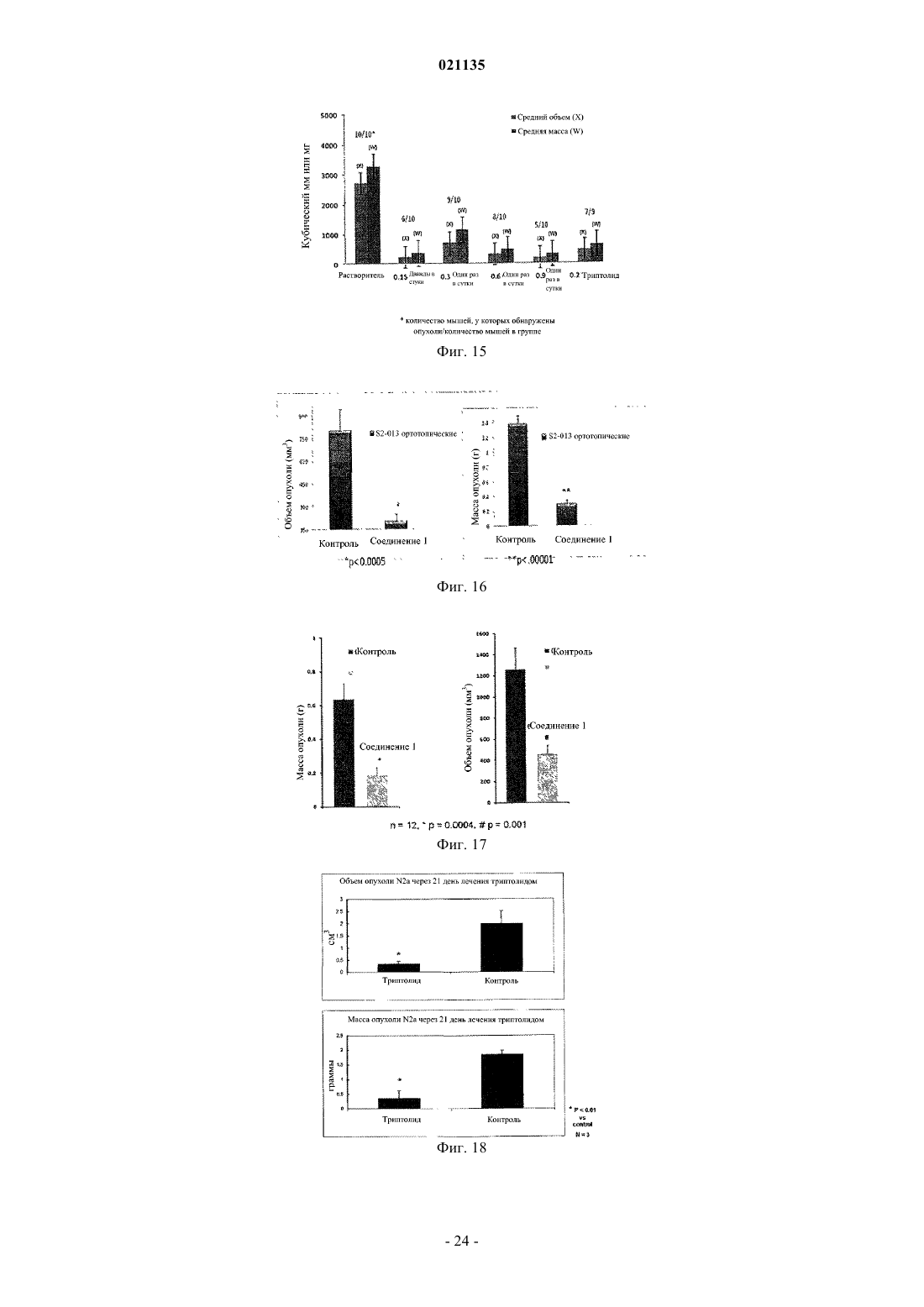
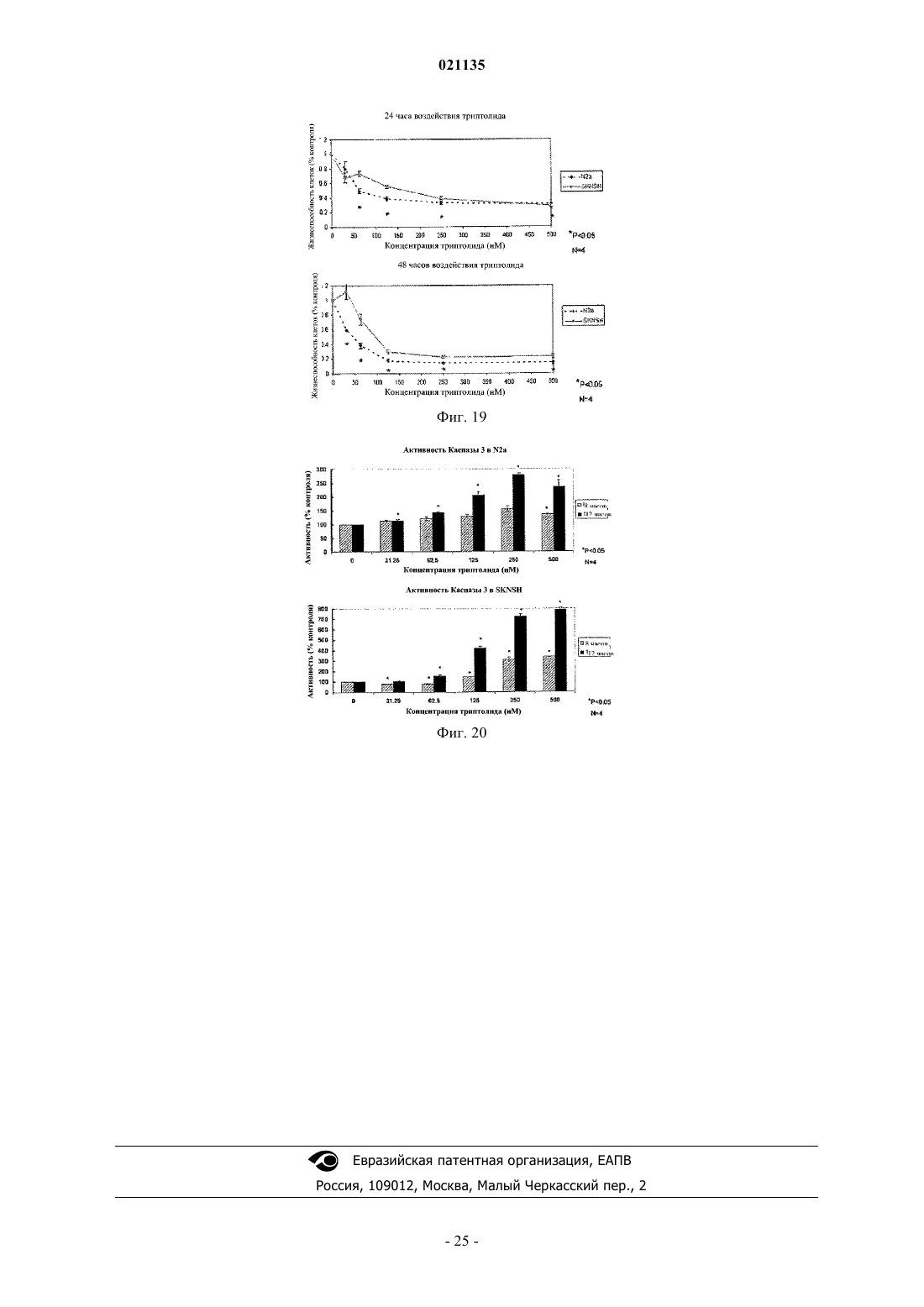
Согласно изобретению предложены соединения формулы I также предложены фармацевтические композиции, содержащие соединение формулы I, и способы лечения с применением соединений формулы I.(71)(73) Заявитель и патентовладелец: РЕДЖЕНТС ОФ ЗЕ ЮНИВЕРСИТИ ОФ МИННЕСОТА (US) Уровень техники Рак поджелудочной железы представляет собой остро протекающее и изнуряющее заболевание, для которого пятилетняя выживаемость составляет менее 5%. В настоящее время отсутствует эффективная лекарственная терапия, способная эффективно повышать выживаемость пациентов. По сообщениям, в 2006 году было выявлено более 35000 новых случаев рака поджелудочной железы и почти столько же составило число умерших от указанного заболевания. В качестве ключевого фактора отсутствия развития у пациентов ответа на терапию для лечения рака поджелудочной железы и других злокачественных опухолей была исследована устойчивость к апоптозу. Триптолид представляет собой встречающееся в природе соединение, получаемое из растения Трхкрыльник Вильфорда (Tripterygium wilfordii). Известно, что триптолид подходит для лечения аутоиммунных заболеваний, отторжения при трансплантации (иммуносупрессии) и обладает противораковым и антифертильным действием, а также другими биологическими действиями (Qui and Kao, 2003,Drugs R.D. 4, 1-18). Триптолид обладает сильным противоопухолевым действием в отношении ксенотрансплантатных опухолей (например, Yang et al. Mol. Cancer Ther, 2003, 2, 65-72). Триптолид представляет собой антиапоптический агент с многочисленными клеточными мишенями, вовлеченными в процесс роста и метастазирования злокачественных опухолей. Триптолид ингибирует активацию NF-kB,индуцирует отщепление bid, блокирует индукцию гена выживания р 21 WAF1/Cip1 (Wang et al. Journal ofMolecular Medicine, 2006, 84, 405-415) и ингибирует функцию фактора транскрипции теплового шока 1(HSF1), тем самым подавляя эндогенную экспрессию гена Hsp70 (Westerheide et al. 2006, Journal of Biological Chemistry, 281, 9616-9622). Триптолид также выполняет функцию мощного ингибитора ангиогенеза опухоли (Не et al. 2010, Int. Journal of Cancer, 126, 266-278). В живых клетках существует несколько механизмов, защищающих от неблагоприятных состояний,включая раковые клетки. Синтез семейства белков, называемых белками теплового шока (HSPs), представляет собой один из указанных защитных механизмов. Основные HSP включают HSP90, HSP70,HSP60, HSP40 и более мелкие HSP. HSP могут присутствовать в большинстве внутриклеточных компартментов, при этом HSP70 главным образом локализован в цитозоле. Известно, что разрегулированная экспрессия HSP70 связана со многими заболеваниями, включая раковые. HSP70 в большом количестве экспрессируется в злокачественных опухолях различного происхождения (например: Hantschel et al. 2000, Cell Stress Chaperones, 5, 438-442), что приводит к резистентности опухолевых клеток к терапии и плохому прогнозу для пациента (Fuqua et al. 1994, Breast CancerRes, Treatment 32, 67-71). Известно, что белок теплового шока 70 (Hsp70) активирован и чрезмерно экспрессирован в клетках рака поджелудочной железы по сравнению с нормальными клетками. Кроме того,HSP70 обладает защитным действием в отношении раковых клеток, ингибируя апоптоз указанных клеток. Было показано, что ингибирование HSP70 в клетках рака поджелудочной железы увеличивает апоптическую гибель данных клеток (см., например, Aghdassi et al., Cancer Research, 67(2) p.616-625 (2007. Было показано, что триптолид ингибирует рост и метастазирование опухоли поджелудочной железы у мышей. Также было показано, что при применении триптолида в комбинации с ионизирующим излучением его терапевтический эффект при лечении рака поджелудочной железы усиливается (Wang et al.Proc. Amer. Assoc. Cancer Res. 2006, 47, abstract 4720 и Wang et al. Clin. Cancer Res. 2007, 13, 4891-4899). Считается, что противораковое действие, связанное с триптолидом, возникает в результате снижения уровней белка HSP70, экспрессируемого в значительных количествах клетками рака поджелудочной железы, по сравнению с нормальными клетками поджелудочной железы. Таким образом, виды терапии с применением триптолида представляют интерес в области медицины в свете потенциального лечения раковых опухолей, которые чрезмерно экспрессируют HSP70, включая рак поджелудочной железы. См.,например, Phillips et al., Cancer Research, 67(19), p.9407-16 (2007). Однако существуют некоторые недостатки, связанные с введением триптолида, и были исследованы различные решения для разрешения данных проблем. Одна из проблем, связанных с природным триптолидом, заключается в том, что он нерастворим в водном растворе. Другая проблема, связанная с природным триптолидом, заключается в плохой биодоступности и токсических побочных эффектах. Известны триптолид, производные триптолида и некоторые пролекарства, обладающие улучшенной растворимостью и сниженной токсичностью. Например, в патенте США 6548537 описаны пролекарства триптолида, обладающие повышенной растворимостью и сниженной токсичностью. В данной области техники, по существу, известна фосфоноксиметильная группа для получения пролекарств некоторых фармацевтических соединений. Например, Krise et al., J. Med. Chem., 42,стр.3094-3100 (1999) описывает получение N-фосфонооксиметильных пролекарств некоторых соединений для улучшения растворимости в воде. Тем не менее, пролекарства должны обладать рядом свойств для того, чтобы быть практически полезными. Например, желаемые пролекарства должны быть стабильными для получения составов и введения. Кроме того, после того, как лекарство введено и присутствует в организме реципиента, оно должно быть успешно активировано. Кроме того, как пролекарство, так и активированное соединение должны быть совместимы с биологическими жидкостями, такими как плазма, и гомогенатами тканей. И наконец,активированное соединение, изначально доставляемое в форме пролекарства, должно обладать необхо-1 021135 димым терапевтическим или фармацевтическим эффектом. Учитывать эти и другие факторы одновременно или совместно сбалансировать с определенными типами соединений может быть непросто. В контексте триптолида и пролекарственных соединений триптолида было трудно достичь улучшенной растворимости в воде, эффективной биодоступности для лекарственных форм для перорального применения, более быстрого высвобождения триптолида in vivo и относительно сниженной или более низкой токсичности в комбинации со значительным ингибированием роста раковых клеток. Например, см. Chassaing et al., Highly Water-Soluble Prodrugs of Anthelminthic Benzimidazole Carbamates: Synthesis, Pharmacodynamics and Pharmacokinetics, J. Med. Chem., 51 (5), стр.1111-1114 (2008). Известны сукцинатные пролекарственные формы триптолида, но им присущи определенные недостатки. См., например, Harrousseau et al., Haematologica 2008, 93(s1), 14 Abstract 0038 и Kitzen et al. European Journal of Cancer 2009, 45, 1764-1772. Наблюдали неполное и неустойчивое превращение сукцинатного пролекарства триптолида. Таким образом, в области медицины и фармацевтики существует потребность в разработке улучшенных лекарственных средств для лечения раковых опухолей, включая агрессивные солидные раковые опухоли, такие как рак поджелудочной железы. Также существует дополнительная потребность в улучшенной доставке или улучшенных фармакокинетических параметрах, или сниженной токсичности таких лекарственных средств. Также существует потребность в разработке пролекарственных форм триптолида, обладающих улучшенной растворимостью или ускоренным высвобождением активного соединения триптолида, или более терапевтически эффективным высвобождением активного соединения триптолида, или в пролекарственных формах триптолида, обладающих улучшенной биодоступностью. Краткое описание изобретения Соответственно, в настоящем изобретении предложено соединение согласно изобретению, представляющее собой соединение формулы In представляет собой 1, 2 или 3; и каждый Х+ представляет собой фармацевтически приемлемый органический катион или неорганический катион. Согласно частному варианту реализации соединение формулы I представляет собой соединение формулы Ia где Х+ представляет собой фармацевтически приемлемый органический катион или неорганический катион. Согласно частному варианту реализации R1 в соединении формулы I представляет собой Н. Согласно частному варианту реализации каждый Х+ в соединении формулы I представляет собой Н. Согласно частному варианту реализации каждый Х+ в соединении формулы I представляет собой литий, натрий, калий, магний, кальций, барий, цинк или алюминий. Согласно частному варианту реализации каждый Х+ в соединении формулы I имеет формулу HY+,где Y представляет собой аммиак, триэтиламин, трометамин, триэтаноламин, этилендиамин, глюкамин,N-метилглюкамин, глицин, лизин, орнитин, аргинин, этаноламин или холин. Согласно частному варианту реализации каждый Х+ в соединении формулы I представляет собойNa . Согласно частному варианту реализации соединение формулы I представляет собой динатриевую соль 14-О-фосфонооксиметилтриптолида, динатриевую соль 14-O-фосфонооксиэтилтриптолида или динатриевую соль 14-O-фосфонооксипропилтриптолида. Согласно частному варианту реализации соединение формулы I представляет собой динатриевую соль 14-O-фосфонооксиметилтриптолида. Согласно настоящему изобретению также предложена фармацевтическая композиция, содержащая соединение формулы I в комбинации с фармацевтически приемлемым носителем. Согласно настоящему изобретению также предложен способ лечения рака (например, рака поджелудочной железы, карциномы желчного протока, нейробластомы, рака толстой кишки, рака молочной железы, миеломы, рака желудка, рака печени, глиобластомы, рака яичников, рака ободочной и прямой кишки, неходжкинской лимфомы, рака легкого, рака предстательной железы, мелкоклеточного рака легкого, крупноклеточного рака легкого, рака почки, рака пищевода, рака желудка, рака шейки матки или лимфомных опухолей) у млекопитающего (например, человека), включающий введение соединения формулы I указанному млекопитающему (например, человеку). Согласно настоящему изобретению также предложен способ ингибирования роста раковых клеток в экспрессирующей HSP70 злокачественной опухоли (например, рак поджелудочной железы, нейробластома, рак молочной железы, рак толстой кишки, рак желудка, рак печени или глиобластома) у млекопитающего (например, человека), включающий введение ингибирующего эффективного количества соединения формулы I или фармацевтически приемлемой соли указанного соединения указанному млекопитающему (например, человеку). Краткое описание чертежей На фиг. 1 показана схема химической реакции для получения соединения 1. На фиг. 2 показана схема химической реакции, демонстрирующая получение производного триптолида, представляющего собой соединение 1, и последующее ферментативное расщепление - химический распад соединения 1 с высвобождением триптолида. На фиг. 3 показано ферментативное превращение in vitro пролекарства триптолида (соединение 1) в триптолид. На фиг. 4 показано сравнительное влияние триптолида и пролекарства триптолида (соединение 1) на жизнеспособность клеток MiaPaca-2 in vitro через 48 ч. На фиг. 5 показано сравнительное влияние триптолида и пролекарства триптолида (соединение 1) на жизнеспособность клеток Panc-1 in vitro как через 24 ч, так и 48 ч. На фиг. 6 показано сравнительное влияние триптолида и пролекарства триптолида (соединение 1) на жизнеспособность клеток S2VP10 in vitro через 24 ч и 48 ч. На фиг. 7 показан рост опухоли в контрольной группе мышей с помощью пяти фотографий in situ и одной фотографии опухолей ex vivo. На фиг. 8 показан рост опухоли в группе мышей, получавшей триптолид, с помощью трех фотографий in situ и одной фотографии опухолей ex vivo. На фиг. 9 показан рост опухоли в группе мышей, получавшей пролекарство триптолида (соединение 1), с помощью четырех фотографий in situ и одной фотографии опухолей ex vivo. Фиг. 10 представляет собой фотографию серии опухолей ex vivo из эксперимента in vivo, демонстрирующую сравнительные размеры опухолей для контрольной группы, группы триптолида и группы пролекарства триптолида (соединение 1). На фиг. 11 показана сравнительная масса (г) опухолей контрольной группы мышей, группы триптолида и группы пролекарства триптолида (соединение 1) из эксперимента in vivo. На фиг. 12 показан сравнительный объем (см 3) опухолей у мышей контрольной группы, группы триптолида и группы пролекарства триптолида (соединение 1) в эксперименте in vivo. На фиг. 13 показан анализ выживаемости мышей, получавших лечение соединением 1, и контрольных мышей. На фиг. 14 показан анализ выживаемости мышей, получавших лечение соединением 1, и контрольных мышей. На фиг. 15 показана опухолевая масса (объем и масса) для мышей, получавших лечение соединением 1 и носителем. На фиг. 16 показана опухолевая масса (объем и масса) для мышей, получавших лечение соединением 1 и носителем. На фиг. 17 показана опухолевая масса (объем и масса) для мышей, получавших лечение соединением 1 и носителем. На фиг. 18 показан объем опухоли для мышей, получавших лечение соединением и носителем. На фиг. 19 показана жизнеспособность клеток (Нейробластома N2a и SKNSH) в присутствии триптолида. На фиг. 20 показана активность Каспазы 3 в присутствии триптолида. Подробное описание изобретения Определения В настоящем описании термин "(C1-C6)алкил" относится к алкильным группам, содержащим от 1 до 6 атомов углерода, которые представляют собой линейные или разветвленные группы. Данныйтермин представлен в качестве примера группами, такими как метил, этил, н-пропил, изопропил, н-бутил, третбутил, изобутил, н-пентил, неопентил и н-гексил, и т.п. В настоящем описании термин "(С 1-С 6)алкокси" относится к группе (С 1-С 6)алкилО-, где (С 1-С 6)алкил является таким, как определено в настоящем описании. Данный термин представлен в качестве примера группами, такими как метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси,пентокси, 3-пентокси или гексилокси и т.п. В настоящем описании термин "(С 3-С 7)циклоалкил" относится к насыщенной или частично ненасыщенной циклической углеводородной кольцевой системе, содержащей от 3 до 7 атомов углерода. Данный термин представлен в качестве примера такими группами, как циклопропил, циклобутил, циклопентил, циклогексил, циклогексен или циклогептан и т.п. В настоящем описании термин "арил" относится к фенильному радикалу или орто-конденсированному бициклическому карбоциклическому радикалу, содержащему примерно от девяти до десяти атомов углерода в кольце, в котором по меньшей мере одно кольцо является ароматическим. Данный термин представлен в качестве примера такими группами, как фенил, инданил, инденил, нафтил, 1,2 дигидронафтил и 1,2,3,4-тетрагидронафтил. В настоящем описании термин "арил(C1-C6)алкил-" относится к группе арил-(С 1-С 6)алкил-, где (C1C6)алкил и арил являются такими, как определено в настоящем описании. Данный термин представлен в качестве примера такими группами, как бензил и фенэтил, и т.п. В настоящем описании термин "содержащий" означает перечисленные элементы или их эквивалент по структуре или функции плюс любой другой элемент (элементы), который не указан. Термины "имеющий" и "включающий" также следует рассматривать как неограничивающие, если из контекста не следует иное. Термины, такие как "примерно", "в целом", "по существу" и т.п. следует рассматривать как модифицирующие термин или значение так, что оно не является абсолютным, но не включает уровень техники. Указанные термины будут определяться обстоятельствами, при этом термины, которые они модифицируют, понятны специалисту в данной области техники. Это включает, по меньшей мере, степень ожидаемой экспериментальной ошибки, технической ошибки и инструментальной ошибки для конкретной методики, применяемой для измерения значения. Термины "терапевтически эффективное количество" и "фармацевтически эффективное количество" употребляются в настоящем описании, например, для обозначения количества, достаточного для уменьшения или ингибирования in vivo роста раковых клеток при введении живому млекопитающему. Данные термины относятся к количеству, определенному как необходимое для оказания физиологического действия, предполагаемого и связанного с данным активным компонентом, измеренному в соответствии с установленными фармакокинетическими способами и технологиями для данного способа введения. Термин "ингибирующее эффективное количество", употребляемый в связи с количеством активного соединения и композиции согласно настоящему изобретению, относится, например, к проявляемым противоопухолевым свойствам, продемонстрированным с применением стандартных методов анализа культуры клеток. В настоящем описании термин "пролекарство" относится к фармацевтическому соединению, для которого необходим дальнейший метаболизм (включая, но не ограничиваясь им, метаболизм в печени) прежде чем оно станет биологически активным. Для специалиста в данной области техники очевидно, что соединения согласно настоящему изобретению, содержащие хиральный центр, могут существовать и быть выделены в оптически активных и рацемических формах. Некоторые соединения могут проявлять полиморфизм. Следует понимать, что настоящее изобретение включает любую рацемическую, оптически активную, полиморфную или стереоизомерную форму соединения согласно настоящему изобретению, или их смеси, обладающие подходящими свойствами, указанными в настоящем описании, при этом в данной области техники хорошо известно как получать оптически активные формы, например, путем разделения рацемической формы с помощью перекристаллизации, путем синтеза из оптически активных исходных веществ, путем хирального синтеза или путем хроматографического разделения с использованием хиральной неподвижной фазы. Соль соединения формулы I может подходить в качестве промежуточного соединения для выделения или очистки соединения формулы I. Кроме того, может быть подходящим введение соединения формулы I в качестве фармацевтически приемлемой кислой или основной соли. Примерами фармацевтически приемлемых солей являются органические соли присоединения кислоты и неорганические соли. Термин "органический катион или неорганический катион" или "катионная органическая или неорганическая соль" включает органические катионы или неорганические катионы (например, соли металлов или аминов), которые хорошо известны в данной области техники, и включает катионные вещества,которые могут образовывать ионную ассоциацию с О-фрагментами в соединении и не оказывают значительного отрицательного влияния на необходимые свойства пролекарства для целей настоящего изобретения. Термин "фармацевтически приемлемые органические катионы или неорганические катионы" или"фармацевтически приемлемая катионная органическая или неорганическая соль" включает "органические катионы или неорганические катионы", которые фармацевтически приемлемы для применения у млекопитающего и хорошо известны в данной области техники. Органические катионы или неорганические катионы включают, но не ограничиваются ими, катионы лития, натрия, калия, магния, кальция, бария, цинка, алюминия и амина. Катионы аминов включают,но не ограничиваются ими, катионы, получаемые из аммиака, триэтиламина, трометамина (TRIS), триэтаноламина, этилендиамина, глюкамина, N-метилглюкамина, глицина, лизина, орнитина, аргинина, этаноламина, холина и т.п. В одном из вариантов реализации катионы аминов представляют собой катионы,где Х+ имеет формулу YH+, где Y представляет собой аммиак, триэтиламин, трометамин (TRIS), триэтаноламин, этилендиамин, глюкамин, N-метилглюкамин, глицин, лизин, орнитин, аргинин, этаноламин,-4 021135 холин и т.п. В одном из вариантов реализации подходящие катионные органические или неорганические соли,которые можно применять, включают катионные фрагменты, которые могут образовывать ионную связь с О-фрагментами в соединении, и не оказывают значительного отрицательного влияния на необходимые свойства пролекарства для целей настоящего изобретения, например, повышенную растворимость, стабильность и быстрое гидролитическое высвобождение активной формы соединения. Предпочтительно, X выбран из Li+, K+ или Na+. Более предпочтительно, X представляет собой Na+, таким образом образуя динатриевую соль. Фармацевтически приемлемые соли также могут включать соли, образованные кислотами, которые образуют физиологически приемлемый анион, например, тозилат, метансульфонат, ацетат, цитрат, малонат, тартрат, сукцинат, бензоат, аскорбат, а-кетоглутарат и -глицерофосфат. Также могут быть образованы подходящие неорганические соли, включая гидрохлоридные, сульфатные, нитратные, бикарбонатные и карбонатные соли. Соли, включая фармацевтически приемлемые соли, могут быть получены с использованием стандартных процедур, хорошо известных в данной области техники, например, путем осуществления взаимодействия достаточно основного соединения, такого как амин, с подходящей кислотой с получением физиологически приемлемого аниона. Настоящее изобретение включает как свободную кислоту (например, -ОР(О)(ОН)2), моносоли (например, -ОР(О)(ОН)(О-Х+, так и двойные соли (например, -ОР(О)О-Х+)2) соединений формулы I. Указанная кислота и соли могут быть очищены с помощью различных методов, хорошо известных в данной области техники, таких как хроматография с последующей лиофилизацией или перекристаллизацией. Для специалиста в данной области техники очевидно, что соединение формулы I, где Х+ представляет собой органический катион или неорганический катион, может быть превращено в соединение формулы I, содержащее один или более разных органических или неорганических катионов. Указанное превращение может быть осуществлено с использованием множества хорошо известных технологий и веществ, включая, но не ограничиваясь ими, ионообменные смолы, ионообменную хроматографию и селективную кристаллизацию. Конкретным значением R1 является Н или (C1-C6)алкил. Другим конкретным значением R1 является Н. Другим конкретным значением R1 является (C1-C6)алкил. Другим конкретным значением R1 является метил или этил. Конкретным значением R2 является Н или (C1-C6)алкил. Другим конкретным значением R2 является Н. Конкретным значением Х+ является Н. Другим конкретным значением Х+ является литий, натрий, калий, магний, кальций, барий, цинк или алюминий. Другая конкретная группа соединений формулы I представляет собой соединения, где Х+ имеет формулу HY+, где Y представляет собой аммиак, триэтиламин, трометамин, триэтаноламин, этилендиамин, глюкамин, N-метилглюкамин, глицин, лизин, орнитин, аргинин, этаноламин или холин. Другим конкретным значением Х+ является Li+, K+ или Na+. Другим конкретным значением Х+ является Na+. Конкретное соединение формулы I представляет собой динатриевую соль 14-O-фосфонооксиметилтриптолида, динатриевую соль 14-O-фосфонооксиэтилтриптолида или динатриевую соль 14-Oфосфонооксипропилтриптолида, или его соль. Конкретная группа соединений формулы I представляет собой соединения формулы Ia: где Х+ представляет собой фармацевтически приемлемый органический катион или неорганический катион. Другая конкретная группа соединений формулы I представляет собой соединения формулы Ia: где Х+ представляет собой фармацевтически приемлемую катионную органическую или неорганическую соль. Способы, которые могут быть использованы для получения соединений формулы I и промежуточных соединений, подходящих для получения соединений формулы I, показаны на схеме 1 и схеме 2. Схема 1 Соединение формулы I может быть получено путем снятия одной или более защитных групп с соединения формулы IA с получением соответствующего соединения формулы I. Таким образом, промежуточное соединение формулы IA подходит для получения соединения формулы I. Соединение формулы I также может быть получено путем превращения группы -SMe соединения формулы IB в группу -ОР(О)(О-Х+)2 с получением соответствующего соединения формулы I. Таким образом,промежуточное соединение формулы IB подходит для получения соединения формулы I. Соединение формулы I также может быть получено путем снятия одной или более защитных групп с соединения формулы IC с получением соответствующего соединения формулы I. Таким образом, промежуточное соединение формулы IC подходит для получения соединения формулы I. Соединение формулы I также может быть получено путем превращения группы -SMe соединения формулы ID в группу -ОР(О)(О-Х+)2 с получением соответствующего соединения формулы I. Таким образом,-6 021135 промежуточное соединение формулы ID подходит для получения соединения формулы I. Соответственно, согласно настоящему изобретению предложен способ:a) получения соединения формулы I, включающий снятие защитных групп с соответствующего соединения формулы IA, содержащего одну или более защитных групп, с получением соединения формулы I;b) получения соединения формулы I, включающий превращение группы -SMe соединения формулыIB в группу -ОР(О)(О-Х+)2 с получением соединения формулы I;c) получения соединения формулы I, включающий снятие защитных групп с соответствующего соединения формулы IC, содержащего одну или более защитных групп, с получением соединения формулыd) получения соединения формулы I, включающий превращение группы -SMe соединения формулыID в группу -ОР(О)(О-Х+)2 с получением соединения формулы I;e) получения соли соединения формулы I, включающий действие на соответствующее соединение формулы I кислотой (например, органической кислотой или неорганической кислотой) или основанием(например, щелочным основанием) с получением соли соединения формулы I;f) превращения соединения формулы I, где один или более Х+ представляет собой катионную органическую или неорганическую соль, в соединение формулы I, где один или более Х+ представляет собой другую органическую или неорганическую соль. Соединение согласно настоящему изобретению может быть также введено в состав фармацевтических композиций путем комбинирования с фармацевтически приемлемым носителем. Фармацевтические композиции могут быть получены на основе хорошо известных соединений и методик, легкодоступных специалисту в области фармацевтики. Для целей настоящего изобретения указанный фармацевтически приемлемый носитель может представлять собой любое традиционное и легкодоступное биологически совместимое или инертное вещество, которое химически совместимо с активным фармацевтическим компонентом и в значительной степени не ослабляет его предполагаемый терапевтический эффект при приготовлении состава или доставке. Фармацевтически приемлемые соли могут быть получены с использованием стандартных процедур и технологий, хорошо известных в данной области техники. Твердая форма соединения согласно настоящему изобретению может представлять собой наночастицу и, таким образом, получена в виде наночастицы. Соответственно, согласно настоящему изобретению предложены наночастицы соединения формулы I и композиции, содержащие наночастицы соединения формулы I. Пролекарственные соединения триптолида согласно настоящему изобретению могут быть получены с использованием множества составов наполнителей и получены в различных лекарственных формах,описанных ниже. Химические свойства и характеристики, связанные с соединениями согласно настоящему изобретению, также могут позволять получать твердые лекарственные формы соединений согласно настоящему изобретению для перорального применения. Соединение согласно настоящему изобретению может быть получено в виде фармацевтических композиций и введено реципиенту в различных формах, подходящих для необходимого конкретного способа введения или системы. Способы введения могут включать, но не ограничиваются ими, пероральные способы, парентеральные способы, внутривенные способы (включая внутривенные способы путем инъекции с помощью помпы), внутримышечные способы, топические способы, включая глазные капли, подкожные способы и способы введения через слизистую. Соединения согласно настоящему изобретению могут быть введены системно, например, перорально, в комбинации с фармацевтически приемлемым носителем, таким как инертный разбавитель или усваиваемый пищевой носитель. Таким образом, фармацевтическая композиция, содержащая соединения согласно настоящему изобретению в качестве активного компонента, может быть получена в разных лекарственных формах. Например, композиции могут быть заключены в твердые или мягкие капсулы (например, желатин или вещества для капсул растительного происхождения). Композиции могут быть спрессованы в форму таблетки для проглатывания или проникновения через слизистую, пастилки, капсулы, эликсиры, суспензии, сиропы, пластины,суппозитории и т.п. Количество активного компонента может варьироваться в соответствии с конкретной необходимой фармацевтически эффективной величиной дозы. Таблетки, пастилки, пилюли, капсулы и т.п. могут содержать дополнительные компоненты, такие как связующие вещества (такие как трагакант, камедь, кукурузный крахмал или желатин); наполнители,такие как дикальцийфосфат; разрыхлители, такие как кукурузный крахмал, картофельный крахмал, альгиновая кислота и т.п.; смазывающие вещества (такие как стеарат магния), которые можно применять для технологий прессования таблеток, например; подсластители, такие как сахароза, фруктоза, лактоза или аспартам; и ароматизаторы, такие как мята перечная, грушанка, вишня и т.п. Дополнительные компоненты, которые могут быть включены в композиции согласно настоящему изобретению, представляют собой маннит, мочевину, декстраны и лактозу, невосстанавливающие сахара. Когда лекарственная форма представляет собой капсулу, она может содержать жидкий носитель,включая полиэтиленгликоль, растительное масло и т.п. Другие вещества, которые могут быть использованы с определенными лекарственными формами, включают желатин, воск, шеллак, сахар и т.п. Формы сиропов или эликсиров могут содержать сахарозу, фруктозу в качестве подсластителей, метил- и пропилпарабены в качестве консервантов, красители и пигменты, и ароматизаторы. При введении внутривенно или интраперитонеально путем инфузии или инъекции, растворы активного компонента и его солей могут быть получены, например, в воде или физиологическом растворе,возможно содержащем нетоксичное поверхностно-активное вещество. Дисперсии могут быть получены в глицерине, жидких полиэтиленгликолях, триацетине и их смесях, и в маслах. Условия хранения также могут потребовать включения консерванта. Фармацевтические лекарственные формы, подходящие для инъекции или инфузии, могут включать стерильные водные растворы или дисперсии, или стерильные порошки, содержащие активный компонент, подходящие для немедленного приготовления стерильных растворов или дисперсий для инъекций или инфузий, возможно заключенные в липосомы. Во всех случаях конечная лекарственная форма должна быть стерильной, текучей и стабильной в условиях получения и хранения. Жидкий носитель или среда может представлять собой растворитель или жидкую дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль, жидкие полиэтиленгликоли и т.п.),растительные масла, нетоксичные глицерил эфиры и подходящие смеси указанных веществ. Необходимая текучесть может быть сохранена, например, путем образования липосом, посредством поддержания требуемого размера частиц в случае дисперсий или путем применения поверхностно-активных веществ. Предотвращение действия микроорганизмов может быть обеспечено различными антибактериальными и противогрибковыми агентами, например, парабенами, хлорбутанолом, фенолом, сорбиновой кислотой,тимеросалом и т.п. Во многих случаях будет предпочтительным включение изотонических агентов, например, сахаров, буферов или хлорида натрия. Пролонгированная абсорбция композиций для инъекций может быть обеспечена применением в указанных композициях агентов, замедляющих абсорбцию, например, моностеарата алюминия и желатина. Стерильные растворы для инъекций получают путем включения активного соединения в необходимом количестве в соответствующий растворитель с различными другими компонентами, перечисленными выше, при необходимости, с последующей стерилизацией путм фильтрации. В случае стерильных порошков для приготовления стерильных растворов для инъекций, предпочтительные способы получения представляют собой технологии вакуумной сушки и сушки методом сублимации, в результате которых получают порошок активного компонента, а также любого необходимого дополнительного компонента, присутствующего в предварительно стерилизованных путем фильтрации растворах. Соответственно, настоящее изобретение включает стерильный препарат соединения согласно настоящему изобретению. Настоящее изобретение также включает нестерильные препараты соединения согласно настоящему изобретению. Фармацевтические лекарственные формы для инъекции или инфузии могут включать стерильные водные растворы или дисперсии, или стерильные порошки, содержащие активные соединения согласно настоящему изобретению, полученные для немедленного приготовления. Жидкие носители могут включать растворители или жидкие дисперсионные среды, содержащие воду, этанол, полиол (например, глицерин, пропиленгликоль, полиэтиленгликоли) и т.п. Могут быть добавлены различные агенты для ингибирования или предотвращения противомикробной активности, такие как парабены, хлорбутанол, фенол,сорбиновая кислота, тимеросал и т.п. Соединения и композиции согласно настоящему изобретению можно вводить в виде однократной дозы или с интервалами в виде нескольких доз. Величина дозы, лекарственная форма, способ введения и компоненты конкретного состава могут варьироваться в соответствии с необходимой концентрации в плазме и задействованной фармакокинетикой. Важный аспект настоящего изобретения заключается в том, что конкретные соединения согласно настоящему изобретению могут обеспечивать улучшенный и эффективный способ введения лекарственной формы для перорального применения в силу характеристик и свойств, связанных со структурой соединения согласно настоящему изобретению и расположением заместителей. Для топического введения, в целом, будет необходимо вводить соединения согласно настоящему изобретению в кожу в виде композиций или составов в комбинации с дерматологически приемлемым носителем, который может представлять собой твердое вещество или жидкость. Подходящие твердые носители включают мелкодисперсные твердые вещества, такие как тальк,глина, микрокристаллическая целлюлоза, диоксид кремния, оксид алюминия и т.п. Подходящие жидкие носители включают воду, спирты или гликоли, или смеси вода-спирт/гликоль, в которых настоящие соединения могут быть растворены или диспергированы на эффективных уровнях, возможно с помощью нетоксичных поверхностно-активных веществ. Адъюванты, такие как отдушки и дополнительные противомикробные агенты, могут быть добавлены для оптимизации свойств для конкретного применения. Полученные жидкие композиции могут быть нанесены с впитывающих прокладок, используемых для пропитки бинтов и других повязок, или могут быть распылены на пораженную область с использованием распылителей насосного типа или аэрозольных распылителей. Загустители, такие как синтетические полимеры, жирные кислоты, соли и эфиры жирных кислот,жирные спирты, модифицированные целлюлозы или модифицированные минеральные вещества, также можно применять с жидкими носителями с образованием паст для нанесения гелей, мазей, мыл и т.п.,для нанесения напрямую на кожу потребителя. Примеры подходящих дерматологических композиций, которые можно применять для доставки соединений формулы I в кожу, известны в данной области техники; например, см., Jacquet et al. (патент США 4608392), Geria (патент США 4992478), Smith et al. (патент США 4559157) и Wortzman (патент США 4820508). Подходящие дозы соединений формулы I могут быть определены путем сравнения их активности invitro и активности in vivo в моделях у животных. Способы экстраполяции эффективных доз у мышей и других животных на людей известны в данной области техники; например, см. патент США 4938949. Количество соединения или его активной соли, или производного, необходимое для применения в лечении, будет варьироваться не только в зависимости от конкретной выбранной соли, но и от способа введения, природы вылечиваемого состояния и возраста, и состояния пациента, и в конечном итоге будет представлено на усмотрение лечащего врача или клинициста. Однако, в целом, подходящая доза будет находиться в диапазоне от примерно 3 до примерно 100 мкг/кг массы тела в сутки (например, от примерно 6 до примерно 96 мкг/кг массы тела в сутки или от примерно 6 до примерно 48 мкг/кг массы тела в сутки, или от примерно 6 до примерно 24 мкг/кг массы тела в сутки, или от примерно 12 до примерно 24 мкг/кг массы тела в сутки). Соединение удобным образом получают в дозированной лекарственной форме; например, содержащей от примерно 80 мкг до примерно 8000 мкг, удобным образом от примерно 480 мкг до примерно 7680 мкг, удобным образом от примерно 480 мкг до примерно 3840 мкг и удобным образом от примерно 960 мкг до примерно 1920 мкг. В одном из вариантов реализации согласно настоящему изобретению предложена композиция, содержащая соединение согласно настоящему изобретению, полученное в указанной дозированной лекарственной форме. Необходимая доза удобным образом может быть представлена в виде однократной дозы или в виде разделенных доз, вводимых через подходящие интервалы, например, в виде двух, трех, четырех или более субдоз в сутки. Сама по себе субдоза может быть дополнительно разделена, например, на некоторое количество отдельных свободно распределенных введений, например, несколько ингаляций из инсуффлятора или путем введения множества капель в глаз. Соединения согласно настоящему изобретению также могут быть введены в комбинации с другими терапевтическими агентами, например, другими агентами, подходящими для лечения рака (например,рака поджелудочной железы, рака яичников, рака ободочной и прямой кишки, неходжкинской лимфомы,лейкоза, острого и хронического миелобластного лейкоза, нейробластомы, рака щитовидной железы,остеосаркомы, рака молочной железы, рака предстательной железы, рака пищевода, рака мочевого пузыря, карциномы желудка, рака уротелия, мультиформной глиобластомы, рака толстой кишки, рака шейки матки, фибросаркомы, плоскоклеточного рака, множественной миеломы, холангиокарциномы, немелкоклеточного рака легкого) в качестве радиосенсибилизатора для раковых клеток, воспалительных заболеваний, ревматических заболеваний, аутоиммунных заболеваний, поликистоза почек, нефрита, выживаемости трансплантата при трансплантации (почка, сердце), легочной гипотензии, воспаления легких, фиброза легких, нейрозащиты, ишемически-реперфузионного повреждения головного мозга, паркинсонизма и язв роговицы. Примеры указанных агентов включают 5-фторурацил, TRAIL (TNF-связанный апоптозиндуцирующий лиганд), активирующие антитела DR-4/5, циклофосфамид (cyclophosphamide), гидроксидаунорубицин (hydroxydaunorubicin) (доксорубицин (doxorubicin, онковин (oncovin) (винкристин (vincristine, паклитаксел (paclitaxel), доцетаксел (doxetaxel), цисплатин (cisplatin), карбоплатин (carboplatin),CPT-11, бортезомиб (bortezimib) и преднизон-преднизолон (prednisone-prednisolone). Соответственно, в одном из вариантов реализации согласно настоящему изобретению также предложена композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль, по меньшей мере один другой терапевтический агент и фармацевтически приемлемый разбавитель или носитель. Согласно настоящему изобретению также предложен набор, содержащий соединение формулы I или его фармацевтически приемлемую соль, по меньшей мере один другой терапевтический агент, упаковочный материал и инструкции по введению соединения формулы I или его фармацевтически приемлемой соли и другого терапевтического агента или агентов животному (например, млекопитающему) для лечения рака (например, рака поджелудочной железы, рака яичников, рака ободочной и прямой кишки, неходжкинской лимфомы, лейкоза, острого и хронического миелобластного лейкоза, нейробластомы, рака щитовидной железы, остеосаркомы, рака молочной железы, рака предстательной железы, рака пищевода, рака мочевого пузыря, карциномы желудка, рака уротелия, мультиформной глиобластомы, рака толстой кишки, рака шейки матки, фибросаркомы, плоскоклеточного рака, множественной миеломы, холангиокарциномы,немелкоклеточного рака легкого), воспалительного заболевания, ревматического заболевания, аутоиммунного заболевания, поликистоза почек, нефрита, выживаемости трансплантата при трансплантации(почка, сердце), легочной гипотензии, воспаления легких, фиброза легких, нейрозащиты, ишемическиреперфузионного повреждения головного мозга, паркинсонизма, язв роговицы или колита. В другом варианте реализации согласно настоящему изобретению также предложен набор, содержащий соединение формулы I или его фармацевтически приемлемую соль, по меньшей мере один другой терапевтический агент, упаковочный материал и инструкции по введению соединения формулы I или его фармацевтически приемлемой соли и другого терапевтического агента или агентов животному (например, млекопитающему) для сенсибилизации раковых клеток, покрытия стентов (выделение лекарственного средства),восстановления спинного мозга или для применения в качестве мужской и женской контрацепции у животных. Следующие документы относятся к триптолиду в комбинации с другими терапевтическими агентами: Важный аспект настоящего изобретения заключается в том, что соединения согласно настоящему изобретению обеспечивают необходимые комбинации фармакокинетических свойств, физических свойств и преимуществ лечения по сравнению с другими пролекарственными формами триптолида. Пролекарственные соединения триптолида согласно настоящему изобретению демонстрируют необходимую комбинацию характеристик, включая химическую стабильность, повышенную растворимость и быстрое метаболическое высвобождение активного триптолида из пролекарственной формы. В совокупности данные свойства обеспечивают улучшенные терапевтические противораковые эффекты. Указанные эффекты включают эффективное ингибирование клеток рака поджелудочной железы путем ингибирования защитных эффектов, обеспечиваемых HSP70 в клетках, и резистентности к апоптозу и видам лечения. Химический путь метаболического и ферментативного расщепления пролекарства триптолида из примера 1 показан на фиг. 2. Исходное природное соединение (не пролекарственная форма) триптолида обладает плохой растворимостью в воде. Полученное соединение из примера 1 демонстрирует высокий уровень растворимости. В ходе ферментативного расщепления и метаболизма соединение из примера 1 в конечном счете высвобождает активную форму соединения триптолида. Соединения и композиции согласно настоящему изобретению можно применять в качестве способа лечения солидных раковых опухолей у млекопитающего, нуждающегося в таком лечении, включающего введение фармацевтически эффективного количества соединения, описанного выше, в качестве активного компонента. При употреблении в контексте способов лечения термин "млекопитающее" включает людей. Соединения и композиции согласно настоящему изобретению могут быть эффективны для ингибирования in vitro и in vivo роста раковых клеток в экспрессирующих HSP70 раковых опухолях. Примеры экспрессирующих HSP70 раковых опухолей включают рак поджелудочной железы, рак молочной железы, рак легкого, рак головного мозга, лейкоз, нейробластому, рак толстой кишки, рак желудка, рак печени и глиобластому. Соответственно, в одном из вариантов реализации настоящее изобретение включает ингибирование популяции раковых клеток, демонстрирующих чрезмерную экспрессию белка теплового шока HSP70,путем введения соединения формулы I. Особенно важное значение для настоящего изобретения имеет эффективное ингибирующее действие на клетки рака поджелудочной железы, экспрессирующие HSP70,такие как клетки Mia-Paca, Panc-1 и S2VP10. Соответственно, в другом варианте реализации согласно настоящему изобретению предложен способ лечения рака S2 (например, рака S2VP10 или S2013) у млекопитающего (например, человека), включающий введение соединения формулы I или его фармацевтически приемлемой соли млекопитающему (например, человеку). Как in vitro, так и живом организме у млекопитающих фермент щелочная фосфатаза превращает соединение из примера 1 в активную форму триптолида, как показано в примерах в настоящем описании. Период полупротекания ферментативного гидролиза (t1/2) для соединения из примера 1 показывает относительно высокую скорость превращения и, следовательно, более быстрое высвобождение активной терапевтической формы соединения. Триптолид применяют для лечения множества заболеваний, таких как воспалительные заболевания. Триптолид также применялся в качестве терапевтического агента для лечения различных заболеваний. Данные заболевания включают рак (например, рак поджелудочной железы, карцинома желчного протока, нейробластома, рак толстой кишки, рак молочной железы, миелома, рак желудка, рак печени, глиоб- 10021135 ластома, рак яичников, рак ободочной и прямой кишки, неходжкинская лимфома, рак легкого, рак предстательной железы, мелкоклеточный рак легкого, крупноклеточный рак легкого, рак почки, рак пищевода, рак желудка, рак шейки матки, лимфомные опухоли), аутоиммунные заболевания, отторжение трансплантата, поликистоз почек, воспалительные заболевания, астму, ревматоидный артрит, системную красную волчанку и нефрит. Триптолид также исследовали в покрытиях стентов (выделение лекарственного средства), восстановлении спинного мозга, колите и контрацепции у самцов и самок. Соответственно, настоящее изобретение включает, но не ограничивается им, применение соединений формулы I для лечения заболеваний, включая рак (например, рак поджелудочной железы, карцинома желчного протока,нейробластома, рак толстой кишки, рак молочной железы, миелома, рак желудка, рак печени, глиобластома, рак яичников, рак ободочной и прямой кишки, неходжкинская лимфома, рак легкого, рак предстательной железы, мелкоклеточный рак легкого, крупноклеточный рак легкого, рак почки, рак пищевода,рак желудка, рак шейки матки, лимфомные опухоли), аутоиммунные заболевания, отторжение трансплантата, поликистоз почек, воспалительные заболевания, астму, ревматоидный артрит, системную красную волчанку и нефрит. Соединения формулы I также можно применять для покрытия стентов (выделение лекарственного средства), восстановления спинного мозга, при колите и для контрацепции у самцов и самок млекопитающих. Следующие документы относятся к триптолиду и раку: Следующие документы относятся к триптолиду и заболеваниям, отличным от рака: Далее настоящее изобретение будет проиллюстрировано следующими неограничивающими примерами: Пример 1. Синтез динатриевой соли 14-О-фосфонооксиметилтриптолида (соединение 1). К раствору дибензилового эфира 14-О-фосфонооксиметилтриптолида (50 мг, 0,08 ммоль) в тетрагидрофуране (5 мл) добавляли палладий на углероде (10%, 10 мг). Смесь перемешивали при комнатной температуре в атмосфере водорода (1 атм) в течение 3 ч. Катализатор удаляли путем фильтрования через ЦЕЛИТ и фильтрат обрабатывали раствором гидрата карбоната натрия (8,9 мг в 3 мл воды, 0,076 ммоль). Тетрагидрофуран выпаривали при пониженном давлении и остаточный водный раствор экстрагировали эфиром (33 мл). Водный слой выпаривали досуха и полученное твердое вещество сушили в течение ночи в вакууме, промывали эфиром и снова сушили в вакууме с получением динатриевой соли 14-О-фосфонооксиметилтриптолида (35 мг, выход 90%) в виде белого порошка. 1 Н ЯМР (400 МГц, D2O)0,81 (d, 3H, J = 6,8 Гц), 1,00 (d, 3H, J=6,8 Гц), 1,03 (s, 3H), 1,35 (m, 1 Н), 1,50 (m, 1 Н), 2,00 (dd, 1H, J1 = 14,7 и J2 = 13,4 Гц), 2,08-2,61 (m, 4 Н), 2,85 (m, 1 Н), 3,63 (d, 1 Н, J = 5,5 Гц), 3,81 (d, 1 Н, J = 3,1 Гц), 3,86 (s,1 Н), 4,12 (d, 1 Н, J = 3,1 Гц), 4,92 (m, 2 Н), 5,07 (m, 2 Н) ррm; 13 С ЯМР (100 МГц, D2O)12,9, 16,0, 16,3,16,5, 22,3, 25,5, 28,9, 35,2, 39,8, 55,4, 56,1, 61,0, 61,5, 65,1, 65,5, 71,9, 77,6, 91,7, 123,8, 164,2, 177,3 ррm; масс-спектрометрия высокого разрешения (HRMS), рассчитанная для (С 21 Н 26 О 10 Р) необходимо m/z[M+1]+ 469,1264, обнаружено m/z 469,1267. Получение дибензилового эфира 14-О-фосфонооксиметилтриптолида. Этап 1. Получали раствор триптолида (100 мг, 0,29 ммоль) в уксусной кислоте (5 мл, 87,5 ммоль) и ангидриде уксусной кислоты (1 мл, 10,5 ммоль) в ДМСО (1,5 мл, 21,4 ммоль) и перемешивали при комнатной температуре в течение 5 дней с получением промежуточного соединения 14-О-метилтиометилтриптолида. Затем реакционную смесь вливали в воду (100 мл) и нейтрализовали твердым NaHCO3, добавляемым порциями. Смесь экстрагировали этилацетатом (50 мл 3) и смешанный органический экстракт сушили над безводным сульфатом натрия и концентрировали с получением продукта в виде масла. В результате колоночной флэш-хроматографии на силикагеле (3:2 гексан/этилацетат) получали 14-О-метилтиометилтриптолид с выходом 52% (60 мг) в виде белой пены. 1 Н ЯМР (400 МГц,CDCl3)0,82 (d, 3H, J = 6,8 Гц), 1,00 (d, 3H, J = 6,8 Гц), 1,09 (s, 3H), 1,20 (m, 1 Н), 1,59 (m, 1 Н), 1,93 (dd,1H, J1 = 14,7 и J2 = 13,4 Гц), 2,19 (s, 3H), 2,10-2,42 (m, 4 Н), 2,68 (m, 1 Н), 3,24 (d, 1 Н, J = 5,5 Гц), 3,51 (d,1 Н, J = 3,1 Гц), 3,67 (s, 1 Н), 3,79 (d, 1 Н, J = 3,1 Гц), 4,68 (m, 2 Н), 4,93 (d, 1 Н, J = 11,8 Гц), 5,07 (d, 1 Н, J =- 12021135 11,8 Гц) ррm; 13 С ЯМР (100 МГц, CDCl3)13,6, 14,8, 16,8, 17,0, 17,1, 23,4, 26,3, 29,5, 35,8, 40,4, 54.5, 55,0,58,0, 61,5, 63,9, 64,4, 69,9, 75,8, 76,7, 125,5, 160,2, 173,2 ppm; HRMS рассчитанная для (C22H28O6SNa) необходимо m/z [M+Na]+ 443,1505, обнаружено m/z 443,1507. Этап 2. Раствор 14-О-метилтиометилтриптолида (50 мг, 0,12 ммоль) в сухом метиленхлориде (2 мл) в атмосфере N2 смешивали с порошкообразными активированными молекулярными ситами 4 (50 мг) с последующим добавлением смеси дибензилфосфата (40 мг, 0,14 ммоль) и N-йодсукцинимида (32 мг, 0,14 ммоль) в тетрагидрофуране (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 5 ч, фильтровали и разбавляли метиленхлоридом (20 мл). Полученный раствор промывали раствором тиосульфата натрия (2 мл, 1 М раствор), насыщенным раствором бикарбоната натрия, солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Маслянистый остаток очищали путем флэш-хроматографии на силикагеле (1:2 гексан/этилацетат) с получением дибензилового эфира 14-О-фосфонооксиметилтриптолида (62 мг, выход 80%) в виде белой пены. 1 Н ЯМР (400 МГц, CDCl3)0,72 (d, 3H, J = 6,8 Гц), 0,89 (d, 3H, J = 6,8 Гц), 1,05 (s, 3H), 1,27 (m, 1 Н), 1,48 (m, 1H), 1,82 Пример 2. Синтез динатриевой соли 14-О-фосфонооксиметилтриптолида (соединение 1). К раствору, содержащему 14-О-метилтиометилтриптолид (50 мг, 0,12 ммоль), фосфорную кислоту(82 мг, 0,84 ммоль) и молекулярные сита (4, 0,45 г) в ТГФ (10 мл), при 0 С добавляли N-йодсукцинимид (41 мг, 0,18 ммоль) и смесь перемешивали при комнатной температуре в течение одного часа. Реакционную смесь фильтровали через Целит и твердые вещества промывали ТГФ. Фильтрат обрабатывали 1 М Na2S2O3 пока он не стал бесцветным, и фильтрат обрабатывали раствором карбоната натрия (13 мг в 3 мл воды, 0,12 ммоль). Фильтрат выпаривали при пониженном давлении и остаточный водный раствор экстрагировали эфиром (33 мл). Водную фазу выпаривали досуха и полученный остаток очищали путем хроматографии (С 18), элюируя градиентом метанола 0-100% в воде, с получением динатриевой соли 14-О-фосфонооксиметилтриптолида (43 мг, выход 70%) в виде бесцветного порошка. Получение 14-О-метилтиометилтриптолида. К раствору триптолида (100 мг, 0,28 ммоль) и метилсульфида (0,16 мл, 2,24 ммоль) в ацетонитриле(10 мл) при 0 С добавляли пероксид бензоила (0,27 г, 1,12 ммоль) четырьмя равными порциями в течение 20 мин, а затем смесь перемешивали при 0 С в течение одного часа, а затем при комнатной температуре в течение одного часа. Смесь разбавляли этилацетатом и промывали 10% Na2CO3, а затем солевым раствором. Органическую фазу сушили над MgSO4, фильтровали и выпаривали. Остаток очищали путем флэш-хроматографии на силикагеле (1:1 гексан/этилацетат) с получением 14-О-метилтиометилтриптолида (63 мг, выход 54%) в виде бесцветного порошка. Пример 3. Синтез динатриевой соли 14-О-фосфонооксиэтилтриптолида.(82 мг, 0,84 ммоль) и молекулярные сита (4, 0,45 г) в ТГФ (10 мл), при 0 С добавляли Nйодсукцинимид (41 мг, 0,18 ммоль) и смесь перемешивали при комнатной температуре в течение одного часа. Реакционную смесь фильтровали через Целит и твердые вещества промывали ТГФ. Фильтрат обрабатывали 1 М Na2S2O3 пока он не стал бесцветным, и фильтрат обрабатывали раствором карбоната натрия(13 мг в 3 мл воды, 0,12 ммоль). Фильтрат выпаривали при пониженном давлении и остаточный водный раствор экстрагировали эфиром (33 мл). Водную фазу выпаривали досуха и полученный остаток очищали путем хроматографии (С 18), элюируя градиентом метанола 0-100% в воде, с получением динатриевой соли 14-О-фосфонооксиэтилтриптолида (46 мг, выход 72%) в виде бесцветного порошка. 1 Н ЯМР(10 мл) при 0 С добавляли пероксид бензоила (0,27 г, 1,12 ммоль) четырьмя равными порциями в течение 20 мин, а затем смесь перемешивали при 0 С в течение одного часа, а затем при комнатной температуре в течение одного часа. Смесь разбавляли этилацетатом и промывали 10% Na2CO3, а затем солевым раствором. Органическую фазу сушили над MgSO4, фильтровали и выпаривали. Остаток очищали путем флэш-хроматографии на силикагеле (1:1 гексан/этилацетат) с получением 14-O-метилтиоэтилтриптолида Пример 4. Синтез динатриевой соли 14-О-фосфонооксипропилтриптолида. К раствору, содержащему 14-О-метилтиопропилтриптолид (54 мг, 0,12 ммоль), фосфорную кислоту(82 мг, 0,84 ммоль) и молекулярные сита (4 А, 0,45 г) в ТГФ (10 мл), при 0 С добавляли Nйодсукцинимид (41 мг, 0,18 ммоль) и смесь перемешивали при комнатной температуре в течение одного часа. Реакционную смесь фильтровали через Целит и твердые вещества промывали ТГФ. Фильтрат обрабатывали 1 М Na2S2O3 пока он не стал бесцветным, и фильтрат обрабатывали раствором карбоната натрия(13 мг в 3 мл воды, 0,12 ммоль). Фильтрат выпаривали при пониженном давлении и остаточный водный раствор экстрагировали эфиром (33 мл). Водную фазу выпаривали досуха и полученный остаток очищали путем хроматографии (С 18), элюируя градиентом метанола 0-100% в воде, с получением динатриевой соли 14-О-фосфонооксипропилтриптолида (43 мг, выход 65%) в виде бесцветного порошка. 1 Н ЯМРppm; 13 С ЯМР (100 МГц, D2O)7,55, 13,5, 16,2, 16,9, 17,2, 20,8, 23,2, 26,1, 28,4, 34,7, 38,5, 54,1, 55,0, 59,0,61,3, 62,5, 63,9, 68,5, 75,4, 76,4, 91,9, 125,7, 160,1, 174,5 ppm; HRMS, рассчитанная для (С 23 Н 29 О 10 Р) необходимо m/z [M+1]+ 497,1294, обнаружено m/z 497,1292. Получение 14-О-метилтиопропилтриптолида. К раствору триптолида (100 мг, 0,28 ммоль) и пропилсульфида (0,32 мл, 2,24 ммоль) в ацетонитриле (10 мл) при 0 С добавляли пероксид бензоила (0,27 г, 1,12 ммоль) четырьмя равными порциями в течение 20 мин и смесь перемешивали при 0 С в течение одного часа, а затем при комнатной температуре в течение одного часа. Смесь разбавляли этилацетатом и промывали 10% Na2CO3, а затем солевым рас- 14021135 твором. Органическую фазу сушили над MgSO4, фильтровали и выпаривали. Остаток очищали путем флэш-хроматографии на силикагеле (1:1 гексан/этилацетат) с получением 14-О-метилтиопропилтриптолида (60 мг, выход 48%) в виде бесцветного порошка. 1 Н ЯМР (400 МГц, CDCl3)0,65 (d, 3H, J = 6,8 Гц),0,67 (d, 3H, J = 6,8 Гц), 0,99 (t, 3H, J = 5,3 Гц), 1,01 (s, 3H), 1,20 (m, 1H), 1,59 (m, 1H), 1,88 (dd, 1H, J1 = 14,7 и J2= 13,4 Гц), 2,18 (s, 3H), 2,01-2,26 (m, 4H), 2,62 (m, 3H), 3,24 (d, 1 Н, J = 5,5 Гц), 3,42 (d, 1 Н, J = 3,1 Гц), 3,70 (d, 1 Н, J = 3,1 Гц), 3,73 (s, 1H), 4,61 (m, 2H), 5,03 (q, 1H, J = 5,3 Гц) ppm; 13 С ЯМР (100 МГц,CDCl3)7,68, 13,5, 14,6, 16,2, 17,0, 17,2, 21,4, 23,2, 26,1, 28,9, 34,7, 39,5, 54,1, 55,6, 59,0, 61,3, 63,5, 64,0,69,5, 75,1, 76,4, 125,1, 160,9, 173,5 ppm; HRMS, рассчитанная для (C24H32O6SNa) необходимо m/z [M+Na]+ 471,1920, обнаружено m/z 471,1918. Пример 5. Химические свойства соединений формулы I. Оценивали химические свойства соединений согласно настоящему изобретению. Измеряли растворимость в воде и химическую стабильность. При применении раствора соединения из примера 1 с рН,доведенным до 7,4, определили, что растворимость при комнатной температуре составляла 61,4 мг/мл. Стабильность соединений согласно настоящему изобретению оценивали в буферном растворе Tris (рН 7,4) и боратном буферном растворе (рН 7,4) при комнатной температуре. По окончании периода, составляющего 1 (один) месяц, не наблюдали распада соединения из примера 1. Результаты приведены в следующей таблице. Таблица 1. Физико-химические свойства соединений формулы I Через один месяц не наблюдали распада. Пример 6. Ферментативное превращение соединения 1 in vitro. Соединение 1 превращается в активную форму триптолида под действием фермента щелочной фосфатазы. Проводили эксперимент in vitro для исследования биологического превращения пролекарственного соединения триптолида согласно настоящему изобретению. Биологическое превращение in vitro имитировали с использованием щелочной фосфатазы (из бычьей слизистой оболочки кишечника, ТипVII-S от Sigma-Aldrich (Сент-Луис, штат Миссури) в глициновом буфере (рН 9,8. Щелочные фосфатазы представляют собой группу ферментов, встречающихся главным образом в печени (изофермент ALP-1) и кости (изофермент ALP-2), и небольшие количества вырабатываются клеточной выстилкой тонкого кишечника (изофермент ALP-3), плаценты и почек. Щелочные фосфатазы отщепляют фосфор с созданием щелочного рН. Другие ферменты помимо щелочной фосфатазы также могут способствовать гидролизу in vivo. На фиг. 3 видно, что уменьшающиеся количества пролекарственной формы триптолида "совпадали" с пропорционально увеличивающимися количествами высвобождаемой активной формы триптолида. Кроме того, наблюдали, что превращение происходило в течение относительно короткого периода времени, при этом большая часть превращения происходила в течение начального периода времени, составляющего 10 мин. Константу распада первого порядка рассчитывали путем подбора оставшейся концентрации относительно периода инкубации. Было определено, что период полураспада (t 1/2) соединения 1 составлял 2 мин. Пример 7. Сравнительное исследование жизнеспособности клеток in vitro. Проводили эксперимент для сравнительной оценки жизнеспособности клеток in vitro с использованием триптолида, пролекарства триптолида (соединение 1) и контроля (без триптолида или пролекарственной формы). Жизнеспособность клеток определяли с использованием набора для подсчета клетокDojindo Cell Counting Kit-8 (от Dojindo Laboratories, Роквилл, штат Мэриленд). Клетки поджелудочной железы высевали в 96-луночный планшет по 2103 клеток на лунку и оставляли на ночь для протекания адгезии. Затем указанные клетки обрабатывали пролекарством триптолида (соединение 1) согласно настоящему изобретению и природным триптолидом в различных концентрациях в течение периодов, составляющих 24 ч и 48 ч. В каждую лунку планшета добавляли тетразолиевый субстрат (10 мкл). Планшеты инкубировали при 37 С в течение 1 ч, после чего измеряли поглощение при 460 нм. Каждый экс- 15021135 перимент проводили в трех повторностях и повторяли независимо три раза. Влияние соединения 1 и триптолида на жизнеспособность клеток рака поджелудочной железы наблюдали после инкубации в среде, содержащей пролекарство триптолида в концентрациях в диапазоне от 50 до 200 нмоль/л через 24 ч и 48 ч. Данные собирали и преобразовывали в графики на фиг. 4 (жизнеспособность клеток Mia-Раса через 48 ч), фиг. 5 (жизнеспособность клеток Рапс-1 через 24 и 48 ч) и фиг. 6 (жизнеспособность клеток S2VP10 через 24 и 48 ч). Как видно из приведенных выше данных и на каждой из фигур, присутствие пролекарства триптолида и щелочной фосфатазы, и природной формы триптолида значительно уменьшало жизнеспособность клеток поджелудочной железы in vitro время- и дозозависимым образом. Пример 8. Сравнительное исследование in vivo соединения 1 на мышах. Тридцать бестимусных мышей линии nude nu/nu были получены из Национального института рака(National Cancer Institute, NCI) (Роквилл, штат Мэриленд) и содержались в центре RAR (Животные ресурсы для исследований, Research Animal Resources). Мышей анестезировали в соответствии с рекомендациями центра RAR с использованием кетамина (ketamine) 75-200 мг/кг и ксилазина (xylazine) 4-8 мг/кг. Делали небольшой разрез 3 мм на левой стороне брюшной стенки и щипцами извлекали селезенку в достаточной степени для того, чтобы обнажить и обеспечить доступ к поджелудочной железе. Получали среду клеток MiaPaca-2 и выдерживали на льду до доставки мышам. Каждой из мышей вводили путем инъекции 1 миллион клеток MiaPaca-2, суспендированных в МАТРИГЕЛЕ (от Becton-Dickinson Corporation, Franklin Lakes, New Jersey), в хвост поджелудочной железы (определяется анатомическим соединением с селезенкой) с использованием шприца Гамильтона. После доставки клеток шприц держали в неизменном положении в течение дополнительных 5-10 с, чтобы позволить МАТРИГЕЛЮ осесть. Селезенку помещали обратно в брюшную полость и брюшную стенку закрывали непрерывным викриловым швом. Кожу соединяли и закрывали с использованием скобок для стягивания краев ран. Затем мышей переносили на грелку-подушку до полного восстановления перед возвращением в клетку. Послеоперационное обезболивающее средство (бупренорфин (buprenonorphine) 0,1 мг/кг) вводили интраперитонеально сразу же после полного восстановления от анестезии для предупреждения угнетения дыхания,а затем вводили каждые 12 ч в течение 2 дней. Скобки для стягивания краев ран снимали у мышей через 7 дней после операции. Мышей рандомизировали на 3 группы, каждая группа включает 10 мышей. Группы были следующие: контрольная группа, группы триптолида и группа пролекарства триптолида (соединение 1). Контрольная группа состояла из мышей, которым вводили интраперитонеально носитель - ДМСО - путем инъекции. Субъектам группы триптолида вводили путем инъекции 0,2 мг/кг триптолида, растворенного в ДМСО и разбавленного фосфатным буферным раствором до объема, равного 100 мкл, при этом интраперитонеальные инъекции осуществляли ежедневно в течение 60 дней. Субъектам группы соединения 1 интраперитонеально вводили путем инъекции ежедневно в течение 60 дней 0,28 мг/кг указанного соединения, растворенного в фосфатном буферном растворе, разбавленном до объема, равного 100 мкл. Мышей подвергали эвтаназии под наркозом по завершении лечения в течение 60 дней. Забирали образцы (кровь, легкое, селезенка, печень, почки и опухолевая ткань) и измеряли объем и массу опухоли,и сравнивали между разными группами. Проводили наблюдения локорегионарного роста и роста раковой опухоли. Фиг. 8 (фотографии контрольной группы), фиг. 9 (фотографии группы триптолида) и фиг. 10 (фотографии группы пролекарства триптолида из Примера 1) представляют собой серию фотографий, демонстрирующих конечный рост опухолей в каждой из групп мышей эксперимента in vivo. Фиг. 11 представляет собой фотографию серии удаленных опухолей, извлеченных у мышей каждой из групп и расположенных в ряд, соответствующий каждой группе. Опухоли, удаленные у мышей контрольной группы, не получавшей лечение, были значительно больше через 60 дней, чем опухоли, удаленные у мышей двух других групп, что демонстрирует продолжающийся агрессивный рост клеток опухоли поджелудочной железы. В отличие от этого, группа соединения из Примера 1 демонстрировала значительное ингибирование роста опухоли поджелудочной железы по сравнению с контрольной группой, не получавшей лечение, и существенное эффективное ингибирование опухолевых клеток по сравнению с природной формой триптолида. Теперь обратимся к фиг. 11 и 12, демонстрирующим сравнительную массу опухоли и объем опухоли среди трех групп соответственно. Опухоли у контрольной группы были значительно больше с точки зрения как массы (г), так и объема(см 3) по сравнению с данными опухолей у группы триптолида и пролекарства триптолида (соединение 1). Таким образом, при введении живому млекопитающему in vivo пролекарственное соединение триптолида согласно настоящему изобретению может эффективно ингибировать рост опухоли, и эффективность ингибирующего действия сопоставима с эффективностью природной не пролекарственной формой триптолида. Как видно из приведенных выше данных и фигур, рост раковой опухоли поджелудочной железы у мышей, получавших лечение природным триптолидом и пролекарством триптолида (соединение 1) в течение 60 дней, демонстрировал значительно уменьшенный объем опухоли по сравнению с контрольной группой, не получавшей лечение. Кроме того, было значимым то, что у субъектов как группы триптолида, так и группы пролекарства триптолида, не наблюдали очевидного значительного влияния на массу тела и не наблюдали очевидных признаков токсичности у указанных субъектов. Таким образом, соединения согласно настоящему изобретению могут обеспечивать ингибирование опухоли и ингибировать рост раковых клеток и, в частности, рост клеток рака поджелудочной железы. Кроме того,соединения согласно настоящему изобретению также могут служить основой для эффективного лечения для ингибирования рака поджелудочной железы с низким уровнем токсических побочных эффектов у живых млекопитающих. Пример 9: Индуцированная соединением 1 регрессия опухоли в ортотопической модели рака поджелудочной железы у мышей (исследование введения доз в течение 60 дней). Клетки MIA РаСа-2 (1106) суспендировали в матригеле и вводили в хвост поджелудочной железы 4-6-недельных самок безтимусных мышей путем инъекции. Через десять дней после имплантации клеток мышам интраперитонеально вводили путем инъекции указанные концентрации соединения 1 (0,1, 0,15, 0,3, 0,6 или 0,9 мг/кг) или 0,2 мг/кг триптолида один раз в сутки в течение 60 дней. Контрольным мышам вводили физиологический раствор путем инъекции дважды в сутки. Лечение прекращали через 60 дней и мышей наблюдали в течение еще 28 дней перед умерщвлением. Забирали образцы опухоли, при наличии, у данных мышей и измеряли массу и объем опухоли. Если опухолевая масса превышала массу, указанную в рекомендациях по уходу за животными Университета Миннесоты (University of Minnesota animal care guidelines), мышей умерщвляли в более ранние моменты времени и их опухоли забирали. Фиг. 13 иллюстрирует повышенную выживаемость мышей, получавших лечение соединением 1 и триптолидом по сравнению с растворителем. Растворитель вводили дважды с сутки: все смерти происходили из-за опухолевой массы; 0,1 мг/кг соединения 1 один раз в сутки: наблюдали 1 смерть из-за опухолевой массы; 0,15 мг/кг соединения 1 дважды в сутки: отсутствие смертей; 0,2 мг/кг соединения 1 один раз в сутки: 1 смерть из-за повреждения, не связана с опухолевой массой или действием лекарственного средства. Фиг. 14 иллюстрирует повышенную выживаемость мышей, получавших лечение соединением 1 по сравнению с растворителем. Растворитель дважды с сутки: все смерти происходили из-за опухолевой массы; 0,3 мг/кг соединения 1 один раз в сутки: 1 смерть из-за повреждения, не связана с опухолевой массой; 0,6 мг/кг соединения 1 один раз в сутки: отсутствие смертей; 0,9 мг/кг соединения 1 один раз в сутки: 1 смерть при инъекции в 6 день исследования, причина не определена. Остальные смерти из-за действия лекарственного средства. На фиг. 15 показана уменьшенная опухолевая масса, измеряемая по объему опухоли или массе опухоли, у мышей, получавших лечение соединением 1, по сравнению с контрольными мышами. Пример 10. Индуцированная соединением 1 регрессия опухоли в ортотопической модели рака поджелудочной железы у мышей (исследование введения доз в течение 21 дня). 1106 клеток S2013, крайне метастатической линии клеток рака поджелудочной железы, суспендировали в матригеле и вводили в хвост поджелудочной железы 4-6-недельных самок безтимусных мышей путем инъекции. Через семь дней после имплантации клеток мышам интраперитонеально вводили путем инъекции 0,42 мг/кг соединения 1 в течение 21 дня. Лечение прекращали через 21 день и мышей умерщвляли. Забирали образцы опухоли, при наличии, у данных мышей и измеряли массу и объем опухоли. Если опухолевая масса превышала массу, указанную в рекомендациях по уходу за животными Университета Миннесоты (University of Minnesota animal care guidelines), мышей умерщвляли и их опухоли забирали в более ранний момент времени. Контрольным мышам вводили физиологический раствор путем инъекции один раз в сутки. На фиг. 16 показана уменьшенная опухолевая масса, измеряемая по объему опухоли или массе опухоли, у мышей, получавших лечение соединением 1, по сравнению с контрольными мышами. Пример 11. Индуцированная соединением 1 регрессия опухоли в модели подкожной холангиокарциномы у мышей. Клетки SkChA-1 (5105) суспендировали в матригеле и вводили подкожно в левый бок 4-6 недельных самок безтимусных мышей путем инъекции. Через семь дней после имплантации клеток мышам интраперитонеально вводили путем инъекции 0,3 мг/кг соединения 1 дважды в сутки в течение 25 дней. Лечение прекращали в данный момент и мышей умерщвляли. Забирали образцы опухоли, при наличии, у данных мышей и измеряли массу и объем опухоли. Если опухолевая масса превышала массу,указанную в рекомендациях по уходу за животными Университета Миннесоты (University of Minnesotaanimal care guidelines), мышей умерщвляли и их опухоли забирали в более ранний момент времени. Контрольным мышам вводили физиологический раствор путем инъекции дважды в сутки. На фиг. 17 показана уменьшенная опухолевая масса, измеряемая по объему опухоли или массе опухоли, у мышей, получавших лечение соединением 1 по сравнению с контрольными мышами. Пример 12. Индуцированная триптолидом регрессия опухоли в ортотопической модели нейробла- 17021135 стомы у мышей. Клетки нейробластомы N2 (1106) суспендировали в матригеле и вводили в левое забрюшинное пространство 4-6-недельных иммунокомпетентных мышей линии A/J путем инъекции. Через три дня после имплантации клеток мышам интраперитонеально вводили путем инъекции 0,4 мг/кг триптолида в течение 21 дня. Лечение прекращали в данный момент и мышей умерщвляли. Забирали образцы опухоли, при наличии, у данных мышей и измеряли массу и объем опухоли. Если опухолевая масса превышала массу, указанную в рекомендациях по уходу за животными Университета Миннесоты (University of Minnesota animal care guidelines), мышей умерщвляли и их опухоли забирали в более ранний момент времени. Контрольным мышам вводили ДМСО путем инъекции в течение 21 дня. На фиг. 18 показана уменьшенная опухолевая масса, измеряемая по объему опухоли и массе опухоли, у мышей, получавших лечение триптолидом по сравнению с контрольными мышами. Пример 13. Индуцированная триптолидом гибель клеток и активация каспазы-3 в клетках нейробластомы. Клетки нейробластомы N2A и SKNSH обрабатывали триптолидом, что приводило к дозо- и времязависимой гибели клеток в N2A, при этом более 50% клеток погибали под действием 62,5 нМ триптолида через 24 ч и около 85% клеток погибали под действием 250 нМ триптолида через 48 ч (фиг. 19). Для подтверждения гипотезы о том, что триптолид приводит к гибели клеток нейробластомы по пути апоптоза, измеряли активность каспазы-3 в качестве маркера апоптоза. В обеих линиях клеток наблюдали повышение активности каспазы при более высоких дозах триптолида и более продолжительной терапии. Лечение триптолидом было связано с дозо- и времязависимыми повышениями уровней активности каспазы-3 (Фиг. 20). Данные результаты показывают, что опосредуемая триптолидом гибель клеток происходит путем индукции апоптоза. Пример 14. Следующие данные иллюстрируют типичные фармацевтические лекарственные формы,содержащие соединение формулы I ('соединение X'), для терапевтического или профилактического применения у людей. Вышеуказанные составы могут быть получены с помощью традиционных процедур, хорошо известных в области фармацевтики. Содержание всех публикаций, патентов и патентных документов включено в настоящее описание посредством ссылки так, как если бы было индивидуально включено посредством ссылки. Настоящее изобретение описано относительно различных конкретных и предпочтительных вариантов реализации и технологий. Однако следует понимать, что возможны различные вариации и модификации в пределах сущности и объема настоящего изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы In представляет собой 1, 2 или 3 и каждый Х+ представляет собой фармацевтически приемлемый органический катион или неорганический катион. 2. Соединение по п.1, представляющее собой соединение формулы Ia где Х+ представляет собой фармацевтически приемлемый органический катион или неорганический катион. 3. Соединение по п.1, где R1 представляет собой Н. 4. Соединение по п.1 или любому из пп.2-3, где каждый Х+ представляет собой Н. 5. Соединение по п.1 или любому из пп.2-3, где каждый Х+ представляет собой литий, натрий, калий, магний, кальций, барий, цинк или алюминий. 6. Соединение по п.1 или любому из пп.2-3, где каждый Х+ имеет формулу HY+, где Y представляет собой аммиак, триэтиламин, трометамин, триэтаноламин, этилендиамин, глюкамин, N-метилглюкамин,глицин, лизин, орнитин, аргинин, этаноламин или холин. 7. Соединение по любому из пп.1-3, где каждый Х+ представляет собой Na+. 8. Соединение, представляющее собой динатриевую соль 14-O-фосфонооксиметилтриптолида, динатриевую соль 14-O-фосфонооксиэтилтриптолида или динатриевую соль 14-Oфосфонооксипропилтриптолида. 9. Соединение, представляющее собой динатриевую соль 14-O-фосфонооксиметилтриптолида. 10. Фармацевтическая композиция, содержащая соединение формулы I по любому из пп.1-9 в комбинации с фармацевтически приемлемым носителем. 11. Способ лечения рака у млекопитающего, включающий введение указанному млекопитающему соединения формулы I по любому из пп.1-9. 12. Способ ингибирования роста раковых клеток в экспрессирующей HSP70 злокачественной опухоли у млекопитающего, включающий введение указанному млекопитающему эффективного для ингибирования количества соединения формулы I по любому из пп.1-9.
МПК / Метки
МПК: A61K 31/665, C07F 9/6561, A61P 35/00
Метки: пролекарства, триптолида
Код ссылки
<a href="https://eas.patents.su/26-21135-prolekarstva-triptolida.html" rel="bookmark" title="База патентов Евразийского Союза">Пролекарства триптолида</a>
Водорастворимые пролекарства противоракового действия

Номер патента: 2400
Опубликовано: 25.04.2002
Авторы: Ю Донг-Фанг, Ли Чун, Янг Дэвид Дж., Уоллейс Сидни
МПК: A61K 35/00, A61M 36/00, A01N 43/02...
Метки: противоракового, действия, водорастворимые, пролекарства
Формула / Реферат:
1. Композиция, включающая противоопухолевое лекарственное средство, конъюгированное с водорастворимым полимером или сополимером, причем указанный водорастворимый полимер или сополимер выбирают из группы, включающей поли-(d-глутаминовую кислоту), поли-(l-глутаминовую кислоту), поли- (dl-глутаминовую кислоту), поли-(d-аспарагиновую кислоту), поли-(l-аспарагиновую кислоту) и поли(dl-аспарагиновую кислоту) и их сополимеры, где используемое...
Пролекарства ингибиторов киназы pi – 3

Номер патента: 12240
Опубликовано: 28.08.2009
Авторы: Сур Роберт Г., Су Дзиндон, Дерден Дональд Л., Паттерсон Мэри, Гарлич Джозеф Р.
МПК: C07D 311/22, A61K 31/5377, A61P 25/00...
Метки: пролекарства, ингибиторов, киназы
Формула / Реферат:
1. Соединение представляет собой соединение формулы или его соль. 2. Применение соединения по п.1 в качестве активного терапевтического средства. 3. Применение соединения по п.1 для производства лекарственного средства, предназначенного для: (a) лечения пациента, страдающего состоянием, ассоциируемым с активностью киназы PI-3; (b) лечения воспалительных заболеваний, причем лекарственное средство необязательно включает один или несколько...
Кристаллические формы пролекарства на основе амида гемцитабина, композиции на их основе и их применение

Номер патента: 14909
Опубликовано: 28.02.2011
Автор: Облезов Александр Евгеньевич
МПК: A61P 31/12, A61K 31/513, A61P 35/00...
Метки: композиции, применение, гемцитабина, основе, пролекарства, формы, амида, кристаллические
Формула / Реферат:
1. Соединение, представляющее собой кристаллический 1-(2,2-дифтор-2-дезокси-b-D-рибофуранозил)-4-(2-пропил-1-оксопентил)аминопиримидин-2-он моно-п-толуолсульфонат или кристаллический полугидрат 1-(2,2-дифтор-2-дезокси-b-D-рибофуранозил)-4-(2-пропил-1-оксопентил)аминопиримидин-2-он гемибензолсульфоната.2. Соединение по п.1, представляющее собой кристаллический 1-(2,2-дифтор-2-дезокси-b-D-рибофуранозил)-4-(2-пропил-1-оксопентил)аминопиримидин-2-он...
Пролекарства

Номер патента: 2647
Опубликовано: 29.08.2002
Авторы: Валльберг Ханс, Сальвадор Лурдес, Линдстрем Стефан, Велинг Хорст, Жоу Ксиао-Ксионг, Йоханссон Нильс Гуннар, Сунд Кристиан
МПК: C07K 5/00, C07D 473/32, C07H 19/16...
Метки: пролекарства
Формула / Реферат:
1. Производное гуанозина, выбранное из 2',3'-дидезокси-3'-фтор-5'-О-[2-(L-валилокси)пропионил]гуанозина или 2',3'-дидезокси-3'-фтор-5'-О-[2,3-бис(L-валилокси)пропаноил]гуанозина, или его фармацевтически приемлемая соль. 2. Соединение по п.1, где 2-(L-валилокси)пропионильный фрагмент молекулы определяет производное L-молочной кислоты. 3. Соединение по п.1, обозначенное как 2',3'-дидезокси-3'-фтор-5'-О-[(R)-2,3-бис(L-валилокси)пропаноил]гуанозин. ...
3,5-дизамещенные и 3,5,7-тризамещенные 3h-оксазоло- и 3h-тиазоло[4,5-d] пиримидин-2-оны и их пролекарства

Номер патента: 13594
Опубликовано: 30.06.2010
Авторы: Леннокс Джозеф Р., Веббер Стефен И., Руден Эрик Дж., Хейли Грегори Дж., Сян Алан Синь
МПК: C07D 495/04, A61K 31/435
Метки: 3,5,7-тризамещенные, пиримидин-2-оны, 3,5-дизамещенные, пролекарства, 3h-оксазоло, 3h-тиазоло[4,5-d
Формула / Реферат:
1. Соединение формулы Iгде X представляет собой атом кислорода или атом серы;Y представляет собой атом кислорода или атом серы;R1 представляет собой атом водорода, алкил, арил, циклоалкил или гетероциклил;R2 представляет собой NH2, -NHC(O)R4, -NHR5, -N=CHNR6R7;R3 представляет собой атом водорода или OR8;R4 представляет собой -C1-C7-алкил или -О(C1-C7-алкил);R5 представляет собой -C1-C7-алкил;R6 и R7независимо представляют собой -C1-C7-алкил или...
Предыдущий патент: Обнаружение газообразных соединений для анализа скважинных текучих сред с использованием микрофлюидных устройств и реагента с оптической регистрацией
Следующий патент: Усовершенствованный метод количественного определения днк в биологическом образце
Случайный патент: Способ и устройство для генерации команд управления мощностью